synthesis of materials within reverse micelles · introduction reverse micelles as nanoscale...
TRANSCRIPT

June 2, 2005 12:33 00700
Surface Review and Letters, Vol. 12, No. 2 (2005) 239–277c© World Scientific Publishing Company
SYNTHESIS OF MATERIALS WITHIN REVERSE MICELLES
VUK USKOKOVIC∗ and MIHA DROFENIK †
Joz e f S t e fan I n s t it u t e , J am ova 38, 1000 Lju b ljan a, S l oven ia†Faculty of Chemistry and Chemical Engineering, Smetanova 17, 2000 Maribor, Slovenia
Received 1 March 2005
Reverse micelles as nanosized aqueous droplets existing at certain compositions of water-in-oil
microemulsions are widely used today in the synthesis of many types of nanoparticles. How-
ever, without a rich conceptual network that would correlate the properties and compositions of
reverse micellar microemulsions to the properties of to-be-obtained particles, the design proce-
dures in these cases usually rely on a trial-and-error approach. As like every other science, what
is presently known is merely the tip of the iceberg compared to the uninvestigated vastness still
lying below. The aim of this article is to present readers with most of the major achievements
from the field of materials synthesis within reverse micelles since the first such synthesis was
performed in 1982 until today, to possibly open up new perspectives of viewing the typical prob-
lems that nowadays dominate the field, and to hopefully initiate the observation and generation
of their actual solutions. We intend to show that by refining the oversimplified representations
of the roles that reverse micelles play in the processes of nanoparticles synthesis, steps toward
a more complex and realistic view of the concerned relationships can be made.The first two sections of the review are of introductory character, presenting the reader with
the basic concepts and ideas that serve as the foundations of the field of reverse micellar syn-thesis of materials. Applications of reverse micelles, other than as media for materials synthesis,as well as their basic structures and origins, together with experimental methods for evalu-ating their structural and dynamic properties, basic chemicals used for their preparation andsimplified explanations of the preparation of materials within, will be reviewed in these twointroductory sections. In Secs. 3 and 4, we shall proceed with reviewing the structural anddynamic properties of reverse micelles, respectively, assuming that knowledge of both staticand dynamic parameters of microemulsions and changes induced thereof, are a necessary stepprior to putting forth any correlations between the parameters that define the properties ofmicroemulsions and the parameters that define the properties of materials synthesized within.Typical pathways of synthesis will be presented in Sec. 5, whereas basic parameters used todescribe correlations between the properties of microemulsion reaction media and materialsprepared within, including reagent concentrations, ionic strength, temperature, aging time andsome of the normally overlooked influences, will be mentioned in Sec. 6. The whole of Sec. 7is devoted to reviewing water-to-surfactant molar ratio as the most often used parameter inmaterials design by performing reverse micellar synthesis routes. The mechanisms of particleformation within precipitation synthesis in reverse micelles is discussed in Sec. 8. Synthesis ofcomposites, with special emphasis on silica composites, is described in Sec. 9. All types of materi-als, classified according to their chemical compositions, that were, to our knowledge, synthesizedby using reverse micelles up-to-date, will be briefly mentioned and pointed to the correspond-ing references in Sec. 10. In Sec. 11, some of the possible future directions for the synthesis ofnanostructured materials within reverse micelles, found in combining reverse micellar syntheses
∗Corresponding author.
239

June 2, 2005 12:33 00700
240 V. Uskokovic & M. Drofenik
and various other synthesis procedures with the aim of reaching self-organizing nanoparticlesystems, will be outlined.
Keywords: Nanomaterials; reverse micelles; review; synthesis.
1. Introduction
Reverse micelles as nanoscale hydrophilic cavi-ties of microemulsions have been known since the1960s, but these diverse multimolecular structureswere for the first time used as nano-templates formaterials synthesis (of monodispersed Pt, Pd, Rhand Ir particles) in 1982.1 After these pioneer-ing researches, many different materials comprisingde-agglomerated and monodispersed particles(Fig. 1) have been prepared2–6 by using reversemicelles. Reverse micellar synthesis of materialsbelongs to the class of wet materials synthesis pro-cedures, and exhibits, in general, all the advantagesthat usually accompany other wet approaches tomaterials synthesis. Excellent control of the finalpowders’ stoichiometries with possibilities of obtain-ing homogeneity and mixing on the atomic scale,narrow particle sizes distributions, negligiblecontamination of the product during the homoge-nization of the starting compounds, low energy con-sumption, low aging times and simple equipment,are some of the ordinary qualities of wet syntheses,especially when compared to high-temperature tra-ditional routes for the synthesis of common ceramicand metallic materials. Improved control of the par-ticle sizes, shapes, uniformity and dispersity areadditional general advantages of reverse micellarsynthesis compared to other, bulk wet approaches.Materials resulting from wet powder preparationmight have extremely small sizes, which implies anumber of potential advantages, such as lower sin-tering temperatures in case of the preparation ofceramic materials. In cases where high-temperaturetreatment cannot be completely avoided by room-temperature aging procedures in reverse micelles,the formation of nanoparticles with high specific-surface area can enable calcination temperaturesto be set lower compared to traditional, solid-stateapproaches.
Organized self-assembled surfactant phases haverecently received a lot of attention as reaction andtemplating media, but have for a long time been used
(a)
(b)
Fig. 1. (a) La–Ni oxalate nanoparticles5 and (b) silicaparticles6 as obtained by reverse micellar synthesis inmicroemulsion.
for many other purposes:
• As wash and cleaning systems — since they facil-itate the solubilization of both hydrophobic andhydrophilic components at the same time and atreduced temperatures.
• As separators — due to selective solubilization ofcertain molecules.
• As lubrication compounds.• As pharmaceuticals.

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 241
• As fuels — due to improved fuel atomization andevaporation of water which increases the heat andtemperature of combustion.
• As a medium in crude oil exploitation — due totheir surface activity.
• As catalyzers7,8 or inhibitors9 of biochemicalenzyme-driven reactions — due to compartmen-talization of reactants and products as well aschanges in activity and substrate specificity ofenzymes due to alteration in solubilized enzymesconformations when accommodating the micellarinterior structure.
• In preparative organic chemistry, in order to over-come reactant solubility problems due to the abil-ity of microemulsions to solubilize both polar andnonpolar substances and to compartmentalize andconcentrate reactants.10
• For storing bioactive chemical reagents.11
• As a cell membrane-mimetic medium for the studyof membrane interactions of bioactive peptides.Encapsulating a protein in a reverse micelle anddissolving it in a low-viscosity solvent can lowerthe rotational correlation time of a protein andthereby provide a novel strategy for studying pro-teins in a variety of contexts.12 Since it wasnoted that denaturation of proteins can be pre-vented in reverse micelles,13 these self-organizedmultimolecular assemblies have been used as life-mimicking systems,14 and as such, have receivedlarge interest after the proposition of hypothe-ses that self-replicating biochemical reactions ofprimordial planetary life were initiated in reversemicelles made of glycerine molecules, palmitic,stearic and oleic acid, which existed at air–waterinterfaces close to the shores of some ancientseas.15 Treating reverse micelles as active and evenas the most basic structures of life was especiallyemphasized after the discovery of the possibility ofinitiating self-replication of reverse micelles due toa reaction ocurring within micellar structures.16,17
2. Basic View of Reverse Micelles,Microemulsions and TheirPotentialities Within MaterialsSynthesis
Reverse micelles exist at certain compositional rangeof water-in-oil microemulsions. Microemulsions — aterm coined by J. H. Schulman in 195918 — aretransparent thermodynamically stable dispersions
of two immiscible liquids containing appropriateamounts of surfactant. Amphiphilic substances, towhich surfactants belong, possess significantly dis-tanced hydrophilic and hydrophobic parts withintheir molecules, and, therefore, each of the twoparts of the surfactant molecule has its preferentsolvent. Two mutually immiscible components areusually water and an alcohol, hydrocarbon or,lately, environmentally-viable supercritical carbondioxide,19,20 which proves to be of extreme con-venience due to solving the problems of difficultseparation and removal of solvent from productsin conventional reverse micellar syntheses. Surfac-tant monolayers separate water and oil domains andhence reduce the unfavorable oil–water contact. Incontrast to macroscopic emulsions which are ther-modynamically unstable, nanosized microemulsiondroplets are formed spontaneously and, althoughthe reverse micellar systems are heterogenous on amolecular scale, they are equilibrium phases and arethus thermodynamically stable. Since the interac-tions between polar head-groups of the surfactantmolecule and the interactions between the nonpolartails favor only aggregates of a very specific size andmolecular configuration, microemulsions typicallyhave narrow droplet size distributions. The dropletuniformity of microemulsions is especially important,having a direct effect on the distribution of resultingparticle sizes during precipitation reactions.
Colloidal particle formation is a complex pro-cess, which involves interplay between nucleation,nanocrystal formation, intermediate growth as wellas eventual coagulation and flocculation, that alldepend on the specific interactions between ionic andmolecular species within microemulsion. The suc-cess of the reverse micellar procedure for materialssynthesis is closely related to the fact that particlenucleation can be initiated simultaneously at a largenumber of locations within reverse micelles, with thenucleation sites well isolated from each other due tothe presence of surfactant films that may act as sta-bilizers of the formed particles. Normally, monodis-perse particles are formed only when the nucleationand growth stages are strictly separated, which is aproperty of reverse micelle material synthesis due tothe uniform nanodroplet structure and specific inter-micellar interactions.
Reverse micelle phases are tiny droplets ofwater, encapsulated by surfactant molecules and thus

June 2, 2005 12:33 00700
242 V. Uskokovic & M. Drofenik
physically separated from the oil phase (Fig. 2).Simplified representation of the reverse micellarpreparation of particles takes that aqueous “pools” ofthe reverse micelles act as nanoreactors for perform-ing simple reactions of synthesis, and that the sizesof the microcrystals of the product are directly deter-mined by the sizes of these pools.21–23 It is possibleto control the sizes of reverse micelles by controllingthe parameter w, defined as molar ratio of water-to-surfactant. The higher the w, the larger the waterpools of the micelles and the nanoparticles formedwithin, and vice versa. Although such a correspon-dence between the size of the synthesized nanopar-ticle and the parameter w was approved in manyexperiments, it has been put into question lately,since within a number of performed syntheses a
(a)
(b)
Fig. 2. A drawing of (a) a reverse micelle and (b) amore realistic model of reverse micelle.35 Blue spheresrepresent surfactant’s head-groups, whereby smalleryellow spheres denote counterions. Note that the surfac-tant head groups do not completely shield the aqueousinterior of the modelled reverse micelle (b).
similar correspondence could not be established.Therefore, the dynamic interaction among micelleshas since lately been generally considered as the mostimportant factor that influences the morphologiesand properties of the final products.24
Particle size control results from regulation ofthe nucleation site size and the particle growth rate.Within reverse micellar materials synthesis, thesetwo factors are often implicitly conveyed to reversemicelle sizes and material exchange between reversemicelles, respectively. It has been observed thatmany properties of the synthesized powders can becontrolled, and thus designed by using proper condi-tions, regarding primarily the composition of a par-ent microemulsion. Control over quantum states ofthe particles or interparticle spacing can lead to novelmesoscopic properties of materials, which are some-times very different from those of their atomic andbulk counterparts.25,26 Not only it is possible to syn-thesize nanosized uniform particles, but the obtainedparticles might also be extremely well dispersed,2
which is a key property for some applications. Eventhough reaction kinetics are neglected in many mod-els since the intermicellar exchange is slow and gov-erns the growth of particles, enhancement of reactionrates is also one of the known possible advantages ofmicellar synthesis routes.24,27
Since nanosized reverse micelles cannot yetbe directly observed in their dynamic interac-tions within a nonpolar medium, indirect tech-niques are usually applied in order to evaluatecertain, both static and dynamic properties of themicelles. In the approximations and different implicitpre-suppositions of various such techniques lie theanswer to the sometimes pronounced impossibilityof matching28 the concluded properties attributedto the same systems by using different experimen-tal methods.
Various indirect experimental methods may beused for the evaluation of the microemulsions’phase diagrams, but generally, transparency mea-surements, conductivity measurements29 and variousdynamic spectroscopy methods are the most oftenused. Scattering methods have in general becomestandard methods for probing microemulsions due tonondestructivity and versatility of such approaches.Since reverse microemulsion systems typically have

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 243
a high degree of optical clarity, the transition fromthe translucent, bright color of microemulsion toa turbid, opaque or viscous white solution mightvisually (with the naked eye) be interpreted as thebreakdown of the microemulsion.30,31 However, itwas shown that the solubilization limits obtained bytitration are different from those obtained by con-tacting the organic phase with an excess aqueousphase,32 and such a difference was ascribed to thefact that in the titration method, the composition ofwater pools is fixed by the composition of the titrant,while in the contacting method, the composition ofthe water pools depends on the exchange of ionsbetween the reverse micelles and the excess aque-ous phase.32 Conductivity measurements are oftenused to routinely characterize microemulsion systemsand determine the maximum amount of water thatcan be introduced into the system with maintainingthe given water-in-oil system33 in stable condition.Upon addition of water into the system, conductiv-ity increases due to percolation of charges throughthe droplet clusters, all until continuous introductionof water into the system makes the microemulsionunstable and eventually induces phase separationresulting in sharp decrease in conductivity.
Since reverse micelles are most often regardedas “nano-templates” or “nano-reactors” for thesynthesis of nanosized particles, special empha-sis in the field of their investigation is, from thepoint of view of materials synthesis, placed onthe attempts to determine their structural param-eters, that is primarily micellar sizes and shapesas well as spatial distributions of these parameters.Beside many attempts to model reverse micellarstructures,34–36 initiated by the pioneering attemptsby Brown and Clarke,37 within indirect experimen-tal methods that are regularly used in order todetermine or evaluate different properties of reversemicelles, are included: light scattering (LS)38 andsmall angle neutron scattering (SANS) studies,39–42
small angle X-ray scattering (SAXS),41 quasi-elasticlight scattering (QELS),43 infrared spectroscopy,including FTIR,44,45 picosecond IR pump-probespectroscopy46 and near-IR spectroscopy,11 dielectricspectroscopy,47,48 fluorescence light and visiblelight scattering measurements,49 X-ray scatteringtechniques,50,51 ESR spectroscopy,52 freeze-fracture
etching transmission electron microscopy,53
NMR,53,54 measurements of diffusion coefficients,55
conductometric measurements29,33,56,57 spec-trophotometric measurements,57,58 shear andextensional viscosity measurements,59–61 photoncorrelation spectroscopy,62 fluorescence quenchingmeasurements,63 dynamic light scattering,46 Ramanspectroscopy,64 and DSC measurements.45 In gen-eral, spectroscopy has been a primary tool for inves-tigating the structures of reverse micelles. Combinedwith time-resolved methods, the spectroscopic meth-ods can also yield dynamic information.35
The principle factors for explaining structuralchanges in microemulsions are surfactant shape,entropy, energy terms, as well as solvent proper-ties such as ionic force and pH. Some of the mostimportant quantities used for defining surfactantsactivities within microemulsion systems are: criti-cal micelle concentration (CMC) — defined as theminimal concentration of surfactant molecules abovewhich micelles are formed; and aggregation number— the number of surfactant molecules per micelle.The phase diagram of CTAB/1-hexanol/watermicroemulsion is shown in Fig. 3.65 The balancebetween electrostatic or polar interactions, geome-try packing factor and topology mostly determinethe geometry of the colloidal structure formed. Theregion denoted as L2 in Fig. 3, typical of its lowwater-to-oil phase ratio, belongs to the phase compo-sitions at which reverse micelles exist as multimolec-ular structures that the microemulsion comprises.Beside reverse micellar structures, membraneousstructures, bilayer (lamellar) and cubic liquid crys-tals, sponge phases, hexagonal rod-like structures,and uninverted (regular) micellar structures can beformed by varying the composition of the given, mostoften three- or four-component microemulsion sys-tem. The general remark is that microemulsion sys-tems are very difficult to treat when the number oftheir components exceeds three. The use of normalmicelles,66–68 bicontinuous structures,69,70 water-in-oil-in-water pseudovesicular structures71 or highlypercolated pearl-like structures72 for the materialssynthesis are also receiving more and more attention.
AOT [sodium bis(2-ethyl hexyl) sulfosuccinate]and CTAB (cetyltrimethylammonium bromide) havebeen two mostly used surfactants for materials

June 2, 2005 12:33 00700
244 V. Uskokovic & M. Drofenik
Fig. 3. Phase diagram of CTAB/1-hexanol/water microemulsion.65
Fig. 4. Molecular structures of some of the most com-monly used surfactants as components of microemulsionsfor materials synthesis.
synthesis (Fig. 4), but surfactants such as dode-cyl penta(oxyethylene) ether (C12E5), n-dodecyloctaoxyethylene glycol monoether (C12E8),cetylbenzyldimethylammonium chloride (CBAC),
didodecyl-dimethylammonium bromide (DDAB),14,73
sorbitan monooleate,74 and sodium dodecylbenzene-sulfonate (NaDBS)75 have all been used often. Sur-factants can be ionic in nature — like anionic AOTor cationic CTAB, or non-ionic (in which case theycan be zwitterionic or dipolar) — like Triton X-100[polyoxyethylene(10)isooctylphenyl ether],76 poly-oxyethylene(4) lauryl ether (known as Brij30),77
pentaoxyethylene-glycol-nonyl-phenyl ether (knownas Igepal-CO520)78 or the various mixtures ofpoly(oxyethylene)5 nonylphenol ether (NP-5),79
poly(oxyethylene)9 nonylphenol ether (NP-9)80 andpoly(oxyethylene)12 nonylphenol ether (NP-12).Cationic surfactants such as CTAB have been shownnot only to be effective in retaining the powder’sproperties that are the direct consequence of de-agglomeration of particles — such as superparamag-netism is, for instance81 — but also to be effectiveat condensing and thereby accelerating the trans-port of genetic material (DNA) across biologicalmembranes.82 Molecular structures of some of thecommonly used surfactants for the reverse micellarmaterials synthesis, are shown in Fig. 4.
Whereas AOT/isooctane/water83 has beenthe major AOT-based microemulsion for thematerials synthesis, AOT/cyclohexane/water, AOT/n-heptane/water42,84 and AOT/toluene/water85
have been used as well. Many different microemul-sions based on CTAB have been used: CTAB/hexanol/water,53,86 CTAB/2-octanol/water,1,87

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 245
CTAB/1-butanol/n-octane/water,29,69,88,89 CTAB/n-pentanol/hexane/water,90,91 and CTAB/n-butanol/isooctane/water.92 It was suggested thatn-pentanol is, in combination with CTAB, a bet-ter co-surfactant compared to n-butanol since it hasa stronger van der Waals interaction with CTAB,which ensures the formation of more compact andstable interfacial film protecting nanoparticles fromaggregation and nonuniform growth.90 Hexane isdenoted as a desirable organic solvent since, dueto its high vapor pressure, it may induce the self-organization of monodisperse monocrystals upondewetting and/or rapid solvent evaporation andeven the formation of energetically unfavorable 1Dstructures.90
Complex processes of particles formation in solu-tion, which involve nucleation, growth, coagulationand flocculation are largely influenced by the pres-ence of surfactant and its ability to compartmentalizereactants as is the case in reverse micellar microemul-sions. This role may largely alter not only propertiesof the final product such as atomic arrangement,morphology, ground transition and product states byaffecting the step velocity and rate of crystal growthin certain preferred directions,93 but by changing thepathway of the reaction it may promote the forma-tion of a product of different identity compared todiluted aqueous solution94 or to the reverse micel-lar medium comprising different surfactants.93 Onlysurfactant as an additional component in the pre-cipitation reaction is often used in order to increasethe specific surface area of the products and stabilizethe colloidal solution,95–98 but using ligand or com-plexing stabilizing agents to inhibit particle growthis a much older procedure than the microemulsiontechnique.99 Surfactant additives have been shownto act like mobile impurities and thus affecting thestep velocity and rate of crystal growth in certainpreferred directions.100 Compared to this approach,reverse micelles present more complex structuresthat medium for the wet synthesis comprises, sincein these systems nanosized droplets of water pos-sess a significant individuality and specific organi-zation, which is supposed to reflect on potentiallyunique and monodispersed properties of the mate-rials synthesized within them. Mixed surfactantshave also been used for the reverse micelle prepa-ration of nanoparticles, and are shown to be espe-cially promising for the formation of silica-coated
nanoparticles101,102 and 1D nanostructures,103,104
such as nanotubes, nanowires, nanorods, nanobeltsand tree-like superstructures, but the effect of anyused mixture is generally unpredictable. The com-ponents in a surfactant mixture could enhance theefficiency of processes due to synergy or even antag-onism among them under different conditions.105 Ingeneral, due to the enormous complexity of physico-chemical influences, the synthesis of materials withinmicroemulsions today relies more on trial-and-errorapproaches and the tendency to reproduce experi-ments than on the formal predictions of morphol-ogy, dispersity and various other properties, priorto the performance of the corresponding synthesisprocedures.
3. Structural Properties of ReverseMicelles
Micellar structures are often presented as beingraspberry-like, with hydrophilic charged head-groupsclosely packed to each other and hydrocarbon chainsstretched towards the center of the micelle. This pic-ture is thought to be wrong for two reasons. First,owing to electrostatic repulsion it is not possible tospontaneously pack up to a hundred charged entitiesclose to each other, even if the counterion bindingis taken into account. Second, the conformation ofall tails being stretched and almost lined up wouldlead to an enormous local pressure. As an example,an NMR study showed that the uninverted micellesformed by SDS have about one-third of their surfacecovered by hydrocarbon tails.49
In an attempt to model reverse micelle by com-puter simulation, nearly all counterions of a reversemicelle comprising 70 AOT molecules, 70 Na+ coun-terions and 525 water molecules, resided at the inter-face. The surfactant head-groups did not completelyshield the aqueous core from the nonpolar exterior,and some water molecules were trapped betweenhead-groups in contact with the nonpolar phase. Thismodel suggests that it should be possible to cat-alyze a reaction of an insoluble probe at the micellarinterface.35
FTIR studies suggest that the water interiorof reverse micelles has a multilayered structure,consisting of the so-called interfacial, intermediateand core water. The interfacial layer is composedof water molecules that are bounded directly to

