sulfur-ligated, oxidative nonheme iron enzymes and related
TRANSCRIPT

Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related ComplexesJesse B Gordon and David P Goldberg, Department of Chemistry, The Johns Hopkins University, Baltimore, MD, United States
© 2020 Elsevier Inc. All rights reserved.
1 Introduction 12 Isopenicillin N Synthase 22.1 Introduction 22.2 Structure and Function 22.3 Mechanism 33 Thiol Dioxygenases 73.1 Introduction 73.2 Cysteine Dioxygenase Structure and Function 73.3 Cysteine Dioxygenase Mechanism 103.4 ADO/3MDO/MSDO—The Other Thiol Dioxygenases 134 Sulfoxide Synthases 154.1 Introduction 154.2 Structure and Function 164.3 Mechanism 175 Persulfide Dioxygenase 195.1 Introduction 195.2 Structure and Function 205.3 Mechanism 216 Synthetic Model Complexes 226.1 Sulfur Oxygenation 236.2 Fe/O2 Intermediates 306.3 Reduced O2 Surrogates 336.4 Models of Thiolate-Ligated Nonheme Iron Enzymes with Other 3d Metals 377 Outlook and Future Work 40References 42
1 Introduction
Nonheme iron oxygenases and oxidases are a structurally and functionally diverse class of metalloenzymes that utilize dioxygen(O2) to catalyze a wide range of oxidative transformations. Although the four-electron reduction of O2 is highly thermodynam-ically favorable, the ability of O2 to oxidize organic molecules is hampered by its remarkable kinetic stability. It has beenhypothesized that the weak OdO s bond confers a significant thermodynamic driving force for O2 reactivity with organicmolecules. However, the triplet ground state of O2 has four resonance structures, which provide a large resonance stabilizationenergy (estimated to be � –100 kcal mol–1), strengthening the O2 p bond and rendering O2 inert towards spontaneous reactionswith organic molecules.1
The kinetic inertness of O2 is circumvented in biological and synthetic catalysis by employing redox active metal ions that canbind and activate dioxygen to unleash its oxidative potency.2 The proposed mechanisms for many nonheme iron enzymes involvesinitial binding of dioxygen to generate an FeIII(superoxo) intermediate, which then proceeds to react with a substrate orco-substrate, leading to a cascade of reactions involving other reduced oxygen species such as Fe((hydro)peroxo) (Fe(OO(H)), orhigh-valent Fe(oxo) (FeIV(O)) intermediates.
One major class of dioxygen activating nonheme iron enzymes are those involving a co-substrate. The archetypical examples ofthis class are the a-ketoglutarate (a-KG) dependent nonheme iron enzymes, which undergo an oxidative decarboxylation of thea-KG co-substrate following binding of O2 to produce a high-valent FeIV(O) species that carries out substrate oxidation. In contrast,a second class of nonheme iron oxygenases does not require a co-substrate.3 A major group of these enzymes function with sulfur-containing substrates, including isopenicillin N synthase (IPNS), the thiol dioxygenases, the sulfoxide synthases, and the persulfidedioxygenases. It is proposed that for each of these enzymes, their respective thiolate/persulfide substrates coordinate to the Fecenters through sulfur, promoting the binding and activation of O2.
The focus of this chapter is the structures, functions, and proposed mechanisms for these oxidative, sulfur-ligated nonheme ironmetalloenzymes, and the related synthetic, small-molecule transition metal complexes that serve as models for these systems.
Comprehensive Coordination Chemistry III https://doi.org/10.1016/B978-0-12-409547-2.14906-6 1

2 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
2 Isopenicillin N Synthase
2.1 Introduction
Isopenicillin N is a member of an important class of molecules known as b-lactams.4 The broad importance of b-lactams stems fromtheir ability to inhibit bacterial cell formation, making them powerful antibiotics used to treat a wide range of bacterial diseases.5
Despite displaying weak antibiotic activity,6 isopenicillin N is a precursor in the biosynthesis of an extensive list of potent antibioticsincluding other penicillins and cephalosporins.4 Isopenicillin N is produced in both bacterial and fungal organisms by a nonhemeiron dependent enzyme, isopenicillin N synthase (IPNS), which uses a mononuclear iron cofactor and dioxygen to convert thelinear tripeptide d-(L-a-aminoadipoyl)-L-cysteinyl-D-valine (ACV) to isopenicillin N, a transformation that entails two oxidativecyclization reactions, each of which necessitates the cleavage of a strong CdH bond (Fig. 1). The significance of IPNS as a key entrypoint into a number of antibiotic biosynthetic pathways and as an Fe-dependent enzyme capable of catalyzing chemicallychallenging reactivity has impelled major research efforts into understanding the function and mechanism of this enzyme.
Fig. 1 Conversion of ACV to isopenicillin N by IPNS.
2.2 Structure and Function
Prior to the first crystal structures of IPNS, the structures of IPNS with and without substrate were examined by sequence alignmentand mutagenesis studies,7–9 as well as spectroscopic techniques such as nuclear magnetic resonance (NMR),10,11 X-ray absorption(XAS),12,13 Mössbauer14,15 and electron paramagnetic resonance spectroscopies.15 Taken together, these studies found that theresting state of IPNS features a six coordinate Fe center ligated by two histidines, an aspartate, a glutamine, and two water molecules.It has been proposed that coordination of the ACV substrate leads to monodentate coordination via the thiolate donor. Nitric oxide(NO) was employed as an O2 surrogate in the presence of the ACV substrate, leading to the formation of an iron nitrosyl ({FeNO}7,S ¼ 3/2) species, which was characterized by EPR, Mössbauer, and optical spectroscopies.14
The first crystallographic evidence for the active site of IPNS from Aspergillus nidulans was obtained in 1995, with Mn substitutedfor Fe in the active site.16 The structure confirmed previous studies suggesting that the metal center was coordinated octahedrally bytwo histidine ligands, one aspartate, and one glutamine residue, and two water molecules. The crystal structure of IPNS containingboth Fe and the ACV substrate revealed a 5-coordinate, trigonal bipyramidal metal ion, in which the ACV thiolate donor haddisplaced a Gln residue, and one water molecule was lost from the metal (Fig. 2).17 The two His and Asp ligands identified in thecrystal represent a structural motif known as the 2-His-1-carboxylate triad, which is prevalent in a number of nonheme iron oxidasesand oxygenases.18 The crystal structure of the {FeNO}7 form confirmed that a small diatomic molecule can bind to the Fe center inthe presence of the ACV substrate.
Fig. 2 Crystal structures of IPNS-ACV complex (PDB: 1BK0) (left) and IPNS-ACV-NO complex (PDB: 1BLZ) (right).

Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 3
2.3 Mechanism
Extensive work in the 1980s and 1990s provided the basis for understanding the mechanism of the four-electron oxidation of ACVcatalyzed by IPNS19 and has been reviewed elsewhere.20 These elegant studies exploited easily derivatized tripeptide substrateanalogues of ACV and the expanded substrate scope of IPNS to observe alternative product distributions, and to prematurely stopthe reaction cycle and intercept intermediates. The observed reactivity patterns led to the mechanistic picture shown in Scheme 1.It was hypothesized that conversion of isopenicillin N from ACV occurs via two sequential two electron oxidation/cyclization steps,b-lactam formation followed by thiazolidine formation. The b-lactam formation requires cleavage of the pro-S-Cys b CdH bond,and thiazolidine formation requires cleavage of the Val b CdH bond. It was proposed that these steps are mediated separately by anFeIII(superoxo) species and an FeIV(oxo) species, respectively, (Scheme 1) and it has been found that both CdH abstraction stepsare partially rate-limiting.21 Although these studies relied upon organic product identification, they did provide a workingmechanistic hypothesis for further analysis.
Scheme 1
Crystallographic methods were developed that involved exposure of anaerobic crystals to high O2 pressures, allowing forstructural characterization of products following oxygenation in the solid state.22 In addition, the ability to crystallize IPNS inthe presence of substrate/substrate analogues has allowed structural “snapshots” to be obtained of the reactions with various IPNSsubstrates. A number of these crystallographic studies expanded on the early reports from the 1980s and 1990s, and provideddefinitive structural evidence for the interaction between substrate analogues as well as oxidation products with the Fe center in theactive site.
The substrate analogue, d-(L-a-aminoadipoyl)-L-cysteinyl-L-S-methylcysteine (ACmC), which replaces D-valine with thethioether, S-methylcysteine, was used in order to prevent the second cyclization from occurring.22 Addition of ACmC to IPNS inthe presence of Fe allowed for crystallographic characterization of the substrate-bound ferrous complex, confirming that thissubstrate analogue can bind to the Fe center as seen for the native substrate ACV. However, in this case, the thioether is bound inthe open coordination site proposed for O2 binding. Addition of dioxygen at high pressure allowed for the formation of amonocyclic b-lactam product in which the thioether moiety was oxygenated to sulfoxide. Crystallographic characterization revealedthat the substrate was bound to Fe via the thiolate donor and sulfoxide O-atom (Fig. 3). Thioether oxidation to sulfoxides byFeIV(oxo) species is well-established;23 therefore, these studies supported previous mechanisms suggesting that b-lactam formationoccurs before thiazolidine formation, and that an FeIV(oxo) species is formed following the first ring closure step.
Fig. 3 (A) Reaction of IPNS with substrate analogues ACmC and (B) crystal structure of oxygenated product bound to IPNS (PDB: 1QJF).

SH
HNAA
O
O
NH
CO2-
O2
S
HN
AAO
O
N
CO2-
O2
SNH
AA
OOHN
CO2-
FeII
NHisOAsp
NHis
OH2
SHN
AA
O
ON
CO2-
FeIV
NHisOAsp
NHis
OH2
O
H
AC-Me-cpG
SHN
AA
O
ON
CO2-
FeIII
NHisOAsp
NHis
OH2
OH
Scheme 2
4 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
The cyclopropyl-containing ACV analogue, d-(l-a-aminoadipoyl)-L-cysteinyl-b-methyl-D-cyclopropylglycine (AC-Me-cpG),undergoes a ring-opening reaction in the presence of IPNS following addition of O2.
24 The observation of ring-opened productswas strongly indicative of the formation of a carbon-based radical. In an earlier report on this work, however, the proposedmechanism of product formation involved formation of an FedC bond prior to ring closure with the metal-bound thiolate. Thecrystal structure of AC-Me-cpG-bound IPNS indicated that the orientation of the cyclopropyl group strongly disfavors formation ofan FedC bond, and instead is more consistent with a radical recombination mechanism in which an FeIV(O) generates the carbon-based radical via H-atom abstraction, which then attacks the metal-bound thiolate, as has been proposed with the native ACVsubstrate (Scheme 2).25
Early studies using other alkyl derivatives of the valine peptide of ACV revealed the possibility for alternate oxidation pathwaysfollowing initial b-lactam formation. Oxidation of d-(L-a-aminoadipoyl)-L-cysteinyl-D-a-aminobutyrate (ACAb) in the presence ofIPNS resulted in the formation of three oxidation products, in a 1: 7: 3 product ratio (Scheme 3). It was hypothesized that this ratiorelated to the ability of the FeIV(O) intermediate to abstract the CdH bond that would result in the formation each of theseproducts.19 Crystallization of ACAb-bound to IPNS with and without a nitrosyl ligand revealed that binding of the substrateanalogue was almost identical to that of ACV with exception of the aminobutyrate moiety, which was highly disordered overmultiple positions. Modeling the relative energies of different conformers of the ACAb substrate revealed three distinct energyminima, which could be correlated with the observed product distribution. These studies suggest that the outcome of the secondring-closure step by IPNS is dictated by positioning of the alkyl chain within the active site.26
SH
HNAA
O
O
NH
CO2-
O2
S
HNAA
O
O
NCO2
-
ACAb
SNHAA
OO
N H
CO2-
SNHAA
OO
N
H
CO2-
1:7:3
Scheme 3
In order to interrupt IPNS prior to b-lactam formation, the substrate analogue, d-(L-a-aminoadipoyl)-L-cysteine D-a-hydroxyisovaleryl ester (ACOV), which replaces the amide linkage of ACV with an ester, was prepared.27 Addition of ACOV toIPNS allowed for crystallization of the substrate-bound complex, which revealed a similar structure to ACV-bound IPNS. However,addition of O2 resulted in the formation of a thiocarboxylate, in which the CdH bond that normally forms the b-lactam is oxidizedto a carbonyl. This observation supports the hypothesis that an initial thioaldehyde intermediate is formed, which, in the absence ofthe amide moiety, reacts either directly with water or with the putative Fe(OOH) intermediate (Scheme 4). Computational studiesaimed at understanding the observed oxygenase reactivity suggested that nucleophilic attack of the thioaldehyde by an FeII(OOH)intermediate was energetically feasible.28
When a slightly altered analogue of ACOV, AmCOV, which contains a methyl group adjacent to the thiolate (Scheme 5), is used,a different reaction product is observed.29 In this case, a thiol-ene type product is formed, and the valine methyl group ishydroxylated. It was proposed that orientation of the valine residue away from the Fe center resulted in the unusual primarycarbon-center hydroxylation. The fact that the radical recombines with the hydroxyl group rather than the thiolate was rationalizedby the absence of the b-lactam, which could position the valine residue in close proximity to the thiolate for the native ACVsubstrate.

S NH AA
OOO
CO2-
FeII
NHisOAsp
NHis
OH2
S
NHAA
OO
O
CO2-
FeII
NHisOAsp
NHis
OH2
OO2
Scheme 5
Scheme 4
Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 5
One study used a substrate analogue that was a hybrid of ACOV and ACmC, d-l-a-aminoadipoyl-l-cysteine (1-(S)-carbox-y-2-thiomethyl)ethyl ester (ACOmC), which incorporates a thioether moiety in place of the valine and an ester in place of the amidenormally found in ACV.30 X-ray crystallography reveals that ACOmC does bind to the Fe center of IPNS. Interestingly, addition ofO2 results in the formation of a new product in which the metal-bound thiolate has been oxygenated to a sulfenate, and thethiomethylcysteine unit has been hydroxylated (Fig. 4). It was proposed that this unusual reactivity is a result of O2 binding trans toHis214, rather than Asp216, and that the methylcysteine hydroxylation step occurs first to generate an FeIV(O) intermediate thatthen S-oxygenates the metal-bound thiolate. An alternate mechanism in which S-oxygenation to form the sulfenate via anFeIII(superoxo) intermediate occurs prior to the CdH hydroxylation step was considered, but was deemed less likely. However,this alternative mechanism is in accordance with the proposed mechanism for cysteine dioxygenase (see Section 3.3).
The extensive organic product analysis and crystallographic studies using ACV and substrate analogues strongly hint at theinvolvement of FeIII(superoxo) and FeIV(oxo) intermediates in the reactivity of IPNS. However, these studies failed to provide directevidence for these Fe/oxygen intermediates, and as a result, little could be learned about their fundamental spectroscopic properties,or their exact involvement in the mechanism of IPNS. Although the O2-bound, Fe
III(superoxo) intermediate in IPNS could not becharacterized, a spectroscopic study employing EPR, circular dichroism (CD), and magnetic circular dichroism (MCD)
Fig. 4 Reaction of ACV substrate analogue ACOmV with IPNS (left) and crystal structure of IPNS-sulfenate complex (PDB: 2VBB).

6 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
spectroscopies on the {FeNO}7 form of IPNS as an analogue of the FeIII(superoxo) intermediate was carried out.31 The spectro-scopic data was accompanied by density functional theory (DFT) calculations, which indicated that the S ¼ 2 state of the FeIII(O2
•−)species should be lowest in energy. The calculations also suggested that the O2
•− unit should be properly oriented to accept theH-atom from the ACV substrate. The binding of O2 is calculated to be exergonic, which provides a possible rationale for why IPNS isable to activate O2 in the absence of a co-substrate or cofactor, as is usually required for other nonheme iron enzymes.18 A numberof other computational studies have also been performed on IPNS, and suggest that mechanisms involving FeIII(O2
•−) and anFeIV(O) intermediates are energetically feasible.28,32–34
In 2016, the first experimental evidence for the Fe/O2 intermediates responsible for each of the CdH cleaving steps in IPNS wasobtained.35 Deuterated substrates were employed to enable the accumulation of intermediates prior to H-atom abstraction.A transient intermediate was observed in the IPNS reaction with the native ACV substrate, exhibiting UV-vis bands at �360 nmand �515 nm. Mössbauer spectroscopy on this species gave d ¼ 0.27 mm s–1 and DEQ ¼ –0.44 mm s–1 (Fig. 5). These parametersare consistent with a high-spin nonheme FeIV(O) species.36 Examination of the deuterated AC[d8-V], in which all eight valineH atoms are substituted with deuterium, showed that this intermediate precedes the second ring closing step in the formation ofisopenicillin N. A kinetic isotope effect (KIE) � 30 was measured for the decay of this intermediate with ACV. Together thesefindings pointed to the FeIV(O) assignment for this intermediate.
A transient intermediate that precedes the formation of the designated FeIV(O) species was observed in low abundance bystopped-flow UV-vis (515, 630 nm) and Mössbauer (d ¼ 0.53 mm s–1, DEQ ¼ 1.02 mm s−1) spectroscopies with the deuteratedsubstrate A[d2-C]V. Preliminary variable-field Mössbauer data indicated an S ¼ 2 or S ¼ 3 spin ground state. These spectralproperties are similar to an FeIII(O2
•−) intermediate characterized in homoprotocatechuate-2,3-dioxygenase (HPCD),37 and togetherthe data point to an FeIII(O2
•−) species. The assignment of the two observed intermediates was supported by DFT calculations. Of themodels tested for the FeIV(O) intermediate, an FeIV(O) (S ¼ 2) species complexed with the 2-His-1-carboxylate triad, the singly-cyclized b-lactam via the thiolate ligand, and one water molecule provided the best match for the observed Mössbauer parameters.For the proposed superoxo intermediate, models with the superoxide ligand coordinated in both a side-on and end-on fashion wereconsidered in the spin states S ¼ 1, 2, or 3, and only the calculated FeIII(O2
•−) structure with a quintet spin ground state provided agood match with the experimental spectroscopic data. Overall, these spectroscopic studies provide support for the mechanismproposed in Scheme 1.
A recent computational study set out to address the origin of the selectivity for CdH activation over S-oxygenation in IPNS.A hypothetical reaction pathway was calculated in which the FeIII(O2
•−) intermediate attacks the coordinated sulfur donor ratherthan the Cys b CdH bond. It was found that the barrier for S-oxygenation is about 4 kcal mol−1 higher than that for CdHactivation, due to an unfavorable steric interaction between the ACV valine alkyl groups and the Fe center on the S-oxygenationpathway. A calculated pathway in which the Val group is frozen lowers the barrier for S-oxygenation relative to H-atom abstrac-tion.38 However, the explanation involving sterics does not address why the significantly less bulky ACV substrate analogues ACGand ACA, which substitute the valine peptide with glycine and alanine, respectively, do not undergo any S-oxygenation reactivity.39
2.0 F
0
Velocity (mm/s)
2 4−2−40
[0–13mT]
[0–53mT]
δ = 0.53 mm/s|ΔEQ| = 1.02 mm/s
Velocity (mm/s)
2 4−2−4
E
D
C
D
A
Ab
sorp
tion
(%)
Ab
sorp
tion
(%)
0.0 0.2
0.0
0.2
0.0
I II
0.20.0
1.00.0
1.0
0.0
1.00.0
1.00.0
4.0
0.0
Fig. 5 (I) (A) Zero-field 4.2 K Mössbauer spectra of IPNS�Fe(II)�ACV complex before adding O2. Zero-field 4.2 K Mössbauer spectra after adding O2 at 5 �C andmonitoring the reaction mixture at different time points trapped by a freeze-quench method: (B) 0.02 s, (D) 0.12 s, (E) 0.45 s, and (F) 90 s. Spectrum C is thedifference spectrum between A and B. (II) Zero-field 4.2 K Mössbauer spectra of samples prepared by reacting IPNS�Fe(II)�A[d2-C]V complex with O2 at 5 �C andfreeze quenching after 0.010 s. The top spectra show overlaid data of the experimental spectrum collected in the absence of a magnetic field (black vertical bars), inthe presence of a 13 mT magnetic field (blue solid line), and in the presence of a 53 mT (red solid line). The middle spectrum shows the difference spectrum for the 0field and 53 mT data, with the green line representing the best fit. The bottom spectrum shows the difference spectrum for the 0 field and 13 mT data. Reprintedwith permission from reference Tamanaha, E.; Zhang, B.; Guo, Y.; Chang, W.-C.; Barr, E. W.; Xing, G.; St. Clair, J.; Ye, S.; Neese, F.; Bollinger, J. M., Jr.; Krebs, C.J. Am. Chem. Soc. 2016, 138, 8862–8874. Copyright 2016 American Chemical Society.

Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 7
3 Thiol Dioxygenases
3.1 Introduction
The thiol dioxygenases are a class of nonheme iron enzymes that are responsible for converting thiol substrates into theircorresponding sulfinic acids. The first discovered thiol dioxygenases were cysteine dioxygenase (CDO),40 which converts cysteine(Cys) to cysteine sulfinic acid (CSA), and cysteamine dioxygenase, or 2-aminoethanethiol dioxygenase (ADO), which is responsiblefor converting cysteamine to hypotaurine (Scheme 6).40,41 Both of these enzymes are found in mammals and play critical roles insulfur amino acid metabolism. The bacterial thiol dioxygenases, namely 3-mercaptopropionoic acid dioxygenase (3MDO) andmercaptosuccinate dioxygenase (MSDO), are also known (Scheme 6).42,43
Scheme 6
Cysteine dioxygenase represents the most well-studied thiol dioxygenase. It is found in mammals, bacteria, and plants, and isknown to generate CSA using O2 as the oxidant and the O-atom source.44–46 The formation of CSA represents the first step in themetabolism of Cys. Further breakdown of CSA eventually leads to the formation of pyruvate, sulfate, and taurine, which areimportant for various signaling pathways and metabolic functions.47,48 The cytotoxicity and neurotoxicity of high levels of cysteineare well-established,49 and evidence suggests that cysteine buildup is associated with a number of neurodegenerative andautoimmune diseases50 such as Parkinson’s disease,51 Alzheimer’s disease,52 rheumatoid arthritis,53 Hallervorden-Spatzsyndrome,54 as well as various cancers.55–57 Cysteine dioxygenases have also been shown to trigger N-terminal cysteine oxidationand initiates an N-degron pathway that controls responses to hypoxic conditions in plants and animals.45,46,58
CDO is most prevalent in hepatic cells where cysteine levels range between 0.01 and 0.1 mmol L−1.59–61 The measured Km forCys consumption by CDO is�0.45 mmol L−1, significantly higher than cellular Cys levels,62 indicating that CDO is able to respondrapidly to changes in Cys concentrations in hepatocytes. CDO activity is regulated by polyubiquitination, which occurs at low levelsof dietary Cys intake, and is inhibited when high levels of Cys are present,60 and as a result, CDO activity in liver tissue is tightlyregulated by dietary cysteine levels. The same responses to dietary Cys intake are not observed for nonhepatic CDO; however,evidence suggests that CDO is able to alter intercellular cysteine levels, indicating that CDO plays a central role in controllingmammalian cysteine levels.49
Cysteamine, which is known to be produced by the catabolism of CoA,63,64 is oxidized by ADO to produce hypotaurine andeventually taurine, which has a wide range of physiological roles including regulating brain and cardiovascular function.65 ADO isfound to be more widespread in various tissues compared to CDO.66 It is thus proposed that, like CDO, cysteamine dioxygenaseplays an important role in maintaining proper human health. It has recently been shown that ADO’s primary role may be to act asan oxygen sensor in plants and animals. A study found that ADO regulates oxygen levels in response to hypoxia by initiatingsulfination of an N-terminal, peptidic cysteine which triggers the eventual decomposition of G protein signaling regulators controlGTPase activity.58
3.2 Cysteine Dioxygenase Structure and Function
Early work on mouse (Mus musculus) and rat (Rattus norvegicus) CDO focused on sequence alignment studies and enzymatic activityassays, which indicated the importance of iron for activity,40,67,68 and that CDO is likely part of the cupin superfamily of enzymes,which includes a number of other nonheme iron enzymes such as homogentisate dioxygenase, quercetin dioxygenase, and thecatechol dioxygenases.69 This superfamily is a diverse class of proteins typically characterized by two conserved peptide chain motifsGX5HXH(X)3–6E(X)6G and G(X)5–7PXG(X)2H(X)3N (where X refers to amino acid residues that are not strictly conserved) thatstructurally leads to the b-barrel shape shared among the superfamily.69 The first crystal structure for CDO was obtained in 2006from Mus musculus.70 This study provided important structural information about the active site of CDO, although it was solvedwith a catalytically inactive nickel ion,40,71 rather than iron, bound in the active site (Fig. 6).
The most striking features from this structure revealed that the metal center in CDO is coordinated to three protein-derived Hisligands (His86, His88, and His140, Fig. 6), forming a 3-His triad, which was an unusual departure from the 2-His-1-carboxylate(Asp/Glu) triad typically found in nonheme iron oxygenases.70 A Cys93-Tyr157 crosslink, which was unprecedented among cupinproteins, was also found within 4.2 Å of the Ni center.

