spin polarized version of dft & accuracy studies of dft
TRANSCRIPT

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 1
Material for these notes follows closely a lecture provided by Prof. G. Ceder
for MIT’s Atomistic Materias Moldeling course.
MIT’s 3.320 course notes (Prof. G. Ceder)
SPIN POLARIZED VERSION OF DFT
& ACCURACY STUDIES OF DFT

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 2
SPIN POLARIZED VERSION OF DFT
In the Hohenberg-Kohn theorem, everything is expressed in terms of the charge
density and the electron spin does not appear explicitly. Electrons have
spin and if we don’t consider any coupling with the angular momentum is just
up or down spin (+) (-) ½ mB , where mB is the Bohr magnetron).
We’ll treat here spin as a scalar quantity so spin is +1/2 or –1/2 in the
appropriate units.
In reality when spin it couples with the angular momentum becomes a vector
quantity. Also when it couples to an external magnetic field it becomes a vector
quantity. But most times that you will use spin you will simply treated it as a
scalar. Spins up and down are often shown with an either up or down arrow.
We need to deal with the spin because of the Pauli exclusion principle -- two
electrons cannot be in exactly the same quantum state.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 3
SPIN POLARIZED VERSION OF DFT
That means that if you have 2 up electrons approaching each other versus an up
and a down electron, these will approach each other differently.
The 2 electrons shown above are in the same spin state so if you bring them
very close, they get the same coordinate as well so they have the same
quantum numbers and the Pauli exclusion principle prevents that. So the
Pauli exclusion principle keeps electrons with parallel spin away from
each other.
On the other hand, if you have electrons with anti-parallel spin, even if these
are at the same position, they don’t have the same spin, so they don’t have the
same quantum numbers so the Pauli exclusion principle is not violated.
The Pauli exclusion principle keeps the electrons away without an explicit
term for it in the Hamiltonian. The spin contribution comes from anti-
symmetrizing the wave function.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 4
HUND’S RULES
Consequences of the Pauli exclusion principle is Hund’s rules. For example, if
you add electrons to e.g. 5 d levels, you are going to add them first with
parallel spin because the Pauli exclusion principle is satisfied and then you will
start filling them with anti-parallel levels.
The same is true with solids. If let’s say 5 d levels remain degenerate (at the
same level or with the split not too large), you are going to fill them according
to Hund’s rule:
In this case you have a strong magnetic moment (5 electron with spins up with
no down spin).
Strong magnetic moment

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 5
SPIN POLARIZED VERSION OF DFT
To fill these levels with 5 electrons with parallel spin you would have to put them
on the top two separated levels but these levels are so much higher.
Instead, you can pay the Hund’s rule penalty and put the 4th and 5th electrons on
a lower orbital with anti-parallel spin.
On the other hand, let’s say, you are in an environment that splits off 2 levels.
Filling of different
orbitals gives the atom
different chemical properties Net spin (magnetic moment)
Levels split

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 6
SPIN POLARIZED VERSION OF DFT
Whether you get a lot of magnetic spin and magnetic moment, depends on how
much your orbital will split in the end.
This can have significant consequences on the physical properties of your
material.
So in the DFT calculation you will carry a magnetic moment locally when the up-
density and the down-density are not the same.
In our DFT calculations, the energy and the potential are functions of the charge
density. So the charge density somehow needs to contain the information
about electron spin and magnetic moment.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 7
LOCAL SPIN DENSITY APPROXIMATION (LSDA)
In principle Exc[ρ] “knows” about this effect, but in practice it is poorly
approximated since only the total charge density is a variable.
For a given charge density there would probably be a certain amount of spin
polarization. This should be in the functional Exc[ρ].
We treat the up and the down densities separately. So you do the local spin
density approximation which is the spin polarized version of the local density
approximation (LSDA). There is also a spin-polarized version of GGA.
You have a separate density for the up electrons and a separate density for the
down electrons and the two will interact differently, i.e. the down will interact
differently with up than the up with down. This comes from the way the
exchange correlation potentials are defined.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 8
LOCAL SPIN DENSITY APPROXIMATION (LSDA)
The idea is similar to restricted and unrestricted Hartree-Fock.
If you have spin polarized materials it’s often much more useful to look at
spin densities than at charge densities. Recall from a figure in an earlier
lecture that you don’t see where the electrons are or any fine structure of
bonding when you just look at charge densities.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 9
Plots of charge density
You see a lots of intensity from the Oxygen layers -- valence electrons only (the
O2-). Co has somewhat less valence electrons on it and you see less intensity.
Li is ionized to Li+ so it has no valence electrons and you see almost nothing.
Here is an example of
Lithium Cobalt Oxide
(LiCoO2) , a transition
metal oxide.
Its a layered material,
with layers of Oxygen
(red), layers of Cobalt
(black) and layers of
Lithium (yellow).
If you plot the charge
density in a plane you
only see big blobs. red is the neutral color in these figures –
i.e. its zero
MIT’s 3.320

