surface-enhanced raman scattering of aromatic sulfides in aqueous gold sol
TRANSCRIPT

378 Volume 54, Number 3, 2000 APPLIED SPECTROSCOPY0003-7028 / 00 / 5403-0378$2.00 / 0
q 2000 Society for Applied Spectroscopy
Surface-Enhanced Raman Scattering of Aromatic Sul® des inAqueous Gold Sol
SANG WOO JOO, SANG WOO HAN, and KWAN KIM*Laboratory of Intelligent Interface, Department of Chemistry and Center for Molecular Catalysis, Seoul National University, Seoul
151-742, Korea
A surface-enhanced Raman scattering (SERS) study has been per-
formed for benzyl phenyl sul® de (BPS) and dibenzyl sul® de (DBS)
in aqueous gold sol to examine the feasibility of the occurrence ofsurface-induced photoreactions of aromatic sul® des. Although BPS
and DBS were reported to be decomposed into mercaptides on the
silver surface by the 514.5 nm radiation, the molecules were foundnot to undergo such reactions on the gold surface. Such a difference
in behavior was speculated to be associated with the different ad-
sorption strength of BPS and DBS on gold and silver. The absorp-tion bands of BPS and DBS seemed to be substantially broadened
on silver so that the low-energy tails of the broadened absorption
bands were in resonance with the 514.5 nm radiation, while theelectronic absorption bands were not broadened suf® ciently on the
gold surface to absorb visible radiation.
Index Headings: SERS; Benzyl phenyl sul ® de; Dibenzyl sul® de; Gold
sol; Photoreaction.
INTRODUCTION
In the past decade, adsorption of molecular monolayerson metal surfaces has attracted tremendous research in-terest.1 In addition to the fundamental interest in suchmetal adsorbate systems, practical considerations such asthe modi® cation of metal surfaces and the preparation oforganic thin ® lms have increased research activity in thisarea.2 The most widely studied and well-characterizedsystems include alkanethiols on gold, silver, and copper, 3
dialkyl sul® des and dialkyl disul® des on gold and sil-ver,4,5 and carboxylic acids on aluminum oxide6 and sil-ver.7
In our earlier surface-enhanced Raman scattering(SERS) study,8,9 aromatic sul® des adsorbed on the sur-faces of colloidal silver sol were found to undergo sur-face reactions involving facile cleavage of C±S bonds by514.5 nm radiation; such a reaction was found to hardlyoccur for the alkyl sul® des such as dimethyl sul® de anddimethyl sul® de adsorbed on the colloidal silver surface.5
Benzyl phenyl sul® de (BPS) was exclusively decom-posed into benzenethiolate on the silver surface, 9 whiledibenzyl sul® de (DBS) turned into benzyl mercaptide bythe 514.5 nm radiation.8,10 (It has to be mentioned thatsuch a decomposition reaction did not take place whenthe silver sol was circulated during the SER spectral mea-surement. Those reactions occurred, however, when theSER spectra were taken in aqueous silver sols in staticconditions.) The dissociation rate constant was found toincrease upon a decrease in the wavelength of the laserlight.8 Since aromatic sul® des had their absorption max-ima at ; 260 nm in free states,11 the C±S bond cleavage
Received 12 July 1999; accep ted 18 October 1999.* Author to whom correspondence should be sent.
on the silver surface was claimed to be due to the ab-sorption of a visible photon in resonance with the low-energy tail of the broadened absorption band for silver.
Considering that gold is more frequently used as thesubstrate for the self-assembly of thiols and sul® des, aquestion is naturally raised whether aromatic sul® des willundergo such surface-induced photoreactions even on agold surface. For this reason, we have attempted to obtainthe SER spectra of BPS and DBS in an aqueous gold sol.Since the 514.5 nm radiation was not effective in obtain-ing the SER spectra in the gold sol, the SER spectra wereexclusively taken with the He/Ne laser at 632.8 nm.Nonetheless, we examined the possibility of the occur-rence of surface reaction by the 514.5 nm as well as632.8 nm radiation; to see the effect of the 514.5 nmradiation, we exposed the sul® de-containing gold sol toan Ar 1 laser for a prolonged time before taking the SERspectra with the He/Ne laser.
