supramolecular control of azobenzene switching on ... · supramolecular control of azobenzene...
TRANSCRIPT

Supramolecular Control of Azobenzene Switching on NanoparticlesZonglin Chu,† Yanxiao Han,‡ Tong Bian,† Soumen De,† Petr Kral,‡,§ and Rafal Klajn*,†
†Department of Organic Chemistry, Weizmann Institute of Science, Rehovot 76100, Israel‡Department of Chemistry, University of Illinois at Chicago, Chicago, Illinois 60607, United States§Department of Physics and Department of Biopharmaceutical Sciences, University of Illinois at Chicago, Chicago, Illinois 60607,United States
*S Supporting Information
ABSTRACT: The reversible photoisomerization of azobenzene has beenutilized to construct a plethora of systems in which optical, electronic,catalytic, and other properties can be controlled by light. However, owing toazobenzene’s hydrophobic nature, most of these examples have been realizedonly in organic solvents, and systems operating in water are relatively scarce.Here, we show that by coadsorbing the inherently hydrophobic azobenzeneswith water-solubilizing ligands on the same nanoparticulate platforms, it is possible to render them essentially water-soluble. Tothis end, we developed a modified nanoparticle functionalization procedure allowing us to precisely fine-tune the amount ofazobenzene on the functionalized nanoparticles. Molecular dynamics simulations helped us to identify two distinctsupramolecular architectures (depending on the length of the background ligand) on these nanoparticles, which can explaintheir excellent aqueous solubilities. Azobenzenes adsorbed on these water-soluble nanoparticles exhibit highly reversiblephotoisomerization upon exposure to UV and visible light. Importantly, the mixed-monolayer approach allowed us tosystematically investigate how the background ligand affects the switching properties of azobenzene. We found that the natureof the background ligand has a profound effect on the kinetics of azobenzene switching. For example, a hydroxy-terminatedbackground ligand is capable of accelerating the back-isomerization reaction by more than 6000-fold. These results pave the waytoward the development of novel light-responsive nanomaterials operating in aqueous media and, in the long run, in biologicalenvironments.
■ INTRODUCTION
The reversible isomerization of azobenzene (1,2-diphenyldia-zene)one of the simplest and most robust photoswitchablecompoundshas been investigated extensively for nearly acentury.1 The trans−cis isomerization process entails asignificant change in the shape of the molecule and a decreasein the distance between two para substituents from ∼9 Å to∼6.5 Å. These changes in various azobenzene derivatives havebeen used to modulate catalysis,2−4 affinity to metal ions,5 andbinding to biological molecules in living organisms6,7 and evento control the conformation of a bound “guest” molecule.8
However, whereas all of these functions have been realized infree molecules, the immediate surroundings of a molecularswitch can have a profound effect on its switching properties. Ithas long been known, for example, that confining azobenzeneswithin small volumes can effectively suppress their switch-ing9−14 and that the success of trans → cis isomerization couldbe correlated15 to the amount of free space available to anazobenzene unit. In contrast, several systems showed evidenceof cooperative azobenzene switching,16−18 whereby denselypacked azobenzenes isomerized very readily in a process mostlikely initiated at the periphery of an ensemble.19,20 Beyondthese single-component systems, it would arguably be moreinteresting to control the properties of molecular switches byother species residing in their proximity.
Inorganic nanoparticles (NPs) functionalized with binary(mixed) monolayers of ligands21−25 provide an attractiveplatform for inducing intermolecular interactions between theadsorbed species and hence for investigating how variousfunctional groups affect the properties of switchable molecules.An additional advantage of the mixed-monolayer approach isthat by decorating NPs with mixtures of functional andsolubilizing ligands, it becomes possible, by tuning thecomposition of the monolayer,26 to effectively solubilizecompounds that are otherwise insoluble in a given solvent.27
For example, whereas azobenzene-functionalized NPs havebeen investigated extensively in various organic solvents,28−32
their properties in aqueous media have remained unknownbecause of the nonpolar nature of azobenzene and,consequently, the very low solubility of azobenzene-function-alized NPs in water. Access to water-soluble, azobenzene-coated NPs could broaden the scope of applications of thesenanomaterials, in particular in the context of biomedicalapplications (such as in photopharmacology33,34).We hypothesized that water-soluble, azobenzene-coated NPs
could be obtained by functionalizing NPs with mixedmonolayers comprising (i) azobenzene and (ii) a ligandterminated with a polar group, such as an oligo(ethylene)
Received: September 6, 2018Published: December 31, 2018
Article
pubs.acs.org/JACSCite This: J. Am. Chem. Soc. 2019, 141, 1949−1960
© 2018 American Chemical Society 1949 DOI: 10.1021/jacs.8b09638J. Am. Chem. Soc. 2019, 141, 1949−1960
Dow
nloa
ded
via
WE
IZM
AN
N I
NST
OF
SCIE
NC
E o
n M
arch
10,
201
9 at
16:
07:0
9 (U
TC
).
See
http
s://p
ubs.
acs.
org/
shar
ingg
uide
lines
for
opt
ions
on
how
to le
gitim
atel
y sh
are
publ
ishe
d ar
ticle
s.
glycol (OEG) chain or a charged group. Such functionalizationis typically achieved by treating NPs capped with weaklybound ligands (e.g., dodecylamine (DDA) for Au NPs) withan excess of ligands having higher affinity (e.g., thiols). Wespeculated that above a critical fractional coverage with polarligands the NPs should be soluble in water despite the
presence of the hydrophobic azobenzene groups. However,cofunctionalization of NPs with mixtures of polar and apolarligands is not an easy task:24 earlier studies showed that suchprocesses typically result in preferential adsorption of only oneof the two ligands. Among numerous other examples,35−40
Bishop and co-workers found that performing ligand exchange
Figure 1. Structural formulas of thiolate ligands used in this study.
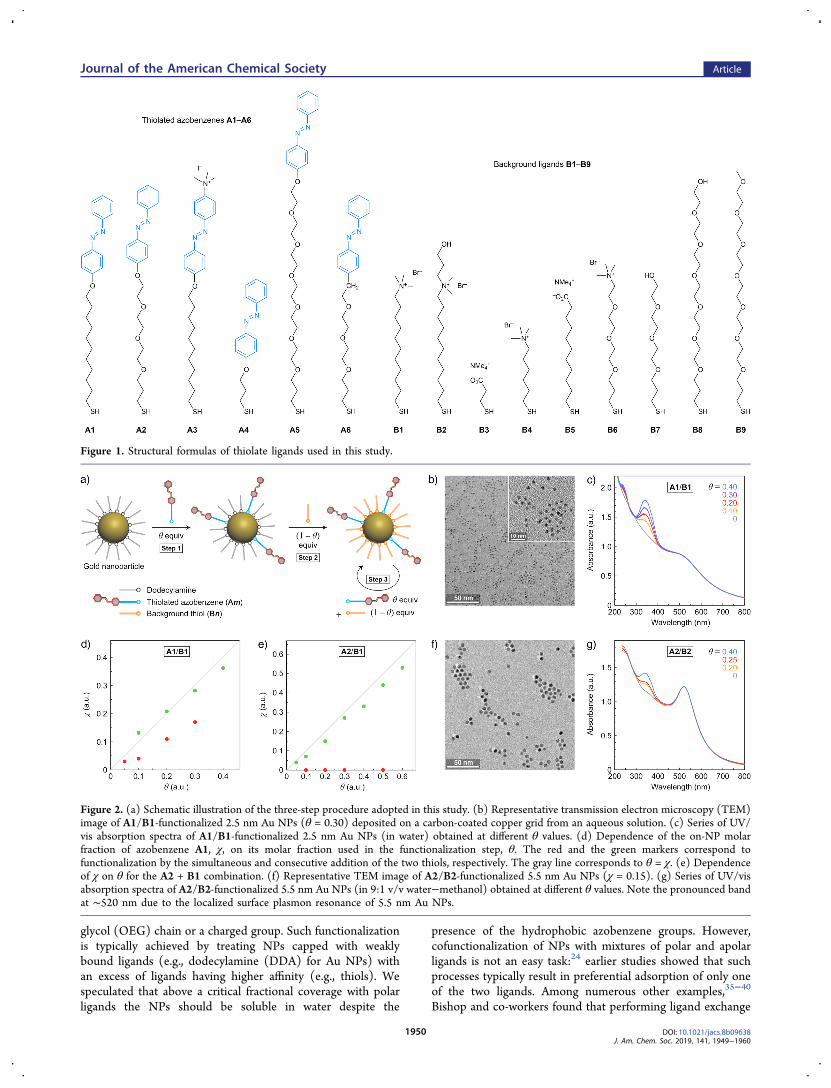
Figure 2. (a) Schematic illustration of the three-step procedure adopted in this study. (b) Representative transmission electron microscopy (TEM)image of A1/B1-functionalized 2.5 nm Au NPs (θ = 0.30) deposited on a carbon-coated copper grid from an aqueous solution. (c) Series of UV/vis absorption spectra of A1/B1-functionalized 2.5 nm Au NPs (in water) obtained at different θ values. (d) Dependence of the on-NP molarfraction of azobenzene A1, χ, on its molar fraction used in the functionalization step, θ. The red and the green markers correspond tofunctionalization by the simultaneous and consecutive addition of the two thiols, respectively. The gray line corresponds to θ = χ. (e) Dependenceof χ on θ for the A2 + B1 combination. (f) Representative TEM image of A2/B2-functionalized 5.5 nm Au NPs (χ = 0.15). (g) Series of UV/visabsorption spectra of A2/B2-functionalized 5.5 nm Au NPs (in 9:1 v/v water−methanol) obtained at different θ values. Note the pronounced bandat ∼520 nm due to the localized surface plasmon resonance of 5.5 nm Au NPs.
Journal of the American Chemical Society Article
DOI: 10.1021/jacs.8b09638J. Am. Chem. Soc. 2019, 141, 1949−1960
1950

on DDA-capped 6 nm Au NPs with an 8:1 mixture ofoctadecanethiol and ω-mercaptoundecanoic acid resulted in a0.35:1 mixture of these two ligands on the functionalizedNPs,41 whereas Meli et al. reported that a 3:1 molar ratio ofdodecanethiol and ω-mercaptoundecanol used to functionalize4 nm NPs was increased up to 32:1 as a result of the ligandexchange reaction.42 The discrepancy between the solutionand the on-NP ratio typically originates from attractive/repulsive interactions between the ligands or when one of theligands induces NP precipitation during the functionalizationprocess. Therefore, we developed a modified NP functionaliza-tion procedure that allowed us to predictably control the molarratio of the two ligands on the NPs. These NPs allowed us tosystematically investigate how the background ligand influ-enced the switching properties of azobenzene in water.
