synthesis and spectral studies of binuclear transition metal complexes of diamidediimine...
TRANSCRIPT

Pergamon Polyhedron Vol. 13, No. 9, pp. 1355-1361, 1994
Copyright 0 1994 Elsevier Science Ltd F’rinted in Great Britain. All rights reserved
0277-5387/W $7.00+0.00
0277%5387(93)EOO6GB
SYNTHESIS AND SPECTRAL STUDIES OF BINUCLEAR TRANSITION METAL COMPLEXES OF DIAMIDEDIIMINE
HEXAAZAMACROCYCLES
MOHAMMAD SHAKIR,* SAJI P. VARKEY and P. SHAHUL HAMEED
Division of Inorganic Chemistry, Department of Chemistry, Aligarh Muslim University, Aligarh-202002, India
(Received 4 November 1993 ; accepted 25 November 1993)
Abstract-The template condensation of alkylacetoacetate with diethylenetriamine resulted in the formation of homodinuclear transition metal complexes of a 20-membered macro- cyclic ligand. The overall geometry and the mode of bonding have been inferred from IR, ‘H NMR, EPR and electronic spectral studies as well as magnetic and molar conductance data. An octahedral geometry around the metal ion is suggested for [M 2L ,X4 and [M 2L’,X2] where M = iron(I1) and cobalt(II), and X = Cl or N03. A distorted square planar and a tetrahedral geometry is proposed for [M2L2]X2 and [M,L’JX, when M = copper(I1) and zinc(H) respectively ; X = Cl or N03. The distortion in the copper complexes is confirmed through EPR investigation.
Much of the current interest in macrocyclic coor- dination chemistry arises from the hope that the unusual geometrical relationship imposed on the metal ions by the macrocyclic donor set, may be transformed into unusual bonding situations. Among polyaza macrocycles, the tetraazacyclo- alkanes are by far the most studied. The hexaaza- macrocycles are known to give mononuclear I and binuclear2 complexes. Dicopper complexes of 20-22 and 24-membered hexaamine macro- cycles have been considered suitable in the acti- vation of small molecules.3 Recently McAuley and co-workers have reported3 the synthesis and characterization of chromium and copper com- plexes of hexaazamacrocycles. The possibility for these macrocycles to bind more than one metal ion in the macrocyclic framework and the recent development of the anion chemistry4,’ have made the investigation of large polyazacycloalkanes of special interest. The study of magnetic interaction between such closely spaced paramagnetic centres is currently receiving some attention, directed at identification of factors which determine the nature
1355
and strength of the interaction. Besides these con- siderations it should be pointed out that dinuclear complexes of synthetic macrocyclic compounds have been extensively studied6s7 mainly using the template technique. 8 These compounds are inter- esting from both theoretical and practical points of views.9-’ ’ Many different synthetic approaches have been followed in order to obtain dinucleating macrocyclic ligands that can be classified as fol- lows : (1) large monocyclic compounds, with many donor atoms of the same or different kinds ; ’ 2 (2) macrocyclic subunits linked together ; ’ 3 (3) poly- cyclic compounds with fused cycles. I 4
*Author to whom all correspondence should be addressed.
We have recently reported’5*‘6 the template syn- thesis of various macrocycles. Many diamide macrocycles have been reported 17-l 9 in the literature by convenient synthetic techniques and most of them isolated as metal free macrocyclic ligands. An amide group offers two potential binding atoms, the oxygen and nitrogen for complexation of metal ions. A number of complexes have been reported with amide group ligands which exhibit diverse coordinating behaviour with different metal ions.20’2’ However, studies on the metal complexes of the derivatives of amide macrocycles are limited. Wiersema and Windle22 on the basis of EPR spectra of copper(I1) salicylamide complexes, have reported

1356 M. SHAKIR et d.
that the salicylamide coordinates through the nitro- gen of the amide group. On the other hand, Pannu et al. have proposed23 the structure for this com- plex with oxygen, but not nitrogen, of the amide group coordinating. Herein we report the synthesis and characterization of dinuclear diamide macro- cyclic complexes [M2L,X2], [M,L’,X,] for M = iron(I1) and cobalt(H); X = Cl or N03, and [M2L2]X2, [M,L;]X, for M = copper(I1) and zinc(I1); X = Cl or NO3 obtained from the tem- plate condensation reaction of diethylenetriamine with alkylacetoacetate.