EXPERIMENTAL
Preparation of Au Sol. The gold sol was prepared byfollowing the documented procedures.12 Namely, 133.5mg of KAuCl4 (Aldrich) was initially dissolved in 250mL of water, and the solution was brought a boil. A so-lution of 1% sodium citrate (25 mL) was then added tothe KAuCl4 solution under vigorous stirring, and boilingwas continued for ; 20 min. The resulting Au sol solutionwas stable for several weeks. To 0.1±1 mL of Au solsolution, 10 2 3 M aqueous solution of benzyl phenyl sul-® de or dibenzyl sul® de was added dropwise to a ® nalconcentration of 10 2 5 M with a micropipette; the purplegold sols became bluish green through the addition ofsul® des. Benzyl phenyl sul® de and dibenzyl sul® de werepurchased from Aldrich and used as received. The chem-icals otherwise speci® ed were reagent grade, and triplydistilled water, of resistivity greater than 18.0 M V ´cm,was used in making aqueous solutions.
Raman Spectral Measurement. Raman spectra wereobtained by using a Renishaw Raman system Model2000 spectrometer equipped with an integral microscope(Olympus BH2-UMA). The 632.8 nm line from a 17 mWair-cooled He/Ne laser (Spectra Physics Model 127) wasused for the excitation source of the Au sol SERS ex-periment. Raman scattering was detected with 180 8 ge-ometry with a Peltier cooled ( 2 70 8 C) charge-coupleddevice (CCD) camera (400 3 600 pixels). A glass cap-illary (KIMAX-51) with an outer diameter of 1.5±1.8 mmwas used as a sampling device. The laser beam was fo-cused onto a spot approximately 2 m m in diameter withan objective microscope on the order of 20 3 . The dataacquisition time was usually 90 s for the gold sols. The

APPLIED SPECTROSCOPY 379
FIG. 1. UV-visible absorption spectra of (a) Ag sol as-prepared and(b) after adding benzyl phenyl sul® de into the sol. UV-vis absorptionspectra of (c) Au sol as-prepared and (d ) after adding benzyl phenylsul® de into the sol.
FIG. 2. (a) OR spectrum of neat benzyl phenyl sul® de. SER spectraof (b) benzyl phenyl sul® de, (c) benzylthiol, (d ) benzenethiol, and (e)1:1 mixture of benzylthiol and benzenethiol in aqueous gold sol. Allthe SER spectra are taken under static conditions with the sample con-centration at ; 10 2 5 M.
holographic grating (1800 grooves/mm) and the slit al-lowed the spectral resolution to be 1 cm 2 1. The Ramanband of a silicon wafer at 520 cm 2 1 was used to calibratethe spectrometer, and the accuracy of the spectral mea-surement was estimated to be better than 1 cm 2 1. TheRaman spectrometer was interfaced with an IBM PC, andthe spectral data were analyzed with Renishaw WiREsoftware v. 1.2 based on the GRAMS/32C suite program(Galactic). To see whether a surface-induced photoreac-tion could take place under 514.5 nm radiation, we ex-posed the sul® de-containing gold sol to an unfocused 20mW air-cooled Ar 1 laser (Spectra Physics Model 163±C4210) for a prolonged time (usually 30 min) before tak-ing the SER spectra with a He/Ne laser at 632.8 nm.
Transmission Electron Microscope and UV-VisibleSpectral Measurements. To estimate the sizes of goldand silver colloidal particles, we obtained their transmis-sion electron microscope (TEM) images with a JEM-200CX transmission electron microscope at 200 kV afterplacing a drop of colloidal solution and a drop of aqueoussul® de solution onto Ni/Cu grids. UV-visible (UV-vis)spectra of the sol solutions were obtained with a SCINCOS-2130 spectrometer.