■ RESULTS AND DISCUSSIONControlling the Fractional Coverage of Azobenzene
on Water-Soluble Nanoparticles. We functionalized goldNPs with different combinations of thiolated azobenzenes Am(m = 1 through 6) and background (“dummy”) thiols Bn (n =1 through 9) (Figure 1 and Supporting Information, Sections2−4) using the following approach. First, we treated DDA-capped NPs with θ (where θ < 1) equivalents of Am (step 1 inFigure 2a; equiv with respect to the number of binding sites onthe NPs, calculated assuming that a single thiolate moietyoccupies an area of 0.214 nm2 on the surface of gold43). After 1h, (1 − θ) equiv of Bn was added and shaking was continuedfor an additional 2 h (Figure 2a, step 2). This two-stepprocedure should lead to a practically quantitative ligandexchange, given alkanethiols’ ∼100 times higher (vs alkylamines) affinity to gold.13 To ensure that the ligand exchangereaction was complete, we then added one extra equivalent ofthiol (i.e., a mixture of θ equiv of Am and (1 − θ) equiv of Bn)(step 3 in Figure 2a). After ∼1 h of mixing, the functionalizedNPs were washed, dried, and dissolved in deionized water (seeSupporting Information, Section 4.1.2, for details).Figure 2b shows a representative transmission electron
microscopy (TEM) image of A1/B1-functionalized 2.5 nmNPs. Comparing TEM images before and after ligand exchangeallowed us to conclude that our functionalization protocol didnot affect the size or size distribution of the NPs. (A validquestion is whether the three-step functionalization procedureyields monolayers of intermixed ligands, as opposed to patchesof azobenzene and background thiol;44 we will address thisissue later, with several observations supporting the formerscenario.) UV/vis absorption spectra of aqueous solutions ofA1/B1-functionalized NPs obtained with increasing values of θdisplayed increasing absorption at ∼350 nm (due to the π →π* transition of trans-azobenzene, Figure 2c). We found thatNPs prepared with as much as θ = 0.4 exhibited excellentaqueous solubility (with no signs of NP aggregation by TEM,dynamic light scattering (DLS), or UV/vis absorptionspectroscopy). When θ > 0.4, however, the NPs could notbe redispersed in water owing to the hydrophobic character ofthe terminal azobenzene groups.To determine the actual molar fraction of A1 on the
functionalized NPs (i.e., χ), we subtracted the spectrum of 2.5nm NPs functionalized with a single-component monolayer ofB1 (gray in Figure 2c) from the spectra of A1/B1-decoratedNPs. Provided that the background ligand B1 exhibits noabsorption in the near-UV and visible range, the operationafforded the optical response of pure A1, based on which its
concentration could be evaluated (see Supporting Information,Section 5). The green markers in Figure 2d denote thedependence of χ on θ for NPs functionalized by theconsecutive addition of the two thiols. With an averagedeviation of ∼11% (based on the four data points), thismethod allows us to predictably control the fractional coverageof azobenzene on Au NPs. In contrast, the simultaneousaddition of A1 and B1 led to a much higher deviation of ∼94%(Figure 2d, red markers).Having demonstrated the ability to control the amount of
A1 on the surfaces of NPs, we sought approaches to further(i.e., beyond χ = 0.36) increase the loading of azobenzene onAu NPs while keeping the particles soluble in water. First, wereplaced A1’s alkyl chain with the hydrophilic tris(ethyleneglycol) to afford A2 (Figure 1). With the same backgroundligand B1, the molar fraction of A2 on 2.5 nm water-solubleNPs could be increased up to χ ≈ 0.53 (Figure 2e). The A2/B1 combination emphasized the usefulness of the three-stepNP functionalization protocol adopted in this work; as the redmarkers in Figure 2e show, treating 2.5 nm NPs simultaneouslywith a mixture of 5 equiv of A2 and 5 equiv of B1 resulted inNPs on which no A2 could be detected. On the other hand,following the modified protocol allowed us to place desiredamounts of A2 on the NPs, with an average deviation (from θ= χ) of only ∼23% (Figure 2e, green markers). We alsosynthesized A3, which contains an extra quaternary ammoniumgroup at the 4′ position of azobenzene. This presence of thishighly polar group allowed us to further increase the molarfraction of azobenzene on Au NPs to χ = 0.84 (see SupportingInformation, Figure S56b).To verify the applicability of our functionalization protocol
to NPs of other sizes, we worked with 5.5 nm, DDA-capped AuNPs. Similar to 2.5 nm NPs, these larger particles could bereadily functionalized with different Am/Bn mixtures such thatthe resulting χ approached the expected θ value (seeSupporting Information, Section 4.2.1, for details). Forexample, the A1:B1 ratio on the NPs differed by only ∼12%from the expected one (average value based on three samples).To further demonstrate the proof-of-concept, we worked witha mixture of A2 and a newly synthesized polar ligand, B2.Figure 2f shows a representative TEM image of A2/B2-functionalized NPs at θ = 0.20 (corresponding to χ = 0.15),and the UV/vis spectra in Figure 2g correspond to NPsdecorated with three different A2:B2 ratios. Based on thesethree samples, we determined the average deviation from θ = χto be ∼14%. Experiments with multiple other Am/Bncombinations confirmed that our functionalization procedureallows us to control the molar fraction of azobenzene on both2.5 and 5.5 nm NPs.
Reversible Isomerization of Azobenzene on Water-Soluble Nanoparticles. It was of critical importance to verifythat azobenzenes adsorbed on the surfaces of gold NPs retaintheir photoswitchable properties. Figure 3a shows changes inthe UV/vis spectra of A1/B1-functionalized 2.5 nm NPs (χ =0.17) resulting from exposure to a low-intensity (∼0.7 mW·cm−2) hand-held UV light source. Within several minutes ofUV irradiation, the band at ∼350 nm decreased nearly to thebackground level, indicative of trans → cis azobenzeneisomerization. Notably, azobenzene switching did not affectthe high colloidal stability of the NPs (no absorption increasein the long-wavelength region, Figure 3a). Therefore, theseNPs behave differently than the azobenzene-coated NPs do inhydrophobic solvents, which, upon UV irradiation, readily
Journal of the American Chemical Society Article
DOI: 10.1021/jacs.8b09638J. Am. Chem. Soc. 2019, 141, 1949−1960
1951
assemble into metastable aggregates to minimize contact withthe nonpolar environment.30,45,46
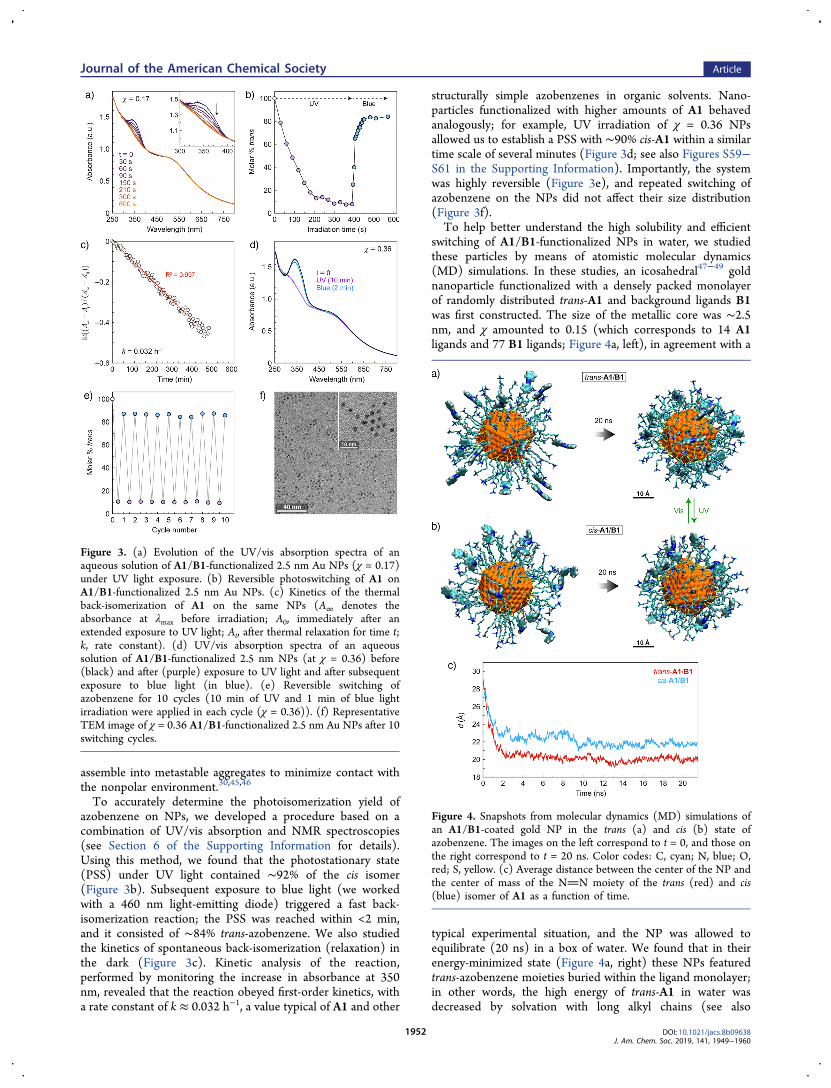
To accurately determine the photoisomerization yield ofazobenzene on NPs, we developed a procedure based on acombination of UV/vis absorption and NMR spectroscopies(see Section 6 of the Supporting Information for details).Using this method, we found that the photostationary state(PSS) under UV light contained ∼92% of the cis isomer(Figure 3b). Subsequent exposure to blue light (we workedwith a 460 nm light-emitting diode) triggered a fast back-isomerization reaction; the PSS was reached within <2 min,and it consisted of ∼84% trans-azobenzene. We also studiedthe kinetics of spontaneous back-isomerization (relaxation) inthe dark (Figure 3c). Kinetic analysis of the reaction,performed by monitoring the increase in absorbance at 350nm, revealed that the reaction obeyed first-order kinetics, witha rate constant of k ≈ 0.032 h−1, a value typical of A1 and other
structurally simple azobenzenes in organic solvents. Nano-particles functionalized with higher amounts of A1 behavedanalogously; for example, UV irradiation of χ = 0.36 NPsallowed us to establish a PSS with ∼90% cis-A1 within a similartime scale of several minutes (Figure 3d; see also Figures S59−S61 in the Supporting Information). Importantly, the systemwas highly reversible (Figure 3e), and repeated switching ofazobenzene on the NPs did not affect their size distribution(Figure 3f).To help better understand the high solubility and efficient
switching of A1/B1-functionalized NPs in water, we studiedthese particles by means of atomistic molecular dynamics(MD) simulations. In these studies, an icosahedral47−49 goldnanoparticle functionalized with a densely packed monolayerof randomly distributed trans-A1 and background ligands B1was first constructed. The size of the metallic core was ∼2.5nm, and χ amounted to 0.15 (which corresponds to 14 A1ligands and 77 B1 ligands; Figure 4a, left), in agreement with a
typical experimental situation, and the NP was allowed toequilibrate (20 ns) in a box of water. We found that in theirenergy-minimized state (Figure 4a, right) these NPs featuredtrans-azobenzene moieties buried within the ligand monolayer;in other words, the high energy of trans-A1 in water wasdecreased by solvation with long alkyl chains (see also
Figure 3. (a) Evolution of the UV/vis absorption spectra of anaqueous solution of A1/B1-functionalized 2.5 nm Au NPs (χ = 0.17)under UV light exposure. (b) Reversible photoswitching of A1 onA1/B1-functionalized 2.5 nm Au NPs. (c) Kinetics of the thermalback-isomerization of A1 on the same NPs (A∞ denotes theabsorbance at λmax before irradiation; A0, immediately after anextended exposure to UV light; At, after thermal relaxation for time t;k, rate constant). (d) UV/vis absorption spectra of an aqueoussolution of A1/B1-functionalized 2.5 nm NPs (at χ = 0.36) before(black) and after (purple) exposure to UV light and after subsequentexposure to blue light (in blue). (e) Reversible switching ofazobenzene for 10 cycles (10 min of UV and 1 min of blue lightirradiation were applied in each cycle (χ = 0.36)). (f) RepresentativeTEM image of χ = 0.36 A1/B1-functionalized 2.5 nm Au NPs after 10switching cycles.