June 2, 2005 12:33 00700
246 V. Uskokovic & M. Drofenik
the surfactant’s polar head-groups; the intermedi-ate layer consists of the next few nearest-neighborwater molecules that can exchange their state withinterfacial water; and the core layer is found atthe interior of the water pool and has the prop-erties of bulk water.44 It was reported that fivewater molecules are tightly bound per one CTABmolecule,52 whereby by using NMR measurementsit was realized that only two molecules of water aretightly bound to one molecule of AOT.106 Thus, ifparameter w < 6− 10, the water which occupies theinterior of a reverse micelle is highly structurized dueto association with the polar head-groups of the sur-factant molecules, and the conditions for a typicaldesign of the particles in reverse micelles are said tobe not available.73,107 On the other hand, if w > 10,micelles have a free water core with bulk water sol-vent characteristics.
Reverse micelles of various surfactants can sol-ubilize different amounts of water. AOT reversemicelles are known for their ability to enclose verylarge amounts of water; parameter w ranges from0 to 70 for many systems.35 Exactly in its abilityto solubilize large amounts of water while retainingspherical micellar shapes in a variety of hydrophobicorganic solvents lies the reason for the often use ofAOT as a surfactant for the application of materialssynthesis.108 However, compared to AOT-based sys-tems, CTAB reverse micellar systems, due to higherflexibility of surfactant film that gives rise to a higherexchange dynamics of the micelles, enable signifi-cantly higher solubilization capacities of high con-centration aqueous salts.28
In contrast to CMC, micellar size varies withvarious factors in a manner which is complex andat present still difficult to predict.28 A large vari-ety of different techniques, including classic lightscattering, viscosity measurements, tracer diffusion,NMR and quasi-elastic light scattering have beenperformed in order to obtain information about thesize distribution of micelles for given parameters suchas w or weight percentage of water or surfactant.Though some of these methods gained very preciseresults according to their performers, a clear crit-icism was raised regarding the reliability of thesemethods.28 For a certain system, NMR measure-ments have clearly indicated that the distributionof micelle size is narrow and somewhat asymmetri-cal around the average value,28 whereby the SANS
measurement results have shown that reversemicelles are spherical and monodisperse at low watercontent, whereas with increase in water content, themicelles increase in size and polydispersity.109
With an increase in water content of a microemul-sion system, the size of the reverse micelles increasesas well. In general, aggregation number together withmicelle radius increases with w. Other influences onthe aggregation number of micellar configurationsare dependent on the whole system used. Thus, it wasreported that the aggregation number showed littledependence on the concentration of the surfactantor the addition of salt (up to 2.3M of sodium azide,the salt consisting of azide ion — N=N=N−, which,due to its small size and high charge is likely to diveinto the core of the investigated reverse micelles),44
whereby it was observed that size of the reversemicelle is largely influenced by the ionic strength ofits aqueous pools. The aggregation number of CTABmicelles increased from 81 to 121 with the additionof NaOH in the concentration of 0.01M, and micelleshapes were found to change from spherical to ellip-tical with the addition of NaOH.110
As expected, micelle size increases with increas-ing alkyl chain length of the amphiphile. In severalcases, the growth in size is small and correspondsto a retention of the same micellar shape, but inothers, shape changes must be invoked to explainthe data. Surfactants with longer tails will accordingto theory have a lower CMC and a larger aggrega-tion number than analogues with shorter tails; also,counterions that are more strongly bound to the sur-factant will induce a lower CMC and a higher aggre-gation number.49 Whereas the CMC depends littleon head-group structure and counterion (ionic sur-factant in a solution dissociates on surfactant ionand counterion), micelle size can vary by orders ofmagnitude.
The aggregation number calculated for linear sur-factants with C12, C14 and C16 atoms by usinga theoretical model49 were 55, 75 and 95, respec-tively. These results are in agreement with experi-mental findings for the corresponding alkylsulphatesurfactants, whereby this model predicts aggrega-tion numbers that are too small for the longestalkyltrimethylammonium surfactant. This is due toa combination of the bulkier head-group of this sur-factant as well as a change in shape of the aggregatefrom a spherical to a more prolate reverse micelle.

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 247
However, the main point of the calculation is stillpresent — within a series of surfactants differingin hydrocarbon chains length only, the aggregationnumber should increase with increasing tail length.
Closely related to the fact that a single entity orvariable cannot be invoked for any scientific expla-nation, the size of the reverse micelles is not depen-dent upon any single variable, but on the complexinteractions which are conditional for their existence.Thus, the microemulsion droplet sizes in CTAB-based reverse micellar microemulsion were in therange of 30–70nm111 at 55◦C, whereby spectro-scopic studies have shown that size of the CTABreverse micelles is in the order of 1–4nm for differ-ent water percentages.63 Reverse micelles based onCTAB ranging in length from 10 to 1000nm werealso found to coexist together.53
Albeit expectations to establishing direct linksbetween thermodynamic stability of reverse micellesin microemulsions and their low polydispersity insize, predictions based on theoretical calculationshave shown that micelles are broadly distributed insize with polydispersity index equal to 2.106 Increasein water content results not only in a nonlinearincrease in the average diameter of the nanodropletsand the synthesized particles within, but in a moreenhanced polydispersity as well.3 For the cases ofgrowth to very long, rod-like micelles, the polydisper-sity becomes large. For many systems, the low degreeof polydispersity is explained by the fact that afterthe increase in association constant up to a certainaggregation number there is a region of marked anti-cooperativity with the equilibrium constant decreas-ing with aggregation number.
Until recently it was thought that only reversemicelles spherical in shape exist. However, ithas been observed that the probe motion withinreverse micelle becomes more anisotropic withincrease in water content.52 It was proposedthat a slight anisotropic rotational motion of theprobe in micelles, as detected, for instance, byelectron spin resonance measurements, might beexplained by the formation of cylindrical aggre-gates. Generally, spherical micelles at low surfac-tant concentration (close to CMC) might, due toaggregate–aggregate interactions, alter their geome-tries at higher monomer concentrations. For micel-lar systems showing the sphere-to-rod transition,the anti-cooperativity, which lies in head-group
repulsions, is partly eliminated this way. As shownby the case of CTAB, the elimination of head-group repulsions can be brought about by coun-terions which may approach the charged groupsclosely or intercalate between them, or by certainsolubilizates which are located in the head-groupregion.28
Linear surfactants tend to form ellipsoidalmicelles more often than branched surfactants, whichalmost always form spherical micelles. Thus, forinstance, a linear surfactant cetyltrimethylammo-nium 4-vinylbenzoate forms viscoelastic solutions inwater containing cylindrical micelles of 4 nm in diam-eter and thousands of nanometers long,40 wherebyAOT-based micelles are known to exist only in spher-ical shapes. In general, the micelles formed by surfac-tants with tails of moderate length (approximatelyC10 − C16; note that both CTAB and AOT are C16
surfactants) are thought to be spherical or nearlyspherical — at least close to CMC.
One of the major challenges within modern sur-factant science is drawing a precise link betweenthe geometry of surfactant self-assemblies and thefinal structures of the synthesized materials.112 Manycases in which acicular particles were unexpectedlyproduced by reverse micellar synthesis were ascribedto the templating effect of worm-shaped reversemicelles.4,66,113 Reverse micelles based on CTAB asthe surfactant are known to produce such effects, soanother CMC at which spherical-to-worm-like tran-sition occurs is ascribed to such microemulsion sys-tems. It is also known that an alkali substance,110
salts66 or co-surfactant,53,114 such as 1-hexanol28 oran alkane,66 might induce a spherical-to-worm-liketransition. Elliptical and spherical CTAB micellesare known to be able to coexist.53,59 However,adding water to the CTAB/water/n-pentanol/n-hexane microemulsion always results in an incrementof the reverse micellar radii, whereby the micelleshapes remains spherical.91
While the formation of rod-like micelles is welldemonstrated for several cases, especially whenCTAB is used as a surfactant, other geome-tries of large non-spherical micelles have not beenobserved yet, which is in agreement with theoreticalpredictions.28 Meanwhile, the computer simulationof a reverse micelle (w = 10) formed from a noveldouble-chained phosphate surfactant in CO2, includ-ing 1616 water molecules, 160 surfactant molecules,

June 2, 2005 12:33 00700
248 V. Uskokovic & M. Drofenik
160 counterions and 6991 CO2 molecules, showedthat the modeled reverse micelle is not restricted toa spherical shape.35 Molecular interactions drive theformation of the reverse micelles, leading to a rangeof shapes.
Sphere-to-rod transitions in CTAB micelles athigher concentrations have been reported in both thepresence and the absence of added salts for surfac-tant concentrations exceeding 0.10M.51,56 The con-centration at which these transitions occur dependson the property measured. By using absolute SAXStechnique it was shown that at c ∼ 0.05M, CTABmicelles are spherical, and that at room tempera-ture the sphere-to-rod transition occurs right above0.05M, at 50◦C at 0.17M and at 70◦C at 0.25M.51
There is a number of evidence indicating a tran-sition from closely spherical to very long rod-likeaggregates for CTAB micelles at a concentration of0.2–0.3M.28 Increasing the temperature from 30to 50◦C, as well as substituting Cl− for Br− ascounterion, eliminates the transition. Addition ofsmall amounts of simple solubilizates, such as ben-zene or a long-chain alcohol (hexanol, octanol, etc.),may markedly facilitate the transition to rod-likemicelles, whereby alkanes have no effect. Decreas-ing the alkyl chain length considerably increasesthe transition concentration or eliminates the tran-sition completely.28 By using SANS studies it wasshown that micelles of CTAB molecules in 0.2M solu-tion were ellipsoidal with semi-minor axis of 2.12nmand semi-major axis of 5.62 nm, whereby aggrega-tion number was 186 and fractional charge 0.09.39
Other SANS studies have concluded that CTABmicelles are ellipsoidal in shape, have an aggrega-tion number of 177, and that in the presence ofhydrotropes the aggregation number increases dra-matically with decrease in the fractional surfacecharge of the micelles.115 Certain calculations relatedwith QELS measurements of CTAB micelles in aque-ous solutions, and the diffusion of mesoscopic opti-cal probes through the same solution have impliedextensive micellar growth and failure of the sphericalmicelle assumption.43 It has also been reported thatCTAB forms rod-shaped micelles in aqueous systemsabove the second CMC of 0.3M and assembles intohexagonal liquid crystals above 1.1M.116
Sizes and shapes of the reverse micelles aredependent on temperature. When the temperature
increases, the aggregation number was found toincrease as well.44 Although the CMC of ionic sur-factants is insensitive to temperature changes, thetendency to form micelles different from the spheri-cal increases with decreasing temperature. The sizeof the reverse micelles comprising nonionic surfac-tants increases with increasing temperature,28 suchthat sometimes at temperatures just above the roomtemperature, radii of the micelle increases togetherwith broadening of the micelle size distributionand increased excluded volume effects in magni-tude, which can be explained by considering thesphere-to-rod or sphere-to-disk transitions of themicelles.55 It is thought that two processes whichdisrupt microemulsions, that is colescence and Ost-wald ripening, accelerate at higher temperatures.Coalescence of reverse micelles is Brownian motion-driven and hence is more present at higher tem-peratures. On the other side, Ostwald ripening, thedisruption of emulsions by the growth of largerdroplets at the expense of smaller ones, is driven byKelvin effect (a high curvature of small droplets cre-ates a high internal Laplace pressure, which conse-quently increases the vapor pressure of the emulsifiedmonomers). The smaller the droplet is, the greaterthe tendency for the droplet to shrink and disappear,since the Laplace pressure increases as droplet diam-eter decreases. It has been shown that L2 range iswidening with increase in temperature.117
One common feature of reverse micellar sys-tems is the relative lack of stability even with-out encapsulated salts. One way of minimizing thisphenomenon is including a co-surfactant in thereverse micellar system. It was even noted thatfor CTAB-based reverse micelles, a co-surfactant isnecessarily required for the formation of the sta-ble microemulsion.118 The incorporation of shortchain alkanols makes reverse micelles more stable andin addition, the presence of alkanols decreases theaggregation number of the surfactant molecules andthe diameter of the reverse micelles.113 N-butanolor SDS (sodium dodecyl sulfate) are often used asco-surfactants, increasing the polarity of the sur-factant and helping to stabilize the reverse micellesolutions.73,119 N-butanol as a co-surfactant is usedtogether with CTAB to help decrease the frac-tion of the micellar head-group that is neutralizedand thereby increase the stability of the micelles.

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 249
Without the addition of a co-surfactant, the amountof free water available to carry on the reactionsis greatly reduced, as most of the water is lockedin the head-groups of surfactant molecules. Whenusing 1-hexanol as a co-surfactant in the systemCTAB/isooctane/water, the encapsulated enzyme’shalf life was increased 45-fold.118 As an oil-phase,a non-branched alcohol has been considered as anoptimal choice, but the use of diesel oil (togetherwith CTAB as a surfactant, 1-butanol, 1-propanol,1-octanol or 1-pentanol as a co-surfactant) — a com-plex mix of aliphatic alkanes with chain lengthsof up to C32 — was also noticed.30 In relationto the application of microemulsions for materialssynthesis, it is generally recommended that surfac-tant and aqueous fluid be each from about 1–30%by weight of the total system and the maximumtotal amount of surfactant and water be up to 50%(preferably to 30%). The amount of co-surfactantis preferably between 25 and 75% by weight ofcomposition.73
Penetrating into the micellar interface, co-surfactant molecules change the mean distancebetween the polar head-groups of surfactantmolecules and thus reduce the electrostatic repulsionbetween the head-groups, promoting the spherical-to-worm-like micelle transition.59,114 This penetra-tion may increase the volume of the micelle core,which is equivalent to increasing effective head-group(hydrophilic) cross-sectional area. It is well-knownthat the curvature of a micellar aggregate (whoseaqueous side is concave rather than convex, beingan essential property of reversed micelles), which isdependent on the interfacial tension between micelleand oil phase,34 is strongly influenced by the ratio ofthe effective head-group cross-sectional area to theeffective cross-sectional area of the aliphatic chain.By decreasing this ratio, the surfactant aggregateshapes should follow the trend: spheroidal micelle→ worm-like (rod-shaped) micelles → bilayer struc-tures → reverse structures.59 When the alcoholicco-surfactant chain length decreases for a givenmicellar radius, the thickness of the penetrated layeris increased and the interaction between micelles istherefore stronger, because attractive intermicellarpotential depends on the volume of the interlappingregion.62 The volume of this region increases with anincrease of micellar radius as well.
4. Dynamic Structure of ReverseMicelles
In order to understand processes in which the syn-thesis of materials within reverse micelles takes place,it is useful to outline the basic dynamic interactionamong reverse micelles. In Fig. 5, a schematic repre-sentation of certain processes ocurring within reversemicellar microemulsion as a medium for the syn-thesis reaction, is shown. On one side, the reversemicellar systems are extremely dynamic in natureand such dynamic character lies at the basis of theirthermodynamical stability. On the other hand, theappearances of reverse micelles which emerge fromnumerous experimental results are one of a labile andsensitive multimolecular configurations.107
Surfactants associate into liquid-like aggregatesthat possess rapid molecular motions often on dif-ferent time scales. The complexity of the system isdue to the highly dispersed and heterogeneous natureof the overall phase. A microemulsion system dur-ing the synthesis procedure comprises a variety ofspecies such as surfactant molecules, solutes in theaqueous phase, species which may be preferentiallydistributed in one of the phases, and the solid parti-cles formed in reaction followed by nucleation. Someof the basic interactions in such a system includethe exchange of surfactant molecules between reversemicelles and the bulk phase, the formation–breakupof dimers, trimers, and lower-level aggregates whichmay be present in relatively small concentrations,intermicellar fusion–fission of reverse micelles, andeffects involving solute species such as intramicellarkinetic reactions, particle nucleation, and growth viaintramicellar attachment and intermicellar exchangeof aqueous phase contents.24
For reactions in microemulsions involving reac-tant species completely confined within the dispersedwater droplets, a necessary step prior to the chemi-cal reaction is the exchange of reactants by the coa-lescence of two droplets.120 Microemulsions have adynamic structure wherein the droplets of the dis-persed phase diffuse through the continuous phaseand collide with each other. These collisions areinelastic because droplets coalesce and temporarilymerge with each other, but subsequently break toform separate droplets again, so that the average sizeand number of the micelles remain the same as afunction of time.120 Surfactant film flexibility is one

June 2, 2005 12:33 00700
250 V. Uskokovic & M. Drofenik
Fig. 5. Schematic representation of entities and dynamic physicochemical effects existing in a reverse micellar system.24
of the key parameters in determining the intermicel-lar exchange and particle growth processes, since theinterdroplet exchange of the particles growing insidethe reverse micelles is inhibited by the inversion ofthe film curvature in the fused dimer which, in turn,depends on the film flexibility.99
The intermicellar exchange of content leads toa distribution of material throughout the system,and a mathematical description of this phenomenonrequires the use of population balances. Partial open-ing of the surfactant layer or interfacial transfer ofaqueous pool solubilizates present close to the micel-lar surface can result in a partial exchange of mate-rial. Under equilibrium conditions, the size and theaggregation number of the reverse micelles are dic-tated by thermodynamics and equilibrium transportbetween micelles and bulk phase. As is evident from
experimental reports, even the molecules of differ-ent coexisting species follow distributions of theiroccupancy numbers, defined as the number of par-ticle species solubilized in the micelle core. For non-interacting and random kinetics, the distribution isknown to be Poissonian, whereas geometric and bino-mial distributions can result under other conditions.This feature clearly leads to a situation different fromcontinuous conditions.
The typical time scales that have been reported24
for different mechanistic effects in polar reversemicellar systems are: 1–10ns for diffusion-controlledintermicellar reaction, 50–100ns for the lifetime ofa pair of reactants confined to the micelle core,300–500ns for the formation of an encounter complexvia diffusion on the micellar surface, 1–100ns for adiffusion-controlled entry of a surfactant molecule

