suementary inrmatin - nature · for organic compounds; low resolution electrospray ionization was...
TRANSCRIPT

NATURE CHEMISTRY | www.nature.com/naturechemistry 1
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
1
Supplementary Information for
Two-Stage Directed Self-assembly of a Cyclic [3]Catenane
Christopher S. Wood1, Tanya K. Ronson1, Ana M. Belenguer1, Julian J. Holstein2†, Jonathan R. Nitschke1*
1 Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
2 Global Phasing Ltd., Sheraton House, Castle Park, Cambridge CB3 0AX, UK
† Current address: GZG, Abteilung Kristallographie, Georg-August-Universität Göttingen, Goldschmidtstr. 1, 37077 Göttingen, Germany
*e-mail: [email protected]
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 2
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
2
Contents
Two-Stage Directed Self-assembly of a Cyclic [3]Catenane ................................................ 1
1 Materials and Methods..................................................................................................... 3
1.1 General Methods ........................................................................................................ 3
1.2 Synthesis of compound A .......................................................................................... 4
1.3 Synthesis of dianiline C ............................................................................................. 6
1.4 Synthesis of diamines for one pot screening .............................................................. 7
1.5 Attempted synthesis of homoleptic species containing ligand LA ........................... 11
1.6 Synthesis of 1 ........................................................................................................... 12
1.7 Investigation into the requirement for coordinating anions ..................................... 16
1.8 Synthesis of a series of derivatives of 1 ................................................................... 17
1.9 Attempted one pot synthesis of metal-organic cyclic [3]catenane 2. ...................... 26
1.10 Synthesis of metal-organic cyclic [3]catenane 2 through dynamic imine exchange. 27
1.11 Attempted synthesis of 2 in the absence of NaBr. ................................................. 31
1.12 Attempted synthesis of 2 using NBu4Br in place of NaBr. .................................... 31
1.13 Synthesis of 2 using LiBr, KBr and RbBr in place of NaBr. ................................. 32
1.14 Preliminary studies on length of ethylene glycol linker suitable for knotting using the subcomponent substitution method. ........................................................................... 32
1.15 Addition of NBu4 Δ-TRISPHAT to 2 .................................................................... 33
1.16 Reduction and demetalation of 2 to form a purely organic cyclic [3]catenane. .... 33
2 LCMS analysis................................................................................................................ 35
2.1 General conditions: .................................................................................................. 35
2.2 LCMS of reduced and demetalated 2 ....................................................................... 36
3 X-ray crystallography .................................................................................................... 38
4 NMR spectra of organic compounds .............................................................................. 44
5 References ...................................................................................................................... 69
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 3
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
3
1 Materials and Methods
1.1 General Methods
All chemicals and solvents, unless otherwise stated, were purchased from commercial suppliers and used as received. Previously published procedures were followed to prepare 6,6′-diformyl-3,3′-bipyridine B1, (6-(dimethoxymethyl)pyridin-3-yl)boronic acid S12 and 5,5'-dibromo-2,2'-bipyridine (S2)3. CH2Cl2, CHCl3 and CH3CN were supplied by Fisher Scientific and diethyl ether (Et2O) and diisopropyl ether (iPr2O) were supplied by Sigma Aldrich. Analytical thin layer chromatography (TLC) was performed on SiO2 60 F254 plates purchased from Merck and visualized under UV light (254 nm). Column chromatography was carried out using silica gel 60F (Merck Grade 9385, 0.040–0.063 mm).
NMR spectra were recorded on a Bruker DPX-400 MHz spectrometer, a Bruker Avance 400 MHz QNP Ultrashield spectrometer, a Bruker Avance III 400 MHz QNP Ultrashield Plus Cryo spectrometer, a Bruker Avance 500 MHz Cryo Ultrashield spectrometer, or a Bruker Avance 500 MHz BroadBand spectrometer using residual non-deuterated solvent as the internal standard. All chemical shifts (δ) are quoted in ppm and coupling constants (J) are expressed in Hertz (Hz). The following abbreviations are used in reporting the multiplicity for NMR resonances: s = singlet, d = doublet, t = triplet, and m = multiplet. Samples were prepared using CDCl3, CD3OD, or CD3CN purchased from EurisoTop or Sigma-Aldrich. The NMR data was processed using Bruker Topspin 3.2. Assignment of all 1H and 13C resonances was achieved using standard 2D NMR techniques as 1H-1H COSY, 1H-1H NOESY, 1H-13C HSQC and 1H-13C HMBC.
For organic compounds; low resolution electrospray ionization was obtained on Waters ZQ with Waters 2795 HPLC – LCMS. The mobile phases are 95% aqueous CH3CN with 0.05% formic acid and 10mM ammonium acetate with 0.1% formic acid. The column is a Phenomenex Kinetex core shell C18 column 50 x 4.6 mm ID. High resolution electrospray ionization mass spectrometry (HiRes-ESI-MS) was performed on a Waters LCT Premier Mass Spectrometer featuring a Z spray source with electrospray ionization and modular LockSpray interface.
For the cationic metal organic complexes; Low resolution electrospray ionization mass spectrometry (LR-ESI-MS) mass spectra were obtained on a Micromass Quattro LC mass spectrometer (cone voltage 20 eV; desolvation temp. 313 K; ionization temp. 313 K) infused from a Harvard syringe pump at a rate of 10 μL min-1. High resolution electrospray ionization mass spectrometry (HR-ESI-MS) was obtained using a Thermofisher LTQ Orbitrap XL hybrid ion trap mass spectrometer (capillary temp. 30 °C; tube lens 40 V).
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 4
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
4
1.2 Synthesis of compound A
Figure 1: Synthetic scheme for compound A; (i) Pd(PPh3)4 , K2CO3, dioxane : H2O (3:1), 100 °C; (ii) p-toluenesulfonic acid, acetone, 120 °C.
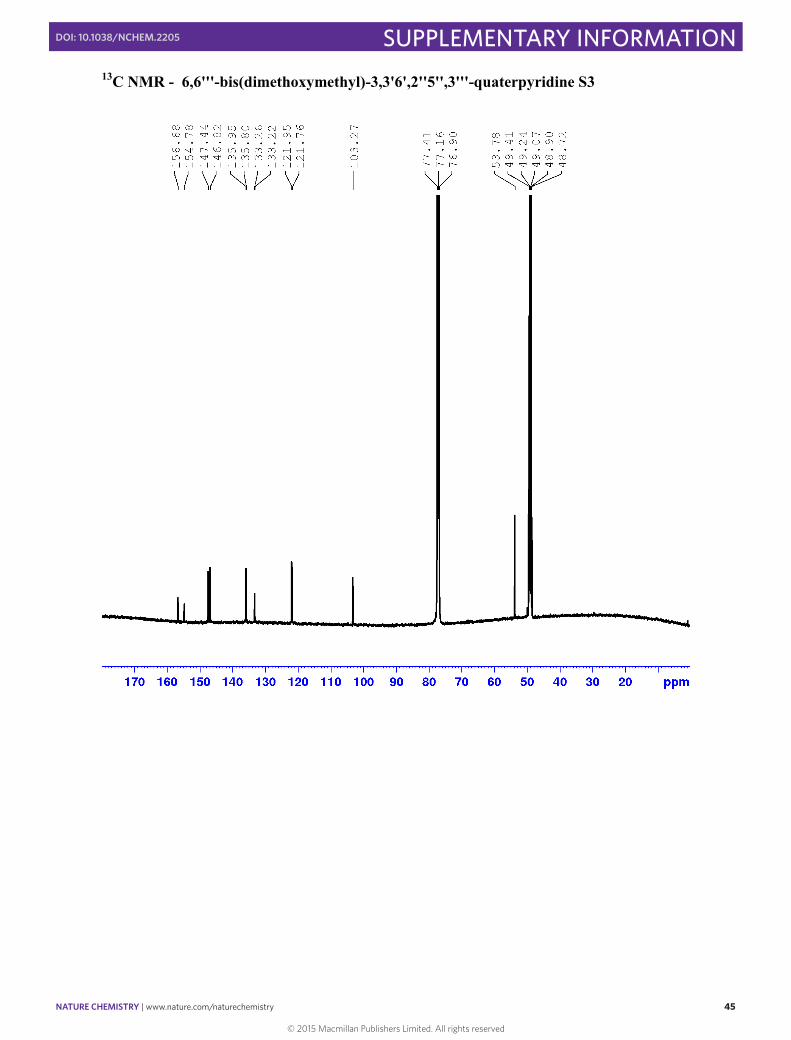
6,6'''-bis(dimethoxymethyl)-3,3'6',2''5'',3'''-quaterpyridine S3
(6-(dimethoxymethyl)pyridin-3-yl)boronic acid (2.02 g, 10.22 mmol, 2.1 equiv.), 5,5'-dibromo-2,2'-bipyridine (1.53 g, 4.87 mmol, 1 equiv.), Pd(PPh3)4 (566.2 mg, 0.49 mmol, 0.1 equiv.) and K2CO3 (3.36 g, 24.4 mmol, 5 equiv.) were dissolved in degassed dioxane : H2O (3:1, 40 ml) and the solution degassed again. The reaction was heated at 100 °C for 6 h where upon a yellow solid precipitated out. The dioxane was removed under reduced pressure and H2O (50 ml) was added. The resulting slurry was extracted with CH2Cl2 (3 × 100 ml), dried and the solvent removed in vacuo. Silica gel chromatography of the resulting orange solid (1-5%, MeOH in CH2Cl2), yielded S3 (r.f. 0.13, 2 % MeOH in CH2Cl2) as a white solid (1.2 g, 2.61 mmol, 53 % yield). 1H NMR (400 MHz, CDCl3 containing a drop of CD3OD): δ 8.89 (m, 2H, H2’) 8.84 (m, 2H, H2), 8.49 (d, J = 8.1 Hz, 2H, H5), 8.07 (dd, J = 8.1, 2.2 Hz, 2H, H4’), 8.03 (dd, J = 8.1, 2,2 Hz, 2H, H4), 7.68 (d, J = 8.1 Hz, 2H, H5’), 5.40 (s, 2H, CH(OMe)2), 3.39 (s, 6H, OMe). 13C NMR (101 MHz, CDCl3 containing a drop of CD3OD): δ 156.68 (C6), 154.78 (C6’), 147.44 (C2’), 146.82 (C2), 135.95 (C4’), 135.80 (C4), 133.22 (C3’), 133.18 (C3), 121.95 (C5), 121.76 (C5’) 103.27 (CH(OMe)2), 53.78(OMe).