Figure 4. Snapshots from molecular dynamics (MD) simulations ofan A1/B1-coated gold NP in the trans (a) and cis (b) state ofazobenzene. The images on the left correspond to t = 0, and those onthe right correspond to t = 20 ns. Color codes: C, cyan; N, blue; O,red; S, yellow. (c) Average distance between the center of the NP andthe center of mass of the NN moiety of the trans (red) and cis(blue) isomer of A1 as a function of time.
Journal of the American Chemical Society Article
DOI: 10.1021/jacs.8b09638J. Am. Chem. Soc. 2019, 141, 1949−1960
1952

Supporting Information, Figure S84). The positively chargedammonium groups of B1, on the other hand, retained theirinitial protruding configuration, which can explain the excellentwater solubility of these NPs despite the presence of thehydrophobic trans-azobenzene groups.We separately considered a cis-A1/B1-coated NP (Figure
4b). Similar to their trans isomers, the cis-azobenzene groupsbecame buried within the nonpolar monolayer, albeit to alesser (by ∼2 Å) extent, which can be visualized by plotting theaverage distance of the center of mass of the NN moiety tothe center of the NP (Figure 4c). This result can berationalized by the more hydrophilic character of the cisform, which can interact with water molecules via its nitrogens’lone electron pairs.50,51 On the basis of these results, wepostulate that azobenzene switching in A1/B1-functionalizedNPs occurs within the nonpolar nanoenvironment of the NP-bound alkyl chains52 (Figure 4, green arrows) rather than inthe aqueous phase.We then proceeded to study azobenzene photoswitching on
NPs functionalized with other Am/Bn combinations. Both A2/B1- and A3/B1-functionalized 2.5 nm NPs behavedanalogously to A1/B1 NPs, with the photoisomerizationreactions completed within ∼10 min for the trans→ cis and ∼2min for the cis → trans reaction, respectively (SupportingInformation, Figures S62−S67 and S85). However, increasingthe loading of azobenzene on the NPs led to poorer PSSsunder the same irradiation conditions (e.g., ∼19% residualtrans-A3 for χ = 0.16 A3/B1-coated NPs, but as much as∼29% trans-A3 for χ = 0.84 NPs). Interestingly, varying χ hadno effect on the kinetics of the back-isomerization reaction: wefound that cis-A2 on A2/B1-coated 2.5 nm NPs relaxed withthe same rate constant of k ≈ 0.024 h−1 irrespective of χ (inthe range 0.15 ≤ χ ≤ 0.49) (Supporting Information, FigureS68). We were also interested in determining whether thekinetics of thermal relaxation are affected by NP curvature. Tothis end, we prepared two batches of differently sized Au NPs(2.5 and 5.5 nm) functionalized with mixtures of A2 and B2 (χ= 0.13 for 2.5 nm and 0.15 for 5.5 nm) and verified that bothcould be switched upon UV light irradiation (these studieswere performed in a 9:1 (v/v) water−methanol mixture as thesolvent; A2/B2-coated 5.5 nm NPs could not be redispersed inpure water). However, back-isomerization of A2 on 5.5 nmNPs proceeded markedly (by approximately 2-fold) slower,compared to 2.5 nm NPs (Supporting Information, FigureS69). This result can be explained by the smaller curvatureassociated with the larger particles,17,53 which can increasemolecular crowding, thus stabilizing the cis isomer.Next, we investigated the effect of ligand length on the
switching properties of NP-bound azobenzene. To this end, wefunctionalized Au NPs with a mixture of a short thiolated-azobenzene A4 and background ligand B3 and found that theresulting NPs were readily soluble in water. However, exposureto UV did not induce any changes in the absorption spectra ofthese NPs, indicating that trans-azobenzene groups residingclose to the gold surface are difficult to photoisomerize. Thiscan be attributed to the quenching of the excited state ofazobenzene by gold, in agreement with previous literaturereports.15,54,55 We therefore considered combining the shortbackground ligand, B3, and the long-chain azobenzene, A1(Figure 5). Similar to A1/B1-coated NPs, azobenzene A1coadsorbed with B3 could be readily switched for many cycles,and the reversible isomerization was not accompanied by NPaggregation.
The good colloidal stability of A1/B3-functionalized NPs inwater may be surprising, given the lack of background ligands’long alkyl chains capable of solvating the azobenzene groups(compare with the right panels of Figure 4a and b). To helpexplain the efficient hydration of these NPs, we performedadditional MD simulations and found that energy-minimizedconfigurations of these NPs in water featured small bundles(aggregates) of azobenzene (Figure 5c,d). As expected, thetrans isomer of A1 exhibited a higher propensity to aggregate,with aggregates of up to five azobenzene units, whereas themore polar cis-A1 afforded a ∼1:1 mixture of free anddimerized azobenzenes. These results indicate that in theabsence of a nonpolar nanoenvironment on the NP surfaces,the surface energy of the NP−water interface is decreased bystacking the azobenzene units.Interestingly, we repeatedly observed that the trans → cis
isomerization of azobenzene within the putative bundles onA1/B3-functionalized NPs proceeds faster (by a factor of ∼2)than on A1/B1- functionalized NPs at the same value of χ. Theseemingly counterintuitive observation that aggregated azo-benzenes isomerize faster than isolated ones can be explainedby a cooperative switching mechanism, whereby isomerizationof an azobenzene unit is facilitated by the switching of its
Figure 5. (a) UV/vis absorption spectra of an aqueous solution ofA1/B3-functionalized 2.5 nm Au NPs before (black) and after(purple) exposure to UV light and after subsequent exposure to bluelight (in blue). (b) Reversible photoswitching of A1 on the same NPs.(c, d) Snapshots from MD simulations of an A1/B3-coated gold NPin the trans (c) and cis (d) state of azobenzene. The images on the leftcorrespond to t = 0, and those on the right correspond to t = 16 ns.
