supporting information - pnas · supporting information hartl et al. 10.1073/pnas.0812101106 si...
TRANSCRIPT

Supporting InformationHartl et al. 10.1073/pnas.0812101106SI TextPromoter Analysis. Bioinformatic analysis of the chicken BASP1regulatory region was performed as described (1–4). To con-struct pLUC-BASP1�SB, pLUC-BASP1 was digested withSmaI/BglI (blunt-ended) and a 376-bp segment was deleted,followed by religation of the vector fragment. For constructionof pLUC-BASP1�DS, a 378-bp PCR fragment encompassingnucleotides 20–378 from the isolated genomic fragment (Gen-Bank accession no. EU888907) was inserted into the KpnI/HindIII sites of pGL3-Basic. In pLUC-BASP1�SB�Sp1, thedeletion of two Sp1 binding regions corresponding to nucleotides436–450 and 461–474 in the genomic fragment was achieved byinsertion of 2 overlapping oligonucleotides (5�-GGGCCG-GGAAATAAATAAATAAGTGCGTGTGGCTTTTGA-ACTGCC-CACAGACTGTCGGGAAGAGAG-3� and 5�-GGGAAGAGAGGAGAGAGAGCGAGAGCG-AGAAAT-GCAGAGGCAGCAGCCGGGC-3�) into the SmaI/XhoIsites of pLUC-BASP1�SB. To construct pRc-Sp1, a 2,397-bpHindIII/EcoRI (blunt-ended) fragment containing the chickenSp1 coding region was excised from the plasmid pH-�GC Sp1 (5)and inserted into the HindIII/XbaI (blunt-ended) sites of thepRc/RSV vector. ChIP analyses were carried out as described byusing sheared extracts derived from formaldehyde-treatedMC29-transformed or normal CEFs or QEFs (3, 4). Immuno-precipitations were performed with specific antibodies followedby PCR amplification of a 158-bp fragment from the chickenBASP1 regulatory region or a 293-bp fragment from the chickenWS5/Mmp115 promoter region by using the BASP1-specificprimer pair 5�-TAAGTGCGTGTGGCTTTTGAAC-3� and 5�-CGGAGTGCCTCGCGCTG-3� or the chicken WS5/Mmp115-specific primer pair 5�-AGACCCTCACGGAGGTCTCAC-3�and 5�-CTGCTGTGCCGTGACCAG-3� encompassing nucleo-tide positions 623–915 of sequence contig NW�001471748.Primer pairs for the quail WS5 promoter were as described (4).The origin of the antisera �-C/EBP� and �-Sp1 has beendescribed (4, 5).
Cell Assays. To assess the viability of cultured avian fibroblasts,cells (1.5 � 106) were seeded onto 60-mm dishes and incubatedovernight at 37 °C. The culture medium was collected andcentrifuged together with the trypsinized adherent cells. The cellpellet was resuspended in 0.5 mL of PBS. An 80-�L aliquot ofthis cell suspension was mixed with 100 �L of a 0.4% (wt/vol)trypan blue (ditolyldisazobis-8-amino-1-naphthol-3,6-disulfon-ate) solution (Sigma) and incubated at room temperature for 5min. Cells from 10 �L of this mixture were counted with ahemacytometer, and the ratio of stained versus unstained cellswas calculated. To measure cell proliferation rates, cells (1.0 �106) were seeded in triplicate onto 60-mm dishes and the mediumwas changed every 2 days. Numbers of trypsinized cells weredetermined every 2 days with a Coulter counter.
For immunofluorescence analysis, cells (1.0 � 105) wereseeded into chamber slide (Nalge/Nunc) wells and incubatedovernight in culture medium. After washing with PBS, cells werefixed in 4% (wt/vol) formaldehyde for 20 min and permeabilizedwith PBS containing 0.2% (wt/vol) Triton X-100 for 5 min.Nonspecific binding was blocked by incubation with PBS con-taining 0.5% (wt/vol) BSA for 30 min. The cells were thenincubated at room temperature for 1 h with rabbit BASP1-specific polyclonal antiserum diluted 1:250. After washing sev-eral times with PBS, the samples were incubated for 45 min withgoat FITC-conjugated anti-IgG rabbit diluted 1:200, followed by
DNA staining for 5 min at room temperature with a solution ofPBS containing 300 nM DAPI (4�,6-diamidino-2-phenylindole)dilactate (Invitrogen). The chamber slide was repeatedly rinsedwith PBS, air-dried, and mounted with Vectashield medium(Vector Lab). Immunofluorescent staining was analyzed with afluorescence microscope (Axiovert 135; Zeiss).