(HiRes-ESI-MS) found: 459.2086 [M+H]+; C26H27N4O4 requires: 459.2027
M.P. >320 °C
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 5
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
5
[3,3'6',2''5'',3'''-quaterpyridine]-6,6'''-dicarbaldehyde A
6,6'''-bis(dimethoxymethyl)-3,3'6',2''5'',3'''-quaterpyridine S3 (892 mg, 1.95 mmol, 2.1 equiv.) and p-toluenesulfonic acid (10 mg, 0.06 mmol, 0.02 equiv.) were dissolved in acetone (50 ml) in a 100 ml ACE® pressure tube and the solution was heated to 120 °C and stirred for 24 h until TLC analysis (5 % MeOH in CH2Cl2) showed the consumption of starting material. The solution was cooled to room temperature, centrifuged and the resulting light pink solid washed with water (1 × 20 ml), acetone (2 × 20 ml) and finally ether (1 × 20 ml). The solid was dried under reduced pressure and recrystallised from hot DMSO to afford a as a light orange solid (559 mg, 1.53 mmol, 78 % yield). 1H NMR (400 MHz, D2O with a few drops of DCl to solubilise): δ 8.56 (s, 2H) 8.49 (s, 2H), 8.31 (d, J = 8.4 Hz, 2H), 8.08 (d, J = 8.3, 2H), 8.00 (d, J = 8.2, 2H), 7.58 (d, J = 8.5 Hz, 2H), 5.62 (s, 2H, CH(OD)2.
The product was insufficiently soluble to obtain 13C NMR.
(HiRes-ESI-MS) found: 367.1218 [M+H]+; C22H15N4O2 requires: 367.1190
M.P. >320 °C
Elemental analysis: C, 71.28; H, 3.98; N, 14.75. Required (C22H15N4O2·0.25 H2O): C, 71.25; H, 3.94; N 15.11.
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 6
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
6
1.3 Synthesis of dianiline C
Figure 2: Synthetic scheme for compound C; (i) Pd/C,H2, MeOH, r.t.
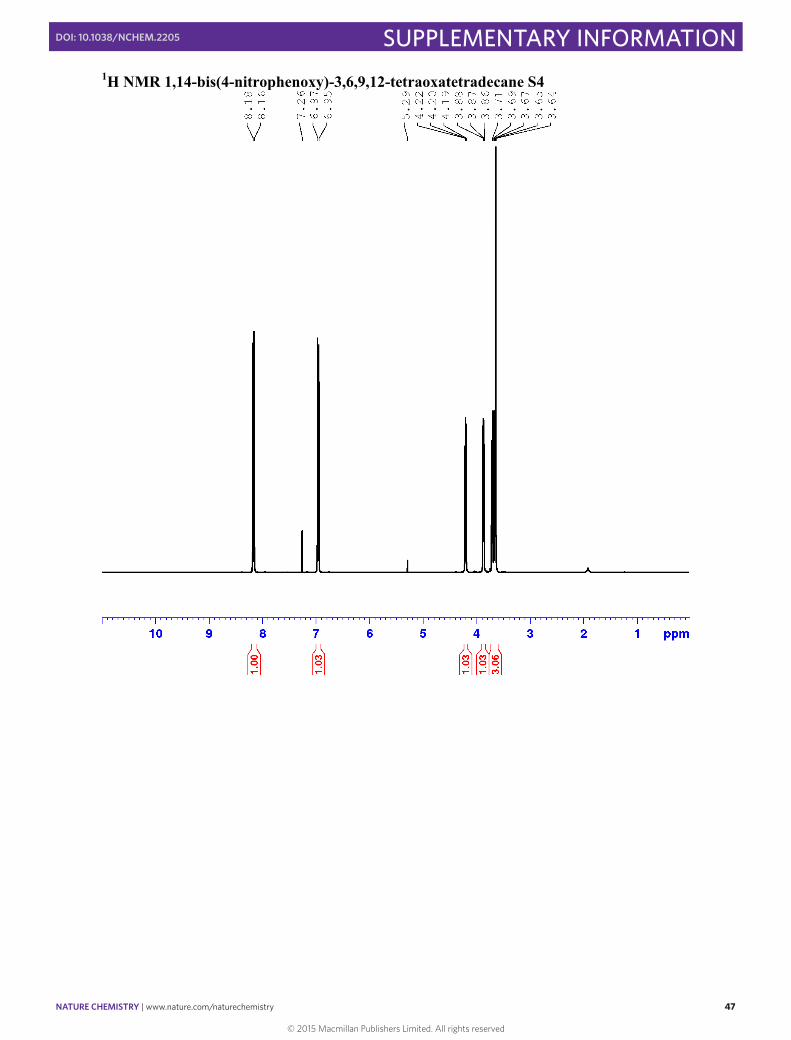
1,14-bis(4-nitrophenoxy)-3,6,9,12-tetraoxatetradecane S4
NaH 60% dispersed in oil (1.06 g, 26.58 mmol, 2.5 equiv.) was added portion wise to a stirred solution of dimethoxyethane (DME) (40 ml) containing pentaethylene glycol (2.3 ml, 10.6 mmol, 1.1 equiv.) and the solution was stirred until the effervescence stopped. 1-Fluoro-4-nitro-benzene (3.00 g, 21.3 mmol, 2 equiv.) was added and the reaction mixture stirred at room temperature for 12 h. The reaction was quenched with H2O (5 ml) and the DME removed under reduced pressure. The resulting brown liquid was dissolved in CHCl3:iPrOH 3:1 (70 ml) and washed with H2O (30 ml). The aqueous layer was extracted with CHCl3:iPrOH 3:1 (2 × 30 ml) and the combined organic layers were dried with MgSO4 and the solvent removed under reduced pressure. The resulting solid was recrystallised from EtOAc yielding S4 as a light brown solid (2.72 g, 53.1 %). 1H NMR (400 MHz, CDCl3) δ 8.14-8.19 (m, 4H, H3), 6.92-6.96 (m, 4H, H2), 4.18-4.22 (m, 4H, Ha), 3.85-3.88 (m, 4H, Hb), 3.62-3.73 (m, 8H, Hc-d), 3.64 (s, 4H, He) 13C NMR (101 MHz, CDCl3): δ 163.95 (C4), 141.69 (C1), 125.95 (C2), 114.69 (C3), 71.03 (Cc/d/e), 70.73 (Cc/d/e), 70.70 (Cc/d/e), 69.49 (Cb), 68.31 (Ca)
(ESI-MS) found: 498.0 [M+NH4 ]+; C22 H32N3O10 requires: 498.51
M.P. 81-84 °C
Elemental analysis: C 54.74, H 5.81, N 5.82, required C 55.00, H 5.87, N 5.83.
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 7
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
7
4,4'-((3,6,9,12-tetraoxatetradecane-1,14-diyl)bis(oxy))dianiline C
1,14-bis(4-nitrophenoxy)-3,6,9,12-tetraoxatetradecane S4 (1.00 g, 2.08 mmol, 1 equiv.) and Palladium/charcoal activated 10 % Pd (200 mg) were sonicated at room temperature in MeOH:CH2Cl2 1:1 (100 ml) until the starting material had dissolved. The reaction mixture was stirred at 30 °C under an atmosphere of nitrogen for 48 h until TLC (r.f. 0.19, 2 % MeOH in CH2Cl2) showed complete consumption of the starting material. The solution was then filtered through Celite® and the solvent removed under reduced pressure. The resulting light brown viscous liquid was used without further purification (0.81 g, 93.3 %) 1H NMR (400 MHz, CDCl3) δ 6.72-6.77 (m, 4H, H3), 6.59-6.64 (m, 4H, H2), 4.04 (m, 4H, Ha), 3.70 (m, 4H, Hb), 3.66-3.72 (m, 8H, Hc-d), 3.65 (s, 4H, He), 3.36 (broad, NH2) 13C NMR (101 MHz; CDCl3): δ 152.09 (C4), 140.29 (C1), 116.49 (C2), 116.02 (C3), 70.90 (Cc-d), 70.75 (Ce), 70.03 (Cb), 68.29 (Ca)
(HiRes-ESI-MS) found: 421.2411 [M+H]+; C22 H33N2O6 requires: 421.2333
Elemental analysis: C, 62.81; H, 7.58; N, 6.52. Required: C, 62.84; H, 7.67; N, 6.66.
1.4 Synthesis of diamines for one pot screening
The following diamines were prepared by the synthetic procedure described above with substitution of the appropriate diol.
4,4'-((oxybis(ethane-2,1-diyl))bis(oxy))dianiline S5
1H NMR (400 MHz, CDCl3) δ 6.72-6.79 (m, 4H, H3), 6.59-6.65 (m, 4H, H2), 4.05-4.09 (m, 4H, Ha), 3.85-3.90 (m, 4H, Hb), 3.27 (broad, NH2) 13C NMR (101 MHz; CDCl3): δ 152.08 (C4), 140.34 (C1), 116.47 (C2), 116.10 (C3), 70.19 (Cb), 68.39 (Ca)
(HiRes-ESI-MS) found: 289.1571 [M+H]+; C16 H21N2O3 requires: 289.1547
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 8
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
8