RESULTS AND DISCUSSION
UV-Visible Spectra of Ag and Au Sols. Figure 1aand 1c show the UV-vis absorption spectra of as-preparedAg and Au sols, respectively. The absorption maximaoccur at 391 nm for Ag sol and at 532 nm for Au sol,in agreement with the literature.13 They must arise froma surface plasmon absorption of metal clusters. The UV-vis absorption spectra changed dramatically upon addi-tion of sul® des into the sols. Figure 1b and 1d show thespectra obtained after the addition of benzyl phenyl sul-® de into the Ag and Au sols, respectively; much the samespectra are obtained after dibenzyl sul® de is added intothe sols. The spectral change can be attributed to the ag-gregation of metal clusters caused by the reduction of thenegative charge of the colloidal particles. Namely, metalparticles in aqueous colloidal dispersions usually bear anegative charge due to adsorbed anions. Aggregation is
induced by the addition of neutral adsorbate moleculessuch as BPS, which displace the adsorbed ions, thus re-ducing the charge on the particles to the point wherecollisions occur as a result of diffusional motion. As canbe seen in Fig. 1b, the position of the maximum absorp-tion shifted to ; 534 nm in Ag sol along with the ab-sorption tail extending up to ; 800 nm. The plasmon ab-sorption of the Au sol similarly shifted toward red by theaddition of the adsorbate molecules. Some irregularhumps seen between 600 and 800 nm in Fig. 1d are pre-sumably due to a wide distribution of gold clusters.
SERS of Benzyl Phenyl Sul® de in Au Sol. Figure 2ashows the ordinary Raman (OR) spectrum of BPS in neatsolid state. Figure 2b shows the SER spectrum of BPStaken in an aqueous gold sol. (It has to be mentioned thatall the gold-sol SER spectra in this work were taken un-der static conditions.) According to the TEM image, thesize of colloidal gold particles was ; 50 nm, whereby theconcentration of BPS used in taking the SER spectrum,10 2 5 M, should be more than a full coverage limit on thecolloidal gold surface. As mentioned in the Introductionsection, the SER spectrum of BPS taken in an aqueoussilver sol under static conditions had to be attributed tobenzenethiolate species adsorbed on colloidal silver sur-faces rather than to BPS. This result contrasts with the

380 Volume 54, Number 3, 2000
TABLE I. Raman spectral data and vibrational assignments ofBPS.a
OR (Neat) SERS (Au sol) Assignmentb,c
308
403485570617695 sh699715801816
10021023109011561185120312441271
15821601
227
415474566614
658698803814
1002102210761156 vw1182 vw12001235
1494 vw15731600
Au±S stretching15 (B)CSC bending7a (P)6a (B), 16b (P)16b (B)6b (B, P)6a (P)CS stretching (B)4 (B)CH2 rocking (B)1 (B)12 (B, P)18a (B, P)1 (P)9b (B, P)9b (B, P)13 (B)CH2 twisting (B)3 (P)19a (B)8a (P)8a (B)
a All data given in cm 2 1; abbreviations: sh 5 shoulder; vw 5 very weak.b Assigned on the basis of Ref. 8.c Abbreviations: B 5 benzylthio moiety; P 5 phenylthio moiety.
SER spectrum of BPS taken in a gold sol. That is, unlikethe case of silver sol, all the SER peaks in gold sol shownin Fig. 2b can be correlated with the OR peaks in Fig.2a; as summarized in Table I, the SERS peaks can beattributed to either the benzylthio or phenylthio moietiesof BPS. The SER spectral pattern of BPS in Fig. 2b isobviously different from the SER spectral patterns ofbenzylthiol and benzenethiol, shown, respectively, in Fig.2c and 2d, both of which are also taken in aqueous goldsol media. This observation implies that BPS should ad-sorb on gold without any C±S bond scission.