Fig. 6 Crystal structure of Ni-substituted CDO from Mus musculus (PDB: 2ATF). Full protein structure shown on left and active site shown on right.
8 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
The first crystal structure of iron-constituted CDO was reported from the rat homolog, which has an identical sequence identitycompared with mouse CDO.72 Like mouse CDO, rat CDO showed the metal center coordinated to a 3-His triad, in close proximityto a Cys–Tyr crosslink. Other conserved residues included Tyr58, Arg60, Ser153, and His155, which are all proposed to be importantfor substrate binding and/or catalysis. The Fe center in CDO appeared to be tetrahedrally coordinated, with water serving as thefourth ligand, an unprecedented geometry among nonheme iron enzymes. The oxidation state of the Fe could not be determined,but it was assumed to be in the ferric state. Attempts to isolate crystals of the substrate (Cys) bound form of rat CDO wereunsuccessful. A separate study described extended X-ray absorption fine structure (EXAFS) measurements on rat CDO, and foundthat the resting state was best modeled as having six N/O ligands, not four, consistent with an FeII(His)3(H2O)3 coordinationsphere.71 Subsequent structural andMössbauer studies onmammalian wild-type (wt) CDO, mutants, and bacterial CDO supportedthat the resting ferrous state is indeed 6-coordinate.73–75
For years the structure of Cys-bound CDO remained controversial. An early EXAFS study suggested that the substrate Cys did notcoordinate via the sulfur donor to the iron center.68 However, the first crystal structure of Cys-bound CDO (human), which exhibits92% sequence homology with mouse and rat CDO, suggested that the Cys coordinates to the Fe via the S/N donors in a distortedtrigonal bipyramidal geometry, with the 3-His triad comprising the rest of the coordination sphere.76 Interestingly, a later studyreexamined these data and found that the electron density can be better modeled by rearranging the active site residues andreplacing the Cys substrate with water, indicating that it is in fact not the Cys-bound structure “with” indicating that the originalstructure was not Cys-bound.77 This same study recrystallized substrate-bound CDO and found that Cys does in fact bind directly toFe via N/S coordination, giving a five-coordinate structure and an FedS distance of 2.35 Å at pH 8 (Fig. 7). The direct coordinationof cysteine was also supported by a number of other structural,77,78 Mössbauer,73 magnetic circular dichroism,79,80 and computa-tional studies.79,80 The Mössbauer data on Cys-bound CDO showed broad features consistent with the presence of two quadrupoledoublets.73,81 One possible explanation was that incomplete Cys–Tyr crosslink formation gives two slightly different iron environ-ments. However, Mössbauer studies on the CDOmutant C93G, which is unable to form the crosslink, also revealed the presence oftwo quadrupole doublets, ruling out this hypothesis. The possibility that Cys-bound CDO exists as a mixture of 5-coordinate and6-coordinate iron, with water serving as the possible sixth ligand, was proposed. This idea was supported by studies on a series ofsynthetic model complexes (see Section 6.1).82
Fig. 7 Crystal structure of Cys-bound human CDO (PDB: 4IEV).

Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 9
The unusual Cys93-Tyr157 crosslink in CDO is also found in the copper-containing metalloprotein galactose oxidase.83 Unlikegalactose oxidase, however, CDO exhibits catalytic activity in both the presence and absence of this crosslink, as seen in studiesinvolving the immature form of the protein (i.e., prior to crosslink formation), and in mutant forms of the protein in which Cys93 issubstituted. It has been demonstrated that the crosslink is a post-translational modification84 that occurs only in the presence ofprotein-bound iron, dioxygen, and the substrate, cysteine, and the enzyme requires �800 turnovers before reaching 50% matura-tion.85 These data suggest that the crosslink forms over the course of catalysis, though relatively inefficiently.85 An early proposedmechanism implicated formation of an FeIII(superoxo) intermediate, which oxidizes Tyr157 prior to insertion of the Cys93 sulfuratom.85 Efforts were made to examine the biogenesis of the Cys–Tyr crosslink by engineering modified CDO variants using fluorineand chlorine substituted tyrosine analogues. Surprisingly, even a mutant containing 3,5-di-fluoro-substituted tyrosine (F2-Tyr157)CDO generated the Cys–Tyr crosslink, suggesting that a potent oxidant capable of cleaving a strong CdF bond is formed duringcrosslink formation (Fig. 8).74 The crystal structures of the Fe–nitrosyl adducts of F2-Tyr157 CDO before and after crosslinkformation were obtained,86 and revealed a strong interaction between F2-Tyr157 and the Cys carboxylate group prior to crosslinkformation, as well as an interaction between the nitrosyl O atom and Cys93. These data led the authors to conclude that anFe(superoxo) species may abstract an H-atom from Cys93 to generate a thiyl radical, which then inserts into the Tyr to form thecrosslink. It has been noted, however, that crosslink formation in vivo may occur via different mechanisms than those occurringin vitro.84
Fig. 8 Crystal structures of engineered CDO mutant with F2-Tyr157 complexed with Cys prior to (left) and after (right) crosslink formation (PDB: 6BPS (left) and6BPV (right)).
The exact role of the crosslink has been subject to intense debate. Initial studies found that the presence of the crosslink increaseswith higher concentrations of Cys, and enhances the catalytic efficiency of the enzyme.85 These results suggest that the crosslink mayserve a regulatory role in response to cellular Cys levels. From a mechanistic perspective, the crosslink is known not to be essentialfor catalysis in mammalian CDOs. Furthermore, bacterial CDOs contain a conserved Gly in place of Cys93 and still exhibit highactivity.87 Comparisons between wt-CDO and C93G-CDO revealed that turnover occurs with similar levels of efficiency, althoughat different optimum pH levels,88 and it was proposed that the crosslink serves to prevent competing reactions at Cys93. Otherproposals have suggested that the crosslink serves a structural function, promoting optimal binding of Fe, substrate, or properlyorienting intermediates formed during catalysis.89–91 In line with these proposals, it has been found that the crosslink may preventoxidative uncoupling during CSA production.92
CDO exhibits a high degree of substrate specificity (Scheme 7). There are numerous reports demonstrating the inability of CDOto oxygenate other sulfhydryl containing molecules, such as cysteamine, 3-mercaptopropionoic acid, homocysteine,2-aminothiophenol, as well as the selenium analogue of Cys, selenocysteine (sec).68,71,80,93,94 One report suggested that CDOcan undergo some activity with homocysteine and L-penicillamine, albeit inefficiently.94 Interestingly, it has been shown byMössbauer spectroscopy that homocysteine can bind to CDO in a similar fashion to Cys, yet does not react with O2.
73 Spectroscopicstudies have demonstrated that selenocysteine (sec) binds to both FeII and FeIII forms of CDO, and sec-bound FeII CDO forms anadduct with nitric oxide. However, sec-bound FeII CDO exhibits no spectral changes upon addition of O2, and there is no evidencefor the formation of oxidized organic products.79,80 The origin of CDO’s substrate specificity was discussed in a combined enzyme/model study, where it was shown by Mössbauer spectroscopy that the non-native substrate 2-aminothiophenol does not chelateproperly to FeII CDO and as a result, no S-oxygenation is observed.82 In contrast, chelation of 2-aminothiophenolate to FeII modelcomplexes leads to efficient S-oxygenation to give the sulfinate product. It was hypothesized that proper S/N substrate chelationto FeII-CDO is likely required for successful S-oxygenation, and this requirement may be a significant contributor to CDO’ssubstrate specificity (see Section 6.1). In addition to oxygenating free cysteine, recent evidence has suggested that cysteinedioxygenation can occur in N-terminal, protein-derived cysteinyl residues mediated by plant cysteine oxidases. The occurrence of

Scheme 7
10 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
O2-derived cysteinyl dioxygenation was supported by 1H NMR studies as well as mass spectrometry using isotopically labeled O2
and water sources.45,46
3.3 Cysteine Dioxygenase Mechanism
Early mechanistic proposals for CDO were aligned with the assumption that Cys does not bind directly to the Fe center, which waslater shown to be incorrect. These proposals have been summarized elsewhere, but will not be discussed here.68 Most currentmechanistic hypotheses include an initial O2 binding step to form an FeIII(superoxo) intermediate, although definitive character-ization of such a species has yet to be obtained (Scheme 8).
Nitric oxide (NO) is commonly used as a surrogate and spectroscopic probe for O2 binding in iron enzymes. Some data on CDOsuggest that Cys must bind first to the Fe center in order to allow for O2 binding.
79,95 Addition of NO to Cys-bound FeII-CDO resultsin the formation of an {FeNO}7 species, which was observed by EPR spectroscopy. Unlike the vast majority of {FeNO}7 species innonheme iron enzymes, which exhibit an S ¼ 3/2 ground state, the {FeNO}7 species of CDO exhibits a low-spin, S ¼ 1/2 groundstate. Density functional theory calculations suggested that the ls-{FeNO}7 should be a six-coordinate iron species with NO boundin the sixth site. It was hypothesized that the O2 may bind in a similar fashion, occupying the sixth site through an “end-on”bonding mode. The unusual spin state of the {FeNO}7 form may arise from the rather different coordination environment of Cys-bound CDO (sulfur ligation, 3-His triad) compared to other mononuclear nonheme Fe enzymes. However, it is unlikely that thepresence of the thiolate donor alone is responsible, because the{FeNO}7 form of IPNS, which is also thiolate-ligated, exhibits themore typical S ¼ 3/2 ground state.31
Attempts to chemically produce the FeIII(superoxo) intermediate involved the addition of O2•− to the ferric form of Cys-bound
CDO, which was proposed to be an FeIII(OH) species from EPR, MCD, and resonance Raman measurements.96 The (Cys)FeIII(OH)form is catalytically inactive; however, addition of O2
•− results in the rapid decay of (Cys)FeIII(OH) CDO (lmax ¼ 630 nm) andformation of a new intermediate with l¼ 485 and 565 nm as seen by UV-vis spectroscopy (Fig. 9).97 This new species has a lifetimeof minutes, and its decay was kinetically matched with the formation of CSA, although CSA production was�200 times slower thanduring native catalysis with O2. Samples of the FeIII enzyme after treatment with superoxide were restored to normal CDO catalyticactivity. EPR characterization of the O2
•− intermediate indicated that it exhibited an integer spin, S ¼ 3 ground state. The authorshypothesized that this intermediate was an FeIII(superoxo). Whether this species lies along the catalytic cycle of CDO is unclear,given its high stability and low rate of CSA formation. This intermediate may be entirely off-pathway, or it may be the result of aslightly altered local environment. Further studies are needed to confirm the identity of this transient intermediate.
Though most mechanisms support formation of an FeIII(superoxo) intermediate, they diverge after this step. These mechanismsprimarily differ in the redox role played by the Fe center in the oxygenation of the thiolate donor. One proposed mechanismsuggests that Fe oxidizes the thiolate via two, sequential mono-oxygenation steps involving initial O-atom transfer from FeIII(su-peroxo) to generate an FeIV(O)(sulfenate) intermediate, followed by a second O-atom transfer from the ferryl to generate asulfinate-bound, ferrous product (Scheme 8, Mechanism I). This mechanism shares some commonalities with the proposedmechanism for a-KG-dependent nonheme iron enzymes such as taurine dioxygenase.18 Other proposals have suggested thatdioxygenation occurs directly on the sulfur donor, in analogy to the proposed mechanisms for the S-oxygenation of Ni–thiolatecomplexes (Scheme 8, Mechanism II).98
Credence for the direct S-attack mechanism was provided by the crystallographic characterization of an Fe(persulfenate) speciesin Cys-bound CDO (Fig. 10), which was generated by soaking crystals of CDOwith excess substrate in air.99 The Cys S atom and theproximal oxygen of the persulfenate moiety were bound to Fe, forming a three-membered ring structure. Because of the stability of

Scheme 8
Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 11
the crystals, it was questioned whether this structure represented a catalytic intermediate or an off-pathway species. The authors laterhypothesized that a structurally similar intermediate (e.g., coordinated persulfenic acid) may represent the functioning, catalyticallyrelevant intermediate.77 The crystalline, persulfenate form of CDO was incapable of generating the expected sulfinate product,suggesting that this species may be off-pathway.100
Computational studies (DFT and QM/MM)101,102 describe a model that involves initial formation of an S ¼ 0 FeIII(superoxo)intermediate, which rapidly undergoes spin state crossing to become the S ¼ 2 FeIII(superoxo) species (5A), which then attacks theS atom of a coordinated Cys via the distal oxygen atom, generating an S ¼ 2, four-membered FedOdOdS ring structure (5B)(Figure 11). Sulfur attack is calculated to be the rate determining step. The FedOdOdS intermediate then undergoes homolyticOdO bond cleavage to produce an FeIV(O)(sulfenate) species (5C). Following S]O bond rotation, the Fe(oxo) species may thenperform an O-atom transfer to the sulfenate to generate the ferrous-bound sulfinate product, which can then be released toregenerate the resting state FeII complex. Calculations on an alternate mechanism (Mechanism II) involving a persulfenateintermediate found that this pathway was significantly higher in energy, suggesting that it might not be a viable mechanism.Other DFT calculations were later performed on these hypothetical intermediates by using computational methods that accuratelymodeled spectroscopic data for the CDO {FeNO}7 species. These calculations indicated that the mechanism in Fig. 11 was viableand suggested that the S ¼ 2 FeIII(superoxo) intermediate is the lowest energy spin state. A significant amount of spin density wasalso calculated to lie on the S atom, suggesting partial thiyl radical character in the superoxo (FeII(O2
•−)(•SR)) and ferryl ((FeIII(O•)(•SR)) intermediates.79

−0.05
0.13
0.31
0.49
0.67
0.85
−0.05 −0.150.000.15
0.0
0.2
0.4
0.6
0.8
1.0
0.07
360
485
565565
630
(A) (B)
-CSA
NHIS
(OH2)O2
•−−O2CH2
NHIS
NHISN
SFeII
NHIS
OH−O2C
H2IrIV
NHIS
NHISN
SFeIII
NHIS
OO•
−O2CH2
NHIS
NHISN
SFeIII
(C)
630
Ab
sorb
ance
468 576 684 792 900λ/nm
360 480 600 720 840 960 0 2 4 6 8 10time/min
Res
idua
lsN
orm
aliz
ed C
once
ntra
tion
λ/nm
0.19
0.31
0.43
0.55
Fig. 9 Proposed formation of FeIII(superoxo) in CDO from reaction of ferric CDO (generated from oxidation with IrIV) with O2•−. (A) UV-vis spectra of (cys)FeIII(OH) CDO
(dashed line, white circle) and after formation of O2•− generated in situ from xanthine oxidase, with dark gray, light gray, and white circles indicating spectra collected
at 0.5, 1.0, and 2 s, respectively. (B) UV-vis spectra of (cys)FeIII(OH) CDO (dashed line, white circle) and following addition of preformed solutions of O2•−, with dark
gray, light gray, and white squares indicating spectra collected at 0.5, 22, and 600 s, respectively. (C) Plot of concentration of CSA (gray circles) versus time andnormalized decay of proposed FeIII(superoxo) species (black circles) versus time. Fits for formation of CSA and decay of FeIII(superoxo) species are shown as dashedand solid lines, respectively. Reprinted with permission reference Crawford, J. A.; Li, W.; Pierce, B. S. Biochemistry 2011, 50, 10241–10253. Copyright 2011American Chemical Society.
Fig. 10 Crystal structure of the CDO Fe(persulfenate) species (PDB: 3ELN).
12 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
Experimental evidence for the mechanism of action of CDO has been hampered by difficulties in trapping catalytically relevantintermediates. However, other methods have provided some information regarding the details of the S-oxygenation mechanism.One study showed that wt-CDO catalysis exhibited a solvent KIE, suggesting that a proton transfer step is important in Cysoxygenation.103 It was proposed that the Tyr moiety may serve in an acid-base role during catalysis.88 However, it is unlikely that theTyr plays a redox role (e.g., serving as an H-atom donor) since addition of radical scavengers does not effect catalytic efficiency, andno spectroscopic evidence for a tyrosyl radical has been observed.95 In 2016, the first report appeared of an intermediate trappedduring native CDO catalysis (Fig. 12).81 This intermediate was stable for only 20 ms, and exhibited two bands at 500 nm and 640nm by UV-vis spectroscopy. It was shown to be on the catalytic pathway. The absorption spectra for possible intermediates werecalculated using DFT and ab initio methods, and the computed spectrum for the proposed FedOdOdS intermediate matchedwell with the spectrum seen for the 20 ms intermediate. However, the calculations also indicated that the spectrum could beconsistent with an FeIII(O2
•−) intermediate. More data are needed to definitively characterize this intriguing CDO Fe/O2
intermediate.

Fig. 11 Reaction energy landscape for singlet, triplet, and quintet spin states for CDO calculated by DFT. Reprinted with permission from reference Aluri, S.; deVisser, S. P. J. Am. Chem. Soc. 2007, 129, 14846–14847. Copyright 2007 American Chemical Society.
Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 13
3.4 ADO/3MDO/MSDO—The Other Thiol Dioxygenases
Compared to CDO, significantly less is known about the other thiol dioxygenases. One major setback in understanding the otherthiol dioxygenases is a lack of structural data for these proteins in the presence of their native substrates.
The mammalian enzyme cysteamine dioxygenase (ADO) converts 2-amino-ethanethiol (cysteamine) to hypotaurine by oxida-tion of the thiol group. This reaction is analogous to Cys oxidation by CDO, and is critical for the production of taurine, abiomolecule that has many important roles and is widespread in various tissues.65 The participation of ADO in hypotaurineproduction was first described in the 1960s, where it was shown to convert cysteamine to hypotaurine.104 However, since theoriginal work on ADO over 40 years ago, there is still relatively little known about the mechanism or active site structure, in partbecause there are still no X-ray structures available for this protein.
Early studies found that ADO, like CDO, exhibits substrate specificity, and other thiols such as cysteine, glutathione, coenzymeA, or lipoic acid were neither substrates nor inhibitors of ADO.105 Some evidence suggested that ADO can S-oxygenate2-mercaptoethanol, homocysteine, N-acetylcysteamine, and 3-mercaptopropionic acid.106 The characterization of human ADOwas first reported in 2007,66 and in this study the previously observed substrate specificity was reiterated, as well as the requirementof iron for proper enzymatic function. This study also found that, in contrast to previous reports, exogenous reductants wereunnecessary for catalysis. It was hypothesized that ADOmay contain the unusual Cys–Tyr crosslink seen in CDO. Efforts to test thishypothesis included genetically encoding the unnatural amino acid 3,5-di-fluoro-tyrosine (F2-Tyr222) in ADO, as done in similarstudies with CDO (see Section 3.2).107 Mass spectral analysis revealed that one of the CdF bonds is cleaved and a new CdS bond isformed with Cys220, confirming crosslink formation. The presence of the crosslink enhances catalytic activity of ADO, and thusmay serve a similar function to that in CDO. The cleavage of a CdF bond suggested that a potent oxidant may be formed duringcrosslink biogenesis. Taken together, these results highlight that ADOmight have similar structure and function compared to CDO,but clearly more work is needed to define the mechanism of ADO. It has recently been shown that ADO can oxygenate N-terminalcysteine during hypoxia to produce the protein-bound sulfinic acid, which was verified with mass spectrometry and isotope labelingstudies.58 Competition experiments revealed that dioxygenation of the peptide cysteinyl residue is inhibited only at concentrationsgreater than 37 mM or 13 mM for free cysteamine or cysteine, respectively. It has thus been suggested that ADO may primarilyfunctions as part of the N-degron pathway in mammals. The mechanism of the dioxygenation of protein-bound cysteine and how itcompares to the oxygenation of free cysteine seen in CDO is not understood.
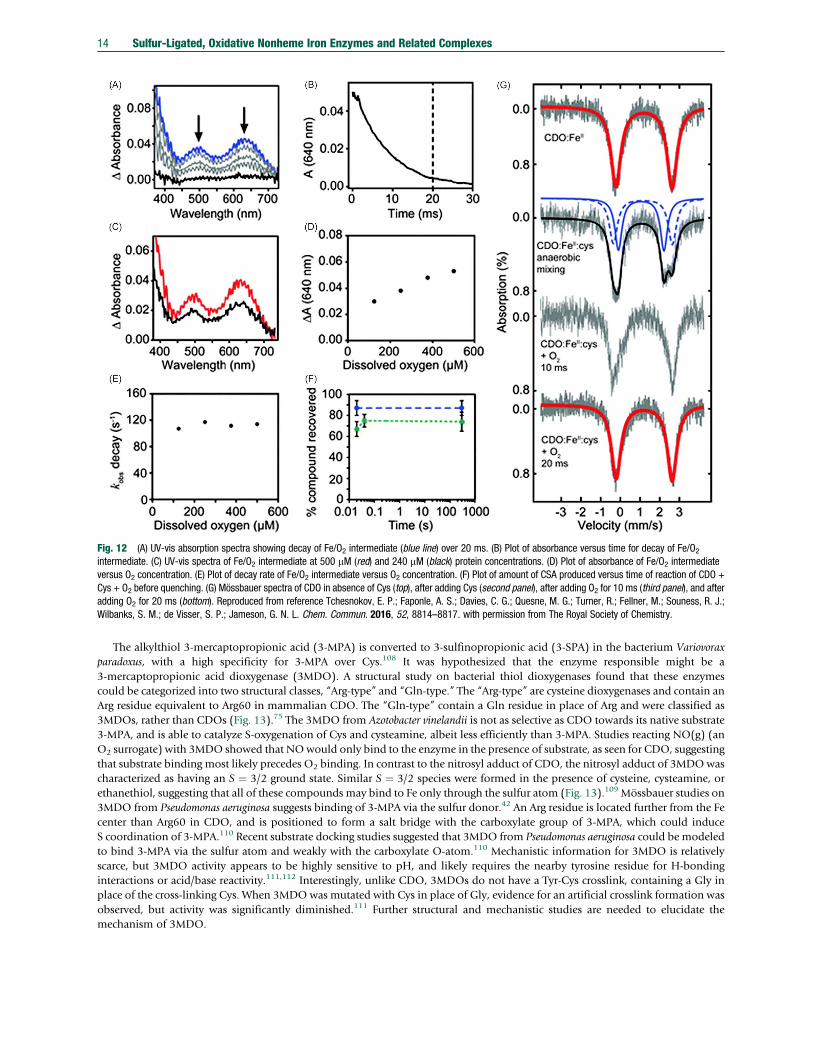
Fig. 12 (A) UV-vis absorption spectra showing decay of Fe/O2 intermediate (blue line) over 20 ms. (B) Plot of absorbance versus time for decay of Fe/O2intermediate. (C) UV-vis spectra of Fe/O2 intermediate at 500 mM (red) and 240 mM (black) protein concentrations. (D) Plot of absorbance of Fe/O2 intermediateversus O2 concentration. (E) Plot of decay rate of Fe/O2 intermediate versus O2 concentration. (F) Plot of amount of CSA produced versus time of reaction of CDO +Cys + O2 before quenching. (G) Mössbauer spectra of CDO in absence of Cys (top), after adding Cys (second panel), after adding O2 for 10 ms (third panel), and afteradding O2 for 20 ms (bottom). Reproduced from reference Tchesnokov, E. P.; Faponle, A. S.; Davies, C. G.; Quesne, M. G.; Turner, R.; Fellner, M.; Souness, R. J.;Wilbanks, S. M.; de Visser, S. P.; Jameson, G. N. L. Chem. Commun. 2016, 52, 8814–8817. with permission from The Royal Society of Chemistry.
14 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
The alkylthiol 3-mercaptopropionic acid (3-MPA) is converted to 3-sulfinopropionic acid (3-SPA) in the bacterium Variovoraxparadoxus, with a high specificity for 3-MPA over Cys.108 It was hypothesized that the enzyme responsible might be a3-mercaptopropionic acid dioxygenase (3MDO). A structural study on bacterial thiol dioxygenases found that these enzymescould be categorized into two structural classes, “Arg-type” and “Gln-type.” The “Arg-type” are cysteine dioxygenases and contain anArg residue equivalent to Arg60 in mammalian CDO. The “Gln-type” contain a Gln residue in place of Arg and were classified as3MDOs, rather than CDOs (Fig. 13).75 The 3MDO from Azotobacter vinelandii is not as selective as CDO towards its native substrate3-MPA, and is able to catalyze S-oxygenation of Cys and cysteamine, albeit less efficiently than 3-MPA. Studies reacting NO(g) (anO2 surrogate) with 3MDO showed that NOwould only bind to the enzyme in the presence of substrate, as seen for CDO, suggestingthat substrate binding most likely precedes O2 binding. In contrast to the nitrosyl adduct of CDO, the nitrosyl adduct of 3MDOwascharacterized as having an S ¼ 3/2 ground state. Similar S ¼ 3/2 species were formed in the presence of cysteine, cysteamine, orethanethiol, suggesting that all of these compounds may bind to Fe only through the sulfur atom (Fig. 13).109 Mössbauer studies on3MDO from Pseudomonas aeruginosa suggests binding of 3-MPA via the sulfur donor.42 An Arg residue is located further from the Fecenter than Arg60 in CDO, and is positioned to form a salt bridge with the carboxylate group of 3-MPA, which could induceS coordination of 3-MPA.110 Recent substrate docking studies suggested that 3MDO from Pseudomonas aeruginosa could be modeledto bind 3-MPA via the sulfur atom and weakly with the carboxylate O-atom.110 Mechanistic information for 3MDO is relativelyscarce, but 3MDO activity appears to be highly sensitive to pH, and likely requires the nearby tyrosine residue for H-bondinginteractions or acid/base reactivity.111,112 Interestingly, unlike CDO, 3MDOs do not have a Tyr-Cys crosslink, containing a Gly inplace of the cross-linking Cys. When 3MDO was mutated with Cys in place of Gly, evidence for an artificial crosslink formation wasobserved, but activity was significantly diminished.111 Further structural and mechanistic studies are needed to elucidate themechanism of 3MDO.

