synthesis of fullerene derivatives with novel structures
TRANSCRIPT

Synthesis of Fullerene Derivatives with Novel Structures
Liquid-Phase versus Solid-State Reactions
Koichi Komatsu* and Yasujiro Murata
Institute for Chemical Research, Kyoto University
Received July 13, 2004
Abstract : Reactions of C60 with various carbon nucleophiles, particularly those with substituted acetylide and fluorenide ions in the liquid phase, give various C60 derivatives. While the reaction with cyanide salts only affords the cyanated C60 in liquid phase, the solid-state mechanochemical reaction selectively gives the dumbbell-shaped dimer. As typically shown by this example, the solid-state reaction was found to be quite advantageous for the formation of various types of fullerene dimers. The reaction of C60 with phthalazine also results in formation of entirely different products depending on the reaction phase. While the solid-state reaction gives a fullerene dimer incorporated in a bicyclic framework, the liquid-phase thermal reaction affords a derivative of an open-cage fullerene. Utilizing this reaction, an attempt has been made for organic synthesis of endohedral fullerene encapsulating molecular hydrogen through a series of proce-dures, i.e., opening a hole on the fullerene cage, insertion of molecular hydrogen, and reduction of the size of the opening.
1. Introduction
Since the achievement of macroscopic production of fullerenes,1 research on this entirely new type of carbon allotropes has been developed explosively in various fields of science covering chemistry, physics, biology, and interdisci-
plinary areas. Particularly, [60]fullerene (C60), which is pro-duced in the largest quantity and is regarded as a representa-tive of all the fullerenes, has attracted researchers not only because of the beauty of the molecular shape with high Ih symmetry but also owing to its great potential as functional materials and from curiosity regarding its fundamental physi-cal and chemical properties.2
Today, it is widely accepted that C60 is not a super-aro-matic compound but should he regarded as a pnlyenyl molecule with 30 double bonds on its surface. With its low-lying triply degenerate LUMO levels, C60 behaves as an electron-deficient olefin.3 Among numerous organic reac-tions reported so far, representative and frequently used reac-tions are summarized in Scheme 1. Among these, the most versatile and widely applied reactions are the "Prato reac-
tion"4 and the "Bingel reaction",5 which can afford C60
derivatives fused with variously substituted pyrrolidine and
cyclopropane ring, respectively. These reactions are typical
examples of the 1,3-dipolar cycloaddition and the nucle-
ophilic addition reactions to C60.
Since fullerenes are clusters of a large number of carbon
atoms, their solubility in common organic solvents is general-
ly quite low, and the requirement of the use of a large
amount of solvent sometimes becomes problematic. In order
to overcome this inconvenience, reactions without the use of
any solvent, or solid-state reactions, have been used in our
group. It has been found that these solvent-free reactions
can sometimes afford products which have not been produced
in liquid-phase reactions and are uniquely obtained under
solid-state reaction conditions. In the present article, our
approach to create fullerene derivatives with novel structures
by the selective use of liquid-phase and solid-state reactions
will be presented.
2. Addition of Carbon Nucleophiles to C60
In the early stage of fullerene research in the 1990s,
Scheme 1
1138 ( 72 ) J. Synth . Org . Chem ., Jpn .
Hirsch and coworkers showed that, under carefully controlled conditions, typical carbon nucleophiles such as organolithi-ums (RLi) and Grignard reagents (RMgX) react with C60 to
give R-C60-H in 47 to 80% yield after acidification.6 Howev-er, curiously there had been no report on the addition of alkynyllithium to C60. We were able to add various alkynyl reagents, R-C C-Li (R= Si(CH3)3,7a C6H5,7a C6H13,7b den.- drimer unit,7c and, terthienyl 7d), to C60 to give the corre-sponding 1,2- or 1,4-fullerene adducts, R-C C-C60-R' (1) after treating the reaction mixture with appropriate elec-trophiles (Scheme 2).7
1
2
3
4 4n 5n
A characteristic feature of the alkynyllithium addition to C60 is that the product yield is fair to good and unfavorable formation of bisadduct issupprcsscd owing to dclocalization of the negative charge in the first formed fullerenyl anion R-C m C-C60.
The dumbbell-like C60 dimer with two fullerene cages connected by an acetylenic linkage 2 was synthesized in order to examine the possible electronic interaction between the two cages, but such interaction was not detected at the
ground state upon electrochemical reduction.8 On the other hand, in the case of a dendrimer having a
fullerene core 3, weak interaction between the C60 core and the dendron units was observed upon laser flash
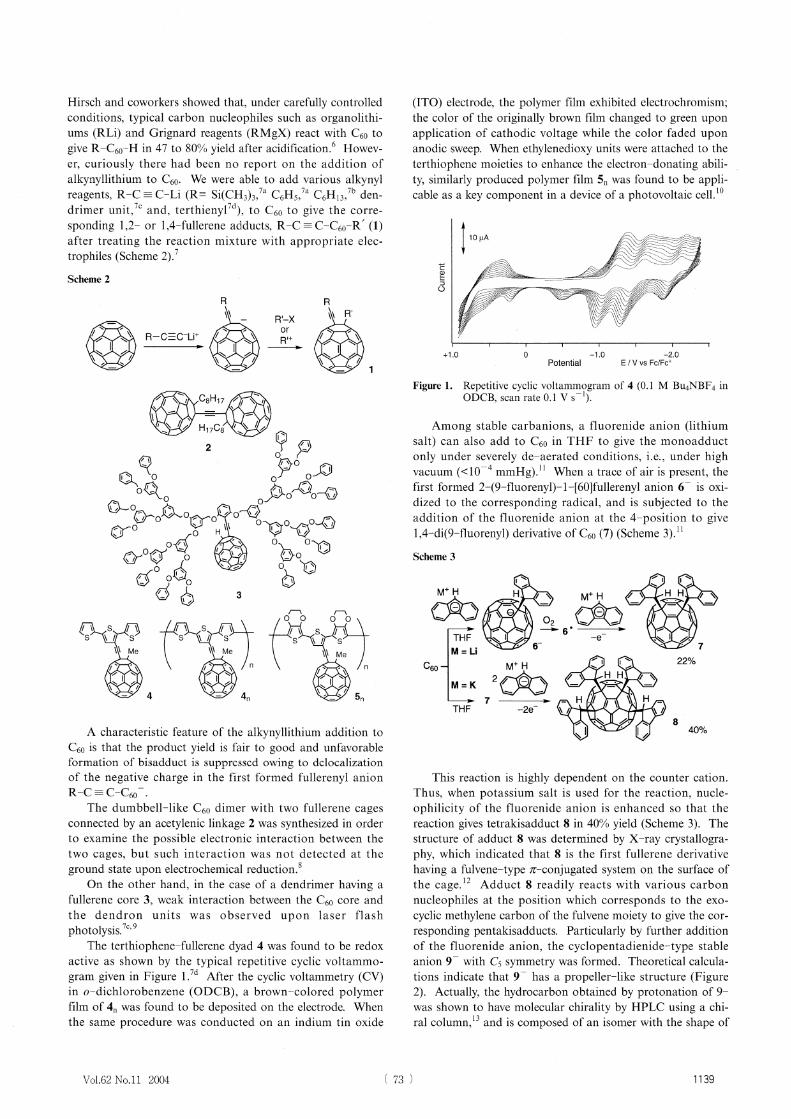
photolysis:7c, 9 The terthiophene-fullerene dyad 4 was found to be redox
active as shown by the typical repetitive cyclic voltammo-
gram given in Figure 1.7d After the cyclic voltammetry (CV) in o-dichlorobenzene (ODCB), a brown-colored polymer film of 4n was found to be deposited on the electrode. When the same procedure was conducted on an indium tin oxide
(ITO) electrode, the polymer film exhibited electrochromism; the color of the originally brown film changed to green upon application of cathodic voltage while the color faded upon anodic sweep. When ethylenedioxy units were attached to the terthiophene moieties to enhance the electron-donating abili-ty, similarly produced polymer film 5n was found to be appli-cable as a key component in a device of a photovoltaic cell.'°