EXPERIMENTAL
The chemicals, methylacetoacetate and ethyl- acetoacetate are obtained from Merck and were used as received. Diethylenetriamine was purchased from Koch-Light. The metal salts, FeC12.4H20, Fe(N03),4H20; CoC1,*6H20, Co(NO3)2*6H2O; CuC12.2H20, CU(NO 3) 2.2H 20, ZnCl 2 and Zn(N0 3) 2 * 6H 20 (all BDH) were commercially pure samples.
Synthesis of dichlorolnitrato (8,18-dimethyl- 10,20- dioxa-1,4,7,11,14,17-hexaazacycloeicosane-7,17- diene) dimetal(I1) [M2L rXd ; w,L’,Xd [M = iron(I1) and cobalt(I1) ; X = Cl or NO31
These complexes are prepared by two different methods given below.
(1) To a warm stirring methanolic solution of (0.02 mol) metal salt was added a warm solution of (0.023 mol) diethylenetriamine in methanol. The mixture was stirred for 30 min and then a methanolic solution of (0.02 mol) methylaceto- acetate was added. The resultant mixture was stirred for 8 h at room temperature and the solid pro- duct obtained [M2L1X2] was recrystallized from methanol and stored in DUCUO. The purity of the final product is checked by TLC of the complexes dissolved in DMF using ethylacetate (85%), meth- anol (10%) and acetic acid (5%) as eluent. Only one spot was observed in each case after developing in an iodine chamber, indicating that the com- pounds were pure.
(2) Here the procedure for [M,L’,X,] is exactly the same as detailed above. In the final step instead of adding methylacetoacetate to the solution of diethylenetriamine-metal mixture, ethylaceto- acetate was added. The analytical data and spec- tral studies show that the compounds [M2LlX2] and [M,L’,X,] are found to be the same. The purity of the compound was tested as mentioned above.
Synthesis of (8,18-dimethyl-10,20-dioxa-1,4,7,11, 14,17-hexaazacycloeicosane-7,17-diene) dimetal(I1) chloride/nitrate, [M2L2]X2; [M2L;lX2 [M = copper (II) and zinc(I1) ; X = Cl and NO31
These complexes were also synthesized by two different methods as in the previous case.
(1) At first the reaction was carried out using methylacetoacetate and the resultant solid product
[M2L21X2 was collected. Here the metal ions employed were copper(I1) and zinc(I1).
(2) For the synthesis of [M,L;]X, the reaction was carried out using ethylacetoacetate instead of methylacetoacetate. The remainder of the pro- cedure is the same as discussed in the above case. The purity of the recrystallized product in both cases was checked by the TLC technique described previously.
Elemental analyses were obtained from the Mic- roanalytical Laboratory of CDRI, Lucknow, India. ‘H NMR spectra in DMSO-d6 using Bruker AC 200 E nuclear magnetic resonance spectrometer with Me,Si as an internal standard was obtained from GNDU, Amritsar, India. Metals and chloride were determined volumetrically24 and gravimetrically,25 respectively. The IR spectra (400&200 cm ‘) were recorded as CsCl discs on a Perkin-Elmer 621 spec- trophotometer. The electronic spectra of com- pounds in DMSO were recorded on a Pye-Unicam 8800 spectrophotometer at room temperature. EPR spectra were recorded on a Jeol JES RE2X EPR spectrometer. Magnetic susceptibility measure- ments were carried out using a Faraday balance at 25°C. The electrical conductivities of 1O-3 M solutions in DMSO were obtained on a systronics type 302 conductivity bridge equilibrated at 25 * 0.01”c.