4,4'-(((ethane-1,2-diylbis(oxy))bis(ethane-2,1-diyl))bis(oxy))dianiline S6
1H NMR (400 MHz, CDCl3) δ 6.72-6.79 (m, 4H, H3), 6.58-6.64 (m, 4H, H2), 4.03-4.06 (m, 4H, Ha), 3.80-3.83 (m, 4H, Hb), 3.73 (m, 4H, Hc), 3.31 (broad, NH2) 13C NMR (101 MHz; CDCl3): δ 152.11 (C4), 140.30 (C1), 116.47 (C2), 116.06 (C3), 70.96 (Cc), 70.09 (Cb), 68.32 (Ca)
(HiRes-ESI-MS) found: 333.1831 [M+H]+; C18H25N2O4 requires: 333.1809
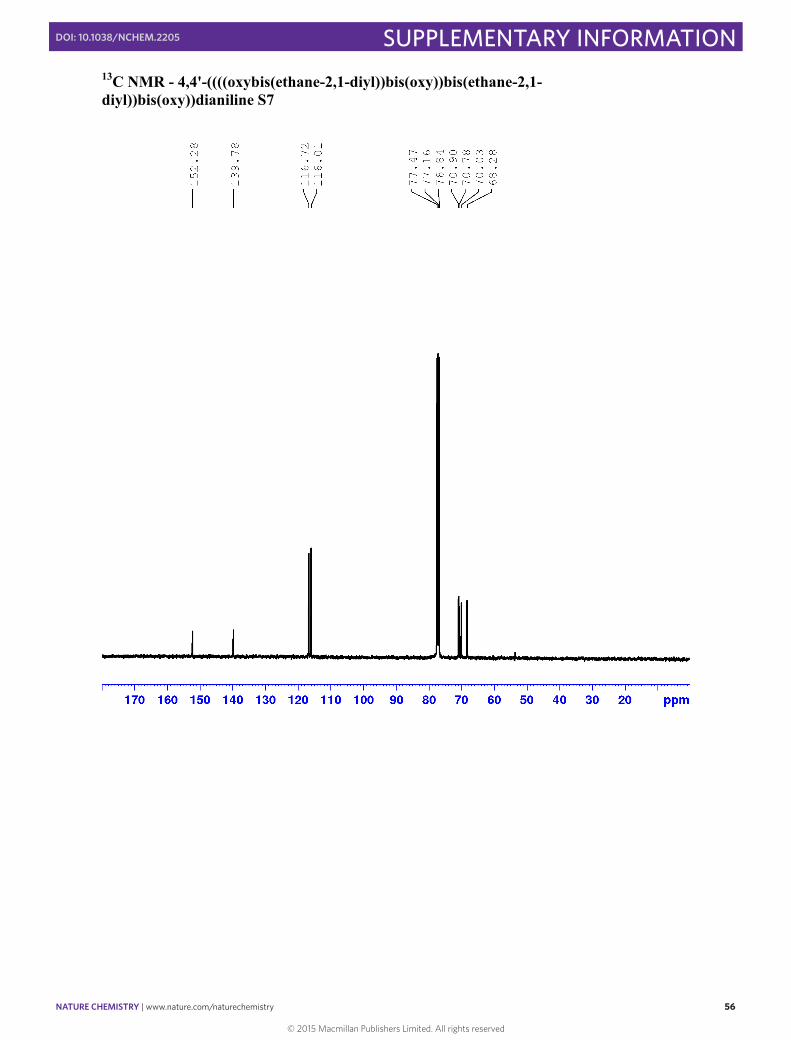
4,4'-((((oxybis(ethane-2,1-diyl))bis(oxy))bis(ethane-2,1-diyl))bis(oxy))dianiline S7
1H NMR (400 MHz, CDCl3) δ 6.71-6.77 (m, 4H, H3), 6.60-6.66 (m, 4H, H2), 4.00-4.05 (m, 4H, Ha), 3.78-3.82 (m, 4H, Hb), 3.65-3.73 (m, 8H, Hc & Hd), 3.47 (broad, NH2) 13C NMR (101 MHz; CDCl3): δ 152.28 (C4), 137.78 (C1), 116.72 (C2), 116.01 (C3), 70.90 (Cc/d), 70.78 (Cc/d), 70.03 (Cb), 68.28 (Ca)
(HiRes-ESI-MS) found: 377.2096 [M+H]+; C20H29N2O5 requires: 377.2071
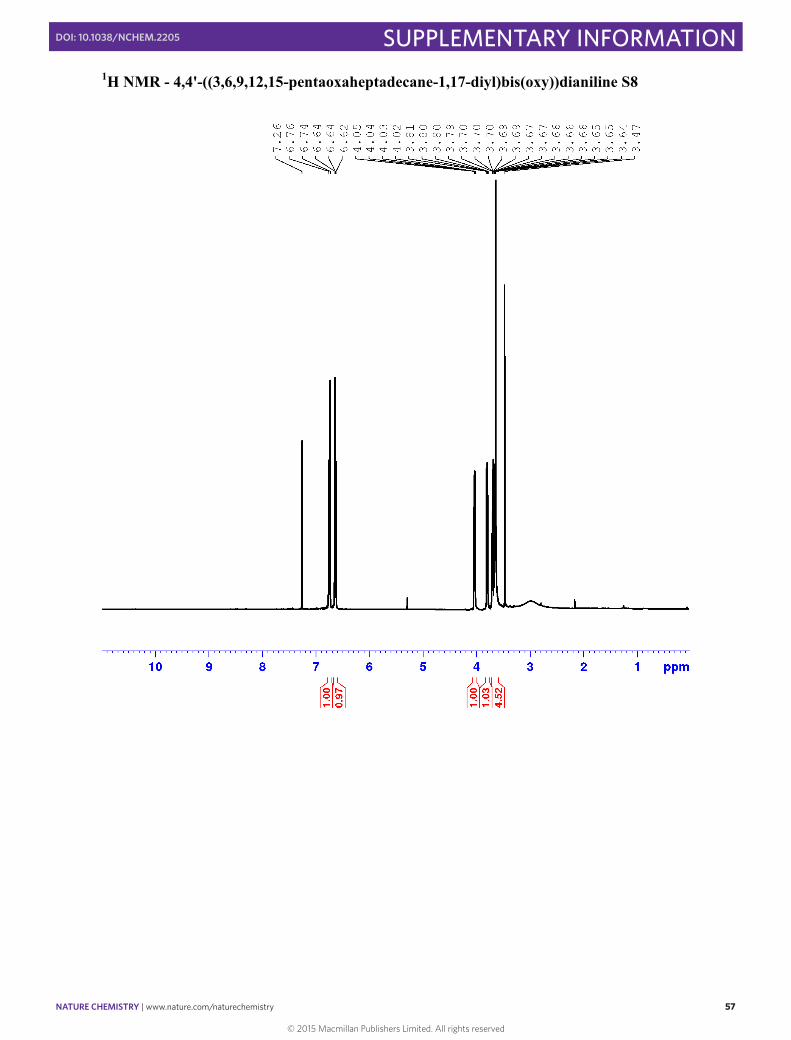
4,4'-((3,6,9,12,15-pentaoxaheptadecane-1,17-diyl)bis(oxy))dianiline S8
1H NMR (400 MHz, CDCl3) δ 6.71-6.77 (m, 4H, H3), 6.60-6.66 (m, 4H, H2), 4.00-4.06 (m, 4H, Ha), 3.77-3.82 (m, 4H, Hb), 3.65-3.73 (m, 8H, Hc-d), 3.64 (s, 8H, He-f) 2.99 (broad, NH2) 13C NMR (101 MHz; CDCl3): δ 152.23 (C4), 139.97 (C1), 116.66 (C2), 116.02 (C3), 70.89 (Cc/d/e/f), 70.74 (Cc/d/e/f), 70.70 (Cc/d/e/f), 70.03 (Cb), 68.29 (Ca)
(HiRes-ESI-MS) found: 465.2647 [M+H]+; C24H37N2O7 requires: 465.2595
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 9
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
9
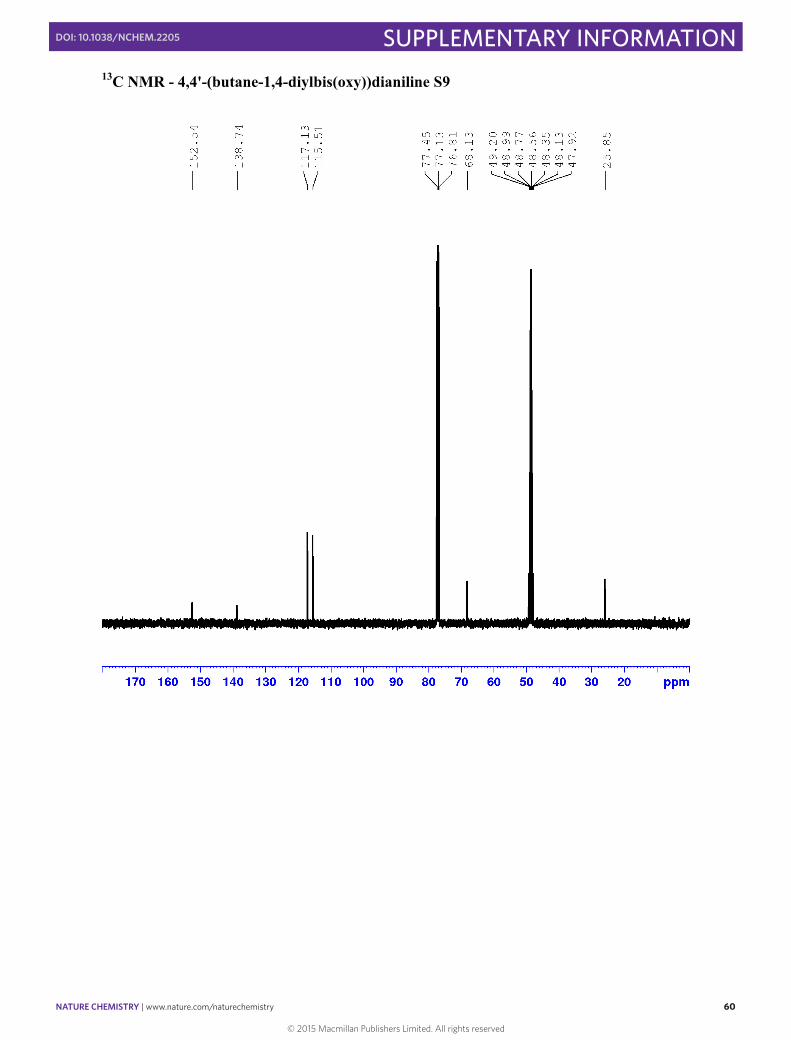
4,4'-(butane-1,4-diylbis(oxy))dianiline S9
1H NMR (400 MHz, 2:1 CDCl3, CD3OD) δ 6.58 (d, J = 8.8 Hz, 4H, H3), 6.54 (J = 8.8 Hz, 4H, H2), 3.79 (m, 4H, Ha), 1.74 (m, 4H, Hb) 13C NMR: (101 MHz; 2:1 CDCl3, CD3OD): δ 152.54 (C4), 138.74 (C1), 117.13 (C2), 115.51 (C3), 68.13 (Ca), 25.85 (Cb)
(HiRes-ESI-MS) found: 273.1590 [M+H]+; C16H21N2O2 requires: 273.1603
4,4'-(pentane-1,5-diylbis(oxy))dianiline S10
1H NMR (400 MHz,2:1 CDCl3, CD3OD) δ 6.68 (d, J = 8.6 Hz, 4H, H3), 6.62 (d, J = 8.7 Hz, 4H, H2), 3.84 (t, J = 6.4 Hz 4H, Ha), 1.74 (q, J= 7.5 Hz, 4H, Hb), 1.53 (q, J = 7.5 Hz, 2H, Hc) 13C NMR: (101 MHz; 2:1 CDCl3, CD3OD): δ 152.73 (C4), 138.81 (C1), 117.14 (C2), 115.70 (C3), 68.54 (Ca), 29.70 (Cb), 22.64 (Cc)
(HiRes-ESI-MS) found: 287.1760 [M+H]+; C17H23N2O2 requires: 287.1754
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 10
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
10
4,4'-(hexane-1,5-diylbis(oxy))dianiline S11
1H NMR (400 MHz, 2:1 CDCl3, CD3OD) δ 6.57-6.36 (m, 8H, H2,3), 3.70-3.60 (m, 4H, Ha), 1.53 (s, br, 4H, Hb), 1.27 (s, br, 4H, Hc) 13C NMR: (101 MHz; 2:1 CDCl3, CD3OD): δ 152.59 (C4), 138.58 (C1), 117.13 (C2), 115.42 (C3), 68.40 (Ca), 28.99 (Cb), 25.53 (Cc)
(HiRes-ESI-MS) found: 301.1910 [M+H]+; C18H25N2O2 requires: 301.1916
4,4'-(heptane-1,5-diylbis(oxy))dianiline S12
1H NMR (400 MHz, 2:1 CDCl3, CD3OD) δ 6.70 (d, J = 8.4 Hz, 4H, H3), 6.63 (d, J = 8.4 Hz, 4H, H2), 3.84 (t, J = 6.5 Hz 4H, Ha), 1.71 (q, J= 6.7 Hz, 4H, Hb), 1.50-1.34 (m, 6H, Hc,d) 13C NMR: (101 MHz; 2:1 CDCl3, CD3OD): δ 152.68 (C4), 139.15 (C1), 116.96 (C2), 115.74 (C3), 68.73 (Ca), 29.35 (Cd), 29.18 (Cb), 26.02(Cc)
(HiRes-ESI-MS) found: 315.2098 [M+H]+; C19H27N2O2 requires: 315.2067
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 11
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
11
4,4'-(octane-1,5-diylbis(oxy))dianiline S13
1H NMR (400 MHz, 2:1 CDCl3, CD3OD) δ 6.58-6.40 (m, 8H, H2,3), 3.70-3.62 (m, 4H, Ha), 1.50 (s, br, 4H, Hb), 1.21 (s, br, 4H, Hc), 1.15 (s, br, 4H, Hd)
13C NMR: (101 MHz; 2:1 CDCl3, CD3OD): δ 152.66 (C4), 138.86 (C1), 117.11 (C2), 115.60 (C3), 68.69 (Ca), 29.21 (Cd), 29.17 (Cb), 25.82 (Cc)
(HiRes-ESI-MS) found: 329.2222 [M+H]+; C20H29N2O2 requires: 329.2229
The following two anilines were obtained from commercial suppliers and were used as received:
2,2'-(ethane-1,2-diylbis(oxy))bis(ethan-1-amine) S14
3,3'-((oxybis(ethane-2,1-diyl))bis(oxy))bis(propan-1-amine) S15
1.5 Attempted synthesis of homoleptic species containing ligand LA [3,3'6',2''5'',3'''-quaterpyridine]-6,6'''-dicarbaldehyde A (4.1 mg, 11 μmol, 1 equiv.) and Fe(BF4)2·6H2O (5.4 mg, 11 μmol, 1 equiv.) were suspended in CH3CN (2.0 ml) and heated at 70 °C in a PTFE stoppered Schlenk tube until all the solids dissolved and a dark red solution was obtained. A solution of 4-chloroaniline in CH3CN (68 μl, 0.34 mM, 2 equiv.) was added and the solution was heated at 70 °C for 3 days. An aliquot was taken and the solids precipitated through the addition of Et2O and isolated through centrifugation. The resulting blue solid was dried under vacuum and analysed by 1H NMR and ESI-MS. No species with charge greater than +1 and an m/z greater than 200 were observed, and the 1H NMR spectrum was broad and poorly-defined in the 12 − 0 ppm diamagnetic region, with no peaks observed in the 200 − -50 ppm paramagnetic region, indicating that no discrete species had formed.