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 251
onto the micellar surface from the bulk phase,0.1–1µs for diffusion-controlled collision of micellaraggregates, and 0.1–1ms for intermicellar exchangeto occur. While micellar re-orientation and themolecular diffusion around the micelle occur onthe nanosecond scale, the local motion of spheri-cal micelles is very fast (1–40ps), compared to re-orientation of rod-shaped aggregates, which is seenas the third contribution to the relaxation times.121
The retention time of water molecules on the surfaceof reverse micelles lies within a nanosecond in orderof magnitude.107 Although reverse micelles have beenstudied in detail and are well characterized, espe-cially on AOT systems, the mechanistic details ofreactions of finite rates, ultrafine particle formation,and growth seem to have not been studied througha single consistent mathematical model.24
There are many unanswered questions and chal-lenges concerning role and behavior of surfactantwithin microemulsion during the materials synthe-sis. Even if a universal relationship or a set ofrelationships are developed between the structure ofsurfactants and their behavior at interfaces, the ques-tion regarding their dynamics when adsorbed layersare perturbed, as in the case of nanoparticles forma-tion, will remain open due to specific interaction withreactants and products of the synthesis procedure.105
It was shown that two populations of reverse micellesexist depending on whether they encapsulate probemolecules or not,13 which is an apparent sign of thefeedback effects of product properties on the struc-ture of reverse micelles, neglected in models thatare built on implicit adoption of chemically inert,nano-templating role of reverse micelles, and nor-mally based on the introduction of parameter w asthe major influence on product’s properties. Refer-ence 99, which is often cited, concerns the indepen-dence of the particle size of the synthesized Pt, Rh,Pd and Ir, always in the range of 2–5nm,1 on thesurfactant, water amount and reactant concentrationused in the experiments. Therefore, particle sizes canbe seen as controllable by solvent stabilization of theparticles, whereas the surfactant acts as a stabilizingagent.10
There is a large attention paid to developinga convenient theoretical framework for correlatingcomplex combinations of compositional, structuraland interactional parameters with morphologicalproperties of synthesized particles. Due to their
nonlinear nature, colloidal spheres are in generaldifficult to model in their dynamic interactions.Contrary to expectations based on DLVO theory122
(according to which, only few static charges oncolloidal particles’ surfaces can cause repulsionsstrong enough to keep them stably separated), ithas been observed that like-charged colloidal par-ticles sometimes attract each other.123 The attrac-tion between aggregates is found to increase whenthe micellar size increases, the alcohol chain lengthdecreases, the polar head-area increases and themolecular volume of oil increases.38,124 Attractionsseem to be favored by highly charged spheres invery low salt concentrations.123 Basically, the bal-ance of forces acting on three energy scales —Van der Waals attraction, randomizing influenceof thermal energy, and hierarchy of electrostaticinteractions among highly-charged colloidal parti-cles and the single-charged simple ions around themdetermines all static and dynamic properties ofmicroemulsions.
It is generally thought that since being con-fined to closed micellar structures, water moleculeshave restricted motions and their diffusion slow andapproximately equal to the diffusion of the droplets.If all the surfactant molecules are located at thewater — oil interface, they will obviously diffusewith equal rates as the droplets too.121 However,from comparing experimental results on the diffusionbehavior of various components in CTAB and water-mediated reverse micellar microemulsion, it can berealized that water has the highest self-diffusion coef-ficient (D = 2×10−9 m2· s−1) — higher than bromideion (around 7 × 10−10 m2 · s−1), surfactant (around8 × 10−11 m2 · s−1) and the least diffusive solubi-lizates with D ranging from 7 × 10−11 m2 · s−1 to7 × 10−12 m2 · s−1 depending on the concentration.Water, bromide ions and surfactant molecules haddiffusion coefficients almost independent on the con-centration above CMC value.125
NMR measurements126,127 have shown thatmobility of the ionic surfactant’s molecular chainprogressively increases from its head-group to theterminal methyl group. However, this is not alwaysthe case since mobility of 2-ethyl side chain in AOTmolecule is very restricted. Anyhow, the generalopinion is that mobility of different parts of long sur-factant molecules is independent on the surfactant’snature and presence of a co-surfactant. As water

June 2, 2005 12:33 00700
252 V. Uskokovic & M. Drofenik
content in a four-component water-in-oil AOT-basedmicroemulsion is increased, mobility of surfactant’spolar head-group progressively decreases. This effectwas ascribed to an increased order in interfacial layeras well as to the decreased freedom for inner move-ment in the case of a larger number of molecules,including the polar head when water content isdecreased. The largest increase in mobility wasobserved at low water content when water moleculeswere engaged in hydratation of sodium ions andsulphonate head, and when the water core with prop-erties of bulk water had not yet been formed.
Beside exchanging molecules of reactants bydirect coalescence of micelles, it is necessary to takeinto account the exchange of molecules betweenphases other than only interiors of aqueous nan-odroplets. This exchange might in the general casebe considered so as to occur between bounded, inter-facial states and free, bulk states. NMR investi-gations have revealed that the exchange time ofNa+ ion between the bounded and the free state inAOT-based water-in-oil microemulsion is less than10−4 s.128 However, the same time was reported tolie in nanosecond range.129 On the other hand, inves-tigations based on electric field jump130 concludedthat the retention time of I− ion in Stern’s layer isabout 10−7 s.
NMR investigations have concluded that the typ-ical time for the exchange of alcohol moleculesbetween the interfacial layer, the continuum phaseand/or the dispersed phase (depending on its affin-ity) is a lot less than 10−4 s.107 The addition of NaCl
in dispersed aqueous phase leads to the decreasein an exchange rate of alcohol molecules,131 prob-ably due to an increase in compactness of surfactantmonolayers induced by an increase in ionic strengthof aqueous phase. The exchange rate of neutralprobe molecules between the interfacial monolayerand the aqueous core of reverse micelle was estimatedto ∼ 107/sec in AOT/heptane/water microemulsionsystem at w = 31.132 The relaxation time of theexchange of surfactant molecules between the inter-facial monolayer and aqueous phase in oil-in-watermicroemulsions was experimentally determined tobe 3 × 10−8 s,133 whereby proof for the existenceof the surfactant’s exchange between two separatephases within water-in-oil microemulsions do notyet exist. NMR researches have shown that move-ment of surfactant is largely limited to the interfa-cial space between the oil and water phases.107 It isalso known that the fluidity of the surfactant’s mono-layer might be increased by decreasing the length ofalcohol co-surfactant’s hydrocarbon chain. Typicaltimes for the rearrangement of surfactant monolay-ers in birefringent microemulsions are estimated to> 0.1µs.134
Two major processes during which exchangeof reactants between reverse micelles takes placeare shown in Fig. 6. Solutes can be exchangedbetween reverse micelles either by processes of coales-cence of reverse micelles — during which temporarymerging of aqueous droplets occur (fusion) —that subsequently fragment (fission), or by partialfragmentation of droplets with consequent loss of
Fig. 6. Processes of exchange of reactants (others than via continuous phase) in reverse micellar microemulsion.107

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 253
aqueous solubilizates, that might later merge withother droplets (coagulation).
There are many indications that the merging ofreverse micelles as well as fast intermicellar exchangeof their aqueous contents are diffusion-controlled andare independent on the surfactant’s nature.135 Thetime needed for single fusion of reverse micelles tooccur is estimated at ∼ 10−6 s, whereby the timeconstant for the collision of reverse micelles withsubsequent merging is between 108 and 109/Msecin respect to the droplet concentration. A modelaccording to which intermicellar material exchangeoccurs through a water channel formed at the pointof temporary merging of reverse micelles (temporarydimer) has been considered.107
It has been discovered that compartmental-ization of reactants in reverse micelles decreasesreaction rates due to the coupling of chemicalreaction and the rate of micellar mergings. How-ever, there are many cases of catalytic effectsderived from the compartmentalization of reagents,including the increase of the rate constant ofthe hydrolysis of acetylsalicylic acid in the pres-ence of imidazole catalyst by 55 times whenthe reaction was performed in AOT/supercriticalethane microemulsion compared to the aqueousbuffer,136 and the reaction of acid hydrolysis ofphthalomohydroxamic acid in AOT/isooctane/watermicroemulsions.27 Kinetic analyses of the bimolec-ular rate constant of exchange ke of reaction
have concluded that rate constant ke is complexsince the reaction of exchange comprises Brown-ian diffusion of reverse micelles leading to collisions,collision of two droplets, water-channel opening(merging), diffusion and chemical reaction betweenreactants, as well as fragmentation of a transientdimer. The slowest step of these basic rate pro-cesses will scale the temporal aspects of the over-all process of synthesis,137 and the second stage ofthis complex reaction is decisive for limiting reac-tion rate. The rate constant ke takes an approximatevalue of 107/sec at room temperature.138 Therefore,approximately one in a thousand collisions resultin a temporary merging of dispersed aqueous nan-odroplets with exchange of reactants. However, such
effectiveness of reactants’ exchange is said to be validonly for rigid surfactant films, such as AOT-formedreverse micelles.10 For flexible films, such as the onesthat CTAB forms, one in ten collisions can giverise to micellar content exchange.10 Large activationentropy of this process is ascribed to the fact thatafter the temporary merging of reverse micelles (thatis, the formation of activated state) some of surfac-tant molecules from the interfacial layer are releasedto one of the separate phases. The lifetime of thetransient dimer cannot be longer than a few µs sincea longer lifetime would lead to the separation ofphases due to the uncontrolled droplets ripening.
Uneven fragmentation of the previously mergeddroplets leads to an increased polydispersity ofreverse micelles. However, modification of the sys-tem ought to result in a decrease in polydisper-sity. In the case of microemulsion systems basedon CTAB and pentanol, large variations in reversemicelles sizes were noticed, but an addition of alkaneled to a decrease in polydispersity, which was mostsignificant for the alkanes with long hydrocarbonchains.107 Since larger ke implies that microemul-sion acts more like a critical system, it is thoughtthat there must exist a correlation between theincrease in ke, the polydispersity of reverse micellesizes, their mutual interaction and the critical behav-ior of microemulsion. It has been observed thata low reaction rate may lead to very large parti-cles irrespective of the exchange protocol of micellarcontent.139 The limiting situations concerning chemi-cal reactions occurring between reactants enclosed inreverse micelles — merging-controlled and reaction-controlled cases are shown in Fig. 7.
Co-surfactant leads to improved chemical reac-tion rates within reverse micellar microemulsions. Ithas been noted that the addition of benzylalcohol asa co-surfactant increased ke by almost 20 times.140
The rate constant ke is dependent on the lengthof alcoholic co-surfactant’s hydrocarbon chain —it increased when 1-hexanol as a co-surfactant wasreplaced by 1-pentanol.140 The value of ke is largelydependent on the oil hydrocarbon chain’s length —decrease in ke by a factor of 10 was noticed whenmoving from dodecane to hexane.107 The micel-lar exchange rates of cyclohexane, heptane, anddecane in AOT-based microemulsions are approxi-mately 106, 107 and 108 M−1 · s−1, respectively.137
In some cases, such as for AOT/n-heptane/water

June 2, 2005 12:33 00700
254 V. Uskokovic & M. Drofenik
Fig. 7. Limiting situations for the kinetics of chemicalreactions taking place within reverse micelles.107
system at w = 33, ke was only slightly smaller(∼ 1.5×109/Msec) than the rate of droplet collisions(∼ 109/Msec), which suggests that in that case reac-tants exchange between reverse micelles is no longerlimited by the rate of interfacial layer opening, butprimarily by the diffusion of droplets.107
Even though kinetic equations describing reac-tions of synthesis include factors such as mergingof micelles which carry reactants, that must hap-pen prior to the reaction, increases in rates of chem-ical reactions taking place within reverse micellarsystems are known phenomena, and have recentlybeen almost taken as the general advantage of thismethod.24 It has been reported that the rate of oxi-dation of Fe2+ and subsequent formation of needle-like FeOOH particles by spontaneous air oxidationis from 100 to 1000 times faster in reverse micellesthan in a bulk solution, regardless of the differencesin surfactant or other conditions.52 In the case ofcertain iron complexes, a two to tenfold increasein the rate of dissociation was measured in com-parison to the pure aqueous solution.121 The cause
of the enhancement of reaction rates is not defi-nitely known, but it is widely accepted that an elec-trostatic effect in the aqueous phase of the reversemicelle is one of the reasons for the acceleration ofreactions. The properties of local reaction media arequite different from those of the bulk solutions as aconsequence of the intense local electric fields, affect-ing all the relevant parameters that modulate thereaction rates.10 Specific intermolecular interactionsat the hydrophilic sides of surfactants surroundingthe aqueous cores and specific water structure inthis region are proposed to have catalytic effects onthe rates of chemical changes.141 However, reactionkinetics are neglected in many models because inter-micellar material exchange is relatively slow, and itplays a major role in particles growth.24
5. General Synthesis Procedures
One way to perform reverse micellar synthesis ofmaterials is to produce one parent microemulsionand then to successively let reactants to diffuse intothe interior of reverse micelles and react. The majorproblem with this so-called single-microemulsionapproach is that all reactants do not react atapproximately same conditions defined by theirphysical surroundings, but significant concentra-tion gradients are involved. The second problem isthat the composition of the microemulsion is grad-ually changed as solutions of different reactantsare successively introduced, which might inducesignificant transitions in micellar or some othermultimolecular structural properties. Therefore, amulti-microemulsion approach, within which sepa-rate microemulsions of the same compositions areprepared for every reactant involved in the syn-thesis, is used most often in order to overcomethe problems of the single-microemulsion approachand achieve a better control over synthesis param-eters. Schematic illustrations of single- and multi-microemulsion approaches to the materials synthesis,are presented in Fig. 8. It has been observed thatthe latter, so-called multi-microemulsion approachyields finer particles when compared to the singlemicroemulsion approach.80,142 The particle size dis-tribution was also found to be different when synthe-sis is performed by single- and double-microemulsionapproaches. The particle size distribution had a

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 255
Fig. 8. Schematic illustration of various stages in the growth of nanosized particles in reverse micelles in multi-microemulsional approach (up) and single-microemulsional approach (down).121
Gaussian symmetrical bell shape for the single-microemulsion approach, whereby bimodal distribu-tion was attributed to the powder synthesized by thedouble-microemulsion approach.142
Whereas the procedures based on separately dis-solving the reagents within dispersed reverse micellesare used most frequently in the context of materialssynthesis, the procedures according to which a typeof ion that is to become incorporated in the finalproduct, is initially introduced as the constituentof the surfactant molecules, are also often followed.CdS nanoparticles were synthesized by using Cd-based surfactant, such as cadmium bis(ethyl-2-hexyl)sulfosuccinate.143,144 Ultrafine ZnS and CdS parti-cles were, as well as CaCO3 particles,145 prepared inreverse micelles by using extractant-metal ion sur-factant complex as a metal ion source.146 In the caseof the preparation of metallic particles, the reduc-tion reactions are initiated, whereby in the case ofthe preparation of ceramic particles, usually a pre-cipitation agent in the form of an acidic or alkaliagent is used and further thermal treatment is per-formed; otherwise, an oxidation reaction is initiatedas well.147,148
Disruption of micelles in the reaction mixturemight be carried out by introducing alcohols inexcess, causing the nanoparticles to precipitate.The major problem of the recovery procedure iscaused by high surface energy of ultrafine parti-cles, which makes them coagulate irreversibly when
reverse micelles are destroyed without any protec-tion treatment,149 such as surface modification of theparticles. Removing the surfactant caps surroundingthe individual, well-dispersed particles most oftenyields highly agglomerated samples even thoughthe particles were well dispersed in their parentmicroemulsion. Centrifugation is mostly used for theseparation of precipitate from the liquid phase, butmagnetic separation by a permanent magnet hasbeen used as well,150,151 just like filtration, freeze-drying or phase separation by cooling the reversemicroemulsion.152 To separate CTAB from sensi-tive polypeptide nanoparticles, gel permeation chro-matography (GPC) was used.111 Washing of theprecipitate in order to remove residual surfactantand oil-phase molecules might be done with water,ethanol, chloroform or the mixture of two. Since mostsurfactants including CTAB and AOT are soluble inthese solvents, their molecules are together with by-products to be separated from the particles of thedesired product.
Even though the calcination is sometimes anecessary step to obtain fine crystalline sam-ples, in situ syntheses of nanocrystalline as-driedpowders have been frequently reported. Nanocrys-talline MnZn-ferrites117,148,153–156 [Fig. 9(a)],NiZn-ferrites147,157,158 [Fig. 9(b)], TiO2
159 weresynthesized in situ at room temperature as wellas a number of various metal particles such as Fe 29
or Bi.160 Metallic particles can be obtained much

June 2, 2005 12:33 00700
256 V. Uskokovic & M. Drofenik
(a)
(b)
Fig. 9. (a) In-situ-obtained nanocrystalline MnZn-ferrite117 and (b) NiZn-ferrite161 in CTAB/1-hexanol/water reverse micellar microemulsion, with the latterhaving saturation magnetization of 50 emu/g, which isapproximately two-thirds of a value that the same stoi-chiometric samples obtained by the traditional solid-stateroute exhibit.
easier in situ by using reverse micelles compared toceramic materials, since they are usually preparedin such a way that they are the direct products ofthe precipitation reaction between the dissociatedcationic species and a reduction agent. The sam-ples synthesized in the reverse micelles are often ofbetter crystallinity when compared to the samplessynthesized in the bulk aqueous phase. The existence
of surfactant molecules acting as cages for growingcrystallites which thereby reduce the average sizeof the particles during the collision and aggregationprocess76 is seen as the major reason for such a dif-ference in particle sizes and crystallinity.
Since many chemicals are included in the ordi-nary microemulsion synthesis (where surfactant, oilphase and co-surfactant are normally environmen-tally degradable162), one of the most important chal-lenges of future researches is to develop a way torecycle163 the components which are included in thesynthesizing procedure, so that literally the samereverse micelles might be used in the repeating cyclesof materials production. Many ‘green’ options arenowadays available, with lecithins, bile salts, dipotas-sium glycerrhizinate, biocompatible solvents likeTranscutol, glycofurol, ethanol and isopropanol, andsurfactants like poloxamers and polysorbates beingroutinely used in microemulsion-assisted synthesesof drug nanoparticles.164 Since the synthesis reac-tions proceed in stable reverse micellar microemul-sions only in the dilute solution of precursors, theresult is a low yield of nanomaterial, which places theproblem on the economic feasibility of the method.
6. General Overview of theInfluences
There are many factors that might be discerned ashaving an effect on the properties of the producedpowders within the synthesis procedure used. Withinthis paragraph, we shall number and discuss someof them.
Besides parameter w (that will be discussed indetail in the following chapter), mentioned earlieras a major variable in the design of materials thatare to be prepared in reverse micelles, particle sizesmight in certain cases be controlled by varying theamount of metal ion in each reverse micelle — moremetal ions will cause larger particles to grow becauseof diffusion.73 It was generally proposed that anincrease of particle size might be induced by increas-ing the reactant concentration, whereas a decreaseof particle size can be induced when one of thereactants is increased in excess until a plateau isreached at high excesses.99 However, one of the majorproblems with microemulsion-assisted material pro-cessing is the effect of the reactants and productson the stability of the microemulsion, particularly