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 10
Spin polarization density
This Figure of charge density does not give you a lots of detail. If you rather plot
the spin polarization density (up minus down), this will indicate how much
magnetic moment there is locally.
MIT’s 3.320

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 11
SPIN POLARIZATION DENSITY
The Figure here is similar to the earlier one but with Nickel and Manganese ions
in it.
Note that you do not see the oxygen at all since oxygen doesn’t have spin (it’s a
filled shell, every time you have filled shells, you don’t have spin).
Plots of spin density often allow you to filter certain ions. For example, the
transitional metals don’t have spin on them and you see that clearly. This is a
useful trick that you should keep in mind.
Oxygen
MIT’s 3.320

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 12
SPIN POLARIZED VERSION OF DFT
Spin polarization can be important (in
particular for transition metal compounds)
If transition metal oxides, most of the time you’ll get the structure right but things
get more subtle. A transition metal has local d states that often have
significant spin polarization. So you need to turn on spin polarization.
It often gets worse. Remember that your spin is a scalar (up or down). Now you
have a spatial degree of freedom of how to organize that spin on the ions. If
you have a bunch of ions, you could put them all with the same direction, that’s a
ferromagnet. Or you could put them with alternating directions as a kind of anti-
ferromagnet.
There are many ways to make them anti-ferromagnetic. And unfortunately in
transition metal oxides it often matters because there is not only a strong spin
polarization effect on the energy but there’s a strong effect of the interaction
between spins on different ions. Lets see an example next.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 13
SPIN POLARIZED VERSION OF DFT
If you do a non spin-polarized calculation, you get a huge error, C2/m is lower in
energy by 250 meV. If you turn on spin-polarization but make them
ferromagnetic, they are degenerate. If you make them anti-ferromagnetic,
Pmmm is the lowest in energy.
This is the
structure of
Lithium
Manganese
Oxide (LiMnO2)
MIT’s 3.320
It is shown here a comparison
between 2 structures labeled by
their symmetry. One is C2/m and
one is Pmmn.
There are rather similar structures
but one is orthorombic and the
other is monoclinic. The correct
real crystal structure is Pmmn.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 14
SPIN POLARIZED VERSION OF DFT
If you take this material at room temperature, it’s paramagnetic and thus you
may think that it does not have any spin polarization, since there’s no net
moment. That’s wrong because a paramagnet still has a local moment, the
ions still have a moment on them that is just randomly oriented. So you still
need to represent that local moment. This is a very common mistake in these
type of calculations.
MIT’s 3.320

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 15
SPIN POLARIZED VERSION OF DFT
It turns out that’s the biggest effect on the energy is the fact that you have that
local moment. It’s not necessarily how they are arranged. How they are
arranged makes you go from the difference between the 2 structures in the Ferro
to the difference in the A-Ferro configuration (which are small). However, by
turning on the local moment makes you go from the difference in the NSP to the
difference e.g. in A-Ferro (which is large).
It is only non-magnetic
materials or diamagnetic
materials for which
you don’t really need
spin polarization.
MIT’s 3.320