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 12
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
12
1.6 Synthesis of 1 [1](BF4)6
Synthesis in methods section of main text.
1H NMR (400 MHz, CD3CN) δ 198.37, 186.43 br, 49.04, 25.68, 19.29, 14.81, 13.66, 13.04, 10.87, 9.32, 8.30, 7.06, 6.60, 5.75, 4.65 19F NMR (376 MHz; CD3CN, reference C6F6): δ 150.99 (BF4
−)
Further assignments were hampered by the paramagnetic nature of the complex
LR-ESI-MS: m/z: 644.0(644.1) [1]6+, 790.2(790.3) [1](BF4)15+, 1009.5(1009.6) [1](BF4)2
4+, 1374.5(1375.1)[1](BF4)3
3+
Elemental analysis: C, 46.10; H, 2.85; N, 9.06. Required [1](BF4)6 ·8H2O : C, 46.14; H, 2.89; N, 9.28.
Figure 3: 1H NMR spectrum (400 MHz, 298 K, CD3CN) of [1](BF4)6
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 13
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
13
Figure 4: LR-ESI-mass spectrum of product [1](BF4)6. Calculated values are in brackets.
[1](BF4)24+
1009.5(1009.6)
[1](BF4)5+
790.2(790.3)
[1](BF4)36+
1374.6(1375.1)
[1]6+
644.0(644.1)
[1](BF4)5+MeCN 799.0(798.6)
[1](BF4)24+MeCN
1020.5(1019.9)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 14
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
14
Figure 5: (a) HR-ESI-mass spectrum of the +5 peak comprising a mixture of [1](BF4) 5+ and [1](Br) 5+. (b) Calculated isotopic distributions for a 1:3 mixture of [1](BF4) 5+ (blue) and [1](Br) 5+ (red)
a
b
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 15
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
15
[1](ClO4)6
[3,3'6',2''5'',3'''-quaterpyridine]-6,6'''-dicarbaldehyde A (30.2 mg, 82.4 μmol, 3 equiv.), [3,3'-bipyridine]-6,6'-dicarbaldehyde B (17.5 mg, 82.4 μmol, 3 equiv.) and Fe(ClO4)2·6H2O (60.4 mg, 164.9 μmol, 6 equiv.) were suspended in CH3CN (20 ml) and heated at 70 °C in a PTFE stoppered Schlenk tube until all the solids dissolved and a dark red solution was obtained. A solution of 4-chloroaniline in CH3CN (5.71 ml, 0.057 mM, 12 equiv.) was added followed by the dropwise addition of tetra-n-butylammonium bromide in CH3CN (1.49 ml, 0.11 mM, 6 equiv.). The reaction mixture was stirred at 70 °C for 36 hours. The solution was filtered through a PTFE syringe filter (4.5 μm pore size) and subsequent addition of Et2O (30 ml) precipitated out [1](ClO4)6 as a green solid that was isolated by centrifugation (106 mg, 86 %). 1H NMR (400 MHz, CD3CN) δ 198.37, 186.43 br, 49.04, 25.68, 19.29, 14.81, 13.66, 13.04, 10.87, 9.32, 8.30, 7.06, 6.60, 5.75, 4.65
LR-ESI-MS: m/z: 644.1(644.1) [1Cl•Br6]6+, 793.0(792.9) [1Cl•Br6](ClO4)5+, 1015.7(1015.9) [1Cl•Br6](ClO4)2
4+, 1388.1(1387.8) [1Cl•Br6](ClO4)33+
Elemental analysis: C, 45.16; H, 2.87; N, 8.85. Required [1](ClO4)6·9H2O : C, 45.20; H, 2.88; N, 9.09.
Figure 6: LR-ESI-mass spectrum of [1](ClO4)6. Calculated values are in brackets.
[1](ClO4)24+
1015.7(1015.9)
[1](ClO4)5+
793.0(792.9)
[1](ClO4)36+
1388.1(1387.8)
[1]6+
644.1 (644.1)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 16
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
16
1.7 Investigation into the requirement for coordinating anions [3,3'6',2''5'',3'''-quaterpyridine]-6,6'''-dicarbaldehyde A (5.3 mg, 14 μmol, 3 equiv.),
[3,3'-bipyridine]-6,6'-dicarbaldehyde B (3.1 mg, 14 μmol, 3 equiv.) and Fe(BF4)2·6H2O (10.7 mg, 28.7 μmol, 6 equiv.) were suspended in CH3CN (5.5 ml) and heated at 70 °C in a PTFE stoppered Schlenk tube until all the solids dissolved and a dark red solution was obtained. A solution of 4-chloroaniline in CH3CN (0.21 ml, 0.27 mM, 12 equiv.) was added and the solution was heated at 70 °C for 7 days. An aliquot was taken and the solids precipitated through the addition of Et2O and isolated through centrifugation. The resulting blue solid was dried under vacuum and analysed by 1H NMR and ESI-MS. No species with charge greater than 1 and an m/z greater than 200 were observed indicating that no discrete species had formed. To the remaining solution NBu4Br in CH3CN (0.38 ml, 0.074 M, 6 equiv.) was added dropwise and the reaction mixture was stirred at 70 °C for 24 hours. A further aliquot was taken and the solids precipitated through the addition of Et2O and isolated through centrifugation. The resulting blue solid was dried under vacuum and analysed by 1H NMR and ESI-MS, which indicated formation of [1](BF4)6.
Figure 7: (a) 1H NMR (400 MHz, 298 K, CD3CN) of aliquot 1 showing the absence of any high spin products. (b) 1H NMR (400 MHz, 298 K, CD3CN) of aliquot 2 showing the expected resonances for [1](BF4)6
a
b
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 17
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
17
Figure 8: LR-ESI-mass spectrum of aliquot 2 with signals indicating the formation of [1](BF4)6. Calculated values are in brackets.
1.8 Synthesis of a series of derivatives of 1
The following series of compounds are variations of anilines and coordinating anions on the metal organic scaffold of 1. For brevity they have been labelled as [1R•X6](Y)6.
Where:
R = substituent on the aniline
X = coordinating anions bound to the three Fe centers in the large circular helicate
Y = non-coordinated anions (derived from the Fe salt used in the reaction)
The following complexes were prepared as described above with substitution of the appropriate para-substituted aniline and tetra n-butyl ammonium salt of the coordinating anion. Fe(ClO4)2·6H2O and in one case Fe(PF6)2·6CH3CN were employed in place of Fe(BF4)2·6H2O as the ClO4
− and PF6− salts of 1 were observed to crystallize more readily
than the analogous BF4− salts.
For complexes formed with chloride as the coordinating anion additional bromide adducts were observed by ESI-MS (Fig. S10, S11 and S13). We hypothesise that these adducts give rise to the additional minor set of peaks in the 1H NMR spectra (Fig. S9, and S12). However, the paramagnetic nature of the complexes hampered further investigation of this phenomenon. For the complexes with bromide or thiocyanate as the coordinating anion, chloride or bromide contamination was observed to lead to the presence of a library of species in the ESI-MS spectra. The source of the contamination is hypothesized to be either from the commercial iron(II) or tetrabutylammonium salts. Bromide and Fe(BF4)2 were
[1](BF4)24+
1009.9(1009.6)
[1](BF4)5+
790.5 (790.3)
[1](BF4)36+
1375.3(1375.1) [1]6 +
644.1(644.1)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 18
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
18
chosen for the subsequent knotting studies as they were found to form 1 with the least contamination. X-ray quality crystals were obtained for three of the combinations ([1Cl•Br6](ClO4)6, [1Me•Cl6](ClO4)6 and [1OMe•(SCN)6](PF6)6 ) through the slow diffusion of diisopropyl ether into separate CH3CN solutions of the complexes.
Table 1: Showing the combinations of aniline used to form the imine in ligands La and Lb and coordinating anion that successfully form the mixed ligand double circular helicate.
[1Cl•Cl6](ClO4)6
LR-ESI-MS: m/z: 599.1(599.7) [1Cl•Cl6]6+, 739.2(739.5) [1Cl•Cl6](ClO4)5+, 949.1(949.3) [1Cl•Cl6](ClO4)2
4+, 1298.3(1298.9)[1Cl•Cl6](ClO4)33+
Figure 9: 1H NMR (400 MHz, 298 K, CD3CN) of [1Cl•Cl6](ClO4)6
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 19
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
19
Figure 10: LR-ESI-mass spectrum of [1Cl•Cl6](ClO4)6 . Calculated values are in brackets.
Figure 11: LR-ESI-mass spectrum of [1Cl•Cl6](ClO4)6 in the range 900-1050 m/z.
Calculated values are in brackets.
[1Cl•Cl6]6+
599.1(599.7)
[NBu4]2(ClO4) 583.5(583.5)
[1Cl•Cl6](ClO4)5+
739.2(739.5)
[1Cl•Cl6](ClO4)24+
949.1(949.3)
[1Cl•Cl6](ClO4)33+
1298.3(1298.9)
[1Cl•Cl6](ClO4)24+
949.1(949.3)
[1Cl•Cl5Br](ClO4)24+
960.0(960.21)
[1Cl•Cl4Br2](ClO4)24+
970.9(971.2)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 20
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
20
[1Me•Cl6](ClO4)6
LR-ESI-MS: m/z: 558.2(558.9) [1Me•Cl6]6+, 690.4(690.5) [1Me•Cl6](ClO4)5+, 887.8(888.0)[1Me•Cl6](ClO4)2
4+, 1217.2(1217.2) [1Me•Cl6](ClO4)33+
Figure 12: 1H NMR (400 MHz, 298 K, CD3CN) of [1Me•Cl6](ClO4)6
Figure 13: LR-ESI-mass spectrum of [1Me•Cl6](ClO4)6. Calculated values are in
brackets.
