sulfonyl chlorides as versatile reagents for chelate ... · ii sulfonyl chlorides as versatile...
TRANSCRIPT

Sulfonyl Chlorides as Versatile Reagents for Chelate-Assisted C–H Bond Functionalizations
by
Elena Dimitrijević
A thesis submitted in conformity with the requirements for the degree of Masters of Science
Graduate Department of Chemistry University of Toronto
© Copyright by Elena Dimitrijević, 2009

ii
Sulfonyl Chlorides as Versatile Reagents for Chelate-Assisted
C–H Bond Funtionalizations
Elena Dimitrijević
Masters of Science
Department of Chemistry
University of Toronto
2009
Abstract
Despite the great abundance of C–H bonds in readily available starting materials, their use in
synthesis of functionalized molecules has been hampered by the high bond strengths, rendering
them inert to common organic reagents. However, recent progress in the field has addressed this
issue, enabling selective C–H bond functionalizations to be performed using catalytic transition
metal mediated processes.
Herein, the use of sulfonyl chlorides as versatile reagents for C–H bond functionalizations is
reported. Using chelation assistance, the regioselective conversion of C–H bonds to either C–S,
C–Cl or C–C bonds was achieved. The methodology development, substrate scope determination
and mechanistic investigations will be discussed.

iii
Acknowledgments
I would like to thank my thesis supervisor, Prof. Vy Dong, for giving me the opportunity to work
in her group and for all the guidance provided throughout my research. The experiences gained
will remain unforgotten, helping to guide me throughout my future career.
I thank Dr. Xiaodan Zhao for initiating the project that we collaborated on together. I greatly
appreciate all the useful and insightful discussions. I sincerely thank Marija Antonic for an
everlasting friendship. I would also like to thank Matthew Coulter, Boni Kim and Thi Phan for
their friendship and helpful discussions. Certainly, I thank the entire rest of the Dong Group,
including Wilmer Alkhas, for all the memories and for creating an enjoyable environment to
work in.
I also have to sincerely thank my family, friends and previous supervisors and co-workers for the
continuous support and encouragement.

iv
Table of Contents
Acknowledgments .......................................................................................................................... iii
Table of Contents ........................................................................................................................... iv
List of Tables ................................................................................................................................. vi
List of Figures .............................................................................................................................. viii
List of Abbreviations ..................................................................................................................... ix
Chapter 1 Introduction ................................................................................................................... 1
1.1 C–H Bond Functionalizations ............................................................................................. 1
1.1.1 Chemoselective C–H Bond Functionalizations ...................................................... 1
1.1.2 Intramolecular C–H Bond Functionalizations ........................................................ 3
1.1.3 Chelate-Assisted C–H Bond Functionalizations ..................................................... 6
1.1.3.1 Phenols as Directing Groups .................................................................... 7
1.1.3.2 Carbonyls as Directing Groups................................................................. 8
1.1.3.3 Amides as Directing Groups .................................................................... 9
1.1.3.4 Pyridines and Quinolines as Directing Groups....................................... 10
1.1.3.5 Other Chelate-Assisted C–H Bond Functionalizations .......................... 11
1.2 Sulfonyl Chlorides: Versatile Reagents in Cross-Coupling Reactions ............................. 12
1.3 Project Objectives: The use of Sulfonyl Chlorides as Versatile Reagents in Chelate-
Assisted C–H Bond Functionalizations ............................................................................ 13
1.4 Sulfones: Importance and Synthesis ................................................................................. 14
1.4.1 Traditional Routes Toward Sulfone Synthesis ..................................................... 15
1.5 Direct Carbon-Sulfur Bond Forming Reactions ............................................................... 16
1.6 Direct Carbon-Chlorine Bond Forming Reactions ........................................................... 17
1.7 Mechanistic Considerations of C–H Bond Functionalizations ......................................... 18
1.8 Previous Work Done By Group ........................................................................................ 21
Chapter 2 Results and Discussion ............................................................................................... 23

v
2.1 C–H Bond Sulfonylation with Sulfonyl Chlorides ........................................................... 23
2.1.1 C–H Bond Sulfonylation: Optimization ............................................................... 23
2.1.2 Substrate Scope for C–H Bond Sulfonylation ...................................................... 28
2.2 Substrate Synthesis ........................................................................................................... 32
2.3 C–H Bond Chlorination with Sulfonyl Chlorides ............................................................. 36
2.4 Mechanistic Considerations .............................................................................................. 38
2.4.1 Mechanism of C–H Bond Sulfonylation ............................................................... 38
2.4.2 Mechanism of C–H Bond Chlorination ................................................................ 45
2.5 C–H Bond Arylation with Sulfonyl Chlorides .................................................................. 53
Chapter 3 Conclusions ................................................................................................................. 65
Chapter 4 Experimental Procedures ............................................................................................ 66
4.1 General Information .......................................................................................................... 66
4.2 Experimental and Characterization Data .......................................................................... 67
4.2.1 General Procedure for Synthesis of 2-cyclohexenylpyridine ............................... 67
4.2.2 General Procedure for Synthesis of (E)-2-styrylpyridine ..................................... 68
4.2.3 General Procedure for Sulfonylation Reactions ................................................... 68
4.2.4 General Procedure for Chlorination Reactions ..................................................... 73
4.2.5 General Procedure for Synthesis of Pd(phpy)2 ..................................................... 74
4.2.6 General Procedure for Synthesis of PdIV
-Complex .............................................. 76
4.2.7 General Procedure for Reductive Elimination from PdIV
-Complex ..................... 76
4.2.8 General Procedure for Direct Arylation (Tables 28 and 29) ................................. 77
4.2.9 General Procedure for Direct Arylation (Tables 33 and 34) ................................. 78
Appendix A NMR Spectra ........................................................................................................... 80
Appendix B Crystallographic Information .................................................................................. 95

vi
List of Tables
Table 1. Sulfonylation under microwave conditions ................................................................... 24
Table 2. Effects of time, work-up and additive on C–H bond sulfonylation ............................... 25
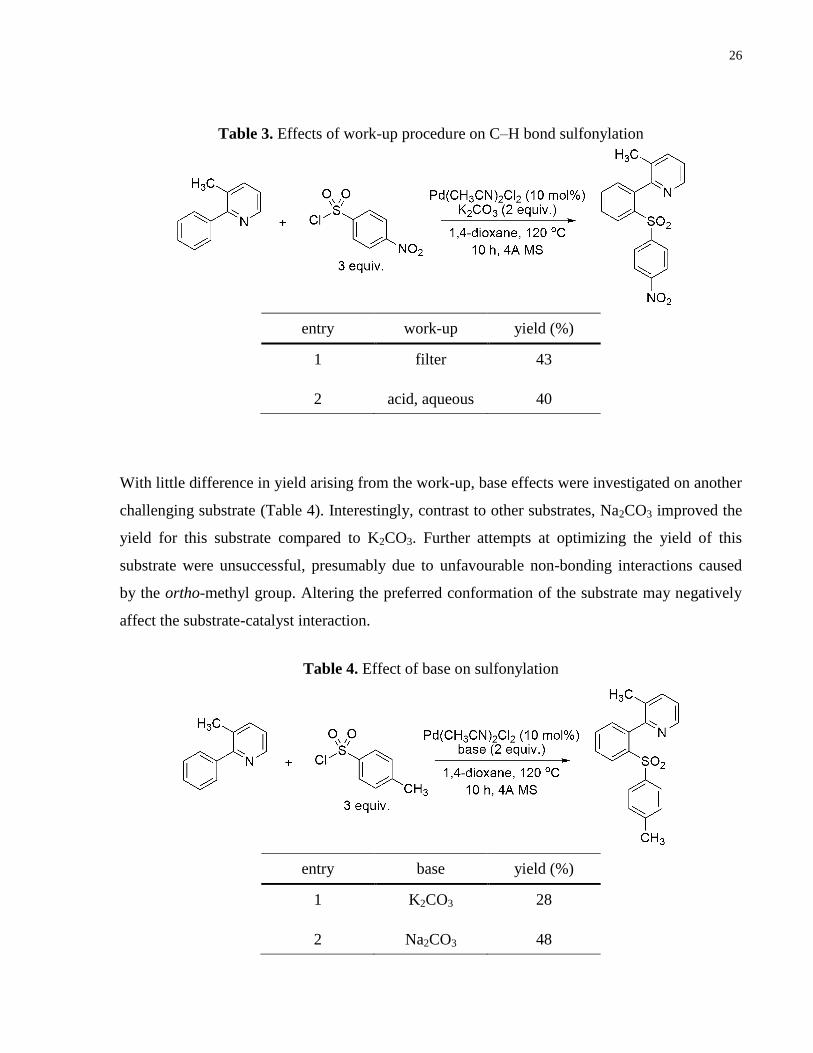
Table 3. Effects of work-up procedure on C–H bond sulfonylation ............................................ 26
Table 4. Effect of base on sulfonylation ...................................................................................... 26
Table 5. Time and base effects of pyrazole-directed C–H bond sulfonylation ............................ 27
Table 6. Effect of arylsulfonyl chloride equivalents on sulfonylation ......................................... 28
Table 7. Substrate scope of C–H bond sulfonylation ................................................................... 30
Table 8. Suzuki cross-coupling conditions in the synthesis of 2-o-tolylpyridine ........................ 32
Table 9. Suzuki cross-coupling conditions in the synthesis of 2-m-tolylpyridine ....................... 33
Table 10. Suzuki cross-coupling conditions in the synthesis of 2-(2-naphthyl)pyridine ............. 34
Table 11. Synthesis of 2-cyclohexenylpyridine ........................................................................... 35
Table 12. Synthesis of (E)-2-styrylpyridine ................................................................................. 35
Table 13. Subtrate scope of C–H bond chlorination .................................................................... 36
Table 14. Oxidative addition of arylsulfonyl chlorides to form PdIV
........................................... 43
Table 15. Reductive elimination from PdIV
to PdII ...................................................................... 45
Table 16. Dimethyl amine hydrochloride involvement in chlorination ....................................... 47
Table 17. Effect of various chloride sources in the chlorination of C–H bonds .......................... 48
Table 18. Chlorination using LiCl in various solvents ................................................................. 49
Table 19. Copper involvement in chlorination ............................................................................. 51

vii
Table 20. Pd involvement in chlorination .................................................................................... 52
Table 21. Effect of radical inhibitors (A) sulfonylation (B) chlorination .................................... 53
Table 22. Ligand screen for C–H arylation .................................................................................. 55
Table 23. Solvent screen for C–H arylation ................................................................................. 56
Table 24. Sulfonyl chloride equivalents screen ........................................................................... 56
Table 25. Base screen for C–H arylation ..................................................................................... 57
Table 26. Ligand equivalents screen ............................................................................................ 58
Table 27. Base equivalents screen ................................................................................................ 58
Table 28. Concentration effects on C–H bond arylation .............................................................. 59
Table 29. Substrates for C–H arylation ........................................................................................ 60
Table 30. Cationic Rh-catalysts for C–H arylation ...................................................................... 61
Table 31. Temperature effects on C–H arylation ......................................................................... 62
Table 32. Microwave conditions for C–H arylation .................................................................... 63
Table 33. Work-up effect on C–H arylation ................................................................................ 64
Table 34. Work-up effect on C–H arylation ................................................................................ 64

viii
List of Figures
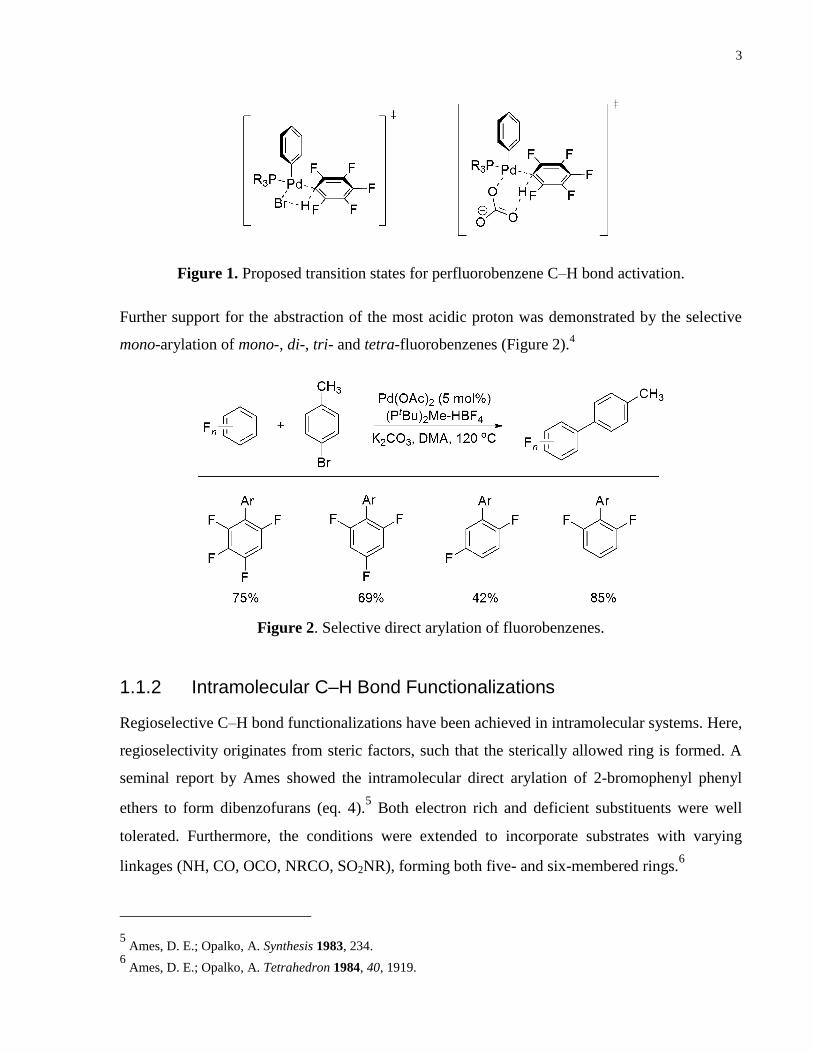
Figure 1. Proposed transition states for perfluorobenzene C–H bond activation. ......................... 3
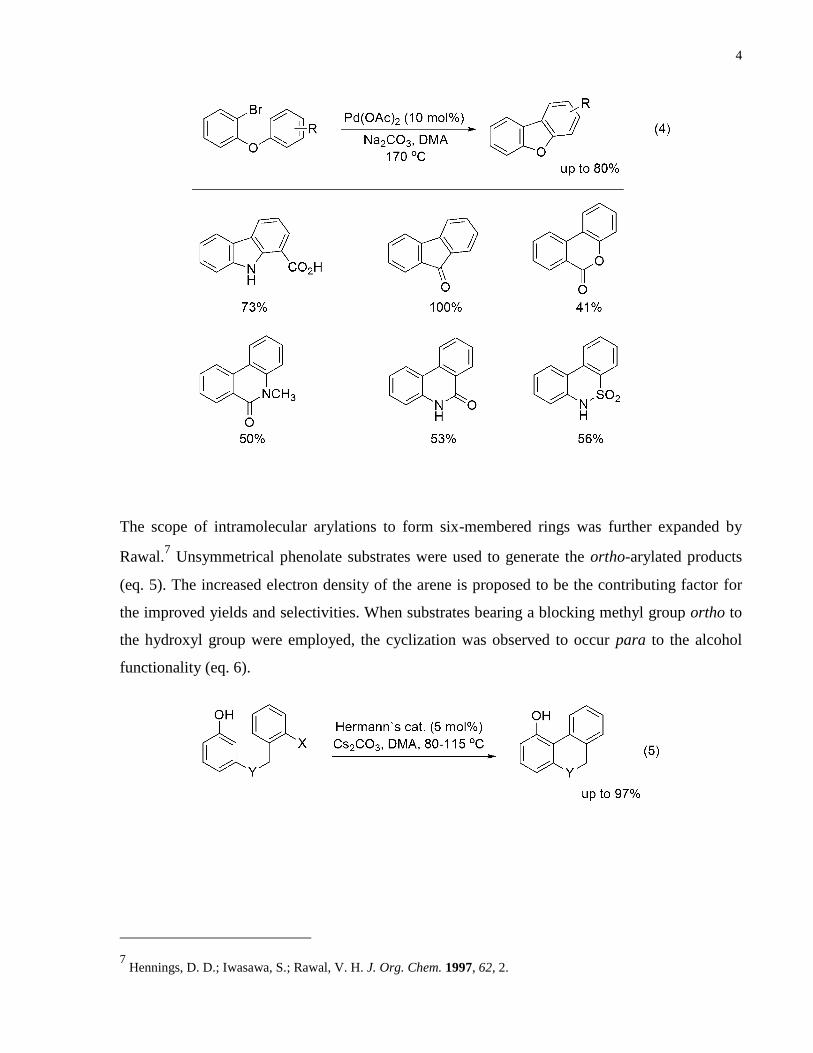
Figure 2. Selective direct arylation of fluorobenzenes. ................................................................. 3
Figure 3. Proposal for use of sulfonyl chlorides to function as sulfur, chlorine and carbon
delivering reagents in C–H bond functionalizations. .................................................................... 14
Figure 4. Sulfone-containing drugs. ............................................................................................. 15
Figure 5. Mechanistic proposal of chelate-assisted C–H bond activation. .................................. 19
Figure 6. Reductive elimination details involving Top: Mechanism A, Middle: Mechanism B
and Bottom: Mechanism C. .......................................................................................................... 21
Figure 7. Crystal structure of Table 7, entry 12 product, indicating that olefin isomerization
occurred. ........................................................................................................................................ 31
Figure 8. Pd0/Pd
II mechanistic proposal of C–H bond sulfonylation. .......................................... 38
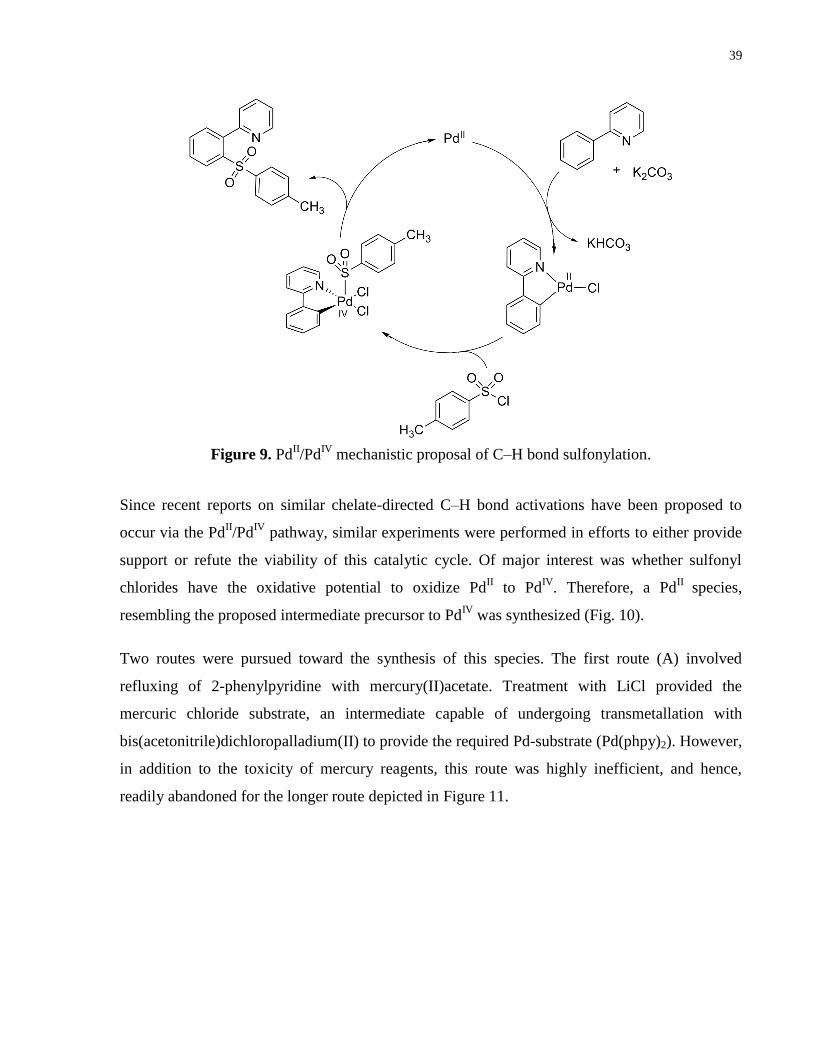
Figure 9. PdII/Pd
IV mechanistic proposal of C–H bond sulfonylation. ........................................ 39
Figure 10. Route A synthesis of PdII(phpy)2. ............................................................................... 40
Figure 11. Route B synthesis of PdII(phpy)2. ............................................................................... 41
Figure 12. Crstal structure of Pd(IV)-complex. ........................................................................... 44
Figure 13. Proposed mechanism of C–H bond chlorination. ....................................................... 46
Figure 14. Byproducts of (a) CuBr and (b) CuI. .......................................................................... 50

ix
List of Abbreviations
1H proton NMR
13C carbon 13 NMR
oC degrees Celsius
Δ heat
aq. aqueous
Ac acetyl
BHT butylhydroxytoluene
Bu butyl
cod 1,5-cyclooctadiene
Cp cyclopendadienyl
d doublet
dba dibenzylideneacetone
DCE 1,2-dichloroethane
DG directing group
DMA dimethylacetamide
DMF N,N-dimethylformamide

x
DMSO dimethyl sulfoxide
dppp 1,3-bis(diphenylphosphino)propane
EI electron impact
Et ethyl
equiv. equivalents
ESI electrospray ionization
FG functional group
GC-FID Gas Chromatography - Flame Ionization Detector
GC-MS Gas Chromatography - Mass Spectrometry
h hours
HRMS high resolution mass spectrometry
IR infrared spectrometry
M molar
m multiplet
M+ parent molecular ion
Me methyl
mg milligram

xi
MHz megahertz
min minutes
mL milliliters
mm millimeters
mmol millimoles
mol mole
MS molecular sieves
NBS N-bromosuccinimide
NHC N-heterocyclic carbene
NMP N-methylpyrrolidone
NMR nuclear magnetic resonance
n.r. no reaction
PhMe toluene
phpy 2-phenylpyridine
ppm parts per million
Pr propyl
r.t. room temperature

xii
s singlet
t triplet
TBAB tetra-N -butylammonium bromide
temp. temperature
Tf triflate
TFA trifluoroacetic acid
THF tetrahydrofuran
TLC thin layer chromatography
TMS trimethylsilyl
Ts tosyl
μL microliter
μw microwave

1
Chapter 1 Introduction
1.1 C–H Bond Functionalizations
Direct and selective elaboration of inert carbon-hydrogen (C–H) bonds to other functional groups
represents an important and long-standing goal in chemistry. Carbon-hydrogen bond
functionalization is an increasingly active area of research due to its broad potential in synthesis
as a result of the great abundance of C–H bonds in a variety of readily available organic
molecules, including natural products and petroleum. In addition to allowing the exploitation of
readily available starting materials, C–H bond functionalizations shorten reaction sequences and
enable the synthesis of compounds otherwise difficult to prepare.
Direct C–H bond functionalizations, however, are associated with two fundamental challenges.
The first challenge arises from the inert nature of C–H bonds. This problem has been addressed
by implementing transition metals as reaction catalysts. Transition metals have been
demonstrated as effective C–H bond activating agents via insertion into C–H bonds to form C–M
bonds. C–M bonds being more reactive than the C–H counterparts may subsequently be
converted to new functional groups under mild conditions.
The second challenge of C–H bond functionalization remains in achieving regioselectivity.
Among many elegant approaches toward addressing this issue, several are outlined below,
including chemoselective, intramolecular, and chelation-assisted approaches.1
1.1.1 Chemoselective C–H Bond Functionalizations
Although uncommon, several reports of intermolecular direct arylation of unfunctionalized
arenes have been achieved for a limited class of substrates. Regioselectivity in these substrates is
controlled via chemoselectivity, which is based on the inequivalent reactivity of the C–H bonds
present. In a report by Dyker, the direct arylation of azulene was achieved with selectivity for the
1 Alberico, D.; Scott, M. E.; Lautens, M. Chem. Rev. 2007, 107, 174.

2
C-1 position (eq. 1).2 This regioselectivity was attributed to the electron-rich character of C-1
and the dipolar nature of azulenes.
Direct arylation of benzene has also been achieved with iodobenzene derivatives under Ir-
catalysis (eq. 2).3 Various substitutions on the aryl iodide were tolerated to form the
corresponding biaryls in moderate yields. In this instance, regioselectivity is not an issue due to
the equivalency of all C–H bonds.
The final example involves the direct arylation of electron-deficient perfluorobenzenes with
various aryl halides (eq. 3).4 In this case, the resultant regioselectivity was attributed to the
acidity of the C–H bonds. This result was corroborated by computational studies, which revealed
that the key C–H bond functionalization occurs via a concerted arene-metallation/proton-
abstraction step. The proton abstraction was proposed to occur either via a ligand bicarbonate
ion or a ligated Br ion (Fig. 1).
2 Dyker, G.; Borowski, S.; Heiermann, J.; Korning, J.; Opwis, K.; Henkel, G; Kockerling, M. J. Organomet. Chem.
2000, 606. 108. 3 Fujita, K.-I.; Nonogawa, M.; Yamaguchi, R. Chem. Commun. 2004, 1926.
4 Lafrance, M.; Rowley, C. N.; Woo, T. K.; Fagnou, K. J. Am. Chem. Soc. 2006, 128, 8754.
3
Figure 1. Proposed transition states for perfluorobenzene C–H bond activation.
Further support for the abstraction of the most acidic proton was demonstrated by the selective
mono-arylation of mono-, di-, tri- and tetra-fluorobenzenes (Figure 2).4
1.1.2 Intramolecular C–H Bond Functionalizations
Regioselective C–H bond functionalizations have been achieved in intramolecular systems. Here,
regioselectivity originates from steric factors, such that the sterically allowed ring is formed. A
seminal report by Ames showed the intramolecular direct arylation of 2-bromophenyl phenyl
ethers to form dibenzofurans (eq. 4).5 Both electron rich and deficient substituents were well
tolerated. Furthermore, the conditions were extended to incorporate substrates with varying
linkages (NH, CO, OCO, NRCO, SO2NR), forming both five- and six-membered rings.6
5 Ames, D. E.; Opalko, A. Synthesis 1983, 234.
6 Ames, D. E.; Opalko, A. Tetrahedron 1984, 40, 1919.
Figure 2. Selective direct arylation of fluorobenzenes.
4
The scope of intramolecular arylations to form six-membered rings was further expanded by
Rawal.7 Unsymmetrical phenolate substrates were used to generate the ortho-arylated products
(eq. 5). The increased electron density of the arene is proposed to be the contributing factor for
the improved yields and selectivities. When substrates bearing a blocking methyl group ortho to
the hydroxyl group were employed, the cyclization was observed to occur para to the alcohol
functionality (eq. 6).
7 Hennings, D. D.; Iwasawa, S.; Rawal, V. H. J. Org. Chem. 1997, 62, 2.

5
Extensive work by the Fagnou Group has widened the scope of intramolecular C–H bond
functionalizations. With their improved conditions, otherwise unreactive substrates underwent
cyclization in the presence of low catalyst loadings. A variety of six-membered ring biaryls were
formed bearing both electron-donating and electron-withdrawing substituents in excellent yields,
including alkyl and nitrogen-bearing tethers. Even more challenging seven-membered rings were
formed in excellent yields (eq. 7).8
This methodology, however, was limited to aryl bromides, with aryl chlorides resulting in no
reaction. However, upon the implementation of an electron-rich NHC ligand, a variety of
functionalized five and six-membered rings were prepared in excellent yields with varying
tethers including ether, amine, amide and alkyl functionalities (eq. 8).9
8 Campeau, L.-C.; Parisien, M.; Fagnou, K. J. Am. Chem. Soc. 2004, 126, 9186.
9 Campeau, L.-C.; Thanssandote, P.; Fagnou, K. Org. Lett. 2005, 7, 1857.

6
Following these studies, the same group developed an efficient, general catalyst system for the
intramolecular direct arylation of a broad scope of aryl bromides, chlorides and iodides to
generate numerous five- and six-membered carbo- and heterocyclic biaryl compounds (eq. 9).10
1.1.3 Chelate-Assisted C–H Bond Functionalizations
The last strategy that will be discussed for controlling regioselectivity of C–H bond
functionalizations involves chelation-assistance. Since this method is based on the coordinating
ability of a tethered organic moiety, common directing groups typically bear a lone pair of
electrons that can coordinate to transition metals, several of which will be outlined below. This
method, through the use of a range of different directing groups, has greatly diversified the
substrate scope of intermolecular C–H bond functionalizations.
The use of a directing group to control the regioselectivity of a transition metal insertion into a
C–H bond was first reported by Kleinman and Dubeck (eq. 10).11
Since their report, this strategy has been widely implemented. The coordinating group aids in
directing the insertion such that either the kinetically or thermodynamically-favoured five- or
six-membered metallacycle is formed. One disadvantage is that both mono- or di-functionalized
products ortho to the directing group can be obtained in symmetrical substrates. However, for
10 Campeau, L.-C.; Parisien, M.; Jean, A.; Fagnou, K. J. Am. Chem. Soc. 2006, 128, 581.
11 Kleinman, J. P.; Dubeck, M. J. Am. Chem. Soc. 1963, 85, 1544.

7
unsymmetrical substrates, sterics become the controlling factor and the resultant
functionalization occurs predominantly at the less hindered ortho-position.
1.1.3.1 Phenols as Directing Groups
Miura reported the selective intermolecular arylation of 2-phenylphenols (eq. 11) and naphthols
(eq. 12), in which hydroxyl group-directed cyclopalladation comprised the key C–H bond
activation step.12
The selectivity of the mono- versus di-arylated products could be controlled by
varying the excess of the iodo coupling partner.
Bedford (eq. 13)13
and Oi (eq. 14)14
independently reported the ortho-arylation of phenols using
Rh-catalysis. In both instances, the selectivity for the mono-functionalized product was achieved
by implementing substrates bearing a blocking group.
12 (a) Satoh, T.; Kawamura, Y.; Miura, M.; Nomura, M. Angew. Chem., Int. Ed. Engl. 1997, 36, 1740. (b) Satoh, T.;
Inoh, J.I.; Kawamura, Y., Miura, M.; Nomura, M. Bull. Chem. Soc. Jpn. 1998, 71, 2239. 13
(a) Bedford, R. B.; Coles, S. J.; Hursthouse, M. B.; Limmert, M. E.; Angew. Chem., Int. Ed. 2003, 42, 112. (b)
Bedford, R. B.; Limmert, M. E. J. Org. Chem. 2003, 68, 8669. 14
Oi. S.; Watanabe, S.-I.; Fukita, S.; Inoue, Y. Tetrahedron Lett. 2003, 44, 8665.

8
1.1.3.2 Carbonyls as Directing Groups
Kakiuchi has shown that ketones may also function as directing groups in C–H bond arylations
in the presence of organometallic coupling partners via Ru-catalysis. Aryl ketones with
phenylboronic esters were successfully coupled, with selectivity for the mono- versus di-arylated
products, and vice-versa, being dependent on steric constraints (eq. 15).15
Isopropyl phenyl
ketone furnished the di-arylated species as the major product, whereas bulkier groups such as
tert-butyl exclusively favoured the formation of the mono-arylated poduct. The steric repulsion
between the tert-butyl group and the phenyl ring are proposed to inhibiting the second C–H bond
cleavage.
More recent use of aldehydes as directing groups has been achieved with benzaldehydes (eq.
16).16
Interestingly, the selectivity for the mono- or di-arylated product was found to be
dependent on the coupling partner employed. Aryl chlorides selectively resulted in the formation
of mono-arylated species, while aryl bromides favoured the formation of di-arylated products.
15 (a) Kakiuchi, F.; Kan, S.; Igi, K.; Chatani, N.; Miura, S. J. Am. Chem. Soc. 2003, 125, 1698. (b) Kakiuchi, F.;
Matsuura, Y.; Kan. S.; Chatani, N. J. Am. Chem. Soc. 2005, 127, 5936. 16
Gurbuz, N.; Ozdemir, I.; Cetinkaya, B. Tetrahedron Lett., 2005, 46, 2273.
9
1.1.3.3 Amides as Directing Groups
As mentioned, Miura devised conditions that were successful in direct arylations using alcohols
and ketones as directing groups and subsequently applied them to amide directing groups.
Benzanilides were successfully converted to the corresponding di-arylated species in good to
excellent yields (eq. 17). 17
Thus far, the majority of reactions utilized aryl halides as coupling partners. Sanford, conversely,
reported an amide-directed C–H bond arylation using hypervalent iodide species as the oxidant
(eq. 18).18
Notably, these conditions are insensitive to air and moisture and do not require the use
of expensive ligands or strong bases. The mechanism, which will be elaborated in latter sections,
was proposed to involve a PdII/Pd
IV catalytic cycle.
17 Kametani, Y.; Satoh, T.; Miura, M.; Nomura, M. Tetrahedron Lett. 2000, 41, 2655.
18 Kalyani, D.; Deprez, N. R.; Desai, L. V.; Sanford, M. S. J. Am. Chem. Soc. 2005, 127, 7330.