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 16
Orbital filling depends on spin polarization
This effect is important because if you spin-polarize an ion you fill different
orbitals. This is shown below for 5 d-orbitals.
This is typically how they split in most oxides. Every time an ion is octahedral,
5d orbitals tend to split as 3 down-2 up, in some cases 2 down-3 up.
MIT’s 3.320

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 17
Orbital filling depends on spin polarization
By spin polarizing you create essentially a different ion. It’s not really an issue of
magnetism because magnetic effects seem to be small in materials. But it’s the
fact that you create chemically a different ion because you fill different levels.
That’s really why these energy differences are so big.
Let’s say you have to put 4
electrons in there. You can put
them on what’s called high
spin or in low spin with no
moment. These 2 ions have
different chemical properties
because the electrons occupy
different d-orbitals. The high
spin orbital points in a
different direction than the
low-spin orbital.
MIT’s 3.320
CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 18
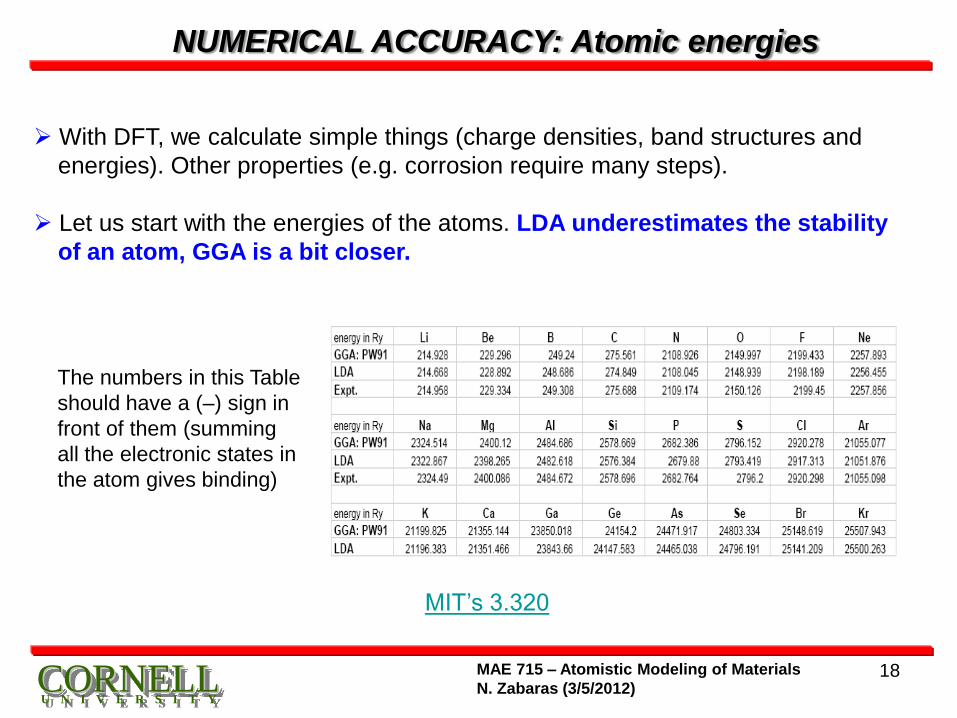
NUMERICAL ACCURACY: Atomic energies
With DFT, we calculate simple things (charge densities, band structures and
energies). Other properties (e.g. corrosion require many steps).
Let us start with the energies of the atoms. LDA underestimates the stability
of an atom, GGA is a bit closer.
MIT’s 3.320
The numbers in this Table
should have a (–) sign in
front of them (summing
all the electronic states in
the atom gives binding)

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 19
ENERGIES OF ATOMS
The Table provides the experimental numbers, the LDA number and then the
GGA with a fairly recent implementation of the exchange correlation function.
There is a very systematic trend in the data. In the LDA, in the atoms, the
electrons are not bound enough, in the GGA they are somewhat closer to
the experiment.
MIT’s 3.320

