srebp-1 mediates angiotensin ii-induced tgf- 1...
TRANSCRIPT

BASIC RESEARCH www.jasn.org
SREBP-1 Mediates Angiotensin II-Induced TGF-b1Upregulation and Glomerular Fibrosis
Tony N. Wang, Xing Chen, Renzhong Li, Bo Gao, Zahraa Mohammed-Ali, Chao Lu,Victoria Yum, Jeffrey G. Dickhout, and Joan C. Krepinsky
Division of Nephrology, McMaster University, Hamilton, Ontario, Canada
ABSTRACTAngiotensin II is an important mediator of CKD of diverse etiology. A common pathologic feature of CKD isglomerular fibrosis, a central mediator of which is the profibrotic cytokine TGF-b. The mechanisms underlyingthe inductionof TGF-b andmatrixbyangiotensin II arenot completelyunderstood.Recent studies showed thatoverexpression of the transcription factor SREBP-1 induces glomerular sclerosis and that angiotensin II canactivate SREBP-1 in tubular cells.We thus studiedwhether SREBP-1 is activated by angiotensin II andmediatesangiotensin II–induced profibrogenic responses in primary rat mesangial cells. Treatment of cells with angio-tensin II induced the upregulation and activation of SREBP-1. Angiotensin II–induced activation of SREBP-1required signaling through the angiotensin II type I receptor and activation of PI3K/Akt in addition to thechaperone SCAP and protease S1P. Notably, angiotensin II-induced endoplasmic reticulum stress was identi-fied as a key mediator of Akt-SREBP-1 activation, and inhibition of endoplasmic reticulum stress or SREBP-1prevented angiotensin II–induced SREBP-1 binding to the TGF-b promoter, TGF-b upregulation, and down-stream fibronectin upregulation. Endoplasmic reticulum stress alone, however, did not induce TGF-b upregu-lationdespiteactivatingSREBP-1.Althoughnot required forSREBP-1activationbyangiotensin II,EGFreceptorsignaling was necessary for activation of the SREBP-1 cotranscription factor Sp1, which provided a requiredsecond signal for TGF-b upregulation. In vivo, endoplasmic reticulum stress and SREBP-1-dependent effectswere induced in glomeruli of angiotensin II-infused mice, and administration of the SREBP inhibitor fatostatinprevented angiotensin II–induced TGF-b upregulation and matrix accumulation. SREBP-1 and endoplasmicreticulum stress thus provide potential novel therapeutic targets for the treatment of CKD.
J Am Soc Nephrol 26: 1839–1854, 2015. doi: 10.1681/ASN.2013121332
Angiotensin II (AngII) is amajor pathogenic factor inthe progression of both diabetic and nondiabeticCKD. Glomerular sclerosis is a characteristic patho-logic featuremarked by extracellular matrix accumu-lation. Mesangial cells (MCs) play a key role in thisprocess. AngII induces MCs to produce the profi-brogenic cytokine TGF-b and matrix proteins, suchas fibronectin and collagen. AngII inhibitors, how-ever, are not completely effective in preventing dis-ease progression and are not tolerated by all patients.Improved understanding of howAngIImediates kid-ney fibrosis will facilitate the development of newtherapeutic agents.
Sterol regulatory element-binding proteins(SREBPs) are transcription factors most extensivelystudied in lipid homeostasis, but data suggest anadditional important role inmatrix regulation. Three
isoforms exist: SREBP-1a/1c, generated from alter-nate transcription start sites, and SREBP-2.1 In itsinactive precursor form, SREBP resides in the endo-plasmic reticulum (ER) membrane. When intracel-lular cholesterol is low, SREBP cleavage-activatingprotein (SCAP) escorts SREBP to the Golgi for se-quential cleavage by proteases S1P and S2P to releasethe N-terminus. This mature transcription factor
Received December 22, 2013. Accepted September 23, 2014.
Published online ahead of print. Publication date available atwww.jasn.org.
Correspondence: Dr. Joan C. Krepinsky, St. Joseph’s Hospital,50 Charlton Avenue East, Room T3311, Hamilton, ON L8N 4A6,Canada. Email: [email protected]
Copyright © 2015 by the American Society of Nephrology
J Am Soc Nephrol 26: 1839–1854, 2015 ISSN : 1046-6673/2608-1839 1839

translocates to the nucleus as a dimer and binds promoters oftarget genes at sterol regulatory element sequences.1,2 Non–sterol-mediated SREBP activation through the same pathwayshas also been described, such as with shear stress, glucose, andgrowth factors.3–6
Renal overexpression of active SREBP-1a or -1c inducedglomerular sclerosis with upregulation of TGF-b, fibronectin,and collagen.7,8 Although SREBP-1 activation was recentlyobserved in tubular epithelium of AngII-infused rats,9
whether it contributes to glomerular fibrosis remains un-known. AngII effects are primarily mediated by the type 1receptor (AT1R) through coupling to G proteins.10 Down-stream signaling molecules include PI3K and Akt, which pos-itively regulate SREBP activation.1 AngIIalso cross-talks with EGFR,10 which weshowed mediates glucose-induced SREBP-1activation.4 In MCs, both PI3K/Akt andEGFR signaling regulate TGF-b1 synthesisby AngII,11,12 but whether SREBP-1 is alsorequired is unknown.
ER stress is emerging as an importantpathophysiologic factor in CKD of variouscauses.13 A response to disruption of ERhomeostasis, it is characterized by activa-tion of three signaling proteins regulated bythe master chaperone GRP78, namelyATF6, IRE1, and PERK. These upregulatechaperones, attenuate translation throughphosphorylation and inhibition of thetranslation-initiating factor eIF2-a, and, ifprolonged, activate proapoptotic signalingmediators.14,15 Interestingly, SREBP-1contains an internal ribosome entry site,allowing its translation despite the overallreduction in protein synthesis.16 AlthoughER stress may activate SREBP and was im-plicated in AngII-induced cardiac pathol-ogy,17–19 whether AngII induces ER stressin MCs and the kidney is unknown.
We thus studied the role of SREBP inAngII profibrotic effects in MCs. Our dataidentify ER stress/Akt-mediated SREBP-1activation by AngII as a novel mediator ofTGF-b1 and matrix upregulation. SREBPinhibition in vivo prevents AngII-inducedglomerular TGF-b and matrix synthesis,providing a promising potential therapeu-tic target for the treatment of CKD.
RESULTS
AngII Activates SREBP-1To determine whether AngII activatesSREBP-1 in MC, we detected appearance
of the mature 68-kD form by immunoblotting. This was seenwithin 1 hour in both nuclear (Figure 1A) and total cell (Sup-plemental Figure 1A) lysates. Subsequent studies assessedSREBP-1 in total cell lysate, with AngII for 3 hours, sinceactivation was most consistent at this time. While increasingconcentrations of AngII further increased SREBP-1 activation(Supplemental Figure 1B), all further studies used 100 nM.Wenext confirmed functionality of SREBP-1 activation by AngIIusing a plasmid in which green fluorescent protein (GFP)expression is under control of the SRE (Figure 1B),20 withcorresponding immunofluorescence images in SupplementalFigure 1C. The well established downstream target fatty acidsynthase (Figure 1C) and lipid synthesis, assessed byOil RedO
Figure 1. AngII activates SREBP-1. (A) MCs were treated with AngII (100 nM) for theindicated times, and SREBP-1 activation was assessed by immunoblotting of nuclearextracts for the cleaved, mature form (mSREBP-1) (†P,0.01 control (con) versus others,n=5). (B) MCs were transfected with the SREBP responsive plasmid SRE-GFP and treatedwith AngII for 1 or 3 hours. GFP production, driven by SREBP activation, was quantifiedby fluorescence measurement (*P,0.05 con versus others, n=8). The immunoblot showscell lysates probed for GFP under the same conditions. (C) Upregulation of the SREBP-1target gene FAS by AngII was assessed after treatment for 6 hours (‡P,0.01 AngII versuscon, n=3). (D) Intracellular lipid accumulation was assessed by Oil Red O (ORO) stainingafter AngII for 24–48 hours (†P,0.01 versus con, n=3).
1840 Journal of the American Society of Nephrology J Am Soc Nephrol 26: 1839–1854, 2015
BASIC RESEARCH www.jasn.org

