t otalenergy mo dels for phasestabilit y studies - ceder...
TRANSCRIPT

Total�energy models for phase�stability studies
in multicomponent oxides
by
Adri�an Favio Kohan
Licenciado en Ciencias F��sicas
Universidad de Buenos Aires� ����
Submitted to the Department of Materials Science and Engineering
in partial ful�llment of the requirements for the degree of
Doctor of Philosophy in Materials Science
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
June ����
c� Massachusetts Institute of Technology ����� All rights reserved�
Author � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �
Department of Materials Science and Engineering
May �� ����
Certi�ed by � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �
Gerbrand Ceder
Associate Professor of Materials Science
Thesis Supervisor
Accepted by � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �
Linn W� Hobbs
John F� Elliott Professor of Materials
Chairman� Departmental Committee on Graduate Students


A el tate�in memoriam


Total�energy models for phase�stability studies in
multicomponent oxides
by
Adri�an Favio Kohan
Submitted to the Department of Materials Science and Engineeringon May �� ����� in partial ful�llment of the
requirements for the degree ofDoctor of Philosophy in Materials Science
Abstract
The computation of thermodynamical properties of oxides from �rst principles in�volves the use of both statistical� and quantum�mechanical models� We show that thesolution of the former models can be considerably simpli�ed with a low�temperatureexpansion of the free energy� This overcomes the numerical problems of standardstatistical�mechanical techniques at low temperatures or for highly�stoichiometriccompounds� We �nd that with only a few terms in the expansion� an accurate de�scription is obtained up to temperatures where Monte Carlo simulations or clustervariation calculations are practical�
The formation energies of a large number of real or hypothetical compounds arean essential input to compute free energies� These values are usually obtained fromsophisticated and time�consuming quantum�mechanical calculations� We show thatthe same accuracy can be obtained from simpler approaches when all the relevantcontributions in the Hamiltonian are properly included� We identify four eects thatare necessary to accurately reproduce formation energies in oxides charge transfer�nonspherical electronic relaxations� ionic breathing� and directional bonding� We suc�cessfully integrated these eects into a tight�binding formulation� The Hamiltonianof the model includes explicit parametrizations of these terms� The parameters are �tto pseudopotential bands and equations of state� From this information� formationenergies can be obtained with an accuracy comparable to the pseudopotential pre�dictions� but at a fraction of the computational cost� On average� the tight�bindingformation energies in the CaO�MgO and CaO�ZrO� systems only dier by ��� fromthe pseudopotential results�
As an example of the applicability of the new tight�binding scheme to complexoxides� we use it to study the dependence of the ionic charge on the atomic environ�ment� We show that the variations in valence state can be important and producesigni�cant errors in the formation energies when they are not properly accounted for�
Thesis Supervisor Gerbrand CederTitle Associate Professor of Materials Science


Total�energy models for phase�stability studies in
multicomponent oxides
by
Adri�an Favio Kohan
Submitted to the Department of Materials Science and Engineeringon May �� ����� in partial ful�llment of the
requirements for the degree ofDoctor of Philosophy in Materials Science
Abstract
The computation of thermodynamical properties of oxides from �rst principles in�volves the use of both statistical� and quantum�mechanical models� We show that thesolution of the former models can be considerably simpli�ed with a low�temperatureexpansion of the free energy� This overcomes the numerical problems of standardstatistical�mechanical techniques at low temperatures or for highly�stoichiometriccompounds� We �nd that with only a few terms in the expansion� an accurate de�scription is obtained up to temperatures where Monte Carlo simulations or clustervariation calculations are practical�
The formation energies of a large number of real or hypothetical compounds arean essential input to compute free energies� These values are usually obtained fromsophisticated and time�consuming quantum�mechanical calculations� We show thatthe same accuracy can be obtained from simpler approaches when all the relevantcontributions in the Hamiltonian are properly included� We identify four eects thatare necessary to accurately reproduce formation energies in oxides charge transfer�nonspherical electronic relaxations� ionic breathing� and directional bonding� We suc�cessfully integrated these eects into a tight�binding formulation� The Hamiltonianof the model includes explicit parametrizations of these terms� The parameters are �tto pseudopotential bands and equations of state� From this information� formationenergies can be obtained with an accuracy comparable to the pseudopotential pre�dictions� but at a fraction of the computational cost� On average� the tight�bindingformation energies in the CaO�MgO and CaO�ZrO� systems only dier by ��� fromthe pseudopotential results�
As an example of the applicability of the new tight�binding scheme to complexoxides� we use it to study the dependence of the ionic charge on the atomic environ�ment� We show that the variations in valence state can be important and producesigni�cant errors in the formation energies when they are not properly accounted for�
Thesis Supervisor Gerbrand CederTitle Associate Professor of Materials Science


Acknowledgments
It would be impossible to acknowledge here all the people that in one way or anothershaped my education and consequently this thesis� I will just mention those directlyinvolved�
I would like to express my deepest gratitude to my advisor� Professor Gerd�Ceder� His guidance� encouragement� and enthusiasm were essential to build thisthesis� But� most of all� I am grateful to Gerd for providing me with the freedom topursue my own ideas with his unconditional support�
I thank the members of my thesis committee� Professor Samuel Allen and ProfessorJohn Joannopoulos for their suggestions� con�dence� and support�
Leaving Argentina to come to MIT was not an easy decision� I am very thankfulto Roberto Perazzo and Alberto Pignotti for their advice and teaching in science andin life� If there had not been for the long talks with Alberto in our way back fromwork� or with Roberto in a rainy Sunday afternoon I would probably have not beenwriting this now�
The pseudopotential calculations in this thesis were performed with a code thatwas generously provided by Professor John Joannopoulos� I am in debt to him andhis former student Kyeongjae Cho for teaching me the nuts and bolts of the pseu�dopotential technique�
Many other people contributed to the research presented here� I thank Markvan Schilfgaarde for providing the LMTO�ASA code� Harold Stockes and PatrickTepesch are acknowledged for the CaO�MgO SSCAD and pair�potential calculations�The General Utility Lattice Program �GULP�� kindly provided by Julian Gale� wasused for the pair�potential calculations in calcia�doped zirconias� Chris Wolvertonsupplied us with the Pd�V ECI�s employed in chapter �� Finally� the vibrationalcontribution to the CaO�MgO ECI�s was computed by Gerardo Garbulsky�
Various �nancial sources helped support my Ph�D� studies� I would like to ac�knowledge the fellowships provided by Roberto Rocca� the Starr Foundation� D� V�Ragone� G� Chin� S� Z� Uram� and N� J� Grant� Additional funding from the NationalScience Foundation �under contract No DMR�������� and the National Institute forHealth �under contract No ��P���ESO�������� is also acknowledged� I thank thePittsburgh Supercomputer Center for the possibility of using their Cray �� computer�where most of the pseudopotential calculations were performed�
I was very fortunate to be part of a highly motivating research group� The numer�ous conversations with Gerd� Patrick� Jerry� Anton� Shuba� Kadri� Axel� Eric� andNie had an invaluable impact on my work�

It is very di�cult to express in words what working with Jerry and Patrick meantto me� I will always miss our long discussions about any possible topic� During thistime is where I learned most of what I know� Their friendship is one of the mostprecious treasures I preserve from these years at MIT�
My frequent trips to Buenos Aires helped mitigate my nostalgia� I always lookedforward to meeting my family and my old friends Faby� Pablin� Guadu� and Gaby�And� when I am in Boston� my new friends Patrick� Kate� Felipe� Flor� Juanpi� Sally�Yair� Jackie� Julio� Vicky� Roby� Sandra� Ale� Daniel� Marcela� Pampa� Analia� andAlan make me feel at home�
Jerry� Marce� Luigi� and Adri were both my friends and family during these years�In the good and in the bad times� they always made me laugh� Our camping experi�ences in the wild�� our ten minutes� hikes� or our long�weekend trips showed methat there was life beyond my thesis� For all these� I thank them very much�
To my older� brother Bele� I have to express thanks not only for his love� butalso for his encouragement and support� I also enjoyed the visits of my in laws��Norma� Jos�e� and Dari� I am sincerely grateful for their support and help in spite ofthe fact I stole they beloved daughter and sister�
When I was at primary school� I could hardly spell the word Mam�a� Without thepatience and dedication of my mom� I would probably still be struggling with this�To her perseverance and love� I owe most of my career� I do not have words to thankher�
But� if there is one person I have to thank the most� that person is Cari� my wife�Without her caring and love� I could have never �nished this thesis� She gave methe strength to face di�cult times� the happiness to enjoy what I was doing� and thewisdom on which to build my dreams� After all� she is my c�omplice en la vida�
The words in this thesis are dedicated to someone that would probably neverread them� To someone that taught me the meaning of love� sweetness� ideals� andinnocence� To someone that would always give and never ask� To someone that wouldtirelessly �ght against windmills� Yes� it is true that according to the laws of physics�on which this thesis are based� he will not read these words� But he doesn�t knowthat� I am sure my dad will smile at the equations I am about to write���

Contents
Abstract �
Acknowledgments �
Contents �
List of Figures ��
List of Tables ��
� Introduction ��
��� Computational experiments � � � � � � � � � � � � � � � � � � � � � � � ������� Computer evolution and the limits of simulations � � � � � � � ������� The need for approximations � � � � � � � � � � � � � � � � � � � ��
��� Phase diagrams � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ����� Total�energy methods � � � � � � � � � � � � � � � � � � � � � � � � � � ����� Oxides � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ����� Thesis outline � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ��
� The computation of phase diagrams ��
��� General formalism � � � � � � � � � � � � � � � � � � � � � � � � � � � � ������� The alloy problem � � � � � � � � � � � � � � � � � � � � � � � � ������� The Ising model � � � � � � � � � � � � � � � � � � � � � � � � � � ������� The cluster expansion � � � � � � � � � � � � � � � � � � � � � � ������� The computation of ECI�s � � � � � � � � � � � � � � � � � � � � ������� Thermodynamic properties � � � � � � � � � � � � � � � � � � � � ��
��� Low�temperature expansion of a thermodynamic potential � � � � � � ������� Formalism � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ������� Implementation � � � � � � � � � � � � � � � � � � � � � � � � � � ��
��� Applications of the low�temperature expansion � � � � � � � � � � � � ������� The MgO�CaO phase diagram � � � � � � � � � � � � � � � � � � ������� The Pd�V phase diagram � � � � � � � � � � � � � � � � � � � � ��
��� Conclusions � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ��
� Total�energy methods ��
��� The computation of total energies � � � � � � � � � � � � � � � � � � � � ��
�

��� Classical models � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ������� Approximations of classical models � � � � � � � � � � � � � � � ������� Pair�potential models � � � � � � � � � � � � � � � � � � � � � � ������� The embedded�atom method � � � � � � � � � � � � � � � � � � ������� Cluster functions for the energy � � � � � � � � � � � � � � � � � ��
��� Quantum�mechanical approaches � � � � � � � � � � � � � � � � � � � � ������� Common approximations in quantum�mechanical approaches � ������� The spherical self�consistent atomic deformation model � � � � ������� The linear mu�n�tin�orbital method � � � � � � � � � � � � � � ������� The full�potential linearized augmented�plane�wave method � ������� The pseudopotential method � � � � � � � � � � � � � � � � � � � ������� The tight�binding method � � � � � � � � � � � � � � � � � � � � ��
��� Time scaling � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ����� Conclusions � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ��
� The eect of approximations in total�energy methods pseudopo�
tentials as a benchmark ��
��� The pseudopotential method � � � � � � � � � � � � � � � � � � � � � � � ������� The plane�wave basis � � � � � � � � � � � � � � � � � � � � � � � ������� Pseudopotential approximation � � � � � � � � � � � � � � � � � ������� Pseudopotential properties � � � � � � � � � � � � � � � � � � � � ������� Pseudopotential generation � � � � � � � � � � � � � � � � � � � � ������� Computational procedure � � � � � � � � � � � � � � � � � � � � ������� Further developments � � � � � � � � � � � � � � � � � � � � � � � ��
��� Applications � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ������� The CaO�MgO system � � � � � � � � � � � � � � � � � � � � � � ������� The CaO�ZrO� system � � � � � � � � � � � � � � � � � � � � � � ���
��� Conclusions � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
� The eect of approximations in total�energy methods relevant terms
in the Hamiltonian ���
��� Eect of the frozen�core approximation � � � � � � � � � � � � � � � � � ������ Eect of the spherical�symmetry approximation for crystal potentials
the SSCAD case � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � �������� The SSCAD CaO�MgO phase diagram � � � � � � � � � � � � � �������� Polymorphic transformations in pure zirconia � � � � � � � � � ���
��� Eect of the pair�potential approximations � � � � � � � � � � � � � � � �������� The pair�potential CaO�MgO phase diagram � � � � � � � � � �������� Pair�potential predictions for pure and doped zirconias � � � � �������� Discussion � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��� Relevant contributions to the total energy of oxides � � � � � � � � � � ������ Conclusions � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
� The tight�binding method ���
��� Formulation of the method � � � � � � � � � � � � � � � � � � � � � � � � ���
��

��� The semiempirical tight�binding method � � � � � � � � � � � � � � � � �������� The Slater�Koster formulation � � � � � � � � � � � � � � � � � � �������� Tight�binding total�energy model � � � � � � � � � � � � � � � � ���
��� Self�consistent tight�binding total�energy model � � � � � � � � � � � � �������� Nonorthogonality contributions � � � � � � � � � � � � � � � � � �������� Intra�atomic electron�electron interactions � � � � � � � � � � � �������� The self�consistent tight�binding Hamiltonian � � � � � � � � � �������� Total energies within the self�consistent formulation � � � � � � �������� Implementation � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��� Computation of the tight�binding parameters � � � � � � � � � � � � � �������� Fitting procedure � � � � � � � � � � � � � � � � � � � � � � � � � �������� Tight�binding parameters for oxide mixtures from end�member
information � � � � � � � � � � � � � � � � � � � � � � � � � � � � ������ Coding the self�consistent tight�binding method � � � � � � � � � � � � ������ Conclusions � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
� Applications of the tight�binding method ���
��� The CaO�MgO system � � � � � � � � � � � � � � � � � � � � � � � � � � �������� Predictions of the universal� model � � � � � � � � � � � � � � �������� Fitting to the CaO�MgO system � � � � � � � � � � � � � � � � �������� Formation energies � � � � � � � � � � � � � � � � � � � � � � � � �������� Phase diagram � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��� Pure and doped zirconia � � � � � � � � � � � � � � � � � � � � � � � � � �������� Pure zirconia � � � � � � � � � � � � � � � � � � � � � � � � � � � �������� Calcia�doped zirconia � � � � � � � � � � � � � � � � � � � � � � ���
��� Tight�binding parameters from �ts to end members � � � � � � � � � � ������ Conclusions � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
� Charge transfer in oxides ���
��� Simple charge model for metallic alloys � � � � � � � � � � � � � � � � � ������ Charge transfer in oxides � � � � � � � � � � � � � � � � � � � � � � � � � ���
����� Charge transfer in calcia�doped zirconia � � � � � � � � � � � � �������� Discussion � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��� Conclusions � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
� Conclusions ���
� Future research ���
A Additional results for the CaO�MgO system ���
A�� Introduction � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���A�� Formation energies � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���A�� Pair�potential model for CaO�MgO � � � � � � � � � � � � � � � � � � � ���
B �k�space integration ���
B�� Introduction � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��

B�� Special �k points � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���B���� Uniform mesh � � � � � � � � � � � � � � � � � � � � � � � � � � � ���B���� The Chadi�Cohen scheme � � � � � � � � � � � � � � � � � � � � ���B���� The Monkhorst�Pack scheme � � � � � � � � � � � � � � � � � � � ���
B�� Superstructures and equivalent� �k points � � � � � � � � � � � � � � � ���B�� Brillouin�zone integrations in metals � � � � � � � � � � � � � � � � � � ���
C The design of lithium batteries � �
C�� Introduction � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���C�� Pseudopotential studies of the battery voltage � � � � � � � � � � � � � ���
C���� Pseudopotential generation � � � � � � � � � � � � � � � � � � � � ���C���� Results � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���C���� Discussion � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
Bibliography � �
��

List of Figures
��� Peak performance of the fastest computer models built as a functionof time� The performance is written in �oating point operations persecond �Flops�� Also shown are the speeds of the latest workstations�Data from reference ��� and from manufacturers� information sheets� � ��
��� General procedure used to compute phase diagrams� The rhombusesrepresent the input information to the model� � � � � � � � � � � � � � ��
��� Stable structure at room temperature of pure zirconia� It can be ob�tained as a deformation of the �uorite structure� The black circlesrepresent zirconium ions while the white circles are the oxygen ions�The corners of the dashed cube are the oxygen positions in an ideal�uorite structure� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ��
��� Experimental phase diagram for the CaO�MgO system� There are nostable compounds in the solid phase� just a miscibility gap� See forexample reference ���� for more details� � � � � � � � � � � � � � � � � � ��
��� Experimental phase diagram for the Pd�V system� The Pd�V phaseis not shown since it is very di�cult to measure its boundaries ex�perimentally� Note that Pd�V and PdV� have a very narrow stabilityregion which makes traditional simulations di�cult to perform� Seereference ���� for experimental information� � � � � � � � � � � � � � � � ��
��� Mapping of dierent microstates j into con�guration space� The ar�rangement of the atoms on the parent lattice is labeled with �� It isassumed that there is a unique mapping from j onto �� � � � � � � � � ��
��� Mapping of a binary alloy con�guration onto an Ising model� If sitei is occupied by the black�white� atomic species� the spinlike variable�i � ������ is assigned to that site� � � � � � � � � � � � � � � � � � � ��
��� Two dimensional binary alloy lattice� where � dierent clusters areshown �� �� �� and �� They represent a point� a pair� a triplet� and aquadruplet cluster respectively� The value of the cluster functions forthese clusters is also indicated� following the convention that blackatoms�� white atoms�� correspond to spinlike variables �i � ������� ��
��� Procedure for computing eective cluster interactions using the Co�nnolly�Williams method� The system of equations is either solved bymatrix inversion� if the number of direct energies and ECI�s is the same�or by �tting the ECI�s� if they are less than the number of structuralenergies� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ��
��

��� Examples of linked and unlinked pair clusters on a two dimensionallattice� The only ECI�s dierent from zero are Vpair
� and Vpair� � � � � � ��
��� The rocksalt crystal structure� The black spheres represent magnesiumor calcium atoms� while the grey ones are the oxygen atoms� � � � � � ��
��� Solubility limits predicted by the low�temperature expansion using justthe single�defect energies� The experimental points are from refer�ence ����� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ��
���� Clusters used in the Pd�V cluster expansion� The pairs are identi�edaccording to their length� and the triplets according to the length oftheir longest side� the other two sides being nearest neighbors� � � � � ��
���� Maximal clusters used in the CVM� The ���point cluster is obtainedby taking a point and all its nearest neighbors on the fcc lattice� andthe ���point cluster corresponds to the conventional fcc unit cell� � � ��
���� Ground states of the Pd�V system predicted by the fcc ECI�s shownin table ���� The solid line represents the fcc ground�state line� Thedashed line corresponds to the change in the ground�state line whenthe A���type phase is included� The A�� structure is not an fcc su�perstructure� Its formation energy was computed by the LMTO�ASAmethod� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ��
���� Phase diagram for an alloy that models the Pd�V system obtained withthe combination of three dierent methods cluster variation method�CVM�� Monte Carlo �MC� simulations� and the low�temperature ex�pansion �LTE�� The solid lines represent low�temperature expansionpredictions and the dashed lines are the results of a Monte Carlo sim�ulation� The dotted lines correspond to phase boundaries computedusing the CVM and the LTE free energies together� Monte Carlo sim�ulations were performed for temperatures above ��� oK� The overlapbetween the methods is not shown for clarity� � � � � � � � � � � � � � ��
��� Illustration of the interaction between two ions in the pair�potentialmodel� Each ion is represented by a massless shell� on which a short�range pairwise interaction acts� and a core �here represented by theshaded and black circles� coupled by a harmonic force to the shell� ACoulombic� long�range interaction� is also assumed to exist betweenthe ions� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ��
��� Mu�n�tin potential often used to approximate the actual crystal po�tential� The sphere radii rspA and rspB �for atoms A and B respectively�are arbitrary� except for the fact that the spheres should not overlap� ��
��� Cubic unit cell showing the Wigner�Seitz overlapping spheres in theASA� The interstitial region �shaded area in the �gure� is usually ne�glected� The sum in the equation is over the spheres in the unit cell� � ��
��

��� Total energy for pure CaO as a function of the ratio between the Caand the O sphere radius in the LMTO�ASA� The volume of the cellwas kept constant and corresponds to the experimental value� Emptyspheres� and mu�n�tin and combined corrections were used to reducethe errors introduced by the ASA� The total energy changes in a scalemuch larger than the scale of the temperature eects and a criterion isneeded to choose the sphere size� � � � � � � � � � � � � � � � � � � � � ��
��� Representation of the pseudo wave function and its corresponding pseu�dopotential �dashed line�� The actual wave function and potential fromwhich they are derived are also shown �solid line�� The point at whichthe real and pseudo values are matched is called rc� � � � � � � � � � � ��
��� Comparison between wave functions obtained by using a norm conserv�ing and an ultrasoft pseudopotential� The solid line is the pseudo wavefunction corresponding to a regular norm�conserving pseudopotentialand the dashed line to an ultrasoft one� � � � � � � � � � � � � � � � � � ��
��� Steps for computing total energies using the ab initio pseudopotentialapproach� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ��
��� Electronic band structures for pure CaO and MgO in the rocksalt struc�ture computed with the pseudopotential method� � � � � � � � � � � � ��
��� Valence electronic density and its contour plot for the ����� plane ofMgO� The Mg ions sit at the corners and center of the region of theplane shown in the picture while the oxygen ions sit at the middle ofthe edges� It is clear from the �gure that almost no valence electronsare close to the Mg ions� � � � � � � � � � � � � � � � � � � � � � � � � � ���
��� Polymorphic structures of zirconia� � � � � � � � � � � � � � � � � � � � ���
��� Experimental CaO�ZrO� phase diagram� See reference ������ � � � � � ���
��� Computed electronic density for the �p and �d orbitals of the Zr�� ion�The wave functions have been normalized to one� The overlap betweenthe densities can clearly be seen� � � � � � � � � � � � � � � � � � � � � � ���
��� Valence electronic density and its contour plot for the ����� plane ofcubic zirconia �shaded plane in the �gure�� �a� Full valence density��b� Contribution from the �d and �s bands� �The density coming fromthe oxygen orbitals is also shown as a reference�� � � � � � � � � � � � � ���
���� Smallest volume Zr�Ca�O structures in the �uorite lattice that pre�serve charge neutrality� The arrows indicate one possible set of primi�tive basis vectors� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
���� Valence electron density for the S� structure� Note that Ca atoms arefully ionized� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��� Total�energy methods and their most commonly used approximations�Also shown are the oxide systems where the eect of these approxima�tions on the formation energies will be tested� � � � � � � � � � � � � � ���
��

��� Eective cluster interactions for the CaO�MgO system computed withthe SSCAD� The pairs are identi�ed according to their length and thetriplets and quadruplets according to their longest side� the other sidesbeing nearest neighbors� � � � � � � � � � � � � � � � � � � � � � � � � � ���
��� CaO�MgO phase diagram� Only the solid solubility limits� computedby a combination of Monte Carlo simulations and the CVM� are shown�The solid and dotted lines were determined with the ECI�s derived fromSSCAD and from empirical potential models respectively� The circlesrepresent experimental values ���� and the dashed line indicates theexperimental eutectic temperature� � � � � � � � � � � � � � � � � � � � ���
��� Formation energies for dierent ordered structures in the CaO�MgOsystem obtained with pseudopotentials �PP�� the SSCAD� and empir�ical potentials �EP�� The number that identi�es each structure corre�sponds to the one used in table ���� � � � � � � � � � � � � � � � � � � � ���
��� Valence electron density for the ����� plane of CaO as computed withthe pseudopotential method� Note that the oxygen ions are not per�fectly spherical� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��� Formation energies in the CaO�ZrO� system predicted by pseudopoten�tials �PP� and empirical potentials �Pot��� The structures are identi�edfollowing the convention in �gure ����� � � � � � � � � � � � � � � � � � ���
��� SSCAD and pair�potential formation energies for the CaO�MgO sys�tem as a function of the pseudopotential predictions� The parametersused in the pair potential are shown in table ���� The closer the pointsto the solid line the better the performance of the potential or theSSCAD method is� � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��� Integrated valence charge density within a sphere centered around oxy�gen� as a function of the sphere radius� The data was computed frompseudopotential calculations� � � � � � � � � � � � � � � � � � � � � � � � ���
��� Nomenclature used to identify the unit cells and atomic positions� �R�
and �R�� point to the origin of the unit cell �two unit cells are markedin the picture with two squares�� �� and �� � indicates the position of theatoms within the unit cell� � � � � � � � � � � � � � � � � � � � � � � � � ���
��� Illustration of the representation of the Hamiltonian integral for a sand a px orbital �Es�x� as a function of two center integrals �tll�m��Note that tsp� is zero by symmetry� � � � � � � � � � � � � � � � � � � � ���
��� Main features of the algorithm used to implement the self�consistenttight�binding total�energy method� The merit function is de�ned asthe sum of the squares of the dierences between the properties pre�dicted by tight binding and the ab initio or experimental results usedin the �t� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��

��� Formation energies for dierent ordered structures in the CaO�MgOsystem obtained with pseudopotentials �PP� and tight binding �TB��The number that identi�es each structure corresponds to the one usedin table ���� The tight�binding parameters were �t only to L�� �struc�ture number � in the plot� and both CaO and MgO� � � � � � � � � � � ���
��� Eective cluster interactions for the CaO�MgO system computed withthe tight�binding formation energies� The pairs are identi�ed accord�ing to their length and the triplets and quadruplets according to theirlongest side� the other sides being nearest neighbors� � � � � � � � � � � ���
��� Formation energies as a function of composition for dierent orderedstructures in the CaO�MgO system� The diamonds correspond totight�binding results and the crosses to the cluster expansion �t� � � � ���
��� CaO�MgO phase diagram� The lines correspond to the solubility lim�its computed with a Monte Carlo simulation and without taking intoaccount the eect of atomic vibrations in the free energy� The pointsrepresent experimental values ����� Only solid phases were calculated� ���
��� Formation energies of structures S� to S� computed with tight binding�TB�� The pseudopotential �PP� and pair�potential �Pot� predictionscomputed in sections ����� and ��� are also shown for comparison� Adescription of the structures can be found in �gure ����� � � � � � � � ���
��� Ionic charges for structures S� to S� computed with tight binding� � � ���
��� Formation energies predicted for dierent mixtures of CaO and ZrO�
for the four cases explained in the text� A description of structures S�to S� can be found in �gure ����� � � � � � � � � � � � � � � � � � � � � ���
��� Charge �in electrons� versus generalized number of neighbors �N � Thestraight line is the prediction of the charge model of equation ��� whentwo neighbor layers are included� The � parameters were �t with�out imposing charge neutrality� The dots correspond to tight�bindinglarge�supercell calculations� � � � � � � � � � � � � � � � � � � � � � � � ���
��� Charge �in electrons� versus generalized number of neighbors �N � Thestraight line is the prediction of the charge model of equation ��� whenfour neighbor layers are included� The � parameters were �t with�out imposing charge neutrality� The dots correspond to tight�bindinglarge�supercell calculations� � � � � � � � � � � � � � � � � � � � � � � � ���
��� Charge �in electrons� versus generalized number of neighbors �N � Thestraight line is the prediction of the charge model of equation ��� whentwo neighbor layers are included� The � parameters were �t imposingcharge neutrality� The dots correspond to tight�binding large�supercellcalculations� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��� Charge �in electrons� at site i versus Coulomb potential at that site�The lines indicate the limiting behavior for small and large chargetransfer� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
C�� Charge and discharge cycle for a lithium battery� � � � � � � � � � � � ���
��

C�� Fermi energy of dierent LiMO� compounds in the ��NaFeO� structure�solid line�� With Ni� a new band starts to be �lled� producing a jumpin the voltage� The highest energy point of the band that was justcompletely �lled in Co is also shown �dashed line�� � � � � � � � � � � � ���
��

List of Tables
��� Eective cluster interactions �ECI�s� used to model the Pd�V system�The clusters are named according to the convention followed in �gure ����� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ��
��� Total�energy methods and their approximations� The bullets indicatethe most common approximations used by each technique� A detaileddescription of the methods and approximations can be found in sec�tion ���� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ��
��� Time scaling of some total�energy methods with respect to the sizeof the system N �number of atoms in the unit cell�� The values inparenthesis represent upper limits according to dierent implementa�tions� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ��
��� Calculated lattice constants and bulk modulus for pure CaO and MgO�The values in parenthesis correspond to experimental measurements� ��
��� Formation energies and cell parameters of dierent ordered structuresin the CaO�MgO system obtained by using the plane�wave pseudopo�tential method� All cell parameters were fully relaxed� including theinternal positions� xi� that are shown here according to the Wyckonotation in reference ������ Lattice constants are expressed in �A andformation energies are in eV ion� A description of each structure canbe found in reference ���� or ����� � � � � � � � � � � � � � � � � � � � � ���
��� Cell parameters and structural energy dierences for the tetragonal andcubic phases of zirconia at zero pressure� The energies are in eV perZrO� formula unit and the lattice constants are in �A� All cell parame�ters were fully relaxed� including the internal position zi �shown hereaccording to the Wycko notation in reference ������� The experimen�tal z value for the tetragonal structure was measured at ���� oC� Allthe other experimental values were interpolated to � oK as explainedin the text� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��� Formation energies and cell parameters of dierent ordered structuresin the CaO�ZrO� system obtained by using the plane�wave pseudopo�tential method� Only cubic volume relaxations were allowed� Thestructures are described in �gure ����� � � � � � � � � � � � � � � � � � ���
��

��� Formation energies and cell parameters for the L�� structure in theCaO�MgO system computed with the pseudopotential �PP� and thefull�potential linearized augmented�plane�wave �FLAPW� methods�The ions in the CaO�MgO system are arranged on two interpenetrat�ing fcc lattices� The L�� nomenclature identi�es the distribution ofthe Ca and Mg ions on the fcc cation sublattice �that corresponds tothe minority species at the corners of the conventional fcc cell� whilethe oxygen fcc sublattice remains fully occupied� Energy values areexpressed in eV ion and the cell parameters are in �A� The values cor�respond to fully relaxed structures� � � � � � � � � � � � � � � � � � � � ���
��� Cell parameters and structural energy dierences for the experimen�tally observed phases in zirconia at zero pressure� The energies are ineV per ZrO� formula unit and the lattice constants are in �A� All cellparameters were fully relaxed� including the internal positions� xi� yi�and zi� that are shown here according to the Wycko notation in refer�ence ������ The experimental z value for the tetragonal structure wasmeasured at ���� oC and the cell parameters are extrapolations to � oK�see chapter ��� PIB calculations for the tetragonal and monoclinicphases of zirconia are not reported in the table since these structuresare not predicted to be stable by this method� � � � � � � � � � � � � � ���
��� Defect formation energies� in eV� of a single MgO �CaO� in an otherwisepure CaO �MgO� lattice� The SSCAD values were computed using a�x�x� fcc supercell� The term rich� denotes the host lattice� � � � � ���
��� Vibrational ECI�s for the CaO�MgO system �dimensionless�� � � � � ���
��� Potential parameters derived in reference ����� The parameters arenamed following the same convention as in section ������ � � � � � � � ���
��� Potential parameters derived in references ���� ����� The parametersare named as in section ������ The �� corresponds to the rigid�ionapproximation� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��� Potential parameters �t to the pseudopotential formation energy ofthe L�� structure� its cell parameters� and the CaO and MgO cellparameters� The �� corresponds to the rigid�ion approximation� � � ���
��� Interatomic matrix elements as proposed by Harrison� The value ofthe rd constants can be found in reference ����� and d is the distancebetween the atoms� � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��� Parameters used with Harrison�s distance dependence� The orbital andintra�atomic parameters were taken from reference ������ � � � � � � � ���
��� Formation energies and cell parameters of dierent ordered arrange�ments in the CaO�MgO system obtained by using the tight�bindingapproach with the coe�cients from table ���� The cells were fully re�laxed� The values in parenthesis are the pseudopotential predictionsfrom table ���� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��

��� Defect formation energies of a single MgO �CaO� on an otherwise pureCaO �MgO� lattice� The values were computed using a �x�x� fccsupercell and are expressed in eV� The term rich� denotes the hostlattice� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��� Cell parameters and structural energy dierences for the experimen�tally observed phases in zirconia at zero pressure� The energies arein eV per ZrO� formula unit and the lattice constants are in �A� Allcell parameters were fully relaxed� including the internal positions� xi�yi� and zi� that are shown here according to the Wycko notation inreference ������ The experimental z value for the tetragonal structurewas measured at ���� oC and the cell parameters are extrapolationsto � oK �see chapter ��� � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��� Cell parameter for structures S� to S� computed by pseudopotential�tight binding� and pair potentials� The structures are described in�gure ����� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
��� Set of hopping integrals that were �t in both CaO�MgO and CaO�ZrO�� The universal parameters found in reference ����� are reproducedfor comparison� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
A�� Formation energies �meV atom� of dierent ordered structures in theCaO�MgO system obtained by using the SSCAD� the pair�potentialin section ������ and the self�consistent tight�binding methods� All thestructures were fully relaxed� A description of the structures can befound in references ���� or ����� The S�x�x� corresponds to the �x�x�supercell used in reference ����� � � � � � � � � � � � � � � � � � � � � � ���
C�� Valence electron con�guration and core radii �rcl� used to generate thepseudopotentials �l� s� p� d�� � � � � � � � � � � � � � � � � � � � � � � ���
C�� Average voltage �in volts� computed for the LiMO� compound in the��NaFeO� structure� The values in parenthesis are experimental� Theagreement is good� The large discrepancy in the nickel case is due tothe fact that the structure is unstable with respect to a Jahn�Tellerdistortion �not taken into account in the calculations�� � � � � � � � � ���
C�� Average voltage �volts� for LiCoX� compounds in the ��NaFeO� struc�ture� � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � � ���
C�� Average voltage �volts� for LiCoO� compound for two dierent struc�tures� For the unrelaxed case� the ideal rocksalt positions were taken� ���
��

��

Chapter �
Introduction
Humans have been trying to unravel the secrets of matter since the origins of civi�lization� Looking for the ideal hunting tools� the deadliest weapons� or the best heartimplants� the synthesis and development of new materials have never stopped� Theimportance of this search is re�ected in the fact that many periods of history arenamed after the materials that were used in them� The Stone Age� the Bronze Age�and the Iron Age were all strongly in�uenced� both socially and culturally� by the useof stone� bronze� and iron respectively�
The concept of matter is a very elusive one� The �rst attempts to apply somerationality to the study of materials can probably be tracked down to the origins ofAlchemy ���� Although most well known as a search for a way to transmutate thebase metals into noble metals in the sixteenth and seventeenth centuries� it producedan enormous amount of information about materials properties� processing� and ex�perimental techniques since the beginnings of the Egyptian era�� Even the brightestminds were seduced by the mysteries and powers of the alchemy knowledge� Forexample� Sir Isaac Newton wrote many alchemical papers in his search for under�standing the structure of matter and how it can be transformed ��� ��� In his Praxis�we can read ���
�����so soon as it beginns to calcine pour upon it � part of ye cold �re extracted
out of Lead ore wth salt of Venus � not yet volatized� � so on till you have
poured on eleven parts � all be calcined� �or ye Saturn will �rst resolve into
water by fusion � then resolve ye quicksilver of the bodies � promote ye action
of ye sympatetic �re������And thus you may understand what the �rst gate of
calcination is � how in the calcination of perfect bodies wth ye �rst menstrue
nothing unclean enters but ye green Lyon � how ye King after his resurrection
is fed with the blood of this Lyon�����
One has to remember that until the chemical elements were identi�ed� the mainalchemy theory �usually ascribed to Aristotle� but widely recognized in Egypt and
�The Egyptians were expert goldsmiths as far back as the year ���� B�C� They were also remark�ably skilled in metallurgy and enameling� The origin of the word �alchemy� can be tracked downto �Al Khem� that is how the Islamic world called �the art of the dark country� ���� Egypt wasknown as �Khem� or the country of �dark soil��
��

India long before his era� was that all bodies were composed of four elements �water�earth� air� and �re� in dierent proportions� One body could be transmutted intoanother by changing these proportions�� Having this theoretical background� mostadvances had to come from purely empirical approaches!
Profound changes occurred at the end of the XVIII and during the XIX centurieswhen the concepts of elements� atoms� and molecules were developed or perfected�The use of chemical compositions� rudimentary crystallography� and new technologies�such as better furnaces� dramatically improved the understanding of matter� Butstill� only a few empirically selected materials were used by the end of last century andonly �� of the �� naturally found elements formed them ���� A major revolution tookplace in the �rst half of the XX century due to the understanding of the interior of theatom� the introduction of new experimental techniques �such as x�ray diraction�� andthe use of accurate mathematical descriptions to formulate the semiempirical relationsalready known� Completely new materials were synthesized during this period and therole of structure and chemistry on their properties was widely investigated� Plastics�semiconductors� and synthetic �bers� are just a few examples� �See reference ��� fora much more detailed description of the evolution of materials science��
But how more advanced is our search for new materials now" What dierentmethods would a scientist use at the verge of the XXI century" The trial and errorprocedure evolved from being purely empirical to a very complex relationship betweensemiempirical laws� mathematical models� and experiments� Current technology pro�vides very advanced tools to characterize and understand materials behavior� makingthe whole process very e�cient� However� the possibilities for combining elements aregigantic� When a material with speci�c properties is sought� one is sometimes leftwith just well educated guesses on which elements to mix and how to process them�
As a hundred years ago� a revolution is starting to take shape� Advances incomputer technology and in the simulation techniques are making possible to justuse the guidance of the laws of physics to tailor the properties of materials to ourneeds ���� These so�called computational experiments are already complementingtraditional experiments and are starting to realize a future where novel materials willbe predicted and developed before they are synthesized�
This thesis is about some of the methods that are helping this revolution to takeplace� Our work is focused on how quantum� and statistical�mechanical techniquescan be used to study and tailor the properties of oxides� Oxides present a wide varietyof properties that make them very useful in many industries� from high�technologycompanies to refractory brick factories� The goal of this thesis is two�fold improvethe methods to compute phase diagrams and the quantum�mechanical inputs to them�For the former� we developed a new approach� based on a low�temperature expan�sion of the free energy� that circumvents the numerical problems that the standardstatistical�mechanical techniques have at low temperatures or for highly stoichiomet�
�This theory counted at those times with some �experimental evidence�� by using �re and waterthe character of matter could be changed actually its properties� For example the properties ofsteels can be changed by precisely using �re and water� Also the preparation of alloys that resemblegold was already known even by the Egyptians e�g� golden brass�
��

ric compounds� For the latter� we studied how the approximations used to computetotal energies worked in oxides� From these studies� we propose a new approach tocompute total energies in these materials� based on the tight�binding method� thatcombines the accuracy of the most complex quantum�mechanical techniques with thespeed of classical treatments�
We start in this chapter by describing what computer simulations are� what theycan do� and how our work relates to this �eld� In the �rst sections� we introduce theidea of computer simulations �section ���� and phase diagrams �section ����� Then� wedescribe the application of total�energy techniques �section ���� to compute oxide�sproperties and we introduce the problems that the complexity of oxide crystals posesfor computer simulations� Finally� we present a brief description of the world of oxidesand their applications �section ���� followed by a summary of the contributions madein this thesis �section �����
��� Computational experiments
Every time we observe a material we can think that nature is solving the fundamentalequations of physics to see how to move the atoms� In the same way� in a computerexperiment� the system under study �atomic nuclei and electrons� is evolved basedon the same equations� but this time in the space of the computer world�
Nature has to be an incredibly fast computer to be able to solve coupled nonlinearequations in more than ���� variables� self�consistently� and in real time�� Realcomputers are of course much slower and calculating the exact state of any relevantsystem in a reasonable time is basically impossible� However� approximations can beintroduced to make some of the problems tractable�
Computer experiments have many advantages over real ones� Extreme environ�mental conditions �e�g�� pressure� temperature� toxicity� are usually as easy to studyas any other condition� An exact control over all the variables of the computer ex�periment is always possible and thus the interpretation of the results can be made asclear as desired� Moreover� materials can be studied under conditions in which theywould not exist in nature� For example� to understand the stability trends of dierentcompounds in a given system any metastable structure can be simulated and not onlythose that can be observed due to sluggish kinetics�
����� Computer evolution and the limits of simulations
The ability to perform atomistic simulations was slow to emerge because of the ex�treme complexity of solving the quantum� and statistical�mechanical equations in�volved� Although most of the currently used approximations were known a long timeago� the calculation power was very limited� In ����� Gordon Moore� co�founder ofIntel� forecasted that the power and complexity of silicon�based integrated circuitswould double every �� to �� months with proportionate reductions in cost� This isknown as Moore�s Law and has proven to be remarkably accurate� For example� the�rst chip introduced by Intel ������ in ���� had ����� transistors and a speed of ����
��

MIPS �millions of instructions per second�� Today� �� years later� the Pentium Proprocessor has ��� million transistors and a speed of ��� MIPS� The fast growth incomputer power made it possible� in the last decades� to perform the �rst realisticatomistic simulations� Moore�s law is expected to be valid for at least �� more yearsusing the current technology� and it may hold for even longer if new advances areintroduced� Figure ��� shows the exponential growth of computer speed since theinvention of the �rst machines�
Correspondingly� computational materials science also had an incredible growth�The size of the systems solved is increasing every day and more accurate predictionsabout macroscopic materials properties can be done� However� even if the rate ofgrowth in computer power is sustained for many more decades� we will not be ableto predict properties exactly� The most accurate� but still approximate� quantum�mechanical techniques available can only deal with just a few atoms� The computertime they require usually scales at least as N� �where N is the number of atoms�� IfMoore�s law is valid for �� more years we will only be able to solve systems containingof the order of ��� atoms� or a cube of material with a side of ���� nanometer� Grainsizes� essential to understand the macroscopic behavior of materials span� according tothe processing� length scales from a few tens of nanometers to many centimeters� Thismeans that performing a brute force� almost exact� quantum�mechanical calculationto study macroscopic systems is out of the question��
����� The need for approximations
The introduction of approximations can dramatically increase the size of the systemsthat can be studied today� Simple classical�potential models can be used to solveproblems containing ��� atoms ���� In �� years� this will be translated into lengthscales of the order of tens of microns �assuming linear scaling with the size of thesystem�� On the other hand� complex quantum�mechanical methods�� can still onlyhandle a few hundreds of atoms� However� they are much more accurate thanpotential models�
There is usually a trade�o between accuracy and speed� The accuracy andspeed that the solution of the problem requires will dictate the necessary approx�imation level� The optimal balance between these two depends on the number ofcalculations that needs to be performed and the scale of the physical phenomenawe are looking at� For example� the study of phase stability usually requires manyquantum�mechanical calculations as input to the statistical models� The accuracyis set by the scale of temperature eects� typically only a few hundreths of an eV�Consequently� only very accurate techniques can be used and long computer runs areneeded� On the other hand� the wealth of information that can be obtained fromthese studies is enormous� and most of this thesis will be devoted to improve the ap�
�Even if the methods scaled linearly the size of the cube of material would be �� nanometers�This is the scale of the so�called nanophase materials that have been studied since ���� and whosegrains are formed by nanometer�size atomic clusters ����
�Using the local�density approximation to density�functional theory� See section ����
��

1940 1950 1960 1970 1980 1990 2000
Pea
k P
erfo
rman
ce (
Flo
ps)
Year
10
10
10
10
10
10
10
10
10
10
10
10
10
1
2
3
4
5
6
7
8
9
10
11
12
13
Eniac
Univac
IBM 701
IBM 704
IBM 7090
IBM StretchCDC 6600
CDC 7600
Cray 1
Intel iPSC/1Cray X-MP
nCUBE-1
CM-2
Intel iPSC/2Cray Y-MP
nCube-2
Intel Paragon, CM-5
Intel/Sandia
IBM RS/600 SP2-66Cray T3E-256
Cray T3D-513Cray C90-16/512
HP C180Alpha 600 5/300
Pentium ProHP9000/735
Sequential machines
At least modestly parallel
Figure ��� Peak performance of the fastest computer models built as a function oftime� The performance is written in �oating point operations per second �Flops��Also shown are the speeds of the latest workstations� Data from reference ��� andfrom manufacturers� information sheets�
��

proximations of current models in order to make them faster and� at the same time�accurate enough�
��� Phase diagrams
Most technologically relevant materials make use of more than one element of theperiodic table� The ability to tailor materials to our needs depends greatly on the un�derstanding of the relationship between properties� structure� and composition whendierent elements are mixed� In this sense� phase diagrams are one of the most im�portant tools to design and process materials e�ciently� We can think of a phasediagram as a map that shows what phases are stable in a temperature�compositionspace �at constant pressure�� This is very valuable information to predict the �nalmicrostructure of an alloy� after processing�
The experimental determination of phase diagrams is usually a di�cult task� Mea�surements are often hampered by sluggish kinetics or extreme temperatures� amongother conditions� On the other hand� by using the laws of physics it is possible� inprinciple� to compute any phase diagram using as only input the atomic numbers andmasses of the elements that are being mixed� This approach to compute materialsproperties is sometimes called ab initio� or from �rst principles��
In spite of the advances in computer power� we can not track the movements andinteractions of the more than ���� nuclei and their electrons that form a macroscopicsolid� All ab initio approaches introduce dierent degrees of approximations� In chap�ter �� we show how the alloy problem can be mapped onto lattice models that arespecially suited for studying the thermodynamic properties of substitutional alloys�From the free energies provided by the model� the phase diagram can be easily com�puted� In a substitutional alloy� the dierent species arrange themselves on the sitesof an underlying parent lattice�� We will focus on the stability of solid phases and wewill not include the liquid or gas portion of the diagrams�
To compute free energies using statistical mechanics� the dependence of the systemenergy on the substitutional arrangement of the ions on the lattice has to be known�Most of the ionic arrangements are not periodic� making these calculations di�cult ifnot impossible� Even for periodic systems� the solution of the quantum�mechanicalproblem is very complex and empirical potential models are often used�
The situation changed dramatically with the introduction of the cluster expansionby Sanchez and collaborators ���� They showed that any property of the alloy thatdepends on the arrangement of the ions on a given lattice can be expanded in clus�ter functions�� These functions are easily computed from the lattice site occupations�When the property that is being expanded only depends on the local environment�the cluster expansion is rapidly convergent and only a few coe�cients need to be
�An alloy is a combination of two or more elements� Note that we do not limit this de�nition tojust metallic alloys but we will also consider a ceramic mixture as an alloy�
�This is not a severe assumption� When two phases that are in equilibrium with each otherarrange themselves on di�erent lattices the free energies can be computed in each of the latticesand then compared to determine the stable phases�
��

determined� The usual procedure is to compute the energy of a few periodic arrange�ments using any total�energy method and from this information �nd the expansioncoe�cients ���� ��� ����
By total energy�� we mean the interaction energy of the whole system of electronsand nuclei that form the solid� Computing this energy from �rst principles is still oneof the most challenging problems in condensed�matter physics and is currently one ofthe limiting factors in ab initio predictions of phase stability� Even for a perfect crys�tal� calculations are very computer�time intensive� particularly when large unit cellsare involved� Consequently� it is very important to understand what approximationscan be introduced to make the methods faster but retaining the required accuracy�This will be the main focus of the present thesis�
The methodology we will follow to compute phase diagrams is summarized in�gure ���� The alloy problem is mapped onto a lattice model� Once a simpli�edHamiltonian is built �i�e�� once we assumed a given set of approximations�� the for�mation energies of many intermediate compounds are computed� The number ofcompounds depends on the system being studied� For example� for the CaO�MgOsystem �� values were enough to obtain the coe�cients used in the cluster expansion�If more than one parent lattice is necessary to describe the full phase diagram� acluster expansion is needed for each lattice� The next step is to compute the phaseboundaries� The regions of phase stability can be obtained by computing free ener�gies for each of the possible competing phases and keeping the ones that have theminimum values �or a combination of them� through the tangent construction���Two techniques are widely used in lattice models the cluster variation method �����CVM� and Monte Carlo ���� �MC� simulations� In its most basic formulation� theMC approach generates equilibrium ensembles using a stochastic procedure and thephase boundaries can be found� for example� by looking for discontinuities in concen�tration vs� temperature instead of computing free energies� Free energies can also beobtained by performing time�consuming path integrations from states where the freeenergy is known� On the other hand� the CVM is a generalized mean �eld techniquethat gives free energies directly�
��� Total�energy methods
The computation of total energies requires the determination of the interaction ener�gies between all the electrons and nuclei that form the material� Even when all thesymmetry of the problem is exploited� the calculations are very involved�
Many dierent techniques have been proposed to compute total energies in thelast seventy years� From classical� approaches to very complex quantum�mechanicalmodels� the whole range of accuracies and speeds has been explored� The reasons forthe existence of this wide variety of schemes lie in the extreme di�culties in solving
�In many cases strong hysteresis may prevent the accurate location of phase boundaries fromdiscontinuities in the sampling averages in a reasonable amount of computer time� Under thesecircumstances free energies have to be computed�
��

