spesifik tedavi gerektiren akut böbrek hasarı olguları tedavi... · 2017-12-18 · bağlanması...
TRANSCRIPT

Spesifik Tedavi Gerektiren Akut Böbrek Hasarı Olguları
Prof. Dr. Nurhan Seyahi
İstanbul Üniversitesi
Cerrahpaşa Tıp Fakültesi

Spesifik Tedavi Gerektiren Akut Böbrek Hasarı Olguları
• Kresentik (İng: Crescent, Tür: yarım ay, hilal)
glomerülonefrit
– Klinik tablo: hızlı ilerleyen glomerülonefrit (Rapidly progressive glomerülonefrit – RPGN-)
• Trombotik mikroanjiopati

Kresentik glomerülonefrit
• Kresentik glomerülonefrit (morfolojik bulgu) –Hızlı ilerleyen glomerülonefrit (klinik tablo)
• Günler veya aylar içinde böbrekfonksiyonlarının bozulması ve patolojikincelemede kresent (yarım ay, hilal) oluşumunun gözlenmesi

Yarım ay oluşumu
• Ağır glomerül hasarı
• Glomerül bariyerinin yırtılması
• Bowman aralığına inflamatuar plazmaürünlerinin geçmesi (FGF, TGF)
• İnflamatuar hücrelerin Bowman aralığınıdoldurması (cellular – hücresel- yarım ay)
• Fibroziz gelişmesi (fibrosellular ve fibröz yarımay)
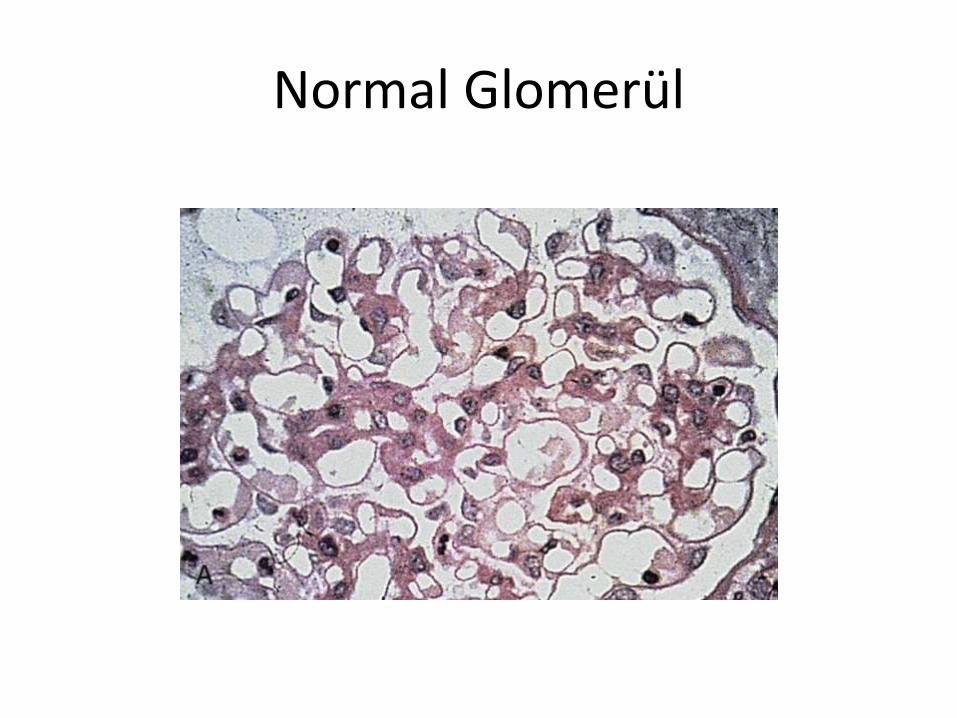
Normal Glomerül
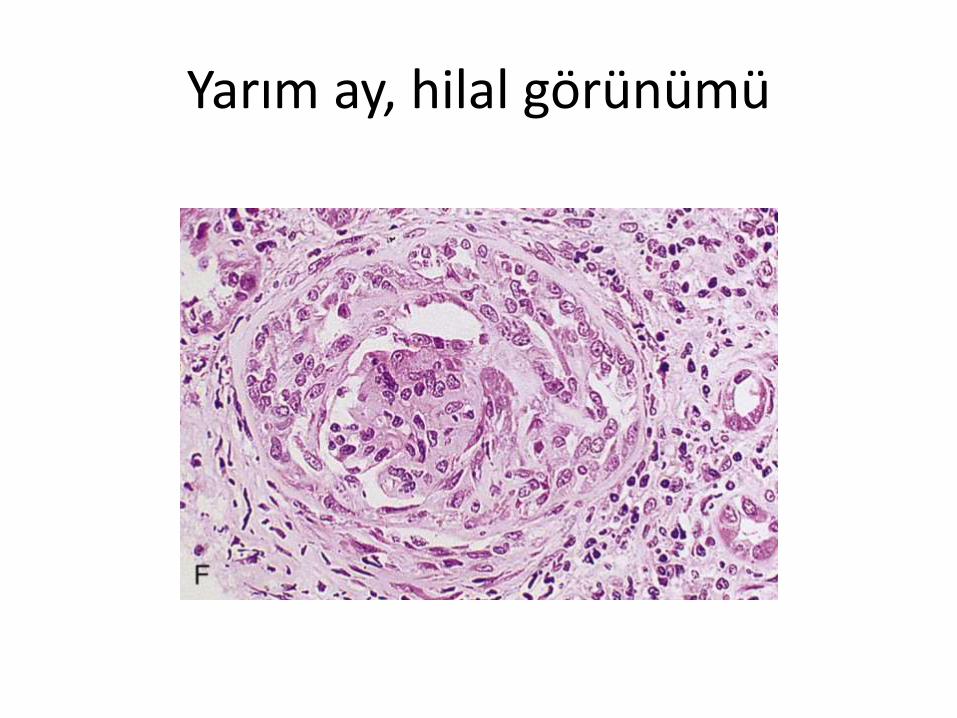
Yarım ay, hilal görünümü

Tipler
• Anti-Glomerüler bazal membran (GBM) antikoru hastalığı
• İmmün-kompleks birikimine bağlı; IgA, postinfeksiyöz, SLE, kriyoglobülinemik
• Pauci-immün-ANCA pozitif; granulomatoziz ve polianjitis (Wegener granülomatözü) veya mikroskopik polianjitis
• İdiyopatik (tipi tespit edilemeyen immun-kompleks hastalığı veya pauci-immun olup ANCA negatif bulunan vakalar)

Böbrek tutulumu bulguları
• Böbrek yetmezliği vakaların çoğunda başvurudan itibaren mevcuttur, kreatiningenellikle 3 mg/dL’nin üstündedir.
• İdrar tahlilinde dismorfik eritrositler, eritrosit silendirleri ve diğer silendir tipleri vardır. Bazı vakalarda hematüri olmayabilir.
• Proteinürinin miktarı değişkendir. Nefrotikdüzeyde proteinüri nadiren ve ancak hafif böbrek yetmezliği olan vakalarda görülebilir.

Böbrek Dışı Tutulım
• Böbrek dışı organ tutulumu ve sistemik şikayetlere pauci-immun vakalarda sık olarak rastlanılır.
– Kas-iskelet sistemi,
– deri,
– sinir sistemi,
– üst solunum yolları ve akciğer tutulumu.
• Anti-GBM antikoru hastalığı olan vakalarda da akciğer kanaması ve hemoptiziye rastlanılabilir..

Glomerülonefrit ve hemoptizibirlikteliği
• Glomerülonefrit ve hemoptizi birlikteliği anti-GBM antikoru hastalığını akla getirse de daha sık rastlanılan diğer nedenler göz ardı edilmemelidir;
• granülomatoziz ve polyanjitis (Wegenergranülomatozu)
• kresentik lupus nefriti
• pulmoner ödem ile komplike olmuş herhangi bir böbrek yetmezliği

Tanı
• Tanı hızlı bir şekilde konmalıdır, serolojiktestler
– ANCA (ayrıca MPO, PR-3)
– anti-GBM antikoru,
– kompleman düzeyleri,
– kriyoglobulin düzeyi,
– ANA
• Renal biyopsi gecikmeden yapılmalıdır.

Tedavi – Genel Prensipler
• Hızlı ilerleyen glomerülonefrit tedavi edilmediği takdirde haftalar veya birkaç ay içinde son dönem böbrek yetmezliğine neden olur.
• Yarım ay oranı daha az olan hastalarda daha yavaş bir seyir gözlenebilir.
• Tedavi başlangıç ve idame olarak iki aşamalı olarak planlanır

Tedavi – Genel Prensipler
• Yoğun immunsüpressif tedavi uygulanan başlangıç tedavisini (3-6 ay )daha ılımlı immunsüpresyonun verildiği idame tedavisi takip eder (>2 yıl)
• Başlangıç tedavisi– pulse metilprednizolon ve takiben oral prednizon– intravenöz siklofosfamid veya rituximab. – Bazı vakalarda tedaviye plazmaferez de eklenebilir
• İdame– Prednizon– Azathioprin veya MMF

Tedavi – Genel Prensipler
• Ağır hastalığı olan vakalarda biyopsi yapılması veya sonuç alınması gecikecek ise ampirik tedaviye hemen başlanabilir.
– Metilprednizolon 500-1000 mg/gün üç gün boyunca uygulanır,
– hemoptizisi olan vakalarda plazmaferez yapılması düşünülebilir.
– Daha spesifik tedavi kesin tanı ortaya konulunca başlanılabilir.