staining (Figure 1D, Supplemental Figure1D), were also increased by AngII.
AT1R/PI3K/Akt Signaling, but NotEGFR, Are Required for SREBP-1Activation by AngIIWe first established that AngII-inducedSREBP-1 activation was mediated by theAT1R (Figure 2A). PI3K/Akt activation isdownstream of the AT1R10 and enableSREBP-1 activation in other settings.4,5 Fig-ures 2, B and C show that PI3K and Akt in-hibition prevented AngII-induced SREBP-1activation. In some cases Akt may signalthrough the mammalian target of rapamy-cin complex-1 to activate SREBP-1.21 Itsinhibitor rapamycin, however, did not pre-vent AngII-induced SREBP-1 activation(Figure 2D). This suggests that Akt may di-rectly regulate SREBP-1, as shown in re-sponse to insulin in hepatocytes,5 or mayfunction through other downstream regu-lators such as GSK-3b.22,23
AngII transactivation of the EGFR me-diates TGF-b upregulation inMC.11,12 Sur-prisingly, two different EGFR inhibitorsdid not affect SREBP-1 activation (Figure2, E–G). Akt activation was also not signif-icantly decreased (Figure 2, E, F, and H).Thus, our data suggest that SREBP-1 acti-vation by AngII depends on PI3K/Akt sig-naling through the AT1R but might beindependent of EGFR transactivation.
Figure 2. AT1R/PI3K/Akt signaling, but not EGFR, is required for SREBP-1 activation.MCs were treated with various inhibitors before AngII, and mature SREBP-1 was as-sessed by immunoblotting. (A) The AT1R inhibitor losartan (10 mM, 30 minutes)
prevented SREBP-1 activation (*P,0.05 ver-sus con, #P,0.05 versus Ang, n=6). (B) Twostructurally distinct PI3K inhibitors—LY294002(20 mM, 30 minutes) or wortmannin (100 nM,60 minutes)—blocked SREBP-1 activation( †P,0.01 versus con; #P,0.01 versus Ang;∧P,0.05 versus Ang, n=5), as did the Akt in-hibitor VIII (10 mM, 1 hour) shown in C (*P,0.01versus con, #P,0.05 versus Ang, n=6). (D)SREBP-1 activation was unaffected by themTORinhibitor rapamycin (20 ng/ml, 1 hour). (E and F)Two EGFR inhibitors—AG1478 (1 mM, 30 mi-nutes) and PD168393 (0.5mM, 30minutes)—didnotblockSREBP-1activation. Immunoblots showeffective inhibitionof EGFRactivation, as assessedby its autophosphorylationonY1068, andminimalinhibition of Akt activation, as assessed by itsphosphorylation on S473. Quantitative changesare shown in the accompanying bar graphs(G and H). For SREBP-1, *P,0.01 versus con(n=6); for pAkt, *P,0.01 versus con only (n=8).
J Am Soc Nephrol 26: 1839–1854, 2015 SREBP in Kidney Fibrosis 1841
www.jasn.org BASIC RESEARCH

AngII-Induced SREBP-1 Activation Requires SCAP andSerine ProteasesCholesterol-regulated activation of SREBP-1 involves SCAP-mediated translocation to the Golgi for cleavage to the maturetranscription factor by proteases S1P/S2P.2 However, SREBPcleavage may also be mediated independently by other en-zymes.24–26 We first tested the SCAP inhibitor fatostatin. ThispreventedAngII-induced SREBP-1 activation (Figure 3A), as did
the S1P inhibitor AEBSF (Figure 3B). As expected, fatostatin alsoprevented SREBP transcriptional activity (Supplemental Figure 2).In vivo, AngII-induced glomerular SREBP-1 induction, withnuclear localization suggesting its activation, was attenuated byfatostatin (Figure 3C). This was confirmed by immunoblottingof renal cortical lysate (Figure 3D). Localization of SREBP-1 toMC was further shown by coimmunofluorescence for SREBP-1and a-smooth muscle actin (Supplemental Figure 3).
Figure 3. SREBP-1 activation requires SCAP and serine proteases. (A) The SCAP inhibitor fatostatin (20 mM, 4 hours) prevented SREBP-1activation by AngII (*P,0.001 versus con, #P,0.001 versus Ang, n=6). (B) SREBP-1 activation was similarly inhibited by the S1P proteaseinhibitor AEBSF (500 mM, 1 hour) (*P,0.001 versus con, #P,0.001 versus Ang, n=5). (C) Cortical sections from AngII-infused mice werestained by IHC for SREBP-1 (at 403 magnification). These show increased glomerular SREBP-1 expression. Nuclear localization supportsSREBP-1 activation. Quantification is shown in the accompanying graph (∧P,0.001 versus Ang, *P,0.001 versus con, #P,0.001 versus Ang).(D) Immunoblotting of protein from kidney cortex confirms that AngII increases SREBP-1 activation, as assessed by appearance of its matureform, and that this is inhibited by fatostatin (∧P,0.001 versus Ang, *P,0.01 versus con, #P,0.05 versus Ang).
1842 Journal of the American Society of Nephrology J Am Soc Nephrol 26: 1839–1854, 2015
BASIC RESEARCH www.jasn.org

ER Stress Mediates SREBP-1 Activation by AngIIAngII induces ER stress in cardiomyocytes and heart diseasemodels,18,19,27 but whether this occurs in MCs is unknownand was first assessed. AngII increased GRP78 expression(3 hours) and eIF2a phosphorylation (30 minutes), twomarkers of ER stress (Figure 4A), both of which required theAT1R (Figure 4B). EGFR inhibitors did not block ER stress,further supporting independence of SREBP-1 activation fromthe EGFR (Supplemental Figure 4). Induction of ER stress byAngII was also seen in vivo in glomeruli (Figures 4, C and D).
As shown in other models,17 ER stress itself led to SREBP-1activation (Figure 4E). Furthermore, ER stress inhibition withthe chemical chaperone 4-PBA or eIF2a phosphatase inhibi-tor salubrinal prevented AngII-induced SREBP-1 activation(Figures 4, F andG) and its transcriptional activity (SupplementalFigure 5). We then determined whether there was crosstalk be-tween PI3K/Akt and ER stress, both required for AngII-inducedSREBP-1 activation. ER stress induced Akt activation, and thiswas PI3K-dependent (Figures 5, A andB, Supplemental Figure 6).Furthermore, AngII-induced Akt activation was attenuated by
Figure 4. ER stress mediates SREBP activation by AngII. (A) ER stress is induced by AngII, as assessed by upregulation of GRP78 andphosphorylation of eIF2a. (B) This is mediated by the AT1R, as both GRP78 induction (AngII, 3 hours) and eIF2a phosphorylation (AngII,30 minutes) were prevented by losartan (10 mM, 30 minutes) (for GRP78, *P,0.01 versus con, #P,0.01 versus Ang, n=5; for pEIF2a,*P,0.01 versus con, #P,0.01 versus Ang, n=3). (C and D) AngII infusion induced glomerular GRP78 upregulation, as assessed by IHC(*P,0.05, 603 magnification) and by immunoblotting of total cortical lysate (*P,0.05). (E) ER stress induction with tunicamycin (Tm,500 ng/ml, 1 hour) or thapsigargin (Tg, 200 nM, 1 hour) induced SREBP-1 activation in MCs (*P,0.001 versus control, n=3). (F and G)SREBP-1 activation by AngII was blocked by two ER stress inhibitors: 4-PBA (5 mM, 2 hours) and salubrinal (30 mM, 1 hour). (F) *P,0.001versus con; #P,0.01 versus Ang, n=6. (G) *P,0.01 versus con, #P,0.01 versus Ang, n=4.
J Am Soc Nephrol 26: 1839–1854, 2015 SREBP in Kidney Fibrosis 1843
www.jasn.org BASIC RESEARCH