Fig. 13 Crystal structure of 3MDO from Pseudomonas aeruginosa (left). Scheme showing formation of {FeNO}7 in 3MDO from Azotobacter vinelandii (right).
Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 15
A proteomics study in 2014 identified a possible thiol dioxygenase in Variovorax paradoxus that may be responsible for thedegradation of mercaptosuccinate.113 This enzyme was found to exhibit high sequence similarity to CDO. Further studiesdemonstrated that Variovorax paradoxus converts mercaptosuccinate (ms) to succinate and sulfite, likely via the intermediacy ofsulfinosuccinate. This enzyme requires Fe for proper function and is highly selective towards ms, suggesting that it is a truemercaptosuccinate dioxygenase (MSDO).43 Mutagenesis studies were performed to identify key residues involved in catalysis.114
The MSDO enzyme contains three conserved His residues like CDO, which upon substitution abolished reactivity. An Arg residue,analogous to Arg60 in CDO, is present and is essential for catalysis, suggesting that it may serve to help bind the succinate, possiblyvia a salt-bridge with the substrate carboxylate group. Mutation of a Gln residue positioned near the active site in 3MDO fromPseudomonas aeruginosa to an Arg leads to MSDO activity.110 It was proposed that this mutation allows for the ms carboxylates toform two salt bridges that favor proper substrate binding/orientation. Though no crosslink is present, a Tyr analogous to Tyr157 inCDO is present in MSDO. However, unlike in CDO, mutation to phenylalanine does not eliminate activity. This observation is asignificant departure from that seen for the other thiol dioxygenases. Further structural and mechanistic studies are needed for thisnewest member of the thiol dioxygenase family.
4 Sulfoxide Synthases
4.1 Introduction
Ovothiol A and ergothioneine are both sulfur containing histidine derivatives that are thought to be important biomolecules due totheir antioxidant properties. Ovothiol A is found in a number of marine organisms and was first isolated from sea urchin eggs.115 Ithas a sulfhydryl pKa of 1.4 and a much more positive redox potential than other sulfur-based antioxidants such as glutathione(GSH). It is thought that ovothiol may scavenge peroxides or serve as a chemical signaling agent; however, its exact biological role isunknown.116 It has been hypothesized that the unique properties of ovothiol derivatives may lead to potential use in therapiestargeting neurodegenerative diseases117 as well as tumor growth.118
Ergothioneine is a related sulfur-containing histidine derivative that exists primarily in its thione form. Like Ovothiol A, theredox potential is relatively high compared to typical thiol antioxidants,119 and as a result is muchmore stable to oxidation. Thoughergothioneine is produced by fungi and bacteria, it is found in both plants and mammals and is obtained through diet. It has beenproposed that ergothioneine may function as an antioxidant and metal chelator and may protect against neurodegenerative andcardiovascular diseases.119,120
In 2010, a series of five genes egtA, egtB, egtC, egtD, and egtE were shown to be responsible for the conversion of histidine toergothioneine in Mycobacterium smegmatis. The EgtB gene produces a protein with a nonheme iron cofactor that is involved in anunusual CdS bond formation and sulfoxidation reaction between g-L-glutamyl-L-cysteine (g-GC) and Na-trimethyl histidine(TMH) (also known as hercynine).121 The enzyme OvoA was discovered around the same time as EgtB, and is responsible for asimilar transformation in the biosynthesis of ovothiol A. Like EgtB, OvoA is a nonheme iron dependent enzyme that catalyzes aCdS bond formation and sulfoxidation reaction; however, OvoA was shown to use histidine and cysteine as its native substratesand performs the CdS bond formation at the imidazole C5 position rather than in the C2 position as seen for EgtB (Fig. 14).122
A related enzyme Etg1 was later discovered in Neurospora crassa, which performed a similar sulfoxide synthase reaction usingcysteine instead of g-GC.123 Further phylogenetic analyses have found that EgtB-like sulfoxide synthases are widespread in a numberof different organisms and can be classified into five different types, each with different structural motifs and substrate require-ments.124 Likewise, OvoA was also found to have over 80 homologues in protobacteria, microalgae, and marine organisms.116,123
The CdS bond formation and sulfoxidation reaction performed by these sulfoxide synthases enzymes are mechanisticallycomplicated and unprecedented among nonheme iron enzymes. This mechanistic uncertainty, as well as the ubiquity and potentialsignificance of ovothiol/ergothioneine biosynthesis makes the sulfoxide synthases an important emerging class of nonheme ironenzymes for structural and mechanistic examination.

Fig. 14 Reactions of EgtB and OvoA in the biosynthesis of ergothioneine and ovothiol A.
16 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
4.2 Structure and Function
Following the discovery that OvoA is an Fe dependent nonheme iron enzyme, it was hypothesized that OvoA contains a2-His-1-carboxylate type coordination geometry based on the protein sequence122; however, it was noted that the proposedmetal-binding residues were unusually close in proximity in the sequence compared to most 2-His-1-carboxylate enzymes.Interestingly, mutation of E176, which was hypothesized to be the possible carboxylate donor, to alanine, significantly diminishedsulfoxide synthase activity.125
No crystal structure for OvoA has been obtained. However, the crystal structure of EgtB from Mycobacterium thermoresistibile(MthEgtB) bound to Fe in the absence of substrates, and EgtB reconstituted with Mn in the presence of g-GC and TMH, were bothobtained in 2015.126 The crystal structure of Fe-bound EgtB in the absence of substrates revealed that Fe is coordinated to threefacially arranged His residues, much like that seen for CDO. Additionally, Tyr377 was shown to be positioned near the Fe center andwas proposed to play a role during catalysis. Attempts to crystallize EgtB with Fe in the presence of g-GC and TMH wereunsuccessful. However, addition of g-GC to the enzyme under aerobic conditions resulted in the formation of a UV-vis absorptionfeature at 565 nm, which could be attributed to an S!Fe charge-transfer transition, indicating that the thiolate likely binds directlyto the Fe center. Further evidence of direct coordination of g-GC came from the structure of the catalytically inactiveMn-reconstituted EgtB. The structure shows the six-coordinate Mn coordinated by g-GC through sulfur, which also forms a saltbridge with Arg87 and Arg90, the TMH imidazole, one water molecule, and the 3-His triad (Fig. 15).
Recent crystal structures of EgtB from Chloracidobacterium thermophilum (CthEgtB) revealed an entirely new structural motif forthe sulfoxide synthases.124 The crystal structure of Fe-bound CthEgtB with and without TMH were both determined, and revealedthat like MthEgtB, the Fe center is coordinated by the 3-His triad. However, CthEgtB differs in its overall protein structure andsequence fromMthEgtB. In addition, CthEgtB was found to contain two tyrosine residues, Tyr93 and Tyr94, in close proximity to theFe active site, rather than the single Tyr residue, Tyr377, seen in MthEgtB. In addition, CthEgtB uses cysteine rather than g-GC as itsnative substrate, and the ability to select for this substrate may come from minor changes in structure. MthEgtB is highly substratespecific and does not accept Cys or histidine as a substrate,127 and likewise, CthEtgB does not oxidize g-GC.
Fig. 15 Crystal structure of Mn-substituted MthEgtB (PDB: 4X8D). Overall dimeric structure (left); active site (right).

Fig. 16 Reactions of mthEgtB and OvoA with native and non-native substrates.
Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 17
OvoA selectively utilizes L-cysteine as the sulfur source, with no activity observed with D-cysteine, N-acetyl-L-cysteine, glutathi-one, or thiophosphate.128 Conflicting results have been reported for the use of g-glutamyl-L-cysteine (g-GC) as a substrate.127,128
The specificity for imidazole substrates is much lower, with reactivity being observed for histamine, L-histidinamide, D-histidine,2-fluoro-L-histidine, and 4-methyl imidazole. The apparent reactivity of 2-fluoro-L-histidine is particularly intriguing because it issignificantly less electron rich than L-histidine, suggesting that the redox potential of the imidazole substrate may not play a largerole in the mechanism of action of OvoA. The regioselectivity of the substitution of imidazole at C2 or C5 is dependent on thedegree of methyl substitution on the histidine amine.128 When the native EgtB substrate, hercynine, is added to OvoA, substitutionof the C2 position is observed as seen in EgtB (Fig. 16). However, the major product in this reaction was cysteine sulfinate,suggesting that OvoA can serve as a cysteine dioxygenase, and the nature of the substrate can steer the selectivity of OvoA towardCDO-type or sulfoxide synthase activity.125 It was also noted that a small amount (10%) of cysteine sulfinate is observed even innative catalysis with histidine. However, when cysteine is added to OvoA in the absence of an imidazole substrate, only the disulfideproduct, cystine, is observed. The formation of cystine is enzymatic; however, it is unclear if an initial S-oxygenated sulfenate speciesis formed, which then could lead to disulfide formation.129
4.3 Mechanism
The differences in stereoselectivity and substrate scope for EgtB and OvoA as well as the observation of both CDO-type reactivity andsulfoxide synthase reactivity have important implications for their mechanisms. The products from these enzymes point to anobvious mechanistic question: does CdS bond formation or sulfoxidation occur first? Two possible mechanistic pathways areshown in Scheme 9. Pathway I involves initial CdS bond formation followed by S-oxygenation, and Pathway II involves initialS-oxygenation, similar to that proposed for CDO, followed by CdS formation. Many of the mechanistic studies on these enzymeshave been performed with this question in mind.
EgtB can be altered to exhibit CDO-like dioxygenation reactivity.130 Mutation of Tyr377 in MthEgtB to phenylalanine results innearly identical values of Km, suggesting that substrate binding is not significantly altered. However, the predominant productobserved is the S-dioxygenated product, with only a small amount of the sulfoxide synthase product being produced. Theobservation of CDO-like reactivity suggests that MthEgtB and CDO may exhibit a common intermediate followed by a branchingpoint that dictates the two reactivity patterns. No KIE was seen upon deuteration of TMH, suggesting that TMH C–H cleavage is notinvolved in the rate determining step. These mutagenesis study suggests that proton movement involving the Tyr residue may play acritical role in the branching point that determines product selectivity.
The observation of two Tyr residues (Tyr93 and Tyr94) in close proximity to the active site of CthEgtB indicates that the role of Tyrin catalysis may be different for the different enzymes.124 Mutagenesis studies of CthEgtB show that when Tyr93 is substituted withPhe, CSA is the major product, but sulfoxide synthase activity is still observed. Substitution of Tyr94 with Phe significantlydiminishes Cys consumption, suggesting that Tyr94 may be involved in O2 binding/activation.
OvoA exhibits both CDO and sulfoxide synthase activity, and an isotope sensitive branching method131 showed that no kineticisotopic effect was observed with a fully deuterated histidine substrate. A small solvent KIE of 1.29 is observed when D2O was usedas the solvent, suggesting that protonation steps may be involved in the mechanism.122,128 Given that no crystal structure for OvoAhas been obtained, it is difficult to determine specifically which residues are involved in catalysis. However, sequence alignmentsuggests that Tyr417 in OvoA may be the equivalent of Tyr377 in EgtB, which is known to play a role in sulfoxide synthase activity.

Scheme 9
18 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
The Tyr417 has been substituted with artificial amino acid tyrosine analogues in order to examine its role in sulfoxide synthaseactivity.131,132 A 2-methylthio-substituted tyrosine analogue (MtTyr) (similar to the Cys–Tyr crosslink in CDO)131 gave a slightincrease in the ratio of CSA:native sulfoxide from 1:9 to 3:7. This observation suggests Tyr417 is involved in the branch pointbetween CDO-like and sulfoxide synthase activity. It must be noted that MtTyr alters both the redox potential and pKa relative totyrosine. Incorporation of another tyrosine analogue, MeOTyr, in which a methoxy group is substituted ortho- to the phenol moiety,provided a second mutant in which the pKa of the unnatural MeOTyr is much closer to that of Tyr, but the redox potential is muchlower than that of Tyr.132 More CSA is produced with MeOTyr-OvoA (26%) as compared to native OvoA (10%). An inverse KIE of0.86 was observed for MeOTyr-OvoA with a fully deuterated His substrate, compared to a KIE of 1.0 for native OvoA. These KIEssuggest that OMe substitution at Tyr417 changes the rate-limiting step for OvoA catalysis. These results together with other findingsled the authors to favor Pathway I. However, if the branching point for CDO-like and sulfoxide synthase activity occurs followingformation of FeIII(O2
•−), it would be expected that incorporation of MeOTyr, would make the H-atom abstraction more favorablerelative to the CDO-like pathway, which should lead to the production of less CSA relative to native catalysis. The observation ofmore CSA with MeOTyr-OvoA contradicts this hypothesis.
Computational studies have been carried out to examine the mechanism of the sulfoxide synthases. Several mechanisms wereproposed, and these mechanisms primarily disagree on the order of sulfoxidation and CdS bond formation. The first computa-tional study on the sulfoxide synthases used DFT to find that formation of the four-membered ring structure along Pathway II is themost likely oxidant to induce oxidation of the histidine ligand via PCET, which would then lead to formation of the CdS bond.However, this mechanism was calculated prior to the first crystal structure of EgtB and utilized a very limited set of residues for thecalculation, and most notably, did not include Tyr377, which is known to play an important role in catalysis.133 A more recentcomputational study of EgtB calculated several mechanistic options along both Pathway I and Pathway II, finding that Pathway II ismore favorable.134 This study proposed the formation of an S ¼ 2 FeIII(superoxo) intermediate that attacks the sulfur via the distaloxygen atom followed by OdO bond cleavage, resulting in the formation of an FeIV(O)(sulfenate) intermediate. It was proposedthat deprotonation of the TMH imidazole NdH followed by coupling of the sulfenate resulted in formation of the final sulfoxideproduct. However, this study found that imidazole NdH deprotonation was the rate determining step, and that there would be asolvent KIE, which is not in line with the experimental solvent KIE near unity.130 This study also predicted the incorrectstereochemistry for the final sulfoxide product.135
An alternate mechanistic pathway was proposed on the basis of QM/MM studies on EgtB (Fig. 17).136 It was proposed thatPathway I is more likely, suggesting that the first step involves an S ¼ 2 FeIII(superoxo) that deprotonates Tyr377 via a Grotthussmechanism, followed by rapid electron transfer from the tyrosinate to generate an FeIII(OOH) intermediate. The next step involvesattack of the coordinated thiolate on the TMHC2 position and simultaneous electron transfer to Fe and proton transfer and electrontransfer from the hydroperoxo ligand to Tyr377 tyrosyl radical, resulting in the formation of an FeII(superoxo) coordinated to thede-aromatized-TMH-g-GC sulfide. It is then proposed that the FeII(superoxo) intermediate abstracts an H-atom from the TMH unit

Fig. 17 Reaction energy landscape for EgtB calculated by QM/MM. Reprinted with permission from reference Faponle, A. S.; Seebeck, F. P.; de Visser, S. P. J. Am.Chem. Soc. 2017, 139, 9259–9270. Copyright 2017 American Chemical Society.
Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 19
to rearomatize the imidazole and produce an FeII(hydroperoxo) intermediate. It is then found that the FeII(hydroperoxo)oxygenates the sulfide to produce the final product with no barrier (Fig. 17).
Alternate mechanisms involving initial FedOdOdS formation were tested and were found to be less energetically accessible.The authors thus propose that proton-coupled electron transfer from Tyr377 is responsible for steering EgtB away from CDO-likeS-dioxygenation reactivity.
No computational studies on OvoA have been carried out. As a result, it is unclear if OvoA follows a similar or entirely differentmechanistic pathway from EgtB. However, the mechanism proposed by Liao and Siegbahn does not line up with the lack of a KIEupon deuteration of the His susbtrate.122 Experimental studies have shown that addition of the thioether formed between histidineand Cys to OvoA results in no oxidation following addition of O2 or H2O2,
125 which is unexpected if Pathway I is operative. It isalso not clear if the other newly discovered EgtB variants could follow alternative mechanisms for sulfoxide synthase reactivity.124
The sulfoxide synthases are a mechanistically complicated class of enzymes. More experimental and computational data areundoubtedly needed to better understand their reactivity patterns.
5 Persulfide Dioxygenase
5.1 Introduction
Persulfide dioxygenases (PDOs) are enzymes that convert persulfides to their corresponding thiol and sulfite (Scheme 10).These enzymes are known in bacteria, plants, and humans, and are proposed to play essential roles in sulfur metabolism.
In mammals, the persulfide dioxygenase, ETHE1, is found in mitochondria and is responsible for the second step in sulfidecatabolism. In the first step, a sulfide:quinone oxidoreductase converts H2S into persulfides, and ultimately pulls from the cellularreservoir of glutathione to generate glutathione persulfide. The second step uses ETHE1 to produce sulfite, regenerating glutathione.The third step involves a sulfur transferase enzyme which reacts persulfides with the formed sulfite to produce thiosulfate, the finalproduct in mitochondrial sulfur oxidation pathway.137
Scheme 10

20 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
Much of the interest in ETHE1 stems from the observation that patients with the rare autosomal recessive disease, ethylmalonicencephalopathy, exhibit defects in the gene that encodes ETHE1.138,139 This disease leads to neurological, cardiovascular, andgastrointestinal defects, and is often lethal.138–140 It has been proposed that dysfunction of ETHE1 is associated with the buildup ofH2S leading to the inhibition of cytochrome c oxidase.138 A number of studies have found that plants contain a homolog of humanETHE1 that is important for plant metabolism.141 Persulfide dioxygenases have also been found in a large number of bacteria, andmay be related to the mechanisms by which pathogens display drug resistence.142–146 Thus, the persulfide dioxygenases represent alarge and important family of enzymes that are widespread in nature.
5.2 Structure and Function
The earliest studies on persulfide dioxygenases were with bacterial forms of the enzyme that were shown to convert elemental sulfurto sulfite in the presence of O2. Very little was known about these enzymes except that many required glutathione (GSH) for properfunction, and that O2 was incorporated into the final sulfite product. In particular, little was known about the nature of the S-atomsource to produce sulfite.147 It was not until a 2003 study that it was shown that the S-atom source in these S-oxidizing bacteria wasglutathione persulfide (GSSH), which was generated in situ from the non-enzymatic reaction between elemental sulfur andglutathione.144
Characterization of human ETHE1 revealed that it binds one equivalent of Fe and has lower or no activity with other persulfidesor thiols, such as cysteine persulfide, glutathione, and thiosulfate compared to GSSH.148 Some activity with CoA persulfide has beenobserved, but much lower than that with GSSH. It has also been shown from studies with plant ETHE1 that the enzyme is selectivetowards GSSH, exhibiting no activity with H2S, Cys, dithiothreitol, or 3-mercaptoethanol. However, it was found that3-mercaptopyruvate can serve to transfer H2S to glutathione. A number of studies have also confirmed that O2 consumption wascorrelated with ETHE1 activity.138,141
The first crystal structure of a persulfide dioxygenase was reported in 2006 from the plant species Arabidopsis thaliana (Fig. 18).149
The protein structure was found to be a member of the metallo b-lactamase superfamily of proteins, which are typically hydrolaseenzymes found in antibiotic-resistant bacteria. However, this structure exhibited high sequence alignment with human ETHE1. Thestructure revealed an octahedral iron center bound by two histidine and one aspartate residue, similar to other nonheme ironoxygenases. However, the exact function of this enzyme was unclear. Further mechanistic studies with Arabidopsis thaliana ETHE1revealed that one equivalent of Fe is bound to the protein and that the two histidines remain bound to the Fe center in solution.150
The structure for human ETHE1 was reported in 2014, and displayed very similar structural features to the plant ETHE1. The proteinexists as a dimer and features Fe bound by two histidines and one aspartate.151 Docking simulations suggested that GSSH should beable to fit into the active site. An unpublished structure of the bacterial protein CstB from Staphylococcus aureus was shown to exhibitsimilar structural motifs to those seen in ETHE1 (PDB: 3R2U). This protein was later found to be a multidomain protein with PDOactivity tightly coupled to persulfide transferase reactivity using a rhodanese domain. Interestingly, this PDO was found to utilizecoenzyme A persulfide and bacillithiol persulfide as substrates.143
The nature of the binding of the glutathione persulfide substrate to PDOs is currently unknown, as no structure for GSS− boundto a PDO has been determined, likely due to the instability of GSS−. Insight into the substrate-bound form of PDOs has beenobtained indirectly. A glutathione-bound structure was obtained from PDO found in Pseudomonas putida.145 This structure revealedthat glutathione binds monodentate via the thiolate donor, replacing one of the resting state water molecules to afford anFe(His)2(Asp)(SR)(H2O) coordination geometry (Fig. 19). It was hypothesized that a similar monodentate sulfur ligation may
Fig. 18 Crystal structure showing overall protein structure of ETHE1 from Arabidopsis thaliana (left) and active site (right) in absence of GSSH substrate(PDB: 4CHL).