Among stable carbanions, a fluorenide anion (lithium
salt) can also add to C60 in THE to give the monoadduct
only under severely de-aerated conditions, i.e., under high
vacuum (•ƒ10-4 mmHg).11 When a trace of air is present, the
first formed 2-(9-fluoreny1)-1-[60]fullerenyl anion 6 is oxi-
dized to the corresponding radical, and is subjected to the
addition of the fluorenide anion at the 4-position to give
1,4-di(9-fluorenyl) derivative of C60 (7) (Scheme 3).11
This reaction is highly dependent on the counter cation. Thus, when potassium salt is used for the reaction, nucle-ophilicity of the fluorenide anion is enhanced so that the reaction gives tetrakisadduct 8 in 40% yield (Scheme 3). The structure of adduct 8 was determined by X-ray crystallogra-
phy, which indicated that 8 is the first fullerene derivative having a fulvene-type 7r-conjugated system on the surface of the cage.12 Adduct 8 readily reacts with various carbon nucleophiles at the position which corresponds to the exo-cyclic methylene carbon of the fulvene moiety to give the cor-responding pentakisadducts. Particularly by further addition of the fluorenide anion, the cyclopentadienide-type stable anion 9- with C5 symmetry was, formed. Theoretical calcula-tions indicate that 9- has a propeller-like structure (Figure 2). Actually, the hydrocarbon obtained by protonation of 9-was shown to have molecular chirality by HPLC using a chi-ral column,13 and is composed of an isomer with the shape of
Scheme 2
Figure 1. Repetitive cyclic voltammogram of 4 (0.1 M Bu4NBF4 inODCB, scan rate 0.1 V s-1).
Scheme 3
Vol.62 No.11 2004 ( 73 ) 1139

a right-handed propeller and its enantiomer. A more selec-
tive and higher-yield synthesis of C5 symmetrical pentak-
isadduct has been developed by Nakamura's group,success-
fully.14
9- 9-
3. Solid-State Reactions of C60
As described above, nucleophilic additions to C60 can give various derivatives of structural interest and possessing
potential functionalities. One major disadvantage in carrying out these reactions is the low solubility of C60 (and some of the products) so that a large amount of the solvents is required for the reaction and for the work-up process. It would be not only convenient but desirable from the view-
point of environmental protection if we can conduct these reactions without the use of any solvent. This consideration has led us to attempt the solid-state reaction of C60.15
For the solid-state reaction to take place, the reactants have to be vigorously mixed by applying external mechanical energy. Thus, the solid-state reaction may be called mechanochemistry. The application of this method to purely organic reactions had not been well developed before the pio-neering and systematic work by Toda and coworkers.16
As the procedures for mechanochemical solid-state reac-tions, the simplest is grinding using a mortar and pestle. When a longer reaction time is required, ball-milling is used." In order to apply much higher mechanical energy for the reaction, we decided to use what we call a "high-speed vibration mill" (HSVM). This is a rapidly vibrating mixer-mill, frequently used in a laboratory for the purpose of milling crystals of KBr into fine powder to be pressed to form a pellet for IR spectral measurement of organic com-
pounds. The main part of this type of mill is essentially a stainless-steel capsule and a milling ball, which are rapidly vibrated at the speed of 3500 rpm. Based on the observation of phase transformation of inorganic materials such as BeF2 and B2O3, pressure as high as 10-20,000 atm is considered to be generated during the milling process using HSVM.18
The chemical reactions are usually driven by the action of external activation such as heat, photo-irradiation, microwave irradiation, and so on. In the case of mechanochemical solid-state reaction, the high local pressure
generated as described above is considered as an important external activator. In addition, the reacting species are free from solvation. These two unusual factors are expected to induce some novel reactions, which can not take place in liq-uid-phase reactions. 3.1 Solid-State Nucleophilic Reaction to C60
The addition of a carbon nucleophile to C60 was the first solid-state reaction we tried. When a mixture of C60, BrCH2CO2C2H5, and Zn was vigorously milled by HSVM for
20 min and treated with acid, the Reformatsky-type reagent
formed in situ was found to add to C60 with satisfactory effi-
ciency as shown in Scheme 4.19
Although similar HSVM treatment of Co with organic bromides (R-Br; R = alkyl, aryl) and magnesium gave a complex mixture, the use of alkali metals afforded the expect-ed C60 derivatives R-C60-H albeit in modest yields from 11 to 24%.20
The versatile cyclopropanation of Co known as the "Bin-
gel reaction" (shown in Scheme 1) is a nucleophilic addition of a carbon nucleophile, generated by deprotonation ofα- halogenated malonic acid esters orα-halogenatedβ-dike-
tones, toCo followed by intramolecular nucleophilic substi-tution.3'5 In typical liquid-phase reactions, the most fre-
quently used base for the deprotonation is DBU (1,8-diazabi-cyclo[5.4.0]undec-7-ene). Although an inorganic base such as NaH was used in early reports, there has been no report as to the use of weaker inorganic bases. However, for the Bingel reaction to proceed in the solid state under the solvent-free HSVM conditions, the organic base (DBU) was found to be not effective, but weak inorganic bases such as Na2CO3, NaHCO3, NaOAc, Ca(OH)2, and Na2B4O7 were to be used. Thus, when a mixture of Co, diethyl bromomalonate, and Na2CO3 in a 1:1:1 molar ratio was vigorously milled under the HSVM conditions for 30 min, cyclopropanated fullerene 10 was obtained in 51% yield (86% based on consumed C60) along with 7% (12% based on consumed C60) of bis-cyclo-