To construct pRc-Src used for transformation assays, thecoding region of the temperature-sensitive v-Src mutant tsLA29was amplified by PCR from the plasmid pRAV29 (6) by usingprimers containing flanking HindIII and XbaI restriction sitesand ligated into the HindIII/XbaI sites of the pRc/RSV vector.
RNA Interference. A lyophilized synthetic siRNA duplex (Qiagen)consisting of sense r(GGUCUCUGCCAAUAAGACA)dTdTand antisense r(UGUCUUAUUGGCAGAGACC)dTdGstrands targeted against the BASP1 mRNA sequence 5�-CAGGUCUCUGCCAAUAAGACA-3� was dissolved in 1 mLof a buffer containing 100 mM potassium acetate, 30 mMHepes-KOH (pH 7.4), and 2 mM magnesium acetate to obtaina 20 �M siRNA solution. The solution was heated at 90 °C for1 min, incubated at 37°C for 1 h, and then stored at �20 °C. Fordelivery of the siRNA into CEFs or the CEF line DF-1 (7), 6 �106 cells pelleted at 50 � g for 5 min at room temperature weresuspended into 100 �L of nucleofector solution V (Lonza/Amaxa) and mixed with 12 �L of siRNA solution containing 3�g of the duplex RNA. The mixture was then subjected toelectroporation (Lonza/Amaxa) using the nucleofector programU-20, and then immediately diluted with 0.5 mL of culturemedium. Transfected cells were seeded onto 60-mm dishescontaining 4 mL of culture medium. Medium was changed thenext day. For Northern analysis, RNA was isolated with theRiboPure Kit (Ambion). Cells were homogenized in a solutioncontaining phenol and guanidine thiocycanate. After addition ofbromochloropropane, RNA was recovered from the aqueousphase by binding to a glass-fiber filter and elution using a low-saltbuffer. For immunoblot analysis, trypsinized cells were directlylysed in protein sample buffer (12,500 cells/�L) as described (8).
In Vitro Mutagenesis. To generate the adapter constructs pA-(G2A), pA-(K7–10A), or pA-(G2A,K7–10A), oligonucleotides(5�-CATGGCAGGCAAACTGAGCAAGAAGAAGAAG-GGG-3�, 5�-CATGGGAGGCAAACTGAGCGCTGCAG-CAGCAGGG-3�, 5�-CATGGCAGGCAAA-CTGAGCGCTG-CAGCAGCAGGG) were ligated together with a 715-bp CviQI/SalI fragment from pA-BASP1 into the NcoI/SalI sites ofpA-CLA12NCO. To construct pRc-(G2A), pRc-(K7–10A), andpRc-(G2A,K7–10A), the NcoI (blunt-ended)/XbaI inserts fromthe adapter constructs were transferred into the HindIII (blunt-ended)/XbaI sites of pRc/RSV. To construct pRCAS-(G2A),pRCAS-(K7–10A), and pRCAS-(G2A,K7–10A), ClaI fragmentsfrom the adapter constructs were inserted into the pRCAS-BPvector as described (2).
Coimmunoprecipitation. Coimmunoprecipitation of protein com-plexes from lysates of 35Smethionine-labeled cells was done asdescribed (4, 9) with the following modifications. Interactingproteins precipitated by the first antibody under native condi-tions were dissociated in 600 �L of RIPA buffer containing 10mM sodium phosphate (pH 7.2), 150 mM NaCl, 1% (vol/vol)Igepal CA-630, 1% (wt/vol) sodium deoxycholate, 0.1% (wt/vol)SDS, and 2 �g/mL aprotinin at 4 °C for 2 h with an overheadshaker. The samples were centrifuged, and the supernatants
Hartl et al. www.pnas.org/cgi/content/short/0812101106 1 of 7

were subjected to immunoprecipitation with the second antibodyunder denaturing conditions as described (4, 9). Antisera against
a recombinant C-terminal fragment of Myc, recombinant Max orBASP1 proteins, or normal rabbit serum were used.
1. Bader AG, Schneider ML, Bister K, Hartl M (2001) TOJ3, a target of the v-Jun transcrip-tion factor, encodes a protein with transforming activity related to human microspher-ule protein 1 (MCRS1). Oncogene 20:7524–7535.
2. Hartl M, Reiter F, Bader AG, Castellazzi M, Bister K (2001) JAC, a direct target ofoncogenic transcription factor Jun, is involved in cell transformation and tumorigen-esis. Proc Natl Acad Sci USA 98:13601–13606.