On the gold and silver surfaces benzylthiol adsorbsmore favorably than benzenethiol.8,14,15 This pattern canbe shown from the SER spectrum in Fig. 2e that is ob-tained after the injection of a 1:1 mixture of benzenethioland benzylthiol into the aqueous gold sol. In fact, theSER spectral pattern in Fig. 2e is much the same as thatin Fig. 2c. The favorable adsorption of benzylthiol canbe attributed to the presence of a methylene group be-tween the phenyl group and the sulfur atom. Due to the¯ exibility of the methylene group, benzylthiol can be ad-sorbed on gold and silver by forming a close-packedstructure in which the benzene rings of the benzylthiolatespecies are aligned normal to the metal surfaces; for thecase of benzenethiol, a close-packed structure is notformed since the benzene rings of the benzenethiolatespecies favor a strongly tilted orientation so that the in-termolecular interaction does not occur ef® ciently on themetal surface.14,15
The fact that the SER spectral pattern of BPS in aque-ous silver sol has to be attributed exclusively to benze-nethiolate species may then imply that it is in fact formedby the rupture of the C±S bond of the benzylthio moietyof BPS on silver. In our previous SERS study on an aque-ous silver sol, such a C±S bond scission was claimed tooccur through the surface-induced photoreaction proceed-
ing via the absorption of a visible photon that was inresonance with the low-energy tail of the broadened ab-sorption band of the adsorbed BPS species;9 in a freestate, the absorption maximum occurs for BPS at 255 nmin the ultraviolet region.11 Considering that the presentSER spectrum of BPS in aqueous gold sol was obtainedwith a He/Ne laser at 632.8 nm while the previous SERspectrum in aqueous silver sol was obtained with an Ar 1
laser at 514.5 nm, the unfeasible occurrence of the C±Sbond scission on gold may be rationalized simply by sur-mising that the low-energy tail of the absorption band ofBPS on gold did not extend up to 632.8 nm. On thesegrounds, we exposed an unfocused 20 mW Ar 1 laser at514.5 nm to the BPS-containing gold sol for a prolongedtime ( ; 30 min), and then obtained the SER spectrum ofthe sol by means of a He/Ne laser at 632.8 nm. Contraryto our expectation, the SER spectrum obtained after theexposure of the Ar 1 laser was hardly different from thatshown in Fig. 2b. Hence, it appeared that the silver sub-strate itself plays a certain role for the C±S bond of BPSto be split by visible radiation. (With a He/Ne laser at632.8 nm, a reliable SER spectrum could not be obtainedfor BPS in a silver sol. We are thus uncertain whetherthe C±S bond scission can occur on silver even with632.8 nm radiation, as likely with 514.5 nm radiation.However, we must mention that photodecomposition ofBPS seemed to hardly occur on silver by 632.8 nm ex-citation. As reported recently,16 2 m m-sized Ag particlesare SERS active not only by 514.5 nm radiation but alsoby 632.8 nm radiation. In this light, we have taken SERspectra of BPS which had been self-assembled on 2 m m-sized Ag particles. In fact, the SER spectrum taken with632.8 nm radiation could be rendered to BPS on silverwhile the SER spectrum taken with 514.5 nm radiationcould be attributed to benzenethiolate on silver.)
It is intriguing that the C±S bond scission of BPS doesnot take place on gold with an Ar 1 laser. One can spec-ulate that the difference between the observations madeon the gold and silver surfaces is associated with thedifferent adsorption strength of BPS on gold and silver.Invoking the fact that organic sul® des as well as organicthiols are generally more strongly chemisorbed on silverthan on gold,1 the absorption band of BPS may not bebroadened enough on gold for the low-energy tail of thebroadened band to be in resonance with the 514.5 nmradiation, while the absorption band is broadened suf® -ciently on the silver surface to absorb visible radiation.On the other hand, the C±S bond scission of BPS onsilver might have occurred because of an instantaneoustemperature increase caused by the plasmon absorption.It has been well documented in the literature that theenergy of nanoparticles gained by plasmon absorptioncan be transferred to the surrounding medium.17 In astudy of the relaxation dynamics of surface plasmon elec-tronic oscillation in gold and silver nanoparticles, Hodaket al. reported that temperature rise in the medium mustbe greater than 100 K, and this heat transfer from nano-particles to nearby dielectric adsorbate molecules occursmore ef® ciently in silver nanoparticles than in gold nano-particles.18 In either case, it will also be desirable to ex-amine whether C±S bond scission can occur on goldwhen the BPS-containing gold sol is exposed to a laserline with a wavelength shorter than 514.5 nm. In this

APPLIED SPECTROSCOPY 381
FIG. 3. SER spectra of benzyl phenyl sul® de in gold sol (a) beforeand (b) after the exposure of a 5 mJ Nd:YAG laser at 355 nm for 30min. SER spectra of (c) benzylthiol and (d ) benzenethiol in gold sol.All the SER spectra are taken under static conditions with a He/Ne laserline at 632.8 nm as the excitation source.
TABLE II. Relative enhancement factors for the SER bands ofBPS.