propanated product 11 (Scheme 5).21
3.2 Solid-State Dimerization of Fullerenes It has been known that NaCN can add to Co in solution
to give cyanated C60 (12) (Scheme 6a).22 Unexpectedly, how-ever, when C60 was treated with KCN or NaCN under solid-state reaction conditions, no cyanated product was formed but an entirely different product, that is, a dumbbell-shaped dimer C120 (13) was selectively obtained (Scheme 6b).23,24a The structure of 13 was unambiguously determined by X-ray crystallography.23 It turned out later that it is not necessary to use such a poisonous compound like KCN as a catalyst but potassium salts such as CH3CO2K, KOH, and K2CO3, metals with high reducing power such as Li, Na, K, Mg, and Al, and also some solid amines such as 4-aminopyridine, 4-(dimethylamino)pyridine, and 3,4-diaminopyridine are as effective as KCN.24a It is to be noted that the yield of the
Figure 2. Calculated structure (AM1) of 9.
Scheme 4
Scheme 5
1140 ( 74 ) J. Synth . Org . Chem . , Jpn .

produced dimer is nearly 30% and unreacted C60 is recovered in almost 70% yield regardless of the catalyst or reaction time employed.24a Based on the effectiveness of the reducing met-als, the mechanism of this reaction is assumed to involve one-electron transfer as a key step as shown in Scheme 6c. Thus, the formation of C60 radical anion, which is stable in solution but should be highly reactive in the solid without any solvation, must play an important role in its reaction with abundant C60 molecules surrounding it. The reverse reaction starting from dimer C120(13) under the same solid-state reaction conditions was also found to give a reac-tion mixture of Co and C120 in a ratio of nearly 7:3, which can thus be regarded as an equilibrium mixture.24a
12
13
When C60 was allowed to react with 4-aminopyridine under solid-state HSVM conditions, not only the fullerene dimer C120 (13) but trimer C180 (14) was also produced albeit in a low yield (4%).25 For trimer 14, theoretically there are eight possible isomers with the linearly connected structures and one isomer with a cyclic structure. The experimentally obtained trimeric product actually consisted of two groups of isomers A and B, which must have structures substantially different from each other as judged from their behaviors on HPLC analysis. Because of the poor solubility, no spectro-scopic identification was possible, but by direct observation with a scanning tunneling microscope (STM), the isomers included in A were found to be those with folded structures while the isomer in B was shown to have a cyclic structure.26 This assignment is in agreement with the results of theoreti-cal calculations on the correlation between structures and energies of each isomer.
C-180
(14)
It was also possible to prepare a fullerene cross-dimer.
Thus, when an equimolar mixture of C60 and C70 was allowed
to react in the presence of 4-aminopyridine under the
solid-state HSVM conditions for 30 min, the cross-dimer C130 (15) was obtained in 3% yield in addition to homo- dimer C120 (10%) and unreacted Co (80%) and C70 (97%).27 The 13C NMR and electronic spectroscopy of produced C130 indicated that C60 reacted selectively at the 1,2-bond of C70, which is known to be the most reactive due to the highest curvature present in this molecule and the resulting largest
pyramidallization in the carbons at the 1,2-positions.3
C130 (15)
The fullerene dimer connected by a single bond and a sili-
con or germanium bridge, 16 or 17, was also synthesized by
the solid-state reaction. When a mixture of C6o,
dichlorodiphenylsilane or -germane, and lithium metal was
vigorously milled with HSVM for 30 min, 16 or 17 was
obtained in 7.5%28 or 9%29 yield, respectively (Scheme 7). A
similar dimer having a methylene bridge or an oxa-bridge
has also been prepared by a solid-phase thermal reaction at
200•Ž.30
16 M = Si 7.5%
17 M = Ge 9%
The fullerene dimers C120 (13) and 0130 (15) as well astrimer C180 (14) are stable in the solid phase but in solution they slowly dissociate into the original fullerene molecules by heating or by photo-irradiation. Also, upon electrochemical reduction these dimers undergo dissociation as soon as they acquire one extra electron.23,27 However, in the case of 1628 and 1729 as well as their oxa analogue,31 the electrochemical reduction did not cause such dissociation, and indicated that there is appreciable electronic interaction between the two fullerene cages. Thus, the two cages are not reduced at the same time but in a stepwise fashion, reflecting the electronic communication between the two C60 cages. 3.3 Solid-State Cycloaddition Reactions of C60
Fullcrcncs arc known to readily undergo cycloaddition reactions with dienes and 1,3-dipolar compounds.3 The most well-known example of the latter is the "Prato reaction",4 which takes place between fullerene and azomethine ylide formed in situ from derivatives of glycine and aldehydes. This reaction can also be conducted under the solid-state conditions. Thus, when the reaction of C60 with N-methyl-or N-ethylglycine in the presence of paraformadehyde or var-ious substituted benzaldehydes was conducted under the HSVM conditions (1 h), it gave a moderate yield (20-30%) of fulleropyrrolidine 18 together with about 50% recovery of C60
(Scheme 8).32 The optimum molar ratio of the reagents, C60, glycine, and aldehyde, was found to be 1:1:1 to 1:2:2.