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 257
the metals concentration in the aqueous phase. Itis mentioned that the ion concentration is, afterwater-to-surfactant ratio, the second most impor-tant parameter to control the particle’s size.21 Ashas been long known in the literature, the introduc-tion of a small amount of a species highly insolublein the continuous phase into the emulsion dropletssubstantially increases the stability of the dropletswith respect to the Ostwald ripening process.165 Asdroplets shrink and lose dispersed monomer, theconcentration of the insoluble species within thedroplet grows. The desire of the droplets to main-tain osmotic equilibrium by maintaining the sameosmotic pressure (same concentration of insolublespecies within each of the droplets) competes againstOstwald ripening. Droplets cannot shrink and dis-appear because of the insoluble species, and so thenumber concentration of droplets, along with thenumber concentration of insoluble species withinthe droplets, remains constant (in the absence of coa-lescence processes). Electrolyte addition in ionic sys-tems in general gives an increased micelle size; inmost cases the effect is rather small, while in othersdramatic changes may occur. Adding [Ru(bpy)3]2+
to small CTAB reverse micelles was found to changethe reverse micelle water pool size distribution frommonodisperse to bimodal.44
The influence of electrolyte addition on thereverse micellar solution phase L2 obviously dependson the interaction of individual components, and todate defies any general conclusions. Some researcheshave demonstrated narrowing the L2 range inDDAB/dodecane/water microemulsion with an in-crease in salinity,166 which is consistent with observ-ed narrowing of the L2 range in CTAB/1-hexanol/water microemulsion.117 There were also resultsaccording to which extensions of one-phase reg-ions in C9H19C6H4O(CH2CH2O)9.7H/cyclohexane/water microemulsion were not affected by the addi-tion of NaCl.167
Salts have been found to affect both aggregationand micelle formation. When the ionic strength ofthe aqueous phase is increased, the water uptakewill decrease dramatically, and the type of ionshas a great effect on the water uptake.53 Aggre-gation number, CMC and critical micelle tem-perature all depend on ionic strength. Studies ofAOT reverse micelles show that introducing elec-trolytes such as NaCl reduces aggregation number
and micelle radius. Others report that at low con-centrations (10−3–10−2 M) ions do not alter reversemicelles appreciably.44 It was reported that addingsalt to an ionic micellar solution will decreasethe CMC and increase the aggregation numberowing to the screened electrostatic repulsion.49
With a decreased electrostatic repulsion between thecharged head-groups of surfactant, it is possible topack the surfactant head-groups closer to each other,with a subsequent increase in aggregation number.
Besides expectations that the higher concentra-tion of salt precursors within reverse micelles tendsto stabilize the micelle structure, because of the sup-pression of surfactant head-groups, which causes thehead-group area to decrease and the packing ratio toincrease, inducing the formation of more stabilizedmicroemulsions, some authors have found out thatexcessively high salt concentrations tend to drivethe alcohol (co-surfactant) into the oil-phase andsome studies even suggested that this would limitthe salt concentration at which microemulsions canform at around 0.4M in the aqueous phase.168 Nev-ertheless, microemulsions, albeit with extremely sol-uble sodium nitrite and alkanes with a variety ofchain lengths comprising the oil phase, were formedat concentrations in the order of ∼ 3.5M.168 Thesolubilization capacity is dependent on the natureof the surfactant as well, since, as has been alreadymentioned, it was shown that CTAB-based reversemicelles have higher solubilization capacities of highconcentration aqueous salt compared to AOT-basedsystems.53
Beside w and pH, the particle size andmorphology is greatly influenced by the ratio of sur-factant to co-surfactant. In the CTAB/n-butanol(co-surfactant)/isooctane/water microemulsion system,the higher the ratio of CTAB to n-butanol was, thesmaller were the particles obtained. Therefore, itwas proposed that the key factor affecting the par-ticle size is the interfacial property rather that thesize of the microemulsion droplets.169 An increasein particle size could be obtained by directing anincrease of the surfactant film flexibility, which mightbe achieved not only by approaching the microemul-sion instability phase boundaries or by changing thedroplet size, but by increasing the amount of co-surfactant (alcohols) or changing the chain lengthof the oil or co-surfactant as well.99 An increasein the chain length of oil results in an increase

June 2, 2005 12:33 00700
258 V. Uskokovic & M. Drofenik
in the value of intermicellar exchange rate coef-ficient due to the fact that as the chain lengthof the oil increases it becomes increasingly coiledand therefore its penetration in the surfactant layerbecomes more difficult, resulting in a stronger mutualinteraction between surfactant tails compared tothe intensity of interaction between surfactant tailsand oil molecules.137 The co-surfactant in a quater-nary microemulsion system may have a more sig-nificant effect on the interfacial properties than theoil phase in a ternary one.169 It was shown that inCTAB/n-hexanol/water microemulsion, n-hexanolacts mainly as the continuous oil phase, although italso affects the interfacial properties. It was observedthat the average size of the nanoparticles increasedwith the increase in the n-butanol/CTAB (co-surfactant/surfactant) weight ratios, except whenthe weight ratio of n-butanol/CTAB was below 0.5(gelation occurred). It was also revealed that thevariation of the weight ratio of n-butanol/CTABaffected only the interfacial properties and not thesize of reverse micelles.169 The presence of alcoholin the CTAB/hexane/pentanol/water microemulsionis shown to be an important factor in regulatingthe size distribution of the synthesized nanoparti-cles, acting on the particles growth by influencingthe flexibility of the interfacial film.170
Different particle morphologies can in somecases be achieved simply by choosing NaOH overNH4OH.76,116 However, in the presence of a largeamount of strong base, the total ionic strength of thewater pool increases, which causes instability in themicroemulsion system.76 The ability of a strong baseto promote the hydrolytic decomposition of an ionicsurfactant will contribute to the destabilization ofthe surfactant aggregates77 and possible subsequentphase separation of the microemulsion system.3 Inorder to keep the pH value of the precipitationat the same level during the process, a computer-controlled constant-pH apparatus was used in someexperiments.171 However, it is important to note thatwater pool properties of reverse micelles — local vis-cosity, local polarity, local acidity — can be substan-tially different compared to the effective macroscopicproperties of an overall measured system.10 Investi-gations of local pH in reverse micelles by using pH-sensitive probes concluded that when NaOH and HClwere used for pH adjustments, an almost constantintensity ratio over a wide pH range was obtained,
suggesting that the water pools of microemulsions,due to a large number of polar surfactant head-groups localized at the oil–water interface, may havebuffer-like action.172
The temperature of the synthesis procedure influ-ences the properties of the synthesized powders. Itwas shown that higher temperatures induce bettercrystallinities of the synthesized powders.154,156 Thereason for this effect might be found not only in anincreased thermal motion of the active species, butby the fact that the micellar structures change aswell. Average radii of AOT-formed micelles increaseby approximately 50% when the temperature isincreased from 20◦C to 38◦C.46 Similar findings wereapplied to CTAB-117 and PEGDE-based173 systems.On the other hand, SANS studies have concludedthat both AOT shell thickness and the inner waterpool in the AOT/n-heptane/water system reversiblydecreased with increasing temperature.42 Contraryto expectations, an increased temperature did notincrease the reduction rate of Pt4+ with H2 in theCTAB/octanol/water system.1
Albeit the existence of many cases where in amatter of moments after the key reaction was initi-ated, particles stable for months were obtained,174
the average size of the particles can sometimes betuned by setting the desired time between the mix-ing of the microemulsions containing the reactingspecies, and their disruption.21 By prolonging thistime from one minute to two hours, the average diam-eter of the particles, as observed by TEM, changedfrom 13 to 35 nanometers. On reducing the reac-tion time, the morphologies of the same particlesevolved from perfect cubic shapes to more spheri-cal ones with rounded edges.21 On the other hand,prolonging the reaction time can sometimes lead toa change from cubic to spherical-shaped particles.175
Within an in situ synthesis of MnZn-ferrite particles,it was observed that the average particle size did notsignificantly change in the aging time range between1 min and 3 h, whereas ∼ 25% increase in size wasdetected after aging of the powder in microemulsionup to 26 h.156
Aging time variations can be applied in order toinduce self-organizing effects on particles assemblyscale. During an aging time of 60 days, the particlesof PANI/TiO2 composite were shown to self-organizein reverse micelles of AOT/iso-octane/water andCTAB/hexanol/water microemulsions into either

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 259
sea urchin-like or needle-like shapes.176 Four-dayaging time of prepared Prussian blue nanoparti-cles in microemulsion led to the formation of highlyordered cubic superlattices.177 By letting the reac-tion microemulsion mixtures age for more than10min in the process of preparation of CaCO3 andBaCO3, aggregation of the already-formed sphericalnanoparticles is initiated, leading to the slow for-mation of networks (after aging of up to 1 h) andsolid wires (after aging of up to 48 h).178 Intro-ducing an external magnetic field to the processof evaporating deposition of cobalt particles pre-pared in reverse micelles, led to the availability ofdesigning the arrangements of the nanocrystals intoeither hexagonal patterns of micrometer-sized dotsor labyrinthine 3D superlattices, depending on theintensity of the applied magnetic field and the evap-oration rate of the previously prepared particles.144
A similar procedure, but without the use of magneticfield, was successfully applied in obtaining “supra-crystals” made of silver nanoparticles self-organizedin 3D superlattices.179
Some of the often skipped influences mightpresent decisive parameters in the synthesis proce-dures. Bunker et al. have found that tripling theamount of microemulsions used in the synthesis pro-cedure leads to significant changes in the obtainedCdS particles absorptivity.180 It was found that theprocedure of passing N2 through the reaction mix-ture not only protects critical oxidation but alsoreduces the particle size by 25% when compared withmethods without removing the oxygen.181 Purging ofinert gases through the reaction mixture is especiallyimportant when easily oxidized elements are present,as is the case for the synthesis of ferrites147,157 andother iron-comprising compounds.182 The matter ofpurity is significant, since any introduction of for-eign substances might induce heterogenous nucle-ation with avoiding the desired monodispersity anduniform morphology of the synthesized particles.Sequence according to which adding of microemul-sion components into the mixture is done is alsoshown to be important.75 It was found that evenstirring microemulsion with a magnetically coupledstir bar during the powder’s aging time can influ-ence crystal quality and in some cases result in adifferent crystal structure as compared with nonmag-netically agitated microemulsions.183 In case of thesynthesis of organic nanoparticles in reverse
micelles,174 the use of a magnetic stirrer led to theformation of nanoparticles larger in size comparedto the particles obtained with using ultrasound bathas a mixer, even though no changes in particle sizewere detected on varying solvent type, parameter w,reactant concentrations and even geometry and vol-ume of the vessel. Particle size can be controlled bydecreasing the exchange of materials between differ-ent micelles, which might be accomplished by lower-ing the temperature or slowing the agitation of thesolution, which will in return slow the intermicellarexchange of materials, giving either smaller particlesor equally-sized particles over a longer time scale.73
All of the processing conditions which affect theformation of a nanocompound, i.e. its microstructureand stoichiometry and therefore its potential appli-cation, may be summarized as follows. Besides theidentities of surfactant (the size of hydrophilic head-group, ionic or neutral, branched or narrow, etc.),oil phase (oil chain length, primarily), co-surfactantif included (its structure, branching, chain length,etc.), metal salts, precipitating agent and/or otherreactants, there are: curvature free energy, deter-mined by the elastic constant and the curvaturesof the surfactant film;99 concentration of reactantswithin reverse micelles; pH; the sequence of introduc-ing the components in the micellar mixture; single- ormultiple-microemulsion synthesis approach; overallcomposition of microemulsion, which is usuallydecoupled to molar ratio of water-to-surfactantdescribed by parameter w and/or surfactant-to-co-surfactant ratio; aging time of the precipitate inmicroemulsion; intensity and mechanism of stirring;a method for separation of precipitated particlesfrom the liquid phase; purity of used substances andincorporation of impurities during the synthesis; andtemperature under which various processes withinmicelles proceed.
However, it is necessary to mention that ten-dencies to outline universal relationships whichgovern the processes of materials synthesis are dis-obeyed by many cases in which changing onlyone ion in the complex microemulsion had dra-matic qualitative effects on the product’s proper-ties. When only Mn2+ ions were replaced by Ni2+
ions,particles of mixed zinc-ferrites had completelydifferent morphologies. Thus, the synthesized MnZn-ferrite117 comprised spherical nanosized particles,whereas particles of NiZn-ferrite were of acicular

June 2, 2005 12:33 00700
260 V. Uskokovic & M. Drofenik
shapes.157,94 When bromide ions of CTAB sur-factant were replaced by chloride ions, the prod-uct’s identity was not the same.87 When 2-octanolwas replaced by octanol, much higher backgroundnoise was observed within XRD measurements ofthe synthesized powders.87 The inability to dis-cern and predict the influences of all the speciesinvolved in the synthesis procedure and providea set of relationships between properties of themicroemulsion used and the potentially designedproperties of the synthesized materials, is the rea-son why the method of synthesis discussed hereinhad relied mostly on the trial-and-error approach inthe past.
7. Influence of Water Content
Over the past two decades, a great deal of researchwithin the reverse micellar method for the mate-rials synthesis was undertaken to discern the var-ious parameters which significantly influence theproperties of the synthesized materials. By preciseevaluation of the effects of all the major influ-ences within the method, through control over pro-cessing parameters the desired material propertiesmight be designed. The parameter, which is in thislight regarded as the most important, is water-to-surfactant molar ratio, described by parameter w.The higher the parameter w, the larger the waterpools of the reverse micelles, and if the micelles areimagined as nanosized templating cages, then theparticle sizes might be seen to increase with theincrease of w value. However, as we shall see, thereare many cases of synthesis for which reversed cor-respondence or no linear correspondence at all hasbeen concluded.
It was found that reverse micelle size generallyincreases linearly with parameter w as well as withwater content of the microemulsion. The relationbetween water core radius r of reverse micelle innanometers and parameter w is in the case of AOT-formed reverse micelles found to be very simple: r =1.5w.22,50 Even though this relation between the sizeof the water pool radius of the reverse micelles and w
was confirmed by SANS and SAXS measurements,50
it is sometimes mistakenly identified121 with allreverse micelles, whereas it was concluded only forcertain types of AOT-based microemulsions. Besideincreasing the volume of the reversed micelles with
the water content when surfactant concentration iskept constant and decreasing the volume of thereversed micelles with the surfactant content whenthe water concentration is kept constant, it is knownthat if both the surfactant and water are increased ina constant ratio, the volume of the reversed micellewill not change, but its numbers will increase.44
In the case of reverse micelles formed by CTABmolecules, it was reported that the reverse micellesize is equal to parameter w.23 There were also indi-cations that parameter w might be taken to influencethe particle size distribution.4
It was reported that at constant surfactantconcentration S [mol/dm3], the radius r [A] ofthe droplets encapsulated by surfactant monolayersincreased linearly with the volume fraction of the dis-persed phase φ and could therefore be readily con-trolled through this parameter184:
r = (4.98 × 103)φ/AsS
where As [A2] is the area occupied by the surfactantat the droplet surface. However, this relation wouldprovide a templating relation for control of particlesize only in cases where the microemulsion struc-ture is not perturbed during the reaction. In caseswhere transfer of the components through inter-micellar diffusion and exchange processes betweenreverse micelles permits fast exchange of both solutesand surfactant between droplets, the result will bean uneven growth from which the structure of thedroplet template may be altered during the reac-tion or be totally destroyed as a result of phaseseparation.
Today, it is known that it is not only synthesizedparticle sizes that do not show in all cases lineardependencies on parameter w, but the very size of thereverse micelles also do not follow the same depen-dency. Experimental results indicate that the size ofreverse micelle depends not only on parameter w,but also on temperature, organic solvent (oil phase),type of surfactant, its concentration and presentelectrolytes. The same SAXS researches which havecome to the conclusion that dependency of reversemicelle radii on w is parametric,50 have shown thatreverse micelle radii in AOT/isooctane/water sys-tem vary upon addition of small amounts of com-pounds solubilized in the microemulsion.22 It wasnoted that the water core radius of microdropletsof water/AOT/toluene microemulsion changes from

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 261
1 to 1.6 nm when parameter w changes from 5to 10.185 As the concentration of the surfactantdecreases, the amount of water that can be solubi-lized decreases too.44 Some studies have shown thatthe smallest Stokes radii, which correspond to thedroplet size, were obtained between 15◦C and 18◦Cin case of AOT-formed microemulsions, and thatthere was no relation between the Stokes radii ofthe microemulsion and AOT content of the system.2
It was shown that the structure and composition ofan elementary water droplet enclosed by surfactantmolecules remained unchanged up to a water volumefraction per composition equal to 0.3, but this obser-vation generally refers only to microemulsions whereattractive forces are not very strong.62
It was very often found that the synthesized par-ticles are of greater average particle size than thesize of their parent reverse micelles as derived fromparameter w.185 Within a synthesis of silica particles,as the parameter w increased from 0.7 to 2.3, the par-ticle size decreased (and the size distribution becamenarrower) not in monotonous manner.6 Within thereverse micelle synthesis of BF2 nanoparticles, theparticle size was larger than the swollen micellesize,87 which demonstrates that the microemulsiondoes not consist of hard spheres that strictly limitparticle growth. In case of microemulsion synthe-sis of AlPO4-5 fibers, the dimensions of the crystalsproduced were many times larger than the typi-cal dimension of microemulsion droplets.183 Directlyproportional correspondence of reverse micelle sizesand average particle size turned to inversely pro-portional at higher water-to-surfactant ratios.186
Within the synthesis of Pd particles in CTAB/n-butanol/isooctane/water microemulsion, the aver-age diameters of the produced particle also decreasedslightly with the increase in water content when theweight ratio of surfactant-to-isooctane was fixed.169
The following explanation was assigned to this phe-nomenon: increase in w pushes the composition ofthe microemulsion toward the solubilization curve,and therefore to increased intermicellar interaction.Increases in intermicellar interaction will promotethe aggregation of the particles contained in theinteracting reverse micelles. Therefore, it can beclearly concluded that in addition to the size ofmicroemulsion droplets (described by parameter w),there must be other factors affecting the size of theproduced nanoparticles.
Today, it is generally known that for the mostsurfactant-mediated synthesis applications, the con-nection between the morphology of the surfac-tant aggregates and the resulting particle structureis much more complex (than the simple connec-tion between the sizes and shapes of the micellesto the sizes and shapes of the produced parti-cles), being affected by hardly reducible conditionsprevailing in the local microenvironment surroundingthe growing particle. These include the pH, compo-sition, and ionic strength, as well as surfactant head-groups spacing. As reaction takes place within thesemicroenvironments, many of these factors are liableto change. The decoupling of each of these effects stillremains a challenge, and provides interesting newopportunities in this field.112
8. Mechanism of Particle Formation
Synthesis of materials in reverse micelles mightbe considered as an ordinary precipitation or(co-)precipitation method of synthesis, but whichtakes place within a very specific environment. Com-pared to many other wet syntheses such as sol-gel,within reverse micelle synthesis, the precursor ionsincorporated in form of a colloidal precipitate andnot a complex compound, must be obtained. One ofthe major problems related to the co-precipitationmethod of synthesis is the inability to control super-saturation of the precipitating system which in turnleads to very poor control over the distribution ofparticle sizes, that is normally much more broadenedthan in the cases of reverse micellar syntheses.29 Dueto narrowly dispersed reverse micelles with template-like properties, the synthesis of materials withinreverse micelles very often overcome these problems.
The particle formation in a microemulsion canoccur either through collisions between micro-droplets which carry reactants within their aque-ous cores (multiple microemulsion approach) orby diffusion of the hydrophilic precipitating reac-tant to the microemulsion droplets containing theto-be-precipitated reactant (single microemulsionapproach). Although the surfactant walls of themicrodroplets act as cages for the growing parti-cles and thereby reduce the average size of the par-ticles, the exchange of the precursor ions betweenthe microdroplets by either a diffusion process or

June 2, 2005 12:33 00700
262 V. Uskokovic & M. Drofenik
a collision of microdroplets affects the particle for-mation, resulting in a similar narrow particle sizedistribution.185 Droplet collisions and rapid intermi-cellar exchange of their water content are processeswhich are mainly controlled by diffusion and are alsodependent on the nature of the surfactant.135
Wide size distribution of the synthesized parti-cles was ascribed to the fact that for reverse micelleswith smaller size, nucleation will occur only in asmall number of micelles at the beginning of theprecipitation reaction because most of them do notcontain enough ions to form a critical nucleus.53
Since the typical ion concentrations per reversemicelle are too small to form critical nuclei, the for-mation of the final precipitated particles requires theaggregation of precursor atoms from several differ-ent reverse micelles. Therefore, due to diffusion, newnuclei will form as a function of time, which will causea different growth rate of particles, and subsequentlya broad size distribution. Size distribution can benarrowed by increasing the concentration of reagents,as was proven in case of the synthesis of Ni nanoparti-cles when increased hydrazine concentration gave riseto enhanced reduction rate that led to the generationof a higher number of nuclei and, therefore, to theformation of smaller and more uniform particles.187
When using a co-surfactant to increase the fluidity ofthe interface and therefore the kinetics of the inter-micellar exchange, a more homogeneous repartitionof reactants among micelles could be ensured, thuspromoting the formation of smaller, but more numer-ous particles.188 By using a Monte Carlo model, Jainet al. showed that as the efficiency of the inter-micellar exchange increased, the resultant particlesize increased,139 whereas within the synthesis of Agnanoparticles in AOT-based reverse micelles, it wasshown that the particle size decreases at higher inter-micellar exchange rates.137 By studying the solventeffects on copper particles prepared in AOT reversemicelles, the authors concluded that growth rate andparticle size are inversely related, so that a decreasein growth rate leads to a larger particle size at aspecific w value.189 From this point of view, thedynamic intermicellar interaction might be consid-ered as the major, but thoroughly complex influenceon the properties of the synthesized particles.
One of the often skipped questions with regardto materials synthesis in reverse micelles is the mat-ter of influence of the nucleation and crystallization
of particles, as well as of their subsequent interac-tion on the structure of reverse micelles. The ten-dency for the particles to agglomerate sometimesexceeds the forces which keep reverse micelles dis-persed within the oil phase and complete or localizedphase separation occurs. Researches based on steady-state and time-resolved emission spectroscopy as afunction of water content came to the conclusionthat encapsulation of a relatively small particle ormolecule (around 1.2 nm) within a large reversemicelle (with diameter of 3 nm and w > 10) doesnot perturb the nanodroplet structure, but on theother hand, an incorporation of a similarly sized par-ticle within a small reverse micelle (with diameterof 1.5 nm and w ∼ 5) results in a redistribution ofsurfactant and water.58 Three possible hypotheseswere proposed in order to explain the final states ofthe particles synthesized within reverse micelles.190
According to the first hypothesis, nanoparticles arein the organic phase and are in direct contact withthe polar heads of the surfactant, whereas the sur-factant tails are in organic phase. According to thesecond hypothesis, nanoparticles are surrounded bya layer of water, whereas according to the thirdhypothesis, nanoparticles are surrounded by surfac-tant tails that point their polar heads toward thewater.
If the microemulsion becomes unstable, mostlyby exceeding the limiting parameter w for givenoil/surfactant ratio, the aggregation of particlestakes place, which leads to the formation of nonuni-form particles regarding their size and shape aswell as to the formation of multicore particles inthe process of composite synthesis.185 It was men-tioned that in the case of AOT/isooctane/watermicroemulsion system, the extreme size limits of5 nm and 30 nm exist, where the size distributionincreases due to the problems in micelle stabilityand crystallization.151 Also, depending on the natureof the surfactant and the type of interaction, theamphiphilic molecule may either stabilize the inor-ganic particles or induce significant aggregation. Ithas been demonstrated, for instance, that positivelycharged particles flocculate upon addition of anionicsurfactants and that, on further addition, the parti-cles re-disperse because of the formation of surfac-tant bilayers.135
When hydrothermal and reverse micellarmethods for producing zincophosphate crystals were

