synthesis and solution-state dynamics of donor–acceptor oligorotaxane foldamers
TRANSCRIPT

Chemical Science
EDGE ARTICLE
Dow
nloa
ded
by U
nive
rsity
of
Mis
sour
i at C
olum
bia
on 0
5 M
arch
201
3Pu
blis
hed
on 0
4 Fe
brua
ry 2
013
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C3S
C00
015J
View Article OnlineView Journal | View Issue
aDepartment of Chemistry, Northwestern Un
Illinois, 60208-3113, USA. E-mail: stodda
1009; Tel: +1-847-491-3793bNanoCentury KAIST Institute, Graduate Sc
Sustainability, World Class University, Ko
Technology (KAIST), 373-1 Guseong Dong,
of KoreacDepartment of Chemistry, University of Read
† Electronic supplementary informa10.1039/c3sc00015j
Cite this: Chem. Sci., 2013, 4, 1470
Received 3rd January 2013Accepted 2nd February 2013
DOI: 10.1039/c3sc00015j
www.rsc.org/chemicalscience
1470 | Chem. Sci., 2013, 4, 1470–148
Synthesis and solution-state dynamics ofdonor–acceptor oligorotaxane foldamers†
Zhixue Zhu,a Carson J. Bruns,ab Hao Li,a Juying Lei,a Chenfeng Ke,a Zhichang Liu,a
Saman Shafaie,a Howard M. Colquhounc and J. Fraser Stoddart*ab
We describe in detail a strategy for creating foldamers in which interactions between mechanically
interlocked components dictate the single-molecule assembly of a folded secondary structure. This
unique folding motif is based on a flexible polyether dumbbell bearing 1,5-dioxynaphthalene (DNP)
donors, which folds its way through a series of cyclobis(paraquat-p-phenylene) (CBPQT4+) acceptor rings
in a serpentine fashion to enable extended donor–acceptor (D–A) stacking between DNP and the
electron-poor 4,40-bipyridinium (BIPY2+) units in CBPQT4+. These oligorotaxanes can be prepared in a
wide range of sizes, with molecular weights up to >15 000 Da, on account of novel one-pot reactions
we developed to generate the necessary oligo-DNP precursors. The product distributions from the final
kinetically controlled stoppering reactions are highly biased towards oligorotaxanes in which
approximately half of the DNP units are encircled by rings, a fact which can be rationalized if the
dominant solution-state structures of the pseudorotaxane precursors reflect the solid-state
superstructures of analogous compounds, which express 50% recognition site occupancy because of
their proclivity to pack into continuous D–A–D–A stacks. The presence of well-defined folded structures
in solution have been confirmed by 1H NMR spectroscopy in CD3CN. Moreover, we discovered an
empirical selection rule forbidding CBPQT4+ rings to occupy adjacent DNP sites, which elegantly explains
both the product distributions and the 1H NMR spectra. Depending on their adherence to this selection
rule, all of the oligorotaxanes belong to one of three families: whereas ‘Confused’ oligorotaxanes adopt
multiple translational isomers that satisfy the rule and ‘Frustrated’ species cannot obey it at all, members
of the ‘Happy’ family each express only one rule-compliant ‘Goldilocks’ isomer. The NMR spectra of
these oligorotaxanes also shed light on their dynamics; rapid 180� rotations of DNP units cause pairs of
heterotopic BIPY2+ protons in the accompanying CBPQT4+ rings to exchange sites, giving rise to time-
averaged signals. This process, which we term ‘superrotation’, will apply much more generally to other
mechanically interlocked systems.
Introduction
The exquisite self-organizing structures of biomolecules andcomplexity of their reactivity networks which comprise ourmolecular biology represent a vast source of inspiration forchemists. Our growing understanding of molecular recognitionand self-assembly, both in the context of biology as well assupramolecular1 and host–guest chemistry,2 has marshaled an
iversity, 2145 Sheridan Road, Evanston,
[email protected]; Fax: +1-847-491-
hool of Energy, Environment, Water, and
rea Advanced Institute of Science and
Yuseong Gu, Daejeon 305-701, Republic
ing, Whiteknights, Reading, RG6 6AD, UK
tion (ESI) available. See DOI:
3
era in which noncovalent bonds demand3 increasingly moreattention. The importance of these relatively weak interactionscannot be overstated, not only because their roles4 in thestructures and functions of DNA, RNA, proteins, andmembranes make life possible, but also because our growingfundamental understanding5 of supramolecular interactionsand their involvement in structure–property relationshipssupports the continual emergence of new materials6 and tech-nologies.7 In particular, since the twists and folds of biomole-cules explicitly underpin their form and function, the task ofimparting synthetic molecules andmacromolecules with foldedsecondary structures should, likewise, have enormous potentialto yield functional materials with novel and emergent proper-ties. Articial molecules that adopt folded secondary structures– widely known8 as foldamers – represent a very rich and activeeld9 of research. At the same time, mechanically interlockedmolecules10 (MIMs) such as catenanes and rotaxanes11 aregaining increasing attention from researchers interested in
This journal is ª The Royal Society of Chemistry 2013

Edge Article Chemical Science
Dow
nloa
ded
by U
nive
rsity
of
Mis
sour
i at C
olum
bia
on 0
5 M
arch
201
3Pu
blis
hed
on 0
4 Fe
brua
ry 2
013
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C3S
C00
015J
View Article Online
developing molecular nanotechnology applications rangingfrom electronics12 to sophisticated diagnostic and therapeuticsystems.13 Although many of the noncovalent bonding interac-tions used to design foldamers are also employed in the tem-plation14 of MIMs, examples15 of foldamers incorporatingmechanical bonds remain very rare indeed, leaving the questionwidely open regarding what might emerge from a synergisticcombination of MIMs and foldamers. This question is partic-ularly interesting in the context of oligo- and polymeric16 MIMsbecause they constitute a link between the molecular world andbulk properties at the macroscopic level, opening the door thatleads out of solution and into functional materials.
One conceptually rational strategy for installing foldedsecondary structures into mechanically interlocked polymers isto design main-chain polyrotaxanes in which noncovalentbonding interactions persuade the long dumbbell componentto fold as it ‘snakes’ its way through the rings. We felt that thetypes of donor–acceptor (D–A) complexes and MIMs which wehave investigated17 extensively, most notably those based onrecognition between p-electron decient 4,40-bipyridinium(BIPY2+) units – expressed in host form as the tetracationiccyclophane cyclobis(paraquat-p-phenylene)17b,f (CBPQT4+) – andthe p-electron rich 1,5-dioxynaphthalene (DNP) donor with itscorresponding 1,5-dinaphtho[38]crown-10 (DNP38C10)host,17a,18 were well-suited for this approach. Indeed, non-interlocked foldamers induced by aromatic D–A interactions –
known19 in some cases as aedamers – have already beenreported19,20 by several groups. In the solid-state, the super-structures of the BIPY2+3DNP38C10 and DNP3CBPQT4+ host–guest complexes and MIMs reveal17b–f a tendency to form stacksof alternating DNP donors and BIPY2+ acceptors in van derWaals contact on account of p–p interactions between donorsand acceptors. Recently, we discovered21 that DNP oligomersand CBPQT4+ can be co-crystallized into folded superstructureshaving continuous D–A–D–A stacks (Fig. 1) with crystallo-graphically invisible defects, making them appear innite inspite of being discrete oligomers. In addition to the p–p andcharge transfer interactions between donors and acceptors,these remarkable superstructures are enabled by [C–H/O]interactions between BIPY2+ protons and the electron-rich poly-ethers that constitute the backbone of the DNP oligomers andimpart them with the exibility needed to adopt their highlyfolded superstructures. We anticipated this type of folding ininterlocked macromolecules years prior when we made ourinitial foray15a into D–A polyrotaxanes. This research revealed
Fig. 1 The apparently infinite folded superstructure of pseudorotaxanes formed betcrystallography.
This journal is ª The Royal Society of Chemistry 2013
that the rotaxanated polymers had greater retention volumes bygel permeation chromatography than their poly-DNP syntheticprecursors, although the polyrotaxanes carry almost twice themass of the non-interlocked DNP polymers. The apparentlyincreased compactness of the polyrotaxanes suggested that ahighly folded structure was at hand, and prompted us toprepare a homologous series of discrete and well-dened oli-gorotaxanes in order to probe the nature of the noncovalentbonding interactions and solution-state secondary structures ina systematic and rational manner by high-eld 1H NMRspectroscopy.
Here we report a general approach to synthesize well-denedoligorotaxanes of arbitrary length in which the mechanicalbond is highly integrated into the folded superstructure. Wehave developed convenient and efficient one-pot reactions thatmake these architectures, which were previously extraordinarilychallenging, more synthetically feasible. Although the oligor-otaxanes possess disulde functional groups attached to thestoppers in order to make them suitable candidates for AFMand surface studies, we focus here rstly on their solution-statesecondary structure and describe a number of interestingproperties the oligorotaxanes possess in solution as probed by1H NMR spectroscopy. In particular, we investigate the effect offolding on the oligorotaxane product distribution, their trans-lational isomerism, and a process – which we deem ‘super-rotation’ – in which constitutionally heterotopic protonsundergo rapid site exchange on the NMR timescale, a conceptwhich we believe can be applied to understand in more detailthe solution-state dynamics of a much wider range of MIMsthan those reported here.
Results and discussionOne-pot synthesis of DNP oligomers
The various approaches to rotaxane formation – threading-fol-lowed-by-stoppering,22 clipping,23 slippage,24 dumbbellcapture,25 threading-followed-by-swelling,26 and threading-fol-lowed-by-shrinking27 – can be extended in principle to side-chain28 or main-chain29 oligo- and polyrotaxanes and performedunder either kinetic30 or thermodynamic31 control, with everyoption having its own set of advantages and drawbacks. Ther-modynamically controlled approaches tend to offer higheryields. For example, we have constructed a family of oligor-otaxanes32 using a thermodynamically controlled clippingprotocol based on a recognition motif33 between secondary
ween DNP oligomers and CBPQT4+ rings in the solid-state as determined21 by X-ray
Chem. Sci., 2013, 4, 1470–1483 | 1471

Scheme 2 One-pot reaction between DNP and excess 1NPE(OTs)2.