Symmetrytypea Normal modeb
Relative enhancement factor(ISERS/IOR)c
Benzyl Phenyl
A1 1126a8a9a
18a
1.431.922.43
16.293.571.00
2.211.92
2.143.571.00
B1 4 2.36B2 13
6b9b
16b
4.542.110.624.21
2.110.622.43
CS stretchingCH 2 rockingCH 2 twisting
7.575.827.43
a Benzyl and phenyl moieties of BPS are independently presumed toassume a C2v symmetry.
b See Table I.c Normalized with respect to the enhancement factor of the ring 18a
mode at 1022 cm 2 1 in the SER spectrum of BPS.
light, we exposed an unfocused 5 mJ Nd:YAG laser beamat 355 nm (Continuum Surelite II-10) to the BPS-con-taining gold sol for a prolonged time ( ; 30 min), afterwhich SER spectra of the sol were obtained with a He/Ne laser at 632.8 nm. Figure 3a and 3b show the SERspectra of BPS before and after the exposure of the Nd:YAG laser line, respectively. For comparative purposes,the SER spectra of benzylthiol and benzenethiol in goldsol are also reproduced in Fig. 3c and 3d, respectively.It can be observed that the SER spectral features of BPSafter the exposure of the 355 nm laser line are compa-rable to those of benzenethiol. As a control experiment,we exposed the Nd:YAG laser line to a 1 mM solutionof BPS in ethanol for up to 1 h, after which an aliquotof the solution was added into gold sol and then SERspectra acquired with the He/Ne laser as an excitationsource. The observed spectra could be rendered exclu-sively to BPS adsorbed on gold. These observations sup-port the presumption that the decomposition of BPS oc-curs via the absorption of a laser line that is in resonancewith the low-energy tail of the broadened absorption bandof BPS caused by the adsorption on the metal surfaces;because we lacked proper light sources, we could notexamine the feasibility of the photoreaction of BPS ongold with the radiation whose wavelength is slightlyshorter than 514.5 nm. Nonetheless, we cannot complete-ly exclude the possibility that the C±S bond scission takesplace via a thermal reaction, although the extent of sur-face plasmon absorption near 355 nm is very low for theaggregated gold particles.
It is intriguing that only the C±S bond of the benzylmoiety of BPS is broken by 355 nm radiation on gold aswell as by 514.5 nm radiation on silver. Recalling thatthe C±S bond dissociation energy of HS±C 6H5 is largerby ; 60.7 kJ/mol than that of HS±C2H5,
19 the cleavage ofthe C±S bond at the phenyl moiety of BPS will be lesslikely to occur in comparison with that at the benzyl moi-ety. The ® nal destiny of the benzyl moiety is, however,a matter of conjecture at the moment.
Without doubt, the main driving force for the adsorp-
tion of BPS on gold must be the formation of the Au±Sbond. This assumption can be veri® ed from the peak shiftin the C±S stretching mode occurring through the surfaceadsorption of BPS on gold. Namely, the n (CS) band ofBPS appears at 699 cm 2 1 in neat solid state (Fig. 2a) butat 658 cm 2 1 in the SER spectrum shown in Fig. 2b. Theelectron donation from sulfur to gold should induce theweakening of the C±S bond(s), resulting in a red shift ofthe C±S stretching band. The exact orientation of BPSon the gold surface is, however, a matter of conjecture,since the set of SERS selection rules 20±22 established sofar is very dependent on the speci® c enhancement mech-anism. Nonetheless, we can see from Table II that theSERS enhancement factor is very dependent on the kindof vibrational mode. On the one hand, the substituentmodes such as CS symmetric stretching and CH 2 rockingand twisting modes exhibited relatively stronger enhance-ment than the ring modes. On the other hand, it can beobserved that the in-plane ring 8a mode of the benzylmoiety at 1600 cm 2 1 is more enhanced than the 8a modeof the phenylthio moiety at 1573 cm 2 1. Invoking the elec-tromagnetic (EM) surface selection rule20±22Ð althoughnot de® nitiveÐ which states that the in-plane ring vibra-tions should be more enhanced than the out-of-plane oneswhen the benzene ring is oriented perpendicularly withrespect to the metal surface, it is tempting to concludethat the benzyl moiety assumes a rather vertical stance,while the phenyl moiety of BPS lies quite ¯ at on the goldsurface. The relatively strong enhancement of the CSsymmetric stretching and the CH 2 twisting modes canalso be understood on the basis of such a structure. Thatis, if the benzyl moiety assumes a parallel orientation, theS±CH2 bond will lie nearly parallel to the gold surface.Then, the CS stretching and the CH 2 twisting vibrationswhose polarizability derivative components are alignedalong the bond direction will be less enhanced than othervibrations. It is also noteworthy that the ring 1 and 8amodes of the phenylthio moiety of BPS are red-shiftedby 14 and 9 cm 2 1, respectively, upon adsorbing on thegold surface, while their counterparts arising from the
382 Volume 54, Number 3, 2000
FIG. 4. Plausible orientation of benzyl phenyl sul® de on gold.