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 263
compared, it was noticed that the crystal growthprocess was about an order of magnitude slowerwithin the reverse micellar procedure, as comparedto the hydrothermal method.191 The surface of thehydrothermally obtained crystals was found to havemultiple layers and terraces, whereas the crystalsobtained from the reverse micellar method wereatomically smooth, which suggests a much finermechanism for the formation of particles withinthe reactions coupled with obviously morphology-regulating intermicellar exchange and surfactant-mediated growth control.
Ultrathin nanowires (Fig. 10) have recently beenproduced by using the so-called catanionic reversemicelles that are formed by mixed cationic–anionicsurfactants, whereby the molar ratio between themixed surfactants is expected to play a key rolein the synthesis of such 1D nanostructures.103 Onlymixtures with an excess of either cationic or anionicsurfactants, when ratio of two oppositely chargedhead-groups is far from 1:1, have been shown toform catanionic micelles.192 The length of V2O5
nanowires was shown to be possible to be tuned bycontrolling the aging time of the synthesized parti-cles in microemulsion,193 which is consistent withthe findings in the case of the formation of ZnSnanowires.194 Parameter w also largely influencedthe process of the formation of ZnS nanowires inC12E9/cyclohexane/water reverse micelles. It wasshown that acicular NiZn-ferrite particles are formedwithin CTAB/1-hexanol/water microemulsion onlywithin the precipitating pH value range of 8–11.94
Colloidal particles with an electric or magneticdipole moment were theoretically predicted to self-assemble into flexible chains, and these assemblieswere recently experimentally observed.195 The pro-duction of such pearl-like microstructures todaysurely presents a significant challenge for materialsdesign by using self-assembly templating amphiphilestructures.
Surfactant aggregates associate (or dissociate)with time scales that can range over several ordersof magnitude. If the time scale associated with theparticle formation is small compared to the timescale of such transformations, intermediate, nonequi-librium aggregate structures might afford additionalopportunities for templated materials synthesis. Fastkinetics associated with several solution-based pre-cipitation processes might be ideally suited for this
(a)
(b)
Fig. 10. BaCrO4 nanowires103 as obtained from catan-ionic reverse micelles consisting of (a) water, decane,undecylic acid and decyl amine and (b) molecularsieve fibers of AlPO4-5
183 as obtained from cetylpyri-dinium chloride/butanol/toluene/water reverse micellarmicroemulsion.
application. The control of solution conditions andsurfactant architecture to exploit nonequilibriumsurfactant morphologies presents an interesting sci-entific challenge.112 The potential of reverse micel-lar microemulsions in the field of nanotectonics —controlled construction of organized matter usingnanoparticle building blocks — is significant and isexpected to bring about new challenges in materialsresearch.
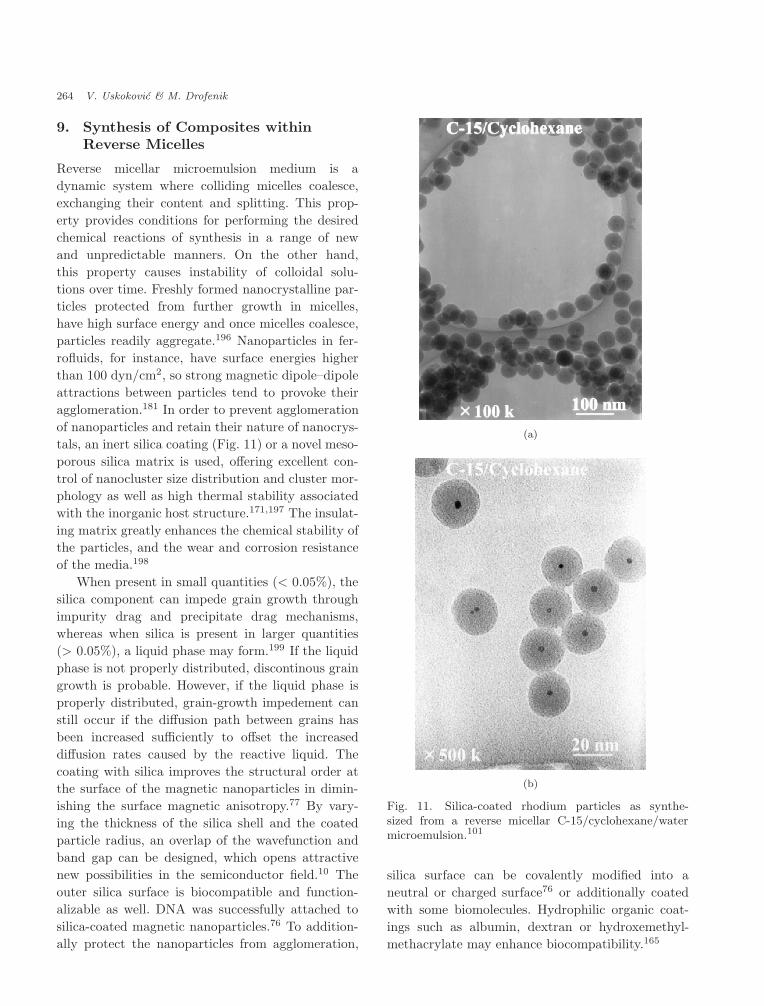
June 2, 2005 12:33 00700
264 V. Uskokovic & M. Drofenik
9. Synthesis of Composites withinReverse Micelles
Reverse micellar microemulsion medium is adynamic system where colliding micelles coalesce,exchanging their content and splitting. This prop-erty provides conditions for performing the desiredchemical reactions of synthesis in a range of newand unpredictable manners. On the other hand,this property causes instability of colloidal solu-tions over time. Freshly formed nanocrystalline par-ticles protected from further growth in micelles,have high surface energy and once micelles coalesce,particles readily aggregate.196 Nanoparticles in fer-rofluids, for instance, have surface energies higherthan 100 dyn/cm2, so strong magnetic dipole–dipoleattractions between particles tend to provoke theiragglomeration.181 In order to prevent agglomerationof nanoparticles and retain their nature of nanocrys-tals, an inert silica coating (Fig. 11) or a novel meso-porous silica matrix is used, offering excellent con-trol of nanocluster size distribution and cluster mor-phology as well as high thermal stability associatedwith the inorganic host structure.171,197 The insulat-ing matrix greatly enhances the chemical stability ofthe particles, and the wear and corrosion resistanceof the media.198
When present in small quantities (< 0.05%), thesilica component can impede grain growth throughimpurity drag and precipitate drag mechanisms,whereas when silica is present in larger quantities(> 0.05%), a liquid phase may form.199 If the liquidphase is not properly distributed, discontinous graingrowth is probable. However, if the liquid phase isproperly distributed, grain-growth impedement canstill occur if the diffusion path between grains hasbeen increased sufficiently to offset the increaseddiffusion rates caused by the reactive liquid. Thecoating with silica improves the structural order atthe surface of the magnetic nanoparticles in dimin-ishing the surface magnetic anisotropy.77 By vary-ing the thickness of the silica shell and the coatedparticle radius, an overlap of the wavefunction andband gap can be designed, which opens attractivenew possibilities in the semiconductor field.10 Theouter silica surface is biocompatible and function-alizable as well. DNA was successfully attached tosilica-coated magnetic nanoparticles.76 To addition-ally protect the nanoparticles from agglomeration,
(a)
(b)
Fig. 11. Silica-coated rhodium particles as synthe-sized from a reverse micellar C-15/cyclohexane/watermicroemulsion.101
silica surface can be covalently modified into aneutral or charged surface76 or additionally coatedwith some biomolecules. Hydrophilic organic coat-ings such as albumin, dextran or hydroxemethyl-methacrylate may enhance biocompatibility.165

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 265
Alkoxides tetraethyl orthosilicate [Si(OC2H5)4 —TEOS, known as tetraethoxysilane as well] orcalcium isopropoxide [Ca(O3H7)2] are mostly used asprecursors for SiO2 or CaO coatings.200–204 Immers-ing TEOS in water and ethanol results in a bubblystructure of solid silicon in a matrix of an organiccompound (HOCH2CH3), which is removed in a sub-sequent step.205 Because this process occurs slowlyat room temperature, acid or base catalyst is addedto the formulation. The amount and type of cata-lyst play key roles in the microstructural propertiesof the final coating. After addition of TEOS into amicroemulsion mixture, the silica shell can be syn-thesized by gradually increasing the pH inside thedroplets with ammonia to catalyze the condensa-tion of TEOS.77 Optimal pH value is found to bebetween 11 and 12.116 When using ammonia as acatalyst, more spherical morphologies are obtainedand therefore, ammonia is often referred to as a mor-phogenic catalyst.116 For higher ammonia concentra-tions, a minimum in particle size was observed atintermediate w values, and it was suggested that thistrend might be associated with the aggregation of thenuclei.206 Ammonia concentration was found to haveimpact on particle sizes, where at relatively low w,the particle size goes to a minimum as ammonia con-centration is increased, but at relatively high w, theparticle size increases monotonically with ammoniaconcentration.6
However, in the presence of a large amountof strong base, the total ionic strength of thewater pool increases, which causes instability ofthe microemulsion system. To avoid this problem,it was suggested76 that a very small amount ofbase be used, that is sufficient for the precipitationreaction followed by the polymerization reaction ofTEOS. Ammonia also provides the particles witha negative, stabilizing surface charge, which doesnot enable aggregation of the particles.207 It wasfound that at low ammonia concentrations, the par-ticle stability is higher. On one hand, an increasein the concentration of ammonia and water willincrease the surface charge through the dissociationof silanol groups and will thus stabilize the parti-cles. On the other hand, increasing the concentrationof ammonia and/or water will increase the concen-tration of NH+
4 and OH− and thereby decrease thedouble layer thickness.207 If the particles are verystable (e.g. at low concentrations of ammonia) the
aggregation stops relatively early and a larger num-ber of silica-coated particles grow to a small finalsize. Because in this case the silica layer that isdeposited after the aggregation is thin, the resultingparticles will not yet be very spherical and the sur-face will be rough. At high ammonia concentrationthe aggregation continues longer and only a relativelysmall number of particles becomes stable.
Silica-coated ceramic particles may be synthe-sized by the double-microemulsion approach, wherethe first microemulsion contains aqueous solutionof the salt precursors and the second microemulsion,that is to be introduced dropwise in the first one,contains an alkali precipitant and TEOS. In this way,mixing and sonication for 2 h together with a day ofaging and subsequent washing with ethanol and cen-trifugation yielded silica-coated magnetic particles.76
By the use of an external magnetic field during thesolidification process, magnetic nanoparticles embed-ded in silica matrix could be preferably orientedin order to obtain an anisotropic macrostructure,known as a magnetically textured system,208 whichhas such properties as anisotropic magnetization andoptical anisotropy.
It was found that the diameter of silica-coatedparticles was highly dependent on the water-to-surfactant molar ratio. Not only did the amountof free water influence the relative rates of hydrol-ysis and condensation reactions, but it also playeda determinant role in colloidal stabilization. Nucle-ation involves the condensation of monomers viaintramicellar or intermicellar exchange polymeriza-tion reactions, whereas particle growth occurredeither by the addition of TEOS to the nuclei or byparticle aggregation.135 The major problem for thereverse micellar preparation of silica-coatings is thatTEOS is an organophilic molecule and is thereforemore readily dissolved in the oil-phase alcohols thanin the aqueous droplets.77 TEOS is also known to beincompatible with strong oxidizing agents.
Upon TEOS addition to the reverse micellar sys-tem, the formation of silica coating over already-formed nanoparticles involves a series of steps,206
which can be identified as:
(i) association of TEOS molecules within thereverse micelles;
(ii) TEOS hydrolysis and formation of monomers;(iii) nucleation;

June 2, 2005 12:33 00700
266 V. Uskokovic & M. Drofenik
(iv) particle growth;(v) nuclei dissolution;(vi) intermicellar exchange of monomers;(vii) ionization of monomers; and(viii) particle surface ionization.
The reaction temperature for hydrolysis and con-densation of TEOS monomers should be kept below25◦C to avoid any complications due to the presenceof a possible thermally induced phase inversion,77
and after 48 h the reaction is normally completed.It was suggested that the synthesis is silica-drivenand that the micelle-initiated nucleation is not thedominant mechanism, but the nucleating site for theporous silica coating is either oligomeric or poly-meric silica.116 In most cases, not all ethoxy groupsfrom TEOS are hydrolyzed and therefore part of thesolvent remain trapped in the silica pores. In fact, itwas proven that several percent of the ethoxy groupsnever leave the TEOS molecules.207
Besides the water-to-surfactant molar ratio andconcentrations of ammonia and water, the ionicstrength can be used to influence the final particlesize. The hydrolysis and subsequent condensation ofTEOS can be affected by the precursor salts intro-duced to the initial solutions of the gels. In the caseof nitrate salts, for instance, due to high solubility ofnitrates, the hydrating water from salts allows a totalhydrolysis of TEOS molecules and as a consequenceof that, the oxygen presence is low. On the contrary,the chloride salts retain the water hydration retard-ing the hydrolysis process and promoting a rich oxy-gen ambient, sometimes convenient for the formationof a different phase.209 The number of ions deter-mines the particle size at which colloidal stability isreached and when the aggregation stops; a smallernumber of growing particles results in a larger finalsilica coating. In a certain experiment, the hydroly-sis rate was unaltered by the addition of LiNO3 salt,while the final radius of synthesized particles was sig-nificantly increased.207
When comparing the number of silica particleswith the number of surfactant aggregates, one silicaparticle was produced out of circa 104 to 106 surfac-tant aggregates, depending on the parameter w. Theminimum number of hydrolyzed TEOS monomersrequired to form a stable nucleus was estimated tobe 2, and the nucleation efficiency factor, i.e. theprobability of effecting nucleation in a reverse micelle
that contains enough monomers to produce a sta-ble nucleus was found to increase with w for rel-atively low ammonia concentrations (1.6 wt%). Adecrease in the apparent nucleation efficiency factorwas observed at high w values with more concen-trated ammonia, and this was attributed to silicanuclei aggregation promoted by enhanced intermi-cellar collisions and intermicellar exchange.206 Thethickness of the SiO2 layer increases with increas-ing hydrolysis time, and therefore it can be designedby controlling the hydrolysis time. In order to ter-minate TEOS hydrolysis, the micellar structure ofthe solution containing SiO2-coated nanoparticlescan be destroyed by adding an alcohol such aspropanol.101
Metallic nanoparticles synthesized in reversemicelles have been coated with gold layer by thereduction of Au3+ from HAuCl4 to Au via excesshydrazine.150 The gold shell on the metallic parti-cles in this case provides the functionality required toform organized arrays on functionalized substrates.Such so-called nano-onion structures comprise a solidcore grown in the first step, which serves as anucleation source for a functionalized shell.119 TheAu:Fe:Au nanocomposites produced by a sequentialreverse micelle technique have been shown to exhibitthe GMR effect.210 Gold has become a favored coat-ing material because of the simple synthetic proce-dure and its chemical functionality.211
Ferrofluids as prosperious magnetic composites212
were prepared by using reverse micellar micro-emulsion.213 Magnetic nanoparticles in ferrofluidcomposites are coated with surfactant monolayersand are immersed within a carrier that might be anorganic solvent such as heptane, xylene, toluene, oran inorganic solvent such as water, a hydrocarbon(synthetic or petroleum) or any ester, polyglycol, orstyrene. Magnetic nanoparticles in such systems areunable to aggregate and thus the superparamagneticnature of the whole system is retained.212 Oleic acidderivatives have proven to be of great benefit in theencapsulation reactions of colloidal magnetic par-ticles through microemulsion polymerization. Thesurface of magnetite colloid was first coated by amonolayer of sodium monooleate and then stabi-lized by adsorption of SDBS surfactant.135 Reversemicellar synthesis is especially convenient for thissort of composites due to capability of tuning vari-ous magnetic properties — such as magnetization or

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 267
coercivity — by control of simple parameters withinthe given procedure of synthesis.
An interesting approach has been found inthe synthesis of ceramic — polymer composites,within which nanosized spherical crystals of theceramic phase are incorporated into micron-sizedpolymer particles. Some of the essential propertiesof magneto–ceramic–polymer composites are its lightweight, good formability toughness and flexibility,typical for plastics, that allow the preparation ofthe products for magnetic circuits with complicatedshapes.214 The remarkable aspect of polymerizationin reverse micelles is the fact that the polymercould form precipitates out from the solution withthe morphology of interconnected submicron-sizedspheres.215 It was claimed that micelles have atemplating effect, to fold chains as they are formedto the resulting spheres. It was proposed that thepolymer chains grow around the micelle interfaceand the collisions of reverse micelles with growingchains result in chain linkages and the formation ofinterconnected spherical particles. As a direct con-sequence of the assembly process of surfactants onmineral surfaces, a hydrophobic interlayer can formon the inorganic particles, into which monomerscan solubilize, and polymerization can subsequentlyproceed. Whereas covalent binding through in situpolymerization techniques allows the formation ofnanocomposite particles with hairy-like, polynuclear,or core-shell morphologies, electrostatic couplingor physicochemical adsorptions preferentially affordheterocoagulated nanostructures with raspberry-like morphologies.135 P-ethylphenol (PEP) is usedas an organic precursor and polymerization ofthese monomers was achieved by adding hydro-gen peroxide with an oxidative enzyme (horseradishperoxidase) in order to polymerize the phenolsencapsulated in the reverse micelles.215 Polymeriza-tion within reverse micelles95 yields oligomers ofcontrolled conjugation lengths and without occur-rence of undesirable nano-oligomers coagulation orinterconnecting, and, therefore, the products ofsuch procedures might find a number of applica-tions in electronics and photonics such as for con-trolled band gaps.216 In this sense, PPV [poly(p-phenylenevinylene)] is an interesting optionbecause it is traditionally made by a base-catalyzedreaction of a water-soluble salt monomer precursorand has a number of applications due to its
electrical, nonlinear optical, electroluminiscent andlasing properties.
The dispersion polymerization or microemulsionpolymerization in the presence of surfactant-coatedparticles carries the risk of incomplete and nonuni-form encapsulation. Reaction conditions must driveall the to-be-coated particles to transfer uniformlyinto the resulting polymer particle, or these parti-cles must provide the only site for the precipita-tion of polymer. One way to potentially guaranteethe uniform loading of core particles into latex wasproposed: directly disperse the magnetic particlesinto the monomer of interest, emulsify, and thenpolymerize.165 Theoretically, the insolubility of thepolymer and magnetic particles in the oil continuumshould guarantee uniform loading of magnetite perlatex; however, no proof has been provided yet. Uponpolymerizing acrylamide monomers microemulsifiedin oil, 40-nm-diameter polyacrylamide latex results.Upon microemulsifying and precipitating iron saltswithin water droplets in microemulsions, magneticnanoparticles result. A combination of the twoapproaches results in magnetic nanoparticles encap-sulated in hydrophilic polymers, with average diam-eters ranging from 80 to 320 nm.185 However, arelatively polydisperse latex in this case containsonly 3.3 wt% (0.6 vol%) of magnetic particles. Amaterial with a greater concentrations of the corephase might arise using the larger droplets in reversemicellar microemulsions, created by using relativelylow amounts of surfactant, as opposed to the nan-odroplets of microemulsions, created with signifi-cantly larger amounts of surfactant.165
It was demonstrated that the presence of thecoupling agent (MPS) was a prerequisite conditionfor the encapsulation reaction within the synthesisof silica/polystyrene colloids. The dispersion of coreparticles, which is a major drawback of encapsula-tion reactions in microemulsion polymerization, canbe significantly improved with the aid of ultrasound.In miniemulsion polymerization, an effective surfac-tant/hydrophobe system was used, and the role ofthe hydrophobe was to prevent or retard coalescenceof the droplets and Ostwald ripening.135 The parti-cle size was shown to be possible to be controlled bywater-soluble cross-linker concentration and surfac-tant/water ratio.217
When using micelle polymerization as a term,surfactant molecules are regarded as monomers,