A reaction of C60 with anthracene can be taken as a typi-cal example of the [4+2] cycloaddition. In a liquid-phase thermal reaction, the produced adduct also dissociates into the starting materials so that the reaction reaches an equilib-rium state as shown in Scheme 9.33
Scheme 6
Scheme 7
Vol.62 No.11 2004 ( 75 ) 1141

18
19 20
When this reaction was conducted under the solid-state reaction conditions using HSVM, the yields of both monoadduct 19 and bisadduct 20 increased to 55% and 19%, respectively, in spite of much shorter reaction time (1 h) as shown in Scheme 9. The time-dependence of the reaction is shown in Figure 3a.34 Apparently, the curve for the forma-tion of 19 reaches a plateau with almost 60% yield at the reaction time of 30 min. The reverse reaction, i.e., the disso-ciation of 19, was also found to take place under the HSVM conditions, again leaving nearly 60% of 19 apparently unchanged as shown in Fig. 3b. Thus, a kind of equilibrium state appears to be attained even under the present heteroge-neous solid-state reaction conditions.
In the case of the reaction of C60 with pentacene, a liquid-phase reaction in refluxing toluene for 72 h gave only
(a) (b)
the monoadduct 21 and bispentacene adducts 22 in 59% and 13% yield, respectively, in accord with the generally known highest reactivity at 6,13-carbons of pentacene.35 In sharp contrast, the solid-state reaction of C60 with an equimolar amount of pentacene using HSVM for 1 h afforded not only the adducts 21 and 22 in 19% and 15% yield, respectively, but the bisfullerene adduct 24 in 11% yield.34 The formation of 24 indicates that C60 trapped the thermodynamically less favored monoadduct 23 formed by the cycloaddition of C60 at 5,14-carbons (Scheme 10).
2122
2324
A quite similar type of solid-state [4+2] reaction was also observed between C60 and an equimolar amount of di(2-pyridy1)-1,2,4,5-tetrazine under the HSVM conditions for 1 h (Scheme 11). The reaction was very clean and the 1H and 13C NMR spectra of the resulting brown powder exhibit-ed signals corresponding to 1:1 adduct 25 as a single product only contaminated by a small amount (5%) of unreacted C60.36 The isolated yield of 25 was larger than 90%, and this is to be compared with 50-60% yield when the same reaction was conducted in refluxing toluene.37
On the other hand, when the HSVM reaction was con-ducted between the same reactants in a 2:1 molar ratio and the resulting solid mixture was heated at 150t for 2 h, fur-ther [4+2] addition of C60 to 25 took place to give bis-fullerene compound 26 with two C60 cages incorporated in a 2,3-diazabicyclo[2.2.2]oct-2-ene framework in 27% yield along with C60 recovered in 40%.38
25
26
As has been described in section 3.2, the mechanochemi-
cal solid-state reaction using HSVM sometimes gives a prod-
uct totally different from that obtained from a liquid-phase
reaction. Another example is the reaction of C60 with phtha-
lazine, i.e., 2,3-diazanaphthalene. This reaction afforded bis-
Scheme 8
Scheme 9
Figure 3. Time dependence of HPLC peak areas under the HSVM conditions; (a) for the reaction of C60 with 1.2 equivalents of anthracene, and (b) for the retro-cycloaddition of C60-anthracene monoadduct 19.
Scheme 10
Scheme 11
1142 ( 76 ) J. Synth . Org . Chem . , Jpn .

27
28
2930
fullerene compound 28 with two C60 cages incorporated in a
benzobicyclo[2.2.2]octene framework under the solid-state
HSVM conditions followed by heating at 200•Ž while the
reaction gave an open-cage benzofullerene derivative 30 when
the starting materials were heated at 255°C in a solution in
1-chloronaphthalene.39 These reactions are considered to
proceed by the mechanism via a common intermediate 27 as
shown in Scheme 12. Thus, the intermediate 27, formed by
the first [4+2] reaction between the starting materials and the
following nitrogen extrusion, is subjected to the second [4+2]
cycloaddition with one of the Co molecules surrounding 27
to give bisfullerene compound 28 in the solid-state reaction
(route a). In contrast, when 27 is formed in solution the
highly reactive butadiene unit of orthoquinodimethane struc-
ture in 27 undergoes the intramolecular formal [4+4] reaction
with the fullerene diene system to give 30 via the retro
[2+2+2] reaction of the second intermediate 29 (route b) in
the liquid phase reaction.39
Since the two C60 cages are rigidly fixed in close proximi-
ty in bisfullerenyl compounds 26 and 28, a facile [2+21 reac-
tion takes place between the two cages upon irradiation of
visible light to their solutions to give the more rigidly con-
nected dimers 3138 and 3239 quantitatively. The electrochemi-
cal measurements indicated that the electronic interaction
between two fullerene cages is stronger in 31 and 32 than in
26 and 28.
31 32
4. Synthesis of Open-Cage Fullerenes
The liquid-phase thermal reaction of Co with phtha-lazine, which afforded a derivative of open-cage fullerene
(Scheme 12b), prompted us to initiate the synthesis of open-cage fullerenes using the reaction with polyazaaromatic compounds.