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 20
BINDING ENERGY FOR SMALL MOLECULES
When you look at small molecules, the binding energy is too high in LDA,
GGA is closer but sometimes the bound too weak. Hartree-Fock is way off.
Here we compute the binding energy for diatomic
molecules, i.e. the energy to pull them apart. We
have a molecule AB with r the vector between them.
We are looking at the well depth in order to
compute the binding energy.
MIT’s 3.320

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 21
ERROR CANCELLATION IN DFT
Suppose you are interested in an oxidation reaction and you want to calculate
the state of an oxide and compare the chemical potential of the Oxygen there to
that of Oxygen gas.
Aluminum + Oxygen Aluminum Oxide
We just showed that there is a big error in the Oxygen gas. How much of that
error carries over to the solid?
If we make exactly the same error in the solid, then the reaction energy is
perfect because we subtract the two.
A lot of practical things you do with density functional theory depend on error
cancellation. You will have more error cancellation as the states that you
subtract are more physically similar.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 22
BINDING ENERGY FOR SMALL MOLECULES
As an example, for H2 the LDA binding is too strong (-4.913). With the GGA,
the binding is too weak (-4.540). The experimental value is -4.753 eV.
Uncorrected Hartree-Fock in also shown here.
For O2, the experimental binding energy is only –5.2 ev. LDA binds it by -7.5 ev
(more than 2 ev off). GGA gets you a little closer. Uncorrected Hartree-Fock is
off.
Uncorrected Hartree-Fock should not be used at all as the binding energy is
way off.
MIT’s 3.320

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 23
ERROR CANCELLATION IN DFT
But let’s say that you look at oxidation of a metal:
Aluminum + Oxygen Aluminum Oxide
The Oxygen in Aluminum Oxide is very different from the Oxygen in the O2
molecule so not all the error will cancel.
Let’s say that we had a 2 ev error in the molecule, this is 1 ev per Oxygen.
That’s an enormous effect.
Note that the chemical potential m (which relates directly to the energy) is given
in terms of the partial pressure of Oxygen as: m = mo + RT log P.
If e.g. you want to calculate the partial pressure of Oxygen at which something
oxidizes, the Oxygen pressure goes exponentially with the energetics. So, if
you have a 1 ev error at room temperature, your error in the Oxygen pressure is
the exponential of 1 ev/RT, which is quite high.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 24
ERROR CANCELLATION IN DFT
When we look at reactions between solids most of the error tends to cancel and
we get much better accuracy. Note that 1eV error in our reactions energy is
very significant.
The point to emphasize again is that we get less error cancellation as the states
we are comparing are different, e.g. going from a gas to a solid is a significant
difference.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 25
Lattice parameters in solids
The GGA results are always bigger. GGA underbinds but generally the GGA
error is more variable – less predictable. In metals you tend to be most of the time
on the high side.
An LDA lattice parameter that agrees with the experiment is usually based on a
wrong calculation!
Lattice parameter prediction for
solids is consistent with what we
see in molecules. The LDA
values are almost always
smaller than the experimental
predictions (by a factor of
1%~2%).
LDA overbinds.
MIT’s 3.320

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 26
BULK MODULUS IN SOLIDS
In almost all cases (except Si), the LDA bulk modulus is too large, so the material
is too stiff. Recall that LDA over-binds. LDA tends to be too stiff. GGA is too soft.
Bulk Modulus in Solids (in GPA)
Remember that when the bonding energy is too high, the lattice parameter is too
small and the material is too stiff.
Bulk modulus effects will also transfer into vibrational frequencies. In materials
when you are too stiff, you’ll have higher vibrational frequecies.
MIT’s 3.320

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 27
LATTICE PARAMETERS IN OXIDES
Here are the lattice parameters for oxides. Note that the errors tend to be
slightly larger.
MIT’s 3.320