June 2, 2005 12:33 00700
268 V. Uskokovic & M. Drofenik
whereas when using microemulsion polymerizationas a term, monomers are thought to be encap-sulated within reverse micelles and polymerizeunder surfactant restrictions. Surfactant monomers(so-called surfmers) are used often in order togain nanoparticle–polymer composites with narrowsize and shape distribution. Since the pioneeringsurfmer synthesis performed by Freedman et al.who reported the first synthesis of vinyl monomerswhich also functioned as an emulsifying agent,218
vast amount of literature has been dedicated to thetopic of polymerization of/in organized amphiphilicassemblies.219 Polymerization of surfmers wasobserved only at concentrations above CMC, provingthat micelle formation is a necessary precondition forpolymerization.219 Surfactants acting as surfmers areCVDAC (cetyl-p-vinylbenzyldimethylammoniumchloride, that might be photopolymerized too),DDM-MEAC [N,N-didodecyl-N-methyl-N-((2-methacryloyloxy)ethyl)ammonium chloride] anddidecyldimethylammonium methacrylate.220 Thefirst synthesis of 2–5-nm-sized latex particles wasperformed by using didecyldimethylammoniummethacrylate, a polymerizable surfactant whichforms reverse micelles in toluene.221
10. Survey of the Types ofMaterials Synthesized by UsingReverse Micelles
Materials of many different chemical composi-tions were synthesized within reverse micelles andwe shall list some of them. Applications of theproduced materials by using reverse micelles arewidespread in paints and surface coatings, catalysis,separation media, drug delivery systems, high-frequency electronic components, etc. The pos-sibility of obtaining materials with high surfaceareas makes reverse micellar methods of syn-thesis viable for the production of catalyticallyactive transition metals, including Pt,222 Au,90
Ag,186 Pd,169 Rh,1,223 Ir,1 PtPd,224 AuAg,225
AuPd,226 PtRu,227 PtCo,228 Zr,142 Si,6 Ni,187 Ti,229
Bi,230 Cu,4,231 Se,84 Ce,232 Ce1-x-ZrxO2,233 Ce–Tbmixed oxides,234 (Ce,Zr)O-x/Al2O3,235 CeZrO4,236
CeZrO4/Al2O3,237 Pd/CexZr1-O-x(2)/Al2O3 three-way catalysts,238 LaMnO3/CdS photocatalyst,239
and ferric sulfide catalyst for direct coal liquefac-tion (DCL).240 The metal nanoparticles produced
within reverse micelles are characterized by a highercatalytic stability and effectiveness than classicalmetal catalysts.108 Nanosized and narrowly size-distributed reverse micelle aqueous cores open away to synthesize narrowly distributed semicon-ductors with marked quantum-size (Q-size) effects,such as: CdS,24,149 PbS,241 ZnS,242 CdE (E = S,Se, Te) semiconductors.243 BaF2 and BaF2:Ce,87
BaF2:Nd,244 AgBr,245 AgI,42,113 AgCl,246 Ag2S,4
Cu2S,247 MoSx,188 CeF3,248 Bi2Te3,249 ZnSe,250 Balloys,251 Ni-, Co- and Fe- borides,252 tungsticacid,253 Cu2[Fe(CN)6],254 amorphous AlOOH255
were also synthesized within reverse micelles.Many composites have been prepared, which
primarily include using silica coatings or matri-ces as additional component. Such materials aresilica-coated Co nanoparticles,256 Pt/silica,257
Pd/SiO2 particles,78 Cu/SiO2,258 Ag/SiO2,259
SiO2-coated Rh nanoparticles,101 SiO2-coatedCeO2 nanoparticles,260 SiO2-coated Co-ferriteand magnetite,102 SiO2-coated Pt, Pd andPtAg particles,261 SiO2-coated NiFe permalloynanoparticles,312 silica-coated Fe2O3,76 silica-coatedZnFe2O4,77 silica-coated CoFe2O4 and MnFe2O4,262
TiO2/SiO2 composites,263 ZrO2/SiO2.264 The use ofgold as an additional component of composites pro-duced in reverse micelles — exemplified by the casesof preparation of gold-coated Fe nanoparticles,211
gold-coated FePtx and CoPtx particles,265 gold-coated Co, CoPt and CoPt3 magnetic alloys266 orsuperparamagnetic magnetite-gold composites267 —is noticed as well as the use of polymers in polymerceramics nanocomposites,86,215,217 including mag-netite polymer composites,185,268 nanocomposites ofpolyaniline with AgCl, BaSO4 and TiO2.86,176 Com-posites of TiO2/Fe2O3,269 CdS particles coated withZnS,270 Co–Ag and Fe–Cu–Fe nanoparticles withantiferromagnetic coupling between the Fe layers271
were also prepared within reverse micelles.Beside using reverse micelles and other
dynamic nanostructures of self-assembling surfac-tant molecules in microemulsions to limit the parti-cle size of dispersed systems formed by precipitationin the case of inorganic particles, various approacheshave been used for lattices gained by polymeriza-tion reactions. Nanoscale polymerization reactionswithin the reverse micellar confinement yield prod-ucts with controlled conjugation lengths and, thus,controlled band gaps for applications to electronics

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 269
and photonics. In addition, control of the lengthof the polymer blocks provides the prospects ofmonodispersity, improved processability, and des-ignable preparation of nanocomposites. Polymericnanosized particles (latexes) were first synthesizedby using the reverse micelle method221 and since thenmany polymers, including poly(p-phenylenevinylene)(PPV),216 were synthesized within reverse micel-lar microemulsions. Many procedures of synthe-sis concerning encapsulation of nanoparticles withpolymers,326 either by extracting the particles fromthe microemulsion or by in situ polymerization, suchas in the case of the preparation of Bi/PMMA,160
CdS/polyacrilamide nanocomposites272 and BaSO4
in polymerized surfactants,273 were also reported inthe literature.
High-Tc superconductors YBa2Cu3O7-x274–276
were prepared by this method as well as manymaterials with exotic morphologies, such as V2O5
nanowires,193 ZnS semiconductor nanowires,194
PbCl2 nanowires,277 Sb2O3 and Sb2O5 nanorods,278
BaCO3 single-crystal nanowires,279 single-crystalSnO2 nanorods,280 CaCO3, BaCO3 and CaSO4
nanowires,178 MnOOH nanorods,281 BaSO4
nanoparticles and nanofilaments,282 star-shapedCdS patterns,283 nanosized fluorescent hybrid sil-ica (NFHS),284 fluorescent europium(III) chelate-doped silica nanoparticles,285 and silica-coatedterbium(III) chelate fluorescent nanoparticles.286
Ca(OH)2,3 CaCO3145 and materials for biomedical
applications, such as calcium hydroxyapatite287,288
and Ca(PO4)3,289 have also been produced withinreverse micelles. The pioneering nanoengineer-ing via biocompatible microemulsion was per-formed by Debuigne et al. and was related tothe preparation of anti-inflammatory drug nime-sulide by direct precipitation in reverse micellarmicroemulsions comprising either Epikuron 170(lecithin)/isopropyl myristate/water/n-butanol orE170/isopropyl myristate/water/n-isopropanol190
and to the synthesis of cholesterol, Rho-vanol and Rhodiarome in AOT/heptane/water,Triton/decanol/water, and CTAB/hexanol/watermicroemulsions.174
Many ceramic materials have been synthesizedby using this method: ZrO2–Y2O3 nanoparticles,53
barium hexaaluminate nanoparticles,152,290 zircono-lite CaZrTi2O7,291 CuHo2O5 and Cu2Er2O5,88
CaSO4,292 ZnAl2O4,89 YVO4,293 perovskite-type
LaNiO3, La2CuO4 and BaPbO3,5 Y2O3: Eu3+,294
Gd2O3 : Eu3+ and Gd2O2S : Eu3+,295
Na2PO4 and K2PO4,296 calcium thiophosphate,297
zincophosphates191,298 as well as many oxides,such as GeO2,299 ZnO,300 EuO,301 SnO,302
TiO2,303 ZrO2,85 CeO2,304 Fe2O3,269 Al2O3,305
iron oxide-doped alumina nanoparticles,306 Cs-doped alumina nanoparticles,307 and barium-stabilized alumina nanoparticles.308 Reverse micelleshave shown to be feasible in producing magneticceramics, such as manganites LaMnO3
89,309,69
and LaSr-manganites,310,311 Li(Mn,Co)2O4,74
ferrites NiFe2O4,79 MnZnFe2O4,80,117,313,314
NiZnFe2O4,147,157,161 MnFe2O4,75 SrFe2O4,31,92
CoFe2O4,67,120 CoCrFe2O4,315,316 BaFe2O4,317
LaFeO3,69 and Fe3O4.318 Many magnetic materi-als, as Fe,29,319 Co,23 FeCo nanocrystals,182 Co,CoPt and CoPt3,266 KMnF3 and NaMnF3 antifer-romagnetic particles,83 were successfully synthesizedwithin reverse micelles.
11. Future Directions
After all that has been said, it useful to take a lookonce again at the major conclusion which emergesfrom this review. On one hand, reverse micelles areuseful multimolecular structures that give significantopportunities for the design of nanostructured mate-rials of well-defined and uniform properties. On theother hand, reverse micelles cannot be consideredas nano-engineering reactor shells because their sizeand permeability cannot yet be tuned with sufficientprecision.108 “Nanoreactor” does not sound as a con-venient term at all for describing the function ofreverse micelles in microemulsion-assisted synthesesof materials, since conventional microreactors implyenabling precise control over the reactions and prop-erties of the resulting nanomaterials, and it is stilldifficult to achieve this aim at the nano level by usingthe reverse micellar technique.
Combinations of the reverse micellar precipita-tion method or any other microemulsion methodwith some other methods of synthesis — such as sol-gel,291,320,321 hydrothermal synthesis,183 ultrasonicirradiation,322 UV irradiation,323 pH-shock wavemethod,324 and flame-spraying325 — that have beenused lately, have presented many new potentialitiesand advantages. Merging of two or more preparationtechniques into one can, due to synergy effects, lead

June 2, 2005 12:33 00700
270 V. Uskokovic & M. Drofenik
to multiple advantages, such as improved control ofthe stoichiometry of the final product (taken fromsol-gel method) and extremely fine and controllablegrain size (taken from reverse micellar synthesis) inthe examples290,327 of the combination of sol-gel andreverse micellar approaches to materials synthesis.Low yields, surfactant-contamination and difficultiesarising from the attempts to separate precipitatedpowders in the form of non-agglomerated particles —serious obstacles of the microemulsion-assistednanoparticle preparation procedures — were over-come by feedstocking flame-spraying apparatuswith nanoparticles together with their parentmicroemulsion,325 at the same time transcendingpoor control of particle size and shape, which isa typical drawback of conventional flame-sprayingmethods of synthesis. Combining reverse micellarsynthesis of cobalt particles with their subsequentevaporating deposition in external magnetic fieldled to the formation of large-scale 3D superlattices(Fig. 12) of cobalt nanocrystals.144 Carbon nan-otubes have as well, for instance, been preparedby direct introduction of in situ prepared catalyt-ically active Co and Mo particles by a reversemicellar method.328,329 Silver nanorods encapsulatedby polystyrene were prepared by a combinationof reverse micellar, gas antisolvent, and ultrasoundtechniques.330 In order to overcome difficulties aris-ing out of the fact that it is not possible to carry outsequential reactions inside the same reverse micellesin order to obtain multilayered composites, layer-by-layer (LbL) techniques comprising adsorption ofoppositely charged polyelectrolytes on a solid surfaceof synthesized particles in reverse micelles have beenused.108
The assemblying of particles formed in theprocesses of reverse micellar and, in general,microemulsion-assisted syntheses into precisely tai-lored, supra-nanocrystalline 3D structures, clearlypresents an important challenge. In situ reactions inwell-organized and immobilized amphiphilic matri-ces present only one approach toward this goal. Theuse of shear on liquid crystalline systems for instance,can be used to produce the alignment of microscopicdomains into macroscopic regions in the directionof shear, or other interesting morphological transi-tions. External stimuli provides great promise, andit is predicted that in the future days, progress in
Fig. 12. Self-organized arrangements of cobalt nanopar-ticles obtained by combining the synthesis of the mate-rial within reverse micelles and evaporating depositionthereof.144
using light, electric and magnetic fields to organizeparticles into 3D matrices will be realized. A moreactive and potentially richer approach is the use ofengineered peptides to direct the self-organization ofnanoparticles into technologically important struc-tures. The real test of self-assembly and materialssynthesis will be the generation of novel functionalstructures with unique and useful properties.112
Future research in the field of the materials syn-thesis within reverse micelles may not only help inthe fabrication of new inorganic materials and thedevelopment of spatially confined and highly con-trollable synthetic approaches, but may also provideus with new understanding of the chemical, physic-ochemical and biochemical processes that occur in aconfined environment at the nanometer scale, whichis significant for the fundamental understanding ofnature and life.

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 271
References1. M. Boutonnet, J. Kizling and P. Stenius, Coll. Surf.
5(3) (1982) 209–225.2. D. O. Yener and H. Giesche, J. Am. Ceram. Soc.
84(9) (2001) 1987–1995.3. A. Nanni and L. Dei, Langmuir 19 (2003) 933–938.4. M. P. Pileni, A. Hammounda, I. Lisiecki, L. Motte,
N. Moumen and J. Tanori, Control of the size andshape of nanoparticles, in Fine Particles Scienceand Technology, ed. E. Pelizzetti (Kluwer AcademicPublishers, 1996), pp. 413–429.
5. L. M. Gan, L. H. Zhang, H. S. O. Chan, C. H. Chewand B. H. Loo, J. Mater. Sci. 31 (1996) 1071–1079.
6. F. J. Arriagada and K. Osseo-Asare, J. Coll. Inter-face Sci. 211 (1999) 210–220.
7. Y. X. Chen, X. Z. Zhang, K. Zheng, S. M. Chen,Q. C. Wang and X. X. Wu, Enzyme Microbial Tech-nol. 23(3–4) (1998) 243–248.
8. P. P. Sun and Y. M. Zhang, Synth. Commun.27(23) (1997) 4173–4179.
9. K. K. B. Lee, L. H. Poppenborg and D. C.Stuckey, Enzyme Microbial Technol. 23(3–4) (1998)253–260.
10. M. A. Lopez-Quintela, C. Tojo, M. C. Blanco,L. Garcia Rio and J. R. Leis, Curr. Opin. Coll.Interface Sci. 9 (2004) 264–278.
11. K. Carlile, G. D. Rees, B. H. Robinson, T. D. Steerand M. Svensson, J. Chem. Soc., Faraday Transact.92(23) (1996) 4701–4708.
12. C. R. Babu, P. F. Flynn and A. J. Wand, J. Biomol.NMR 25(4) (2003) 313–323.
13. E. P. Melo, S. M. B. Costa, J. M. S. Cabral, P. Fojanand S. B. Petersen, Chem. Phys. Lipids 124 (2003)37–47.
14. G. G. Chang, T. M. Huang and H. C. Hung,Proc. Natl. Sci. Counc. ROC B24(3) (1999)89–100.
15. A. Tuck, Surv. Geophys. 23 (2002) 379–409.16. P. A. Bachmann, P. Walde, P. L. Luisi and J. Lang,
J. Am. Chem. Soc. 113(22) (1991) 8204–8209.17. P. A. Bachmann, P. L. Luisi and J. Lang, Chimia
45(9) (1991) 266–268.18. J. H. Schulman, W. Stoeckenius and L. M. Prince,
J. Phys. Chem. 63 (1959) 1677–1680.19. J. Liu, Y. Ikushima and Z. Shervani, Curr. Opin.
Solid State Mater. Sci. 7 (2003) 255–261.20. M. C. McLeod, R. S. McHenry, E. J. Beckman and
C. B. Roberts, J. Phys. Chem. B107(12) (2003)2693–2700.
21. C. J. O’Connor, V. Kolesnichenko, E. Carpenter,C. Sangregorio, W. Zhou, A. Kumbhar, J. Sims andF. Agnoli, Synth. Met. 122 (2001) 547–557.
22. M. P. Pileni, T. Zemb and C. Petit, Chem. Phys.Lett. 118(4) (1985) 414–420.
23. E. E. Carpenter, C. T. Seip and C. J. O’Connor,J. Appl. Phys. 85(8) (1999) 5184–5186.
24. U. Natarajan, K. Handique, A. Mehra, J. R. Bellareand K. C. Khilar, Langmuir 12 (1996) 2670–2678.
25. R. H. Kodama, J. Magn. Magn. Mater. 200 (1999)359–372.
26. P. Tartaj and C. J. Serna, Chem. Mater. 14 (2002)4396–4402.
27. K. K. Ghosh and L. K. Tiwary, J. Dispersion Sci.Technol. 22(4) (2001) 343–348.
28. B. Lindman and H. Wennerstrom, Micelles.Amphiphile Aggregation in Aqueous Solution,Topics in Current Chemistry, Vol. 87 (Springer-Verlag, Berlin, 1980).
29. F. Li and C. Vipulanandan and K. K. Moharty,Coll. Surf. A223 (2003) 103–112.
30. S. L. Watt, D. Tunaley and S. Biggs, Coll. Surf.A137 (1998) 25–33.
31. J. Fang, J. Wang, L. M. Gan, S. C. Ng, J. Dingand X. Liu, J. Am. Ceram. Soc. 83(5) (2000)1049–1055.
32. H. R. Rabie, M. E. Weber and J. H. Vera, J. Coll.Interface Sci. 174 (1995) 1–9.
33. F. Li and C. Vipulanandan, Conductivity of water-in-oil microemulsion system, retrieved from Uni-versity of Houston website at http://gem1.cive.uh.edu.
34. D. Brown and J. H. R. Clarke, J. Phys. Chem. 92(1988) 2881–2888.
35. N. E. Levinger, Science 298 (2002) 1722–1723.36. S. Senapati and M. L. Berkowitz, J. Chem. Phys.
118(4) (2003) 1937–1944.37. D. Brown and J. H. R. Clarke, J. Phys. Chem.
92(10) (1988) 2881–2888.38. S. Brunetti, D. Roux, A. M. Bellocq, G. Fourche
and P. Bothorel, J. Phys. Chem. 87(6) (1983)1028–1034.
39. V. K. Aswal and P. S. Goyal, Physica B245 (1998)73–80.
40. C. C. Co, E. W. Kaler and S. R. Kline, Microstruc-ture transformation during microemulsion andmicellar polymerizations, Research Highlights inthe monthly review paper of NIST Center for Neu-tron Research, world wide web.
41. V. K. Aswal, Bhabha Atom. Res. Centre Newslett.237 (2003).
42. S. Tamura, K. Takeuchi, G. Mao, R. Csencsits,L. Fan, T. Otomo and M. L. Saboungi, J. Elec-troanal. Chem. 559 (2003) 103–109.
43. N. V. Sushkin, Ph.D. dissertation, Worcester Poly-technic Institute (1997).
44. Q. Zhong, D. A. Steinhurst, E. E. Carpenter andJ. C. Owrutsky, Langmuir 18 (2002) 7401–7408.
45. G. Y. Xu, L. Zhang, S. L. Yuan, X. R. Huang andG. Z. Li, J. Dispersion Sci. Technol. 22(6) (2001)563–567.
46. T. Patzlaff, M. Janich, G. Seifert and H. Graener,Chem. Phys. 261 (2000) 381–389.