We considered that this might lead to a "molecular surgery" approach 40 to the organic synthesis of endohedral fullerenes. Although the endohedral fullerenes are expected
to have great potential to be used as functional materials in
molecular electronics and also in the medical field, their pro-
duction still relies on the physical method, which has not
made much progress since the early days of fullerene
research. Thus, the preparation and isolation of the endohe-
dral fullerenes require special techniques and tremendously
laborious work. It is a truly challenging task for organic
chemists to try for the organic synthesis of endohedral
fullerenes in macroscopic amounts. Conceptually, this will be
attained by the following steps, i.e., opening a large enough
hole on the fullerene cage, inserting a small molecule or atom
through this hole into the cage, and closeing the hole, all
using organic reactions.
4.1 Opening a Hole on C60
The typical examples of open-cage fullerenes having a
hole larger than an 11-membered ring are shown in Figure 4.
The first example of an open-cage fullerene having an
11-membered-ring hole (33) was synthesized by Wud1.41
This compound containing the smallest stable atom, helium,
in a ratio of about 1/1000 was once prepared, and an attempt
was made to discharge the helium atom by heating. Howev-
er, it was found that a helium-3 atom could not pass through
the opening upon heating at the temperature higher than
100•Ž.42 On the other hand, Rubin reported a derivative hav-
ing a cobalt metal coordinated just above an 11-membered-
ring opening of his open-cage compound, but it was not pos-
sible to push the metal atom inside the fullerene cage.43
However, he later synthesized an open-cage fullerene with a
14-membered-ring hole 34.44 He succeeded in incorporation
of a helium atom or a hydrogen molecule in this compound
under the high temperature and high pressure conditions
(288-305•Ž/475 atm or 400•Ž/100 atm, respectively), but the
incorporation rate was only 1.5% and 5%, respectively.40
Quite recently, Iwamatsu and Murata reported a reaction of
33 34 35 36
Scheme 12
Figure 4. Examples of open-cage C60 derivatives.
Vol.62 No.11 2004 ( 77 ) 1143

diaminobenzene with an open-cage fullerene 35, which they
prepared previously,45 to widely open the hole into a 20-membered ring(36).46 This compound with an amazingly large hole was found to readily incorporate a molecule of water into the opened cage in a ratio of about 75%.46
Following the previous experience described above
(Scheme 12b), we attempted a liquid-phase thermal reaction of C60 with 4,6-dimethyl-1,2,3-triazine at 180°C, and obtained a C60 derivative with an 8-membered-ring open-ing,47 which was further enlarged to a 12-membered-ring by
photochemical oxidation. However, because of the low yield (7%) and extreme instability of the first obtained product, we decided to search for a better reaction.
We next examined a reaction of C60 with 3-(2-pyridy1)- 5,6-dipheny1-1,2,4-triazine in the hope of introducing a nitrogen atom at the rim of the hole, which was expected to be advantageous for further functionalization of the product. In addition, the aromatic substituents are favorable for enhancement of the solubility of the C60 derivatives to be formed. When an equimolar mixture of C60 and the 1,2,4-triazine was heated at 180°C in ODCB, there was obtained the expected open-cage C60 derivative 37 in 50% yield (85% based on consumed C60) together with unchanged C60 recov-ered in 41% (Scheme 13).48 The mechanism for this reaction is considered to be essentially the same as that for the forma-tion of 30 in Scheme 12, that is, by (1) [4+2] cycloaddition of Co with the 3,6-positions of the triazine, (2) extrusion of nitrogen, (3) intramolecular formal [4+4] reaction, and (4) retro [2+2+2] reaction. In addition to the full spectroscopic data, the structure of 37 was unambiguously determined by X-ray crystallography as shown in Figure 5. Upon a close look at the structure, it is noticed that the double bond C2-C3 on the rim of the orifice is considerably twisted with a dihedral angle for Z Cl-C2-C3-C4 being 39°. The HOMO coefficients of the corresponding olefinic carbons are also rel-atively higher than those of other sp2-carbons. These facts
37
imply the high reactivity of these double bonds at the rim, which is advantageous for the further ring-enlargement reac-tion as will be described below. 4.2 Enlargement of a Hole on C60
In agreement with the previous reports,45,49 the enlarge-ment of an eight-membered-ring hole on C60 in 37 by air-oxidation under irradiation of visible light proceeded smoothly to give mainly two isomers of ring-opened dike-tones 38 and 39 (Scheme 14) in good total yield.48
37 38 39
It is well known that Co itself is readily reduced to a
tetraanion state upon cyclic voltammetry (CV) at room tem-
perature and to a hexaanion state at low temperature, reflect-
ing the presence of low-lying triply-degenerate LUMOs.3
Then it was of interest to examine the redox behaviors of the
open-cage fullerenes 37, 38, and 39 with considerably rup-
tured ƒÎ-conjugated systems. Surprisingly, when we conduct-
ed CV on these open-cage fullerenes they were found to
exhibit four reversible one-electron reduction waves similarly
to C60 in spite of such structural deformation. Particularly,
the first reduction potentials of compounds 38 and 39
(-0.97 and -0.98 V vs Fc/Fc+, respectively) are even lower
than that of C60 (-1.17 V). This is attributed to the pres-
ence of two carbonyl groups, which causes the lowering of
their LUMOs, and, thus, 38 and 39 can be regarded as good
electron acceptors.
So, for the purpose of further enlargement of the 12-
membered-ring hole on these compounds, insertion of a sul-
fur atom to the rim of the opening was attempted in the pres-
ence of tetrakis(dimethylamino)ethylene (TDAE) as a typical
π-electron donor. As shown in Scheme 15, this reaction on
38 proceeded smoothly by heating in ODCB to give aCo
derivative with a 13-membered-ring hole 40 as a single prod-
uct in 77% yield."