several ER stress inhibitors (Figure 5C). Taken together, thesedata suggest that AngII-induced ER stress mediates SREBP-1activation at least partially through Akt.
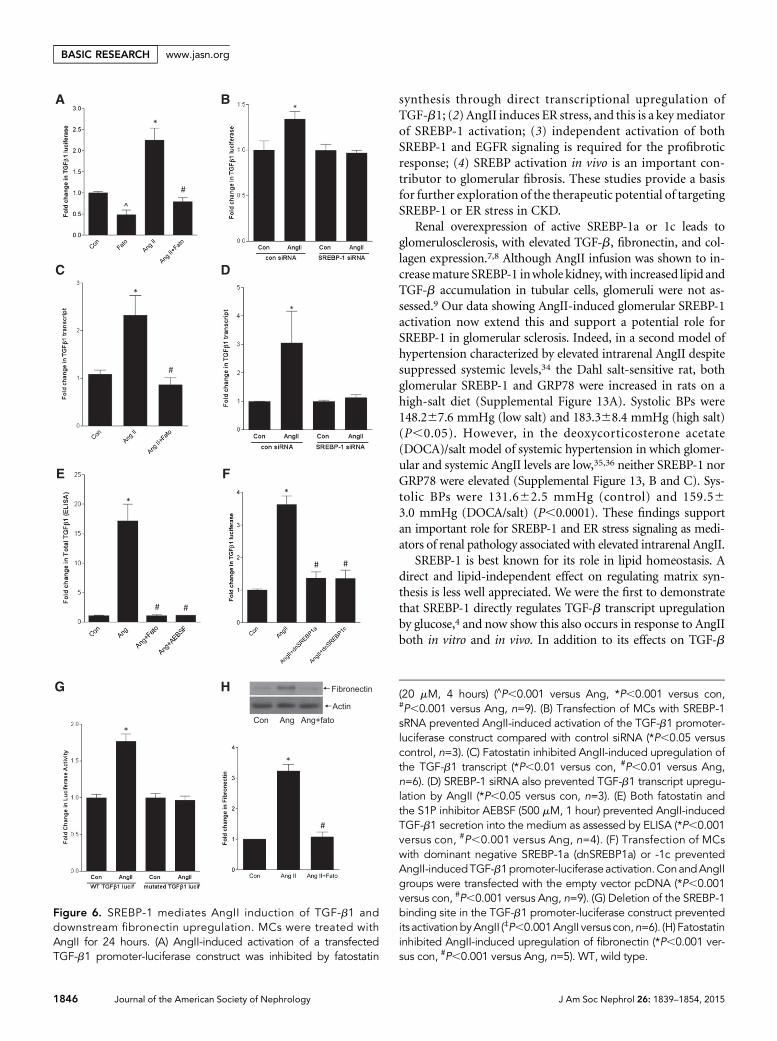
SREBP-1 Mediates AngII Upregulation of TGF-b1 andFibronectin SynthesisWenext askedwhether SREBP-1mediates AngIIupregulation ofthe well established target TGF-b1.11,12 Both SCAP and S1P in-hibition prevented activation of a TGF-b1 promoter-luciferasereporter by AngII (Figure 6A, Supplemental Figure 7A), as diddownregulation of SREBP-1 with siRNA (Figure 6B). Supple-mental Figure 7B shows efficacy of SREBP-1 siRNA. TGF-b1transcript upregulation and its secretion into the medium alsorequired SREBP activation (Figure 6, C–E). Furthermore, over-expression of dominant negative SREBP1a-Y335A or SREBP1c-Y321A prevented promoter activation (Figure 6F). Finally,deletion of the SREBP-1 binding site in the TGF-b1 promoterreporter prevented its activation by AngII (Figure 6G).
It is well known that AngII activates Smad3, the canonicaldownstream mediator of TGF-b1 signaling. Although early
activation (within minutes) is TGF-b indepen-dent, requiring Erk and p38, later activation isTGF-b dependent.28,29 Because we showed thatSREBP-1 mediates TGF-b1 upregulation byAngII at 24 hours, it should alsomediate down-stream signaling. To verify this, we assessedSmad3 transcriptional activity using theCAGA12-luciferase reporter after 24 hours ofAngII. Supplemental Figure 8 confirms its inhi-bition by fatostatin. Figure 6H shows that upre-gulation of fibronectin, an important matrixprotein component of sclerotic glomeruli in-ducedbyTGF-b, was also preventedby fatostatin.
ER Stress Is Required for TGF-bUpregulation by AngIIFigure 7A shows that ER stress inducers alonecannot activate the TGF-b promoter. Theirtendency to reduce baseline promoter activitymay be due to translational inhibition by ERstress.30 Indeed, cycloheximide (5 mg/ml) at-tenuated promoter activity to a similar degree(not shown). Blocking ER stress with variousagents, however, inhibited AngII-induced pro-moter activation, TGF-b secretion into themedium, and fibronectin upregulation (Fig-ures 7, B–D). These data support an importantrole for ER stress–induced SREBP-1 activationin matrix upregulation by AngII but demon-strate that ER stress alone is insufficient for thisresponse. Because ER stress does, however, ac-tivate SREBP-1, we postulated that AngII sig-naling provides an important second signalthat enables TGF-b upregulation.
EGFR, but Not ER Stress, Regulates Activation of Sp1As a relatively weak transcription factor, SREBP-1 functions incoordination with cofactors, most commonly Sp1.31,32 A po-tential SREBP-binding sequence lies close to an Sp1 site within2100 bp of the start codon in the human, mouse, and ratTGF-b promoters (J04431, M57902, NM021578). Using its in-hibitor mithramycin, we first established that Sp1 was requiredfor AngII-induced TGF-b promoter upregulation (Figure 8A).Furthermore, AngII, but not ER stress, induced activation of anSp1-responsive luciferase plasmid (Figure 8B). The attenuationof baseline Sp1 transcriptional activity by ER stress may also berelated to translational inhibition because cycloheximide simi-larly decreased Sp1-luciferase activity (not shown). Finally, AngII-induced Sp1 activationwas prevented by EGFR inhibitors, butnot ER stress blockers (Figure 8, C and D), suggesting thatalthough EGFR is not required for SREBP-1 activation, it pro-vides an important second signal for TGF-b upregulationthrough its effects on Sp1. Indeed, deletion of the Sp1 bindingsite close to the SREBP-1 site in the TGF-b1 promoter preventedthe AngII response (Figure 8E).
Figure 4. Continued.
1844 Journal of the American Society of Nephrology J Am Soc Nephrol 26: 1839–1854, 2015
BASIC RESEARCH www.jasn.org