10
1.1.3.4 Pyridines and Quinolines as Directing Groups
In addition to using the above developed conditions on amide directing groups, Sanford has also
shown that arylation with hypervalent iodine species is possible with pyridine and quinoline
directing groups (eq. 19).18
Again, the extent of mono- and di-arylation was steric-dependent.
However, when this methodology was applied to alternative arylating agents, such as
iodobenzene, no desired product was obtained.
Conditions devised by the Daugulis Group have demonstrated that pyridine-directed arylations
with aryl iodides can be achieved with Pd-catalysis in the presence of silver acetate salts for
substrates such as 2-phenylpyridine and 7,8-benzoquinoline (eq. 20).19
The above mentioned directing groups are only representative examples of the wide range that
are used. Imines, oxazolines, imidazolines and pyrazoles, among many others, are also
commonly employed. Moreover, as shown, a variety of transition metal catalysts have also been
successfully utilized. Therefore, this form of intermolecular C–H bond functionalization enables
the incorporation of a range of substrates. Despite the above examples focusing solely on
arylations, which were chosen to exhibit the variety of directing groups that may be employed,
19 Shabashov. D.; Daugulis, O.; Org. Lett. 2005, 7, 3657.
11
this form of C–H bond functionalization is amenable to other reactions, further expanding the
usefulness of this methodology.
1.1.3.5 Other Chelate-Assisted C–H Bond Functionalizations
Yu has demonstrated that alkylations can also be achieved with boroxines and boronic acids as
coupling partners (eq. 21).20
Sanford has also extended the use of hypevalent iodides as coupling partners to include direct C–
H bond oxygenations (eq. 22).21
Furthermore, Sanford has also displayed the halogenation of C–H bonds using N-
halosuccinimide reagents (eq. 23).22
20 Chen, X.; Goodhue, C. E.; Yu, J.-Q. J. Am. Chem. Soc. 2006, 128, 12634.
21 Dick, A. R.; Hull, K. L.; Sanford, M. S. J. Am. Chem. Soc. 2004, 126, 2300.
22 Kalyani, D.; Dick, A. R.; Anani, W. Q.; Sanford, M. S. Org. Lett. 2006, 8, 2523.

12
1.2 Sulfonyl Chlorides: Versatile Reagents in Cross-Coupling Reactions
While there has been great progress in the field of chelate-assisted C–H bond functionalizations,
versatile and economical coupling partners remain limited. In efforts to develop systems
incorporating these criteria, it was observed that sulfonyl chlorides are inexpensive reagents that
have the ability to perform as three independent functional group delivering agents in cross
coupling reactions. They have been implemented in sulfonylation, chlorination and arylation
reactions.
Sulfonyl chlorides have been used in the synthesis of biaryl sulfones via Suzuki-type couplings
with boronic acids (eq. 24). 23
The Yu Group has shown the ability of sulfonyl chlorides, when combined with phenylsilane, to
act as chlorinating agents in cobalt-catalyzed hydrochlorinations (eq. 25).24
Furthermore, Vogel has achieved desulfitative arylation of a number of substrates using sulfonyl
chlorides. Arylation with sulfonylchlorides has gained the most attention, as it has successfully
been applied to a number of cross-coupling reactions, including, but not limited to, Stille (eq.
26),25
Suzuki (eq. 27)26
and Sonogashira (eq. 28)27
cross-couplings.
23 Bandgar, B. P.; Bettigeri, S. V.; Phopase, J. Org. Lett. 2004, 6, 2105.
24 Gaspar, B.; Carreira, E. M. Angew. Chem. Int. Ed. 2008, 47, 5758.
25 Dubbaka, S. R.; Vogel, P. J. Am. Chem. Soc. 2003 125, 15292.
26 Dubbaka, S. R.; Vogel, P. Org. Lett. 2004, 6, 95.

13
1.3 Project Objectives: The use of Sulfonyl Chlorides as Versatile Reagents in Chelate-Assisted C–H Bond Functionalizations
Based on this literature precedence, Dr. Xiaodan Zhao, a postodoctoral fellow in the group,
envisioned that sulfonyl chlorides can be used as versatile reagents in C–H bond
functionalization reactions. He proposed that with this sole reagent, C–H bonds can selectively
be transformed to C–S, C–Cl or C–C bonds (Fig. 3). This approach would enable the use of
readily available, cost-effective cross-coupling reagents capable of performing a number of
useful transformations, as well as enabling the use of unfunctionalized substrates. This overall
process would enable efficient synthesis of synthetically and medicinally relevant molecules
with minimal steps, greater yields and minimal waste production.
27 Dubbaka, S. R.; Vogel, P. Adv. Synth. Catal. 2004, 346, 1793.

14
Chelate-assisted C–H bond functionalization was chosen as the method of choice due to its wide
applicability to a number of substrates and catalysts. Also, the organic directing group
component chosen for initial screening was pyridine, as it represents a motif prevalent in
biologically significant molecules.
1.4 Sulfones: Importance and Synthesis
The first of the above outlined reactions involves the synthesis of sulfones. One of the main
reasons as to why it would be of importance to develop an efficient and catalytic synthesis of
sulfones is that they represent a structural motif prevalent in many medicinal targets due to their
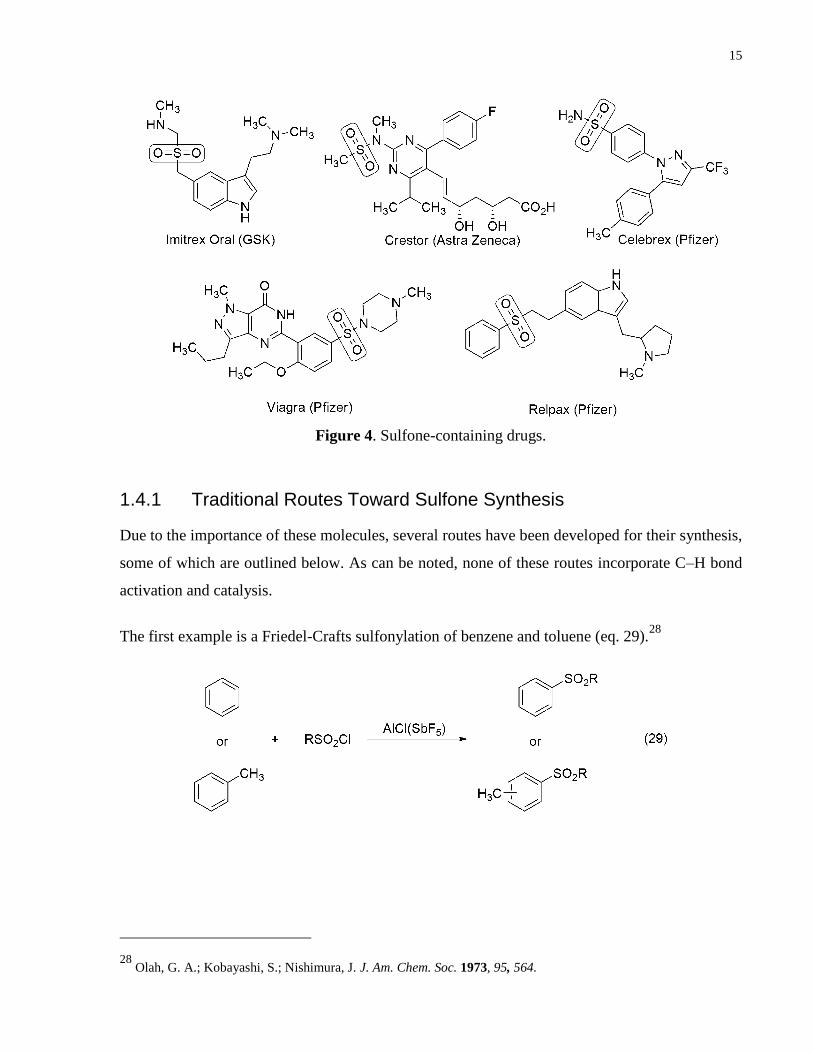
biological activities. Several of the Top 200 Drugs containing sulfone moieties are illustrated
below (Fig. 4).
Figure 3. Proposal for use of sulfonyl chlorides to function as sulfur, chlorine and
carbon delivering reagents in C–H bond functionalizations.
15
1.4.1 Traditional Routes Toward Sulfone Synthesis
Due to the importance of these molecules, several routes have been developed for their synthesis,
some of which are outlined below. As can be noted, none of these routes incorporate C–H bond
activation and catalysis.
The first example is a Friedel-Crafts sulfonylation of benzene and toluene (eq. 29).28
28 Olah, G. A.; Kobayashi, S.; Nishimura, J. J. Am. Chem. Soc. 1973, 95, 564.
Figure 4. Sulfone-containing drugs.

16
The following two examples illustrate Pd-catalyzed routes toward sulfone synthesis using pre-
functionalized substrates (eq. 30 and 31).29,30
1.5 Direct Carbon-Sulfur Bond Forming Reactions
Therefore, in addition to providing a novel, more efficient route toward sulfones, the above
proposed reaction sequence creates a new approach toward the direct transformation of C–H to
C–S bonds. In order to assess the novelty of this transformation, it is of interest to consider
analogous processes.
Inamoto has developed conditions for the intramolecular direct C–H to C–S bond forming
processes to form five-membered sulfur-containing heterocycles such as phenylbenzothiophenes
(eq. 32)31
and phenylbenzothiazoles (33).32
29 Cacchi, S.; Fabrizi, G.; Gogiamani, A.; Parisi, M. N. Org. Lett. 2002, 4, 4719.
30 Bandagar, B. P.; Bettigeri, S. V.; Phopase, J. Org. Lett. 2004, 6, 2105.
31 Inamoto, K.; Arai, Y.; Hiroya, K.; Doi, T. Chem. Commun. 2008, 5529.
32 Inamoto, K.; Hasegawa, C.; Hiroya, K.; Doi, T. Org. Lett. 2008, 10, 5147.
17
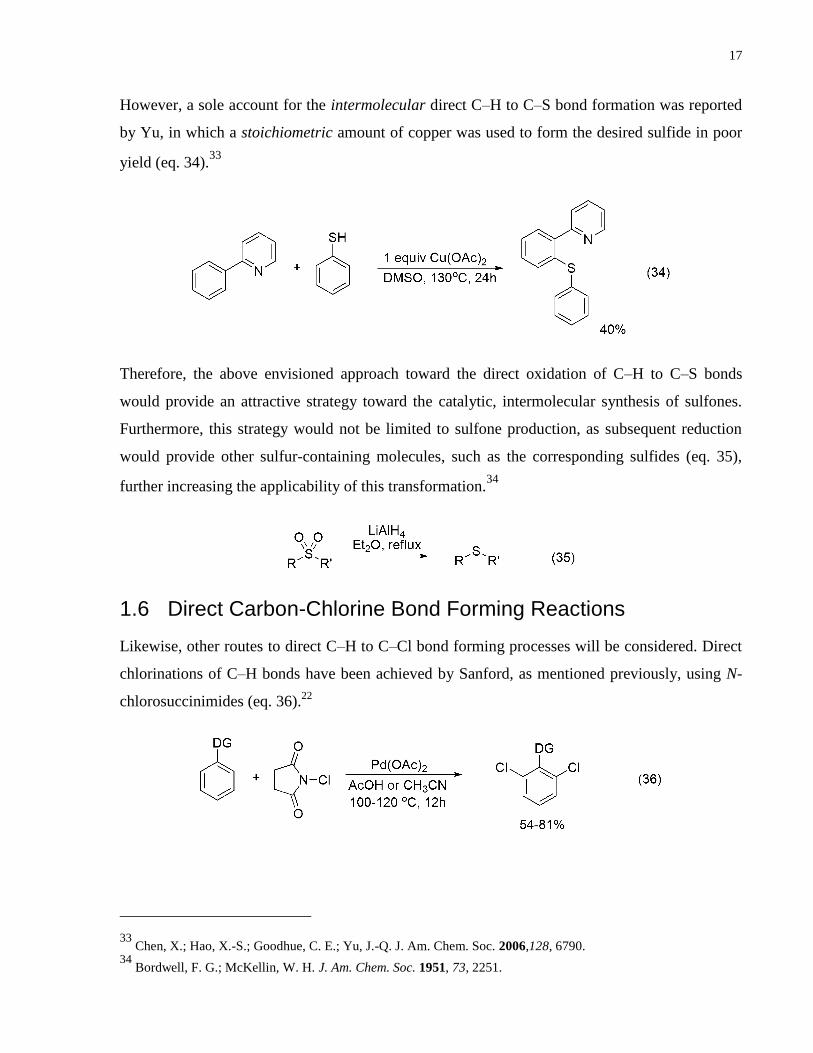
However, a sole account for the intermolecular direct C–H to C–S bond formation was reported
by Yu, in which a stoichiometric amount of copper was used to form the desired sulfide in poor
yield (eq. 34).33
Therefore, the above envisioned approach toward the direct oxidation of C–H to C–S bonds
would provide an attractive strategy toward the catalytic, intermolecular synthesis of sulfones.
Furthermore, this strategy would not be limited to sulfone production, as subsequent reduction
would provide other sulfur-containing molecules, such as the corresponding sulfides (eq. 35),
further increasing the applicability of this transformation.34
1.6 Direct Carbon-Chlorine Bond Forming Reactions
Likewise, other routes to direct C–H to C–Cl bond forming processes will be considered. Direct
chlorinations of C–H bonds have been achieved by Sanford, as mentioned previously, using N-
chlorosuccinimides (eq. 36).22
33 Chen, X.; Hao, X.-S.; Goodhue, C. E.; Yu, J.-Q. J. Am. Chem. Soc. 2006,128, 6790.
34 Bordwell, F. G.; McKellin, W. H. J. Am. Chem. Soc. 1951, 73, 2251.

18
Yu has also displayed that 1,1,2,2-tetrachloroethane can act as a chlorinating agent of 2-
phenylpyridine derivatives with copper-catalysis (eq. 37).35
Therefore, it was envisioned that sulfonylchlorides, which are economically competitive to these
reagents, could act as complementary chlorinating agents.
1.7 Mechanistic Considerations of C–H Bond Functionalizations
Reaction condition development is greatly facilitated by mechanistic insights of similar
reactions. Therefore, previously proposed mechanistic details of chelate-assisted C–H bond
functionalizations will be considered. The below diagram schematically depicts the sequence of
steps proposed to occur in these types of reactions (Fig. 5). Upon oxidative addition, substrate
complexation to the metal center occurs through the chelating group. Such substrate-catalyst
binding selectively brings one C–H bond to close proximity, thus enabling metal-assisted C–H
bond activation. Finally, reductive elimination affords the product, regenerating the active
catalyst. As mentioned, various metals and substrates have been implemented, and thus, the
order of oxidative addition and coordination may be reversed, depending on the metal and
substrate combination used.
35 Chen, X.; Hao, X.-S.; Goodhue, C. E.; Yu, J.-Q. J. Am. Chem. Soc. 2006, 128, 6790.

19
Due to the great success of palladium catalysis in chelate-assisted C–H bond activations, it
represents the catalyst chosen for initial condition screening. Hence, literature precedence of
mechanistic elucidation of palladium-catalyzed reactions will be of main focus. From the above
cycle, it can be noted that the oxidation state of the metal varies by two throughout the cycle. For
palladium, this implies that the cycle involves either Pd0/Pd
II or Pd
II/Pd
IV intermediates. Work by
the Sanford Group has pioneered the delineation between these two possible pathways, and
hence, will be considered in detail.
Upon substrate binding, functional group deliverance may occur in either of the three
possibilities proposed (eq. 38). Palladium(II) may either directly be oxidized to PdIV
, direct
electrophilic cleavage of the PdII–C bond may occur without change in the oxidation state,
similarly to SE2 processes, or a single electron mechanism involving PdIII
may be involved.36
Generally, the PdII/Pd
IV sequence is proposed when strong oxidants such as hypervalent iodide
species are used as the coupling partners. This proposal is supported by the observation that
when hypervalent iodide is replaced by other oxidants, such as PhI, PhOTf, benzoquinone or
Cu(OAc)2, which are common oxidants for Pd0 to Pd
II, no reaction occurs.
37,18
36 Whitfield, S. R.; Sanford, M. S. J. Am. Chem. Soc. 2007, 129, 15142.
37 Dick, A. R.; Hull, K. L.; Sanford, M. S. J. Am. Chem. Soc. 2004, 126, 2300.
Figure 5. Mechanistic proposal of chelate-assisted C–H bond activation.

20
For further study on the plausibility of PdIV
intermediate likelihood, analogous species syntheses
were sought. However, the low stability of PdIV
complexes, arising from their propensity to
undergo side reactions such as C–C bond forming reductive elimination and intermolecular alkyl
ligand exchange, required the rational design of intermediates that would overcome these
undesired pathways. Therefore, cyclometallated pyridine compounds with aryl versus alkyl
ligands were envisioned as optimal at stabilizing PdIV
, preventing ligand exchange and C–C
bond forming reductive elimination, on account of system rigidity.38
PdII(phpy)2 was found to incorporate the necessary criteria to stabilize high oxidation states,
containing a rigid phenylpyridine metallacycle with electron-donating nitrogen ligands. This PdII
intermediate was found to react with numerous oxidants, including several of the previously
mentioned ones (PhICl2 and NCS), to form stable PdIV
intermediates. Further studies indicated
that upon subsequent subjection of these complexes to reductive elimination conditions, the
desired products were generated.36
The reductive elimination step was considered in more detail. Either of the three proposals of
Figure 6 were considered.38
Because studies indicated that the reductive elimination rate was
independent of solvent polarity, the ionic pathway of Mechanism A was discarded, as ionic
reductive eliminations typically show a great dependence on solvent polarity. Moreover, to
distinguish between the final two mechanisms, it was noted that if dissociation of one of the
bidentate ligands was required, such as in Mechanism C, the exchange of 2-phenylpyridine for a
more rigid system, such as with benzo[h]quinoline, the reaction rate would decrease. This was
indeed observed, supporting Mechanism C. Furthermore, the final product dissociation step has
been proposed to either occur through intramolecular C–X bond forming reductive elimination
from the metal center or via attack by an external nucleophile in an SN2-like reaction.
38 Dick. A. R.; Kampf, J. W.; Sanford, M. S. J. Am. Chem. Soc. 2005, 127, 12790.

21
Therefore, the overall cycle is proposed to occur via chelate-assisted C–H bond activation to
form a cyclopalladated intermediate, which upon oxidation forms a PdIV
intermediate that can
undergo C–heteroatom bond forming reductive elimination to furnish the desired product.
1.8 Previous Work Done By Group
With the above disclosed research goals, the initial aim was in developing a regioselective
sulfone synthesis. Using 2-phenylpyridine and p-toluenesylfonyl chloride, optimization reactions
were performed by Dr. Zhao by varying reaction parameters such as catalyst, solvent,
temperature, base and additives. The use of Pd2(dba)3 and Pd(OAc)2 as catalyst precursors
resulted in only trace sulfone production. However, using Pd(CH3CN)2Cl2, in the presence of
K2CO3 in 1,4-dioxane at 120 oC for 6 hours, the ortho-sulfonylated product was isolated in 82%
yield (eq. 39).39
Therefore, the first intermolecular transition metal catalyzed C–H bond
activation to form a C–S bond was achieved.
39 Zhao, X.; Dimitrijević, E.; Dong, V. M. J. Am. Chem. Soc. 2009, 131, 3466.
Figure 6. Reductive elimination details involving Top: Mechanism A, Middle:
Mechanism B and Bottom: Mechanism C.
22
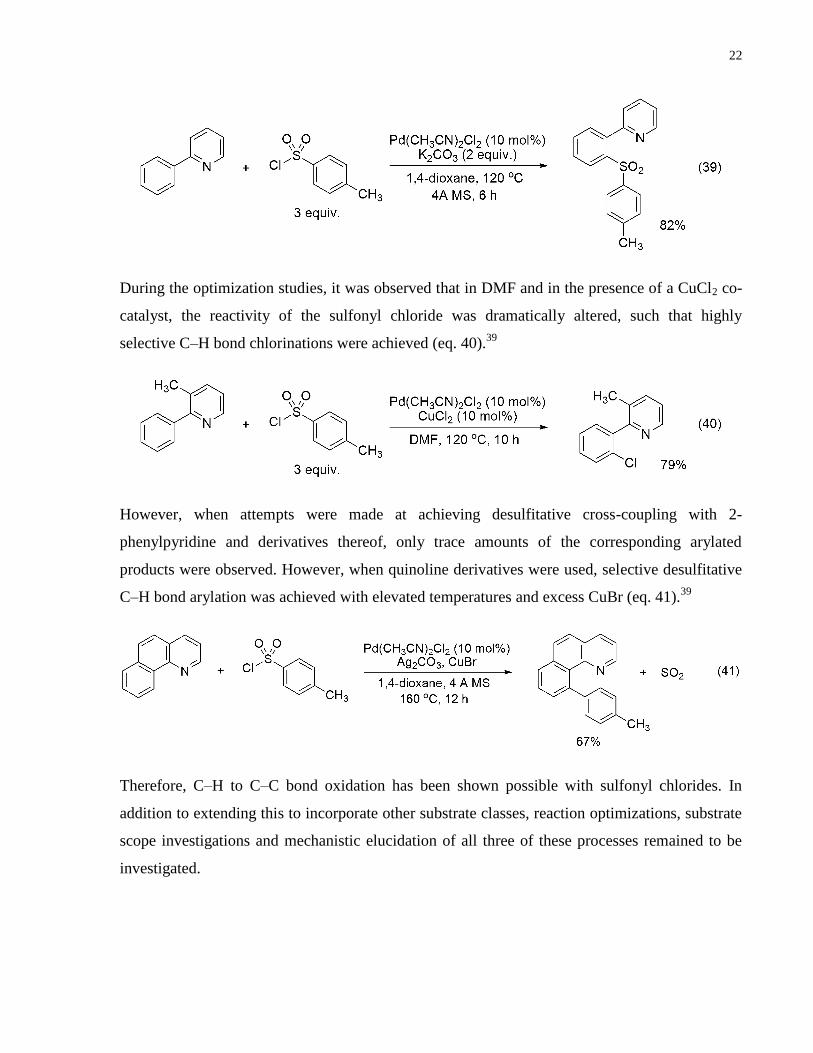
During the optimization studies, it was observed that in DMF and in the presence of a CuCl2 co-
catalyst, the reactivity of the sulfonyl chloride was dramatically altered, such that highly
selective C–H bond chlorinations were achieved (eq. 40).39
However, when attempts were made at achieving desulfitative cross-coupling with 2-
phenylpyridine and derivatives thereof, only trace amounts of the corresponding arylated
products were observed. However, when quinoline derivatives were used, selective desulfitative
C–H bond arylation was achieved with elevated temperatures and excess CuBr (eq. 41).39
Therefore, C–H to C–C bond oxidation has been shown possible with sulfonyl chlorides. In
addition to extending this to incorporate other substrate classes, reaction optimizations, substrate
scope investigations and mechanistic elucidation of all three of these processes remained to be
investigated.

23
Chapter 2 Results and Discussion
2.1 C–H Bond Sulfonylation with Sulfonyl Chlorides
As mentioned previously, Pd-catalyzed C–H bond sulfonylation of 2-phenylpyridine with
arylsulfonyl chlorides has been developed in our group. In efforts to expand this methodology,
initial focus has been on further condition optimization. Efficiency improvement was targeted by
increasing yield, decreasing reaction time, lowering catalyst loading, and importantly, increasing
the scope, to encompass other directing groups, sulfonyl chlorides and electronically and
sterically demanding substrates.
2.1.1 C–H Bond Sulfonylation: Optimization
In efforts to reduce reaction time, microwave conditions were explored (Table 1). Palladium(II)
and palladium(0) sources were explored in the forms of PdCl2 (entries 1-5) and Pd(PPh3)4 (entries
6-9). At 120 °C, for 30 minutes, in a variety of solvents, no product was obtained.

24
entry [Pd] solvent yield (%)
1 PdCl2 THF n.r.
2 PdCl2 toluene n.r.
3 PdCl2 1,4-dioxane n.r.
4 PdCl2 o-dichlorobenzene n.r.
5 PdCl2 NMP n.r.
6 Pd(PPh3)4 THF n.r.
7 Pd(PPh3)4 toluene n.r.
8 Pd(PPh3)4 1,4-dioxane n.r.
9 Pd(PPh3)4 o-dichlorobenzene n.r.
Since the attempted microwave conditions only resulted in recovery of starting materials,
emphasis was redirected to optimize the previously devised conditions. In efforts to optimize
these, the effect of the work-up procedure, catalyst and additive effects were briefly considered
as potential areas of improvement (Table 2). Entries 1 and 2 indicate that a basic work-up
resulted in only a marginal yield improvement. Decreasing the reaction time under these
conditions had a negative effect on the reactivity (entry 3). Addition of a copper co-catalyst did
not improve the yield (entry 4) and other Pd(II) sources, such as Pd(OAc)2, resulted in much
lower yields (entry 5).
Table 1. Sulfonylation under microwave conditions
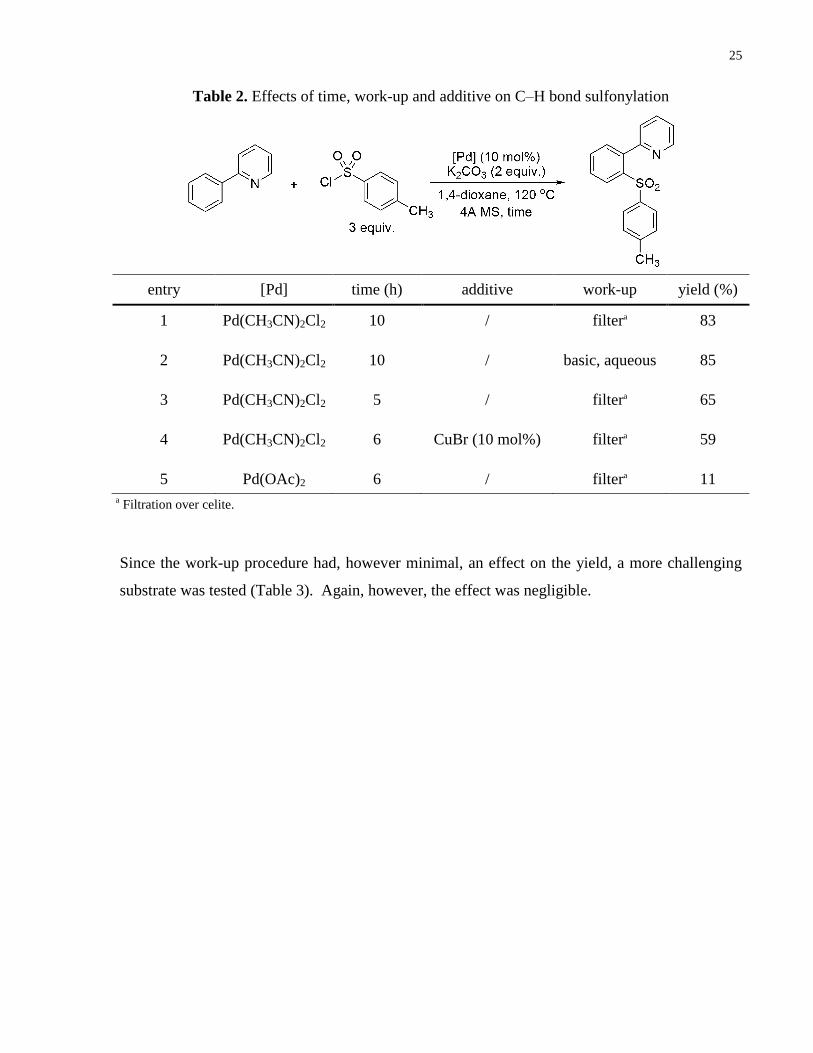
25
entry [Pd] time (h) additive work-up yield (%)
1 Pd(CH3CN)2Cl2 10 / filtera 83
2 Pd(CH3CN)2Cl2 10 / basic, aqueous 85
3 Pd(CH3CN)2Cl2 5 / filtera 65
4 Pd(CH3CN)2Cl2 6 CuBr (10 mol%) filtera 59
5 Pd(OAc)2 6 / filtera 11
Since the work-up procedure had, however minimal, an effect on the yield, a more challenging
substrate was tested (Table 3). Again, however, the effect was negligible.
Table 2. Effects of time, work-up and additive on C–H bond sulfonylation
a Filtration over celite.
26
entry work-up yield (%)
1 filter 43
2 acid, aqueous 40
With little difference in yield arising from the work-up, base effects were investigated on another
challenging substrate (Table 4). Interestingly, contrast to other substrates, Na2CO3 improved the
yield for this substrate compared to K2CO3. Further attempts at optimizing the yield of this
substrate were unsuccessful, presumably due to unfavourable non-bonding interactions caused
by the ortho-methyl group. Altering the preferred conformation of the substrate may negatively
affect the substrate-catalyst interaction.
entry base yield (%)
1 K2CO3 28
2 Na2CO3 48
Table 3. Effects of work-up procedure on C–H bond sulfonylation
Table 4. Effect of base on sulfonylation

27
Expanding this methodology to include directing groups other than pyridine was also of interest.
Therefore, a substrate bearing a pyrazole directing group was tested under the devised conditions
(Table 5, entry 1). Although the sulfone was produced, the reaction was low yielding. Only slight
improvement was observed by prolonged reaction times (entry 2). Exchange of base to Na2CO3
was ineffective, as reduced yields were obtained (entry 3).
entry time (h) base yield (%)
1 6 K2CO3 41
2 10 K2CO3 46a
3 10 Na2CO3 39
The final attempts at optimization were directed at determining whether such an excess of 3
equivalents of the sulfonyl chloride was necessary (Table 6). A decrease and an increase of the
equivalents by a factor of two resulted in greatly diminished yields (entries 1 and 3). Therefore, 3
equivalents of the sulfonyl chloride were found to be optimal. This can be attributed to the
byproduct observed as a result of sulfonyl chloride homocoupling. As a result, an excess is
required, such that sufficient amounts remain to couple to 2-phenylpyridine. However, a greater
excess may result in the preferential rate acceleration of the byproduct formation over the desired
product.
Table 5. Time and base effects of pyrazole-directed C–H bond sulfonylation
a Combined yield of the mono- and di-functionalized products produced in a 5.3:1
ratio, respectively.

28
entry Tos-Cl equiv. yield (%)
1 1.5 54
2 3 81a
3 6 53
2.1.2 Substrate Scope for C–H Bond Sulfonylation
A compiled series of substrates used for the herein developed C–H bond sulfonylation (Table 7)
is provided below. The selected substrates were chosen to vary sterically and electronically and
to contain different directing groups. Furthermore, various sulfonyl chloride coupling partners
were explored.
2-Phenylpyridine coupled with p-toluenesulfonyl chloride resulted in the production of the
sulfone in an isolated yield of 85% (entry 1). Substrates bearing both meta- and para-methyl
substituents were successfully coupled to p-tosyl chloride to furnish the corresponding products
in good yields of 91% and 81%, respectively (entries 3 and 4). In both of these instances the
reactions proceeded to form both the mono- and di-substituted products in 6.6:1 and 9.7:1 ratios,
respectively, in favour of the mono-substituted product. However, a limitation to this
methodology was uncovered with a more sterically hindered ortho-methyl substituted substrate
that resulted in the formation of the sulfone in a modest yield of 48% (entry 2). The use of 3-
methyl-2-phenylpyridine proved to be most effective at balancing the required steric strain
Table 6. Effect of arylsulfonyl chloride equivalents on sulfonylation
a Combined yield of the mono- and di-functionalized products
produced in a 9.7:1 ratio, respectively.

