synthesis of de-crosslinkable polyamide having ... polymer 158 (2018...in a mixed solvent of ethanol...
TRANSCRIPT

Contents lists available at ScienceDirect
Polymer
journal homepage: www.elsevier.com/locate/polymer
Synthesis of de-crosslinkable polyamide having hexaarylbisimidazolylmoietiesMasato Takasakia, Takeru Iwamuraa,b,∗
a Department of Chemistry and Energy Engineering, Graduate School of Engineering, Tokyo City University, 1-28-1 Tamazutsumi, Setagaya-ku, Tokyo 158-8857, JapanbDepartment of Chemistry and Energy Engineering, Faculty of Engineering, Tokyo City University, 1-28-1 Tamazutsumi, Setagaya-ku, Tokyo 158-8557, Japan
H I G H L I G H T S
• The ε-caprolactam monomer having a triphenylimidazole moiety was synthesized by Heck reaction.
• The copolymer having a triphenylimidazole moiety was prepared at 250 °C by using H2O as an initiator.
• The crosslinking reaction of the obtained copolymer proceeded with K3[Fe(CN)6] and KOH.
• The resulting crosslinked polymer was de-crosslinked under irradiation with visible light.
A R T I C L E I N F O
Keywords:CrosslinkingDe-crosslinkingTriphenylimidazole
A B S T R A C T
The ε-caprolactam monomer having a triphenylimidazole moiety such as DL-3-[α-4-(4,5-diphenyl-1H-imidazole-2-yl)-phenylacryloyl-amine]-ε-caprolactam (1) was synthesized by the Heck reaction of 2-(4-bromophenyl)-1-(methoxymethyl)-4,5-diphenylimidazole and the ε-caprolactam derivative having an acryloyl group in a goodyield. The copolymerization of 1 and ε-caprolactam was carried out at 250 °C by using H2O as an initiator to givecopolymer (4) in a good yield. The crosslinking reaction of the obtained 2 proceeded with K3[Fe(CN)6] and KOHin a mixed solvent of ethanol and phenol. The resulting crosslinked polymer (5) was de-crosslinked under ir-radiation with visible light. Consequently, the thiol-capped de-crosslinked polymer (6) was obtained in a goodyield.
1. Introduction
Polyamides are crystalline thermoplastic polymers that are alsoimportant as molding materials. Ever since Carothers’ development ofpolymers at DuPont, polyamides have become an excellent material.Polyamides are synthesized by condensation of dichloride and diamineor ring-opening polymerization of lactams. The strong intermolecularinteractions caused by the hydrogen bonding of amide groups conferspolyamides with a high melting point despite being soft. In addition,these polymers have excellent heat resistance and strength. Moreover,polyamides have been used as fibers because they have tenacity andwear resistance. Polyamide fibers are generally manufactured by meltspinning, which is the most widely used method for producing com-mercial synthetic fibers [1,2]. In addition to fibers, polyamides havebeen applied to various industries such as films and engineering plas-tics. This is because they have superior performance such as excellentheat resistance, mechanical properties, chemical resistance, excellent
abrasion resistance and processability [3]. The synthesis of polyamidesand its mechanical properties have been extensively studied by manyresearchers [4–7]. Furthermore, in recent years, polyamides have at-tracted attention as automotive parts [8–11]. In recent years, cross-linked polymers have been extensively studied because various physicalproperties are expected to be improved [12–18]. However, on the otherhand, material recycling or chemical recycling of crosslinked polymersis generally very difficult due to their insolubility and infusible nature.To overcome these limitations, we proposed a possible approach thatconsists of the introduction of a de-crosslinking system through the useof weak covalent bonding that would allow for the chemical recyclingof crosslinked polymers.
We reported a novel de-crosslinking system utilizing pressure or irra-diation with visible light [19,20]. The crosslinking reaction of a copolymerhaving a 2,4,5-triphenylimidazole moiety in the side chain of the polymerwhich was prepared by radical copolymerization proceeded with K3[Fe(CN)6] and KOH. In this research, polymers consisting of monomers having
https://doi.org/10.1016/j.polymer.2018.10.070Received 31 August 2018; Received in revised form 23 October 2018; Accepted 30 October 2018
∗ Corresponding author. Department of Chemistry and Energy Engineering, Graduate School of Engineering, Tokyo City University, 1-28-1 Tamazutsumi, Setagaya-ku, Tokyo 158-8857, Japan.
E-mail address: [email protected] (T. Iwamura).
Polymer 158 (2018) 270–278
Available online 30 October 20180032-3861/ © 2018 Elsevier Ltd. All rights reserved.
T

a 2,4,5-triphenylimidazole moiety were synthesized. The radical poly-merization of these monomers was examined in detail. In the crosslinkingreaction, the hexaarylbisimidazole (HABI) moiety as a crosslinking pointwas included in the crosslinked polymers. In addition, the de-crosslinkingproperties of the obtained de-crosslinkable polymers were also investigated.
In recent years, apart from the findings of our group, several reportsexist. Honda et al. reported that non-liquid network polymers liquefiedupon UV irradiation and produced liquid star-shaped polymers with2,4,5-triphenylimidazoryl radical end groups that reverted to non-li-quid network polymers again by recoupling of the generated 2,4,5-tri-phenylimidazoryl radicals immediately after terminating UV irradiation[21]. Sijbesma et al. reported that the HABI moiety can cleave to tri-phenylimidazolyl radicals when incorporated into a polymer matrixunder mechanical stress [22].
It would be extremely difficult to recycle crosslinked polymersprepared from polyamides with excellent properties. However, it isexpected that a reversible crosslinking network polymer can be ob-tained by introducing a triphenylimidazole moiety into the side chain ofa polyamide and crosslinking this moiety. In this article, we synthesizedpolyamides having a triphenylimidazole moiety. Furthermore, the ob-tained polyamides were crosslinked with K3[Fe(CN)6] and KOH and thecrosslinking properties of the obtained network polymers were eval-uated.