38 40
Again, the structure of 40 was determined by X-ray crys-
tallography as shown in Figure 6. The size of the opening
measures 3.8 •~ 5.6•ð by the distance between carbon atoms
although the actual hole should be much smaller when real
atomic sizes are taken into consideration. On the other
hand, the shape of the opening in 40 is rather circular com-
pared with that of 34 having a 14-membered-ring hole,
Scheme 13
Figure 5. X-ray structure of open-cage fullerene derivative 37.
Scheme 14
Scheme 15
1144 ( 78 ) J. Synth . Org . Chem . , Jpn .
which is somewhat more elliptic. Thus, the energy for inser-
tion of hydrogen as a representative of a small molecule into
40 was estimated to be 30.1 kcal mol-1,50 which is 11.3 kcal
mol-1 lower than that for 34.40
4.3 Hydrogen Insertion through a 13-Membered-ring
Hole on 40
Insertion of a hydrogen molecule into 40 was conducted
simply by applying high pressure of hydrogen gas (800 atm)
to the powdery sample of 40 heated at 200°C in an autoclave
for 8 hours. There is no change or decomposition observed
in the recovered sample, and it exhibited 1H and 13C NMR
spectra which are the same as those of 40 itself except a
sharp singlet signal appearing at -7.25 ppm in 11-1 NMR.
This signal is assigned to the hydrogen molecule encapsulated
in the fullerene cage, which is known to exert a strong shield-
ing effect.51 The integrated intensity of this signal corre-
sponded to 2.00 H, thus indicating that 100% encapsulation
was attained to give the endohedral open-cage fullerene
derivative, [email protected]
Although H2@40 was quite stable at room temperature
under air or under vacuum, it slowly released hydrogen when
heated in an ODCB solution at 160 to 190•Ž. The rate of
release followed the first-order kinetics. The activation ener-
gy for release of the encapsulated hydrogen was found to be
34.3 kcal mol-1, which is in fairly good agreement with the
calculated value, 28.7 kcal mol-1.
The identity of H2@40 was confirmed also by MALDI
TOF MS spectrometry, which exhibited a clear molecular-
ion peak for 112g40 as shown in Figure 7(a). When the
(a) (b)
spectrum was taken under the irradiation of higher laser
power, the peak intensity for H2@40 decreased, but instead peaks at m/z 720 and 722 appeared as shown in Figure 7(b).50These peaks apparently correspond to C60 and hydrogen con-taining fullerene, i.e., H2@C60, indicating that the structure of C60 is energetically so favorable that it can be reconstructed even from such a complicated structure as 40 by a series of rearrangements involving removal of the benzene and pyri-dine rings and extra carbon, oxygen, and nitrogen atoms. It is to be noted that about 1/3 of the produced C60 still retains hydrogen inside, based on the comparison of the peak inten-sities.
5. Conclusion
Among numerous organic reactions reported for fullerene C60, nucleophilic reactions and cycloadditions, those which
give rise to fullerene derivatives with structures of particular interest were selected in the present article. The advantage in the use of the solid-state, solvent-free reactions under the mechanochemical conditions has clearly been shown in the selective synthesis of the [2+2] type fullerene dimers, C120 and 0130, in addition to other dimers of various structures. On the other hand, the liquid-phase thermal reaction of C60 with a 1,2,4-triazine derivative was found to be a versatile method for the synthesis of an open-cage fullerene. A series of trans-formations including opening a cage on the fullerene surface, enlargement of the opening, and insertion of hydrogen gas into the cage through the opening have afforded a fullerene derivative containing molecular hydrogen in 100% for the first time. Gradual reduction of the size of the opening and final removal of the extra organic addends are expected to lead us to reach the first organic synthesis of endohedral fullerene in the near future. Acknowledgments The present research was supported by a Grant-in-Aid for COE Research on Elements Science (No. 12CE2005) and by a Grant-in-Aid for Creative Scientific Research Collaboratory on Electron Correlation Toward a New Research Network between Physics and Chemistry (No. 13NP0201) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
References
1) Kratschmer, W; Lamb, L. D.; Fostiropoulos, K.; Huffman, D. R. Nature 1990, 347, 354.
2) (a) McLafferty, F. W, Eds. Acc. Chem. Res. 1992, 25(3), specialissue on buckminsterfullerenes. (b) The Fullerenes; Kroto, H. W., Fischer, J. E., Cox, D. E., Eds.; Pergamon Press: Oxford, 1993. (c) Electronic Properties of Fullerenes; Kuzmany, H., Fink, J., Mehring, M., Roth, S., Eds.; Springer Series in Solid-State Sciences 117; Springer-Verlag: Berlin, 1993. (d) Physics and Chemistry of the Fullerenes; Prassides, K., Eds.; NATO ASI Series, Series-C: Mathematical and Physical Science-Vol. 443; Kluwer Academic Publishers: Dordrecht, 1994. (e) Progress in Fullerene Research; Kuzmany, H., Fink, J., Mehring, M., Roth, S., Eds.; World Scientific: Singapore, 1994. (f) The Chemistry of Fullerenes; Taylor, R., Eds.; World Scientific: Singapore, 1995. (g) The Chemical Physics of Fullerenes 10 (and 5) Years Later; Andreoni, W, Eds.; NATO ASI Series, Series-E: Applied Sci-ence-Vol. 316; Kluwer Academic Publishers: Dordrecht, 1996. (h) Taylor, R. Lecture Notes on Fullerene Chemistry: A Handbook for Chemists; World Scientific: Singapore, 1999. (i) Diederich, F.; GOmez-LOpez, M. Chem. Sos, Rev. 1999, 28, 263. (j) Guldi, D. M.; Prato, M. Acc. Chem. Res. 2000, 33, 695. (k) Braun, T.; Schubert, A. P.; Kostoff, R. N. Chem. Rev. 2000, 100, 23. (1) Fullerenes: From Synthesis to Optoelectronic Properties (Develop-ments in Fullerene Science, V 4); Guldi, D. M., Martin, N., Eds.;
Figure 6. X-ray structure of open-cage fullerene derivative 40.