29
necessary to preclude difunctionalization, thereby ensuring full selectivity for the mono-
functionalized product, meanwhile permitting sulfonylation to occur in good yields. When this
substrate was coupled to m-toluenesulfonyl chloride, the resultant product was obtained in 85%
yield with the selective formation of the mono-substituted product (entry 5).
The effect of substrate electronics on reactivity were assessed by the use of a naphthyl
substituent, the result of which indicated a dramatic decrease in the yield (38%, entry 6). This
was attributed to the decreased arene aromaticity, impacting the electronic nature of the C–H
bond. To further investigate this effect, two alkenyl substrates were synthesized (entries 11 and
12). The cyclohexenyl substrate underwent sulfonylation to produce an inseperable mixture of
isomers (entry 11), whereas (E)-2-styrylpyridine produced the unexpected trans product over cis
(entry 12, Fig. 740
). Both of these data suggest that sulfonylation of alkene C–H bonds occurs not
via direct C–H bond activation, but perhaps via a Heck type mechanism, thereby accounting for
the observed bond isomerizations. In light of the probability that a different mechanism is
involved for this class of substrates, the discrepancy of the yield of the naphthyl substituent
compared to other substrates is less surprising.
Although these reaction conditions were optimized for pyridine-directed C–H bond
sulfonylations, other directing groups were also explored. Pyrazole, as mentioned, directed the
sulfonylation, resulting in the formation of the product in a moderate yield of 46% (entry 7).
However, when an amide directing group was implemented, no reactivity was observed (entry
8).
As can be noted from the introduction (Chapter 1, Fig. 4), sulfones are a prevalent motif among
pharmaceutically relevant agents. Moreover, it can also be noted that a major class of these
sulfones are of the sulfonamide form. Hence, several preliminary reactions were run using this
methodology to test whether it can be extended to sulfamoyl chlorides (entries 9 and 10).
Unfortunately, neither of these resulted in the production of the desired sulfamides. This may be
a result of catalyst deactivation in the presence of these specific reagents. Other, less basic
materials would be worth investigating in the future.
40 Crystal structure obtained by Dr. Zhao.
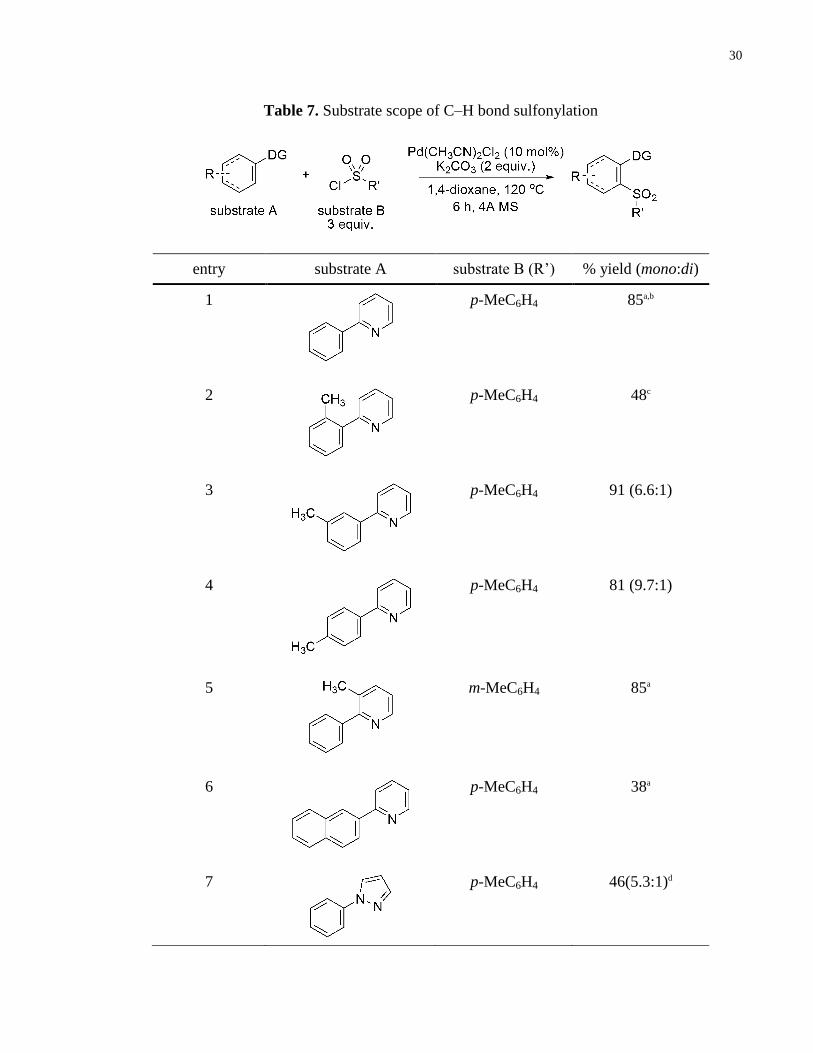
30
entry substrate A substrate B (R’) % yield (mono:di)
1
p-MeC6H4 85a,b
2
p-MeC6H4 48c
3
p-MeC6H4 91 (6.6:1)
4
p-MeC6H4 81 (9.7:1)
5
m-MeC6H4 85a
6
p-MeC6H4 38a
7
p-MeC6H4 46(5.3:1)d
Table 7. Substrate scope of C–H bond sulfonylation
31
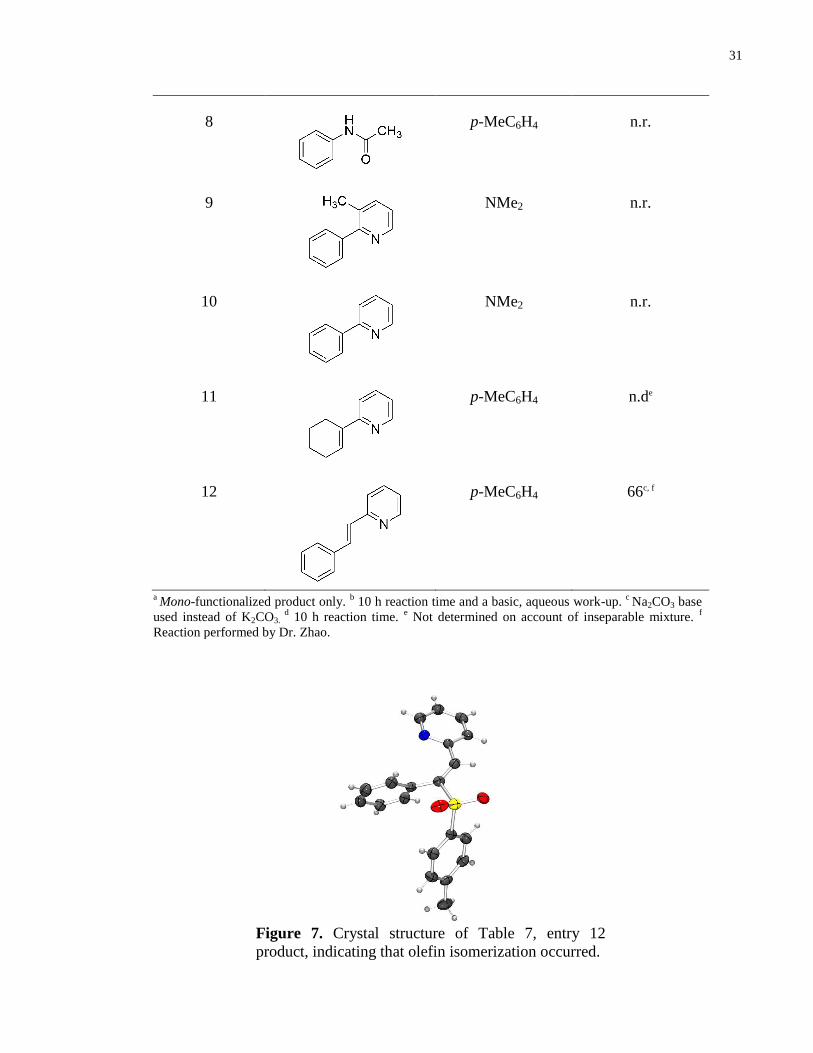
8
p-MeC6H4 n.r.
9
NMe2 n.r.
10
NMe2 n.r.
11
p-MeC6H4 n.de
12
p-MeC6H4 66c, f
a Mono-functionalized product only.
b 10 h reaction time and a basic, aqueous work-up.
c Na2CO3 base
used instead of K2CO3. d 10 h reaction time.
e Not determined on account of inseparable mixture.
f
Reaction performed by Dr. Zhao.
Figure 7. Crystal structure of Table 7, entry 12
product, indicating that olefin isomerization occurred.

32
2.2 Substrate Synthesis
The majority of the substrates depicted above were synthesized via disconnection of the two
arenes, the coupling of which was envisioned to be possible under Suzuki cross-coupling
conditions. However, this was found to be difficult for the sterically encumbered boronic acid
coupling partners. The retrosynthetic analysis of 2-o-tolylpyridine is shown below (eq. 42).
Halogen-lithium exchange of the brominated precursor, followed by trapping and hydrolysis of
the boronic ester would produce the corresponding boronic acid. The coupling of the boronic
acid to 2-phenylpyridine was attempted under several Suzuki conditions, none of which provided
the substrate in excellent yields (Table 8). However, over the course of trials, enough material
was obtained for the test reaction (Table 7, entry 2).
entry conditions overall yield (%)
1 Pd2(dba)3 (1 mol%), PCy3 (2 mol%)
KF, THF, 40 oC
2.4
2 Pd(OAc)2 (1 mol%), PPh3 (4 mol%)
KF, THF, 75 oC
10
3 Pd(OAc)2 (0.5 mol%), TBAB
K2CO3, THF, 50 oC
n.r.
Table 8. Suzuki cross-coupling conditions in the synthesis of 2-o-tolylpyridine
33
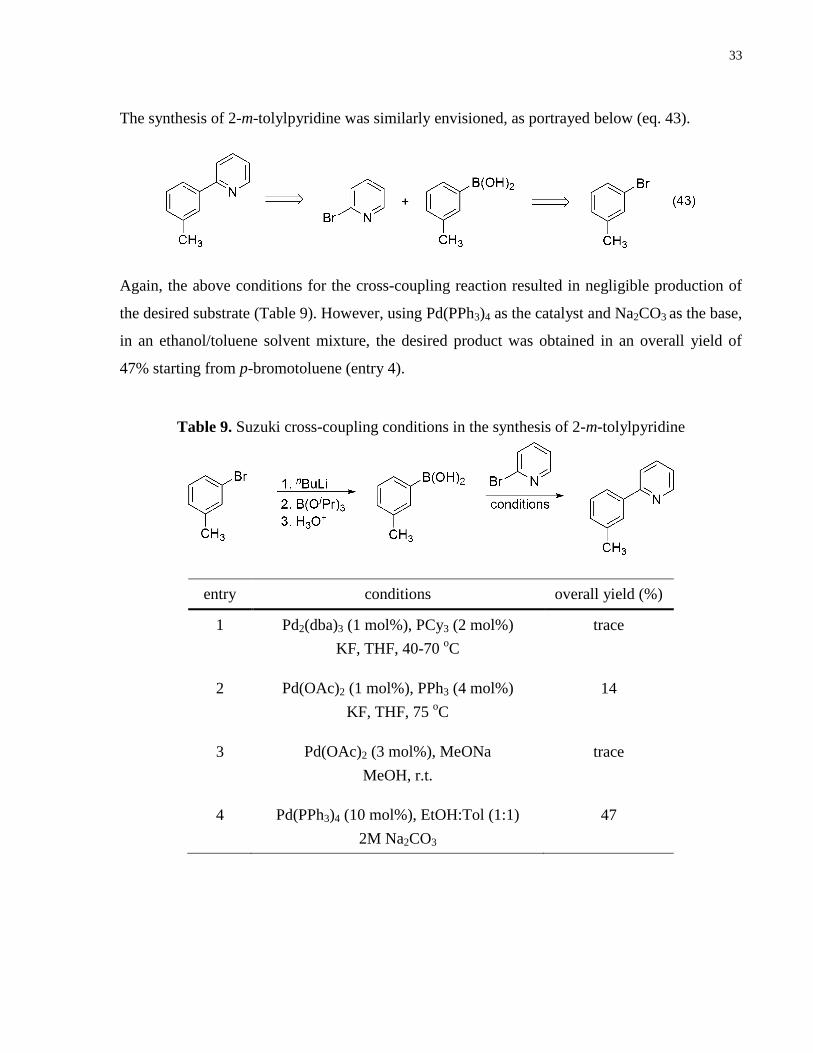
The synthesis of 2-m-tolylpyridine was similarly envisioned, as portrayed below (eq. 43).
Again, the above conditions for the cross-coupling reaction resulted in negligible production of
the desired substrate (Table 9). However, using Pd(PPh3)4 as the catalyst and Na2CO3 as the base,
in an ethanol/toluene solvent mixture, the desired product was obtained in an overall yield of
47% starting from p-bromotoluene (entry 4).
entry conditions overall yield (%)
1 Pd2(dba)3 (1 mol%), PCy3 (2 mol%)
KF, THF, 40-70 oC
trace
2 Pd(OAc)2 (1 mol%), PPh3 (4 mol%)
KF, THF, 75 oC
14
3 Pd(OAc)2 (3 mol%), MeONa
MeOH, r.t.
trace
4 Pd(PPh3)4 (10 mol%), EtOH:Tol (1:1)
2M Na2CO3
47
Table 9. Suzuki cross-coupling conditions in the synthesis of 2-m-tolylpyridine
34
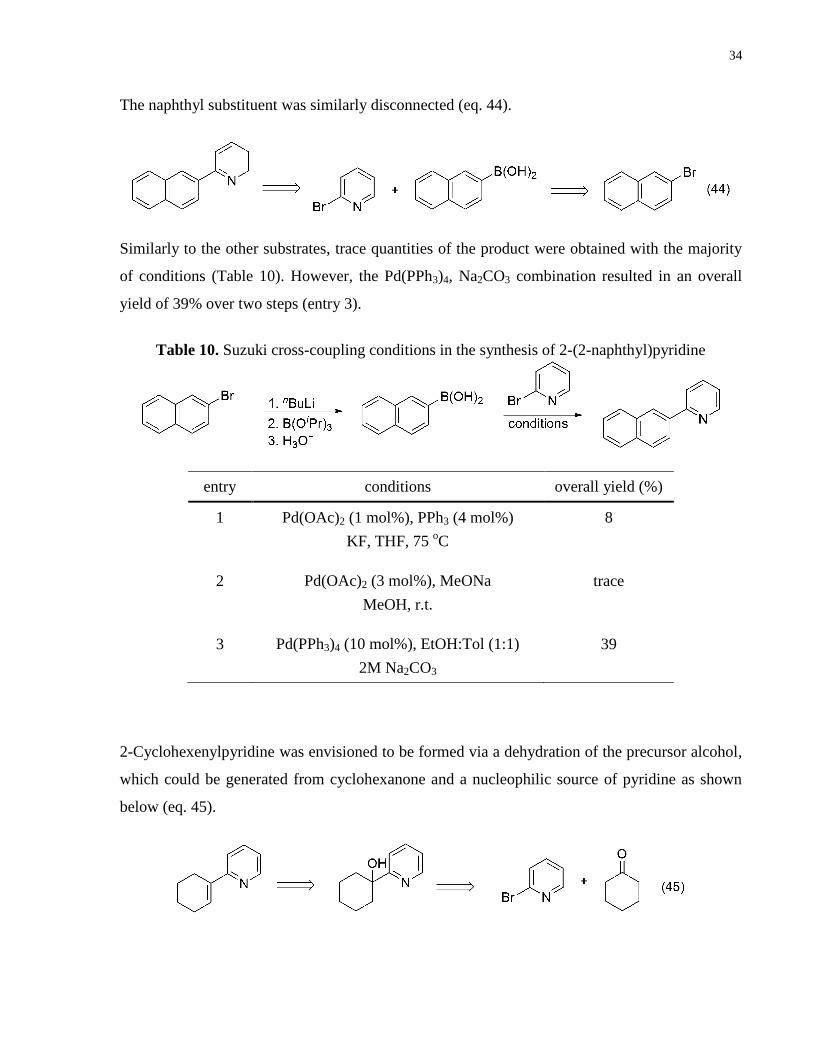
The naphthyl substituent was similarly disconnected (eq. 44).
Similarly to the other substrates, trace quantities of the product were obtained with the majority
of conditions (Table 10). However, the Pd(PPh3)4, Na2CO3 combination resulted in an overall
yield of 39% over two steps (entry 3).
entry conditions overall yield (%)
1 Pd(OAc)2 (1 mol%), PPh3 (4 mol%)
KF, THF, 75 oC
8
2 Pd(OAc)2 (3 mol%), MeONa
MeOH, r.t.
trace
3 Pd(PPh3)4 (10 mol%), EtOH:Tol (1:1)
2M Na2CO3
39
2-Cyclohexenylpyridine was envisioned to be formed via a dehydration of the precursor alcohol,
which could be generated from cyclohexanone and a nucleophilic source of pyridine as shown
below (eq. 45).
Table 10. Suzuki cross-coupling conditions in the synthesis of 2-(2-naphthyl)pyridine
35
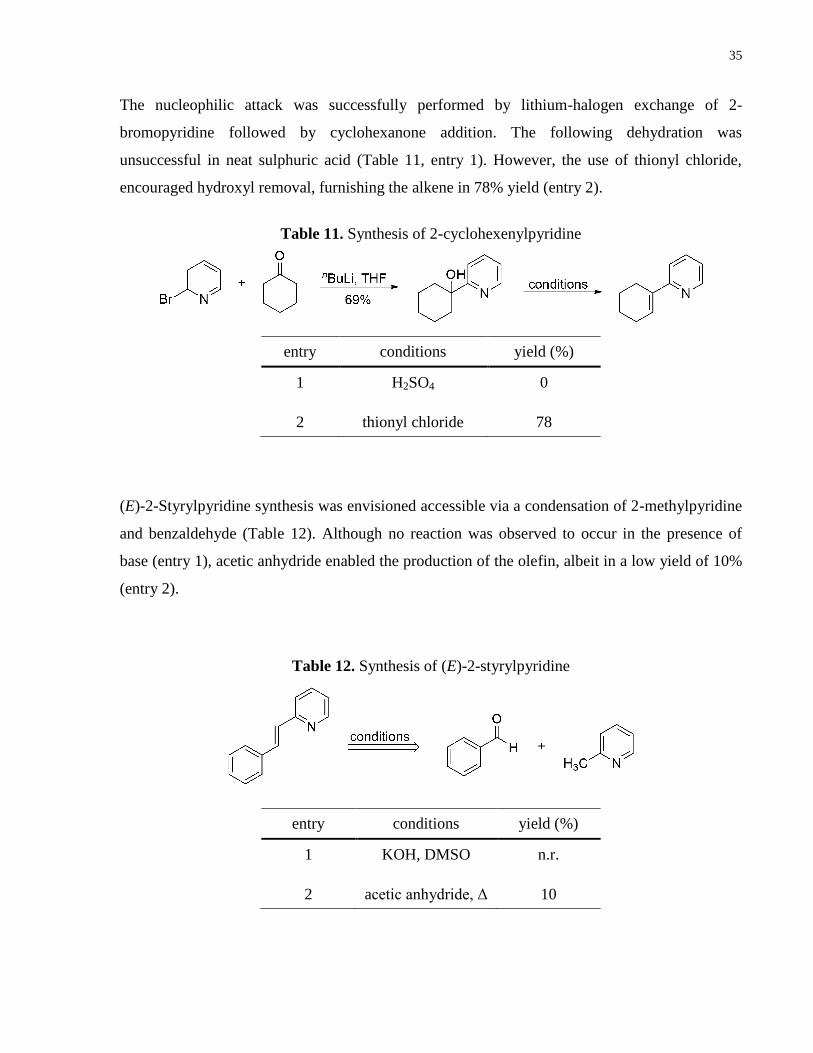
The nucleophilic attack was successfully performed by lithium-halogen exchange of 2-
bromopyridine followed by cyclohexanone addition. The following dehydration was
unsuccessful in neat sulphuric acid (Table 11, entry 1). However, the use of thionyl chloride,
encouraged hydroxyl removal, furnishing the alkene in 78% yield (entry 2).
entry conditions yield (%)
1 H2SO4 0
2 thionyl chloride 78
(E)-2-Styrylpyridine synthesis was envisioned accessible via a condensation of 2-methylpyridine
and benzaldehyde (Table 12). Although no reaction was observed to occur in the presence of
base (entry 1), acetic anhydride enabled the production of the olefin, albeit in a low yield of 10%
(entry 2).
entry conditions yield (%)
1 KOH, DMSO n.r.
2 acetic anhydride, Δ 10
Table 11. Synthesis of 2-cyclohexenylpyridine
Table 12. Synthesis of (E)-2-styrylpyridine

36
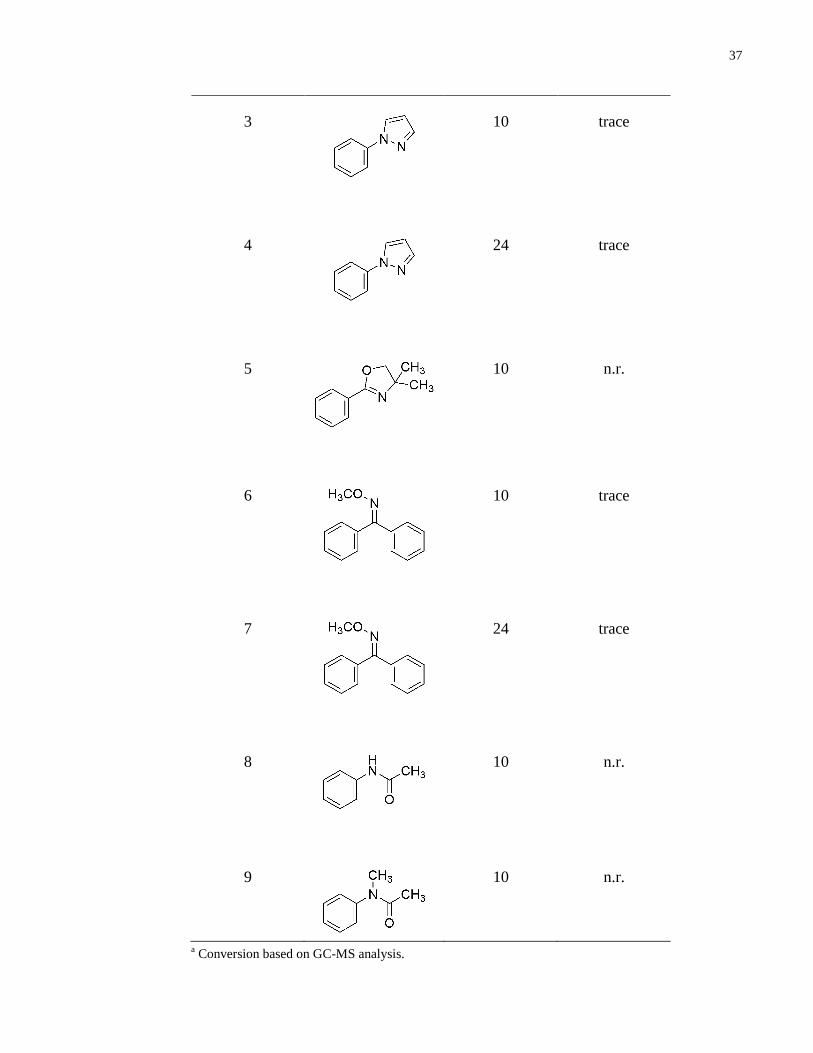
2.3 C–H Bond Chlorination with Sulfonyl Chlorides
Carbon-hydrogen bond chlorination conditions, developed by Dr. Zhao, were tested on the above
mentioned synthesized substrates, as well as on a wide range of directing groups (Table 13). The
m-methyl substituted substrate underwent chlorination in a good yield of 71% (entry 1). The
naphthyl substrate, similarly to the sulfonylation, was obtained in a moderate yield of 59% (entry
2), likely due to the same reasons mentioned previously. Pyrazole-directed chlorinations were
observed to only provide trace amounts of the product (entry 3). Even prolonged reaction times
did not increase the yield (entry 4).
Furthermore, an oxazole directing group was found to be ineffective, as it resulted in only
recovery of starting material (entry 5). An oxime directing group enabled trace formation of the
product (entry 6). Despite prolonged reaction times, no increase in product formation was
observed (entry 7).
Again, amide directing groups provided no reaction. An unprotected amide (entry 8) as well as a
methyl protected amide (entry 9) were tested, neither furnishing the product.
entry substrate time (h) yield (%)
1
10 71
2
10 59a
Table 13. Subtrate scope of C–H bond chlorination
37
3
10 trace
4
24 trace
5
10 n.r.
6
10 trace
7
24 trace
8
10 n.r.
9
10 n.r.
a Conversion based on GC-MS analysis.
38
2.4 Mechanistic Considerations
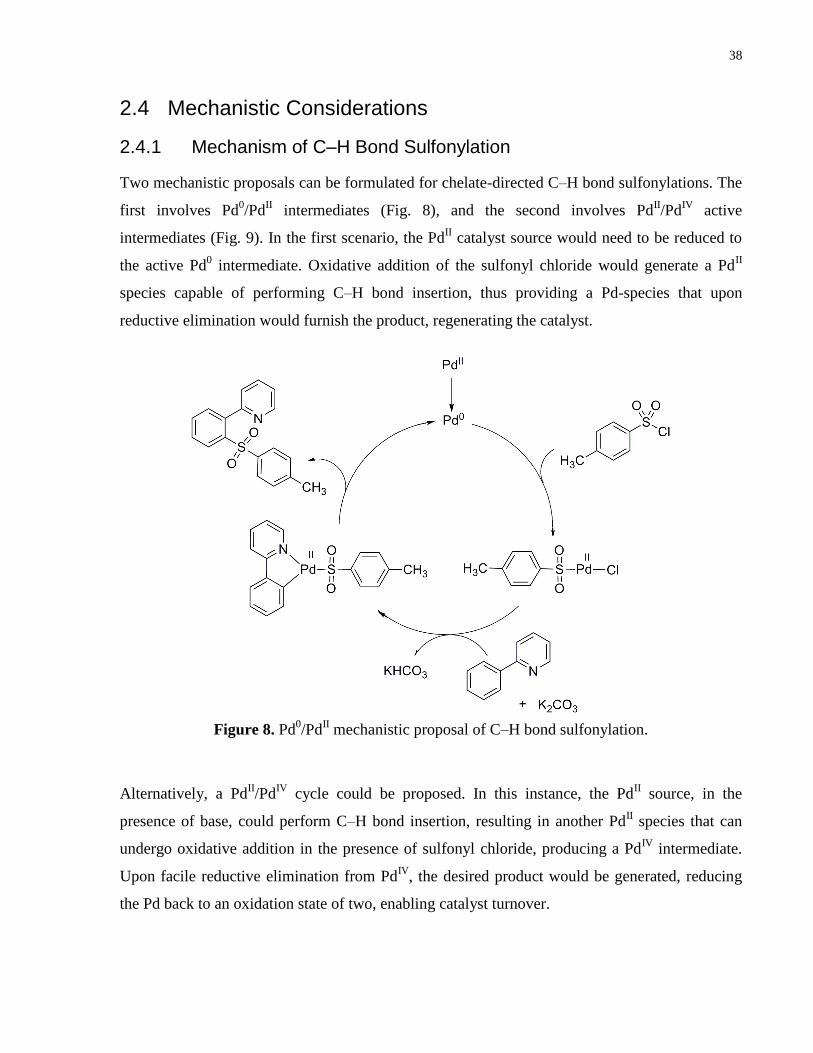
2.4.1 Mechanism of C–H Bond Sulfonylation
Two mechanistic proposals can be formulated for chelate-directed C–H bond sulfonylations. The
first involves Pd0/Pd
II intermediates (Fig. 8), and the second involves Pd
II/Pd
IV active
intermediates (Fig. 9). In the first scenario, the PdII catalyst source would need to be reduced to
the active Pd0 intermediate. Oxidative addition of the sulfonyl chloride would generate a Pd
II
species capable of performing C–H bond insertion, thus providing a Pd-species that upon
reductive elimination would furnish the product, regenerating the catalyst.
Alternatively, a PdII/Pd
IV cycle could be proposed. In this instance, the Pd
II source, in the
presence of base, could perform C–H bond insertion, resulting in another PdII species that can
undergo oxidative addition in the presence of sulfonyl chloride, producing a PdIV
intermediate.
Upon facile reductive elimination from PdIV
, the desired product would be generated, reducing
the Pd back to an oxidation state of two, enabling catalyst turnover.
Figure 8. Pd0/Pd
II mechanistic proposal of C–H bond sulfonylation.
39
Since recent reports on similar chelate-directed C–H bond activations have been proposed to
occur via the PdII/Pd
IV pathway, similar experiments were performed in efforts to either provide
support or refute the viability of this catalytic cycle. Of major interest was whether sulfonyl
chlorides have the oxidative potential to oxidize PdII to Pd
IV. Therefore, a Pd
II species,
resembling the proposed intermediate precursor to PdIV
was synthesized (Fig. 10).
Two routes were pursued toward the synthesis of this species. The first route (A) involved
refluxing of 2-phenylpyridine with mercury(II)acetate. Treatment with LiCl provided the
mercuric chloride substrate, an intermediate capable of undergoing transmetallation with
bis(acetonitrile)dichloropalladium(II) to provide the required Pd-substrate (Pd(phpy)2). However,
in addition to the toxicity of mercury reagents, this route was highly inefficient, and hence,
readily abandoned for the longer route depicted in Figure 11.
Figure 9. PdII/Pd
IV mechanistic proposal of C–H bond sulfonylation.
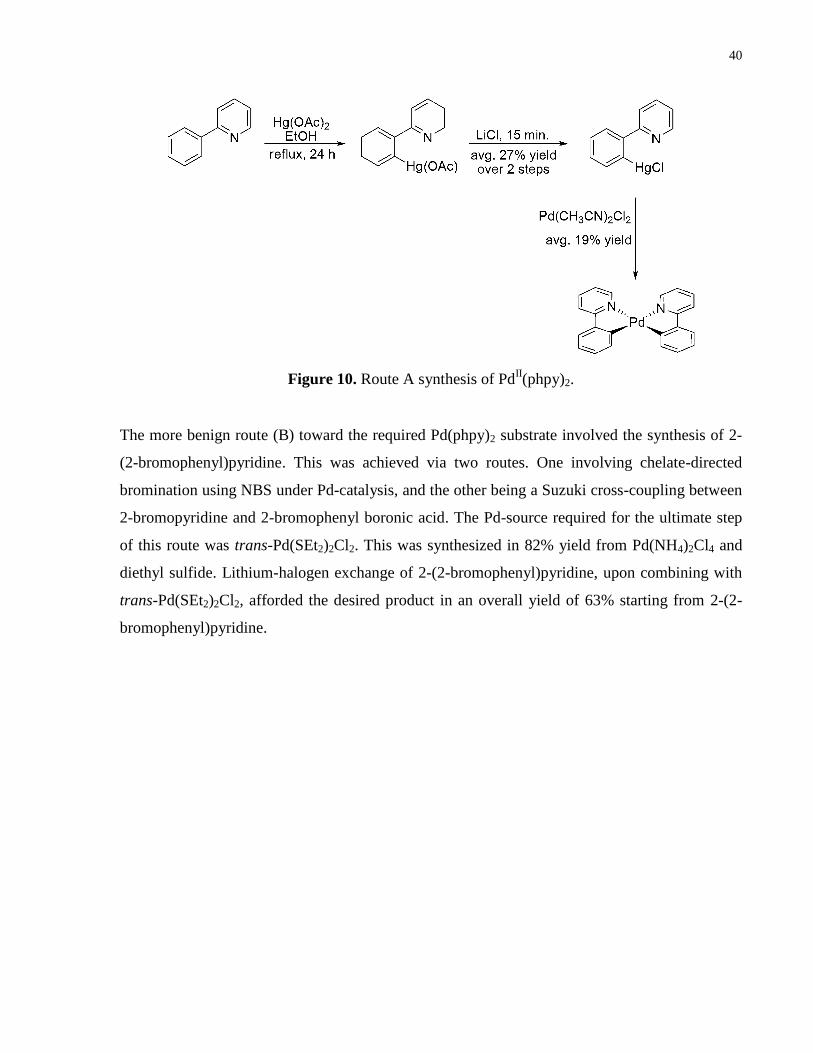
40
The more benign route (B) toward the required Pd(phpy)2 substrate involved the synthesis of 2-
(2-bromophenyl)pyridine. This was achieved via two routes. One involving chelate-directed
bromination using NBS under Pd-catalysis, and the other being a Suzuki cross-coupling between
2-bromopyridine and 2-bromophenyl boronic acid. The Pd-source required for the ultimate step
of this route was trans-Pd(SEt2)2Cl2. This was synthesized in 82% yield from Pd(NH4)2Cl4 and
diethyl sulfide. Lithium-halogen exchange of 2-(2-bromophenyl)pyridine, upon combining with
trans-Pd(SEt2)2Cl2, afforded the desired product in an overall yield of 63% starting from 2-(2-
bromophenyl)pyridine.
Figure 10. Route A synthesis of PdII(phpy)2.

41
The initial test of this substrate was to determine whether it was a plausible intermediate of the
cycle. Therefore, it was subjected to the typical sulfonylation conditions in the absence of 2-
phenylpyridine and Pd(CH3CN)2Cl2. As shown in equation 46, this indeed resulted in sulfone
production in 30% yield, thus indicating that Pd(phpy)2 is a plausible active species. However,
the yield, in conjunction with the results of equation 47, indicate that it is not catalytically active.
This is not of great surprise, since the resultant Pd-species following reductive elimination would
not resemble a proposed intermediate, and hence may not be able to undergo further oxidative
addition with the sulfonyl chloride.
Figure 11. Route B synthesis of PdII(phpy)2.