Fig. 19 Crystal structure of active site of ETHE1 in the presence of glutathione (PDB: 4YSL).
Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 21
occur for the native substrate, GSS−. The monodentate coordination is consistent with structures of cysteine persulfide and3-mercaptopropionic acid persulfide fortuitously crystallized in CDO, which exhibit similar monodentate coordination.100
5.3 Mechanism
Some specific residues in ETHE1 that are mutated in patients with ethylmalonic encephalopathy have been identified.139,152
Understanding the mechanisms of action, especially with relation to these mutations, has been a major focus of studies involvingPDOs. One study found that mutation of R163 in human ETHE1 results in a lowered redox potential of the Fe center withoutsignificantly altering the protein structure.153 Another study has found that that same mutation leads to significantly loweredcatalytic activity in the presence of substrates.154 The implications and relevance of these results to the mechanism of S-oxygenationare unclear, however. Mutants involving substitution of residues T152 and D196 resulted in a significant decrease in the stabilityand Fe binding affinity of the protein,148 but detailed mechanistic studies involving these mutations were not reported. Mutationsto L55, C161, and T136 have also been shown to exhibit significant drops in catalytic activity.154
Based on the known S-oxygenation reactivity and the structure of ETHE1, the earliest proposed mechanism was in analogy tothe proposed mechanisms of S-oxygenation by CDO.148 This mechanism invokes coordination of the GSS– to Fe, followed bybinding of O2 to generate an FeIII(superoxo) intermediate. The distal oxygen atom attacks the coordinated sulfur, andsubsequent OdO bond cleavage gives an FeIV(O) species bound to an S-oxygenated persulfide. Oxygen atom transfer fromthe ferryl species leads to formation of thiosulfate (RSSO2
- ), which is expected to undergo hydrolysis to produce glutathione andsulfite (Scheme 11).
A similar mechanism was tested in a computational QM/MM study,155 which found glutathione persulfide binds bidentate tothe Fe center via S-amide ligation. Binding of glutathione was followed by a series of O2 activation steps that mirror the reactivityproposed computationally for CDO156 (see Section 3.3). However, this study utilized a relatively limited set of protein residues andassumed bidentate coordination of the substrate, which seems less likely than monodentate coordination based on otherstudies.38,100,145
Scheme 11

Scheme 12
22 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
One intriguing study used g-glutamyl-homocysteinyl-glycine (GSHcySH) as a substrate analogue of GSSH.154 They hypothe-sized that that GSHcySH was topologically similar enough to GSSH to enable proper binding of the substrate and O2 activation, butthe lack of a cleavable SdS bond would allow for the dioxygenated product prior to hydrolysis to be observed. Indeed, addition ofGSHcySH resulted in single turnover and formation of the sulfinate product. This species remains bound to the Fe and preventscatalysis, indicating that the hydrolysis step in native catalysis is essential for turnover (Scheme 12). Spectroscopic studies indicatedthat GSHcyS− binds to the ferric center of ETHE1, as seen by the visible absorption band at 600 nm. Interestingly, glutathionegenerates a similar feature when added to ferric ETHE1, suggesting that it also binds to Fe. However, addition of O2 to ETHE1incubated with GSH results only in the formation of disulfide (GSSG). It is possible that ETHE1 is capable of performing a singleS-oxygenation of GSH to generate a sulfenate, which then can easily form disulfide in the presence of excess thiol. Alternatively,uncoupled autoxidation pathways could lead to the same product. Distinguishing these pathways would be interesting and relevantto the mechanism of S-oxygenation by PDO.
MCD studies on human ETHE1 revealed significant changes to the ferrous active site upon binding of the persulfide substrate.38
The MCD spectrum of the persulfide bound Fe center was consistent with the presence of two species, most likely a 5-coord and6-coord species. It was thus proposed that there is a mixture of 5-coordindate FeII(ON2)(SSR)(H2O) and 6-coordintate FeII(ON2)(SSR)(H2O)2 centers, similar to what has been proposed for CDO.82 In this study NO was used as a probe for the initial O2 bindingstep in PDOs. Addition of NO led to the formation of a new EPR signal consistent with an S ¼ 3/2 {FeNO}7 species. DFT studieswere able to reproduce spectral features of this {FeNO}7 and the same DFT model was used to predict the most likely bondingmode of the persulfide donor, the electronic structure of the hypothetical Fe(superoxo) intermediate expected to form uponaddition of O2 to the ferrous center, and a possible mechanism for S-oxygenation (Fig. 20).
The proposed mechanism for S-oxygenation by ETHE1 mirrors that proposed for CDO38,101 and is similar to that depicted inScheme 11. In the first step, O2 binds to the ferrous center ligated to GSS− as a monodentate ligand to form an S¼ 2 FeIII(superoxo)species. Of note, docking simulations indicated that only monodentate coordination of the persulfide could be accommodated inthe active site. The next step of the reaction is electrophilic attack of the persulfide S via the distal oxygen, and is rate limiting. Theresulting FedOdOdS intermediate could then undergo a barrierless OdO bond cleavage step to generate an FeIV(O) which couldthen undergo rearrangement and O-atom transfer to generate the dioxygenated product. Hydrolysis releases thiol and produce thesulfite product. The same computational method was applied to CDO and predicts a similar reaction mechanism.101,102 More workis still needed to experimentally validate or provide alternatives to this proposed mechanism.
6 Synthetic Model Complexes
Studies involving synthetic analogues of the active sites of metalloenzymes provide the opportunity to directly correlate spectro-scopic properties and reactivity with the local environment surrounding the metal center.157 The amenability of model complexestowards synthetic modification allows their reactivity to be rationally tuned as a function of steric, geometric, and electronicproperties. The feasibility of proposed enzymatic mechanisms can be assessed by determining the reactivity patterns of biomimeticmodel complexes. If proposed bond-making or bond-breaking events at the metal can be mimicked in a model system, the modelscan provide satisfying support for hypothetical mechanistic steps that are challenging, or sometimes impossible to probe directly inthe biological systems. Similarly, a complete failure to reproduce a proposed enzymatic step in a well-defined model system mayreveal that one or more hypothetical mechanistic steps are ill-conceived, and provide strong motivation for revising the mechanistic
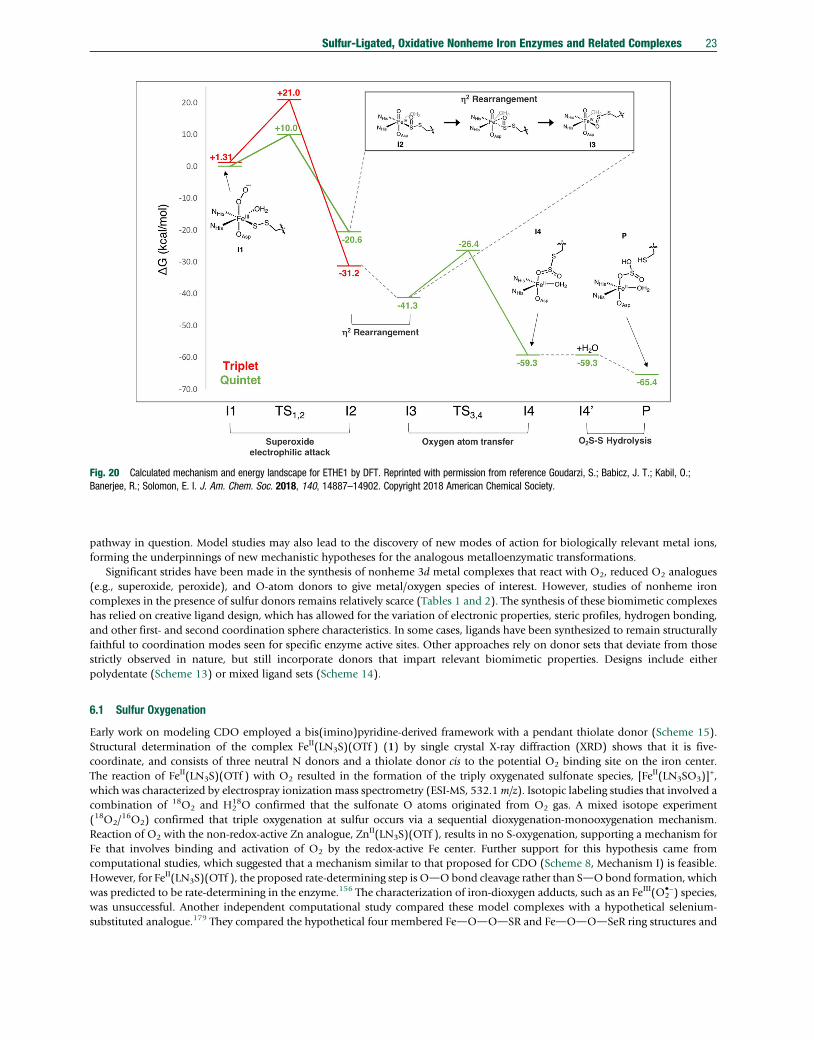
Fig. 20 Calculated mechanism and energy landscape for ETHE1 by DFT. Reprinted with permission from reference Goudarzi, S.; Babicz, J. T.; Kabil, O.;Banerjee, R.; Solomon, E. I. J. Am. Chem. Soc. 2018, 140, 14887–14902. Copyright 2018 American Chemical Society.
Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 23
pathway in question. Model studies may also lead to the discovery of new modes of action for biologically relevant metal ions,forming the underpinnings of new mechanistic hypotheses for the analogous metalloenzymatic transformations.
Significant strides have been made in the synthesis of nonheme 3d metal complexes that react with O2, reduced O2 analogues(e.g., superoxide, peroxide), and O-atom donors to give metal/oxygen species of interest. However, studies of nonheme ironcomplexes in the presence of sulfur donors remains relatively scarce (Tables 1 and 2). The synthesis of these biomimetic complexeshas relied on creative ligand design, which has allowed for the variation of electronic properties, steric profiles, hydrogen bonding,and other first- and second coordination sphere characteristics. In some cases, ligands have been synthesized to remain structurallyfaithful to coordination modes seen for specific enzyme active sites. Other approaches rely on donor sets that deviate from thosestrictly observed in nature, but still incorporate donors that impart relevant biomimetic properties. Designs include eitherpolydentate (Scheme 13) or mixed ligand sets (Scheme 14).
6.1 Sulfur Oxygenation
Early work on modeling CDO employed a bis(imino)pyridine-derived framework with a pendant thiolate donor (Scheme 15).Structural determination of the complex FeII(LN3S)(OTf ) (1) by single crystal X-ray diffraction (XRD) shows that it is five-coordinate, and consists of three neutral N donors and a thiolate donor cis to the potential O2 binding site on the iron center.The reaction of FeII(LN3S)(OTf ) with O2 resulted in the formation of the triply oxygenated sulfonate species, [FeII(LN3SO3)]
+,which was characterized by electrospray ionization mass spectrometry (ESI-MS, 532.1m/z). Isotopic labeling studies that involved acombination of 18O2 and H2
18O confirmed that the sulfonate O atoms originated from O2 gas. A mixed isotope experiment(18O2/
16O2) confirmed that triple oxygenation at sulfur occurs via a sequential dioxygenation-monooxygenation mechanism.Reaction of O2 with the non-redox-active Zn analogue, ZnII(LN3S)(OTf ), results in no S-oxygenation, supporting a mechanism forFe that involves binding and activation of O2 by the redox-active Fe center. Further support for this hypothesis came fromcomputational studies, which suggested that a mechanism similar to that proposed for CDO (Scheme 8, Mechanism I) is feasible.However, for FeII(LN3S)(OTf ), the proposed rate-determining step is OdObond cleavage rather than SdObond formation, whichwas predicted to be rate-determining in the enzyme.156 The characterization of iron-dioxygen adducts, such as an FeIII(O2
•−) species,was unsuccessful. Another independent computational study compared these model complexes with a hypothetical selenium-substituted analogue.179 They compared the hypothetical four membered FedOdOdSR and FedOdOdSeR ring structures and

Table 1 Structural and spectroscopic properties of selected nonheme, sulfur-ligated ferrous complexes.
Coord. sphere d(FedS) Spin state d (│DEQ│) E1/2 (FeIII/FeII) Reference
(Å) mm s–1 V vs. Fc+/Fc
FeII(LN3S)(OTf ) (1) N3SO 2.2942(8) S ¼ 2 n.r.a n.r. [158][FeII(LN3S)(py)]OTf (2) N4S 2.3246(18) S ¼ 2 n.r. n.r. [159][FeII(LN3S)(DMAP)]OTf (3) N4S 2.3246(18) S ¼ 2 0.87 (2.70)b n.r. [159][FeII(LN3S)(DMAP)]
0 (4) N4S 2.2179(5) S ¼ 1/2 0.33 (2.04)b n.r. [159](iPrBIP)FeII(SPh)(Cl) (7) N3SCl 2.3305(9) S ¼ 2 0.78 (2.62)b –0.173c [159,160](iPrBIP)FeII(SPh)(OTf ) (8) N3SO 2.2795(9) S ¼ 2 n.r. –0.372c [160][FeII(N3PyS)(CH3CN)]BF4 (9a) N5S 2.3018(4) S ¼ 0 0.14 (0.26)d –0.226e [161][FeII(N3PyS)(MeOH)]BF4 (9b) N4SO n.r. S ¼ 2 1.05 (2.77)d –0.583f [161,162]FeII(N3PySO2))(SCN) (10) N5SO 2.1812(9) S ¼0 n.r. –0.001e [161][TpMe,PhFeCysOEt] (12) N4S 2.3122(9) S ¼ 2 n.r. n.r. [163]TpMe,PhFeCysAm (13) N4S 2.3175(6) S ¼ 2 n.r. n.r. [164][Fe(Ph2TIP)(CysOMe)]BPh4 (14) N4S 2.3107(6) S ¼ 2 n.r. n.r. [165][Fe(Ph2TIP)(CysAm)]BPh4 (15) N4S 2.3051(5) S ¼ 2 n.r. n.r. [165]FeII(Me3TACN)(abt)(OTf ) (16) N4SO 2.4352(4) S ¼ 2 1.07 (3.55)d n.r. [82]FeII(Me3TACN)(abt
CF3)(OTf ) (17) N4SO 2.4343(4) S ¼ 2 1.08 (3.20)d n.r. [82][FeII(iPr3TACN)(abt)](OTf ) (18) N4S 2.3573(4) S ¼ 2 0.93 (1.97)d n.r. [82][FeII(iPr3TACN)(abt
CF3)](OTf ) (19) N4S 2.3753(6) S ¼ 2 0.95 (2.06)d n.r. [82][FeII(SMe2N4(tren))](PF6) (20) N4S 2.328(1) S ¼ 2 n.r. –0.482e [166][FeII(SMe2N4(tren-Et4))](PF6) (22) N4S 2.317(1) S ¼ 2 n.r. 0.028e,f [166]NiIILFeII(CH3CN)(Z-C5Me5)]BPh4 (23) C3S2N 2.2850(6) S ¼ 0 n.r. n.r. [167]
2.2863(6)NiIILFeII(EtCN)(Z-C5Me5)]BPh4 (24) C3S2N 2.2867(6) S ¼ 0 0.55 (2.10)d −0.57e 167
2.2839(6)[Fe(TpMe2)(2-ATP)] (26) N4S 2.3107(4) S ¼ 2 n.r. n.r. [168][FeII(S2
Me2N3(Pr,Pr))] (28) N3S2 2.3306(5) S ¼ 1 n.r. n.r. [169]2.3263(5)
FeII(Me3TACN)(S2SiMe2) (33) N3S2 2.371(2) S ¼ 2 0.93 (2.30)g,h –0.6e [170,171]2.410(2)
[FeII(N3PyamideSR)](BF4)2 (37) N4S(thioether)O 2.2911(6) S ¼ 0 0.44 (0.77)g n.r. [172][FeII(N3PyamideS(O)R)]2+(BF4)2 (39) N4S(thioether)O 2.254(13) n.r. n.r. n.r. [172][FeII(TMCS)](PF6) (40) N4S 2.297(3) S ¼ 2 0.90 (3.0)g 0.148e [173,174][FeII(cyclam-PrS)]+ (47) N4S 2.286(1) S ¼ 2 n.r. n.r. [175][FeII([15]aneN4)(SC6H5)]BF4 (50a) N4S 2.3316(11) S ¼ 2 n.r. 0.076c,f [176,177][FeII([15]aneN4)(SC6H4Cl)]BF4 (50b) N4S 2.3197(12) S ¼ 2 n.r. 0.156c,f [176][FeII([15]aneN4)(SC6H4OMe)]BF4 (50c) N4S 2.3240(17) S ¼ 2 n.r. 0.021c,f [176][FeII([15]aneN4)(SC6F4SC6F5)]BF4 (50d) N4S 2.3426(8) S ¼ 2 n.r. 0.388c,f [176][FeII([15]aneN4)(SC6F4NO2)]BF4 (50e) N4S 2.3305(15) S ¼ 2 n.r. 0.223c,f [176][FeII(N3PySR)(CH3CN)](BF4)2 (52) N5S (thioether) 2.2848(4) S ¼ 0 0.44 (0.55)h n.r. [162,178]
an.r. indicates not reported.bData collected as a solid at 5 K in the presence of a 47 mT magnetic field.cCV data collected in CH2Cl2.dData collected with zero field as a solid at 5 K.eCV data collected in CH3CN.fAnodic peak potential reported.gData collected in CH3CN at 5 K in the presence of a 47 mT magnetic field.hData collected with zero field at 80 K.
24 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
found that the -SR donor was better at donating electron density necessary to cleave the OdO bond than -SeR. The authorshypothesized that this ability to cleave the OdO bond is why selenocysteine is not oxygenated by CDO.
Addition of either pyridine or 4-dimethylaminopyridine (DMAP) to FeII(LN3S)(OTf ) generates a new high-spin ferrouscomplex, [FeII(LN3S)(py)]OTf (2) or [FeII(LN3S)(DMAP)]OTf (3), each with an [N4S]
1− coordination environment.180 Reductionof 3 with Na/Hg amalgam produced a new S ¼ 1/2 species [FeII(LN3S)(DMAP)]0 (4), best described as an S ¼ 1 Fe centerantiferromagnetically coupled with a ligand-based radical. The reactivity of 2, 3, and 4 were compared, and it was found that 2and 3 performed S-oxygenation, producing [FeII(LN3SO3)]
+, as determined by mass spectrometry. However, 4 performed both Feand S oxygenation, producing a mixture of S-oxygenates and m-oxo dimer products, as seen by mass spectrometry.

Table 2 Structural and spectroscopic properties of selected nonheme sulfur-ligated Fe/oxygen complexes.
Complex Coordination Spin lmax, (e) n(OdO) n(FedO) d (│DEQ│) EPRSphere State nm (M–1 cm–1) cm–1 cm–1 mm s–1
Superoxo[Fe(O2)(Tp
Me2)(2-ATP)] (27)a N4SO S � 1 490 (1200), 655 (1800),860 (2200)
1135,1105
504 n.r.b Silent
[FeIII(S2Me2N3(Pr,Pr))(O2)] (29)
c N3S2O S ¼ 0 409, 520, 707 1122,1093
n.r. n.r. Silent
Peroxo[NiIILFeIV(Z2-O2)(Z
5-C5Me5)]BPh4(25)d
C3S2O2 S ¼ 0 410 940 n.r. 0.42 (0.33) n.r.
[FeIII(O2- )(TMCS)] (49)e N4SO S¼ 5/2 460 (6100), 610 (1200) n.r. n.r. 0.71 (-1.9) 7.9, 5.75
Fe2(O2)(Me3TACN)2(S2SiMe2)2 (34)f N3S2O n.r. 300 (9000), 390 (5000) 849 n.r. 0.53 (0.77) Silent
530 (2050), 723 (3600)Hydroperoxo[FeIII(cyclam-PrS)(OOH)]PF6 (48)
g N4SO S¼ 5/2 530 (1400) 891 419 n.r. 7.72, 5.40,4.15
[FeIII(OOH)(N3PyamideSR)](BF4)2(54a)h
N4S(thioether)O
S¼ 1/2 567 (900) 800 612 n.r. 2.17, 2.16,1.95
[FeIII(OOH)(N3PySR)](BF4)2 (53a)h N4S(thioether)
OS¼ 1/2 542 (1000) 809, 787 615 –
664n.r. 2.17, 2.11,
1.97[FeIII(S2
Me2N3(Pr,Pr))(OOH)] (31)c N3S2O S¼ 1/2 696 n.r. n.r. n.r. 2.23, 2.15,
2.00[FeIIISMe2N4(tren)(OOH)]
+ (45)i N4SO S¼ 1/2 452 (2780) 788, 781 n.r. n.r. 2.14, 1.97Alkylperoxo[FeIII([15]aneN4)(SC6F4NO2)(OO
tBu)]BF4 (51e)
jN4SO S¼ 1/2 522 (�3100) 800 615 n.r. n.r.
[FeIII([15]aneN4)(SC6F4SC6F5)(OOtBu)]BF4 (51d)
jN4SO S¼ 1/2 508 (�3000) 799 623 n.r. n.r.
[FeIII([15]aneN4)(SC6H4Cl)(OOtBu)]BF4
(51b)jN4SO S¼ 1/2 524 (�2100) 803 612 n.r. 2.19, 1.97
[FeIII([15]aneN4)(SC6H4OMe)(OOtBu)]
BF4 (51c)j
N4SO S¼ 1/2 514 (�1900) 802 608 n.r. n.r.
[FeIII([15]aneN4)(SC6H5)(OOtBu)]BF4
(51a)jN4SO S¼ 1/2 526 (2150) 803 612 n.r. 2.20, 1.97
[FeIII(OOtBu)(N3PyamideSR)](BF4)2(54b)k
N4SO S¼ 1/2 620 (2000) 796 700 n.r. 2.17, 2.11,1.96
[FeIII(OOtBu)(N3PySR)](BF4)2 (53b)k N4SO S¼ 1/2 600 (1670) 796 691 n.r. 2.14, 2.08,
1.96Oxo[FeIV(O)(N3PyamideSR)](BF4)2 (38)
l N4S(thioether)O
S ¼ 1 750 (400) – n.r. 0.04 (0.80) Silent
FeIV(O)(Me3TACN)(S2SiMe2) (35)f N3S2O S ¼ 1 385 (2600), 460 (1500),
890 (390)– 735 0.21 (1.54) Silent
[FeIV(O)(TMCS)](PF6) (41)m N4SO S ¼ 1 460 (1300), 570 (1100),
850 (230)– n.r. 0.19 (-
0.22)Silent
aFrom reference [168].bn.r. indicates not reported.cFrom reference [169].dFrom reference [167].eFrom reference [192].fFrom reference [171].gFrom reference [175].hFrom reference [178].iFrom reference [166].jFrom references [176, 177].kFrom reference 172.lFrom reference [172].mFrom reference [174].
Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 25

Scheme 13
26 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
Structurally related thioether complexes [FeII(LN3SMe)(H2O)3](OTf )2 (5), and [FeII(LN3SMe)Cl2] (6) were also prepared andwere shown to perform S-oxygenation reactivity.181 In this case, high temperatures (up to 60 �C) were required to initiate O2
reactivity. Oxygenation of 5 resulted in the formation of the sulfoxide product, [Fe(LN3S(O)Me)(OTf )]2+ and oxygenation of 6generated the sulfone, [Fe(LN3S(O2)Me)Cl]2+, as determined by mass spectrometry and by 1H NMR spectroscopic analysis of thehydrolyzed organic products. The mechanism of S-oxygenation was not discussed, but these results suggest that the nature of thesupporting ligands can control the extent of S-oxygenation (i.e., mono- versus di-oxygenation).
The related complexes (iPrBIP)FeII(SPh)(Cl) (7) and (iPrBIP)FeII(SPh)(OTf ) (8) featured a similar bis(imino)pyridine frame-work but with an untethered arylthiolate donor. These complexes were structurally characterized and both contain 5-coordinatehs-FeII centers; however, a striking difference between these related complexes is the orientation of the thiolate donor with respect tothe potential open coordination site for binding of O2. In (iPrBIP)FeII(SPh)(Cl), the phenylthiolate ligand is trans to the possible O2
binding site, whereas in (iPrBIP)FeII(SPh)(OTf ), the phenylthiolate ligand is cis. For the trans-thiolate ligated complex, addition ofO2 leads to the formation of disulfide. Laser desorption ionization mass spectrometry (LDIMS) analysis of the reaction mixturerevealed a dominant peak consistent with the FeIV(O) formulation, [(iPrBIP)Fe(O)(Cl)]+, which shifted by +2 units upon 18O2
substitution. Reaction of this species with PPh3 gave OPPh3 in good yield, consistent with the oxidized FeIV(O) formulation.In contrast, addition of O2 to the OTf complex with a thiolate donor cis to the open binding site leads to the formation ofbenzenesulfonic acid, based on organic product analysis by 1H NMR spectroscopy and reversed phase high performance liquid

Scheme 14
N
NN
S
M
TfO
N
NNAr
S
Fe
TfO
OO
OO2
H218O
CH2Cl223 °C
O2
CH2Cl223 °C
M = FeM = Zn
No Reaction
1: M = Fe
Scheme 15
Scheme 16
Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 27
chromatography (HPLC). These studies suggest that the orientation of the thiolate donor in the FeII starting material plays anessential role in determining the ultimate selectivity of Fe versus S-oxygenation in these model complexes (Scheme 16).A computational study182 found that OTf must bind trans to the open coordination site in 8 due to an unfavorable steric interactionwith the BIP ligand framework, whereas Cl is able to bind either cis or trans. Optimizing the four putative and hypotheticalFe(superoxo) structures (iPrBIP)FeIII(SPh)(X)(O2) (where X ¼ Cl and OTf ligated cis or trans) indicated that the thiolate liganddissociates as a thiyl radical when bound trans to the iron center. These calculations support the observed experimental requirementfor cis ligation of the thiolate donor for S-oxygenation reactivity.