Scheme 3 One-pot reaction between 1NPE(OTs)2 and excess DNP.
Chemical Science Edge Article
Dow
nloa
ded
by U
nive
rsity
of
Mis
sour
i at C
olum
bia
on 0
5 M
arch
201
3Pu
blis
hed
on 0
4 Fe
brua
ry 2
013
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C3S
C00
015J
View Article Online
dialkylammonium salts and crown ethers, and found that evenvery large oligorotaxanes (with up to 20 components) can beproduced32 in >90% yields. We also found that positive coop-erativity between adjacent rings in p–p contact facilitate theself-assembly and dene32b,c the rigid-rod secondary structure ofthe corresponding oligorotaxanes. On the other hand, rotaxanesassembled under thermodynamic control exist in a dynamicequilibrium that is sensitive to experimental conditions.Dynamic ammonium-binding rotaxanes dissociate33c in highlypolar solvents like Me2SO and at elevated temperatures, forexample, which may raise problems in the context of polymericmaterials where kinetically stable and robust compounds aredesired. Ideally, it should be possible to leverage positivecooperativity in a kinetic regime to form highly robustmechanically interlocked polymers with well-dened secondarystructures in high yields. Although we have been able to syn-thesize15b small DNP3CBPQT4+ oligomers consisting of up tofour interlocked components under kinetic control by the Cu(I)-catalyzed Huisgen34 azide–alkyne 1,3-dipolar cycloaddition(‘click’ chemistry35) stoppering protocol, the task of synthe-sizing well-dened linear DNP oligomers that are long enoughto truly bridge the gap between small molecules and polymerswas a challenge of rigorous dimensions that limited our abilityto either probe for positive cooperativity in a kineticallycontrolled synthesis or fully rationalize the behavior of foldedD–A polyrotaxanes based on DNP and CBPQT4+.
Here we report our solution to the oligomerization challenge– namely, a one-pot synthesis of DNP oligomers of wide-ranginglengths that are separable by conventional chromatographytechniques and allow efficient access to well-dened oligomerswith up to 15 repeating units. We sought an alternativesynthesis of DNP oligomers aer consideration of the legion ofiterative steps and purications that would be involved in thestepwise approach.15b Scheme 1 summarizes the stepwise routeto a DNP oligomer with 15 repeating units, 15NPE(OTs)2, fromthe ditosylated DNP monomer 1NPE(OTs)2. Although thestrategy requires only straightforward substitutions and tosy-lations, the 14 steps of synthesis – indeed, 18 steps whenstarting from commercially available compounds – make itrather impractical, especially when considering the requiredquantity of building block 1NPEOH, which is a compound that
Scheme 1 The stepwise approach to the synthesis of DNP oligomers. Encirclednumbers denote the number of iterative steps.
1472 | Chem. Sci., 2013, 4, 1470–1483
is not easy to make15b on account of its asymmetry. Hence, webegan experimenting with one-pot approaches starting from1NPE(OTs)2 which, by way of comparison, can be prepared18 inhigh yields and large quantities without having to resort to ashcolumn chromatography (FCC) to obtain suitably pure material.
Schemes 2 and 3 show the outcomes of one-pot reactionsbetween 1NPE(OTs)2 and 1,5-dihydroxynaphthalene. When1NPE(OTs)2 is used (Scheme 2) in excess, 3NPE(OH)2 and5NPE(OH)2 are isolated in appreciable yields; 7NPE(OH)2 and9NPE(OH)2 were detected by mass spectrometry but could notbe isolated in pure form by FCC because of their highly polarcharacteristics, i.e., low Rf values and poor solubility, with atendency to crystallize in the column. Similar yields for trimersand pentamers were observed (Scheme 3) when 1,5-dihydroxy-naphthalene was used in excess, and on large scales, we couldrecover sufficient quantities (�500 mg) of lower-yielding (�1%)large oligomers such as 9NP and 11NP in high purity. Ourexperience with these one-pot reactions culminated in theprocedure summarized in Scheme 4; we employ an excess of3NPE(OTs)2 in place of 1NPE(OTs)2 to bias the products towardlarger oligomers, and immediately react the crude productswith tosyl chloride in CH2Cl2 to reduce their polarity andimprove their solubility, rendering them separable by FCC. On a700 mg scale with respect to 1,5-dihydroxynaphthalene, we haveisolated 7NPE(OTs)2, 11NPE(OTs)2, and 15NPE(OTs)2 in 6, 7,and 3% yields, respectively. To conrm the monodispersity ofthe compounds, 11NPE(OTs)2 was selected as a representativesample, which was analyzed by gel permeation chromatography(Fig. S1 in the (ESI†)), from which we calculated the smallpolydispersity index of 1.01. Although the yields are low, it islikely that they are comparable to the overall yield that would be
This journal is ª The Royal Society of Chemistry 2013

Scheme 4 One-pot reaction between DNP and excess of 3NPE(OTs)2, imme-diately followed by a reaction with tosyl chloride to afford the tosylated DNPoligomers.
Edge Article Chemical Science
Dow
nloa
ded
by U
nive
rsity
of
Mis
sour
i at C
olum
bia
on 0
5 M
arch
201
3Pu
blis
hed
on 0
4 Fe
brua
ry 2
013
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C3S
C00
015J
View Article Online
achieved from the stepwise approach (Scheme 1), meaning thatthis two-step/one-purication route vastly exceeds the stepwiseapproach in terms of efficiency and still affords the oligomers insufficient (200–700 mg) quantities.
Synthesis of donor–acceptor oligorotaxane foldamers
With a range of well-dened DNP oligomers in hand, we pro-ceeded with their conversion to the corresponding oligorotax-anes with CBPQT4+. Since click chemistry has proven36 to be anefficient means of stoppering pseudorotaxanes under kineticcontrol, we selected the copper-catalyzed azide–alkyne cyclo-addition (CuAAC) reaction in order to transform the DNP olig-omers into oligorotaxanes. We converted the ditosylatedoligomers 1NPE(OTs)2–15NPE(OTs)2 to the corresponding dia-zides 1NPE(N3)2–15NPE(N3)2 with sodium azide in excellent(>90%) yields, and reacted them with the alkyne-terminateddithiolane-functionalized stopper DS in the presence of cupricsulfate to form the dumbbells (Scheme 5) in good yields(76–87%) as a result of triazole formation. The dumbbellsynthesis indicated that the click chemistry conditions weselected were efficient enough for stoppering pseudorotaxanesderived from the same DNP oligomers, despite the potential ofthe dithiolane group on DS to disrupt the click reaction bycoordinating with Cu(I). Encouraged by these results, the
Scheme 5 Synthesis of DNP oligomer dumbbells by transformation of tosylated oalized alkyne stopper by click chemistry.
This journal is ª The Royal Society of Chemistry 2013
diazides were mixed with CBPQT4+ (0.9 equiv. per DNP unit) toform the corresponding pseudorotaxanes and stoppered withDS (Table 1) using a similar CuAAC protocol, albeit at lowerinitial temperatures to facilitate self-assembly of the pseudor-otaxanes balanced by longer reaction times, since lowertemperatures, as well as steric and electrostatic constraintsimparted by threaded CBPQT4+ rings near the reactive terminalazide groups, can reduce reaction rates. The neutral productswere extracted into CH2Cl2, leaving behind a sticky residue ofcharged products that were converted to their TFA salts bycounterion exchange and puried by preparative scale reversed-phase high performance liquid chromatography (RP-HPLC),eluting with H2O–MeCN. The product distributions from eachof the reactions are summarized in Table 1. All of the oligor-otaxanes we isolated were characterized by high-resolutionmass spectrometry (HRMS) with an electrospray ionization (ESI)source (Table 2), and the purity of each compound was veriedby analytical RP-HPLC.