2. Experimental procedure
2.1. Materials
2-(4-Bromo-phenyl)-4,5-diphenyl-1H-imidazol was prepared by Siddiqui'sprocedure [23]. Triethylamine, N,N-dimethylformamide (DMF), 1,4-dioxane,pyridine, N,N-dimethyl-4-aminopyridine, and tri-o-tolylphosphine were pur-chased from Wako Pure Chemicals Ltd. (extra pure grade). ε-Caprolactamwas purchased from Tokyo Chemical Industry Co., Ltd. (extra pure grade).Triethylamine, N,N-dimethylformamide (DMF) and 1,4-dioxane were driedover CaH2, distilled under reduced pressure, and stored under nitrogen.Pyridine was dried over KOH, distilled, and stored under a nitrogen atmo-sphere. N,N-Dimethyl-4-aminopyridine, tri-o-tolylphosphine and ε-capro-lactam were recrystallized from hexane. Other solvents and reagents wereused as supplied.
2.2. Measurements
1H NMR and 13C NMR spectra were recorded on a JEOL JNM-EPC300 spectrometer at 300 MHz and 75 MHz, respectively. FT-IR spectrawere measured on a JASCO FT/IR-4200 spectrometer. Thermal ana-lyses were performed on Seiko Instruments, Inc., TG/DTA200 andDSC210. Melting points were determined by differential scanning ca-lorimetry (DSC) at a heating rate of 5 °C/min under a nitrogen atmo-sphere. A 10% weight loss temperature (Td10) was determined bythermogravimetric analysis (TGA) at a heating rate of 10 °C/min in air.Viscosity of the polymer was measured in 90% formic acid at 25 °C withan Ostwald viscometer. Fast atom bombardment mass spectra (FAB/MS) were recorded by using a JEOL JMS-700 spectrometer, whereby amixture of a sample and m-nitrobenzyl alcohol on a standard FAB targetwas subjected to a beam of xenon atoms produced at 6 keV and 2 mA.
2.3. Synthesis of α-(N-acryloyl-amino)-ε-caprolactam
Acryloyl chloride (0.129 g, 1.42 mmol) was added dropwise to astirred solution of DL-α-amino-ε-caprolactam (0.140 g, 1.09 mmol),pyridine (0.108 g, 1.69 mmol) and N,N-dimethyl-4-aminopyridine(13.3 mg, 0.11 mmol) in anhydrous DMF (22 mL). After stirring at 60 °Cfor 24 h, the reaction mixture was evaporated under reduced pressureto remove DMF. The residue was purified by chromatography on silicagel with chloroform/methanol (99/1, v/v) as an eluent to isolate α-(N-acryloyl-amino)-ε-caprolactam. Yield: 52%.
Mp. 173.9 °C; IR (KBr): 3310, 2918, 2857, 1671, 1651, 1621, 1607,1524, 1480, 1455, 1434, 1370, 1332, 1283, 1232, 1116, 1070, 992,952, 879, 827, 805, 743 cm−1; 1H NMR (CDCl3, 300 MHz) δ: 1.16–1.65(m, -CH2-CH2-NH-, -CH2-C3H6-NH-, 2H), 1.70–2.26 (m, -C3H6-CH2-NH-,4H), 3.12–3.44 (m, -C3H6-CH2-NH-, 2H), 4.46–4.76 (m, -NH-C(C4H8)H-CO-, 1H), 5.66 (dd, CH2=CH- cis, J= 9.87 and 1.65 Hz, 1H), 6.18 (dd,CH2=CH-, J= 17.03 and 9.87 Hz, 1H), 6.31 (dd, CH2=CH- trans,J= 17.04 and 1.92 Hz, 1H), 6.34 (s, CH2=CH-NH-, 1H), 7.28 (brs, -CO-NH-C4H8-, 1H) ppm; 13C NMR (CDCl3/TFA = 4/1 v/v, 75 MHz) δ:27.70, 27.89, 31.04, 43.23, 53.68, 128.77, 131.11, 168.95,178.39 ppm.
2.4. Synthesis of DL-3-[α-4-(4,5-diphenyl-1H-imidazole-2-yl)-phenylacryloyl-amine]-ε-caprolactam (1)
2-(4-Bromo-phenyl)-4,5-diphenyl-1H-imidazole (79.1mg, 0.21mmol), α-(N-acryloyl-amino)-ε-caprolactam (47.3mg, 0.26mmol), triethylamine(67.4mg, 0.65mmol), tri-o-tolylphosphine (67.4 mg, 0.65mmol) and palla-dium acetate (0.5mg, 0.02mmol) were dissolved in anhydrous 1,4-dioxane(2.6mL). The mixture was refluxed for 60h under a nitrogen atmosphere,and then evaporated under reduced pressure. The residue was purified bychromatography on silica gel with chloroform/methanol (from 95/5 to 4/1,v/v) as an eluent to isolate a monomer (1). Yield: 80%.
Mp. 156.8 °C; IR (KBr): 3433, 3063, 2933, 2857, 2802, 2739, 2678,2492, 1655, 1619, 1561, 1544, 1525, 1510, 1491, 1476, 1437, 1399,1340, 1278, 1213, 1171, 1122, 1073, 1037, 1003, 973, 835, 768,698 cm−1; 1H NMR (CDCl3/TFA = 4/1 v/v, 300 MHz) δ: 1.41–2.27 (m,-C3H6-CH2-, 6H), 3.30–3.58 (m, -C3H6-CH2-NH-, 2H), 4.81–5.01 (m,-NH-C(C4H8)H-CO-, 1H), 6.75 (d, -CO-CH=CH- trans, J= 15.93 Hz,1H), 7.38–7.56 (m, Ph-H, 10H), 7.61–7.84 (m, -NH-CO-CH = , 1H, -CO-CH=CH- trans, 1H), 7.73 (d, -CH=CH-C6H4-, J= 8.52 Hz, 2H), 7.94(d, -C6H4-C3N2H-, J= 8.52 Hz, 2H), 8.00 (brs, -CO-NH-C4H8-, 1H) ppm;13C NMR(DMSO‑d6, 75 MHz) δ: 27.62, 28.77, 31.19, 40.61, 51.54,122.55, 125.44, 127.19, 127.70, 127.95, 128.38, 130.83, 134.67,138.04, 144.92, 163.90, 174.15 ppm; High-resolution FAB-MS [M+H]+: found, 477.2299; calcd for C30H29N4O2, 477.2291.