Figure 7. MALDI-TOF mass spectra of H2@40 obtained in thenegative ionization mode. (a) Spectrum taken with a laser power slightly above the threshold for ion formation. (b) Spectrum taken with a higher laser power. Insets show the expanded spectra.
Vol.62 No.11 2004 ( 79 ) 1145

Kluwer Academic Publishers: Dordrecht, 2002. (m) Perspec-tives of Fullerene Nanotechnology, Osawa, E., Eds.; Kluwer Aca-demic Publishers: Dordrecht, 2002. (n) Fullerene-Based Materi-als, Prassides, K., Eds.; Springer Series in Structure and Bond-ing 109; Springer-Verlag: Berlin, 2004.
3) Hirsch, A. The Chemistry of the Fullerenes; Thime: New York, 1994.
4) Maggini, M.; Scorrano, G.; Prato, M. J Am. Chem. Soc. 1993, 115, 9798.
5) Bingel, C. Chem. Ber. 1993, 126, 1957. 6) (a) Hirsch, A.; Soi, A.; Karfunkel, H. R. Angew. Chem. Mt. Ed.
Engl. 1992, 31, 766. (b) Hirsch, A.; GrOsser, T.; Skiebe, A.; Soi, A. Chem. Ber. 1993, 126, 1061.
7) (a) Komatsu, K.; Murata, Y; Takimoto, N.; Mori, S.; Sugita, N.; Wan T. S. M. J Org. Chem. 1994, 59, 6101. (b) Murata, Y; Motoyama, K.; Komatsu, K.; Wan, T. S. M. Tetrahedron 1996, 52, 5077. (c) Murata, Y; Ito, M.; Komatsu, K. J. Mater. Chem. 2002, 12, 2009. (d) Murata, Y; Suzuki, M.; Komatsu, K. Org. Biomol. Chem. 2003, 1, 2624.
8) Komatsu, K.; Takimoto, N.; Murata, Y; Wan, T. S. M.; Wong, T. Tetrahedron Lett. 1996, 37, 6153.
9) Kunieda, R.; Fujitsuka, M.; Ito, O.; Ito, M.; Murata, Y.; Komatsu, K..1 Phys. Chem. B 2002, 106, 7193.
10) (a) Furukawa, K.; Morita, M.; Nakashima H.; Kashimura, Y; Torimatsu, K.; Murata, Y.; Yamazaki T.; Komatsu, K.; Maruyama, N.; Yamao, T.; Fujita, S. Proceedings of the 206th Electrochemical Society Meeting (Honolulu), 2004, in press. (b) Yamazaki, T.; Murata, Y; Komatsu, K.; Furukawa, K.; Mori-ta, M.; Maruyama, N.; Yamao, T.; Fujita, S. Org. Lett., submit-ted for publication.
11) Murata, Y; Komatsu, K. Tetrahedron Lett. 1996, 37, 7061. 12) Murata, Y.; Shiro, M.; Komatsu, K. I Am. Chem. Soc. 1997,
119, 8117. 13) Murata, Y; Komatsu, K. to be published. 14) (a) Sawamura, M.; Iikura, H.; Nakamura, E. J Am. Chem. Soc.
1996, 118, 12850. (b) Nakamura, E.; Isobe, H. Acc. Chem. Res. 2003, 36, 807.
15) Komatsu, K. Top. Cure Chem. in press. 16) (a) Toda, F. Synlett 1993, 303. (b) Toda, F. Acc. Chem. Res.
1995, 28, 480. (c) Tanaka, K.; Toda, F. Chem. Rev. 2000, 100, 1025.
17) (a) Rowlands, S. A.; Hall, A. K.; McCormick, P. G.; Street, R.; Hart, R. J.; Ebell, G. F.; Donecker, P. Nature 1994, 367, 223. (b) Field, L. D.; Sternhell, S.; Wilton, H. V. Tetrahedron 1997, 53, 4051.
18) Dacheille,F.; Rustum, R. Nature 1960, 186, 34. 19) Wang,,G-W; Murata, Y; Komatsu, K.; Wan, T. S. M. Chem.
Commun. 1996, 2059. 20) Tanaka, T.; Komatsu, K. Synth. Commun. 1999, 29, 4397. 21) Wang, C. W; Zhang, T. H.; Li, Y J.; Lu, P.; Zhan, II.; Liu,
Y.-C.; Murata, Y.; Komatsu, K. Tetrahedron Lett. 2003, 44, 4407.
22) Keshavarz-K. M.; Knight, B.; Srdanov, G.; Wudl, F. J Am. Chem. Soc. 1995, 117, 11371.
23) Wang, G.-W.; Komatsu, K.; Murata, Y; Shiro, M. Nature 1997, 387, 583.
24) (a) Komatsu, K.; Wang, G-W; Murata, Y; Tanaka, T.; Fuji-wara, K.; Yamamoto, K.; Saunders, M. J Org. Chem. 1998, 63, 9358. (b) Lebedkin, S.; Gromov, A.; Giesa, S.; Gleiter, R.; Renker, B.; Rietschel, H.; Krdtschmer, W. Chem. Phys. Lett. 1998, 285, 210.
25) (a) Komatsu, K.; Fujiwara, K.; Murata, Y. Chem. Lett. 2000, 1016. (b) Fujitsuka, M.; Fujiwara, K.; Murata, Y.; Uemura,
S.; Kunitake, M.; Ito, O.; Komatsu, K. Chem. Lett. 2001, 384. 26) Kunitake, M.; Uemura, S.; Ito, O.; Fujiwara, K.; Murata, Y;
Komatsu, K. Angew. Chem. Mt. Ed. 2002, 41, 969. 27) Komatsu, K.; Fujiwara, K.; Murata, Y. Chem. Commun. 2000,
1583. 28) Fujiwara, K.; Komatsu, K. Org. Lett. 2002, 4, 1039. 29) Murata, Y.; Han, A.; Komatsu, K. Tetrahedron Lett. 2003, 44,
8199. 30) (a) Smith III, A. B.; Tokuyama, H.; Strongin, R. M.; Furst, G.