Despite our use of kinetically controlled click reactions, it isapparent that the product distribution obtained from ourstoppering protocol is not a statistical one, selecting instead forrotaxanes in which approximately half of the DNP units areencircled by CBPQT4+ rings, providing important clues aboutthe nature of the pseudorotaxane precursors. In general, the[0.5(n � 1) + 2]rotaxanes (terms in square brackets denote thetotal number of interlocked components and n is the number ofDNP units in the dumbbell) were obtained in the highest yields,which ranged from 55% for the [2]rotaxane [2]1NPR4+ to 10%for [9]rotaxane [9]15NPR32+. The stoppering of the trimericpseudorotaxane to form [3]3NPR8+ was the only exception,where a [0.5(n � 1) + 1]rotaxane [2]3NPR4+ was preferentiallyformed (44%) instead. Although the [0.5(n � 1)+2]rotaxanes –
hereaer referred to as the ‘Happy’ rotaxanes – were the domi-nant products, in many cases, we could also isolate the‘Confused’ [0.5(n � 1) + 1]rotaxanes and ‘Frustrated’ [0.5(n � 1) +3]rotaxanes from the product mixtures in low yields. Oligor-otaxanes with components numbering beyond the boundariesof [0.5(n � 1) + 1] and [0.5(n � 1) + 3] could not be isolated. Theorigin of the Confused, Happy, and Frustrated classications liesin certain characteristics that typify the molecules within each
ligomers to the corresponding diazides and reaction with a dithiolane-function-
Chem. Sci., 2013, 4, 1470–1483 | 1473
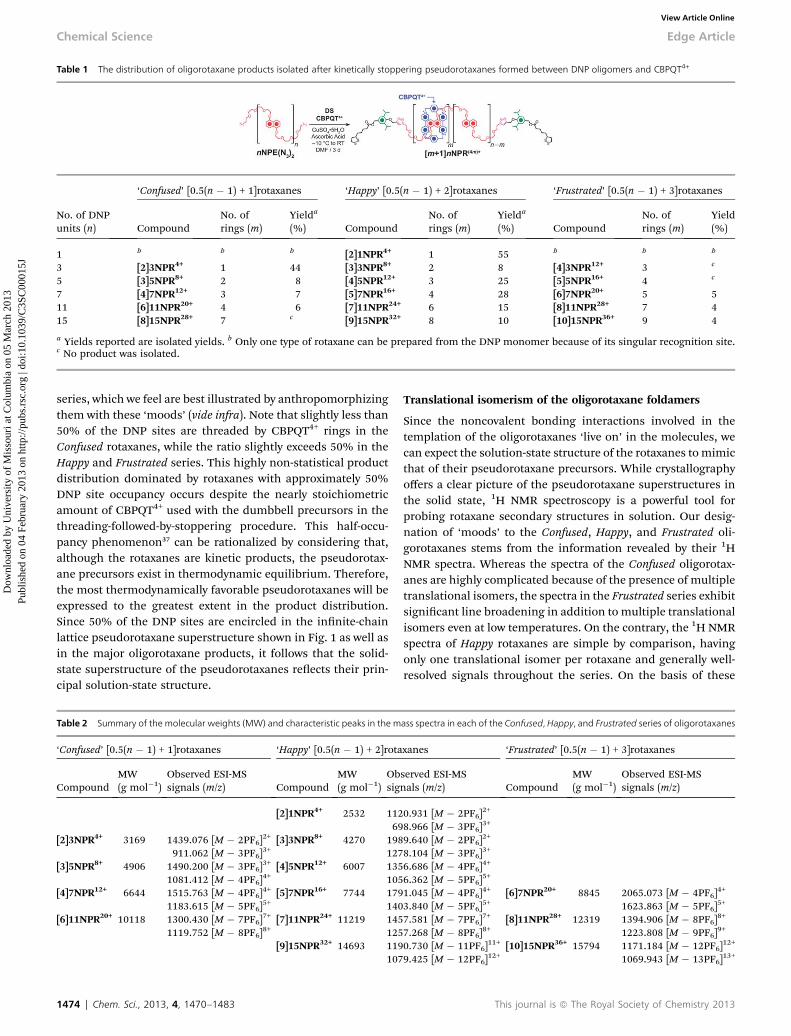
Table 1 The distribution of oligorotaxane products isolated after kinetically stoppering pseudorotaxanes formed between DNP oligomers and CBPQT4+
No. of DNPunits (n)
‘Confused’ [0.5(n � 1) + 1]rotaxanes ‘Happy’ [0.5(n � 1) + 2]rotaxanes ‘Frustrated’ [0.5(n � 1) + 3]rotaxanes
CompoundNo. ofrings (m)
Yielda
(%) CompoundNo. ofrings (m)
Yielda
(%) CompoundNo. ofrings (m)
Yield(%)
1 b b b [2]1NPR4+ 1 55 b b b
3 [2]3NPR4+ 1 44 [3]3NPR8+ 2 8 [4]3NPR12+ 3 c
5 [3]5NPR8+ 2 8 [4]5NPR12+ 3 25 [5]5NPR16+ 4 c
7 [4]7NPR12+ 3 7 [5]7NPR16+ 4 28 [6]7NPR20+ 5 511 [6]11NPR20+ 4 6 [7]11NPR24+ 6 15 [8]11NPR28+ 7 415 [8]15NPR28+ 7 c [9]15NPR32+ 8 10 [10]15NPR36+ 9 4
a Yields reported are isolated yields. b Only one type of rotaxane can be prepared from the DNP monomer because of its singular recognition site.c No product was isolated.
Chemical Science Edge Article
Dow
nloa
ded
by U
nive
rsity
of
Mis
sour
i at C
olum
bia
on 0
5 M
arch
201
3Pu
blis
hed
on 0
4 Fe
brua
ry 2
013
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C3S
C00
015J
View Article Online
series, which we feel are best illustrated by anthropomorphizingthem with these ‘moods’ (vide infra). Note that slightly less than50% of the DNP sites are threaded by CBPQT4+ rings in theConfused rotaxanes, while the ratio slightly exceeds 50% in theHappy and Frustrated series. This highly non-statistical productdistribution dominated by rotaxanes with approximately 50%DNP site occupancy occurs despite the nearly stoichiometricamount of CBPQT4+ used with the dumbbell precursors in thethreading-followed-by-stoppering procedure. This half-occu-pancy phenomenon37 can be rationalized by considering that,although the rotaxanes are kinetic products, the pseudorotax-ane precursors exist in thermodynamic equilibrium. Therefore,the most thermodynamically favorable pseudorotaxanes will beexpressed to the greatest extent in the product distribution.Since 50% of the DNP sites are encircled in the innite-chainlattice pseudorotaxane superstructure shown in Fig. 1 as well asin the major oligorotaxane products, it follows that the solid-state superstructure of the pseudorotaxanes reects their prin-cipal solution-state structure.
Table 2 Summary of the molecular weights (MW) and characteristic peaks in the m
‘Confused’ [0.5(n � 1) + 1]rotaxanes ‘Happy’ [0.5(n � 1) + 2]rota
CompoundMW(g mol�1)
Observed ESI-MSsignals (m/z) Compound
MW(g mol�1)
Obsig
[2]1NPR4+ 2532 11269
[2]3NPR4+ 3169 1439.076 [M � 2PF6]2+ [3]3NPR8+ 4270 198
911.062 [M � 3PF6]3+ 127
[3]5NPR8+ 4906 1490.200 [M � 3PF6]3+ [4]5NPR12+ 6007 135
1081.412 [M � 4PF6]4+ 105
[4]7NPR12+ 6644 1515.763 [M � 4PF6]4+ [5]7NPR16+ 7744 179
1183.615 [M � 5PF6]5+ 140
[6]11NPR20+ 10118 1300.430 [M � 7PF6]7+ [7]11NPR24+ 11219 145
1119.752 [M � 8PF6]8+ 125
[9]15NPR32+ 14693 119107
1474 | Chem. Sci., 2013, 4, 1470–1483
Translational isomerism of the oligorotaxane foldamers
Since the noncovalent bonding interactions involved in thetemplation of the oligorotaxanes ‘live on’ in the molecules, wecan expect the solution-state structure of the rotaxanes to mimicthat of their pseudorotaxane precursors. While crystallographyoffers a clear picture of the pseudorotaxane superstructures inthe solid state, 1H NMR spectroscopy is a powerful tool forprobing rotaxane secondary structures in solution. Our desig-nation of ‘moods’ to the Confused, Happy, and Frustrated oli-gorotaxanes stems from the information revealed by their 1HNMR spectra. Whereas the spectra of the Confused oligorotax-anes are highly complicated because of the presence of multipletranslational isomers, the spectra in the Frustrated series exhibitsignicant line broadening in addition to multiple translationalisomers even at low temperatures. On the contrary, the 1H NMRspectra of Happy rotaxanes are simple by comparison, havingonly one translational isomer per rotaxane and generally well-resolved signals throughout the series. On the basis of these
ass spectra in each of the Confused, Happy, and Frustrated series of oligorotaxanes
xanes ‘Frustrated’ [0.5(n � 1) + 3]rotaxanes
served ESI-MSnals (m/z) Compound
MW(g mol�1)
Observed ESI-MSsignals (m/z)
0.931 [M � 2PF6]2+
8.966 [M � 3PF6]3+
9.640 [M � 2PF6]2+
8.104 [M � 3PF6]3+
6.686 [M � 4PF6]4+
6.362 [M � 5PF6]5+
1.045 [M � 4PF6]4+ [6]7NPR20+ 8845 2065.073 [M � 4PF6]
4+
3.840 [M � 5PF6]5+ 1623.863 [M � 5PF6]
5+
7.581 [M � 7PF6]7+ [8]11NPR28+ 12319 1394.906 [M � 8PF6]
8+
7.268 [M � 8PF6]8+ 1223.808 [M � 9PF6]
9+
0.730 [M � 11PF6]11+ [10]15NPR36+ 15794 1171.184 [M � 12PF6]
12+
9.425 [M � 12PF6]12+ 1069.943 [M � 13PF6]
13+
This journal is ª The Royal Society of Chemistry 2013

Fig. 2 A Goldilocks effect in donor–acceptor oligorotaxanes, exemplified by the7NP-based compounds. The [0.5(n � 1) + 1]rotaxane [4]7NPR12+ has up to sixtranslational isomers that satisfy the selection rule forbidding CBPQT4+ rings toencircle adjacent DNP sites, eliciting the ‘Confused’ designation. On the otherhand, it is impossible to draw a structure that obeys the rule for the [0.5(n � 1) +3]rotaxane [6]7NPR20+, hence it is ‘Frustrated’. Only the [0.5(n � 1) + 2]rotaxane[5]7NPR16+ has a single ‘Happy’ translational isomer that satisfies the rule.
Edge Article Chemical Science
Dow
nloa
ded
by U
nive
rsity
of
Mis
sour
i at C
olum
bia
on 0
5 M
arch
201
3Pu
blis
hed
on 0
4 Fe
brua
ry 2
013
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C3S
C00
015J
View Article Online
observations, we can infer the following empirical selectionrule: the most stable co-conformation38 in any of the oligorotaxaneswill be one in which no two CBPQT4+ rings reside on adjacent DNPsites. This rule simultaneously explains the presence of multipletranslational isomers in the Confused series and the linebroadening observed in the Frustrated series, since multiple co-conformations can be identied to satisfy the rule in the former,while no translational isomers will satisfy the selection rule inthe case of the latter. Thus, we use the term Confused to imply arotaxane that expresses multiple stable translational isomersthat are slow to equilibrate on the 1H NMR timescale. A Frus-trated rotaxane, on the other hand, equilibrates more rapidlybetween multiple higher-energy states. It seems that the Happyseries exhibits a ‘Goldilocks’39 effect, where the number of ringsand recognition units are perfectly matched so as to make asingle ‘happy’ translational isomer far more stable than any
Fig. 3 Molecular structures of the oligomers in the Happy series of donor–accepto
This journal is ª The Royal Society of Chemistry 2013
alternative. These anthropomorphic concepts are illustratedgraphically using the 7NP oligorotaxanes (the smallest set weisolated with members in all three ‘moods’) as examples inFig. 2. A selection rule that forbids the CBPQT4+ rings to encircleadjacent DNP sites also agrees well with a secondary structurethat is stabilized by continuous D–A–D–A stacking, asobserved21 in the donor–acceptor pseudorotaxanes with 50%recognition-site occupancy in the solid state.