2.5. Synthesis of 2
A 2% aqueous solution of potassium ferricyanide (5 mL) was addedslowly to a solution of 1 (10 mg) dissolved in a solvent of ethanol (1 mL)containing potassium hydroxide (70 mg). The reaction mixture wasstirred by a stream of oxygen for 1.5 h at 5–10 °C. The precipitateddimer was separated by centrifugation at 3000 rpm for 10 min.Inorganic regents were removed by centrifugation/washing three timeswith distilled water. The obtained dimer (2) was dried in vacuo. Yield:81%.
2.6. Copolymerization of 1 and ε-caprolactam
Monomer (1) (0.477 g, 1.01 mmol), ε-caprolactam (1.02 g,9.06 mmol) and distilled water (7.5 μL, 1.5 wt%) were placed in a testtube that was then sealed. After 3 h at 250 °C, the mixture was heatedfor 4 h at 250 °C in air. The reaction mixture was purified with boilingchloroform by a Soxhlet extractor for 48 h, and the obtained copolymer(4) was dried in vacuo at 50 °C. Yield: 77%.
Mv = 8000 (determined by viscosity); IR (KBr): 3300, 3087, 2934,2861, 1701, 1639, 1559, 1545, 1510, 1499, 1492, 1474, 1459, 1450,1439, 1374, 1281, 1262, 1201, 1172, 1118, 1072, 1027, 966, 926, 831,753, 730, 698 cm−1; 1H NMR (CF3COOD, 300 MHz) δ: 1.25–2.33 (m,-C3H6-CH2-NH-, 6H×0.92, -C3H6-CH2-NH-, 6H×0.08), 2.48–2.99 (m,-CO-CH2-CH2-, 2H×0.92), 3.40–3.90 (m, -CH2-CH2-NH-, 2H×0.92,-CH2-CH2-NH-, 2H×0.08), 4.82 (s, -CO-C(R)H-CH2-, 1H×0.08),6.88–8.38 (m, Ph-H, 14H×0.08, -CO-CH=CH-, 1H×0.08, -CO-CH=CH-, 1H×0.08, -NH-CO-, 1H×0.08) ppm; 13C NMR (CF3COOD,75 MHz) δ: 27.51, 27.94, 29.36, 35.84, 44.51, 126.25, 127.82, 129.64,
M. Takasaki, T. Iwamura Polymer 158 (2018) 270–278
271

Scheme 1. Synthesis of 1.
Scheme 2. Dimerization of 1.
Scheme 3. Photochromism of 2.
Scheme 4. Copolymerization of 1 and ε-caprolactam.
M. Takasaki, T. Iwamura Polymer 158 (2018) 270–278
272
130.28, 131.63, 132.20, 132.96, 139.97, 140.30, 145.43, 146.13,146.48, 147.54, 162.67, 181.58 ppm.
2.7. Crosslinking reaction of 4
A 1% aqueous solution of potassium ferricyanide (3.4 mL) was
added slowly to a solution of 4 (40.7 mg) dissolved in a mixed solvent ofethanol (1 mL) and phenol (0.5 g) containing potassium hydroxide(0.105 g). The reaction mixture was stirred by a stream of oxygen for48 h at room temperature. The precipitated crosslinked polymer wasseparated by centrifugation at 3000 rpm for 10 min. Inorganic regentswere removed by centrifugation/washing three times with distilled
Fig. 1. 1H NMR (CF3COOD) spectrum of 4.
Fig. 2. 13C NMR (CF3COOD) spectrum of 4.
M. Takasaki, T. Iwamura Polymer 158 (2018) 270–278
273
water and ethanol. The obtained crosslinked polymer (5) was dried invacuo. Yield: 70%.
IR (KBr): 3299, 3083, 2935, 2863, 1638, 1543, 1500, 1492, 1475,1460, 1450, 1439, 1419, 1374, 1263, 1200, 1171, 1119, 1072, 1028,961, 929, 835, 768, 729, 697 cm−1.
2.8. De-crosslinking of crosslinked polymer under visible light irradiation(typical procedure)
Crosslinked polymer (5) (10.5 mg) was dispersed in a mixed solventof ethanol (1 mL) and phenol (0.5 g). The dispersion of the crosslinkedpolymer was irradiated for 24 h under visible light using a desk lamp(HITACHI RS3205, 34 W). After irradiation with visible light, etha-nethiol (21 μL, 50 eq.) was added to a stirred solution of the reactionmixture. The stirred reaction mixture was poured into methanol(20 mL) and the precipitated polymer was separated by centrifugationat 3000 rpm for 10 min. The obtained thiol-capped de-crosslinkedpolymer (6) was purified with methanol and ether by a solid-liquidextraction, and dried in vacuo at 50 °C. Yield: 80%.