Anti-GBM hastalığı
• Plazmaferez
• Prednizon
• Siklofosfamid / Rituximab
• Tedaviye erken başlanması tedavi şansını artıran bir faktördür.

Plazmaferez
• Plazmaferez günlük veya gün aşırı 4 litrelik değişimler iki veya üç hafta boyunca uygulanır.
• Genellikle replasman sıvısı olarak albümin verilse de yeni biyopsi yapılmış olan veya pulmoner kanaması olan hastalarda işlemin son kısmında bir iki litre kadar taze donmuş plazma ile replasman yapılabilir.

Plazmaferez
• Taze donmuş plazma verilmesi sonucu (hacminin %14’ü kadar sitrat içerir) sitratınbikarnonat oluşturmasına bağlı olarak metabolik alkaloz gelişebilir.
• Hasta iki-üç haftalık seansın sonunda tekrar değerlendirilmelidir, aktif pulmoner hastalık devam ediyorsa veya antikor titresi belirgin olarak düşmemiş ise plazmafereze devam edilmelidir.

İmmunosupressif
• Siklofosfamid alan hastalara trimetoprim-sulfometaksazol ile proflaksi yapılması uygun olur.
• Metilprednizolon (15-30 mg/kg – maksimum 1000 mg /20dk’da) intravenöz olarak üç gün ardı ardına uygulanır takiben oral prednizon (1mg/kg/gün –maksimum 60-80 mg/gün) ile tedaviye devam edilir.
• Siklofosfamid oral (2mg/kg 60 yaş üstünde maksimum 100mg/gün) veya intravenoz (500-1000mg/2-4 haftada bir) olarak uygulanabilir.

Rituximab
• Rituximab uygulandıktan sonra 48 saat süre ile plazmaferez yapılmamalıdır (molekülü uzaklaştıracağı için) bu nedenle ilk yedi plazmaferez seansı sonrası ilk rituximabdozunun uygulanması düşünülebilir.

İdame
• Tedavinin optimal süresi tam olarak bilinmektedir,
• ancak antikor oluşumu en az 6 ay kadar devam edebilir bu nedenle başlangıç tedavisinden sonra daha az toksikilaçlar ile idame tedavisi uygulanmalıdır.
• Başlangıç immunosupressif tedavisi antikor düzeyleri de göz önüne alınarak üç dört ay kadar devam edebilir.
• Bu süre sonrası antikor düzeyleri yüksek ise siklofosfamit yerine azatioprin eklenerek (1-2 mg/kg/gün) altı-dokuz ay kadar tedaviye devam edilebilir.

Pauci-immun ANCA pozitif vakalar
• Tedavi prensipleri temel olarak Anti-GBM antikoru hastalığında uygulanan tedavi yaklaşımına benzer.
• Önce yoğun immunusüpresyonun uygulandığı indüksiyon dönemi takiben daha az toksikilaçlar ile idame tedavisi düzenlenir.

Pauci-immun ANCA pozitif vakalar
• İki büyük çalışmada (RAVE ve RITUXVAS) indüksiyonda rituximab’ın siklofosfamitealternatif olarak kullanılabileceği belirtilmiştir.
• Siklofosfamitin intravenöz uygulanmasında oral yola göre daha az lökopeni ve enfeksiyon görülmektedir ancak remisyon oluşturma oranları eşit olsa de relaps intravenöz yolla daha sık görülebilir.

Pauci-immun ANCA pozitif vakalar
• ANCA pozitif vakalarda plazmaferezin yeri Anti-GBM antikoru hastalığındaki kadar açık değildir – böbrek fonksiyonları hızla bozulan
– diyaliz gereken olgular
– pulmoner hemorajisi olan olgular
– eşlik eden anti-GBM antikoru
• olan olgular olmak üzere belli hasta gruplarında uygulanması önerilmektedir.

• İmmun kompleks hastalığı (IgA nefropatisi, lupusnefropatisi, membranöz nefropati, kriyoglobulinemi) Diğer hızlı ilerleyen glomerülonefritlere benzer şekilde tedavi edilirler.
• Poststreptokoksik glomerülonefrit tipik olarak spontan olarak düzelir ancak özellikle bazen erişkinlerde düzelme tam olmayabilir, yarım ay oluşumu fazla olan vakalarda steroid tedavisi uygulanabilir

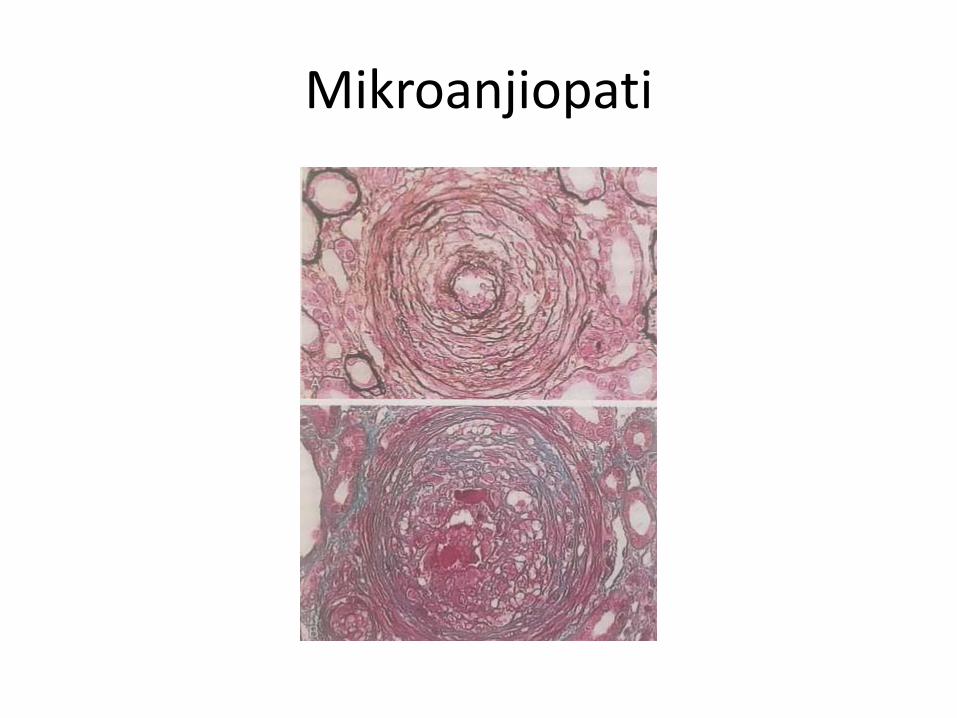
Trombotik Mikroanjiopati
• Kapiller veya arteriol duvarındaki anomalinin mikrovasküler tromboza yol açması sonucu ortaya çıkan patolojik tablo
• Hemen hemen tüm TMA vakalarında mikroanjiopatik(non-immun) hemolitik anemiye (MAHA) rastlanır.
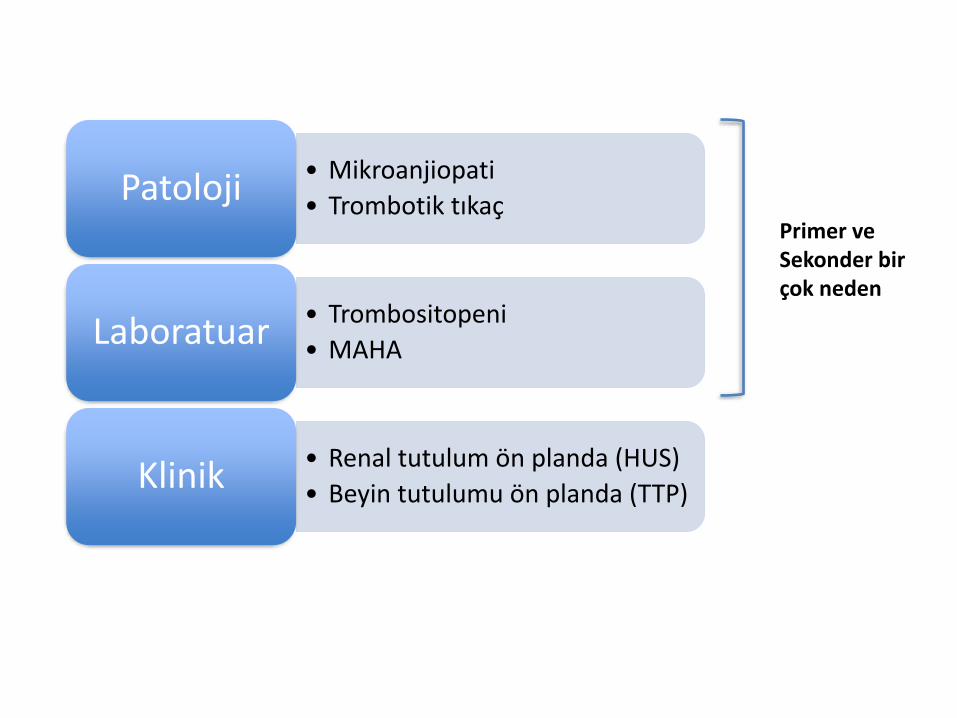
• Mikroanjiopati
• Trombotik tıkaçPatoloji
• Trombositopeni
• MAHALaboratuar
• Renal tutulum ön planda (HUS)
• Beyin tutulumu ön planda (TTP)Klinik
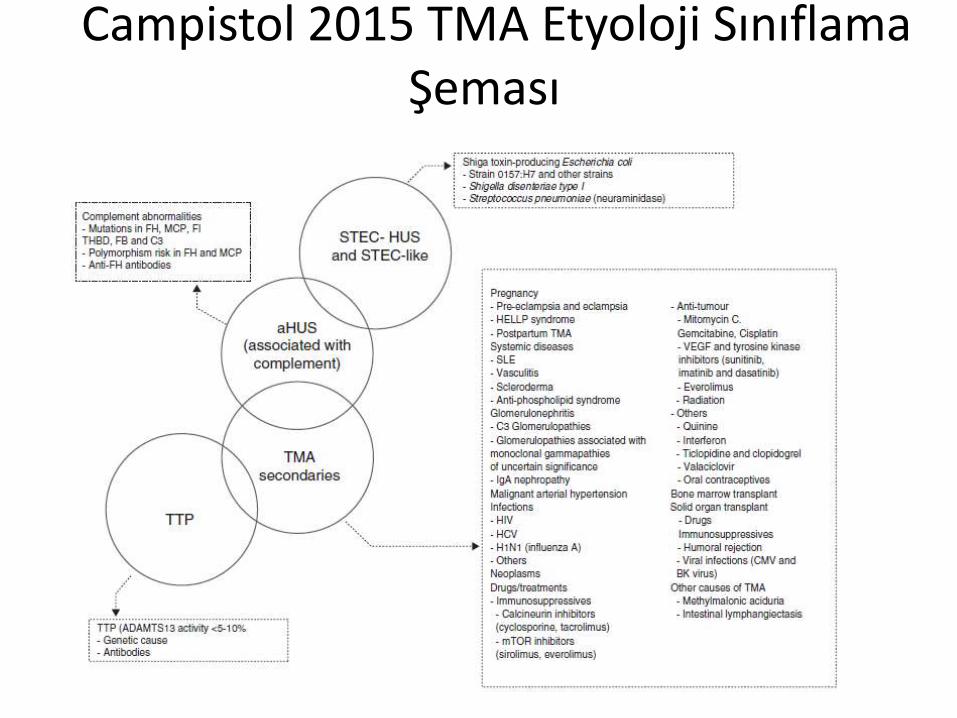
Primer veSekonder birçok neden
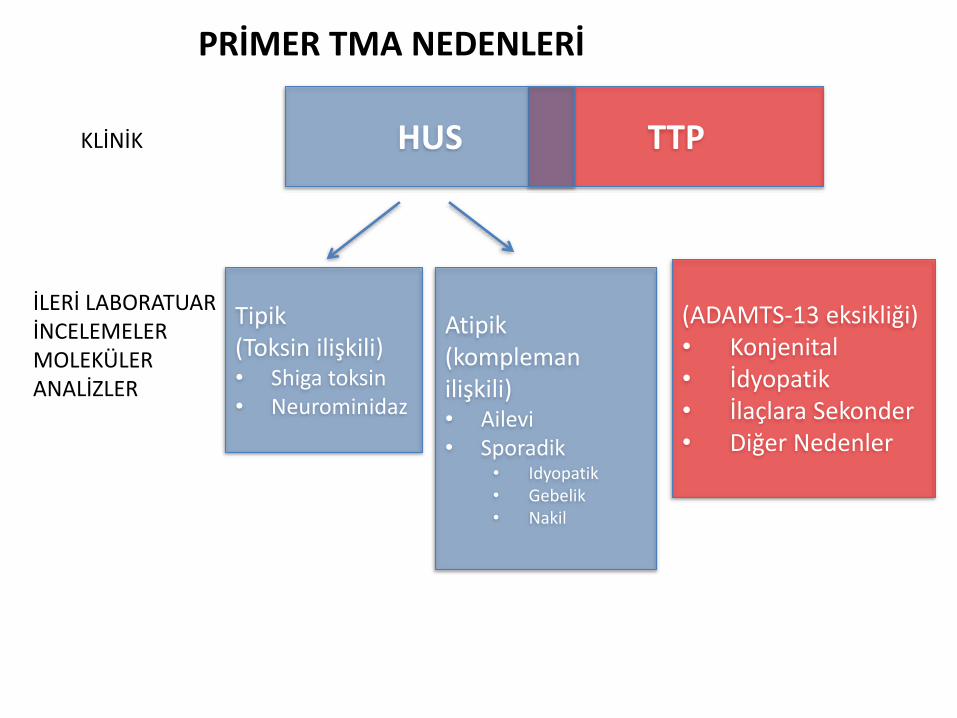
TTPHUSKLİNİK
İLERİ LABORATUAR İNCELEMELERMOLEKÜLER ANALİZLER
(ADAMTS-13 eksikliği)• Konjenital• İdyopatik• İlaçlara Sekonder• Diğer Nedenler
Tipik(Toksin ilişkili)• Shiga toksin• Neurominidaz
Atipik(komplemanilişkili)• Ailevi• Sporadik
• Idyopatik• Gebelik• Nakil
PRİMER TMA NEDENLERİ
Mikroanjiopati
Hemolitik anemi ve miğfer hücreleri