T.; Romanow, W J. J. Am. Chem. Soc. 1995, 117, 9359. (b) Lebedkin, S.; Ballenweg, S.; Gross, J.; Taylor, R.; Kratschmer, W. Tetrahedron Lett. 1995, 36, 4971.
31) Balch, A, L.; Costa, D. A.; Fawcett, W. R.; Winkler, K. J Phys. Chem. 1996, 100, 4823.
32) Wang, G.-W; Zhang, T.-H.; Hao, E.-H.; Jiao, L.-J.; Murata, Y; Komatsu, K. Tetrahedron 2003, 59, 55.
33) (a) Komatsu, K.; Murata, Y; Sugita, N.; Takeuchi, K.; Wan, T. S. M. Tetrahedron Lett. 1993, 34, 8473. (b) Schlueter, J. A.; Sea-man, J. M.; Taha, S.; Cohen, H.; Lykke, K. R.; Wang, H. H.; Williams, J. M. J Chem. Soc., Chem. Commun. 1993, 972. (c) Tsuda, M.; Ishida, T.; Nogami, T.; Kurono, S.; Ohashi, M. J Chem. Soc., Chem. Commun. 1993, 1296. (d) Cruz, P.; Hoz, A.; Langa, F.; Illescas, B.; Martin, N. Tetrahedron 1997, 53, 2599.
34) Murata, Y; Kato, N.; Fujiwara, K.; Komatsu, K. J. Org. Chem. 1999, 64, 3483.
35) Mack, J; Miller, G. P. Fullerene Sci. Technol. 1997, 5, 607. 36) (a) Komatsu, K.; Murata, Y; Kato, N. Fullerenes 2000-Volume
9. Functionalized Fullerenes; N. Matrin, M. Maggini, and D. M. Guldi Eds.; Electrochemical Society: Pennington, N. J., 2000, 20. (b) Murata, Y; Suzuki, M.; Rubin, Y; Komatsu, K. Bull. Chem. Soc. Jpn. 2003, 76, 1669.
37) Miller, G. P.; Tetreau, M. C. Org. Lett. 2000, 2, 3091. 38) Murata, Y.; Suzuki, M.; Komatsu, K. Chem. Commun. 2001,
2338. 39) Murata, Y.; Kato, N.; Komatsu, K. J. Org. Chem. 2001, 66,
7235. 40) Rubin, Y.; Jarrosson, T.; Wang, G.-W.; Bartberger, M. D.;
Houk, K. N.; Schick, G.; Saunders, M.; Cross, R. J. Angew. Chem. Int. Ed. 2001, 40, 1543.
41) Hummelen, J. C.; Prato, M.; Wudl, F. J Am. Chem. Soc. 1995, 117, 7003.
42) Private communication from H. A. Jimenez-Vazquez, R. J. Cross, and M. Saunders. The authors are grateful to Professor R. J. Cross and Professor M. Saunders of Yale University for this information.
43) Ace, M.-J.; Viado, A. L.; An, Y.-Z.; Khan, S. I.; Rubin, Y. J Am. Chem. Soc. 1996, 118, 3775.
44) Schick, G.; Jarrosson, T.; Rubin, Y Angew. Chem. Mt. Ed. 1999, 38, 2360.
45) Inouc, II.; Yamaguchi, II.; Iwamatsu, S.; Uozaki, T.; Suzuki, T.; Akasaka, T.; Nagase, S.; Murata, S. Tetrahedron Lett. 2001, 42, 895.
46) Iwamatsu, S.; Uozaki, T.; Kobayashi, K.; Re, S.; Nagase, S.; Murata, S. J Am. Chem. Soc. 2004, 126, 2668.
47) Murata, Y.; Murata, M.; Komatsu, K. J. Org. Chem. 2001, 66, 8187.
48) Murata, Y; Murata, M.; Komatsu, K. Chem. Eur. J. 2003, 9, 1600.
49) Murata, Y; Komatsu, K. Chem. Lett. 2001, 896. 50) Murata, Y; Murata, M.; Komatsu, K. .J. Am. Chem. Soc. 2003,
125, 7152. 51) Saunders, M.; Cross, R. J.; Jimenez-Vazquez, H. A.; Shimshi,
R.; Khong, A. Science 1996, 271, 1693.
1146 ( 80 ) J. Synth. Org. Chem., Jpn.

PROFILE
Koichi Komatsu is Professor of organic chemistry at the Institute for Chemical Research, Kyoto University. He received his B. Eng. (1966) and Ph. D. degrees (1974) from Kyoto University. He joined the Department of Hydrocarbon Chemistry, Kyoto University, as Instruc-tor in 1971, right after he finished his graduate study in the same university. He worked as a postdoctoral fellow in the group of Professor Robert West of the University of Wisconsin in 1975-76. He was promoted to Lecturer in 1984 and to Associate Professor in 1989 in the same department, and to Professor at the present institute of Kyoto Univer-sity in 1995. Professor Komatsu received the Divisional Award of the Chemical Society of Japan (organic chemistry) in 1998 and the Alexander von Humboldt Research Award in 2002. His research interests include the synthe-sis, structures, and properties of novelπ-cor巾gated systems havingσ-π conju-
gation and also three-dimensional
π-conjugation, i.e., fullerene derivatives.
Yasujiro Murata is an Assistant Profes-sor at the Institute for Chemical Research, Kyoto University. He was born in Kanazawa in 1970 and received his B. Eng. (1993) and Ph. D. degrees (1998) from Kyoto University. During that time he joined Professor Fred Wudl's group as a summer student (1995). After working as a postdoctoral fellow at Kyoto University under Profes-sor Koichi Komatsu, he joined the Insti-tute for Chemical Research, Kyoto Uni-versity, as an Assistant Professor. In 2004, he received The Chemical Society of Japan Award for Young Chemists. His research interests include the synthe-sis of fullerene derivatives having novel structures and properties.
Vol.62 No.11 2004 ( 81 ) 1147