Although the Confused oligorotaxanes adopt multiple trans-lational isomers, the species with the longest D–A stackingmotif will be the most highly stabilized and therefore thedominant isomer. In the simplest Confused rotaxane [2]3NPR4+,for example, the most stable translational isomer – that with theCBPQT4+ ring encircling the central DNP unit – is adopted by80% of the molecular population (see Section 3B of the ESI†).The remaining 20% of the molecules adopt a co-conformationwith the CBPQT4+ on the periphery, where the stabilizing D–Ainteractions are inevitably weaker on account of the ring'saccess to half as many ‘alongside’ DNP units. This 4 : 1 distri-bution indicates that there is only a small difference betweenthe free energies (<1 kcal mol�1) of the various rule-compliantisomers of the Confused oligorotaxanes. There is no indicationthat Happy oligorotaxanes are signicantly more stabilized thanthe dominant Confused rotaxanes, however, despite the bias infavor of Happy oligorotaxanes in the product distributions.Rather, the preference for Happy oligorotaxanes during thestoppering protocol is most likely a result of using CBPQT4+ innearly 1 : 1 stoichiometry with the DNP recognition units,which amounts to an excess with respect to the half-occupancyfoldamers. Indeed, the entropic penalty for adding more ringsonto the dumbbells of Happy rotaxanes may compete with theenthalpic gains earned from the extra D–A interactions enabledby the BIPY2+ units of the terminal cyclophanes. In any event,only the Happy family of oligorotaxanes (Fig. 3) are conducive todetailed solution-state analysis by high eld 1H NMR
r oligorotaxanes.
Chem. Sci., 2013, 4, 1470–1483 | 1475

Fig. 4 The symmetry and expected number of a and b 1H NMR signals forcomplexes between CBPQT4+ hosts and DNP guests with various symmetryelements, assuming the structures are static on the timescale of a 1H NMRexperiment.
Chemical Science Edge Article
Dow
nloa
ded
by U
nive
rsity
of
Mis
sour
i at C
olum
bia
on 0
5 M
arch
201
3Pu
blis
hed
on 0
4 Fe
brua
ry 2
013
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C3S
C00
015J
View Article Online
spectroscopy. Whereas the larger Confused and Frustratedrotaxanes produce intractably complicated spectra, the 1H NMRresonances of the single-isomer Happy rotaxanes can beassigned in full.
Fig. 5 The 1H NMR spectrum (CD3CN, 600 MHz, 233 K) of [3]3NPR8+.
1476 | Chem. Sci., 2013, 4, 1470–1483
Solution-state structure and dynamics of the Happyoligorotaxanes
We turn our attention now to an analysis of the 1H NMR spectraof the Happy series of oligorotaxanes. The symmetry of theDNP3CBPQT4+ complexes are important with respect to howwe interpret their 1H NMR spectra, especially in regards to thetimescale of the molecular dynamics, i.e., which protons areundergoing rapid site exchange on the NMR timescale versuswhich environments are ‘frozen out’ in a slow exchange regime.Consider the conceptual structures in Fig. 4. When a DNP unitis locked within a CBPQT4+ ring and does not undergo anyrearrangements on the NMR timescale, its C2 symmetry isimposed on the more symmetrical (D2h) CBPQT4+ ring andcauses the a and b signals – each single resonances in the 1HNMR spectrum of CBPQT4+– to separate into pairs. When DNPis further desymmetrized by appending inequivalent R groupsat the 1- and 5-positions, the complex is reduced to Cs
symmetry, where a plane of reection is the highest remainingsymmetry operation, and the number of a and b signals aredoubled once again. This Cs scenario applies to, minimally, theperipheral DNP3CBPQT4+ subcomplexes in any oligorotaxanewith two or more CBPQT4+ rings, since their DNP units connectto the stoppers on only one side. Finally, if the two glycol chainsthat commonly accompany DNP units each wrap aroundopposite BIPY2+ subunits by way of their characteristic [C–H/O] hydrogen bonding interactions (denoted with dashed lines inFig. 4), the plane of reection is broken and the symmetry isreduced to the C1 point group. With no symmetry operationsthat can equate any of the a or b protons of CBPQT4+, a total ofeight a and eight b signals would be expected in this nal case.The literature suggests17e that, at least inmonomeric complexes,the C1 scenario does not apply to NMR interpretation becausethe glycol chains exchange sites rapidly. It was unclear at theoutset, however, if the same behavior would arise in oligor-otaxanes stabilized by extended D–A stacking. Inspection of thea and b signals in the 1H NMR spectra at 233 K in CD3CN of anyof the Happy oligorotaxane reveals the answer. Taking the
This journal is ª The Royal Society of Chemistry 2013

Fig. 6 Polyrotaxane co-conformational analysis, exemplified by the simplestoligomer [3]3NPR8+. This interlocked system can access up to eight folded co-conformations in the superrotation network by iteratively executing 180� rota-tions (about a ‘vertical’ axis) on either the DNP3CBPQT4+ subcomplexes, whichexchanges the outside (‘O’) and alongside (‘A’) BIPY2+ units, or the centralunencircled DNP unit. Because these processes occur rapidly on the NMR time-scale, the corresponding resonances give rise to time-averaged signals thatsimplify the corresponding 1H NMR spectra.
Edge Article Chemical Science
Dow
nloa
ded
by U
nive
rsity
of
Mis
sour
i at C
olum
bia
on 0
5 M
arch
201
3Pu
blis
hed
on 0
4 Fe
brua
ry 2
013
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C3S
C00
015J
View Article Online
simplest oligorotaxane [3]3NPR8+ (Fig. 5) as a representativeexample, we observe only four a and four b signals. Thisobservation obliges us to conclude that the 1H NMR spectrumrepresents either (1) the linear, fully unfolded co-conformationin which the reection plane is maintained, (2) a static, foldedco-conformation in which all heterotopic a and b protonsresonate in pairs with equal frequencies by coincidence,40 or (3)a dynamic series of folded co-conformations undergoing rapidsite exchange, giving pairs of a and b protons equivalent time-averaged resonances. We argue in favor of scenario (3) (illus-trated graphically in Fig. 6) on the basis of a detailed analysis
Fig. 7 The 1H NMR spectrum (CD3CN, 600 MHz, 233 K) of [4]5NPR12+.
This journal is ª The Royal Society of Chemistry 2013
(vide infra) of the collective behavior of the oligorotaxanes asprobed by 1H NMR spectroscopy at 233 K in CD3CN.
Fig. 6 shows a co-conformational analysis of [3]3NPR8+. Wedistinguish between ‘outside’ (labeled ‘O’) and ‘alongside’(labeled ‘A’) BIPY2+ subunits in the folded co-conformation of[3]3NPR8+ to help with visualization of the exchange processes,which are somewhat reminiscent of the pseudorotationphenomenon known41 to take place in cyclopentane. Corre-spondingly, the [3]3NPR8+ rotaxane can pass through up toeight folded co-conformations by iteratively executing 180�
turns on either the DNP3CBPQT4+ subcomplexes or theunencircled central DNP unit. Borrowing from conformationalanalysis of ve-membered ring systems, we propose the term‘superrotation’ to describe pseudorotation-like processes inmechanically interlocked systems. Superrotation exchanges Oand A BIPY2+ sites rapidly on the NMR timescale and rendersthem equivalent for the purposes of 1H NMR spectroscopicinterpretation. In other words, the protons that can beexchanged by a reection plane in the unfolded co-conforma-tion are averaged in the folded co-conformation by super-rotation. Note that the number of possible foldedco-conformations in an oligorotaxane scales with 2n as moreDNP units and DNP3CBPQT4+ subcomplexes are introduced,since each of the n DNP (including DNP3CBPQT4+) units canaccess two states in the D–A stack by performing a 180� rotation.Any given oligorotaxane co-conformation has n connectionpoints – where n is, again, the total number of DNP units – inthe superrotational network. The degree to which the rotaxaneswill unfold and access alternative intermediate co-conforma-tions – unfolded or globular co-conformations, for example – tocarry out superrotation is unclear at this time and will requirerigorous computational studies.
So far, we have supported the superrotation hypothesis onlyon the basis of the number of observed a and b proton signals inthe 1H NMR spectra of the oligorotaxanes. These observationsrule out the conclusion of a single static folded co-conforma-tion, since it would be highly unlikely to always have pairs ofheterotopic protons coincidentally resonating at the same
Chem. Sci., 2013, 4, 1470–1483 | 1477

Fig. 8 The partial 1H NMR spectrum (CD3CN, 600 MHz, 233 K) of [7]11NPR24+.
Fig. 9 Stacked plot of the partial 1H NMR spectra (CD3CN, 600 MHz, 233 K) ofHappy oligorotaxanes [2]1NPR4+–[9]15NPR32+, showing the aromatic region inwhich the a and b protons resonate. The a and b resonances of interior cyclo-phanes (distinguished with progressively darker shades of blue) always resonateat the lowest frequencies.
Fig. 10 The averaged chemical shifts of each CBPQT4+ (A) and DNP (B) reso-nance, plotted against the number of components in the oligorotaxanes.