TGF-b1 Promoter Binding by SREBP-1 Is Regulated byER Stress, but Not the EGFRWeconfirmed that AngII induces SREBP-1 binding to theTGF-bpromoter by chromatin immunoprecipitation, using primersencompassing the region2230–0. This was blocked by fatostatin(Figure 9A), with no binding detected in IgG immunoprecipi-tates. ER stress induction effected a similar degree of SREBP-1binding (Figure 9B). Conversely, ER stress inhibition blockedAngII-induced SREBP-1binding, in agreementwith ourdata show-ing that ER stress is required for SREBP-1 activation (Figure 9C).
Binding was unaffected by EGFR inhibition(Figure 9D). In keeping with a coactivatorrole for Sp1, AngII, but not ER stress, alsoinduced its binding to the TGF-b1 promoter(Figure 9E). Sp1 binding was prevented byEGFR inhibition (Figure 9F). In aggregate,these data support an important role forER stress–mediated SREBP-1 activation inTGF-b upregulation by AngII, in cooperationwith EGFR-mediated Sp1 activation.
Fatostatin Attenuates AngII-InducedRenal FibrosisWe next assessed the importance of SREBP-1activation to glomerular fibrosis induced byAngII in vivo.Mice infusedwithAngII developglomerular fibrosis characterized by TGF-bupregulation and matrix deposition.33 Thesemice developed significant hypertensionthat was not attenuated by fatostatin (systolicBP: control, 129.868.3 mmHg; fatostatin,129.061.7 mmHg; AngII, 208.8615.5 mmHg,AngII/fatostatin, 210.2624.8 mmHg). Pro-teinuria was attenuated by fatostatin, althoughthere was no effect on serum creatinine(Supplemental Figures 9, A and B).
Fatostatin inhibited AngII-induced TGF-bupregulation, which colocalized to MCs (Fig-ure 10A, Supplemental Figure 10). TGF-b1transcript induction was similarly preventedby fatostatin (Supplemental Figure 11A), aswas TGF-b signaling as assessed by Smad3 ac-tivation (Figure 10, A andB). The lack of com-plete inhibition to basal levels may represent asmall contribution of TGF-b–independentactivation of Smad3 in vivo.29
TGF-b is a major profibrotic regulator.Figure 10C shows that AngII-induced matrixaccumulation, identified by periodic acid-Schiff staining, was also inhibited by fatosta-tin. Treatment similarly prevented collagenand fibronectin upregulation, determined byimmunohistochemistry (IHC), immunoblot-ting (Figure 10, C–G), and real-time PCR(Supplemental Figure 11, B and C). Supple-
mental Figure 12 shows that fatostatin also attenuated tubuloin-terstitial fibrosis. Thus, fatostatin inhibits AngII-induced SREBP-1activation in vivo, and this is associated with inhibition of TGF-bupregulation and activity and downstream matrix accumulation.
DISCUSSION
Our studies present several novel observations in MCs: (1)AngII induces SREBP-1 activation and mediates matrix
Figure 5. PI3K/Akt activation upstream of SREBP-1 activation is also regulated by ER stress.(A) ER stress induction with thapsigargin (Tg, 200 nM, 30 minutes) or tunicamycin (Tm,500 ng/ml, 30 minutes) activated Akt, as assessed by its phosphorylation on S473 (*P,0.01versus con, n=6). (B) ER stress–induced Akt activation was blocked by PI3K inhibition withLY294002 (20 mM, 30 minutes) or wortmannin (100 nM, 60 minutes). (C) AngII (30 minutes)–induced Akt activation was attenuated by the ER stress inhibitors 4-PBA (5 mM, 2 hours),TUDCA (300 mg/ml, 1 hour), and salubrinal (30 mM, 1 hour) (n=5; *P,0.01 versus con,#P,0.05 versus Ang).
J Am Soc Nephrol 26: 1839–1854, 2015 SREBP in Kidney Fibrosis 1845
www.jasn.org BASIC RESEARCH
synthesis through direct transcriptional upregulation ofTGF-b1; (2) AngII induces ER stress, and this is a key mediatorof SREBP-1 activation; (3) independent activation of bothSREBP-1 and EGFR signaling is required for the profibroticresponse; (4) SREBP activation in vivo is an important con-tributor to glomerular fibrosis. These studies provide a basisfor further exploration of the therapeutic potential of targetingSREBP-1 or ER stress in CKD.
Renal overexpression of active SREBP-1a or 1c leads toglomerulosclerosis, with elevated TGF-b, fibronectin, and col-lagen expression.7,8 Although AngII infusion was shown to in-creasemature SREBP-1 inwhole kidney, with increased lipid andTGF-b accumulation in tubular cells, glomeruli were not as-sessed.9 Our data showing AngII-induced glomerular SREBP-1activation now extend this and support a potential role forSREBP-1 in glomerular sclerosis. Indeed, in a second model ofhypertension characterized by elevated intrarenal AngII despitesuppressed systemic levels,34 the Dahl salt-sensitive rat, bothglomerular SREBP-1 and GRP78 were increased in rats on ahigh-salt diet (Supplemental Figure 13A). Systolic BPs were148.267.6 mmHg (low salt) and 183.368.4 mmHg (high salt)(P,0.05). However, in the deoxycorticosterone acetate(DOCA)/salt model of systemic hypertension in which glomer-ular and systemic AngII levels are low,35,36 neither SREBP-1 norGRP78 were elevated (Supplemental Figure 13, B and C). Sys-tolic BPs were 131.662.5 mmHg (control) and 159.563.0 mmHg (DOCA/salt) (P,0.0001). These findings supportan important role for SREBP-1 and ER stress signaling as medi-ators of renal pathology associated with elevated intrarenal AngII.
SREBP-1 is best known for its role in lipid homeostasis. Adirect and lipid-independent effect on regulating matrix syn-thesis is less well appreciated. We were the first to demonstratethat SREBP-1 directly regulates TGF-b transcript upregulationby glucose,4 and now show this also occurs in response to AngIIboth in vitro and in vivo. In addition to its effects on TGF-b
Figure 6. SREBP-1 mediates AngII induction of TGF-b1 anddownstream fibronectin upregulation. MCs were treated withAngII for 24 hours. (A) AngII-induced activation of a transfectedTGF-b1 promoter-luciferase construct was inhibited by fatostatin
(20 mM, 4 hours) (∧P,0.001 versus Ang, *P,0.001 versus con,#P,0.001 versus Ang, n=9). (B) Transfection of MCs with SREBP-1sRNA prevented AngII-induced activation of the TGF-b1 promoter-luciferase construct compared with control siRNA (*P,0.05 versuscontrol, n=3). (C) Fatostatin inhibited AngII-induced upregulation ofthe TGF-b1 transcript (*P,0.01 versus con, #P,0.01 versus Ang,n=6). (D) SREBP-1 siRNA also prevented TGF-b1 transcript upregu-lation by AngII (*P,0.05 versus con, n=3). (E) Both fatostatin andthe S1P inhibitor AEBSF (500 mM, 1 hour) prevented AngII-inducedTGF-b1 secretion into the medium as assessed by ELISA (*P,0.001versus con, #P,0.001 versus Ang, n=4). (F) Transfection of MCswith dominant negative SREBP-1a (dnSREBP1a) or -1c preventedAngII-inducedTGF-b1promoter-luciferaseactivation.ConandAngIIgroups were transfected with the empty vector pcDNA (*P,0.001versus con, #P,0.001 versus Ang, n=9). (G) Deletion of the SREBP-1binding site in the TGF-b1 promoter-luciferase construct preventedits activationbyAngII (‡P,0.001AngII versus con,n=6). (H) Fatostatininhibited AngII-induced upregulation of fibronectin (*P,0.001 ver-sus con, #P,0.001 versus Ang, n=5). WT, wild type.
1846 Journal of the American Society of Nephrology J Am Soc Nephrol 26: 1839–1854, 2015
BASIC RESEARCH www.jasn.org

synthesis, SREBP-1 may also affect matrix accumulation in al-ternate ways, such as its regulation of collagen 6a1 transcriptionin 3T3 cells.37 Plasminogen activator inhibitor-1, which inhibitsmatrix breakdown,was also identified as an SREBP-1c transcrip-tional target in adipocytes.38 These data suggest that SREBP-1 isan important regulator of organ fibrosis.
TheEGFR is awell describedmediatorofAngII-induced TGF-b upregulation.11,39
We previously showed that SREBP-1 acti-vation by glucose requires the EGFR.4 Sur-prisingly, this is not the case with AngII,despite a requirement for both the EGFRand SREBP-1 in regulating TGF-b synthe-sis. This suggested the presence of twocomplementary signals for TGF-b induc-tion. Indeed, SREBPs are knownweak tran-scription factors requiring cooperationwith other coactivators.31,32 Our studiesnow show that Sp1, downstream of theEGFR, is required for TGF-b upregulation.These data are consistent with functionalinput from both SREBP-1 and EGFR/Sp1to induce TGF-b synthesis.
AngII induces ER stress in heart andcardiomyocytes.18,19,27 Our data now iden-tify that this also occurs inMCs and glomer-uli and is an importantmediator of SREBP-1activation. Several different mechanisms bywhich ER stress activates SREBP have beenproposed, depending on cell and stimulus:17
(1) Caspase activation may lead to SREBPcleavage at a site different from S1P. How-ever, in conditions that cause ER stress with-out apoptosis, SREBP is cleaved throughS1P/S2P as we found for AngII; (2) ER stressmay decrease Insig through eIF2a-mediatedtranslational inhibition, thereby releasingSCAP/SREBP for activation; and (3) GRP78may retain SCAP/SREBP-1 in the ER throughdirect interaction, with complex release uponER stress.17 We now describe an additionalmechanism, showing that AngII-inducedER stress regulates PI3K/Akt activation. Thismay enable SREBP activation at different lev-els, including facilitating its ER-to-Golgitranslocation and protecting nuclear SREBPfrom degradation.5,22,23 How ER stress leadsto Akt activation, however, remains to beelucidated.
Recent studies suggested that ER stressmay lead to the production of extracellularmatrixandorganfibrosis in somesettings.40,41
Interestingly, alleviation of ER stress with4-PBA attenuated AngII-induced cardiacfibrosis and TGF-b upregulation.19 Our
data support an important role for ER stress in AngII-induced matrix synthesis through activation of SREBP-1.ER stress alone, however, was not profibrotic in our cells despiteits induction of SREBP-1 binding to the TGF-b promoter.This is in contrast to increased fibronectin synthesis by ER stressin renal tubular cells42 but agrees with the lack of fibrotic
Figure 7. ER stress alone does not induce TGF-b upregulation but mediates TGF-b andfibronectin upregulation by AngII. (A) MCs were transfected with a TGF-b1 promoter-luciferase construct. AngII, but not ER stress inducers tunicamycin (Tm, 500 ng/ml) orthapsigargin (Tg, 200 nM), induced promoter activation after 24 hours (*P,0.01 versuscon, #P,0.01 versus Ang, n=12). (B) ER stress inhibition with TUDCA (300 mg/ml, 1 hour)or salubrinal (30 mM, 1 hour) prevented TGF-b1 promoter activation by AngII (*P,0.01versus con, #P,0.01 versus Ang, n=9). (C) Increased TGF-b1 secretion into the medium inresponse to AngII (24 hours) was prevented by the ER stress inhibitors TUDCA and sa-lubrinal (*P,0.001 versus con, #P,0.01 versus Ang; &P,0.001 versus Ang, n=4). (D) TheER stress inhibitors salubrinal, 4-PBA (5 mM, 2 hours), and TUDCA prevented AngII(24 hours)-induced fibronectin upregulation (*P,0.001 versus con, #P,0.01 versus Ang;&P,0.001 versus Ang, n=3).
J Am Soc Nephrol 26: 1839–1854, 2015 SREBP in Kidney Fibrosis 1847
www.jasn.org BASIC RESEARCH