Journal of the American Chemical Society Article
DOI: 10.1021/jacs.8b09638J. Am. Chem. Soc. 2019, 141, 1949−1960
1953
neighbor. This mechanism has previously been used torationalize the accelerated isomerization of azobenzene in thecrystalline state18 and within molecules self-assembled onplanar substrates.16,17,19,20
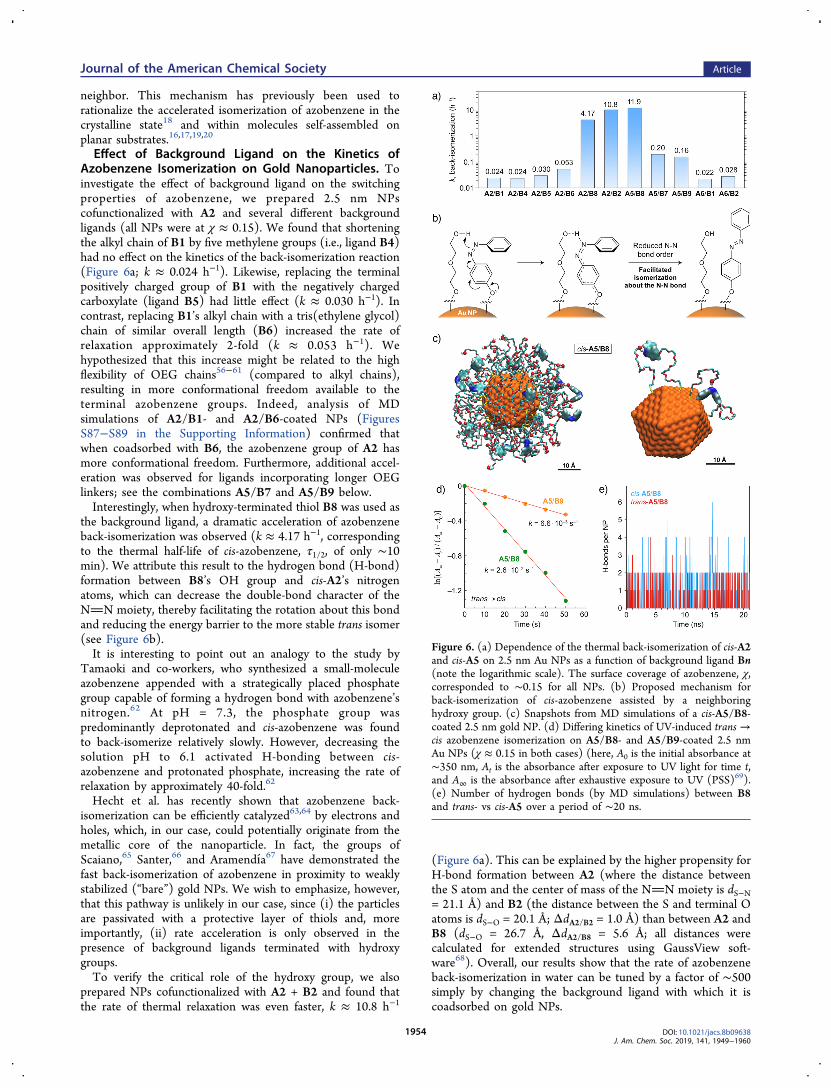
Effect of Background Ligand on the Kinetics ofAzobenzene Isomerization on Gold Nanoparticles. Toinvestigate the effect of background ligand on the switchingproperties of azobenzene, we prepared 2.5 nm NPscofunctionalized with A2 and several different backgroundligands (all NPs were at χ ≈ 0.15). We found that shorteningthe alkyl chain of B1 by five methylene groups (i.e., ligand B4)had no effect on the kinetics of the back-isomerization reaction(Figure 6a; k ≈ 0.024 h−1). Likewise, replacing the terminalpositively charged group of B1 with the negatively chargedcarboxylate (ligand B5) had little effect (k ≈ 0.030 h−1). Incontrast, replacing B1’s alkyl chain with a tris(ethylene glycol)chain of similar overall length (B6) increased the rate ofrelaxation approximately 2-fold (k ≈ 0.053 h−1). Wehypothesized that this increase might be related to the highflexibility of OEG chains56−61 (compared to alkyl chains),resulting in more conformational freedom available to theterminal azobenzene groups. Indeed, analysis of MDsimulations of A2/B1- and A2/B6-coated NPs (FiguresS87−S89 in the Supporting Information) confirmed thatwhen coadsorbed with B6, the azobenzene group of A2 hasmore conformational freedom. Furthermore, additional accel-eration was observed for ligands incorporating longer OEGlinkers; see the combinations A5/B7 and A5/B9 below.Interestingly, when hydroxy-terminated thiol B8 was used as
the background ligand, a dramatic acceleration of azobenzeneback-isomerization was observed (k ≈ 4.17 h−1, correspondingto the thermal half-life of cis-azobenzene, τ1/2, of only ∼10min). We attribute this result to the hydrogen bond (H-bond)formation between B8’s OH group and cis-A2’s nitrogenatoms, which can decrease the double-bond character of theNN moiety, thereby facilitating the rotation about this bondand reducing the energy barrier to the more stable trans isomer(see Figure 6b).It is interesting to point out an analogy to the study by
Tamaoki and co-workers, who synthesized a small-moleculeazobenzene appended with a strategically placed phosphategroup capable of forming a hydrogen bond with azobenzene’snitrogen.62 At pH = 7.3, the phosphate group waspredominantly deprotonated and cis-azobenzene was foundto back-isomerize relatively slowly. However, decreasing thesolution pH to 6.1 activated H-bonding between cis-azobenzene and protonated phosphate, increasing the rate ofrelaxation by approximately 40-fold.62
Hecht et al. has recently shown that azobenzene back-isomerization can be efficiently catalyzed63,64 by electrons andholes, which, in our case, could potentially originate from themetallic core of the nanoparticle. In fact, the groups ofScaiano,65 Santer,66 and Aramendıa67 have demonstrated thefast back-isomerization of azobenzene in proximity to weaklystabilized (“bare”) gold NPs. We wish to emphasize, however,that this pathway is unlikely in our case, since (i) the particlesare passivated with a protective layer of thiols and, moreimportantly, (ii) rate acceleration is only observed in thepresence of background ligands terminated with hydroxygroups.To verify the critical role of the hydroxy group, we also
prepared NPs cofunctionalized with A2 + B2 and found thatthe rate of thermal relaxation was even faster, k ≈ 10.8 h−1
(Figure 6a). This can be explained by the higher propensity forH-bond formation between A2 (where the distance betweenthe S atom and the center of mass of the NN moiety is dS−N= 21.1 Å) and B2 (the distance between the S and terminal Oatoms is dS−O = 20.1 Å; ΔdA2/B2 = 1.0 Å) than between A2 andB8 (dS−O = 26.7 Å, ΔdA2/B8 = 5.6 Å; all distances werecalculated for extended structures using GaussView soft-ware68). Overall, our results show that the rate of azobenzeneback-isomerization in water can be tuned by a factor of ∼500simply by changing the background ligand with which it iscoadsorbed on gold NPs.
Figure 6. (a) Dependence of the thermal back-isomerization of cis-A2and cis-A5 on 2.5 nm Au NPs as a function of background ligand Bn(note the logarithmic scale). The surface coverage of azobenzene, χ,corresponded to ∼0.15 for all NPs. (b) Proposed mechanism forback-isomerization of cis-azobenzene assisted by a neighboringhydroxy group. (c) Snapshots from MD simulations of a cis-A5/B8-coated 2.5 nm gold NP. (d) Differing kinetics of UV-induced trans →cis azobenzene isomerization on A5/B8- and A5/B9-coated 2.5 nmAu NPs (χ ≈ 0.15 in both cases) (here, A0 is the initial absorbance at∼350 nm, At is the absorbance after exposure to UV light for time t,and A∞ is the absorbance after exhaustive exposure to UV (PSS)69).(e) Number of hydrogen bonds (by MD simulations) between B8and trans- vs cis-A5 over a period of ∼20 ns.
Journal of the American Chemical Society Article
DOI: 10.1021/jacs.8b09638J. Am. Chem. Soc. 2019, 141, 1949−1960
1954

Extending the OEG chain of A2 by three EG units (i.e.,ligand A5) again resulted in a fast back-isomerization reaction(k ≈ 11.9 h−1 on A5/B8-functionalized 2.5 nm NPs,corresponding to a τ1/2 value of 3.5 min). Although this resultmay appear surprising given the relatively large ΔdA5/B8 = 5.5 Å(dS−N in A5 = 32.2 Å), it can be explained by the highflexibility of long OEG chains, which can facilitate interactionsbetween H-bond donors and acceptors. When, however, thedistance between the donor and acceptor sites was increased toΔd = 16.3 Å (using the A5 + B7 combination; dS−O in B7 =15.9 Å), the rate of back-isomerization dropped substantially(k ≈ 0.20 h−1; see Figure 6a).An ideal control experiment verifying the importance of
hydrogen bonding between coadsorbed azobenzene- andhydroxy-terminated thiols would be based on replacing theacidic hydrogen with a methyl group. To this end, wesynthesized ligand B9 and indeed found that the rate of back-isomerization decreased from 11.9 h−1 (for A5/B8) to 0.16 h−1
(A5/B9). This on-nanoparticle H-bonding was furtherinvestigated by MD simulations, which revealed the presenceof H-bonds between cis-A5 and B8 residing on the same NPs(Figure 6c). In sharp contrast, the terminal methoxy groups ofB9 were exposed to the solvent; see Supporting Information,Figure S91a.We separately simulated a mixture of trans-A5 and B8
coadsorbed on 2.5 nm Au NPs. These simulations showed thatthe trans isomer of A5 may also be capable of forming H-bondswith B8’s hydroxy group (Supporting Information, FigureS91b), thus possibly reducing the double-bond character of theNN group and increasing the rate of the trans→ cis forward-isomerization. To verify this hypothesis, we exposed NPsfunctionalized with trans-A5/B8 and trans-A5/B9 to UV lightfor increasing periods and found that, indeed, isomerizationproceeded faster with the OH-terminated B8 as the back-ground ligand (Figure 6d). The difference in rates, however,was much smaller than in the case of back-isomerization (∼4and ∼74, respectively), which can be accounted for by thelower percentage of trans-A5 vs cis-A5 engaged in H-bondingwith B8 (estimated as 0.7% and 1.9%, respectively, over aperiod of of 20 ns; Figure 6e) and by the lower strength of H-bonds formed by trans-A5 (a significant distortion fromplanarity in the O−H···N moiety; see Figure S91c in theSupporting Information).To verify the mechanism shown in Figure 6b (specifically,
the importance of the oxygen atom at the para position ofazobenzene), we also synthesized thiolated azobenzene A6with an alkylas opposed to alkoxysubstituent at the paraposition. Interestingly, this small change in the structure ofazobenzene had a profound effect on the kinetics of back-isomerization, with nearly no acceleration observed in thepresence of hydroxy-terminated background ligand B2(compare k ≈ 0.028 h−1 for A6/B2-coated NPs with k ≈10.8 h−1 for A2/B2-coated NPs; Supporting Information,Figures S71 and S73). These results are in agreement with ourearlier results on light-induced self-assembly of NPs innonpolar solvents, where we showed that the surface polarityof NPs coated with 4,4′-dialkoxysubstituted cis-azobenzenes ismuch higher compared to NPs functionalized with cis-azobenzenes having only one substituent at the paraposition.13,70 In other words, removing the electron-donatingalkoxy substituent in A6 decreased the electron density on thenitrogen atoms, thus decreasing the hydrogen-bond-acceptingcharacter of the cis-azobenzene group.
Next, we investigated the effect of solvent on theintermolecular interactions between azobenzene A5 andbackground ligands. To this end, we prepared concentratedsolutions of A5/B8-functionalized 2.5 nm Au NPs in water anddiluted them with 24 volumes of various organic solvents (i.e.,volume fractions of water in the resulting solutions = 4%). Wehypothesized that organic solvents can solvate azobenzeneand/or hydroxy groups, thus weakening the H-bondingbetween them. Indeed, the presence of organic solventsdecreased the rate of thermal relaxation of cis-A5 from k ≈ 11.9h−1 (pure water) all the way to k ≈ 0.025 h−1 (for 24:1dioxane−water) (Figure 7a; see also Supporting Information,Figure S74). Notably, the back-isomerization rates in DMF-,acetone-, and dioxane-rich solutions were similar to those offree A5 in organic solvents, suggesting excellent solvation of
Figure 7. (a) Rates of thermal back-isomerization of cis-A5 on A5/B8-functionalized 2.5 nm Au NPs in different 1:24 water−solventmixtures, where the solvent is indicated on the left. (b) Dependenceof the back-isomerization rate of cis-A5 on A5/B8-functionalized 2.5nm Au NPs as a function of the composition of the acetonitrile−watermixture. (c) Rates of the thermal back-isomerization of cis-A5 on A5/B9-functionalized 2.5 nm Au NPs in different 1:24 water−solventmixtures, where the solvent is indicated on the left (HCl solution inpure water).