[1Me•Cl6]6+
559.0(558.9)
[1Me•Cl6](ClO4)5+
690.4(690.5)
[1Me•Cl6](ClO4)24+
887.8(888.0)
[1Me•Cl6](ClO4)33+
1298.3(1217.2)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 21
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
21
Figure 14: LR-ESI-mass spectrum of [1Me•Cl6](ClO4)6 in the range 840-990 m/z. Calculated values are in brackets. [1Me•Br6](ClO4)6
LR-ESI-MS: m/z: 603.4(603.3) [1Me•Br6]6+, 743.9(743.9) [1Me•Br6](ClO4)15+, 954.5(954.7)
[1Me•Br6](ClO4)14+, 1306.1(1306.1) [1Me•Br6](ClO4)3
3+
Figure 15: 1H NMR (400 MHz, 298 K, CD3CN) of [1Me•Br6](ClO4)6
[1Me•Cl6](ClO4)24+
887.8(888.0)
[1Me•Cl5Br](ClO4)24+
898.97(898.88)
[1Me•Cl4Br2](ClO4)24+
910.06(909.87)
[1Me•Cl3Br3 ](ClO4)24+
921.14(921.11)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 22
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
22
Figure 16: LR-ESI-mass spectrum of [1Me•Br6](ClO4)6. Calculated values are in
brackets.
[1Me•(SCN)6](ClO4)6
LR-ESI-MS: m/z: 581.4(581.5) [1Me•(SCN)6]6+, 717.4(717.7) [1Me•(SCN)6](ClO4)15+,
921.9(922.0) [1Me•(SCN)6](ClO4)24+, 1262.1(1262.4) [1Me•(SCN)6](ClO4)3
3+
Figure 17: 1H NMR (400 MHz, 298 K, CD3CN) of [1Me•SCN6](ClO4)6
[1Me•Br6]6+
603.4(603.3)
[1Me•Br6](ClO4)5+
743.9(743.9)
[1Me•Br5Cl](ClO4)24+
943.3(943.7)
[1Me•Br6](ClO4)33+
1306.1(1036.1)
[1Me•Br6](ClO4)24+
954.5(954.7)
[1Me•Br5Cl](ClO4)5+
734.5(735.0)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 23
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
23
Figure 18: LR-ESI-mass spectrum of [1Me•SCN6](ClO4). Calculated values are in
brackets.
[1OMe•Br6](ClO4)6
LR-ESI-MS: m/z: 635.2(635.3) [1OMe•Br6]6+, 782.4(782.3) [1OMe•Br6](ClO4)5+, 1002.9(1002.7) [1OMe•Br6](ClO4)2
4+, 1370.1(1370.1) [1OMe•Br6](ClO4)33+.
Figure 19: 1H NMR (400 MHz, 298 K, CD3CN) of [1OMe•Br6](ClO4)6
[1Me•SCN6]6+
581.4(581.5)
[1Me•SCN6](ClO4)5+
717.4(717.7)
[1Me•SCN6](ClO4)33+
1262.1(1262.4)
[1Me•SCN6](ClO4)24+
921.9(922.0)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 24
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
24
Figure 20: LR-ESI-mass spectrum of [1OMe•Br6](ClO4)6. Calculated values are in
brackets.
Figure 21: LR-ESI-mass spectrum of [1OMe•Br6](ClO4)6 in the range of 940-1020 m/z. Calculated values are in brackets.
[1OMe•Br6]6+
635.2(635.3)
[1OMe•Br6](ClO4)5+
782.4(782.3)
[1OMe•Br6](ClO4)33+
1370.1(1370.1)
[1OMe•Br6](ClO4)24+
1002.9(1002.7)
[1OMe•Br5 Cl2](ClO4)24+
980.3(980.3)
[1OMe•Br6Cl](ClO4)24+
991.5(991.6)
[1OMe•Br4 Cl3](ClO4)24+
969.7(969.1)
[1OMe•Br6](ClO4)24+
1002.9(1002.7)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 25
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
25
[1OMe•(SCN)6](ClO4)6
LR-ESI-MS: m/z: 613.4(613.5)[1OMe•(SCN)6]6+, 755.9(755.6)[1OMe•(SCN)6](ClO4)5+, 969.9(970.0) [1OMe•(SCN)6](ClO4)2
4+, 1326.2(1326.4) [1OMe•(SCN)6](ClO4)33+.
Figure 22: 1H NMR (400 MHz, 298 K, CD3CN) of [1OMe•SCN6](ClO4)6
Figure 23: LR-ESI-mass spectrum of [1OMe•SCN6](ClO4)6. Calculated values are in brackets.
[1OMe•SCN6]6+
613.4(613.5)
[1OMe•SCN6](ClO4)5+
755.9(756.1)
[1OMe•SCN6](ClO4)33+
1326.2(1326.4)
[1OMe•SCN6](ClO4)24+
969.9(970.0)
© 2015 Macmillan Publishers Limited. All rights reserved
NATURE CHEMISTRY | www.nature.com/naturechemistry 26
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
26
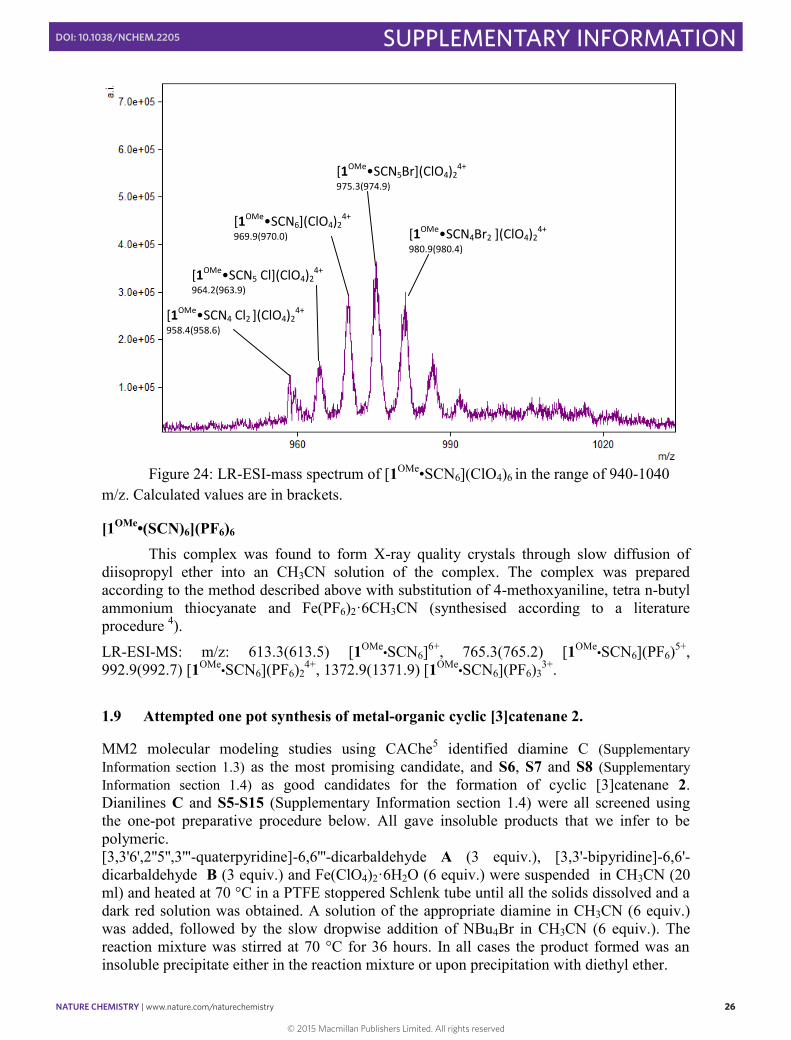
Figure 24: LR-ESI-mass spectrum of [1OMe•SCN6](ClO4)6 in the range of 940-1040
m/z. Calculated values are in brackets.
[1OMe•(SCN)6](PF6)6 This complex was found to form X-ray quality crystals through slow diffusion of diisopropyl ether into an CH3CN solution of the complex. The complex was prepared according to the method described above with substitution of 4-methoxyaniline, tetra n-butyl ammonium thiocyanate and Fe(PF6)2·6CH3CN (synthesised according to a literature procedure 4).
LR-ESI-MS: m/z: 613.3(613.5) [1OMe•SCN6]6+, 765.3(765.2) [1OMe•SCN6](PF6)5+, 992.9(992.7) [1OMe•SCN6](PF6)2
4+, 1372.9(1371.9) [1OMe•SCN6](PF6)33+.
1.9 Attempted one pot synthesis of metal-organic cyclic [3]catenane 2. MM2 molecular modeling studies using CAChe5 identified diamine C (Supplementary Information section 1.3) as the most promising candidate, and S6, S7 and S8 (Supplementary Information section 1.4) as good candidates for the formation of cyclic [3]catenane 2. Dianilines C and S5-S15 (Supplementary Information section 1.4) were all screened using the one-pot preparative procedure below. All gave insoluble products that we infer to be polymeric. [3,3'6',2''5'',3'''-quaterpyridine]-6,6'''-dicarbaldehyde A (3 equiv.), [3,3'-bipyridine]-6,6'-dicarbaldehyde B (3 equiv.) and Fe(ClO4)2·6H2O (6 equiv.) were suspended in CH3CN (20 ml) and heated at 70 °C in a PTFE stoppered Schlenk tube until all the solids dissolved and a dark red solution was obtained. A solution of the appropriate diamine in CH3CN (6 equiv.) was added, followed by the slow dropwise addition of NBu4Br in CH3CN (6 equiv.). The reaction mixture was stirred at 70 °C for 36 hours. In all cases the product formed was an insoluble precipitate either in the reaction mixture or upon precipitation with diethyl ether.
[1OMe•SCN5 Cl](ClO4)24+
964.2(963.9)
[1OMe•SCN6](ClO4)24+
969.9(970.0)
[1OMe•SCN4 Cl2 ](ClO4)24+
958.4(958.6)
[1OMe•SCN5Br](ClO4)24+
975.3(974.9)
[1OMe•SCN4Br2 ](ClO4)24+
980.9(980.4)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 27
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
27
1.10 Synthesis of metal-organic cyclic [3]catenane 2 through dynamic imine exchange.
[2](BF4)6
Synthesis in methods section of main text.