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 28
SUMMARY OF GEOMETRY PREDICTION
LDA under-predicts bond lengths
GGA error is less systematic though over-prediction is common.
errors are in many cases (e.g. semi-conductor metals) < 1%,
for transition metal oxides < 5%.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 29
STRUCTURE PREDICTION AND REQUIRED ENERGY
You often want to know if you put energy in a particular arrangement is that
lower in energy than some other arrangement? This way you can predict what
the most stable structure. This could be applicable for crystal structure or
surfaces.
For Vanadium, the atomic energy (the energy of all its electrons and not just the
valence electrons) in Rydbergs is given below assuming FCC and BCC
configurations.
The first line is the atom (not in a solid),
the 2nd line is the fcc ion, and the 3nd
line is the bcc ion.
Look at the differences. If you are going from the atom to the solid, your first
4 digits don’t even change!
The Fcc-Bcc difference is 0.02 Ry,
only 0.001% of the total energy.
In many cases we are going to work with
energy differences that are 10-6 or 10-7
times the total energy. This is telling you
how much numerical accuracy we need.
Mixing energies are also of order 10-6
fraction of the total E

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 30
COHESIVE ENERGY
Cohesive energy for V is 0.638 Ry (0.03% of total E)
The cohesive energy is only 0.03% of the total energy. If you are calculating the
cohesive energy by first calculating the total energy of a solid and then
calculating the atomic energy, you will need to do these things very accurately
because you are going to subtract them and most of what you subtract is nearly
the same.
To get any significance in the results, you need to have high numerical
accuracy.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 31
MIXING ENERGIES
Mixing energies are also of order of 10-6 fraction of the total E
Suppose you mix Vanadium with Platinum and you want to know what is the
mixing entropy (that sets the whole temperature scale for mixing and the phase
diagram topology). The mixing energy tends to be a 10-6 to 10-7 times of the
total energy.
Unfortunately, the total energy is not accurate up to a fraction of 10-6. The only
reason we can get physical behavior right is because a lot of the error DFT
makes in LDA and GGA is systematic. A lots of it cancels away when you take
energy differences.
When we do fcc and bcc Vanadium, we may have an error of 10-4 in the energy
but most of it cancels away when we take the energy differences. The less
cancellation we have, the bigger the error on the result.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 32
ELEMENT CRYSTAL STRUCTURE PREDICTIONS: bcc or fcc?
The comparison here is done in GGA with standard pseudopotential method. In
red we have metals that are experimentally fcc and in green we have metals
that are experimentally bcc.
We show the calculated
energy difference
between bcc and fcc.
When that is positive the
bcc energy is higher than
fcc so fcc is preferred
over bcc. If it’s negative
then bcc is preferred over
fcc.
All results are correct.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 33
ELEMENT CRYSTAL STRUCTURE PREDICTIONS: hcp or fcc?
You could do a more
subtle comparison,
look at the difference
between hcp and
fcc. hcp and fcc are
much more alike --
they are all close
packed. It’s just a
difference in
stacking: abab
versus abcabc.
Again you see that all results are correct. The red ones are the fcc ones
(first line is positive). The yellow ones have the 1st number negative.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 34
REACTION ENERGIES
Lets discuss reaction energies.
For example, a metal Lithium bcc with another metal Aluminum forming a Li-Al
compound. The LDA is 10% of from the experimental reaction energy. Metallic
reaction energies are somewhere in the range 5 to 15%.
The one that is next is Copper-Gold. This is a notable exception where
you are over 50% off.
Most of them it’s much simpler and in metals you tend to get very good
reaction energies.
MIT’s 3.320

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 35
COMPARING ENERGY OF STRUCTURES
For most elements, both LDA and GGA seem to predict the correct
structure for a material
Notable exceptions: Fe is FCC in LDA, not BCC. In GGA, that is correct.
Materials with substantial electron correlation effects (e.g. Pu)
High Throughput studies are now possible.
http://datamine.mit.edu/htap/alloys.jsp
If you go deep down in the periodic table, especially f-electron metals you start to
see failures of LDA and GGA. An important ion is Plutonium.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 36
REDOX REACTIONS: Reaction of an oxide with a metal
The errors become bigger in redox reactions. The 3 reactions shown are
essentially a reaction between an oxide (phosphate) with a metal.
The reaction energies are not considerably off.
GGA gives you 2.8 eV for the first reaction, the experiment is 3.5. The third
reaction which is very similar gives an error of 30%. You get 3.3 eV, the
experiment is 4.6. Why is that?
MIT’s 3.320