Journal of the American Chemical Society Article
DOI: 10.1021/jacs.8b09638J. Am. Chem. Soc. 2019, 141, 1949−1960
1955
the terminal groups of A5 and B8 and no strong interactionsbetween them. Surprisingly, however, the addition ofacetonitrile (MeCN) led to a pronounced increase in thereaction rate (k ≈ 79.2 h−1), indicating efficient stabilization ofthe intermolecular interactions between A5 and B8 in thissolvent. To confirm that the >3000-fold difference (0.025 h−1
vs 79.2 h−1) in relaxation kinetics in dioxane- and MeCN-richsolutions is due to intermolecular interactions on NPs andinteractions of azobenzene with solvent molecules, weprepared solutions of small-molecule A5 in dioxane andMeCN and found only a small (∼1.2-fold) difference in theback-isomerization rates (Supporting Information, FigureS72). By studying the behavior of A5/B8-functionalized NPsin several different MeCN−water mixtures, we found that thetransition between the fast and the very fast regime occursbetween 87.5% and 90% v/v MeCN contents (Figure 7b; seealso Supporting Information, Figure S75). We thereforespeculate that when present at ≥10%, water can to someextent interfere with the on-NP H-bond formation; however at≤7.5%, no such competition exists.As expected, replacing B8’s OH group with OMe decreased
the kinetics of back-isomerization in all solvents (Figure 7c).The extent of decrease was most striking for the 24:1 MeCN−water mixture, where the relaxation rate of cis-A5 decreased bya factor of ∼6100 simply by replacing background ligand B8with B9 (as illustrated in the abstract graphic). We also studiedthe back-isomerization of cis-A5 coadsorbed with B9 in thepresence of a strong acid, potentially capable of reducing thebond order of the NN moiety, thus facilitating back-isomerization. Indeed, reducing a solution’s pH from 7 to ∼1.6increases the rate from ∼0.16 h−1 to ∼9.43 h−1 (23 mM HClin Figure 7c). Quite remarkably, k ≈ 9.43 h−1, observed forA5/B9-functionalized NPs at a low pH, was still lower than k≈ 11.9 h−1 found for A5/B8-coated NPs at pH = 7,emphasizing the importance of preorganization/high effectivemolarity on hydrogen bond formation.Having discussed the properties of azobenzenes on the
surfaces of water-soluble NPs, we revisit the issue of ligandmixing on NP surfaces. We argue that, with the exception ofNPs decorated with the very short background ligand B3(Figure 5c,d), thiolated azobenzenes Am and backgroundligands Bn are intermixed and do not phase-separate on NPs.This argument is supported by the following observations.First, NPs featuring patches of azobenzene would have apropensity to aggregate into small clusters by hydrophobicinteractions; no such clusters could be detected by TEM.Second, replacing the background ligand on azobenzene-coated NPs would have little effect on the switching propertiesof azobenzenes if Am and Bn were phase-separated; however,we found that the properties of azobenzene are greatly affectedby the background ligand. In particular, all the azobenzeneunits on NPs cofunctionalized with Am and hydroxy-terminated background ligands back-isomerize rapidly, suggest-ing that each has at least one OH-donating neighbor. Third,the back-isomerization rate was independent of χ, asdemonstrated for A2/B1-functionalized 2.5 nm gold NPs,suggesting that a good mixing of the two ligands exists. It isimportant to point out that the first step of our ligand-exchange protocol (i.e., treating DDA-coated NPs with asubstoichiometric amount of azobenzene) most likely doeslead to patches of azobenzene; it is the subsequent addition ofan excess of thiols that apparently results in equilibration tomonolayers of intermixed ligands.
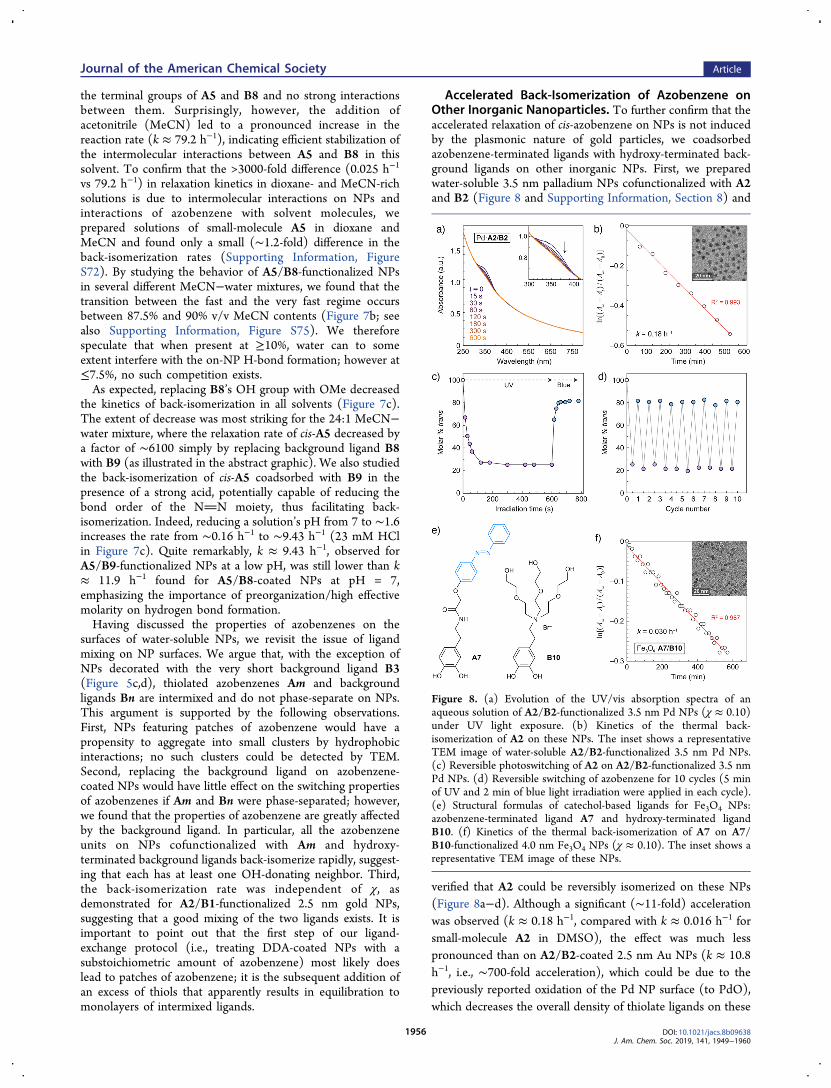
Accelerated Back-Isomerization of Azobenzene onOther Inorganic Nanoparticles. To further confirm that theaccelerated relaxation of cis-azobenzene on NPs is not inducedby the plasmonic nature of gold particles, we coadsorbedazobenzene-terminated ligands with hydroxy-terminated back-ground ligands on other inorganic NPs. First, we preparedwater-soluble 3.5 nm palladium NPs cofunctionalized with A2and B2 (Figure 8 and Supporting Information, Section 8) and
verified that A2 could be reversibly isomerized on these NPs(Figure 8a−d). Although a significant (∼11-fold) accelerationwas observed (k ≈ 0.18 h−1, compared with k ≈ 0.016 h−1 forsmall-molecule A2 in DMSO), the effect was much lesspronounced than on A2/B2-coated 2.5 nm Au NPs (k ≈ 10.8h−1, i.e., ∼700-fold acceleration), which could be due to thepreviously reported oxidation of the Pd NP surface (to PdO),which decreases the overall density of thiolate ligands on these
Figure 8. (a) Evolution of the UV/vis absorption spectra of anaqueous solution of A2/B2-functionalized 3.5 nm Pd NPs (χ ≈ 0.10)under UV light exposure. (b) Kinetics of the thermal back-isomerization of A2 on these NPs. The inset shows a representativeTEM image of water-soluble A2/B2-functionalized 3.5 nm Pd NPs.(c) Reversible photoswitching of A2 on A2/B2-functionalized 3.5 nmPd NPs. (d) Reversible switching of azobenzene for 10 cycles (5 minof UV and 2 min of blue light irradiation were applied in each cycle).(e) Structural formulas of catechol-based ligands for Fe3O4 NPs:azobenzene-terminated ligand A7 and hydroxy-terminated ligandB10. (f) Kinetics of the thermal back-isomerization of A7 on A7/B10-functionalized 4.0 nm Fe3O4 NPs (χ ≈ 0.10). The inset shows arepresentative TEM image of these NPs.