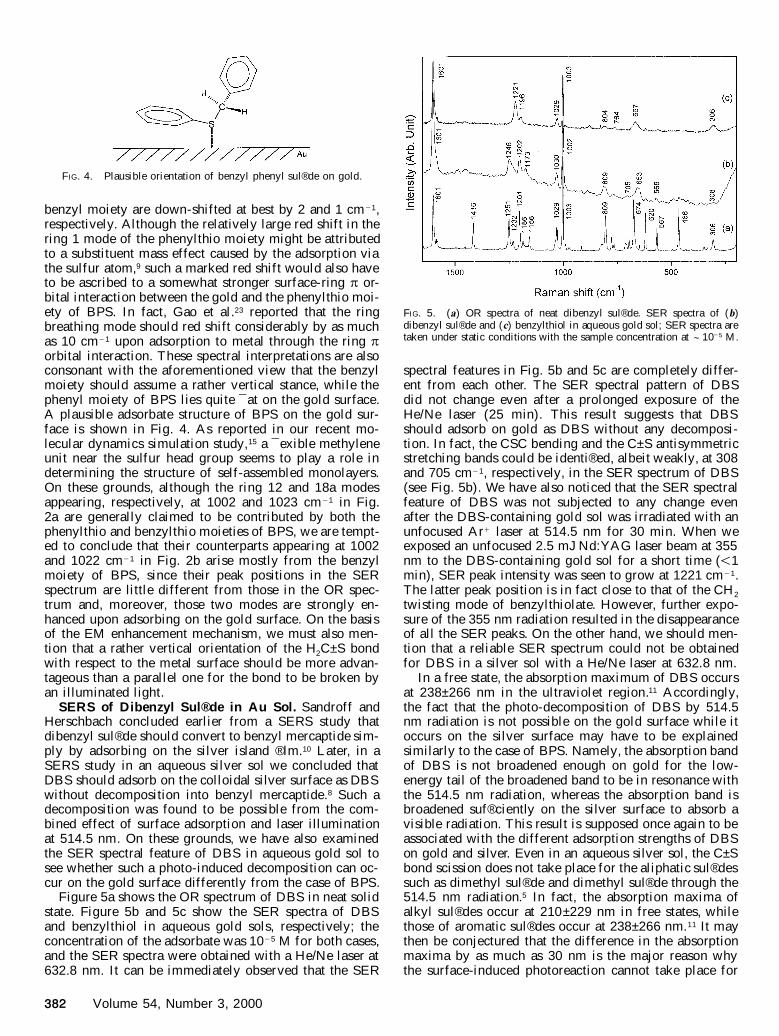
FIG. 5. (a) OR spectra of neat dibenzyl sul® de. SER spectra of (b)dibenzyl sul® de and (c) benzylthiol in aqueous gold sol; SER spectra aretaken under static conditions with the sample concentration at ; 10 2 5 M.
benzyl moiety are down-shifted at best by 2 and 1 cm 2 1,respectively. Although the relatively large red shift in thering 1 mode of the phenylthio moiety might be attributedto a substituent mass effect caused by the adsorption viathe sulfur atom,9 such a marked red shift would also haveto be ascribed to a somewhat stronger surface-ring p or-bital interaction between the gold and the phenylthio moi-ety of BPS. In fact, Gao et al.23 reported that the ringbreathing mode should red shift considerably by as muchas 10 cm 2 1 upon adsorption to metal through the ring porbital interaction. These spectral interpretations are alsoconsonant with the aforementioned view that the benzylmoiety should assume a rather vertical stance, while thephenyl moiety of BPS lies quite ¯ at on the gold surface.A plausible adsorbate structure of BPS on the gold sur-face is shown in Fig. 4. As reported in our recent mo-lecular dynamics simulation study,15 a ¯ exible methyleneunit near the sulfur head group seems to play a role indetermining the structure of self-assembled monolayers.On these grounds, although the ring 12 and 18a modesappearing, respectively, at 1002 and 1023 cm 2 1 in Fig.2a are generally claimed to be contributed by both thephenylthio and benzylthio moieties of BPS, we are tempt-ed to conclude that their counterparts appearing at 1002and 1022 cm 2 1 in Fig. 2b arise mostly from the benzylmoiety of BPS, since their peak positions in the SERspectrum are little different from those in the OR spec-trum and, moreover, those two modes are strongly en-hanced upon adsorbing on the gold surface. On the basisof the EM enhancement mechanism, we must also men-tion that a rather vertical orientation of the H2C±S bondwith respect to the metal surface should be more advan-tageous than a parallel one for the bond to be broken byan illuminated light.