response in glial and alveolar cells andin vivo in lungs.43,44 With a second insult(bleomycin), however, mice previously ex-posed to ER stress developed more severelung fibrosis. This was postulated to occurthrough increased apoptotic cells stimulat-ing matrix production in neighboringcells.44,45 Our data, however, show thatAngII activates two parallel but necessarysignals for TGF-b promoter activation,namely ER stress-induced SREBP-1 activa-tion and EGFR-mediated activation of thecotranscription factor Sp1.
Taken together, our studies have identifiedER stress and SREBP-1 activation as novelmediators of AngII-induced TGF-b1 upre-gulation and matrix synthesis in MCs.SREBP inhibition prevented glomerular fi-brosis in vivo. Future studies will determinewhether alleviation of ER stress can similarlyinhibit AngII-induced glomerular fibrosis,and whether there is an effect on renal func-tion using a more sensitive means than cre-atinine for assessment. Given the importantrole of AngII in the development of renalfibrosis, these also represent promising po-tential therapeutic targets that should be eval-uated for their efficacy in the prevention ofdiabetic and nondiabetic glomerular fibrosis.
CONCISE METHODS
Cell CulturePrimary MCs were obtained from glomeruli of
Sprague-Dawley rats by differential sieving and
cultured in DMEM with 20% FCS (Invitrogen),
streptomycin (100 mg/ml), and penicillin
(100 units/ml). Experiments used cells between
passages 6 and 15. MCs were serum deprived for
24 hours before treatment with AngII (100 nM,
Sigma-Aldrich). Inhibitorswere addedbeforeAngII
as follows: AEBSF 500 mM, 1 hour (Calbiochem);
Figure 8. EGFR, but not ER stress, regulates activation of the coregulatory transcriptionfactor Sp1 required for TGF-b upregulation. (A) AngII-induced activation of a transfectedTGF-b1 promoter-luciferase construct was inhibited by the Sp1 inhibitor mithramycin(100 nM, 1 hour) (*P,0.05 Ang versus con; ∧P,0.01 versus Ang, #P,0.001 versus Ang,n=12). (B) AngII, but not the ER stress inducers tunicamycin (Tm, 500 ng/ml) or thapsi-gargin (Tg, 200 nM), induced activation of an Sp1-responsive luciferase construct after24 hours (*P,0.001 versus con, #P,0.001 versus Ang, n=9). (C) ER stress inhibitors 4-PBA(5 mM, 2 hours) and salubrinal (30 mM, 1 hour) did not prevent AngII-induced Sp1 luciferase
activation (*P,0.01 versus con, n=6). (D) Twodistinct EGFR inhibitors—AG1478 (AG, 1 mM,30 minutes) and PD168393 (PD, 0.5 mM,30 minutes)—prevented AngII-induced Sp1luciferaseactivation (*P,0.01versuscon, #P,0.01versus Ang, n=6). (E) Deletion of the Sp1 bindingsite close to the SREBP-1 binding site in theTGF-b1 promoter-luciferase construct preventedits activationbyAngII (*P,0.001versus con,n=6).WT, wild type.
1848 Journal of the American Society of Nephrology J Am Soc Nephrol 26: 1839–1854, 2015
BASIC RESEARCH www.jasn.org

fatostatin 20mM, 4 hours (Chembridge); AG1478
1 mM, 30 minutes (Sigma-Aldrich); PD168393
0.5mM, 30 minutes (Calbiochem); LY294002
20 mM, 30 minutes (Sigma-Aldrich); wortmannin
100 nM, 1 hour (Sigma-Aldrich); Akt inhibitor
VIII 10 mM, 1 hour (EMD Millipore); rapamycin
20 ng/ml, 1 hour (Sigma-Aldrich); 4-PBA 5 mM,
2 hours (Sigma-Aldrich); tauroursodeoxycholic
acid (TUDCA)300mg/ml, 1 hour (Sigma-Aldrich);
salubrinal 30 mM, 1 hour (Selleckchem); and
mithramycin 100 nM, 1 hour (AG Scientific).
ER stress inducers were tunicamycin 500 ng/ml
and thapsigargin 200 nM (both Sigma-Aldrich).
Protein AnalysisCells were lysed and protein extracted as pub-
lished, with the addition of ALLN to the lysis
buffer at 25mg/ml (Calbiochem).46 Lysates were
centrifuged at 4°C, 14,000 rpm for 10 minutes.
Supernatant (50 mg) was separated on SDS-
PAGE, and Western blotting was performed.
Antibodies used were SREBP-1 (mouse; Santa
Cruz Biotechnology), SREBP-2 (mouse; BD
Biosciences Pharmingen), GFP (rabbit; Cell Sig-
naling Technology), FAS (rabbit monoclonal,
Cell Signaling), pEGFR Y1068 (mouse; Cell Sig-
naling Technology), pAkt S473 (rabbit; Cell Sig-
naling Technology), peIF2a S51 (rabbit; Cell
Signaling Technology), GRP78 (mouse; BD
Transduction Laboratories), and fibronectin
(mouse; BD Transduction Laboratories). Equal-
ity of loading was assessed by immunoblotting
Figure 9. SREBP-1 binding to the TGF-b1 promoter is regulated by ER stress, but not theEGFR. Chromatin immunoprecipitation (ChIP) was performed as described in the ConciseMethods to detect association of SREBP-1 or Sp1 with the TGF-b1 promoter after treat-ment of MCs with AngII for 24 hours. (A) AngII induced SREBP-1 binding to the TGF-b1promoter. This was inhibited by pretreatment with fatostatin (20 mM, 4 hours) (*P,0.001
versus con, #P,0.05 versus Ang, n=6). (B) ERstress inductionwith tunicamycin (Tm,500ng/ml)or thapsigargin (Tg, 200 nM) similarly inducedSREBP-1 binding to the TGF-b1 promoter.(C) AngII-induced SREBP-1 binding to theTGF-b1 promoter was inhibited by the ERstress blockers salubrinal (30 mM, 1 hour) andTUDCA (300 mg/ml, 1 hour) (*P,0.01 versuscon, #P,0.01 versus Ang, n=3). (D) AngII-induced SREBP-1 binding to the TGF-b1 pro-moter was unaffected by the EGFR inhibitorsAG1478 (1 mM, 30 minutes) and PD168393(0.5 mM, 30 minutes). (E) ER stress inductionwith tunicamycin (Tm, 500 ng/ml) or thapsigargin(Tg, 200 nM) did not affect Sp1 binding tothe TGF-b1 promoter (*P,0.01 versus con,#P,0.01 versus Ang, n=3). (F) Two distinct EGFRinhibitors—AG1478 (AG, 1 mM, 30 minutes)and PD168393 (PD, 0.5 mM, 30 minutes)—prevented AngII-induced Sp1 binding tothe TGF-b1 promoter (*P,0.01 versus con,#P,0.01 versus Ang, n=3).
J Am Soc Nephrol 26: 1839–1854, 2015 SREBP in Kidney Fibrosis 1849
www.jasn.org BASIC RESEARCH