Journal of the American Chemical Society Article
DOI: 10.1021/jacs.8b09638J. Am. Chem. Soc. 2019, 141, 1949−1960
1956
NPs, thereby increasing the average distance between hydro-gen bond donors (OH groups) and acceptors (azobenzene).71
We also synthesized 4.0 nm Fe3O4 (magnetite) NPscofunctionalized with the previously reported45 azobenzeneA7 and hydroxy-terminated background ligand B10 (Figure8e). Despite considering several hydroxy-terminated back-ground ligands, we did not succeed in preparing water-soluble,azobenzene-coated Fe3O4 NPs and studied their properties in2:1 v/v toluene−methanol. Azobenzene A7 on these NPsback-isomerized with k ≈ 0.030 h−1 (Figure 8f), correspondingto a minor acceleration effect (∼3-fold; k ≈ 0.0097 h−1 for freeA7 in the same solvent mixture; Supporting Information,Figure S78), which could be attributed to the relatively largefootprint of catechol-based ligands45,72 and consequentlydecreased intermolecular interactions between A7 and B10.Nevertheless, the accelerated reaction on both Pd and Fe3O4NPs, together with the other experimental observations andcomputer simulations, indicate that fast thermal relaxation ofcis-azobenzene on nanoparticulate platforms is predominantly(if not exclusively) due to intermolecular coupling to thenearby hydroxy-terminated ligands.Wetting Properties of Aqueous Solutions of Azo-
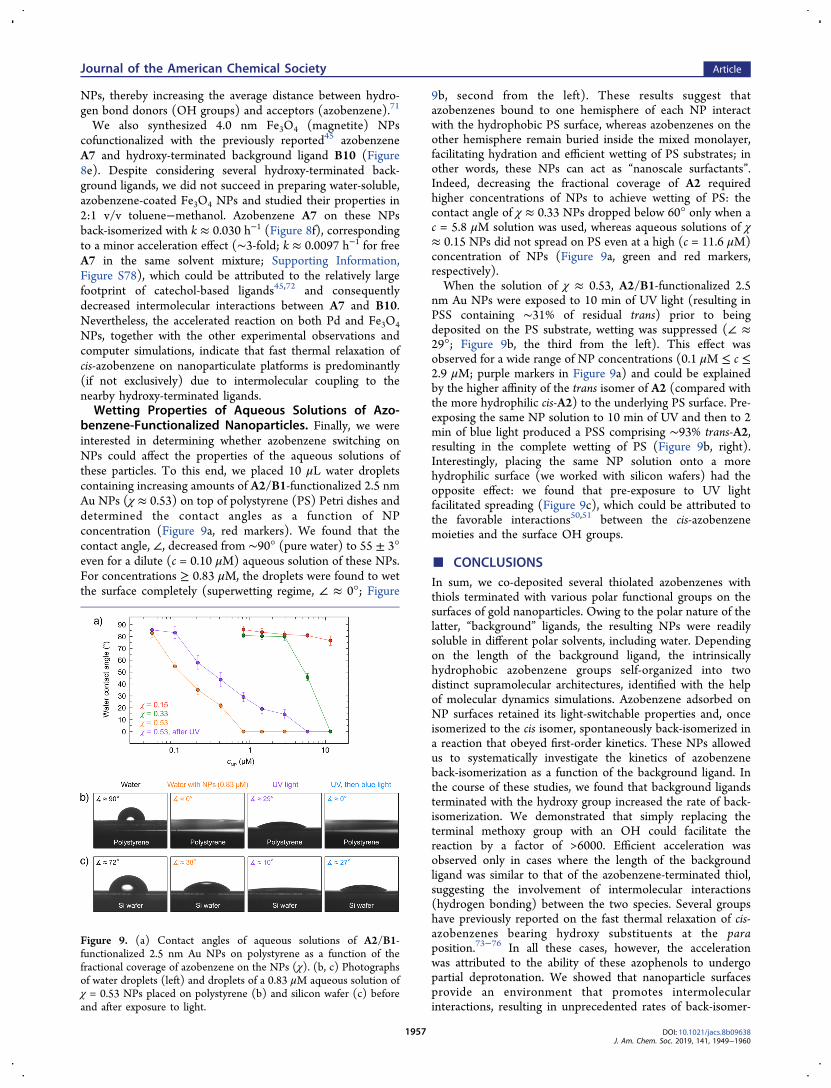
benzene-Functionalized Nanoparticles. Finally, we wereinterested in determining whether azobenzene switching onNPs could affect the properties of the aqueous solutions ofthese particles. To this end, we placed 10 μL water dropletscontaining increasing amounts of A2/B1-functionalized 2.5 nmAu NPs (χ ≈ 0.53) on top of polystyrene (PS) Petri dishes anddetermined the contact angles as a function of NPconcentration (Figure 9a, red markers). We found that thecontact angle, ∠, decreased from ∼90° (pure water) to 55 ± 3°even for a dilute (c = 0.10 μM) aqueous solution of these NPs.For concentrations ≥ 0.83 μM, the droplets were found to wetthe surface completely (superwetting regime, ∠ ≈ 0°; Figure
9b, second from the left). These results suggest thatazobenzenes bound to one hemisphere of each NP interactwith the hydrophobic PS surface, whereas azobenzenes on theother hemisphere remain buried inside the mixed monolayer,facilitating hydration and efficient wetting of PS substrates; inother words, these NPs can act as “nanoscale surfactants”.Indeed, decreasing the fractional coverage of A2 requiredhigher concentrations of NPs to achieve wetting of PS: thecontact angle of χ ≈ 0.33 NPs dropped below 60° only when ac = 5.8 μM solution was used, whereas aqueous solutions of χ≈ 0.15 NPs did not spread on PS even at a high (c = 11.6 μM)concentration of NPs (Figure 9a, green and red markers,respectively).When the solution of χ ≈ 0.53, A2/B1-functionalized 2.5
nm Au NPs were exposed to 10 min of UV light (resulting inPSS containing ∼31% of residual trans) prior to beingdeposited on the PS substrate, wetting was suppressed (∠ ≈29°; Figure 9b, the third from the left). This effect wasobserved for a wide range of NP concentrations (0.1 μM ≤ c ≤2.9 μM; purple markers in Figure 9a) and could be explainedby the higher affinity of the trans isomer of A2 (compared withthe more hydrophilic cis-A2) to the underlying PS surface. Pre-exposing the same NP solution to 10 min of UV and then to 2min of blue light produced a PSS comprising ∼93% trans-A2,resulting in the complete wetting of PS (Figure 9b, right).Interestingly, placing the same NP solution onto a morehydrophilic surface (we worked with silicon wafers) had theopposite effect: we found that pre-exposure to UV lightfacilitated spreading (Figure 9c), which could be attributed tothe favorable interactions50,51 between the cis-azobenzenemoieties and the surface OH groups.
■ CONCLUSIONSIn sum, we co-deposited several thiolated azobenzenes withthiols terminated with various polar functional groups on thesurfaces of gold nanoparticles. Owing to the polar nature of thelatter, “background” ligands, the resulting NPs were readilysoluble in different polar solvents, including water. Dependingon the length of the background ligand, the intrinsicallyhydrophobic azobenzene groups self-organized into twodistinct supramolecular architectures, identified with the helpof molecular dynamics simulations. Azobenzene adsorbed onNP surfaces retained its light-switchable properties and, onceisomerized to the cis isomer, spontaneously back-isomerized ina reaction that obeyed first-order kinetics. These NPs allowedus to systematically investigate the kinetics of azobenzeneback-isomerization as a function of the background ligand. Inthe course of these studies, we found that background ligandsterminated with the hydroxy group increased the rate of back-isomerization. We demonstrated that simply replacing theterminal methoxy group with an OH could facilitate thereaction by a factor of >6000. Efficient acceleration wasobserved only in cases where the length of the backgroundligand was similar to that of the azobenzene-terminated thiol,suggesting the involvement of intermolecular interactions(hydrogen bonding) between the two species. Several groupshave previously reported on the fast thermal relaxation of cis-azobenzenes bearing hydroxy substituents at the paraposition.73−76 In all these cases, however, the accelerationwas attributed to the ability of these azophenols to undergopartial deprotonation. We showed that nanoparticle surfacesprovide an environment that promotes intermolecularinteractions, resulting in unprecedented rates of back-isomer-
Figure 9. (a) Contact angles of aqueous solutions of A2/B1-functionalized 2.5 nm Au NPs on polystyrene as a function of thefractional coverage of azobenzene on the NPs (χ). (b, c) Photographsof water droplets (left) and droplets of a 0.83 μM aqueous solution ofχ = 0.53 NPs placed on polystyrene (b) and silicon wafer (c) beforeand after exposure to light.
Journal of the American Chemical Society Article
DOI: 10.1021/jacs.8b09638J. Am. Chem. Soc. 2019, 141, 1949−1960
1957

ization even in azobenzenes lacking the hydroxy group. Ourresults pave the way toward the development of novel light-responsive materials operating in aqueous media and, in thelong run, in biological environments. Importantly, these studiescan readily be extended to other switchable molecules77−79 andbackground ligands, as well as to nanoparticles of other sizesand compositions, including upconversion NPs.80
■ ASSOCIATED CONTENT*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/jacs.8b09638.
Figures S1−S92; Schemes S1−S9; detailed descriptionof the synthesis and analysis of ligands, preparation,functionalization, and analysis of nanoparticles, molec-ular dynamics simulations; description of supportingmovies; supporting references (PDF)Movie (MPG)Movie (MPG)Movie (MPG)Movie (MPG)
■ AUTHOR INFORMATIONCorresponding Author*[email protected] De: 0000-0003-4674-9183Petr Kral: 0000-0003-2992-9027Rafal Klajn: 0000-0002-6320-8875NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSThis work was supported by the European Research Council(grant #336080 to R.K.), the Israel Ministry of Science(China−Israel cooperation, grant 3-13555 to R.K.), and theNSF (Division of Materials Research, grant #1506886 to P.K.).Z.C. acknowledges support from the Planning and BudgetingCommittee of the Council for Higher Education, the KoshlandFoundation, and a McDonald−Leapman grant. Anton I.Hanopolskyi is acknowledged for donating crude compound22.
■ REFERENCES(1) Hartley, G. S. The Cis-Form of Azobenzene. Nature 1937, 140,281.(2) Peters, M. V.; Stoll, R. S.; Kuhn, A.; Hecht, S. Photoswitching ofBasicity. Angew. Chem., Int. Ed. 2008, 47, 5968−5972.(3) Imahori, T.; Yamaguchi, R.; Kurihara, S. Azobenzene-TetheredBis(Trityl Alcohol) as a Photoswitchable Cooperative Acid Catalystfor Morita-Baylis-Hillman Reactions. Chem. - Eur. J. 2012, 18, 10802−10807.(4) Osorio-Planes, L.; Rodríguez-Escrich, C.; Pericas, M. A.Photoswitchable Thioureas for the External Manipulation of CatalyticActivity. Org. Lett. 2014, 16, 1704−1707.(5) Shinkai, S.; Minami, T.; Kusano, Y.; Manabe, O. Photo-responsive Crown Ether. 2. Photocontrol of Ion Extraction and IonTransport by a Bis(crown Ether) with a Butterfly-Like Motion. J. Am.Chem. Soc. 1983, 105, 1851−1856.(6) Laprell, L.; Repak, E.; Franckevicius, V.; Hartrampf, F.; Terhag,J.; Hollmann, M.; Sumser, M.; Rebola, N.; DiGregorio, D. A.;Trauner, D. Optical Control of NMDA Receptors with a DiffusiblePhotoswitch. Nat. Commun. 2015, 6, 8076.