Chemical Science Edge Article
Dow
nloa
ded
by U
nive
rsity
of
Mis
sour
i at C
olum
bia
on 0
5 M
arch
201
3Pu
blis
hed
on 0
4 Fe
brua
ry 2
013
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C3S
C00
015J
View Article Online
frequency without exception. On this evidence alone, however,an unfolded co-conformation is no less plausible than a foldedone undergoing rapid site exchange. To rule out the possibilitythat the linear co-conformation dominates in solution, we turnto the trends that emerge in the chemical shis of all DNP andCBPQT4+ signals in the Happy series. Conveniently, the 1H NMRsignals could be unambiguously assigned using 1H–1H gradientselected correlation spectroscopy (gCOSY) and 1H–1H gradientselected nuclear Overhauser effect spectroscopy (gNOESY) toidentify important through-bond and through-space correla-tions, respectively. A detailed explanation of how the signalswere assigned is provided in Section 3 of the ESI.† Fig. 7 showsthe 1H NMR spectrum of [4]5NPR12+. In this case, we distin-guish between the ‘outside’ and ‘alongside’ BIPY2+ units with ‘a’and ‘o’ prexes, respectively, on the a and b protons of theBIPY2+ units for the sake of demonstrating that their signals areaveraged, though this distinction is formally unnecessary. Note
1478 | Chem. Sci., 2013, 4, 1470–1483
that the innermost CBPQT4+ ring is constitutionally inequiva-lent to the outermost rings and also has an inversion center ofsymmetry. Moreover, its a and b signals uniformly resonate atlower frequencies than their peripherally located counterparts.The 1H NMR spectrum of [5]7NPR16+ is very similar to that of [4]5NPR12+ because it has two equivalent interior rings instead ofonly one, but the trend also continues in the 1H NMR spectrum(Fig. 8) of [7]11NPR24+. In [7]11NPR24+, the protons in theadditional pair of CBPQT4+ rings located in the inner depths ofthe oligomer resonate again at slightly lower frequencies. In thiscase, the extraneous ‘a’ and ‘o’ distinctions are removed, andmore interior proton environments are colored with progres-sively darker shades of blue in the structural diagrams for
This journal is ª The Royal Society of Chemistry 2013

Fig. 11 The difference between the chemical shifts (Dd) of protons in the oli-gorotaxanes and the appropriate corresponding uncomplexed dumbbell orcyclophane analogue, plotted as a function of the number of interlockedcomponents in each oligorotaxane.
Edge Article Chemical Science
Dow
nloa
ded
by U
nive
rsity
of
Mis
sour
i at C
olum
bia
on 0
5 M
arch
201
3Pu
blis
hed
on 0
4 Fe
brua
ry 2
013
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C3S
C00
015J
View Article Online
clarity. DNP protons are also iteratively labeled with ‘i’ prexesto distinguish them from one another; protons with more ‘i’prexes are located more ‘inward’ in the molecules. The stackedplot in Fig. 9 demonstrates that, as more repeating units areintroduced into the interior of the oligomer backbone, theinnermost a and b protons always resonate at the lowestfrequencies. This apparent shielding effect on the BIPY2+ unitscan be explained by the accumulated aromatic ring-current shithat would be expected in the folded co-conformation and notin a linear one.
The BIPY2+ protons of CBPQT4+ are not the only specieswhose signals migrate to lower frequencies as the oligomerbackbone is extended. Most importantly, one would expectnoticeable changes in the chemical shis of unencircled DNPprotons if they are indeed incorporated into a continuous D–Astack; ‘alongside’ DNP protons should resonate at lowerfrequencies relative to protons in DNP units that do notparticipate in a D–A stack, such as those in the free dumbbells1NPD–15NPD. The graphs in Fig. 10 plot the averaged chemicalshis of the BIPY2+ and DNP protons against the number ofcomponents in the corresponding oligorotaxanes, where the
Fig. 12 The partial 1H NMR spectrum (CD3CN, 600 MHz, 233 K) of [9]15NPR32+.
This journal is ª The Royal Society of Chemistry 2013
one-component data points represent the appropriate signals inuncomplexed dumbbell or cyclophane species. These data showthat all DNP protons (both included and alongside) follow thesame trend observed in the case of the BIPY2+ protons, wherethe innermost species resonate at the lowest frequencies, sinceintroducing more repeating units to the oligomers continuouslyshis their averaged signals to lower frequencies. The specic(non-averaged) frequencies of every DNP and BIPY2+ signal istabulated in Tables S1 and S2 in the ESI,† and show that theinnermost protons uniformly resonate at the lowest frequen-cies. We used Dd to quantify the changes in chemical shis ofDNP and CBPQT4+ protons in Fig. 11. The value Dd representsthe difference in chemical shi between the specied signals(averaged where applicable) of the oligorotaxanes and those inthe corresponding uncomplexed dumbbells and cyclophanes.Thus, the magnitude of Dd quanties the extent to which eachproton environment is affected by the secondary structure.Remarkably, certain alongside DNP protons (a4 and a8) exhibiteven greater aromatic ring-current shis than one set ofincluded DNP protons (i2 and i6), and all alongside protons aremore affected than the a CBPQT4+ protons. One would expectthe resonances of unencircled DNP protons to be the leastdisplaced if the unfolded co-conformation dominates, sinceaccumulated aromatic ring-current shis are not sensitive42 todistances beyond �12 A. The included 4 and 8 DNP protons areremoved from these plots because they experience suchdramatic shis on account of their highly shielded positions inthe CBPQT4+ rings that they marginalize the remaining shis bycomparison. Note also that the only way to obtain an alongsideDNP resonance in a two-component system is with a non-Happyoligorotaxane, so data from the dominant translational isomerof [2]3NPR4+ were used in that particular case. Inspection of thetrends in Fig. 10 and 11 show that they approach an asymptoticlimit as the oligomers grow longer, where further extension ofthe longest oligomers no longer has an appreciable effect on theaveraged chemical shis of the signals. Thus, it is appropriateto classify the longest oligomers [7]11NPR24+ and [9]15NPR32+
Chem. Sci., 2013, 4, 1470–1483 | 1479

Chemical Science Edge Article
Dow
nloa
ded
by U
nive
rsity
of
Mis
sour
i at C
olum
bia
on 0
5 M
arch
201
3Pu
blis
hed
on 0
4 Fe
brua
ry 2
013
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C3S
C00
015J
View Article Online
as monodisperse polymers, since their behavior is relativelyinsensitive to the precise number of repeating units.Completing the series, the partial 1H NMR spectrum of [9]15NPR32+ is presented in Fig. 12. Some line broadening typicalof high-MW polymers is apparent, but not at the cost ofassigning the majority of signals.
In addition to the trends in chemical shi that have beenobserved in the oligorotaxanes, we should expect to observethrough-space correlations between alongside DNP units andthe BIPY2+ units of the CBPQT4+ rings – if they do indeedparticipate in a continuous D–A stack – by way of the nuclearOverhauser effect. Many of these expected through-spacecorrelations are indeed observable in the 1H–1H NOESY spectraof the Happy oligorotaxanes, especially for the innermost DNPand BIPY2+ units in any given compound, which are presentedin detail in the ESI.†
Conclusion
We have reported a novel class of foldamers in which the foldedsuperstructures are enabled by stabilizing donor–acceptor andhydrogen bonding interactions among mechanically inter-locked components. By developing efficient one-pot reactions toaccess well-dened DNP oligomers of variable length, we wereable to access three series of homologous donor–acceptor oli-gorotaxanes by a stoppering protocol employing click chem-istry. Despite being in a kinetically controlled regime, theproduct distribution is highly biased in favor of rotaxanes inwhich approximately half of the DNP sites are occupied byCBPQT4+ rings, indicating that positive cooperativity fromextended donor–acceptor stacking in the folded secondarystructure plays a role in kinetic selection. The largest oligomerswe report have 15 DNP units in their dumbbells and areunprecedented in terms of their size for well-dened andmonodisperse rotaxanes formed under kinetic control, withmolecular weights extending above 15 000 Da that put them inthe category of genuine macromolecules. The 1H NMR spectraof the oligorotaxanes revealed that the DNP : CBPQT4+ ratiocontrols certain behaviors in the NMR spectra, which we char-acterize by the anthropomorphic ‘moods’ of Confused, Happy,and Frustrated. Whereas multiple translational isomers areobservable in Confused and Frustrated rotaxanes, a Goldilockseffect is apparent in the Happy rotaxanes, since only one kind ofstable translational isomer is observed for any oligorotaxanewithin the series. Detailed analysis of the 1H NMR spectra of theHappy rotaxanes suggests that these compounds equilibratebetween multiple folded co-conformations rapidly on the 1HNMR timescale. This ‘superrotation’ process, as we deem it inour co-conformational analysis, is likened to the well-knownpseudorotation phenomenon from conformational analysis ofcyclopentane and other ve-membered ring systems. We ndthe superrotation hypothesis to be a more appropriate expla-nation of the NMR data than the supposition of a linear(unfolded) co-conformation as the dominant solution-statesecondary structure on the basis of the chemical shis of allDNP and BIPY2+ signals in the series, which are shied tosignicantly lower frequencies with respect to their non-
1480 | Chem. Sci., 2013, 4, 1470–1483
oligomeric and non-interlocked counterparts. The fundamentalknowledge gained from these investigations can help guide thedevelopment of interlocked polymers with novel and emergentproperties governed by well-dened secondary structures, andperhaps even shed further light on the origins of secondarystructure in biomolecules and biopolymers.
Acknowledgements
This work was supported by the Non-Equilibrium EnergyResearch Centre (NERC), which is an Energy Frontier ResearchCentre (EFRC) funded by the U.S. Department of Energy, Officeof Basic Sciences (DOE-BES) under Award DE-SC0000989. J.F.S.and C.J.B. were supported by theWCU Program (NRF R-31-2008-000- 10055-0) funded by the Ministry of Education, Science andTechnology, Korea. C.J.B. acknowledges the National ScienceFoundation for the award of a Graduate Research Fellowship.
References
1 J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112.2 D. J. Cram, Angew. Chem., Int. Ed. Engl., 1988, 27, 1009–1020.3 (a) D. Philp and J. F. Stoddart, Synlett, 1991, 445–458; (b)G. M. Whitesides, J. P. Mathias and C. T. Seto, Science,1991, 254, 1312–1319; (c) J. S. Lindsey, New J. Chem., 1991,15, 153–180; (d) M. C. T. Fyfe and J. F. Stoddart, Acc. Chem.Res., 1997, 30, 393–401; (e) G. A. Ozin, Chem. Commun.,2000, 419–432; (f) J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A.,2002, 99, 4763–4768; (g) J. F. Stoddart and H.-R. Tseng,Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4797–4800; (h)D. N. Reinhoudt and M. Crego-Calama, Science, 2002, 295,2403–2407; (i) G. Whitesides and B. Grzybowski, Science,2002, 295, 2418–2421; (j) I. W. Hamley, Angew. Chem., Int.Ed., 2003, 42, 1692–1712; (k) J. A. A. W. Elemans,A. E. Rowan and R. J. M. Nolte, J. Mater. Chem., 2003, 13,2661–2670; (l) J. F. Stoddart, Nat. Chem., 2009, 1, 14–15.