Figure 10. AngII-induced TGF-b and matrix upregulation are inhibited by fatostatin. (A) IHC of cortical sections showed a significantincrease in TGF-b1 expression, quantified in the graph (*P,0.01 versus con, ∧P,0.01 versus Ang, #P,0.01 versus Ang). Activation ofSmad3, a canonical downstream mediator of TGF-b1, was also seen, as assessed by IHC for its C-terminal S423/S425 phosphorylatedform (pSmad3) (*P,0.001 versus con, ∧P,0.001 versus Ang, #P,0.001 versus Ang). (B) Smad3 activation, as assessed by its phos-phorylation, was also confirmed by immunoblotting (*P,0.05 versus con, ∧P,0.05 versus Ang, #P,0.05 versus Ang). (C) AngII inducedmatrix upregulation as seen by increased periodic acid-Schiff staining. Increased collagen deposition was assessed by both Picrosiriusred, visualized under polarized light, as well as by IHC for collagen I. These were quantified in (D) (*P,0.001 versus con, ∧P,0.001versus Ang, #P,0.001 versus Ang). (E) Inhibition of AngII-induced collagen I upregulation by fatostatin was confirmed by immuno-blotting (*P,0.01 versus con, ∧P,0.05 versus Ang, #P,0.05 versus Ang). (F) Increased fibronectin upregulation by AngII, assessed byIHC, was prevented by fatostatin (*P,0.05 versus con, ∧P,0.05 versus Ang, #P,0.05 versus Ang), and this was confirmed by immu-noblotting as seen in (G) (*P,0.001 versus con, ∧P,0.001 versus Ang, #P,,0.01 versus Ang). All images are taken at 403 magnifi-cation.
1850 Journal of the American Society of Nephrology J Am Soc Nephrol 26: 1839–1854, 2015
BASIC RESEARCH www.jasn.org

for actin (mouse; Sigma-Aldrich) and for nuclear lysates for CREB
(rabbit; Cell Signaling Technology).
GFP QuantificationMCs plated to 85% confluence were transfected with 1 mg of an SRE-
GFP plasmid (kindly provided by Dr. R. Austin20) in six-well plates
using Effectene (Qiagen). MCs were serum-deprived overnight
24 hours after transfection, then exposed to AngII. GFP was quantified
as inBradburne et al.47: Cells werewashed in cold PBS, lysed in 0.2NHCl,
and centrifuged to remove cell debris. GFP fluorescence in the su-
pernatant was measured in a fluorometer (Gemini EM, Molecular
Device) at excitation of l 475 and emission of l 510. Readings were
normalized to protein concentration.
Oil Red O StainingAfter treatment,MCswerefixed in formaldehyde and incubated inOil
RedO (Sigma-Aldrich) for 1 hour. Following awashwithwater, lipids
were recovered by incubationwith 60% isopropanol and quantified at
510 nm. Readings were normalized to protein concentration.
Luciferase AssayMCsplated to85%confluencewere transfectedwith0.5mg of aTGF-b1
promoter-luciferase construct (kindly provided by Dr. N. Kato),
CAGA12-luc with 12 copies of the Smad3-binding element (kindly pro-
vided by Dr. M. Bilandzic), or 3xSp1-luciferase construct (kindly pro-
vided by Dr. Y.P. Di48) and 0.05 mg pCMV-b-galactosidase (b-gal)
(Clontech) using Effectene (Qiagen). The TGF-b1 promoter-luciferase
plasmid was also used as a template to construct new plasmids with
deletion of the SREBP-1 or the Sp1 binding site
using overlapping primer pairs. Primer sequences
will be providedon request.Mutant constructswere
sequenced for confirmation.
Where indicated, cells were also transfected
with empty vector pcDNA or dominant negative
SREBP-1a (Y335A) or -1c (Y321A), kindly provided
by Dr. A. Schulze.49MCs were serum-deprived over-
night 24 hours after transfection, then exposed to
AngII for 24 hours. Lysis was achieved with reporter
lysis buffer (Promega, Madison, WI) using one
freeze-thaw cycle, and luciferase and b-gal activities
were measured on clarified lysate using specific kits
(Promega) with a Berthold luminometer and a plate
reader (420 nm), respectively. b-gal activity was
used to adjust for transfection efficiency.
Quantitative Real-Time PCRTotal RNA was extracted using Trizol according to
manufacturer’s instructions (Life Technologies).
Reverse transcription was performed using stan-
dard methods and cDNA analyzed by real-time
PCR using a SYBRGreen PCRMaster Mix kit (Ap-
plied Biosystems). Amplification using specific pri-
mers for TGF-b1, SREBP-1a and 1c, fibronectin
and collagen Ia1, with 18S as an internal control,
was measured continuously using an ABI 7500 Se-
quence Detector (Applied Biosystems). Fold changes over the control
values were calculated using the DDCt method.
TGF-b ELISATo quantify TGF-b secretion, media were collected after treatment with
AngII for 24 hours. After heat activation of the samples, total TGF-b1
was determined according to kit instructions (R&D Systems).
Small Interfering RNAOn-target plus SMART pool small interfering RNA (siRNA) for
SREBP-1 and control nontargeting siRNA were obtained from
Dharmacon. MCs were transfected with 100nM siRNA using Lip-
ofectamine RNAiMAX (Life Technologies) at 60% confluence. Pro-
tein expressionwas used to assess efficacy of downregulation byRNAi.
Chromatin ImmunoprecipitationAfter treatment, MCs were cross-linked with 1% formaldehyde, then
washed and scraped into cold PBS with protease inhibitors. After
centrifugation, the cell pellet was resuspended in buffer (20 mM
HEPES [pH, 7.9], 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2,
0.2 mM EDTA, protease inhibitors), incubated on ice for 20 minutes
and centrifuged. The pellet (nucleus) was resuspended in breaking buffer
(50 mM Tris-HCl [pH, 8.0], 1 mM EDTA, 150 mM NaCl, 1% SDS,
2% Triton X-100, protease inhibitors), sonicated 5310s, and Triton
buffer added (50 mM Tris-HCl [pH, 8.0], 1 mM EDTA, 150 mM
NaCl, 0.1% Triton X-100). Samples were precleared with blocked
protein G Sepharose beads, an aliquot reserved as the input, and
the remainder divided to immunoprecipitate with control rabbit
Figure 10. Continued.
J Am Soc Nephrol 26: 1839–1854, 2015 SREBP in Kidney Fibrosis 1851
www.jasn.org BASIC RESEARCH

IgG (Jackson ImmunoResearch), SREBP-1 (sc-89843; Santa Cruz Bio-
technology), or Sp1 (Abcam, Inc.) antibodies followed by incubation
with protein G beads. Samples were washed three times in Triton buffer
and SDS buffer added (62.5 mM Tris HCl [pH, 6.8], 200 mM NaCl,
2% SDS, 10 mMDTT, 2 ml of proteinase K [40 mg/ml]), then vortexed
and incubated at 65°C overnight to reverse crosslinking. DNA was
isolated using phenol/chloroform extraction and resuspended in
dH2O. PCR was performed using the rat TGF-b1 promoter primers
59-ATCCCGGTGGCATACTGAG (F) and 59-CACGGAACTTCGGA-
GAGC (R), at 60°C annealing temperature for 35 cycles.
Animal StudiesAll animal studies were carried out in accordance with McMaster
University and Canadian Council on Animal Care guidelines. Male
129Sv/Evmice (Taconic), 7–8 weeks old, underwent a left nephrectomy
followed by infusion with AngII (1000ng/kg/min (Sigma-Aldrich) or
saline by osmotic minipump (Alzet). Fatostatin was given to vehicle- or
AngII-infused mice at 30 mg/kg per day intraperitoneally as described
previously.50Control groups received vehicle alone (5%DMSO inPBS).
There were seven mice in each AngII group, five in the control group,
and three in the fatostatin alone group. Tail cuff BP wasmeasured using
tail cuff plethysmography (CODA 2 system; Kent Scientific). Mice were
euthanized after 8 weeks. Before euthanasia, urine was collected in a
metabolic cage, and albumin and creatinine were measured using com-
mercially available kits (Exocell for albumin, Crystal Chem for creati-
nine) to yield an albumin-to-creatinine ratio. Serum creatinine was also
measured as per kit instructions (Crystal Chem).
Male Dahl salt-sensitive rats (Charles River Laboratories) were
maintained on a regular chow diet until 12 weeks of age, when diets
were changed to normal salt (AIN-76A; 0.4% NaCl, n=4) or high salt
(AIN-76A; 8% NaCl, n=4; Research Diets) available ad libitum. After
4 weeks, BPs were measured as described earlier, after which kidneys
were perfused with Hank’s balanced salt solution and collected for
IHC.
For theDOCA-saltmodel, 10-week-oldmaleC57BL/6mice (Charles
River Laboratories) underwent a right uninephrectomy. After 1 week of
recovery, a 50 mg 21-day release DOCA pellet (Innovative Research of
America)was implanted subcutaneously.Micewere thengiven1%NaCl
in their drinking water (n=6). These animals were compared with a
uninephrectomized group that did not receive a DOCA pellet and
were maintained on normal drinking water (n=5). After 3 weeks, BPs
weremeasured as above and kidneys perfused withHank’s balanced salt
solution and collected for IHC.
IHCwas performedondeparaffinized 4-mmparaffin sections after
heat-induced epitope retrieval, with the exception of fibronectin
IHC, for which antigen retrieval was not used. Picrosirius red staining
Figure 10. Continued.
1852 Journal of the American Society of Nephrology J Am Soc Nephrol 26: 1839–1854, 2015
BASIC RESEARCH www.jasn.org