TMA’nın Klinik Ölçümleri
Azalmış Trombosit Sayısı
• Trombüs oluşumu trombositleri tüketir.
• Hastalarda trombositopeni görülebilir.
– Trombosit sayısı normal değerin altına iner<150,000 /mm3
• Hastalarda gittikçe azalan trombosit sayısı bu hücrelerin tüketiminin devam ettiğini akla getirir.
• Periferik yaymada dev trombositler görülebilir.
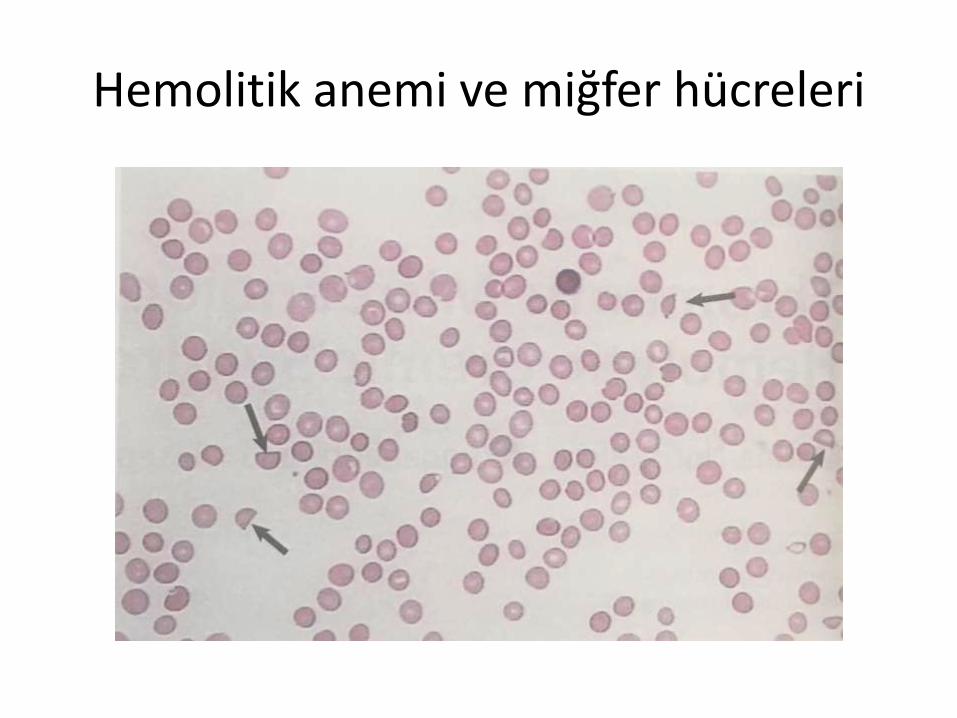
Mikroanjiyopatik Hemoliz
Pek çok vaka mikroanjiyopatik hemolizile kendini gösterir (daha önceden MAHA denirdi).
Coombs testi negatiftir
Hemoglobin miktarında azalma
LDH yüksek.
Düşük / tespit edilemeyen haptoglobinkonsantrasyonu
Şistositler