(7) Velema, W. A.; van der Berg, J. P.; Hansen, M. J.; Szymanski, W.;Driessen, A. J. M.; Feringa, B. L. Optical Control of AntibacterialActivity. Nat. Chem. 2013, 5, 924−928.(8) Muraoka, T.; Kinbara, K.; Aida, T. Mechanical Twisting of aGuest by a Photoresponsive Host. Nature 2006, 440, 512−515.(9) Jaschke, M.; Schonherr, H.; Wolf, H.; Butt, H. J.; Bamberg, E.;Besocke, M. K.; Ringsdorf, H. Structure of Alkyl and PerfluoroalkylDisulfide and Azobenzenethiol Monolayers on Gold(111) Revealedby Atomic Force Microscopy. J. Phys. Chem. 1996, 100, 2290−2301.(10) Yu, H. Z.; Wang, Y. Q.; Cheng, J. Z.; Zhao, J. W.; Cai, S. M.;Inokuchi, H.; Fujishima, A.; Liu, Z. F. Electrochemical Behavior ofAzobenzene Self-Assembled Monolayers on Gold. Langmuir 1996, 12,2843−2848.(11) Wang, R.; Iyoda, T.; Jiang, L.; Tryk, D. A.; Hashimoto, K.;Fujishima, A. Structural Investigation of Azobenzene-Containing Self-Assembled Monolayer Films. J. Electroanal. Chem. 1997, 438, 213−219.(12) Kondo, T.; Kanai, T.; Uosaki, K. Control of the Charge-Transfer Rate at a Gold Electrode Modified with a Self-AssembledMonolayer Containing Ferrocene and Azobenzene by Electro- andPhotochemical Structural Conversion of Cis and Trans Forms of theAzobenzene Moiety. Langmuir 2001, 17, 6317−6324.(13) Klajn, R.; Bishop, K. J. M.; Grzybowski, B. A. Light-ControlledSelf-Assembly of Reversible and Irreversible Nanoparticle Supra-structures. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 10305−10309.(14) Samanta, D.; Gemen, J.; Chu, Z.; Diskin-Posner, Y.; Shimon, L.J. W.; Klajn, R. Reversible Photochromism of EncapsulatedAzobenzenes in Water. Proc. Natl. Acad. Sci. U. S. A. 2018, 115,9379−9384.(15) Klajn, R. Immobilized Azobenzenes for the Construction ofPhotoresponsive Materials. Pure Appl. Chem. 2010, 82, 2247−2279.(16) Pace, G.; Ferri, V.; Grave, C.; Elbing, M.; von Hanisch, C.;Zharnikov, M.; Mayor, M.; Rampi, M. A.; Samorì, P. CooperativeLight-Induced Molecular Movements of Highly Ordered AzobenzeneSelf-Assembled Monolayers. Proc. Natl. Acad. Sci. U. S. A. 2007, 104,9937−9942.(17) Moldt, T.; Brete, D.; Przyrembel, D.; Das, S.; Goldman, J. R.;Kundu, P. K.; Gahl, C.; Klajn, R.; Weinelt, M. Tailoring the Propertiesof Surface-Immobilized Azobenzenes by Monolayer Dilution andSurface Curvature. Langmuir 2015, 31, 1048−1057.(18) Lai, C. Y.; Raj, G.; Liepuoniute, I.; Chiesa, M.; Naumov, P.Direct Observation of Photoinduced Trans-Cis Isomerization onAzobenzene Single Crystal. Cryst. Growth Des. 2017, 17, 3306−3312.(19) Zheng, Y. B.; Pathem, B. K.; Hohman, J. N.; Thomas, J. C.;Kim, M.; Weiss, P. S. Photoresponsive Molecules in Well-DefinedNanoscale Environments. Adv. Mater. 2013, 25, 302−312.(20) Pathem, B. K.; Claridge, S. A.; Zheng, Y. B.; Weiss, P. S.Molecular Switches and Motors on Surfaces. Annu. Rev. Phys. Chem.2013, 64, 605−630.(21) Hostetler, M. J.; Templeton, A. C.; Murray, R. W. Dynamics ofPlace-Exchange Reactions on Monolayer-Protected Gold ClusterMolecules. Langmuir 1999, 15, 3782−3789.(22) Liu, X.; Yu, M.; Kim, H.; Mameli, M.; Stellacci, F.Determination of Monolayer-Protected Gold Nanoparticle Ligand-Shell Morphology using NMR. Nat. Commun. 2012, 3, 1182.(23) Kay, E. R. Dynamic Covalent Nanoparticle Building Blocks.Chem. - Eur. J. 2016, 22, 10706−10716.(24) Yang, Y.; Serrano, L. A.; Guldin, S. A. Versatile AuNP SyntheticPlatform for Decoupled Control of Size and Surface Composition.Langmuir 2018, 34, 6820−6826.(25) Chu, Z. L.; Han, Y. X.; Kral, P.; Klajn, R. ″Precipitation onNanoparticles″: Attractive Intermolecular Interactions StabilizeSpecific Ligand Ratios on the Surfaces of Nanoparticles. Angew.Chem., Int. Ed. 2018, 57, 7023−7027.(26) Edwards, W.; Marro, N.; Turner, G.; Kay, E. R. ContinuumTuning of Nanoparticle Interfacial Properties by Dynamic CovalentExchange. Chem. Sci. 2018, 9, 125−133.(27) Klajn, R.; Fang, L.; Coskun, A.; Olson, M. A.; Wesson, P. J.;Stoddart, J. F.; Grzybowski, B. A. Metal Nanoparticles Functionalized
Journal of the American Chemical Society Article
DOI: 10.1021/jacs.8b09638J. Am. Chem. Soc. 2019, 141, 1949−1960
1958

with Molecular and Supramolecular Switches. J. Am. Chem. Soc. 2009,131, 4233−4235.(28) Manna, A.; Chen, P. L.; Akiyama, H.; Wei, T. X.; Tamada, K.;Knoll, W. Optimized Photoisomerization on Gold NanoparticlesCapped by Unsymmetrical Azobenzene Disulfides. Chem. Mater.2003, 15, 20−28.(29) Raimondo, C.; Reinders, F.; Soydaner, U.; Mayor, M.; Samorì,P. Light-Responsive Reversible Solvation and Precipitation of GoldNanoparticles. Chem. Commun. 2010, 46, 1147−1149.(30) Chovnik, O.; Balgley, R.; Goldman, J. R.; Klajn, R. DynamicallySelf-Assembling Carriers Enable Guiding of Diamagnetic Particles byWeak Magnets. J. Am. Chem. Soc. 2012, 134, 19564−19567.(31) Kohntopp, A.; Dabrowski, A.; Malicki, M.; Temps, F.Photoisomerisation and Ligand-Controlled Reversible Aggregationof Azobenzene-Functionalised Gold Nanoparticles. Chem. Commun.2014, 50, 10105−10107.(32) Lee, J.-W.; Klajn, R. Dual-Responsive Nanoparticles thatAggregate under the Simultaneous Action of Light and CO2. Chem.Commun. 2015, 51, 2036−2039.(33) Velema, W. A.; Szymanski, W.; Feringa, B. L. Photo-pharmacology: Beyond Proof of Principle. J. Am. Chem. Soc. 2014,136, 2178−2191.(34) Broichhagen, J.; Frank, J. A.; Trauner, D. A Roadmap toSuccess in Photopharmacology. Acc. Chem. Res. 2015, 48, 1947−1960.(35) Bidoggia, S.; Milocco, F.; Polizzi, S.; Canton, P.; Saccani, A.;Sanavio, B.; Krol, S.; Stellacci, F.; Pengo, P.; Pasquato, L. Fluorinatedand Charged Hydrogenated Alkanethiolates Grafted on Gold:Expanding the Diversity of Mixed-Monolayer Nanoparticles forBiological Applications. Bioconjugate Chem. 2017, 28, 43−52.(36) Kalsin, A. M.; Kowalczyk, B.; Wesson, P.; Paszewski, M.;Grzybowski, B. A. Studying the Thermodynamics of SurfaceReactions on Nanoparticles by Electrostatic Titrations. J. Am. Chem.Soc. 2007, 129, 6664−6665.(37) Gao, J. H.; Zhang, O.; Ren, J.; Wu, C. L.; Zhao, Y. B.Aromaticity/Bulkiness of Surface Ligands to Promote the Interactionof Anionic Amphiphilic Gold Nanoparticles with Lipid Bilayers.Langmuir 2016, 32, 1601−1610.(38) Rao, A.; Roy, S.; Unnikrishnan, M.; Bhosale, S. S.; Devatha, G.;Pillai, P. P. Regulation of Interparticle Forces Reveals ControlledAggregation in Charged Nanoparticles. Chem. Mater. 2016, 28, 2348−2355.(39) Lee, H. Y.; Shin, S. H. R.; Drews, A. M.; Chirsan, A. M.; Lewis,S. A.; Bishop, K. J. M. ACS Nano 2014, 8, 9979−9987.(40) Pillai, P. P.; Kowalczyk, B.; Pudlo, W. J.; Grzybowski, B. A.Electrostatic Titrations Reveal Surface Compositions of Mixed, On-Nanoparticle Monolayers Comprising Positively and NegativelyCharged Ligands. J. Phys. Chem. C 2016, 120, 4139−4144.(41) Lee, H. Y.; Shin, S. H. R.; Abezgauz, L. L.; Lewis, S. A.; Chirsan,A. M.; Danino, D. D.; Bishop, K. J. M. Integration of GoldNanoparticles into Bilayer Structures via Adaptive Surface Chemistry.J. Am. Chem. Soc. 2013, 135, 5950−5953.(42) Bradford, S. M.; Fisher, E. A.; Meli, M. V. Ligand ShellComposition-Dependent Effects on the Apparent Hydrophobicityand Film Behavior of Gold Nanoparticles at the Air-Water Interface.Langmuir 2016, 32, 9790−9796.(43) Strong, L.; Whitesides, G. M. Structures of Self-AssembledMonolayer Films of Organosulfur Compounds Adsorbed on GoldSingle Crystals: Electron Diffraction Studies. Langmuir 1988, 4, 546−558.(44) Kim, H.; Carney, R. P.; Reguera, J.; Ong, Q. K.; Liu, X.;Stellacci, F. Synthesis and Characterization of Janus Gold Nano-particles. Adv. Mater. 2012, 24, 3857−3863.(45) Das, S.; Ranjan, P.; Maiti, P. S.; Singh, G.; Leitus, G.; Klajn, R.Dual-Responsive Nanoparticles and their Self-Assembly. Adv. Mater.2013, 25, 422−426.(46) Manna, D.; Udayabhaskararao, T.; Zhao, H.; Klajn, R.Orthogonal Light-Induced Self-Assembly of Nanoparticles usingDifferently Substituted Azobenzenes. Angew. Chem., Int. Ed. 2015,54, 12394−12397.