SERS of Dibenzyl Sul® de in Au Sol. Sandroff andHerschbach concluded earlier from a SERS study thatdibenzyl sul® de should convert to benzyl mercaptide sim-ply by adsorbing on the silver island ® lm.10 Later, in aSERS study in an aqueous silver sol we concluded thatDBS should adsorb on the colloidal silver surface as DBSwithout decomposition into benzyl mercaptide.8 Such adecomposition was found to be possible from the com-bined effect of surface adsorption and laser illuminationat 514.5 nm. On these grounds, we have also examinedthe SER spectral feature of DBS in aqueous gold sol tosee whether such a photo-induced decomposition can oc-cur on the gold surface differently from the case of BPS.
Figure 5a shows the OR spectrum of DBS in neat solidstate. Figure 5b and 5c show the SER spectra of DBSand benzylthiol in aqueous gold sols, respectively; theconcentration of the adsorbate was 10 2 5 M for both cases,and the SER spectra were obtained with a He/Ne laser at632.8 nm. It can be immediately observed that the SER
spectral features in Fig. 5b and 5c are completely differ-ent from each other. The SER spectral pattern of DBSdid not change even after a prolonged exposure of theHe/Ne laser (25 min). This result suggests that DBSshould adsorb on gold as DBS without any decomposi-tion. In fact, the CSC bending and the C±S antisymmetricstretching bands could be identi® ed, albeit weakly, at 308and 705 cm 2 1, respectively, in the SER spectrum of DBS(see Fig. 5b). We have also noticed that the SER spectralfeature of DBS was not subjected to any change evenafter the DBS-containing gold sol was irradiated with anunfocused Ar 1 laser at 514.5 nm for 30 min. When weexposed an unfocused 2.5 mJ Nd:YAG laser beam at 355nm to the DBS-containing gold sol for a short time ( , 1min), SER peak intensity was seen to grow at 1221 cm 2 1.The latter peak position is in fact close to that of the CH 2
twisting mode of benzylthiolate. However, further expo-sure of the 355 nm radiation resulted in the disappearanceof all the SER peaks. On the other hand, we should men-tion that a reliable SER spectrum could not be obtainedfor DBS in a silver sol with a He/Ne laser at 632.8 nm.
In a free state, the absorption maximum of DBS occursat 238±266 nm in the ultraviolet region.11 Accordingly,the fact that the photo-decomposition of DBS by 514.5nm radiation is not possible on the gold surface while itoccurs on the silver surface may have to be explainedsimilarly to the case of BPS. Namely, the absorption bandof DBS is not broadened enough on gold for the low-energy tail of the broadened band to be in resonance withthe 514.5 nm radiation, whereas the absorption band isbroadened suf® ciently on the silver surface to absorb avisible radiation. This result is supposed once again to beassociated with the different adsorption strengths of DBSon gold and silver. Even in an aqueous silver sol, the C±Sbond scission does not take place for the aliphatic sul® dessuch as dimethyl sul® de and dimethyl sul® de through the514.5 nm radiation.5 In fact, the absorption maxima ofalkyl sul® des occur at 210±229 nm in free states, whilethose of aromatic sul® des occur at 238±266 nm.11 It maythen be conjectured that the difference in the absorptionmaxima by as much as 30 nm is the major reason whythe surface-induced photoreaction cannot take place for

APPLIED SPECTROSCOPY 383
the aliphatic sul® des even in the silver sol medium: thetail of the absorption band would not extend up to 514.5nm for the aliphatic sul® des adsorbed on the silver sur-face.