Scheme 17
28 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
Following the O2 chemistry of the BIP complexes was significantly hampered by the intense absorption bands for BIP-FeII species(e.g., lmax ¼ 715 nm (e � 4000 M−1 cm−1) for (iPrBIP)FeII(SPh)(OTf )), making it very difficult to identify any putative O2
intermediates. Several of these BIP-derived model complexes also did not incorporate the complete [N4S]1− coordination environ-
ment present in substrate-bound CDO. Such a coordination environment is presented to the metal by the N3PyS ligand shown inScheme 17. Reaction of N3PyS– and [FeII(H2O)6](BF4)2, or metalation of the sulfur-protected N3PySEtCN followed by deprotec-tion183 with added base results in the formation of [FeII(N3PyS)(CH3CN)]BF4 (9a) (Scheme 17), which was characterized by XRD.The crystal structure of [FeII(N3PyS)(CH3CN)]BF4 reveals a six-coordinate Fe center with a potentially labile solvent ligand bound cisto the thiolate donor. This FeII complex is low-spin (S ¼ 0) in CH3CN, and does not react with O2 in this solvent. However, upondissolution in methanol, the FeII complex undergoes a color change indicating conversion to a high-spin (S ¼ 2) species. Additionof O2 in CH3OH leads to the consumption of the FeII complex and formation of a new green species. Addition of KSCN to this greenspecies leads to single crystals of FeII(N3PySO2)(SCN) (10) (XRD), a doubly oxygenated, FeII(sulfinate). The reaction of high-spin[FeII(N3PyS)(CH3OH)]+ (9b) with O2 provides a rare functional model of the thiol dioxygenase enzymes, in which the sulfur centerin FeII(SR) is preferentially oxygenated over the iron center. Unlike the BIP complexes, the N3PyS ligand allows for the observationof a new green species with lmax ¼ 674 nm upon oxygenation. However, this species could not be identified.
Nitric oxide was employed as an O2 surrogate with [FeII(N3PyS)(CH3CN)]BF4. Addition of NO results in the formation of a new{FeNO}7 species (11), which was characterized by EPR, Mössbauer, XAS, UV-vis, and NMR spectroscopies.184 Complex 11 exhibitsa low-spin (S ¼ ½) ground state, the same as that seen for the NO adduct of CDO, but which contrasts the S ¼ 3/2 ground statetypically observed for other nonheme {FeNO}7 enzymes.95,185,186 This {FeNO}7 species releases NO upon illumination with whitelight, and under aerobic conditions, leads to nitric oxide oxidation to give an FeIII(nitrite) complex. In addition, [FeII(N3PyS)(NO)]+
could be interconverted between {FeNO}6 and {FeNO}8 species by one electron, outer-sphere oxidation and reduction, respec-tively (Scheme 18).183,187 These complexes represent a rare series of {FeNO}6–8 complexes stabilized within the same ligandenvironment, and provide a unique example of such a series with thiolate ligation.185,186,188 The oxidized {FeNO}6 complex readilyundergoes photolytic, thermal, or solvent-mediated NO dissociation.187 The reduced {FeNO}8 complex provides a rare example ofa nitric oxide adduct that spontaneously produces N2O upon thermal decay,183 which has implications for possible mechanisms ofthe nitric oxide reductases (NORs). These reactivity patterns suggest the inclusion of the unique thiolate donor has an influence onpromoting unusual nonheme iron transformations with nitric oxide.
A complex utilizing the facially-coordinating N3 ligand, hydrotris(pyrazolyl)borate, and cysteine ester, TpMe,PhFeCysOEt (12),was prepared as a model of CDO.163 Addition of O2 to 12 in CH2Cl2 resulted in slow decomposition and the formation of the
Scheme 18

Fig. 21 S-oxygenation reactivity by Tp and TIP cysteine ester and cysteamine complexes and time-resolved UV-vis spectra for reaction of 14 with O2. Plot adaptedwith permission from reference Fischer, A. A.; Stracey, N.; Lindeman, S. V.; Brunold, T. C.; Fiedler, A. T. Inorg. Chem. 2016, 55, 11839–11853. Copyright 2016American Chemical Society.
Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 29
sulfinate product following workup and analysis by 1H NMR spectroscopy (Fig. 21). Mass spectrometry studies with mixtures of16O2/
18O2 clearly indicated that both O-atoms from O2 were incorporated into the sulfinate product. Though the Fe(sulfinate)product could not be definitively characterized by XRD, XAS data and DFT calculations were consistent with a model involvingformation of an Z2-O,O bound Fe(sulfinate) product. Further studies showed that the analogous cysteamine complex, TpMe,
PhFeCysAm (13), could be prepared as a model of ADO. As with cysteine ethyl ester, dioxygenation of the cysteamine to taurine wasobserved upon workup of the reaction of 13 with excess O2. However, no Fe/oxygen intermediates or Fe-containing products werereported.
Related iron(II) complexes containing the neutral N3 ligand tris(imidazolyl)phosphine (TIP) and cysteine methyl ester wereprepared, with formulas [Fe(Ph2TIP)(CysOMe)]BPh4 (14) and [Fe(Ph2TIP)(CysAm)]BPh4 (15) (Fig. 21).165 These complexescontain high spin ferrous centers with the expected bidentate coordination of the S/N ligands. As was seen for the former Tpcomplexes, addition of O2 results in slow decay (k1(20 �C)¼ 1.9(4)� 10−4 s−1 for 14), of the FeII starting material and formation ofsulfinate products. Kinetics studies of the oxygenation reaction indicated that the N3 ligand has only minor effects on the rate of thereaction. Calculations on the reaction trajectories for both the Tp and TIP complexes suggested that similar mechanisms wereoperative, with a rate-determining step involving SdO bond formation via an FeIII(superoxo) intermediate. A larger degree of thiylradical character for the TIP versus the Tp complexes was calculated by DFT, leading to the hypothesis that the neutral N donors inthe enzyme (3 His) may be important in promoting thiyl character and a lower barrier for SdO bond formation. As with the Tpcomplexes, no Fe/O2 intermediates were experimentally observed. Thus NO was added as an O2 surrogate. Formation of high-spin{FeNO}7 (S ¼ 3/2) species was observed, in contrast to the low-spin (S¼1/2) ground state FeNO adducts seen in CDO. However,the binding of NO suggests that O2 may bind directly to the Fe center in the model complexes prior to S-oxygenation.
The iron(II) 1,4,7-triazacyclononane (TACN) derivatives [FeII(Me3TACN)(CH3CN)3](OTf )2 and FeII(iPr3TACN)(OTf )2 wereemployed to synthesize the [FeII(N4S(thiolate))] complexes shown in Fig. 22.82 These complexes have the advantage of labile sites
Fig. 22 (A) Syntheses of complexes 16–19. (B) Zero-field 57Fe Mössbauer spectra of 16–19 collected at 5 K. Plot adapted with permission from referenceGordon, J. B.; McGale, J. P.; Prendergast, J. R.; Shirani-Sarmazeh, Z.; Siegler, M. A.; Jameson, G. N. L.; Goldberg, D. P. J. Am. Chem. Soc. 2018, 140,14807–14822. Copyright 2018 American Chemical Society.

30 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
on the metal for chelation of bidentate NS donors similar to cysteine. Addition of [Et3NH][abt] or [Et3NH][abtCF3] (abt ¼2-aminobenzenethiolate) to the FeII(TACN) precursors led to the isolation and structural characterization of FeII(Me3TACN)(abt)(OTf ) (16), FeII(Me3TACN)(abtCF3)(OTf ) (17), [FeII(iPr3TACN)(abt)](OTf ) (18), and [FeII(iPr3TACN)(abtCF3)](OTf ) (19) ingood yields. The two Me3TACN complexes are six-coordinate (6-coord) FeII(N4SO) centers, with a triflate anion occupying thesixth site, and the iPr3TACN complexes are five-coordinate (5-coord) FeII(N4S) centers with the triflate counterion sitting outside thecoordination sphere. Analysis by 57Fe Mössbauer spectroscopy on solid samples of complexes 16–19 reveals an interesting trend forthese high-spin ferrous centers. The 5-coordinate complexes 18–19 exhibit a quadrupole splitting of |DEQ|�2 mm s–1, whereas the6-coordinate complexes 16–17 exhibit a much larger |DEQ|> 3 mm s–1. Trends between coordination number and the quadrupolesplitting of structurally well-defined Fe complexes are difficult to establish. The complexes in Fig. 22 show that 5- versus6-coordinate iron(II) complexes can be readily distinguished by their |DEQ| values in a series of structurally related complexes.
The spectroscopic trends exhibited by the series of complexes in Fig. 22 were useful in helping to assign the different species seenin the Mössbauer spectrum of the substrate-bound CDO enzyme. There are two closely overlapping doublets observed in theMössbauer spectrum of Cys-bound FeII CDO, with parameters modeled as d¼ 1.03mm s–1, |DEQ|¼ 2.28mm s–1 (species A) and d¼ 1.10 mm s–1, |DEQ|¼ 3.14 mm s–1 (species A0) (Section 3.2). Early computational modeling (DFT) predicted that A0 was 6-coordwith H2O in the open site (FeII(His)3(Cys)(H2O)), while A was the 5-coord center, FeII(His)3(Cys). However, comparison with thetrends established for the model complexes combined with further DFT calculations indicated that the two quadrupole doubletsobserved for Cys-bound CDO likely correspond to water bound and unbound forms of the Fe center, with A assigned as the5-coordinate, water-bound complex and A0 as the 6-coordinate Fe center.
Reaction of 16 with excess O2 at −95 �C results in a transient blue species that converts to a red species upon warmup to 23 �C.Analysis by 1H, 13C, and 1H–
13C HSQC NMR spectroscopies and electron ionization mass spectrometry (EI-MS) revealed theformation of the S-oxygenated product methyl 2-aminobenzenesulfinate (42%). An initial sulfinate species likely forms, followedby esterification in MeOH to give the final sulfinate ester. Employing 18O2 showed that one oxygen of the sulfinate derivativeoriginates fromO2. Adding O2 to the CF3-substituted 17 at −95 �C also results in S-oxygenation. In this case, however, the Fe-boundsulfinate product is isolated as a (m-oxo)diferric complex with bridging sulfinate ligands, characterized by XRD (Scheme 19). Massspectrometry studies confirmed that the four sulfinate O-atoms in the diferric product were derived from O2.
Addition of the non-native abt substrate followed by O2 to a mutant of the enzyme, C93G CDO, led to disulfide as the majorproduct. Very small changes were observed in the Mössbauer spectra of C93G CDO upon addition of the abt substrate, suggestingonly weak (or no) binding to the Fe in the enzyme. Thus, unlike in the model complexes, proper chelation of abt does not occur inthe enzyme, preventing S-oxygenation. This combined enzyme/model study with abt suggests that N/S substrates need to bechelated properly to the Fe center in CDO to undergo S-oxygenation. This requirement likely contributes to the substrate specificityobserved for CDO (see Section 3.3).
6.2 Fe/O2 Intermediates
The 5-coordinate, N4S-ligated ferrous complex [FeII(SMe2N4(tren))](PF6) (20) reacts with O2 to produce a m-oxo dimer,[FeIII(SMe2N4(tren))]2(m-O)(PF6)2•3MeCN (21) (Scheme 20), which was characterized by X-ray crystallography.189 The reactionrequired 0.25 equiv of O2, a stoichiometry that is consistent with mechanisms observed for the formation of Fe(porphyrin) m-oxodimer complexes involving initial FeIII(superoxo), FeIII2 (peroxo) and FeIV(oxo) intermediates.190 An intermediate with an absorp-tion feature at 460 nm was observed in MeOH when O2 is added to 20 at −78 �C. The authors did not elaborate further on theidentity of this intermediate. The electronic properties and reactivity of the (m-oxo) dimer complex 21 were examined. It was foundthat the ferric centers were weakly anti-ferromagnetically coupled with J ¼ −28 cm−1. Addition of acids led to cleavage of the oxo-bridged dimer, which could be reformed upon addition of a hydroxide source. To prevent the formation of an oxo-bridged dimer, amore sterically encumbered complex, [FeII(SMe2N4(tren-Et4))](PF6) (22) was prepared. However, 22 did not react with O2 (Scheme20). Cyclic voltammetry of 20 and 22 showed that 20 exhibits a reversible redox couple (E1/2 ¼ −100 mV versus SCE), although 22exhibited only an irreversible wave at a much higher potential (+410 mV versus SCE). It was hypothesized that the FeIII center could
Scheme 19

Scheme 20
Fig. 23 Catalytic reduction of O2 to H2O by NiIILFeII(RCN)(Z-C5Me5)]BPh4.
Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 31
not be accessed in the latter complex because it would produce shorter MdL bond lengths which would create unfavorable stericinteractions with the bulky ethyl groups.
A dithiolate-ligated Fe/O2 adduct, assigned as an FeIV(peroxo), was prepared by addition of O2 to NiIILFeII(RCN)(Z-C5Me5)]BPh4 (23–24) (L¼N,N0-diethyl-3,7-diazanonane-1,9-dithiolato, R¼CH3 (23) or Et (24)) at –40 �C in acetonitrile (Fig. 23).191 Theproduct [NiIILFeIV(Z2-O2)(Z
5-C5Me5)]BPh4 (25) was crystallographically characterized, and the OdO bond distance (1.381(3) Å),as well as the n(OdO) of 940 cm–1 (Dn(18O2–
16O2) ¼ –53 cm–1) as seen by IR spectroscopy, are in the range for metal peroxospecies. Furthermore, addition of H2
18O2 resulted in exchange of the peroxo core, consistent with the peroxo assignment. Magneticsusceptibility measurements on 25 indicated an S ¼ 0 ground state. Analysis of 25 by 57Fe Mössbauer spectroscopy revealed adoublet with d ¼ 0.42 mm s–1 and |DEQ| ¼ 0.33 mm s–1. It was found that 23 and 24 were capable of reducing O2 to water uponaddition of a reductant and proton source, regenerating ferrous starting material.
The ferrous complex, [Fe(TpMe2)(2-ATP)] (26) was prepared as a structural model of the active site of CDO, using the non-nativesubstrate 2-aminobenzenethiolate (abt or 2-ATP).168 Addition of O2 in THF at −80 �C resulted in the formation of a new purple,metastable, EPR-silent species (27) with UV-vis features at 490, 655, and 860 nm (Scheme 21). Interestingly, the presence of the twobands with lmax > 500 nm resembles those seen for Fe/O2 intermediates trapped in CDO81 and IPNS.35 This intermediate had ahalf-life of 10 min, and gradually decayed to a new EPR active species. The resonance Raman spectrum of this intermediate revealedO2-sensitive peaks at 1105 and 1135 cm−1 (18O2, 1055 cm−1). An additional feature at 504 cm−1 (Dn(18O2–
16O2)¼ –16 cm−1) wasalso observed. The bands at 1105 and 1135 cm−1 are in the typical range seen for the n(OdO) of metal-superoxo species, and thefeature at 504 cm−1 was assigned as n(FedO), leading to the assignment of 27 as a mononuclear FeIII(superoxo) species. Time-dependent DFT (TD-DFT) calculations supported the assignment. A caveat is that the Mössbauer spectrum for 27 was not reported,which would help determine the purity and yield of the superoxo adduct. Decay of 27 resulted in the formation of an EPR active
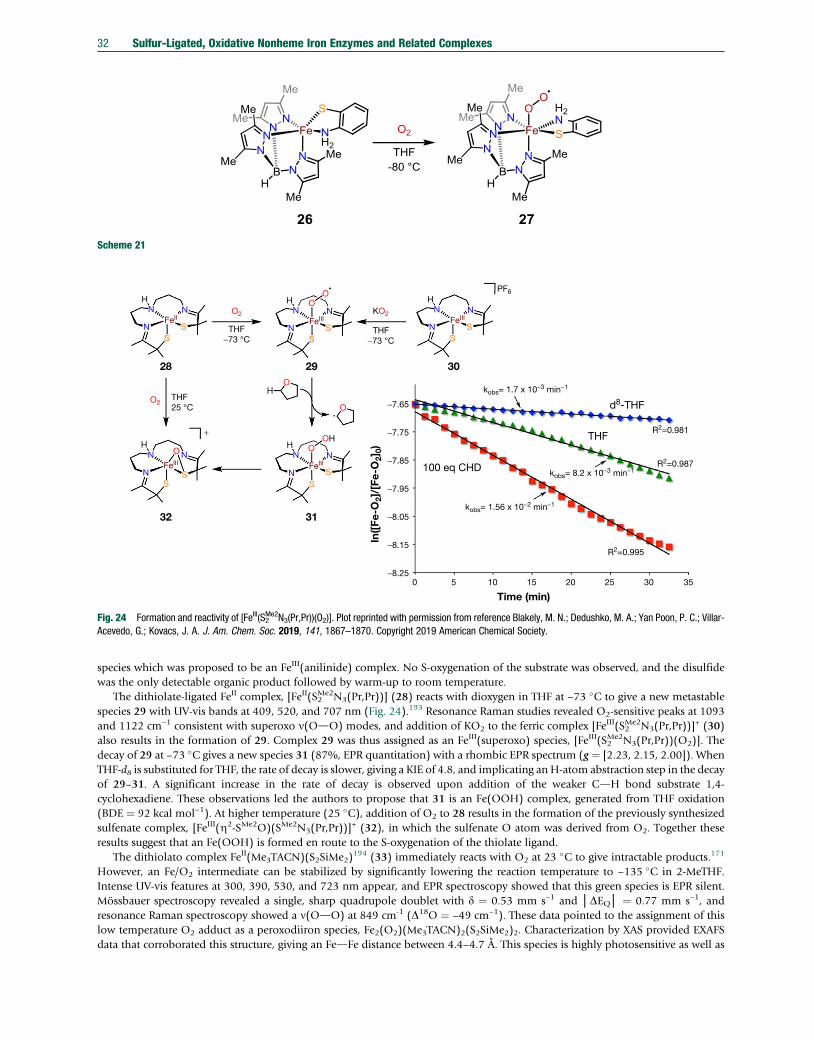
Scheme 21
H
28
32 31
29 30
N
N
N
SS
FeII
HN
N
N
SS
FeIII
HPF6
THF−73 °C
THF−73 °C
+
THF25 °C
N
N
N
SS
FeIII
H
H
N
N
N
SS
FeIII
O
O2
O2
KO2
O
OH
−8.250 5 10 15 20 25 30 35
R2=0.995
R2=0.987
R2=0.981THF
100 eq CHD
Time (min)
In([
Fe-O
2]/[
Fe-O
2]0)
d8-THF
kobs= 1.56 x 10−2 min−1
kobs= 1.7 x 10−3 min−1
kobs= 8.2 x 10−3 min−1
−8.15
−8.05
−7.95
−7.85
−7.75
−7.65
N
N
N
SS
FeIII
OOH
O
O
Fig. 24 Formation and reactivity of [FeIII(S2Me2N3(Pr,Pr))(O2)]. Plot reprinted with permission from reference Blakely, M. N.; Dedushko, M. A.; Yan Poon, P. C.; Villar-
Acevedo, G.; Kovacs, J. A. J. Am. Chem. Soc. 2019, 141, 1867–1870. Copyright 2019 American Chemical Society.
32 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
species which was proposed to be an FeIII(anilinide) complex. No S-oxygenation of the substrate was observed, and the disulfidewas the only detectable organic product followed by warm-up to room temperature.
The dithiolate-ligated FeII complex, [FeII(S2Me2N3(Pr,Pr))] (28) reacts with dioxygen in THF at −73 �C to give a new metastable
species 29 with UV-vis bands at 409, 520, and 707 nm (Fig. 24).193 Resonance Raman studies revealed O2-sensitive peaks at 1093and 1122 cm–1 consistent with superoxo n(OdO) modes, and addition of KO2 to the ferric complex [FeIII(S2
Me2N3(Pr,Pr))]+ (30)
also results in the formation of 29. Complex 29 was thus assigned as an FeIII(superoxo) species, [FeIII(S2Me2N3(Pr,Pr))(O2)]. The
decay of 29 at –73 �C gives a new species 31 (87%, EPR quantitation) with a rhombic EPR spectrum (g ¼ [2.23, 2.15, 2.00]). WhenTHF-d8 is substituted for THF, the rate of decay is slower, giving a KIE of 4.8, and implicating an H-atom abstraction step in the decayof 29–31. A significant increase in the rate of decay is observed upon addition of the weaker CdH bond substrate 1,4-cyclohexadiene. These observations led the authors to propose that 31 is an Fe(OOH) complex, generated from THF oxidation(BDE ¼ 92 kcal mol–1). At higher temperature (25 �C), addition of O2 to 28 results in the formation of the previously synthesizedsulfenate complex, [FeIII(Z2-SMe2O)(SMe2N3(Pr,Pr))]
+ (32), in which the sulfenate O atom was derived from O2. Together theseresults suggest that an Fe(OOH) is formed en route to the S-oxygenation of the thiolate ligand.
The dithiolato complex FeII(Me3TACN)(S2SiMe2)194 (33) immediately reacts with O2 at 23 �C to give intractable products.171
However, an Fe/O2 intermediate can be stabilized by significantly lowering the reaction temperature to −135 �C in 2-MeTHF.Intense UV-vis features at 300, 390, 530, and 723 nm appear, and EPR spectroscopy showed that this green species is EPR silent.Mössbauer spectroscopy revealed a single, sharp quadrupole doublet with d ¼ 0.53 mm s–1 and │DEQ│ ¼ 0.77 mm s–1, andresonance Raman spectroscopy showed a n(OdO) at 849 cm-1 (D18O ¼ –49 cm–1). These data pointed to the assignment of thislow temperature O2 adduct as a peroxodiiron species, Fe2(O2)(Me3TACN)2(S2SiMe2)2. Characterization by XAS provided EXAFSdata that corroborated this structure, giving an FedFe distance between 4.4–4.7 Å. This species is highly photosensitive as well as