RESULTS AND DISCUSSION
All the complexes are crystalline powders, freely soluble in water, acetonitrile, DMF and DMSO and slightly soluble in ethanol, benzene and dioxane. The analytical data (Table 1) suggest their proposed 1 : 2 ligand: metal stoichiometry as shown in Scheme 1. The molar conductance values of the iron(I1) and cobalt(I1) complexes in DMSO at room temperature support the non-ionic nature of these complexes while the copper(I1) and zinc(I1) com- plexes show high conductivity values suggesting their 1: 2 electrolytic nature26 (Table 1). However, we could not grow any single crystal suitable for X- ray crystallographic studies. The successful cycliz- ation at the terminal amine position seems to be due to the difference in the reactivities between the
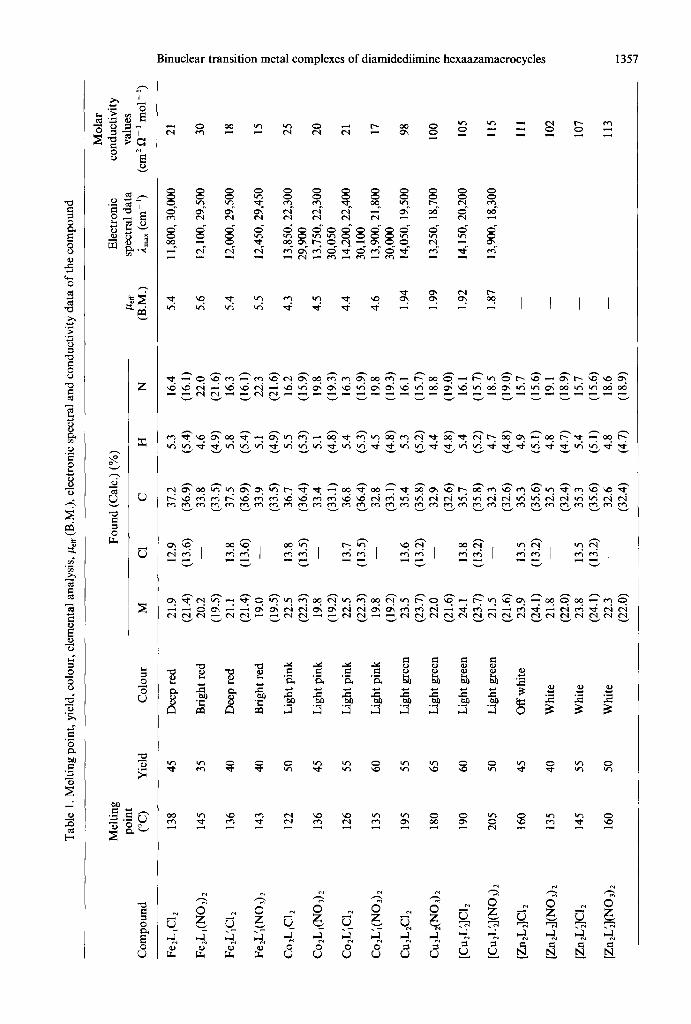
Tab
le
1. M
eltin
g po
int,
yiel
d,
colo
ur,
elem
enta
l an
alys
is,
pcfl
(B.M
.),
elec
tron
ic
spec
tral
an
d co
nduc
tivity
da
ta
of t
he
com
poun
d
Com
poun
d
Fe,L
,C
l,
FezL
I(N
OJz
Fe2L
’,C
l,
Fe2L
’0Q
)2
co
ZL
Cl*
Coz
L,(
NO
A
Co,
L’,
Cl,
Co,
L;(
NO
,),
Cu2
L*C
l,
Cu*
L,(
NO
,)*
[CU
~L;lC
I,
ICu,
L’z
l(N
OJ,
[ZnI
LK
1,
Pn,L
zl(N
O,)
,
Pn*L
;IC
l*
[znJ
-‘~I
WJ,
h
Mel
ting
poin
t
(“C
)
138
145
136
143
122
136
126
135
195
180
190
205
160
135
145
160
Yie
ld
45
35
40
40
50
45
55
60
55
65
60
50
45
40
55
50
Col
our
Dee
p re
d
Bri
ght
red
Dee
p re
d
Bri
ght
red
Lig
ht
pink
Lig
ht
pink
Lig
ht
pink
Lig
ht
pink
Lig
ht
gree
n
Lig
ht
gree
n
Lig
ht
gree
n
Lig
ht
gree
n
Off
whi
te
Whi
te
Whi
te
Whi
te
Foun
d (C
alc.