IR (KBr): 3300, 3086, 2937, 2866, 1726, 1719, 1701, 1639, 1543, 1509,1499, 1476, 1464, 1450, 1439, 1419, 1375, 1237, 1157, 1118, 1075, 1025,983, 959, 930, 879, 836, 730, 694 cm−1; 1H NMR (CF3COOD, 300MHz) δ:
1.22–2.25 (m, -C3H6-CH2-NH-, 6H×0.92, -C3H6-CH2-NH-, 6H×0.08, CH3-CH2-S-, 0.24H [=3H×0.08]), 2.43–3.06 (m, -CO-CH2-CH2-, 2H×0.92),3.34–4.01 (m, -CH2-CH2-NH-, 2H×0.92, CH2-CH2-NH-, 2H×0.08, CH3-CH2-S-, 0.16H [=2H×0.08]), 4.82 (s, -CO-C(R)H-CH2-, 1H×0.08), 6.85–8.38(m, Ph-H, 14H×0.08, -CO-CH=CH-, 1H×0.08, -CO-CH=CH-, 1H×0.08,-NH-CO-, 1H×0.08) ppm; 13C NMR (CF3COOD, 75MHz) δ: 3.72, 14.85,23.66, 27.51, 27.94, 29.37, 31.68, 35.88, 44.47, 125.15, 129.01, 130.28,130.52, 130.80, 131.12, 131.66, 132.20, 133.12, 133.26, 136.64, 138.23,146.39, 159.12, 181.60ppm.
3. Results and discussion
3.1. Synthesis of 1
Monomer (1) was synthesized by a two-step reaction. First, α-(N-acryloyl-amino)-ε-caprolactam was prepared by DL-α-amino-ε-capro-lactam and acryloyl chloride in moderate yield (Scheme 1). Then, thecoupling reaction of α-(N-acryloyl-amino)-ε-caprolactam with 2-(4-bromophenyl)-4,5-diphenyl-1H-imidazole was catalyzed by Pd(OAc)2in dioxane under reflux conditions [24]. The obtained 1 was wellcharacterized by 1H NMR, 13C NMR, elemental analysis and FT-IRspectroscopy.
3.2. Synthesis of 2
The dimerization reactions of 1 were carried out by a previouslyreported procedure (Scheme 2) [19,20]. To a solution of 1 and KOH in asolvent of ethanol, a 1% K3[Fe(CN)6] aqueous solution was added withstirring by a stream of oxygen at 5–10 °C for 1.5 h. The precipitateddimer (2) was repeatedly washed with distilled water. Consequently, 2was obtained in 81% yield. Maeda et al. reported that a benzene so-lution of triphenylimidazole photo-dimer changed from pale yellow toreddish-purple following irradiation at room temperature [25]. Incontrast, the chloroform solution of 2 changed from pale green to darkgreen under visible light irradiation using a desk lamp. This dark greenmay be considered to result from the triphenylimidazolyl radical (3)(Scheme 3). The triphenylimidazolyl radical was indicated as beingreddish purple, whereas 3 was dark green because a double bond existsat the para position of the benzene ring possessed by triphenylimida-zole, i.e., the conjugation is extended. A change in color of the imida-zolyl radical might be due to an extension of conjugation.
Fig. 3. FT-IR spectrum of 4.
Scheme 5. Crosslinking reaction of 4.
M. Takasaki, T. Iwamura Polymer 158 (2018) 270–278
274

3.3. Copolymerization of 1 and ε-caprolactam
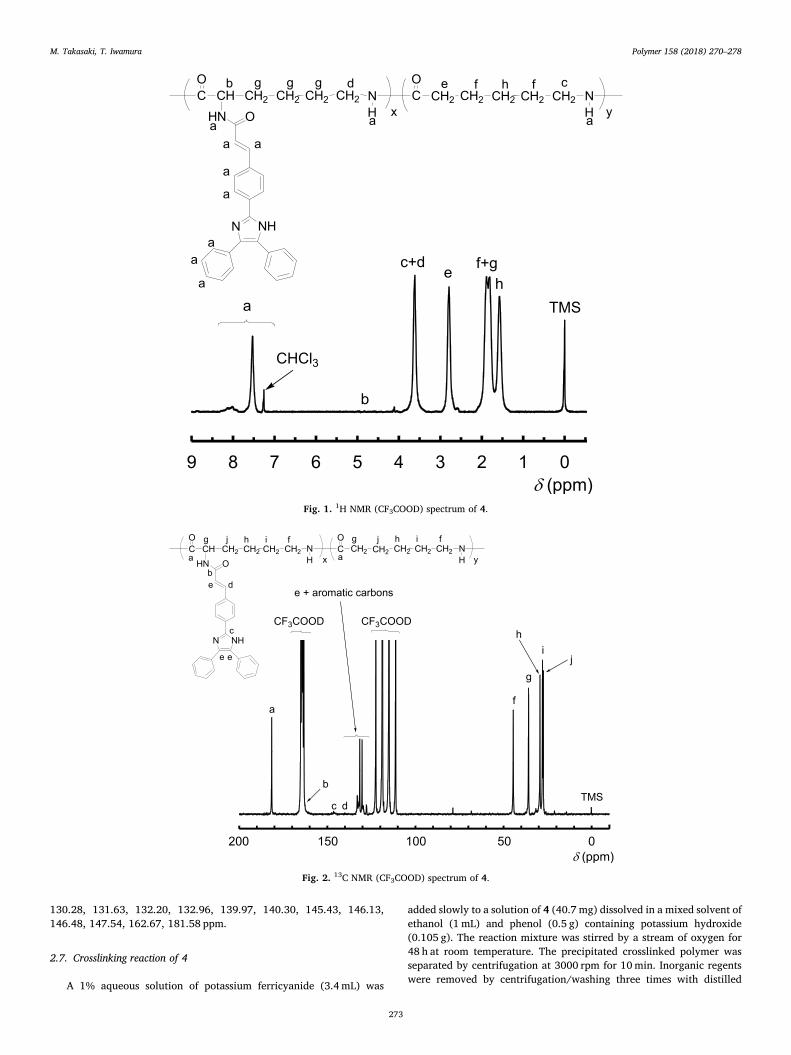
The copolymerization of 1 and ε-caprolactam was carried out at250 °C for 3 h using H2O as an initiator. The corresponding randomcopolymer (4) was obtained in 77% yield (Scheme 4). The 1H NMRspectrum of 4 is shown in Fig. 1. The aromatic protons resulting fromthe triphenylimidazole moieties, the amide protons in the main chain ofthe polymer, and the alkene protons adjacent to the amide group in the
side chain of the polymer were observed at δ 6.87–8.47 ppm (a). Ad-ditionally, peaks due to the methylene proton in the main chain of thepolymer were observed at δ 3.32–3.95 ppm (c, d), δ 2.52–3.00 ppm (e),and δ 1.29–2.18 ppm (f, g, h), respectively (Fig. 1). The copolymercomposition was determined by its 1H NMR spectra. The unit ratio wasdetermined to be x:y = 1:9 from the integral ratio between the aromaticprotons (a) and methylene protons (c and d). The 13C NMR spectrum of4 is shown in Fig. 2. The peaks assignable to the carbonyl carbons were
Table 1Crosslinking and de-crosslinking behavior of 4.