4 (a) J. W. Steed and J. L. Atwood, in Supramolecular Chemistry,Wiley, West Sussex, UK, 2nd edn, 2009, pp. 49–103; (b)Supramolecular Chemistry: From Molecules to Nanomaterials,ed. J.W.SteedandP.A.Gale,Wiley,2012, vol. 4, pp. 1381–1963.
5 (a) H.-J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977; (b) J. N. Israelachvili, Intermolecular and SurfaceForces, Elsevier, Oxford, 3rd edn, 2011.
6 (a) A. Ulman, Chem. Rev., 1996, 96, 1533–1554; (b) J. Y. Ying,C. P. Mehnert and M. S. Wong, Angew. Chem., Int. Ed., 1999,38, 56–77; (c) H.-A. Klok and S. Lecommandoux, Adv. Mater.,2001, 13, 1217–1229; (d) B. Moulton and M. J. Zaworotko,Chem. Rev., 2001, 101, 1629–1658; (e) D. E. Discher andA. Eisenberg, Science, 2002, 297, 967–973; (f) S. Zhang, Nat.Biotechnol., 2003, 21, 1171–1178; (g) E. Katz and I. Willner,Angew. Chem., Int. Ed., 2004, 43, 6042–6108; (h) G. A. Ozinand A. A. Arsenault, Nanochemistry: A Chemical Approach toNanomaterials, The Royal Society of Chemistry, Cambridge,2005; (i) A. B. Descalzo, R. Martınez-Ma~nez, F. Sancenon,K. Hoffmann and K. Rurack, Angew. Chem., Int. Ed., 2006,45, 5924–5948; (j) T. Kato, N. Mizoshita and K. Kishimoto,Angew. Chem., Int. Ed., 2006, 45, 38–68; (k) Y. Wang, H. Xu
This journal is ª The Royal Society of Chemistry 2013

Edge Article Chemical Science
Dow
nloa
ded
by U
nive
rsity
of
Mis
sour
i at C
olum
bia
on 0
5 M
arch
201
3Pu
blis
hed
on 0
4 Fe
brua
ry 2
013
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C3S
C00
015J
View Article Online
and X. Zhang, Adv. Mater., 2009, 21, 2849–2864; (l) D. Astruc,E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959; (m) B. Rybtchinski, ACS Nano, 2011, 5, 6791–6818; (n)Y.-F. Song, D.-L. Long, C. Ritchie and L. Cronin, Chem.Rec., 2011, 11, 158–171; (o) T. Aida, E. W. Meijer andS. I. Stupp, Science, 2012, 335, 813–817.
7 For discussions on the role of noncovalent chemistry invarious emerging technologies, see: (a) R. Haag, Angew.Chem., Int. Ed., 2004, 43, 278–282; (b) D. Fiedler,D. H. Leung, R. G. Bergman and K. N. Raymond, Acc.Chem. Res., 2005, 38, 349–358; (c) N. L. Rosi andC. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562; (d) C. OrtizMellet, J. M. Benito and J. M. Garcıa Fernandez, Chem.–Eur. J., 2010, 16, 6728–6742; (e) F. Wurthner andK. Meerholz, Chem.–Eur. J., 2010, 16, 9366–9373; (f) Z. Li,J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem.Soc. Rev., 2012, 41, 2590–2605; (g) Applications ofSupramolecular Chemistry, ed. H.-J. Schweider, CRC Press,Boca Raton, FL, 2012.
8 S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180.9 (a) D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes andJ. S. Moore, Chem. Rev., 2001, 101, 3893–4012; (b)G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933–5941.
10 (a) J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820; (b)J.-C. Olsen, K. E. Griffiths and J. F. Stoddart, in From Non-Covalent Assemblies to Molecular Machines, ed. J.-P. Sauvageand P. Gaspard, Wiley-VCH, Weinheim, 2011, pp. 67–139;(c) C. J. Bruns and J. F. Stoddart, Top. Curr. Chem., 2012,323, 19–72.
11 (a) G. Schill, Catenanes, Rotaxanes, and Knots, AcademicPress, New York, 1971; (b) C. O. Dietrich-Buchecker andJ.-P. Sauvage, Chem. Rev., 1987, 87, 795–810; (c)D. B. Amabilino and J. F. Stoddart, Chem. Rev., 1995, 95,2725–2829; (d) A. C. Benniston, Chem. Soc. Rev., 1996, 25,427–435; (e) R. Jager and F. Vogtle, Angew. Chem., Int. Ed.Engl., 1997, 36, 930–944; (f) J.-P. Sauvage, Acc. Chem. Res.,1998, 31, 611–619; (g) M. Fujita, Acc. Chem. Res., 1999, 32,53–61; (h) G. A. Breault, C. A. Hunter and P. C. Mayers,Tetrahedron, 1999, 55, 5265–5293; (i) Molecular Catenanes,Rotaxanes, and Knots: A Journey Through the World ofMolecular Topology, ed. J.-P. Sauvage and C. Dietrich-Buchecker, Wiley-VCH, Weinheim, 1999; (j) K. Kim, Chem.Soc. Rev., 2002, 31, 96–107; (k) E. R. Kay and D. A. Leigh,Top. Curr. Chem., 2005, 262, 133–177; (l) G. Wenz,B.-H. Han and A. Muller, Chem. Rev., 2006, 106, 782–817;(m) M. S. Vickers and P. D. Beer, Chem. Soc. Rev., 2007, 36,211–225; (n) J. E. Beves, B. A. Blight, C. J. Campbell,D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed.,2011, 50, 9260–9327.
12 A. Coskun, J. M. Spruell, G. Barin, W. R. Dichtel, A. H. Flood,Y. Y. Botros and J. F. Stoddart, Chem. Soc. Rev., 2012, 41,4827–4859.
13 M. W. Ambrogio, C. R. Thomas, Y.-L. Zhao, J. I. Zink andJ. F. Stoddart, Acc. Chem. Res., 2011, 44, 903–913.
14 (a) D. H. Busch and N. A. Stephenson, Coord. Chem. Rev.,1990, 100, 119–154; (b) S. Anderson, H. L. Anderson and
This journal is ª The Royal Society of Chemistry 2013
J. K. M. Sanders, Acc. Chem. Res., 1993, 26, 469–475; (c)R. Hoss and F. Vogtle, Angew. Chem., Int. Ed. Engl., 1994,33, 375–384; (d) Templated Organic Synthesis, ed. F. Dietrichand P. J. Stang, Wiley-VCH, Weinheim, 1999; (e)T. J. Hubin and D. H. Busch, Coord. Chem. Rev., 2000, 200–202, 5–52; (f) F. Arico, J. D. Badjic, S. J. Cantrill,A. H. Flood, K. C.-F. Leung, Y. Liu and J. F. Stoddart, Top.Curr. Chem., 2005, 249, 203–259; (g) J. D. Crowley,S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney,Chem. Soc. Rev., 2009, 38, 1530–1541; (h) C. A. Schalley,T. Weilandt, J. Bruggemann and F. Vogtle, Top. Curr.Chem., 2004, 248, 141–200.
15 (a) W. Zhang, W. R. Dichtel, A. Z. Stieg, D. Benıtez,J. K. Gimzewski, J. R. Heath and J. F. Stoddart, Proc. Natl.Acad. Sci. U. S. A., 2008, 105, 6514–6519; (b) S. Basu,A. Coskun, D. C. Friedman, M. A. Olson, D. Benıtez,E. Tkatchouk, G. Barin, J. Yang, A. C. Fahrenbach,W. A. Goddard III and J. F. Stoddart, Chem.–Eur. J., 2011,17, 2107–2119; (c) K.-D. Zhang, X. Zhao, G.-T. Wang,Y. Liu, Y. Zhang, H.-J. Lu, X.-K. Jiang and Z.-T. Li, Angew.Chem., Int. Ed., 2011, 50, 9866–9870; (d) Q. Gan,Y. Ferrand, C. Bao, B. Kauffmann, A. Grelard, H. Jiang andI. Huc, Science, 2011, 331, 1172–1175; (e) K.-D. Zhang,X. Zhao, G.-T. Wang, Y. Liu, Y. Zhang, H.-J. Lu, X.-K. Jiangand Z.-T. Li, Tetrahedron, 2012, 68, 4517–4527.
16 (a) H. W. Gibson, M. C. Bheda and P. T. Engen, Prog. Polym.Sci., 1994, 19, 843–945; (b) G. J. Clarkson, D. A. Leigh andR. A. Smith, Curr. Opin. Solid State Mater. Sci., 1998, 3,579–584; (c) F. M. Raymo and J. F. Stoddart, Chem. Rev.,1999, 99, 1643–1663; (d) T. Takata, N. Kihara andY. Furusho, Adv. Polym. Sci., 2004, 171, 1–75; (e) F. Huangand H. W. Gibson, Prog. Polym. Sci., 2005, 30, 982–1018; (f)L. Fang, M. A. Olson, D. Benıtez, E. Tkatchouk,W. A. Goddard III and J. F. Stoddart, Chem. Soc. Rev., 2010,39, 17–29; (g) A. Harada, A. Hashidzume, H. Yamaguchiand Y. Takashima, Chem. Rev., 2009, 109, 5974–6023.