) (%
) E
lect
roni
c sp
ectr
al
data
M
Cl
C
H
N
(B.M
.)
i,,,
(cm
‘)
21.9
(2
1.4)
20
.2
(19.
5)
21.1
(2
1.4)
19
.0
(19.
5)
22.5
(2
2.3)
19
.8
(19.
2)
22.5
(2
2.3)
19
.8
(19.
2)
23.5
(2
3.7)
22
.0
(21.
6)
24.1
(2
3.7)
21
.5
(21.
6)
23.9
(24.
1)
21.8
(2
2.0)
23
.8
(24.
1)
22.3
(2
2.0)
12.9
(1
3.6)
- 13
.8
(13.
6)
- 13.8
(13.
5)
- 13.7
(1
3.5)
- 13
.6
(13.
2)
13.8
(1
3.2)
- 13
.5
(13.
2)
13.5
(1
3.2)
-
37.2
(3
6.9)
33
.8
(33.
5)
37.5
(3
6.9)
33
.9
(33.
5)
36.7
(3
6.4)
33
.4
(33.
1)
36.8
(3
6.4)
32
.8
(33.
1)
35.4
(3
5.8)
32
.9
(32.
6)
35.7
(3
5.8)
32
.3
(32.
6)
35.3
(3
5.6)
32
.5
(32.
4)
35.3
(3
5.6)
32
.6
(32.
4)
5.3
(5.4
)
(Z) 5.8
(5.4
)
(Z) 5.5
(5.3
)
(::A
)
(:::,
(Z)
(Z)
(Z)
(Z)
(Z)
(Z)
(Z)
(:::)
(Z)
16.4
(1
6.1)
22
.0
(21.
6)
16.3
(1
6.1)
22
.3
(21.
6)
16.2
(1
5.9)
19
.8
(19.
3)
16.3
(1
5.9)
19
.8
(19.
3)
16.1
(1
5.7)
18
.8
(19.
0)
16.1
(1
5.7)
18
.5
(19.
0)
15.7
(1
5.6)
19
.1
(18.
9)
15.7
(1
5.6)
18
.6
(18.
9)
5.4
5.6
5.4
5.5
4.3
4.5
4.4
4.6
1.94
1.99
1.92
1.87
11,8
00,
30,0
00
21
12,1
00,
29,5
00
30
12,0
00,
29,5
00
18
12,4
50,
29,4
50
15
13,8
50,
22,3
00
29,9
00
13,7
50,
22,3
00
30,0
50
14,2
00,2
2,40
0 30
,100
13
,900
,21,
800
30,0
00
14,0
50,
19,5
00
25
20
21
17
98
13,2
50,
18,7
00
14,1
50,
20,2
00
13,9
00,
18,3
00
100
105
115
Mol
ar
cond
uctiv
ity
valu
es
(cm
’R_’
m
ol-‘
)
111
102
107
113
1358 M. SHAKIR et al.
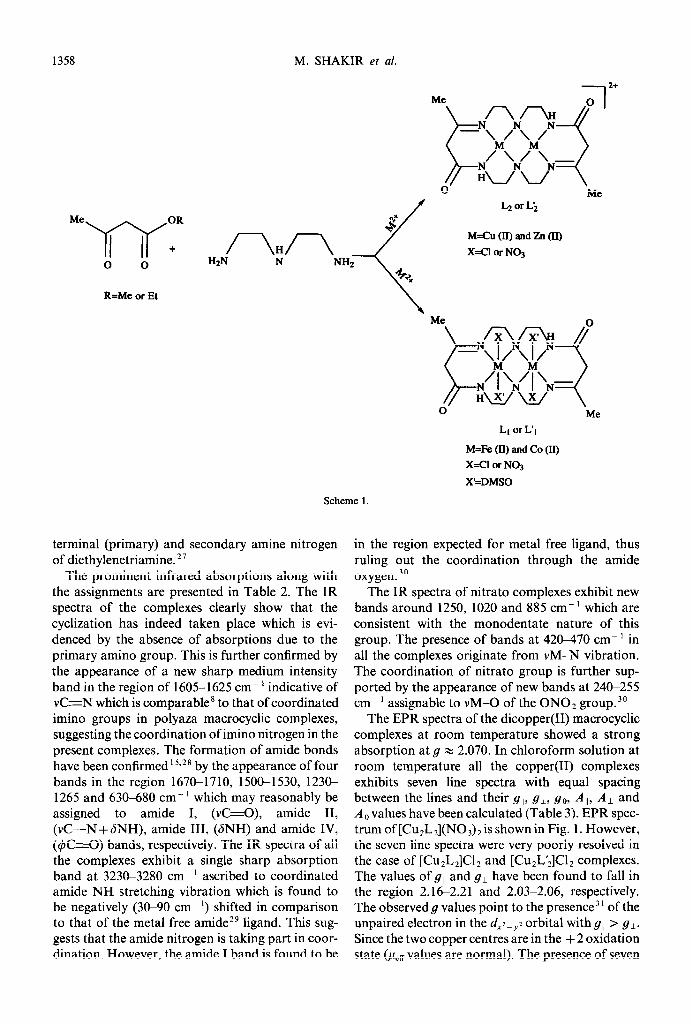
R=Me or Et
Me
Me
M=Cu (II) and Zn (II)
X=CI or NO,
Me
LI or L’I
M=Fe (II) and Co (II)
X=CI or NO3
X’=DMSO
Scheme 1.