was performed according to the manufacturer’s protocol (Polyscien-
ces Inc.) and viewed under polarized light. Positive staining for all
imaging including Picrosirius red was quantified using ImagePro 6.2
from five different fields at a magnification of 203, and expressed as a
ratio to total image area. These were normalized to control samples to
yield fold-change values. Immunofluorescence was conducted on
cortical cryosections (6 mm) as previously described.51 Primary anti-
sera used were FITC-conjugated a-smooth muscle actin (Abcam,
Inc.), rabbit SREBP-1 (Abcam, Inc.), and rabbit TGF-b1 (R&D Sys-
tems). An AF568-conjugated secondary antibody (Invitrogen) was
used. For protein and RNA assessment, snap-frozen kidney cortex
was homogenized in lysis buffer or Trizol respectively, and samples
analyzed as described above.
Statistical AnalysesStatistical analyses were performed with SPSS software, version 21 for
Windows (IBM,Chicago, IL), using one-wayANOVAandTukeyhonest
significant difference for post hoc analysis. A t test was used for experi-
ments with only two groups. A P value,0.05 (two-tailed) was consid-
ered to represent a statistically significant difference. Data are presented
as the mean6SEM, and number of repetitions are denoted as “n=”.
ACKNOWLEDGMENTS
J.K. gratefully acknowledges the support of the Canadian Diabetes As-
sociation and St. Joseph’s Healthcare for their support of nephrology
research. We thank Dr. N. Kato (University of Tokyo, Japan) for pro-
viding the TGF-b1 promoter-luciferase construct, Dr. M. Bilandzic
(PrinceHenry’s Institute, Australia) for providingCAGA12-luc,Dr. Y.P.Di
(University of Pittsburgh, PA) for providing the 3xSp1-Luc construct,
Dr. R. Austin (McMaster University, Hamilton, ON, Canada) for pro-
viding the pSRE-GFP plasmid and Dr. A. Schulze (London Research
Institute, UK) for providing the SREBP-1a Y335A and SREBP-1c Y321A
constructs.
DISCLOSURESNone.
REFERENCES
1. Eberlé D, Hegarty B, Bossard P, Ferré P, Foufelle F: SREBP transcriptionfactors:Master regulatorsof lipidhomeostasis.Biochimie86:839–848,2004
2. Bengoechea-Alonso MT, Ericsson J: SREBP in signal transduction: Cho-lesterol metabolism and beyond. Curr Opin Cell Biol 19: 215–222, 2007
3. Liu Y, Chen BP, Lu M, Zhu Y, Stemerman MB, Chien S, Shyy JY: Shearstress activation of SREBP1 in endothelial cells is mediated by integrins.Arterioscler Thromb Vasc Biol 22: 76–81, 2002
4. Uttarwar L, Gao B, Ingram AJ, Krepinsky JC: SREBP-1 activation byglucose mediates TGF-b upregulation in mesangial cells. Am J PhysiolRenal Physiol 302: F329–F341, 2012
5. Yellaturu CR, Deng X, Cagen LM, Wilcox HG, Mansbach CM 2nd, SiddiqiSA, Park EA, Raghow R, Elam MB: Insulin enhances post-translationalprocessing of nascent SREBP-1c by promoting its phosphorylation andassociation with COPII vesicles. J Biol Chem 284: 7518–7532, 2009
6. Zhou RH, Yao M, Lee TS, Zhu Y, Martins-Green M, Shyy JY: Vascularendothelial growth factor activation of sterol regulatory elementbinding protein: A potential role in angiogenesis.Circ Res 95: 471–478,2004
7. Sun L, Halaihel N, ZhangW, Shimomura I, Horton JD, Rogers T, Levi M:Role of sterol regulatory element-binding protein 1 in regulation ofrenal lipid metabolism and glomerulosclerosis in diabetes mellitus.J Biol Chem 277: 18919–18927, 2002
8. Ishigaki N, Yamamoto T, Shimizu Y, Kobayashi K, Yatoh S, Sone H,Takahashi A, Suzuki H, Yamagata K, Yamada N, Shimano H: In-volvement of glomerular SREBP-1c in diabetic nephropathy. BiochemBiophys Res Commun 364: 502–508, 2007
9. Saito K, IshizakaN, HaraM,Matsuzaki G, SataM,Mori I, OhnoM,NagaiR: Lipid accumulation and transforming growth factor-beta upregula-tion in the kidneys of rats administered angiotensin II.Hypertension 46:1180–1185, 2005
10. Mehta PK, Griendling KK: Angiotensin II cell signaling: Physiologicaland pathological effects in the cardiovascular system.Am J Physiol CellPhysiol 292: C82–C97, 2007
11. Perlman A, Lawsin LM, Kolachana P, Saji M, Moore J Jr, Ringel MD: An-giotensin II regulationof TGF-beta inmurinemesangial cells involvesbothPI3 kinase and MAP kinase. Ann Clin Lab Sci 34: 277–286, 2004
12. Uchiyama-Tanaka Y, Matsubara H, Nozawa Y, Murasawa S, Mori Y,Kosaki A, Maruyama K, Masaki H, Shibasaki Y, Fujiyama S, Nose A, IbaO, Hasagawa T, Tateishi E, Higashiyama S, Iwasaka T: Angiotensin IIsignaling and HB-EGF shedding via metalloproteinase in glomerularmesangial cells. Kidney Int 60: 2153–2163, 2001
13. Dickhout JG, Krepinsky JC: Endoplasmic reticulum stress and renaldisease. Antioxid Redox Signal 11: 2341–2352, 2009
14. Kitamura M: Endoplasmic reticulum stress and unfolded protein re-sponse in renal pathophysiology: Janus faces. Am J Physiol RenalPhysiol 295: F323–F334, 2008
15. Tabas I, Ron D: Integrating the mechanisms of apoptosis induced byendoplasmic reticulum stress. Nat Cell Biol 13: 184–190, 2011
16. Damiano F, Alemanno S, Gnoni GV, Siculella L: Translational control ofthe sterol-regulatory transcription factor SREBP-1 mRNA in response toserum starvation or ER stress is mediated by an internal ribosome entrysite. Biochem J 429: 603–612, 2010
17. Colgan SM, Hashimi AA, Austin RC: Endoplasmic reticulum stress andlipid dysregulation. Expert Rev Mol Med 13: e4, 2011
18. Xu J, Wang G, Wang Y, Liu Q, Xu W, Tan Y, Cai L: Diabetes- and angio-tensin II-induced cardiac endoplasmic reticulum stress and cell death:metallothionein protection. J Cell Mol Med 13[8A]: 1499–1512, 2009
19. Kassan M, Galán M, Partyka M, Saifudeen Z, Henrion D, Trebak M,Matrougui K: Endoplasmic reticulum stress is involved in cardiacdamage and vascular endothelial dysfunction in hypertensive mice.Arterioscler Thromb Vasc Biol 32: 1652–1661, 2012
20. Colgan SM, Tang D, Werstuck GH, Austin RC: Endoplasmic reticulumstress causes the activation of sterol regulatory element binding pro-tein-2. Int J Biochem Cell Biol 39: 1843–1851, 2007
21. PorstmannT, SantosCR,Griffiths B,CullyM,WuM, Leevers S,Griffiths JR,Chung YL, Schulze A: SREBP activity is regulated by mTORC1 and con-tributes to Akt-dependent cell growth. Cell Metab 8: 224–236, 2008
22. Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J,Harper JW, Ericsson J: Control of lipidmetabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors bySCF(Fbw7). Cell Metab 1: 379–391, 2005
23. Bengoechea-Alonso MT, Ericsson J: A phosphorylation cascade controlsthe degradation of active SREBP1. J Biol Chem 284: 5885–5895, 2009
24. Higgins ME, Ioannou YA: Apoptosis-induced release of mature sterolregulatory element-binding proteins activates sterol-responsive genes.J Lipid Res 42: 1939–1946, 2001
25. Pastorino JG, Shulga N: Tumor necrosis factor-alpha can provokecleavage and activation of sterol regulatory element-binding protein inethanol-exposed cells via a caspase-dependent pathway that is cho-lesterol insensitive. J Biol Chem 283: 25638–25649, 2008
J Am Soc Nephrol 26: 1839–1854, 2015 SREBP in Kidney Fibrosis 1853
www.jasn.org BASIC RESEARCH