42
With the reactivity determined as plausible, efforts were set out to isolate the PdIV
species in
order to determine whether sulfonyl chlorides have the capacity to oxidize PdII to Pd
IV. Various
conditions were tried with a number of sulfonyl chlorides (Table 14). With p-tosyl chloride,
several solvents and temperatures were tried, none of which formed any product (entries 1-3).
However, after stirring in dichloromethane, at room temperature for 15 minutes, followed by
sitting in chloroform at room temperature for 3 days, a precipitate formed in 36% yield (entry 4).
It was insoluble in nearly all solvents, however, gratifyingly, in DMSO-d6 a 1H NMR was
successfully obtained which indicated the possible formation of the sought PdIV
species. Further
spectroscopic methods were used to support the formation of this species. However, an X-ray
structure would have provided unambiguous proof. However, due to the high insolubility,
crystallization proved to be difficult. THF, acetonitrile, DMSO, toluene, benzene, acetone,
dichloromethane and 1,4-dioxane were all tried as solvents for recrystallization, none of which
were deemed suitable, mainly due to insolubility. However, a recrystallization from DMF with
slow diffusion of diethyl ether resulted in crystals, which upon X-ray crystallography proved to
be of the expected PdIV
-complex (Fig. 12).41
In the meantime, two other sulfonyl chlorides were used with efforts towards increasing the
solubility of the resultant product. Trifluoromethyl and naphthaline sulfonyl chlorides (entries 5-
41 Crystal structure solved by Dr. Alan Lough.
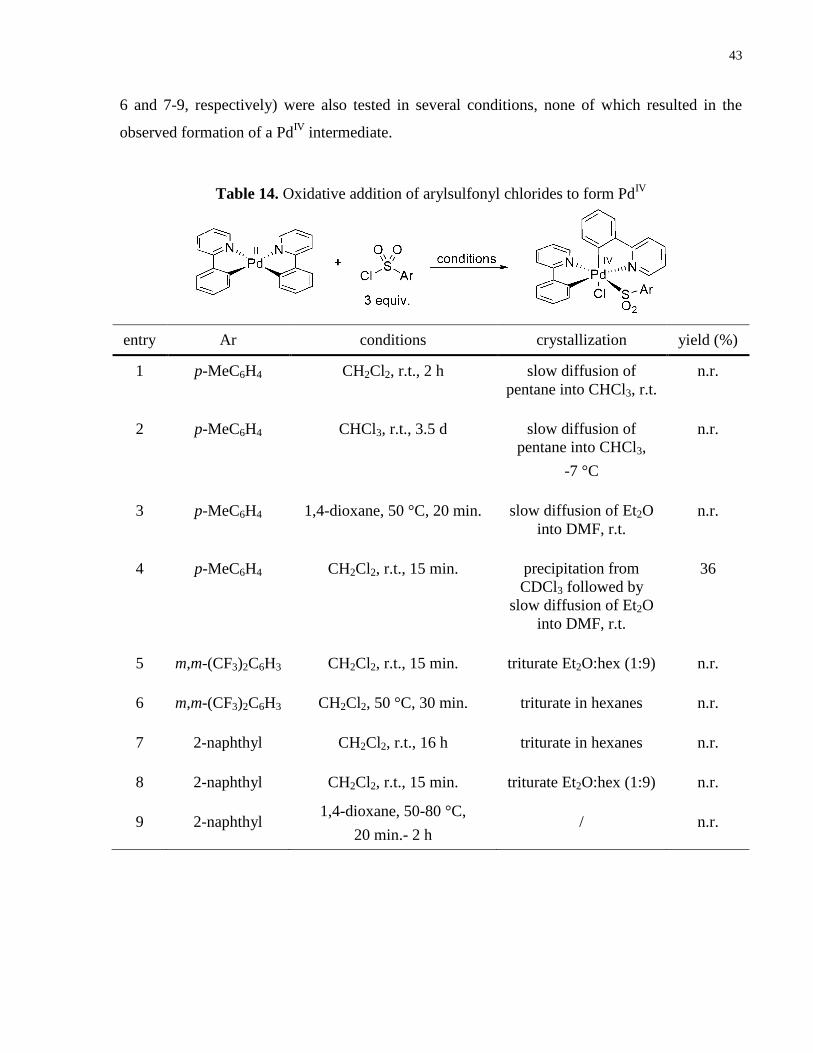
43
6 and 7-9, respectively) were also tested in several conditions, none of which resulted in the
observed formation of a PdIV
intermediate.
entry Ar conditions crystallization yield (%)
1 p-MeC6H4 CH2Cl2, r.t., 2 h slow diffusion of
pentane into CHCl3, r.t.
n.r.
2 p-MeC6H4 CHCl3, r.t., 3.5 d slow diffusion of
pentane into CHCl3,
-7 °C
n.r.
3 p-MeC6H4 1,4-dioxane, 50 °C, 20 min. slow diffusion of Et2O
into DMF, r.t.
n.r.
4 p-MeC6H4 CH2Cl2, r.t., 15 min. precipitation from
CDCl3 followed by
slow diffusion of Et2O
into DMF, r.t.
36
5 m,m-(CF3)2C6H3 CH2Cl2, r.t., 15 min. triturate Et2O:hex (1:9) n.r.
6 m,m-(CF3)2C6H3 CH2Cl2, 50 °C, 30 min. triturate in hexanes n.r.
7 2-naphthyl CH2Cl2, r.t., 16 h triturate in hexanes n.r.
8 2-naphthyl CH2Cl2, r.t., 15 min. triturate Et2O:hex (1:9) n.r.
9 2-naphthyl 1,4-dioxane, 50-80 °C,
20 min.- 2 h / n.r.
Table 14. Oxidative addition of arylsulfonyl chlorides to form PdIV

44
The isolation of a PdIV
species indicated that certain sulfonyl chlorides can oxidize PdII to Pd
IV.
However, to further illustrate that this PdIV
intermediate is a kinetically viable catalytic
intermediate, it was necessary to show that it was capable of undergoing reductive elimination to
yield the sulfone product over several other possible species, including regeneration of the
sulfonyl chloride, homocoupling of two 2-phenylpyridines or chlorination of 2-phenylpyridine.
Therefore, this complex was heated to 120 oC for 7 hours in 1,4-dioxane and DMF (Table 15).
No reaction was observed to occur in 1,4-dioxane, and this was attributed to the lack of solubility
of the substrate in the solvent even at these elevated temperatures (entry 1). Since the product
was soluble in DMF, it was also tried (entry 2). This reaction indeed resulted in the formation of
the sulfone in 57% isolated yield. Therefore, it has been shown that not only is the production of
a PdIV
species possible, but it can reductively eliminate to form sulfones, thus indicating that it is
a likely catalytic intermediate under these specific conditions.
Figure 12. Crstal structure of Pd(IV)-complex.

45
entry solvent yield (%)
1 1,4-dioxane n.r.
2 DMF 57
2.4.2 Mechanism of C–H Bond Chlorination
Insights of the chlorination mechanism were simultaneously being investigated with the
sulfonylation mechanism. The mechanism of the chlorination was proposed to involve two
conjoint catalytic cycles, each involving a transition metal co-catalyst (Fig. 13). The PdII source
is believed to perform C–H bond activation much like in the sulfonylation mechanism.
Following reductive elimination, the chlorinated product would be formed, as well as a Pd0
intermediate. In the presence of CuII, however, this Pd
0 species could readily be oxidized to Pd
II.
Since substoichiometric CuII was used, and the oxidation of Pd would reduce the copper to an
oxidation state of one, the various oxidants in the system capable of replenishing the supply of
CuII were considered. Since the chlorination reactions were conducted in the presence of air,
either atmospheric oxygen or the sulfonyl chloride, or both, may be necessary for the oxidation.
Reactions that were previously performed by Dr. Zhao indicated that inert atmosphere resulted in
significantly lower yields.
Table 15. Reductive elimination from PdIV
to PdII

46
Therefore, the potential roles of the sulfonyl chloride were explored. The first, based on literature
precedence stating that DMF reacts with sulfonyl chlorides to produce amidonium ions was
investigated (eq. 48).42
In the presence of dimethylamine, a byproduct of DMF decomposition at
elevated temperatures, an immonium ion is produced, as well as an arylsulfonate and
dimethylamine hydrochloride. In the presence of copper(I), the generated hydrochloride salt
could serve as a chlorine source, which in the presence of an oxidant, enables the formation of
CuCl2 (eq. 49).
To test whether dimethylamine hydrochloride is required, a series of experiments were
performed (Table 16). In 1,4-dioxane, no reaction was observed (entry 1). 1,4-Dioxane was
initially used in order to eliminate any other interactions that may arise from DMF. However,
since no reaction was observed, it was noted that the salts may not be as soluble as in DMF.
Hence, a control in DMF was performed (entry 2). This also yielded no product formation. As
42 Hall, H. K. Jr. J. Am. Chem. Soc. 1956, 78, 2717.
Figure 13. Proposed mechanism of C–H bond chlorination.

47
these reactions were only performed so that O2 was the only present oxidant in the system, other
oxidants were tried as additives. Benzoquinone (entry 3) and toluene-4-sulfonic acid, an oxidant
resembling the sulfonylchloride oxidant proposed to be involved (entry 4), were tested. Again,
neither of these resulted in product formation. Since the production of these amidonium ions was
reported to be reversible and the equilibrium disfavouring the ionic species, it was postulated that
large quantities of dimethylamine may be detrimental to the catalyst. As a result, other chloride
sources were investigated (Table 17).
entry solvent additive (3 equiv.) yield (%)
1 1,4-dioxane / n.r.
2 DMF / n.r.
3 DMF benzoquinone n.r.
4 DMF toluene-4-sulfonic acid n.r.
Tetrabutylammonium chloride, sodium chloride and lithium chloride were tested (Table 17).
None of these resulted in chlorination. Again, in these systems, no oxidant other than O2 was
present. Hence, a reaction with excess lithium chloride, in the presence of ethyl-p-
toluenesulfonate was performed (eq. 50).
Table 16. Dimethyl amine hydrochloride involvement in chlorination
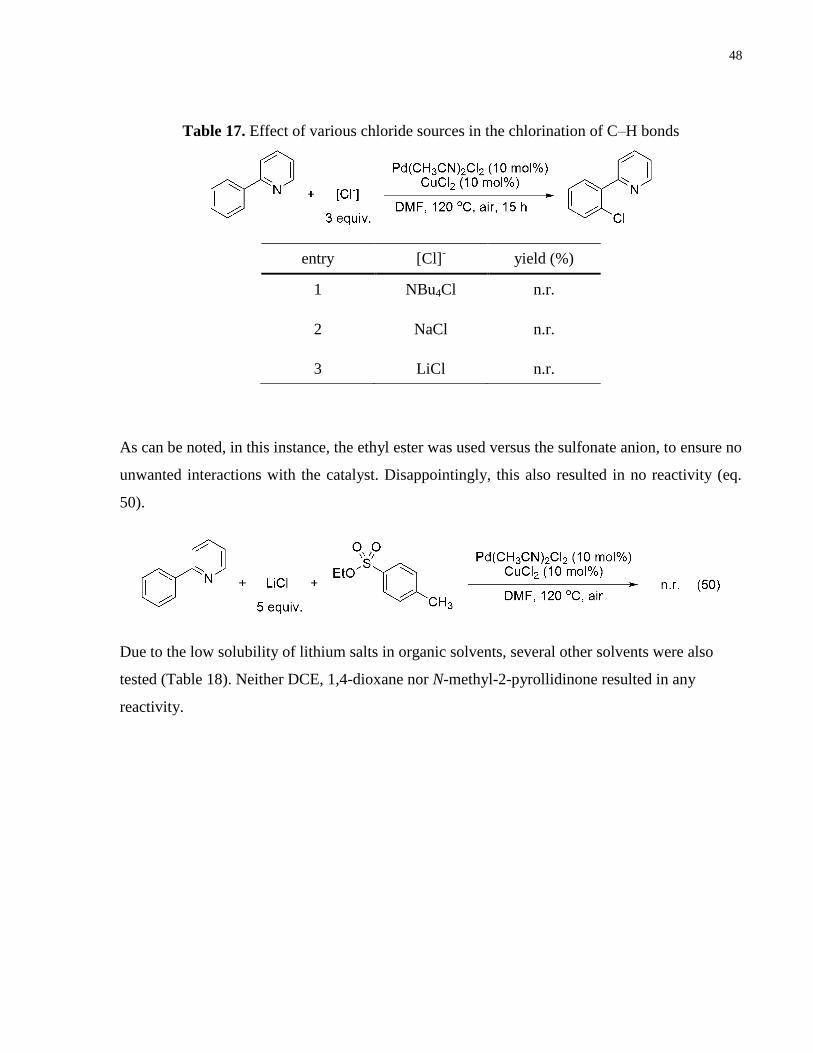
48
entry [Cl]-
yield (%)
1 NBu4Cl n.r.
2 NaCl n.r.
3 LiCl n.r.
As can be noted, in this instance, the ethyl ester was used versus the sulfonate anion, to ensure no
unwanted interactions with the catalyst. Disappointingly, this also resulted in no reactivity (eq.
50).
Due to the low solubility of lithium salts in organic solvents, several other solvents were also
tested (Table 18). Neither DCE, 1,4-dioxane nor N-methyl-2-pyrollidinone resulted in any
reactivity.
Table 17. Effect of various chloride sources in the chlorination of C–H bonds
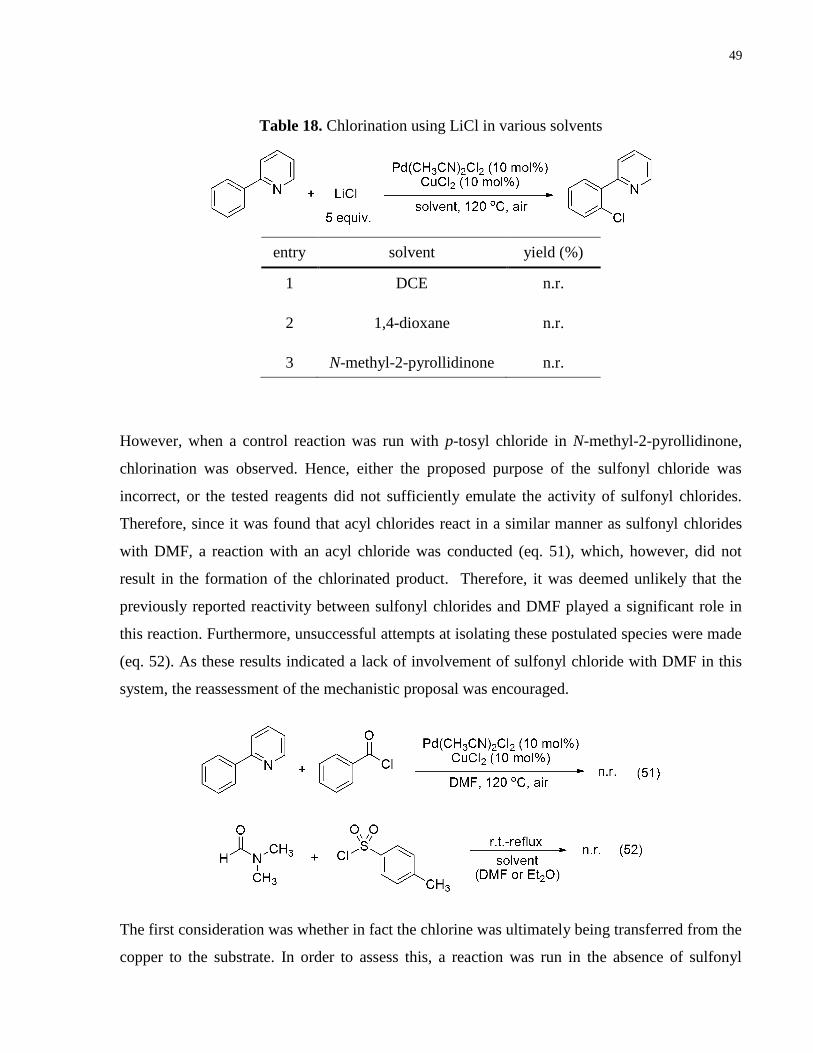
49
entry solvent yield (%)
1 DCE n.r.
2 1,4-dioxane n.r.
3 N-methyl-2-pyrollidinone n.r.
However, when a control reaction was run with p-tosyl chloride in N-methyl-2-pyrollidinone,
chlorination was observed. Hence, either the proposed purpose of the sulfonyl chloride was
incorrect, or the tested reagents did not sufficiently emulate the activity of sulfonyl chlorides.
Therefore, since it was found that acyl chlorides react in a similar manner as sulfonyl chlorides
with DMF, a reaction with an acyl chloride was conducted (eq. 51), which, however, did not
result in the formation of the chlorinated product. Therefore, it was deemed unlikely that the
previously reported reactivity between sulfonyl chlorides and DMF played a significant role in
this reaction. Furthermore, unsuccessful attempts at isolating these postulated species were made
(eq. 52). As these results indicated a lack of involvement of sulfonyl chloride with DMF in this
system, the reassessment of the mechanistic proposal was encouraged.
The first consideration was whether in fact the chlorine was ultimately being transferred from the
copper to the substrate. In order to assess this, a reaction was run in the absence of sulfonyl
Table 18. Chlorination using LiCl in various solvents

50
chloride, but in the presence of a stoichiometric amount of CuCl2 (eq. 53). The chlorinated
product was produced in a ratio of 69:31 (starting material:product), as determined by GC-MS
analysis. This indicated some involvement of chlorine transfer from copper to the substrate.
Subsequently, other copper sources were tested to determine whether the oxidative potential of
the copper species used had an effect on the reaction (Table 19).
Entries 1 and 2 of Table 19 indicate that both CuI and Cu
II react with equal efficacy, thus
indicating that CuI is readily oxidized in the environment such that it is capable of oxidizing Pd
back to the active species. Interestingly, CuBr (entry 3) and CuI (entry 4) catalyzed not only the
formation of the chlorinated species, but also a difunctionalized, chloro-arylated product and a
mono-arylated product, respectively (Fig. 14). The reactivity of CuBr and CuI, therefore, could
not be attributed to the oxidation differentials on account of the byproducts observed only with
these additives.
Figure 14. Byproducts of (a) CuBr and (b) CuI.

51
entry [Cu] % conversiona (mono:di)
1 CuCl2 93 (1.7:1)
2 CuCl 92 (2.5:1)
3 CuBr 91 (1:3)
4 CuI 14
With the requirement of copper established, it was necessary to also test the role of Pd (Table
20). The catalytic activity of Pd was ensured to be critical to the reactivity by observing that only
traces of the chlorinated products were obtained in the absence of Pd, even when stoichiometric
amounts of copper halide species were used.
Table 19. Copper involvement in chlorination
a Determined by GC-MS analysis

52
entry [Cu] conversiona (%)
1 CuCl2 3
2 CuBr 15
3 CuI 3
Therefore, Pd was found to be crucial for reactivity and the chloride source required for the
regeneration of the active Pd-species was found to arise from the copper. However, since the
oxidation of copper could not be conclusively attributed to sulfonyl chloride reactivity with
DMF, the current working hypothesis, particularly in light of the byproducts observed with CuBr
and CuI, invokes the copper functioning as a disproportionating reagent of Tos-Cl as depicted in
eq. 54.
This hypothesis could be used to explain the different reactivity of the various copper sources.
For instance, CuI may preferentially eliminate to arylate the activated C–H bond instead of
chlorinate. In attempts to obtain support for this proposal, radical inhibitors were used to test
whether they would dampen this disproportionation by quenching the formed radicals.
Moreover, since the sulfonylation chemistry was not proposed to undergo a radical mechanism,
as it is performed in the absence of Cu, it was used as the control (Table 21).
Table 20. Pd involvement in chlorination
a Determined by GC-MS analysis
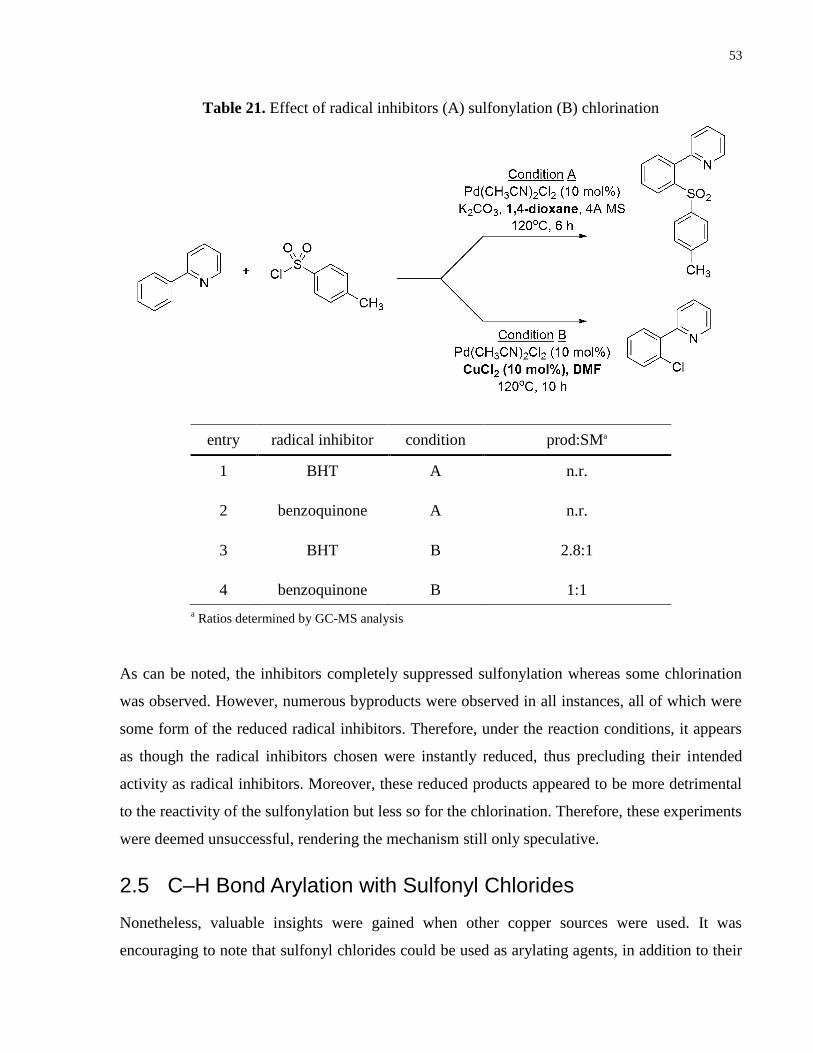
53
entry radical inhibitor condition prod:SMa
1 BHT A n.r.
2 benzoquinone A n.r.
3 BHT B 2.8:1
4 benzoquinone B 1:1
As can be noted, the inhibitors completely suppressed sulfonylation whereas some chlorination
was observed. However, numerous byproducts were observed in all instances, all of which were
some form of the reduced radical inhibitors. Therefore, under the reaction conditions, it appears
as though the radical inhibitors chosen were instantly reduced, thus precluding their intended
activity as radical inhibitors. Moreover, these reduced products appeared to be more detrimental
to the reactivity of the sulfonylation but less so for the chlorination. Therefore, these experiments
were deemed unsuccessful, rendering the mechanism still only speculative.
2.5 C–H Bond Arylation with Sulfonyl Chlorides
Nonetheless, valuable insights were gained when other copper sources were used. It was
encouraging to note that sulfonyl chlorides could be used as arylating agents, in addition to their
Table 21. Effect of radical inhibitors (A) sulfonylation (B) chlorination
a Ratios determined by GC-MS analysis

54
proven ability to perform as competitive sulfonylating and chlorinating agents. However, a series
of Pd/Cu catalyzed reactions were conducted, all of which resulted in the formation of mixtures
of chlorinated and arylated products at best. However, Dr. Zhao showed that for a different
substrate class, benzo[h]quinoline, in the presence of excess CuBr, the arylated species was
formed in 67% yield (eq. 41). Therefore, to expand the substrate class scope, other transition
metals were considered as potential catalysts. Due to the literature precedence of Rh performing
decarbonylative arylations, desulfitative arylation with Rh was considered as a starting point.
The initial set of conditions (eq. 55) formed the arylated species in a promising yield of 9%, with
no observed formation of the competing sulfonylation or chlorination.
To these initial conditions, a phosphine ligand screen was performed (Table 22). Bidentate and
monodentate alkyl phosphines resulted in no or trace reactivity (entries 1 and 2). Aryl
phosphines resulted in no reactivity (entry 3). A phosphate ligand resulted in 16% conversion
(entry 4). The greatest yield was obtained by tri-2-furyl phosphine, which resulted in the
formation of the mono and di-arylated products in a combined yield of 23% in a 6:1 ratio,
favouring the mono-arylated product.
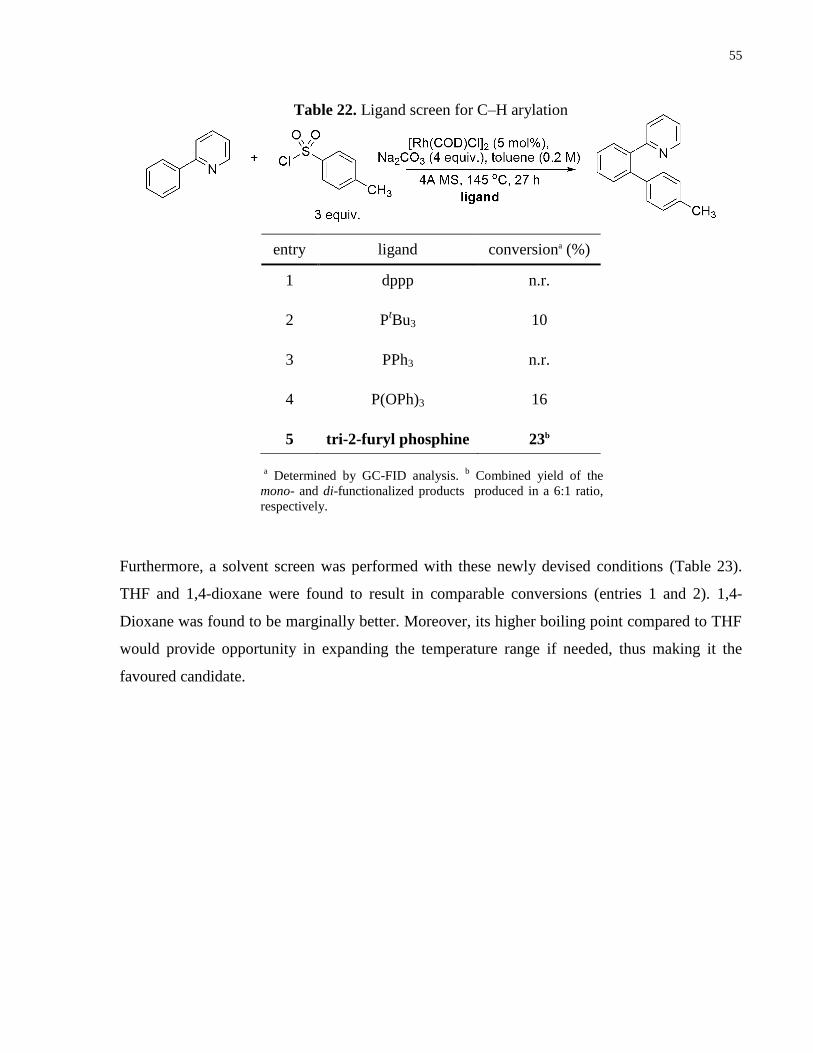
55
entry ligand conversiona (%)
1 dppp n.r.
2 PtBu3 10
3 PPh3 n.r.
4 P(OPh)3 16
5 tri-2-furyl phosphine 23b
Furthermore, a solvent screen was performed with these newly devised conditions (Table 23).
THF and 1,4-dioxane were found to result in comparable conversions (entries 1 and 2). 1,4-
Dioxane was found to be marginally better. Moreover, its higher boiling point compared to THF
would provide opportunity in expanding the temperature range if needed, thus making it the
favoured candidate.
Table 22. Ligand screen for C–H arylation
a Determined by GC-FID analysis.
b Combined yield of the
mono- and di-functionalized products produced in a 6:1 ratio,
respectively.
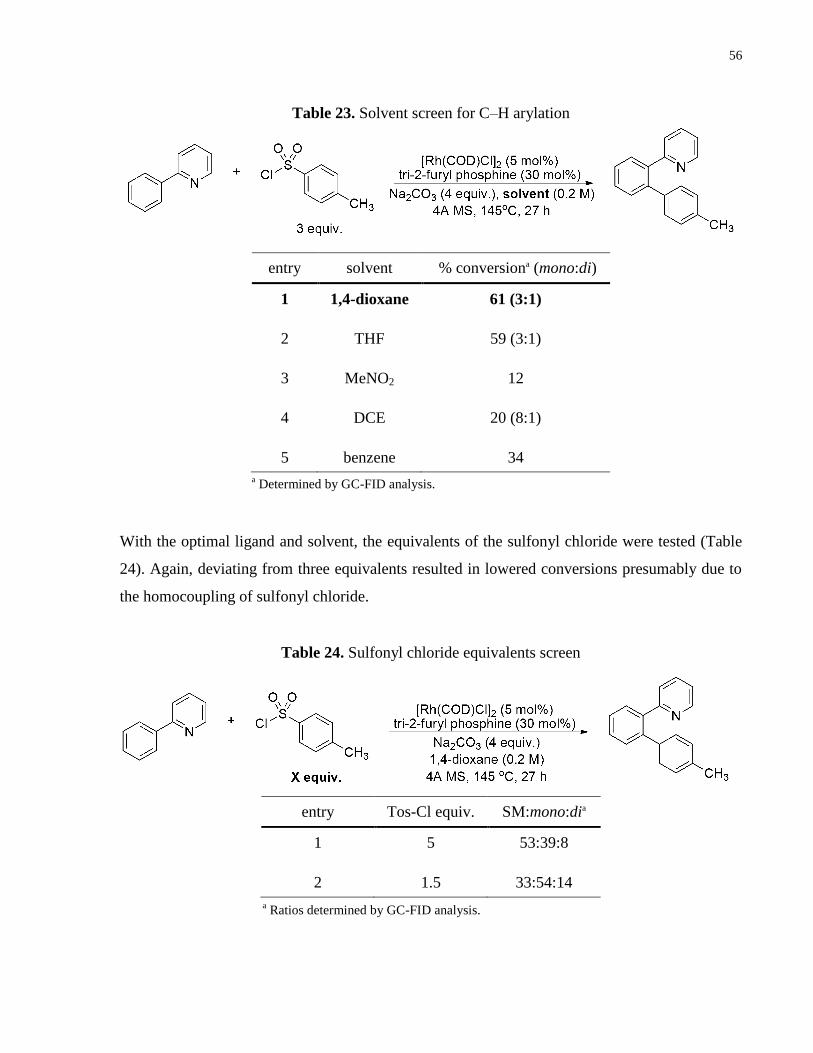
56
entry solvent % conversiona (mono:di)
1 1,4-dioxane 61 (3:1)
2 THF 59 (3:1)
3 MeNO2 12
4 DCE 20 (8:1)
5 benzene 34
With the optimal ligand and solvent, the equivalents of the sulfonyl chloride were tested (Table
24). Again, deviating from three equivalents resulted in lowered conversions presumably due to
the homocoupling of sulfonyl chloride.
entry Tos-Cl equiv. SM:mono:dia
1 5 53:39:8
2 1.5 33:54:14
Table 23. Solvent screen for C–H arylation
Table 24. Sulfonyl chloride equivalents screen
a Determined by GC-FID analysis.
a Ratios determined by GC-FID analysis.
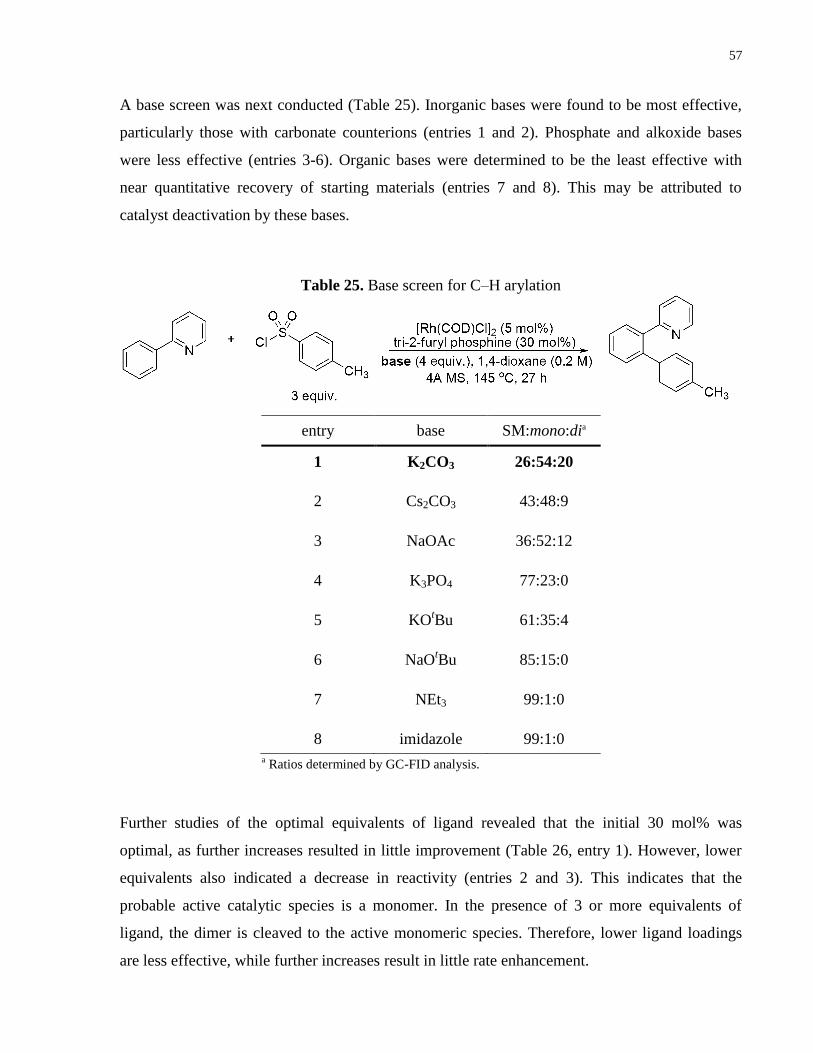
57
A base screen was next conducted (Table 25). Inorganic bases were found to be most effective,
particularly those with carbonate counterions (entries 1 and 2). Phosphate and alkoxide bases
were less effective (entries 3-6). Organic bases were determined to be the least effective with
near quantitative recovery of starting materials (entries 7 and 8). This may be attributed to
catalyst deactivation by these bases.
entry base SM:mono:dia
1 K2CO3 26:54:20
2 Cs2CO3 43:48:9
3 NaOAc 36:52:12
4 K3PO4 77:23:0
5 KOtBu 61:35:4
6 NaOtBu 85:15:0
7 NEt3 99:1:0
8 imidazole 99:1:0
Further studies of the optimal equivalents of ligand revealed that the initial 30 mol% was
optimal, as further increases resulted in little improvement (Table 26, entry 1). However, lower
equivalents also indicated a decrease in reactivity (entries 2 and 3). This indicates that the
probable active catalytic species is a monomer. In the presence of 3 or more equivalents of
ligand, the dimer is cleaved to the active monomeric species. Therefore, lower ligand loadings
are less effective, while further increases result in little rate enhancement.
Table 25. Base screen for C–H arylation
a Ratios determined by GC-FID analysis.
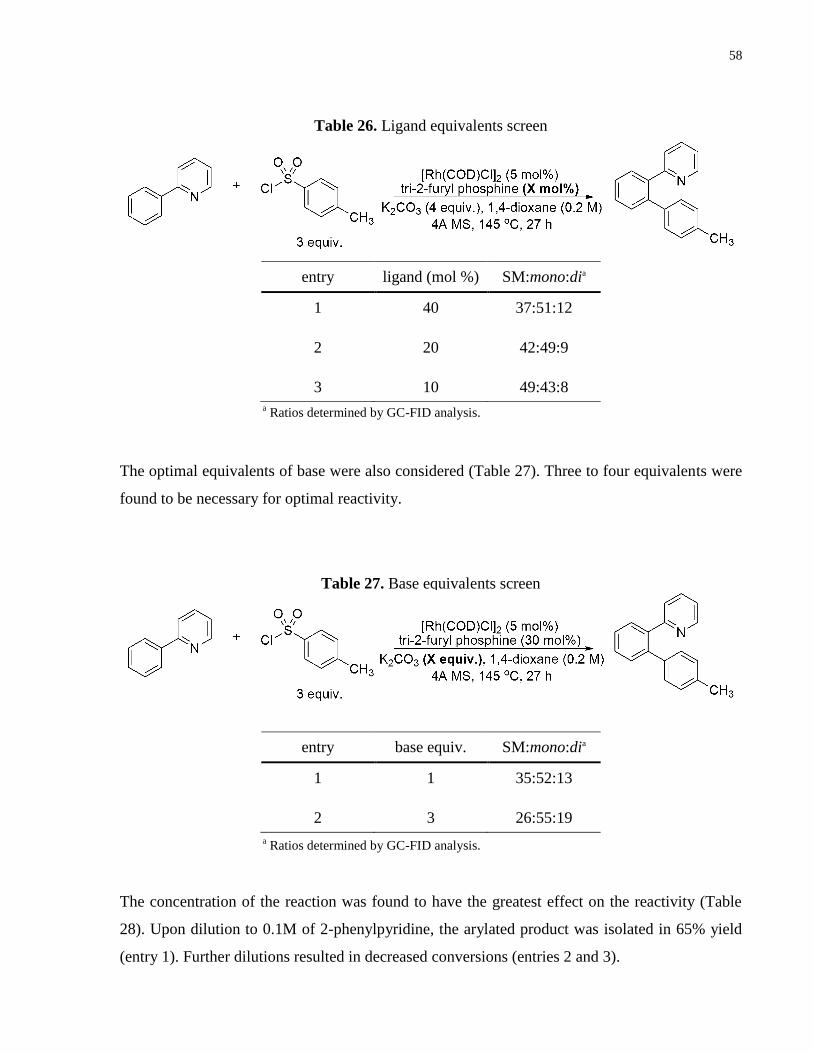
58
entry ligand (mol %) SM:mono:dia
1 40 37:51:12
2 20 42:49:9
3 10 49:43:8
The optimal equivalents of base were also considered (Table 27). Three to four equivalents were
found to be necessary for optimal reactivity.
entry base equiv. SM:mono:dia
1 1 35:52:13
2 3 26:55:19
The concentration of the reaction was found to have the greatest effect on the reactivity (Table
28). Upon dilution to 0.1M of 2-phenylpyridine, the arylated product was isolated in 65% yield
(entry 1). Further dilutions resulted in decreased conversions (entries 2 and 3).
Table 26. Ligand equivalents screen
Table 27. Base equivalents screen
a Ratios determined by GC-FID analysis.
a Ratios determined by GC-FID analysis.
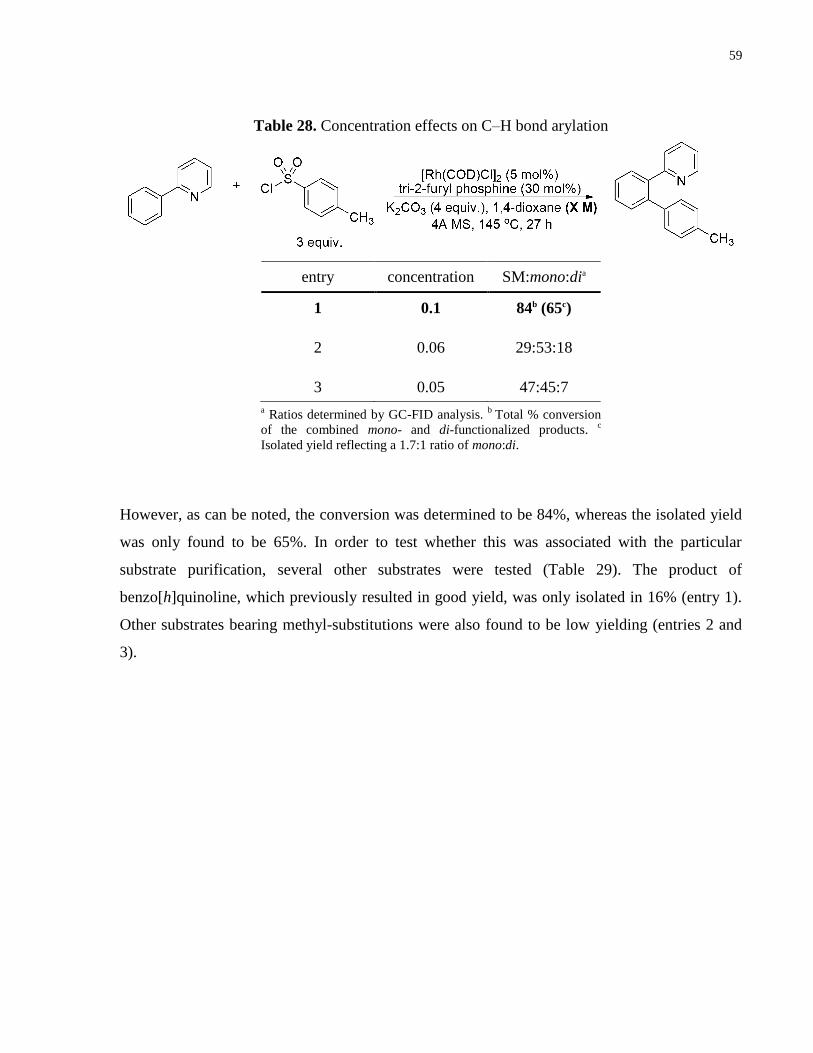
59
entry concentration SM:mono:dia
1 0.1 84b (65c)
2 0.06 29:53:18
3 0.05 47:45:7
However, as can be noted, the conversion was determined to be 84%, whereas the isolated yield
was only found to be 65%. In order to test whether this was associated with the particular
substrate purification, several other substrates were tested (Table 29). The product of
benzo[h]quinoline, which previously resulted in good yield, was only isolated in 16% (entry 1).
Other substrates bearing methyl-substitutions were also found to be low yielding (entries 2 and
3).
Table 28. Concentration effects on C–H bond arylation
a Ratios determined by GC-FID analysis.
b Total % conversion
of the combined mono- and di-functionalized products. c
Isolated yield reflecting a 1.7:1 ratio of mono:di.
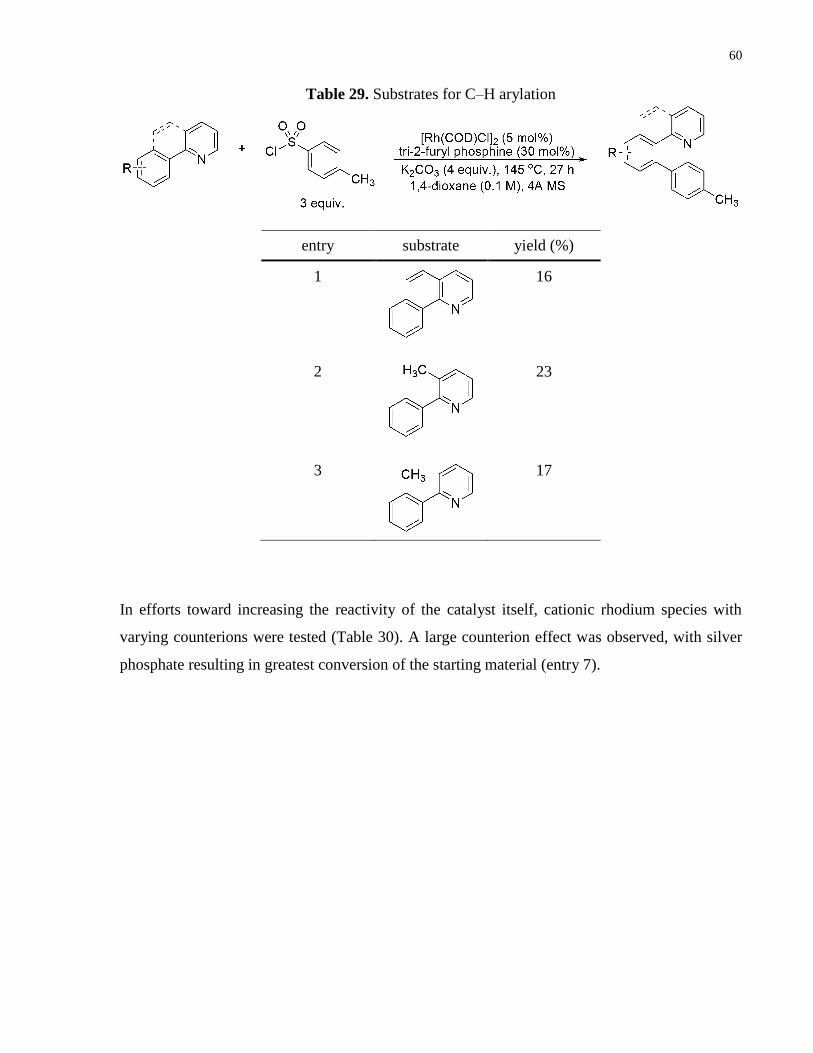
60
entry substrate yield (%)
1
16
2
23
3
17
In efforts toward increasing the reactivity of the catalyst itself, cationic rhodium species with
varying counterions were tested (Table 30). A large counterion effect was observed, with silver
phosphate resulting in greatest conversion of the starting material (entry 7).
Table 29. Substrates for C–H arylation