TMA
HÜS (tipik & atipik)
• ADAMTS13 YetersizliğiTTP
• Böbrek Yetmezliği veya Renal Arter StenozuMalign hipertansiyon
• Coombs(+), anti-dsDNA, anti-smDNA, ANA(+)Sistemik Lupus Eritematozus
• Antikorlar (Lupus Antikoagülan, Anti Kardiyolipin)Antifosfolipid antikor sendromu
• PT ve TT uzar, F V ve XIII azalır, fibrinojen azalırDissemine İntravasküler Koagülopati
• Enfeksiyon, kan gazı, fibrin yıkım ürünleri, lökositozSepsis
• Karaciğer fonksiyon testleri ve klinik bulgular HELLP Sendromu / Preeklampsi
• Spesifik belirteçlerMetastatik Adenokarsinom
• ELISAHIV
• ANA, anti-ENA, anti-ScL-70, direkt grafi, EKOSkleroderma
• MMA+Homosisteinüri, MMACHC gen mutasyonuKobalamin C Hastalığı
• Mitomisin-C, siklosporin, kinin ve tiklopidinİlaca Bağlı
TMA: vücuttaki küçük kan damarlarının, trombositopeni ve hemoliz ile karakterize tromboz, enflamasyon ve oklüzyonudur.
Campistol 2015 TMA Etyoloji Sınıflama Şeması
Sessiz
Klinik Seyir
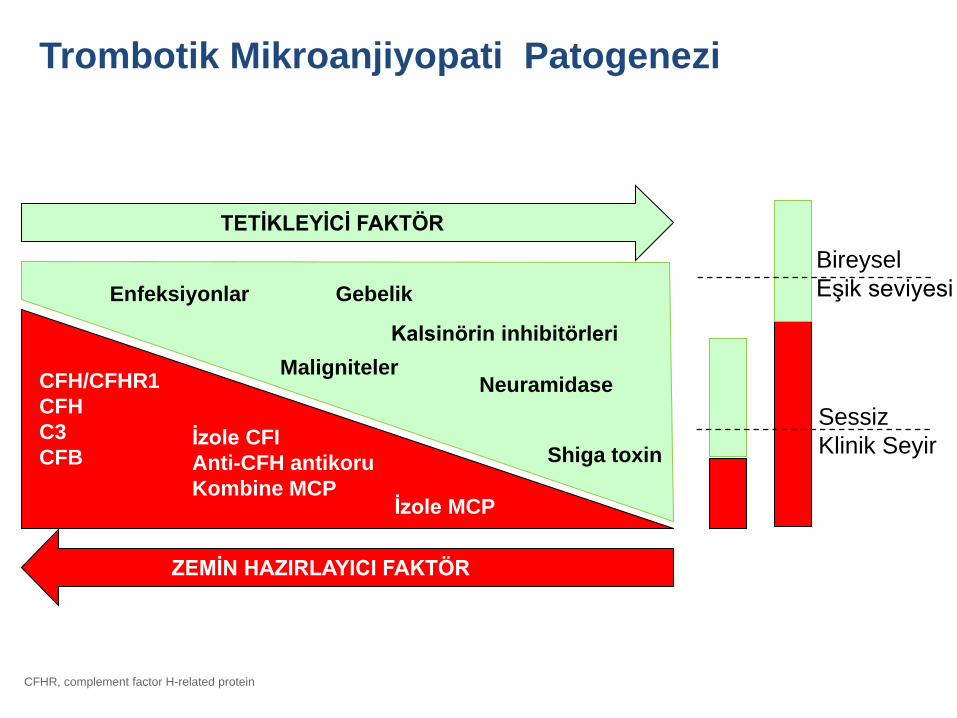
CFH/CFHR1
CFH
C3
CFBİzole CFI
Anti-CFH antikoru
Kombine MCPİzole MCP
TETİKLEYİCİ FAKTÖR
Enfeksiyonlar Gebelik
Maligniteler
Shiga toxin
Kalsinörin inhibitörleri
Neuramidase
Bireysel
Eşik seviyesi
ZEMİN HAZIRLAYICI FAKTÖR
CFHR, complement factor H-related protein
Trombotik Mikroanjiyopati Patogenezi
Progresif Organ Hasarı
eritrosit trombosit aktive trombosit lökosit aktive lökosit vWF Protrombotik faktörler şistosit
Klinik TMA ve İskemiEndotel hasarı ve kompleman aktivite
artışı
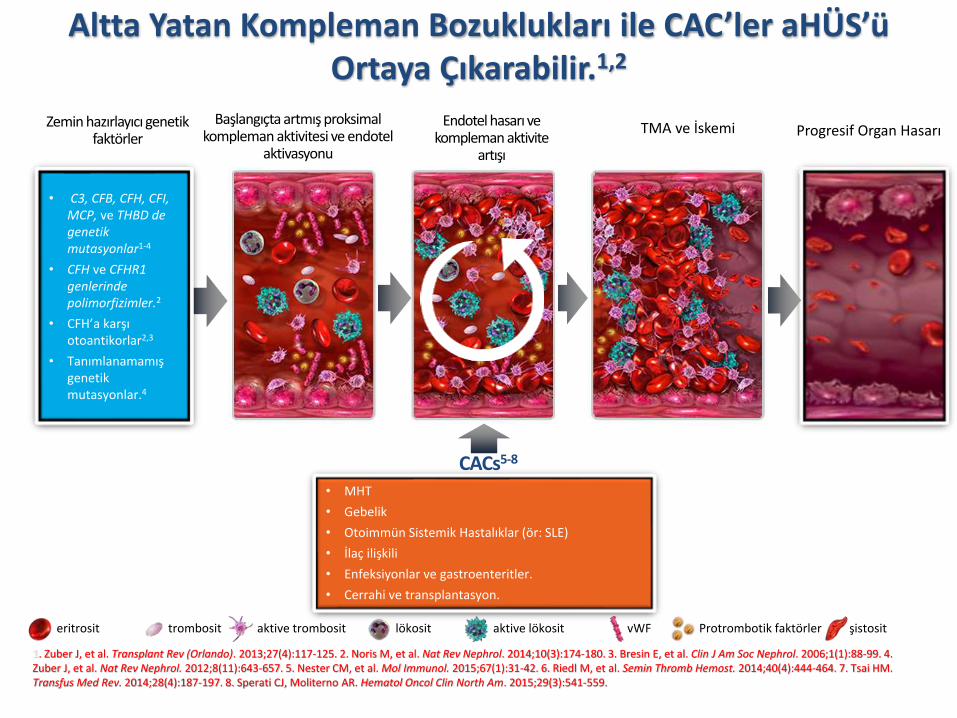
Altta Yatan Kompleman Bozuklukları ile CAC’ler aHÜS’üOrtaya Çıkarabilir.1,2
Başlangıçta artmış proksimalkompleman aktivitesi ve endotel
aktivasyonu
• C3, CFB, CFH, CFI, MCP, ve THBD de genetik mutasyonlar1-4
• CFH ve CFHR1genlerinde polimorfizimler.2
• CFH’a karşı otoantikorlar2,3
• Tanımlanamamış genetik mutasyonlar.4
Zemin hazırlayıcı genetik faktörler
• MHT
• Gebelik
• Otoimmün Sistemik Hastalıklar (ör: SLE)
• İlaç ilişkili
• Enfeksiyonlar ve gastroenteritler.
• Cerrahi ve transplantasyon.
CACs5-8
1. Zuber J, et al. Transplant Rev (Orlando). 2013;27(4):117-125. 2. Noris M, et al. Nat Rev Nephrol. 2014;10(3):174-180. 3. Bresin E, et al. Clin J Am Soc Nephrol. 2006;1(1):88-99. 4. Zuber J, et al. Nat Rev Nephrol. 2012;8(11):643-657. 5. Nester CM, et al. Mol Immunol. 2015;67(1):31-42. 6. Riedl M, et al. Semin Thromb Hemost. 2014;40(4):444-464. 7. Tsai HM. Transfus Med Rev. 2014;28(4):187-197. 8. Sperati CJ, Moliterno AR. Hematol Oncol Clin North Am. 2015;29(3):541-559.

Trombotik Mikroanjiopati
• Primer trombotik mikroanjiopati (TMA) vakaları – trombotik trombositopenik purpura (TTP;
herediter veya edinsel),
– Shiga-toksin ilişkili HUS (ST-HUS),
– ilaç ilişkili TMA (DITMA),
– kompleman ilişkili TMA (herediter veya kazanılmış)
– vitamin B12 metabolizmasının nadir hastalıkları olarak ayrılabilir.

Trombotik trombositopenik purpura(TTP)
• Edinsel (otoantikora bağlı) veya konjenitalADAMTS13 eksikliği (<10%) olması ile tanınır.
• Sistemik organ tutulumu belirgin olmasına rağmen böbrek tutulumu ön planda değildir. – Merkezi sinir sistemi, – kalp, – pankreas, – tiroid, – adrenal, – intestinal mukoza
• Akciğer tutulumu olmaması tipiktir.