Run Cross-linking reaction of 4a. De-crosslinking reaction of 5
Solvent Temp. Time Yield Td10 Solvent Temp. Time Yield Degree of hydrolysisb Modification ratiob
(°C) (h) (%) (°C) (°C) (h) (%) (%) (%)
1 EtOH/Phenol 5–10 1.5 59 – EtOH/Phenol rt 1 59 15 02 EtOH/Phenol rt 24 79 380.4 EtOH/Phenol rt 1 73 10 483 EtOH/Phenol rt 48 70 374.4 EtOH/Phenol rt 24 80 49 744 EtOH/THF rt 48 69 378.6 THF rt 24 88 27 395 EtOH/DMF rt 48 75 – DMF rt 24 63 17 12
a Td10 of 4= 365.7 °C.b Determined by 1H NMR spectrum.
Fig. 4. FT-IR spectra of 4 and 5 (Table 1, Run 3).
Scheme 6. De-crosslinking reaction of 5.
M. Takasaki, T. Iwamura Polymer 158 (2018) 270–278
275
observed at δ 181.58 ppm. The signals ascribable to the aryl imidazolemoiety (x unit) were observed at δ 126.56–148.63 ppm. The signalsascribable to the ε-caprolactam moiety (y unit) were observed at δ44.51, 35.84, 29.36, 27.94 and 27.51 ppm. As a result, the 13C NMRspectrum of 4 supported the fact that 1 underwent copolymerization.
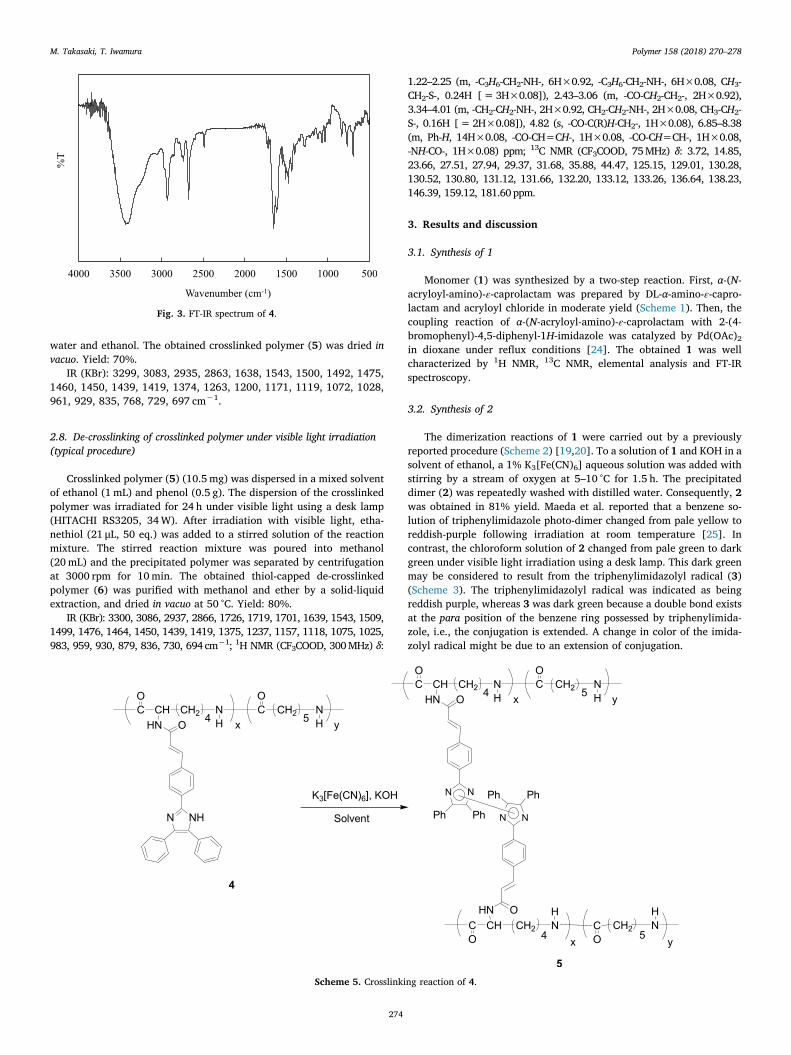
In Fig. 3, the FT-IR spectrum of 4 showed an absorption band at1639 cm−1 based on the amide carbonyl group (vC=O) and 1545 cm−1
based on the amide N-H bending vibration. Moreover, the absorptionbands of the imidazole ring observed at 1492 cm−1 and 1450 cm−1
were assigned to the C=C stretching frequency and C=N-C=C sym-metric stretch frequency, respectively. The viscosity average molecularweight (Mv) of 4 was estimated to be 8000 by an Ostwald viscometer.