61
entry AgX SM:mono:dia
1 AgOTf 14:52:34
2 AgNO3 30:53:17
3 AgOAc 44:46:10
4 AgTFA 44:46:10
5 AgTs 36:49:14
6 Ag2CO3 11:45:44
7 Ag3PO4 8:50:42
In attempts to determine whether such elevated temperatures were necessary, slightly less
forcing conditions were tried (Table 31). However, the yields were observed to suffer at
diminished temperatures.
Table 30. Cationic Rh-catalysts for C–H arylation
a Ratios determined by GC-FID analysis.

62
entry temperature (°C) SM:mono:dia
1 120 46:45:9
2 130 44:44:12
In efforts to improve efficacy of the reaction, microwave conditions were explored in attempts to
reduce reaction time (Table 32). Although, compared to other microwave conditions, the reaction
times were not effectively decreased, the monitoring of the reaction progression was more
feasible.
It was observed that for each of the below listed conditions, the reaction plateaued after the
initial thirty minutes, such that the mono-arylated product formation equilibrated and only the di-
arylated product was increasing, albeit slowly. This series of experiments initiated the set of
investigations to follow. In attempts to offset the equilibrium, substrates with distinctly opposing
electronics were tested, in hopes of providing mechanistic insight.
Table 31. Temperature effects on C–H arylation
a Ratios determined by GC-FID analysis.
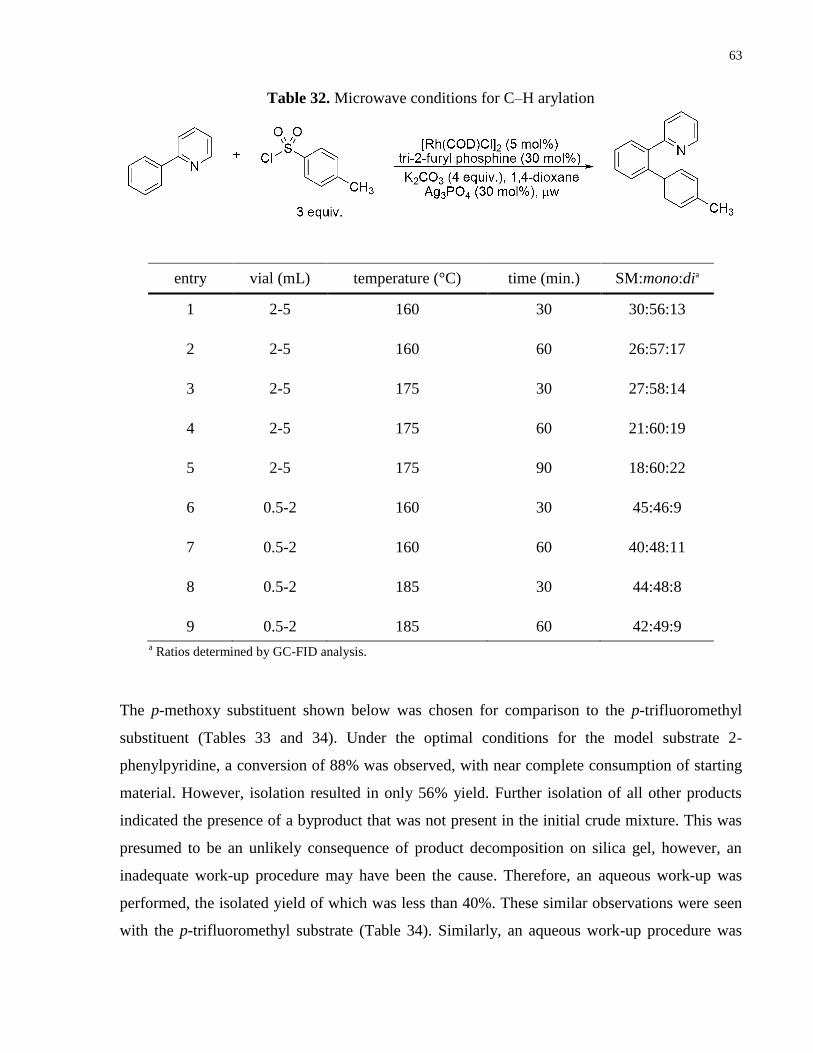
63
entry vial (mL) temperature (°C) time (min.) SM:mono:dia
1 2-5 160 30 30:56:13
2 2-5 160 60 26:57:17
3 2-5 175 30 27:58:14
4 2-5 175 60 21:60:19
5 2-5 175 90 18:60:22
6 0.5-2 160 30 45:46:9
7 0.5-2 160 60 40:48:11
8 0.5-2 185 30 44:48:8
9 0.5-2 185 60 42:49:9
The p-methoxy substituent shown below was chosen for comparison to the p-trifluoromethyl
substituent (Tables 33 and 34). Under the optimal conditions for the model substrate 2-
phenylpyridine, a conversion of 88% was observed, with near complete consumption of starting
material. However, isolation resulted in only 56% yield. Further isolation of all other products
indicated the presence of a byproduct that was not present in the initial crude mixture. This was
presumed to be an unlikely consequence of product decomposition on silica gel, however, an
inadequate work-up procedure may have been the cause. Therefore, an aqueous work-up was
performed, the isolated yield of which was less than 40%. These similar observations were seen
with the p-trifluoromethyl substrate (Table 34). Similarly, an aqueous work-up procedure was
Table 32. Microwave conditions for C–H arylation
a Ratios determined by GC-FID analysis.
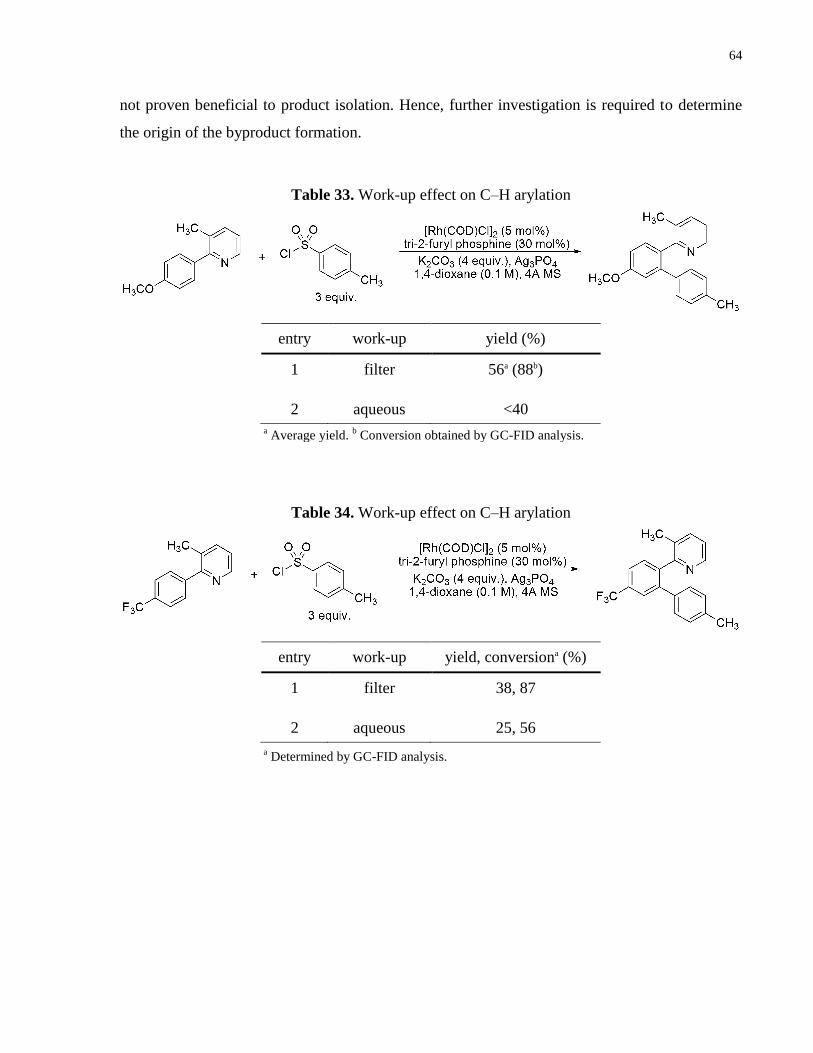
64
not proven beneficial to product isolation. Hence, further investigation is required to determine
the origin of the byproduct formation.
entry work-up yield (%)
1 filter 56a (88b)
2 aqueous <40
entry work-up yield, conversiona (%)
1 filter 38, 87
2 aqueous 25, 56
Table 33. Work-up effect on C–H arylation
Table 34. Work-up effect on C–H arylation
a Average yield.
b Conversion obtained by GC-FID analysis.
a Determined by GC-FID analysis.

65
Chapter 3 Conclusions
Therefore, the use of arylsulfonyl chlorides as versatile reagents has been highlighted. Under Pd-
catalysis, C–H bond functionalizations were performed to form C–S and C–Cl bonds.
Sulfonylation reactions were achieved in 38-91% yield and chlorination reactions were
performed in a range of 59-71% yield. The indicated processes are presumed to occur via C–H
bond activation involving either Pd0/II
or PdII/IV
intermediates. Support in favour of a catalytic
cycle involving PdII/IV
intermediates was obtained for the sulfonylation reactions via synthesis
and isolation of a PdIV
species analogous to the proposed intermediates. Successful reductive
elimination to furnish the desired sulfone products provided further support for the plausibility of
this mechanism. Furthermore, using Rh-catalysis, the oxidation of C–H bonds to C–C bonds was
achieved in yields up to 65% for the model substrate 2-phenylpyridine.
Future work will be focused on providing further scope and mechanistic insight of these
processes, as well as optimization of the Rh-catalyzed C–H to C–C bond formation to increase
efficiency.

66
Chapter 4 Experimental Procedures
4.1 General Information
Purification of reaction products was carried out by flash column chromatography using EMD
Silica Gel 60 (particle size 0.040-0.063 mm, 230-400 mesh ASTM) or by preparative thin layer
chromatography (EMD Silica Gel 60 F254 precoated plates, 2mm). Analytical thin layer
chromatography (TLC) was performed on glass pre-coated with silica gel (EMD Silica Gel 60
F254), cut to size. Visualization was accomplished with UV light followed by treatment with
potassium permanganate solution and heating. Microwave reactions were run in a Biotage
Initiator instrument. GC-MS analysis was performed on an Agilent Technologies 7890A (GC)
and 5975C inert XL EI/CI MSD system with an HP-5 column (0.320 mm, 0.25 μm film). GC-
FID analysis was performed on an Agilent Technologies (7890A) system.
1H NMR and
13C NMR spectra were recorded on Varian 400 or 300 MHz and 100 or 75 MHz
spectrometers, respectively, at ambient temperature. Spectral data were reported in ppm using
TMS or the NMR solvent peak as the reference (CDCl3 at 7.26 ppm, DMSO-D6 at 2.50 ppm or
TMS at 0.00 ppm for 1H NMR and CDCl3 at 77.0 ppm, DMSO-d6 at 39.43 ppm for
13C NMR).
1H NMR was reported as: multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q =
quartet, m = multiplet), integration and coupling constant(s) in Hz. All the melting points were
uncorrected and obtained on a Gallenkamp melting point apparatus. Infrared (IR) spectra were
obtained on a Perkin-Elmer 1000 FT-IR spectrometer. Mass spectra (MS) were recorded on a
Sciex Qstar Mass Spectrometer. High-resolution mass spectra (HRMS) were recorded on a
micromass 70S-250 spectrometer (EI) or an ABI/Sciex Qstar mass spectrometer (ESI).
Unless otherwise noted, all reagents and catalysts were commercially available and reactions
were carried out under inert atmosphere. 1,4-Dioxane was distilled from sodium and
benzophenone under argon and degassed by three freeze-pump-thaw cycles prior to storage in
the glovebox. Remaining solvents were dried through two columns of activated alumina. The
inorganic bases were dried in the oven at 120 °C overnight, and 4A molecular sieves were
activated under vacuum at 150 °C for 8 h, and then cooled to ambient temperature before use. p-
Tolylsulfonyl chloride was recrystallized using hexanes. All other commercially available

67
reagents and solvents were used directly from the bottle without further purification.
4.2 Experimental and Characterization Data
4.2.1 General Procedure for Synthesis of 2-cyclohexenylpyridine
1-(pyridin-2-yl)cyclohexanol. Literature procedure used.43
Reagents were treated with 4A MS
overnight prior to use. To an oven dried Schlenk flask equipped with a magnetic stirring bar was
added THF (10 mL). The flask was cooled to -95 oC.
nBuLi (6.2 mmol, 1.08 equiv.) was added
followed by slow addition of a mixture of THF (10 mL) and 2-bromopyridine (15.0 mmol, 1.00
equiv.). The solution was stirred for 45 minutes and cyclohexanone (16.2 mmol, 1.08 equiv.)
with THF (10 mL) was added. The solution was stirred overnight. The solution was diluted with
water and extracted with chloroform. The collected organic phases were dried over MgSO4,
filtered and concentrated under reduced pressure. Isolated 1.84 g (69% yield) as a yellow oil
after column chromatography (2-20% EtOAc/Hex). TLC Rf 0.27 (10% EtOAc/Hex). NMR data
were found to be in good agreement with those in literature.44
2-cyclohexenylpyridine. Literature procedure used.45
Pyridine solvent was treated with 4A MS
overnight prior to use. To a round bottom flask equipped with a magnetic stirring bar was added
pyridine (15 mL) and the above prepared alcohol (10.4 mmol, 1.0 equiv). The solution was
cooled to -10 oC. Thionyl chloride (40.6 mmol, 4.0 equiv.) was slowly added and solution stirred
43 Zhao, X.; Ivanova, N.; Hadzovic, A.; Zimmer-De Iuliis, M.; Lough, A. J.; Morris, R. H. Organometallics, 2008,
27, 503. 44
Herrmann, W. A.; Lobmaier, G. M.; Priermeier, T.; Mattner, M. R.; Scharbert, B. J. Molec. Catal. A. 1997, 117,
455. 45
Kaiho, T.; Sannohe, K.; Kajiya, S.; Suzuki, T.; Otsuka, K.; Ito, T.; Kamiya, J.; Maruyama, M. J. Med. Chem.
1989, 32, 351.