Kompleman aracılıklı TMA
• Kalıtsal olarak alternatif kompleman yolunu kısıtlayan regülatör proteinlerin eksikliği (Kompleman Faktör H ve I gibi)
• Bu yolun hızlanmasına yol açan proteinlerin aktive olması
• Faktör H ve I edinsel olarak da otoantikorlarabağlı olarak inhibe olabilirler
• kontrolsüz kompleman aktivasyonu
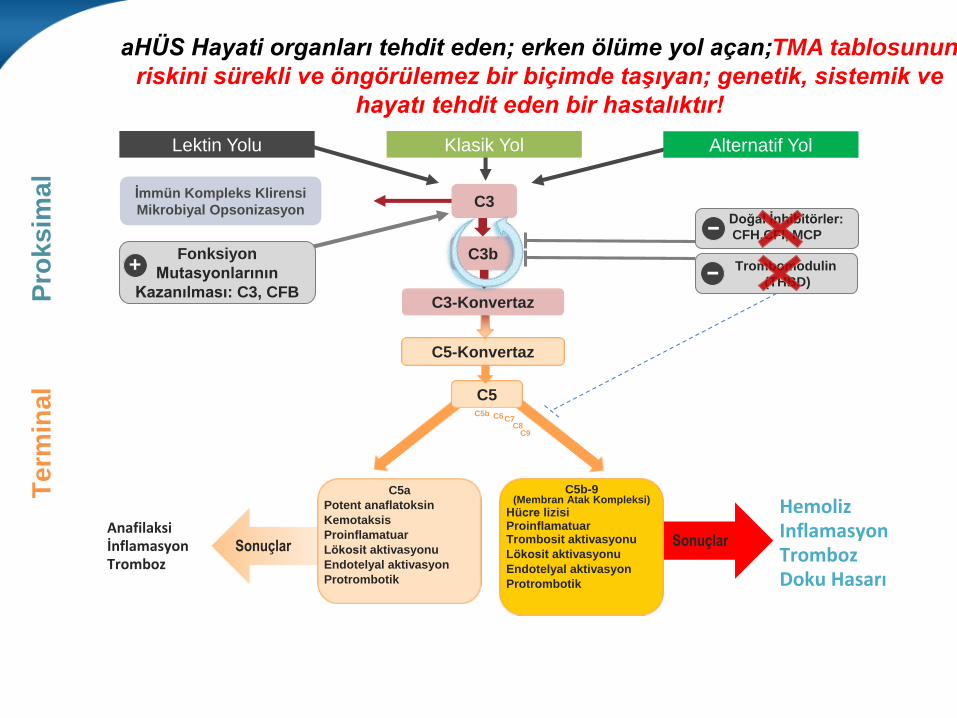
aHÜS Hayati organları tehdit eden; erken ölüme yol açan;TMA tablosunun
riskini sürekli ve öngörülemez bir biçimde taşıyan; genetik, sistemik ve
hayatı tehdit eden bir hastalıktır!
SonuçlarSonuçlarAnafilaksi İnflamasyonTromboz
C5b-9 (Membran Atak Kompleksi)
Hücre lizisiProinflamatuarTrombosit aktivasyonu
Lökosit aktivasyonu
Endotelyal aktivasyon
Protrombotik
C5a
Potent anaflatoksin
Kemotaksis
Proinflamatuar
Lökosit aktivasyonu
Endotelyal aktivasyon
Protrombotik
C5
C5-Konvertaz
Pro
ksim
al
Lektin Yolu Alternatif YolKlasik Yol
Term
inal
C3-Konvertaz
C3b
İmmün Kompleks Klirensi
Mikrobiyal OpsonizasyonC3
Doğal İnhibitörler:
CFH,CFI, MCP
Trombomodulin
(THBD)
C5b C6C7C8
C9
HemolizInflamasyonTrombozDoku Hasarı
Fonksiyon
Mutasyonlarının
Kazanılması: C3, CFB
+
−
−
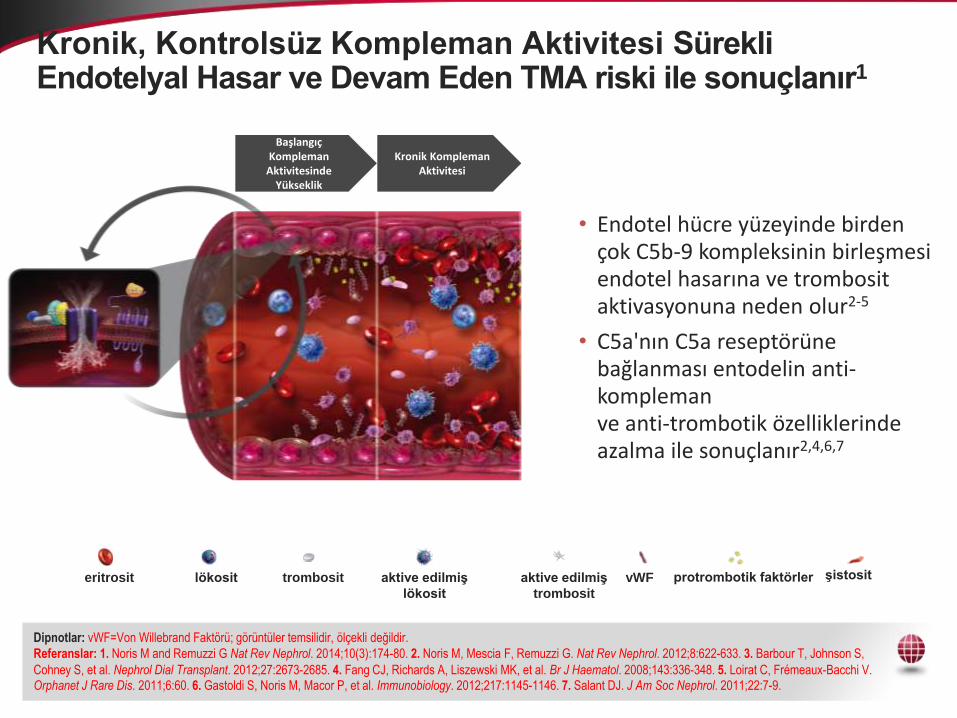
Kronik, Kontrolsüz Kompleman Aktivitesi Sürekli Endotelyal Hasar ve Devam Eden TMA riski ile sonuçlanır1
eritrosit lökosit trombosit aktive edilmiş
lökosit
aktive edilmiş
trombosit
vWF protrombotik faktörler şistosit
Dipnotlar: vWF=Von Willebrand Faktörü; görüntüler temsilidir, ölçekli değildir.
Referanslar: 1. Noris M and Remuzzi G Nat Rev Nephrol. 2014;10(3):174-80. 2. Noris M, Mescia F, Remuzzi G. Nat Rev Nephrol. 2012;8:622-633. 3. Barbour T, Johnson S,
Cohney S, et al. Nephrol Dial Transplant. 2012;27:2673-2685. 4. Fang CJ, Richards A, Liszewski MK, et al. Br J Haematol. 2008;143:336-348. 5. Loirat C, Frémeaux-Bacchi V. Orphanet J Rare Dis. 2011;6:60. 6. Gastoldi S, Noris M, Macor P, et al. Immunobiology. 2012;217:1145-1146. 7. Salant DJ. J Am Soc Nephrol. 2011;22:7-9.
Başlangıç Kompleman
Aktivitesinde Yükseklik
Kronik Kompleman Aktivitesi
• Endotel hücre yüzeyinde birden çok C5b-9 kompleksinin birleşmesi endotel hasarına ve trombosit aktivasyonuna neden olur2-5
• C5a'nın C5a reseptörüne bağlanması entodelin anti-komplemanve anti-trombotik özelliklerinde azalma ile sonuçlanır2,4,6,7
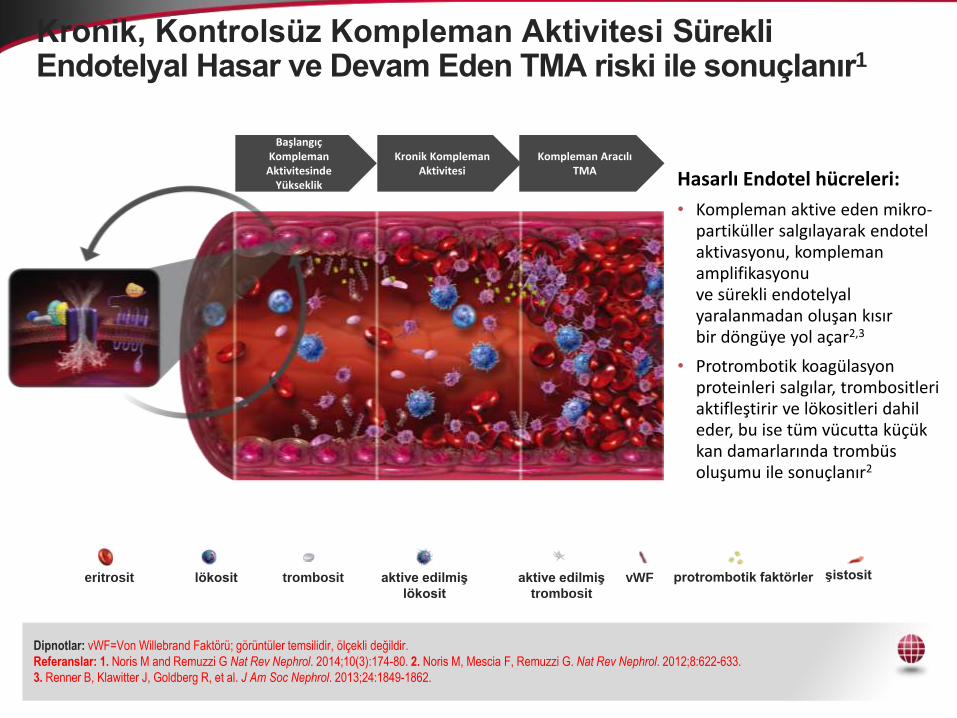
Kronik, Kontrolsüz Kompleman Aktivitesi Sürekli Endotelyal Hasar ve Devam Eden TMA riski ile sonuçlanır1
eritrosit lökosit trombosit aktive edilmiş
lökosit
aktive edilmiş
trombosit
vWF protrombotik faktörler şistosit
Dipnotlar: vWF=Von Willebrand Faktörü; görüntüler temsilidir, ölçekli değildir.
Referanslar: 1. Noris M and Remuzzi G Nat Rev Nephrol. 2014;10(3):174-80. 2. Noris M, Mescia F, Remuzzi G. Nat Rev Nephrol. 2012;8:622-633.
3. Renner B, Klawitter J, Goldberg R, et al. J Am Soc Nephrol. 2013;24:1849-1862.
Başlangıç Kompleman
Aktivitesinde Yükseklik
Kronik Kompleman Aktivitesi
Kompleman Aracılı TMA
Hasarlı Endotel hücreleri:
• Kompleman aktive eden mikro-partiküller salgılayarak endotel aktivasyonu, kompleman amplifikasyonuve sürekli endotelyal yaralanmadan oluşan kısırbir döngüye yol açar2,3