3.4. Crosslinking reaction of 4
The crosslinking reactions of 4 were carried out with reference to apreviously reported procedure [18,19]. Polymer 4 can be dissolved byusing phenol. Consequently, phenol was employed because the cross-linking reaction can proceed sufficiently. To a solution of 4 and KOH ina mixed solvent of ethanol and phenol, a 1% K3[Fe(CN)6] solution wasadded with vigorous stirring at room temperature for 48 h (Scheme 5,Table 1). The precipitated crosslinked polymer was repeatedly washedwith distilled water and ethanol. Consequently, 4 was obtained in 70%yield. The FT-IR spectra of 4 and 5 are shown in Fig. 4. In these spectra,a C=C stretching vibration and a C=N-C=C symmetric stretching
Fig. 5. FT-IR spectra of 4 and 6 (Table 1, Run 3).
Fig. 6. 1H NMR (CF3COOD) spectrum of 6 (Table 1, Run 3).
M. Takasaki, T. Iwamura Polymer 158 (2018) 270–278
276

vibration were observed at 1499 and 1450 cm−1, respectively. Thesepeaks were derived from the imidazole ring. In the case of 5, it is ex-pected that the HABI moiety, which was a crosslinking point, was in-cluded in the network polymer structure. For that reason, the data re-ported by White et al. and Tanino et al. were cited. In the data reportedby White et al., the 1,2’-isomer of HABI absorbed at 1600 (m), 1554(m), and 1480 (m) cm−1. They also reported that the 4,4’-isomer ofHABI absorbed at 1605 (m), 1562 (s), and 1489 (s) cm−1 [26]. In thereported data by Tanino et al., the 1,4’-isomer and 2,4’-isomer absorbedat 1610 (s) and 1613 (vs), respectively [27]. As mentioned above, thepeaks derived from HABI should be detected at around 1600 cm−1.However, in the FT-IR spectrum of 5, since the peaks derived from HABIwere buried in a strong peak derived from the amide group (δNH), itcould not be clearly confirmed. Overall, the spectra of 4 and 5 weresimilar. This is because these structures were very similar since thestructure of the crosslinked polymer was almost identical to the struc-ture of the linear polymer and only a small number of crosslinking siteswere present in the crosslinked polymer. The thermal stability ofcrosslinking polymer 5 was measured from TGA analysis in air. In thecase of 4, Td10 was observed at 365.7 °C. In the case of the crosslinkingpolymer (5), Td10 shifted to a higher temperature (Table 1, runs 2–4).
3.5. De-crosslinking of 5
During the de-crosslinking reaction of 5 under visible light irra-diation, 5 was dispersed in a mixed solvent of ethanol and phenol. Thedispersion of 5 was irradiated under visible light irradiation using a
desk lamp. Subsequently, to isolate the de-crosslinked polymer, etha-nethiol was added to the reaction mixture after the de-crosslinkingreaction (Scheme 6). The corresponding 6 was obtained as a methanol-insoluble part in a good yield. The results are summarized in Table 1.
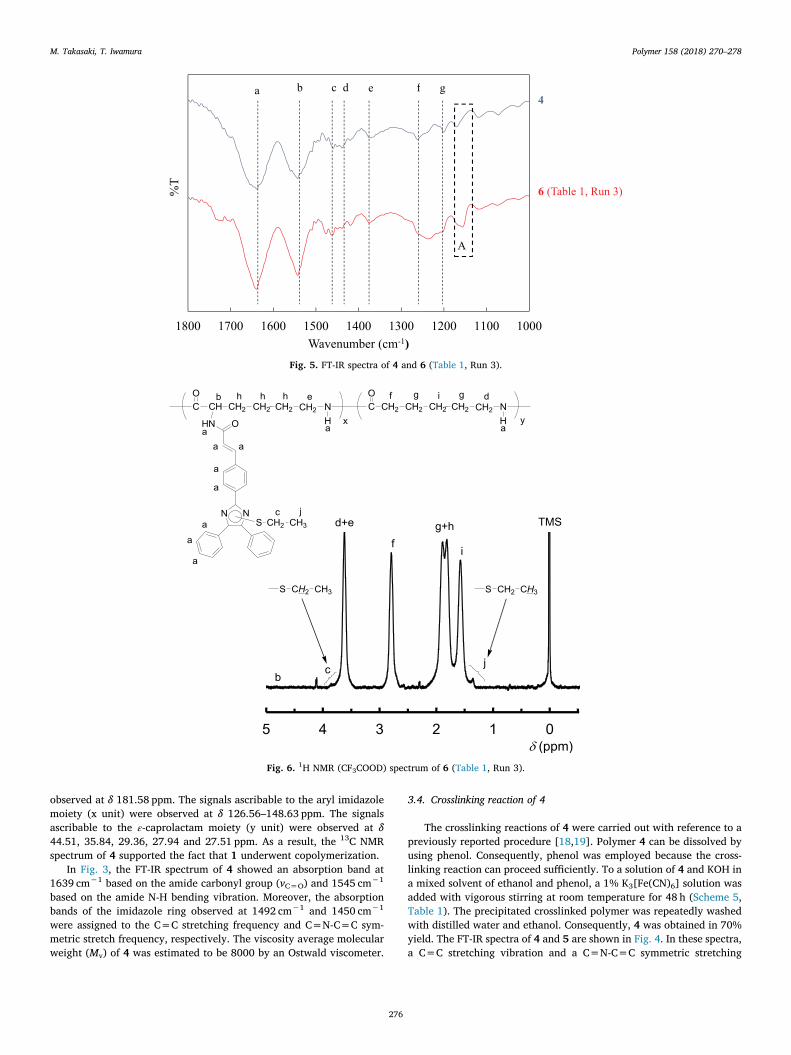
The IR spectra of 4 and 6 are shown in Fig. 5. In particular, the FT-IR spectra of 4 and 6 were compared in the 1800-1000 cm−1 region.The peaks a-g of 4 are peaks that can mostly all be found in 6 althoughone of these peaks was buried beneath other peaks. This result showsthat the structure of 6 is almost maintained in 4. In contrast, in the caseof 6, the peaks not observed in 4 were observed at 1167 cm−1 (regionof A). Iwamura et al. reported the FT-IR spectra of a thiol-capped de-crosslinked polymer, suggesting that this peak was assigned to the S-alkyl group of the imidazole ring [20]. The 1H NMR spectrum of 6 isshown in Fig. 6. The 1H and 13C NMR spectra of the thiol-capped de-crosslinked polymer which was reported by Iwamura et al. were cited[20]. In the case of the 1H NMR spectrum, a peak caused by the methylgroup of the S-alkyl group was observed at around δ 1.36 ppm. Ad-ditionally, in the 13C NMR spectrum of 6, a peak due to the methylgroup of the S-alkyl group was observed at around δ 14.9 ppm, and themethylene group of the S-alkyl group was observed at around δ31.7 ppm in Fig. 7.