terminal (primary) and secondary amine nitrogen of diethylenetriamine.”
The prominent infrared absorptions along with the assignments are presented in Table 2. The IR spectra of the complexes clearly show that the cyclization has indeed taken place which is evi- denced by the absence of absorptions due to the primary amino group. This is further confirmed by the appearance of a new sharp medium intensity band in the region of 16051625 cm-’ indicative of vC=N which is comparable* to that of coordinated imino groups in polyaza macrocyclic complexes, suggesting the coordination of imino nitrogen in the present complexes. The formation of amide bonds have been confirmed ’ S-2 8 by the appearance of four bands in the region 167G1710, 1500-1530, 1230- 1265 and 630-680 cm- ’ which may reasonably be assigned to amide I, (vC=O), amide II, (vC-N+ 6NH), amide III, (6NH) and amide IV, (&?.=O) bands, respectively. The IR spectra of all the complexes exhibit a single sharp absorption band at 3230-3280 cm-’ ascribed to coordinated amide NH stretching vibration which is found to be negatively (30-90 cm- ‘) shifted in comparison to that of the metal free amide*’ ligand. This sug- gests that the amide nitrogen is taking part in coor- dination. However, the amide I band is found to be
in the region expected for metal free ligand, thus ruling out the coordination through the amide oxygen. 3 O
The IR spectra of nitrato complexes exhibit new bands around 1250, 1020 and 885 cm-’ which are consistent with the monodentate nature of this group. The presence of bands at 420-470 cm-’ in all the complexes originate from vM-N vibration. The coordination of nitrato group is further sup- ported by the appearance of new bands at 240-255 cm-’ assignable to vM-0 of the ON02 group.30
The EPR spectra of the dicopper(I1) macrocyclic complexes at room temperature showed a strong absorption at g w 2.070. In chloroform solution at room temperature all the copper(I1) complexes exhibits seven line spectra with equal spacing between the lines and their gl,, gl, go, A,,, Al and A ,, values have been calculated (Table 3). EPR spec- trum of [Cu,L,](NO,), is shown in Fig. 1. However, the seven line spectra were very poorly resolved in the case of [Cu,L,]Cl, and [CU~L’~]CI~ complexes. The values of g,, and g1 have been found to fall in the region 2.162.21 and 2.03-2.06, respectively. The observed g values point to the presence3’ of the unpaired electron in the dX~_Y~ orbital with gll > gl. Since the two copper centres are in the + 2 oxidation state (pefi values are normal). The presence of seven

Binuclear transition metal complexes of diamidediimine hexaazamacrocycles
Table 2. IR spectral data (cm- ‘) of the complexes
1359
Amide bands
Compound vNH vCH I II III IV vC=N KH vM-N
Fe2L,C12 3240 FezL,(NOJ, 3260 Fe,L’,Cl, 3270
Fe,L;(NOJ, 3230
Co2L,Cl, 3235
CozL,(NO,), 3240 co,L’,c1, 3250 Co ,L’,(NO 3) z 3280
D&1c1* 3230
PM4wo3)* 3245
Ku *L>W 2 3260
F%L’dW% 3280