3. Hartl M, Karagiannidis AI, Bister K (2006) Cooperative cell transformation by Myc/Mil(Raf) involves induction of AP-1 and activation of genes implicated in cell motilityand metastasis. Oncogene 25:4043–4055.
4. Reiter F, Hartl M, Karagiannidis AI, Bister K (2007) WS5, a direct target of oncogenictranscription factor Myc, is related to human melanoma glycoprotein genes and hasoncogenic potential. Oncogene 26:1769–1779.
5. Chamboredon S, et al. (2003) v-Jun down-regulates the SPARC target gene by bindingto the proximal promoter indirectly through Sp1/3. Oncogene 22:4047–4061.
6. Welham MJ, Wyke JA (1988) A single point mutation has pleiotropic effects onpp60v-src function. J Virol 62:1898–1906.
7. Himly M, Foster DN, Bottoli I, Iacovoni JS, Vogt PK (1998) The DF-1 chicken fibroblastcell line: Transformation induced by diverse oncogenes and cell death resulting frominfection by avian leukosis viruses. Virology 248:295–304.
8. Oberst C, Hartl M, Weiskirchen R, Bister K (1999) Conditional cell transformation bydoxycycline-controlled expression of the MC29 v-myc allele. Virology 253:193–207.
9. Hartl M, et al. (2003) Cell transformation by the v-myc oncogene abrogates c-Myc/Max-mediated suppression of a C/EBP�-dependent lipocalin gene. J Mol Biol 333:33–46.
Hartl et al. www.pnas.org/cgi/content/short/0812101106 2 of 7

Fig. S1. Analysis of the chicken BASP1 promoter. (A) Nucleotide sequence of the BASP1 regulatory region. The transcription start site (arrow), potential binding sitesfor Sp1 and TBP (bold), and binding sites for 5� and 3� ChIP primers (underlined) are indicated. (B) Dissection of the regulatory region. Aliquots (4.0 �g) of the indicatedpLUC-BASP1 reporter construct or of deletion mutants and of the pSV-�-galactosidase plasmid (2.0 �g) were transfected into normal fibroblasts (QEFs) and QEFstransformed by the v-myc oncogene (QEF/MC29). Luciferase activities were determined as described in Fig. 2D. (C) Samples of pLUC-BASP1-�SB (4.0 �g) containing theminimal BASP1 regulatory region were used for transfection into QEFs together with pRc-derived expression constructs (2.0 �g) carrying the coding regions of v-Mycor Sp1. (D) ChIP using chromatin from CEFs or CEF/MC29 cells transformed by the v-Myc protein. Antisera directed against Myc, BASP1, Sp1, or C/EBP� were used forprecipitation, followed by PCR amplification of a 158-bp fragment from the BASP1 regulatory region containing 2 active Sp1 binding sites or a 293-bp fragment fromthe WS5 promoter containing an E-box cluster and a C/EBP�-binding site. Reactions with normal rabbit serum (NRS) or total chromatin (Input) were used as controls.
Hartl et al. www.pnas.org/cgi/content/short/0812101106 3 of 7

Fig. S2. Specificity of transformation inhibition by BASP1. QEFs were transfected with RCAS or RCAS-BASP1 and passaged 5 times, and then supertransfectedwith pRc-Myc or pRc-Src, containing the v-myc or v-src oncogene, respectively. Foci were recorded as described in Fig. 4B.
Hartl et al. www.pnas.org/cgi/content/short/0812101106 4 of 7

Fig. S3. Effects of overexpression or siRNA knockdown of BASP1 in avian fibroblasts. (A) Micrographs showing the morphologies of QEFs or QEFs transfectedwith the RCAS constructs described in Fig. 3. (B) Viability assay of cells shown in A using trypan blue staining of dead cells. (C) Proliferation rates of cell culturesshown in A. A total of 1 � 106 cells were seeded onto 60-mm dishes, and cell numbers were determined at the indicated time points. (D) Subcellular localizationof BASP1. Immunofluorescent staining of QEFs and QEF/RCAS-BASP1 using a polyclonal antiserum directed against recombinant BASP1 protein and a secondaryFITC antibody. Nuclei are counterstained with DAPI. (E) BASP1 knockdown in DF-1 chicken cells. DF-1 cells were transiently nucleofected with a synthetic siRNA(BASP1-SIR3) directed against the chicken BASP1 mRNA. Total RNA was isolated 2 days after siRNA delivery and analyzed by Northern hybridization using DNAprobes specific for chicken BASP1, c-myc, or quail GAPDH. Protein extracts were prepared after 3 days and analyzed by immunoblotting using a polyclonalBASP1-specific antiserum. The morphology of DF-1 cells and cells treated with the BASP-1-specific siRNA is shown in the micrographs taken after 3 days.