CONCLUSION
Using SERS, we examined whether aromatic sul® desadsorbed on the colloidal gold surface could decomposeinto mercaptides by 514.5 nm as well as 632.8 nm ra-diation. Although BPS and DBS were readily decom-posed into mercaptides on the colloidal silver surface bythe 514.5 nm radiation, such reactions were found tohardly occur on the colloidal gold surface. Although theC±S bond scission on silver might have occurred becauseof an instantaneous temperature increase caused by theplasmon absorption, the difference between the obser-vations made on the gold and silver surfaces could alsobe associated with the different adsorption strengths ofBPS and DBS on gold and silver. That is, the absorptionbands of BPS and DBS were assumed not to be broad-ened enough on gold for the low-energy tails of thebroadened bands to be in resonance with the 514.5 nmradiation, while the absorption bands were broadened suf-® ciently on the silver surface to absorb visible radiation.We could also con® rm in this work that a ¯ exible meth-ylene unit near the sulfur headgroup plays an importantrole in discriminating the structures of self-assembledmonolayers on metal surfaces; here, the benzyl moiety ofBPS appeared to assume a rather vertical stance, whilethe phenyl moiety of BPS lies ¯ at on the gold surface.In any event, for a better understanding of the C±S bondscission on silver, a more systematic investigation has tobe performed considering several factors such as the ef-fect of laser power, excitation wavelength, temperature,and controlled sol circulation. Our future work will befocused on such considerations.
ACKNOWLEDGMENTS
This work was supported in part by the Korea Science and Engi-neering Foundation through the Center for Molecular Catalysis at SeoulNational University (SNU) and by the Korea Research Foundationthrough the Research Institute of Basic Sciences at SNU. S.W.J. andS.W.H. acknowledge the Korea Research Foundation for providingBK21 fellowships.
1. A. Ulman, An Introduction to Ultrathin Organic Films from Lang-muir± Blodgett to Self-Assembly (Academic Press, San Diego, Cal-ifornia, 1991).
2. L. H. Dubois and R. G. Nuzzo, Annu. Rev. Phys. Chem. 43, 437(1992).
3. P. E. Laibinis, G. M. Whitesides, D. L. Allara, Y.-T. Tao, A. N.Parikh, and R. G. Nuzzo, J. Am. Chem. Soc. 113, 7152 (1991).
4. R. G. Nuzzo, B. R. Zegarski, and L. H. Dubois, J. Am. Chem. Soc.109, 733 (1987).
5. T. H. Joo, K. Kim, and M. S. Kim, J. Mol. Struct. 162, 191 (1987).6. O. Klug, G. Parlagh, and W. Forsling, J. Mol. Struct. 410, 183
(1997).7. S. H. Kim, S. J. Ahn, and K. Kim, J. Phys. Chem. 100, 7174 (1996).8. T. H. Joo, Y. H. Yim, K. Kim, and M. S. Kim, J. Phys. Chem. 93,
1422 (1989).9. Y. H. Yim, K. Kim, and M. S. Kim, J. Phys. Chem. 94, 2552 (1990).
10. C. J. Sandroff and D. R. Herschbach, J. Phys. Chem. 86, 3277(1982).
11. E. A. Fehnel and M. Carmack, J. Am. Chem. Soc. 71, 84 (1949).12. P. C. Lee and D. Meisel, J. Phys. Chem. 86, 3391 (1982).13. J. A. Creighton, C. G. Blatchford, and M. G. Albrecht, J. Chem.
Soc. Faraday Trans. 75, 790 (1979).14. Y.-T. Tao, C.-C. Wu, J.-Y. Eu, W.-L. Lin, K.-C. Wu, and C.-H. Chen,
Langmuir 13, 4018 (1997).15. H. H. Jung, Y. D. Won, S. Shin, and K. Kim, Langmuir 15, 1147
(1999).16. H. S. Han, S. W. Han, C. H. Kim, and K. Kim, Langmuir, paper
in press.17. T. S. Ahmadi, S. L. Logunov, and M. A. El-Sayed, J. Phys. Chem.
100, 8053 (1996).18. J. H. Hodak, I. Martini, and G. V. Hartland, J. Phys. Chem. B 102,
6958 (1998).19. S. W. Benson, Chem. Rev. 78, 23 (1978).20. S. H. Cho, H. S. Han, D.-J. Jang, K. Kim, and M. S. Kim, J. Phys.
Chem. 99, 10594 (1995).21. M. Moskovits, Rev. Mod. Phys. 57, 783 (1985).22. J. A. Creighton, Surf. Sci. 124, 209 (1983).23. X. Gao, J. P. Davies, and M. J. Weaver, J. Phys. Chem. 94, 6858
(1990).