~Zn2WA 3270
Pd4NW2 3240
L%LW2 3250
PW4Wd2 3265
2895 1690 1530 1240 2910 1680 1510 1265 2900 1700 1500 1230 2920 1705 1525 1240
2930 1710 1520 1245 2915 1670 1520 1230 2910 1690 1510 1240 2940 1680 1520 1230
2930 1680 1515 1260 2910 1685 1530 1240 2925 1690 1540 1230 2920 1685 1540 1240
2925 1690 1510 1230 2915 1685 1520 1225 2910 1695 1530 1240 2925 1690 1525 1225
650 1615 640 1625 665 1610 670 1605
630 1620 660 1615 640 1620 635 1605
665 1610 680 1605 645 1615 640 1610
660 1605 640 1615 670 1610 635 1620
1425 440 1440 420 1430 460 1445 465
1410 450 1405 470 1425 445 1415 465
1430 460 1410 470 1435 455 1415 460
1410 450 1415 460 1420 465 1410 455
Table 3. EPR spectral” parameters for the dinuclear copper(H) complexes
Compound
DM41c~, 2.160 2.041 2.080 90.00 12.50 38.33 4.0 240 KJ.&l(NO,)~ 2.171 2.030 2.076 86.60 13.00 37.53 5.6 250 [c~*L;Ic~, 2.210 2.050 2.103 81.51 18.51 39.84 4.2 267 DW&NO,), 2.201 2.042 2.093 100.50 12.00 41.50 5.0 218
“go = :(g,,+%J; Ao = &4,+2A,); G = hi-‘MgL-2).
equally spaced lines implies that the unpaired electrons are interacting equally with two copper nuclei and suggests weak interaction between the copper(I1) centres. 8
The isotropic nuclear hyperhne constant, A,,, of these complexes in solution is much lower than
I I I I I
2700 2900 3100 3300 3500
Fig. 1. EPR spectrum of [Cu,L&NO,),.
those of planar copper species, but slightly higher than the values of tetrahedral species. The lowering of A0 value is an indication of the distortion of the copper environments from planar to pseudo- tetrahedral. 32 Often the quotient g,,/A,, is empir- ically treated 3 3 as an index of tetrahedral distortion. The g,,/A,, values are found to be in the range 105-135 and 150-250 cm for square planar and tetrahedrally distorted complexes. In the present case the value is found to be in the range 218-250 cm, clearly suggesting the presence of tetrahedral distortion from D,h symmetry. Furthermore, the tendency for g,, to increase and A,, values to decrease (Table 3) is again suggestive34,35 of tetrahedral dis- tortion of the complexes. Proctor and co-workers have postulated36 the magnitude of the ratio, G = (g,, -2)/(gl - 2) indicating the possibility of exchange interaction in the copper(I1) complexes. Thus when G > 4 the exchange interaction is neg- ligible and vice versa. In the present case the value

1360 M. SHAKIR et nl.
Table 4. ‘H NMR spectroscopic data“ of the complexes
Compound CO-NH CH,--C=N CH,--N---CH* CHrN=C C-CH,-C
lZnzLXl2 8.41(s) 2.45(s) 2.68(t) 3.64(t) 2.32(s)
[zn aL &NO 3) 2 8.38(s) 2.40(s) 2.75(t) 3.52(t) 2.19(s)
[zn &ICl Z 8.49(s) 2.51(s) 2.87(t) 3.61(t) 2.02(s)
tznJ-~I(NW2 8.45(s) 2.52(s) 2.71(t) 3.62(t) 2.25(s)
a Chemical shift (6/ppm) with multiplicities in parentheses. s = singlet; t = triplet.
ranges from 4.0-5.6 which indicates the exchange interaction is negligible. However, we have tried to get EPR spectra of the dicobalt complexes at room temperature without success.