• Protrombotik koagülasyon proteinleri salgılar, trombositleri aktifleştirir ve lökositleri dahil eder, bu ise tüm vücutta küçük kan damarlarında trombüs oluşumu ile sonuçlanır2
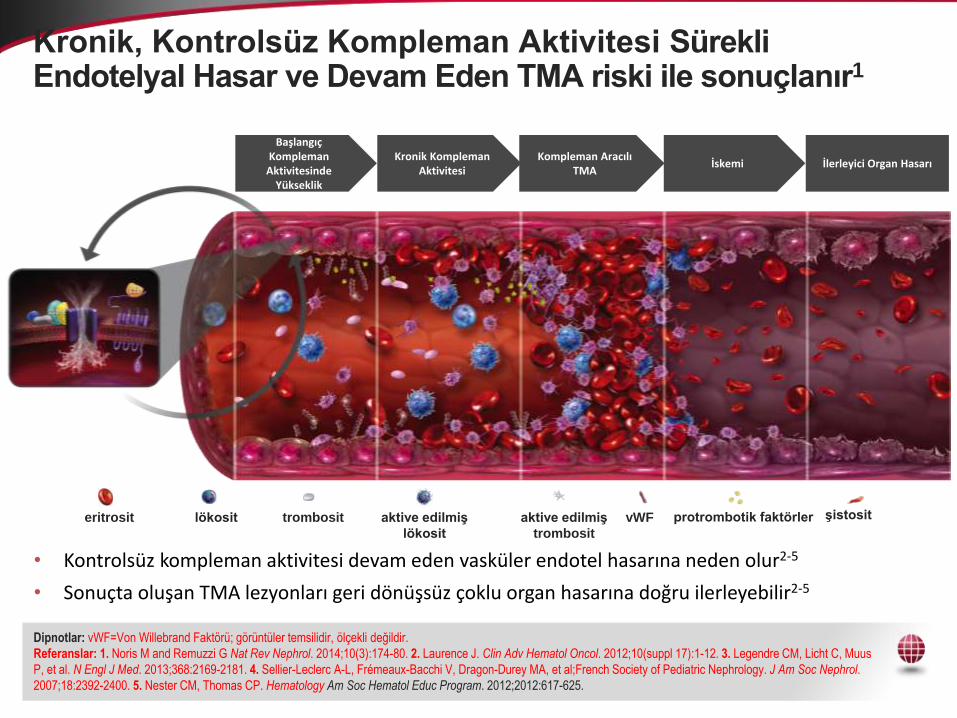
Kronik, Kontrolsüz Kompleman Aktivitesi Sürekli Endotelyal Hasar ve Devam Eden TMA riski ile sonuçlanır1
eritrosit lökosit trombosit aktive edilmiş
lökosit
aktive edilmiş
trombosit
vWF protrombotik faktörler şistosit
Dipnotlar: vWF=Von Willebrand Faktörü; görüntüler temsilidir, ölçekli değildir.
Referanslar: 1. Noris M and Remuzzi G Nat Rev Nephrol. 2014;10(3):174-80. 2. Laurence J. Clin Adv Hematol Oncol. 2012;10(suppl 17):1-12. 3. Legendre CM, Licht C, Muus
P, et al. N Engl J Med. 2013;368:2169-2181. 4. Sellier-Leclerc A-L, Frémeaux-Bacchi V, Dragon-Durey MA, et al;French Society of Pediatric Nephrology. J Am Soc Nephrol.
2007;18:2392-2400. 5. Nester CM, Thomas CP. Hematology Am Soc Hematol Educ Program. 2012;2012:617-625.
Başlangıç Kompleman
Aktivitesinde Yükseklik
Kronik Kompleman Aktivitesi
Kompleman Aracılı TMA
İskemi İlerleyici Organ Hasarı
• Kontrolsüz kompleman aktivitesi devam eden vasküler endotel hasarına neden olur2-5
• Sonuçta oluşan TMA lezyonları geri dönüşsüz çoklu organ hasarına doğru ilerleyebilir2-5
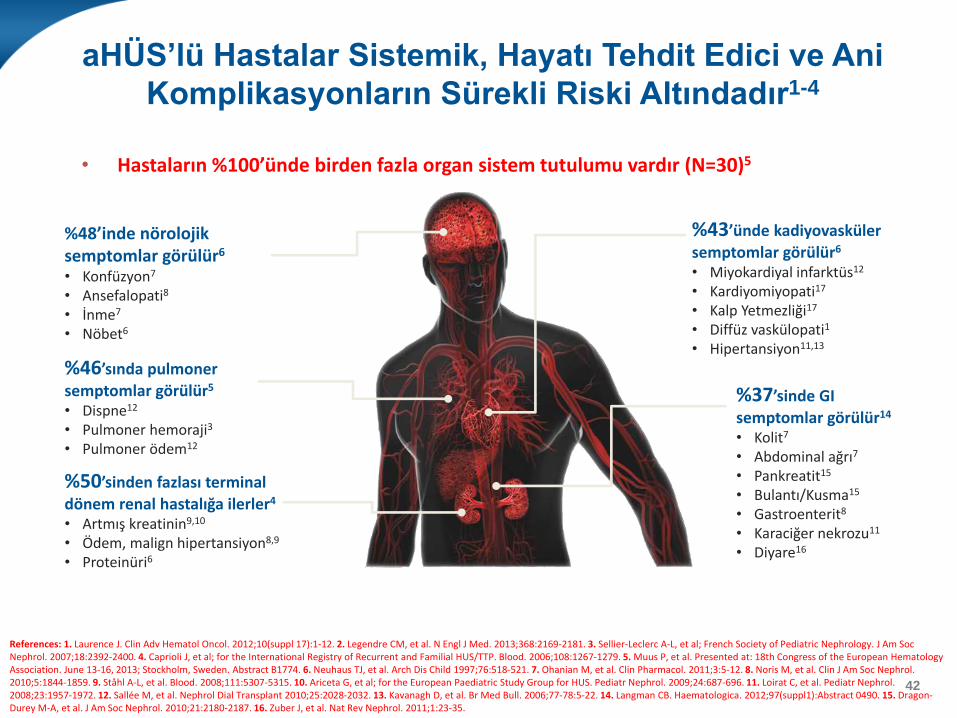
aHÜS’lü Hastalar Sistemik, Hayatı Tehdit Edici ve Ani
Komplikasyonların Sürekli Riski Altındadır1-4
• Hastaların %100’ünde birden fazla organ sistem tutulumu vardır (N=30)5
42
%48’inde nörolojik semptomlar görülür6
• Konfüzyon7
• Ansefalopati8
• İnme7
• Nöbet6
%43’ünde kadiyovasküler
semptomlar görülür6
• Miyokardiyal infarktüs12
• Kardiyomiyopati17
• Kalp Yetmezliği17
• Diffüz vaskülopati1
• Hipertansiyon11,13
%37’sinde GI
semptomlar görülür14
• Kolit7
• Abdominal ağrı7
• Pankreatit15
• Bulantı/Kusma15
• Gastroenterit8
• Karaciğer nekrozu11
• Diyare16
%50’sinden fazlası terminal
dönem renal hastalığa ilerler4
• Artmış kreatinin9,10
• Ödem, malign hipertansiyon8,9
• Proteinüri6
%46’sında pulmoner
semptomlar görülür5
• Dispne12
• Pulmoner hemoraji3
• Pulmoner ödem12
References: 1. Laurence J. Clin Adv Hematol Oncol. 2012;10(suppl 17):1-12. 2. Legendre CM, et al. N Engl J Med. 2013;368:2169-2181. 3. Sellier-Leclerc A-L, et al; French Society of Pediatric Nephrology. J Am SocNephrol. 2007;18:2392-2400. 4. Caprioli J, et al; for the International Registry of Recurrent and Familial HUS/TTP. Blood. 2006;108:1267-1279. 5. Muus P, et al. Presented at: 18th Congress of the European Hematology Association. June 13-16, 2013; Stockholm, Sweden. Abstract B1774. 6. Neuhaus TJ, et al. Arch Dis Child 1997;76:518-521. 7. Ohanian M, et al. Clin Pharmacol. 2011;3:5-12. 8. Noris M, et al. Clin J Am Soc Nephrol. 2010;5:1844-1859. 9. Ståhl A-L, et al. Blood. 2008;111:5307-5315. 10. Ariceta G, et al; for the European Paediatric Study Group for HUS. Pediatr Nephrol. 2009;24:687-696. 11. Loirat C, et al. Pediatr Nephrol. 2008;23:1957-1972. 12. Sallée M, et al. Nephrol Dial Transplant 2010;25:2028-2032. 13. Kavanagh D, et al. Br Med Bull. 2006;77-78:5-22. 14. Langman CB. Haematologica. 2012;97(suppl1):Abstract 0490. 15. Dragon-Durey M-A, et al. J Am Soc Nephrol. 2010;21:2180-2187. 16. Zuber J, et al. Nat Rev Nephrol. 2011;1:23-35.