The salts NaBr, NaBF4, KBr, KBF4 MgBr2, Mg(ClO4)2, RbBF4 were screened as additives to the mother liquor in order to aid crystallisation. The addition of KBr was found to produce crystals that gave the best resolution X-ray data. In addition, [2](ClO4)6 was synthesised from [1](ClO4)6 using the same procedure but was not found to produce crystals of sufficient quality. 1H NMR (400 MHz, CD3CN) δ 191.98, 179.98 br, 48.36, 25.68, 17.83, 13.80, 13.01, 8.76, 8.11, 7.16, 6.67, 6.59, 6.15, 5.46, 5.30, 5.07, 4.74, 4.59, 4.43, 4.34, 4.14, 3.65, 3.58, 1.15 19F NMR (376 MHz; CD3CN, reference C6F6): δ 151.04 (BF4
-)
Further assignments were hampered by the paramagnetic nature of the complex.
LR-ESI-MS: m/z: 809.48(809.51)( [2•Br6]6+, 988.78(988.77) [2•Br6](BF4)15+,
1257.98(1257.66) [2•Br6](BF4)24+
Elemental analysis: C, 50.23; H, 4.34; N, 7.23. Required [2](BF4)6 12H2O : C, 50.24; H, 4.65; N, 7.51.
Figure 25: 1H NMR (400 MHz, 298 K, CD3CN) of [2](BF4)6
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 28
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
28
Figure 26: LR-ESI-MS of [2](BF4)6. Calculated values are in brackets.
Figure 27: LR-ESI-MS of [2](BF4)6 in the range of 940-1040 m/z. Calculated values are in brackets. Peaks surrounding the principal 6+ and 4+ cations are assigned similarly.
[2•Br6]6+
809.48(809.51)
[2•Br6](BF4)24+
1257.98(1257.66)
[2•Br6](BF4)5+
988.78(988.77)
[2](BF4)NaBr5+ 1009.36(1009.35) [2](BF4)Na2BrCl5+
1019.11(1018.94)
[2•Br6](BF4)5+
988.78(988.77)
[2•Br6](Cl)5+
978.44(978.36)
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 29
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
29
Figure 28: (a) HR-ESI-mass spectrum of the [2]6+ ion. (b) Calculated isotopic distribution of [2]6+
a
b
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 30
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
30
Figure 29: (a) HR-ESI-mass spectrum of [2](Br)NaBr5+, [2](BF4)NaBr5+, [2](BF4)NaBF4
5+. (b) Calculated isotopic distribution of [2](Br)NaBr5+(blue), [2](BF4)NaBr5+(red), [2](BF4)NaBF4
5+(grey) in a 1:4:4 ratio.
a
b
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 31
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
31
1.11 Attempted synthesis of 2 in the absence of NaBr.
A 1.86 mM solution of [1](BF4)6 in CH3CN (2.33 ml, 4.33 mmol, 1 equiv.) was added to a stirred solution of C (11.9 mg, 28.0 μmol, 6.5 equiv.) in 2 ml CH3CN. The solution was degassed by the freeze pump thaw method 3 times and then heated at 70 °C. The reaction progress was monitored by LR-ESI-Mass spectrometry. After one day species were observed in the ESI-MS with m/z values corresponding to substitution of one or two C diamines for 1-4 4-chloroanilines. Subsequent spectra recorded after 2 days showed no distinct species suggesting decomposition of the complex under these conditions.
Figure 30: LR-ESI-MS after 1 day. Signals for partially substituted 1 are labelled indicating the number of dianiline C added and number of 4-chloroanilines removed.
1.12 Attempted synthesis of 2 using NBu4Br in place of NaBr. A 1.7 mM solution of [1](BF4)6 in CH3CN (1 ml, 1.7 μmol, 1 equiv.) was added to a
stirred solution of C (4.6 mg, 11 μmol, 6.5 equiv.) in 2 ml CH3CN and a 0.3 mM solution of NBu4Br in CH3CN (5.5 μl, 1.7 μmol, 1 equiv.). The solution was degassed by the freeze pump thaw method 3 times and then heated at 70 °C. The reaction progress was monitored by LR-ESI-Mass spectrometry. After one day some low intensity signals were observed in the ESI-MS (Figure 31) with m/z values corresponding to substitution of one or two C diamines for 1-4 4-chloroanilines, along with other fragments. Subsequent spectra recorded after 2 days showed no assignable signals, suggesting decomposition of the complex under these conditions.
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 32
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
32
Figure 31: LR-ESI-MS after 1 day. Tentatively assigned signals for partially substituted
1 are labelled indicating the number of dianiline C added and number of 4-chloroanilines removed.
1.13 Synthesis of 2 using LiBr, KBr and RbBr in place of NaBr.
LiBr, KBr and RbBr were screened using the same conditions as those used for NaBr as outlined in the methods section of the main text. The reactions were followed by LR-ESI-Mass spectrometry. After 5 days each reaction showed the full formation of 2 and the related alkali cation adducts, indicating that these cations also serve as competent templates.
1.14 Preliminary studies on length of ethylene glycol linker suitable for knotting using the subcomponent substitution method.
The MM2 molecular modeling studies mentioned earlier had identified C and to a lesser extent S6, S7 and S8 as the most promising candidates for the formation of cyclic [3]catenane 2. These were screened using the reaction conditions detailed for the synthesis of 2 in the methods section of the main text. Of these diamines only C and S7 (the tetraethylene and pentaethylene glycol derivatives) showed formation of the related [3]catenane although S7 produced a significant amount of insoluble polymer. Only partial substitution was observed for S8 even with extended reaction times (>21 days). S6 produced an insoluble polymer.
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 33
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
33
1.15 Addition of NBu4 Δ-TRISPHAT to 2
2 (8.12 mg, 1.5 μmol) was dissolved in 0.5 ml CD3CN and the solution was filtered. Aliquots (10 μl, 48 mmol, 0.3 equiv.) of a solution of [Tetrabutylammonium] [Δ-tris(tetrachloro-1,2 benzenediolato)phosphate(V)] TBA Δ-TRISPHAT in CD3CN were added and the solution mixed. After each addition the sample was analysed by 1H NMR spectroscopy. No changes were observed in the 200 – 10 ppm region of the spectrum. After the addition of 4 aliquots of Δ-TRISPHAT solution (i.e. 1.2 equiv. with respect to 2) a significant amount of precipitate was observed to form precluding further investigation.
Figure 32: 1H NMR (400 MHz, 298 K, CD3CN) of [2](BF4)6 after the addition of 1 equiv. (30 μl of a 48 mmol solution) of NBu4 Δ-TRISPHAT in CD3CN.
1.16 Reduction and demetalation of 2 to form a purely organic cyclic [3]catenane.
[2•Br6](BF4)6 (15.0 mg, 2.6 μmol, 1 equiv.) was dissolved in dry CH3CN (5 ml) and added dropwise to a stirred suspension of sodium borohydride (23.6 mg, 630 μmol, 250 equiv.) in CH3CN (5 ml) and the mixture was stirred at room temperature for 20 minutes. The reaction was quenched through the addition of a saturated solution of disodium EDTA in water (3 ml) and the resulting biphasic mixture was stirred at room temperature for 1 hour. The phases were separated and the aqueous fraction extracted with CHCl3:iPrOH 3 : 1 (3 × 10 mL). The organic fractions were combined dried with MgSO4 and the solvent removed in vacuo. The resulting reddish brown solid was quickly dissolved in CH2Cl2 with a drop of CH3OH, filtered and subjected to LCMS analysis.
The reaction produced a significant amount of insoluble precipitate both before and
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 34
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
34
after work up. Filtration of the final CH2Cl2:MeOH solution produced a solid that was insoluble in all solvents tried, including DMF, DMSO, acetone, ethyl acetate, methanol CHCl3, CH2Cl2 and 2 M HCl (aq). This is similar to what was observed by Stoddart et al. upon borohydride reduction of the imine-based Boromean rings.6 The presence of this solid, combined with poor separation obtained using both flash chromatography and preparative HPLC, hampered the isolation of the organic [3]catenane for further characterisation or metal ion reinsertion.
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 35
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
35
2 LCMS analysis
2.1 General conditions: CH3CN (Merck, LiChrosolv hyper grade for LCMS), water (H2O) (Rathburn, HPLC
grade), formic acid (FA) (Fluka, eluent additive for LCMS) and ammonium acetate (NH4OAc) (Fluka, eluent additive for LCMS) were used without further purification. Analytical separations were achieved by injecting 5 µl aliquots of a 8 mg/ml solution of the crude product from the reduction of 2 onto a reverse-phase Waters Symmetry Shield RP18 column (75 x 4.6 mm, 3.5 µm particle size and 100 pore size). UV/Vis absorbance was monitored at 261nm (16nm band width) and 318 nm (32nm band with) with a reference of 700nm (100nm band width). Separations were achieved by running through the HPLC column a linear gradient using CH3CN with 0.05%v/v of formic acid as solvent B and aqueous buffer (10 mM ammonium acetate and 0.1% v/v of formic acid, pH unadjusted) as solvent A at a flow rate of 2.0 ml min-1 and the HPLC column heated to 45 °C.
Time/min H2O (NH4OAc 10 mM) (0.1 % FA) CH3CN (0.05 % FA) 0 90 % 10 % 20 0 % 100 %
Table S2: Eluent gradient for LCMS
LC-MS was carried out on an Agilent 1100 LC/MSD trap XCT system composed of a high pressure mixing, binary pump with a low-volume static mixer, a micro vacuum degasser, an autosampler, a Peltier controlled thermostated column department with 3uL heat-exchange preheater, a diode array detector (1024 diodes from 190nm to 800 nm) with a 5ul, 6mm path semimicro flowcell and a MSD trap XCT with MSn capabilities fitted with an ElectroSpray Ion (ESI) source designed with orthogonal spray orientation. A divert switching valve was installed on the MS to direct the eluent to the MS or to waste, as required. The HPLC system was connected with low volume flexible stainless steel tubing to reduce the gradient delay and minimise diffusion of the sample once injected. A zero-volume splitter was installed as close as possible to the inlet of the ion source nebulising needle to ensure that the influx of analyte solution into the ESI ion source was maintained at a flow rate of around 50 µl/min and so that the delay between the UV and MS signal was minimised. ESI-MS spectra (positive ion) were acquired in Standard Enhanced mode (8100 s) in the m/z range of 200-2200 and the ionization parameters were set to operate with a drying temperature of 325 °C, nebulizer pressure of 12 psi and a drying gas flow of 5 l/min. Capillary voltage was set to -3500 V (high voltage end place offset of -500V) and an Ion Charge Control (ICC) target of 30,000 ions or accumulation time of 200 ms and an average taken every 5 spectra. Using the Agilent user-friendly SMART mode facility, the target mass was set to 2032 m/z, the compound stability to 20% and Trap Drive Level to 100% which equates to setting the Ion Optic voltages with a Capillary Exit of +121.5V, Octopole 1DC to +12V, Octopole 2DC to +4.5V, Octopole RF amplitude of 212Vpp, Lens 1 voltage of -5V, Lens 2 voltage of -60V, skimmer of 40V, a partition of 8.2 V and the Trap Drive to 171.3. The ions with m/z of 778.32, 813.78, 904.09, 1017.2 and 1355.64 were selected for MS/MS fragmentation with a fragmentation amplitude of 1.0 V (start at 20% and end at 100%). The first 1.2 minute of all LCMS eluents were directed to waste to avoid contaminating the ion source with strongly ionic compound eluting from the solvent front. HPLC with UV-Vis data was processed using HP ChemStation software and LCMS data was processed using the LC/MSD trap Data Analysis software from Bruker.