(47) Heaven, M. W.; Dass, A.; White, P. S.; Holt, K. M.; Murray, R.W. Crystal Structure of the Gold Nanoparticle [N(C8H17)4][Au25(SCH2CH2Ph)18]. J. Am. Chem. Soc. 2008, 130, 3754−3755.(48) Zhu, M.; Lanni, E.; Garg, N.; Bier, M. E.; Jin, R. KineticallyControlled, High-Yield Synthesis of Au25 Clusters. J. Am. Chem. Soc.2008, 130, 1138−1139.(49) Mori, T.; Hegmann, T. Determining the Composition of GoldNanoparticles: A Compilation of Shapes, Sizes, and Calculations usingGeometric Considerations. J. Nanopart. Res. 2016, 18, 295.(50) Zhao, H.; Sen, S.; Udayabhaskararao, T.; Sawczyk, M.;Kucanda, K.; Manna, D.; Kundu, P. K.; Lee, J.-W.; Kral, P.; Klajn,R. Reversible Trapping and Reaction Acceleration within DynamicallySelf-Assembling Nanoflasks. Nat. Nanotechnol. 2016, 11, 82−88.(51) Moldt, T.; Przyrembel, D.; Schulze, M.; Bronsch, W.; Boie, L.;Brete, D.; Gahl, C.; Klajn, R.; Tegeder, P.; Weinelt, M. DifferingIsomerization Kinetics of Azobenzene-Functionalized Self-AssembledMonolayers in Ambient Air and in Vacuum. Langmuir 2016, 32,10795−10801.(52) Taguchi, T.; Isozaki, K.; Miki, K. Enhanced Catalytic Activity ofSelf-Assembled-Monolayer-Capped Gold Nanoparticles. Adv. Mater.2012, 24, 6462−6467.(53) Zdobinsky, T.; Maiti, P. S.; Klajn, R. Support Curvature andConformational Freedom Control Chemical Reactivity of Immobi-lized Species. J. Am. Chem. Soc. 2014, 136, 2711−2714.(54) Zhang, J.; Whitesell, J. K.; Fox, M. A. Photoreactivity of Self-Assembled Monolayers of Azobenzene or Stilbene DerivativesCapped on Colloidal Gold Clusters. Chem. Mater. 2001, 13, 2323−2331.(55) Thomas, K. G.; Kamat, P. V. Chromophore-FunctionalizedGold Nanoparticles. Acc. Chem. Res. 2003, 36, 888−898.(56) Ahmed, S. A.; Tanaka, M. Synthesis of Oligo(ethylene glycol)toward 44-mer. J. Org. Chem. 2006, 71, 9884−9886.(57) Han, J.-M.; Pan, J.-L.; Lei, T.; Liu, C.; Pei, J. Smart MacrocyclicMolecules: Induced Fit and Ultrafast Self-Sorting Inclusion Behaviorthrough Dynamic Covalent Chemistry. Chem. - Eur. J. 2010, 16,13850−13861.(58) Prest, P.-J.; Prince, R. B.; Moore, J. S. SupramolecularOrganization of Oligo(m-phenylene ethynylene)s in the Solid-State.J. Am. Chem. Soc. 1999, 121, 5933−5939.(59) Aykac, A.; Martos-Maldonado, M. C.; Casas-Solvas, J. M.;Quesada-Soriano, I.; García-Maroto, F.; García-Fuentes, L.; Vargas-Berenguel, A. β-Cyclodextrin-Bearing Gold Glyconanoparticles for theDevelopment of Site Specific Drug Delivery Systems. Langmuir 2014,30, 234−242.(60) Chen, X.; Zhang, Z.; Liu, J.; Wang, L. A polymer ElectronDonor based on Isoindigo units Bearing Branched Oligo(ethyleneGlycol) Side Chains for Polymer Solar Cells. Polym. Chem. 2017, 8,5496−5503.(61) May, F.; Marcon, V.; Hansen, M. R.; Grozemac, F.; Andrienko,D. Relationship between Supramolecular Assembly and Charge-Carrier Mobility in Perylenediimide Derivatives: TheIimpact of SideChains. J. Mater. Chem. 2011, 21, 9538−9545.(62) Ochi, R.; Perur, N.; Yoshida, K.; Tamaoki, N. Fast Thermalcis−trans Isomerization Depending on pH and Metal Ions of Water-Soluble Azobenzene Derivatives Containing a Phosphate Group.Tetrahedron 2015, 71, 3500−3506.(63) Goulet-Hanssens, A.; Utecht, M.; Mutruc, D.; Titov, E.;Schwarz, J.; Grubert, L.; Bleger, D.; Saalfrank, P.; Hecht, S.Electrocatalytic Z → E Isomerization of Azobenzenes. J. Am. Chem.Soc. 2017, 139, 335−341.(64) Goulet-Hanssens, A.; Rietze, C.; Titov, E.; Abdullahu, L.;Grubert, L.; Saalfrank, P.; Hecht, S. Hole Catalysis as a GeneralMechanism for Efficient and Wavelength-Independent Z → EAzobenzene Isomerization. Chem. 2018, 4, 1740−1755.(65) Hallett-Tapley, G. L.; D’Alfonso, C.; Pacioni, N. L.; McTiernan,C. D.; Gonzalez-Bejar, M.; Lanzalunga, O.; I, A. E.; Scaiano, J. C.Gold Nanoparticle Catalysis of the cis−trans Isomerization ofAzobenzene. Chem. Commun. 2013, 49, 10073−10075.
Journal of the American Chemical Society Article
DOI: 10.1021/jacs.8b09638J. Am. Chem. Soc. 2019, 141, 1949−1960
1959

(66) Titov, E.; Lysyakova, L.; Lomadze, N.; Kabashin, A. V.;Saalfrank, P.; Santer, S. Thermal Cis-to-Trans Isomerization ofAzobenzene-Containing Molecules Enhanced by Gold Nanoparticles:An Experimental and Theoretical Study. J. Phys. Chem. C 2015, 119,17369−17377.(67) Simoncelli, S.; Aramendía, P. F. Mechanistic Insight into theZ−E Isomerization Catalysis of Azobenzenes Mediated by Bare andCore−Shell Gold Nanoparticles. Catal. Sci. Technol. 2015, 5, 2110−2116.(68) Dennington, R.; Keith, T. A.; Millam, J. M. GaussView 5,version 5.0.8; Semichem Inc.: Shawnee Mission, 2009.(69) Dobbelin, M.; Ciesielski, A.; Haar, S.; Osella, S.; Bruna, M.;Minoia, A.; Grisanti, L.; Mosciatti, T.; Richard, F.; Prasetyanto, E. A.;De Cola, L.; Palermo, V.; Mazzaro, R.; Morandi, V.; Lazzaroni, R.;Ferrari, A. C.; Beljonne, D.; Samorì, P. Light-Enhanced Liquid-PhaseExfoliation and Current Photoswitching in Graphene-AzobenzeneComposites. Nat. Commun. 2016, 7, 11090.(70) Klajn, R.; Wesson, P. J.; Bishop, K. J. M.; Grzybowski, B. A.Writing Self-Erasing Images using Metastable Nanoparticle ″Inks″.Angew. Chem., Int. Ed. 2009, 48, 7035−7039.(71) Sharma, S.; Kim, B.; Lee, D. Water-Soluble Pd NanoparticlesCapped with Glutathione: Synthesis, Characterization, and MagneticProperties. Langmuir 2012, 28, 15958−15965.(72) Amstad, E.; Gillich, T.; Bilecka, I.; Textor, M.; Reimhult, E.Ultrastable Iron Oxide Nanoparticle Colloidal Suspensions UsingDispersants with Catechol-Derived Anchor Groups. Nano Lett. 2009,9, 4042−4048.(73) Garcia-Amoros, J.; Sanchez-Ferrer, A.; Massad, W. A.; Nonell,S.; Velasco, D. Kinetic Study of the Fast Thermal cis-to-transIsomerisation of para-, ortho- and Polyhydroxyazobenzenes. Phys.Chem. Chem. Phys. 2010, 12, 13238−13242.(74) Garcia-Amoros, J.; Velasco, D. Understanding the Fast ThermalIsomerisation of Azophenols in Glassy and Liquid-CrystallinePolymers. Phys. Chem. Chem. Phys. 2014, 16, 3108−3114.(75) Poutanen, M.; Ahmed, Z.; Rautkari, L.; Ikkala, O.; Priimagi, A.Thermal Isomerization of Hydroxyazobenzenes as a Platform forVapor Sensing. ACS Macro Lett. 2018, 7, 381−386.(76) Dunn, N. J.; Humphries, W. H.; Offenbacher, A. R.; King, T.L.; Gray, J. A. pH-Dependent Cis → Trans Isomerization Rates forAzobenzene Dyes in Aqueous Solution. J. Phys. Chem. A 2009, 113,13144−13151.(77) Bleger, D.; Hecht, S. Visible-Light-Activated MolecularSwitches. Angew. Chem., Int. Ed. 2915, 54, 11338−11349.(78) Klajn, R. Spiropyran-based dynamic materials. Chem. Soc. Rev.2014, 43, 148−184.(79) Qian, H.; Pramanik, S.; Aprahamian, I. PhotochromicHydrazone Switches with Extremely Long Thermal Half-Lives. J.Am. Chem. Soc. 2017, 139, 9140−9143.(80) Haase, M.; Schaefer, H. Upconverting Nanoparticles. Angew.Chem., Int. Ed. 2011, 50, 5808−5829.
Journal of the American Chemical Society Article
DOI: 10.1021/jacs.8b09638J. Am. Chem. Soc. 2019, 141, 1949−1960
1960