68
for 4 hours at 0oC. The reaction was poured onto crushed ice and extracted with CH2Cl2. The
aqueous layer was basified and extracted with CH2Cl2. The organic phases were combined, dried
over MgSO4 and concentrated under reduced pressure. Isolated 2.1 g (78% yield) as a yellow oil
after column chromatography (5-10% EtOAc/Hex). TLC Rf 0.42 (10% EtOAc/Hex). NMR data
were found to be in good agreement with those in literature.46
4.2.2 General Procedure for Synthesis of (E)-2-styrylpyridine
(E)-2-styrylpyridine. Literature procedure used.47
To a Schlenk flask equipped with a magnetic
stirring bar was added benzaldehyde (30 mmol, 1 equiv.) and 2-methylpyridine (30 mmol, 1
equiv.). Acetic anhydride (10 mL) was added and the solution heated to reflux overnight. The
reaction was poured over crushed ice and NaOH pellets added until solution was basified. The
resultant mixture was extracted three times with EtOAc, dried over Na2SO4 and concentrated
under reduced pressure. Isolated 0.55 g (10% yield) as a yellow solid after column
chromatography (100% Hex-10% EtOAc/Hex). TLC Rf 0.19 (1:5 EtOAc/Hex). NMR data were
found to be in good agreement with those in literature.48
4.2.3 General Procedure for Sulfonylation Reactions
In a glovebox, to a 20-mL vial equipped with a magnetic stirring bar were successively added the
substrate (0.2 mmol, 1 equiv.), Pd(CH3CN)2Cl2 (0.02 mmol, 0.1 equiv), K2CO3 (0.4 mmol, 2
equiv.), 4A molecular sieves (100 mg), arylsulfonyl chloride (0.6 mmol, 3 equiv.) and 1,4-
dioxane (1 mL). The vial was sealed with a Teflon cap and removed from the glovebox. The
mixture was stirred on a heating block at 120 °C for 6 h. Upon cooling to ambient temperature,
46 Kang, D. M.; Kang, J.-W.; Park, J. W.; Jung, S. O.; Lee, S.-H.; Park, H. D.; Kim, Y. H.; Shin, S. C.; Kim, J.-J.;
Kwon, S.-K. Adv. Mater. 2008, 20, 2003. 47
McKennis H. Jr.; Turnbull, L. B.; Bowman, E. R.; Tamaki, E. J. Org. Chem. 1963, 28, 387. 48
(a) Kaliappan, R.; Kaanumalle, L. S.; Natarajan, A.; Ramamurthy, V. Photochem. Photobiol Sci. 2006, 5, 925. (b)
Shi, X.-F.; Xing, Z.-Y.; Wu, L.; Zhang, W.-Q. Inorganica Chemica Acta 2006, 359, 603.
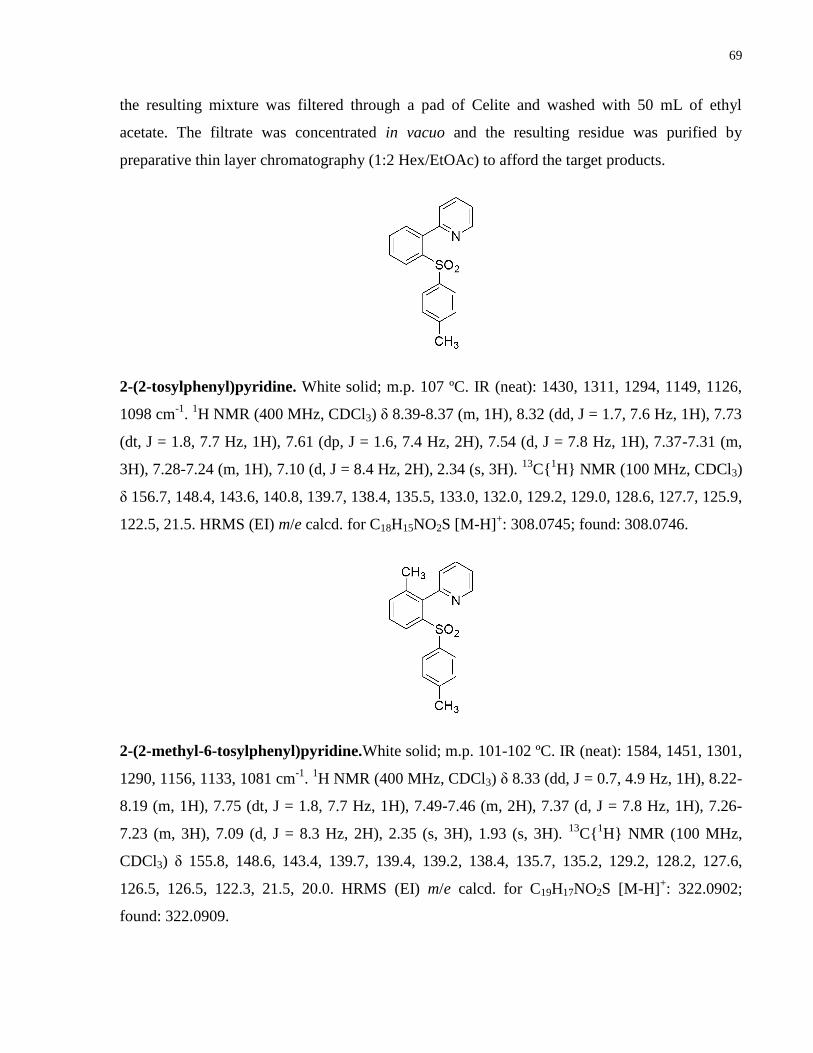
69
the resulting mixture was filtered through a pad of Celite and washed with 50 mL of ethyl
acetate. The filtrate was concentrated in vacuo and the resulting residue was purified by
preparative thin layer chromatography (1:2 Hex/EtOAc) to afford the target products.
2-(2-tosylphenyl)pyridine. White solid; m.p. 107 ºC. IR (neat): 1430, 1311, 1294, 1149, 1126,
1098 cm-1
. 1H NMR (400 MHz, CDCl3) δ 8.39-8.37 (m, 1H), 8.32 (dd, J = 1.7, 7.6 Hz, 1H), 7.73
(dt, J = 1.8, 7.7 Hz, 1H), 7.61 (dp, J = 1.6, 7.4 Hz, 2H), 7.54 (d, J = 7.8 Hz, 1H), 7.37-7.31 (m,
3H), 7.28-7.24 (m, 1H), 7.10 (d, J = 8.4 Hz, 2H), 2.34 (s, 3H). 13
C{1H} NMR (100 MHz, CDCl3)
δ 156.7, 148.4, 143.6, 140.8, 139.7, 138.4, 135.5, 133.0, 132.0, 129.2, 129.0, 128.6, 127.7, 125.9,
122.5, 21.5. HRMS (EI) m/e calcd. for C18H15NO2S [M-H]+: 308.0745; found: 308.0746.
2-(2-methyl-6-tosylphenyl)pyridine.White solid; m.p. 101-102 ºC. IR (neat): 1584, 1451, 1301,
1290, 1156, 1133, 1081 cm-1
. 1H NMR (400 MHz, CDCl3) δ 8.33 (dd, J = 0.7, 4.9 Hz, 1H), 8.22-
8.19 (m, 1H), 7.75 (dt, J = 1.8, 7.7 Hz, 1H), 7.49-7.46 (m, 2H), 7.37 (d, J = 7.8 Hz, 1H), 7.26-
7.23 (m, 3H), 7.09 (d, J = 8.3 Hz, 2H), 2.35 (s, 3H), 1.93 (s, 3H). 13
C{1H} NMR (100 MHz,
CDCl3) δ 155.8, 148.6, 143.4, 139.7, 139.4, 139.2, 138.4, 135.7, 135.2, 129.2, 128.2, 127.6,
126.5, 126.5, 122.3, 21.5, 20.0. HRMS (EI) m/e calcd. for C19H17NO2S [M-H]+: 322.0902;
found: 322.0909.
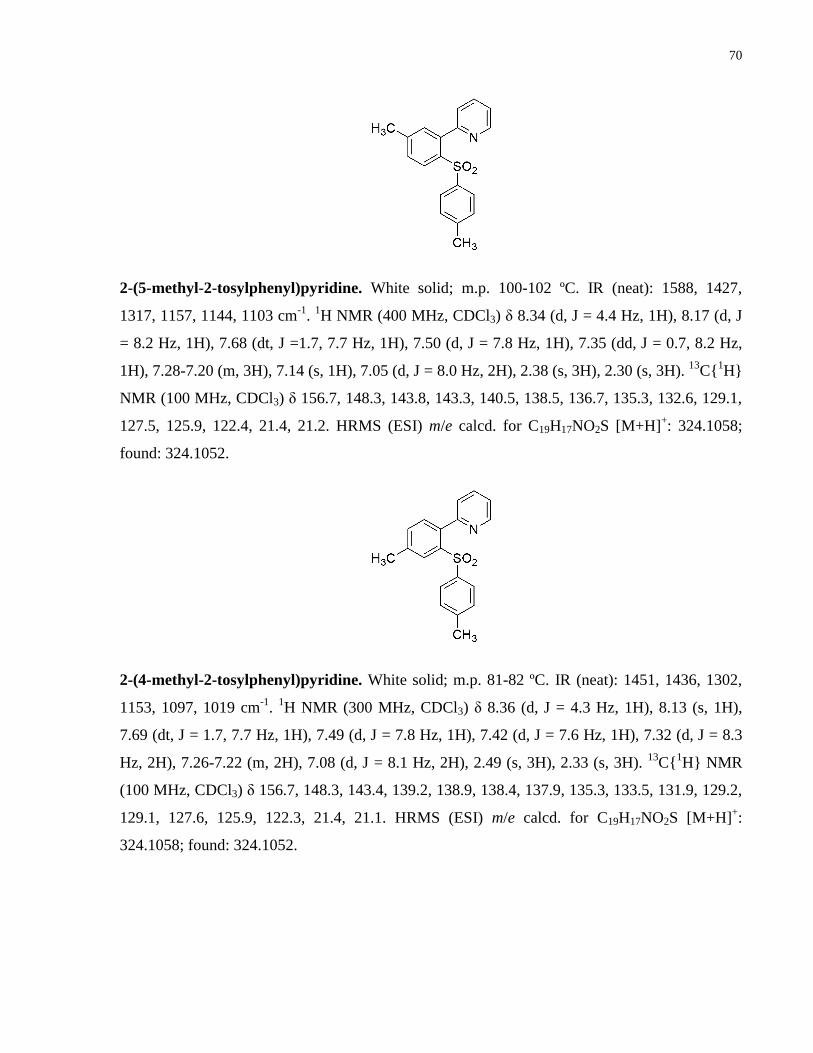
70
2-(5-methyl-2-tosylphenyl)pyridine. White solid; m.p. 100-102 ºC. IR (neat): 1588, 1427,
1317, 1157, 1144, 1103 cm-1
. 1H NMR (400 MHz, CDCl3) δ 8.34 (d, J = 4.4 Hz, 1H), 8.17 (d, J
= 8.2 Hz, 1H), 7.68 (dt, J =1.7, 7.7 Hz, 1H), 7.50 (d, J = 7.8 Hz, 1H), 7.35 (dd, J = 0.7, 8.2 Hz,
1H), 7.28-7.20 (m, 3H), 7.14 (s, 1H), 7.05 (d, J = 8.0 Hz, 2H), 2.38 (s, 3H), 2.30 (s, 3H). 13
C{1H}
NMR (100 MHz, CDCl3) δ 156.7, 148.3, 143.8, 143.3, 140.5, 138.5, 136.7, 135.3, 132.6, 129.1,
127.5, 125.9, 122.4, 21.4, 21.2. HRMS (ESI) m/e calcd. for C19H17NO2S [M+H]+: 324.1058;
found: 324.1052.
2-(4-methyl-2-tosylphenyl)pyridine. White solid; m.p. 81-82 ºC. IR (neat): 1451, 1436, 1302,
1153, 1097, 1019 cm-1
. 1H NMR (300 MHz, CDCl3) δ 8.36 (d, J = 4.3 Hz, 1H), 8.13 (s, 1H),
7.69 (dt, J = 1.7, 7.7 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.42 (d, J = 7.6 Hz, 1H), 7.32 (d, J = 8.3
Hz, 2H), 7.26-7.22 (m, 2H), 7.08 (d, J = 8.1 Hz, 2H), 2.49 (s, 3H), 2.33 (s, 3H). 13
C{1H} NMR
(100 MHz, CDCl3) δ 156.7, 148.3, 143.4, 139.2, 138.9, 138.4, 137.9, 135.3, 133.5, 131.9, 129.2,
129.1, 127.6, 125.9, 122.3, 21.4, 21.1. HRMS (ESI) m/e calcd. for C19H17NO2S [M+H]+:
324.1058; found: 324.1052.
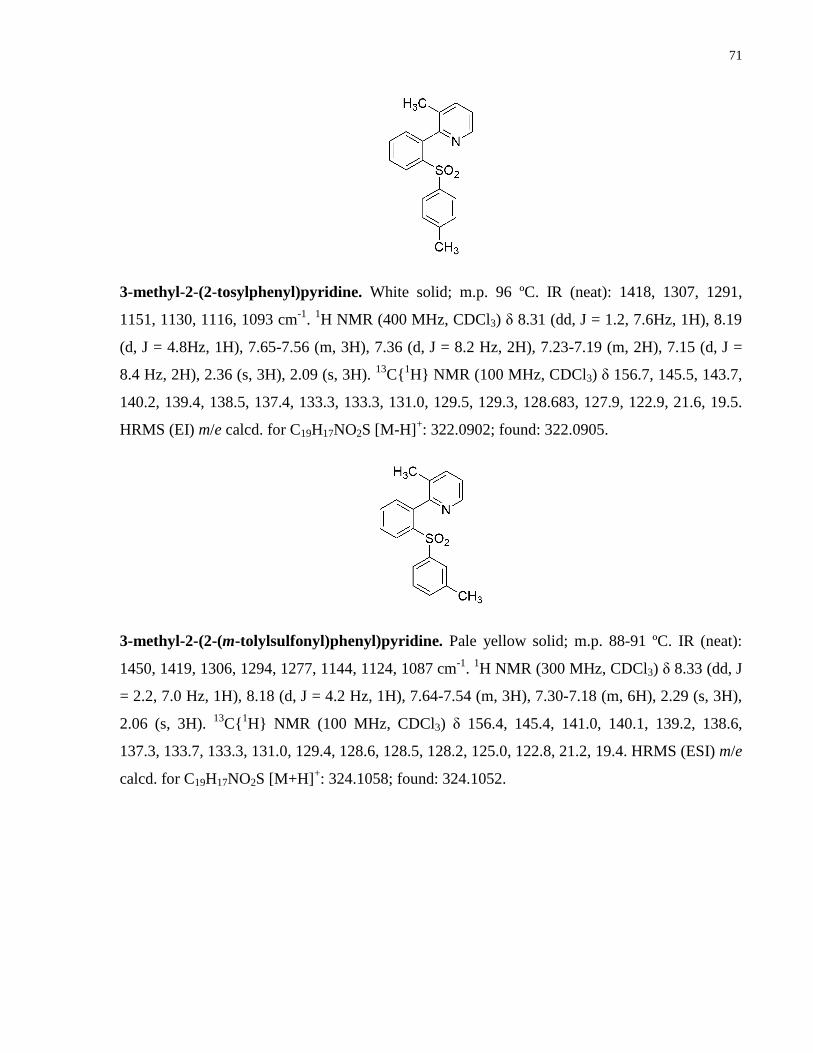
71
3-methyl-2-(2-tosylphenyl)pyridine. White solid; m.p. 96 ºC. IR (neat): 1418, 1307, 1291,
1151, 1130, 1116, 1093 cm-1
. 1H NMR (400 MHz, CDCl3) δ 8.31 (dd, J = 1.2, 7.6Hz, 1H), 8.19
(d, J = 4.8Hz, 1H), 7.65-7.56 (m, 3H), 7.36 (d, J = 8.2 Hz, 2H), 7.23-7.19 (m, 2H), 7.15 (d, J =
8.4 Hz, 2H), 2.36 (s, 3H), 2.09 (s, 3H). 13
C{1H} NMR (100 MHz, CDCl3) δ 156.7, 145.5, 143.7,
140.2, 139.4, 138.5, 137.4, 133.3, 133.3, 131.0, 129.5, 129.3, 128.683, 127.9, 122.9, 21.6, 19.5.
HRMS (EI) m/e calcd. for C19H17NO2S [M-H]+: 322.0902; found: 322.0905.
3-methyl-2-(2-(m-tolylsulfonyl)phenyl)pyridine. Pale yellow solid; m.p. 88-91 ºC. IR (neat):
1450, 1419, 1306, 1294, 1277, 1144, 1124, 1087 cm-1
. 1H NMR (300 MHz, CDCl3) δ 8.33 (dd, J
= 2.2, 7.0 Hz, 1H), 8.18 (d, J = 4.2 Hz, 1H), 7.64-7.54 (m, 3H), 7.30-7.18 (m, 6H), 2.29 (s, 3H),
2.06 (s, 3H). 13
C{1H} NMR (100 MHz, CDCl3) δ 156.4, 145.4, 141.0, 140.1, 139.2, 138.6,
137.3, 133.7, 133.3, 131.0, 129.4, 128.6, 128.5, 128.2, 125.0, 122.8, 21.2, 19.4. HRMS (ESI) m/e
calcd. for C19H17NO2S [M+H]+: 324.1058; found: 324.1052.
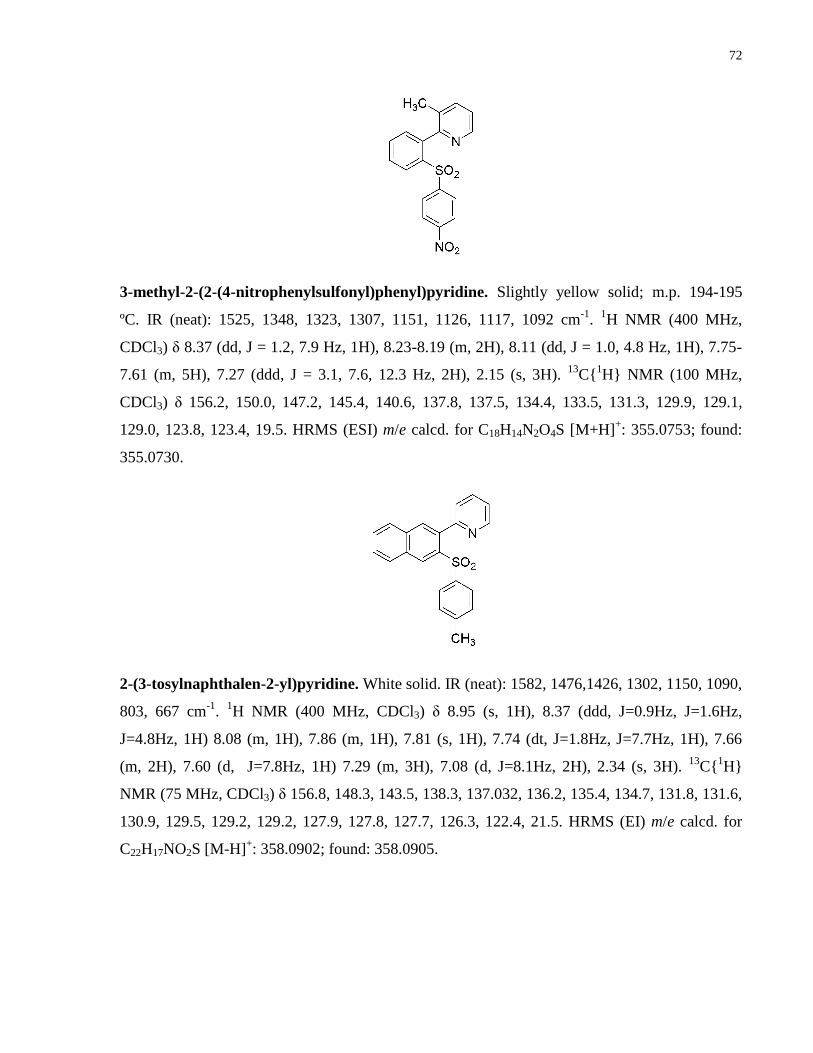
72
3-methyl-2-(2-(4-nitrophenylsulfonyl)phenyl)pyridine. Slightly yellow solid; m.p. 194-195
ºC. IR (neat): 1525, 1348, 1323, 1307, 1151, 1126, 1117, 1092 cm-1
. 1H NMR (400 MHz,
CDCl3) δ 8.37 (dd, J = 1.2, 7.9 Hz, 1H), 8.23-8.19 (m, 2H), 8.11 (dd, J = 1.0, 4.8 Hz, 1H), 7.75-
7.61 (m, 5H), 7.27 (ddd, J = 3.1, 7.6, 12.3 Hz, 2H), 2.15 (s, 3H). 13
C{1H} NMR (100 MHz,
CDCl3) δ 156.2, 150.0, 147.2, 145.4, 140.6, 137.8, 137.5, 134.4, 133.5, 131.3, 129.9, 129.1,
129.0, 123.8, 123.4, 19.5. HRMS (ESI) m/e calcd. for C18H14N2O4S [M+H]+: 355.0753; found:
355.0730.
2-(3-tosylnaphthalen-2-yl)pyridine. White solid. IR (neat): 1582, 1476,1426, 1302, 1150, 1090,
803, 667 cm-1
. 1H NMR (400 MHz, CDCl3) δ 8.95 (s, 1H), 8.37 (ddd, J=0.9Hz, J=1.6Hz,
J=4.8Hz, 1H) 8.08 (m, 1H), 7.86 (m, 1H), 7.81 (s, 1H), 7.74 (dt, J=1.8Hz, J=7.7Hz, 1H), 7.66
(m, 2H), 7.60 (d, J=7.8Hz, 1H) 7.29 (m, 3H), 7.08 (d, J=8.1Hz, 2H), 2.34 (s, 3H). 13
C{1H}
NMR (75 MHz, CDCl3) δ 156.8, 148.3, 143.5, 138.3, 137.032, 136.2, 135.4, 134.7, 131.8, 131.6,
130.9, 129.5, 129.2, 129.2, 127.9, 127.8, 127.7, 126.3, 122.4, 21.5. HRMS (EI) m/e calcd. for
C22H17NO2S [M-H]+: 358.0902; found: 358.0905.
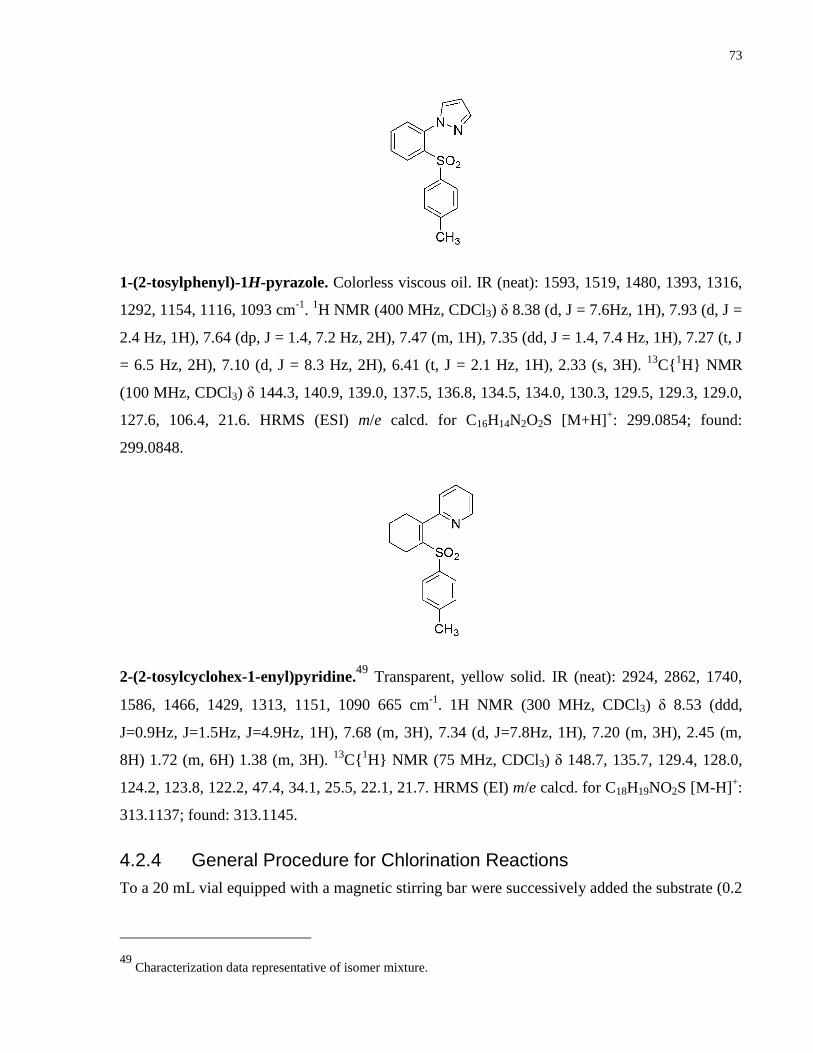
73
1-(2-tosylphenyl)-1H-pyrazole. Colorless viscous oil. IR (neat): 1593, 1519, 1480, 1393, 1316,
1292, 1154, 1116, 1093 cm-1
. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 7.6Hz, 1H), 7.93 (d, J =
2.4 Hz, 1H), 7.64 (dp, J = 1.4, 7.2 Hz, 2H), 7.47 (m, 1H), 7.35 (dd, J = 1.4, 7.4 Hz, 1H), 7.27 (t, J
= 6.5 Hz, 2H), 7.10 (d, J = 8.3 Hz, 2H), 6.41 (t, J = 2.1 Hz, 1H), 2.33 (s, 3H). 13
C{1H} NMR
(100 MHz, CDCl3) δ 144.3, 140.9, 139.0, 137.5, 136.8, 134.5, 134.0, 130.3, 129.5, 129.3, 129.0,
127.6, 106.4, 21.6. HRMS (ESI) m/e calcd. for C16H14N2O2S [M+H]+: 299.0854; found:
299.0848.
2-(2-tosylcyclohex-1-enyl)pyridine.49
Transparent, yellow solid. IR (neat): 2924, 2862, 1740,
1586, 1466, 1429, 1313, 1151, 1090 665 cm-1
. 1H NMR (300 MHz, CDCl3) δ 8.53 (ddd,
J=0.9Hz, J=1.5Hz, J=4.9Hz, 1H), 7.68 (m, 3H), 7.34 (d, J=7.8Hz, 1H), 7.20 (m, 3H), 2.45 (m,
8H) 1.72 (m, 6H) 1.38 (m, 3H). 13
C{1H} NMR (75 MHz, CDCl3) δ 148.7, 135.7, 129.4, 128.0,
124.2, 123.8, 122.2, 47.4, 34.1, 25.5, 22.1, 21.7. HRMS (EI) m/e calcd. for C18H19NO2S [M-H]+:
313.1137; found: 313.1145.
4.2.4 General Procedure for Chlorination Reactions
To a 20 mL vial equipped with a magnetic stirring bar were successively added the substrate (0.2
49 Characterization data representative of isomer mixture.

74
mmol, 1 equiv.), Pd(CH3CN)2Cl2 (0.02 mmol, 0.2 mmol), arylsulfonyl chloride (0.6 mmol, 3
equiv.) and DMF (1 mL). The mixture was stirred at 120 °C for 10 h under an atmosphere of air.
After cooling to ambient temperature, the resulting mixture was added to saturated Na2S (aq., 10
mL) and extracted with ether (3 x 20 mL). The combined organic layers were washed with brine
(2 x 15 mL) and dried over anhydrous Na2SO4. The organic phase was filtered and all volatiles
were removed under reduced pressure. The resulting residue was purified by preparative thin
layer chromatography (5% EtOAc/Hex, eluted 7 times) to afford the target products.
2-(2-chloro-5-methylphenyl)pyridine. Colorless oil. IR (neat): 1586, 1564, 1457, 1427, 1100,
1089, 1050, 1027 cm-1
. 1H NMR (400 MHz, CDCl3) δ 8.72 (ddd, J = 0.8, 1.5, 4.8 Hz, 1H), 7.75
(dt, J = 1.8, 7.7 Hz, 1H), 7.65 (d, J = 7.9 Hz, 1H), 7.42 (d, J = 2.0 Hz, 1H), 7.35 (d, J = 8.2 Hz,
1H), 7.27 (m, 1H), 2.37 (s, 3H). 13
C{1H} NMR (100 MHz, CDCl3) δ 156.9, 149.5, 138.7, 136.9,
135.8, 135.8, 132.1, 132.1, 130.4, 130.3, 129.8, 129.0, 124.9, 122.3, 122.3, 20.8. HRMS (ESI)
m/e calcd. for C12H10ClN [M+H]+: 204.0580; found: 204.0571.
2-(3-chloronaphthalen-2-yl)pyridine. NMR data were found to be in good agreement with
those in literature.22
4.2.5 General Procedure for Synthesis of Pd(phpy)2
Procedure generously provided by the Ritter Group based on literature precedence.50,51
50 Mann, F. G.; Purdie, D. J. Chem. Soc. 1935, 1549.
51 Jolliet, P.; Gianini, M.; vonZelewsky, A.; Bernardinelli, G.; StoeckliEvans, H. Inorg. Chem. 1996, 35, 4883.

75
Pd(Et2S)2Cl2. (NH4)2PdCl4 was dissolved in a minimum amount of water and Et2S (836 µl, 7.74
mmol, 2.2 equiv.) was added in 2 portions (5 min interval) and the mixture stirred for 30 min at
room temperature. The bright orange solid was filtered off and washed with water (5 x 20 ml).
The solid was dried in a vacuum desiccator for 48 h to give 1.12 g (81% yield). Used without
further characterization.
2-(2-bromophenyl)pyridine. Literature procedure used.22
To a round bottomed flask equipped
with a magnetic stirring bar was added 2-phenylpyridine (6.5 mmol, 1.0 equiv.), N-
bromosuccinimide (7.8 mmol, 1.2 equiv.), Pd(OAc)2 (0.32 mmol, 0.05 equiv.) and CH3CN (92
mL) and the resultant mixture was refluxed for 12 h. Upon cooling the solvent was evaporated
under reduced pressure. Isolated 0.8 g (53% yield) as an orange oil after column chromatography
(5% EtOAc/Hex). TLC Rf 0.33 (10% EtOAc/Hex). NMR data were found to be in good
agreement with those in literature.22
cis-Bis(2-phenylpyridine)palladium(II). 2-(2-Bromophenyl)pyridine (0.839 mmol, 3 equiv.)
was dissolved in anhydrous diethylether (8 ml) and the solution cooled to -78°C. nBuLi (0.699
mmol, 2.5 M in hexanes, 2.5 equiv.) was added drop wise and the mixture stirred for 30 min at
what time the solution had formed a pale yellow suspension. Pd(Et2S)2Cl2 (0.28 mmol, 1 equiv.)
was dissolved in anhydrous Et2O (8 ml) in a second flask. The solution was added to the lithiated
species drop wise via syringe over a period of 60 min at -78°C. The resulting mixture was stirred
for additional 30 min and H2O (2 ml) added at -78°C, the cooling bath removed and the mixture
warmed to room temperature. The mixture was transferred to a separating funnel and H2O added.
The mixture was extracted with CH2Cl2 three times and the combined organic extracts were
dried over MgSO4, filtered and concentrated under reduced pressure. The resulting solid was

76
triturated with diethylether-hexanes (3 mL, 1/9 (v/v)), filtered off and washed with diethylether-
hexanes (1/9 (v/v)) and hexanes. The resulting pale yellow solid was dried to a constant weight
of 92.8 mg (80% yield). NMR data were found to be in good agreement with those in literature.51
4.2.6 General Procedure for Synthesis of PdIV-Complex
To a 1 DR vial equipped with a magnetic stirring bar was added Pd(phpy)2 (0.046 mmol, 1.0
equiv.), p-toluenesulfonyl chloride (0.092 mmol, 2 equiv.) and CH2Cl2 (3 mL) and the resultant
mixture was stirred for 15 min. The solvent was evaporated under reduced pressure, dissolved in
CDCl3 and allowed to precipitate after 3 days. Isolated 10.1 mg (36% yield) as clear solid cubes
after recrystallization via slow diffusion of Et2O into DMF. Decomposed at 221-223 oC. IR
(neat): 1650, 1601, 1568, 1483, 1442, 1387, 1316, 1240, 1060, 1058, 1006, 767 cm-1
. 1H NMR
(300 MHz, DMSO-d6) δ 8.93 (dd, J=0.9Hz, J=5.5Hz, 1H) 8.74 (d, J=8.3Hz, 1H) 8.25 (dd,
J=5.5Hz, J=7.8Hz, 2H) 8.15 (dt, J=1.5Hz, J=7.8Hz, 1H) 8.05 (dd, J=1.6Hz, J=7.8Hz, 1H) 7.91
(m, 2H) 7.66 (m, 1H) 7.47 (ddd, J=0.9Hz, J=6.4Hz, J=8.3Hz, 2H) 7.12 (dt, J=1.0Hz, J=7.5Hz,
2H) 6.95 (m, 6H) 6.43 (dd, J=0.8Hz, J=8.1Hz, 1H), 2.22 (s, 3H). 13
C{1H} NMR (75 MHz,
DMSO-d6) δ 166.2, 163.5, 160.4, 158.4, 147.5, 145.7, 145.3, 141.6, 141.4, 140.6, 140.5, 140.3,
140.2, 132.4, 131.8, 131.6, 131.2, 128.0, 126.6, 126.6, 126.4, 126.1, 125.8, 125.7, 124.5, 124.4,
121.1, 120.7, 20.7. HRMS (ESI) m/e calcd. for C29H23N2O2SPd [M-Cl]+: 569.0509; found:
569.0494 and (ESI) m/e calcd. for C29H23N2O2NaSClPd [M+Na]+: 627.0095; found: 627.0062.
4.2.7 General Procedure for Reductive Elimination from PdIV-Complex
To a 1DR vial equipped with a magnetic stirring bar was added the above PdIV
-complex (0.0165
mmol) and dissolved in DMF (200 μL). The resultant mixture was heated to 120 oC for 7 h.
Upon cooling to ambient temperature, the resulting residue was purified by preparative thin layer
chromatography (40 % EtOAc/Hex) to afford the target sulfone product (2.9 mg, 57% yield).

77
4.2.8 General Procedure for Direct Arylation (Tables 28 and 29)
In a glovebox, to a 20-mL vial equipped with a magnetic stirring bar were successively added tri-
2-furylphosphine (0.03 mmol, 0.3 equiv.), [Rh(cod)Cl]2 (0.005 mmol, 0.05 equiv) and 1,4-
dioxane (200 μL) and stirred for 15 mins. The substrate (0.1 mmol, 1 equiv.), p-toluenesulfonyl
chloride (0.3 mmol, 3 equiv.), K2CO3 (0.4 mmol, 4 equiv.), 4A molecular sieves (100 mg) and
1,4-dioxane (0.8 mL) were subsequently added. The vial was sealed with a Teflon cap and
removed from the glovebox. The mixture was stirred on a heating block at 145 °C for 27 h. Upon
cooling to ambient temperature, the resulting mixture was filtered through a pad of Celite and
washed with ethyl acetate. The filtrate was concentrated in vacuo and the resulting residue was
purified by preparative thin layer chromatography (7% Et2O/Hex) to afford the target products.
2-(4'-methylbiphenyl-2-yl)pyridine. NMR data were found to be in good agreement with those
in literature.18
10-p-tolylbenzo[h]quinoline. NMR data were found to be in good agreement with those in
literature.52
52 Jin, W.; Yu, Z.; He, W.; Ye, W.; Xiao, W.-J. Org. Lett. 2009, 11, 1317.

78
3-methyl-2-(4’-methylbiphenyl-2-yl)pyridine. NMR data were found to be in good agreement
with those in literature.18
2-(3,4'-dimethylbiphenyl-2-yl)pyridine. Yellow oil. IR (neat): 2922, 1585, 1459, 1422, 1025,
823, 784, 748 cm-1
. 1H NMR (300 MHz, CDCl3) δ 8.63 (d, J=4.1Hz, 1H), 7.46 (dt, J=1.8Hz,
J=7.7Hz, 1H), 7.34 (m, 1H), 7.27 (m, 2H), 7.10 (ddd, J=1.1Hz, J=4.9Hz, J=7.6Hz, 1H), 6.91 (m,
5H), 2.25 (s, 3H), 2.17 (s, 3H). 13
C{1H} NMR (75 MHz, CDCl3) δ 159.8, 148.8, 141.1, 139.3,
138.7, 136.7, 135. 8, 135.7, 129.5, 129.2, 128.3, 128.0, 127.6, 125.6, 121.2, 21.0, 20.5. HRMS
(EI) m/e calcd. for C19H17N [M-H]+: 528.1283; found: 258.1284.
4.2.9 General Procedure for Direct Arylation (Tables 33 and 34)
In a glovebox, to a 20-mL vial equipped with a magnetic stirring bar were successively added tri-
2-furylphosphine (0.03 mmol, 0.3 equiv.), [Rh(cod)Cl]2 (0.005 mmol, 0.05 equiv) and 1,4-
dioxane (200 μL) and stirred for 15 mins. Ag3PO4 (0.003 mmol, 0.03 equiv.) and 1,4-dioxane
(200 μL) were added and the resultant mixture stirred for an additional 15 mins. The substrate
(0.1 mmol, 1 equiv.), p-toluenesulfonyl chloride (0.3 mmol, 3 equiv.), K2CO3 (0.4 mmol, 4
equiv.), 4A molecular sieves (100 mg) and 1,4-dioxane (0.6 mL) were subsequently added. The
vial was sealed with a Teflon cap and removed from the glovebox. The mixture was stirred on a
heating block at 145 °C for 27 h. Upon cooling to ambient temperature, the resulting mixture
was filtered through a pad of Celite and washed with ethyl acetate. The filtrate was concentrated
in vacuo and the resulting residue was purified by preparative thin layer chromatography (7%
Et2O/Hex) to afford the target products.

79
2-(5-methoxy-4'-methylbiphenyl-2-yl)-3-methylpyridine. Yellow solid. IR (neat): 2924, 1606,
1444, 1297, 1222, 1176, 1018, 825, 792 cm-1
. 1H NMR (300 MHz, CDCl3) δ 8.48 (dd, J=1.1Hz,
J=4.7Hz, 1H), 7.30 (m, 2H), 7.08 (dd, J=4.8Hz, J=7.6Hz, 1H), 6.96 (m, 6H), 3.87 (s, 3H), 2.27
(s, 3H), 1.73 (s, 3H). 13
C{1H} NMR (75 MHz, CDCl3) δ 159.5, 159.4, 146.5, 141.9, 138.2,
137.4, 136.4, 132.2, 131.9, 131.1, 129.0, 128.6, 121.8, 114.9, 112.7, 55.4, 21.1, 18.9. HRMS (EI)
m/e calcd. for C20H19NO [M-H]+: 288.1388; found: 288.1382.
3-methyl-2-(4'-methyl-5-(trifluoromethyl)biphenyl-2-yl)pyridine. Orange oil. IR (neat): 2925,
1335, 1286, 1167, 1128, 1083. 1H NMR (300 MHz, CDCl3) δ 8.51 (d, J=3.8Hz, 1H), 7.68 (d,
J=10.8Hz, 2H), 7.51 (d, J=7.8Hz, 1H), 7.34 (d, J=7.6Hz, 1H), 7.26 (s, 1H), 7.14 (dd, J=4.8Hz,
J=7.7Hz, 1H), 7.00 (s, 3H), 2.28 (s, 3H), 1.75 (s, 3H). 13
C{1H} NMR (75 MHz, CDCl3) δ 146.8,
141.4, 137.7, 137.1, 136.7, 131.5, 130.6, 129.0, 128.8, 126.6, 126.5, 123.9, 123.8, 123.6, 122.6,
21.1, 18.7. HRMS (EI) m/e calcd. for C20H16F3N [M-H]+: 327.1235; found: 327.1226.