Scheme 22
Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 33
thermally sensitive, and exposure to light or warming of 34 above –120 �C leads to the isosbestic decay of 34 and formation of asecond intermediate with UV-vis bands at 385, 460 and 890 (e�390) nm. The relatively weak absorbance at 890 nm is characteristicof an FeIV(O) (S ¼ 1) species, suggesting that 34 undergoes OdO bond homolysis to produce an FeIV(O) complex, FeIV(O)(Me3TACN)(S2SiMe2) (35). Mössbauer spectroscopy of 35 revealed a quadrupole doublet with d¼0.21 mm s–1 and│DEQ│¼ 1.54mm s–1, and EPR spectroscopy indicated that 35 is EPR-silent, both consistent with a ferryl species. Analysis by XAS confirmed thepresence of a short FedO distance of 1.687(6) Å, consistent with the ferryl assignment. Resonance Raman spectroscopy on 35revealed a band at 735 cm-1 assigned to the n(Fe¼O) (D18O¼ –32 cm–1). Such a low n(Fe¼O) is unprecedented among previouslyvibrationally characterized FeIV(O) species, which typically exhibit n(Fe]O) between 800–900 cm-1.195 The highly activated Fe]Obond likely arises from the strong donation from the dithiolato donors, which is expected to weaken the Fe]O bond. Unlike otherFeIV(O) complexes,196 35 is unreactive towards OAT reactions with either the internal thiolate donors, or exogenous O-atomacceptors (e.g., phosphines, sulfides). However, 35 reacts readily with H-atom donors containing OdH bonds, including 2,2,6,6-tetramethylpiperidin-1-ol (TEMPOH) derivatives and phenols, to produce the expected FeIII(OH)(Me3TACN)(S2SiMe2) (36). BothEPR and Mössbauer spectroscopy (d¼ 0.50 mm s–1, │DEQ│¼ 1.09 mm s–1) corroboratea 36 as an FeIII(OH) complex, and the XASdata were consistent with the expected geometry, showing an elongation of the FedO bond to 1.907(12) Å which is expectedfollowing HAT to 35. In addition, 36 could be independently synthesized by one-electron outer-sphere oxidation and addition ofOH- to 33. Complex 33 is a rare example of an iron(II) complex that activates O2 to produce a series of detectable Fe/oxygenintermediates (Scheme 22). This work also shows that each of these intermediates can be formed in the presence of cis-ligatedthiolate donors.171
6.3 Reduced O2 Surrogates
A number of FeIV(O) complexes have been prepared from O-atom transfer reagents (e.g., ArIO) in the absence of sulfur ligands.197
The ligand in Scheme 23 incorporates a thioether moiety and a potential amide, H-bond donor, and was employed to generate arare sulfur-ligated FeIV(O) species. Addition of iodosylbenzene (PhIO) to the low-spin ferrous complex [FeII(N3PyamideSR)](BF4)2(37) at −40 �C resulted in the formation of an FeIV(O) complex, [FeIV(O)(N3PyamideSR)](BF4)2 (38). Mössbauer spectroscopyrevealed a quadrupole doublet with d ¼ 0.04 mm s–1 and │DEQ│ ¼ 0.80 mm s–1, and the low isomer shift observed is typical ofFeIV(O) complexes.197 Complex 38 is stable at −40 �C, and no sulfur oxidation is seen. However, addition of the exogenous sulfursubstrate, thioanisole (PhSMe) results in the rapid reformation of the FeII complex 37 (82% yield) and PhS(O)Me (80% yield). The
Scheme 23

34 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
same reaction could be performed with the FeII complex as catalyst, leading to 40 turnovers at −40 �C. In contrast, addition of thestronger oxidant meta-chloroperoxybenzoic acid (mCPBA), results in direct attack of the thioether ligand to give the sulfoxidecomplex [FeII(N3PyamideS(O)R)]2+(BF4)2 (39). Taken together these observations indicate that there is a barrier to direct intramo-lecular O-atom transfer from the ferryl to the coordinated sulfur ligand, but not to exogenous thioether substrates. The origins ofthis inter- versus intramolecular discrimination were not determined.
Reaction of mCPBA and [FeII(TMCS)](PF6) (40) in the presence of a base, KOtBu, at −40 �C produces the thiolate-ligatedFeIV(O) complex, [FeIV(O)(TMCS)](PF6) (41). This species gave near-infrared bands at 850 nm and 1050 nm, which are charac-teristic of nonheme FeIV(O) species. Mössbauer spectroscopy revealed parameters of d ¼ 0.19 mm s–1 and |DEQ| ¼ 0.22 mm s–1,and variable field Mössbauer studies confirmed the expected integer-spin nature of 41. X-ray absorption spectroscopy and DFTstudies suggested that 41 had the expected Fe(O)(N4S) coordination sphere, with the sulfur ligated trans to the oxo ligand.Interestingly, in contrast to the non-thiolate ligated FeIV(O) complex, [FeIV(O)(TMC)(CH3CN)]2+, which reacts with the O-atomacceptor PPh3 to generate OPPh3 and the two electron reduced ferrous complex, 41 is inert towards PPh3. However, 41 is a reactiveH-atom abstractor, reacting with 9,10-dihydroanthracene (DHA) to produce a new species distinct from the FeII starting material byUV-vis spectroscopy (lmax ¼ 514 nm). The authors hypothesized that the expected one electron reduced FeIII(OH) product wasformed on the basis of mass spectrometry analysis, which showed a peak consistent with [FeIII(TMCS)(OMe)]+. In contrast, [FeIV(O)(TMC)(CH3CN)]+, which does not contain a thiolate ligand, reacts extremely slowly with DHA (Fig. 25).
When [FeII(TMCS)](PF6) is reacted with mCPBA in the absence of a base, a new species is formed. The new species 42 is anO-bound FeII(sulfinate) complex, [FeII(TMCSO2)](PF6) based on UV-vis, Mössbauer, and X-ray absorption spectroscopies andmassspectrometry (Fig. 26).198 Further mCPBA treatment results in the formation of an FeIV(O) complex, [FeIV(O)(TMCSO2)](PF6) (43).
Fig. 25 Comparison of reactivity between [FeIV(O)(TMCS)]+ and [FeIV(O)(TMC)(CH3CN)]2+.
Fig. 26 Reactivity of [FeII(TMCS)](PF6) with mCPBA in the absence of base and Mössbauer spectra following addition of (A) 0 equiv, (B) 1 equiv, (C) 2 equiv, (D) 3equiv of mCPBA to [FeII(TMCS)](PF6). Reprinted with permission from reference McDonald, A. R.; Bukowski, M. R.; Farquhar, E. R.; Jackson, T. A.; Koehntop, K. D.;Seo, M. S.; De Hont, R. F.; Stubna, A.; Halfen, J. A.; Münck, E.; Nam, W.; Que, L., Jr. J. Am. Chem. Soc. 2010, 132, 17118–17129. Copyright 2010 AmericanChemical Society.

Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 35
Complex 43 exhibits a low intensity band at 830 nm, characteristic of nonheme FeIV(O) (S ¼ 1) complexes, and was characterizedby resonance Raman spectroscopy, exhibiting n(FedO) ¼ 831 cm–1, and Mössbauer spectroscopy exhibiting d ¼ 0.19 mm s–1 and|DEQ|¼ 1.28mm s–1. EXAFS analysis revealed a short Fe–Odistance of 1.64 Å, expected for an FeIV(O) complex. Unlike the thiolateligated 41, complex 43 was able to transfer an O atom to PPh3. Complex 43 is also able to abstract an H-atom from anthracene at arate comparable to that of 41. The differences in reactivity for 41 and 43 are not obvious. The formation of 41 versus 43 based on thepresence of base suggests that Fe versus S oxygenation is dependent on the presence of a proton. Addition of acid to 41 results information of a new FeIV species in 38% yield, in which it was proposed that the thiolate is protonated and no longer bound to the Fecenter. It was further proposed that intermolecular O-atom transfer to the thiol results in the formation of 42.
Another example of sulfur-ligated iron reacting with strong O-atom donors comes from the reaction of the dithiolate-ligatedcomplex [FeIII(S2
Me2N3(Pr,Pr))](PF6) (30), giving the FeIII(sulfenate) complex [FeIII(Z2-SMe2O)(SMe2N3(Pr,Pr))](PF6) (32) as shown
in Fig. 27.199 In contrast, reaction of PhIO with the azide-bound, 6-coordinate complex [FeIII(S2Me2N3(Pr,Pr))N3] resulted in no
reaction, suggesting that S-oxygenation occurred via a metal mediated process rather than direct sulfur attack. Running the reactionof PhIO with 30 at –73 �C resulted in the formation of a new intermediate (lmax ¼ 677 nm), which was hypothesized to be eitherFeV(O) or an iron(III)-iodosylarene adduct. The reactivity of 30 was compared with that of the more electron-rich analoguecontaining carboxamido donors, [Et4N][FeIIIS2
Me2NMeN2amide(Pr,Pr)] (44) (Fig. 27). In contrast to 30, 44 does not readily bind a
sixth ligand, and thus undergoes no reaction with O-atom donors. However, DFT calculations comparing 30 and 44 indicate thatsignificantly more electron density is located on the S donors in 44, indicating that they should be more easily oxidized. Theseresults suggest that oxidation of a sulfur donor, as seen for 30, occurs via the formation of an Fe-based oxidant.
Addition of KO2 to a solution of [FeII(SMe2N4(tren))]+ (20) in the presence of trace water at ambient temperature results in the
formation of [FeIIISMe2N4(tren)(MeCN)]2+, which was characterized crystallographically. The formation of this ferric complexsuggested that 20 is able to reduce superoxide.200 When the same reaction is carried out in MeOH at −90 �C, a transient intermediate45 is formed, but decays to [FeIIISMe2N4(tren)(OMe)]+ (46) within 10 min.201 Complex 45 (lmax ¼ 452 nm) gives an EPR spectrumwith g ¼ [2.14, 1.97], indicative of a low-spin ferric center. Resonance Raman spectroscopy showed an isotopically sensitive Fermidoublet (788, 781 cm–1) that collapsed to a single peak at 784 cm–1 with MeOD, and substitution of K18O2 resulted in a down-shiftto 753 cm–1. These vibrational data are consistent with a peroxo ligand, and 45was assigned as low-spin [FeIIISMe2N4(tren)(OOH)]+
(Fig. 28). Further evidence for this assignment came from XAS studies, which indicated that 45 was a 6-coordinate Fe(N4SO)complex, consistent with an Fe–O(peroxo) bond length of 1.86(3) Å. Decay of 45 proceeds to give an FeIII(OMe) complex.202 Nofurther substrate reactivity for 45 was reported.
The ferrous complex [FeII(cyclam-PrS)]+ (47) with an open coordination site trans to an alkylthiolate donor, reacts with KO2 inthe presence of a proton donor (MeOH) to produce a metastable intermediate [FeIII(cyclam-PrS)(OOH)]+ (48) (lmax ¼ 530 nm)(Scheme 24).175 EPR analysis gave g ¼ [7.72, 5.40, 4.15], indicative of a high-spin ferric center. Resonance Raman spectroscopy on48 reveals a n(OdO) band at 891 cm–1 and n(FedO) at 419 cm–1, which downshift to 856 and 400 cm–1, respectively, with K18O2.
Fig. 27 (A) Formation of FeIII(sulfenate) from [FeIII(S2Me2N3(Pr,Pr))]PF6. (B) Reaction of [Et4N][Fe
IIIS2Me2NMeN2
amide(Pr,Pr)] with PhIO. (C) Crystal structure of 32.Reprinted with permission from reference Villar-Acevedo, G.; Lugo-Mas, P.; Blakely, M. N.; Rees, J. A.; Ganas, A. S.; Hanada, E. M.; Kaminsky, W.; Kovacs, J. A.J. Am. Chem. Soc. 2017, 139, 119–129. Copyright 2017 American Chemical Society.

N
N
N
NH2 NH2
H2
FeIIN
N
NH2
FeIII
S S
NH2N
N
NH2
FeIII
S
++ +
KO2
OOH
OMe
MeOH−90 °C
(A)(B)
Decay
0800 790 780 770
cm−1
% T
rans
.
760 750
753
740 730
20
40
60
80
100
20 45 46
Fig. 28 (A) Reactivity of [FeII(SMe2N4(tren))]+ with KO2 in MeOH. (B) IR spectrum of 45 prepared from K16O2 (dashed line) and K
18O2 (solid line). Reprinted withpermission from reference Shearer, J.; Scarrow, R. C.; Kovacs, J. A. J. Am. Chem. Soc. 2002, 124, 11709–11717. Copyright 2002 American Chemical Society.
Scheme 24
36 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
Addition of HOAc results in the release of H2O2 and formation of [FeIII(cyclam-PrS)(OAc)]+, which could be reduced to 47 usingCoII(Cp)2, indicating that 47 should perform catalytic superoxide reduction.
Addition of excess (>50 equiv) KO2 to a solution of [FeII(TMCS)](PF6) at −90 �C results in the formation of a new species 49,192
which decayed over time to re-form [FeII(TMCS)](PF6), suggesting a possible equilibrium of superoxide binding. EPR andMössbauer spectroscopy were consistent with the formation of an S ¼ 5/2 ferric species, giving g ¼ 7.9, 5.75, and d ¼ 0.71 mms–1, DEQ ¼ −1.9 mm s–1, respectively. XAS analysis was consistent with an Fe(N4SO) coordination environment, leading to theassignment of 49 as an end-on peroxide anion complex (Scheme 25), [FeIII(O2
- )(TMCS)]. Addition of excess acid resulted in theformation of a new species tentatively assigned as the hydroperoxo complex, [FeIII(OOH)(TMCS)]+. However, no further charac-terization was carried out. Complex 49 was shown to be unreactive towards CdH bonds, but reacted with electrophiles such asmenadione to produce the corresponding epoxide, and 2-phenylpropionaldehyde to produce acetophenone and formate.
Scheme 25
The tetraazamacrocycle complexes with tethered alkyl thiolate donors have been shown to stabilize Fe/oxygen intermediates.To access a series of FeII(thiolate) complexes in which the thiolate donor strength can be rationally tuned as a function of itselectronic properties, a series of ferrous complexes coordinated to the tetraazamacrocycle, [15]aneN4, were prepared with differentaxial thiolate donors (Fig. 29).203 The reaction of [FeII([15]aneN4)(SPh)]BF4 (50a) with tBuOOH or cumenyl hydroperoxide(CmOOH) in CH2Cl2 at −78 �C resulted in the formation of the low-spin alkylperoxo complexes [FeIII([15]aneN4)(SPh)(OOR)]BF4 (51a, R¼ tBu, 51f: R¼ Cm).177 These complexes exhibited UV-vis spectral features at lmax¼ 526 nm (2150M−1 cm−1) and lmax
¼ 527 nm (1650 M−1 cm−1) for 51a and 51f, respectively, which were assigned to alkylperoxo-to-Fe(III) ligand to metal chargetransfer bands. These complexes were further characterized by EPR spectroscopy, giving g ¼ 2.20 and 1.97, and resonance Ramanspectroscopy, which showed n(OdO) features � 800 cm−1 (Dn(tBu16O2H–
tBu18O2H) ¼ −46 cm−1 for 51a) and n(FedO) � 615cm−1 (Dn(tBu16O2H–
tBu18O2H) ¼ –28 cm−1 for 51a). The Fe(OOR) complexes for each of the thiolate donors shown in Fig. 29exhibited UV-vis features between 500 and 550 nm, n(OdO) features �800 cm−1, and n(FedO) between 600 and 635 cm−1.203
Further characterization by XAS in conjunction with density functional theory calculations were in accordance with the assignmentsof these species as low-spin alkyl peroxo complexes.204 Interestingly, the n(FedO) increases as the electron donating ability

Fig. 29 (A) Formation of [FeIII([15]aneN4)(SPh)(OOR)]BF4 complexes. (B) Plot of n(OdO) and n(FedO) versus redox potential for a series of FeII(thiolate)complexes.
Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 37
decreases, suggesting a weakening of the FedO bond with increasing electron density from the axial donor, with a nearly lineartrend observed in the plot of n(FedO) versus the redox potential of the starting ferrous complex (Fig. 29). However, the n(OdO)remains relatively invariant across the series of complexes. These results indicate that the trans thiolate donor can tune the FedObond strength without significantly altering the OdO bond strength. The rates of decay of these complexes were measured andrevealed that the rates increased as the FedO bond strength decreased. These observations are consistent with FedO bond cleavagebeing involved in the decay process. However, attempts to identify the decomposition products were unsuccessful.204 The decay ofthe series was accelerated in the presence of acids or aldehydes, suggesting these complexes may exhibit nucleophilic character.However, no products were identified. No reactivity with weak O-atom acceptors or H-atom donors was observed.
The structurally related FeII complexes 37 and 52, with and without a potential H-bonding amide group, react with H2O2 ortBuOOH to generate a series of FeIII(OOR) (S¼½) complexes (53–54) (R¼ tBu, H), characterized by resonance Raman, UV-vis, andEPR spectroscopies (Fig. 30).178 The presence of the H-bondingmoiety shifted the UV-vis feature associated with the alkylperoxo-to-Fe(III) ligand to metal charge transfer bands, but only minor changes in the n(OdO) and n(FedO) vibrational modes wereobserved, indicating that H-bonding to the peroxo unit does not significantly perturb the Fe/O2 bond strengths. Interestingly, asignificant decrease in n(OdO) and increase in n(FedO) were observed when compared with the all-nitrogen-ligated [FeIII(N4Py)(OOH)]2+, suggesting that the cis-sulfur ligation weakens the OdO bond while strengthening the FedO bond.205
6.4 Models of Thiolate-Ligated Nonheme Iron Enzymes with Other 3d Metals
Though the structure and reactivity of Fe–thiolate complexes provide the most direct comparison with enzymatic reactivity,analogous studies using other metals are also germane to our understanding of how O2 can be activated at a metal center in the
Fig. 30 (A) Reactions of [FeII(N3PySR)]2+ complexes with ROOH (R]H, tBu). (B) Resonance Raman spectra of FeIII(OOtBu) complexes. Reproduced from referenceWidger, L. R.; Jiang, Y.; McQuilken, A. C.; Yang, T.; Siegler, M. A.; Matsumura, H.; Moënne-Loccoz, P.; Kumar, D.; de Visser, S. P.; Goldberg, D. P. Dalton Trans.2014, 43, 7522–7532, with permission from The Royal Society of Chemistry.

Scheme 26
38 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
presence of thiolate donors. Studies using Co as a surrogate for Fe, in particular, have a long history in the field of bioinorganicchemistry in helping to understand fundamental aspects of O2 binding.
206
The crystallographically characterized Cr(superoxo) species, [CrIII(O2)(TMC)(Cl)]+ (55-O2) was shown to undergo a singleO-atom transfer to thioanisole to generate [CrIV(O)(TMC)(Cl)]+ (55-O) (Scheme 26). This report provided evidence that a metal-superoxo species could directly oxidize a sulfur substrate, as proposed for CDO.
A series of MnII–thiolate complexes react with O2 to generate unsupported MnIII oxo-bridged dimers, which were characterizedcrystallographically.207 Reaction of [MnII(SMe2N4(6-Me-DPEN))](BPh4) (56) with O2 at low temperature (−80 �C) produced abinuclear MnIII2 -trans-1,2-peroxo complex (57) that was characterized structurally by XRD (Fig. 31), and, upon warmup, led toformation of the oxo-bridged dimer.208 This complex gave n(OdO) ¼ 819 cm–1 (D18O ¼ −47 cm–1) and n(MndO) ¼ 611 cm–1
(D18O¼ −25 cm–1). Magnetic susceptibility measurements indicated that the complex is uncoupled (J¼ 0 cm-1). Prior to formationof the peroxo species, a transient intermediate with lmax ¼ 515 nm was observed by stopped-flow UV-vis spectroscopy. Thisintermediate was proposed to be aMn(superoxo) species, which rapidly converted to the Mn2
III(m-O2) complex. It was proposed thatthe formation of the Mn2
III(mdO) complex occurred via homolytic OdO bond cleavage of 57 to generate a MnIV(O) species, whichthen goes on to give the final Mn2
III(mdO) complex. This hypothesis is supported from the observation that PPh3 and 2,4-di-tert-butylphenol are both oxidized in the reaction of 56 with O2. However, there was no direct evidence for the formation of any high-valent MnIV(O) species. A recent study using a more electron-rich and less sterically bulky ligand derivative found that several otherintermediates, assigned as MnIII(O2
•−), MnIV2 (O)2, and MnIIIMnIV(O)(OH) species, could be observed by stopped-flow UV-visspectroscopy. A detailed kinetic analysis of a series of related MnII complexes revealed that metal Lewis acidity as well as ligandsteric properties play key roles in the binding of dioxygen and subsequent OdO bond cleavage steps.209
Reaction of the tris(thiolato)-ligated MnI complex [PPN][Mn(CO)3(P(C6H3-3-R-2-S)2(C6H3-3-R-2-SH))] (R]SiMe3) (58) withexcess O2 leads to a mononuclear S ¼ 3/2 side-on Mn(O2) adduct, [PPN][MnIV(TMSPS3)(O2)] (59), that was structurallycharacterized by XRD, which indicated an OdO bond length of 1.379(3) Å. Vibrational analysis by infrared spectroscopy gaven(OdO) ¼ 903 cm–1 (D18O ¼ –42 cm–1), which is in the range typically seen for peroxo species, leading the authors to formulate59 as a MnIV(peroxo) species.210 Protonation of this complex is proposed to lead to the formation of a putative MnIV(OOH)complex that can rapidly release hydroperoxyl radical, which could be scavenged by PPh3. However, no direct evidence of theMnIV(OOH) complex was obtained.211 Addition of 59 to the MnII complex [PPN][MnII(TMSPS3)(DABCO)] (60) (DABCO ¼ 1,4-diazabicyclo[2.2.2]octane) results in the formation of a MnIII2 (mdO) complex (61) (Scheme 27), which was proposed to form via anintermediate MnIII2 (O2) dimer, though this intermediate could not be detected.212
The MnII dimer, [MnII2(LS)(LSH)]+ (62) (where LS2−¼2,20-(2,20-bipyridine-6,60-diyl)bis(1,10-diphenylethanethiolate)) containsa bound thiol moiety and reacts with air to perform the four-electron reduction of O2, producing a dimeric MnIII2 (m–OH) complex(63) (Scheme 28). It was proposed that this intermediate is formed via an undetected dimeric peroxo intermediate prior to eventualformation of 63. In contrast, addition of O2 to 62 in the presence of an acid source leads to two-electron reduction of dioxygen and
Fig. 31 Crystal structure of complex 57. Reprinted with permission from reference Coggins, M. K.; Sun, X.; Kwak, Y.; Solomon, E. I.; Rybak-Akimova, E.;Kovacs, J. A. J. Am. Chem. Soc. 2013, 135, 5631–5640. Copyright 2013 American Chemical Society.