Mikronajiopatik hemolitik anemi ve trombositopeni: Ayırıcı Tanı
• gebelik komplikasyonu (ağır preeklempsi ve HELLP sendromu),
• ağır hipertansiyon, • sistemik enfeksiyonlar (bakteriyel endokardit, HIV, CMV,
malarya gibi), • maligniteler, • romatolojik hastalıklar (SLE, skleroderma, antifosfolipid
sendromu gibi), • kök hücre veya organ nakli (genellikle kullanılan ilaçlara
bağlı, akut graft rejeksiyonu), • dissemine intravasküler koagülasyon, • ağır B12 eksikliği

Hastanın Değerlendirilmesi
• Yaş– Erişkinler
• edinsel TTP• DITMA • kompleman aracılıklı TMA
• Böbrek yetmezliğinin derecesi ve süresi– minimal böbrek hasarı TTP, – akut böbrek hasarı ve anüri DITMA, – günler içinde gelişen karın ağrısı ve diyarenin eşlik ettiği tedricen
ortaya çıkan böbrek hasarı ST-HUS veya kompleman aracılıklıHUS,
– haftalar veya aylar içinde ortaya çıkan böbrek hasarı kemoterapi veya immunosupressif ilaçlara bağlı TMA olduğunu

Laboratuar testleri• Tüm hastalardan
– tam kan sayımı, LDH, kreatinin günlük takipleri yapılmalıdır. – ADAMTS13 aktivitesi TTP olasılığını ortaya koymak için
istenmelidir.– Karın ağrısı ve diyare olan tüm hastalardan mutlaka
enterohemorajik E. Coli için kültür alınmalıdır ayrıca Shiga Toksin için immun test yapılabilir
– ADAMTS13 seviyesi normal olan ve ST-HUS bulgusu olmayan hastalarda homosistein ve metilmalonik asit seviyeleri ölçülmelidir
– Kompleman seviyelerinin normal olması kompleman aracılıklıTMA dışlamada kullanılamayacağı için rutin ölçümün yeri tartışmalıdır.
– Rutin tetkiklerde tanıya gidilemeyen vakalarda komplemanproteinlerini kodlayan genlerdeki mütasyonlar incelenebilir. Diğer mütasyonlar için inceleme rutin olarak önerilmemektedir

Hastaya Yaklaşım
• Böbrek biyopsisi TMA’nın tipini ortaya koymak için ilave bir katkı sağlamaz ancak böbrek hasarının TMA’ya bağlı olup olmadığını ortaya koyabilir.

Tipik HUS
Atipik HUS
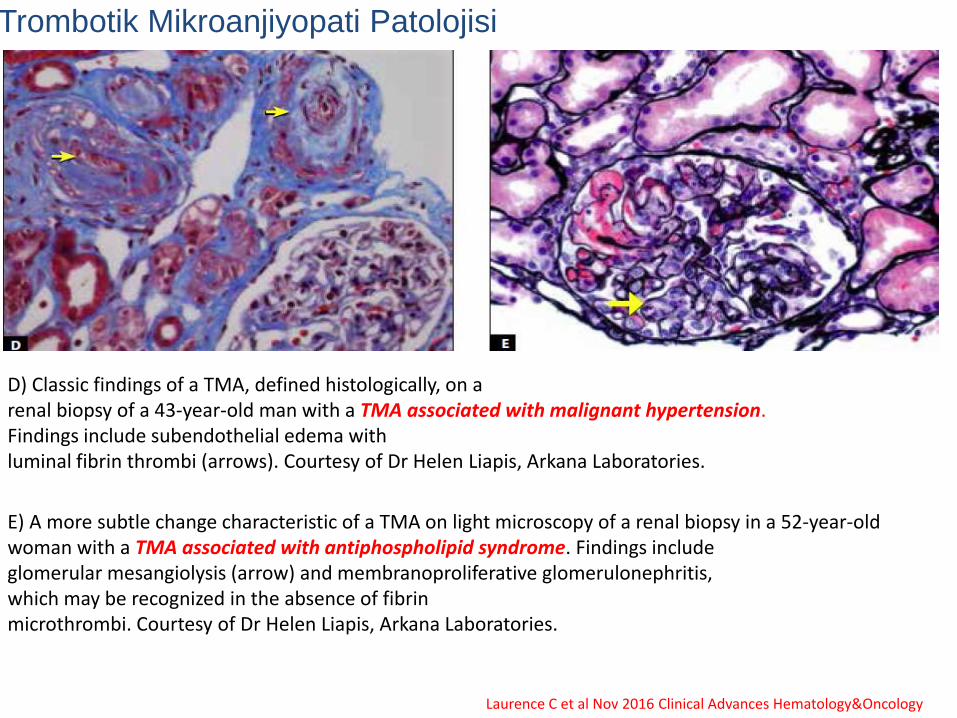
Trombotik Mikroanjiyopati Patolojisi
D) Classic findings of a TMA, defined histologically, on arenal biopsy of a 43-year-old man with a TMA associated with malignant hypertension. Findings include subendothelial edema withluminal fibrin thrombi (arrows). Courtesy of Dr Helen Liapis, Arkana Laboratories.
E) A more subtle change characteristic of a TMA on light microscopy of a renal biopsy in a 52-year-old woman with a TMA associated with antiphospholipid syndrome. Findings includeglomerular mesangiolysis (arrow) and membranoproliferative glomerulonephritis, which may be recognized in the absence of fibrinmicrothrombi. Courtesy of Dr Helen Liapis, Arkana Laboratories.
Laurence C et al Nov 2016 Clinical Advances Hematology&Oncology

TMA'lar için Ayırıcı Tanı: aHUS, TTP ve STEC-HUS
Bu yol, sağlık uzmanları için eğitici bilgi sunma amacını taşımaktadır. Sağlık uzmanının profesyonel kararının veya klinik tanısının yerini tutmaz.
Dipnotlar: *Shiga-toksin/EHEC testi, GI semptomları geçmişi/mevcudiyeti ile teminat altına alınır. Referanslar: 1. Dosyaya ilişkin veriler. Alexion Pharmaceuticals, Inc. 2. Caprioli J et al. Blood.
2006;108(4):1267-1279. 3. Noris M et al. NEJM. 2009;361:1676-1687. 4. Neuhaus et al. Arch Dis Chilid. 1997;76:518-521. 5. Noris M et al. JASN. 2005;16:1177-1183. 6. Dragon-Durey et al. J Am
Soc Nephrol. 2010;21:2180-2187. 7. Davin et al. Am J Kid Dis. 2010;55:708-777; 8. Bianchi et al. Blood. 2002;110:710-713. 9. Al-Akash et al. Pediatr Nephrol. 2011;26:613-619. 10. Sellier-Leclerc
AL. JASN. 2007;18:2392-2400. 11. Benz et al. Curr Opin Nephrol Hypertens. 2010;19:242-247. 12. Noris M et al. Clin J Am Soc Nephrol. 2010;5:1844-1859. 13. Tsai H-M. Int J Hematol.
2010;91:1-19; 14. Barbot et al. Br J Haematol. 2001;113:649. 15. Bitzan M. Semin Thromb Hemost. 2010;36:594-610. 16. Zuber J et al. Nat Rev Nephrol. 2012;8:643-657.
≤ %5 ADAMTS13 Aktivitesi8,13,14 > %5 ADAMTS13 Aktivitesi12 Shiga-toksin/EHEC Pozitif14
STEC-HUSTTP aHüS
ADAMTS13 Aktivitesi ve Shiga-toksin/EHEC* Testi değerlendirmesi8,13-15
ADAMTS13 sonuçlarını beklerken, > 30.000 mm3'lük bir trombosit sayımı veya > 150-200 μmol/l'lik bir serum
kreatinin (> 1,7-2,3 mg/dl), şiddetli ADAMTS13 eksikliği (TTP) tanısın dışlar16
Ek olarak şunlardan biri veya birkaçı:
Trombositopeni1,2
Trombosit Sayımı < 150.000 veya
Başlangıca göre > %25 Azalma1
Böbrek yetmezliği2,9,10
Yüksek kreatinin10 ve/veyaDüşük eGFR2,10 ve/veya
Yüksek kan basıncı11 ve/veyaAnormal idrar analizi9
Nörolojik Semptomlar4-7
Karışıklık4,5 ve/veyaNöbetler6,8 ve/veya
Diğer serebral anormallikler5
Gastrointestinal Semptomlar2,612
Diyare +/– kan12 ve/veyaMide bulantısı/kusma6 ve/veya Karın
ağrısı6 ve/veyaGastroenterit2,12
Mikroanjiyopatik Hemoliz2,3
Şistositler2,3 ve/veya
Yüksek LDH2 ve/veya
Düşük haptoglobin2 ve/veya
Düşük hemoglobin2
VE

Tedavi
• Plazmaferez TTP’nin öncelikle ve yaşam kurtarıcı tedavisidir
• TMA şüphesi olan her hastada en öncelikli olarak TTP tanısı ile plazmafereze başlanıp başlanmayacağına karar verilmelidir.
• Primer TMA tipi kesin olarak ortaya konmayan vakalarda klinik bulgular TTP düşündürüyorsa ADAMTS13 sonucu beklenmeden plazmaferezbaşlanmalıdır.

Tedavi
• TTP tanısı olan ve plazmaferezin gecikebileceği hastalarda bu tedaviye başlanılana kadar taze donmuş plazma verilebilir.
• TTP tanısı ile plazmaferez uygulanan hastalarda steroidtedavisi de rutin olarak başlanır. Plazmaya alerjisi olan nadir hastalarda yüksek düzeyde ADAMTS13 iceren Faktör VIII konsantresi verilebilir. Plazmafereze hastanın trombosit sayımı iki gün boyunca normalize olana kadar devam edilir.