80
Appendix A NMR Spectra
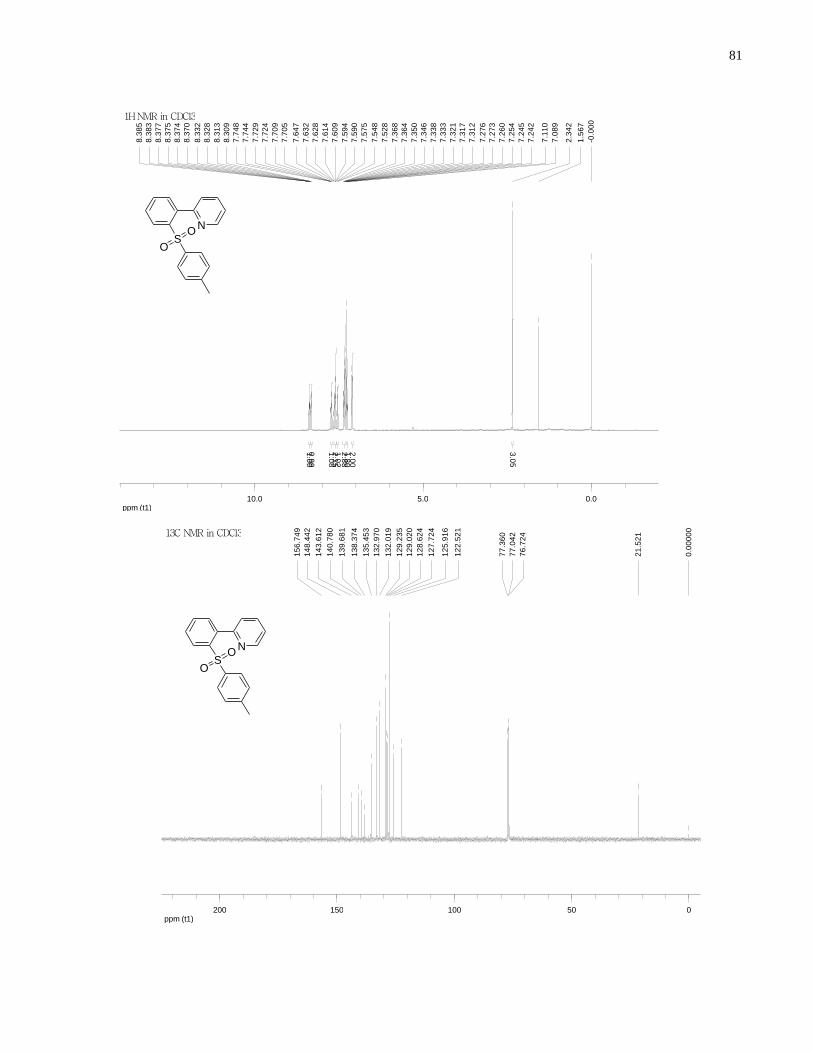
81
ppm (t1)0.05.010.0
8.3
85
8.3
83
8.3
77
8.3
75
8.3
74
8.3
70
8.3
32
8.3
28
8.3
13
8.3
09
7.7
48
7.7
44
7.7
29
7.7
24
7.7
09
7.7
05
7.6
47
7.6
32
7.6
28
7.6
14
7.6
09
7.5
94
7.5
90
7.5
75
7.5
48
7.5
28
7.3
68
7.3
64
7.3
50
7.3
46
7.3
38
7.3
33
7.3
21
7.3
17
7.3
12
7.2
76
7.2
73
7.2
60
7.2
54
7.2
45
7.2
42
7.1
10
7.0
89
2.3
42
1.5
67
-0.0
00
1.0
00
.99
1.0
82
.15
1.0
22
.88
1.8
02
.00
3.0
5
1H NMR in CDCl3
N
SO
O
ppm (t1)050100150200
15
6.7
49
14
8.4
42
14
3.6
12
14
0.7
80
13
9.6
81
13
8.3
74
13
5.4
53
13
2.9
70
13
2.0
19
12
9.2
35
12
9.0
20
12
8.6
24
12
7.7
24
12
5.9
16
12
2.5
21
77
.36
0
77
.04
2
76
.72
4
21
.52
1
0.0
00
0013C NMR in CDCl3
N
SO
O
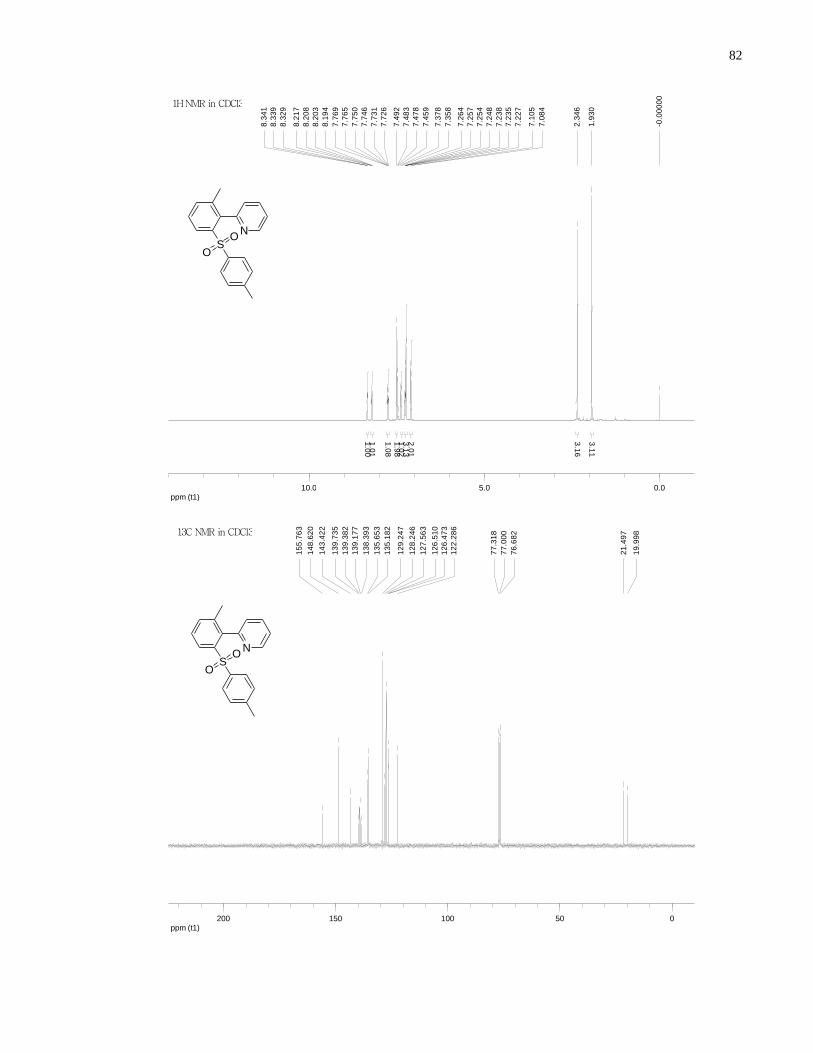
82
ppm (t1)0.05.010.0
8.3
41
8.3
39
8.3
29
8.2
17
8.2
08
8.2
03
8.1
94
7.7
69
7.7
65
7.7
50
7.7
46
7.7
31
7.7
26
7.4
92
7.4
83
7.4
78
7.4
59
7.3
78
7.3
58
7.2
64
7.2
57
7.2
54
7.2
48
7.2
38
7.2
35
7.2
27
7.1
05
7.0
84
2.3
46
1.9
30
-0.0
00
00
1.0
01
.01
1.0
81
.98
1.0
23
.13
2.0
1
3.1
6
3.1
1
1H NMR in CDCl3
N
SO
O
ppm (t1)050100150200
15
5.7
63
14
8.6
20
14
3.4
22
13
9.7
35
13
9.3
82
13
9.1
77
13
8.3
93
13
5.6
53
13
5.1
82
12
9.2
47
12
8.2
46
12
7.5
63
12
6.5
10
12
6.4
73
12
2.2
86
77
.31
8
77
.00
0
76
.68
2
21
.49
7
19
.99
813C NMR in CDCl3
N
SO
O
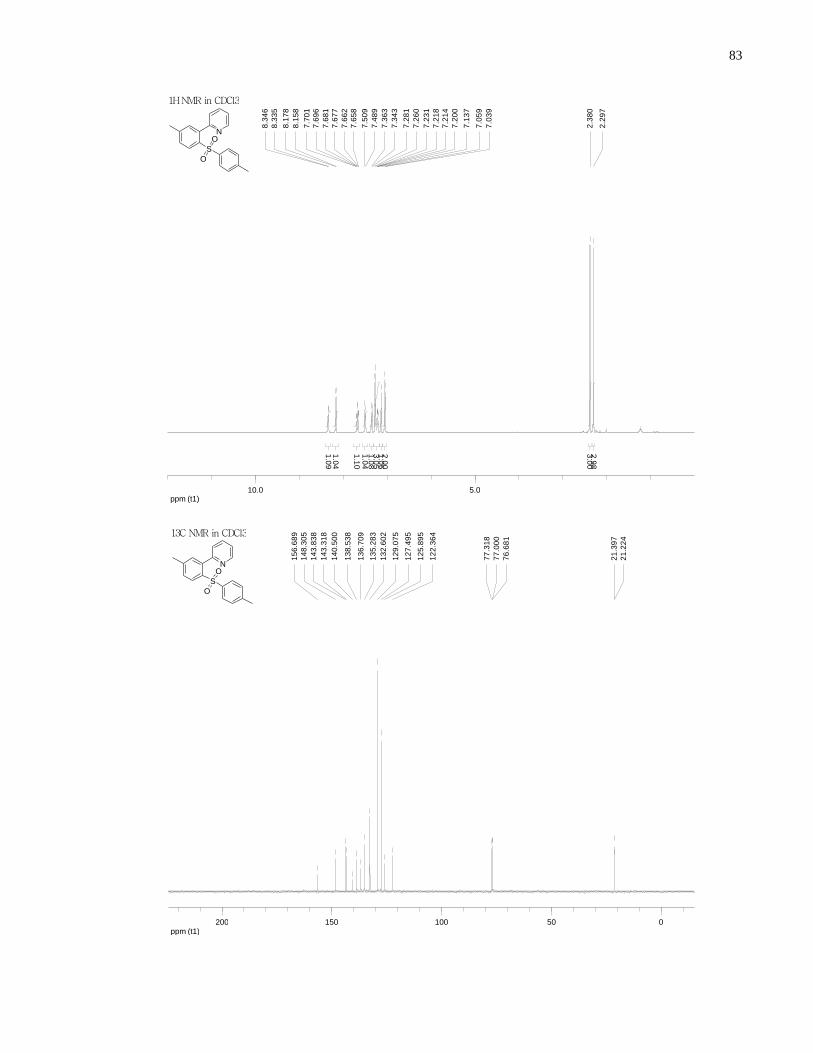
83
ppm (t1)5.010.0
8.3
46
8.3
35
8.1
78
8.1
58
7.7
01
7.6
96
7.6
81
7.6
77
7.6
62
7.6
58
7.5
09
7.4
89
7.3
63
7.3
43
7.2
81
7.2
60
7.2
31
7.2
18
7.2
14
7.2
00
7.1
37
7.0
59
7.0
39
2.3
80
2.2
97
1.0
4
1.1
01
.04
1.0
83
.05
1.0
62
.00
3.0
02
.98
1.0
9
1H NMR in CDCl3
N
S
O
O
ppm (t1)050100150200
15
6.6
89
14
8.3
05
14
3.8
38
14
3.3
18
14
0.5
00
13
8.5
38
13
6.7
09
13
5.2
83
13
2.6
02
12
9.0
75
12
7.4
95
12
5.8
95
12
2.3
64
77
.31
8
77
.00
0
76
.68
1
21
.39
7
21
.22
4
13C NMR in CDCl3
N
S
O
O
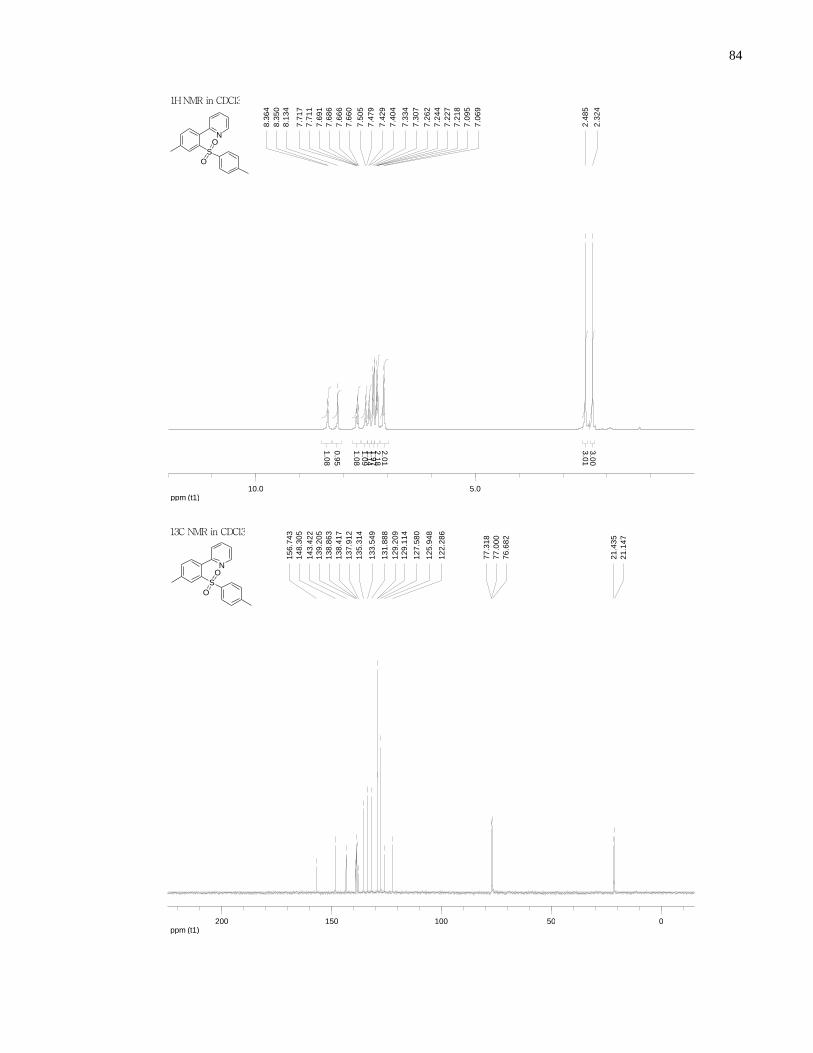
84
ppm (t1)5.010.0
8.3
64
8.3
50
8.1
34
7.7
17
7.7
11
7.6
91
7.6
86
7.6
66
7.6
60
7.5
05
7.4
79
7.4
29
7.4
04
7.3
34
7.3
07
7.2
62
7.2
44
7.2
27
7.2
18
7.0
95
7.0
69
2.4
85
2.3
24
1.0
8
1.0
8
0.9
5
1.0
91
.14
1.9
72
.18
2.0
1
3.0
13
.00
1H NMR in CDCl3
N
S
O
O
ppm (t1)050100150200
15
6.7
43
14
8.3
05
14
3.4
22
13
9.2
05
13
8.8
63
13
8.4
17
13
7.9
12
13
5.3
14
13
3.5
49
13
1.8
88
12
9.2
09
12
9.1
14
12
7.5
80
12
5.9
48
12
2.2
86
77
.31
8
77
.00
0
76
.68
2
21
.43
5
21
.14
7
13C NMR in CDCl3
N
S
O
O
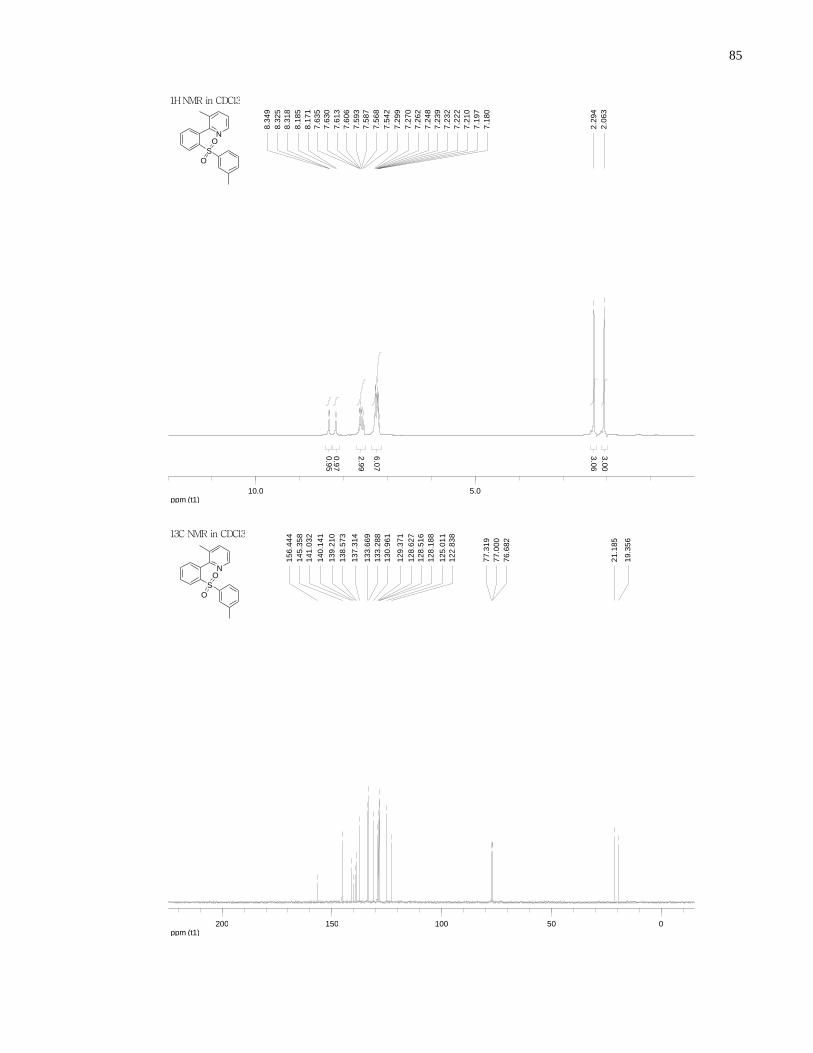
85
ppm (t1)5.010.0
8.3
49
8.3
25
8.3
18
8.1
85
8.1
71
7.6
35
7.6
30
7.6
13
7.6
06
7.5
93
7.5
87
7.5
68
7.5
42
7.2
99
7.2
70
7.2
62
7.2
48
7.2
39
7.2
32
7.2
22
7.2
10
7.1
97
7.1
80
2.2
94
2.0
63
0.9
50
.97
2.9
9
6.0
7
3.0
6
3.0
0
1H NMR in CDCl3
N
S
O
O
ppm (t1)050100150200
15
6.4
44
14
5.3
58
14
1.0
32
14
0.1
41
13
9.2
10
13
8.5
73
13
7.3
14
13
3.6
69
13
3.2
88
13
0.9
61
12
9.3
71
12
8.6
27
12
8.5
16
12
8.1
88
12
5.0
11
12
2.8
38
77
.31
9
77
.00
0
76
.68
2
21
.18
5
19
.35
6
13C NMR in CDCl3
N
S
O
O
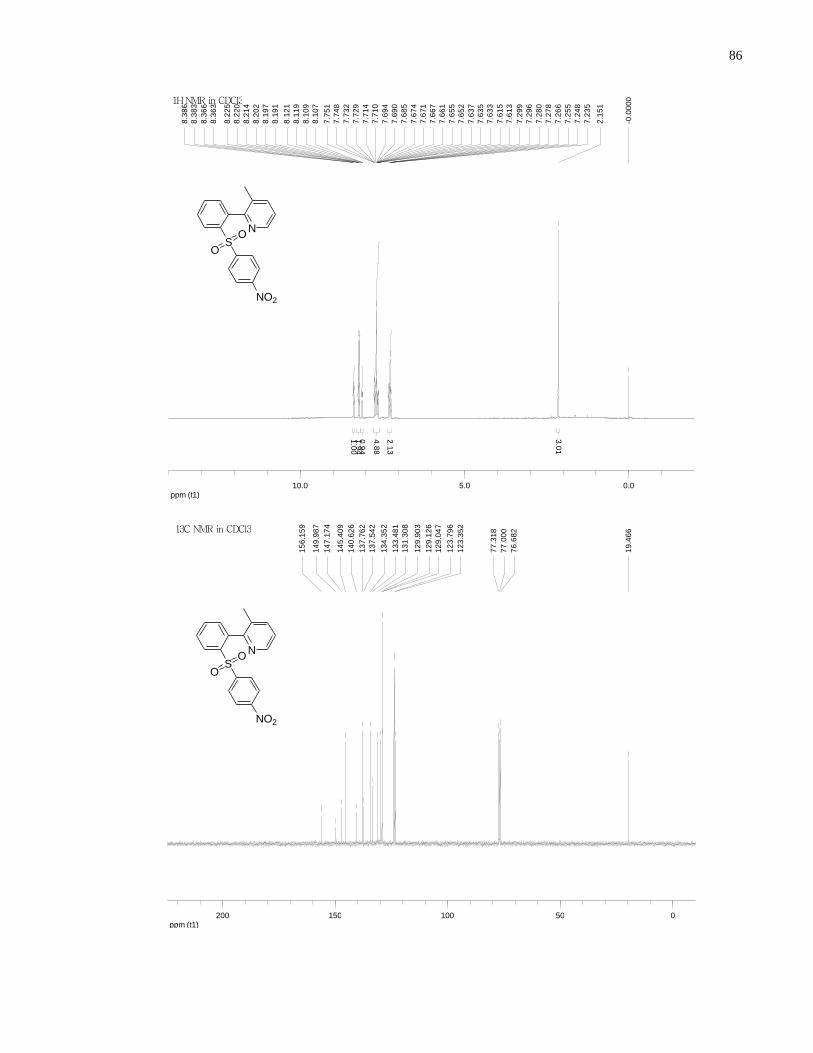
86
ppm (t1)0.05.010.0
8.3
86
8.3
83
8.3
66
8.3
63
8.2
25
8.2
20
8.2
14
8.2
02
8.1
97
8.1
91
8.1
21
8.1
19
8.1
09
8.1
07
7.7
51
7.7
48
7.7
32
7.7
29
7.7
14
7.7
10
7.6
94
7.6
90
7.6
85
7.6
74
7.6
71
7.6
67
7.6
61
7.6
55
7.6
52
7.6
37
7.6
35
7.6
33
7.6
15
7.6
13
7.2
99
7.2
96
7.2
80
7.2
78
7.2
66
7.2
55
7.2
48
7.2
35
2.1
51
-0.0
00
0
1.0
01
.94
0.9
4
4.8
8
2.1
3
3.0
1
1H NMR in CDCl3
N
SO
O
NO2
ppm (t1)050100150200
15
6.1
59
14
9.9
87
14
7.1
74
14
5.4
09
14
0.6
26
13
7.7
62
13
7.5
42
13
4.3
52
13
3.4
81
13
1.3
08
12
9.9
03
12
9.1
26
12
9.0
47
12
3.7
96
12
3.3
52
77
.31
8
77
.00
0
76
.68
2
19
.46
613C NMR in CDCl3
N
SO
O
NO2
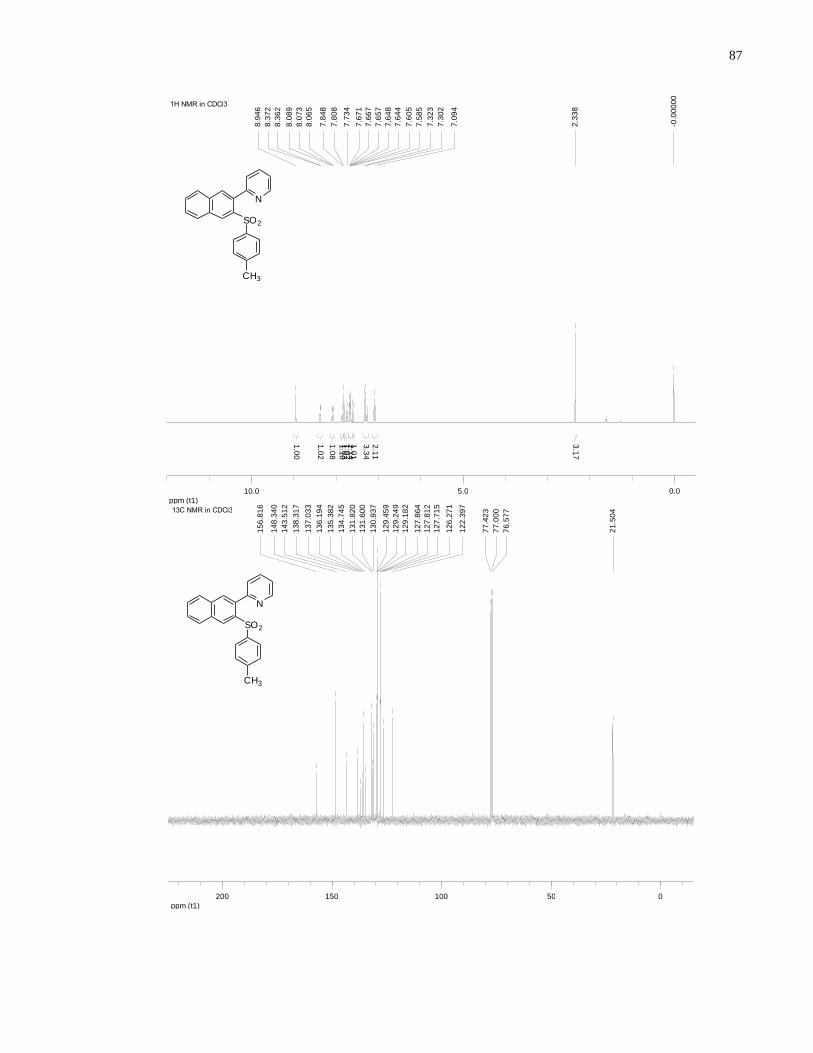
87
ppm (t1)
0.05.010.0
8.9
46
8.3
72
8.3
62
8.0
89
8.0
73
8.0
65
7.8
48
7.8
08
7.7
34
7.6
71
7.6
67
7.6
57
7.6
48
7.6
44
7.6
05
7.5
85
7.3
23
7.3
02
7.0
94
2.3
38
-0.0
00
00
1.0
0
1.0
2
1.0
8
2.1
13
.34
1.1
01
.03
1.0
72
.14
1.0
1
3.1
7
1H NMR in CDCl3
N
SO2
CH3
ppm (t1)
050100150200
15
6.8
16
14
8.3
40
14
3.5
12
13
8.3
17
13
7.0
33
13
6.1
94
13
5.3
82
13
4.7
45
13
1.8
20
13
1.6
00
13
0.9
37
12
9.4
59
12
9.2
49
12
9.1
82
12
7.8
64
12
7.8
12
12
7.7
15
12
6.2
71
12
2.3
97
77
.42
3
77
.00
0
76
.57
7
21
.50
413C NMR in CDCl3
N
SO2
CH3
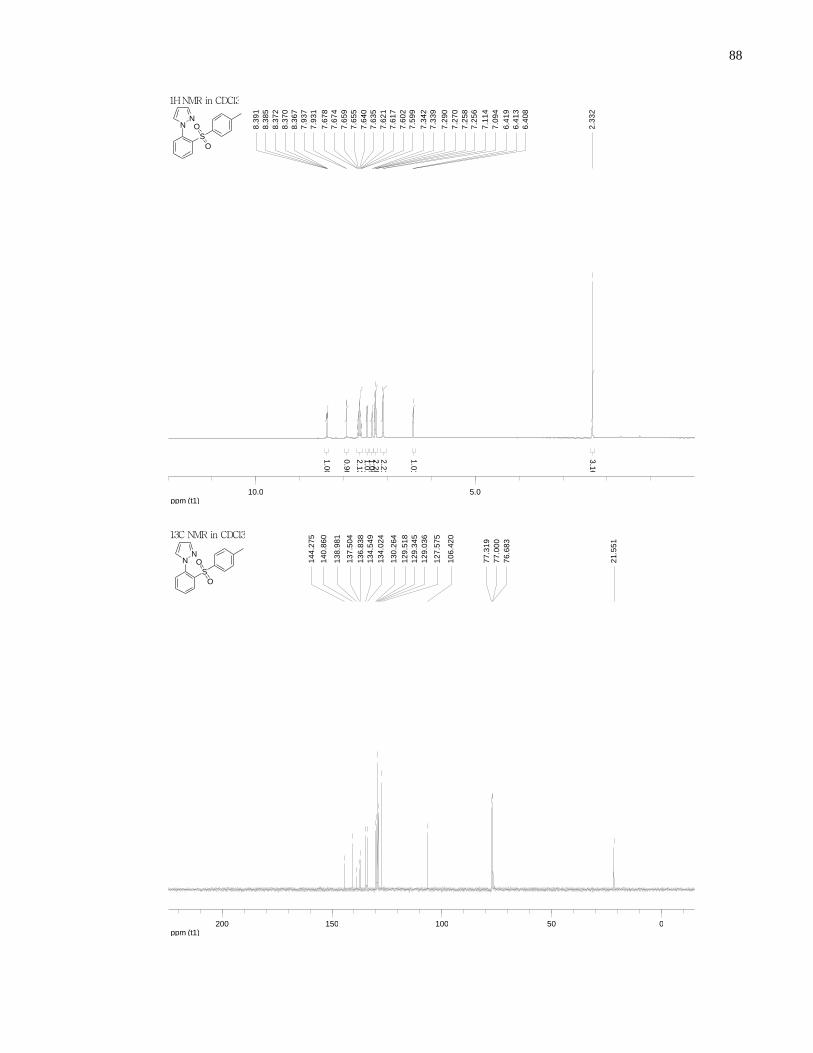
88
ppm (t1)5.010.0
8.3
91
8.3
85
8.3
72
8.3
70
8.3
67
7.9
37
7.9
31
7.6
78
7.6
74
7.6
59
7.6
55
7.6
40
7.6
35
7.6
21
7.6
17
7.6
02
7.5
99
7.3
42
7.3
39
7.2
90
7.2
70
7.2
58
7.2
56
7.1
14
7.0
94
6.4
19
6.4
13
6.4
08
2.3
32
1.0
0
0.9
6
2.1
31
.01
1.0
42
.28
2.2
1
1.0
1
3.1
6
1H NMR in CDCl3
N
S
O
O
N
ppm (t1)050100150200
14
4.2
75
14
0.8
60
13
8.9
81
13
7.5
04
13
6.8
38
13
4.5
49
13
4.0
24
13
0.2
64
12
9.5
18
12
9.3
45
12
9.0
36
12
7.5
75
10
6.4
20
77
.31
9
77
.00
0
76
.68
3
21
.55
1
13C NMR in CDCl3
N
S
O
O
N
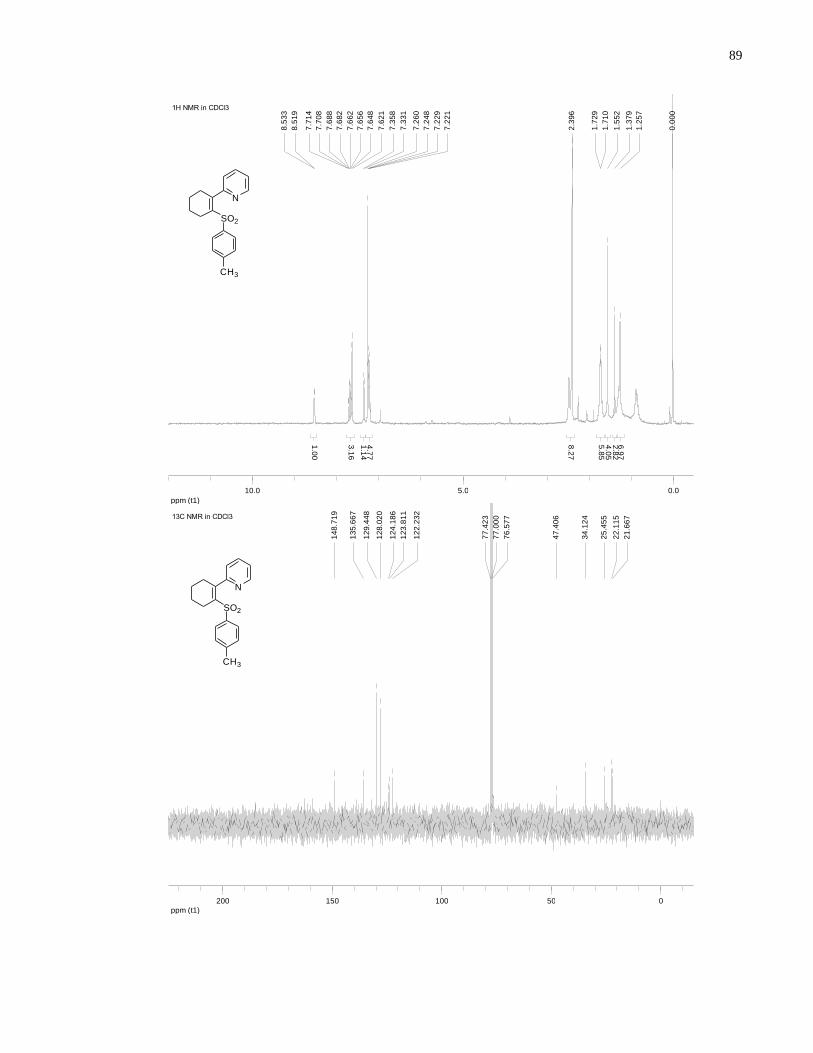
89
ppm (t1)
0.05.010.0
8.5
33
8.5
19
7.7
14
7.7
08
7.6
88
7.6
82
7.6
62
7.6
56
7.6
48
7.6
21
7.3
58
7.3
31
7.2
60
7.2
48
7.2
29
7.2
21
2.3
96
1.7
29
1.7
10
1.5
52
1.3
79
1.2
57
0.0
00
1.0
0
3.1
6
1.1
44
.77
8.2
7
5.8
54
.05
2.8
26
.97
1H NMR in CDCl3
N
SO2
CH3
ppm (t1)
050100150200
14
8.7
19
13
5.6
67
12
9.4
48
12
8.0
20
12
4.1
86
12
3.8
11
12
2.2
32
77
.42
3
77
.00
0
76
.57
7
47
.40
6
34
.12
4
25
.45
5
22
.11
5
21
.66
713C NMR in CDCl3
N
SO2
CH3
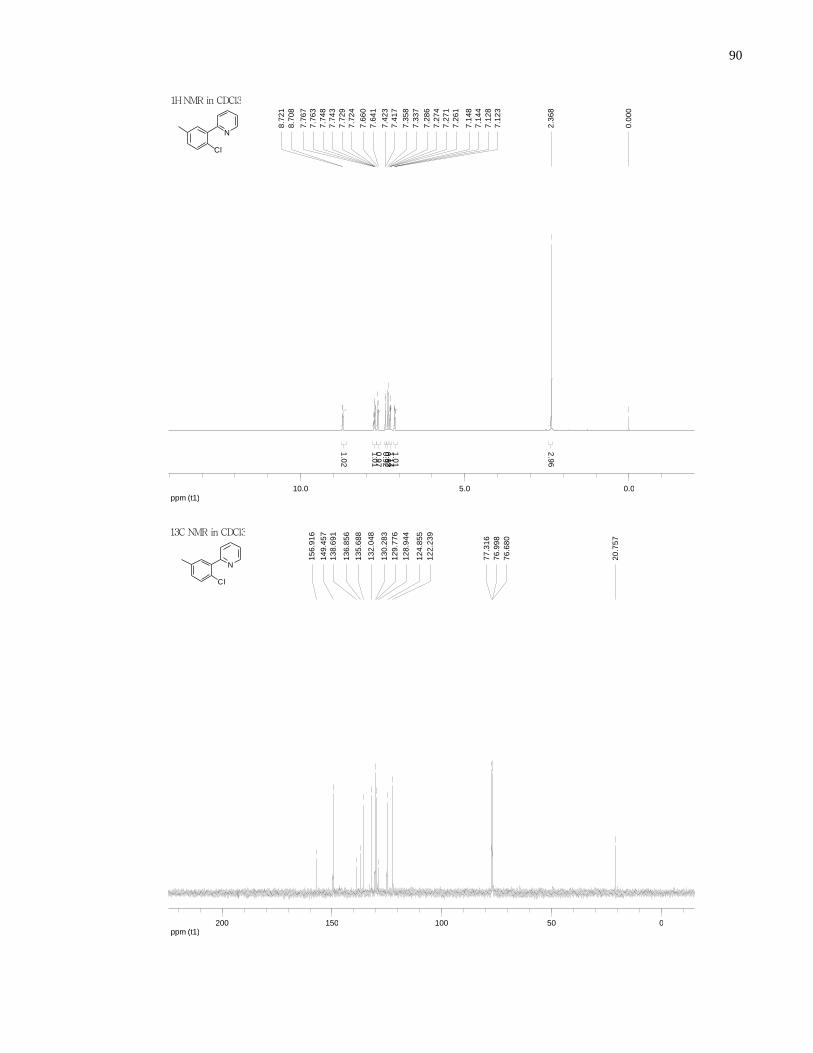
90
ppm (t1)0.05.010.0
8.7
21
8.7
08
7.7
67
7.7
63
7.7
48
7.7
43
7.7
29
7.7
24
7.6
60
7.6
41
7.4
23
7.4
17
7.3
58
7.3
37
7.2
86
7.2
74
7.2
71
7.2
61
7.1
48
7.1
44
7.1
28
7.1
23
2.3
68
0.0
00
1.0
2
1.0
10
.97
1.0
1
0.9
20
.92
1.1
4
2.9
6
1H NMR in CDCl3
N
Cl
ppm (t1)050100150200
15
6.9
16
14
9.4
57
13
8.6
91
13
6.8
56
13
5.6
88
13
2.0
48
13
0.2
83
12
9.7
76
12
8.9
44
12
4.8
55
12
2.2
39
77
.31
6
76
.99
8
76
.68
0
20
.75
7
13C NMR in CDCl3
Cl
N
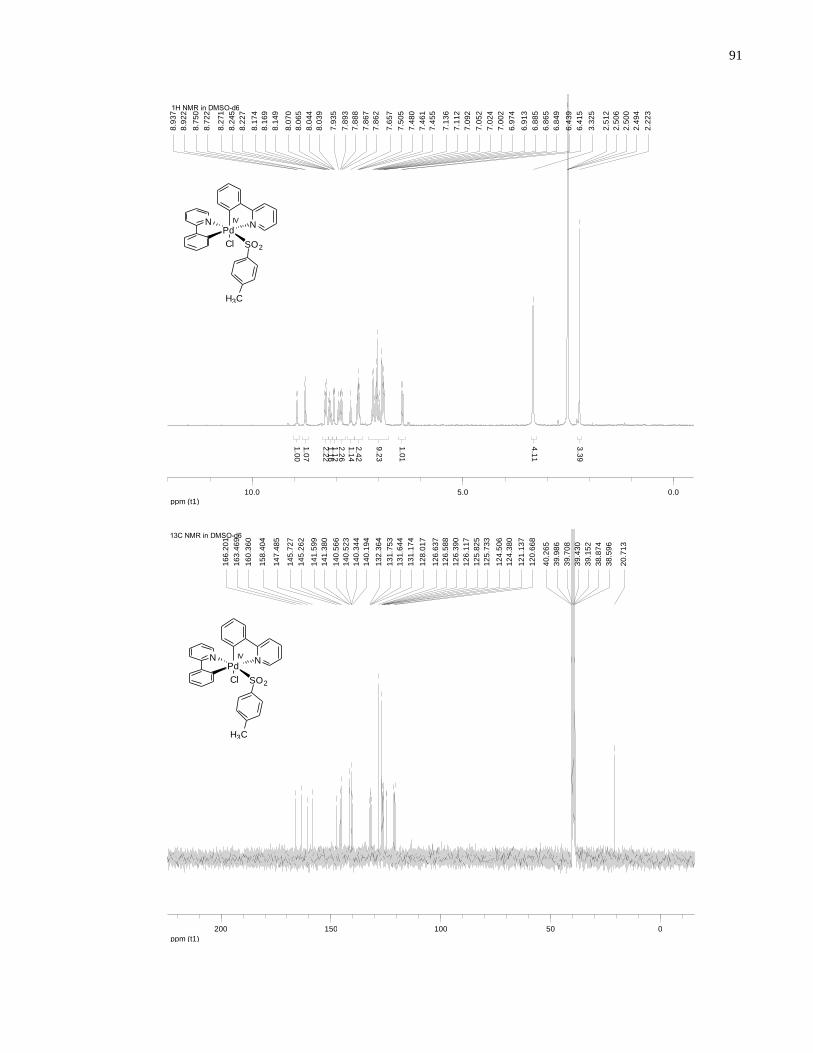
91
ppm (t1)
0.05.010.0
8.9
37
8.9
22
8.7
50
8.7
22
8.2
71
8.2
45
8.2
27
8.1
74
8.1
69
8.1
49
8.0
70
8.0
65
8.0
44
8.0
39
7.9
35
7.8
93
7.8
88
7.8
67
7.8
62
7.6
57
7.5
05
7.4
80
7.4
61
7.4
55
7.1
36
7.1
12
7.0
92
7.0
52
7.0
24
7.0
02
6.9
74
6.9
13
6.8
85
6.8
65
6.8
49
6.4
39
6.4
15
3.3
25
2.5
12
2.5
06
2.5
00
2.4
94
2.2
23
1.0
0
1.0
7
2.2
21
.16
1.1
22
.26
1.1
42
.42
9.2
3
1.0
1
4.1
1
3.3
9
1H NMR in DMSO-d6
NPd
SO2
N
Cl
H3C
IV
ppm (t1)
050100150200
16
6.2
01
16
3.4
69
16
0.3
60
15
8.4
04
14
7.4
85
14
5.7
27
14
5.2
62
14
1.5
99
14
1.3
80
14
0.5
66
14
0.5
23
14
0.3
44
14
0.1
94
13
2.3
64
13
1.7
53
13
1.6
44
13
1.1
74
12
8.0
17
12
6.6
37
12
6.5
88
12
6.3
90
12
6.1
17
12
5.8
25
12
5.7
33
12
4.5
06
12
4.3
80
12
1.1
37
12
0.6
68
40
.26
5
39
.98
6
39
.70
8
39
.43
0
39
.15
2
38
.87
4
38
.59
6
20
.71
3
13C NMR in DMSO-d6
NPd
SO2
N
Cl
H3C
IV
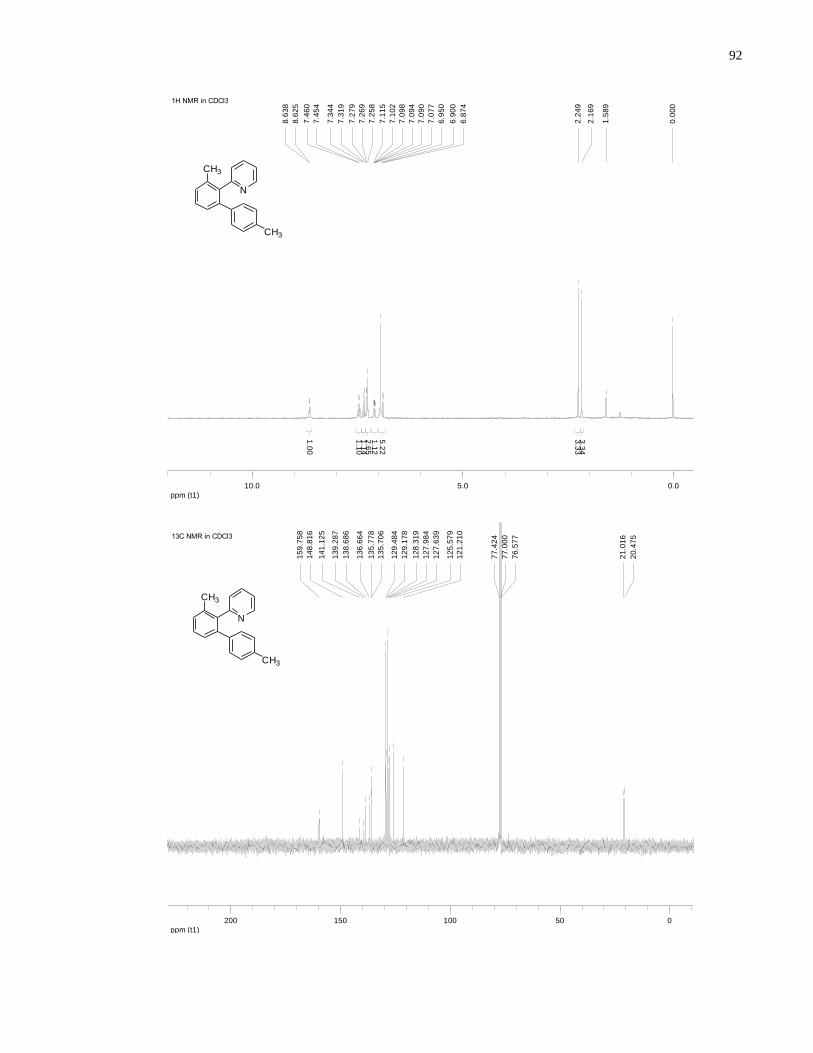
92
ppm (t1)
0.05.010.0
8.6
38
8.6
25
7.4
60
7.4
54
7.3
44
7.3
19
7.2
79
7.2
69
7.2
58
7.1
15
7.1
02
7.0
98
7.0
94
7.0
90
7.0
77
6.9
50
6.9
00
6.8
74
2.2
49
2.1
69
1.5
89
0.0
00
1.0
0
1.1
01
.14
2.6
51
.12
5.2
2
3.3
33
.34
1H NMR in CDCl3
N
CH3
CH3
ppm (t1)
050100150200
15
9.7
58
14
8.8
16
14
1.1
25
13
9.2
87
13
8.6
86
13
6.6
64
13
5.7
78
13
5.7
06
12
9.4
84
12
9.1
78
12
8.3
19
12
7.9
84
12
7.6
39
12
5.5
79
12
1.2
10
77
.42
4
77
.00
0
76
.57
7
21
.01
6
20
.47
513C NMR in CDCl3
N
CH3
CH3
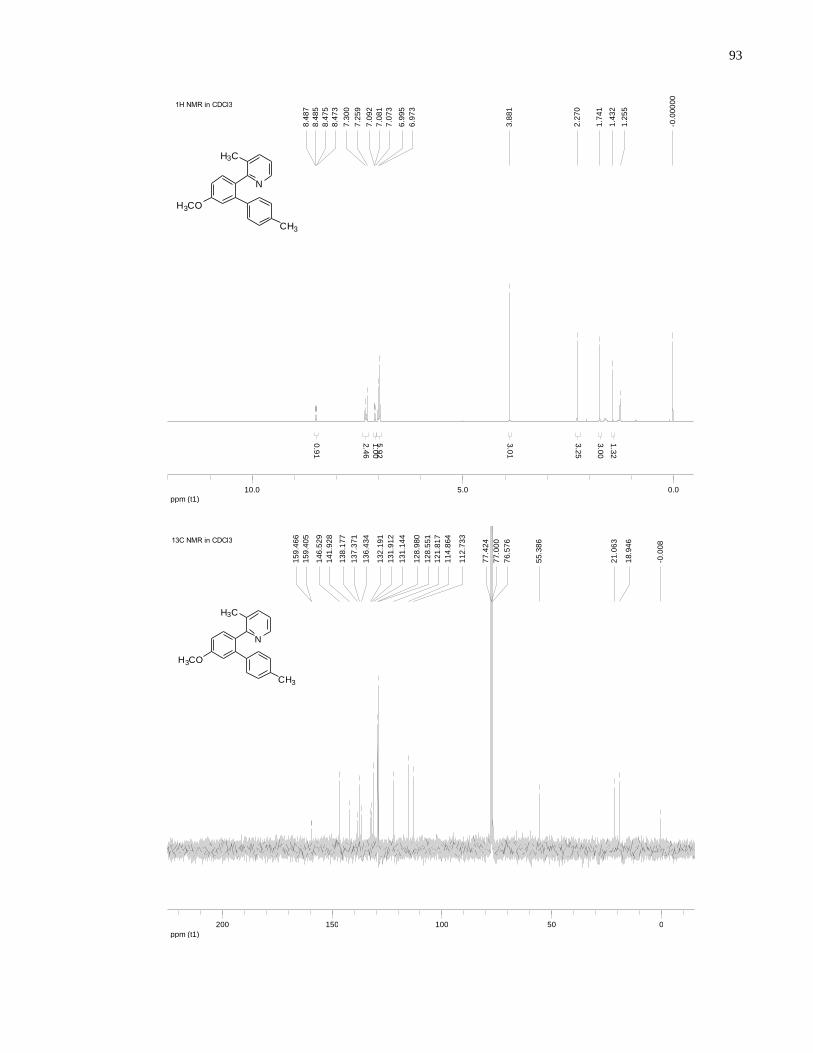
93
ppm (t1)
0.05.010.0
8.4
87
8.4
85
8.4
75
8.4
73
7.3
00
7.2
59
7.0
92
7.0
81
7.0
73
6.9
95
6.9
73
3.8
81
2.2
70
1.7
41
1.4
32
1.2
55
-0.0
00
00
0.9
1
2.4
6
5.9
2
3.0
1
1.0
0
3.2
5
3.0
0
1.3
2
1H NMR in CDCl3
N
CH3
H3C
H3CO
ppm (t1)
050100150200
15
9.4
66
15
9.4
05
14
6.5
29
14
1.9
28
13
8.1
77
13
7.3
71
13
6.4
34
13
2.1
91
13
1.9
12
13
1.1
44
12
8.9
80
12
8.5
51
12
1.8
17
11
4.8
64
11
2.7
33
77
.42
4
77
.00
0
76
.57
6
55
.38
6
21
.06
3
18
.94
6
-0.0
0813C NMR in CDCl3
N
CH3
H3C
H3CO
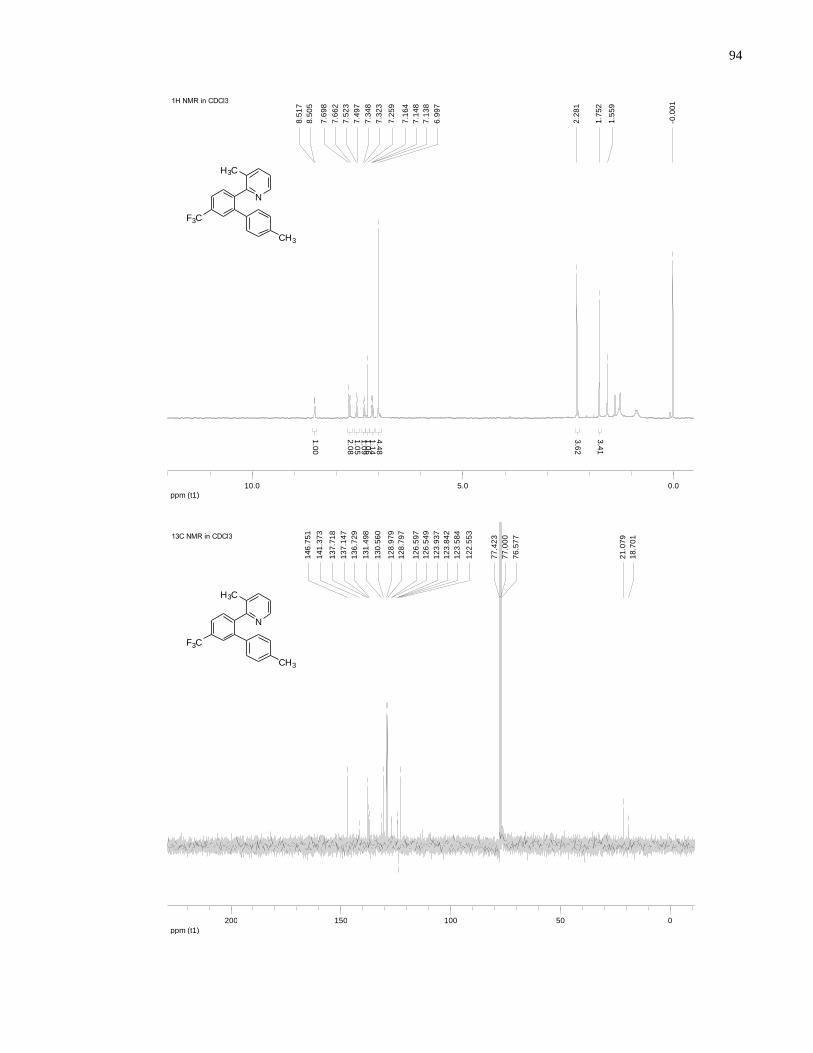
94
ppm (t1)
0.05.010.0
8.5
17
8.5
05
7.6
98
7.6
62
7.5
23
7.4
97
7.3
48
7.3
23
7.2
59
7.1
64
7.1
48
7.1
38
6.9
97
2.2
81
1.7
52
1.5
59
-0.0
01
1.0
0
2.0
81
.05
1.0
91
.06
1.1
44
.48
3.6
2
3.4
1
1H NMR in CDCl3
N
CH3
H3C
F3C
ppm (t1)
050100150200
14
6.7
51
14
1.3
73
13
7.7
18
13
7.1
47
13
6.7
29
13
1.4
98
13
0.5
60
12
8.9
79
12
8.7
97
12
6.5
97
12
6.5
49
12
3.9
37
12
3.8
42
12
3.5
84
12
2.5
53
77
.42
3
77
.00
0
76
.57
7
21
.07
9
18
.70
113C NMR in CDCl3
N
CH3
H3C
F3C