Scheme 28
Scheme 27
Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 39
release of H2O2. In the presence of an exogenous reductant, 62 could perform the reduction of O2 to H2O2 catalytically.213
Deprotonation of 62 leads to the formation of [MnII2(LS)2] (64), which reacts with O2 to produce the high-valent MnIV2 (O)2 dimer(65). Protonation of this species gives MnIV2 (O)(OH) (66). It was found that addition of 62–66 leads to a comproportionationreaction to generate 63, implicating 66 as an intermediate in the four-electron reduction of O2 by 62.214
The CoII complex [CoII(SMe2N4(tren))](PF6) (67) was shown to bind O2, generating a diamagnetic, binuclear CoIII2 -trans-1,2-peroxo complex (68) that was characterized crystallographically (Scheme 29). Complex 68 exhibited a CodO and OdO bondlengths of 1.899(2) Å and 1.482(4) Å, respectively. The OdO bond is slightly longer than a Mn2(peroxo) species, 57, with a similarligand set. DFT calculations suggested that the long OdObondmay arise from the lack of donation from the p�(OdO) orbital intothe filled cobalt d orbitals.
The Tp-ligated CoII complexes, [Co(TpR2)(CysOEt)] (R]Ph (69), Me (70)), were prepared and their reactivity with O2 wasexamined (Scheme 30). Addition of O2 to 69 resulted in no reaction, possibly due to the steric profile of the ligand, or high CoIII/CoII redox potentials. Addition of O2 to 70 at −70 �C leads to the formation of [Co(O2)(Tp
Me2)(CysOEt)] (71), which was identifiedas a CoIII(O2
•−) complex by its characteristic EPR signature with small 59Co hyperfine coupling, and a n(OdO) mode at 1152 cm−1
(D18O¼ 61 cm−1) from resonance Raman spectroscopy. In contrast to the analogous Fe complex, no S-oxygenation is observed andinstead, release of O2 and reformation of the CoII starting complex is seen upon warmup.
The CoII(silanedithiolate) (S ¼ 3/2) complex CoII(Me3TACN)(S2SiMe2) (72), which is the Co analogue of the Fe complex, 33,was prepared in order to stabilize a Co(superoxo) species bound to thiolate donors.215 Addition of excess O2(g), or one equivalentof O2 in an O2-saturated 2-MeTHF solution to 72 at −80 �C resulted in the formation of 73, which exhibits UV-vis spectral features at

Scheme 29
Scheme 30
40 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
340, 360, 455, and 555 nm (Fig. 32). Complex 73 is stable at −80 �C, and does not revert back to 72 upon sparging with argon gas orunder reduced pressure. Characterization by EPR spectroscopy gave g � 2.11, 2.02, 1.98 and A(59Co) ¼ 30 G, typical of an S ¼ 1/2cobalt superoxo complex. The assignment of 73 was confirmed by resonance Raman spectroscopy, with a peak at 1133 cm−1 thatdownshifts to 1070 cm−1 upon 18O2 substitution, which was in line with the O–O stretch for a superoxide complex. Additional O2-sensitive features at 520 cm-1 (Dn(18O2–
16O2) ¼ –29 cm−1) and 367 cm–1 (Dn(18O2–16O2) ¼ –9 cm–1) were assigned to CodO
stretching and Co–O2 bending modes, respectively. Analysis by EXAFS was consistent with the proposed Co(O2)(Me3TACN)(S2SiMe2) structure, and DFT calculations were in accordance with the experimental data. Interestingly, analysis of the Co K-edgeXAS spectrum revealed that the pre-edge and rising edge energies for 72 and 73 are nearly identical. These observations suggestedthat upon binding of O2, redox chemistry is not occurring exclusively at the Co center. DFT calculations on 73 revealed significanthole character on the thiolate donors, suggesting that the entire [CoS2] unit is acting as the redox active moiety and providing theelectron for reducing O2 to superoxide.
The reactivity of 73 was tested with a variety of O-atom acceptors, including phosphines, sulfides, and alkylthiolates, exhibitingno reactivity with these compounds. In addition, 73 was stable against intramolecular attack of the bound thiolate donors,indicating that the superoxide moiety in 73 is relatively inert toward sulfur oxidation. In contrast, reaction of 73 with theH-atom donor 4-methoxy-2,2,6,6-tetramethylpiperidin-1-ol (4-OMe-TEMPOH) or the un-substituted analogue TEMPOH, resultedin rapid decay of the features associated with 73 and formation of the corresponding nitroxyl radicals (Fig. 32). A KIE of 8.8 for thereaction with TEMPOH(D) was obtained at −105 �C. An Eyring analysis gave activation parameters DH{¼ 3.6 kcal mol–1 and DS{¼−46.2 cal mol–1 K–1, indicating a highly ordered transition state. The bond dissociation free energy (BDFE) of the putative formedCo(OOdSH) bond was estimated from the reactivity of 73 with separated acid/reductant pairs of various strengths (e.g., Et3NH/Fe(Cp�)2 effective BDFE ¼ 70 kcal mol–1). The reactivity pattern gave an upper limit BDFE of 70 kcal mol–1 for the hydroperoxoOdH bond. This estimate was matched nicely with a DFT calculated OdH bond strength of 67 kcal mol–1. These results indicatethat 73 is only a weak H-atom abstractor, but lends support to the feasibility of a metal(superoxo) complex preferentially oxidizingan OdH bond over a sulfur ligand, as proposed in the mechanism of EgtB/OvoA.
7 Outlook and Future Work
The burgeoning class of O2 activating nonheme iron enzymes containing thiolate ligation has emerged as an important andmechanistically complex group of enzymes. These enzymes, IPNS, the thiol dioxygenases, the sulfoxide synthases, and thepersulfide dioxygenases, are all functionally unique yet share structural and mechanistic similarities. However, the chemistry carriedout by these enzymes following the binding of O2 diverges markedly, involving either H-atom abstraction, S-oxygenation, orpossibly both, via poorly understood mechanisms. Elucidating these different mechanisms and trapping relevant intermediatesremains a challenging and important goal for future mechanistic studies.

Fig. 32 (A) Formation and reactivity of Co(O2)(Me3TACN)(S2SiMe2). (B) UV-vis, EPR, and (C) X-ray absorption (XANES) spectra for Co(O2)(Me3TACN)(S2SiMe2). Blacklines in B and C corresponds to data for 72 and red lines correspond to data for 73. Adapted with permission from reference Gordon, J. B.; Vilbert, A. C.;Siegler, M. A.; Lancaster, K. M.; Moënne-Loccoz, P.; Goldberg, D. P. J. Am. Chem. Soc. 2019, 141, 3641–3653. Copyright 2019 American Chemical Society.
Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 41
A key question surrounding these mechanisms is why and how their reactivities diverge. Though themechanism for IPNS is fairlywell established, it is unclear how the iron center in IPNS preferentially abstracts the Cys b H-atoms over S-oxygenation of thebound sulfur ligand. Conversely, it is unclear why the iron/oxygen pathway in CDO does not lead to attack of the Cys b H-atom ofthe chelated Cys substrate. Perhaps one of the most interesting mechanistic observations for these enzymes is that their reactivitiescan be interconverted. For example, addition of an ACV substrate analogue (ACOmV) to IPNS leads to S-oxygenation, generating asulfenate species, which is a proposed intermediate in CDO.30 The sulfoxide synthases EgtB and OvoA can be converted into thioldioxygenases with appropriate mutations or substrates.216 The persulfide dioxygenase ETHE1 was shown to undergoes singleturnover S-dioxygenation reactivity analogous to the thiol dioxygenases.154 These observations suggest that these enzymes are allmechanistically related, and studies of each of these enzymes can lend important information for the entire class.
As structural data are obtained for the enzymes with and without substrates bound, our understanding of sulfur-ligatednonheme iron enzymes will be refined, and more mechanistic studies guided by structure can be carried out. Though several ofthese sulfur-ligated enzymes have been known for decades, the recent discovery of the sulfoxide synthases121,122,124 as well as newthiol dioxygenases such as MSDO43 and plant cysteine oxidase,45 suggests that there are many fascinating new related enzymes to bediscovered.
Many of these intriguing questions can be addressed by examination of synthetic model complexes. The study of modelcomplexes provides fundamental spectroscopic signatures of Fe/oxygen intermediates in the presence of sulfur donors, which canbe essential in the assignment of intermediates observed in enzymatic studies. It is apparent from studies with well-defined syntheticmetal–thiolate complexes that the key factors controlling the preference for S-oxygenation or H-atom abstraction are not yetunderstood. However, in the last two decades, we have seen interesting examples of both reactivity patterns. The ability to exertprecise control over the coordination sphere of synthetic Fe–thiolate complexes provides significant promise for understanding theguiding principles that determine these two pathways. Ultimately, the insights developed from studies of these enzymes andsynthetic model complexes can be directed towards the rational design of new synthetic catalysts that can bind and activate O2 forthe controlled, selective oxidation of organic substrates.

42 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
References
1. Borden, W. T.; Hoffmann, R.; Stuyver, T.; Chen, B. J. Am. Chem. Soc. 2017, 139, 9010–9018.2. Feig, A. L.; Lippard, S. J. Chem. Rev. 1994, 94, 759–805.3. Peck, S. C.; van der Donk, W. A. J. Biol. Inorg. Chem. 2017, 22, 381–394.4. Cohen, G.; Shiffman, D.; Mevarech, M.; Aharonowitz, Y. Trends Biotechnol. 1990, 8, 105–111.5. Tipper, D. J.; Strominger, J. L. Proc. Natl. Acad. Sci. U. S. A. 1965, 54, 1133–1141.6. Martín, J. F.; Gutiérrez, S.; Fernández, F. J.; Velasco, J.; Fierro, F.; Marcos, A. T.; Kosalkova, K. Antonie Van Leeuwenhoek 1994, 65, 227–243.7. Borovok, I.; Landman, O.; Kreisberg-Zakarin, R.; Aharonowitz, Y.; Cohen, G. Biochemistry 1981-1987, 1996, 35.8. Tiow-Suan, S.; Tan, D. S. H. FEMS Microbiol. Lett. 1994, 120, 241–247.9. Kreisberg-Zakarin, R.; Borovok, I.; Yanko, M.; Frolow, F.; Aharonowitz, Y.; Cohen, G. Biophys. Chem. 2000, 86, 109–118.
10. Ming, L. J.; Que, L., Jr.; Kriauciunas, A.; Frolik, C. A.; Chen, V. J. Biochemistry 1991, 30, 11653–11659.11. Ming, L. J.; Que, L., Jr.; Kriauciunas, A.; Frolik, C. A.; Chen, V. J. Inorg. Chem. 1990, 29, 1111–1112.12. Randall, C. R.; Zang, Y.; True, A. E.; Que, L., Jr.; Charnock, J. M.; Garner, C. D.; Fujishima, Y.; Schofield, C. J.; Baldwin, J. E. Biochemistry 1993, 32, 6664–6673.13. Scott, R. A.; Wang, S.; Eidsness, M. K.; Kriauciunas, A.; Frolik, C. A.; Chen, V. J. Biochemistry 1992, 31, 4596–4601.14. Chen, V. J.; Orville, A. M.; Harpel, M. R.; Frolik, C. A.; Surerus, K. K.; Münck, E.; Lipscomb, J. D. J. Biol. Chem. 1989, 264, 21677–21681.15. Orville, A. M.; Chen, V. J.; Kriauciunas, A.; Harpel, M. R.; Fox, B. G.; Munck, E.; Lipscomb, J. D. Biochemistry 1992, 31, 4602–4612.16. Roach, P. L.; Clifton, I. J.; Fülöp, V.; Harlos, K.; Barton, G. J.; Hajdu, J.; Andersson, I.; Schofield, C. J.; Baldwin, J. E. Nature 1995, 375, 700–704.17. Roach, P. L.; Clifton, I. J.; Hensgens, C. M. H.; Shibata, N.; Schofield, C. J.; Hajdu, J.; Baldwin, J. E. Nature 1997, 387, 827–830.18. Kal, S.; Que, L., Jr. J. Biol. Inorg. Chem. 2017, 22, 339–365.19. Blackburn, J. M.; Sutherland, J. D.; Baldwin, J. E. Biochemistry 1995, 34, 7548–7562.20. Baldwin, J. E.; Bradley, M. Chem. Rev. 1990, 90, 1079–1088.21. Baldwin, J. E.; Abraham, E. Nat. Prod. Rep. 1988, 5, 129–145.22. Burzlaff, N. I.; Rutledge, P. J.; Clifton, I. J.; Hensgens, C. M. H.; Pickford, M.; Adlington, R. M.; Roach, P. L.; Baldwin, J. E. Nature 1999, 401, 721–724.23. Nam, W.; Lee, Y.-M.; Fukuzumi, S. Acc. Chem. Res. 2014, 47, 1146–1154.24. Baldwin, J. E.; Adlington, R. M.; Domayne-Hayman, B. P.; Knight, G.; Ting, H.-H. J. Chem. Soc. Chem. Commun. 1987, 1661–1663.25. Howard-Jones, A. R.; Elkins, J. M.; Clifton, I. J.; Roach, P. L.; Adlington, R. M.; Baldwin, J. E.; Rutledge, P. J. Biochemistry 2007, 46, 4755–4762.26. Long, A. J.; Clifton, I. J.; Roach, P. L.; Baldwin, J. E.; Schofield, C. J.; Rutledge, P. J. Biochem. J. 2003, 372, 687–693.27. Ogle, J. M.; Clifton, I. J.; Rutledge, P. J.; Elkins, J. M.; Burzlaff, N. I.; Adlington, R.; Roach, P. L.; Baldwin, J. E. Chem. Biol. 2001, 8, 1231–1237.28. Brown-Marshall, C. D.; Diebold, A. R.; Solomon, E. I. Biochemistry 2010, 49, 1176–1182.29. Daruzzaman, A.; Clifton, I. J.; Adlington, R. M.; Baldwin, J. E.; Rutledge, P. J. ChemBioChem 2006, 7, 351–358.30. Ge, W.; Clifton, I. J.; Stok, J. E.; Adlington, R. M.; Baldwin, J. E.; Rutledge, P. J. J. Am. Chem. Soc. 2008, 130, 10096–10102.31. Brown, C. D.; Neidig, M. L.; Neibergall, M. B.; Lipscomb, J. D.; Solomon, E. I. J. Am. Chem. Soc. 2007, 129, 7427–7438.32. Lundberg, M.; Siegbahn, P. E. M.; Morokuma, K. Biochemistry 2008, 47, 1031–1042.33. Wirstam, M.; Siegbahn, P. E. M. J. Am. Chem. Soc. 2000, 122, 8539–8547.34. Lundberg, M.; Morokuma, K. J. Phys. Chem. B. 2007, 111, 9380–9389.35. Tamanaha, E.; Zhang, B.; Guo, Y.; Chang, W.-c.; Barr, E. W.; Xing, G.; Clair, J., St.; Ye, S.; Neese, F.; Bollinger, J. M., Jr.; Krebs, C. J. Am. Chem. Soc. 2016, 138, 8862–8874.36. Krebs, C.; Galoni�c Fujimori, D.; Walsh, C. T.; Bollinger, J. M., Jr. Acc. Chem. Res. 2007, 40, 484–492.37. Mbughuni, M. M.; Chakrabarti, M.; Hayden, J. A.; Bominaar, E. L.; Hendrich, M. P.; Münck, E.; Lipscomb, J. D. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 16788–16793.38. Goudarzi, S.; Babicz, J. T.; Kabil, O.; Banerjee, R.; Solomon, E. I. J. Am. Chem. Soc. 2018, 140, 14887–14902.39. Long, A. J.; Clifton, I. J.; Roach, P. L.; Baldwin, J. E.; Rutledge, P. J.; Schofield, C. J. Biochemistry 2005, 44, 6619–6628.40. Yamaguchi, K.; Hosokawa, Y. Cysteine dioxygenase. In Methods in Enzymology, 143; Academic Press, 1987; pp 395–403.41. Cavallini, D.; Scandurra, R.; Duprè, S. [200] Cysteamine oxygenase (Horse Kidney). In Methods in Enzymology, 17; Academic Press, 1971; pp 479–483.42. Tchesnokov, E. P.; Fellner, M.; Siakkou, E.; Kleffmann, T.; Martin, L. W.; Aloi, S.; Lamont, I. L.; Wilbanks, S. M.; Jameson, G. N. L. J. Biol. Chem. 2015, 290, 24424–24437.43. Brandt, U.; Schürmann, M.; Steinbüchel, A. J. Biol. Chem. 2014, 289, 30800–30809.44. Lombardini, J. B.; Singer, T. P.; Boyer, P. D. J. Biol. Chem. 1969, 244, 1172–1175.45. Weits, D. A.; Giuntoli, B.; Kosmacz, M.; Parlanti, S.; Hubberten, H.-M.; Riegler, H.; Hoefgen, R.; Perata, P.; van Dongen, J. T.; Licausi, F. Nat. Commun. 2014, 5, 3425.46. White, M. D.; Klecker, M.; Hopkinson, R. J.; Weits, D. A.; Mueller, C.; Naumann, C.; O’Neill, R.; Wickens, J.; Yang, J.; Brooks-Bartlett, J. C.; Garman, E. F.; Grossmann, T. N.;
Dissmeyer, N.; Flashman, E. Nat. Commun. 2017, 8, 14690.47. Dominy, J. E.; Hirschberger, L. L.; Coloso, R. M.; Stipanuk, M. H. Biochem. J. 2006, 394, 267–273.48. Sarkar, B.; Kulharia, M.; Mantha, A. K. Int. J. Exp. Pathol. 2017, 98, 52–66.49. Stipanuk, M. H.; Ueki, I.; Dominy, J. E.; Simmons, C. R.; Hirschberger, L. L. Amino Acids 2009, 37, 55–63.50. Gordon, C.; Emery, P.; Bradley, H.; Waring, R. H. The Lancet 1992, 339, 25–26.51. Jameson, G. N. L. Monatshefte für Chemie - Chemical Monthly 2011, 142, 325–329.52. Heafield, M. T.; Fearn, S.; Steventon, G. B.; Waring, R. H.; Williams, A. C.; Sturman, S. G. Neurosci. Lett. 1990, 110, 216–220.53. Emery, P.; Bradley, H.; Arthur, V.; Tunn, E.; Waring, R. Rheumatology 1992, 31, 449–451.54. Perry, T. L.; Norman, M. G.; Yong, V. W.; Whiting, S.; Crichton, J. U.; Hansen, S.; Kish, S. J. Ann. Neurol. 1985, 18, 482–489.55. Brait, M.; Ling, S.; Nagpal, J. K.; Chang, X.; Park, H. L.; Lee, J.; Okamura, J.; Yamashita, K.; Sidransky, D.; Kim, M. S. PLoS One 2012, 7e44951.56. Jeschke, J.; Hagan, H. M.; Zhang, W.; Vatapalli, R.; Calmon, M. F.; Danilova, L.; Nelkenbrecher, C.; Van Neste, L.; Bijsmans, I. T. G. W.; Van Engeland, M.; Gabrielson, E.;
Schuebel, K. E.; Winterpacht, A.; Baylin, S. B.; Herman, J. G.; Ahuja, N. Clin. Cancer Res. 2013, 19, 3201.57. Ueno, K.; Kumagai, T.; Kijima, T.; Kishimoto, T.; Hosoe, S. Hum. Genet. 1998, 102, 63–68.58. Masson, N.; Keeley, T. P.; Giuntoli, B.; White, M. D.; Puerta, M. L.; Perata, P.; Hopkinson, R. J.; Flashman, E.; Licausi, F.; Ratcliffe, P. J. Science 2019, 365, 65–69.59. Lee, J.-I.; Londono, M.; Hirschberger, L. L.; Stipanuk, M. H. J. Nutr. Biochem. 2004, 15, 112–122.60. Stipanuk, M. H.; Hirschberger, L. L.; Londono, M. P.; Cresenzi, C. L.; Yu, A. F. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E439–E448.61. Yu, A. F.; Lee, J.-I.; Hu, M.; Londono, M.; Stipanuk, M. H. J. Nutr. 2002, 132, 3369–3378.62. Bagley, P. J.; Hirschberger, L. L.; Stipanuk, M. H. Anal. Biochem. 1995, 227, 40–48.63. Besouw, M.; Masereeuw, R.; van den Heuvel, L.; Levtchenko, E. Drug Discov. Today 2013, 18, 785–792.64. Fraser-Pitt, D. J.; Mercer, D. K.; Smith, D.; Kowalczuk, A.; Robertson, J.; Lovie, E.; Perenyi, P.; Cole, M.; Doumith, M.; Hill, R. L. R.; Hopkins, K. L.; Woodford, N.; O’Neil, D. A.
Infect. Immun. 2018, 86. e00947-17.65. Huxtable, R. J. Physiol. Rev. 1992, 72, 101–163.66. Dominy, J. E.; Simmons, C. R.; Hirschberger, L. L.; Hwang, J.; Coloso, R. M.; Stipanuk, M. H. J. Biol. Chem. 2007, 282, 25189–25198.67. Sörbo, B.; Ewetz, L. Biochem. Biophys. Res. Commun. 1965, 18, 359–363.

Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 43
68. Joseph, C. A.; Maroney, M. J. Chem. Commun. 2007, 3338–3349.69. Stipanuk, M. H.; Simmons, C. R.; Andrew Karplus, P.; Dominy, J. E. Amino Acids 2011, 41, 91–102.70. McCoy, J. G.; Bailey, L. J.; Bitto, E.; Bingman, C. A.; Aceti, D. J.; Fox, B. G.; Phillips, G. N. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 3084–3089.71. Chai, S. C.; Bruyere, J. R.; Maroney, M. J. J. Biol. Chem. 2006, 281, 15774–15779.72. Simmons, C. R.; Liu, Q.; Huang, Q.; Hao, Q.; Begley, T. P.; Karplus, P. A.; Stipanuk, M. H. J. Biol. Chem. 2006, 281, 18723–18733.73. Tchesnokov, E. P.; Wilbanks, S. M.; Jameson, G. N. L. Biochemistry 2012, 51, 257–264.74. Li, J.; Griffith, W. P.; Davis, I.; Shin, I.; Wang, J.; Li, F.; Wang, Y.; Wherritt, D. J.; Liu, A. Nat. Chem. Biol. 2018, 14, 853–860.75. Driggers, C. M.; Hartman, S. J.; Karplus, P. A. Protein Sci. 2015, 24, 154–161.76. Ye, S.; Wu, X. A. J. Biol. Chem. 2007, 282, 3391–3402.77. Driggers, C. M.; Cooley, R. B.; Sankaran, B.; Hirschberger, L. L.; Stipanuk, M. H.; Karplus, P. A. J. Mol. Biol. 2013, 425, 3121–3136.78. Fellner, M.; Siakkou, E.; Faponle, A. S.; Tchesnokov, E. P.; de Visser, S. P.; Wilbanks, S. M.; Jameson, G. N. L. J. Biol. Inorg. Chem. 2016, 21, 501–510.79. Blaesi, E. J.; Gardner, J. D.; Fox, B. G.; Brunold, T. C. Biochemistry 2013, 52, 6040–6051.80. Gardner, J. D.; Pierce, B. S.; Fox, B. G.; Brunold, T. C. Biochemistry 2010, 49, 6033–6041.81. Tchesnokov, E. P.; Faponle, A. S.; Davies, C. G.; Quesne, M. G.; Turner, R.; Fellner, M.; Souness, R. J.; Wilbanks, S. M.; de Visser, S. P.; Jameson, G. N. L. Chem. Commun.
2016, 52, 8814–8817.82. Gordon, J. B.; McGale, J. P.; Prendergast, J. R.; Shirani-Sarmazeh, Z.; Siegler, M. A.; Jameson, G. N. L.; Goldberg, D. P. J. Am. Chem. Soc. 2018, 140, 14807–14822.83. Ito, N.; Phillips, S. E. V.; Stevens, C.; Ogel, Z. B.; McPherson, M. J.; Keen, J. N.; Yadav, K. D. S.; Knowles, P. F. Nature 1991, 350, 87–90.84. Kleffmann, T.; Jongkees, S. A. K.; Fairweather, G.; Wilbanks, S. M.; Jameson, G. N. L. J. Biol. Inorg. Chem. 2009, 14, 913–921.85. Dominy, J. E.; Hwang, J.; Guo, S.; Hirschberger, L. L.; Zhang, S.; Stipanuk, M. H. J. Biol. Chem. 2008, 283, 12188–12201.86. Li, J.; Koto, T.; Davis, I.; Liu, A. Biochemistry 2019, 58, 2218–2227.87. Dominy, J. E.; Simmons, C. R.; Karplus, P. A.; Gehring, A. M.; Stipanuk, M. H. J. Bacteriol. 2006, 188, 5561.88. Davies, C. G.; Fellner, M.; Tchesnokov, E. P.; Wilbanks, S. M.; Jameson, G. N. L. Biochemistry 2014, 53, 7961–7968.89. Driggers, C. M.; Kean, K. M.; Hirschberger, L. L.; Cooley, R. B.; Stipanuk, M. H.; Karplus, P. A. J. Mol. Biol. 2016, 428, 3999–4012.90. Arjune, S.; Schwarz, G.; Belaidi, A. A. Amino Acids 2015, 47, 55–63.91. Blaesi, E. J.; Fox, B. G.; Brunold, T. C. Biochemistry 2015, 54, 2874–2884.92. Li, W.; Blaesi, E. J.; Pecore, M. D.; Crowell, J. K.; Pierce, B. S. Biochemistry 2013, 52, 9104–9119.93. Morrow, W. P.; Sardar, S.; Thapa, P.; Hossain, M. S.; Foss, F. W.; Pierce, B. S. Arch. Biochem. Biophys. 2017, 631, 66–74.94. Li, W.; Pierce, B. S. Arch. Biochem. Biophys. 2015, 565, 49–56.95. Pierce, B. S.; Gardner, J. D.; Bailey, L. J.; Brunold, T. C.; Fox, B. G. Biochemistry 2007, 46, 8569–8578.96. Blaesi, E. J.; Fox, B. G.; Brunold, T. C. Biochemistry 2014, 53, 5759–5770.97. Crawford, J. A.; Li, W.; Pierce, B. S. Biochemistry 2011, 50, 10241–10253.98. Grapperhaus, C. A.; Darensbourg, M. Y. Acc. Chem. Res. 1998, 31, 451–459.99. Simmons, C. R.; Krishnamoorthy, K.; Granett, S. L.; Schuller, D. J.; Dominy, J. E.; Begley, T. P.; Stipanuk, M. H.; Karplus, P. A. Biochemistry 2008, 47, 11390–11392.
100. Souness, R. J.; Kleffmann, T.; Tchesnokov, E. P.; Wilbanks, S. M.; Jameson, G. B.; Jameson, G. N. L. Biochemistry 2013, 52, 7606–7617.101. Aluri, S.; de Visser, S. P. J. Am. Chem. Soc. 2007, 129, 14846–14847.102. Kumar, D.; Thiel, W.; de Visser, S. P. J. Am. Chem. Soc. 2011, 133, 3869–3882.103. Crowell, J. K.; Li, W.; Pierce, B. S. Biochemistry 2014, 53, 7541–7548.104. Cavallini, D.; Scandurra, R.; De Marco, C. J. Biol. Chem. 1963, 238, 2999–3005.105. Richerson, R. B.; Ziegler, D. M. Cysteamine dioxygenase. In Methods in Enzymology, 143; Academic Press, 1987; pp 410–415.106. Cavallini, D.; Federici, G.; Ricci, G.; Duprè, S.; Antonucci, A.; De Marco, C. FEBS Lett. 1975, 56, 348–351.107. Wang, Y.; Griffith, W. P.; Li, J.; Koto, T.; Wherritt, D. J.; Fritz, E.; Liu, A. Angew. Chem. Int. Ed. 2018, 57, 8149–8153.108. Bruland, N.; Wübbeler, J. H.; Steinbüchel, A. J. Biol. Chem. 2009, 284, 660–672.109. Pierce, B. S.; Subedi, B. P.; Sardar, S.; Crowell, J. K. Biochemistry 2015, 54, 7477–7490.110. Aloi, S.; Davies, C. G.; Karplus, P. A.; Wilbanks, S. M.; Jameson, G. N. L. Biochemistry 2019.111. Fellner, M.; Aloi, S.; Tchesnokov, E. P.; Wilbanks, S. M.; Jameson, G. N. L. Biochemistry 2016, 55, 1362–1371.112. Crowell, J. K.; Sardar, S.; Hossain, M. S.; Foss, F. W.; Pierce, B. S. Arch. Biochem. Biophys. 2016, 604, 86–94.113. Brandt, U.; Waletzko, C.; Voigt, B.; Hecker, M.; Steinbüchel, A. Appl. Microbiol. Biotechnol. 2014, 98, 6039–6050.114. Brandt, U.; Galant, G.; Meinert-Berning, C.; Steinbüchel, A. Enzyme Microb. Technol. 2019, 120, 61–68.115. Turner, E.; Hager, L.; Shapiro, B. Science 1988, 242, 939–941.116. Castellano, I.; Seebeck, F. P. Nat. Prod. Rep. 2018, 35, 1241–1250.117. Vamecq, J.; Maurois, P.; Bac, P.; Bailly, F.; Bernier, J.-L.; Stables, J. P.; Husson, I.; Gressens, P. Eur. J. Neurosci. 2003, 18, 1110–1120.118. Russo, L. G.; Russo, M.; Castellano, I.; Napolitano, A.; Palumbo, A. Mar. Drugs 2014, 12.119. Halliwell, B.; Cheah, I. K.; Tang, R. M. Y. FEBS Lett. 2018, 592, 3357–3366.120. Cheah, I. K.; Halliwell, B. Biochim. Biophys. Acta. Mol. Basis Dis. 2012, 1822, 784–793.121. Seebeck, F. P. J. Am. Chem. Soc. 2010, 132, 6632–6633.122. Braunshausen, A.; Seebeck, F. P. J. Am. Chem. Soc. 2011, 133, 1757–1759.123. Hu, W.; Song, H.; Sae Her, A.; Bak, D. W.; Naowarojna, N.; Elliott, S. J.; Qin, L.; Chen, X.; Liu, P. Org. Lett. 2014, 16, 5382–5385.124. Stampfli, A. R.; Goncharenko, K. V.; Meury, M.; Dubey, B. N.; Schirmer, T.; Seebeck, F. P. J. Am. Chem. Soc. 2019, 141, 5275–5285.125. Song, H.; Her, A. S.; Raso, F.; Zhen, Z.; Huo, Y.; Liu, P. Org. Lett. 2014, 16, 2122–2125.126. Goncharenko, K. V.; Vit, A.; Blankenfeldt, W.; Seebeck, F. P. Angew. Chem. Int. Ed. 2015, 54, 2821–2824.127. Song, H.; Leninger, M.; Lee, N.; Liu, P. Org. Lett. 2013, 15, 4854–4857.128. Mashabela, G. T. M.; Seebeck, F. P. Chem. Commun. 2013, 49, 7714–7716.129. Allison, W. S. Acc. Chem. Res. 1976, 9, 293–299.130. Goncharenko, K. V.; Seebeck, F. P. Chem. Commun. 2016, 52, 1945–1948.131. Chen, L.; Naowarojna, N.; Song, H.; Wang, S.; Wang, J.; Deng, Z.; Zhao, C.; Liu, P. J. Am. Chem. Soc. 2018, 140, 4604–4612.132. Chen, L.; Naowarojna, N.; Chen, B.; Xu, M.; Quill, M.; Wang, J.; Deng, Z.; Zhao, C.; Liu, P. ACS Catal. 2019, 9, 253–258.133. Bushnell, E. A. C.; Fortowsky, G. B.; Gauld, J. W. Inorg. Chem. 2012, 51, 13351–13356.134. Wei, W.-J.; Siegbahn, P. E. M.; Liao, R.-Z. Inorg. Chem. 2017, 56, 3589–3599.135. Vit, A.; Mashabela, G. T.; Blankenfeldt, W.; Seebeck, F. P. ChemBioChem 2015, 16, 1490–1496.136. Faponle, A. S.; Seebeck, F. P.; de Visser, S. P. J. Am. Chem. Soc. 2017, 139, 9259–9270.137. Hildebrandt, T. M.; Grieshaber, M. K. FEBS J. 2008, 275, 3352–3361.138. Tiranti, V.; Viscomi, C.; Hildebrandt, T.; Di Meo, I.; Mineri, R.; Tiveron, C.; Levitt, M.; Prelle, A.; Fagiolari, G.; Rimoldi, M.; Zeviani, M. Nat. Med. 2009, 15, 200.

44 Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes
139. Tiranti, V.; D’Adamo, P.; Briem, E.; Ferrari, G.; Mineri, R.; Lamantea, E.; Mandel, H.; Balestri, P.; Garcia-Silva, M.-T.; Vollmer, B.; Rinaldo, P.; Hahn, S. H.; Leonard, J.;Rahman, S.; Dionisi-Vici, C.; Garavaglia, B.; Gasparini, P.; Zeviani, M. Am. J. Hum. Genet. 2004, 74, 239–252.
140. Grosso, S.; Mostardini, R.; Farnetani, M. A.; Molinelli, M.; Berardi, R.; Dionisi-Vici, C.; Rizzo, C.; Morgese, G.; Balestri, P. J. Neurol. 2002, 249, 1446–1450.141. Krüßel, L.; Junemann, J.; Wirtz, M.; Birke, H.; Thornton, J. D.; Browning, L. W.; Poschet, G.; Hell, R.; Balk, J.; Braun, H.-P.; Hildebrandt, T. M. Plant Physiol. 2014, 165,
92–104.142. de Lira, N. P. V.; Pauletti, B. A.; Marques, A. C.; Perez, C. A.; Caserta, R.; de Souza, A. A.; Vercesi, A. E.; Paes Leme, A. F.; Benedetti, C. E. Sci. Rep. 2018, 8, 3508.143. Shen, J.; Keithly, M. E.; Armstrong, R. N.; Higgins, K. A.; Edmonds, K. A.; Giedroc, D. P. Biochemistry 2015, 54, 4542–4554.144. Rohwerder, T.; Sand, W. Microbiology 1699-1710, 2003, 149.145. Sattler, S. A.; Wang, X.; Lewis, K. M.; DeHan, P. J.; Park, C.-M.; Xin, Y.; Liu, H.; Xian, M.; Xun, L.; Kang, C. J. Biol. Chem. 2015, 290, 18914–18923.146. Liu, H.; Xin, Y.; Xun, L. Appl. Environ. Microbiol. 2014, 80, 1799–1806.147. Suzuki, I. Method Enzymol. 1994, 243, 455–462.148. Kabil, O.; Banerjee, R. J. Biol. Chem. 2012, 287, 44561–44567.149. McCoy, J. G.; Bingman, C. A.; Bitto, E.; Holdorf, M. M.; Makaroff, C. A.; Phillips, G. N., Jr. Acta Crystallogr. Sect. D 2006, 62, 964–970.150. Holdorf, M. M.; Bennett, B.; Crowder, M. W.; Makaroff, C. A. J. Biol. Chem. 2008, 102, 1825–1830.151. Schofield, C. J.; Pettinati, I.; Brem, J.; McDonough, M. A. Hum. Mol. Genet. 2015, 24, 2458–2469.152. Tiranti, V.; Briem, E.; Lamantea, E.; Mineri, R.; Papaleo, E.; Gioia, L. D.; Forlani, F.; Rinaldo, P.; Dickson, P.; Abu-Libdeh, B.; Cindro-Heberle, L.; Owaidha, M.; Jack, R. M.;
Christensen, E.; Burlina, A.; Zeviani, M. J. Med. Genet. 2006, 43, 340.153. Henriques, B. J.; Lucas, T. G.; Rodrigues, J. V.; Frederiksen, J. H.; Teixeira, M. S.; Tiranti, V.; Bross, P.; Gomes, C. M. PLoS One 2014, 9e107157.154. Kabil, O.; Motl, N.; Strack, M.; Seravalli, J.; Metzler-Nolte, N.; Banerjee, R. J. Biol. Chem. 2018, 293, 12429–12439.155. Lin, B.; Ma, G.; Liu, Y. ACS Catal. 2016, 6, 7010–7020.156. Kumar, D.; Sastry, G. N.; Goldberg, D. P.; de Visser, S. P. J. Phys. Chem. A 2012, 116, 582–591.157. Karlin, K. Science 1993, 261, 701–708.158. Jiang, Y.; Widger, L. R.; Kasper, G. D.; Siegler, M. A.; Goldberg, D. P. J. Am. Chem. Soc. 2010, 132, 12214–12215.159. Widger, L. R.; Jiang, Y.; Siegler, M. A.; Kumar, D.; Latifi, R.; de Visser, S. P.; Jameson, G. N. L.; Goldberg, D. P. Inorg. Chem. 2013, 52, 10467–10480.160. Badiei, Y. M.; Siegler, M. A.; Goldberg, D. P. J. Am. Chem. Soc. 2011, 133, 1274–1277.161. McQuilken, A. C.; Jiang, Y.; Siegler, M. A.; Goldberg, D. P. J. Am. Chem. Soc. 2012, 134, 8758–8761.162. Confer, A. M.; Vilbert, A. C.; Dey, A.; Lancaster, K. M.; Goldberg, D. P. J. Am. Chem. Soc. 2019, 141, 7046–7055.163. Sallmann, M.; Siewert, I.; Fohlmeister, L.; Limberg, C.; Knispel, C. Angew. Chem. Int. Ed. 2012, 51, 2234–2237.164. Sallmann, M.; Braun, B.; Limberg, C. Chem. Commun. 2015, 51, 6785–6787.165. Fischer, A. A.; Stracey, N.; Lindeman, S. V.; Brunold, T. C.; Fiedler, A. T. Inorg. Chem. 2016, 55, 11839–11853.166. Theisen, R. M.; Shearer, J.; Kaminsky, W.; Kovacs, J. A. Inorg. Chem. 2004, 43, 7682–7690.167. Kishima, T.; Matsumoto, T.; Nakai, H.; Hayami, S.; Ohta, T.; Ogo, S. Angew. Chem. Int. Ed. Engl. 2016, 55, 724–727.168. Fischer, A. A.; Lindeman, S. V.; Fiedler, A. T. Chem. Commun. 2018, 54, 11344–11347.169. Blakely, M. N.; Dedushko, M. A.; Poon, P. C. Y.; Villar-Acevedo, G.; Kovacs, J. A. J. Am. Chem. Soc. 2019, 141, 1867–1870.170. Komuro, T.; Matsuo, T.; Kawaguchi, H.; Tatsumi, K. Inorg. Chem. 2003, 42, 5340–5347.171. Gordon, J. B.; Vilbert, A. C.; DiMucci, I. M.; MacMillan, S. N.; Lancaster, K. M.; Moënne-Loccoz, P.; Goldberg, D. P. J. Am. Chem. Soc. 2019, 141, 17533–17547.172. Widger, L. R.; Davies, C. G.; Yang, T.; Siegler, M. A.; Troeppner, O.; Jameson, G. N. L.; Ivanovi�c-Burmazovi�c, I.; Goldberg, D. P. J. Am. Chem. Soc. 2014, 136, 2699–2702.173. Fiedler, A. T.; Halfen, H. L.; Halfen, J. A.; Brunold, T. C. J. Am. Chem. Soc. 2005, 127, 1675–1689.174. Bukowski, M. R.; Koehntop, K. D.; Stubna, A.; Bominaar, E. L.; Halfen, J. A.; Münck, E.; Nam, W.; Que, L., Jr. Science 2005, 310, 1000–1002.175. Kitagawa, T.; Dey, A.; Lugo-Mas, P.; Benedict, J. B.; Kaminsky, W.; Solomon, E.; Kovacs, J. A. J. Am. Chem. Soc. 2006, 128, 14448–14449.176. Namuswe, F.; Kasper, G. D.; Sarjeant, A. A. N.; Hayashi, T.; Krest, C. M.; Green, M. T.; Moënne-Loccoz, P.; Goldberg, D. P. J. Am. Chem. Soc. 2008, 130, 14189–14200.177. Krishnamurthy, D.; Kasper, G. D.; Namuswe, F.; Kerber, W. D.; Narducci Sarjeant, A. A.; Moënne-Loccoz, P.; Goldberg, D. P. J. Am. Chem. Soc. 2006, 128, 14222–14223.178. Widger, L. R.; Jiang, Y.; McQuilken, A. C.; Yang, T.; Siegler, M. A.; Matsumura, H.; Moënne-Loccoz, P.; Kumar, D.; de Visser, S. P.; Goldberg, D. P. Dalton Trans. 2014, 43,
7522–7532.179. Che, X.; Gao, J.; Liu, Y.; Liu, C. J. Biol. Chem. 2013, 122, 1–7.180. Widger, L. R.; Jiang, Y.; Siegler, M. A.; Kumar, D.; Latifi, R.; de Visser, S. P.; Jameson, G. N. L.; Goldberg, D. P. Inorg. Chem. 2013, 52, 10467–10480.181. Widger, L. R.; Siegler, M. A.; Goldberg, D. P. Polyhedron 2013, 58, 179–189.182. Gonzalez-Ovalle, L. E.; Quesne, M. G.; Kumar, D.; Goldberg, D. P.; de Visser, S. P. Org. Biomol. Chem. 2012, 10, 5401–5409.183. Confer, A. M.; McQuilken, A. C.; Matsumura, H.; Moënne-Loccoz, P.; Goldberg, D. P. J. Am. Chem. Soc. 2017, 139, 10621–10624.184. McQuilken, A. C.; Ha, Y.; Sutherlin, K. D.; Siegler, M. A.; Hodgson, K. O.; Hedman, B.; Solomon, E. I.; Jameson, G. N. L.; Goldberg, D. P. J. Am. Chem. Soc. 2013, 135,
14024–14027.185. Kupper, C.; Rees, J. A.; Dechert, S.; DeBeer, S.; Meyer, F. J. Am. Chem. Soc. 2016, 138, 7888–7898.186. Serres, R. G.; Grapperhaus, C. A.; Bothe, E.; Bill, E.; Weyhermüller, T.; Neese, F.; Wieghardt, K. J. Am. Chem. Soc. 2004, 126, 5138–5153.187. Dey, A.; Confer, A. M.; Vilbert, A. C.; Moënne-Loccoz, P.; Lancaster, K. M.; Goldberg, D. P. Angew. Chem. Int. Ed. 2018, 57, 13465–13469.188. Speelman, A. L.; White, C. J.; Zhang, B.; Alp, E. E.; Zhao, J.; Hu, M.; Krebs, C.; Penner-Hahn, J.; Lehnert, N. J. Am. Chem. Soc. 2018, 140, 11341–11359.189. Theisen, R. M.; Shearer, J.; Kaminsky, W.; Kovacs, J. A. Inorg. Chem. 2004, 43, 7682–7690.190. Balch, A. L.; Chan, Y. W.; Cheng, R. J.; La Mar, G. N.; Latos-Grazynski, L.; Renner, M. W. J. Am. Chem. Soc. 1984, 106, 7779–7785.191. Kishima, T.; Matsumoto, T.; Nakai, H.; Hayami, S.; Ohta, T.; Ogo, S. Angew. Chem. Int. Ed. 2016, 55, 724–727.192. McDonald, A. R.; Van Heuvelen, K. M.; Guo, Y.; Li, F.; Bominaar, E. L.; Münck, E.; Que, L., Jr. Angew. Chem. Int. Ed. 2012, 51, 9132–9136.193. Blakely, M. N.; Dedushko, M. A.; Yan Poon, P. C.; Villar-Acevedo, G.; Kovacs, J. A. J. Am. Chem. Soc. 2019, 141, 1867–1870.194. Komuro, T.; Matsuo, T.; Kawaguchi, H.; Tatsumi, K. Inorg. Chem. 2003, 42, 5340–5347.195. Klein, J. E.; Que, L., Jr. Biomimetic High-Valent Mononuclear Nonheme Iron-Oxo Chemistry. In Encyclopedia of Inorganic and Bioinorganic Chemistry; Scott, R. A., Ed.; John
Wiley: Chichester, 2016; pp 1–22.196. Guo, M.; Corona, T.; Ray, K.; Nam, W. ACS Cent. Sci. 2019, 5, 13–28.197. McDonald, A. R.; Que, L., Jr. Coord. Chem. Rev. 2013, 257, 414–428.198. McDonald, A. R.; Bukowski, M. R.; Farquhar, E. R.; Jackson, T. A.; Koehntop, K. D.; Seo, M. S.; De Hont, R. F.; Stubna, A.; Halfen, J. A.; Münck, E.; Nam, W.; Que, L., Jr. J. Am.
Chem. Soc. 2010, 132, 17118–17129.199. Villar-Acevedo, G.; Lugo-Mas, P.; Blakely, M. N.; Rees, J. A.; Ganas, A. S.; Hanada, E. M.; Kaminsky, W.; Kovacs, J. A. J. Am. Chem. Soc. 2017, 139, 119–129.200. Shearer, J.; Nehring, J.; Lovell, S.; Kaminsky, W.; Kovacs, J. A. Inorg. Chem. 2001, 40, 5483–5484.201. Shearer, J.; Scarrow, R. C.; Kovacs, J. A. J. Am. Chem. Soc. 2002, 124, 11709–11717.202. Nam, E.; Alokolaro, P. E.; Swartz, R. D.; Gleaves, M. C.; Pikul, J.; Kovacs, J. A. Inorg. Chem. 2011, 50, 1592–1602.203. Namuswe, F.; Hayashi, T.; Jiang, Y.; Kasper, G. D.; Sarjeant, A. A. N.; Moënne-Loccoz, P.; Goldberg, D. P. J. Am. Chem. Soc. 2010, 132, 157–167.204. Stasser, J.; Namuswe, F.; Kasper, G. D.; Jiang, Y.; Krest, C. M.; Green, M. T.; Penner-Hahn, J.; Goldberg, D. P. Inorg. Chem. 2010, 49, 9178–9190.

Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes 45
205. Roelfes, G.; Vrajmasu, V.; Chen, K.; Ho, R. Y. N.; Rohde, J.-U.; Zondervan, C.; la Crois, R. M.; Schudde, E. P.; Lutz, M.; Spek, A. L.; Hage, R.; Feringa, B. L.; Münck, E.; Que, L.,Jr. Inorg. Chem. 2003, 42, 2639–2653.
206. Woodruff, W. H.; Spiro, T. G.; Yonetani, T. Proc. Natl. Acad. Sci. U. S. A. 1974, 71, 1065–1069.207. Coggins, M. K.; Toledo, S.; Shaffer, E.; Kaminsky, W.; Shearer, J.; Kovacs, J. A. Inorg. Chem. 2012, 51, 6633–6644.208. Coggins, M. K.; Sun, X.; Kwak, Y.; Solomon, E. I.; Rybak-Akimova, E.; Kovacs, J. A. J. Am. Chem. Soc. 2013, 135, 5631–5640.209. Yan Poon, P. C.; Dedushko, M. A.; Sun, X.; Yang, G.; Toledo, S.; Hayes, E. C.; Johansen, A.; Piquette, M. C.; Rees, J. A.; Stoll, S.; Rybak-Akimova, E.; Kovacs, J. A. J. Am.
Chem. Soc. 2019, 141, 15046–15057.210. Lee, C.-M.; Chuo, C.-H.; Chen, C.-H.; Hu, C.-C.; Chiang, M.-H.; Tseng, Y.-J.; Hu, C.-H.; Lee, G.-H. Angew. Chem. Int. Ed. 2012, 51, 5427–5430.211. Lee, C.-M.; Sankaralingam, M.; Chuo, C.-H.; Tseng, T.-H.; Chen, P. P. Y.; Chiang, M.-H.; Li, X.-X.; Lee, Y.-M.; Nam, W. Dalton Trans. 2019, 48, 5203–5213.212. Lee, C.-M.; Wu, W.-Y.; Chiang, M.-H.; Bohle, D. S.; Lee, G.-H. Inorg. Chem. 2017, 56, 10559–10569.213. Gennari, M.; Brazzolotto, D.; Pécaut, J.; Cherrier, M. V.; Pollock, C. J.; DeBeer, S.; Retegan, M.; Pantazis, D. A.; Neese, F.; Rouzières, M.; Clérac, R.; Duboc, C. J. Am. Chem.
Soc. 2015, 137, 8644–8653.214. Brazzolotto, D.; Cantú Reinhard, F. G.; Smith-Jones, J.; Retegan, M.; Amidani, L.; Faponle, A. S.; Ray, K.; Philouze, C.; de Visser, S. P.; Gennari, M.; Duboc, C. Angew. Chem.
Int. Ed. 2017, 56, 8211–8215.215. Gordon, J. B.; Vilbert, A. C.; Siegler, M. A.; Lancaster, K. M.; Moënne-Loccoz, P.; Goldberg, D. P. J. Am. Chem. Soc. 2019, 141, 3641–3653.216. Naowarojna, N.; Cheng, R.; Chen, L.; Quill, M.; Xu, M.; Zhao, C.; Liu, P. Biochemistry 2018, 57, 3309–3325.