17 (a) P. R. Ashton, E. J. T. Chrystal, J. P. Mathias, K. P. Parry,A. M. Z. Slawin, N. Spencer, J. F. Stoddart andD. J. Williams, Tetrahedron Lett., 1987, 28, 6367–6370; (b)B. Odell, M. V. Reddington, A. M. Z. Slawin, N. Spencer,J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed.Engl., 1988, 27, 1547–1550; (c) P. R. Ashton, C. L. Brown,E. J. T. Chrystal, T. T. Goodnow, A. E. Kaifer, K. P. Parry,D. Philp, A. M. Z. Slawin, N. Spencer, J. F. Stoddart andD. J. Williams, J. Chem. Soc., Chem. Commun., 1991, 634–639; (d) P. L. Anelli, P. R. Ashton, N. Spencer,A. M. Z. Slawin, J. F. Stoddart and D. J. Williams, Angew.Chem., Int. Ed. Engl., 1991, 30, 1036–1039; (e)D. B. Amabilino, P.-L. Anelli, P. R. Ashton, G. R. Brown,E. Cordova, L. Godınez, W. Hayes, A. E. Kaifer, D. Philp,A. M. Z. Slawin, N. Spencer, J. F. Stoddart, M. S. Tolley andD. J. Williams, J. Am. Chem. Soc., 1995, 117, 11142–11170;(f) M. Asakawa, W. Dehaen, G. L'abbe, S. Menzer,J. Nouwen, F. M. Raymo, J. F. Stoddart and D. J. Williams,J. Org. Chem., 1996, 61, 9591–9595; (g) G. Barin, A. Coskun,M. M. Foudah and J. F. Stoddart, ChemPlusChem, 2012, 77,159–185.
Chem. Sci., 2013, 4, 1470–1483 | 1481

Chemical Science Edge Article
Dow
nloa
ded
by U
nive
rsity
of
Mis
sour
i at C
olum
bia
on 0
5 M
arch
201
3Pu
blis
hed
on 0
4 Fe
brua
ry 2
013
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C3S
C00
015J
View Article Online
18 C. J. Bruns, S. Basu and J. F. Stoddart, Tetrahedron Lett., 2010,51, 983–986.
19 (a) R. S. Lokey and B. L. Iverson, Nature, 1995, 375, 303–305;(b) J. Q. Nguyen and B. L. Iverson, J. Am. Chem. Soc., 1999,121, 2639–2640.
20 (a) P. Hodge, P. Monvisade, G. J. Owen, F. Heatley andY. Pang, New J. Chem., 2000, 24, 703–709; (b) A. J. Zych andB. L. Iverson, J. Am. Chem. Soc., 2000, 122, 8898–8909; (c)G. J. Gabriel and B. L. Iverson, J. Am. Chem. Soc., 2002, 124,15174–15175; (d) S. Ghosh and S. Ramakrishnan, Angew.Chem., Int. Ed., 2004, 43, 3264–3268; (e) H. M. Colquhoun,Z. Zhu, C. J. Cardin and Y. Gan, Chem. Commun., 2004,2650–2652; (f) H. M. Colquhoun and Z. Zhu, Angew. Chem.,Int. Ed., 2004, 43, 5040–5045; (g) X. Zhao, M.-X. Jia,X.-K. Jiang, L.-Z. Wu, Z.-T. Li and G.-J. Chen, J. Org. Chem.,2004, 69, 270–279; (h) S. Ghosh and S. Ramakrishnan,Angew. Chem., Int. Ed., 2005, 44, 5441–5447; (i) S. Ghoshand S. Ramakrishnan, Macromolecules, 2005, 38, 676–686;(j) G. J. Gabriel, S. Sorey and B. L. Iverson, J. Am. Chem.Soc., 2005, 127, 2637–2640; (k) H. M. Colquhoun, Z. Zhu,C. J. Cardin, Y. Gan and M. G. B. Drew, J. Am. Chem. Soc.,2007, 129, 16163–16174; (l) A. Petitjean, L. A. Cuccia,M. Schmutz and J.-M. Lehn, J. Org. Chem., 2008, 73, 2481–2495; (m) H. M. Colquhoun, Z. Zhu, C. J. Cardin,M. G. B. Drew and Y. Gan, Faraday Discuss., 2009, 143,205–220; (n) S. De and S. Ramakrishnan, Macromolecules,2009, 42, 8599–8603; (o) Z. Zhu, C. J. Cardin, Y. Gan andH. M. Colquhoun, Nat. Chem., 2010, 2, 653–660; (p)S. G. Ramkumar and S. Ramakrishnan, Macromolecules,2010, 43, 2307–2312; (q) Z. Zhu, C. J. Cardin, Y. Gan,C. A. Murray, A. J. P. White, D. J. Williams andH. M. Colquhoun, J. Am. Chem. Soc., 2011, 133, 19442–19447.
21 Z. Zhu, H. Li, Z. Liu, J. Lei, H. Zhang, Y. Y. Botros, C. L. Stern,A. A. Sarjeant, J. F. Stoddart and H. M. Colquhoun, Angew.Chem., Int. Ed., 2012, 51, 7231–7235.
22 (a) C. Wu, P. R. Lecavalier, Y. X. Shen and H. W. Gibson,Chem. Mater., 1991, 3, 569–572; (b) P. R. Ashton, D. Philp,N. Spencer and J. F. Stoddart, J. Chem. Soc., Chem.Commun., 1992, 1124–1128; (c) J.-C. Chambron, V. Heitzand J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1992,1131–1133; (d) F. Diederich, C. Dietrich-Buchecker,J.-F. Nierengarten and J.-P. Sauvage, J. Chem. Soc., Chem.Commun., 1995, 781–782; (e) D. J. Cardenas, P. Gavi~na andJ.-P. Sauvage, Chem. Commun., 1996, 1915–1916; (f)Y.-M. Jeon, D. Whang, J. Kim and K. Kim, Chem. Lett.,1996, 503–504; (g) S. Anderson, T. D. W. Claridge andH. L. Anderson, Angew. Chem., Int. Ed. Engl., 1997, 36,1310–1313; (h) S. J. Loeb and J. A. Wisner, Chem. Commun.,1998, 2757–2758; (i) T. Takata, H. Kawasaki, S. Asai,Y. Furusho and N. Kihara, Chem. Lett., 1999, 223–224; (j)D. W. Zehnder and D. B. Smithrud, Org. Lett., 2001, 3,2485–2487; (k) M. J. Gunter, N. Bampos, K. D. Johnstoneand J. K. M. Sanders, New J. Chem., 2001, 25, 166–173; (l)J. S. Hannam, T. J. Kidd, D. A. Leigh and A. J. Wilson, Org.Lett., 2003, 5, 1907–1910; (m) H. Sasabe, N. Kihara,Y. Furusho, K. Mizuno, A. Ogawa and T. Takata, Org. Lett.,2004, 6, 3957–3960; (n) C. Gaeta, M. O. Vysotsky, A. Bogdan
1482 | Chem. Sci., 2013, 4, 1470–1483
and V. Bohmer, J. Am. Chem. Soc., 2005, 127, 13136–13137;(o) W. R. Dichtel, O. S. Miljanic, J. M. Spruell, J. R. Heathand J. F. Stoddart, J. Am. Chem. Soc., 2006, 128, 10388–10390; (p) C.-C. Hsu, C.-C. Lai and S.-H. Chiu, Tetrahedron,2009, 65, 2824–2829; (q) T. Matsumura, F. Ishiwari,Y. Koyama and T. Takata, Org. Lett., 2010, 12, 3828–3831.
23 (a) P. R. Ashton, M. Grognuz, A. M. Z. Slawin, J. F. Stoddartand D. J. Williams, Tetrahedron Lett., 1991, 32, 6235–6238;(b) A. G. Johnston, D. A. Leigh, A. Murphy, J. P. Smart andM. D. Deegan, J. Am. Chem. Soc., 1996, 118, 10662–10663;(c) P. T. Glink, A. I. Oliva, J. F. Stoddart, A. J. P. White andD. J. Williams, Angew. Chem., Int. Ed., 2001, 40, 1870–1875;(d) J. A. Wisner, P. D. Beer, M. G. B. Drew andM. R. Sambrook, J. Am. Chem. Soc., 2002, 124, 12469–12476; (e) A. F. M. Kilbinger, S. J. Cantrill, A. W. Waltman,M. W. Day and R. H. Grubbs, Angew. Chem., Int. Ed., 2003,42, 3281–3285; (f) E. Arunkumar, C. C. Forbes, B. C. Nolland B. D. Smith, J. Am. Chem. Soc., 2005, 127, 3288–3289;(g) A.-M. L. Fuller, D. A. Leigh and P. J. Lusby, Angew.Chem., Int. Ed., 2007, 46, 5015–5019.
24 (a) I. T. Harrison, J. Chem. Soc., Chem. Commun., 1972, 231–232; (b) I. T. Harrison, J. Chem. Soc., Perkin Trans. 1, 1974,301–304; (c) G. Schill, W. Beckmann, N. Schweikert andH. Fritz, Chem. Ber., 1986, 119, 2647–2655; (d) P. R. Ashton,M. Belohradsky, D. Philp and J. F. Stoddart, J. Chem. Soc.,Chem. Commun., 1993, 1269–1274; (e) D. H. Macartney, J.Chem. Soc., Perkin Trans. 2, 1996, 2775–2778; (f) M. Handel,M. Plevoets, S. Gestermann and F. Vogtle, Angew. Chem.,Int. Ed. Engl., 1997, 36, 1199–1201; (g) C. Heim, A. Affeld,M. Nieger and F. Vogtle, Helv. Chim. Acta, 1999, 82, 746–759; (h) J. W. Lee, K. Kim and K. Kim, Chem. Commun.,2001, 1042–1043; (i) S.-Y. Hsueh, C.-C. Lai, Y.-H. Liu,Y. Wang, S.-M. Peng and S.-H. Chiu, Org. Lett., 2007, 9,4523–4526; (j) M. A. Bolla, J. Tiburcio and S. J. Loeb,Tetrahedron, 2008, 64, 8423–8427; (k) H. W. Gibson,N. Yamaguchi, Z. Niu, J. W. Jones, C. Slebodnick,A. L. Rheingold and L. N. Zakharov, J. Polym. Sci., Part A:Polym. Chem., 2010, 48, 975–985; (l) A. J. McConnell andP. D. Beer, Chem.–Eur. J., 2011, 17, 2724–2733.