June 2, 2005 12:33 00700
272 V. Uskokovic & M. Drofenik
47. R. H. Cole, S. Mashimo and P. Winson, J. Chem.Phys. 84(7) (1980) 786–793.
48. B. Gestblom and E. Noreland, J. Chem. Phys.81(8) (1977) 782–788.
49. J. van Stam, S. Depaemelaere and F. C. DeSchryver, J. Chem. Edu. 75(1) (1998) 93–98.
50. M. P. Pileni, J. Phys. Chem. 97 (1993) 6961–6973.51. F. Reiss-Husson and V. Luzzati, J. Phys. Chem.
68(12) (1964) 3504–3511.52. F. Li, G. Z. Li, H. Q. Wang and Q. J. Xue, Coll.
Surf. A127 (1997) 89–96.53. X. Fang and C. Yang, J. Coll. Interface Sci. 212
(1999) 242–251.54. G. Lindblom, B. Lindman and L. Mandell, J. Coll.
Interface Sci. 34(2) (1970) 262–271.55. T. Kato and T. Seimiya, J. Phys. Chem. 90 (1986)
3159–3167.56. L. Wang and R. E. Verrall, J. Phys. Chem. 98
(1994) 4368–4374.57. I. Benito, M. A. Garcia, C. Monge, J. M. Saz
and M. L. Marina, Coll. Surf. A125 (1997)221–224.
58. J. J. Rack, T. M. McCleskey and E. R. Birnbaum,J. Phys. Chem. B106 (2002) 632–636.
59. Z. Lin, J. J. Cai, L. E. Scriven and H. T. Davis,J. Phys. Chem. 98 (1994) 5984–5993.
60. T. Kinugasa, A. Kondo, S. Nishimura, Y. Miyauchi,Y. Nishii, K. Watanabe and H. Takeuchi, Coll. Surf.A204 (2002) 193–199.
61. G. Montelvo, M. Valiente and E. Rodenas, Lang-muir 12 (1996) 5202–5208.
62. D. Roux, A. M. Bellocq and P. Bothorel, Prog. Coll.Polymer Sci. 69 (1984) 1–11.
63. E. Rodenas and M. Valiente, Coll. Surf. 62 (1992)289–295.
64. J. Kim and M. Lee, J. Phys. Chem. A103(18)(1999) 3378–3382.
65. P. Ekwall, L. Mandell and P. Solyom, J. Coll. Inter-face Sci. 35(2) (1970) 267–272.
66. Y. Liu, W. Wang, Y. Zhan, C. Zheng and G. Wang,Mater. Lett. 56 (2002) 496–501.
67. C. Liu, A. J. Rondinone and Z. J. Zhong, Pure Appl.Chem. 72(1–2) (2000) 37–45.
68. C. R. Vestal and Z. J. Zhang, J. Solid State Chem.175 (2003) 59–62.
69. A. E. Giannakas, A. K. Ladavos and P. J. Pomonis,Appl. Catal. B49 (2004) 147–158.
70. D. Walsh and S. Mann, Chem. Mater. 8(8) (1996)1944–1953.
71. T. Hirai, S. Hariguchi, I. Komasawa and R. J.Davey, Langmuir 13(25) (1997) 6650–6653.
72. H. Zhang and A. I. Cooper, Chem. Mater. 14 (2002)4017–4020.
73. J. C. Linehan, J. L. Fulton and R. M. Bean, Pro-cess of forming compounds using reverse micelleor reverse microemulsion systems, US Patent5,170,172 (1998).
74. C. H. Lu and H. C. Wang, J. Eur. Ceram. Soc. 23(2003) 865–871.
75. C. Liu, B. Zuo, A. J. Rondinone and Z. J. Zhang,J. Phys. Chem. 104(6) (2000) 1141–1145.
76. S. Santra, R. Tapec, N. Theodoropoulou,J. Dobson, A. Hebard and W. Tan, Langmuir 17(2001) 2900–2906.
77. F. Grasset, N. Labhsetwar, D. Li, D. C. Park,N. Saito, H. Haneda, O. Cador, T. Roisnel,S. Mornet, E. Duguet, J. Portier and J. Etourneau,Langmuir 18(21) (2002) 8209–8216.
78. D. S. Bae, K. S. Han and J. H. Adair, J. Mater.Chem. 12(10) (2002) 3117–3120.
79. J. Fang, N. Shama, L. D. Tung, E. Y. Shin, C. J.O’Connor, K. L. Stokes, G. Caruntu, J. B. Wiley,L. Spinu and J. Tang, J. Appl. Phys. 93(10) (2003)7483–7485.
80. J. Wang, P. F. Chong, S. C. Ng and L. M. Gan,Mater. Lett. 30 (1997) 217–221.
81. T. Liu, L. Guo, Y. Tao, Y. B. Wang andW. D. Wang, Nanostruct. Mater. 11(4) (1999)487–492.
82. Y. Y. Luk and N. L. Abbott, Curr. Opin. Coll.Interface Sci. 7 (2002) 267–275.
83. E. E. Carpenter, C. Sangregorio and C. J.O’Connor, Mol. Cryst. Liq. Cryst. 334 (1999)641–649.
84. J. A. Johnson, M. L. Saboungi, P. Thiyagarajan,R. Csecsits and D. Meisel, J. Phys. Chem. B103(1999) 59–63.
85. X. Li, C. K. Loong, P. Thiyagarajan, G. A. Lagerand R. Miranda, J. Appl. Crystalog. 33(1) (2000)628–631.
86. X. M. Sui, Y. Chu, S. X. Xing and C. Z. Liu, Mater.Lett. 58(7–8) (2004) 1255–1259.
87. R. Hua, C. Zang, C. Shao, D. Xie and C. Shi,Nanotechnology 14 (2003) 588–591.
88. F. Porta, C. Bifulco, P. Fermo, C. L. Bianchi,M. Fadoni and L. Prati, Coll. Surf. A160 (1999)281–290.
89. A. E. Giannakas, T. C. Vaimakis, A. K. Ladavos,P. N. Trikalitis and P. J. Pomonis, J. Coll. InterfaceSci. 259 (2003) 244–253.
90. F. Chen, G. Q. Xu and T. S. A. Hor, Mater. Lett.57 (2003) 3282–3286.
91. G. Palazzo, F. Lopez, M. Giustini, G. Colafemminaand A. Ceglie, J. Phys. Chem. B107 (2003)1924–1931.
92. D. H. Chen and Y. Y. Chen, J. Coll. Interface Sci.236 (2001) 41–46.
93. J. P. Wilcoxon and P. P. Provencio, J. Phys. Chem.B103 (1999) 9809–9812.
94. V. Uskokovic and M. Drofenik, Coll. Surf. A, sub-mitted, 2005.
95. Y. Teraoka, S. Nanri, I. Moriguchi, S. Kagawa,K. Shimanoe and N. Yamazoe, Chem. Lett. 10(2000) 1202–1203.

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 273
96. M. Mamak, G. S. Metraux, S. Petrov, N. Coombs,G. A. Ozin and M. A. Green, J. Am. Chem. Soc.125(17) (2003) 5161–5175.
97. K. Kurumada, S. Itakura, S. Nagamine andM. Tanigaki, Langmuir 13(14) (1997) 3700–3705.
98. M. P. Pileni, N. Moumen, J. F. Hochepied,P. Bonville and P. Veillet, J. Phys. IV France 7,C1-505-8 (1997).
99. M. A. Lopez-Quintela, Curr. Opin. Coll. InterfaceSci. 8 (2003) 137–144.
100. B. R. Smith and A. E. Alexander, J. Coll. InterfaceSci. 34(1) (1970) 81.
101. T. Tago, Y. Shibata, T. Hatsuta, K. Miyajima,M. Kishida, S. Tashiro and K. Wakabayashi,J. Mater. Sci. 37 (2002) 977–982.
102. T. Tago, T. Hatsuta, K. Miyajima, M. Kishida,S. Tashiro and K. Wakabayashi, J. Am. Ceram.Soc. 85(9) (2002) 2188–2194.
103. H. Shi, L. Qi, J. Ma, H. Cheng and B. Zhu, Adv.Mater. 15(19) (2003) 1647–1651.
104. B. A. Simmons, S. Li, V. T. John, G. L. McPherson,A. Bose, W. Zhou and J. He, Nano Lett. 2(4) (2002)263–268.
105. P. Somasundaran, J. Coll. Interface Sci. 256 (2002)3–15.
106. G. Porte, D. Roux, J. F. Berret, S. Lerouge,J. P. Decruppe, P. Lindner and L. Noirez, Rheologyof wormlike micelles, ITP Complex Fluid Program3/27/02 (CNRS-Rhodia, 2002).
107. R. Zana and J. Lang, Dynamics of Microemulsions,in Microemulsions: Structure and Dynamics, eds.S. E. Friberg and P. Bothorel (CRC Press: BocaRaton, 1987), pp. 153–172.
108. D. G. Shchukin and G. B. Sukhorukov, Adv. Mater.16(8) (2004) 671–682.
109. P. van Beurten and A. Vrij, J. Chem. Phys. 74(5)(1981) 2744–2748.
110. F. G. Sanchez and C. C. Ruiz, J. Lumin. 69 (1996)179–186.
111. Z. Cui and R. J. Mumper, Pharm. Res. 19(7)(2002) 939–946.
112. V. T. John, B. Simmons, G. L. McPherson andA. Bose, Curr. Opin. Coll. Interface Sci. 7 (2002)288–295.
113. S. Xu, H. Zhou, J. Xu and Y. Li, Langmuir 18(2002) 10503–10504.
114. J. Yang, Curr. Opin. Coll. Interface Sci. 7 (2002)276–281.
115. M. Bhat, Studies in surfactant science andhydrotropy, Ph.D. Thesis, University Institute ofChemical Technology, Mumbai, India.
116. R. I. Nooney, D. Thirunavukkarasu, Y. Chen,R. Josephs and A. E. Ostafin, Chem. Mater. 14(2002) 4721–4728.
117. D. Makovec, A. Kosak and M. Drofenik, Nanotech-nol. 15 (2004) S160–S166.
118. A. M. Goncalves, A. P. Serro, M. R. Aires-Barrosand J. M.S. Cabral, Biochim. Biophys. Acta 1480(2000) 92–106.
119. E. E. Carpenter, Ferrites: Proc. 8th Int. Conf. Fer-rites (ICF 8), Kyoto/Tokyo, Japan (2000).
120. V. Pillai and D. O. Shah, J. Magn. Magn. Mater.163 (1996) 243–248.
121. J. Sjoblom, R. Lindberg and S. E. Friberg, Adv.Coll. Interface Sci. 95 (1996) 125–287.
122. S. Bhattacharjee, M. Elimelech andM. Borkovec, Croatica Chem. Acta 71(4) (1998)883–903.
123. D. G. Grier, Nature 393(6686) (1998) 621.124. T. Dichristina, D. Roux, A. M. Bellocq and
P. Bothorel, J. Phys. Chem. 89(8) (1985)1433–1437.
125. B. Lindman and P. Stilbs, in Microemulsions:Structure and Dynamics, eds. S. E. Friberg andP. Bothorel (CRC Press, Boca Raton, 1987),119–152.
126. J. R. Hansen, J. Phys. Chem. 78 (1974) 256.127. H. F. Eicke and P. E. Zinsli, J. Coll. Interface Sci.
65 (1978) 131.128. M. Wong, J. K. Thomas and T. Nowak, J. Am.
Chem. Soc. 99 (1977) 4730.129. B. Lindman and H. Wennerstrom, in Solution
Behavior of Surfactants, Vol. 1, eds. K. L. Mittaland E. J. Fendler (Plenum Press, New York, 1982).
130. H. Grunhagen, J. Coll. Interface Sci. 53 (1975) 282.131. J. Biais, B. Clin, P. Lalanne and B. Lemanceau,
J. Chim. Phys. 74 (1977) 1197.132. H. Yoshioka, J. Coll. Interface Sci. 95 (1983) 81.133. J. Lang, A. Djavanbakht and R. Zana, J. Phys.
Chem. 84 (1980) 145.134. J. M. DiMeglio, M. Dvolaitzky, R. Ober and
C. Taupin, J. Phys. Lett. 44 (1983) L229.135. E. Bourgeat–Lami, J. Nanosci. Nanotechnol. 2(1)
(2002) 1–24.136. Z. Shervani and Y. Ikushima, J. New Chem. 26
(2002) 1257–1260.137. R. P. Bagwe and K. C. Khilar, Langmuir 16 (2000)
905–910.138. B. H. Robinson and P. D. Fletcher, Ber. Bunsenges.
Phys. Chem. 85 (1981) 863.139. R. Jain and A. Mehra, Langmuir 20 (2004)
6507–6513.140. S. Atik and J. K. Thomas, J. Phys. Chem. 85
(1981) 3921.141. K. Inouye, R. Endo, Y. Otsuka, K. Miyashiro,
K. Kaneko and T. Ishikawa, J. Phys. Chem. 86(8)(1982) 1465–1469.
142. J. Fang, J. Wang, S. C. Ng, C. H. Chewand L. M. Gan, Nanostruct. Mater. 8(4) (1997)499–505.
143. L. Motte, C. Petit, L. Boulanger, P. Lixon and M. P.Pileni, Langmuir 8(4) (1992) 1049–1053.

June 2, 2005 12:33 00700
274 V. Uskokovic & M. Drofenik
144. J. Legrand, A. T. Ngo, C. Petit and M. P. Pileni,Adv. Mater. 13(1) (2001) 58–62.
145. K. Kandori, K. Kon-No and A. Kitahara, J. Coll.Interface Sci. 122(1) (1988) 78–82.
146. H. Sato and I. Komasawa, J. Chem. Eng. Jpn.33(2) (2000) 262–266.
147. V. Uskokovic and M. Drofenik, J. Magn. Magn.Mater. 284 (2004) 294–302.
148. A. Kosak, D. Makovec, A. Znidarsicand M. Drofenik, J. Eur. Ceram. Soc. 24 (2004)959–962.
149. A. Agostiano, M. Catalano, M. L. Curri,M. Della Monica, L. Manna and L. Vasanelli,Micron 31 (2000) 253–258.
150. C. T. Seip and C. J. O’Connor, Nanostruct. Mater.12 (1999) 183–186.
151. C. J. O’Connor, C. T. Seip, E. E. Carpenter, S. Liand V. T. John, Nanostruct. Mater. 12 (1999)65–70.
152. A. J. Zarur and J. Y. Ying, Nature 403 (2000)65–67.
153. A. Kosak, D. Makovec and M. Drofenik, Phys. Sta-tus Solidi. C1(12) (2004) 3521–3524.
154. A. Kosak, D. Makovec and M. Drofenik, J.Metastable Nanocryst. Mater. 23 (2005) 251–254.
155. A. Kosak, D. Makovec, M. Drofenik andA. Znidarsic J. Magn. Magn. Mater. 272–276(2004) 1542–1544.
156. A. Kosak, D. Makovec and M. Drofenik, Mater. Sci.Forum 453–454 (2004) 219–224.
157. V. Uskokovic and M. Drofenik, Mater. Sci. Forum453–454 (2004) 225–230.
158. V. Uskokovic and M. Drofenik, Mater. Technol.37(3–4) (2003) 129–131.
159. D. B. Zhang, L. M. Qi, H. M. Cheng and J. M. Ma,Chin. Chem. Lett. 14(1) (2003) 100–103.
160. J. Fang, L. Stokes, J. Wiemann and W. Zhou,Mater. Lett. 42 (2000) 113–120.
161. V. Uskokovic and M. Drofenik, Surf. Rev. Lett. 12(2005) 97–100.
162. M. C. Moran, A. Pinazo, L. Perez, P. Clapes,M. Angelet, M. T. Garcia, M. P. Vinardelland M. R. Infante, Green Chem. 6 (2004)233–240.
163. Nano-Crystalline Materials Research, The Newslet-ter of the New York State Center for AdvancedCeramic Technology 11.3 (1999).
164. A. A. Date and V. B. Patravale, Curr. Opin. Coll.Interface Sci. 9 (2004) 222–235.
165. K. Wormuth, J. Coll. Interface Sci. 241 (2001)366–377.
166. J. Sjoblom, R. Skurtveit, J. O. Saeten andB. Gestblom, J. Coll. Interface Sci. 141(2) (1991)329–337.
167. K. Shinoda and H. Takeda, J. Coll. Interface Sci.32(4) (1970) 642.
168. S. L. Watt, D. Tunaley and S. Biggs, Coll. Surf.A137 (1998) 25–33.
169. C. C. Wang, D. H. Chen and T. C. Huang, Coll.Surf. A189 (2001) 145–154.
170. M. L. Curri, A. Agostiano, F. Mavelli andM. Della Monica, Mater. Sci. Eng. C22 (2002)423–426.
171. L. Zhang, G. C. Papaefthymiou and J. Y. Ying,J. Appl. Phys. 81(10) (1997) 6892–6900.
172. M. Hasegawa, Langmuir 17 (2001) 1426–1431.173. J. Kizling and P. Stenius, J. Coll. Interface Sci.
118(2) (1987) 482–492.174. F. Debuigne, L. Jeunieau, M. Wiame and J. B.
Nagy, Langmuir 16 (2000) 7605–7611.175. E. E. Carpenter, Synthesis of nanoparticles
using reverse micelles, www.darpa.mil/dso/future/metamaterials/presentations/carpenter.pdf.
176. X. M. Sui, Y. Chu, S. X. Xing, M. Yu and C. Z.Liu, Coll. Surf. A251(1–3) (2004) 103–107.
177. S. Vaucher, M. Li and S. Mann, Angew. Chem. Int.Ed. 39 (2000) 1793–1796.
178. D. Kuang, A. Xu, Y. Fang, H. Ou and H. Liu,J. Cryst. Growth 244 (2002) 379–383.
179. A. Courty, C. Fermon and M. P. Pileni, Adv. Mater.13(4) (2001) 254–258.
180. C. E. Bunker, B. A. Harruff, P. Pathak, A. Payzant,L. F. Allard and Y. P. Sun, Langmuir 20 (2004)5642–5644.
181. D. K. Kim, Y. Zhang, W. Voit, K. V. Rao andM. Muhammed, J. Magn. Magn. Mater. 225 (2001)30–36.
182. N. C. Tansil, R. V. Ramanujan and H. F. Li, Trans.Ind. Inst. Met. 56(5) (2003) 509–512.
183. J. C. Lin, J. T. Dipre and M. Z. Yates, Chem.Mater. 15 (2003) 2764–2773.
184. D. C. Steytler, A. Gurgel and R. Ohly, Langmuir20 (2004) 3509–3512.
185. P. A. Dresco, V. S. Zaitsev, R. J. Gambino andB. Chu, Langmuir 15 (1999) 1945–1951.
186. P. Barnickel, A. Wokaun, W. Sager and H. F.Eickel, J. Coll. Interface Sci. 148(1) (1992)80–90.
187. D. H. Chen and S. H. Wu, Chem. Mater. 12 (2000)1354–1360.
188. K. E. Marchand, M. Tarret, J. P. Lechaire,L. Normand, S. Kasztelan and T. Cseri, Coll. Surf.A214 (2003) 239–248.
189. C. Kitchens, M. C. McLeod and B. Roberts,J. Phys. Chem. B107 (2003) 11331–11338.
190. F. Debuigne, J. Cuisenaire, L. Jeunieau,B. Masereel and J. B. Nagy, J. Coll. Interface Sci.243 (2001) 90–101.
191. R. Singh, J. Doolittle, M. A. George and P. K.Dutta, Langmuir 18(21) (2002) 8193–8197.
192. H. Chakraborty and M. Sarkar, Langmuir 20 (2004)3551–3558.