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 37
REDOX REACTIONS
It has to do with a lack of error cancellation.
You need to look in detail what happens to the electronic structure in these
materials. These are redox reactions. If you do the math on the valences (lets
say for the first reaction), the iron on FePO4 is Fe3+, Phosphorous is P5+ and
Oxygen is O2-. Iron in LiFePO4 is Fe2+ and Lithium is Li1+. What has happened
in this reaction?
MIT’s 3.320

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 38
Transferring electrons from a delocalized to a localized state
All these reactions involve the transfer of an electron from a
delocalized state in Li metal to a localized state in the transition
metal oxide (phosphate)
You’ve taken essentially an electron from Lithium in its metallic state (and in the
process ionize Lithium) , and put it on the Iron Fe3+ to make Iron Fe2+.
That electron in Lithium (alkaline metal) is in a wide delocalized orbital and you
are transferring it to a localized d-state on the iron. That electron is essentially
being transferred between extremely different states. You essentially transfer
between such different states that you start losing the cancellation of errors that
you need in DFT.
In particular, the error here comes from what we call self-interaction error.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 39
SELF INTERACTION ERROR
Remember that in DFT, we reduce the problem to 1-electron equations where you
have the kinetic energy , the nuclear potential and then the effective
potential Veff(ri)

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 40
Self-interaction effect in DFT
The effective potential has the exchange correlation in it but also has the Hartree
field i.e. the Coulombic field from all the electrons. That field includes the
electron itself.
When you calculate the charge density that’s the charge density of all the
electrons. When you calculate the potential coming from that charge density,
that’s the potential coming from all the electrons but you operate that now on a
single electron.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 41
SELF INTERACTION ERROR IN DFT
The electron is feeling its own potential. Part of the exchange correlation term
corrects for that but not all of it. And the problem is that the correction does not
operate as well on different forms of the charge density.
In a metal you have a small self-interaction error and the reason is that a state
in a metal is sort of very delocalized -- has very spread out charge density. If
you want to think of it, part of the electron in one of the locations marked in the
figure below doesn’t feel much of the potential coming from the other marked
piece of the charge density because they are very far away.
X X

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 42
SELF INTERACTION ERROR IN DFT
However, if you consider a very localized state, the potential from the electron is
very high where the electron itself is sitting because it’s all very close. If you put
an electron in a Delta function, if you could, if you didn’t have an uncertainty
principle and you calculated its potential it’s basically sitting on top of itself.
Then you would have an infinite self-interaction.
So, the more local the state is, the more self-interaction you have and the
exchange correlation functional can’t quite correct these 2 in the same
way.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 43
SPIN POLARIZED VERSION OF DFT
Remember that the exchange correlation correction comes from homogeneous
charge densities. So it tends to correct the metallic state better than the
localized state.
This is why the redox reactions showed a big error because we were
transferring from a state that was metallic, the electron went from the Lithium
state to the transitional metal state. Somehow the self-interaction error
doesn’t cancel.
You will see things like that whenever you transfer electrons between
quite different states.

CCOORRNNEELLLL U N I V E R S I T Y
MAE 715 – Atomistic Modeling of Materials
N. Zabaras (3/5/2012) 44
LDA Vs. GGA: Summary of results for property calculation
Summary (LDA)
Lattice constants: 1-3% too small
Cohesive Energies: 5-20% too strongly bound
Bulk Modulus: 5-20% (largest errors for late TM)
Bandgaps: too small
GGA gives better cohesive energies. Effect on lattice
parameters is more random. GGA important for magnetic systems
When you work in metals and semi-conductors (which tend to be fairly
delocalized states) LDA and GGA do quite well for the lattice parameters and
even cohesive energies.
In more complicated materials, we have learned more about the errors of LDA
and GGA.
There is a series of methods under development to deal better with the
correlation energy and the self-interaction energy to solve these problems of
both energetics and also electronic structure.