26. Wang X, Zelenski NG, Yang J, Sakai J, Brown MS, Goldstein JL:Cleavage of sterol regulatory element binding proteins (SREBPs) byCPP32 during apoptosis. EMBO J 15: 1012–1020, 1996
27. Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, TakashimaS, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K,Ogawa S, Tomoike H, Hori M, Kitakaze M: Prolonged endoplasmicreticulum stress in hypertrophic and failing heart after aortic constric-tion: Possible contribution of endoplasmic reticulum stress to cardiacmyocyte apoptosis. Circulation 110: 705–712, 2004
28. Wang W, Huang XR, Canlas E, Oka K, Truong LD, Deng C, BhowmickNA, Ju W, Bottinger EP, Lan HY: Essential role of Smad3 in angiotensinII-induced vascular fibrosis. Circ Res 98: 1032–1039, 2006
29. Yang F, Chung AC, Huang XR, Lan HY: Angiotensin II induces con-nective tissue growth factor and collagen I expression via transforminggrowth factor-beta-dependent and -independent Smad pathways: therole of Smad3. Hypertension 54: 877–884, 2009
30. Inagi R: Endoplasmic reticulum stress as a progression factor for kidneyinjury. Curr Opin Pharmacol 10: 156–165, 2010
31. Reed BD, Charos AE, Szekely AM, Weissman SM, Snyder M: Genome-wide occupancy of SREBP1 and its partners NFY and SP1 reveals novelfunctional roles and combinatorial regulation of distinct classes ofgenes. PLoS Genet 4: e1000133, 2008
32. Seo YK, Chong HK, Infante AM, Im SS, Xie X, Osborne TF: Genome-wide analysis of SREBP-1 binding in mouse liver chromatin reveals apreference for promoter proximal binding to a new motif. Proc NatlAcad Sci U S A 106: 13765–13769, 2009
33. Eckel J, Lavin PJ, Finch EA, Mukerji N, Burch J, Gbadegesin R, Wu G,Bowling B, Byrd A, Hall G, Sparks M, Zhang ZS, Homstad A, Barisoni L,Birbaumer L, Rosenberg P, Winn MP: TRPC6 enhances angiotensin II-induced albuminuria. J Am Soc Nephrol 22: 526–535, 2011
34. Kobori H, Nishiyama A, Abe Y, Navar LG: Enhancement of intrarenalangiotensinogen in Dahl salt-sensitive rats on high salt diet. Hyper-tension 41: 592–597, 2003
35. Kobori H, Nishiyama A, Harrison-Bernard LM, Navar LG: Urinary an-giotensinogen as an indicator of intrarenal Angiotensin status in hy-pertension. Hypertension 41: 42–49, 2003
36. Pinto YM, Paul M, Ganten D: Lessons from rat models of hypertension:From Goldblatt to genetic engineering. Cardiovasc Res 39: 77–88, 1998
37. Ferrari A, Maretto S, Girotto D, Volpin D, Bressan GM: SREBP con-tributes to induction of collagen VI transcription by serum starvation.Biochem Biophys Res Commun 313: 600–605, 2004
38. Le Lay S, Lefrère I, Trautwein C, Dugail I, Krief S: Insulin and sterol-regu-latory element-binding protein-1c (SREBP-1C) regulation of gene ex-pression in3T3-L1adipocytes. IdentificationofCCAAT/enhancer-bindingproteinbetaasanSREBP-1C target. JBiolChem277:35625–35634,2002
39. Lautrette A, Li S, Alili R, Sunnarborg SW, Burtin M, Lee DC, FriedlanderG, Terzi F: Angiotensin II and EGF receptor cross-talk in chronic kidneydiseases: A new therapeutic approach. Nat Med 11: 867–874, 2005
40. Tanjore H, LawsonWE, Blackwell TS: Endoplasmic reticulum stress as apro-fibrotic stimulus. Biochim Biophys Acta 1832: 940–947, 2013
41. Lenna S, Trojanowska M: The role of endoplasmic reticulum stress andthe unfolded protein response in fibrosis. Curr Opin Rheumatol 24:663–668, 2012
42. Pallet N, Bouvier N, Bendjallabah A, Rabant M, Flinois JP, Hertig A,LegendreC, Beaune P, Thervet E, AnglicheauD: Cyclosporine-inducedendoplasmic reticulum stress triggers tubular phenotypic changes anddeath. Am J Transplant 8: 2283–2296, 2008
43. Natori T, Nagai K: Endoplasmic reticulum stress upregulates thechondroitin sulfate level which thus prevents neurite extension in C6glioma cells and primary cultured astrocytes. Cell Mol Neurobiol 28:857–866, 2008
44. Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC,Newcomb DC, Jones BR, Roldan J, Lane KB, Morrisey EE, Beers MF,Yull FE, Blackwell TS: Endoplasmic reticulum stress enhances fibroticremodeling in the lungs. Proc Natl Acad Sci U S A 108: 10562–10567,2011
45. Tamaki N, Hatano E, Taura K, Tada M, Kodama Y, Nitta T, Iwaisako K,Seo S, Nakajima A, Ikai I, Uemoto S: CHOP deficiency attenuatescholestasis-induced liver fibrosis by reduction of hepatocyte injury. AmJ Physiol Gastrointest Liver Physiol 294: G498–G505, 2008
46. Krepinsky JC, Ingram AJ, Tang D,Wu D, Liu L, Scholey JW: Nitric oxideinhibits stretch-induced MAPK activation in mesangial cells throughRhoA inactivation. J Am Soc Nephrol 14: 2790–2800, 2003
47. Bradburne C, Robertson K, Thach D: Assessment of methods andanalysis of outcomes for comprehensive optimization of nucleofection.Genet Vaccines Ther 7: 6, 2009
48. Di YP, Zhao J, Harper R: Cigarette smoke induces MUC5AC proteinexpression through the activation of Sp1. J Biol Chem 287: 27948–27958, 2012
49. Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR,Downward J, Schulze A: PKB/Akt induces transcription of enzymes in-volved in cholesterol and fatty acid biosynthesis via activation ofSREBP. Oncogene 24: 6465–6481, 2005
50. Kamisuki S, Mao Q, Abu-Elheiga L, Gu Z, Kugimiya A, Kwon Y,Shinohara T, Kawazoe Y, Sato S, Asakura K, Choo HY, Sakai J, Wakil SJ,Uesugi M: A small molecule that blocks fat synthesis by inhibiting theactivation of SREBP. Chem Biol 16: 882–892, 2009
51. Wu D, Peng F, Zhang B, Ingram AJ, Gao B, Krepinsky JC: Collagen Iinduction by high glucose levels is mediated by epidermal growthfactor receptor and phosphoinositide 3-kinase/Akt signalling in mes-angial cells. Diabetologia 50: 2008–2018, 2007
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2013121332/-/DCSupplemental.
1854 Journal of the American Society of Nephrology J Am Soc Nephrol 26: 1839–1854, 2015
BASIC RESEARCH www.jasn.org