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 275
193. N. Pinna, M. Willinger, K. Weiss, J. Urban andR. Schlogl, Nano Lett. 3(8) (2003) 1131–1134.
194. Q. S. Wu, N. W. Zheng, Y. P. Ding and Y. D. Li,Inorg. Chem. Commun. 5(9) (2002) 671–673.
195. S. A. Safran, Nature Mater. 2 (2003) 71–72.196. J. Sims, A. Kumbhar, J. Lin, F. Agnoli,
E. Carpenter, C. Sangregorio, C. Frommen,V. Kolesnichenko and C. J. O’Connor, Mol. Cryst.Liq. Crys. 379 (2002) 121–130.
197. L. P. Li and G. S. Li, Hyperfine Interact. 128 (2000)437–442.
198. S. H. Liou and C. L. Chien, Appl. Phys. Lett. 52(6)(1988) 512–514.
199. C. R. Hendricks, V. W. R. Amarakoon andD. Sullivan, Ceram. Bull. 70(5) (1991)817–823.
200. Y. S. Cho, D. Schaffer, V. L. Burdick and V. R.W. Amarakoon, Mater. Res. Bull. 34(14/15) (1999)2361–2368.
201. K. Mandal, S. P. Mandal, P. Agudo and M. Pal,Appl. Surf. Sci. 182 (2001) 386–389.
202. K. G. Brooks and V. R. W. Amarakoon, J. Am.Ceram. Soc. 74(4) (1991) 851–853.
203. F. A. Selmi and V. R. W. Amarakoon, J. Am.Ceram. Soc. 71(11) (1988) 934–937.
204. A. Chatterjee, D. Das, S. K. Pradhan andD. Chakravorty, J. Magn. Magn. Mater. 127 (1993)214–218.
205. G. Beckingham, Aerogels: their history, struc-ture and application, http://members.tripod.com/∼geobeck/frontier/aerogels.html.
206. F. J. Arriagada and K. Osseo-Asare, Coll. Surf.A154 (1999) 311–326.
207. A. Van Blaaderen, J. Van Geest and A. Vrij, J. Coll.Interface Sci. 154(2) (1992) 481–501.
208. F. Bentivegna, J. Ferre, M. Nyvlt, J. P. Jamet,D. Imhoff, M. Canva, A. Brun, P. Veillet,S. Visnovsky, F. Chaput and J. P. Boilot, J. Appl.Phys. 83(12) (1998) 7776–7788.
209. S. Ponce-Castaneda, J. R. Martinez, F. Ruiz,S. Palomares-Sanchez and O. Dominguez, J. Sol-Gel Sci. Technol. 25 (2002) 29–36.
210. J. Wiggins, E. E. Carpenter and C. J. O’Connor,J. Appl. Phys. 87(9) (2000) 5651–5653, Part 2.
211. J. Lin, W. Zhou, A. Kumbhar, J. Wiemann,J. Fang, E. E. Carpenter and C. J. O’Connor,J. Solid State Chem. 159 (2001) 26–31.
212. K. Raj, B. Moskowitz and R. Casciari, J. Magn.Magn. Mater. 149 (1995) 174–180.
213. J. A. Lopez-Perez, M. A. Lopez-Quintela, J. Miraand J. Rivas, IEEE Trans. Magn. 33(5) (1997)4359–4362.
214. A. Kosturiak, J. Polavka, L. Valko, J. Slama,A. Gruskova and M. Miglierini, J. Magn. Magn.Mater. 153 (1996) 184–188.
215. N. S. Kommareddi, M. Tata, V. T. John,G. L. McPherson, M. F. Herman, Y. S. Lee,
C. J. O’Connor, J. A. Akkara, and D. L. Kaplan,Chem. Mater. 8 (1996) 801–809.
216. M. Lal, N. D. Kumar, M. P. Joshi and P. N. Prasad,Chem. Mater. 10 (1998) 1065–1068.
217. Y. Deng, L. Wang, W. Yang, S. Fu and A. Elaissari,J. Magn. Magn. Mater. 257 (2003) 69–78.
218. H. H. Freedman, J. P. Mason and A. I. Medalia,J. Org. Chem. 23 (1958) 76–82.
219. M. Summers and J. Eastoc, Adv. Coll. InterfaceSci. 100–102 (2003) 137–152.
220. T. Hirai, T. Watanabe and I. Komasawa, J. Phys.Chem. B104 (2000) 8962–8966.
221. A. Hammouda, T. Gulik and M. P. Pileni, Lang-muir 11 (1995) 3656–3659.
222. O. P. Yadav, A. Palmqvist, N. Cruise andK. Holmberg, Coll. Surf. A221 (2003) 131–134.
223. T. Hanaoka, T. Hatsuta, T. Tago, M. Kishidaand K. Wakabayashi, Appl. Catal. A190 (2000)291–296.
224. R. Touroude, P. Girard, G. Maire, J. Kizling,M. Boutonnet, M. Kizling and P. Stenius, Coll.Surf. 67 (1992) 9–19.
225. D. H. Chen and C. J. Chen, J. Mater. Chem. 12(2002) 1557–1562.
226. M. L. Wu, D. H. Chen and T. C. Huang, Langmuir17 (2001) 3877–3883.
227. X. Zhang and K. Y. Chan, Chem. Mater. 15 (2003)451–459.
228. X. Zhang and K. Y. Chan, J. Mater. Chem. 12(2002) 1203–1206.
229. W. E. Stallings and H. H. Lamb, Langmuir 19(2003) 2989–2994.
230. J. Fang, K. L. Stokes, J. A. Wiemann, W. L. Zhou,J. Dai, F. Chen and C. J. O’Connor, Mater. Sci.Eng. B83 (2001) 254–257.
231. J. P. Cason and C. B. Roberts, J. Phys. Chem.B104 (2000) 1217–1221.
232. S. Patil, S. C. Kuiry, S. Seal and R. Vanfleet,J. Nanoparticle Res. 4(5) (2002) 433–438.
233. J. A. Rodriguez, J. C. Hanson, J. Y. Kim andG. Liu, J. Phys. Chem. B107 (2003) 3535–3543.
234. A. B. Hungria, A. Martinez-Arias, M. Fernandez-Garcia, A. Iglesias-Juez, A. Guerrero-Ruiz, J. J.Calvino, J. C. Conesa and J. Soria, Chem. Mater.15(22) (2003) 4309–4316.
235. M. Fernandez-Garcia, A. Martinez-Arias, A. B.Hungria, A. Iglesias-Juez, J. C. Conesa andJ. Soria, Phys. Chem. Chem. Phys. 4(11) (2002)2473–2481.
236. A. Martinez-Arias, M. Fernandez-Garcia, A. B.Hungria, J. C. Conesa and G. Munuera, J. Phys.Chem. B107(12) (2003) 2667–2677.
237. A. Martinez-Arias, M. Fernandez-Garcia, A. B.Hungria, J. C. Conesa and J. Soria, J. Alloy. Com-pound. 323 (2001) 605–609.
238. M. Fernandez-Garcia, A. Martinez-Arias,A. Iglesias-Juez, A. B. Hungria, J. A. Anderson,

June 2, 2005 12:33 00700
276 V. Uskokovic & M. Drofenik
J. C. Conesa and J. Soria, Appl. Catal. B31(1)(2001) 39–50.
239. T. Kida, G. Q. Guan and A. Yoshida, Chem. Phys.Lett. 371(5–6) (2003) 563–567.
240. D. B. Dadyburjor, T. E. Fout and J. W. Zondlo,Catal. Today 63(1) (2000) 33–41.
241. A. J. I. Ward, E. C. O’Sullivan, J. C. Rang,J. Nedeljkovic and R. C. Patel, J. Coll. InterfaceSci. 161(2) (1993) 316–320.
242. T. Hirai, H. Sato and I. Komasawa, Ind. Eng.Chem. Res. 33 (1994) 3262.
243. C. B. Murray, D. J. Norris and M. G. Bawendi,J. Am. Chem. Soc. 115(19) (1993) 8706–8715.
244. C. M. Bender, J. M. Burtlich, D. Baber et al.,Chem. Mater. 12(7) (2000) 1969–1976.
245. M. I. Freedhoff, W. Chen, J. M. Rehm, C. Meyers,A. Marchetti and G. McLendon, in Fine ParticlesScience and Technology, ed. E. Pelizzetti (1996),pp. 281–293.
246. T. Sugimoto and K. Kimijima, J. Phys. Chem.B107 (2003) 10753–10759.
247. S. G. Dixit, A. R. Mahadeshwar and S. K. Haram,Coll. Surf. A133(1–2) (1998) 69–75.
248. S. Q. Qiu, J. X. Dong and G. X. Chen, PowderTechnol. 113(1–2) (2000) 9–13.
249. E. E. Foos, R. M. Stroud and A. D. Berry, NanoLett. 1(12) (2001) 693–695.
250. F. T. Quinlan, J. Kuther, W. Tremel, W. Knoll,S. Risbud and P. Stroeve, Langmuir 16(8) (2000)4049–4051.
251. J. B. Nagy, I. Bodart-Ravet and E. G. Derouane,Faraday Discuss. 87 (1989) 189–198.
252. J. B. Nagy, Coll. Surf. 35(2–4) (1989) 201–220.253. A. K. Panda, S. P. Moulik, B. B. Bhowmik and
A. R. Das, J. Coll. Interface Sci. 235 (2001)218–226.
254. S. P. Moulik, G. C. De, A. K. Panda, B. B.Bhowmik and A. R. Das, Langmuir 15 (1999)8361–8367.
255. Y. Berkovich, A. Aserin, E. Wachtel and N. Garti,J. Coll. Interface Sci. 245 (2000) 58–67.
256. M. Wu, Y. D. Zhang, S. Hui, T. D. Xiao, S. Ge,W. A. Hines and J. I. Budnick, J. Appl. Phys. 92(1)(2002) 491–495.
257. D. S. Bae, K. S. Han and J. H. Adair, J. Am.Ceram. Soc. 85(5) (2002) 1321–1323.
258. D. S. Bae, K. S. Han and J. H. Adair, J. Mater.Sci. Lett. 21(1) (2002) 53–54.
259. D. S. Bae, S. W. Park, K. S. Han and J. H. Adair,Met. Mater. Int. 7(4) (2001) 399–402.
260. T. Tago, S. Tashiro, Y. Hashimoto, K. Wakabayashiand M. Kishida, J. Nanoparticles Res. 5 (2003)55–60.
261. K. M. K. Yu, C. M. Y. Yeung, D. Thompsettand S. C. Tsang, J. Phys. Chem. B107 (2003)4515–4526.
262. C. Vestal and Z. J. Zhang, Nano Lett. 3 (2003)1739–1743.
263. S. S. Hong, M. S. Lee, S. S. Park and G. D. Lee,Catal. Today 87(1–4) (2003) 99–105.
264. P. Tartaj and L. C. De Jonghe, J. Mater. Chem.10(12) (2000) 2786–2790.
265. C. J. O’Connor, J. A. Sims, A. Kumbhar, V. L.Kolesnichenko, W. L. Zhou and J. A. Wiemann,J. Magn. Magn. Mater. 226–230 (2001)1915–1917.
266. A. Kumbhar, L. Spinu, F. Agnoli, K. Y. Wang,W. L. Zhou and C. J. O’Connor, IEEE Trans.Magn. 37(4) (2001) 2216–2218.
267. T. Kinoshita, S. Seino, K. Okitsu, T. Nakayama,T. Nakagawa and T. A. Yamamoto, J. Alloy. Com-pound. 359 (2003) 46–50.
268. S. A. Gomez-Lopera, R. C. Plaza and A. V.Delgado, J. Coll. Interface Sci. 240 (2001) 40–47.
269. T. Hirai, J. Y. Mizumoto, S. Shiojiri andI. Komasawa, J. Chem. Eng. Jpn. 30(5) (1997)938–943.
270. L. M. Qi, J. M. Ma, H. M. Cheng and Z. G. Zhao,Chin. Chem. Lett. 6(11) (1995) 1013–1016.
271. M. A. Lopez-Quintela, J. Rivas, M. C. Blancoand C. Tojo, in Nanoscale Materials, eds. L. M.L. Marzan and P. V. Kamat, (Kluwer AcademicPlenum Publ, 2003), pp. 135–155.
272. Y. Ni, X. Ge, H. Liu, Z. Zhang and Q. Ye, Chem.Lett. 9 (2001) 924–925.
273. M. Summers, J. Eastoe and S. Davis, Langmuir 18(2002) 5023–5026.
274. P. Ayyaub, A. N. Maitra and D. O. Shah, PhysicaC168(5–6) (1990) 571–579.
275. P. Ayyub and M. S. Multani, Mater. Lett. 10(9–10)(1991) 431–436.
276. F. Li and C. Vipulanandan, IEEE Trans. Appl.Supercond. 13(2) (2003) 3196–3198.
277. Q. S. Wu, N. W. Zheng and Y. P. Ding, Chem. J.Chin. Uni. 22(6) (2001) 898–900.
278. L. Guo, Z. Wu, T. Liu, W. Wang and H. Zhu, Chem.Phys. Lett. 318 (2000) 49–52.
279. L. M. Qi, J. M. Ma, H. M. Cheng and Z. G. Zhao,J. Phys. Chem. B101(18) (1997) 3460–3463.
280. Y. Liu, C. Zheng, W. Wang, Y. Zhan and G. Wang,J. Cryst. Growth 233 (2001) 8–12.
281. X. Y. Dong, X. T. Zhang, G. Cheng, Y. C. Li andZ. L. Du, Acta Chim. Sin. 62(24) (2004) 2441–2443.
282. J. D. Hopwood and S. Mann, Chem. Mater. 9(8)(1997) 1819–1828.
283. X. Mo, C. Wang, L. Hao, M. You, Y. Zhu,Z. Chen and Y. Hu, Mater. Res. Bull. 36 (2001)1925–1930.
284. H. H. Yang, H. Y. Qu, P. Lin, S. H. Li, M. T. Dingand J. G. Xu, Analyst 128(5) (2003) 462–466.
285. Z. Q. Ye, M. Q. Tan, G. L. Wang and J. L. Yuan,J. Mater. Chem. 14(5) (2004) 851–856.

June 2, 2005 12:33 00700
Synthesis of Materials within Reverse Micelles 277
286. Z. Q. Ye, M. Q. Tan, G. L. Wang and J. L. Yuan,Anal. Chem. 76(3) (2004) 513–518.
287. S. Bose and S. K. Saha, Chem. Mater. 15 (2003)4464–4469.
288. J. Z. Liu, B. D. Gou, S. J. Xu and K. Wang, Prog.Natural Sci. 12(8) (2002) 615–617.
289. S. Sarda, M. Heughebaert and A. Lebugle, Chem.Mater. 11(10) (1999) 2722–2727.
290. A. J. Zarur, H. H. Hwu and J. Y. Ying, Langmuir16 (2000) 3042–3049.
291. C. J. Barbe, R. Graf, K. S. Finnie, M. Blackford,R. Trautman and J. R. Bartlett, J. Sol-Gel Sci.Technol. 26 (2003) 457–462.
292. G. D. Rees, R. G. Evans, S. J. Hammond and B. H.Robinson, Langmuir 15(6) (1999) 1993–2002.
293. L. Sun, Y. Zhang, J. Zhang, C. Yan, C. Liao andY. Lu, Solid State Commun. 124 (2002) 35–38.
294. M. H. Lee, S. G. Oh and S. C. Yi, J. Coll. InterfaceSci. 226(1) (2000) 65–70.
295. T. Hirai, T. Hirano and I. Komasawa, J. Coll. Inter-face Sci. 253(1) (2002) 62–69.
296. B. Delfort, L. Normand, P. Dascotte and L. Barre,J. Coll. Interface Sci. 207(2) (1998) 218–227.
297. B. Delfort, A. Chive and L. Barre, J. Coll. InterfaceSci. 186(2) (1997) 300–306.
298. K. S. N. Reddy, L. M. Salvati, P. K. Dutta, P. B.Abel, K. I. Suh and R. R. Ansari, J. Phys. Chem.100(23) (1996) 9870–9880.
299. K. Kon-no, in Fine Particles Science and Technol-ogy, ed. E. Pelizzetti (1996), pp. 431–42.
300. J. Y. Liang, GuoLin, X. H. Bin, L. Jing, L. X.Dong, W. Z. Hua, W. Z. Yu and J. Weber, J. Cryst.Growth 252 (2003) 226–229.
301. G. Wakefield, H. A. Keron and P. J. Dobson et al.J. Coll. Interface Sci. 215(1) (1999) 179–182.
302. K. C. Song and J. H. Kim, J. Coll. Interface Sci.212(1) (1999) 193–196.
303. M. S. Lee, G. D. Lee and S. S. Hong, J. Indust.Eng. Chem. 9(4) (2003) 412–418.
304. Z. Wu, J. Zhang, R. E. Benfield, Y. Ding,D. Garndjean, Z. Zhang and X. Ju, J. Phys. Chem.B106 (2002) 4569–4577.
305. Y. X. Pang and X. Bao, J. Mater. Chem. 12 (2002)3699–3704.
306. P. Tartaj and J. Tartaj, Chem. Mater. 14 (2002)536–541.
307. Z. You, I. Balint and K. Aika, Chem. Lett. 11 (2002)1090–1091.
308. I. Balint, Z. You and K. Aika, Phys. Chem. Chem.Phys. 4 (2002) 2501–2503.
309. M. Hayashi, H. Uemura, K. Shimanoe, N. Miuraand N. Yamazoe, Electrochem. Solid State Lett.1(6) (1998) 268–270.
310. V. Uskokovic, D. Makovec and M. Drofenik, Mater.Sci. Forum 494 (2005) 155–160.
311. V. Uskokovic and M. Drofenik, J. Mater. Sci., sub-mitted (2005).
312. I. Ban, M. Drofenik and D. Makovec, Mater. Sci.Forum 494 (2005) 161–166.
313. S. A. Morrison, C. L. Cahill, E. E. Carpenter,S. Calvin and V. G. Harris, J. Appl. Phys. 93(10)(2003) 7489–7491.
314. M. Drofenik, D. Lisjak and D. Makovec, Mater. Sci.Forum 494 (2005) 129–136.
315. M. Han, R. Vestal and Z. J. Zhang, J. Phys. Chem.B108 (2004) 583–587.
316. C. R. Vestal and Z. J. Zhang, Chem. Mater. 14(9)(2002) 3817–3822.
317. M. Han, R. Vestal and Z. J. Zhang, J. Phys. Chem.B108 (2004) 583–587.
318. S. Bandow, K. Kimura, K. Kon-no and A. Kitahara,Jpn. J. Appl. Phys. 26(5) (1987) 713–717.
319. C. Y. Wang, W. Q. Jiqng, Y. Zhou, Y. N. Wang andZ. Y. Chen, Mater. Res. Bull. 35(1) (2000) 53–58.
320. F. Meyer, A. Dierstein, C. Beck, W. Hartl,R. Hempelmann, S. Mathur and M. Veith, Nanos-truct. Mater. 12 (1999) 71–74.
321. P. Kluson, P. Kacer, T. Cajthaml and M. Kalaji,J. Mater. Chem. 11(2) (2001) 644–651.
322. J. Huang, Y. Xie, B. Li, Y. Liu, J. Lu and Y. Qian,J. Coll. Interface Sci. 236 (2001) 382–384.
323. K. Mallick, Z. L. Wang and T. Pal, J. Photochem.Photobiol. A140(1) (2001) 75–80.
324. G. C. Koumoulidis, A. P. Katsouilidis, A. K.Ladavos, P. J. Pomonis, C. C. Trapalis, A. T.Sdoukos and T. C. Vaimakis, J. Coll. Interface Sci.259(2) (2003) 254–260.
325. M. Bonini, U. Bardi, D. Berti, C. Neto andP. Baglioni, J. Phys. Chem. B106 (2002)6178–6183.
326. H. S. Wu and E. W. Kaler, Microemulsion polymer-ization systems and coated materials made there-from, US Patent 5,539,047 (1996).
327. H. Althues and H. Kaskel, Langmuir 18 (2002)7428–7435.
328. H. Ago, S. Ohshima, K. Uchida, T. Komatsu andM. Yumura, Physics B323 (2002) 306–307.
329. H. Ago, S. Ohshima, K. Tsukuagoshi, M. Tsujiand M. Yumura, Curr. Appl. Phys. 5(2) (2005)128–132.
330. J. L. Zhang, Z. M. Liu, B. X. Han, T. Jiang, W. Z.Wu, J. Chen, Z. H. Li and D. X. Liu, J. Phys. Chem.B108(7) (2004) 2200–2204.