25 (a) W. L. Mock, T. A. Irra, J. P. Wepsiec and M. Adhya, J. Org.Chem., 1989, 54, 5302–5308; (b) D. Tuncel andJ. H. G. Steinke, Chem. Commun., 1999, 1509–1510; (c)D. Tuncel and J. H. G. Steinke, Chem. Commun., 2002, 496–497; (d) P. Ghosh, O. Mermagen and C. A. Schalley, Chem.Commun., 2002, 2628–2629; (e) N. Kameta, K. Hiratani andY. Nagawa, Chem. Commun., 2004, 466–467; (f) S. Saito,E. Takahashi and K. Nakazono, Org. Lett., 2006, 8, 5133–5136; (g) V. Aucagne, K. D. Hanni, D. A. Leigh, P. J. Lusbyand D. B. Walker, J. Am. Chem. Soc., 2006, 128, 2186–2187;(h) K. Hirose, K. Nishihara, N. Harada, Y. Nakamura,D. Masuda, M. Araki and Y. Tobe, Org. Lett., 2007, 9, 2969–2972; (i) J. D. Crowley, K. D. Hanni, A.-L. Lee andD. A. Leigh, J. Am. Chem. Soc., 2007, 129, 12092–12093; (j)J. Berna, J. D. Crowley, S. M. Goldup, K. D. Hanni,A.-L. Lee and D. A. Leigh, Angew. Chem., Int. Ed., 2007, 46,5709–5713; (k) J. Berna, S. M. Goldup, A.-L. Lee,D. A. Leigh, M. D. Symes, G. Teobaldi and F. Zerbetto,
This journal is ª The Royal Society of Chemistry 2013

Edge Article Chemical Science
Dow
nloa
ded
by U
nive
rsity
of
Mis
sour
i at C
olum
bia
on 0
5 M
arch
201
3Pu
blis
hed
on 0
4 Fe
brua
ry 2
013
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C3S
C00
015J
View Article Online
Angew. Chem., Int. Ed., 2008, 47, 4392–4396; (l) S. M. Goldup,D. A. Leigh, P. J. Lusby, R. T. McBurney and A. M. Z. Slawin,Angew. Chem., Int. Ed., 2008, 47, 3381–3384; (m) J. D. Crowley,K. D. Hanni, D. A. Leigh and A. M. Z. Slawin, J. Am. Chem.Soc., 2010, 132, 5309–5314; (n) S. M. Goldup, D. A. Leigh,P. R. McGonigal, V. E. Ronaldson and A. M. Z. Slawin, J.Am. Chem. Soc., 2010, 132, 315–320; (o) H. M. Chean,D. A. Leigh, F. Maffei, P. R. McGonigal, A. M. Z. Slawin andJ. Wu, J. Am. Chem. Soc., 2011, 133, 12298–12303; (p) C. Ke,R. A. Smaldone, T. Kikuchi, H. Li, A. P. Davis andJ. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 381–387; (q)D. Tuncel, O. Unal and M. Artar, Isr. J. Chem., 2011, 51,525–532.
26 (a) C.-W. Chiu, C.-C. Lai and S.-H. Chiu, J. Am. Chem. Soc.,2007, 129, 3500–3501; (b) A. M. Cagulada andD. G. Hamilton, J. Am. Chem. Soc., 2009, 131, 902–903; (c)J.-L. Ko, S.-H. Ueng, C.-W. Chiu, C.-C. Lai, Y.-H. Liu,S.-M. Peng and S.-H. Chiu, Chem.–Eur. J., 2010, 16, 6950–6960.
27 (a) I. Yoon, M. Narita, T. Shimizu and M. Asakawa, J. Am.Chem. Soc., 2004, 126, 16740–16741; (b) K. Hirose,Y. Shiba, K. Ishibashi, Y. Doi and Y. Tobe, Chem.–Eur. J.,2008, 14, 981–986; (c) S.-Y. Hsueh, J.-L. Ko, C.-C. Lai,Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Angew. Chem., Int.Ed., 2011, 50, 6643–6646.
28 (a) I. T. Harrison and S. Harrison, J. Am. Chem. Soc., 1967, 89,5723–5724; (b) M. Born and H. Ritter, Angew. Chem., Int. Ed.Engl., 1995, 34, 309–311; (c) I. Yamaguchi, K. Osakada andT. Yamamoto, Macromolecules, 1997, 30, 4288–4294; (d)T. Takata, H. Kawasaki, N. Kihara and Y. Furusho,Macromolecules, 2001, 34, 5449–5456; (e) T. J. Kidd,T. J. A. Loontjens, D. A. Leigh and J. K. Y. Wong, Angew.Chem., Int. Ed., 2003, 42, 3379–3383; (f) I. Tomatsu,A. Hashidzume and A. Harada, Angew. Chem., Int. Ed.,2006, 45, 4605–4608; (g) M. Bria, J. Bigot, G. Cooke,J. Lyskawa, G. Rabani, V. M. Rotello and P. Woisel,Tetrahedron, 2009, 65, 400–407; (h) M. Born and H. Ritter,Makromol. Chem. Rapid Commun., 1991, 12, 471–476.
29 (a) A. Harada, J. Li and M. Kamachi, Nature, 1992, 356, 325–327; (b) Y. X. Shen and H. W. Gibson, Macromolecules, 1992,25, 2058–2059; (c) S. S. Zhu, P. J. Carroll and T. M. Swager, J.Am. Chem. Soc., 1996, 118, 8713–8714; (d) D. Whang andK. Kim, J. Am. Chem. Soc., 1997, 119, 451–452; (e)W. Herrmann, M. Schneider and G. Wenz, Angew. Chem.,Int. Ed. Engl., 1997, 36, 2511–2514; (f) H. W. Gibson,P. T. Engen and S. H. Lee, Polymer, 1999, 40, 1823–1832;(g) P. N. Taylor, M. J. O'Connell, L. A. McNeill, M. J. Hall,R. T. Aplin and H. L. Anderson, Angew. Chem., Int. Ed.,2000, 39, 3456–3460; (h) T. Oku, Y. Furusho and T. Takata,J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 119–123; (i)D. Tuncel and J. H. G. Steinke, Macromolecules, 2004, 37,288–302; (j) Y.-G. Lee, Y. Koyama, M. Yonekawa andT. Takata, Macromolecules, 2010, 43, 4070–4080; (k)Y. Kohsaka, Y. Koyama and T. Takata, Angew. Chem., Int.Ed., 2011, 50, 10417–10420.
30 (a) J.-P. Sauvage, Acc. Chem. Res., 1990, 23, 319–327; (b)W. R. Dichtel, O. S. Miljanic, W. Zhang, J. M. Spruell,
This journal is ª The Royal Society of Chemistry 2013
K. Patel, I. Aprahamian, J. R. Heath and J. F. Stoddart, Acc.Chem. Res., 2008, 41, 1750–1761.
31 (a) L. Raehm, D. G. Hamilton and J. K. M. Sanders, Synlett,2002, 1743–1761; (b) R. L. E. Furlan, S. Otto andJ. K. M. Sanders, Proc. Natl. Acad. Sci. U. S. A., 2002, 99,4801–4804; (c) M. J. Gunter, Eur. J. Org. Chem., 2004, 1655–1673; (d) K. C.-F. Leung and K.-N. Lau, Polym. Chem., 2010,1, 988–1000.
32 (a) J. Wu, K. C.-F. Leung and J. F. Stoddart, Proc. Natl. Acad.Sci. U. S. A., 2007, 104, 17266–17271; (b) M. E. Belowich,C. Valente and J. F. Stoddart, Angew. Chem., Int. Ed., 2010,49, 7208–7212; (c) M. E. Belowich, C. Valente,R. A. Smaldone, D. C. Friedman, J. Thiel, L. Cronin andJ. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 5243–5261.
33 (a) P. R. Ashton, P. J. Campbell, E. J. T. Chrystal, P. T. Glink,S. Menzer, D. Philp, N. Spencer, J. F. Stoddart, P. A. Taskerand D. J. Williams, Angew. Chem., Int. Ed. Engl., 1995, 34,1865–1869; (b) A. G. Kolchinski, D. H. Busch andN. W. Alcock, J. Chem. Soc., Chem. Commun., 1995, 1289–1291; (c) P. R. Ashton, E. J. T. Chrystal, P. T. Glink, S. Menzer,C. Schiavo, N. Spencer, J. F. Stoddart, P. A. Tasker,A. J. P.WhiteandD. J.Williams,Chem.–Eur. J., 1996,2, 709–728.
34 (a) R. Huisgen, Angew. Chem., 1963, 75, 604–637; (b)R. Huisgen, G. Szeimies and L. Mobius, Chem. Ber., 1967,100, 2494–2507.
35 H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int.Ed., 2001, 40, 2004–2021.
36 K. Hanni and D. Leigh, Chem. Soc. Rev., 2010, 39, 1240–1251.37 The 50% recognition-site occupancy phenomenon has also
been observed in certain polypseudorotaxane systemsbased on poly-hydroquinol ethers and CBPQT4+. See: (a)G. J. Owen and P. Hodge, Chem. Commun., 1997, 11–12; (b)Ref. 20a.
38 Since ‘conformation’ applies strictly to discrete molecules,we use the term ‘co-conformation’ to describe the three-dimensional spatial arrangements of the components insupramolecular and mechanically interlocked systems. SeeM. C. T. Fyfe, P. T. Glink, S. Menzer, J. F. Stoddart,A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed.Engl., 1997, 36, 2068–2070.
39 (a) R. D. Schmidt, D. A. Shultz, J. D. Martin and P. D. Boyle, J.Am. Chem. Soc., 2010, 132, 6261–6273; (b) V. Maheshwari,P. A. Marzilli and L. G. Marzilli, Inorg. Chem., 2011, 50,6626–6636.
40 We acknowledge that, in a previous analysis (ref. 15b) ofthese types of donor–acceptor oligomers, we made theassumption that the folded co-conformation was static onthe timescale of an NMR experiment, forcing us to adoptconclusion (2). In light of our multidimensional NMRanalysis, we nd that conclusion (3) is a more appropriateinterpretation of the NMR spectra, forcing us toretrospectively negate the a and b proton assignments wemade previously.
41 J. E. Kilpatrick, K. S. Pitzer and R. Spitzer, J. Am. Chem. Soc.,1947, 69, 2483–2488.
42 S. Klod and E. Kleinpeter, J. Chem. Soc., Perkin Trans. 2, 2001,1893–1898.
Chem. Sci., 2013, 4, 1470–1483 | 1483