The ‘H NMR spectra (Table 4) of all the zinc(I1) complexes show a broad signal in the region of 8.38-8.49 ppm which may be assigned” to amide (HN-CO ; 2H) protons. A sharp signal observed at 2.40-2.52 ppm for the macrocyclic complexes corresponds to imine methyls (CH +Z=N- ; 6H) protons3’ All the macrocyclic zinc(I1) complexes gave two signals at 2.68-2.87 and 3.52-3.64 ppm as triplets due to inequivalent methylene proton of the amine linkage. The former signal is assigned to the methylene protons adjacent to secondary nitrogen atom (CH,-N-CH,; 8H) and the latter, more downfield signal, is for the methylene protons adjac- ent to the imine (CH,--N=C; 4H) groups. In all the complexes a singlet observed in the region 2.02-2.32 ppm may be assigned to methylene (-C-CH2-C ; 4H) protons of alkylacetoacetate moiety. However, no band could be assigned either
for -OCH,, -OCH2CH3 or -NH2 protons which can be suggested for the formation of the proposed macrocyclic framework.
The magnetic susceptibility measurements (Table 1) for all the binuclear macrocyclic complexes are
comparable to their mononuclear metal complexes indicative of non-interacting metal centres. This was further confirmed’ from the EPR spectra of copper complexes, which displays a poorly resolved four line pattern in the g,, signal with a coupling constant of A,, values around 100 G.
The electronic spectra (Table 1) of diiron(II) com- plexes exhibit a weak intensity band in the 11,800- 12,450 cm- ’ region, which may reasonably be assigned to ‘T, + ‘E, transition consistent with38,39 a high spin octahedral environment around the iron(I1) ion. The magnetic susceptibility measurements confirm the above geometry. How- ever, the electronic spectra of the dicobalt(I1) com- plexes gave two ligand field bands in the region 13,750-14,200 and 21,80&22,400 cm- ‘, which may
be assigned to 4T1,(fl ---f 4A2g(F) and 4T,,(F) +
4T,,(P) transition, respectively corresponding to the octahedral geometry3’ around the cobalt(I1) ion. Here electronic spectral data suggestive of octa- hedral geometry which is an indication that the solvent molecule occupied the sixth coordination site of the metal ions. The possibility of such coor- dination has been reported4’ previously in the pres- ence of a strong coordinating solvent like DMSO. The band below 11,111 cm- ’ could not be recorded as it is beyond the range of the instrument used. The magnetic susceptibility measurements support their proposed high spin octahedral geometry of cobalt(I1) ion (Table 1).
The electronic spectra of dicopper(I1) macro- cyclic complexes exhibit two bands in the region 13,25&14,150 and 18,3OO-20,200 cm-’ which are in close agreement 3 8,4 ’ with those expected for a pseudo tetrahedral structure. The magnetic sus- ceptibility measurements (Table 1) of these com- plexes are observed in the range 1.87-1.99 B.M. which fall in the border line region of the square planar and tetrahedral types. This suggests slight distortion from square planar towards tetrahedral
symmetry, 3 5 which is confirmed by EPR spectral studies. All the complexes exhibit a strong absorp- tion band around 30,000 cm- ’ which may be due to a charge-transfer band.
Acknowledgements-The authors thank the Chairman, Department of Chemistry, A.M.U., Ahgarh, Dr Istiaque Ahmad of GNDU, Amritsar and Dr S. I. Khan of CDRI, Lucknow, are gratefully acknowledged for providing ‘H NMR, IR and elemental analysis facilities. Mr Saji P.V. is thankful to CSIR, New Delhi for the award of SRF.
REFERENCES
1. A. Bencini, A. Bianchi, E. G. Espana, M. Micheloni and P. Paoletti, Znorg. Chem. 1989, 28,248O.
2. J. E. Bulkowski, U.S. Patent 4545937 (1985). 3. S. Chandrasekhar, D. G. Fortier and A. McAuley,
Znorg. Chem. 1993, 32, 1424.

Binuclear transition metal complexes of diamidediimine hexaazamacrocycles 1361
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20. 21.