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 36
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
36
2.2 LCMS of reduced and demetalated 2
a: Reverse-phase HPLC trace of the mixture obtained upon reduction and demetalation of 2. Absorbance was recorded at 318 nm (32 nm bandwidth).
b: MS of reduced [3]catenane
c: MS/MS of reduced [3]catenane
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 37
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
37
d: MS of reduced [2]catenane
e: MS/MS of reduced [2]catenane
f: MS of macrocycle
Figure 33: (A) Reverse-phase HPLC trace of reduced and demetalated cyclic [3]catenane 2 recorded at 318 nm (32 nm band width) against a reference of 700 nm(100 nm band width), (B) MS of reduced [3]catenane 3, (C) MS/MS of reduced [3]catenane 3, (D) MS of reduced [2]catenane, (E) MS/MS of reduced [2]catenane, (F) MS of reduced macrocycle.
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 38
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
38
3 X-ray crystallography Data were collected at Beamline I19 of Diamond Light Source7 employing silicon double crystal monochromated synchrotron radiation (0.6889 Å) with ω scans at 100(2) K8. Data integration and reduction were undertaken with SAINT and XPREP9. Subsequent computations were carried out using the WinGX10 or ShelXle11 graphical user interfaces. Multi-scan empirical absorption corrections were applied to the data using SADABS9. Structures were solved by direct methods using SUPERFLIP12 then refined and extended with SHELXL-97 or SHELXL-201313. In general, non-hydrogen atoms with occupancies greater than 0.5 were refined anisotropically. Carbon-bound hydrogen atoms were included in idealised positions and refined using a riding model. Disorder was modeled using standard crystallographic methods including constraints, restraints and rigid bodies where necessary. Crystallographic data along with specific details pertaining to the refinement (inclusively addressing CheckCIF alerts) follow. Crystallographic data have been deposited with the CCDC (numbers 1022470-1022473).
[1Cl•Br6]·5ClO4·0.5FeBr4·10CH3CN·H2O (referred to as 1 in the main text).
CCDC 1022470
The elemental analysis of the isolated bulk material was not consistent with the presence of FeBr4
2− counterions. The single crystal subjected to diffraction was obtained from the crude reaction mixture of a small scale reaction. We infer the observation of FeBr4
2− anions in this structure was due to an excess of FeII and Br− present in this particular sample.
Figure 34: (a) Alternative view of the crystal strucure of [1] with LA coloured blue
and LB coloured red for clarity, (b) inner Fe3LB3 helicate, (c) outer Fe3LA
3 helicate. Non-coordinating anions, hydrogen atoms and solvent are omitted for clarity (Fe: purple, Br: brown).
Formula C194H146Br8Cl17Fe6.5N40O21, M 4978.45, Triclinic, space group P-1 (#2), a
16.0089(7), b 25.8622(10), c 29.8790(11) Å, 89.880(2), 89.852(2), 72.396(2)º, V
11791.3(8) Å3, Dc 1.402 g cm-3, Z 2, crystal size 0.01 by 0.01 by 0.01 mm, colour blue,
habit block, temperature 100(2) Kelvin, (synchrotron) 0.6889 Å, (synchrotron) 0.697 mm-
1, T(SADABS)min,max 0.6019, 0.7452, 2max 51.07, hkl range -19 20, -32 32, -37 37, N
124553, Nind 46520(Rmerge 0.0402), Nobs 28726(I > 2(I)), Nvar 2515, residuals* R1(F)
0.0751, wR2(F2) 0.2426, GoF(all) 1.053, min,max -1.440, 1.819 e- Å-3.
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 39
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
39
*R1 = ||Fo| - |Fc||/|Fo| for Fo > 2(Fo); wR2 = (w(Fo2 - Fc
2)2/(wFc2)2)1/2 all
reflections, w=1/[2(Fo2)+(0.1557P)2] where P=(Fo
2+2Fc2)/3
Specific refinement details: Despite appearing visually to be of good quality the crystals employed in this study proved to be weakly diffracting and rapidly suffered solvent loss. Rapid (<1 min) handling prior to flash cooling in the cryostream was required to collect data. Despite these measures and the use of synchrotron radiation few reflections at greater than 0.88 Å resolution were observed. There is a significant amount of thermal motion in the extremities of the molecule resulting in some higher than ideal thermal parameters and high Uiso min/max ratios. The perchlorate anions show substantial disorder and three were modelled as disordered over two locations with a number of bond length and thermal parameter restraints. One anion lattice site was modelled as a disordered mixture of perchlorate and FeBr4
2-, each with half occupancy. One CH3CN molecule was also modelled as disordered over two locations. The hydrogen atoms of the disordered water molecules were not modelled. Further reflecting the solvent loss and poor diffraction properties there is a significant amount of void volume in the lattice containing smeared electron density from disordered solvent. This area of diffuse electron density could not be successfully modelled and the SQUEEZE 14 function of PLATON 15 was employed. The remaining areas of electron density (up to 1.819 e- Å-3) are all close to bromine atoms, indicative of some minor unresolved disorder. CheckCIF gives one level B alert resulting from the high thermal motion of one chloride substituent.
[1Me•Cl6]·6ClO4·5CH3CN CCDC 1022471
Figure 35: X-ray crystal structure of [1Me•Cl6]·6ClO4. Non-coordinating anions,
hydrogen atoms and solvent are omitted for clarity (C: gray, N: blue, Fe: purple, Cl: green).
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 40
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
40
Formula C196H165Cl12Fe6N35O24, M 4155.13, Monoclinic, space group P21/n (#14), a
33.0443(19), b 19.1840(11), c 41.840(2) Å, 111.339(2), V 24705(2) Å3, Dc 1.117 g cm-3, Z 4, crystal size 0.01 by 0.01 by 0.01 mm, colour purple, habit block, temperature 100(2) Kelvin, (synchrotron) 0.68890 Å, (synchrotron) 0.533 mm-1, T(SADABS)min,max 0.2879, 0.7448, 2max 36.50, hkl range -30 30, -14 17, -38 38, N 107897, Nind
19387(Rmerge 0.1215), Nobs 12695(I > 2(I)), Nvar 2343, residuals* R1(F) 0.0919,
wR2(F2) 0.2713, GoF(all) 1.020, min,max -0.752, 1.065 e- Å-3. *R1 = ||Fo| - |Fc||/|Fo| for Fo > 2(Fo); wR2 = (w(Fo
2 - Fc2)2/(wFc
2)2)1/2 all
reflections, w=1/[2(Fo2)+(0.1929P)2] where P=(Fo
2+2Fc2)/3
Specific refinement details: The crystals employed in this study proved to be weakly diffracting and rapidly suffered solvent loss. Rapid (<1 min) handling prior to flash cooling in the cryostream was required to collect data. Despite these measures and the use of synchrotron radiation few reflections at greater than 1.1 Å resolution were observed. Nevertheless, the quality of the data is more than sufficient to establish the connectivity of the structure. There is a significant amount of thermal motion in the extremities of the molecule and a number of restraints were required for the realistic modelling of the toluidine groups. Even with these restraints the thermal parameters of these groups are larger than ideal and combined with the poor diffraction resulted in high hydrogen and carbon Uiso min/max ratios. The perchlorate anions show substantial disorder and four were modelled as disordered over two locations with a number of bond length and thermal parameter restraints. One CH3CN molecule was also modelled as disordered over two locations. Further reflecting the solvent loss and poor diffraction properties there is a significant amount of void volume in the lattice containing smeared electron density from disordered solvent. This area of diffuse electron density could not be successfully modelled and the SQUEEZE14 function of PLATON15 was employed. CheckCIF gives 2 A and 2 B level alerts. These alerts result from the large amount of thermal motion (high/low Ueqs and ratios) and the poor diffraction properties (low resolution) which have been described above.
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 41
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
41
[1OMe•(SCN)6]·4PF6 CCDC 1022472
Figure 36: X-ray crystal structure of [1OMe•(SCN)6]·4PF6. Non-coordinating anions and hydrogen atoms are omitted for clarity (C: gray, N: blue, O: red, Fe: purple, S: yellow). Formula C192H150F24Fe6N36O12P4S6, M=4260.81 g/mol, Monoclinic, space group Cc
(#9), a=42.114(3) Å, b=54.516(3) Å, c=14.0878(9) Å, 107.763(4), V=30802(3) Å3, Dc
0.919 g cm-3, Z 4, crystal size 0.05 by 0.04 by 0.02 mm3, colour purple, habit block, temperature 100(2) Kelvin, (synchrotron) 0.68890 Å, (synchrotron) 0.360 mm-1, T(SADABS)min,max 0.2730 0.4267, 2max 45.00, hkl range -46 39, -60 60, -15 15, N 154080, Nind 38792 (Rmerge= 0.2327, Rpim= 0.1256), Nobs= 22362 (I > 2(I)),
Nrestraints= 7373, Nvar= 2521, residuals* R1(F)= 0.1411, wR2(F2)= 0.3353, GoF(all)=
1.231, min,max= -0.899, 2.488 e Å-3. *R1 = ||Fo| - |Fc||/|Fo| for Fo > 2(Fo); wR2 = (w(Fo
2 - Fc2)2/(wFc
2)2)1/2 all
reflections, w=1/[2(Fo2)+(0.2P)2+0.0000P] where P=(Fo
2+2Fc2)/3
Specific refinement details: The crystals employed in this study proved to be weakly diffracting and rapidly suffered solvent loss. Rapid (<1 min) handling prior to flash cooling in the cryostream was required to collect data. Despite these measures and the use of synchrotron radiation few reflections at greater than 0.9 Å resolution were observed. Nevertheless, the quality of the data is more than sufficient to establish the connectivity of the structure. There is a significant amount of thermal motion in the extremities of the molecule. As in other cases of large and complicated supramolecular structures with high solvent content,16-18 the application of geometric restraint dictionaries for organic ligands and counter ions generated with the Grade Web Server (http://grade.globalphasing.org) helped build a complete molecular model. Atoms of ligand a (Q2I) and ligand b (T2I) as well as thiocyanate (SCN) and hexafluorophosphate (PF6) anions were grouped into residues. The corresponding residue identifiers employed are
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 42
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
42
given in brackets. Local structural similarity restraints (LSSR) exploiting non-crystallographic symmetry (NCS)19 were applied to additionally enhance similar geometries of each occurrence of ligand a and ligand b. Rigid bond restraints (RIGU) 20 and isotropic restraints (ISOR) were applied to the anisotropic displacement parameters of carbon, nitrogen, oxygen, fluorine, sulfur and phosphorus atoms. Even with these restraints the ADP ratio of some atoms near to disordered solvent/anion areas were not ideal. Disordered solvent/anion areas occupy 46% of the unit cell volume, which further reflects the solvent loss and poor diffraction properties. This area of diffuse electron density could not be successfully modelled with discrete atomic positions and the SQUEEZE14 function of PLATON15 was employed. Two PF6
− counter ions are expected to lie in the squeezed area, which is why there is no charge balance in the above given formula, as it only reflects the modelled part of the unit cell. Residual density peaks remaining near positions of iron ions and low bond precision on carbon-carbon bonds can be accounted for by high mosaicity and poor overall crystal quality, which is reflected by the high value of the (precision-indicating) R value of merged intensities (see above). CheckCIF gives 3 A and 8 B level alerts. These alerts (both A and B level) result from the large amount of thermal motion (high/low Ueqs and ratios, short contacts) and the poor diffraction properties (high residuals, low resolution, low bond precision) which have been described above.