95
Appendix B Crystallographic Information
Table 1. Crystal data and structure refinement for k09146a.
Identification code k09146a
Empirical formula C32 H30 Cl N3 O3 Pd S
Formula weight 678.50
Temperature 150(2) K
Wavelength 0.71073 Å
Crystal system Monoclinic
Space group P 21/c
Unit cell dimensions a = 16.5610(5) Å = 90°.
b = 13.8095(5) Å = 91.897(2)°.
c = 12.7616(3) Å = 90°.
Volume 2916.97(15) Å3
Z 4
Density (calculated) 1.545 Mg/m3
Absorption coefficient 0.838 mm-1
F(000) 1384
Crystal size 0.20 x 0.15 x 0.14 mm3
Theta range for data collection 2.87 to 27.50°.
Index ranges -20<=h<=21, -17<=k<=17, -16<=l<=16
Reflections collected 23075

96
Independent reflections 6657 [R(int) = 0.0871]
Completeness to theta = 27.50° 99.3 %
Absorption correction Semi-empirical from equivalents
Max. and min. transmission 0.894 and 0.748
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 6657 / 0 / 374
Goodness-of-fit on F2 1.060
Final R indices [I>2sigma(I)] R1 = 0.0572, wR2 = 0.1157
R indices (all data) R1 = 0.1146, wR2 = 0.1416
Largest diff. peak and hole 2.265 and -1.008 e.Å-3
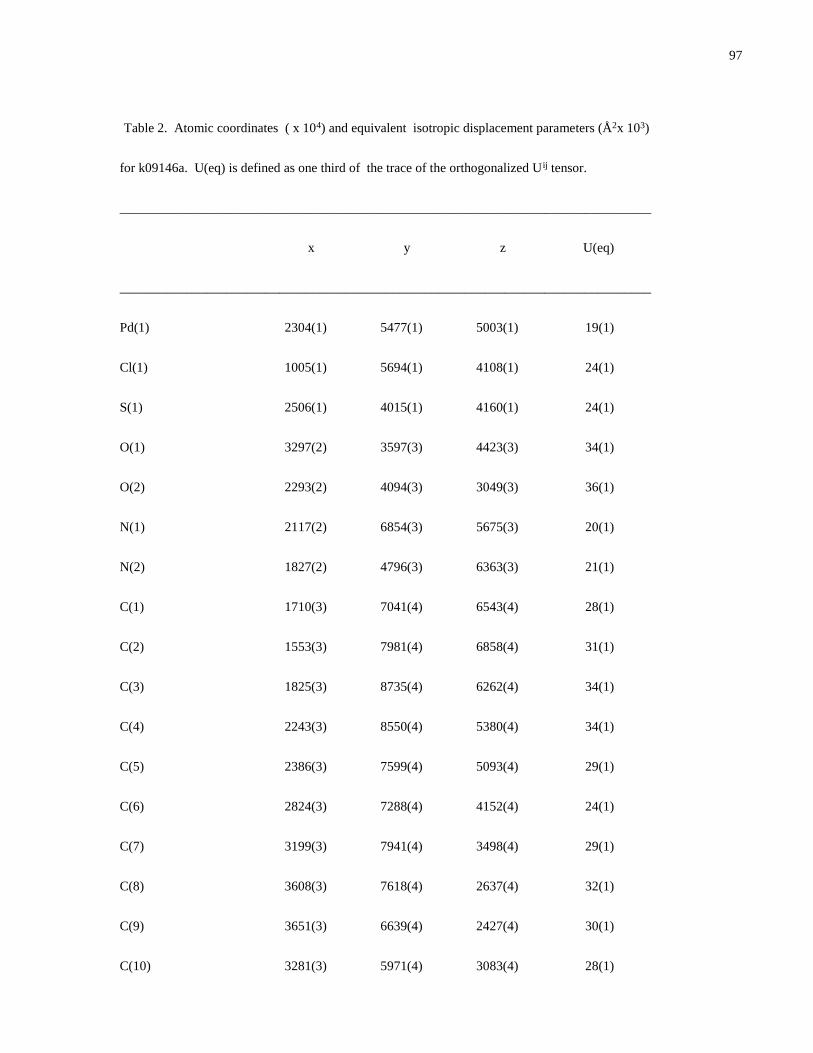
97
Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2x 103)
for k09146a. U(eq) is defined as one third of the trace of the orthogonalized U ij tensor.
________________________________________________________________________________
x y z U(eq)
________________________________________________________________________________
Pd(1) 2304(1) 5477(1) 5003(1) 19(1)
Cl(1) 1005(1) 5694(1) 4108(1) 24(1)
S(1) 2506(1) 4015(1) 4160(1) 24(1)
O(1) 3297(2) 3597(3) 4423(3) 34(1)
O(2) 2293(2) 4094(3) 3049(3) 36(1)
N(1) 2117(2) 6854(3) 5675(3) 20(1)
N(2) 1827(2) 4796(3) 6363(3) 21(1)
C(1) 1710(3) 7041(4) 6543(4) 28(1)
C(2) 1553(3) 7981(4) 6858(4) 31(1)
C(3) 1825(3) 8735(4) 6262(4) 34(1)
C(4) 2243(3) 8550(4) 5380(4) 34(1)
C(5) 2386(3) 7599(4) 5093(4) 29(1)
C(6) 2824(3) 7288(4) 4152(4) 24(1)
C(7) 3199(3) 7941(4) 3498(4) 29(1)
C(8) 3608(3) 7618(4) 2637(4) 32(1)
C(9) 3651(3) 6639(4) 2427(4) 30(1)
C(10) 3281(3) 5971(4) 3083(4) 28(1)

98
C(11) 2865(2) 6288(4) 3931(4) 22(1)
C(12) 3354(3) 5328(3) 5867(3) 20(1)
C(13) 4128(3) 5598(3) 5588(4) 23(1)
C(14) 4763(3) 5486(4) 6293(4) 25(1)
C(15) 4649(3) 5101(4) 7271(4) 29(1)
C(16) 3882(3) 4819(4) 7565(4) 26(1)
C(17) 3225(3) 4935(4) 6861(4) 22(1)
C(18) 2396(3) 4650(3) 7135(4) 22(1)
C(19) 2178(3) 4317(4) 8114(4) 27(1)
C(20) 1377(3) 4103(4) 8278(4) 32(1)
C(21) 805(3) 4244(4) 7482(4) 29(1)
C(22) 1048(3) 4589(3) 6528(4) 23(1)
C(23) 1773(3) 3220(4) 4707(4) 22(1)
C(24) 981(3) 3255(3) 4306(4) 22(1)
C(25) 409(3) 2675(4) 4804(4) 28(1)
C(26) 621(3) 2092(4) 5659(4) 31(1)
C(27) 1415(3) 2073(4) 6012(4) 31(1)
C(28) 1997(3) 2633(4) 5543(4) 28(1)
C(29) -18(3) 1495(5) 6182(5) 52(2)
O(3) 3571(2) 313(3) 5144(3) 46(1)
N(3) 4139(3) 1642(3) 5899(4) 35(1)
C(30) 4120(4) 2361(5) 6733(5) 61(2)

99
C(31) 4711(3) 1776(5) 5096(5) 53(2)
C(32) 3618(3) 910(4) 5837(4) 34(1)
________________________________________________________________________________

100
Table 3. Bond lengths [Å] and angles [°] for k09146a.
_____________________________________________________
Pd(1)-C(11) 2.019(5)
Pd(1)-C(12) 2.038(4)
Pd(1)-N(1) 2.113(4)
Pd(1)-N(2) 2.147(4)
Pd(1)-S(1) 2.3169(13)
Pd(1)-Cl(1) 2.4210(11)
S(1)-O(2) 1.455(4)
S(1)-O(1) 1.461(4)
S(1)-C(23) 1.794(5)
N(1)-C(1) 1.341(6)
N(1)-C(5) 1.353(6)
N(2)-C(22) 1.346(6)
N(2)-C(18) 1.354(6)
C(1)-C(2) 1.387(7)
C(2)-C(3) 1.373(7)
C(3)-C(4) 1.365(7)
C(4)-C(5) 1.386(7)
C(5)-C(6) 1.486(7)
C(6)-C(7) 1.390(7)
C(6)-C(11) 1.411(7)

101
C(7)-C(8) 1.383(7)
C(8)-C(9) 1.381(8)
C(9)-C(10) 1.401(7)
C(10)-C(11) 1.374(7)
C(12)-C(13) 1.392(6)
C(12)-C(17) 1.402(7)
C(13)-C(14) 1.369(7)
C(14)-C(15) 1.376(7)
C(15)-C(16) 1.393(7)
C(16)-C(17) 1.397(6)
C(17)-C(18) 1.482(6)
C(18)-C(19) 1.390(7)
C(19)-C(20) 1.383(7)
C(20)-C(21) 1.380(7)
C(21)-C(22) 1.379(7)
C(23)-C(28) 1.382(7)
C(23)-C(24) 1.394(6)
C(24)-C(25) 1.408(7)
C(25)-C(26) 1.393(7)
C(26)-C(27) 1.376(7)
C(26)-C(29) 1.515(7)
C(27)-C(28) 1.385(7)

102
O(3)-C(32) 1.210(7)
N(3)-C(32) 1.329(7)
N(3)-C(31) 1.431(7)
N(3)-C(30) 1.456(7)
C(11)-Pd(1)-C(12) 91.09(18)
C(11)-Pd(1)-N(1) 81.48(17)
C(12)-Pd(1)-N(1) 90.31(16)
C(11)-Pd(1)-N(2) 168.74(17)
C(12)-Pd(1)-N(2) 81.31(16)
N(1)-Pd(1)-N(2) 90.21(15)
C(11)-Pd(1)-S(1) 95.30(15)
C(12)-Pd(1)-S(1) 91.71(13)
N(1)-Pd(1)-S(1) 176.24(11)
N(2)-Pd(1)-S(1) 93.24(11)
C(11)-Pd(1)-Cl(1) 91.90(12)
C(12)-Pd(1)-Cl(1) 175.30(13)
N(1)-Pd(1)-Cl(1) 86.56(10)
N(2)-Pd(1)-Cl(1) 95.19(10)
S(1)-Pd(1)-Cl(1) 91.61(4)
O(2)-S(1)-O(1) 116.1(2)
O(2)-S(1)-C(23) 106.3(2)

103
O(1)-S(1)-C(23) 106.4(2)
O(2)-S(1)-Pd(1) 110.70(17)
O(1)-S(1)-Pd(1) 112.31(15)
C(23)-S(1)-Pd(1) 104.06(16)
C(1)-N(1)-C(5) 119.4(4)
C(1)-N(1)-Pd(1) 126.3(3)
C(5)-N(1)-Pd(1) 114.0(3)
C(22)-N(2)-C(18) 120.1(4)
C(22)-N(2)-Pd(1) 126.9(3)
C(18)-N(2)-Pd(1) 112.8(3)
N(1)-C(1)-C(2) 121.6(5)
C(3)-C(2)-C(1) 118.8(5)
C(4)-C(3)-C(2) 120.0(5)
C(3)-C(4)-C(5) 119.4(5)
N(1)-C(5)-C(4) 120.9(5)
N(1)-C(5)-C(6) 113.7(5)
C(4)-C(5)-C(6) 125.4(5)
C(7)-C(6)-C(11) 119.3(5)
C(7)-C(6)-C(5) 122.5(5)
C(11)-C(6)-C(5) 118.2(4)
C(8)-C(7)-C(6) 120.5(5)
C(9)-C(8)-C(7) 120.0(5)

104
C(8)-C(9)-C(10) 120.2(5)
C(11)-C(10)-C(9) 120.0(5)
C(10)-C(11)-C(6) 120.0(4)
C(10)-C(11)-Pd(1) 127.6(4)
C(6)-C(11)-Pd(1) 112.4(3)
C(13)-C(12)-C(17) 120.3(4)
C(13)-C(12)-Pd(1) 127.7(4)
C(17)-C(12)-Pd(1) 112.0(3)
C(14)-C(13)-C(12) 119.7(5)
C(13)-C(14)-C(15) 121.0(4)
C(14)-C(15)-C(16) 120.4(4)
C(15)-C(16)-C(17) 119.5(5)
C(16)-C(17)-C(12) 119.1(4)
C(16)-C(17)-C(18) 121.7(4)
C(12)-C(17)-C(18) 119.1(4)
N(2)-C(18)-C(19) 120.6(4)
N(2)-C(18)-C(17) 114.6(4)
C(19)-C(18)-C(17) 124.7(4)
C(20)-C(19)-C(18) 119.1(5)
C(21)-C(20)-C(19) 119.8(5)
C(22)-C(21)-C(20) 119.0(4)
N(2)-C(22)-C(21) 121.4(4)

105
C(28)-C(23)-C(24) 121.9(4)
C(28)-C(23)-S(1) 119.5(4)
C(24)-C(23)-S(1) 118.5(4)
C(23)-C(24)-C(25) 117.0(5)
C(26)-C(25)-C(24) 121.8(5)
C(27)-C(26)-C(25) 118.8(5)
C(27)-C(26)-C(29) 121.3(5)
C(25)-C(26)-C(29) 119.9(5)
C(26)-C(27)-C(28) 121.2(5)
C(23)-C(28)-C(27) 119.3(4)
C(32)-N(3)-C(31) 119.9(5)
C(32)-N(3)-C(30) 122.1(5)
C(31)-N(3)-C(30) 117.8(5)
O(3)-C(32)-N(3) 126.0(5)
_____________________________________________________________
Symmetry transformations used to generate equivalent atoms:
106
Table 4. Anisotropic displacement parameters (Å2x 103) for k09146a. The anisotropic
displacement factor exponent takes the form: -22[ h2 a*2U11 + ... + 2 h k a* b* U12 ]
______________________________________________________________________________
U11 U22 U33 U23 U13 U12
______________________________________________________________________________
Pd(1) 17(1) 22(1) 18(1) 1(1) 0(1) -1(1)
Cl(1) 18(1) 32(1) 22(1) 5(1) -3(1) -2(1)
S(1) 24(1) 25(1) 24(1) -4(1) 4(1) -4(1)
O(1) 23(2) 31(2) 48(2) -8(2) 6(2) -3(2)
O(2) 43(2) 41(2) 22(2) -7(2) 4(2) -14(2)
N(1) 21(2) 22(2) 17(2) 0(2) -3(2) 0(2)
N(2) 19(2) 27(2) 18(2) -4(2) 1(2) 0(2)
C(1) 20(2) 30(3) 32(3) -2(2) 0(2) 1(2)
C(2) 27(3) 32(3) 33(3) -9(3) -2(2) 4(2)
C(3) 35(3) 23(3) 44(4) -11(3) -3(3) 2(2)
C(4) 36(3) 25(3) 39(3) -2(3) -8(2) 0(2)
C(5) 21(3) 32(3) 33(3) -2(3) -6(2) -5(2)
C(6) 22(2) 27(3) 22(3) 1(2) -7(2) 4(2)
C(7) 25(3) 31(3) 31(3) 4(2) -1(2) -2(2)
C(8) 26(3) 38(4) 32(3) 15(3) 1(2) -4(2)
C(9) 24(3) 40(4) 27(3) 7(3) 2(2) 1(2)
C(10) 23(3) 33(3) 30(3) 0(3) 5(2) -2(2)

107
C(11) 11(2) 29(3) 25(3) 7(2) -1(2) -3(2)
C(12) 22(2) 25(3) 13(2) -7(2) -7(2) 10(2)
C(13) 21(2) 28(3) 20(3) -2(2) 7(2) 2(2)
C(14) 20(2) 28(3) 29(3) -11(2) 1(2) -3(2)
C(15) 23(3) 35(3) 30(3) -3(3) -5(2) 5(2)
C(16) 28(3) 26(3) 23(3) -2(2) -4(2) 5(2)
C(17) 24(2) 18(3) 23(3) -2(2) -2(2) 0(2)
C(18) 23(2) 21(3) 21(3) 1(2) 2(2) 0(2)
C(19) 28(3) 31(3) 23(3) 4(2) -4(2) -2(2)
C(20) 40(3) 34(3) 21(3) 6(2) 8(2) -5(2)
C(21) 26(3) 38(3) 24(3) 3(2) 3(2) -5(2)
C(22) 23(2) 23(3) 23(3) -4(2) -1(2) 0(2)
C(23) 24(3) 21(3) 22(3) -7(2) 4(2) -1(2)
C(24) 24(3) 19(3) 24(3) -5(2) -4(2) 1(2)
C(25) 24(3) 25(3) 37(3) -8(3) -1(2) -1(2)
C(26) 32(3) 30(3) 32(3) -8(3) 8(2) -5(2)
C(27) 38(3) 26(3) 28(3) 5(2) 1(2) -1(2)
C(28) 26(3) 28(3) 30(3) -4(2) -7(2) 3(2)
C(29) 47(4) 52(4) 56(4) 6(3) 19(3) -13(3)
O(3) 55(3) 43(3) 40(3) -7(2) 0(2) -16(2)
N(3) 30(2) 37(3) 37(3) -4(2) -7(2) 4(2)
C(30) 50(4) 57(5) 73(5) -35(4) -23(3) 23(3)

108
C(31) 42(4) 58(5) 59(5) -1(4) -5(3) -10(3)
C(32) 36(3) 41(4) 24(3) 7(3) -4(2) 5(3)
______________________________________________________________________________
109
Table 5. Hydrogen coordinates ( x 104) and isotropic displacement parameters (Å2x 10 3)
for k09146a.
________________________________________________________________________________
x y z U(eq)
________________________________________________________________________________
H(1A) 1524 6516 6951 33
H(2A) 1263 8102 7474 37
H(3A) 1723 9384 6464 41
H(4A) 2433 9069 4966 40
H(7A) 3174 8615 3643 35
H(8A) 3859 8070 2191 39
H(9A) 3932 6418 1835 37
H(10A) 3319 5297 2942 34
H(13A) 4216 5858 4912 27
H(14A) 5289 5678 6103 30
H(15A) 5097 5028 7749 35
H(16A) 3806 4549 8239 31
H(19A) 2575 4238 8662 33
H(20A) 1220 3860 8938 38
H(21A) 251 4105 7589 35
H(22A) 656 4683 5977 28
110
H(24A) 833 3653 3723 27
H(25A) -137 2682 4549 34
H(27A) 1568 1669 6588 37
H(28A) 2543 2612 5794 34
H(29A) 113 1447 6935 77
H(29B) -35 844 5875 77
H(29C) -546 1806 6075 77
H(30A) 3674 2215 7194 91
H(30B) 4040 3006 6426 91
H(30C) 4632 2346 7140 91
H(31A) 4797 1160 4736 80
H(31B) 5224 2004 5411 80
H(31C) 4504 2257 4590 80
H(32) 3249 851 6387 40
________________________________________________________________________________