Hartl et al. www.pnas.org/cgi/content/short/0812101106 5 of 7
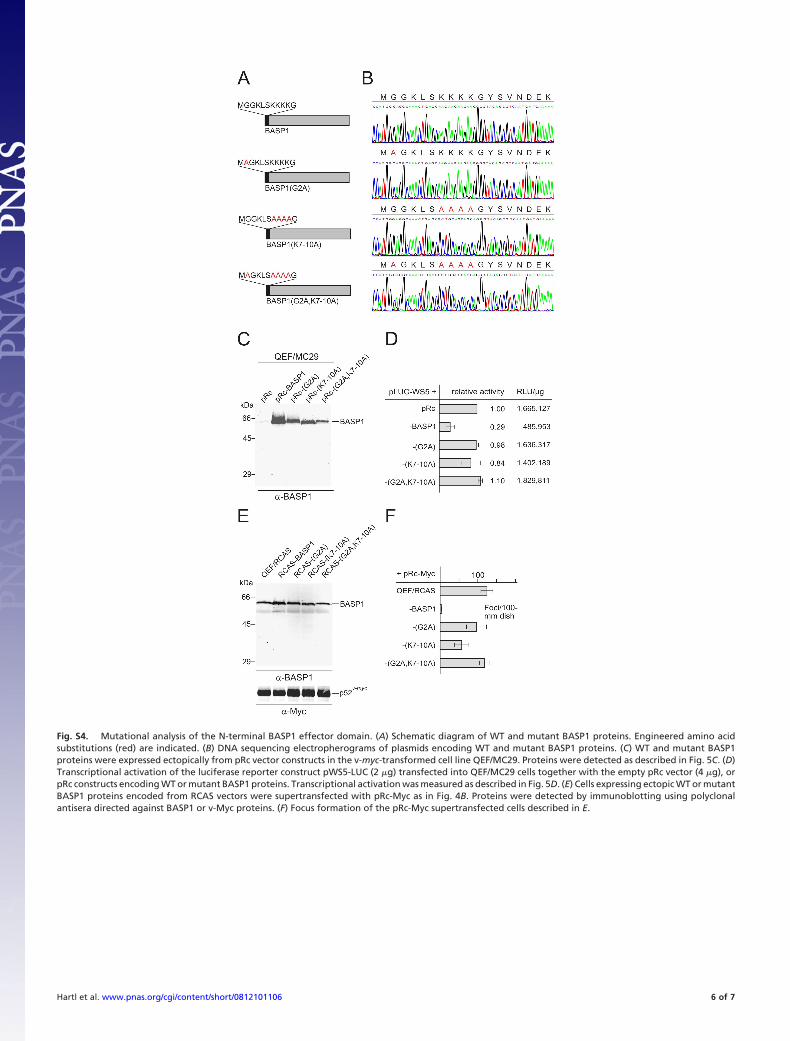
Fig. S4. Mutational analysis of the N-terminal BASP1 effector domain. (A) Schematic diagram of WT and mutant BASP1 proteins. Engineered amino acidsubstitutions (red) are indicated. (B) DNA sequencing electropherograms of plasmids encoding WT and mutant BASP1 proteins. (C) WT and mutant BASP1proteins were expressed ectopically from pRc vector constructs in the v-myc-transformed cell line QEF/MC29. Proteins were detected as described in Fig. 5C. (D)Transcriptional activation of the luciferase reporter construct pWS5-LUC (2 �g) transfected into QEF/MC29 cells together with the empty pRc vector (4 �g), orpRc constructs encoding WT or mutant BASP1 proteins. Transcriptional activation was measured as described in Fig. 5D. (E) Cells expressing ectopic WT or mutantBASP1 proteins encoded from RCAS vectors were supertransfected with pRc-Myc as in Fig. 4B. Proteins were detected by immunoblotting using polyclonalantisera directed against BASP1 or v-Myc proteins. (F) Focus formation of the pRc-Myc supertransfected cells described in E.
Hartl et al. www.pnas.org/cgi/content/short/0812101106 6 of 7

Fig. S5. Coimmunoprecipitation analysis. Extracts of QEFs prepared under native conditions were incubated with normal rabbit serum (NRS), �-Myc, �-Max,or �-BASP1 antisera. Precipitated proteins were dissociated and subsequently immunoprecipitated under denaturing conditions using NRS, �-Max, or �-Myc asthe second antibody. Immunoprecipitates were analyzed by SDS/PAGE (12%, wt/vol).
Hartl et al. www.pnas.org/cgi/content/short/0812101106 7 of 7