B. Dietrich, M. W. Hosseini, J. M. Lehn and R. B. Sessions, J. Am. Chem. Sot. 1981, 103, 1282. E. Kimura and A. Sakonaka, J. Am. Chem. Sot. 1982,104,4984. M. Yamashita, H. Ito and T. Ito, Znorg. Chem. 1983, 22,210l. J. Comarmond, J. Plumere, J. M. Lehn, Y. Angus, R. Louis, R. Weiss, 0. Kahn and I. Badaran, J. Am. Chem. Sac. 1982,104,6330. J. Nelson, B. P. Murphy, M. G. B. Drew, P. C. Yates and S. M. Nelson, J. Chem. Sot., Dalton Trans. 1988, 1001. N. Herron, W. P. Schammel, S. C. Jackels, J. J. Grzybowski, L. L. Zimmer and D. H. Busch, Znorg. Chem. 1983,22, 1433. P. J. Hay, J. C. Thilbeault and R. J. Hoffmann, J. Am. Chem. Sot. 1975,97,4884. C. Chanvel, J. J. Girerd, Y. Jeannin, 0. Kahn and G. Lavigne, Znorg. Chem. 1979, 18, 3015. U. Casellato, P. A. Vigato, D. E. Fenton and M. Vidali, Chem. Sot. Rev. 1979,8, 199. A. Buttafava, L. Fabbrizzi, A. Periotti and B. Seghi, J. Chem. Sot., Chem. Commun. 1982, 1166. J. M. Lehn, S. H. Pine, E. Watanabe and A. K. Willard, J. Am. Chem. Sot. 1977,99,6766. M. Shakir, S. P. Varkey and P. S. Hameed, Poly- hedron 1993, 12,277s. M. Shakir, S. P. Varkey and P. S. Hameed, J. Chem. Res. (S) 1993, 11, 442. J. F. Carvalho, S. H. Kim and C. A. Chang, Znorg. Chem. 1992,31,4065. S. C. Rawle, A. J. Clarke, P. Moore and N. W. Alcock, J. Chem. Sot., Dalton Trans. 1992, 2755. E. Kimura, Y. Lin, R. Machda and H. Zenda, J. Chem. Sot., Chem. Commun. 1986, 1020. H. Sigel and R. B. Martin, Chem. Rev. 1982,82,385. R. L. Dutta and A. K. Sarkar, J. Znorg. Nucl. Chem. 1981,43, 57.
22.
23.
24.
25.
26. 27. 28.
29.
30.
31.
32. 33.
34.
35.
36.
37.
38.
39.
40.
41.
A. K. Wiersema and J. J. Windle, J. Phys. Chem. 1964,68,2316. B. S. Pannu, S. L. Chopra and S. S. Parmer, Indian J. Chem. 1971,9, 1396. C. N. Reilley, R. W. Schmid and F. A. Sadak, J. Chem. Educ. 1959,36,555. A. I. Vogel, A Text Book of Quantitative Inorganic Analysis, p. 433. Longmans, London (1961). W. J. Geary, Coord. Chem. Rev. 1971,7, 81. B. L. Shaw, J. Am. Chem. Sot. 1975,97,3857. D. L. Arora, K. Lal, S. P. Gupta and S. K. Sahni, Polyhedron 1986, 5, 1499. I. Tabushi, H. Okino and Y. Kuroda, Tetrahedron Lett. 1976, 48, 4339. K. Nakamoto, Znfrared Spectra of Inorganic and Coordination Compounds. Wiley Intersciences, New York (1970). R. C. Agarwal, N. K. Singh and R. P. Singh, Znorg. Chem. 1981,20,2794. D. J. Hodgson, Znorg. Chim. Acta 1983,75, 225. U. Sakaguchi and A. W. Addison, J. Chem. Sot., Dalton Trans. 1979,600. H. Yokoi and A. W. Addison, Znorg. Chem. 1977, 16, 1341. T. Blaanwen and G. W. Canters, J. Am. Chem. Sot. 1993,115, 1121. I. M. Proctor, B. J. Hathaway and P. Nicholls, J. Chem. Sot. A 1968, 1678. R. W. Hay, M. A. Ali and B. Jeragh, J. Chem. Sot., Dalton Trans. 1988, 2763. A. B. P. Lever, Inorganic Electronic Spectroscopy. Elsevier, Amsterdam (1984). A. T. Baker, P. Singh and V. Vignevich, Aust. J. Chem. 1991,44, 1041. D. Lexa and J. M. Lhoste, Experimentia 1971, 18, 395. T. K. Chandhekar and D. D. Khanolkar, Indian J. Chem. 1986,25A, 868.