The crosslinking reaction of 4 was carried out in a mixed solvent ofethanol and phenol at 5–10 °C for 1.5 h, and the crosslinked polymer 5was obtained in 59% yield (Table 1, run 1). Similarly, the crosslinkingreaction of 4 was carried out in a mixed solvent of ethanol and phenolat room temperature for 24 h, and 5 was obtained in 79% yield(Table 1, run 2). Furthermore, when the reaction time was prolonged,
Fig. 7. 13C NMR (CF3COOD) spectrum of 6 (Table 1, Run 3).
M. Takasaki, T. Iwamura Polymer 158 (2018) 270–278
277

the yield of 5 decreased to 70% (Table 1, run 3). The crosslinkingpolymers 5 obtained by the reaction of runs 1–3 were irradiated undervisible light at room temperature for 1 h, and then ethanethiol wasadded to a solution of 5. In the case of run 1, 5 was obtained in 59%yield. The hydrolysis ratio and the thiol-modification ratio were esti-mated by 1H NMR measurements. As a result, it was clarified that theside chain of 5 was hydrolyzed by 15% and the thiol-modification ratiowas 0%. In the case of run 2, 5 was obtained in 73% yield. From 1HNMR measurements, the side chain of 5 was hydrolyzed by 10% and thethiol-modification ratio was 48%. In the case of run 3, 5 was obtained in80% yield. From 1H NMR measurements, the side chain of 5 was hy-drolyzed by 49% and the thiol-modification ratio was 74%. These re-sults indicate that the thiol-modification ratio was improved when thereaction time was prolonged. From runs 2 and 3, it was clarified thatthe hydrolysis of the side chain of polymers proceeded further when thecrosslinking reaction of 4 was prolonged. The side chain of the polymerwill be hydrolyzed by KOH, which was employed in the crosslinkingreaction. In the crosslinking reaction of 4 (Table 1, runs 4 and 5), hy-drolysis could be suppressed by changing from a mixed solvent ofethanol and phenol to THF or DMF. However, the thiol-modificationratios also decreased.
4. Conclusions
A monomer (1) was prepared from 2-(4-bromophenyl)-1-(methox-ymethyl)-4,5-diphenyl-imidazole and an ε-caprolactam derivativehaving an acryloyl group in a good yield. Copolymerizarion of 1 and ε-caprolactam was performed at 250 °C by ring-opening polymerization.Consequently, copolymer (4) was obtained in a good yield. The cross-linking reaction of 4 proceeded with potassium ferricyanide and po-tassium hydroxide, and the corresponding polymer (5) was obtained in70% yield. The de-crosslinking reaction of 5 proceeded by visible light,and the corresponding yield of polymer (6) was 80%.
From the viewpoint of environmental science, it is very importantthat crosslinked polymers can de-crosslink to linear polymers capable ofmaterial recycling, i.e., that polymers can be dissolved in solvents.Therefore, a crosslinked polymer having a HABI moiety is an importantpolymer in terms of polymer recycling. Such a de-crosslinkable poly-amide is expected to be a material having excellent heat resistance,mechanical properties, chemical resistance, excellent abrasion re-sistance and processability. Furthermore, the de-crosslinkable poly-amide can be an important material for automobiles.
Acknowledgment
This work was supported by a Grant-in-Aid for Scientific Research(C) (Grant Number JP 16K00659) from Japan Society for the Promotionof Science.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.polymer.2018.10.070.
References
[1] V.G. Bankar, J.E. Spruiell, J.L. White, Melt-spinning dynamics and rheologicalproperties of nylon 6, J. Appl. Polym. Sci. 21 (1977) 2135–2155 https://doi.org/10.1002/app.1977.070210812.
[2] A. Ziabicki, K. Kedzierska, Studies on the orientation phenomena by fiber formationfrom polymer melts. Part I. Preliminary investigations on polycapronamide, J. Appl.Polym. Sci. 2 (1959) 14–23 https://doi.org/10.1002/app.1959.070020403.
[3] M.I. Kohan, Nylon Plastics Handbook, Hanser/Gardner Publications Inc, Cincinnati,
1995, pp. 293–360.[4] P.H. Hermans, D. Heikens, P.F. van Velden, On the mechanism of the polymeriza-
tion of ε–caprolactam. II. The polymerization in the presence of water, J. Polym.Sci. 30 (1958) 81–104 https://doi.org/10.1002/pol.1958.1203012108.504.
[5] V.G. Bankar, J.E. Spruiell, J.L. White, Melt spinning of nylon 6: structure devel-opment and mechanical properties of as-spun filaments, J. Appl. Polym. Sci. 21(1977) 2341–2358 https://doi.org/10.1002/app.1977.070210905.
[6] T.F. Meyabadi, M.R.M. Mojtahedi, S.A.M. Shoushtari, Melt spinning of reused nylon6: structure and physical properties of as-spun, drawn, and textured filaments, J.Text. Inst. 101 (2010) 527–537 https://doi.org/10.1080/00405000802561085.