Tedavi
• TTP dışındaki TMA tipleri için plazmaferezinetkinliği bu kadar açık değildir.
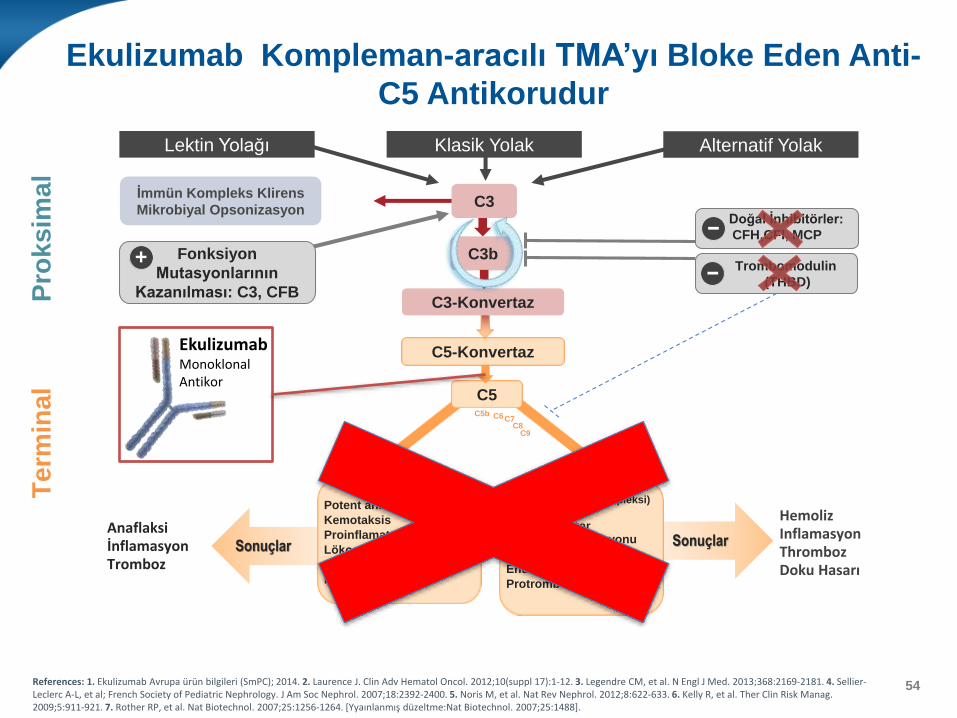
Ekulizumab Kompleman-aracılı TMA’yı Bloke Eden Anti-
C5 Antikorudur
54References: 1. Ekulizumab Avrupa ürün bilgileri (SmPC); 2014. 2. Laurence J. Clin Adv Hematol Oncol. 2012;10(suppl 17):1-12. 3. Legendre CM, et al. N Engl J Med. 2013;368:2169-2181. 4. Sellier-Leclerc A-L, et al; French Society of Pediatric Nephrology. J Am Soc Nephrol. 2007;18:2392-2400. 5. Noris M, et al. Nat Rev Nephrol. 2012;8:622-633. 6. Kelly R, et al. Ther Clin Risk Manag. 2009;5:911-921. 7. Rother RP, et al. Nat Biotechnol. 2007;25:1256-1264. [Yyaınlanmış düzeltme:Nat Biotechnol. 2007;25:1488].
SonuçlarSonuçlarAnaflaksiİnflamasyonTromboz
C5b-9 (Membran Atak Kompleksi)
Hücre lizisiProinflamatuarTrombosit aktivasyonu
Lökosit aktivasyonu
Endotelyal aktivasyon
Protrombotik
C5a
Potent anaflatoksin
Kemotaksis
Proinflamatuar
Lökosit aktivasyonu
Endotelyal aktivasyon
Protrombotik
C5
C5-Konvertaz
Pro
ksim
al
Lektin Yolağı Alternatif YolakKlasik Yolak
Term
inal
C3-Konvertaz
C3b
İmmün Kompleks Klirens
Mikrobiyal OpsonizasyonC3
Doğal İnhibitörler:
CFH,CFI, MCP
Trombomodulin
(THBD)
C5b C6C7C8
C9
HemolizInflamasyonThrombozDoku Hasarı
Fonksiyon
Mutasyonlarının
Kazanılması: C3, CFB
+
−
−
EkulizumabMonoklonal Antikor

Tedavi
• Anti-kompleman tedavi (eculizumab) kompleman aracılıklıTMA düşünülen vakalarda uygulanmalıdır.
• Yüksek klinik şüphe tedaviye başlamak için yeterlidir zira kompleman sistemi ilgili hızlı sonuç verecek kesin tanı testleri mevcut değildir. – Kanlı diyaresi olmayan TMA ve böbrek yetmezliği olan çocuklarda
– doğum sonrası TMA ve böbrek yetmezliği olan vakalarda antikompleman tedavinin geciktirilmemesi önemlidir.
– Bunların dışındaki vakalarda tanı teyit edilene değin tedaviye plazmaferez ile başlanılabilir.
• spesifik bir TMA nedeni oraya konmazsa veya sekonder bir sistemik hastalık yoksa tedaviye eculizumab ile devam edilmesi uygun olabilir.
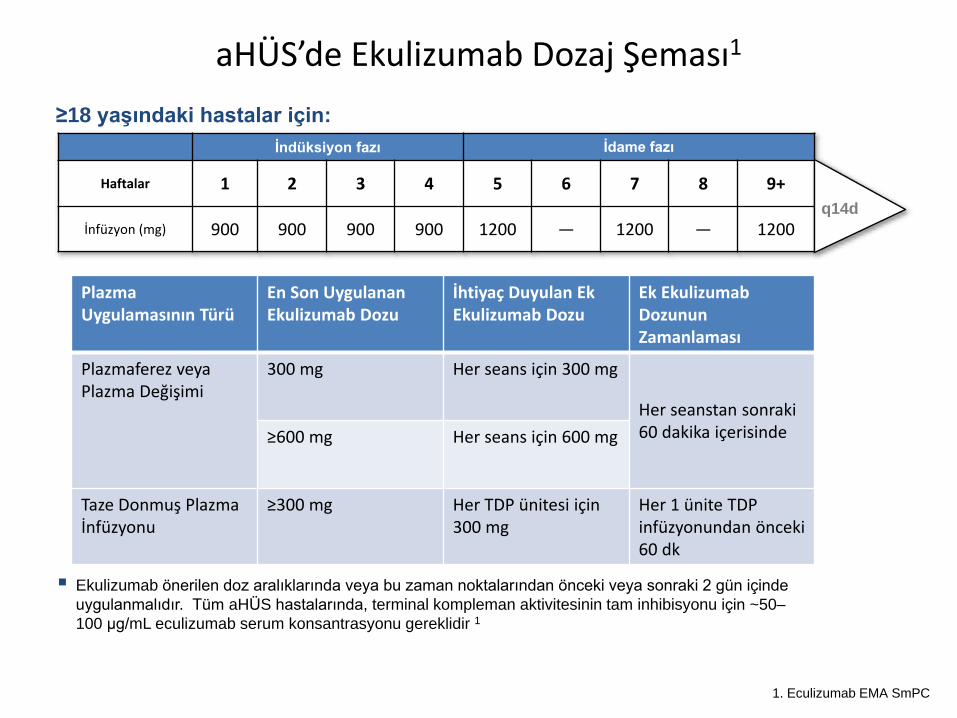
aHÜS’de Ekulizumab Dozaj Şeması1
Ekulizumab önerilen doz aralıklarında veya bu zaman noktalarından önceki veya sonraki 2 gün içinde
uygulanmalıdır. Tüm aHÜS hastalarında, terminal kompleman aktivitesinin tam inhibisyonu için ~50–
100 μg/mL eculizumab serum konsantrasyonu gereklidir 1
≥18 yaşındaki hastalar için:
İndüksiyon fazı İdame fazı
Haftalar 1 2 3 4 5 6 7 8 9+
İnfüzyon (mg) 900 900 900 900 1200 — 1200 — 1200q14d
aHÜS, atipik Hemolitik Üremik Sendrom;
q14d, her 14 günde bir 1. Eculizumab EMA SmPC
Plazma Uygulamasının Türü
En Son Uygulanan Ekulizumab Dozu
İhtiyaç Duyulan Ek Ekulizumab Dozu
Ek EkulizumabDozunun Zamanlaması
Plazmaferez veya Plazma Değişimi
300 mg Her seans için 300 mg
Her seanstan sonraki 60 dakika içerisinde≥600 mg Her seans için 600 mg
Taze Donmuş Plazma İnfüzyonu
≥300 mg Her TDP ünitesi için 300 mg
Her 1 ünite TDP infüzyonundan önceki 60 dk

Meningokok Aşılaması ve antibiyotikproflaksisi
• Tüm hastalara meninkokok aşısı yapılmalı (ACWY ve B)
• Aşılama sonrası hemen tedavi başlanmış ise en az 15 gün bazıönerilere göre tedavi boyunca antibiyotik proflaksisiyapılmalıdır.– 2 hafta siprofloksasin
– Takiben Pen-V veya ertirtomisin (veya infeksiyon hastalıklarınınönerisine göre)
CHRISTENSEN, H., MAY, M., BOWEN, L., HICKMAN, M. & TROTTER, C. L. 2010. Meningococcal carriage by age: a systematic review and meta-analysis. Lancet Infect Dis, 10, 853-61.

Tedavi
• Eculizumab genellikle iyi tolere edilir.
• Hastalarda meningokok infeksiyonu riskinin artacağı için rutin olarak aşılama ve aşılama sonrası en az iki hafta boyunca antibiyoterapiile proflaksi yapılması önerilmektedir.
• Eculizumab’ın plazmaferez ile uzaklaşacağı göz önüne alınarak kombine tedavi alan hastalarda uygulama zamanlamasına göre ek doz gerekebileceği düşünülmelidir.

• Kaynakça
– UPTODATE internet sitesi (ilgili bölümler)
– Comprehensive Nephrology (5th edition)
• Teşekkür
– Alexion firmasının görselleri için