[2]·6Br·0.75KBr·6.25H2O CCDC 1022473
Formula C234H234Br12.75Fe6K0.75N30O42.25, M 5525.78, Monoclinic, space group C 2/c
(#15), a 59.465(5), b 16.8956(15), c 62.436(5) Å, 116.189(3), V 56289(8) Å3, Dc 1.304 g
cm-3, Z 8, crystal size 0.031 by 0.025 by 0.011 mm, colour dark green, habit block, temperature 100(2) Kelvin, (synchrotron) 0.6889 Å, (synchrotron) 2.019 mm-1, T(SADABS)min,max 0.5461, 0.7440, 2max 32.98, hkl range -47 47, -13 13, -49 49, N
159461, Nind 15975(Rmerge 0.1276), Nobs 10664(I > 2(I)), Nvar 2605, residuals* R1(F)
0.1243, wR2(F2) 0.3586, GoF(all) 1.392, min,max -0.841, 0.945 e- Å-3. *R1 = ||Fo| - |Fc||/|Fo| for Fo > 2(Fo); wR2 = (w(Fo
2 - Fc2)2/(wFc
2)2)1/2 all reflections
w=1/[2(Fo2)+(0.2000P)2] where P=(Fo
2+2Fc2)/3
Specific refinement details: The crystals employed in this study proved to be weakly diffracting and rapidly suffered solvent loss. Rapid (<1 min) handling prior to flash cooling in the cryostream was required to collect data. Despite these measures and the use of synchrotron radiation few reflections at greater than 1.2 Å resolution were observed. Nevertheless, the quality of the data is more than sufficient to establish the connectivity of the structure. There is a significant amount of thermal motion in the structure and extensive thermal parameter and bond length restraints were required to facilitate realistic modelling for the organic parts of the structure. Bond lengths and angles within pairs of ligands in the rigid central core of the complex were restrained to be similar to each other. Two of the ethylene glycol chains were modelled as
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 43
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
43
disordered over two locations and all atoms in the ethylene glycol chains were refined with isotropic thermal parameters; bond length and anti-bumping restraints were applied where necessary to achieve a reasonable refinement. Even with these restraints the thermal parameters of these groups are larger than ideal and combined with the poor diffraction resulted in a high hydrogen and carbon Uiso min/max ratios. The thermal motion results in part from the presence of dynamic disorder in these groups which was not modelled; consequently there are some close contacts between a number of symmetry-generated ethylene glycol chains. One of the ethylene glycol chains is coordinated to a potassium ion; this potassium and the bromide coordinated to it were modelled with a fractional occupancy of 0.75. The bromide counterions were modelled as disordered over 16 partial occupancy sites and further areas of electron density were modelled as disordered water molecules, the hydrogen atoms of these disordered water molecules were not modelled. Further reflecting the solvent loss and poor diffraction properties there is a significant amount of void volume in the lattice containing smeared electron density from further disordered solvent. This area of diffuse electron density could not be successfully modelled and the SQUEEZE14 function of PLATON15 was employed. CheckCIF gives 5 A and 9 B level alerts. These alerts (both A and B level) result from the large amount of thermal motion (high/low Ueqs), poor diffraction properties (high residuals, low resolution, isotropic modelling, low bond precision, poor data/parameter ratio) and the disordered water molecules (short contacts, isolated oxygen atoms) which have been described above.
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 44
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
44
4 NMR spectra of organic compounds 1H NMR - 6,6'''-bis(dimethoxymethyl)-3,3'6',2''5'',3'''-quaterpyridine S3
© 2015 Macmillan Publishers Limited. All rights reserved
NATURE CHEMISTRY | www.nature.com/naturechemistry 45
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
45
13C NMR - 6,6'''-bis(dimethoxymethyl)-3,3'6',2''5'',3'''-quaterpyridine S3
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 46
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
46
1H NMR [3,3'6',2''5'',3'''-quaterpyridine]-6,6'''-dicarbaldehyde A
© 2015 Macmillan Publishers Limited. All rights reserved
NATURE CHEMISTRY | www.nature.com/naturechemistry 47
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
47
1H NMR 1,14-bis(4-nitrophenoxy)-3,6,9,12-tetraoxatetradecane S4
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 48
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
48
13C NMR - 1,14-bis(4-nitrophenoxy)-3,6,9,12-tetraoxatetradecane S4
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 49
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
49
1H NMR - 4,4'-((3,6,9,12-tetraoxatetradecane-1,14-diyl)bis(oxy))dianiline C
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 50
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
50
13C NMR - 4,4'-((3,6,9,12-tetraoxatetradecane-1,14-diyl)bis(oxy))dianiline C
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 51
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
51
1H NMR - 4,4'-((oxybis(ethane-2,1-diyl))bis(oxy))dianiline S5
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 52
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
52
13C NMR - 4,4'-((oxybis(ethane-2,1-diyl))bis(oxy))dianiline S5
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 53
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
53
1H NMR - 4,4'-(((ethane-1,2-diylbis(oxy))bis(ethane-2,1-diyl))bis(oxy))dianiline S6
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 54
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
54
13C NMR - 4,4'-(((ethane-1,2-diylbis(oxy))bis(ethane-2,1-diyl))bis(oxy))dianiline S6
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 55
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
55
1H NMR - 4,4'-((((oxybis(ethane-2,1-diyl))bis(oxy))bis(ethane-2,1-diyl))bis(oxy))dianiline S7
© 2015 Macmillan Publishers Limited. All rights reserved
NATURE CHEMISTRY | www.nature.com/naturechemistry 56
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
56
13C NMR - 4,4'-((((oxybis(ethane-2,1-diyl))bis(oxy))bis(ethane-2,1-diyl))bis(oxy))dianiline S7
© 2015 Macmillan Publishers Limited. All rights reserved
NATURE CHEMISTRY | www.nature.com/naturechemistry 57
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
57
1H NMR - 4,4'-((3,6,9,12,15-pentaoxaheptadecane-1,17-diyl)bis(oxy))dianiline S8
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 58
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
58
13C NMR - 4,4'-((3,6,9,12,15-pentaoxaheptadecane-1,17-diyl)bis(oxy))dianiline S8
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 59
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
59
1H NMR - 4,4'-(butane-1,4-diylbis(oxy))dianiline S9
© 2015 Macmillan Publishers Limited. All rights reserved
NATURE CHEMISTRY | www.nature.com/naturechemistry 60
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
60
13C NMR - 4,4'-(butane-1,4-diylbis(oxy))dianiline S9
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 61
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
61
1H NMR - 4,4'-(pentane-1,5-diylbis(oxy))dianiline S10
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 62
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
62
13C NMR - 4,4'-(pentane-1,5-diylbis(oxy))dianiline S10
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 63
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
63
1H NMR - 4,4'-(hexane-1,5-diylbis(oxy))dianiline S11
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 64
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
64
13C NMR - 4,4'-(hexane-1,5-diylbis(oxy))dianiline S11
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 65
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
65
1H NMR - 4,4'-(heptane-1,5-diylbis(oxy))dianiline S12
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 66
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
66
13C NMR - 4,4'-(heptane-1,5-diylbis(oxy))dianiline S12
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 67
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
67
1H NMR - 4,4'-(octane-1,5-diylbis(oxy))dianiline S13
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 68
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
68
13C NMR - 4,4'-(octane-1,5-diylbis(oxy))dianiline S13
© 2015 Macmillan Publishers Limited. All rights reserved

NATURE CHEMISTRY | www.nature.com/naturechemistry 69
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.2205
69
5 References
1 Riddell, I. A. et al. Five discrete multinuclear metal-organic assemblies from one ligand: deciphering the effects of different templates. J. Am. Chem. Soc. 135, 2723-2733, (2013).
2 Sørensen, A., Castilla, A. M., Ronson, T. K., Pittelkow, M. & Nitschke, J. R. Chemical signals turn on guest binding through structural reconfiguration of triangular helicates. Angew. Chem. Int. Ed. 52, 11273-11277, (2013).
3 Ayme, J. F. et al. A synthetic molecular pentafoil knot. Nature Chem. 4, 15-20, (2012). 4 Clegg, J. K. et al. A stimuli responsive system of self-assembled anion-binding Fe4L68+ cages.
Chem. Sci. 4, 68-76, (2013). 5 v. CAChe WorkSystem Pro (CAChe WorkSystem Pro, Version 7.5.0.85 ed., Fujitsu Limited,
2000 – 2006). 6 Peters, A. J., Chichak, K. S., Cantrill, S. J. & Stoddart, J. F. Nanoscale Borromean links for real.
Chem. Commun., 3394-3396, (2005). 7 Nowell, H., Barnett, S. A., Christensen, K. E., Teat, S. J. & Allan, D. R. J. Synchrotron Rad. 19,
435, (2012). 8 CrystalClear v. 2.0 (Rigaku Americas and Rigaku Corporation., 9009 TX, USA 1997-2009). 9 Bruker-Nonius. APEX, SAINT and XPREP. (Bruker AXS Inc., 2013). 10 Farrugia, L. J. WinGX and ORTEP for Windows: an update. J. Appl. Crystallogr. 45, 849-854,
(2012). 11 Hubschle, C. B., Sheldrick, G. M. & Dittrich, B. ShelXle: a Qt graphical user interface for
SHELXL. J. Appl. Crystallogr. 44, 1281-1284, (2011). 12 Palatinus, L. & Chapuis, G. SUPERFLIP - a computer program for the solution of crystal
structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 40, 786-790, (2007).
13 Sheldrick, G. A short history of SHELX. Acta Crystallogr. A64, 112-122, (2008). 14 van der Sluis, P. & Spek, A. L. BYPASS: an effective method for the refinement of crystal
structures containing disordered solvent regions. Acta Crystallogr. A46, 194-201, (1990). 15 PLATON: A Multipurpose Crystallographic Tool (Utrecht University, Utrecht, The
Netherlands, 2008). 16 Ronson, T. K. et al. Size-selective encapsulation of hydrophobic guests by self-assembled
M4L6 cobalt and nickel cages. Chem. Eur. J. 19, 3374-3382, (2013). 17 Pascu, M. et al. Anionic bipyridyl ligands for applications in metallasupramolecular
chemistry. Chem. Eur. J. 20, 5592-5600, (2014). 18 Schouwey, C. et al. Self-Assembly of a Giant Molecular Solomon Link from 30
Subcomponents. Angew. Chem. Int. Ed., 1-6, (2014). 19 Smart, O. S. et al. Exploiting structure similarity in refinement: automated NCS and target-
structure restraints in BUSTER. Acta Crystallogr. D68, 368-380, (2012). 20 Thorn, A., Dittrich, B. & Sheldrick, G. M. Enhanced rigid-bond restraints. Acta Crystallogr.
A68, 448-451, (2012).
© 2015 Macmillan Publishers Limited. All rights reserved