[7] N.S. Murthy, R.G. Bray, S.T. Correale, R.A.F. Moore, Drawing and annealing ofnylon-6 fibres: studies of crystal growth, orientation of amorphous and crystallinedomains and their influence on properties, Polymer 36 (1995) 3863–3873 https://doi.org/10.1016/0032-3861(95)99780-X.
[8] H.-D. Nguyen-Tran, V.-T. Hoang, V.-T. Do, D.-M. Chun, Y.-J. Yum, Effect ofMultiwalled carbon nanotubes on the mechanical properties of carbon fiber-re-inforced polyamide-6/polypropylene composites for lightweight automotive parts,Materials 11 (2018) 429 https://doi.org/10.3390/ma11030429.
[9] T. Terashima, T. Nomura, Polyamide resin composition and a shaped article, USPatent (1991) 4,981,920.
[10] C. E. Koning, J. Tijssen, Car parts made from a polyamide composition, US Patent(2001), 6,172,178.
[11] D. S. Kim, H. S. Chang, B. H. Oh, Polyamide resin composition, US Patent (2012), 8,114,941.
[12] C.O. Sánchez, F.R. Díaz, N. Gatica, C. Bustos, K. Espiñeira, D. Huaquimilla,Synthesis and study of the effect of carboxyvinyl group position on the properties ofpolyamides. Crosslinked polymers, Polym. Bull. 67 (2011) 29–43 https://doi.org/10.1007/s00289-010-0356-0.
[13] J. Xu, Z. Wang, X. Wei, S. Yang, J. Wang, S. Wang, The chlorination process ofcrosslinked aromatic polyamide reverse osmosis membrane: new insights from thestudy of self-made membrane, Desalination 313 (2013) 145–155 https://doi.org/10.1016/j.desal.2012.12.020.
[14] T. Wang, Y. Yang, J. Zheng, Q. Zhan, S. Zhang, A novel highly permeable positivelycharged nanofiltration membrane based on a nanoporous hyper-crosslinked poly-amide barrier layer, J. Membr. Sci. 488 (2013) 180–189 https://doi.org/10.1016/j.memsci.2013.08.012.
[15] H. Bouchékif, D. Tunc, C.L. Coz, A. Deffieux, P. Desbois, S. Carlotti, Controlledsynthesis of crosslinked polyamide 6 using a bis-monomer derived from cyclizedlysine, Polymer 55 (2014) 5991–5997 https://doi.org/10.1016/j.polymer.2014.09.050.
[16] C.O. Sánchez, P. Sobarzo, N. Gatica, D. Mac-leod-carey, New fluorescent crosslinkedaromatic polyamides containing thiophene and furane in their backbone, J. Chil.Chem. Soc. 60 (2015) 3040–3044 https://doi.org/10.4067/S0717-97072015000300014.
[17] S.K. Lim, L. Setiawan, T.-H. Bae, R. Wang, Polyamide-imide hollow fiber mem-branes crosslinked with amine-appended inorganic networks for application insolvent-resistant nanofiltration under low operating pressure, J. Membr. Sci. 501(2016) 152–160 https://doi.org/10.1016/j.memsci.2015.11.016.
[18] C. Leisen, M. Wolf, D. Drummer, Influence of the mold temperature on the materialproperties and the vibration welding process of crosslinked polyamide 66, Polym.Eng. Sci. 58 (2018) E207–E214 https://doi.org/10.1002/pen.24636.
[19] T. Iwamura, M. Sakaguchi, A novel de-cross-linking system from cross-linkedpolymer to linear polymer utilizing pressure or visible light irradiation,Macromolecules 41 (2008) 8995–8999 https://doi.org/10.1021/ma801298e.
[20] T. Iwamura, S. Nakamura, Synthesis and properties of de-cross-linkable acrylatepolymers based on hexaarylbiimidazole, Polymer 54 (2013) 4161–4170 https://doi.org/10.1016/j.polymer.2013.05.066.
[21] S. Honda, T. Toyota, Photo-triggered solvent-free metamorphosis of polymericmaterials, Nat. Commun. 8 (2017) 502 https://doi.org/10.1038/s41467-017-00679-1.
[22] F. Verstraeten, R. Gostl, R.P. Sijbesma, Stress-induced colouration and crosslinkingof polymeric materials by mechanochemical formation of triphenylimidazolyl ra-dicals, Chem. Commun. 52 (2016) 8608–8611 https://doi.org/10.1039/c6cc04312g.
[23] S.A. Siddiqui, U.C. Narkhede, S.S. Palimkar, T. Daniel, R.J. Lahoti, K.V. Srinvasan,Room temperature ionic liquid promoted improved and rapid synthesis of 2,4,5-triaryl imidazoles from aryl aldehydes and 1,2-diketones or α-hydroxyketone,Tetrahedron 61 (2005) 3539–3546 https://doi.org/10.1016/j.tet.2005.01.116.
[24] R.F. Heck, Palladium-catalyzed Vinylation of Organic Halides, Organic Reactions,John Wiley & Sons, Inc., New York, 1982, pp. 345–390.
[25] K. Maeda, T. Hayashi, The mechanism of photochromism, thermochromism andpiezochromism of dimers of triarylimidazolyl, Bull. Chem. Soc. Jpn. 43 (1970)429–438 https://doi.org/10.1246/bcsj.43.429.
[26] D.M. White, J. Sonnenberg, Oxidation of triarylimidazoles. structures of the pho-tochromic and piezochromic dimers of triarylimidazyl radicals, J. Am. Chem. Soc.88 (1966) 3825–3829 https://doi.org/10.1021/ja00968a027.
[27] H. Tanino, T. Kondo, K. Okada, T. Goto, Structures of three isomeric dimers of2,4,5-triphenylimidazolyl, Bull. Chem. Soc. Jpn. 45 (1972) 1474–1480 https://doi.org/10.1246/bcsj.45.1474.
M. Takasaki, T. Iwamura Polymer 158 (2018) 270–278
278