Classical/quantummodel for atomic
interactions
SimplifiedHamiltonian
Formationenergies
atomic data
Free energies
Phase diagram
Parent lattice
Latticemodel
Clusterexpansion
Figure ��� General procedure used to compute phase diagrams� The rhombusesrepresent the input information to the model�
��

the problem at hand that force the introduction of dierent approximations� Therevolutionizing changes that computational materials science is producing are duenot only to the advances in computer technology but also� in a comparable extent� tothe improvements in the computational techniques�
In general� two dierent approaches can be taken to compute total energies we canassume a model for the electronic density and solve for the energy� or we can directlysolve Schr#odinger�s equation in the solid self�consistently� In the �rst case� quantum�mechanical equations are not necessarily used� For example� in empirical potentials�the electronic density and ionic charges are replaced by point charges centered atthe ions� The energy is computed as the sum of the electrostatic interaction and ashort�range repulsive term that represents the overlap of the electronic clouds� Theparameters of these models are usually �t to reproduce experimental or ab initiodata� On the other hand� in the self�consistent quantum�mechanical methods theelectronic wave function� and therefore the interaction potential between the electrons�is not known before the problem is solved� Dierent quantum�mechanical methodstypically dier in the basis functions they use to expand this electronic wave function�A priori assumptions� such as spherical symmetry for the electrostatic potentials�are introduced to speed up the calculations� A more detailed description of themethods and the eects of the approximations used can be found in chapters � and �respectively�
A major step in the development of total�energy techniques was the introductionof the density�functional theory ���� for the treatment of the many�body electronicproblem� Currently� most ab initio quantum�mechanical models are based on a local�density approximation ���� �LDA� to this theory or on improvements over the LDAby using gradient corrections ���� ���� Other methods� based on the Hartree�Fockapproximation are also used �����
Not all quantum�mechanical methods are ab initio� One particularly useful tech�nique is the semiempirical tight�binding formalism� Originally proposed by J� C�Slater and G� F� Koster ���� as a way of interpolating bands� it can be reformulatedas a very eective total�energy technique ���� ��� ���� We will use this techniqueextensively in chapters � and � since it will allow us to introduce the relevant phys�ical eects for computing total�energy dierences in oxides while still retaining thesimplicity and speed of the semiempirical approaches�
Within the appropriate model for the system� most of the materials properties canbe related to dierences between total energies� Lattice parameters� elastic constants�pressure�induced transitions� and defect concentrations are just a few examples ofmaterials aspects that can be analyzed using total�energy methods� We will mainlyfocus on their use to compute phase diagrams� However� other applications� such asthe engineering of new cathode materials for batteries� will also be discussed�
We will use total�energy methods to compute formation energies� which are im�portant inputs for the statistical�mechanical models� The formation energy of acompound is the dierence between its total energy and the total energy of its con�stitutive compounds �or elements�� A large number of formation energies are usuallyrequired to compute phase diagrams� Oxides present a di�cult challenge to total�energy techniques since they have very complex crystal structures with low symmetry
��

that make accurate methods very computer�time intensive�
��� Oxides
Oxide materials are everywhere in our daily life� From glass products and bricks toelectronic components� from enamels and clay to computer screens and nuclear fuels�the applications of these materials are countless� As our knowledge of their propertiesincrease� new applications are being found every day� In ����� J� G� Bednorz and K�A� Muller discovered superconductivities above �� oK� in oxygen�de�cient compoundsin the Ba�La�Cu�O system ����� These temperatures were higher than previouslyknown cases� One year later� the critical temperature was raised again� to �� oK�in YBa�Cu�Oz ���� and even higher values have been observed in recent years� TheYBa�Cu�Oz case represents an example of the interplay between experiments andsimulations we mentioned earlier� D� de Fontaine� G� Ceder� and M� Asta studiedthe oxygen ordering in this superconductor� They developed a phase diagram thatpredicted new ordered phases that were later observed experimentally �����
In the following chapters� we will focus on high�technology applications of oxidessuch as lithium�transition�metal oxides and doped zirconias for battery cathodesand oxygen sensors respectively� These are usually called advanced materials andtheir use is growing at a fast pace� The world market for high�technology ceramics�not only oxides� in the year ���� is estimated to be $� billion ����� Although it isstill small compared to the $��� billion steel market� its growth rate of ��� is tentimes larger than for steel ����� Oxides have many uses not only because oxygen isvery abundant� but also because they form numerous and complex crystal structures�Typical oxide crystals may contain more than four elements of the periodic table�have low symmetry� and large unit cells� This complexity is re�ected in the widerange of properties that oxides exhibit making them very attractive to the materialsengineer� The insight that computational tools can oer will be fundamental to fullytake advantage of the properties of complex oxides�
The simplest metal�oxide structures are based on a close packing of the oxygenions� with the cations �lling the available interstices� For example� �gure ��� shows thecrystal structure of the stable phase of pure zirconia �ZrO�� at room temperature�This structure is monoclinic� with �� atoms at symmetry�distinct positions in theunit cell and with �� degrees of freedom� The computation of the total energy ofthis structure is already a formidable task and no calculation with state�of�the�artmethods has been reported so far� To determine the phase diagram of zirconia whenit is mixed with CaO or Y�O�� the formation energies of many even more complicatedcompounds would have to be computed� seriously pushing the limits of the methodscurrently available� Any technique� such as the one presented in chapter �� that allows
�Oxygen accounts for ��� of the elements in the earth crust ������These degrees of freedom refer to all the possible displacement of the atoms that do not violate
the symmetry of the structure� They include both volume and internal relaxations� To computeformation energies the total energies of the di�erent structures are minimized with respect to thesedegrees of freedom�
��

Figure ��� Stable structure at room temperature of pure zirconia� It can be obtainedas a deformation of the �uorite structure� The black circles represent zirconium ionswhile the white circles are the oxygen ions� The corners of the dashed cube are theoxygen positions in an ideal �uorite structure�
��

the accurate interpolation of complex calculations is fundamental if we want to makeany progress in predicting this type of phase diagrams ab initio�
Any material property is markedly dependent on small deviations from ideal struc�tures� Atomic defects� interfaces� and grain boundaries are examples of such devia�tions that add a new dimension to the problem of predicting materials properties from�rst principles� Oxides are no exception and we are far from any rigorous treatmentwith the techniques currently available� For this reason� the tight�binding formula�tion presented here can play a fundamental role in the understanding of oxides in thefuture�
��� Thesis outline
New technological advances are imposing stronger demands than ever before on ma�terials performance� To help the materials engineer meet these challenges� new toolsare being introduced every day� Among them� there is a particular set of techniquesthat do not respond to the traditional concept for developing novel materials� Usuallycalled atomistic simulations� or computer experiments�� they are revolutionizingthe way in which materials science and engineering works� Both the increase incomputer power and the improvement in the numerical methods are making thisrevolution possible�
The increase in computer power alone� will not allow us to accurately model com�plex materials or macroscopic properties in the near future� New and faster numericaltechniques have to be developed� In this thesis� we focus on ways of improving themethods to compute phase diagrams in complex oxides� In recent decades� there hasbeen an increased interest in oxides� From simple applications in glasses to high�technology tiles for the aerospace industry� their wide variety of properties has beenused in almost any industry� This �exibility correlates with the complexity of thematerials� The complexity pushes the limits of the computational methods that areused to understand� and eventually design new oxides� With this work we hope tocontribute to a future where these novel materials can be created from purely com�putational experiments�
Phase diagrams are one of the most important tools to develop new materials�The ab initio study of phase stability involves solving complicated quantum� andstatistical�mechanical equations� In the statistical�mechanical �eld� we will intro�duce a new methodology to compute phase diagrams at low temperatures or forhighly�stoichiometric compounds� In chapter �� we will explain the details of a low�temperature expansion technique of the thermodynamical potentials that overcomesthe numerical problems that more traditional methods� such as Monte Carlo simula�tions or the cluster variation method� have in that temperature regime� The generalformalism of lattice models will also be described�
In the quantum�mechanical �eld� we will focus on how to compute formationenergies in a fast and accurate way� Formation energies are the main input to thestatistical�mechanical models� The complexity of oxides makes the computation oftotal energies very computer�time consuming� In chapters �� �� and �� we will show
��

that although it is possible to accurately compute formation energies in oxides� onlythe very complex quantum�mechanical techniques can accomplish the task� Thesemethods are very slow and many approximations need to be introduced�
In chapter �� we will also analyze the eects of the approximations used in dier�ent total�energy methods and we will determine the relevant terms that should beincluded in a simpli�ed Hamiltonian in order to reproduce formation energies accu�rately� This simpli�ed Hamiltonian will be then formalized within a self�consistenttight�binding model in chapters � and �� We will demonstrate that within the tight�binding framework� formation energies can be computed very accurately and withoutan excessive computational burden�
Most oxides are highly ionic� The majority of the simple total�energy modelsassume �xed point charges to compute the inter�atomic Coulomb interaction� Inchapter �� we will use the self�consistent tight�binding technique to demonstrate thatlarge charge variations between the same ionic species can exist� The actual chargewill depend on the particular atomic arrangement around the ion �e�g�� dierentstructures� dierent site symmetries within a given unit cell� etc��� We will show thatassuming constant charges can introduce large errors in the total energy� Simple waysof modeling the charge transfer will be evaluated�
Finally� we will summarize the conclusions of this work and future research in the�eld of computational materials science in chapters � and �� respectively�
Along the process� we will also discuss applications of these techniques to spe�ci�c materials systems� The low�temperature expansion will be tested in the Pd�Vphase diagram� Although this particular alloy is not an oxide� it oers a remarkableillustration of the di�culties occurring at low temperatures� The CaO�MgO systemwill be also extensively analyzed from dierent perspectives not only because of itstechnological applications �refractory ceramics� but also because of its relative sim�plicity� In chapters �� �� and �� its phase diagram and energetics will be discussed�We will also study doped zirconias� that are widely used as oxygen sensors� fuel cells�and coatings� Their intricate structures and behavior will be an ultimate test for oursimpli�ed tight�binding Hamiltonian �chapter ��� Finally� the power of the ab initioapproach and total�energy techniques will also be illustrated in the design of cathodematerials for lithium�transition�metal oxide batteries �Appendix C��
��

��

Chapter �
The computation of phase
diagrams
The stability regions of the dierent phases �or their coexistence regions� in a phasediagram can be computed by minimizing �tangent construction� the free energies ofall the possible phases in the system� A brief review of the methods to implement thisprocedure will be done in section ���� In some of these approaches� the free energiesare not even evaluated and the stable phases are obtained by simply observing� thearrangement of the atoms during a simulation�
At low temperatures� equilibration times become unaordably long in some of thesimulations� These problems are aggravated in systems with highly ordered stoichio�metric phases since most of the standard statistical�mechanical techniques� such asMonte Carlo or the cluster variation method� are only practical if at least a minimumamount of disorder is present� In section ���� we develop a low�temperature ex�pansion �LTE� of the thermodynamic potentials that overcomes the above problems�Our work is an extension to complex alloy Hamiltonians of the formalism previouslydeveloped for the Ising model with pair interactions �����
The LTE complements MC simulations and the CVM extremely well as it providesanalytical expressions for the free energy in the situations where the latter methodsbecome ine�cient highly stoichiometric phases with little disorder� In section ����we apply this combined� approach to compute the phase diagrams of the CaO�MgOand the Pd�V systems� The former presents a simple miscibility gap� as shown in�gure ���� Although any technique can be used in this case� we are including thissystem as a simple example to illustrate the formalism� On the other hand� the Pd�V alloy is very complex at low temperatures� The experimental phase diagram isshown in �gure ���� Bellow ��� oC� measurements are hampered by sluggish kinetics�However� Cheng and Ardell were able to detect a new ordered phase� Pt�Ti �withstoichiometry Pd�V�� in this region ����� Note that some of the compounds presentin the phase diagram stay stoichiometric up to very high temperatures�
��

Figure ��� Experimental phase diagram for the CaO�MgO system� There are no sta�ble compounds in the solid phase� just a miscibility gap� See for example reference ����for more details�
��

Figure ��� Experimental phase diagram for the Pd�V system� The Pd�V phase isnot shown since it is very di�cult to measure its boundaries experimentally� Notethat Pd�V and PdV� have a very narrow stability region which makes traditionalsimulations di�cult to perform� See reference ���� for experimental information�
��

��� General formalism
A direct simulation of the equilibrium solid phases with a single technique is not possi�ble since the phenomena being observed spans many dierent time scales� The energyand entropy contributions to the free energy involve� for example� electronic excita�tions� atomic vibrations� and con�gurational disorder� While atomic vibrations havetypical periods of ������ seconds� the changes in con�guration �related to atomicjumps� have time scales that are many orders�of�magnitude longer� A combinationof dierent techniques� such as molecular dynamics ����� for treating the atomic vi�brations� and Monte Carlo simulations� for dealing with the con�gurational disorder�is one option to study dissimilar time scales�
In this thesis� we follow a dierent approach� We integrate out the faster degreesof freedom and only retain the substitutional degrees of freedom� which can then bemapped onto a lattice Hamiltonian� The cluster expansion mentioned in chapter �can be thought of as a generalized Ising Hamiltonian within this model� Finally�we use standard statistical�mechanical lattice�model techniques to study the systemcon�guration space�
����� The alloy problem
The free energy of any alloy can be computed from the partition function� Z�
Z�T P � �Xj
e��Ej � �����
The sum is over all possible quantum�mechanical eigenstates j of the Hamiltonian ofthe system� T is the absolute temperature� Ej is the eigenvalue corresponding to j�and � � �
KBT� where KB is the Boltzmann constant� We assume the pressure� P � to
be constant and equal to zero�� We will not explicitly write the pressure in the restof this work�
For the temperatures at which the solid phases are stable� the contribution ofthe noncrystalline states to the partition function is negligible� We can then onlyconsidered the crystalline states in the sum in equation ���� These states can begrouped according to the parent lattice L� �e�g�� fcc� on which the atoms order�Dierent atomic arrangements on a given lattice will be labeled by �� We will assumethat there is a unique mapping from the microstates j of equation ��� onto ��� Notethat these microstates not only correspond to states with the atoms at the ideal latticepositions� but also states with small deviations from them and in general� any excitedstate� such as atomic vibrations or electronic excitations� that can be mapped onto�� This procedure is illustrated in �gure ����
The sum in equation ��� can be arranged so that it is written as an explicitfunction of the dierent arrangements of the atoms at the sites of all the possible
�In this thesis we will not address pressure�induced transitions��This is true for most systems of interest�
��

microstates
configurations
j=n j=n+1 j=n+2 j=n+3 j=n+4
σ= σ=kσ k+1σ
Figure ��� Mapping of dierent microstates j into con�guration space� The arrange�ment of the atoms on the parent lattice is labeled with �� It is assumed that there isa unique mapping from j onto ��
parent lattices of the system�
Z�T � �XL
X��L
Xj��
e��Ej � �����
The last sum is now over all quantum�mechanical eigenstates that correspond to agiven distribution � of atoms on the lattice L�� Many phase diagrams can be describedas arrangements of atoms on a single underlying lattice� In this case� the �rst sum inequation ��� has only one term� For simplicity� we will assume that this is the case�If more than one lattice is present� the procedure has to be repeated for each lattice�
Since the time scales of the atomic jumps are orders�of�magnitude larger thanthe time scales of the other excitations� we can assume that at each con�guration�the system will ergodically sample all the possible j microstates� In this way� it ispossible to de�ne a free energy� F �� T �� for every con�guration on the lattice�� Thus�equation ��� can be expressed in the following way�
Z�T � �X�
e��F ���T � �����
where
F �� T � � � �
�ln
��Xj��
e��Ej
�A � �����
�Note that the only condition for equation ��� to be exact is that there is a unique mapping fromj onto ��
�This assumption is not always correct but for most systems of interest the error it introducesin the phase boundaries is negligible� See reference ���� for a detailed discussion on this problem�
��

Note that in equation ���� only the con�guration dependence appears explicitly� allthe other degrees of freedom have been integrated out�� This procedure is usuallyreferred to as the coarse�graining� of the partition function and it allows us tocompute the free energy with a single simulation technique�
Equation ��� can be thought of as the partition function of a system with Hamil�tonian F �� T �� This Hamiltonian accounts for all the nonsubstitutional degrees offreedom� It can be explicitly written as
F �� T � � Eo��� � F exc�� T � �����
where Eo��� corresponds to the eigenstate with the lowest energy for a given � andF exc�� T � represents the free energy of the dierent excitations available to the sys�tem for that �� The lowest energy state does not necessarily correspond to the atomssitting at their ideal lattice sites�� It is common practice to use Eo��� as an approx�imation to F �� T �� eectively neglecting F exc�� T �� In this case� the Hamiltonianis temperature independent� However� in recent years� an argument has been madefor the importance of the dierent nonsubstitutional excitations for the determina�tion of phase diagrams� For some systems� changes in transition temperatures of theorder of ��� can be expected if they are not included� �See for example references���� ��� ��� ��� and references therein��
Dierent contributions to F exc�� T � should be considered� Among them� atomicvibrations and electronic excitations are the most relevant ���� ���� However� Gar�bulsky argued that the former are the most important for determining alloy phasestability ����� For insulators� as many oxides are� it can safely be assumed that this isthe case� Consequently� we will only take into account the eect of atomic vibrations�Equation ���� is then reduced to�
F �� T � � Eo��� � F vibr�� T �� �����
In the following chapters� we will focus on dierent ways of computing Eo����Since most of the con�gurations � are not periodic� a cluster expansion is used�Before introducing this concept� it is convenient to map the alloy problem onto anIsing lattice model�
����� The Ising model
The Ising model was introduced in ���� by Lenz and Ising�� It was developed to studymagnetic ordering and consists of a �xed lattice with a spin variable �i de�ned ateach site i� They also de�ned a simple nearest�neighbor coupling interaction betweenthe spins�
In alloy theory� the value of the spin variables is not associated with a particularmagnetic moment but with the identity of an atom� For example� for a binary alloyA�B� �i takes the value ������ when site i is occupied by an A�B� atom� In this
�For a given � the atoms will relax to their lowest energy position at T� � oK��The history of the model can be found in reference �����
��

Alloy configuration Ising Model
σ = +1i
σ = -1i
Convention:
+1
+1
+1
+1 +1
+1
+1
+1
-1 -1
-1-1
-1-1
-1-1
Figure ��� Mapping of a binary alloy con�guration onto an Ising model� If site iis occupied by the black�white� atomic species� the spinlike variable �i � ������ isassigned to that site�
way� any arrangement � of the atoms on a lattice can be labeled with the set formedby the �i at each lattice site� This mapping is illustrated in �gure ��� for a binarysystem on a two dimensional lattice�
In this chapter� we have been using � to denote a speci�c arrangement of the ionson a lattice� In terms of the Ising model� � is the set of all the spinlike variables �i�In other words�
� � ��� �� �� ���� �N � �����
where N is the total number of lattice sites� The Ising model is a convenient way tolabel the possible atomic arrangements on a lattice� It should not be confused withatoms actually sitting at �xed atomic positions� As we mentioned earlier� a completeensemble of microstates is associated with a given con�guration and most of themcorrespond to atoms displaced from their ideal lattice sites�
In the following section� we will generalize the simple Ising nearest�neighbor pairHamiltonian by expanding F �� T � in cluster functions�
�Contrary to the crystallographic de�nition it is customary in the �eld of lattice models to referto a �lattice� as any periodic structure of sites instead of just one site per unit cell�
��

α
β
γ
δ
= -1
= +1
= -1
σβ
σγ
σδ
= +1σα
Figure ��� Two dimensional binary alloy lattice� where � dierent clusters are shown�� �� �� and �� They represent a point� a pair� a triplet� and a quadruplet clusterrespectively� The value of the cluster functions for these clusters is also indicated�following the convention that black atoms�� white atoms�� correspond to spinlikevariables �i � �������
����� The cluster expansion
A cluster� �� is a set of sites on a lattice� A cluster function� ��� on the other hand�corresponds to the product of all the spin variables �i at each site of the cluster�
����� �Yi��
�i��� �����
where we have used the convention of naming the clusters with Greek subindices andthe sites with Roman subindices� In �gure ���� we show dierent clusters and theirassociated cluster functions for a two dimensional lattice�
Given an appropriate de�nition of an internal product� it can be shown that thecluster functions form an orthonormal basis of functions in con�guration space ��� ����For two arbitrary scalar functions� f��� and g���� the scalar product can be de�nedas�
hf gi � �
NX�
f���g���� �����
The sum is over all possible N con�gurations of the lattice� Note that for a binaryalloy� N � �N � where N is the number of sites on the lattice�
Consequently� any function f��� of the con�guration of the lattice can be expressedas a linear combination of cluster functions
f��� �X�
C������ ������
��

whereC� � hf ��i� ������
Equation ���� is usually called the cluster expansion of f���� The energy and theelastic or lattice constants are a few examples of the properties that can be clusterexpanded� In particular� since F �� T � is a function of the con�guration it can alsobe cluster expanded�
F �� T � �X�
V��T ������ ������
where the expansion coe�cients� V��T �� are generally referred to as the e�ectivecluster interactions �ECI�s��
Expansion ����� can be thought of as a generalization of the Ising Hamiltonian�where not only are nearest�neighbor pair interactions used but also more distant pair�triplet� quadruplet� and larger cluster interactions�� Not all the ECI�s are indepen�dent� but they can be related by the symmetry of the lattice� Equation ���� can bewritten as�
F �� T � � NX�
�m�V��T �h��i���� ������
The prime in the sum indicates that the addition is over the non symmetry�equivalentclusters �� The coe�cient m� is the number of clusters equivalent to cluster � persite� and h��i��� is the average of the cluster functions over the equivalent clusters�These averages are often called correlation functions�
Even with the transformation used in the last equation� the number of ECI�s di�verges in the thermodynamic limit� This implies that in order to compute phasediagrams an in�nite number of coe�cients should be computed� This is similar tothe original situation since in any simulation an in�nite number of total�energy cal�culations should� in principle� be done� However� the bene�ts of performing a clusterexpansion of the lattice Hamiltonian become clear when one notes that in general� theECI�s decay very fast with respect to the cluster size� In metallic alloys� for example�the energy of the system depends mostly on the local atomic arrangements� Whenthis is the case� a few clusters� with sites close together are enough to accurately de�scribe the dependence of the Hamiltonian on �� Even in systems in which the exactECI�s are long�range� as is the case of oxides �due to the Coulombic interactions�� itcan be shown that short�range ECI�s can be used as an accurate approximation �����Consequently� expression ���� can be truncated after a maximal cluster �max�
F �� T � �X
���max
V��T ������� ������
The number and type of clusters that are actually needed in expansion ���� dependon the system� Convergence of the cluster expansion should always be carefully
�The ECI�s should not be confused with �real� interactions� For example a pair V�T is notthe actual interaction between two atoms but it is just a coe�cient in an expansion�
�Finite�size simulations are used to approximate the thermodynamic limit�
��

checked� There are dierent techniques to accelerate this convergence���
The application of the cluster formalism we just described is computationallycumbersome when more than two dierent species are present ��� ���� However� thereare some cases where the problem can be reduced to two binary problems that arecoupled through the ECI�s� For example� in oxides� the cation and anion sublatticesdo not interchange ions��� When there are at most two dierent species on eachsublattice� the problem can be treated as two binary systems that interact throughcoupling ECI�s �����
����� The computation of ECI�s
The temperature dependence of the ECI�s arises from the noncon�gurational ex�citations in equation ���� As we already mentioned� we will just consider atomicvibrations since they are the largest contributions in most oxide systems� In the har�monic approximation� the temperature dependence can be linearized above the Debyetemperature �����
V� � V chem� � kbTV
vibr� � ������
The �rst term in the right�hand side of equation ���� represents the chemical con�tribution to the ECI coming from Eo��� in equation ���� The second term is thecontribution from the vibrational free energy� When only con�gurational excitationsare considered� the last term is zero� In the rest of the thesis� we will focus on e�cientways for computing the chemical ECI�s��� There are three well established approachesto compute these ECI�s the Connolly�Williams method� the direct con�gura�tional averaging technique� and the coherent�potential approximation withthe generalized�perturbation method�
The Connolly�Williams method �CWM� takes advantage of the fact that thecluster expansion can be truncated at some maximal cluster� By computing totalenergies for a �nite set of ordered structures� for which the ���s are easily computed� aset of linear equations on the ECI�s is formed based on equation ����� In the originalformulation ����� the same number of structures and ECI�s were used� However�working with more structures than unknown ECI�s usually gives more stable ECI�sthrough a least�square �t���
In the direct con�gurational average technique� equation ���� is followed asclose as possible ���� � For example� for a pair interaction in a binary alloy A�B� this
�See for example reference ���� where the cluster expansion is recast in reciprocal space orreference ���� where the composition�dependent but con�guration�independent long�range elasticenergy is analyzed separately from the remaining local contributions�
��In most systems the size mismatch and a large increase in the electrostatic energy prevent theseinterchanges from happening�
��For the vibrational ECI we use the method recently developed by Garbulsky ����� The ECI�sare �t to the vibrational free energy of a few structures� These free energies are computed withinthe harmonic approximation by diagonalizing the dynamical matrix�
��Since the cluster expansion is truncated a direct inversion does not provide the exact ECI�s�Contributions from clusters larger than the maximal cluster are folded�back into the smaller clusters�Using more structures than ECI�s tends to minimize these errors�
��

equation is equivalent to
Vpair ��
��hEAAi� hEBBi � hEABi � hEBAi� ������
where hEiji corresponds to the energy when the pair cluster is occupied by atoms iand j� averaged over all possible occupations of the other sites� In practice� a �nitecrystal is built around the pair whose ECI we want to compute� and the occupation ofthat cluster is held �xed� Then� the energy is averaged over all possible con�gurationsof the other sites��� This average has to be repeated for the dierent con�gurationsof the pair� Tight�binding and recursion techniques are used in the calculations�
The coherent�potential approximation ���� is a mean �eld theory to computethe electronic structure of the disordered alloy� This state is then considered as a refer�ence state for the generalized�perturbation ���� method to produce concentrationdependent ECI�s�
The last two techniques have the advantage that the ECI�s are not �t but the exactde�nition is used� On the other hand� up to date� no atomic relaxations away fromthe ideal lattice sites have been included in their formalism� The eect of relaxationsis very important in oxides since size mismatch of the ions is a common featureof these materials� The Connolly�Williams method can naturally incorporate theseeects since the structural energies used as inputs �usually called direct energies� canalways be the ones from relaxed structures� Furthermore� any total�energy technique�from very simple classical approaches to very complex quantum�mechanical methods�can be used to compute the direct energies��� Consequently� the Connolly�Williamsmethod seems to be the best choice in oxides�
The Connolly�Williams procedure is illustrated in �gure ���� The direct energiesusually correspond to formation energies of periodic structures with small unit cells�The number of ECI�s required to obtain a converged cluster expansion depends onthe system and determines the number of total�energy calculations that are needed�Due to the characteristic long range of the interactions in oxides� many more ECI�sthan in metallic systems have to be computed�
None of these procedures indicates what clusters should be used to obtain a con�vergent cluster expansion� Since the degree of convergence is not known� the cluster�expanded energies should always be checked against the direct energies� Some of thesedirect energies can be left out of the �t to be later compared with the predictions ofthe cluster expansion� More complex schemes can also be used to check the clusterschosen and their ECI�s� by carefully watching the eect of adding or taking out themduring the �t �����
It is important to realize that the Connolly�Williamsmethod is a �t to the absolutevalue of the formation energies� while most of the phase diagram features dependon subtle di�erences of these values� Furthermore� it can not guarantee that thedirect ground states�� be predicted by the computed ECI�s� Whenever there are close
��Usually �� di�erent con�gurations are enough to obtain a good convergence �������This is also true for the DCA though for complex methods it is very computer intensive���Ground states are arrangements of atoms on the lattice that minimize the energy of the system�
��

Correlation functionsfor each structure
, ,....,
ii
Inversion Least-squares fit
Number of structures > Number of ECI’s
Number of structures =Number of ECI’s
Direct energies fromtotal-energy calculations
EEE , k21 , ,.......
k +σ1 + km αm
1k
E1 = ++ +m
11 1m α................σ
11 σ
21
+ + +m1E2
= 2 2m α................σ12
.................
.................
m21 σα
1
m22 σ
22 σα
2
σαk+m
2k ................kσ
2Ek =
Test ECI
(convergence, accuracy)
σ2
< >σ1i< > iσ α< >
< >
< >
< >< >
< >
V1 V2 Vα
< >
< > < >< >
V1 V2 Vα
V1
V2 Vα
Figure ��� Procedure for computing eective cluster interactions using the Conno�lly�Williams method� The system of equations is either solved by matrix inversion�if the number of direct energies and ECI�s is the same� or by �tting the ECI�s� if theyare less than the number of structural energies�
��

metastable structures� they can became stable during the �t� Finally� as we alreadymentioned� there is no systematic way of truncating the expansion� These di�cultiescan be overcome by a linear programming technique recently developed ����� in whichnot only are the direct energies �t to� but also the ground�state line� and the relativevalues of the formation energies�
����� Thermodynamic properties
Once the lattice Hamiltonian has been determined� thermodynamic properties of thesystem� such as phase diagrams� can be obtained� Two traditional approaches inlattice models have been used in the past years Monte Carlo �MC� simulationsand the cluster variation method �CVM��
For any large lattice model� the total number of possible con�gurations is astro�nomically large and sampling these states is not straightforward� The Monte Carlo
method is a way of generating statistical trajectories� so that averages over thesestates correspond to the equilibrium ensemble averages� Many good articles havebeen written about the method �see ���� and references therein� and is not our inten�tion to review them here� We would just like to point out to a few limitations of themethod when applied to phase diagrams�
As we mentioned� MC simulations provide a way of obtaining thermodynamically�averaged properties� such as the internal energy� Free energies can be computed inMC by performing a time�consuming integration from states where their value isknown �i�e�� ordered state at absolute zero or disordered state at in�nite tempera�ture�� This procedure is not trivial and in general� a simpler approach is adoptedto compute phase boundaries� One can� for example� look for discontinuities in thetemperature dependence of the concentration �at constant chemical potential� or evensimply inspect� the structures during the simulation�
Even when free energies are not computed� MC simulations present problems�specially at low temperatures or for highly stoichiometric compounds� Under theseconditions� averaging times become very large and complex phase boundary featuresmight be impossible to determine �hysteresis eects become important��
The cluster variation method is a generalized mean �eld technique that treatscorrelations exactly between sites within a maximal cluster� with all other correlationsimplicitly approximated by a superposition over smaller clusters� The CVM has theadvantage that it gives a precise value for the free energies� These free energiesare written in a variational form with respect to the probabilities of all possibleoccupations on all the clusters within the maximal cluster�
They correspond to the stable phases at � oK� If the right ground states are not reproduced by thecluster expansion the topology of the phase diagram at low temperature will be wrong� The problemof �nding all the ground�state structures for a general Ising�like Hamiltonian is nontrivial� Manyapproaches have been developed to �nd the ground�state structures ��� ��� and their predictionsare limited by the current computer power� For a review of all these techniques see reference ����and �����
��The ground�state line is the line formed by the ground�state structures in an energy vs� com�position plot�
��

At low temperatures or near stoichiometric phases� some of these variational pa�rameters are very close to zero and numerical instabilities appear in the minimizationprocess� Another disadvantage of the method is that only a limited range of ECI�scan be used� In the CVM� the maximal cluster should contain at least one clusterthat corresponds to each non�zero ECI� Having a large maximal cluster increasesthe computational burden considerably�� and MC simulations are preferred over theCVM when long�range interactions are necessary to model the system� The CVM�though widely applied to metallic alloys� is not usually used in oxides since the elec�trostatic contributions to the lattice Hamiltonian make the ECI�s long�ranged� Moredetails on the CVM can be obtained in references ���� ��� ��� ����
��� Low�temperature expansion of a thermody�
namic potential
Low�temperature expansions for the free energy of the Ising model have been usedfor a long time ����� Systematic methods for deriving the expansion coe�cients havebeen studied for the Ising Hamiltonian with pair interactions ���� ��� ��� ��� ���� Here�we apply the LTE to the generalized Ising Hamiltonian we developed in section �����expressing the energy in cluster functions �not necessarily pairs�� By expanding thegrand potential around dierent ground states� the low�temperature region of anyphase diagram can be computed regardless of its complexity�
The coe�cients of the expansion are computed by a direct counting scheme� takingadvantage of the symmetry of each structure� This is very similar to the standardprocedures used to compute the input data for the CVM ����� As we will show�only a few terms need to be considered in the LTE� making the construction of theexpansion relatively straightforward� The low�temperature expansion is well behavedfor highly stoichiometric phases with little disorder� This condition naturally arisesfor all nondegenerate ground states as the temperature is lowered�
����� Formalism
For a binary A�B alloy� the grand potential per mole� � can be written as�
�T �� � u� Ts� �
��xA � xB� ������
where u� s� and x are the molar energy� entropy� and atomic fraction respectivelyand T is the temperature� The chemical potential � is de�ned as the dierence in
��The computational requirements of the CVM increase rapidly as the symmetry is lowered or thenumber of di�erent species is more than two� For example the number of variational parametersincreases from ��� on the fcc lattice to ���� in the simple L�� ordering when the ����� point clustercombination see �gure ���� is used as the maximal cluster�
��See reference ����� Also recall that we assume the pressure to be equal to zero�
��

chemical potentials between species A and B
� � �A � �B� ������
Equation ���� can be rewritten as
�T �� � u� Ts� �xB � �
�� ������
The last term in the right�hand side of equation ���� can be omitted when computingphase diagrams as it only contributes a constant amount to � The grand potentialcan then be obtained from the grand partition function� Z�T ��� in the followingway��
�T �� � � �
�Nln �Z�T ��� � � �
�Nln
�X�
e���E�����NB ����
�� ������
Here� N is the total number of lattice sites and NB��� is the number of sites occupiedby species B for con�guration ����
For a given ground state� the energy� Eo� can be factored out from equation �����
�T �� �Eo
N� �xoB �
�
�Nln
�� �
X�
�e��� E�����n����
�� ������
In this equation� xoB is the atomic fraction of species B in the ground state� n���is the number of spins in con�guration � that are overturned with respect to theground�state arrangement� and the prime in the sum indicates that we sum over allpossible con�gurations except for the ground state� We also de�ne the excitationenergy %E��� as�
%E��� � E���� Eo� ������
In equation ����� the logarithm can be expanded in a Taylor series�
�T �� �Eo
N� �xoB �
�
�N
�Xj��
�������j���
j
�X�
�e��� E�����n����
�j�� � ������
Using the linked cluster theorem��� equation ���� can be rewritten in a simpler form�
�For simplicity we will assume that F exc�� T � � in equation ������One should note that � plays the role of an external magnetic �eld in the Ising spin language�
This chemical potential selects the di�erent ground states so that � can be expanded at low tem�peratures around any of them�
��This theorem states that it is su�cient to keep the terms proportional to N in the expansion oflnZ� The other contributions cancel out� This can be seen from the fact that �T� � is an intensive
property� The theorem applies to various types of expansions and a formal proof can be found inreference ����� See also reference �����
��

�T �� �Eo
N� �xoB �
�
�N
X�
��e��� E�����n���� ������
in which the sum is performed by grouping all the terms with the same number ofoverturned spins and the same excitation energy� The double prime means that onlythe resulting terms with coe�cients linear in N are summed over� At low enoughtemperatures� usually only a few terms need to be considered in the sum� We willshow that these low temperatures are high enough to get reliable free energies up tovalues where Monte Carlo simulations become practical�
In the present form� the LTE requires that a �nite number of ground states bepresent in the system except for a trivial degeneracy�� �i�e�� dierent ground�statecon�gurations that transform into each other through trivial symmetry operations ofthe lattice�� For the system and range of interactions used in this work� it is possibleto �nd all the fcc ground�state structures in the Pd�V alloy ����� In the CaO�MgOsystem� the only stable structures are the pure oxides�
����� Implementation
The practical implementation of equation ���� involves two main tasks the compu�tation of %E��� and the determination of the dierent con�gurations that shouldbe summed over� The excitation energy of overturning spins from the ground�statecon�guration can be computed using the cluster expansion� When just one spin� atsite i� is overturned�
%E��i� �X�
V�����i�� V�����
o�
� ��X��i
V�����o�
� �� X���i
ai�V�����o�� ������
�i is the con�guration of the lattice when the spin at site i is �ipped� �o is thecon�guration of the ground state� and ai� is the number of � clusters that containthe site i� This last quantity can be computed as��
ai� �m�
mi
mi� ������
where m� and mi are the multiplicities of the cluster � and the site i respectively�and mi� is the number of sites of the same type as i in cluster �� Note that in
��If ngs is the number of trivially degenerate structures of a given ground state a term of theform � �
�Nlnngs should be added to the grand potential� However since N � � this term goes
to zero in the thermodynamic limit and consequently can be safely ignored���Note that �m�mi�� overcounts the number of i�type sites �mi� because each i�type site
belongs to �ai�� � clusters� Consequently ai�mi � m�mi��
��

L
L
U
L: linked.
U: Unlinked.
V2pair
V1pair
Figure ��� Examples of linked and unlinked pair clusters on a two dimensional lattice�The only ECI�s dierent from zero are Vpair
� and Vpair� �
equation ����� the sum is over all the dierent sets of clusters� &�� Each of thesesets is formed by all the clusters � that are related by symmetry operations of thelattice� Equation ���� can be used iteratively to compute the energy of overturningmore than one spin by replacing �o with �i and so on�
The dierent con�gurations that should be summed over in equation ���� aregrouped together according to their excitation energy� This is a trivial operationto do when �ipping just one spin� The dierent excitation energies correspond tooverturning spins on the symmetry�distinct sites of the ground state� Consequently�the coe�cients of the terms in the sum are the multiplicity of each site�
In con�gurations with multiple spins overturned� the excited spins can form alinked or an unlinked cluster� A linked cluster is one in which the overturned spinsare interconnected through the interactions of the cluster expansion� All the linkedclusters related by symmetry have the same excitation energy� The number of theseclusters on the lattice scales linearly with N � On the other hand� an unlinked clusterhas one or more spins that are not connected with any of the other overturned spinsthrough any of the ECI�s �see �gure ����� From the unlinked terms� only thosecontributions coming from excluded volume eects� �the same spin can not be �ippedtwice� and counterterms� from linked clusters �the linked clusters are not available�as unlinked excitations� need to be retained��� See examples in section ����
The LTE can be built simply from the information that is already required for the
��For example if two spins are being overturned on a cubic lattice with N sites there areN�
�
NN���� ways of picking them� If only nearest neighbor interactions are de�ned in the Hamiltonian
there are �N linked pair clusters� Consequently the number of unlinked clusters is NN���� � �N �
Since only terms linear in N should be kept the coe�cient in the expansion corresponding to theunlinked clusters is �N
� � �N ��N� � comes from the excluded volume e�ect and ���N� is the
�counterterm� from the linked clusters�
��

Figure ��� The rocksalt crystal structure� The black spheres represent magnesiumor calcium atoms� while the grey ones are the oxygen atoms�
construction of the CVM functional� The linked clusters� their multiplicity� and themi� are automatically found for each ground state with a computer code that uses adirect enumeration procedure combined with simple group�theoretical concepts �����The same code is used for computing the CVM free energy expressions� From thesedata� the LTE coe�cients are generated� This procedure is very fast and needs to beperformed once for a given ground state and set of cluster interactions���
��� Applications of the low�temperature expan�
sion
����� The MgO�CaO phase diagram
MgO and CaO crystallize in the rocksalt structure shown in �gure ���� This structurecan be thought of as two interpenetrating fcc lattices� When they are mixed� thecalcium and magnesium ions distribute themselves on the fcc cation sublattice whilethe oxygen sublattice remains fully occupied� Consequently� we can regard this systemas a substitutional binary alloy with occupation variables �i only de�ned on the sitesof the cation sublattice� We do not assume that the ions occupy the ideal positionson the lattice but only that there is a one�to�one correspondence between ions andlattice sites� Note that� although the anion sublattice is fully occupied� the oxygenions are still allowed to move away from their ideal lattice sites�
��Note that we assumed temperature�independent ECI�s� The use of temperature�dependentECI�s through the incorporation of noncon�gurational excitations is straightforward�
��

The substitutional species �Mg and Ca�� have the same valence charge �isovalentsystem� and consequently� no charge�compensating defects need to be introduced�This considerably simpli�es the calculations� Moreover� the above assumption thatthe oxygen sublattice remains fully occupied is likely to be valid� Furthermore� thefact that both Ca and Mg ions have the same valence charge� implies that the de�pendence of the long�range interactions on con�guration is very small� resulting in arapidly convergent cluster expansion�
The CaO�MgO phase diagram is very simple� and any of the methods alreadymentioned can be used to compute its boundaries� The only purpose of introducingit here is to clarify the concepts of the previous section before dealing with the complexPd�V phase diagram�
We will use expression ���� to compute the grand potential around the CaO andMgO ground states� We will start with the terms corresponding to overturning onespin� This is equivalent to the exchange of one Ca �Mg� with one Mg �Ca� on anotherwise pure Ca �Mg� sublattice� The energy of this exchange corresponds to theformation energy of a single substitutional defect� Since all the sites on the cationsublattice are equivalent for the CaO and MgO ground states� equation ���� reducesto�
CaO�T �� � � �
�e��� ECaO��� ������
MgO�T �� � �� �
�Ne��� EMgO��� ������
where %ECaO �%EMgO� is the energy of replacing a Ca �Mg� by a Mg �Ca� in pureCaO �MgO�� and CaO and MgO are the one �ip� low�temperature expansions forthe CaO and MgO phases respectively�
The phase boundaries can be obtained by requiring CaO�T �� � MgO�T ��and solving for � at each desired temperature ����� The concentration of the phaseboundaries is given by�
xCaO ��CaO��
� e��� ECaO��� ������
xMgO ��MgO
��� �� e��� EMgO���� ������
Since � � � when T � �� if %ECaO %EMgO the miscibility gap will be tilted�towards the MgO�rich side� with a larger solubility of MgO in CaO than vice versa���
In section ������ we compute these energies and �nd %ECaO %EMgO� in agreementwith the experimental phase diagram of �gure ���� Using these values� we computed
��In what follows we identify CaO with the �A� species and MgO with the �B� species of equa�tion ����� Consequently xoB is equal to �� for the CaO MgO phase� Since the energy is de�nedexcept for a constant we can safely assume that Eo is zero for both CaO and MgO� Once this choiceis made the zero of any mixture is �xed�
��This is in agreement with the fact that if �ECaO � �EMgO it �costs� more at the sametemperature to add a Ca on the cation sublattice of MgO than vice versa�
��

0
500
1000
1500
2000
2500
3000
3500
4000
0 20 40 60 80 100
Tem
pera
ture
(C
)
Composition (%MgO)
LTEExp.
Figure ��� Solubility limits predicted by the low�temperature expansion using justthe single�defect energies� The experimental points are from reference �����
the solubility limits �see �gure ����� The low�temperature expansion correctly pre�dicts the asymmetry of the miscibility gap� Also� the agreement with the experimentalpoints is good up to relatively high temperatures��
If the energy of overturning spins were independent of the atomic con�guration�equations ���� and ���� would be exact� However� this is not true� and a correctiondue to the fact that the spins are actually linked� has to be introduced� Thesecorrection terms are the remaining contributions in expansion ����� We will showhow to incorporate their eect in the next section�
����� The Pd�V phase diagram
To illustrate the application of the LTE and demonstrate how it can be combinedwith the CVM and MC simulations� we computed the phase diagram of a systemthat models the Pd�V substitutional alloy� The Pd�V system has been extensivelystudied both theoretically ���� ��� ��� ��� ��� ��� ��� ��� and experimentally �seereference ���� and references therein�� In the Pd�rich side of the phase diagram� thestable phases are based on the fcc lattice while on the V�rich side PdV� is stable in the
��In chapter � we compute the phase diagram using Monte Carlo simulations� For this case theprediction of the simulation and the experiments coincide� This is why it is fair to compare the LTEprediction with the experiments and not to a Monte Carlo calculation to assess the accuracy of themethod�
��

cluster ECI �meV atom�VPair� �����
VPair� �����
VPair� ����
VPair� ����
VTriplet� ����
VTriplet� �����
VTriplet� �����
VTriplet� �����
VQuadr�� �����
Table ��� Eective cluster interactions �ECI�s� used to model the Pd�V system� Theclusters are named according to the convention followed in �gure �����
A�� structure �which is not an fcc superstructure� see reference ���� for a descriptionof the structure�� Pure vanadium is bcc� To test the LTE� we computed the fcc phasediagram of the Pd�rich side�
In order to compute the ECI� total�energy calculations for dierent structures inthe Pd�V system need to be performed� We used the so�called linear mu�n�tin�orbital method in the atomic�sphere approximation ���� �LMTO�ASA�� Details ofthis technique are given in chapter �� For this system� the accuracy of the method iscomparable to the most accurate quantum�mechanical approach� In each calculation�the total energy was minimized with respect to a hydrostatic volume deformation�Internal relaxations were neglected� in accord with other calculations which foundthem to be negligible ����� Details of these computations and the resulting formationenergies were reported in reference �����
We performed a Connolly�Williams �t to the energy of these structures� Theresulting ECI�s are shown in table ���� We used � dierent ECI�s pairs up to fourthnearest neighbors� � triplets� and the nearest�neighbor quadruplet as shown in �g�ure ����� Going beyond this range of interactions implies using a prohibitively largeCVM maximal cluster� We used the ����� point cluster combination shown in �g�ure ���� as the maximal cluster�
By minimizing equation ���� under the constraints that the probabilities on the��� and ���point clusters be between zero and one ����� we computed the groundstates predicted by these ECI�s� They are shown in �gure ����� A description of thedierent ground�state structures can be found in references ���� and ����� To clarifythe relation with the experimental Pd�V phase diagram� we also show the calcu�lated energy of the A�� structure in �gure ����� At stoichiometry� PdV� is so stable�that its tie�line with the MoPt� structure on the Pd�rich side� makes the L�� struc�ture metastable� in accordance with experimental information ����� Consequently�although it is not an fcc superstructure� we decided to include the A�� structure inthe calculations since it clearly aects the fcc Pd�rich side of the phase diagram�
��
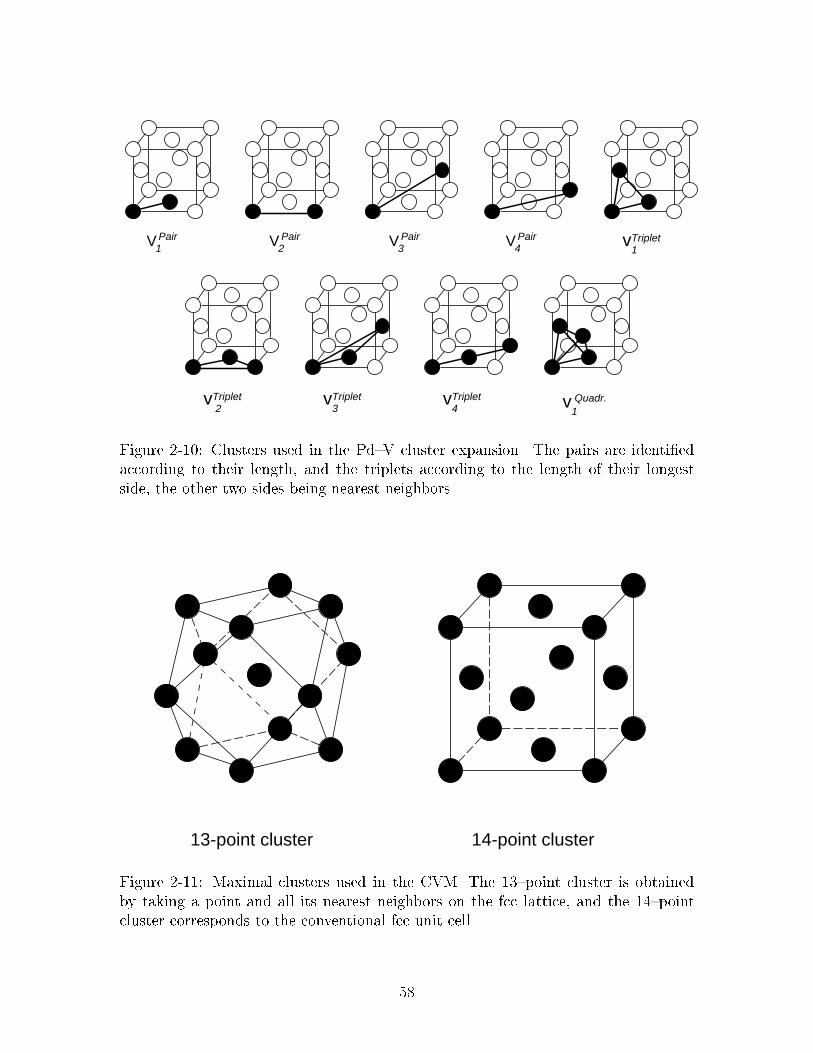
v Quadr.
V Pair1 V Pair
2 V Pair3 V Pair
4
vTriplet2
vTriplet4
vTriplet1
1vTriplet
3
Figure ���� Clusters used in the Pd�V cluster expansion� The pairs are identi�edaccording to their length� and the triplets according to the length of their longestside� the other two sides being nearest neighbors�
14-point cluster13-point cluster
Figure ���� Maximal clusters used in the CVM� The ���point cluster is obtainedby taking a point and all its nearest neighbors on the fcc lattice� and the ���pointcluster corresponds to the conventional fcc unit cell�
��
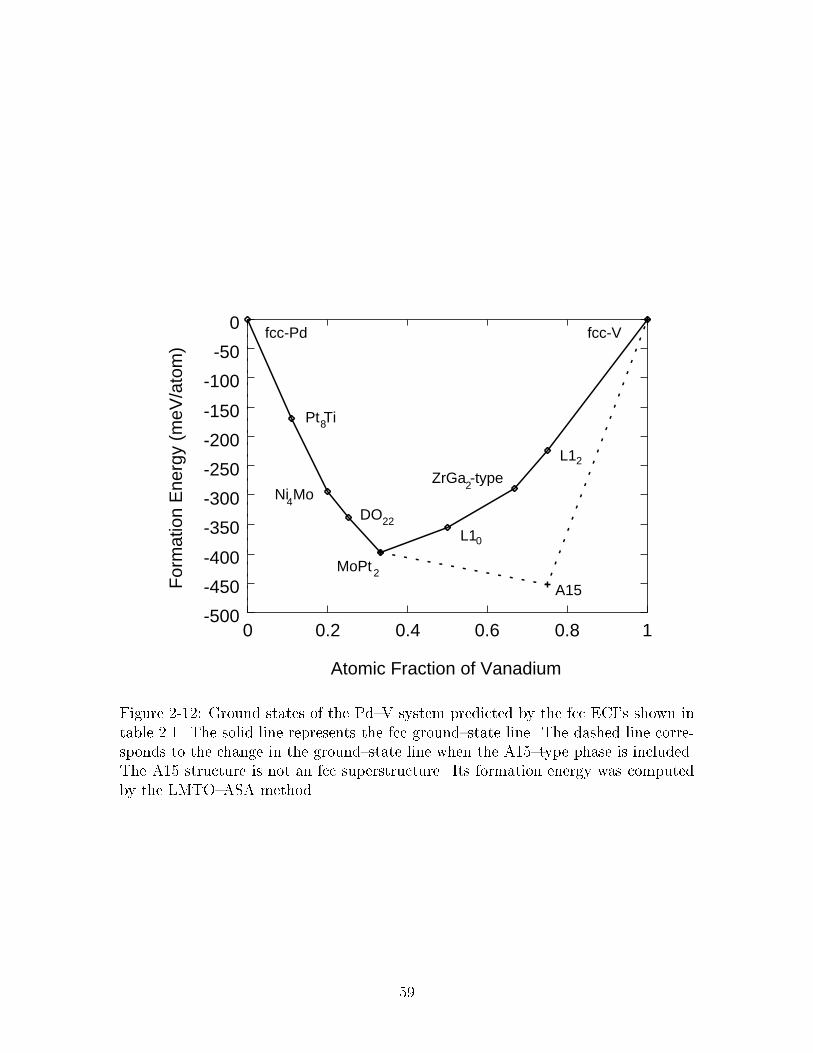
-500
-450
-400
-350
-300
-250
-200
-150
-100
-50
0
0 0.2 0.4 0.6 0.8 1
For
mat
ion
Ene
rgy
(meV
/ato
m)
Atomic Fraction of Vanadium
Pt Ti
Ni MoDO
MoPt
L1
A15
ZrGa -type
L1
8
4
2
0
2
2
22
fcc-Pd fcc-V
Figure ���� Ground states of the Pd�V system predicted by the fcc ECI�s shown intable ���� The solid line represents the fcc ground�state line� The dashed line corre�sponds to the change in the ground�state line when the A���type phase is included�The A�� structure is not an fcc superstructure� Its formation energy was computedby the LMTO�ASA method�
��

For simplicity� we treated the A�� phase as a line compound with no con�gurationaldisorder� The only phase boundary this approximation will aect is the MoPt��A��two�phase region�
The MoPt�� DO��� and Pt�Ti structures have all been observed experimentally �������� It is a remarkable success of this ab initio method that they are all correctlypredicted� given the strong ground�state competition in this system ����� The Ni�Mostructure� predicted by the cluster expansion in table ���� has never been reported�This structure is metastable if one uses the direct LMTO�ASA energies �to which thecluster expansion was �t�� The appearance of the Ni�Mo as a ground state is thereforea direct consequence of truncating the cluster expansion at the �th nearest�neighbordistance� Using the linear programming techniques mentioned in section ������ it canbe demonstrated that a cluster expansion with only pairs and multiplet interactions upto the �th nearest�neighbor distance always makes Ni�Mo a ground state when Pt�Ti�MoPt�� and DO�� are ground states ����� Although this problem can be resolved byincluding the �th or �th nearest�neighbor pair interactions� we limited ourselves hereto the interactions in table ���� Longer�ranged interactions would make the CVMintractable and deprive us of a tool to benchmark the LTE� which is the primarypurpose of this section�
In �gure ����� we show the computed phase diagram� The high�temperature part�dashed line� was obtained using a Monte Carlo simulation on a ��x��x�� lattice withperiodic boundary conditions� Sampling was done in the grand canonical ensembleover typically ���� MC passes� after ���� MC equilibration passes� To speed upcalculations� a divided local environment sampling approach ���� ��� was used to tab�ulate excitation energies from overturning spins� The phase boundaries were foundby observing discontinuities in concentration vs� temperature �at constant chemicalpotential�� At these high temperatures little or no hysteresis is observed� At tem�peratures around ���� oK the equilibration times become larger and problems withmetastable states start to appear during the simulation�
Some phases� such as DO��� remain stoichiometric up to very high temperaturesresulting in very poor sampling in a MC simulation� However� this persistent stoi�chiometry also allows the LTE �solid line in �gure ����� to be used with only a fewterms in the expansion� For example� the LTE with two spin��ip excitations con�verges well up to at least ���� oK for the DO�� phase and up to ���� oK for theMoPt� phase� The convergence of the LTE was tested by recomputing the phaseboundaries with terms in the expansion corresponding to up to three spin �ips� Nosigni�cant changes occurred for temperatures bellow the ones previously mentioned�As can be seen from �gure ����� the results of the MC simulation and the LTE ex�pansion join smoothly� The use of the LTE was indispensable for determining theshape of the phase diagram around ���� oK and for �nding the peritectoid reactionof the Pt�Ti�type phase�
Only for the disordered fcc phase is the LTE problematic� Large solubility limitsare present at very low temperatures and consequently� many terms need to be kept inthe free energy expansion of equation ����� On the other hand� the free energy of thisphase can be most easily computed with the CVM as it has a higher symmetry thanthe ordered phases� Consequently� for temperatures higher than ��� oK we replaced
��

0 10 20 30 40 50 60 70 80
Concentration (%V)
Tem
pera
ture
(ke
lvin
)
0
500
1000
1500
2000
A15
(FCC)N
i Mo
Pt Ti8
MoPt2
DO22
4
MCCVM +LTE
LTE
Figure ���� Phase diagram for an alloy that models the Pd�V system obtained withthe combination of three dierent methods cluster variation method �CVM�� MonteCarlo �MC� simulations� and the low�temperature expansion �LTE�� The solid linesrepresent low�temperature expansion predictions and the dashed lines are the resultsof a Monte Carlo simulation� The dotted lines correspond to phase boundaries com�puted using the CVM and the LTE free energies together� Monte Carlo simulationswere performed for temperatures above ��� oK� The overlap between the methods isnot shown for clarity�
��

the LTE free energy of the fcc phase by the CVM free energy� The phase boundariescomputed with this combination of free energies are shown in �gure ���� as dottedlines�
Although dierent approximations are involved in the LTE� CVM� and MC sim�ulations� all phase boundaries in the Pd�V system join very smoothly�
��� Conclusions
In this chapter� we showed how we can map the alloy problem onto a lattice model�By coarse�graining the system partition function to the partition function of a latticemodel� an eective Hamiltonian for the space of substitutional excitations was con�structed� This Hamiltonian can account for other entropic eects such as vibrationsand electronic excitations� We do not assume that the ions occupy the ideal positionson the lattice� but only that there is a one�to�one correspondence between ions andlattice sites�
The generalized Ising Hamiltonian was explicitly constructed by expanding it inan orthonormal basis set of cluster functions� In practice� this expansion is truncatedat some maximal cluster and its coe�cients �ECI�s� are obtained from total�energycalculations� In principle� one only needs as many ordered arrangements as thereare nonzero ECI�s� However� many more are used to obtain a stable least�squares�t� Finding these energies is the computing�intensive part of ab initio models forobtaining phase diagrams� Especially in oxides� that have large and complex unitcells� current accurate methods are not fast enough� Solving this problem will be thefocus of the following chapters�
Monte Carlo simulations or the cluster variation method are usually used to com�pute phase boundaries from the lattice Hamiltonian we de�ned� However� both meth�ods present numerical problems at low temperature� We overcame these di�culties byextending the low�temperature expansion formalism of the grand potential energy tothe alloy Hamiltonian with multi�site interactions� With this expanded free energy�we were able to determine complex temperature�composition phase diagrams underconditions in which the use of MC or the CVM is nontrivial or even impossible tocomplete in a reasonable amount of computer time� We showed that with just a fewterms in the expansion� the LTE is an accurate and simple tool to compute the freeenergies up to temperatures where the other more traditional methods can be used�
As an illustration� we computed the solubility limits of CaO in MgO and vice versaand the phase diagram of a system that models the fcc Pd�rich side of the Pd�V alloy�We demonstrated that the LTE� MC simulation� and CVM predictions join smoothlyand can be used as complimentary techniques� Furthermore� the coe�cients of theLTE can be obtained from the same information used to build the CVM free energieswhich makes this approach very easy to implement�
��

Chapter �
Total�energy methods
As described in chapter �� the formation energies of many compounds in a system areone of the key inputs to the models for computing phase diagrams� The formationenergies are obtained from dierences in the total energies of the compounds and ofthe end members��� These total energies are determined by the interactions betweenthe electrons and nuclei that form the solid�
The equations that govern these interactions have been well known for almost�� years� but their exact solution for a solid is far beyond the current or foreseeablecomputer power� Starting from Dirac�s equation� a hierarchy of approximations needsto be introduced�� In this chapter� we will describe the dierent techniques thatcan be used to compute total energies� We start in section ��� by summarizing theapproximations that lie at the core of most modern quantum�mechanical and classicalmethods�
Section ��� deals with the classical techniques� The word classical� can be mis�leading� Here� it will be used to identify those total�energy methods in which noexplicit calculation of the electron wave function is performed� However� they usu�ally account approximately for some quantum�mechanical eects� For example� thelargest contribution to the total energy in an ionic solid is the inter�ionic Coulombinteraction� This interaction is balanced by a short�range core�core repulsion thatprevents the crystal from collapsing� The repulsion originates in the Pauli exclusionprinciple� Consequently� even the simplest classical model for an ionic solid has toaccount for this quantum�mechanical eect in order to provide meaningful results�
In section ���� we introduce the quantum�mechanical approaches and their mostrelevant approximations� Dierent materials have dierent types of bonding betweenthe constituent atoms� In general� some of the approximations are better suited forone type of bonding leading to total�energy models applicable to speci�c materials�In all of them� there is a balance between accuracy and simplicity or computationalspeed� From this point of view� oxides present a specially di�cult case since they
�The �end members� are the elements or compounds that are mixed and whose phase diagramwe want to determine�
�Quantum mechanics is essential to study the electrons in a solid since the scale of the periodicityof the ionic potential ���A is comparable to the size of the typical de Broglie wavelength for theseparticles�
��

usually have a mixed type of ionic and covalent bonding� In general� approximatetechniques can accurately deal with one type of bonding but not with both� Themethods that will be described here seem to oer the best compromise between speedand accuracy in oxides�
The time scaling of the methods with the number of atoms �size of the system� isalso a concern� Most of the accurate formulations scale at least as N� lnN � where Nis the total number of atoms� Since the �nal goal is to be able to predict macroscopicproperties� considerable work is being done to develop techniques that would scalelinearly with N � In section ���� we compare the time performance of dierent total�energy techniques and brie�y summarize the advances in the area of order�N methods�i�e�� linear time scaling��
��� The computation of total energies
Dirac�s equation can be approximated by Schr#odinger�s equation plus perturbation�terms that account for relativistic eects such as the variation of the electron mass�the spin�orbit coupling� the coupling between the proton and the electron spin��
etc� ���� ���� The corrections to the total energy introduced by these terms are usuallysmall� and� for simplicity� we will neglect them� Most of the systems studied here arenonmagnetic�� For a review on how to incorporate magnetic eects� see reference ����and references therein�
Due to the large dierence in masses between the electrons and nuclei� it is commonpractice to assume that the nuclei are frozen� In this way� the electronic and nuclearcoordinates can be treated separately in the solid wave function� This is the so�called Born�Oppenheimer approximation and is equivalent to consider that theelectronic wave functions respond instantaneously� to the positions of the nuclei�These nuclei are treated as classical particles� The dependence of the total energy ontheir positions plays the role of a potential energy of interaction between the nuclei�
Under these assumptions� the total energy of the system can be regarded as afunction of the position �RI of the nuclei so that
E � E��R� �R� �������� �RN� �����
where N is the number of atoms in the material� Classical and quantum�mechanicalmethods dier on how to determine this function� The latter techniques will com�pute it explicitly by solving the Schr#odinger equation� On the other hand� classicalapproaches will assume some functional form whose parameters are �t to reproduceexperimental or ab initio results�
�The electronic spin appears naturally when both the postulates of quantum mechanics andspecial relativity are imposed�
�The order of magnitude of these corrections is typically of a few meV�atom ��meV�atom for
terms that depend on the proton spin ������Even for magnetic systems the di�erences in total energies between a magnetic and a nonmag�
netic treatment are usually small ��� ����
��

In the following sections� we will describe both formulations�
��� Classical models
As mentioned in the previous section� the Born�Oppenheimer approximation allows usto write the interaction energy of the atoms in a solid as a function of only the atomiccoordinates� Classical models suggest dierent expressions for this dependence �����The simplest and most commonly used one is the pair potential� in which the totalenergy is a sum of pair�interaction terms between the atoms� An improvement overthe pair�potential approximation is obtained if terms that depend on the position ofmore than two atoms are also included �cluster potential�� Finally� pair and clusterfunctionals can also be used to work with a more accurate representation of the atomicinteraction� In this case� nonlinear functions are applied to pair and cluster interactionterms respectively� Here� we will discuss pair�potentials and pair�functional models�
Ionic crystals are good examples of systems that can be modeled with pair poten�tials� The solid is regarded as an arrangement of charged spheres �ions�� They areassumed to be classical particles placed at the equilibrium crystal lattice positions�These positions are determined by the competition between the attraction of thepositively and negatively charged spheres �simple Coulombic electrostatic force� andthe repulsion caused by the Pauli exclusion principle�� The repulsion interaction isrepresented as an eective pair potential that acts between the ions� The coe�cientsand sometimes the ionic charges of these potential functions are �t to ab initio orexperimental results�
Since a large contribution to the dierences in total energies comes from purelyelectrostatic eects� pair�potential models are sometimes good enough to study thestructural stability of highly ionic crystals� However� they can not be used in metals�A pair potential assumes that the total energy of a solid is a sum over independentbonds �i�e�� the strength of the bond does not depend on the existence of other bonds��This is only correct if the nature of the bond is purely ionic� In metals� the cohesiveenergy of a series of crystal structures that only dier on the atomic coordinationz scales more as �z �
� than as �z �which is the result one expects for independentbonds� �����
In order to correct for this problem� a pair�functional method is generally used�see for example reference ������ The total energy of the crystal is regarded as a sumof an embedding energy� for the atoms and a pair�potential term that accounts forelectrostatic contributions� The embedding energy� is the energy associated withplacing an atom in the electronic environment of the solid� It not only incorporatessome many�body eects but also the dependence of bonding with coordination� Here�
�Due to this principle the electrons are prevented from occupying the same energy levels whenthey overlap and since the ions in these crystals usually have closed shells they need to occupyhigher orbitals� As a result it costs too much energy to overlap the ions and they e�ectively repeleach other�
�Most frequently however the ionic charge is not �t but the corresponding chemical oxidationstate is used instead�
��

we will discuss a particular implementation of this approach� called the embedded�atom method �EAM� since it has recently been applied to oxides ���� ����
Both the pair�functional and the pair�potential models lack a good descriptionof directional bonds� This is specially important in semiconductors and covalentmaterials in general� Cluster potential and functionals should be used in this case�We will come back to this point later� in section ������
����� Approximations of classical models
The major approximation of the classical methods is to assume a given functional formfor the energy in equation ���� Although sometimes inspired in the actual physics ofbonding� only a self�consistent formulation of Schr#oedinger�s equation is capable ofcapturing all the details of the problem�
But even when the potential or functional form for the energy is adequate� mostpopular versions of these models introduce other approximations that can be partic�ularly important in oxides� For example� it is common practice to assume constantcharges for the ions� independently of the environment� We will show in chapter �that this can produce large errors in the formation energies� Also� it is important inoxides to account for the oxygen breathing�� Recently� some schemes to correct forthese problems have been introduced� and will be described in section ������
The transferability of the parameters of classical models depends on the accuracyof the function chosen to describe the energy� These parameters are usually �t toab initio or experimental information for a few structures� When applied to otherstructures with dierent atomic arrangements large errors can be introduced� Mixingstudies of multicomponent systems may suer from a similar problem� When the �tis only done to the end members� the properties of mixtures are not necessary wellreproduced�
����� Pair�potential models
Due to the complexity of oxides� modeling in these materials has been mainly donewith simple empirical potential models� To date� most potentials for oxides have beenbased on Coulomb and pairwise short�range interactions� with ionic polarizabilitytreated by the shell model of Dick and Overhauser ����� See ���� ��� ��� ��� andreferences therein for a review� These models have been successful in predictingdefect energies� lattice constants� and some elastic properties ���� ���� However� theyare inadequate to make quantitative predictions when computing phase diagrams ����as we will show in chapter ��
With these potentials� the total energy is expressed as the sum of the short�rangeinteractions� Coulomb terms� and the polarization of the ions� The electronic cloudof the i ion is simulated by a massless shell of charge Yi and the nucleus by a core of
�Oxygen breathing is the change in the size of the oxygen due to changes in the Madelung �eldsee section ������
��

Massless shell
Short rangeinteraction
Harmonic interactionCore
Figure ��� Illustration of the interaction between two ions in the pair�potentialmodel� Each ion is represented by a massless shell� on which a short�range pairwiseinteraction acts� and a core �here represented by the shaded and black circles� coupledby a harmonic force to the shell� A Coulombic� long�range interaction� is also assumedto exist between the ions�
charge Xi� The total charge� qi� is
qi � Xi � Yi� �����
The core and the shell of the ion are coupled by a harmonic force� with spring constantki� The polarization energy is given by
Vi ��
�kid
�i � �����
The distance di is the relative displacement of the core and the shell� The short�range interaction is assumed to act between the shells� The situation is illustrated in�gure ����
Dierent functional forms have been proposed to model the short�range pair in�teractions� The simplest one is represented by hard spheres�
V �rij� �
� � rij � a� rij � a
� �����
�Within this model the free�ion electronic polarizability �i is equal toY �
i
ki�
��

Here� a is the ionic radius and rij is the distance between ions i and j� The two�bodypotential used in this work has the following functional form
V �rij� � Aije
��rij�ij
� Cij
r�ij �����
where Aij� �ij� and Cij are parameters that depend on the identity of the ions���
The functional form in equation ��� is known as the Buckingham potential� Theexponential term represents the repulsion due to the electronic cloud overlap and ther�� term is related to induced dipole�induced dipole interactions� These potentialsare usually truncated beyond a distance corresponding to a few lattice constants�
The Ewald summation method ���� is used to compute the electrostatic energy ofthe point charges qi� The value of the charges is �xed and does not depend on theenvironment��� For simplicity� formal charges equal to the valence of the atom areusually assumed� Partial charges� that correspond to nonintegral values� have beenused to study some systems such as alumina ����� Unless they are used in conjunctionwith a charge transfer model� their transferability to structures other than the onesused in the �t is questionable�
The parameters of the potential� Aij� �ij� Cij� Yi� and Ki� are typically obtainedfrom �tting to lattice parameters� elastic and dielectric constants� phonon frequen�cies� etc� All these properties sample a system close to equilibrium� To enhance thetransferability of the potential to dierent environments �e�g�� dierent crystal struc�tures�� where they are not necessarily evaluated at the same distance range� �ts todefect properties� ab initio equations of state� and dierent structures should also beincluded�
Pair�potential models present serious limitations� They can neither account formultibody eects �angular�dependent interactions� oxygen breathing� charge transfer�etc�� nor for the fact that the strength of the bond may depend on the actual numberof bonds� Their parameters are environment independent� Furthermore� for cubicmaterials at zero pressure� they predict relationships between elastic constants thatare not necessarily valid �for example� c�� � c��� see reference ������
Some of these issues have been addressed by pair functionals or by models thatinclude breathing and variation of charge� We will discuss some of these models inthe next sections�
Charge transfer
Recently� Streitz and Mintmire have proposed a method to model the charge transferbetween the ions ����� It can be used within a potential or functional formalism� Thecharges are obtained by minimizing the electrostatic energy Ees�
��
The electrostatic energy is written as a sum of atomic energies Ei�qi� and inter�
�Typical values for these parameters are� Aij � ��� eV �ij � ��� �A and Cij � ��eV �A����This is a strong assumption and it is often not valid as we will show in chapter ����Note that there is no variational principle that justify this procedure�
��

action energies between all pairs of atoms� Using their notation
Ees �Xi
Ei�qi� ��
�
Xi��j
Vij��rij qi qj� �����
where Vij��rij qi qj� is the Coulomb pair interaction between ions i and j and qi arethe charges to be computed�
The atomic energy represents the intra�electrostatic interaction between electronson the same atom� It is written as a Taylor expansion on the ionic charge qi� Thecoe�cients of this expansion are determined during the �t of the potential or thefunctional form��� The Vij��rij qi qj� can be computed by simply assuming pointcharges or an atomic charge�density distribution�
Finally� the values qi are obtained by minimizing Ees subject to the constraintthat the sum of all the charges in the solid should be zero �charge neutrality�� Thismodel was used to study metal�oxide interfaces �aluminum ��alumina� �����
Compressible�ion model
The eect of the change in size of the ions with the Madelung �eld can be modeledby computing the energy to create the ionic charge density appropriate to a given setof lattice parameters� Dierent ways of implementing this procedure exist� Here� wewill brie�y describe the scheme by Wilson and coworkers since they integrated theirformalism within a pair�potential method ����� Other approaches will be presentedin section ������
The electronic charge density of the ion is computed by solving for the self�consistent wave function within a potential that simulates the crystal� This potentialis the sum of two contributions a long�range Madelung �eld �assuming the ions to bepoint charges� and an electrostatic repulsion from the neighboring charge densities�The energy to obtain a given charge distribution �or its dierence with respect toisolated ions� is then incorporated into a potential as a function of generalized ioniccoordinates�
The generalized coordinates include not only the positions of the ions but also adescription of their state�� The state� is speci�ed by assigning a radius to the ionsthat would depend on the environment� A term is then added to the short�rangepotential� that depends on this radius and represents the energy to rearrange theionic charge density� For a �xed set of ionic positions� the total energy should beminimized with respect to the radius coordinates before evaluating the energy�
The term that represents the charge rearrangement at the ions is given the form
F ��� � c�e��� � e��� �����
where � represents the change in the ionic radius with respect to a reference valueand � and c are coe�cients to be �t to ab initio results�
��The �t is performed as explained in the previous section�
��

Note that the overlap within the ionic wave functions also depend on �� Con�sequently� within this model� the pair potential short�range repulsion is a functionof the ionic radius� For more details about the method� see reference ����� Thecompressible�ion model has recently been used to study MgO� CaO� Al�O�� andZrO� ���� ��� ����
����� The embedded�atom method
Up to now we have assumed that the total energy of the solid was given by a sumover independent bonds� As already mentioned� this is only true for a perfectly ioniccrystal� A solution for this problem can be found by using pair functionals such as itis done in the embedded�atom method �EAM��
The basis of the EAM consists in expressing the energy of a solid in terms of embedding� energies and electrostatic interactions� Each atom is assumed to be embedded� into the electron gas created by the other atoms� The cohesive energyEcoh is then written as�
Ecoh �Xi
Fi
��Xj ��i
�j�Rij�
�A�
�
�
Xi�ji��j
Vij�Rij�� �����
Here� Fi is the embedding energy for atom i and is applied to the superpositionof the spherically averaged electron atomic�density tails ��j�Rij�� from other atomsevaluated at the position of atom i� Vij�Rij� represents electrostatic� two�atom inter�actions�
Equation ���� and correspondingly Fi and Vij�Rij�� can be derived from density�functional theory��� However� they are generally �t to bulk properties such as latticeand elastic constants and vacancy formation energies� The embedding energy is non�linear �convex�� re�ecting the saturation of the bonds with the number of neighbors�
Analytic functions or tables are used to express Fi� The Vij�Rij� are representedas decaying exponentials �due to electrostatic screening�� The spherical electron den�sities can also be parametrized with simple decaying exponentials� For more detailson the EAM� see reference �����
The embedded�atom method has been recently applied to oxides� yielding reason�able results for alumina ����� To appropriately account for the long�range Coulombinteraction� Streitz and Mintmire added Ees �see equation ���� to the cohesive energyin equation ��� ����� They did not eliminate the Vij�Rij� terms in equation ��� butkept them so that their coe�cients could be used as free parameters� Baskes alsoreported EAM calculations in oxides �����
��Two assumptions should be made� One is that the electron density of the solid can be computedas a linear superposition of the atomic electronic densities� The other one is that the kinetic exchange and correlation energies see section ����� can be written in terms of local electron densitiesand its derivatives� See reference ���� for more details�
��

����� Cluster functions for the energy
Both the pair�potential and the embedded�atom methods do not work well for sys�tems with directional bonding� Cluster potentials and functionals should be usedinstead�
It is common practice to add �� and ��body interactions to potential modelswhen semiconductors are modeled� These new terms allow for the inclusion of bond�bending and torsion eects� For example� computational simulations in silicon areusually performed with multibody potentials such as the Stillinger�Weber one �����
The EAM can also be modi�ed to accommodate directional bonding eects� Themodi�ed embedded�atom method �MEAM� is similar to the EAM except that nowthe electronic density contributions from the neighbor atoms can include angulardependencies� See reference ���� ��� for more details�
��� Quantum�mechanical approaches
Quantum mechanics can be used to explicitly �nd the dependence of the total energyon the nuclear coordinates in equation ���� In the remainder of this chapter� we willstudy dierent techniques to perform this operation�
The methods dier on the dierent approximations used to solve the Schr#odingerequation� Within the assumptions made in section ���� this equation can be writtenas��
H' �
��� NeX
i��
(h�
�me
r�i �
NXI��
NeXi��
ZIe�����ri � �RI
��� ��
�
NXI
NXJ ��I
ZIZJe�����RI � �RJ
����
�
�
NeXi
NeXj ��i
e�
j�ri � �rjj
�A' � E'� �����
Here� ' is the solid wave function� E is its eigenvalue� Ne is the number of electrons�N is the number of atomic nuclei� Z is the atomic number� e is the electron charge�and �R and �r are the nuclear and electron coordinates respectively�
The dierent terms in the Hamiltonian of equation ��� represent the kinetic energyand the electron�ion� ion�ion� and electron�electron electrostatic interaction operatorsrespectively� The solid wave function '� is a function of the electronic and ioniccoordinates�
Even after decoupling the nuclear and electronic coordinates� solving equation ���is unfeasible since there are practically an in�nite number of electrons that interactwith each other� We will show that this problem can be surmounted by using theperiodicity of the crystal and assuming that equation ��� can be transformed into aset of single�electron equations coupled through the electronic interactions� �Thus�the solution of Schr#odinger�s equations has to be found self�consistently��
The interaction of a single electron with the nuclei and the other electrons has
��We use capital and small letters to denote nuclear and electron coordinates respectively�
��

the periodicity of the crystal� This allows the use of Bloch�s theorem to express theelectron wave function � as��
�nk��r� � eik�runk��r� ������
where unk is a function with the periodicity of the Bravais lattice� �k is a wave vector�
and n is known as the band index� Not all the possible �k points are allowed but theyare restricted by imposing appropriate boundary conditions to the bulk solid� namelythat ����
���r ��Xi��
Ni�ai� � ���r�� ������
The �ai are the three primitive vectors of the unit cell and Ni are positive integernumbers such that the total number of primitive cells in the solid be N�N�N��
� Inthis way� the problem of �nding the wave functions of an in�nite number of electronsis transformed into �nding the wave functions of a �nite number of electrons at anin�nite number of �k points� At each �k point� only a �nite number of levels areoccupied�
The advantage of this procedure becomes clear when we realize that solutions at�k points that are close together are very similar� Consequently� we can representdierent regions in �k space by a �nite number of �k points� Thus� the electronicpotential and total energies can be obtained from computing the electronic states atthis limited set of points� See appendix B on how the �k points are chosen�
Defects and atomic vibrations usually break the periodicity� In what follows� wewill always assume crystals that have a perfect periodicity� Nonperiodic con�gurationscan be dealt with by using a supercell approximation� In a supercell approximation�a particular unit cell containing the defect� in the arrangement of atoms is repeatedperiodically throughout space so that a perfect regular con�guration is recovered�The defect in question is thus also arti�cially repeated throughout space and thefurther apart its images are �i�e�� the larger the unit cell�� the more accurate theapproximation becomes� It is common practice to evaluate how the defect propertiesvary with the size of the supercell in order to assess the importance of the errorsintroduced by the interaction� of the images�
Although the original problem seems to have been greatly simpli�ed by the aboveapproximations� more work needs to be done to deal with the many�body electron�electron interactions and to make the computational burden more aordable� Thesenew approximations are more speci�c to the particular methods and are dealt within the next sections� In table ���� we summarize the methods we use in this workand their most common approximations� We have chosen these techniques since theyprovide the best compromise between computational speed and accuracy in oxides�
��See for example reference �������We are using a generalization of the Born�von Karman periodic boundary condition ����� The
surface of the solid is eliminated by replicating the solid throughout space� An electron coming toa surface simultaneously reenters the solid on the opposite surface� An in�nite solid has an in�nitenumber of �k vectors� We will assume the size of the solids to be in�nite�
��

LDAa Spherical symmetry Frozen coreFLAPW �LMTO�ASA � �Pseudopotentials � �SSCAD � �Tight binding � �
Table ��� Total�energy methods and their approximations� The bullets indicate themost common approximations used by each technique� A detailed description of themethods and approximations can be found in section ����
aLocal�density approximation to density�functional theory
In section ������ we will describe the dierent approximations mentioned in ta�ble ���� The details of the particular implementation of the techniques will be dis�cussed starting in section ������ The major dierence between them is the basisfunctions used to expand the solutions of the quantum�mechanical equations� Somefunctions are more suited than others for solving certain problems �such as the cal�culation of forces� or work with speci�c types of bonds�
����� Common approximations in quantum�mechanical ap�
proaches
Dierent choices exist for breaking equation ��� into single�electron equations andexpressing the interaction between the electrons� In this work� we will apply thedensity�functional theory �DFT� which is an exact transformation of equation ���into single�electron equations� but valid only for computing ground�state properties�Unfortunately� DFT does not oer a recipe to determine the potential felt by a singleelectron� Extrapolations from limiting cases where the solution is known have to betaken� Another commonly used approach is the Hartree�Fock method� This methodwill not be described here since its implementation is very computer intensive anddoes not provide signi�cant bene�ts over DFT� �A detailed explanation of this methodcan be found in reference ������
Once the problem is described by one�electron equations� other approximationsare still introduced to reduce the computational burden of the methods� They rangefrom assuming special geometries or symmetries for the electronic potential� to freez�ing� the core electrons�
Density�functional theory and its approximations
Many reviews have been written about DFT �see for example references ���� and ������Here� we will only describe its main features� The focus of this theory is more onthe electronic density rather than on the wave functions� Hohenberg and Kohn ����
��

proved that the total energy of a system is a unique functional of the electron densityand that the minimum value of the functional corresponds to the actual ground�stateenergy and the corresponding electronic density to its actual density� The next stepwas taken by Kohn and Sham ���� who showed that equation ��� can be replacedby a set of noninteracting single�electron equations such that the ground�state totalelectronic density is the sum of the single�electron ground�state densities� Thus�DFT provides a general method for calculating the energy and the electronic densityof the lowest energy state �and all the properties that can be obtained from them��
The Kohn�Sham equations are written as���
HKS�i �
��� (h�
�me
r� �NXI��
ZIe�����r � �RI
��� � eZ n��r������r� � �r
���d�r� � Vxc
�A�i � �i�i� ������
HKS is the Kohn�Sham Hamiltonian� Vxc is the exchange and correlation potential��
�i is the single�electron wave function� and �i its eigenvalue� The total electronicdensity n is given by
n � eNeXi
j�ij� � ������
Although the Kohn�Sham equations are an exactmapping of the many�electron prob�lem onto a single�electron one� the �i are not the real single�particle eigenvalues �����
The exchange and correlation potential that the electrons face is not known anddierent approximations are used to determine it� The most popular one is the local�density approximation �LDA� ����� In the LDA� the exchange�correlation energyper electron at point �r� �xc� is replaced by the exchange�correlation energy of anhomogeneous electron gas with the same density as the electron gas at point �r� TheLDA assumes that �xc is local and ignores all the eects of inhomogeneities around �r�
Several parameterizations exist for the exchange�correlation energy in a uniformelectron gas� They are frequently expressed as interpolations between the high andlow electron�density limits� See for example references ���� and ������ Most of themlead to very similar total�energy results�
In recent years� the generalized�gradient approximation �GGA� ���� ��� has beendeveloped as a way to improve over the LDA� In this approach� the �xc is expressedas a local function of not only the density� but also its gradient� In this way� one triesto capture the eects of inhomogeneities in the electronic density�
Spherical approximations for crystal potentials
Shape approximations are introduced to accelerate the solution of Schr#odinger�s equa�tion� The most widely used approximation was �rst applied by Slater ������ and is
��For simplicity the electrostatic interaction between the ionic cores is not written� It will bedirectly added to the total energy�
��The Vxc potential captures all the electronic exchange and correlation e�ects� It is computed
from the exchange�correlation energy per electron xc as Vxc � ��n�xc n
where n is the electronicdensity�
��

A B
Vo
spAr B
spr
Figure ��� Mu�n�tin potential often used to approximate the actual crystal poten�tial� The sphere radii rspA and rspB �for atoms A and B respectively� are arbitrary�except for the fact that the spheres should not overlap�
called the mu�n�tin approximation �MTA��In the MTA� the crystal potential�� is assumed to be spherically symmetric inside
spheres centered at each atomic site� and constant outside these spheres� If we placea sphere of radius rspi at site Ri� the mu�n�tin potential U��r� is
U��r� � V �����r � �Ri
���� when����r � �Ri
��� rspi
� Vo�rspi � when
����r � �Ri
��� � rspi � ������
The shape of this potential is shown in �gure ���� Since U��r� is spherically symmetric�only the radial components need to be speci�ed� The only constraint to �x theradii of the spherical regions is that the spheres do not overlap� The nonsphericalcontributions to the crystal potential and the eect of assuming a constant value in theinterstitials can frequently be included using some sort of perturbation or correction�As we will mention in the next sections� these eects can be taken into account veryaccurately�
The spherical�symmetry approximation is generally correct as long as the spheresare centered at high�symmetry sites� Consequently� the MTA gives accurate resultsfor closed packed materials �for example� fcc or ideal hcp� and is increasingly lessreliably as the site symmetry decreases and the coordination number lowers�
�The crystal potential is usually de�ned as the potential experienced by an electron that originatesin the electrostatic interactions with the atomic nuclei and the other electrons�
��

ri
3Unit cell volume= 43ΠΣ ri
i
Figure ��� Cubic unit cell showing the Wigner�Seitz overlapping spheres in the ASA�The interstitial region �shaded area in the �gure� is usually neglected� The sum inthe equation is over the spheres in the unit cell�
In some cases� an even simpler approximation than the MTA� called the atomic�
sphere approximation �ASA�� is taken ���� ����� In the ASA� the unit cell ismapped onto the space of Wigner�Seitz overlapping spheres generally centered atatoms� All integrals over the cell are substituted by integrals over these spheres andthe electronic density is spherically averaged within each of them� The potential isthen a superposition of spherical contributions from each sphere� The situation isdescribed in �gure ����
Note that in the ASA case� the errors would come not only from nonsphericalcontributions but also from the overlap and the neglect of the interstitial regions forthe integrals��� In open structures� some of the overlap errors can be alleviated bythe use of empty spheres to reduce the degree of overlap�
Frozen�core approximation
In any material� there is often a clear distinction between core and valence electrons�Core electrons are usually strongly bound to the atomic nucleus and do not respondsigni�cantly to the motion of the valence electrons� Consequently� when atoms arebrought together to form a solid� the changes in the core electron wave functionsare very small� They can be regarded as essentially frozen� This is known as thefrozen�core approximation�
To take advantage of this behavior� some total�energy methods compute onlythe valence wave functions� The eect of the core electrons on them is taken intoaccount as a potential added on top of the nuclear potential� Considerable savings incomputational time may be obtained when this approximation is used�
The contribution of the core electrons to the total energy is very large and cancels
��Corrections for the overlap regions and nonspherical contributions can be used� See sec�tion ������
��

out when formation energies are taken� When the frozen�core approximation is used�this contribution is not computed and the formation energies can be determined asdierences between small total�energy numbers requiring a smaller relative accuracy�
The frozen�core approximation is very accurate as long as the valence and the coreelectron wave functions do not overlap signi�cantly� If this is not the case� some coreelectrons have to be treated as part of the valence electrons� As we discuss in thefollowing chapters� some total�energy methods incorporate special treatments thatcorrect these problems while still retaining the so�called semicore states as part ofthe core�
����� The spherical self�consistent atomic deformation
model
The spherical self�consistent atomic deformation �SSCAD� model ����� is a �rst�principles method for computing total energies in ionic crystalline solids� It is basedon density�functional theory in the local�density approximation� The SSCAD takesadvantage of the characteristics of the ionic materials to make simplifying approxi�mations that allow fast calculations without sacri�cing too much accuracy�
The electronic density is assumed to be localized about each ion and the totalelectron density n to be the sum of these individual densities�
n��r� �NXi
ni�ri�� ������
In equation ����� ni is the density corresponding to the ith ion and ri the distancefrom �r to that ion� The total energy of the crystal can be written as a functional ofthis total electron density as
E�n� � T ��
�
NXI
NXJ ��I
ZIZJe�����RI � �RJ
��� �NXI��
ZIe�Rn��r�����r � �RI
��� d�r
��
�e�Z Z n��r��n��r�����r� � �r
��� d�r�d�r �
Z�XC��r�n��r�d
�r ������
where T is the kinetic energy of the electrons �the ions are assumed to be �xed��The kinetic energy is approximated as a sum of an intra�ionic term plus a correctioncoming from the overlap of the electronic clouds �computed from the Fermi�Thomasenergy for a gas of interacting electrons with uniform density� ������
Applying density�functional theory to the energy� as in reference ����� a one�particle Schr#odinger�like equation is obtained for each ion�
� �
�r�� � Vi� � ��� ������
Here� Vi and � are the Kohn�Sham potential and eigenfunctions for ion i respectively�As in reference ����� the potential Vi depends on the total density� and the equation
��

is solved self�consistently starting from the densities of isolated ions� To reduce thecomputational time� Vi is spherically averaged about ion i� After imposing sphericalsymmetry� equation ���� becomes an ordinary radial� Schr#odinger equation andcan be solved either by �nite dierences or by expanding the solution in a set ofbasis functions� Usually� the Slater basis�set expansions of the Roothaan�Hartree�Fock wave functions are used in this expansion ����� ����� Once equation ���� issolved� the total�electron density is computed and the total energy is obtained fromequation �����
The SSCAD has many similarities with the Gordon�Kim electron�gas model �����for calculating the interaction energy in closed�shell ions� However� two major im�provements have been introduced in the former ionic breathing and self�consistency�In the Gordon�Kim scheme� free�ion electron densities were used� This can introducelarge errors since these densities can change considerable when the solid is formed�This phenomena is commonly known as the breathing of the ions��� In the SSCAD�the dependence of the ionic electron density on the Madelung �eld is incorporatedthrough the potential Vi in equation ����� The self�consistency in the SSCAD modelallows the eect of the nearest�neighbor electronic cloud to be taken into accountwhen computing the electronic density in the ion�
The major limitation for the use of the SSCAD in oxides is the imposed sphericalsymmetry and the approximated treatment of the electron kinetic energy� The elec�tronic density in these materials is not necessarily spherically symmetric and a largeerror in the total energy can be introduced as we will show in chapter �� Nonsphericalversions of the SSCAD are currently under development�
����� The linear mun�tin�orbital method
It is not our intention to review here all the complex details of the linear mu�n�tin�orbital �LMTO� method in the atomic�sphere approximation �ASA�� The readeris referred to the many review articles and books that have been published on thistopic ���� ���� ����� Instead� we will brie�y mention the main approximations thatare incorporated into the LMTO� Due to these approximations� the solution of theKohn�Sham equations can be performed much faster than with other less approximatemethods�
In the LMTO�ASA� space is divided into overlapping Wigner�Seitz spheres� gen�erally centered at the atomic positions� The solutions are computed in the atomic�sphere approximation �see section ������� The basis functions used to expand the solu�tion of the Kohn�Sham equations in an ASA potential are computed as the solutionsof the Laplace equation �i�e�� Schr#odinger�s equation with zero kinetic energy� out�side each sphere which are augmented by the solutions for the spherically�symmetric
��For example O�� is unstable as a free ion� It can only exist in a solid due to the stabilizatione�ect of the Madelung �eld� Since the Gordon�Kim scheme could not be applied to this ion Boyer and coworkers developed the potential�induced breathing PIB model ���� ����� PIB usesa spherical shell of charge Watson sphere ����� to simulate the e�ect of the solid environment onthe ion� Thus the O�� ion is made stable and its electronic density can be computed�
��

potential within each sphere� These functions are usually called mu�n�tin orbitals�in the ASA�� Both the wave functions and their spatial derivatives are required tobe continuous at the sphere boundary���
The mu�n�tin orbitals have a complicated energy dependence that can be elimi�nated by expanding around a �xed energy E to �rst order in �E �E��
�� The basisfunctions thus obtained are called linear mu�n�tin orbitals and are used to solve theeigenvalue problem� Only a reduced number of basis orbitals are needed to obtainconverged results��� For example� most atoms require only nine orbitals �correspond�ing to the s� p� and d quantum�angular momenta�� The small number of basis orbitalsincreases the computational e�ciency of the method considerably� Other methods�such as the plane�wave pseudopotential technique �see section ������� may requireorders�of�magnitude more basis functions�
There is no unique way to choose the radii of the atomic spheres� The onlycondition is that the sum of the volumes of all the spheres in the unit cell be equalto the volume of the cell itself� This is enough to determine the radii only whenone atom per cell is present in the solid� For all other cases� a criterion for choosingthe radii is needed� Four dierent criteria are usually used� The �rst criterion isto take equal�size spheres for all the atoms in the crystal� The second one is tomake the atomic spheres charge neutral �in this way� the Madelung energy error inthe ASA vanishes�� The third one is to minimize the total energy of the compoundwith respect to the radii of the spheres� though there is no variational principle thatjusti�es this procedure� Finally� the last criterion� proposed by Andersen ������ givesan explicit formula for the sphere radii in terms of Wigner�Seitz radii� bulk moduli�and elemental volumes�
For close�packed solids� the LMTO�ASA method is very accurate� Correctionsare sometimes used to compensate for the sphere overlap �usually called combinedcorrections� ����� ���� ����� and the symmetrization of the potential �usually called mu�n�tin corrections� ������� In more open structures� the method gives less ac�curate energies and empty spheres �not centered at atoms� are used to reduce theoverlap� A full�potential version of the LMTO� where no shape approximations aremade� has been developed ����� but it will not be discussed here�
Formation energies in oxides are poorly predicted by the LMTO�ASA even inclose�packed solids� As we will show in the next section� total energies are verysensitive to the sphere radii chosen� and the consistent use of any of the mentionedfour criteria along many compounds in the same system does not provide accurateformation energies���
��The mu�n�tin orbitals we just de�ned decay slowly in space specially for low quantum�angularmomentum� Short�range basis functions can be de�ned by combining linearly independent solutionsof Laplace equation through what is usually known as a screening transformation ������
��E� is usually the value corresponding to the center of the occupied part of the band� Seereference ���� for more details�
��The linear mu�n�tin orbitals in the ASA form a complete basis set for the wave functions ofan electron in a crystalline ASA potential and provide a very good description of them�
��In metallic alloys these four criteria usually coincide and LMTO�ASA formation energies arecomparable to the values of the most accurate methods� In oxides on the other hand some of the
��

Eect of the ASA in oxides
We performed numerous LMTO�ASA calculations in CaO�MgO� ZrO�� Na�O� andLi�transition�metal oxides� In all cases� we found an important dependence of thetotal energies on the size of the atomic spheres even when the combined� and the mu�n�tin� corrections were used� We will illustrate this behavior in the CaO�MgOsystem�
An s�p�d basis was used for the calculations in CaO�MgO� The number of self�consistent iterations and �k points were selected so that the variation of the total energywas less than �meV cell� This resulted in a ��� �k space uniform grid �we consideredcrystal symmetry�� The von Barth�Hedin form for the exchange�correlation potentialwas used ������
In �gure ���� we show the total energy versus sphere radius for the CaO structure�The variation of the total energy is almost � eV cell�� Since formation energies areof the order of tens of meV� their value can be switched from positive to negative bychoosing dierent points on this curve�
There is a clear need for a criterion to determine the sphere sizes� Unfortunately�our results show that it is not possible to use the same criteria in oxides as in othermaterials� Here� it does not make sense to make the spheres charge neutral� Ingeneral� we also found that even the consistent use of any of the criteria for allcompounds in the same system does not provide accurate formation energies whencompared to FLAPW or pseudopotentials� For example� we found that both the Ca�and Mg�rich L�� structures in the CaO�MgO system have negative formation energieswhen using equal�size spheres while all the other total�energy methods predict themto be positive� Using a smaller radii for the cations than for the anions makes theformation energies of the L�� structures positive but we could not �nd a consistentway of setting the sphere sizes that provided reasonable results for all compounds�We observed the same behavior in the other oxide systems we studied �ZrO��CaO�Na�O� and Li�transition metal oxides�� In metals� the error introduced by the ASAis less critical and it is usually very small when using neutral spheres�
The dependence of the lattice constants and the shape of the band structures onthe sphere radii is much smaller� For example� the lattice constant for a cubic cell�such as the rocksalt structure� may only change by a few percent�
The LMTO�ASA method on the other hand� is orders�of�magnitude faster thanmore accurate total�energy techniques� However� the errors introduced by an incor�rect treatment of the overlap region and the spherical approximation can be verylarge in oxides� Consequently� we will not make use of the LMTO�ASA formalism�A full�potential non�ASA LMTO method can overcome these di�culties but at theexpense of computational speed���
criteria can not even be applied� For example it does not make sense to make the spheres chargeneutral�
��The sphere radii range in �gure ��� was chosen taking into account that Ca ions are usuallysmaller than O ones and that very dissimilar radii will considerably increase the overlap betweenthe atomic spheres�
��For example the formation energy of the CaMg�O� compound in the L�� structure was predicted
��

Tot
al E
nerg
y (R
y/ce
ll)
r /r Ca O
-1507.31
-1507.3
-1507.29
-1507.28
-1507.27
-1507.26
-1507.25
-1507.24
-1507.23
-1507.22
0.8 0.85 0.9 0.95 1 1.05
Figure ��� Total energy for pure CaO as a function of the ratio between the Ca andthe O sphere radius in the LMTO�ASA� The volume of the cell was kept constant andcorresponds to the experimental value� Empty spheres� and mu�n�tin and combinedcorrections were used to reduce the errors introduced by the ASA� The total energychanges in a scale much larger than the scale of the temperature eects and a criterionis needed to choose the sphere size�
��

����� The full�potential linearized augmented�plane�wave
method
The full�potential linearized augmented�plane�wave �FLAPW� method has its originsin the augmented�plane�wave �APW� technique introduced by Slater ������ Thebasis functions on this approach are quite similar to those of the LMTO� Again�space is divided using spheres�� Plane waves are used in the interstitials whichare then augmented by the solutions for the spherical radial equation �multiplied byspherical harmonics� within the spheres� These basis functions are also made energyindependent in a similar fashion to the LMTO�
The FLAPW method is one of the most accurate techniques that are currentlyavailable� The only major approximation in�uencing the results is the LDA� TheFLAPW method will not be used here� However� we will take results from the liter�ature as benchmarks for some of our calculations� More details of this technique canbe found in references ����� ���� ���� �����
����� The pseudopotential method
The pseudopotential approach will be described in detail in chapter �� Contraryto the previous methods� plane waves are used to describe the solid wave functionseverywhere in space �and not only in an interstitial region�� Only the solutions for thevalence electrons are computed �frozen�core approximation�� The eect of the coreelectrons on the valence states is taken into account by a potential that is added tothe nuclear potential� This pseudopotential is chosen so that its valence pseudo wavefunctions are equal to the actual all�electron wave functions beyond a given coreradius and so that they do not have nodes in the core region� Since only the outerelectrons participate in bonding� using pseudopotentials is an excellent approximationin most cases�
The use of the pseudopotential approximation reduces not only the number ofplane�wave basis functions but also the number of orbitals to be determined� Thecore�electron wave functions are not computed� All�electron techniques� on the otherhand� need to accurately treat these core electrons that constitute a large portion ofthe total energy� while generally their eects are canceled out when energy dierencesare taken� Consequently� the pseudopotential energy is much smaller and requiresa smaller relative accuracy than all�electron methods when computing formationenergies�
Plane waves greatly simplify the solution of the Kohn�Sham equation shapeapproximations are not necessary and Pulay forces are zero ������ Furthermore� verye�cient fast Fourier transforms are available�
The computational speed of the method depends on the chemical identity of the
to be ����� meV�ion with a full�potential version of the LMTO ������ This is within ��� of thevalues shown in table ��� which were computed with the most accurate techniques that are available�
��As in the LMTO�ASA case spheres centered at atomic positions are used� However in theAPW these spheres do not overlap�
��

elements that form the material� Sharply peaked valence states� as in �rst row non�metals such as oxygen� or in transition metals� require a large number of plane wavesto be expanded� However� in the last years� new developments such as Car�Parrinellomolecular dynamics ������ conjugate�gradients ������ and optimized ����� and ultra�soft ����� pseudopotentials have shown that plane�wave pseudopotential calculationscan be e�ciently performed for any element in the periodic table ������
For a review on the pseudopotential method� see references ����� ���� ���� ����and references therein�
���� The tight�binding method
Descriptions of the tight�binding method can be found in references ����� ���� andreferences therein� In this technique� the one�electron wave functions are expanded ina set of atomiclike local wave functions� This choice of basis makes the computationof the Hamiltonian matrix very involved� Consequently� two popular ways of thinkingabout this technique were developed as an ab initio method or as a semiempiricalapproach� In the early days��� the latter was the only way to avoid the calculation ofvery time�consuming interaction integrals��� The basis functions remained unspeci��ed and the Hamiltonian matrix elements were �t to reproduce physical propertiesobtained from other methods or from experiments �e�g�� electronic band structure��
Due to the increase in computer power� the �rst�principles tight�binding �TB�approach has been gaining popularity in recent years� specially to compute electronicproperties of ceramic materials �particularly oxides� ������ In this work� we willuse the semiempirical tight�binding �SETB� method ���� in the self�consistent formsuggested in references ���� and ������ Contrary to empirical potential models� anexplicit treatment of the electronic structure is made and all the relevant terms forcomputing total�energy dierences in systems with charge transfer �such as oxides�are incorporated� A detailed description of the method will be given in chapter ��
The SETB model is not limited to a given type of bonding and no shape approxi�mations for the ionic potentials need to be made� Furthermore� it is particularly fastsince the interaction integrals are �t to ab initio results� In this sense� the SETBrepresents a much more sophisticated and accurate interpolation tool than potentialmodels and is still orders�of�magnitude faster than ab initio quantum�mechanicaltechniques�
��� Time scaling
The time it takes for the dierent total�energy methods to perform a certain calcula�tion is assumed to be given by cN i where N is the number of atoms in the unit cell�
�The use of a linear combination of atomic orbitals to solve the quantum�mechanical equationsin a solid was originally proposed by Bloch in ���� ������
��These integrals correspond to the �hopping� and �overlap� terms we will describe in chapter ��
��

Method Time scalingPotentials N �N��SSCAD N �N��Tight binding N �N��Pseudopotentials N� ln�N� �N��LMTO�ASAa N�
FLAPW N�
Hartree�Fock N����N
Table ��� Time scaling of some total�energy methodswith respect to the size of the systemN �number of atomsin the unit cell�� The values in parenthesis representupper limits according to dierent implementations�
aIt can be order�N in TB form�
c is a prefactor� and i is a number that corresponds to the order of the algorithm���
It is generally the case that a low order corresponds to a large prefactor� so whenevertwo techniques with dierent orders are compared� the crossover point �where bothtake the same computational time� corresponds to systems with large unit cells�
Ideally� if we are interested in exploring larger and larger systems� the order ofthe time scaling should be as small as possible� Most traditional electronic structurecalculation methods have a cubic scaling� This cubic scaling re�ects the need to keepthe wave functions orthonormal� Recently� several propositions have been made toprovide methods that scale with N �the so�called order�N methods�� This is a veryactive �eld of research ����� ���� ���� ���� ����� The crossover point varies frommethod to method and goes from just a few atoms to a few hundred atoms�
The scaling of the methods mentioned in this chapter are shown in table ����The techniques with the smallest number of approximations have at least a cubicor N� ln�N� scaling��� Potential models can be of order�N or N� according to howthey deal with the Coulomb interaction� The same applies to the SSCAD��� Thetraditional tight�binding method scales as N�� but order�N algorithms have recentlybeen developed ����� �����
From this point of view� the tight�binding technique is very promising since it can
��Actually this is the leading term in the expression� The �time� equation is usually much morecomplex since di�erent parts of the algorithm scale with di�erent i and c� It is conventional tocharacterize the method by the leading power i�
��This re�ects the fact that an orthogonalization step is required in these methods� Although thisstep scales as N� its prefactor can be made very small and many people call them order N� lnNmethods�
��The calculation of the Ewald sum is of order�N� but the prefactor is very small compared withthe total calculation time specially in the SSCAD� Furthermore this part of the calculation onlyneeds to be done once�
��

be framed as an order�N method� with a relatively small prefactor� As we will showin chapter �� it is also very accurate�
The prefactors of the methods shown in table ��� have dierent values� A typicalcalculation for a two�atom cell may take a few milliseconds with Potentials� a fewseconds with the SSCAD or the SETB� close to a minute with the LMTO�ASA� anda few minutes with pseudopotentials�
��� Conclusions
We used many dierent approximations to transform Dirac�s original equation intoa tractable Schr#odinger�s equation� The wide variety of paths taken re�ects thecomplexity of the problem at hand� There is no hope� at least in the near future� tosolve this problem exactly�
In this chapter� we reviewed the dierent total�energy methods that can be usedto compute formation energies� Only those that are relevant in oxides were men�tioned� We extensively described the dierent approximations commonly introducedby these techniques� In the following chapters� they will play a fundamental role inunderstanding the failure of some methods to produce accurate results�
We already showed that the ASA can introduce large errors when computingformation energies in oxides� Consequently� we will not further pursue this approach�We expect that all shape approximations will in general cause large errors since theyare only suited for closed packed materials� Oxides� on the other hand� usually havelow symmetry�
Another important characteristic of oxides is that they present a combination ofan ionic and a covalent type of bonding� Classical models will need to incorporateterms to deal with directional bonding� nonspherical electron�density relaxations� andcharge transfer� Furthermore� the SSCAD was designed to be used in ionic materials�Its performance will have to be carefully tested when this condition is not fully met�
The pseudopotential method seems to be the best suited technique to computeformation energies in oxides� In the next chapter� we will evaluate its performance�
When choosing a method� both the accuracy and speed should be taken intoaccount� If our �nal goal is to simulate macroscopic behavior from the atomisticpoint of view� order�N methods are going to prevail� In the next chapters� we willshow that the semiempirical tight�binding method is a very good approach not onlybecause it can be formulated as an order�N method with a low prefactor� but alsobecause it is a very accurate technique for oxides�
��

��

Chapter �
The e�ect of approximations in
total�energy methods�
pseudopotentials as a benchmark
At this point� after so many approximations have been introduced in both the quan�tum� and the statistical�mechanical treatment of the solid� the skeptical reader mayjusti�ably have some doubts about the applicability of our models� It is the purposeof the next sections to clarify these points and illustrate the power of the ab initioapproach to predict and give insight on the behavior of materials�
We will study the eects of approximations in total�energy methods in two stages�In this chapter� we will show that the plane�wave pseudopotential technique can beused to accurately compute oxide properties� In the next chapter� we will use theseresults as a benchmark to evaluate the other� more approximate� methods�
To properly test the performance of the dierent total�energy techniques� it isessential to investigate materials systems that exhibit dierent types of bonding andbehavior� We will mainly focus on three cases CaO�MgO� ZrO�� and CaO�ZrO��The �rst one� is one of the simplest oxide systems� Its properties are very well knownand is an excellent case to test the accuracy of the methods against experimentalresults� It is also a typical example of a highly ionic material�
Zirconia �ZrO�� and calcia�doped zirconia �CaO�ZrO��� on the other hand� presenta mixture of a covalent and an ionic bonding� Pure zirconia undergoes structuraltransformations from a low�symmetry monoclinic into a tetragonal and a cubic phase�They are an excellent case to test shape approximations of the crystal potentials�
Finally� since Ca and Zr are aliovalent�� the doping of zirconia by calcia is ac�companied by the presence of charge�compensating vacancies� It is expected that inthis situation� the cation and the anion will exhibit a variation in their charge thatdepends on the particular atomic arrangements ����� ����� Consequently� it will bean ideal test case for those formalisms in which �xed charges are assumed�
Apart from the FLAPW method� the pseudopotential technique makes the leastnumber of simplifying assumptions� The simplicity of the plane�wave basis and the
�Aliovalent means that the two cations have di�erent valence�
��

ability of easily controlling the convergence and accuracy of the results have made thepseudopotential approach one of the most popular choices for performing ab initiocalculations� Furthermore� as we mentioned in chapter �� the use of the frozen�coreapproximation considerably simpli�es the computation of formation energies �whichis one of our main goals��
In the following sections� we will describe the use of pseudopotentials and latticemodels for predicting materials properties� We will also study the eects of theapproximations introduced by the former� and how they may aect its predictions�Applications of these techniques to dierent oxide systems will be discussed� Wewill show that they provide very reliable results� This will demonstrate that we areapproaching a situation where computer experiments are becoming a reality�
We will start in section ��� by introducing the main features of the pseudopotentialmethod� In section ������ we will apply this approach to the CaO�MgO system�Finally� in section ����� calcia�doped zirconia and the polymorphic transformationsof zirconia will be studied�
Some of these applications push the limits of what can be computed in a reasonabletime� The need for faster� but still accurate methods should be clear by the end ofthis chapter� Ful�lling this need is the objective of the rest of this work�
��� The pseudopotential method
The pseudopotential theory was introduced in ���� by Phillips and Kleinman ������Very useful reviews can be found in references ����� ���� ���� ���� ���� and in thebook by Singh ������
In the pseudopotential method� the solution of the Kohn�Sham equation ���� isobtained by expanding the one�electron wave functions in plane waves� This is oneof the easiest choices for a basis� which is re�ected in the simplicity of the underlyingideas and in the implementation of the method� The valence wave functions areexpanded poorly with a truncated plane�wave basis� Even by using the frozen�core approximation they have rapid oscillations near the core�� A pseudopotential isde�ned so that the valence wave functions have no nodes and consequently� are easierto expand� These pseudo wave functions are identical to the actual wave functionsbeyond a given core radius�
In the next sections� these dierent approximations and the actual implementationof the plane�wave technique are discussed�
�The origin of this behavior can be understood from the fact that the core and valence electronwave functions are orthogonal�
��

����� The plane�wave basis
The cell�periodic part� unk��r�� of the electron wave function in equation ���� can beexpanded in terms of a discrete plane�wave basis set�
unk��r� �
XG
cnk G
eiG�r� �����
The sum is over all reciprocal lattice vectors �G� Equation ���� thus transforms into�
�nk��r� �XG
cnk Gei�k� G��r� �����
Even though the plane�wave basis set is discrete� an in�nite number of reciprocallattice vectors is needed� in principle� to expand the valence wave functions in greatdetail� However� the coe�cients cnk G corresponding to the large kinetic energy plane�wave contributions are small and the expansion is truncated beyond some particularcuto energy Ecut�
�
Plane waves form a complete set of basis functions� However� this is not truefor a truncated expansion� leading to an error in the description of the valence wavefunctions� The magnitude of the error can be controlled by adjusting the value of thecuto energy�
In principle� Ecut should be increased until the total energy has converged withinthe desire error� However� di�erences in energy converge faster than total energieswith respect to Ecut� The high kinetic energy components of the expansion are usedto reproduce the details of the rapidly varying parts of the valence wave functions�These parts are located close to the core and they usually remain unchanged when theatom is placed under dierent environments� Consequently� quantities that dependon di�erences of this electron�density distribution� such as the formation energy�converge faster with respect to Ecut than the total energy�
To avoid large cuto energies with unmanageable large basis sets� the pseudopo�tential approximation needs to be used�
����� Pseudopotential approximation
The physical properties of a material are mostly determined by the behavior of thewave functions beyond the core region� The pseudopotential approximation takes ad�vantage of this fact by replacing the actual valence orbitals by pseudo wave functions�The core orbitals� on the other hand� are assumed to be frozen �frozen�core approx�imation�� Their eect on the valence electrons is taken into account as a potentialadded to the nuclear potential� The idea is then to replace these two contributionsby a pseudopotential with the same scattering properties as the original potential but
�The kinetic energy corresponds to �h�
�m
����k � �G����� The reciprocal vectors that are used in the
expansion are determine by the relationship �h�
�m
����k � �G���� � Ecut�
��

r
V
V
ϕϕ
rc
ps
ps
Figure ��� Representation of the pseudo wave function and its corresponding pseu�dopotential �dashed line�� The actual wave function and potential from which theyare derived are also shown �solid line�� The point at which the real and pseudo valuesare matched is called rc�
chosen so that the pseudo�valence wave functions do not have nodes inside the coreregion�
The situation is illustrated in �gure ���� Outside the core� the pseudo and actualpotentials are identical� The strong potential near the core is replaced by a muchweaker pseudopotential� The resulting valence pseudo wave function is smootherthan the real one in the core region� Consequently� a much lower Ecut is needed toexpand this orbital�
The all�electron valence wave functions have dierent number of nodes accord�ing to their angular momentum� The pseudopotential should be able to introducedierent phase shifts for the dierent angular components of the pseudo wave func�tions� A pseudopotential that depends on the angular momentum is called a nonlocalpseudopotential �and local otherwise���
�Though it is possible to introduce arbitrary shifts for each angular momentum with a localpotential there is not a very good control over its smoothness�
��

����� Pseudopotential properties
Usually three properties are required of a pseudopotential norm conservation�transferability� and smoothness�
It is very important that the pseudopotential reproduce the valence electron den�sity outside the core regions since the total energy critically depends on its value�This is guaranteed by requiring not only that the spatial dependence of the real andthe pseudo wave function be equal beyond the core but also that the charge withinthe core be identical� This last condition corresponds to
Z�ps�ps�dv �
Z���dv� �����
The integrals in this equation are over the core volume� A pseudopotential whose wavefunctions satisfy equation ��� is said to be norm conserving� The idea was originallyintroduced by Topp and Hop�eld ����� and Startklo and Joannopoulos ������
A pseudopotential should produce accurate results for dierent valence�electroncon�gurations� This property is known as transferability� Hamman and coworkersshowed that� by imposing norm conservation� the �rst order energy dependence ofthe scattering properties of the core and its pseudo counterpart are the same ������This guarantees that the phase shifts of the pseudopotential are accurate over a widerange of energies� Also� it has to be noted that the smaller the value of rc �where thepseudo� and all�electron values match�� the more transferable the potential� However�in this last case the potential will become stronger�
Finally� since the only two conditions imposed on the pseudo wave function insidethe core are to be nodeless and norm�conserving� there is still some freedom to chooseits actual shape� In order to speed up the calculations� the shape should be smooth
so that a minimum number of plane waves is needed� For example� Rappe and collab�orators optimized the convergence with basis size by minimizing the kinetic energy ofthe high Fourier components of the pseudo wave function ������ The correspondingpseudopotentials are called optimized pseudopotentials�
Various research groups introduced dierent variants for these nonlocal� norm�conserving pseudopotentials� They are described in references ����� ���� �����
It is not necessary to impose norm conservation to make the all�electron and thepseudo wave functions equal outside the core� This condition can be imposed whileletting the pseudo wave function be as smooth� �or soft�� as possible� Consequently�while the actual and pseudo density outside the core still coincide� the same is not trueinside the core regions� The pseudopotentials that are used to generate these wavefunctions are called ultrasoft and were recently introduced by Vanderbilt ������ Theidea is illustrated in �gure ���� The high kinetic energy plane waves serve to expandthe pseudo wave function mainly within the core� If the normalization requirement isrelaxed� strongly peaked functions can be softened� and considerably smaller cutoenergies can be used�
The use of ultrasoft pseudopotentials results in considerable time savings due tothe reduction in the size of the basis set� On the other hand� since the pseudo wavefunction is no longer normalized� there is no correct change of the phase shift with
��

ϕ
ϕ
r
Ps
Pssoft
c
Figure ��� Comparison between wave functions obtained by using a norm conservingand an ultrasoft pseudopotential� The solid line is the pseudo wave function corre�sponding to a regular norm�conserving pseudopotential and the dashed line to anultrasoft one�
energy and extra time is required to correct for this� Also� new terms need to beadded when computing electron densities ������
����� Pseudopotential generation
Originally� the pseudopotential method was a semiempirical approach� The pseu�dopotential was represented by some simple function �e�g�� a few terms in a Fourierexpansion� whose coe�cients were adjusted to �t experimental results� such as somefeatures of the band structure ������
Here� we use a completely ab initio approach to �nd the pseudopotential� Anall�electron calculation is performed on an atom� and a parametrized form of thepseudopotential is adjusted so that it reproduces the eigenvalues and eigenfunctionsbeyond the core radius rc�
� A strict lower limit for rc is the position of the outermostnode in the actual wave function� In general� rc is chosen so that it lies between thislower value and the position of the outermost peak of the all�electron wave function�
Note that dierent pseudopotentials are computed from dierent atomic con�gu�rations� Though the transferability properties of a norm�conserving pseudopotentialare very good� a con�guration close to the one present in the solid should be used�e�g�� instead of using a neutral atomic con�guration� ionized or excited con�gurationsare taken to avoid bumpy� pseudo wave functions �������
�It is common practice to use di�erent rc for di�erent angular momenta i�e� rcl where l speci�esthe angular momentum�
��

The procedure just outlined provides a pseudopotential whose predictions areequivalent to those from all�electron calculations for the reference atom� but thatrepresent a linearization of the exchange�correlation energy when the valence electroncon�guration is changed�� When the valence and core densities are well separated�the errors introduced are small� On the other hand� when there is a signi�cant overlapbetween them� the transferability is greatly reduced and the errors in the total energycan be important� This problem can be avoided by using the so�called core correctionsthat explicitly treat the nonlinear exchange and correlation interaction between thecore and the valence electrons ������
The Kleinman�Bylander transformation
The computational speed is signi�cantly improved by using local pseudopotentials�However� there are certain cases in which nonlocal pseudopotentials have to be used toobtain accurate results� To make this as e�cient as possible� only a partial projectionof the nonlocal components of the wave functions is usually performed� Kleinmanand Bylander suggested using a single basis state pslm �the wave functions of thepseudoatom� for each angular momentum component ������ The Kleinman�Bylanderpseudopotential is then written as
VKB � Vloc �Xlm
jpslm�Vl � �Vlpslmj
pslm j�Vljpslm � �����
where Vloc is an arbitrary local potential used to produce an accurate and transferablepseudopotential and �Vl � VNL�l � Vloc� VNL�l being the real l component of thenonlocal pseudopotential� For the reference atomic state� both VKB and the realnonlocal potential produce the same result� By carefully choosing Vloc�
a high�qualitypseudopotential can be obtained��
����� Computational procedure
The general procedure for computing the total energy of a solid using the pseudopo�tential approach is summarized in �gure ���� It starts by computing the pseudopoten�tials for the elements that form the solid and by de�ning the size of the basis by meansof the cuto energy� A starting electronic density �or the coe�cients of the electronwave functions� is guessed to build the potential in the Hamiltonian� Its eigenvaluesand eigenfunctions can be computed using methods such as conventional diago�nalization� molecular�dynamics� or conjugate gradient minimization� From
�The interaction between the valence and core electrons is transferred to the pseudopotentialand consequently linearized� This is a source of error since the kinetic and exchange�correlationenergy are explicitly nonlinear� The contribution of these terms is only important when there is asigni�cant overlap between the core and valence orbitals�
�Usually the l � � component is taken as the local potential��The application of the Kleinman�Bylander transformation can introduce problems such as the
existence of �ghost� states that can be avoided by a proper choice of the local potential or bychanging the core radius ������
��

Conjugate gradients
Rep
eat n
tim
es
or u
ntil
conv
erge
d
Next band
done
Conventional Diagonalization
Molecular dynamics or
Build the Hamiltonian
(eq. 3.12)
Update coefficients
of wave functions
Recompute n
Self Consistency
achieved?
Compute energy
Build pseudopotentialfrom atomic data
Define basis(E )
cut
Pick initial density(Wave functions)
Build the Hamiltonian
(eq. 3.12)
Conjugate gradient step
for band
New trial wave function
Were all bands scanned?
Is energy converged?
Choose
yes
yes
no
no
no
yes
indirect directminimization minimization
Figure ��� Steps for computing total energies using the ab initio pseudopotentialapproach�
��

the resulting eigenfunctions� the density is recomputed and the process repeated untilself�consistency is achieved� At each iteration� a linear combination of the new� and old� electronic density is used to speed up convergence�
Conventional diagonalization procedures are straightforward to use but theycan only handle Hamiltonian matrices of limited size in a reasonable time� The Hamil�tonian matrix can be very large �of the order of thousands or more basis functions�and increases with the number of atoms in the unit cell� Consequently� this method islimited to those cases where only a few atoms are present� The time scaling is also aproblem since the computational cost of diagonalization increases as the third powerof the number of plane waves used�
Alternative approaches to minimize the Kohn�Sham energy have been suggested�For example� the Car�Parrinello method to perform molecular dynamics on theelectronic degrees of freedom can be used ����� ����� The Kohn�Sham energy func�tional is a function of the coe�cients of the plane�wave basis set� These coe�cientsare regarded as the coordinates of classical particles�� Starting from a set of trialwave functions� these classical particles are given an initial kinetic energy and thesystem is cooled down until the set of coordinates reaches the minimum� After eachmolecular�dynamics step the Hamiltonian is recomputed� The positions of the ionsand the electronic degrees of freedom can be optimized at the same time� This pro�cedure scales as NbNpw lnNpw where Npw is the number of plane waves and Nb isthe number of bands �generally orders of magnitude less than Npw�� This method isusually much faster than the conventional diagonalization�
The two techniques just described constitute an indirect way of minimizing theenergy functional by �nding the self�consistent Kohn�Sham Hamiltonian� This indi�rect procedure can lead to instabilities when performing calculations on large systemswith large energy cutos ������ This problem is avoided with the conjugate gradientprocedure that allows for a direct minimization of the Kohn�Sham energy functional�To avoid using large amounts of computer memory� the conjugate gradient steps areusually performed one band at a time� Each conjugate gradient iteration requirestwice as many operations than a molecular�dynamics step does for a single band�but usually less iterations are needed� More details of this approach can be found inreference ������
���� Further developments
The use of plane waves to expand the solutions of the Kohn�Sham equation has manyadvantages� They are very simple to implement� e�cient fast Fourier transforms canbe used� and an excellent control of convergence is obtained by changing the energycuto� Furthermore� plane waves provide a uniform and unbiased resolution in space�Physical quantities can thus be computed accurately and with little complication� Forexample� forces are determined using the Hellmann�Feynman theorem ����� without
�The Kohn�Sham energy functional takes the place of the potential energy of a conventionalmolecular dynamics procedure and a �ctitious mass � is introduced so that the kinetic energy is de��ned as � � j �� The electronic wave functions are constrained by orthonormality requirements�
��

corrections from Pulay forces ����� since plane waves do not depend on the ionicpositions�
On the other hand� these same characteristics make their use in all�electron cal�culations� or for elements with hard pseudopotentials� very di�cult� Solutions of theKohn�Sham equations vary rapidly close to the ions and decay smoothly in the spacebetween them� Increasing the cuto energy increases the resolution not only in thecore regions but in all space� Thus� a large part of the computational eort is spentin describing the interstitial regions with unnecessary precision� In order to reducethese eorts� a nonuniform resolution can be used���
Recently� some schemes have been proposed to modify the plane�wave approachso that the simplicity of the basis is maintained but its size greatly reduced� Gigygeneralized the use of plane waves to a non�Euclidean curvilinear space ����� ���� �����As a result� an eective energy cuto that varies in the unit cell in an unbiased way isobtained� Other approaches have been developed to go beyond the use of plane waveswhile retaining their advantages� They employ a wavelet formalism that provides asystematically improvable basis and a nonuniform resolution ������
In Gigy�s approach� the curvilinear coordinates themselves are treated as a varia�tional parameters� The coordinate system is thus allowed to adapt to the variations inthe potential� eectively increasing the plane�wave resolution in those regions wherethe inhomogeneities are larger �for the same cuto�� Consequently� a much smallerenergy cuto is required��� considerably increasing the speed of the method� Theresulting basis set does not depend explicitly on the ionic positions� therefore Pulaycorrections are not needed ������
The use of plane waves in adaptative curvilinear coordinates do not necessarilyrequire the use of pseudopotentials� Due to the considerable reduction in the cut�o energy� all�electron calculations can be carried out� as shown by Devenyi andcollaborators ������
The wavelet basis functions on the other hand� are localized both in real andreciprocal space� This allows the description of functions with spatially variableresolution� These bases do not depend on the positions of the ions and thus Pulaycorrections are not necessary� More details can be found in reference ������
��� Applications
The plane�wave pseudopotential method will be applied to three cases CaO�MgO�ZrO�� and CaO�ZrO�� Various properties such as formation energies� lattice con�stants� and electron densities will be computed� From this information� we will be ableto show that reliable predictions can be obtained from the pseudopotential method�Furthermore� in the next chapter� these properties will be used as a benchmark toassess the accuracy of dierent approximations in the total�energy methods shown
�Localized wave functions such as atomic orbitals can be used to obtain nonuniform resolutionin space� However it is usually di�cult to systematically improve the basis� Furthermore theydepend on the ionic positions so that Pulay forces need to be computed�
��Usually four times smaller ������
��

in table ���� Whenever possible� we will also use the results to gain insight in thebehavior of these oxides�
In appendix C� we describe another application of pseudopotentials� We showthat this method can be used to compute voltages of lithium batteries� The voltageis directly related to the formation energy of a lithium�transition�metal oxide from ametallic lithium and a transition�metal oxide� The insight pseudopotential providesin understanding the factors that determine the voltage will lead to the design ofbetter batteries�
Some characteristics of the pseudopotential calculations are common to the threecases� We will use norm�conserving Kleinman�Bylander�type pseudopotentials� Theywill be optimized to reduce the energy cuto� This is very important since all thecompounds have oxygen atoms �and some of them even transition metals�� We willminimize the Kohn�Sham energy functional using the conjugate�gradients technique�
In all cases� we will use the same oxygen pseudopotential� Its core radius is ���� �Afor the �s and �p valence orbitals� The s component is taken to be the local one inthe Kleinman�Bylander transformation� The Perdew�Zunger parametrization of theexchange and correlation energy ���� will be used in all the calculations�
����� The CaO�MgO system
The CaO�MgO system was described in section ������ It can be thought of as twointerpenetrating fcc sublattices� One of them is fully occupied by oxygen atoms whilethe other one is occupied by dierent arrangements of calcium and magnesium atoms�Since one of the main goals is to compute the phase diagram� the formation energiesof many dierent structures �or arrangements of Ca and Mg in one of the sublattices�have to be computed� At least �� structures are required to obtain a convergedcluster expansion� pushing the limits of what can be done with pseudopotentials in areasonable time�
Instead of computing the phase diagram� we constructed ten of the smallest pos�sible structures in the system and we computed their formation energies and cellparameters� This information was then used to evaluate faster� more approximatetotal�energy methods� We will also use a subset of these results in the next chaptersto �t pair�potential models and a tight�binding Hamiltonian� The end members�of the phase diagram� CaO and MgO� were also studied extensively�
We identi�ed each ordered structure according to its cation arrangement on the fccsublattice� The assigned names correspond to either their Strukturbericht notation�their prototype structure� or a conventional name� A description of each of the atomicarrangements can be found in references ���� and �����
Pseudopotential generation
Both the Ca and the Mg pseudopotentials were generated for a neutral atom with a�Ar��s� and a �Ne��s� con�guration respectively �the core is marked between brackets��The core radius was ���� �A and ���� �A for Ca and Mg respectively�
There is a slight overlap between the core �p and the valence �s orbitals in
��

CaO MgO
Lattice constant ��A� ��� �����a� ���� ������b�Bulk Modulus �Mbar� ���� �����a� ���� �����c�����d�
Table ��� Calculated lattice constants and bulk modulus for pure CaO and MgO�The values in parenthesis correspond to experimental measurements�
aReference ������bReference ������cNeutron scattering data ������dUltrasonic interferometry ������
Mg ������ Core corrections can be used to compensate for nonlinearities in theexchange�correlation functional� We did not include these corrections since theireect is only important at very high pressures as shown in the FLAPW calculationsof reference ������ �See also reference �������
Details of the calculation
We used �� special Chadi�Cohen �k points �see appendix B� for Brillouin zone in�tegrations for the rocksalt structure� resulting in a total�energy convergence betterthan � meV per atom� We also added extra points when computing bands �with zero
weight for the integrations�� For all the other superstructures� we took a set of �kpoints equivalent to these Chadi�Cohen points���
Convergence of formation energies with respect to basis size �number of planewaves� was carefully veri�ed within �meV per atom� A ��� eV energy cuto wasused for all the superstructures���
All the structures were fully relaxed with respect to their symmetry�allowed de�grees of freedom� This means that the total energies were minimized by letting theatoms move in the directions compatible with the symmetry of the structure�
Results for pure CaO and MgO
Both CaO and MgO have the rocksalt structure shown in �gure ���� The side ofthe cubic unit cell is the only degree of freedom than can be relaxed according tothe symmetry� In table ���� we compare the experimental and the computed latticeconstants and bulk moduli for these structures�
The electronic band structures are shown in �gure ���� We �nd CaO and MgO to
��The equivalent Chadi�Cohen �k points in the Brillouin zone were determined by applying all thefcc symmetry operations� These points were then reduced back to a smaller set by applying thesymmetry operations of the corresponding superstructures� See appendix B�
��This is an extremely high cuto� value� By increasing the accepted error in the energy to a fewmeV the cuto� energy can be reduced to ��� eV�
��

-20
-15
-10
-5
0
5
10
15
-20
-15
-10
-5
0
5
10
15
20
Γ ΓX W K L
Ene
rgy
(eV
)E
nerg
y (e
V)
CaO
MgO
Figure ��� Electronic band structures for pure CaO and MgO in the rocksalt struc�ture computed with the pseudopotential method�
��

Figure ��� Valence electronic density and its contour plot for the ����� plane ofMgO� The Mg ions sit at the corners and center of the region of the plane shown inthe picture while the oxygen ions sit at the middle of the edges� It is clear from the�gure that almost no valence electrons are close to the Mg ions�
be insulators with a direct )�) gap of ���� eV and ���� eV respectively� The experi�mentally measured band gaps are ���� eV for CaO ����� and ���� eV for MgO ������
The total electron valence density for MgO is shown in �gure ���� The density forCaO �not shown� is very similar�
Results for CaO�MgO compounds
The lattice constants and formation energies of all the computed structures are shownin table ����
Discussion
The lattice constants calculated for CaO and for MgO are predicted within less than���� of the experimental values� One has to remember that the only inputs to themethod were the atomic numbers and the structure��� In general� the experimentaland computed lattice constants coincide within a few percent for LDA methods� TheLDA consistently overestimates the stability of the bonds leading to lattice constantssmaller than the experimental ones ����� The errors in the bulk modulus predictedby the LDA are usually larger� The dierence can be as large as ��� with respect toexperimentally measured values� For CaO and MgO� they were within ���
��The structural information is not required but when used it considerably speeds up thecalculations�
���

Structure Composition Cell Parameters %Epseudo
�� L�� CaMgO� a�b������* c������ ������� L�� CaMgO� a�b������* c������� ������� A�B� CaMgO� a�b������* c������* �����
x� � ������� L�� Ca�MgO� a�b�c������ ������� L�� CaMg�O� a�b�c������ ������� DO�� Ca�MgO� a�b������* c������* �����
x� � ������� DO�� CaMg�O� a�b������* c������* �����
x� � ������� MoPt� Ca�MgO� a������* b������* c������* �����
x� � �����* x� � ������� MoPt� CaMg�O� a������* b������* c������* �����
x� � �����* x� � �������� Ni�Mo Ca�MgO� a�b������* c������* �����
x� � �����* y� � �����*x� � �����* y� � �����
Table ��� Formation energies and cell parameters of dierent ordered structures inthe CaO�MgO system obtained by using the plane�wave pseudopotential method�All cell parameters were fully relaxed� including the internal positions� xi� that areshown here according to the Wycko notation in reference ������ Lattice constants areexpressed in �A and formation energies are in eV ion� A description of each structurecan be found in reference ���� or �����
���

The errors in the band structure are larger� The LDA usually underpredictsthe band gaps by ��� �as in our case�� However� we have to be careful with theinterpretation of the computed bands� The bands shown in �gure ��� are a plot of theKohn�Sham single electron eigenvalues for dierent �k points� These are not the quasi�particle energies although they represent a good approximation of them� particularlyfor the occupied states� Strictly speaking� these energies are the derivatives of thetotal energy with respect to the occupation numbers of the corresponding states �������
The electronic density in �gure ��� shows no evidence of any concentration ofvalence charge between the ions� The great majority of the charge resides in theoxygen atom� the valence orbitals of the magnesium atoms being almost empty� Thesecharacteristics agree well with the picture of ionic bonding �with almost perfect formalcharges� that is used to describe CaO and MgO� However� the charge does not havea perfect spherical symmetry around the oxygen atoms and their size changes withthe environment� This will be carefully analyzed in chapter ��
All the formation energies of intermediate compounds in the CaO�MgO systemwere found to be positive� This agrees well with the fact that no stable orderedstructure has been observed in the system except for the pure end members� Thenumber of computed formation energies is not enough to obtain a converged clusterexpansion� However� if the SSCAD ECI�s are rescaled according to the dierencebetween the pseudopotential and the SSCAD formation energies the solid part of theCaO�MgO phase diagram can be estimated� The resulting phase boundaries are veryclose to the experimental ones� We will come to this point later in chapter ��
Though not rigorous� the above process shows the power of the �rst�principlesapproach to study phase stability if an accurate and fast total�energy technique isdeveloped� The rest of this work is dedicated towards that goal�
����� The CaO�ZrO� system
Zirconia is stable at room temperature in a monoclinic structure �baddeleyite� andtransforms into a tetragonal phase �space group P���nmc� at approximately���� oC ������ Before melting� it transforms again� but now into a cubic phase��uorite�type structure� at approximately ���� oC ������ In �gure ���� we describethese three phases� The addition of impurities such as CaO� MgO� or Y�O�� canstabilize the cubic and tetragonal phases to room temperatures� In �gure ���� weshow the zirconia rich part of the CaO�ZrO� phase diagram� Apart from the purezirconia structures� three other structures have been detected in this portion of thediagram with compositions CaZr�O� Ca�Zr�O��� and CaZrO�� They correspond tospace groups C��c� R(�c� and Pcmn respectively� Note that CaZr�O and Ca�Zr�O��
are not stable at low temperatures� For a detailed description of these structures� seereferences ����� and ������
��The highest occupied eigenvalue gives the ionization energy of the system work function inmetals if the exact density�functional formalism is used� For most approximate functionals it iscommon practice to keep that interpretation though it is not completely correct ������
���

a
a
a
Cubic
a
a
c
Tetragonal
a
b
c
Monoclinic
Zr
O
Figure ��� Polymorphic structures of zirconia�
���

0 10 20 30 40 50
500
1000
1500
2000
2500
3000
Atomic percent CaO
Fluorite phase
Monoclinic phase
Tetragonalphase
Liquid phase
CaZrO3
Ca Zr O6 19 44
CaZr O4 9
Tem
pera
ture
( C
)o
Figure ��� Experimental CaO�ZrO� phase diagram� See reference ������
���

The stabilized zirconias present excellent ionic conductivity�� and thermomechan�ical properties�� Consequently� zirconia has many applications from refractory orstructural materials to oxygen sensors� solid state electrolytes� high�temperature fuelcells� and resistance heating elements�
Ceramics are ideal to be used in environments with extremely high temperaturessince their melting point is above the one for most common structural metals� How�ever� their brittleness sometimes prevents this kind of applications� The low fracturetoughness implies little resistance to crack propagation� Consequently� there is a con�siderably interest in understanding the toughening mechanisms in doped zirconia andspecially the stabilization eects that lead to them� The toughening process has itsorigins in the stress�induced martensitic transformation of tetragonal particles to themonoclinic phase� This transformation causes a �� increase in particle volume whoseresidual strain �eld tends to limit the crack opening ������
Many experimental and theoretical studies have been conducted to understandthe stabilization mechanisms in zirconia ���� ���� ���� ���� ����� Measurements aredi�cult to perform since they must be done at extremely high temperatures� Thequality of the samples is also a concern �defects and impurities�� This seems to bethe ideal scenario for the use of computational experiments� However� the extremelycomplex crystal structure of monoclinic zirconia prevents the use of accurate total�energy methods� Most quantum�mechanical calculations have been performed on thetetragonal and cubic phases�
Even potential models are problematic in this system� not because of the com�plexity or size of the cell but because oxygen breathing� electron polarizability� andcovalency play an important role in determining the formation energies �as we explainin the next chapter�� These eects are not well represented in such simple energymodels� However� very useful information has been obtained about the defect struc�ture ����� and the identi�cation of possible stabilization mechanisms ������ Recently�Wilson and collaborators went beyond the standard potential and shell model �����They introduced a breathing mechanism for the oxygen atoms �see section ������ andincluded the dipolar and quadrupolar induction eects� Although they adopted for�mal ionic charges in their model� the right ordering of the energies of the dierentpossible structures in pure zirconia was predicted���
Many ab initio calculations have been reported for the tetragonal and cubic phases�Almost every total�energy technique has been used in zirconia FLAPW ����� �����ab initio Hartree�Fock ����� ����� full potential LMTO ����� linear combination ofatomic orbitals �LCAO� ����� ����� and PIB ������ However� to our knowledge� thisis the �rst time pseudopotential studies were performed�
Our goal is to use the pseudopotential calculations as both an input and a test
��The ionic conductivity is produced by the numerous charge�compensating vacancies originatedin the replacement of Zr�� by Ca�� Mg�� Y�� etc� See references ����� and ������
��Stabilized zirconia presents extremely high strength toughness and thermal�shock resistance���Partial charges are not physically well de�ned� However we will show in chapter � that there is
a strong dependence of the ionic charge on the environment when we adopt a consistent de�nitionfor the charge� This e�ect needs to be taken into account�
���

0
0.005
0.01
0.015
0.02
0.025
0.03
0.035
0 1 2 3 4 5 6 7
r (a.u.)
sd
4r
πϕϕ
2*
Figure ��� Computed electronic density for the �p and �d orbitals of the Zr�� ion�The wave functions have been normalized to one� The overlap between the densitiescan clearly be seen�
to our tight�binding Hamiltonian� We will compute the structural energy dierencefor the cubic�tetragonal transition and the formation energies of several CaO�ZrO�
ordered structures� We will also analyze the electronic charge densities to understandthe approximations of simpler methods�
Pseudopotential generation
The electronic con�guration of zirconium is �Kr��d��s�� One would like to treat�Kr� as the atomic core� However� we detected a signi�cant overlap between the �dand the �p orbitals� This is illustrated in �gure ���� Furthermore� zirconium atomsare usually highly ionized in oxide environments� Consequently� we generated thepseudopotential for the valence con�guration �s��p��d��s� corresponding to Zr���
We generated an optimized pseudopotential� with the Kleinman�Bylander trans�formation� The s angular component was taken as the local potential� with the p andd components as the nonlocal ones� The core radii were ����� �A� ���� �A� and ���� �Afor the s� p� and d orbitals respectively�
Results for pure ZrO�
We calculated the energy dierence between cubic and tetragonal zirconia� The mon�oclinic phase was not computed since it is very complex� with �� atoms in the unitcell and �� variables to optimize� Performing these calculations with pseudopoten�tials requires a large number of supercomputer hours� A single point calculation may
���

Experimentsa Pseudopotentials
Cell Parameter
Cubic ZrO�
a�b�c ����� �����Tetragonal ZrO�
a�b ����� �����c ���� �����z�O� ����� �����Energy dierences
Ecubic�Etetragonal ����� �����
Table ��� Cell parameters and structural energy dierences for the tetragonal andcubic phases of zirconia at zero pressure� The energies are in eV per ZrO� formula unitand the lattice constants are in �A� All cell parameters were fully relaxed� including theinternal position zi �shown here according to the Wycko notation in reference �������The experimental z value for the tetragonal structure was measured at ���� oC� Allthe other experimental values were interpolated to � oK as explained in the text�
aSee references ����� ����� and ������
take about �� Cray�� cpu hours �see �gure ����� On the other hand� a single�pointcalculation of the tetragonal phase takes about �� minutes and has � relaxation pa�rameters� The cubic phase has just one relaxation parameter and takes about ��minutes�
Ten special Chadi�Cohen �k points were chosen for the cubic phase and an equiv�alent set for the tetragonal phase ��� �k points�� The energy cuto was ��� eV� Thetwo structures were fully relaxed�
The results are presented in table ���� The tetragonal phase is predicted to havea lower energy than the cubic one� as one would expect from the experimental in�formation� The dierence in energy between these two phases agrees well with theestimated enthalpy of the transition measured at ���� oC in reference ������ The cellparameters for the cubic and tetragonal phases also agree well with experiment� Thereported experimental values for the tetragonal a and c� and the cubic a parameterare linear extrapolations to � oK using the data from reference ����� �since the twophases are very similar we assumed the same temperature dependence in both cases��
In �gure ���� we show the charge density along the ����� plane of cubic zirconia�Contrary to the CaO�MgO case� it is not clear from the total valence density if theorbitals �d and �s are fully ionized� Consequently� we also plot in the �gure� thecontributions coming from these orbitals� They show a signi�cant presence of chargein them� The value of the integrated electronic charge density within a sphere of
���

Zr
O
(a)
(b)
Figure ��� Valence electronic density and its contour plot for the ����� plane of cubiczirconia �shaded plane in the �gure�� �a� Full valence density� �b� Contribution fromthe �d and �s bands� �The density coming from the oxygen orbitals is also shown asa reference��
���

radius� ��� �A centered at the zirconium ion is �� electrons�� �i�e�� � electrons wereremoved from the valence�� When the contribution from the bands with �s and�p character is left out of the integration� the number of electrons in the sphere wasreduced to ����� This indicates that part of the � electrons that are being transferredto the oxygen ions come from the �s� and �p�type bands���
Results for CaO�ZrO�
The ordering of oxygen vacancies and the cations in CaO�ZrO� produce very com�plex structures� Except for the high�temperature phases of pure zirconia� the abinitio treatment of these compounds requires computer runs beyond any reasonabletime� The computation of the phase diagram is out of the question� Using a simplerapproach� such as the tight�binding technique presented in this thesis� is the onlyfeasible alternative for studying phase stability in this system from �rst principles�
However� �tting and testing the tight�binding Hamiltonian will require more in�formation about the observed compounds than what is available experimentally� Con�sequently� we generated new compounds� simpler than the experimentally observedones� by replacing some zirconium atoms in the �uorite structure by calcium atomsand adding the right number of vacancies to preserve charge balance��� In �gure �����we show the �ve structures �S� to S�� with the smallest unit cells that were generatedusing this procedure�
The pseudopotential formation energies and lattice parameters of these structuresare shown in table ���� For simplicity� only volume relaxations were allowed� Theenergy cuto was ��� eV� We used a uniform mesh in the Brillouin zone for the �k�space integrations� Only the symmetry independent �k points were kept� A �x�x� anda �x�x� mesh were used for structures S� to S� and S� to S� respectively� With theseparameters� the formation energies were converged within a few meV atom�
In �gure ����� we show the valence electron density corresponding to the S� struc�ture� As in CaO�MgO� the Ca atoms are fully ionized� We found the same situationin all the �ve structures�
The charge of an ion is not a well�de�ned quantity� However� we have seen thatclassical models make an extensive use of this concept� One of the approximationsof these formulations is that the charge is �xed �i�e�� independent of the particulararrangements of the ions in the lattice�� To test these assumptions� we integrated thecharge density within spheres centered at the ions of structures S� to S�� The sameradius was taken for all the ions of the same species� The results detected dierences
��The nearest neighbor distance between zirconium and oxygen is ���� �A� The ��� �A value cor�responds to the minimum of the spherically�averaged density along the axis that joins a zirconiumand an oxygen ion�
�Recall that the we took �s��p��d��s� as the valence of zirconium���If we had taken the �s and �p orbitals as part of the pseudopotential core this e�ect would
have not been properly accounted for� The large overlap between these and the �d orbitals is a clearindication that the semicore states had to be included in the valence�
��For this we assumed the ions to have the integral formal charges� i�e� �� �� and �� for Zr Ca and O respectively�
���

Ca
Zr
O
S1 S3
S4 S5
S2
Figure ���� Smallest volume Zr�Ca�O structures in the �uorite lattice that preservecharge neutrality� The arrows indicate one possible set of primitive basis vectors�
Structure Composition Cell Parameter ��A� %E �meV atom�S� ZrCaO� a����� ���S� ZrCaO� a����� ���S� ZrCaO� a������ ���S� Zr�CaO a����� ���S� ZrCa�O� a����� ���
Table ��� Formation energies and cell parameters of dierent ordered structures inthe CaO�ZrO� system obtained by using the plane�wave pseudopotential method�Only cubic volume relaxations were allowed� The structures are described in �gure �����
���

Zr
O
Ca
Figure ���� Valence electron density for the S� structure� Note that Ca atoms arefully ionized�
as large as ��� electrons between Zr �or O� spheres� This is an indication that thecharge associated with the ions of the same species may change considerably withinand along the � structures���
Discussion
The computed structural energy dierence between the cubic and the tetragonalphases� �� meV ZrO�� is in very good agreement with the estimated enthalpy of thetransition measured at ���� oC ���meV ZrO���
As in the CaO�MgO case� the comparison between the pseudopotential and theexperimental cell parameters is excellent� The calculated lattice constants of the cubicand tetragonal phases and the experimentally measured values extrapolated to � oKagree within less than ���
The charge�density plots consistently show that zirconium is not fully ionized�Not all the �d and �s electrons are transferred to the oxygen atoms� A small amountof charge lies in between the ions indicating some degree of covalency�
Although the charge of the ions is not a well�de�ned physical quantity� our resultsindicate that there seems to be an important dependence of its value on the atomicenvironment� This dependence can introduce large errors when computing formationenergies with potential models� The situation will be further analyzed in chapter ��
��Note that other e�ects such as ionic breathing may be responsible for this variation in theintegrated electron densities� We regard these results as an indication and we will come back tothis point later in chapter � where the uniquely de�ned tight�binding charges will be used to studythese claims�
���

��� Conclusions
In this chapter� we showed that in spite of its approximations� the pseudopotentialmethod can be used to compute useful materials properties� Many technologicallyrelevant questions can be answered faster and at a smaller cost by performing com�putational rather than real experiments�
However� only simple systems can be studied with accurate �rst�principles ap�proaches� Calcium stabilized zirconias� more complex arrangements of the Li andmetal ions in the batteries� or even the phase diagram of one of the simplest oxidesystems �CaO�MgO� can not be analyzed by using the pseudopotential approach� Itis clear that a faster method is necessary if we want to study more complex systems�
Faster methods make use of many simplifying assumptions at a risk of reducingaccuracy� Thus� it is essential to analyze the eect of the approximations employedto speed up the calculations� In the next chapter� the pseudopotential results will beused to assess the accuracy of these simpler methods� At the same time� we will beable to determine the relevant physical eects that should be accounted for in anytotal�energy calculation for oxides�
���

Chapter �
The e�ect of approximations in
total�energy methods� relevant
terms in the Hamiltonian
With the currently available total�energy methods� the study of even relatively simpleoxides is beyond the computational capability of today�s fastest computers� Whenstudying other materials� many approximations are introduced to obtain the desiredspeeds� Unfortunately� these same approximations produce large errors in oxides�
In the following sections� we will study the eects on oxides of the approximationsintroduced in chapter �� The goal is to analyze their performance and determinethe causes of their failure to predict accurate formation energies� There is a wide�spread belief that only the state�of�the�art methods should be able to accurately�
compute those values� Here� we will follow a dierent path� By studying the eects ofthe dierent approximations� we will identify the relevant terms in the Hamiltonianneeded to compute formation energies� This information will then be used in chapter �to build a tight�binding Hamiltonian that will allow us to compute energy dierencesfast and accurately�
Our plan consists of two stages� In the �rst one� we showed that pseudopotentials�within the LDA� produce reliable results in a wide variety of oxides� Now� in the sec�ond stage� we will use these calculations as a benchmark to test other methods� and atthe same time� identify the important contributions to the Hamiltonian� In �gure ����we summarize both the main approximations used by the dierent total�energy meth�ods and the systems where their eects will be tested� The techniques chosen representthe most popular methods currently used in oxides� On the other hand� the oxideswere selected for their dissimilar characteristics� As previously explained� CaO�MgOis the prototype system for an ionic material with highly symmetric structures andcompletely ionized cations� These aspects make it an ideal candidate for shape andnon�self�consistent approximations� Pure and doped zirconias have low�symmetry
�Recall that the accuracy of a calculation depends on the sensitivity to changes in the total�energy value of the physical property we want to compute� Since we look at energies of the order ofthermal e�ects the energy di�erences have to be computed within a few meV�atom�
���

LDASemiempiricalE=E(R ,.....,R )
1 N
Nonself-consistency
Sphericalsymmetry
Frozencore
FLAPW
PP
SSCAD
Pair
Potentials
HighSymmetry
FixedCharge
CaO-MgO
Compatible
with:
Applies
Approximately applies
ZrO 2
ZrO 2CaO-
Figure ��� Total�energy methods and their most commonly used approximations�Also shown are the oxide systems where the eect of these approximations on theformation energies will be tested�
���

Ca�MgO� CaMg�O� CaO MgOPP Formation energy ����� ����� � �FLAPW Formation energy �����a �����a � �PP Lattice constant ����� ����� ��� ����FLAPW Lattice constant �����b �����b �����c �����c
Table ��� Formation energies and cell parameters for the L�� structure in the CaO�MgO system computed with the pseudopotential �PP� and the full�potential lin�earized augmented�plane�wave �FLAPW� methods� The ions in the CaO�MgO sys�tem are arranged on two interpenetrating fcc lattices� The L�� nomenclature identi�esthe distribution of the Ca and Mg ions on the fcc cation sublattice �that correspondsto the minority species at the corners of the conventional fcc cell� while the oxygenfcc sublattice remains fully occupied� Energy values are expressed in eV ion and thecell parameters are in �A� The values correspond to fully relaxed structures�
aFrom reference �����bFrom reference ������cFrom reference ������
phases and a combination of a covalent� and an ionic�type of bonding� Consequently�they represent a major challenge for approximate total�energy methods�
We will start by studying the eects of the frozen�core approximation by compar�ing plane�wave pseudopotential calculations with all�electron FLAPW results �sec�tion ����� Although the frozen�core approximation is used in many other methods�this will be a last test of the results we intend to use as a benchmark� We will thenproceed to relax the accuracy of the calculations by allowing shape approximations tothe potential �section ���� and non�self�consistency including a classical� represen�tation of the dependency of the total energy on the ionic coordinates �section �����Finally� we will analyze which contributions should be included in the Hamiltonianto accurately compute formation energies in oxides �section ������
��� E�ect of the frozen�core approximation
We analyze the eect of the frozen�core approximation by comparing the pseudopo�tential and the all�electron FLAPW results� The dierences between them are verysmall in the CaO�MgO system� as shown in table ���� The formation energies agreewell within the convergence errors of the calculation� These errors were � meV atom
�Recall that we use the word �classical� to denote those techniques that do not explicitly solvethe Schr!odinger equation to �nd the dependence of the total energy on the coordinates of the ions�
�In this chapter we will not test the accuracy of the local�density approximation� The onlyproof of the accuracy of the LDA is the excellent agreement with experiments of the FLAPW andpseudopotential results shown in chapter ��
���

and �meV atom in our calculations and in reference ����� respectively� The latticeconstants are slightly dierent� Part of this dierence can be attributed to the use of adierent exchange and correlation potential �Hedin�Lundqvist ����� in reference �����and Perdew�Zunger ���� in our case�� Other possible source of error is the overlap ofthe valence orbitals with the core� However� the predicted MgO lattice constant inreference ����� coincides within less than ���� with our values although they includedcore corrections� This seems to indicate that the disagreement is due to dierent LDAformulations and dierent convergence criteria rather than the frozen�core approxi�mation�
On the other hand� for the predictions of the energy dierences between the tetrag�onal and cubic phases of zirconia� the agreement is not good �see table ����� However�we do not think this dierence comes from the pseudopotential approximations� Thepseudopotential predictions are not only much closer to the experimental values butalso the FLAPW calculations ����� were not fully converged� In reference ������ alimited number of basis functions was used� which introduced relative errors of theorder of ��meV cell� compared to �meV cell in our case� and the relaxation of thetetragonal phase was not thoroughly performed�
When there is an appreciable overlap between the valence and core orbitals thefrozen�core approximation can become an important source for error�� When this isthe case� some of the core orbitals should be taken as valence orbitals reducing thesize of the core or a correction term should be introduced� If a pseudopotential isbeing used� a smaller core results in a deeper potential� considerably increasing thenumber of plane waves and the computational demands of the method� Consequently�core corrections are usually implemented�
��� E�ect of the spherical�symmetry approxima�
tion for crystal potentials the SSCAD case
The SSCAD method was specially designed for systems with interacting closed�shellions and highly�symmetric structures� A highly�ionic system� such as CaO�MgO�meets all these requirements and should be an ideal case for the method to work well�The major approximation of the SSCAD is the spherical symmetrization of the ionicpotentials�
We will extensively study the CaO�MgO system� in particular its phase diagram�Computed phase diagrams are very sensitive to small changes in the formation en�ergies� Consequently� a comparison between the measured and the predicted phaseboundaries is an excellent test of the accuracy of the SSCAD� We will also directlycompare the SSCAD and the pseudopotential formation energies�
On the other hand� more open structures can present an important challenge
�For example in reference ����� the lattice constant of bcc lithium is computed with and withoutcore corrections using the pseudopotential method� The reported values were ����� �A and ����� �Arespectively� Note that there is more than a �� di�erence between them� Furthermore the experi�mental result ����� �A is closer to the former values�
���

Expt�a Pot�b PIBc PP FLAPWd
Cell Parameter
Cubic ZrO�
a�b�c ����� ����� ���� ����� �����Tetragonal ZrO�
a�b ����� ����� ����� �����c ���� ����� Not ����� �����z�O� ����� ���� stable ����� �����Monoclinic ZrO�
a ������ �����b ������ �����c ������ ������ ������ ����x��Zr� ������ ����y��Zr� ������ ��� Notz��Zr� ������ ������ stablex��O� ������ ������y��O� ������ ������z��O� ������ ������x��O� ������ ������y��O� ������ ������z��O� ������ ������Energy dierences
Ecubic�Etetragonal ����� ����� ����� �����Etetragonal�Emonoclinic ����� �����
Table ��� Cell parameters and structural energy dierences for the experimentallyobserved phases in zirconia at zero pressure� The energies are in eV per ZrO� formulaunit and the lattice constants are in �A� All cell parameters were fully relaxed� includingthe internal positions� xi� yi� and zi� that are shown here according to the Wyckonotation in reference ������ The experimental z value for the tetragonal structurewas measured at ���� oC and the cell parameters are extrapolations to � oK �seechapter ��� PIB calculations for the tetragonal and monoclinic phases of zirconia arenot reported in the table since these structures are not predicted to be stable by thismethod�
aSee references ���� ��� �����bSee references ���� �����cSee reference ������dSee reference ������
���

EC
I (eV
)
Pairs Triplets Quadruplets
-0.02
-0.015
-0.01
-0.005
0
0.005
1 2 3 4 5 6 1 2 3 1 2
Figure ��� Eective cluster interactions for the CaO�MgO system computed withthe SSCAD� The pairs are identi�ed according to their length and the triplets andquadruplets according to their longest side� the other sides being nearest neighbors�
to the spherical�symmetry approximation� We will analyze its eects on the totalenergy by studying the polymorphic transformations of zirconia� Speci�cally� we willtest if the SSCAD predicts the correct energy dierences between the dierent ZrO�
structures�
����� The SSCAD CaO�MgO phase diagram
The formation energy of �� compounds was computed to determine the eectivecluster interactions of the system� Their values can be found in appendix A� TheECI�s are shown in �gure ���� Convergence of the cluster expansion was veri�edby eliminating the six structures with the largest unit cells from the �t� The ECI�swere �t again and were used to predict the energy of these six structures� The RMSdierence between the direct and the predicted values was only � meV atom� Theseresults clearly show the predictive power of the expansion�
A further test involved predicting the defect energies of inserting a molecule of CaO�MgO� in an otherwise pure host material� As explained in section ������ these energiescorrespond to the single atom spin �ip and are directly related to the solubility limitsin the end�member phases� In table ���� we show the defect energies predicted by thecluster expansion and by the SSCAD� The agreement is excellent� indicating a wellconverged cluster expansion�
For the CaO�MgO system� we also took into account the eect of atomic vibra�
���

Cluster expansion SCPIBMgO rich ����� �����CaO rich ����� �����DierenceMgO rich � CaO rich ����� �����
Table ��� Defect formation energies� in eV� of a single MgO �CaO� in an other�wise pure CaO �MgO� lattice� The SSCAD values were computed using a �x�x� fccsupercell� The term rich� denotes the host lattice�
Nearest neighbor pair Nearest neighbor tripletV vibr� �������� ��������
Table ��� Vibrational ECI�s for the CaO�MgO system �dimensionless��
tions by using two vibrational ECI�s �V vibr� in equation ������ They were �t to the
vibrational free energies of CaO� MgO� and the two L�� structures� The vibrationalfree energies were computed within the harmonic approximation ����� The vibrationalECI�s are shown in table ����
The phase diagram was computed using a combination of Monte Carlo simula�tions and the CVM� The CVM entropy expression was used in the ��� and ���pointmaximal�cluster approximation �see �gure ������ The probabilities of the dierentarrangements in that cluster� the energy� and the composition were obtained from agrand canonical Monte Carlo simulation in a ��x��x�� fcc supercell� These valueswere averaged for ���� steps� after another ���� equilibration passes� The solubilitylimits can then be found by looking at the intersections of the grand canonical freeenergy� More details of this hybrid technique can be found in reference �����
The computed phase diagram is shown in �gure ���� Note that no experimentalinput has been used except for the atomic numbers and the lattice�
Discussion
The agreement between the experimental and predicted solubility limits in �gure ���is very good� Errors of more than ���� in transition temperatures are not uncommonfor �rst�principles techniques� This is an indication of the good performance of theSSCAD to compute formation energies�
However� since we are including all the relevant contributions to the free energy�the dierences between the predicted and experimental solubility limits are likelyto come from errors in the SSCAD formation energies� In �gure ���� we comparethe pseudopotential formation energies of the �� compounds in table ��� against theSSCAD predictions� The latter are systematically lower than the pseudopotential
���

o
0.250 0.5 0.75 1
0
1000
2000
3000
4000
5000
Mole fraction MgO
Tem
pera
ture
( K
)
Figure ��� CaO�MgO phase diagram� Only the solid solubility limits� computed bya combination of Monte Carlo simulations and the CVM� are shown� The solid anddotted lines were determined with the ECI�s derived from SSCAD and from empiricalpotential models respectively� The circles represent experimental values ���� and thedashed line indicates the experimental eutectic temperature�
���

For
mat
ion
Ene
rgy
(eV
/ato
m)
Structure Number
PPSSCAD
MgO concentration increases
0
0.05
0.1
0.15
0.2
0.25
0.3
10 6 4 8 1 2 3 9 7 5
EP
Figure ��� Formation energies for dierent ordered structures in the CaO�MgO sys�tem obtained with pseudopotentials �PP�� the SSCAD� and empirical potentials �EP��The number that identi�es each structure corresponds to the one used in table ����
���

Ca O
Ca
Ca
Ca
OO
O
Figure ��� Valence electron density for the ����� plane of CaO as computed withthe pseudopotential method� Note that the oxygen ions are not perfectly spherical�
results� The average error is of the order of ����Note that the dierences between the methods are larger in the MgO�rich than
in the CaO�rich side of �gure ���� This indicates that the SSCAD will underpredict�in particular� the defect energy of introducing a CaO in an otherwise pure MgO host�As we previously explained this will result in an excessively large solubility limit inthe MgO�rich side of the phase diagram�
Figure ��� clearly shows this behavior� with a much larger overprediction of thesolubility limits in the MgO�rich than in the CaO�rich side� Furthermore� if theSSCAD formation energies were to be rescaled to be close to the pseudopotentialpredictions� the resulting ab initio phase diagram will lie on top of the experimentalpoints� Unfortunately� computing the pseudopotential phase diagram is beyond thecurrent computer power�
The main source of errors in the SSCAD is the spherical symmetry imposed on thedensity� Even for these highly ionic oxides the assumption is not correct� In �gure ����we show the contour plot of the valence electron density for the ����� plane of CaO�The oxygen atoms are clearly not spherical� The SSCAD will fail to reproduce thesenonspherical relaxations of the valence charge� introducing an error in the energy�
In order to assess the accuracy of the SSCAD in oxides� systems in which non�spherical relaxations are important should be tested� This is the objective of the nextsection�
���

����� Polymorphic transformations in pure zirconia
At low temperature� the stable zirconia structure is monoclinic� which has a lowsymmetry� The structural transitions in the system were studied by Cohen� Mehl� andBoyer ����� using a non�self�consistent version of the SSCAD �called the potential�induced breathing �PIB� method ������� Their results are reproduced in table ����The tetragonal and monoclinic phases were found to be unstable at zero pressure�Cubic zirconia was predicted to have the lowest energy when compared to the rutile�the orthorhombic� and the cotunnite structures��
The approximations introduced in the method clearly break down for the zirconiasystem� The non�self�consistent SSCAD predicts the cubic structure to be the stablephase in contradiction with the experimental �ndings� Furthermore� the method failsto reproduce the stable low�symmetry phases� Cohen et al� ����� suggested thatthese discrepancies were related to nonspherical charge relaxations not included inthe model�
Since many oxides have covalent bonding� signi�cant errors can be expected dueto this approximation� It is possible to avoid the spherical approximation in theSSCAD but at the expense of an increase in the computational burden� Consequently�the SSCAD can only be used to compute formation energies in systems with highsymmetry� But even in this case� some error should be expected as shown in theCaO�MgO system�
��� E�ect of the pair�potential approximations
In this section� we will analyze the two remaining approximations in �gure ��� the useof non�self�consistent formulations and the use of semiempirical functions to modelthe dependence of the energy on the ionic coordinates� To perform these tasks� we willuse a simple pair�potential formulation� Actually� this model also assumes sphericalsymmetry for the crystal potential and the frozen�core approximation�� However�in the previous sections� we showed that although the last two approximations canintroduce signi�cant errors� they are very small in CaO�MgO� Thus� we will startinvestigating the phase diagram of this system and analyzing any additional errorsthat non�self�consistency and the use of semiempirical functions introduce�
We will complete the analysis by performing calculations in pure and calcia�dopedzirconias�
����� The pair�potential CaO�MgO phase diagram
We computed the CaO�MgO phase diagram using an empirical pair�potential model�It consisted of a Buckingham potential �see equation ���� combined with a shell
�It is unlikely that a self�consistent treatment will change the results qualitatively��Actually no explicit mention to the electrons is made within a pair�potential model� We can
assume that the core electrons are frozen and that the valence ones are only allowed to deform withinthe polarization model described in section ������
���

O���O�� Ca���O�� Mg���O��
Aij �eV� ����� ������ �������ij ��A� ����� ������ ������Cij �eV�A
�� ����� ��� ���
O Ca MgYi �e� ���� ����� �����Xi �e� ���� ������ �����
ki �eV�A
� � ���� ����� �����
Table ��� Potential parameters derived in reference ����� The parameters are namedfollowing the same convention as in section ������
model �see section ������� The parameters were obtained from reference ���� and arereproduced in table ���� They were �t to experimental structural information �cellparameters�� elastic and dielectric constants� and cohesive energies�
The same cluster as in section ����� were used here for the cluster expansion�Their coe�cients were �t to formation energies of the potential model� The predictedsolubility limits are shown in �gure ���� There is a clear underestimation of theirvalues�
In �gure ���� we show the formation energies for some compounds in the CaO�MgO system� The potential model predicts almost twice as large formation energiesthan the pseudopotential method� Consequently� the eect on the phase diagram willbe to raise the transition temperature� in agreement with �gure ����
����� Pair�potential predictions for pure and doped zirco�
nias
Here� we will also use a Buckingham potential combined with a shell model� Itsparameters were �t to reproduce structural and dielectric constants of the tetragonalzirconia phase ����� �the O�O and Ca�O parameters were taken from reference �����and are reproduced in table ����
The potential model incorrectly predicts the energy dierences for the polymorphictransformations in zirconia �see table ����� The monoclinic phase is stable whencompared to the tetragonal and cubic phases�
Problems are also present in calcia�doped zirconias� In �gure ���� we compare thepseudopotential and the empirical potential predictions for the formation energies ofthe structures S� to S� of section ������ Again� the latter model overestimates theformation energies�
���

O���O�� Ca���O�� Zr���O��Aij �eV� ����� ������ ��������ij ��A� ����� ������ �����Cij �eV�A
�� ����� ��� ���
O Ca ZrYi �e� ������ ��� ����Xi �e� ����� ��� ����
ki �eV�A
� � ����� � �������
Table ��� Potential parameters derived in references ���� ����� The parameters arenamed as in section ������ The �� corresponds to the rigid�ion approximation�
0
0.5
1
1.5
2
2.5
3
3.5
S1 S2 S3 S4 S5
Structure
For
mat
ion
ener
gy (
meV
/ato
m) PPPot.
Figure ��� Formation energies in the CaO�ZrO� system predicted by pseudopoten�tials �PP� and empirical potentials �Pot��� The structures are identi�ed following theconvention in �gure �����
���

O���O�� Ca���O�� Mg���O��
Aij �eV�A�� ������ ������� �����
O Ca MgYi �e� ��� ��� ���Xi �e� ����� ��� ���
ki �eV�A
� � � � �
Table ��� Potential parameters �t to the pseudopotential formation energy of theL�� structure� its cell parameters� and the CaO and MgO cell parameters� The ��corresponds to the rigid�ion approximation�
����� Discussion
The use of potential models is widespread since they combine simplicity with com�putational speed� Although they have been successfully used to study many oxideproperties ���� ���� they only render qualitative agreement for phase diagrams� Inthe CaO�MgO system� the pair�potential model predicts a miscibility gap with theright asymmetry� Also the correct energy ordering of the zirconia polymorphs isreproduced� However� their quantitative predictions can be o by more than �����
Most published potentials have been �t to energies on the scale of eV� whiletemperature eects are determined in the scale of several meV� This can be improvedby actually �tting to formation energies and lattice parameters of dierent structuresin the system� For example� we studied the CaO�MgO system using a potential witha simple repulsive term of the form
V �rij� �Aij
r�ij �����
�t to the pseudopotential formation energy of the L�� structure and the lattice pa�rameters of the rocksalt MgO and CaO� and the L�� structure�
� No polarization termswere included� The parameters used are shown in table ���� The resulting formationenergies are compared to pseudopotential and SSCAD predictions in �gure ���� Acomplete list of formation energy values can be found in appendix A� It is clear fromthe results that the agreement has improved� and the potential formation energies arecloser to the SSCAD predictions� However� other properties such as the elastic anddielectric constants� or the vibrational properties are much more poorly reproduced
�The distance dependence is derived from a very popular formulation of the semiempirical tight�binding Hamiltonian� Its value is di�erent from zero only for the �rst nearest�neighbor O�Ca andO�Mg interactions and for the second nearest�neighbor O�O interaction� See appendix A for moredetails�
�Note that since the system presents a miscibility gap this �t can not be done to experimental
results� The L� structure is not thermodynamically stable� This is an important advantage of�tting potentials to ab initio values�
���

0.04
0.06
0.08
0.1
0.12
0.14
0.16
0.04 0.06 0.08 0.1 0.12 0.14 0.16
For
mat
ion
ener
gy (
meV
/ato
m)
PP formation energy (meV/atom)
PotentialsSSCADPP
Figure ��� SSCAD and pair�potential formation energies for the CaO�MgO systemas a function of the pseudopotential predictions� The parameters used in the pairpotential are shown in table ���� The closer the points to the solid line the better theperformance of the potential or the SSCAD method is�
than when using the potentials in reference �����
A potential model can thus be regarded as an interpolation tool with not muchphysical content� The mapping of the many physical eects that determine the en�ergy onto a simple pair�potential form is not accurate� This is supported by thefact that when formation energies are well reproduced other properties are worsen�Furthermore� pair�potential values do not agree with the SSCAD or pseudopotentialpredictions� It seems unlikely that a single set of parameters will be able to reproduceall the properties�
Most of the energy eects that cause problems in pair�potential models havea many�body nature� For example� the size of the oxygen ion is very dependenton the nature of the ionic environment� The free O�� ion is unstable with respectto decomposition to O� �plus one electron�� In a crystal� it is only stabilized by theeect of the Madelung �eld ������ Consequently� its size and polarizability can changeconsiderably from one structure to the other� As we already explained in section ������this eect is known as the breathing of the oxygen ion� In �gure ���� we show theintegrated valence electron charge within an sphere centered around the oxygen ionas a function of the sphere radius and for two dierent compounds CaO and MgO�It is clear from the picture that the size of the oxygen atoms is quite dierent inboth cases� The result indicates that the oxygen volume will be variable in dierent
�Even by using a shell model the results do not improve �����
���

5
5.5
6
6.5
7
7.5
8
8.5
0.8 0.9 1 1.1 1.2 1.3 1.4 1.5 1.6 1.7
Num
ber
of e
lect
rons
Sphere Radius (A)o
MgO
CaO
Figure ��� Integrated valence charge density within a sphere centered around oxygen�as a function of the sphere radius� The data was computed from pseudopotentialcalculations�
mixtures of CaO and MgO�We estimated the eect of the oxygen volume change on the formation energies by
freezing the oxygen wave functions to their SSCAD values for CaO and MgO respec�tively ����� When the formation energies predicted in the two cases were compared�the use of the dierent wave functions accounted for changes of the order of ����
The nonspherical deformation of the oxygen shape can account for at least another��� dierence in the formation energy values� This can be seen by comparing thepseudopotential and the SSCAD formation values� The major dierence between themethods is precisely the nonspherical electronic relaxations�
A potential model that accounts for both the volume change and the polarizabilityeects introduced by the breathing of the oxygen ion has recently been applied to theenergetics of zirconia ����� However� even when these eects can properly be accountedfor �either mapping them into a potential of the form shown in equation ��� or bythe approach in reference ������ potentials can not be always used� Charge transfermechanisms have to be considered�
In the case of the CaO�MgO system� the ionic charge does not change considerablyfrom one structure to the other and a potential could be found that reproduces theformation energies relatively well� However� the same does not apply to calcia�dopedzirconias� The charge will change when dopants and vacancies are introduced in thesystem as shown in section ����� �see also references ����� ������ This seriously limitsthe use of potentials in these materials�
It is clear from �gures ��� and ��� that the potential predictions are worse in
���

CaO�ZrO� than in CaO�MgO� Relative errors of more than ���� in the predictionof formation energies are present in the former system� The larger errors correlatewell with those structures where the variation of the charge in the Zr and O ions islarger� We will come back to this point later� in chapter ��
��� Relevant contributions to the total energy of
oxides
Four mechanisms seem to seriously limit the applicability of the current total�energymethods in oxides breathing� nonspherical electronic relaxations� chargetransfer� and directional bonding � For some simple systems� such as CaO�MgO� some of these eects do not play an important role� However� if one wants toaccurately describe the energetics of many oxides� a proper treatment of all them isdesirable�
There are signi�cant computer�time bene�ts of using simple methods such aspotential models� However� it is not straightforward to include all the above eectsin their formalism�
Of the four mechanisms� directional bonding is probably the simplest to in�corporate to a potential model� Although there is always the problem of �ndingthe right functional dependence� many�body potentials have been widely used �seesection �������
The breathing of the ions �specially the oxygen ions� can be approximatelyaccounted for by the compressible�ion model developed by Wilson and collabora�tors ����� They represented this many�body eect in a pairwise form� The nearest�neighbor interaction between the ions is considered as the sum of a rearrangementenergy �breathing� and an overlap between the ion wave functions �see section �����for more details��
Nonspherical electronic relaxations can be incorporated through the shellmodel explained in section ������ A quadrupole polarizability of the ions has beenrecently developed �����
Still� all potential models assume ionic charges that are independent of the en�vironment �either partial� or full�valence charges�� There is no systematic way todeal with the charge transfer mechanisms� Magri et al� and lately Wolverton et al�suggested that the charge on a given ion is a linear function of the number of unlikeclose neighbors ����� ����� This model has proven to be very accurate for metallicalloys� However� its use in highly�ionic materials is questionable since long�rangecontributions are expected� We will come back to this point later in chapter �� An�other possibility of including charge transfer is the model developed by Streitz andMintmire ���� �see section �������
Up to now� there has not been a simple way of including all these four mechanismsin a potential model� Also� note that part of the simplicity of the model is lost� notonly because more complex terms need to be included but also because ab initiocalculations are needed to �t some of the parametrizations� The incorporation of
���

charge transfer and breathing may also imply performing additional minimizationswith respect to the ionic radius or charge�
In chapter �� we will show that a self�consistent tight�binding formulation canincorporate all these mechanisms in a very accurate and simple way� Furthermore�since it provides a quantum�mechanical description of the system� a more realisticrepresentation of bonding in the crystal is obtained� Within this framework� there isno need to map physical eects onto simple functional forms� The fact that a tight�binding Hamiltonian can account for all the relevant physical mechanisms providesexcellent transferability properties�
The �tting of a potential model that incorporates all these mechanisms is becom�ing as complex and demanding as the determination of the tight�binding parame�ters� However� potential models are still faster than any semiempirical tight�bindingmethod� Still� the latter is many orders of magnitude faster than any ab initio methodwith comparable accuracy�
��� Conclusions
We showed that full�potential quantum�mechanical methods are reliable tools tocompute total�energy dierences on a scale relevant to the study of phase transitions�When taken to their full capabilities� their predictions compare well with experiments�For example� the agreement between the computed and measured CaO�MgO phasediagrams is excellent� Unfortunately� these techniques are also the most computer�time demanding of all �specially when needed as input to the statistical�mechanicalmodels��
Approximations such as spherical averages of potentials� non�self�consistency� orreplacing complex ion�ion interactions by simple pair potentials can dramaticallyspeed up total�energy calculations� However� we showed that� even for the relativelysimple CaO�MgO system� most of these approximations break down�
We have identi�ed four mechanisms that are necessary to accurately reproduceformation energies in oxides charge transfer� nonspherical electronic relaxations� ionicbreathing� and directional bonding� These eects should be incorporated into anyenergy model to capture the relevant physics of the problem� Unfortunately� all themethods that are currently used either do not take into account all these eects orare slow�
In the next chapter� we propose the use of a self�consistent tight�binding modelthat is capable of capturing the relevant physics of the problem while retaining a lowcomputational cost�
���

Chapter �
The tight�binding method
The sophistication of total�energy methods has reached a level in which it is nowpossible to accurately predict materials properties with no experimental input� Inthe previous chapters� we showed that the use of density�functional theory in thelocal�density approximation combined with lattice models is a very powerful toolto design new materials� Unfortunately� the methods are very time demanding andsimpler approaches are necessary�
LDA techniques� such as pseudopotentials or FLAPW� do not provide a simplebasis to develop an understanding of the relation between physical intuition� such ascharge� electronegativities� or atomic sizes� and structural properties� This intuitionis hidden in their complex formulation� However� understanding this relationship isfundamental if we would like to develop simple methods to account for the relevantcontributions in the Hamiltonian for oxides� These physical eects were identi�edin chapter �� We also illustrated how their incorporation in potential models is farfrom being trivial� In general� detailed quantum�mechanical information is needed toproperly deal with them�
In this chapter� we will demonstrate that the semiempirical tight�binding �SETB�method� is ideal to incorporate all the relevant physics to compute total energies inoxides� The SETB provides a simpli�ed framework in which a clear identi�cationbetween physical concepts and the resulting materials behavior can be achieved� Thisallows all the eects found in chapter � to be easily included�
The concept of the tight�binding approach was introduced by Bloch in ���� ������It is based on the use of combinations of atomic orbitals to express the electronicwave functions in a solid� This is appropriate when there is a relatively small overlapbetween neighboring atoms as in the case of the d�transition metals or the alkalihalides� Originally� the tight�binding method was regarded as an empirical approachto qualitatively interpret the electronic structure of a solid� Later� Slater and Kosterproposed to use it as an interpolation tool to be combined with other methods such
�The use of the tight�binding name is sometimes limited only to its empirical formulation� Theab initio approach is then called linear combination of atomic orbitals LCAO ������ In this the�sis we will not make a distinction between the two and tight binding and LCAO will be usedinterchangeable�
���

as APW ����� This was the origin of what we have called the semiempirical tight�binding technique� where the various interaction integrals are parametrized accordingto the results of these other techniques � However� in recent years� there has been arenaissance in the use of the ab initio tight�binding method due to the growth in thecomputing capabilities and the improvement in the numerical techniques�
The use of the SETB approach in oxides requires that one carefully incorporatesthe important physical eects identi�ed in the previous chapter into the Hamiltonian�The ionic breathing and the charge transfer mechanisms force the introduction of self�consistency into the tight�binding framework� Only when all these terms are properlyaccounted for� will a good description of the energetics be obtained� Furthermore�under these conditions� the parameters of the model will be transferable under changesin the atomic environment�
The transferability of the tight�binding parameters is essential if we want to usethe method to calculate formation energies of dierent compounds� For example�when computing temperature�composition phase diagrams we will �t the SETB pa�rameters to ab initio calculations for a few simple ordered structures� From thisinformation the formation energies of many more complex ordered structures will becomputed to determine the ECI� Thus� the tight�binding method will replace thetime�consuming ab initio approaches when dealing with compounds that have largeunit cells�
There exists an extensive literature about the tight�binding method� A generalreview of the dierent formulations can be found in references ����� ���� ���� and thecombination with recursion techniques in references ����� ����� The semiempiricalapproach was introduced in great detail in references ���� ��� ���� ����� Applicationsin ceramic materials of the �rst�principles tight�binding technique are described inreference ����� and references therein�
We start in sections ��� and ��� by brie�y describing the �rst�principles and thesemiempirical formulation of the tight�binding approach� Section ����� is mainly con�cerned with the self�consistent tight�binding model used in this thesis� In section ����we deal with the computation of the parameters of this method from �rst�principlescalculations� Finally� in section ������ we discuss the computer implementation of thelatter technique�
�� Formulation of the method
Solutions of the Kohn�Sham equation ���� are sought by expanding the one�electronwave functions� �n� in a basis of atomiclike orbitals� i�� centered at site i�
�n �Xi��
cni�i�� �����
���

The indices n and � identify the band and the orbital type respectively� Thus�equation ���� becomes equivalent to a set of linear equations��
Xj��
�Hi��j� � �nSi��j�� cnj� � � �����
whereHi��j� �
Z�i�Hj�d
�r �����
andSi��j� �
Z�i�j�d
�r� �����
Hi��j� and Si��j� de�ne the elements of the Hamiltonian and overlap matrices respec�tively �for the atomic basis functions we are using�� The integrals are over all space�The quantity Hi��j� is sometimes called a hopping integral� The eigenvalues �n arethe solutions of the secular equation��
det jH� �Sj � �� �����
The translational symmetry of a crystal can be exploited by using Bloch�s theorem�see section ����� The basis functions are rede�ned as a combination of atomic orbitalscentered at dierent sites� The new basis functions� �k���� � are
�k���� ��r� ��pN
XR�
eik�R�������r � �R� � �� � �����
where �R� denotes the location of the origin of the unit cell and �� is the position of aparticular atom within that unit cell�� Note that the sum is over atoms on equivalentpositions �i�e�� same ��� on the N dierent unit cells �i�e�� dierent �R�� and for a givenorbital �� If we replace i� in equation ��� by �k���� �
S����� ���k� �Z��k���� ��r��k���� �d
�r
��
N
XR�� R��
eik�R���R����� ��
Z����r � �R� � �� ����r � �R�� � �� ��d�r� �����
We can de�ne �R � �R�� � �R� �see �gure ���� so that equation ��� can be written as
S���� � �
��k� ��
N
XR
eik�R�� ����
Z����r � �� ����r � �R� �� ��d�r� �����
�This set of equations is obtained by multiplying each ��i� to the left of HKSn and integratingover all space� In what follows HKS will be simply called H �
�This equation is the mathematical condition under which equation ��� has nontrivial answers��For clarity we do not use the i index in � since we are implicitly indicating the location of the
basis functions with �R� and �� �
���

y
x
ττ
R’
R
R’’
’
Figure ��� Nomenclature used to identify the unit cells and atomic positions� �R� and�R�� point to the origin of the unit cell �two unit cells are marked in the picture withtwo squares�� �� and �� � indicates the position of the atoms within the unit cell�
Similarly�
H���� � �
��k� ��
N
XR
eik�R�� ����
Z����r � ���H���r � �R� �� ��d�r� �����
Note that the use of Bloch�s condition reduced the dimension of the secular equationto the number of basis functions in a single unit cell��
If the atomiclike basis functions � are su�ciently localized� the integrals in equa�tions ��� and ��� are negligible for atoms that are far apart� The sum over the directlattice vectors �R is thus limited to those terms that involve atoms that are close neigh�bors �for a given �� and �� ��� Consequently� the sum only has a few terms� However�the computation of the overlap and Hamiltonian matrices is very time consumingsince these terms involve the calculation of two� and three�center integrals��
Up to now� we have not speci�ed the basis functions �� Atomic orbitals wereproposed in the original formulation ������ This basis is incomplete unless the contin�uum of unbound states is included� However� as long as the neighboring potentialsdo not overlap strongly� the low�valence states form an adequate expansion� �Whenthe potentials overlap signi�cantly� the bound states can be expected to liberate the
�However we will now have an in�nite number of secular equations one for each �k point��For example the integrals can involve the product of an atomic function centered at one atom
another atomic function centered at a second atom and a potential centered on a third atom�
���

electrons into free�electron�like states��The atomic orbital basis is not orthogonal and problems of overcompletness can
arise� These problems can make the overlap matrices ill conditioned� They areusually solved by using orthonormalized orbitals�
Modern formulations of the tight�binding method replace the atomic wave func�tions by Gaussian�type orbitals and spherical harmonics for the radial and angularparts respectively ������ These orbitals have the advantage that the three�center inte�grals can be easily computed by the Gaussian transformation technique ������ On theother hand� the use of several Gaussian�type functions per orbital can make the secu�lar determinant very large� This can be improved by using a �xed linear combinationof Gaussians� chosen to mimic the orbitals of the free atoms� as basis functions��
It is common practice to use the term minimal basis for one in which only thecore orbitals and all orbitals in the valence shell are used� Correspondingly� a fullbasis is formed by the minimal basis plus excited orbitals�
The secular equation has to be solved self�consistently since the Kohn�ShamHamiltonian depends on the electronic density� The computation of multicenter in�tegrals takes a large percentage of the calculation time and only in recent years hasthe method started to be used from �rst principles ������ In the next section� wewill describe the semiempirical approach� which replaces these costly calculations bysimple parametrizations of the matrix elements�
�� The semiempirical tight�binding method
The use of Gaussians as basis functions considerably improves the e�ciency of thetight�binding method and the speed of evaluating the various integrals� However�this is achieved at the expense of an increase in computer storage requirements�As systems get larger� more of these terms need to be computed� Consequently�signi�cant time savings can be obtained from evaluating the integrals in equations ���and ��� approximately�
The computation of these integrals requires the knowledge of both the basis func�tions and the potential in the Hamiltonian� However� they always appear together inthe formalism outlined in section ���� The semiempirical tight�binding method takesadvantage of this fact by developing parametrizations of the elements of the hoppingand overlap matrices without specifying an explicit form for the basis functions orthe Hamiltonian potential separately� This is the basis of the SETB formulation� Itis particularly fast since the time�consuming hopping and overlap integrals are �t toab initio results� In this sense� this method represents a much more sophisticated
�As we already mentioned the atomic orbitals centered at a single site are already complete whenunbounded states are included� Our basis functions are formed from orbitals at many sites whichcauses the overcompletness problem�
�In self�consistent calculations the basis can be reoptimized at each step� Also it is oftenconvenient to eliminate the core states by using the frozen�core approximation particularly whenwe are dealing with heavy atoms�
�No frozen�core approximation is used�
���

and accurate interpolation tool than potential models and is still orders of magnitudefaster than ab initio quantum�mechanical techniques�
Although it has often been used to interpret results from ab initio quantum�mechanical calculations� the SETB has also provided semiquantitative answers instudies of structural stability in a wide variety of systems such as sp�bonded non�metals ������ semiconductors ������ pd�bonded transition metals ������ etc� Recently� ageneral tight�binding total�energy formalism that accurately reproduces total�energydierences� defect energies� and elastic constants for transition and noble metals hasalso been introduced ���� ��� �����
���� The Slater�Koster formulation
Slater and Koster proposed the use of the tight�binding method as an interpolationprocedure by treating the various integrals as constants chosen to �t �rst�principlescalculations ����� Note that the integrals are �k independent� thus by �tting to ab initio
results at a few high�symmetry �k points� they were able to extend their calculationsthroughout the Brillouin zone� The values of the integrals are not independent butare interrelated by the symmetry of the crystal and the basis orbitals�
In the original formulation� the basis functions were assumed to be orthogonal�In this way� the number of parameters to �t was reduced in half as no overlap in�tegrals need to be determined� However� atomic orbitals centered at dierent sitesare not orthogonal� Lowdin showed that it is always possible to perform an orthog�onalization procedure that preserves the original symmetry properties of the atomicorbitals ������ Slater and Koster assumed that they always worked with this orthogo�nalized basis� The resulting functions are not necessary localized� producing non�zerohopping integrals between very distant atoms �and thus requiring more parameters��Many authors investigated the construction of highly localized� but yet small� basissets��� See references ����� �����
Another approximation usually made is to neglect the three�center integrals� Thepotential energy in the Hamiltonian is approximated as the sum of spherical potentialscentered at the atomic sites� The only part of the potential energy that is retained isthe one coming from the sum of the spherical potentials at the sites of the two atomswhere the basis orbitals are located� Thus� the Hamiltonian integrals have the sameangular dependence as those in a diatomic molecule� The basis functions can then beexpressed as sums of space quantized functions with respect to the axis that join thecenter of the atoms� For example� a p�type basis orbital is a linear combination of ap� and a p� function with respect to that axis���
�The idea of these theories is to be able to exactly represent the solid single�electron wave functionas a combination of localized orbitals that do not need to be recalculated for each system� In thisway they can still provide a simple chemical picture of bonding� The orthogonality constraint isgenerally removed� However the overlap matrix does not appear in the formalism at the expense ofhaving a nonhermitian secular matrix�
��A p� and a p orbital are oriented along and perpendicular to the axis joining the two atomsrespectively�
���

= +
E(s,x) t t sin θspπ= +
θ
cos θσsp
+-
+
++
-
-
++x
y
z
Figure ��� Illustration of the representation of the Hamiltonian integral for a s anda px orbital �Es�x� as a function of two center integrals �tll�m�� Note that tsp� is zeroby symmetry�
The dierent Hamiltonian integrals can then be expressed in terms of the two�center integrals between these quantized functions usually called tll�m �e�g�� tsp��� Thisis illustrated in �gure ���� for the Hamiltonian integral between a s and a px orbital�Es�x� Note that some two center tll�m integrals� such us tsp� in the �gure� are zero bysymmetry� Tables for dierent energy integrals can be found in reference ���� for s�p� and d orbitals� and reference ����� for f and g orbitals���
The neglected three�center contributions are not necessary small as already poin�ted out by Slater and Koster� For some structures� the number of tll�m integrals isthe same as the E integrals� In this case� no savings are made regarding the numberof parameters when using the two�center approximation�
The values of the two�center integrals tll�m depend on the actual distance betweenthe atoms� By relating the tight�binding bands to free electron bands� Harrison andcollaborators predicted not only what this distance dependence should be for s�pmaterials� but also universal inter�atomic coe�cients for this parametrization ������They later obtained general expressions for other bonding types ����� ���� ����� Their�ndings are summarized in table ���� It should be noted that these universal param�eters and distance dependencies are valid for nearest�neighbors integrals� For largerdistances� the basis functions decay exponentially� This eect is not taken into ac�count in table ����
Other authors have proposed dierent distance expressions� whose parameters are
��There are some typographical errors in both references� See references ���� ���� for the correc�tions� Reference ����� presents a general formalism to express the hopping integrals as a function oftwo�center integrals valid for any angular�momentum quantum numbers�
���

s�s� p�p p�d d�d
tll�m � �ll�m�h�
med�tldm � �ldm
�h�rd��
med��
tddm � �ddm�h�r�
d
med�
�ss� � ����� �sd� � ����� �dd� � ������sp� � ����� �pd� � ����� �dd� � ������pp� � ����� �pd� � ����� �dd� � ������pp� � �����
Table ��� Interatomic matrix elements as proposed by Harrison� The value of the rdconstants can be found in reference ����� and d is the distance between the atoms�
�t to the particular systems� For example� Mehl and coworkers have used
tll�m�d� ��c��ll�m � c��ll�md� c��ll�md
��
�� � e�d�do�
lo �e�c�
��ll�md ������
where d is the distance between atoms� ci�ll�m are the coe�cients to be �t to ab initioresults and lo and do are taken as ��� a�u� and �� a�u� respectively ������ On the otherhand� Mercer and Chou ����� proposed using
tll�m �c��ll�mdc��ll�m
ec��ll�md� ������
The same expressions are usually used for the overlap integrals if the basis is nonorthog�onal�
The diagonal elements of the Hamiltonian matrix are usually called on�site terms�These terms correspond to the hopping integrals computed for identical orbitals� Ifatomic orbitals were used as basis functions� the on�site terms would represent theirfree�atom eigenvalues changed by the solid environment� Since in most cases ofinterest the tight�binding model is applied to a dierent environment than the onein which it was �t� it is necessary to incorporate this environmental dependence�Dierent functional forms have been proposed to relate the on�site terms to thedistance to� and the number of� nearest neighbors� See for example references ���������� We will come back to this point in section ������
It is common practice to �t the hopping and overlap terms to the bands producedby ab initio methods�
���� Tight�binding total�energy model
The tight�binding formalism explained in section ����� is sometimes referred to as aband model� No total�energy expression has been presented� Thus� the Slater�Kostermodel can be regarded as a procedure to interpolate the bands to all points in theBrillouin zone from calculations for a few high�symmetry �k points� Since our aim
���

is to compute formation energies� it is essential to develop a way of extracting totalenergies from this technique�
We start from the density�functional expression in the LDA for the total en�ergy ������� For simplicity� we disregard any reference to �k�space�
E�n��r�� �occXi
�i � �
�
Z Zn��r�n��r��d�rd�r� �
Zn��r� ��xc�n��r�� Vxc�n��r��� d
�r
��
�
NXI
NXJ ��I
ZIZJe�����RI � �RJ
���� Ebs � F �n��r��� ������
Here� �i are the Kohn�Sham eigenvalues and Ebs �Pocc
i �i is the so�called band�structure energy �the sum is over occupied electronic states�� F �n��r�� representsthe overcounting of the electron�electron interaction in the Ebs term� the parts ofthe exchange and correlation not included in the eigenvalue sum� and the core�coreelectrostatic energy�
The F �n��r�� term is essentially a short�range repulsive interaction if there is nosigni�cant charge transfer between the atoms� In this case� the core�core interac�tion tends to cancel out with the electron�electron interaction��� Furthermore� theexchange and correlation part is usually small ����� �����
Many tight�binding total�energy methods compute total energies by using theSlater�Koster band model to obtain the band�structure energy and a sum of pairpotentials to evaluate F �n��r����� Consequently� the tight�binding total energy isgiven by
E � Ebs � Epp� ������
The pair potential� Epp� is repulsive and is a function of the bond length rij� Forexample� Sigalas and Papaconstantopoulos suggested using ����
Epp ��
�
Xi�j
j ��i
�Xn
An
rnij
�e�rij � ������
The second sum represents a polynomial in �rij� An and � are coe�cients that are
�t� together with the hopping and overlap terms� to �rst�principles band and total�energy vs� volume curves for dierent structures� Instead� other authors chose to �tto phonon frequencies� bulk modulus� or other experimental measurements� How�ever� the Sigalas�Papaconstantopoulos approach provides more accurate results forcomputing formation energies since we are directly �tting to the quantities of interest�
There are severe approximations in going from the LDA equation ���� to the
��The procedure we will follow varies according to the treatment of the many�body electronproblem� However the �nal form of the result equation ���� is always the same�
��If charge transfer is allowed there can be a long�range contribution in the form of a Madelungsum�
��See for example references ��� ��� ���� and references therein�
���

tight�binding expression ����� In the latter� �i is not the result of a self�consistentapproximation� There is also the question of whether it is possible to replace F �n��r��by a pairwise short�range interaction� These issues are discussed in detail in refer�ence �������� See also reference ������
There are also practical problems in the implementation of equation ���� aspointed out by Cohen and coworkers ����� The Kohn�Sham eigenvalues can be shiftedby an arbitrary constant without changing the results� since its contribution to thetotal energy is canceled out by F �n��r�� in a self�consistent calculation� If the twoterms in equation ���� are parametrized independently� extra care should be taken tohandle this constant�
On the other hand� the introduction of a shift in the potential can be used toeliminate the Epp term altogether ����� The shift� So� can be de�ned to be
So �F �n��r��
Ne
������
where Ne is the total number of electrons� Thus� equation ���� is transformed into�
E�n��r�� �occXi
��i �
F �n��r��
Ne
�� ������
A set of Slater�Koster parameters that simultaneously �ts the band�structure andthe total�energy vs� volume curves will solve the total�energy problem without theneed of a pair�potential term� This model has been successfully applied to computeelastic constants� vacancy formation energies� and other physical properties for manyalkaline�earth� transition� and noble metals ���� �����
Unfortunately� these simple models can not be applied in oxides� As it was alreadymentioned� when there is a signi�cant charge transfer between the atoms in the solid�Epp is no longer short�ranged� Furthermore� the transfer of charge between atomswill considerably shift the on�site terms� This transfer can not be accurately capturedwith the standard environment dependence of the diagonal Hamiltonian terms� Thislong�range eect can only be represented by a proper description of the charge transfermechanisms�
The explicit use of the atomic charge in the tight�binding Hamiltonian� forcesthe reinstatement of self�consistency into the calculations� We are now ready toincorporate into the formalism all the relevant physical contributions to the totalenergy we mentioned in chapter �� This will be accomplished in the next section�
�� Self�consistent tight�binding total�energy
model
The tight�binding formalism and its localized basis provide an intuitive picture of
��The procedure is justi�ed by showing that the tight�binding model can be understood as astationary approximation to self�consistent density�functional theory�
���

how the solid can be viewed as a set of interacting atoms� We will take advantage ofthis to build an appropriate Hamiltonian model for oxides�
When the atoms are brought together to form a solid� the electrons are attractednot only to their own cores but also to the neighboring ones� The total energy is re�duced by this electronic sharing between the ions� In a tight�binding formulation� thepresence of nonzero hopping integrals reproduces this eect� The hopping integralshave a fundamental role in determining the shape of the bands�
A second eect of bringing the atoms together is the compression of the atomicclouds that causes a repulsive interaction due to an increase in the kinetic energy� Itis identi�ed with the overlap of the basis functions and is taken into account throughthe overlap matrix ����� ����� This was also pointed out by Gordon and Kim �����and is an integral part of the SSCAD formalism� as mentioned in section ������
It should be clear then that of the four mechanisms identi�ed in chapter �� only theionic breathing and the charge transfer eects are missing in the total�energy modelof section ������ The displacement of charge between the atoms is accompanied bythe appearance of a long�range electrostatic interaction� The eect of this �eld �thecrystal �eld� is to shift the on�site terms� For example� in CaO or MgO� this shiftmoves the oxygen p levels downward� stabilizing the O�� state� The electrostaticinteraction between the cores should also be taken into account� It is usually lumpedtogether with the electron�electron interatomic interactions�
Given a certain charge distribution between the ions� the eect of the ionic breath�ing can be taken into account by adding the crystal �eld to the on�site terms� The�eld is easily computed in the form of a Madelung sum��
The occupation of the atomic orbitals is changed by the rearrangements of chargewhen the atoms are compressed to form the solid� The changes in the intra�atomicinteractions between the electrons also modify the electronic levels and should betaken into account�
Both the eect of the crystal �eld and the change in the intra�atomic interac�tions require the knowledge of the charge density which� in turn� depends on theHamiltonian matrix �since it determines the one�electron wave function from whichthe density is computed�� Consequently� the charge density has to be determinediteratively� This is the origin of the required self�consistency of the method�
It should be noted that the last two contributions only aect the diagonal terms�In the following� sections we will give details of how these dierent contributions canbe computed and incorporated into the tight�binding Hamiltonian� We will followreference ����� closely�
��The procedure can be compared to the PIB model where the isolated oxygen density is computedwithin Watson spheres� These spheres reproduce the crystal �eld that the atom feels when placedin the solid�
���

���� Nonorthogonality contributions
As it was already mentioned� the atomiclike basis� ji� � is usually not orthogonal���
However� it is more convenient to work with localized orthogonal functions� Theoriginal basis can be transformed into an orthogonal one� j�i� �� in the following way
j�i� �� ji� � ��
�
Xj ��i�� ���
Sj��i�jj� � � ������
The sum is over all the elements in the basis �except for ji� ���In the new basis� the overlap matrix S is zero to �rst order in the old overlap
matrix� except for the diagonal terms �that are equal to one�� Furthermore� the newHamiltonian matrix elements are now
ti��j� � �i�jHj�j� �� i�jHjj� � ��
�
Xi� ��j��� ���
Si����j� i�jHji��� � �
�
�
Xj� ��i��� ���
Sj����i� j���jHjj� � ���S��� ������
Since the nondiagonal terms are directly �t to ab initio results� there is no need toincorporate S terms in them� They are implicitly taken into account during the �t�On the other hand� the on�site terms are formed by dierent contributions which willbe �t independently� Consequently� we need to write the eects of nonorthogonalitiesexplicitly�
ti��i� � i�jHji� � � X� ����j ��i
Si��j� i�jHjj� � ���S��
� i�jHji� � �Fi�� ������
In what follows� we will assume that the unspeci�ed basis we use is orthogonal�Furthermore we will correct the on�site terms by Fi�� Thus� for example� if weassume that our starting� nonorthogonal basis functions are the free atomic orbitals�equation ���� will translate into�
ti��i� � �i� � Fi� ������
where �i� are the atomic eigenvalues��
��In what follows we will adopt Dirac�s bra�ket notation to make the mathematical manipulationsmore clear� See for example reference ����� Recall that we use the symbol i and � to identify theionic site and angular momentum respectively�
��Note that to compute Fi� we need the hopping integrals in the nonorthogonal basis� However the �tting process will provide these matrix elements in the orthogonal basis� We can safely replaceone by the other to �rst order in S ������
���

���� Intra�atomic electron�electron interactions
An Anderson Hamiltonian ����� model is used to describe the eect on the orbitalenergies of the Coulomb repulsion between electrons on the same atom� The levelsin a free atom� �i�� are described as a function of the orbitals occupation� Qi�� in thefollowing way
�i� � �oi� �X�
Qi�Ui��i�� ������
Here� � are the set of quantum numbers that identify the orbitals and Ui��i� are theintra�atomic repulsion parameters computed as�
Ui��i� � e�Z �i���r�i���r�
�i���r
��i���r��d�rd�r
j�r � �r�j � ������
This model assumes that the atomic energies are linear functions of the occupationof the orbitals� As shown in reference ������ this approximation is a very reasonableone�
The formulation of the electron�electron interaction in equation ���� correspondsto a Hartree�type approximation��� These direct contributions are usually much largerthan the exchange terms ������ For the case of open d�shells� it may be necessaryto include the exchange energy explicitly within a Hartree�Fock�type approxima�tion ������ For simplicity� we treat equation ���� as a phenomenological model� whoseparameters are �t to ab initio LDA results or Hartree�Fock calculations���
Hartree�Fock calculations usually provide a more accurate representation of anisolated atom� Consequently� it is common practice to obtain the parameters of themodel� Ui��i� and �o� from them ������ Dierences between the experimental ionizationenergy and the electron a�nity� or between the �rst and second ionization energies arealso used ������ In this last case� a single Ui��i� is generally employed independentlyof the orbitals � and �� In reference ������ a table is provided for sp�con�gurationatoms�
The change in the atomic eigenvalues with respect to the occupation of the orbitalscan also be obtained from the so�called chemical hardness matrix ������ AssumingQi� to be continuous� the total energy is expanded in a Taylor series� and the eigen�values are obtained as the derivative of the total energy with respect to the orbitaloccupations� The coe�cients of the expansion are the elements of the chemical hard�ness matrix and can be computed from ab initio atomic calculations� This can beconsidered as a generalization of our simple model to any order on the variation oforbital occupancy� For more details� see reference ������
�Ui��i� is formally the exchange self�energy of the state �i� as well as the Hartree�type Coulombinteraction ������
��Harrison has argued that the principal e�ect of the Coulomb interactions is to add a correctionproportional to the occupation of the orbital with the proportionality constant depending on theelement ������
���

���� The self�consistent tight�binding Hamiltonian
We are now ready to write the one�electron tight�binding Hamiltonian �HTB� byadding the charge transfer eects to the Slater�Koster formalism� In the orthogonal�ized basis of section ������ HTB can be written as�
HTB �Xi��
j�i� � �i� �i�j�Xi���j��
i��j
j�i� � t�� �j�j� ������
The �rst term in the equation corresponds to the on�site or diagonal Hamiltonianmatrix elements� while the second term represents the nondiagonal or hopping terms�t�� will be �t to ab initio results as explained in section ������
The extra contributions to the Slater�Koster Hamiltonian can be incorporatedinto �i��
�i� � �oi� � Fi� �X�
U intrai��i� Qi� �
Xj ��i��
U interi��j�Qj� � Vi�� ������
�oi� represents the kinetic energy and the interaction with its own ionic core �at sitei� of an electron at orbital �� Fi� is the shift in the eigenvalues due to the orthogo�nalization process �see section ������� The �rst sum represents the electron�electroninteraction within atom i� while the other sum is between electrons at dierent atoms�Finally� Vi� represents the crystal �eld at site i�
Similarly to U intrai��i� � U
interi��j� is computed as
U interi��j� �
Z �i���r�i���r��j���r
��j���r��d�rd�r
j�r � �r�j � ������
The electron�electron interatomic interactions can be computed as Madelung sumsover point charges��� Note that this is an important approximation since the Coulombinteraction can not be meaningfully divided into strictly inter� and intra�atomic termsfor overlapping charges� A simple way of solving this is to interpolate the electron�electron interaction between a e��d� potential for large distances and a constant U�for short distances� See reference ����� for more details�
The eigenvalues� �n��k�� and eigenvectors� j n�k ���� of the Hamiltonian matrixHTB
at each �k have to be computed self�consistently since its diagonal elements dependon Qi� which is obtained as
Qi� �occXn�k
j �i� j n�k �j� � ������
This sum is over all occupied states�Variants of the Hamiltonian in equation ���� have been successfully used by Rob�
��Combined with the core charges���Note that j n�k � is written as a function of Bloch�s sums of the orthogonalized basis functions
j�i� �� See section ��� for more details�
���

bins and Falicov ������ Schilfgaarde and coworkers ������ Harrison ������ and Majewskiand Vogl ����� among others� Within this model� atomic forces can be computed verysimply as shown in reference ������
���� Total energies within the self�consistent formulation
In section ������ we showed that the total energy can be written as �see equation �����
E�n��r�� � Ebs � F �n��r��� ������
The band�structure contribution is easily computed by �nding the eigenvalues ofHTB�
Ebs �occXn�k
�n��k� ������
where the sum is over the single�particle energies of the occupied states with bandindex n and wave vector �k�
The second contribution to the total energy in equation ����� F �n��r��� can be ex�plicitly computed within the self�consistent formalism� There is neither a need to addextra parameters in a pair�potential form nor a concern about arti�cial shifts in theeigenvalues��� F �n��r�� incorporates the eect of the core�core repulsion and correctsfor the double�counted electron�electron interactions� Thus� it can be computed as
F �n��r�� � ��
�
Xi�
��X
�
U intrai��i� Qi� �
Xj ��i��
U interi��j�Qj�
�AQi�
��
�
NXI
NXJ ��I
ZIZJe�����RI � �RJ
��� � ������
Note that this contribution is not an electrostatic interaction between the atoms� Theelectron�electron contribution ��rst term in equation ����� has a minus sign�
���� Implementation
The model Hamiltonian in equation ���� describes all the physical eects we foundto be important to compute formation energies in oxides� By solving for its eigenval�ues and eigenvectors self�consistently� total energies can be computed using expres�sion ����� Signi�cant time savings are obtained compared to ab initio methods sincethe various complex integrals have been replaced by adequate parametrizations� andthe intra�atomic electron�electron interactions by a phenomenological model�
Before the tight�binding model can be used� its numerous parameters have tobe determined� It is common practice to �t the hopping and overlap integrals toab initio or experimental results� Since charge�transfer mechanisms were included in
��The electron�electron parameters that are used to compute F n�r should be the same onesused to determine Ebs�
���

the formalism� we also need to compute the intra�atomic electron�electron interactioncoe�cients�
Dierent approximations can be used to reduce the number of unknown parame�ters of the model� For example� some authors �see references ����� ����� make use ofthe extended H#uckel theory ����� to relate the overlap and hopping integrals�
ti��j� � Ki��j���i� � �j��Si��j�� ������
In the simplest approximation� Ki��j� is regarded as a constant for each row of theperiodic table �or a geometric average for cases that involve atoms from dierentrows� ������ We did not �nd this approximation accurate in oxides� Consequently� we�t to both types of integrals independently but with the same distance dependence�
If H#uckel�s theory is combined with the set of universal parameters in table ���� no�tting is necessary within the Slater�Koster formalism� Furthermore� the electron�electron intra�atomic interaction parameters can be obtained from ionization energiesand electron a�nities� and the atomic levels from published Hartree�Fock calcula�tions ����� ����� This opens the possibility for the self�consistent formulation to alsobe a �tting�free� model� The results thus obtained are usually qualitatively cor�rect� As already mentioned� the values and distance dependence of the parametersin table ��� are not known exactly� They are usually �t to results for many dierentsystems� Moreover� the computation of the intra�atomic interaction coe�cients alsoinvolves many approximations�
Many authors have been successful in predicting general trends in materials prop�erties by performing �ts to dierent systems ����� ���� ����� However� our aim willbe to reproduce very accurately the properties of a speci�c system� For example� wewill �t the overlap and hopping integrals to ab initio calculations for pure CaO� MgO�and CaMgO� �L�� structure�� and then predict formation energies for many mixturesof the end members� The values of the intra�atomic interactions and the electroniclevels will be taken from the tables��� The distance dependence of the integrals willbe assumed to be the ones in table ������
We expect the �t parameters to be transferable within dierent structures in agiven oxide system since our tight�binding Hamiltonian incorporates all the relevanteects to compute total energies� Of course� the transferability of the parameters canbe reduced by the approximate treatment of some of these terms or by other eectsnot accounted for that can be mapped onto them� However� we will show in chapter �that the formulation used here is an adequate representation of the energetics of oxidesand that no signi�cant transferability problems arise�
��When the intra�atomic parameters were taken as �tting coe�cients they stayed very close tothe values in the tables�
��Note that there are tremendous savings in the number of parameters to be �t when simpledistance dependencies such as the ones in table ��� are used� Polynomial dependencies will requireat least twice as many parameters�
���

�� Computation of the tight�binding parame�
ters
As already mentioned at the beginning of this chapter� the computation of the hoppingand overlap integrals is very time�consuming� Consequently� semiempirical formula�tions �t those parameters to ab initio calculations or experimental properties� The�tting process is generally very involved� A large number of parameters should bedetermined and the optimization space is nonlinear� with many local minima�
The number of parameters to �t varies with the size of the basis set� For a typicaloxide� with � dierent species in the unit cell and a s�p�d basis� it is around ���This number can be � times larger if a more complex distance dependence of theparameters are used� The parameters are adjusted so that the tight�binding modelreproduces a set of dierent physical properties� A distance� or merit function�usually proportional to the square of the dierence between the predicted and the direct� values� is de�ned� The parameters are evolved to minimized this distance�by using a nonlinear least�squares algorithm such as the Marquardt procedure ������
However� a dierent approach in which the coe�cients are directly computedfrom a �rst�principles method can be taken� Andersen and Jepsen showed that thereexists an exact transformation of the mu�n�tin�orbital basis into a tight�bindingbasis ������� Within the ASA� they were able to cast their Hamiltonian into a Slater�Koster tight�binding Hamiltonian in a two�center form� See references ����� ���� formore details�
Unfortunately� as it was shown in chapter �� the LMTO�ASA approximations inoxides do not yield accurate results� The choice of sphere radii was critical and nocriterion to properly set the size could be found� Consequently� in this work we will�t the parameters to results from pseudopotential calculations�
Signi�cant time savings would be achieved if all the necessary parameters of themodel could be obtained from �ts to the end members� These compounds are usu�ally simpler than any of their mixtures� A fast optimization procedure can then beperformed� We will come back to this point at the end of the chapter�
���� Fitting procedure
We will use the tight�binding model mainly to reproduce structural energy dier�ences� Consequently� following reference ����� the �tting will consist in computingbands and equations of state for a given set of parameters� compare these predictionsto LDA calculations� and change the parameters to improve the matching� This is avery di�cult procedure since the function to be optimized is nonlinear in a large mul�tidimensional space� Many local� minima exist� and a straightforward �t frequentlydoes not �nd the absolute minimum�
The starting point in parameter space is very important to converge to the right�minimum� Many universal values exist based on the LMTO�TB transformation or
��Since the transformation is exact all the results obtained with mu�n�tin orbitals can be repro�duced by the tight�binding method�
���

on plain �tting to many elements across the periodic table� However� they are notalways close to the optimum values� Many dierent results for the parameters canstill be obtained from the process� A more restrictive criteria during the optimizationis necessary�
We imposed constraints during the �tting so that many local minima are removed�By analyzing the physics of the problem� it is possible to identify properties that arerelevant and that should be reproduced correctly� For example� in the CaO�MgOsystem� the pseudopotential results clearly show that Ca and Mg are fully ionized� Byusing this information we penalized� any signi�cant departure from this condition�eectively reducing the size of the space where the optimization search is performed�This allowed us to easily obtain the parameters used in chapter ��
In other cases� the situation is less clear� For example� although Ca is fully ionizedin CaO�ZrO�� neither Zr nor O has its formal charge� Still� an estimation of theircharge can be made so that it can then be used during the �t���
For all the systems studied� we found that imposing these extra constraints allowsthe optimization process to select the right� minimum� As in any minimizationtechnique� it is never sure that the resulting parameter set is not a local minimum�Checking new structures� not included in the �t� is always a good idea�
It is important that the equation of state scan all the relevant relaxations� Acorrect distance dependence of the integrals can only be captured if the volume ofthe cell and internal position of the ions used in the �t are varied over a wide rangeof values�
The more species involved in the �t the more di�cult it becomes� The optimizationspace is not only larger but also the structures are bigger� which makes the calculationslower� We are usually interested in formation energies resulting from a combinationof two oxides� Fitting them independently is simpler than �tting to a mixture of them�However� the end members do not provide all the required parameters� Some cation�cation integrals will be missing� In the next section� we will explore the possibilitiesof computing these missing values by just using the information provided by theend members� If successful� not only will the �tting process be simpler but once theparameters for several oxides are obtained� total�energy calculations can be performedfor any mixture of them�
���� Tight�binding parameters for oxide mixtures from
end�member information
The CaO�MgO is the simplest oxide system we will deal with� Even in this case��tting to the end members does not provide enough information to compute all theoverlap and hopping integral parameters� It is clear that the Ca�Mg interactionparameters can only be obtained from �ts to properties of the mixtures�
��From equation ���� F n�r � En�r � Ebs� If the intra�atomic repulsion parameters areknown F n�r only depends on the values of the charges� These values can be assigned so that thetight�binding representation of F n�r best matches the ab initio predictions for the En�r�Ebs
di�erence as a function of the distance between the ions�
���

Many attempts have been made to guess� these coe�cients� Most of them wereapplied to binary alloys with elements of similar electronic structure ������ A simplegeometric average of the self cation�cation interactions of the type�
t�i��j� � tj��j�ti��i�� ������
is often suggested� The oxide cations do not necessarily have a similar electronicstructure� Furthermore� the use of equation ���� has no formal justi�cation other thanits simplicity� However� it is important to note that the cation�cation interactionsare usually small since they are not nearest neighbors�
One is tempted to take them as zero� as a �rst approximation� and just use theparameters computed from the end members� The problem is that many dierentparameter sets �t the end members properly but result in variations of properties ofthe mixtures of more than ����� Without using �tting information from the mixture�it does not seem possible to predict correct mixing properties�
In the next chapter� we will further analyze these problems and suggest possiblesolutions�
�� Coding the self�consistent tight�binding
method
We have implemented the self�consistent tight�binding total�energy method in acomputer� A very simple description of the algorithm can be found in �gure ����
The two�center approximation is used to compute the Hamiltonian and overlapmatrix elements� Following the procedure suggested by Sharma ����� ����� we con�struct the Slater�Koster integrals for arbitrary angular momentum quantum numbers�For the Hamiltonian diagonal terms� we usually assume an initial orbital occupationcorresponding to their chemical formal valence� These occupations are then changedself�consistently�
The short�range electron�electron interaction is computed in the Hartree approx�imation� For the Madelung �eld and the long�range electron�electron interactions�we consider the electronic clouds and the cores as point charges� The Ewald sums areevaluated with the algorithm developed by Monkenbusch ������
There are several possibilities to carry out the self�consistent iterations �see refer�ence ����� and references therein�� We implemented both a simple mixing procedureand the Steensen or Aitken�s �� method ������ At each iteration� the diagonalizationof the Hamiltonian is performed by using the LAPACK libraries ������
The scheme to sample the Brillouin zone is based on special �k points� For metals�we also use the high�precision sampling technique proposed by Methfessel ������ Seeappendix B�
The �tting is performed by minimizing the merit function� This function is de�nedas the sum of the squares of the dierences between physical properties computed bytight binding and any �rst�principles method� The physical properties can range fromHamiltonian eigenvalues �bands� and equations of state� to lattice constants and bulk
���

Set basis vectors
Assign initial orbital occupation
Fitting?
Diagonalization of theHamiltonian
Is chargeself-consistent?
Relaxations?
Fitting?
Done!
Compute Hamiltonian inthe two-center approximation
Assign initialvalues to parameters
Change cell andionic positions
Is the total
its minimum?energy at
yes
no
yes
no
no
function?Minimum of merityes
yes
no
yes
no
yes
no
Change valuesof parameters
Figure ��� Main features of the algorithm used to implement the self�consistenttight�binding total�energy method� The merit function is de�ned as the sum of thesquares of the dierences between the properties predicted by tight binding and theab initio or experimental results used in the �t�
���

moduli� These values can be added in the merit function with dierent weights� Theminimization was carried out using the algorithm proposed by Marquardt ������
This implementation of tight binding allows us not only to compute total energiesand relax structures but also to obtain the adequate parametrizations of the hoppingand overlap integrals�� In the next chapter� we will evaluate the accuracy of theself�consistent tight�binding total�energy formulation by comparing its predictionsto experimental and �rst�principles results�
� Conclusions
The tight�binding formulation presents a physically transparent picture to understandthe formation of a solid from isolated atoms� The dierent contributions to the totalenergy can be clearly identi�ed within the model� This is critical in oxides� wherefailure to account for mechanisms such as oxygen breathing or charge transfer canresult in poor predictions of formation energy values�
In this chapter� we developed a semiempirical tight�binding Hamiltonian thatis suitable for use in oxides� The Hamiltonian matrix elements were described asdistance dependent parametrizations �t to several physical properties� Since some ofthese terms depend on the atomic charges� solutions for this model have to be foundself�consistently�
The semiempirical approach results in signi�cant time savings to compute ma�terials properties� The various time�consuming integrals of the �rst�principles tightbinding are replaced by suitable parametrizations� The name semiempirical can bemisleading� In principle� no experimental results other than the atomic numbers arenecessary as input to the method� However� the parameters of the model have to be�t to ab initio calculations� It is in this sense that the word semiempirical is usedhere�
Note that no radical assumptions have been made about the nature of the bonds�Our approach will deal with ionicity and covalency on equal footing� Furthermore�neither shape approximations for the potential nor arti�cial constraints to the amountof charge assigned to the atoms were made�
There is still the question of accuracy of this approach� The parameters of themodel will be transferable as long as the correct physical eects are included in theHamiltonian� In the next chapter� we will discuss this in detail�
��Di�erent distance dependencies can be used�
���

���

Chapter
Applications of the tight�binding
method
In chapter �� we introduced a tight�binding model that accounts for the key mecha�nisms involved in studies of structural energy dierences in oxides� By parametrizingthe various hopping and overlap integrals� the computational time requirements ofthe method are greatly reduced� Now� we will assess the accuracy of this scheme todetermine formation energies of mixed compounds� Our �nal goal is the applicationof the method to the calculation of phase diagrams�
We will start by revisiting the CaO�MgO system� The simplicity of the systemand the abundance of theoretical and experimental results make it an ideal choice� Byapplying our self�consistent formalism� we will be able to extend the pseudopotentialresults and compute the phase diagram with an accuracy comparable to the mostsophisticated LDA calculations� As far as we know� this is the most accurate calcu�lation ever made on this system� This application clearly demonstrates the power ofour approach�
The transferability of the tight�binding parameters is an important issue andCaO�MgO only represents a limited test of this property since all the structureshave the same underlying lattice� We will therefore also explore the application ofour model Hamiltonian to cases where the atomic arrangements are very dierentfrom the ones used during the �t� Zirconia is ideally suited for this analysis due toits polymorphic transformations from a high�temperature� highly�symmetric �uoritestructure to less symmetric arrangements at lower temperatures� It is also an impor�tant test of our approach since only the most accurate LDA methods provide correctanswers in this system ����� �����
Finally� the ultimate challenge to the self�consistent tight�binding approach is acase where covalency and ionicity are both important in bonding and where chargetransfer is signi�cant� The CaO�ZrO� system is such a case� The formation ener�gies and the charge transfers of many intermediate compounds will be compared topseudopotential predictions�
Section ��� deals with the CaO�MgO system� The phase diagram and single�defect energies will be computed� Section ��� is mainly concerned with the polymor�phic transitions in pure zirconia and the charge transfers and formation energies of
���

mixtures of CaO and ZrO�� The chapter concludes� in section ���� with an evaluationof the possibility of combining parameters of single�metal oxides to predict propertiesof mixtures�
��� The CaO�MgO system
The calculation of a simple phase diagram� such as that of the CaO�MgO system�is currently at the computational time limits of the pseudopotential or FLAPW ap�proach� It is the purpose of this section to show that the tight�binding method canreduce the computational burden of these techniques by interpolating their resultsfor simple structures to more complex cases� As a result� a converged cluster expan�sion is obtained that can be used to determine the phase diagram with an accuracycomparable to the more complex LDA techniques but at a fraction of the time theywould require�
In the following sections� we will assess the accuracy of the tight�binding formationenergies and compute the CaO�MgO phase diagram� At the same time� we willevaluate dierent aspects of the model such as the distance dependence of the hoppingand overlap integrals� the accuracy of universal parameters� the H#uckel theory� andthe �tting process�
����� Predictions of the �universal model
As explained in section ������ universal� coe�cients and distance dependencies canbe assigned to the parametrization of the hopping and overlap integrals� The advan�tage of using them is that no parameters need to be determined from �tting� On theother hand� since they try to reproduce the properties of many dierent systems theirpredictions are not very accurate�
We will use the same distance dependencies proposed by Harrison but the co�e�cients will be taken from references ����� ����� These parameters were carefully�t to reproduce the valence and conduction bands of sp�bonded solids� as is thecase of CaO�MgO� They are reproduced in table ��� together with the intra�atomiccoe�cients and orbital energies� The overlap matrix elements were computed usingH#uckel�s theory with Ki��j� �see equation ����� from reference ������
The formation energies and cell parameters predicted by this model are shown intable ���� The same �k points as in the pseudopotential calculations were used� For allstructures� the solid wave function was expanded in a s�p basis� Although the resultsare qualitatively correct� the formation energies are well below their actual values� Assuggested in reference ����� �tting to the bands is not enough to accurately computestructural energy dierences� The equation of state should also be used�
In what follows� we will not use the universal� parameters� They will only betaken as a guide to select the starting point for the �tting process�
���

a�c c�a a�a c�c Ca Mg O�ss� ����� ����� ����� ����� + + +�pp� ����� ����� ���� ���� + + +�pp� ����� ����� ���� ���� + + +�sp� ����� ����� ���� ���� + + +s�orbital �eV� + + + + ����� ����� ������p�orbital �eV� + + + + ����� ����� ������Ui��i� �eV� + + + + ����� ����� ������
Table ��� Parameters used with Harrison�s distance dependence� The orbital andintra�atomic parameters were taken from reference ������
Cell parameters ��A� Formation energy �eV ion�CaO a����� ������ +MgO a����� ������ +L�� a�b����� ������ * c����� ������ ����� �������L�� �Ca�rich� a����� ����� ����� �������L�� �Mg�rich� a����� ������ ����� �������
Table ��� Formation energies and cell parameters of dierent ordered arrangementsin the CaO�MgO system obtained by using the tight�binding approach with thecoe�cients from table ���� The cells were fully relaxed� The values in parenthesis arethe pseudopotential predictions from table ����
���

����� Fitting to the CaO�MgO system
The semiempirical tight�binding method was originally used to interpolate electronicband structures ������ Consequently� the si��j� and ti��j� matrix elements were usu�
ally �t to ab initio bands at selected points in �k space� To study elastic propertiesand crystal stability� a �t to other properties such as the equation of state is alsonecessary� In this work� we �t to bands and equations of state of CaO� MgO� and theL�� compound �the conduction bands were assigned a lower weight in the �t thanthe valence bands�� As expected� the resulting tight�binding parameters are verydependent on the initial guess and it is not immediately clear where the �t should bestarted�
In multicomponent oxides� charge transfer is one of the key mechanisms involvedin the energetics� Therefore it is very important to have a good description of thecharge density in order to accurately predict formation energies and make the pa�rameters more transferable to other ordered structures� Since one of the main sim�pli�cations of this semiempirical tight�binding model is that the basis functions arenot parametrized independently but as part of the si��j� and ti��j� matrix elements�we can only check that the occupations Qi� resemble the pseudopotential results� In�gure ���� we showed an electronic valence density plot corresponding to the MgO����� plane obtained from the pseudopotential calculations� From this plot� we expectthe cations to transfer most of their valence electrons to the oxygen atoms� Usingperturbation theory� it can be shown that by setting the hopping matrix elementsbetween the orbitals of the anions and the cations to small values� the model willgive occupation values according to the pseudopotential results for the CaO�MgOsystem�� Consequently� we choose the initial parameters to re�ect this fact�
In H#uckel�s theory� the si��j� and ti��j� matrix elements are assumed to be propor�tional� While doing this considerably reduces the number of parameters of the model�we found that for the CaO�MgO system� only a separate parametrization for S andthe o�diagonal terms of H could accurately reproduce the bands and the equationof state simultaneously� We also found that the �t was more stable whenever a penal�ization function was introduced to restrict Qi� to values close to the pseudopotentialones�
The �t tight�binding Hamiltonian reproduces the main features of the three struc�tures used in the procedure� The lattice constants and bulk moduli are� on average�within �� and �� respectively of the pseudopotential values� The valence bands arealso well represented� showing a RMS dierence of a few hundredths of an eV� Seesection ��� for the �t parameters and a comparison of their values across dierentoxide systems�
We tested dierent distance scaling laws for the hopping integrals and the overlapmatrix elements� We tried both the the usual d�� distance dependence for the s�pbonded materials as well as expression ����� This implied a four�fold increase in thenumber of parameters to be determined during the �tting process and considerably
�The softening of the ions the reduction in absolute value of the cation and anion charges is
proportional to�
Hij
Hii�Hjj
�see reference ����� for more details�
���

0.05
0.06
0.07
0.08
0.09
0.1
0.11
0.12
0.13
0.14
0.15
0 2 4 6 8 10
For
mat
ion
Ene
rgy
(eV
/ato
m)
Structure Number
PPTB
Figure ��� Formation energies for dierent ordered structures in the CaO�MgOsystem obtained with pseudopotentials �PP� and tight binding �TB�� The numberthat identi�es each structure corresponds to the one used in table ���� The tight�binding parameters were �t only to L�� �structure number � in the plot� and bothCaO and MgO�
augmented its complexity� Although the �t to bands and total�energy curves im�proved� the predicted formation energies only changed by a few meV� Consequently�for the CaO�MgO system� the simpler d�� scaling law gives enough accuracy�
����� Formation energies
With the parameters obtained in section ������ we computed the tight�binding for�mation energies of �� ordered structures in the CaO�MgO system relaxing all theirstructural degrees of freedom compatible with their symmetry� We used �� specialChadi�Cohen �k points for Brillouin zone integrations for the rocksalt structure� re�sulting in a total�energy convergence better than �meV atom� For all the other
superstructures� we took a set of �k points equivalent to these �� Chadi�Cohen points�see appendix B��
In �gure ���� we compare the results obtained using the pseudopotential andthe tight�binding methods for the ten structures listed in table ���� The agreementbetween both techniques is excellent if we note that� except for L��� none of the
���

structures was used in the �t� The RMS dierence between the two methods is �meV� Consequently� we can expect the tight�binding cluster expansion obtained fromthese values to be very close to the actual pseudopotential expansion�
The relative stability of the dierent ordered structures is also well representedwithin the tight�binding model� From �gure ���� it can be clearly seen that thedi�erences between the predicted formation energies follow the same tendencies as inthe pseudopotential calculations� This is important if the asymmetry of the phasediagram is to be well reproduced�
The tight�binding formation energies for the �� ordered structures can be foundin appendix A� This information will be used in the next section to compute and testthe ECI�s�
����� Phase diagram
The tight�binding formation energies of all the ordered structures studied in theCaO�MgO system are positive� This is consistent with the fact that the experimentalphase diagram shows a miscibility gap� Two main features characterize any miscibilitygap the consolute temperature and the asymmetry of the solubility limits� Beforecomputing the actual tight�binding phase diagram� and as a last test of the accuracyof the model we will make sure that both features are reproduced according to thepseudopotential predictions�
The consolute temperature is mainly determined by the energy scale and� as weshowed in section ������ there is a very good agreement between the methods� Thesolubility limits depend� at low temperatures� on the energy cost of substituting aCaO�MgO� by a MgO�CaO� on an otherwise perfect CaO�MgO� lattice �see sec�tion ������� This energy corresponds to the energy necessary to invert one of theoccupation variables on our lattice model from ������ to ������� Consequently�the solubility is determined� at low temperatures� by the single�defect energy� whilethe asymmetry is driven by the dierence in this value at both end�sides of the phasediagram�
We computed the single�defect energy using a �x�x� fcc supercell�� Using acomplex LDA technique on this cell is prohibitively time consuming and consequently�we compared the tight�binding results to the SSCAD calculations from table ���� Intable ���� we show the single�defect energies for both a pure CaO and a pure MgO hostlattice� The tight�binding defect energies are larger� in agreement with the tendencyof the SSCAD to underpredict formation energies by ������ �see section ����� Notethat there is an important dierence between the defect energy at the MgO� and theCaO�rich sides� This translates into a lower solubility of CaO in MgO than for MgOin CaO �at the same temperature�� This asymmetry is also observed experimentally�see �gure �����
To determine the phase diagram� we computed the ECI�s from the tight�bindingformation energies reported in section ������ We used pairs� triplets� and quadruplets
�In reference ���� we showed that a �x�x� supercell is big enough to avoid signi�cant self�interactions between the defect and its images�
���

Tight�binding SCPIBMgO rich ����� �����CaO rich ����� �����DierenceMgO rich � CaO rich ����� �����
Table ��� Defect formation energies of a single MgO �CaO� on an otherwise pureCaO �MgO� lattice� The values were computed using a �x�x� fcc supercell and areexpressed in eV� The term rich� denotes the host lattice�
as clusters to expand the energy� and obtained a RMS dierence in our cluster expan�sion �t of about � meV per atom� The computed ECI�s are shown in �gure ���� Theagreement between the tight�binding input and the results reproduced by the clusterexpansion is very good� as shown in �gure ���� As a test� we left out a few structuresout of the Connolly�Williams �t� and made sure the cluster expansion predicted theirformation energy within a few meV�
The phase diagram was computed by performing a Monte Carlo simulation� Theresult is shown in �gure ���� The consolute temperature is higher than the extrapo�lated experimental one and the one calculated with the SSCAD method in section ����by approximately ���� The tight�binding results do not include the eect of vibra�tional entropy� Perfect agreement with experiments should therefore not be expected�
An estimation of the eect of vibrations can be obtained from the SSCAD vibra�tional ECI�s of table ���� When they are combined with the tight�binding chemicalECI�s� the resulting phase diagram coincides almost perfectly with the experimentalpoints� Although this might be somewhat fortuitous� it is a clear indication of the corrective� eect vibrations will have�
��� Pure and doped zirconia
Pure zirconia undergoes two transformations from its low�temperature monoclinicphase to its high�temperature tetragonal and cubic arrangements before becominga liquid� The high�temperature phases can be stabilized at lower temperature bythe addition of dopants� such as calcia� As already mentioned in section ������ thereis considerable technological interest in studying these stabilization eects� Experi�ments are usually di�cult to perform due to the high temperatures or the presence ofimpurities and defects� Unfortunately� a good description of the energetics of the sys�tem with ab initio LDA calculations is also out of the question due to the complexityof the crystal structures of the doped zirconias�
The only known mixture of CaO and ZrO� that is stable at room temperatureis CaZrO� �Pcmn structure�� Although this is the simplest superstructure in the
�See section ������
���

-0.025
-0.02
-0.015
-0.01
-0.005
0
0.005
0 2 4 6 8 2 4 21 3 5 7 9 1 3 1
Pairs Triplets Quadruplets
EC
I (eV
)
Figure ��� Eective cluster interactions for the CaO�MgO system computed with thetight�binding formation energies� The pairs are identi�ed according to their lengthand the triplets and quadruplets according to their longest side� the other sides beingnearest neighbors�
���

0
0.02
0.04
0.06
0.08
0.1
0.12
0.14
0.16
0 0.2 0.4 0.6 0.8 1
For
mat
ion
ener
gy (
eV/a
tom
)
Mole fraction MgO
Figure ��� Formation energies as a function of composition for dierent orderedstructures in the CaO�MgO system� The diamonds correspond to tight�binding re�sults and the crosses to the cluster expansion �t�
���
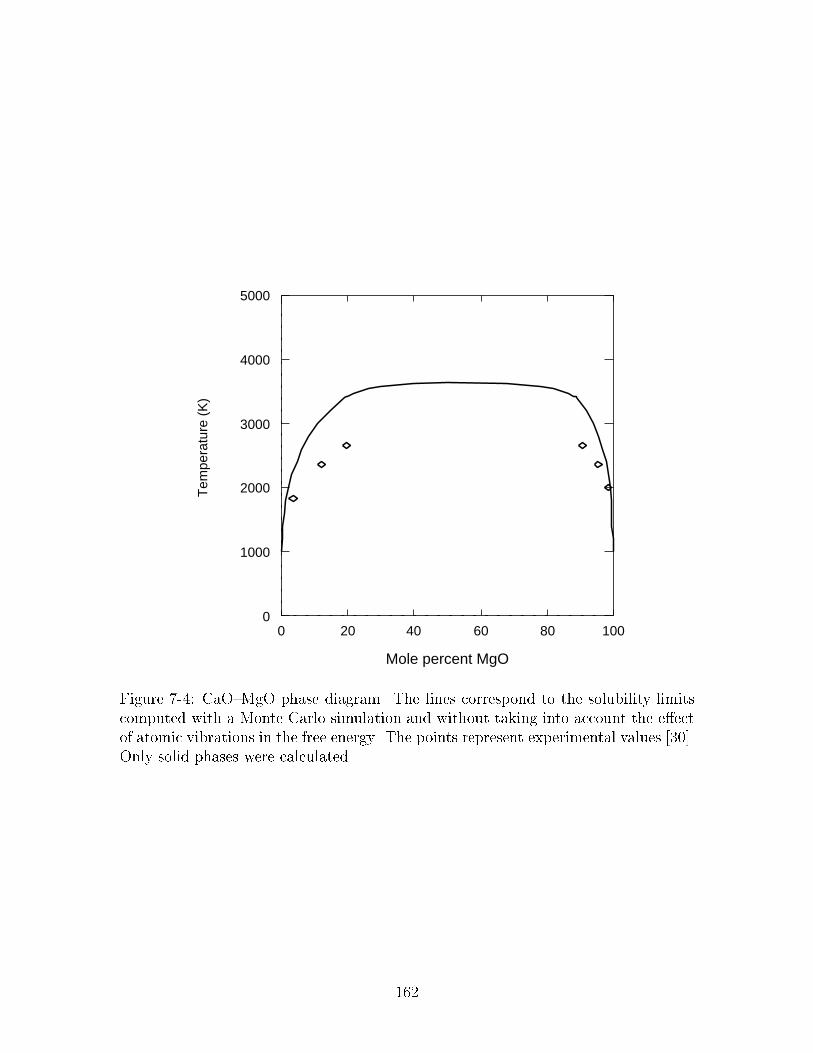
0
1000
2000
3000
4000
5000
0 20 40 60 80 100
Tem
pera
ture
(K
)
Mole percent MgO
Figure ��� CaO�MgO phase diagram� The lines correspond to the solubility limitscomputed with a Monte Carlo simulation and without taking into account the eectof atomic vibrations in the free energy� The points represent experimental values �����Only solid phases were calculated�
���

system� it has a large primitive unit cell with �� atoms� Other compounds� detectedat high temperatures� have even larger cells �for example� Ca�Zr�O���� Fully relaxedpseudopotential calculations for the number of compounds necessary to determine acluster expansion� can not be performed in a reasonable amount of time�
The tight�binding method can be used to overcome these di�culties� We will showthat accurate formation energies can be predicted by this technique in the CaO�ZrO�
system� We will study mixtures of calcia and zirconia within the �uorite lattice so thata direct comparison with the pseudopotential results can be made� The parametersof the model will be �t to the structure S� �see section ������ and both pure CaO andZrO�� The quality and transferability of their values will be tested by computing theformation energies of all the S� structures�
Possible mechanisms for the dramatic failure of potential models in this system�see section ���� will also be explored� This will lead us to investigate how charge isdistributed between the ions�
Before analyzing the energetics of the CaO�ZrO� system we will study the purephases of zirconia� The purpose of this is to evaluate the transferability of the tight�binding parameters under lattice distortions and to assess the accuracy of these pa�rameters when directly used in mixtures �i�e�� without �tting to intermediate com�pounds�� The last point will be discussed later� in section ����
����� Pure zirconia
In section ������ we performed a detailed pseudopotential study of the tetragonalphase� Here� we will use that information to �t the tight�binding parameters� Wecan then predict the lattice constants of the �uorite �cubic� phase and compare theenergy dierence between these structures to the pseudopotential results� This willhelp us to assess the accuracy of our model Hamiltonian under lattice distortions�We will also explore the monoclinic phase though no ab initio calculations have beenreported up to date for this structure�
Rosenbauer and Jansen performed a very accurate Slater�Koster interpolation ofthe tetragonal zirconia bands based on FLAPW calculations� They showed that thevariation of the Slater�Koster integrals with the bond length could not be describedby a simple function ������ These problems are solved within the self�consistentformulation as demonstrated in the next section�
Fitting of pure zirconia
The elements of the overlap matrix and the hopping integrals were �t to the valencebands of pseudopotential calculations for tetragonal ZrO�� The conduction bandswere included with a smaller weight � �
��times� than the valence bands� Signi�cant
departures from the pseudopotential lattice constants were penalized during the �t�
�Note that the coupled cluster expansion mentioned in chapter � should be use here� The cationand anion sublattices have two species each Zr and Ca for the cation one and vacancies and O forthe anion one�
���

In all the calculations� s�p�d and s�p bases were used for zirconium and oxygenrespectively�
To study the problems reported by Rosenbauer and Jansen in their Slater�Kosterinterpolation we �t the parameters in two dierent ways we either kept the cellvolume constant or we changed the lattice constants in a ��� range around theenergy minimum� In this last case� the distance dependence of the hopping andoverlap terms was assumed to be the one proposed by Harrison �see table �����
The RMS of the dierence between the pseudopotential and the tight�bindingbands was �mRy per �k point when the cell volume was kept constant against �mRyper �k point reported in reference ������ Their smaller value is a consequence of theuse of three�center integrals��
When the cell was allowed to change volume� the dierence between the tight�binding and the pseudopotential bands had a RMS value of �mRy per �k point� Thisis a very reasonable value� specially if we take into account that a simple distancedependence was assumed� A polynomial �t will certainly reduce this dierence� Onthe other hand� the error incurred by neglecting charge transfer and only using theSlater�Koster formalism made a good� �t impossible �unfortunately no numberswere given in reference �������
In what follows� we assume the distance dependence proposed by Harrison� Forthe tetragonal and cubic phases� the same �k points as in the pseudopotential case wereused� For the monoclinic structure� a �x�x� uniform grid �plus symmetry� was used�The intra�atomic zirconium parameters were obtained by applying the procedure inreference ����� �see section ������� Similar to the CaO�MgO case� the same Ui��i� wasassumed for all the orbitals �i�e�� an average over the relevant orbitals�� The valueused was ����� eV�
Pure�zirconia results
The computed cell parameters for the dierent structures and the correspondingenergy dierences are shown in table ���� The experimental� pseudopotential� andpair�potential results from table ��� are also reproduced here for comparison� Thecubic and tetragonal structures were fully relaxed� Since only the tetragonal bandswere used in the �t� the energy dierence between the tetragonal and cubic phases is aprediction� This prediction is in excellent agreement with the pseudopotential values�The lattice constants and internal cell parameters are also in very good agreement�
Note that the monoclinic structure has �� crystallographic degrees of freedom�We optimized all of them except for the cell angle not �xed by symmetry which washeld constant at ��o� The experimental ��o angle will place the atoms so that theintegrals will have to be evaluate beyond the region of validity of the �t parameters�Furthermore� the simple Harrison distance dependence would have to be replacedby a polynomial function times an exponential decay� We decided not to pursue this�tting since we would need � times the number of parameters currently used and con�
�The typical error in the total�energy �t is much smaller than for the bands� It is usually of theorder of ���mRy�atom�
���

Expt�a Pot�b TB PP
Cell Parameter
Cubic ZrO�
a�b�c ����� ����� ����� �����Tetragonal ZrO�
a�b ����� ����� ����� �����c ���� ����� ����� �����z�O� ����� ���� ����� �����Monoclinic ZrO�
a ������ ����� ����b ������ ����� ����c ������ ����� ����� ������ ���� ����c
x��Zr� ������ ���� �����y��Zr� ������ ��� �����z��Zr� ������ ������ �����x��O� ������ ������ �����y��O� ������ ������ �����z��O� ������ ������ �����x��O� ������ ������ �����y��O� ������ ������ �����z��O� ������ ������ �����Energy dierences
Ecubic�Etetragonal ����� ����� ����� �����Etetragonal�Emonoclinic ����� ����� �����
Table ��� Cell parameters and structural energy dierences for the experimentallyobserved phases in zirconia at zero pressure� The energies are in eV per ZrO� formulaunit and the lattice constants are in �A� All cell parameters were fully relaxed� includingthe internal positions� xi� yi� and zi� that are shown here according to the Wyckonotation in reference ������ The experimental z value for the tetragonal structurewas measured at ���� oC and the cell parameters are extrapolations to � oK �seechapter ���
aSee reference ���� ��� ����bSee reference ���� ����cThe � angle of the monoclinic structure was �xed� See text�
���

sequently more pseudopotential calculations for the tetragonal phase� However� theresults are important� since the monoclinic phase� even not fully relaxed� is predictedto have a lower energy than the tetragonal phase in agreement with the experimentalobservations�
����� Calcia�doped zirconia
Fitting to the CaO�ZrO� system
The solid wave functions are expanded in atomiclike �s and �p orbitals localized atthe oxygen ions� �s and �p centered at the calcium ions� and �s� �p� and �d for thezirconium ions� The same �k points as in the pseudopotential calculations were usedhere�
The initial parameters for the �tting process were taken as the CaO and ZrO� setsfound in sections ����� and ����� �the O�O being an average of both results�� For theCa�Zr second nearest neighbors coe�cients� only the tsd� integrals were included��
The integrals were assumed to obey Harrison�s distance dependencies of table ����We independently �t the overlap and hopping integrals �no H#uckel�s approxima�
tion� to the bands and lattice constants of the S� structure and pure CaO and ZrO��Both the valence and the lower levels of the conduction band were used� For thelatter� a much lower weight ������ in the merit function was given�
The root�mean square dierence between the pseudopotential and the tight�binding bands was of the order of a few hundreths of an eV� On the other hand�the lattice constants diered in less than �����
Tight�binding predictions for the CaO�ZrO� system
In �gure ���� the formation energies of structures S� to S� computed by the tight�binding approach are shown� As a reference� the pseudopotential and pair�potentialpredictions are also displayed� To be able to relate these results� only volume re�laxations were allowed� The average dierence between the pseudopotential and thetight�binding values is approximately ���� Although the bands for the structureS� were used in the �tting process� its formation energy can be considered as a realprediction� since neither this value nor the S� equation of state was used in the �t�
The predicted lattice constants are also in good agreement� Table ��� shows thecomputed values for structures S� to S��
Note that the self�consistent calculations considerably stabilize the structurescompared to the pair�potential model� The formation energies predicted by the latterare always higher� For example� for the S� structure it is more than three times theself�consistent values� These dierences are much larger than the ones previouslyobserved in the CaO�MgO system �see section ����� We believe this is caused by thevariation of ionic charges with dierent atomic environments� This extra degree offreedom is absent in the potential models�
The situation is clearly observed in �gure ��� where we show the charges predicted
�We also tested adding s�s and s�p integrals but the results did not improve appreciably�
���

0
0.5
1
1.5
2
2.5
3
3.5
0 1 2 3 4 5 6
For
mat
ion
Ene
rgy
(eV
/ato
m)
Structure Number
PPTBPot
Figure ��� Formation energies of structures S� to S� computed with tight binding�TB�� The pseudopotential �PP� and pair�potential �Pot� predictions computed insections ����� and ��� are also shown for comparison� A description of the structurescan be found in �gure �����
Structure Cell Parameter ��A�Pseudopotentials Tight binding Pair potentials
S� ����� ����� �����S� ����� ����� �����S� ����� ����� �����S� ����� ����� �����S� ����� ����� �����
Table ��� Cell parameter for structures S� to S� computed by pseudopotential� tightbinding� and pair potentials� The structures are described in �gure �����
���
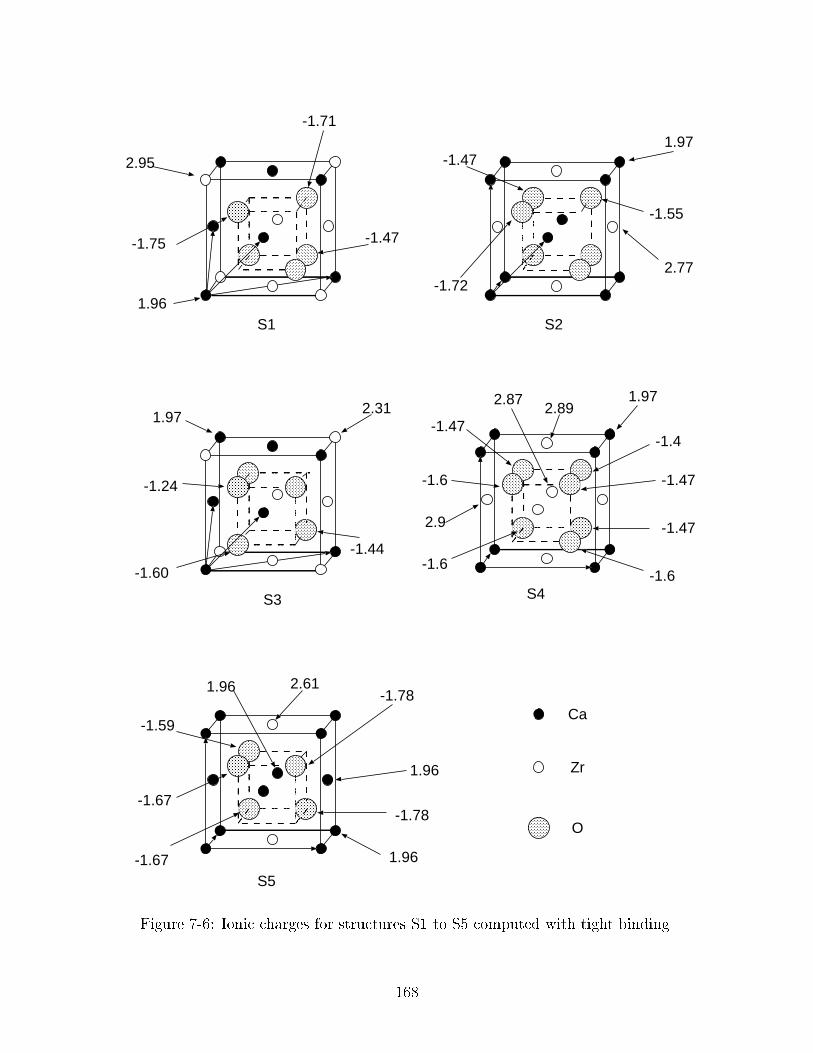
Ca
Zr
O
S2
1.97
2.77
-1.55
-1.47
-1.72
S11.96
2.95
-1.47
-1.71
-1.75
S4
1.97
-1.4
-1.6
-1.47
-1.47
-1.47
-1.6
-1.6
2.9
2.872.89
S3
1.972.31
-1.60
-1.24
-1.44
S5
-1.78
-1.78
-1.59
-1.67
-1.67
2.61
1.96
1.96
1.96
Figure ��� Ionic charges for structures S� to S� computed with tight binding�
���

by tight�binding for dierent structures in the system� In agreement with the pseu�dopotential results� calcium is fully ionized in all the structures� On the other hand�zirconium and oxygen considerably change their charge from one structure to another�Furthermore� within the same compound� they have dierent values according to thesymmetry of the site�
In the CaO�MgO case� both calcium and magnesium kept their formal charge inall the dierent atomic arrangements in the structures� Here� oxygen and zirconiumchange their charges according to these arrangements� The larger the variations�the larger the errors in the pair�potential predictions� For example� structure S�presents the worst agreement between pair potentials and the self�consistent methods�Correspondingly� the oxygen and zirconium ions have the largest charge variationswithin dierent cell sites ����� electrons��
As a check of the tight�binding results we integrated the pseudopotential electronicdensity within spheres centered at the dierent oxygen sites �the sphere radii werethe same in all cases�� Again� signi�cant dierences were detected� The maximumvariations were of the order of ��� electrons�
For a potential model to work properly� there is a clear need for a way of intro�ducing charge transfer mechanisms� This will be analyzed in chapter ��
��� Tight�binding parameters from �ts to end
members
In this section� we will determine the accuracy of tight�binding parameters �t onlyto physical properties of the end members� If two binary oxides AOx and BOy aremixed� all the hopping and overlap two�center integrals can be obtained from the pureoxides except for the ones involving the cations A and B� As explained in section ������we can either disregard the missing cation�cation interaction or approximate it usingdierent assumptions such us the one suggested in reference ����� �see equation ������
Here� we will focus on binary oxides mixtures� We will start by studying theeects of the dierent approximations in the CaO�ZrO� system� Four cases will beanalyzed in detail
Case � The parameters of the model are taken from the �ts to CaO�MgO andZrO� in sections ����� and ������ Since the O�O integrals are �t twice� once for eachsystem� we use the one from CaO� No Zr�Ca overlap or hopping terms are considered�
Case � The same as case � but now the O�O parameters are taken from the ZrO�
�t�
Case � The same as case � and case � but an average of the two available sets ofO�O integrals is used�
�Another concern is the transferability to the mixtures of the anion�anion and anion�cationparameters �t to the end members�
���

Universal CaO�MgO CaO�ZrO�
�pO�pO�� ����� ����� ������sO�sCa�� ����� ����� ������sO�pCa�� ����� ����� ������pO�sCa�� ����� ����� �����
Table ��� Set of hopping integrals that were �t in both CaO�MgO and CaO�ZrO��The universal parameters found in reference ����� are reproduced for comparison�
Case The same as case � except that the Zr�Ca parameters are not assumed tobe zero� The s�d integrals are approximated by the Zr�Zr ones� We also tested theZr p�d values and geometrical averages of Ca s�s and Zr d�d with similar results�
Note that cases � to � not only test the eects of absent Zr�Ca integrals but alsothe transferability of the O�O parameters� In a perfect situation� there should not beany dierence between them� However� table ��� shows how the parameters changewhen they are �t to dierent systems �in this case CaO�MgO and CaO�ZrO��� Case�� evaluates the eect of dierent approximations for the Zr�Ca hopping integrals�
The results are shown in �gure ���� The predicted formation energies are comparedto the pseudopotential �PP� values� The pair�potential �Pot� estimations are alsoincluded since it is the only practical method that can be currently used to computeformation energies of many large cells in the system� This will provide a standard towhich the tight�binding predictions can be fairly compared�
In all four cases� the tight�binding predictions are much closer to the pseudopo�tential values than the pair�potential models� In this sense� by building a table of twocenter integrals from �ts to binary oxides� the tight�binding method can be almost assimple to use as a potential model� Fits to binary oxides are usually straightforward�
However� cases � to � also show that the transferability of the parameters is notoptimum� By using the O�O integral from dierent �ts� dierences of the order of ���are obtained� Although the dierence is large� it is still of the order of the errors in theformation energies introduced by neglecting the cation�cation parameters and muchbetter than the potential predictions� We further explored this issue by re�tting bothpure oxides with a common oxygen�oxygen set of parameters �we took the averageused in case ��� This worsened the predictions of the end members by a few percentwithout signi�cantly changing the results for the mixtures�
By �tting to more systems it might be possible to obtain a better set of O�Oparameters� However� approximations of the tight�binding formalism� such as theuse of two�center integrals� certainly map their eects onto the parameters duringa �t� As it was shown in the previous sections� this does not constitute a problemwhen the �t is done to a particular system� Furthermore� only if a better descriptionof the missing cation�cation integrals were used would it be worthy to correct these
���

S1 S2 S3 S4 S5
StructureF
orm
atio
n en
ergy
(eV
/ato
m)
S1 S2 S3 S4 S5
Structure
For
mat
ion
ener
gy (
eV/a
tom
)
S1 S2 S3 S4 S5
Structure
For
mat
ion
ener
gy (
eV/a
tom
)
S1 S2 S3 S4 S5
Structure
For
mat
ion
ener
gy (
eV/a
tom
)
PP
Case 1
PP
Case 2
PP
Case 3 Case 4
PP
0
0.5
1
1.5
2
2.5
3
3.5
0
0.5
1
1.5
2
2.5
3
3.5
Pot
0
0.5
1
1.5
2
2.5
3
3.5
Pot
0
0.5
1
1.5
2
2.5
3
3.5
Pot Pot
Figure ��� Formation energies predicted for dierent mixtures of CaO and ZrO� forthe four cases explained in the text� A description of structures S� to S� can be foundin �gure �����
���

approximations� The error introduced by not considering these integrals is still larger�Case � indicates that the dierent estimations of the Zr�Ca integrals do not im�
prove the results� However� our results show that their eect on the formation energiescan be important� When compared to pseudopotentials� they can introduce errors ofthe order of ����
It is important to note that the parameters used in the � cases do not correspondto straightforward �ts to the end members� The Ca�O and Ca�Ca parameters forexample� were obtained from a �t to the CaO�MgO system and not just CaO� Thisrestricted the space of possible parameters considerably� When CaO and MgO are �tindependently� many dierent sets reproduce the pseudopotential results with similaraccuracy� It is only when the L�� structure is used that one set was selected� whichwas the one used here� Consequently� enough restrictions� in the �ts should be used�By restrictions� we mean either �tting to mixtures or using dierent deformationsof the unit cell��
Finally� even when a detailed �t to a given system is pursued� the combinationof the end�member parameters can be used as a starting point� They have provenduring the CaO�ZrO� �t to be much closer to the �nal parameters than the universalvalues�
��� Conclusions
We have shown that the semiempirical self�consistent tight�binding formalism repre�sents an intermediate method between simple potential models and highly�accurateLDA techniques for studying the energetics of oxides� It retains the high computa�tional speed of the former� but at the same can correctly interpolate very accurateLDA results� The computation of the CaO�MgO phase diagram is an excellent exam�ple of this� In this system� the pseudopotential results for only three structures wereemployed to predict formation energies of many more compounds� This informationwas then used to compute a converged cluster expansion from which the solubilitylimits of the system were obtained�
Therefore� the semiempirical tight�binding method oers a clear advantage forinvestigations in which the energies of many ordered structures� in the same system�have to be computed� Such is the case in the ab initio prediction of multicomponentoxide phase diagrams� Although a few computing�intensive LDA calculations haveto be performed to �t the tight�binding parameters� the result is a transferable total�energy Hamiltonian that can be used on complex and large unit cells� This makes itan excellent extrapolation tool� considerably reducing the computational burden forphase stability calculations�
The excellent transferability properties of the tight�binding Hamiltonian weredemonstrated not only in the CaO�MgO system but also in the more demanding
�The deformations of the unit cell do not necessarily have to be compatible with the symmetry�For example atomic displacements that allow a sampling of many di�erent integrals as a functionof the full range of distances between ions can be used�
���

CaO�ZrO� system� Most of the approximate total�energy methods assume eithercovalent or ionic bonding� Consequently� systems such as CaO�ZrO� present anextremely di�cult case for them� For example� the SSCAD and pair potentials failedto reproduce nonspherical electronic relaxations and charge transfers respectively�On the other hand� the tight�binding model predicts very accurately the energydierences between the pure tetragonal and cubic phases and the formation energiesof calcia�doped zirconias� The incorporation of the relevant physical mechanismsfound in chapter � was essential for the Hamiltonian to be transferable�
The eectiveness of the method depends critically on the �tting process� This isclearly a crucial and time�consuming step� Therefore� physical insight should be usedto obtain reliable results� Fitting the end members of the system is usually simplerthan dealing with the mixtures� Using just these parameters� disregarding some ofthe cation�cation hopping and overlap integrals� proved to be more accurate thanemploying pair potentials� However� the overall accuracy ����� error� is too low tobe used for quantitative predictions and a full �t should be performed�
The general accuracy of the tight�binding formalism can be improved by using amore complex distance dependence in the parametrization of the hopping and overlapterms� In all this chapter� we have just used the one�coe�cient Harrison distancedependence� To deal with major distortions of the structures �with respect to theones �t to� a more complex function should be used� This will result in a signi�cantincrease in the number of parameters to �t� In the CaO�MgO case� this produceda better description of the bands but only a marginal improvement in the formationenergies� Total energies are integrated quantities that do not depend on the exactdetails of the bands� However� in cases such as the monoclinic ZrO� phase� usingmore parameters seems to be unavoidable�
Other improvements of the tight�binding formalism� such as the use of three�center integrals or a Hartree�Fock instead of a Hartree description of the electron�electron interactions� should also be considered�
The doped�zirconia results showed a signi�cant variation of the ionic chargesbetween and within dierent structures� The speed of the tight�binding formulationwill let us� in the next chapter� perform large cell calculations to further understandthis behavior�
���

���

Chapter
Charge transfer in oxides
Many approximate total�energy methods work with point charges instead of the fullelectronic density� Small changes in their values can have an important eect on theenergy� in the scale we are interested in�
While in sophisticated quantum�mechanical techniques the electron density iscomputed by solving Schr#oedinger�s equation self�consistently� a simple formulationto �nd the point charges within approximate methods is still lacking for oxides� Tradi�tionally� the latter methods overcame this di�culty by simply assuming �xed chargesfor the ions� independently of their atomic environments� Unfortunately� as it wasshown in this work� the above simpli�cation can introduce large errors in the forma�tion energy �see chapter ���
In this chapter� we will describe how the ionic charge in an oxide changes withdierent atomic arrangements on a lattice� Using this information� simple rules tomodel the variation of the charge will be tested� We will perform these studies incalcia�doped zirconias� As shown in chapter �� they exhibit a strong dependence of theionic charge on the environment� The charges will be extracted from a self�consistentcalculation in a large �uorite supercell� We chose this structure to maximize thenumber of dierent atomic neighborhoods in a single calculation�
In order to relate point charge models to actual self�consistent electronic densities�a procedure for extracting point charges from them should be de�ned� The electroniccharge density is usually partitioned into domains of dierent shapes �e�g�� spheres�polyhedra� etc�� and integrated within each of them to obtain the correspondingcharges� This procedure is clearly not unique� However� if a consistent way of de�ningthe charges is adopted� di�erences in charge transfer can be studied� Here� we willuse either the mu�n�tin approximation or the projection of the solid wave functionsonto atomiclike basis functions to de�ne the ionic charges as explained in the nextsections�
In metallic alloys� point charges extracted from LDA calculations can be very wellreproduced with a very simple analytic model ����� ����� In this scheme� the chargeon a given ion only depends on the number of unlike neighbors close to that ion�Here� we will explore if a similar set of rules can be applied to oxide systems� wherelarge charge transfers and long�range interactions are present� This will involve thestudy of the relationship between the charge on a given site and the local environment
���

and Madelung �eld for that site� If successful� a simple formulation such as the oneused in metallic alloys can considerably improve the performance of potential models�specially if they are used in conjunction with the compressible�ion model describedin chapter � to account for the ionic breathing�
The self�consistent tight�binding method described in chapter � will be used tocompute the charges� It is the only quantum�mechanical technique capable of han�dling� in a reasonable time� the large supercell calculation �more than ��� atoms�required in this study�� We already showed that this method accurately reproducedformation energies in doped zirconias� Since the accuracy of these predictions criti�cally depends on the correct description of the ionic charge� it is reasonable to assumethat the tight�binding model will also provide correct values for the charges�
We will start� in section ���� summarizing the results obtained for binary metallicalloys� Then� in section ��� we will use the self�consistent tight�binding formulationto study the variation of charge in oxides� Finally� in section ������ we will discussour �ndings and evaluate the accuracy of simple point�charge models�
�� Simple charge model for metallic alloys
Mott suggested early on that the charge of an atom in a solid depends on the numberof chemically similar atoms by which it is surrounded ������ The charge is expectedto have the smallest absolute value when all the neighbors of the ion are of its samespecies� The justi�cation is that if the system is screened enough� an atom surroundedby like neighbors will not know� that it is not part of a pure elemental solid but ofan alloy and consequently� its charge will be close to zero�
The idea was used by Magri and collaborators to express the charge in a binaryalloy as a linear function of the number of unlike atoms on the nearest neighborshell ������ This was later extended to any number of shells by Wolverton et al� ������They expressed the charge� qi� at site i as�
qi � ��Xs
�s��
Xks
�Si � Sks � �����
where �s are constants and the �rst sum goes over the desired number of shells� Si is���� if an A�B� atom is located at site i and the last sum is over all sites in the shells� Note that the latter sum counts the number of unlike neighbors in the s shell� Ageneralized number of neighbors� �N is de�ned so that qi is proportional to it�
�N �Xs
�s��
Xks
�Si � Sks� � �����
It was shown in reference ����� that the same �s are extracted from small� and large�
�Another option would be to perform many small cell calculations� Still to have a similar numberof di�erent atomic neighborhoods to the one provided by a large supercell a prohibitively large setof small structures should be used�
���

unit�cell �with a random distribution of the atoms� LDA calculations� This is a clearindication that similar mechanisms underlay the transfer of charge for both orderedand random alloys� These coe�cients depend on the volume �and thus implicitly onthe composition� for lattice mismatched alloys�� See reference ����� for more details�
Expression ��� was tested in large supercell LDA calculations �������� atoms� forthe Cu��xZnx random alloy ������ The total charge at site i was de�ned in terms ofthe mu�n�tin approximation used in the evaluation of the Coulomb �eld�
qi � ��Z Ri
MT
��iMT �r�r
�dr � ��
��i � ��
��Ri
MT ���� Zi �����
where RiMT is the mu�n�tin radius used for the atom at site i� �iMT is the spherically�
symmetric electronic density within the mu�n�tin sphere� �� is the constant electronicdensity in the interstitial� �i is the volume of the Wigner�Seitz cell surrounding eachsite� and Zi is the nuclear charge�
The accuracy of equation ��� depends on the number of shells considered� Takingthe �rst two �three� nearest neighbors shells was enough to reproduce the LDA resultsfor a fcc �bcc� lattice very closely�
The supercell calculations also showed a remarkably linear relation between thecharges associated with a given site and the long�range electrostatic potential at thatsite ������ This behavior can be predicted from the simple charge model when enoughneighboring shells are considered ������
These results show that even for systems where small charge transfers are ex�pected� the charges on dierent crystallographic sites will dier according to theatomic environment� In the next sections� we will study these mechanisms whenlarge charge transfers are expected�
�� Charge transfer in oxides
The variation of the ionic charge in oxides within and along dierent structures wasclearly demonstrated in calcia�doped zirconia �see section ������� Here� we will in�vestigate if the simple model described in section ��� is still valid for this system� inspite of the fact that large charge dierences are expected between dissimilar crystal�lographic sites�
Similar to what was described in section ���� we performed a large �uorite supercellcalculation� The zirconia and calcium atoms were randomly distributed on the cationsublattice� Vacancies and oxygen atoms were randomly placed on the the anionsublattice� A similar number of calcium and zirconium ions was used� In this way�many dierent crystallographic sites and local environments could be sampled in asingle calculation� The number of vacancies was selected to ensure charge neutralitywhen formal charges are assigned to the ions�
The point charge model� with two �rst neighboring shells� in equation ��� can beextended to this system in the following way�
���

qi�Zr� � ��NCa � ��NO � CZr
qi�Ca� � ���NZr � ��NO � CCa
qi�O� � ���NZr � ��NCa � ��NO � CO �����
where �i and C� are constants� and N� is the number of � neighbors �� � Ca� Zr� orO�� To preserve charge neutrality the coe�cients C� should be related by�
xZrCZr � xCaCCa � xOCO � �� �����
Here� xZr� xCa� and xO indicate the atomic fraction of zirconium� calcium� and oxygenions respectively� Equations ��� can be written to include any number of shells�
Both a �x�x� ���� sites� and a �x�x� ��� sites� �uorite supercell were used in thestudy� If the charge on a given site is mostly determined by local eects the resultsshould be similar in both cases�
The same tight�binding parameters as in section ����� were used in the calcula�tions� For �k�space integrations� we tested both a �x�x� uniform grid and the ) pointin the �x�x� supercell calculation� The charges were already converged within ����electrons in the latter case� Consequently� all the calculations were done with the )point�
The charge was de�ned �equation ����� as the projection of the electron wavefunction in the solid onto the basis orbitals� Note that it is the same charge used tocompute the Madelung energy within the tight�binding model�
����� Charge transfer in calcia�doped zirconia
A generalized number of neighbors for each ion can be de�ned from equations ����
qi�Zr� � ��
�NCa �
����NO
�� CZr
� �� �NZr � CZr
qi�Ca� � ����NZr � ��
��NO
�� CCa
� ��� �NCa � CCa
qi�O� � ����NZr �
����NCa � ��
��NO
�� CO
� ��� �NO � CO� �����
The charges for the dierent ions can then be expressed as a function of these �N �If we do not enforce charge neutrality� each of the equations ��� can be �t inde�
pendently providing the best linear match to the data set� The results are shown in�gure ��� and ��� as a function of �N for two and four neighbor shells respectively�i�e�� one cation and one anion shells in the �rst case� and two anion and two cation
���

shells in the second�� Relation ��� predicts the straight line plot in the �gures�Clearly� even by �tting the equations ��� independently� the dispersion in the valuesis very important� The variation is of the order of the change in the ionic chargereported in section ������ As we mentioned� this variation introduced large errors inthe formation energies�
The �t � parameters change from the �x�x� to the �x�x� �uorite supercell calcu�lations� although the qualitative behavior shown in �gures ��� and ��� is the same�For example� �� changed from ����� ���� to ������ ����� from the smallest to thelargest supercell� This is an indication that the eects that determine the chargeare not entirely local or that the dependence on the local environment goes beyondsimply counting the number of unlike neighbors��
When the charge neutrality constraint is imposed� all the coe�cients in equa�tion ��� have to be determined simultaneously� The �t is clearly worse� as shown in�gure �����
The dependence of the charge at a given site on the Coulomb potential at thatsite is shown in �gure ����
����� Discussion
The results of section ����� clearly show a strong dependence of the ionic chargeon its environment� Dierences of more than one electron occur in our supercells�Consequently� potential models that assume �xed charges are bound to fail sincelarge errors in the Coulomb energy can be expected� The variation of charge withstructure in zirconias has also been suggested by other experimental and theoreticalwork ����� �����
The calcium ions are shown to be almost completely ionized� independently of thestructure or the symmetry of the site that they occupy� This behavior was con�rmedby the pseudopotential calculations� Electronic density plots with virtually zero elec�trons in the Ca ions� such us the one shown in �gure ����� can be obtained for eachof the S structures�
Unfortunately� �gures ��� and ��� show that a simple description of the variationof the charge such as the one used in metallic alloys is not possible� The dispersionof the charges around a linear behavior on the number of unlike neighbors exceeds��� electrons even when � shells of neighbors are accounted for� If the scheme wereto be used to predict ionic charges within a total�energy model� large errors in theformation energies should be expected�
The charge shows a perfectly linear behavior with respect to the Coulomb potentialfor small values on the oxygen and zirconium ions� The calculations for the Cu��xZnxrandom alloy� mentioned in section ���� also showed this behavior �only small charges
�One possible improvement is to look at the particular arrangements of the atoms around thesite where the charge is computed�
�The predictions for the oxygen charges are closer to the actual values� This is just an artifact ofthe �tting process� There is more than twice the number of oxygen ions than the number of calciumor zirconium ions in the unit cell� Since equal weights were given to all data points the oxygen dataset was implicitly weighted more�
���

0
0.5
1
1.5
2
2.5
3
3.5
4
3 4 5 6 7 8 9 10 11
Ca
N~
Cha
rge
-4
-3.5
-3
-2.5
-2
-1.5
-1
-0.5
0
1 2 3 4 5 6 7 8
O
N~
Cha
rge
0
0.5
1
1.5
2
2.5
3
3.5
4
2 3 4 5 6 7 8 9 10
Zr
N~
Cha
rge
Figure ��� Charge �in electrons� versus generalized number of neighbors �N � Thestraight line is the prediction of the charge model of equation ��� when two neighborlayers are included� The � parameters were �t without imposing charge neutrality�The dots correspond to tight�binding large�supercell calculations�
���

0
0.5
1
1.5
2
2.5
3
3.5
4
5 6 7 8 9 10 11 12 13
Ca
Cha
rge
N~
-4
-3.5
-3
-2.5
-2
-1.5
-1
-0.5
0
2 3 4 5 6 7 8
O
Cha
rge
N~
0
0.5
1
1.5
2
2.5
3
3.5
4
2 3 4 5 6 7 8 9
Zr
Cha
rge
N~
Figure ��� Charge �in electrons� versus generalized number of neighbors �N � Thestraight line is the prediction of the charge model of equation ��� when four neighborlayers are included� The � parameters were �t without imposing charge neutrality�The dots correspond to tight�binding large�supercell calculations�
���

0
0.5
1
1.5
2
2.5
3
3.5
4 6 8 10 12 14 16
Ca
N~
Cha
rge
-4
-3.5
-3
-2.5
-2
-1.5
-1
-0.5
0
2.5 3 3.5 4 4.5 5 5.5 6 6.5 7 7.5
N~
Cha
rge
O
0
0.5
1
1.5
2
2.5
3
3.5
4
3 4 5 6 7 8 9 10 11 12
N~
Cha
rge
Zr
Figure ��� Charge �in electrons� versus generalized number of neighbors �N � Thestraight line is the prediction of the charge model of equation ��� when two neighborlayers are included� The � parameters were �t imposing charge neutrality� The dotscorrespond to tight�binding large�supercell calculations�
���

0
0.5
1
1.5
2
2.5
33.5
4
-30-25-20-15-10
Cha
rge
Coulomb Potential
Ca
-4
-3.5
-3
-2.5
-2
-1.5
-1-0.5
04 6 8 10 12 14 16 18 20 22 24 26
Cha
rge
Coulomb potential
O
0
0.5
1
1.5
22.5
33.5
4
-40-35-30-25-20-15-10-505
Cha
rge
Coulomb potential
Zr
Figure ��� Charge �in electrons� at site i versus Coulomb potential at that site� Thelines indicate the limiting behavior for small and large charge transfer�
���

were involved in these studies�� For large �elds� the charge levels o at the formalvalence charge� i�e�� �� and �� for Zr and O ions respectively� This is expected sincethose values correspond to electronic shell levels completely full or empty��
Figure ��� also shows a much smaller dispersion of the ionic charge as a functionof the Madelung �eld than as a function of �N � There is almost a one�to�one cor�respondence between charge and Coulomb potential for the zirconium atoms� Thesame is true for oxygen up to an absolute charge of ����� For larger values� thedispersion starts increasing considerably� The second electron that is added to theoxygen atom is not stable if the ion is isolated� It is stabilized in the solid by theMadelung �eld� Consequently� we can expect that this loosely bounded electron willbe more dependent on the particular local environment of the ions� We believe thiseect is responsible for the increase in the dispersion of the values of the oxygencharges beyond the ��� electron absolute charge�
�� Conclusions
The self�consistent supercell calculations performed in this chapter clearly show thedependence of the charge of an ion on its environment� Our results indicate thatthe simple local rules used in metallic alloys to describe this dependence can not bedirectly applied to oxides� Even by considering four neighbor shells in the analysis�the dispersion in the predicted charges can introduce large errors if they are used tocompute formation energies�
A combination of both a local and a long�range formulation may be necessary toproperly describe charge variations in oxides� A more careful treatment of the localrules can also be implemented� As suggested in reference ������ the speci�c atomicarrangements around the ion� or nonlinear dependencies with the number of unlikeneighbors can be considered�
An almost perfect one�to�one dependence between Coulomb potential and chargewas found for the zirconium ions� For small charges� this relation is linear� in agree�ment with previous calculations in metallic alloys ������ The same holds for theoxygen ions� though a wider dispersion in the values was found� We believe the lattereect is related to the instability of the O�� ion� Calcium ions were fully ionizedindependently of the particular environments�
Apart from the simple point�charge model described in section ���� there is an�other non�self�consistent formalism in the literature �due to Streitz and Mintmire�that incorporates charge transfer eects� It was described in section ��� and is basedonly on the minimization of the electrostatic energy� While the point�charge model is�t to reproduce self�consistent LDA results� quantum�mechanical eects are absentin the Streitz�Mintmire approach� We have not tested this last scheme since ourtight�binding formulation is more general and not much more complex to implement�
In summary� the relation between the charge of an ion and the arrangement of its
�The Coulomb potential would have to be increased decreased considerably more in order tostart adding removing electrons to from the following previous electronic shell�
���

neighbor atoms can not be captured with simple local rules in oxides� The variation ofthe charge can be large and it is essential to incorporate these changes into any total�energy formalism� Compared to other models in the literature� the self�consistenttight�binding formalism oers the best alternative to account for these eects withoutusing complex quantum�mechanical methods�
���

���

Chapter �
Conclusions
The period of time we are now living will likely be recognized as the ComputerAge� Although materials will probably no longer be used directly to name the agesof history� the innovations that a deeper knowledge of their properties bring� willcertainly be� This shift from materials to applications is tied to the fact that we donot need to take the properties of materials as given but that we can shape them toour needs�
Actually� there is a fruitful relationship between materials and computers� Notonly can more powerful computers be developed from a better control and under�standing of materials properties� but also computer simulations can now help to de�sign better materials� In this thesis� we focused on how to improve the computationaltechniques in order to make their predictions more reliable in oxides�
At the basis of most computational investigations lies a model to describe theenergetics of the system since nearly all materials properties can be related to dif�ferences in total energies� Energy values can be determined from the solution ofcomplex quantum�mechanical equations� In principle� the only input they require isthe atomic numbers of the species that form the system to be studied� By couplingthese methods to statistical�mechanical approaches� the thermodynamical propertiesof materials can be assessed� From these properties� essential information to processmaterials e�ciently can be obtained�
The complexity of oxides has prevented the widespread use of state�of�the�arttechniques to solve the quantum�mechanical equations� A tradition to use very sim�ple total�energy models in these systems exists� One conclusion of this work is thatthese simple models do not incorporate all the relevant terms to accurately predictformation energies on the scale of thermodynamical eects �i�e�� tens of meV atom��For example� the CaO�MgO phase diagram we computed using pair�potential ap�proaches agrees only qualitatively with the experimental observations� A miscibilitygap with the correct asymmetry was predicted using these rudimentary models� butthe solubility limits were considerably o�
On the other hand� we showed that very complex and computationally demandingmethods� such as pseudopotentials or FLAPW can provide an accurate description ofthe energetics of oxides� We found very good agreement between their predictions andexperiments whenever the local�density approximation to density�functional theory
���

was employed�Still� the use of these sophisticated methods is limited to very simple cases� The
computation of phase diagrams requires the determination of formation energies formany dierent mixtures of the end members� Some of these compounds� such as indoped zirconias� can have more than �� atoms in a unit cell with very low symmetry� Afew of these calculations can be performed with the help of supercomputers� However�the computational resources necessary to obtain all the essential energies to determinethermodynamical information are not currently available�
Even in �� years� at the current rate of improvement in computer power� thecomputational simulation of many technologically relevant oxide systems will stillbe out of reach� Moreover� the study of surfaces or some macroscopic propertiessuch as fracture opens a new dimension to the complexity of the problem� Unless abreakthrough is achieved in both the quantum� and statistical�mechanical models�simplifying assumptions will always have to be made in the foreseeable future�
The main conclusion of this work is that it is possible to compute formationenergies in oxides very accurately without using sophisticated quantum�mechanicalmethods� They can be replaced by simple models where all the main contributionsto the Hamiltonian are accounted for� However� approximations currently used inmany energy techniques are not adequate to deal with the relevant eects in oxides�We showed that charge transfer mechanisms� oxygen breathing� and nonsphericalrelaxations of the electronic density have to be incorporated into the Hamiltonian inorder to capture the essential physics of the problem�
As part of this thesis� we successfully integrated into a tight�binding formulationmost of the eects mentioned above� The Hamiltonian of the model includes explicitparametrizations of these terms� The parameters are �t to the pseudopotential bandsand equations of state of the end members of a speci�c system and one mixture ofthem� From this information� the formation energy of any other mixture can becomputed with an accuracy comparable to the pseudopotential predictions but ata fraction of the time� Thus� the tight�binding method can be regarded as a veryaccurate interpolation tool� The predicted formation energies in the CaO�MgO andCaO�ZrO� system only diered ��� on average from the pseudopotential results�
Most of the novel terms in the tight�binding Hamiltonian depend on the chargeof the ions� which is taken as variable within this model� This forces us to �nd theHamiltonian eigenvalues and eigenvectors self�consistently� The procedure providesa very accurate way to treat charge transfer within highly ionic systems� We demon�strated that total energies can be considerably aected by charge transfer betweenthe ions� The variation of the charge is easily described in metallic alloys in terms oflocal environments� Unfortunately� we showed that the same is not possible in oxides�Long�range interactions and local eects are needed� Tight binding was shown to bea very good option to deal with these eects�
The tight�binding formulation oers a full quantum�mechanical description ofbonding and a natural way of incorporating charge transfer� oxygen breathing� andnonspherical relaxations� Even though some of these eects have recently been in�cluded within a potential or an embedded atommodel� there was no simple integrationof all of them into a single approximate approach� Furthermore� even if they are in�
���

corporated� they will still lack other quantum�mechanical eects that tight�bindingprovides �e�g�� band eects��
The use of the tight�binding method to interpolate bands and compute totalenergies is not new� However� this is the �rst time that a self�consistent formulationhas been used to predict formation energies in multicomponent oxide alloys� Thisapproach proved to be not only fast� but very reliable�
Even when a good and fast description of the energetics of the system is provided�the evaluation of the thermodynamical properties is a very di�cult task� To computephase diagrams an eective lattice Hamiltonian is usually constructed� Monte Carlosimulations or the cluster variation method are then used to determine the phaseboundaries� We showed the predictive power of this approach by computing the tight�binding CaO�MgO phase diagram� The agreement with experiments is very good�At low temperatures� equilibration times in Monte Carlo become unaordably longand numerical instabilities can appear in the CVM� We developed a low�temperatureexpansion of the free energy that overcomes these di�culties� Its prediction can besmoothly joined with the other techniques at temperatures where the three approachescan be used�
As computers become more powerful and the simulation techniques more accurate�computational experiments will have a great impact in the development of novel ma�terials� Although the scales of every day problems are still intractable in most cases�the insight computer simulations can provide is invaluable� Approximate methods�that correctly treat the relevant physical phenomena certainly extend the limits ofwhat can be studied today�
���

���

Chapter ��
Future research
Computational experiments are a new and powerful tool in materials science and en�gineering� In this thesis� we have described how the combination of quantum� andstatistical�mechanical techniques can be used to study phase stability in multicom�ponent oxides� For the predictions of the models to be more accurate� improvementsin the methods are necessary�
The development of total�energy methods is a very active �eld of research� Workis being carried on both new and existent techniques� For example� the speed of theplane�wave approach was considerably increased by the use of ultrasoft pseudopoten�tials� which require a much smaller energy cuto� Also� nonspherical relaxations ofthe electronic densities are being incorporated into the SSCAD model�
Novel formulations are focusing on both the speed and the accuracy of the models�Most of the calculations in this thesis have been done within the local�density ap�proximation to density�functional theory� LDA is known to be overbinding in solids�leading to an underprediction of lattice constants �typically �� to ��� ����� Errors aslarge as ��� are also common for elastic constants� The generalized�gradient approx�imation �see section ������ was proposed as a way to go beyond LDA� and is constantlybeing improved �see for example ������� It tends to provide better structural energydierences but it sometimes overestimates lattice constants ����� ����� For systemswith strong Coulomb correlations� such as Mott insulators� the LDA was recentlycombined with a Hubbard�type model� This is the so�called LDA�U approach ������The LDA�U method has successfully been applied to transition�metal oxides� Seereference ����� for a review of the method and its applications�
The accuracy of the calculations is not the only factor to be considered� The speedof the total�energy methods is crucial to compute the formation energies needed asinput to phase diagram calculations� Massively parallel computers have started to beused� Most methods are being adapted to exploit the advantages of these computers�As mentioned in chapter �� some techniques are also being reformulated so that theyscale linearly with the number of atoms in the unit cell�
To speed up the calculations� new basis sets that provide a description of a func�tion with spatially variable resolution have been introduced� This is ideal for oxides�since oxygen atoms have strongly peaked orbitals� In some cases� the simplicity ofthe plane�wave basis set is retained while generalizing it to a non�Euclidean curvi�
���

linear space to reduce the size of the set ������ In other cases� the multiresolutionproperties of wavelets are combined with the smooth interpolation characteristics of�nite element methods ������
More accurate semiempirical approaches for oxides are also being investigated�Recently� a charge transfer scheme and a compressible ion model which includes bothdipole and quadrupolar polarizability were introduced ���� ��� ���� However� a wayof integrating all these eects into a potential model is still lacking� On the otherhand� we demonstrated that a self�consistent tight�binding formulation can easilyincorporate these contributions�
A critical step in the formulation of the tight�binding Hamiltonian is the deter�mination of an adequate set of parameters for a given system� It would be interestingto study alternative ways to perform the �tting process� One possibility is to usedierent atomic displacements that would independently sample dierent regions ofthe optimization space� This will also provide a more reliable determination of thedistance dependence of the parametrizations� At the same time� forces can be com�puted and �t to� A good description of the forces is essential if vibrational ECI�s willbe computed�
The tight�binding Hamiltonian used in this work can be improved in many ways�Both the inter� and intra�atomic interaction can be treated more accurately� Forexample� a Hartree�Fock instead of a Hartree formulation can be implemented� Thiswill be important to treat transition�metal oxides� Also� a more accurate distancedependence in the parametrizations of the hopping and overlap integrals can be used�
The computation of alloy phase diagrams needs not only a fast and accuratedescription of the energetics of the system but also a methodology to account for allthe important contributions to the free energy� We described how the Ising latticemodel can be used to accommodate both substitutional and nonsubstitutional degreesof freedom� For the latter� a coarse�graining of the alloy Hamiltonian was done�Recently� a formalism to include lattice vibrations within this model Hamiltonianin the harmonic approximation was proposed ����� A way to accurately deal withanharmonic eects is still lacking� Electronic excitations can also be included �����
The thermodynamic properties of binary alloys have been explored in many sys�tems� However� most technologically important oxides are multicomponent alloys� Inthe CaO�MgO system� the anion sublattice is fully occupied� This allowed us to treatthis problem as a binary ordering on the cation sublattice� However� in the CaO�ZrO�
system the anion lattice accommodates not only oxygen ions but also vacancies� �Thevacancies are introduced to compensate for the dierence in charge between the Zrand Ca ions�� A coupled�sublattice cluster expansion is required here� It would bevery interesting to use this approach� in combination with our tight�binding Hamil�tonian� to compute the CaO�ZrO� phase diagram� The available experimental phasediagram is incomplete� There are doubts about many of its regions due to kineticeects� the presence of defects and impurities� or just because the measurements aredi�cult at extreme temperatures�
If more than one type of cation dopant is added to zirconia� the system canno longer be treated as two binary problems� A ternary model seems unavoidable�and much more eort needs to be spent� It will certainly be worth developing new
���

approximate models to deal with the complexity of these systems�We have investigated techniques to determine the solid portion of a phase diagram�
In order to compute the full stability map� the liquid phase has to be accounted for�An accurate atomistic model for the thermodynamics of liquids is needed�
In this thesis� we have mainly been concerned with equilibrium properties andperfect crystalline systems� Although a great wealth of information can be obtainedfrom these studies� real materials are aected by kinetic processes� grain boundaries�and other defects� The modeling of kinetic processes in oxides is very complex� Itwould be very interesting to apply the tight�binding approach developed in this thesisin conjunction with molecular�dynamics methods and lattice models �see for examplereference ����� to study this problem� Parallel supercomputer technology is startingto allow ab initio calculations on systems big enough to explore the interactionsamong dislocations ������ Furthermore� combinations of �rst�principles approachesand simple energy models can be used to build a bridge between the dierent lengthscales of the problem�
It should be clear by now that the modeling of materials requires a combinationof both sophisticated quantum�mechanical methods and simple energy models withdynamical and statistical�mechanical approaches� There is still plenty of room forimprovements in all these �elds� As computers become more powerful� and the meth�ods more sophisticated the dream of designing real materials from �rst principles isturning into a reality�
���

���

Appendix A
Additional results for the
CaO�MgO system
A�� Introduction
In this appendix� we provide the computed formation energies for dierent structuresin the CaO�MgO system� These values were used to �t the ECI�s shown in �gures ���and ����
A derivation of the potential used in section ����� will also be described�
A�� Formation energies
In table A��� we show the formation energies computed using the SSCAD� the poten�tial in section ������ and the self�consistent tight�binding method for the CaO�MgOsystem�
A�� Pair�potential model for CaO�MgO
In section ������ we used the following form for the short�range repulsive interactionin the CaO�MgO system�
V �rij� �Aij
r�ij� �A���
It can be directly derived from the tight�binding Hamiltonian in equation ���� if weassume that there is no softening of the ions�
The softening of the ions is the reduction� in absolute value� of the cation andanion charges� It is proportional to
�Hij
Hii �Hjj
��
�A���
where Hij is the hopping matrix element between the ions �i and j� that are reducing
���

Structure Concentration SSCAD Pair Potential Tight�BindingRocksalt CaO ����� ����� �����Rocksalt MgO ����� ����� �����L�� MgCaO� ����� ����� �����L�� MgCa�O� ����� ����� �����L�� Mg�CaO� ����� ����� �����A�B� MgCaO� ����� ����� �����L�� MgCaO� ����� ����� �����MoPt� MgCa�O� ����� ����� �����MoPt� Mg�CaO� ����� ����� �����A�B MgCa�O� ����� ����� �����A�B Mg�CaO� ����� ����� �����Ni�Mo MgCa�O� ����� ����� �����Ni�Mo Mg�CaO� ����� ����� �����Pt�Ti MgCa�O ����� ����� �����Pt�Ti Mg�CaO ����� ����� �����A�B� MgCa�O� ����� ����� �����A�B� Mg�CaO� ����� ����� �����Do�� MgCa�O� ����� ����� �����Do�� Mg�CaO� ����� ����� �����S�x�x� MgCa��O�� ����� ����� �����S�x�x� Mg��CaO�� ����� ����� �����
Table A�� Formation energies �meV atom� of dierent ordered structures in theCaO�MgO system obtained by using the SSCAD� the pair�potential in section ������and the self�consistent tight�binding methods� All the structures were fully relaxed�A description of the structures can be found in references ���� or ����� The S�x�x�corresponds to the �x�x� supercell used in reference �����
���

their charge� See reference ����� for more details�If we assume formal values for the charges� the calcium and magnesium s orbitals
have a higher energy than the oxygen p orbitals within the tight�binding model�Consequently� if all the nondiagonal terms Hij are set close to zero � hard� ions��the ionic charges will stay at those formal values� No self�consistency is needed�The Hamiltonian matrix is basically diagonal and an expression for the total energy�E�n��r��� can be written directly as the sum of the Hamiltonian eigenvalues �Ebs�plus the Madelung energy and the corrections for the double�counting of the electron�electron interaction �F �n��r����
E�n��r�� � Ebs � F �n��r��� �A���
Apart from the Coulomb terms� only Fi� in Ebs �see equation ����� depends on theionic distances� As we already mentioned� Fi� is a repulsive contribution that comesfrom the overlap of the nonorthogonal basis� For sp�bonded materials� its distancedependence can be computed from Harrison�s work �table ���� and the use of H#uckel�stheory as explained in reference ������ The result is the �
d�dependence in equation A���
The Aij values were �t to the pseudopotential lattice constant of CaO and MgO�and the formation energy and lattice constant of CaMgO� �L�� structure��
���

���

Appendix B
�k�space integration
B�� Introduction
Most solid�state total�energy methods require the evaluation of integrals of periodicfunctions in �k space to compute quantities such as the electronic density or single�particle energy sums� Since the calculation time scales linearly with the number of �kpoints� the smallest possible number of them should be used�
Many systematic ways have been proposed to choose small sets of �k points that arenormally called special points� Usually they provide a more accurate integration� witha smaller number of points� than the ordinary tetrahedron�integration method �������
Here� we will brie�y review the most popular ways of choosing the special �k points�
B�� Special �k points
B���� Uniform mesh
A uniform grid is used in �k space� The accuracy of the integrations can be improved byincreasing the number of grid points� The symmetry of the crystal may be employedto eliminate those points that are equivalent� When this is done� the weight� of theremaining �k vectors has to be adjusted� �The origin of the mesh can be shifted tooptimize the use of symmetry��
B���� The Chadi�Cohen scheme
The �k points are chosen so that the integrals are computed exactly for those functionsthat contain up to a certain Fourier component� By increasing this component� ahierarchy of points can be obtained�
The point�group symmetry of the crystal is normally used to reduce the numberof �k points to the irreducible part of the Brillouin zone� See reference ����� for more
�For a comparison between the tetrahedron and the special�points integration methods see ref�erence ������
���

details�
B���� The Monkhorst�Pack scheme
The method is similar in spirit to the Chadi�Cohen scheme� Again� the �k points arechosen so that the integrals are computed exactly for those functions that contain upto a certain Fourier component� However� they dier on how to actually pick thesepoints� Furthermore� a dierent number of special points and order of the termscanceled in the Fourier expansion is predicted� A comparison between the Chadi�Cohen and the Monkhorst�Pack scheme can be found in references ����� ����� Seereference ����� for more details�
This method was later generalized by Froyen ������
B�� Superstructures and �equivalent� �k points
In many applications� such as the computation of formation energies or forces� it isnecessary to compare results for supercells of dierent geometry� In order to minimizesystematic errors in the evaluation of Brillouin�zone integrals� it is convenient to usean equivalent set of �k points� By equivalent� we mean using the same points inreciprocal space�
The equivalent points can be obtained from any set of points� such as the ones insection B��� The procedure consists in expanding all the �k points used for the parentlattice by means of the symmetry of that lattice� Once all the equivalent points areidenti�ed in the �rst Brillouin zone� the symmetry of the supercell can be used tokeep the independent ones �the weights have to be adjusted��
B�� Brillouin�zone integrations in metals
The functions to be integrated in metals are discontinuous due to the partial �llingof the energy bands� While the integrals of in�nitely dierentiable functions� such asthe ones obtained in insulators� converge exponentially with the spacing of the grid�discontinuous functions have a very low convergence� In order to be able to performBrillouin�zone integrations in metals with high precision but without a prohibitivelylarge set of �k points� dierent methods have been proposed �see reference ����� andreferences therein��
In one of these methods� the convergence is improved by a smearing of the valuesat each �k point into Gaussians� In this way� an exponential convergence with respectto the grid spacing is recovered� However� there is no guarantee that the integral willconverged to the correct value�
Methfessel and Paxton showed that� with a more careful choice of the broadeningfunction� an independent control of both the �k convergence �i�e�� number of points�and the integral value convergence can be achieved� Thus� an exponential convergencewith the number of sampling points is recovered� but without a loss in accuracy� A
���

hierarchy of smooth approximations to the step and � functions was developed� Seereference ����� for more details�
���

���

Appendix C
The design of lithium batteries
C�� Introduction
The objective of this appendix is to illustrate a dierent application of total�energymethods� The pseudopotential formation energies will be directly linked to the voltageof a battery� Ab initio calculations allow the systematic investigation of the eectsof chemistry and structure on the voltage� This is an excellent example of a casewhere computer experiments can help to develop materials with properties tailoredto speci�c needs�
The voltage is one of the most important parameters when designing a battery�We will only give a brief introduction to this problem by focusing on the computationof the average voltage of lithium�transition�metal batteries� A much broader perspec�tive and interpretation of the results can be found in references ����� ���� ���� �����
Figure C�� illustrates how a lithium battery works� During discharge� Li� ionsare transferred from the anode to the cathode through the electrolyte� The charge ofLi� is compensated in the cathode by absorbing an electron from the external circuit�By applying an external voltage the process can be reversed��
The anode is usually a solution of lithium in carbon� However� for simplicity� wewill assume a pure metallic lithium anode� Although dierent materials can be usedas cathodes� we will investigate the lithium�transition�metal oxides �LixMO�� whereM�Ti� V� Mn� Co� Ni� Cu� or Zn��� Depending on the structure� the ratio of lithiumto metal ions� x� can be varied between � and ��
For the discharge process to proceed� the following reaction should be thermody�namically favored�
MO� � Li �� LiMO�� �C���
Using thermodynamical arguments� it is possible to compute the voltage of the cellfrom the lithium chemical potential ��Li� on both sides of expression C��� In general�the battery is discharged between Lix�MO� and Lix�MO�� The battery voltage� V � is
�This is assuming that lithium can be reversibly cycled in and out of the cathode without majorstructural changes�
�Composite materials are actually used� The transition metals are combined with carbon toprovide a conducting medium for the electrons�
���

Ano
de
Cat
hode
Electrolyte
Li
e
+
-
Discharge
Ano
de
Cat
hode
Electrolyte
Li
e
+
-
Charge
Figure C�� Charge and discharge cycle for a lithium battery�
related to the lithium content� x� in LixMO� as �����
V �x� � ��cathodeLi�x� � �anodeLi
ZF� �C���
Z is the number of electrons in the external circuit for each Li ion �in our case Z � ���and F is the Faraday constant�
The average voltage of the battery �between x � � and x � ��� hV i� dependson the formation energy of LiMO�� This can be shown by writing the total electricalenergy of the battery as
hV ie � eZ �
�V �x�dx � �e
Z �
�
�cathodeLi�x� � �anodeLi
Fdx � �%G
Fe� �C���
Here� %G is the Gibbs free energy dierence �per mole� of reaction C�� and is com�puted as
%G � %E � P%V � T%S� �C���
The last two terms on the right�hand side of equation C�� are very small compared to%E� which is approximated by its value at � oK�� �P � V � T � and S are the pressure�volume� temperature� and entropy respectively�� Finally�
hV i � %E
F� �C���
�The term P�V is ����� eV T�S is ����� eV and �E is ���� eV�
���

Valence electron con�guration rcs ��A� rcp ��A� rcd ��A�Li �s���p��� ���� ���� �Ti �s���p����d��� ���� ���� ����V �s���p����d��� ���� ���� ����Mn �s����p�����d��� ���� ���� ����Co s����p�����d�� ���� ���� ����Ni s����p�����d��� ���� ���� ����Cu s����p�����d��� ���� ���� ����Zn s����p�����d���� ���� ���� ����S s����p���� ���� ���� �Se s����p�����d���� ���� ���� ����
Table C�� Valence electron con�guration and core radii �rcl� used to generate thepseudopotentials �l� s� p� d��
We would like to stress the importance of equation C�� since it directly relates theaverage voltage of the battery to the formation energy of LiMO�� This voltage canbe simply determined by computing the total energy of LiMO�� MO�� and Li� Thus�ab initio approaches are particularly suited to design new cathode materials since thevoltages can easily be computed� Furthermore� the eects of metal chemistry andstructure on hV i can be studied independently� without being constrained to thosecombinations that can be actually synthesized�
We will perform two dierent computational experiments We will �x the structureand change the metal or anion chemistry� and we will change the structure whilekeeping the identity of the elements in the cathode� For the �rst type of experiments�we chose to use the structure of the commercially employed LiCoO� ������ This is the��NaFeO� arrangement� The same structure corresponds to LiVO� ������
C�� Pseudopotential studies of the battery volt�
age
C���� Pseudopotential generation
Nonlocal� optimized� Kleinman�Bylander type pseudopotentials were used� The sangular component was taken as the local potential� The atomic con�gurations andcore radii used in the generation are shown in table C��� There is a considerable over�lap between the lithium �s and �s orbitals� Core corrections are needed to accuratelytreat the bcc lithium metal� However� they are not expected to be important in theLi� ion� Consequently� they will only aect the pure lithium energy� shifting all theformation energies by a �xed amount� For simplicity� we will not include these correc�tions here since we are primarily interested in computing the di�erences in voltages
���

Ti V Mn Co Ni Cu ZnVoltage ���� ���� ���� ���� ���� ���� ����
�����a �����b �����c
Table C�� Average voltage �in volts� computed for the LiMO� compound in the��NaFeO� structure� The values in parenthesis are experimental� The agreement isgood� The large discrepancy in the nickel case is due to the fact that the structureis unstable with respect to a Jahn�Teller distortion �not taken into account in thecalculations��
aExperimental averaged voltage computed from reference ������ The error in the value ���� V�bExperimental averaged voltage computed from reference ������ The error in the value ���� V�cExperimental averaged voltage computed from reference ������ The error in the value ���� V�
O S SeVoltage ���� ���� ����
Table C�� Average voltage �volts� for LiCoX� compounds in the ��NaFeO� structure�
between dierent structures and chemistries��
C���� Results
A ��� eV energy cuto was used in all the calculations� The �k points were chosenusing the Monkhorst�Pack scheme ������ Convergence within tens of meV in theformation energies was obtained�
In table C��� we show the average voltages computed for dierent transition metalsin the ��NaFeO� structure�
The voltages obtained for dierent anion species� X� in LiCoX� ���NaFeO� struc�ture� are shown in table C��� Finally� in table C��� we show the average voltage forLiCoO� in two dierent structures ��NaFeO� �structure , �� and a variant of thisone with Pearson symbol tI�� ����� �structure , ����
C���� Discussion
Table C�� indicates that there is a strong tendency for the voltage to increase withthe nuclear charge� As explained in references ����� ����� this can be qualitativelyunderstood from the fact that the electron coming from Li populate band levels
�By comparing to FLAPW results we showed the e�ect of core corrections in the overall voltageto be small less than ��� eV ������
�Here the Co and Li ions are intermixed in the cation layers�
���

structure ,� structure ,�Voltage �relaxed� ���� ����Voltage �unrelaxed� ���� ����
Table C�� Average voltage �volts� for LiCoO� compound for two dierent structures�For the unrelaxed case� the ideal rocksalt positions were taken�
-10
-9.8
-9.6
-9.4
-9.2
-9
-8.8
-8.6
-8.4
-8.2
-8
Ti V Mn Co Ni Cu Zn
Ene
rgy
(eV
)
Figure C�� Fermi energy of dierent LiMO� compounds in the ��NaFeO� structure�solid line�� With Ni� a new band starts to be �lled� producing a jump in the voltage�The highest energy point of the band that was just completely �lled in Co is alsoshown �dashed line��
that are systematically shifted down by the increase in the nuclear charge� This isillustrated in �gure C��� where we show the decrease in Fermi energy when the nuclearcharge increases� Consequently� the electron coming from the external circuit is placedat a lower energy in the transition�metal oxide� eectively raising the voltage� Notethat the jump in the nickel voltage is due to the fact that the Li electron has to startpopulating a new band at a higher energy�
The eect of structural changes is small for the two arrangements we studied�However� the eects of relaxations are signi�cant� They can introduce voltage changesof the order of ��� volts� Even more important is the eect of replacing the anion�that can change hV i by more than � volts�
The predicted voltages are in good agreement with the measured values when�ever they can be compared� This shows the predictive power of the pseudopotentialmethod� The advantage of performing computational experiments to determine the
���

voltage is clear� There is no need to synthesize new compounds� Many dierent pos�sibilities can be tested without a costly and laborious processing� At the same time�the ab initio approach gave insight on the mechanisms that lead to the determinationof voltages� which in turn provides guidance to choose new compounds�
���

Bibliography
��� J� Read� Prelude to Chemistry An Outline of Alchemy� The M�I�T� Press�Cambridge� �����
��� B� J� Teeter Dobbs� The Foundations of Newton�s Alchemy or �The Hunting ofthe Greene Lyon � Cambridge University Press� Cambridge� �����
��� B� J� Teeter Dobbs� The Janus faces of genius� Cambridge University Press�Cambridge� �����
��� M� Kranzberg and C� Stanley Smith� Materials in history and society� InM� Cohen� editor� Materials Science and Engineering Its Evolution� Practiceand Prospects� pages ����� Lausanne� ����� Elsevier�
��� M� L� Cohen� Predicting useful materials� Science� ����������� �����
��� G� C� Fox and P� D� Coddington� An overview of high performance comput�ing for the physical sciences� In D� A� Browne� J� Callaway� J� P� Draayer�R� W� Haymaker� R� K� Kalia� J� E� Tohline� and P� Vashishta� editors� Highperformance computing and its applications in the physical sciences proceed�ings of the Mardi Gras ��� conference� February ������ ����� Louisiana StateUniversity� pages ����� Singapore� ����� World Scienti�c�
��� R� W� Siegel� Creating nanophase materials� Scienti�c American� ��������������
��� F� F� Abraham� D� Brodbeck� R� A� Rafey� and W� E� Rudge� Instabilitydynamics of fracture A computer simulation investigation� Physical ReviewLetters� ���������� �����
��� J� M� Sanchez� F� Ducastelle� and D� Gratias� Generalized cluster descriptionof multicomponent systems� Physica� ���A������� �����
���� J� W� D� Connolly and A� R� Williams� Density�functional theory applied tophase transformations in transition�metal alloys� Physical Review B� ������������ �����
���� Z� W� Lu� S� H� Wei� A� Zunger� S� Frota�Pessoa� and L� G� Ferreira� First�principles statistical mechanics of structural stability of intermetalic com�pounds� Physical Review B� ���������� �����
���

���� G� D� Garbulsky and G� Ceder� Linear�programming method for obtaining ef�fective cluster interactions in alloys from total�energy calculations Applicationto the fcc Pd�V system� Physical Review B� �������� �����
���� R� Kikuchi� A theory of cooperative phenomena� Physical Review� ����������������
���� K� Binder and D� W� Heermann� Monte Carlo simulation in statistical physics�Springer�Verlag� Berlin� �����
���� P� Hohenberg and W� Kohn� Inhomogeneous electron gas� Physical Review������������ �����
���� W� Kohn and L� J� Sham� Self�consistent equations including exchange andcorrelation eects� Physical Review� ������������� �����
���� D� C� Langreth and M� J� Mehl� Beyond the local�density approximation incalculations of ground�state electronic properties� Physical Review B� ������������ �����
���� A� D� Becke� Density�functional exchange�energy approximation with correctasymptotic behavior� Physical Review A� ������������ �����
���� C� Pisani� R� Dovesi� and C� Roetti� Hartree�Fock Ab Initio Treatment ofCrystalline Systems� Springer�Verlag� Berlin� �����
���� J� C� Slater and G� F� Koster� Simpli�ed LCAO method for the periodic po�tential problem� Physical Review� ������������ �����
���� M� M� Sigalas and D� A� Papaconstantopoulos� Transferable total�energyparametrizations for metals Applications to elastic�constant determination�Physical Review B� ������������ �����
���� R� E� Cohen� M� J� Mehl� and D� A� Papaconstantopoulos� Tight�binding total�energy method for transition and noble metals� Physical Review B� �������������� �����
���� A� F� Kohan and G� Ceder� Tight�binding calculation of formation energies inmulticomponent oxides Application to the MgO�CaO phase diagram� PhysicalReview B� ���������� �����
���� J� G� Bednorz and K� A� Muller� Possible high Tc superconductivity in the Ba�La�Cu�O system� Zeitschrift fur Physik B �Condensed Matter�� ���������������
���� M� K� Wu� J� R� Ashburn� C� J� Torng� P� H� Hor� R� L� Meng� L� Gao� Z� J�Huang� Y� Q� Wang� and C� W� Chu� Superconductivity at �� K in a new mixed�phase Y�Ba�Cu�O compound system at ambient pressure� Physical ReviewLetters� ���������� �����
���

���� D� de Fontaine� G� Ceder� and M� Asta� Low�temperature long�range oxygenorder in YBa�Cu�Oz� Nature� ����������� �����
���� T� W� Eagar� The future of metals� Welding Journal� �������� �����
���� W� D� Kingery� H� K� Bowen� and D� R� Uhlmann� Introduction to Ceramics�John Wiley - Sons� New York� �����
���� J� L� Martin� Computer techniques for evaluating lattice constants� In C� Domband M� S� Green� editors� Phase Transition and Critical Phenomena� volume ��pages ������� London� ����� Academic Press Inc�
���� Z� Jin and Y� Du� A reassessment of the CaO�MgO system� CALPHAD��������� �����
���� J� F� Smith� The Pd�V �palladium vanadium� system� Journal of Alloy PhaseDiagrams� ����� �����
���� J� Cheng and A� J� Ardell� On the stability of the ordered Pd�V phase in aproton�irradiated Pd��� at�� V alloy� Journal of the Less�Common Metals���������� �����
���� M� P� Allen and D� J� Tildesley� Computer simulation of liquids� Oxford Uni�versity Press� New York� �����
���� G� D� Garbulsky� Ground�state structures and vibrational free energy in �rst�principles models of substitutional�alloy thermodynamics� PhD thesis� Mas�sachusetts Institute of Technology� �����
���� G� D� Garbulsky and G� Ceder� Contribution of the vibrational free energy tophase stability in substitutional alloys Methods and trends� Physical ReviewB� ������������ �����
���� G� D� Garbulsky and G� Ceder� Eect of lattice vibrations on the orderingtendencies in substitutional binary alloys� Physical Review B� �����������������
���� C� Wolverton and A� Zunger� First�principles theory of short�range order�electronic excitations� and spin polarization in Ni�V and Pd�V alloys� PhysicalReview B� ������������ �����
���� H� E� Stanley� Introduction to Phase Transitions and Critical Phenomena�Clarendon Press� Oxford� �����
���� G� Ceder� G� D� Garbulsky� and P� D� Tepesch� Convergent real�space clusterexpansion for con�gurational disorder in ionic systems� Physical Review B��������������� �����
���� D� B� Laks� L� G� Ferreira� S� Froyen� and A� Zunger� E�cient cluster expansionfor substitutional systems� Physical Review B� �������������� �����
���

���� L� G� Ferreira� A� A� Mbaye� and A� Zunger� Chemical and elastic eects onisostructural phase diagrams the ��G approach� Physical Review B� �������������� �����
���� R� McCormack� D� de Fontaine� C� Wolverton� and G� Ceder� Nonempiricalphase equilibria in the W�Mo�Cr system� Physical Review B� �������������������
���� P� D� Tepesch� G� D� Garbulsky� and G� Ceder� Model for con�gurationalthermodynamics in ionic systems� Physical Review Letters� ������������ �����
���� H� Dreyss�e� A� Berera� L� T� Wille� and D� de Fontaine� Determination ofeective�pair interactions in random alloys by con�gurational averaging� Phys�ical Review B� ������������ �����
���� F� Ducastelle� Order and Phase Stability in Alloys� North�Holland� Amsterdam������
���� P� D� Tepesch� Atomistic Modeling of Ceramic Materials Predicting CrystalStructures� Thermodynamic Properties� and Di�usion Behavior� PhD thesis�Massachusetts Institute of Technology� �����
���� G� D� Garbulsky� P� D� Tepesch� and G� Ceder� Ground state analysis on the fcclattice with four pair interactions� In J� Broughton� P� Bristowe� and J� Newsam�editors� Materials Research Society Symposium Proceedings� volume ���� pages�������� Pittsburgh� ����� Mater� Res� Soc�
���� K� Binder� Ordering of the face�centered�cubic lattice with nearest�neighborinteraction� Physical Review Letters� ���������� �����
���� J� A� Barker� Methods of approximation in the theory of regular mixtures�Proceedings of the Royal Society� London� ��������� �����
���� G� Ceder� Alloy theory and its applications to long period superstructure or�dering in metalic alloys and high temperature superconductors� PhD thesis�University of California at Berkeley� �����
���� C� Domb� Graph theory and embeddings� In C� Domb and M� S� Green� editors�Phase Transitions and Critical Phenomena� volume �� pages ����� London������ Academic Press Inc�
���� C� Domb� On the theory of cooperative phenomena in crystals� Advances inPhysics� ��������� �����
���� J� L� Lebowitz� M� K� Phani� and D� F� Styer� Phase diagram of Cu�Au�typealloys� Journal of Statistical Physics� ���������� �����
���� M� F� Sykes� J� W� Essam� and D� S� Gaunt� Derivation of low temperatureexpansions for the ising model of a ferromagnet and an antiferromagnet� Journalof Mathematical Physics� ��������� �����
���

���� D� de Fontaine� The cluster variation method and the calculation of alloy phasediagrams� In G� M� Stocks and A� Gonis� editors� Alloy Phase Stability� pages�������� Dordrecht� ����� Kluwer Academic Publishers�
���� M� Wortis� Linked cluster expansion� In C� Domb and M� S� Green� editors�Phase Transitions and Critical Phenomena� volume �� pages �������� London������ Academic Press Inc�
���� G� Parisi� Statistical Field Theory� Addison Wesley� California� �����
���� C� Wolverton� G� Ceder� D� de Fontaine� and H� Dreyss�e� Ab�initio ground�statestudy with fourth�nearest�neighbor cluster interactions for fcc Pd�V alloys�Physical Review B� �������������� �����
���� C� Wolverton� A� Zunger� and Z� W� Lu� Long�range versus short�range orderin Ni�V and Pd�V alloys� Physical Review B� �������������� �����
���� P� E� A� Turchi� G� M� Stocks� W� H� Butler� D� M� Nicholson� and A� Gonis�First�principles study of ordering properties of substitutional alloys using thegeneralized perturbation method� Physical Review B� ������������ �����
���� G� Ceder� P� D� Tepesch� C� Wolverton� and D� de Fontaine� Ab initio com�putation of the fcc Pd�V phase diagram� In A� Gonis� editor� Statistics andDynamics of Alloy Phase Transformations� page ���� New York� ����� PlenumPress�
���� C� Wolverton� G� Ceder� D� de Fontaine� and H� Dreyss�e� Ab�initio determina�tion of structural stability in fcc�based transition�metal alloys� Physical ReviewB� ���������� �����
���� P� D� Tepesch� G� Ceder� C� Wolverton� and D� de Fontaine� An ab�initiocalculation of the Pd�V fcc superstructure phase diagram with fourth nearestneighbor cluster interactions� In J� Broughton� P� Bristowe� and J� Newsam�editors� Materials Research Society Symposium Proceedings� volume ���� pages�������� Pittsburgh� ����� Mater� Res� Soc�
���� C� Wolverton and A� Zunger� Comparison of two cluster�expansion methods forthe energetics of transition�metal alloys� Physical Review B� �������������������
���� H� L� Skriver� The LMTO Method� Springer� Berlin� �����
���� J� Kanamori and Y� Kakehashi� Conditions for the existence of ordered struc�tures in binary alloy systems� Journal de Physique� ��C������ �����
���� C� Cohen�Tannoudji� B� Diu� and F� Lalo#e� Quantum Mechanics� John Wiley- Sons� New York� �����
���� L� I� Schi� Quantum Mechanics� McGraw�Hill� New York� �����
���

���� D� J� Singh� W� E� Pickett� and H� Krakauer� Gradient�corrected density func�tionals Full�potential calculation for iron� Physical Review B� �������������������
���� D� Singh� D� P� Clougherty� J� M� MacLaren� R� C� Albers� and C� S� Wang�In�uence of the local�spin�density correlation functional on the stability of bccferromagnetic iron� Physical Review B� ������������ �����
���� H� J� F� Jansen� Electronic structure calculations for magnetically orderedsystems� Physics Today� �������� �����
���� A� E� Carlsson� Beyond pair potentials in elemental transition metals andsemiconductors� In H� Ehrenreich and D� Turnbull� editors� Solid State Physics�volume ��� pages ����� Boston� ����� Academic Press�
���� M� S� Daw� S� M� Foiles� and M� I� Baskes� The embedded�atom method areview of theory and applications� Materials Science Reports� ��������� �����
���� T� J� Raeker and A� E� Depristo� Theory of chemical bonding based on theatom�homogeneous electron gas system� International Reviews in PhysicalChemistry� ������� �����
���� F� H� Streitz and J� W� Mintmire� Electrostatic potentials for metal�oxidesurfaces and interfaces� Physical Review B� �������������� �����
���� M� I� Baskes� Modi�ed embedded atom method calculations of interfaces� San�dia Report ������ pages ����� �����
���� B� G� Dick Jr� and A� W� Overhauser� Theory of the dielectric constants ofalkali halide crystals� Physical Review� ���������� �����
���� I� M� Torrens� Interatomic Potentials� Academic Press� New York� �����
���� G� V� Lewis and C� R� A� Catlow� Potential models for ionic oxides� Journal ofPhysics C Solid State Physics� ������������ �����
���� V� Vitek� Pair potentials in atomistic computer simulations� MRS Bulletin��������� �����
���� M� Stoneham� J� Harding� and T� Harker� The shell model and interatomicpotentials for ceramics� MRS Bulletin� �������� �����
���� R� E� Watson� S� C� Parker� and A� Wall� Molecular dynamics simulation of�uoride�perovskites� J� Phys� Condens� Matter�� ����������� �����
���� S� C� Parker and G� D� Price� Computer modelling of phase transitions inminerals� Adv� Solid�State Chem� ����� �����
���

���� P� D� Tepesch� A� F� Kohan� G� D� Garbulsky� G� Ceder� C� Coley� H� T� Stokes�L� L� Boyer� M� J� Mehl� B� P� Burton� K� Cho� and J� Joannopoulos� A modelto compute phase diagrams in oxides with empirical or �rst�principles energymethods and application to the solubility limits in the CaO�MgO system� Jour�nal of the American Ceramic Society� ������������ �����
���� P� P� Ewald� Die berechnung optischer und elektrostatischer gitterpotentiale�Annalen der Physic� ���������� �����
���� J� D� Gale� C� R� A� Catlow� and W� C� Mackrodt� Periodic ab initio deter�mination of interatomic potentials for alumina� Modelling and simulation inmaterials science and engineering� ������� �����
���� J� F� Nye� Physical Properties of Crystals� Oxford University Press� New York������
���� M� Wilson� P� A� Madden� N� C� Pyper� and J� H� Harding� Molecular dynamicssimulations of compressible ions� Journal of Chemical Physics� ������������������
���� M� Wilson� U� Sch#onberger� and M� Finnis� Transferable atomistic model todescribe the energetics of zirconia� Physical Review B� ������������ �����
���� M� Wilson� M� Exner� Y� M� Huang� and M� W� Finnis� Transferable model forthe atomistic simulation of Al�O�� Physical Review B� �������������� �����
���� M� S� Daw� Model of metallic cohesion the embedded�atom method� PhysicalReview B� ������������ �����
���� F� H� Stillinger and T� A� Weber� Computer simulation of local order in con�densed phases of silicon� Physical Review B� ������������ �����
���� M� I� Baskes� Application of the embedded�atom method to covalent materialsa semiempirical potential for silicon� Physical Review Letters� �����������������
���� M� I� Baskes� Modi�ed embedded�atom potentials for cubic materials and im�purities� Physical Review B� ������������ �����
���� N� W� Ashcroft and N� D� Mermin� Solid State Physics� Saunders CollegePublishing� Fort Worth� �����
���� U� von Barth� Density functional theory for solids� In P� Phariseau and W� M�Temmerman� editors� The Electronic Structure of Complex Systems� pages ������� New York� ����� Plenum Press�
���� R� O� Jones and O� Gunnarsson� The density functional formalism� its applica�tions and prospects� Reviews of Modern Physics� ���������� �����
���

���� J� F� Janak� Proof that �e�ni
� �i in density�functional theory� Physical ReviewB� ������������ �����
���� J� P� Perdew and A� Zunger� Self�interaction correction to density�functionalapproximations for many�electron systems� Physical Review B� �����������������
����� L� Hedin and B� I� Lundqvist� Explicit local exchange�correlation potentials�Journal of Physics C� ����������� �����
����� J� C� Slater� Wave functions in a periodic potential� Physical Review� ���������� �����
����� O� K� Andersen� Linear methods in band theory� Physical Review B� ������������ �����
����� H� T� Stokes� L� L� Boyer� and M� J� Mehl� Spherical self�consistent atomicdeformation model for �rst�principles energy calculations in ionic crystallinesolids� Physical Review B� ������������ �����
����� E� Clementi and C� Roetti� Roothaan�Hartree�Fock atomic wavefunctions�basis functions and their coe�cients for ground and certain excited states ofneutral and ionized atoms� Z� ��� Atomic Data and Nuclear Data Tables����������� �����
����� A� D� McLean and R� S� McLean� Roothaan�Hartree�Fock atomic wave func�tions Slater basis�set expansions for z������� Atomic Data and Nuclear DataTables� ���������� �����
����� R� G� Gordon and Y� S� Kim� Theory for the forces between closed�shell atomsand molecules� The Journal of Chemical Physics� ������������ �����
����� L� L� Boyer� M� J� Mehl� J� L� Feldman� J� R� Hardy� J� W� Flocken� and C� Y�Fong� Beyond the rigid�ion approximation with spherically symmetric ions�Physical Review Letters� ������������ �����
����� M� J� Mehl� R� J� Hemley� and L� L� Boyer� Potential�induced breathing modelfor the elastic moduli and high�pressure behavior of the cubic alkaline�earthoxides� Physical Review B� ������������ �����
����� R� E� Watson� Analytic Hartree�Fock solutions for O�� Physical Review�������������� �����
����� O� K� Andersen� O� Jepsen� and D� Gl#otzel� Canonical description of the bandstructures of metals� In F� Bassani� F� Fumi� and M� P� Tosi� editors� Proceedingsof the International School of Physics �Enrico Fermi�� Course LXXXIX� High�lights of Condensed�Matter Theory� pages ������� Amsterdam� ����� NorthHolland�
���

����� O� K� Andersen and O� Jepsen� Explicit� �rst�principles tight�binding theory�Physical Review Letters� ������������ �����
����� O� K� Andersen� O� Jepsen� and M� Sob� Linearized band structure methods� InM� Yussou� editor� Electronic band structure and its applications proceedingsof the International School on Electronic Band Structure and its ApplicationsKanpur� India� October ���November �� ����� volume �� pages ����� Berlin������ Springer�Verlag�
����� A� M� Bratkovsky and S� Y� Savrasov� On the calculation of combined correc�tions in the LMTO method� Journal of Computational Physics� ���������������
����� N� E� Christensen and S� Satpathy� Pressure�induced cubic to tetragonal tran�sition in CsI� Physical Review Letters� ���������� �����
����� K� H� Weyrich� Full�potential linear mu�n�tin�orbital method� Physical Re�view B� �������������� �����
����� U� von Barth and L� Hedin� A local exchange�correlation potential for the spinpolarized case� I� Journal of Physics C� ����������� �����
����� A� van de Walle� Private communication�
����� E� Wimmer� H� Krakauer� M� Weinert� and A� J� Freeman� Full�potential self�consistent linearized�augmented�plane�wave method for calculating the elec�tronic structure of molecules and surfaces O� molecule� Physical Review B����������� �����
����� H� J� F� Jansen and A� J� Freeman� Total�energy full�potential linearizedaugmented�plane�wave method for bulk solids Electronic and structural prop�erties of tungsten� Physical Review B� ���������� �����
����� M� J� Mehl� R� E� Cohen� and H� Krakauer� Linearized augmented plane waveelectronic structure calculations for MgO and CaO� J� Geophys� Res�� ������������ �����
����� D� J� Singh� Planewaves� Pseudopotentials and the LAPW Method� KluwerAcademic Publishers� Boston� �����
����� P� Pulay� Ab initio calculation of force constants and equilibrium geometries inpolyatomic molecules� I� Theory� Molecular Physics� ���������� �����
����� R� Car and M� Parrinello� Uni�ed approach for molecular dynamics and density�functional theory� Physical Review Letters� ������������ �����
����� M� C� Payne� M� P� Teter� D� C� Allan� T� A� Arias� and J� D� Joannopoulos� Iter�ative minimization techniques for ab initio total�energy calculations moleculardynamics and conjugate gradients� Reviews of Modern Physics� �����������������
���

����� A� M� Rappe� K� M� Rabe� E� Kaxiras� and J� D� Joannopoulos� Optimizedpseudopotentials� Physical Review B� ������������ �����
����� D� Vanderbilt� Soft self�consistent pseudopotentials in a generalized eigenvalueformalism� Physical Review B� ������������ �����
����� W� E� Pickett� Pseudopotential methods in condensed matter applications�Computer Physics Reports� ��������� �����
����� P� J� H� Denteneer and W� van Haeringen� The pseudopotential�density�functional method in momentum space details and test cases� J� Phys� C�������������� �����
����� J� Ihm� Total energy calculations in solid state physics� Rep� Prog� Phys������������ �����
����� W� A� Harrison� Electronic Structure and the Properties of Solids� Dover Pub�lications� New York� �����
����� D� G� Pettifor� The tight binding approximation Concepts and predictions� InD� G� Pettifor and A� H� Cottrell� editors� The Electron Theory in Alloy Design�pages ������� London� ����� The Institute of Materials�
����� F� Bloch� #Uber die quantenmechanik der elektronen in kristallgittern� Z� Physik����������� �����
����� W� Y� Ching� Theoretical studies of the electronic properties of ceramic mate�rials� Journal of the American Ceramic Society� ������������ �����
����� J� A� Majewski and P� Vogl� Crystal stability and structural transition pressuresof sp�bonded solids� Physical Review Letters� ������������ �����
����� G� Galli and M� Parrinello� Large scale electronic structure calculations� Phys�ical Review Letters� ������������ �����
����� S� Baroni and P� Giannozzi� Towards very large�scale electronic�structure cal�culations� Europhysics Letters� ���������� �����
����� X� P� Li� R� W� Nunes� and D� Vanderbilt� Density�matrix electronic�structuremethod with linear system�size scaling� Physical Review B� �������������������
����� L� W� Wang and M� P� Teter� Simple quantum�mechanical model of covalentbonding using a tight�binding basis� Physical Review B� �������������� �����
����� S� Goedecker� Low complexity algorithms for electronic structure calculations�Journal of Computational Physics� ����������� �����
����� S� Goedecker and L� Colombo� E�cient linear scaling algorithm for tight�binding molecular dynamics� Physical Review Letters� ���������� �����
���

����� E� V� Stefanovich� A� L� Shluger� and C� R� A� Catlow� Theoretical study of thestabilization of cubic�phase ZrO� by impurities� Physical Review B� �������������� �����
����� R� H� French� S� J� Glass� F� S� Ohuchi� Y� N� Xu� and W� Y� Ching� Exper�imental and theoretical determination of the electronic structure and opticalproperties of three phases of ZrO�� Physical Review B� ������������ �����
����� J� C� Phillips and L� Kleinman� New method for calculating wave functions incrystals and molecules� Physical Review� ����������� �����
����� M� L� Cohen� V� Heine� and J� C� Phillips� The quantum mechanics of materials�Scienti�c American� ���������� �����
����� W� C� Topp and J� J� Hop�eld� Chemically motivated pseudopotential forsodium� Physical Review B� ����������� �����
����� T� Starklo and J� D� Joannopoulos� Local pseudopotential theory for transitionmetals� Physical Review B� ������������ �����
����� D� R� Hamann� M� Schluter� and C� Chiang� Norm�conserving pseudopotentials�Physical Review Letters� ������������ �����
����� G� B� Bachelet� D� R� Hamann� and M� Schl#uter� Pseudopotentials that workFrom H to Pu� Physical Review B� ������������ �����
����� G� P� Kerker� Non�singular atomic pseudopotentials for solid state applications�Journal of Physics C� ��L�������� �����
����� M� L� Cohen and V� Heine� The �tting of pseudopotentials to experimental dataand their subsequent application� In H� Ehrenreich� F� Seitz� and D� Turnbull�editors� Solid state physics advances in research and applications� volume ���pages ������� London� ����� Academic�
����� S� G� Louie� S� Froyen� and M� L� Cohen� Nonlinear ionic pseudopotentials inspin�density�functional calculations� Physical Review B� ������������ �����
����� L� Kleinman and D� M� Bylander� E�cacious form for model pseudopotentials�Physical Review Letters� ������������ �����
����� X� Gonze� P� K#ackell� and M� Sche.er� Ghost states for separable� norm�conserving� ab initio pseudopotentials� Physical Review B� �������������������
����� R� P� Feynman� Forces in molecules� Physical Review� ���������� �����
����� F� Gygi� Adaptive Riemannian metric for plane�wave electronic�structure cal�culations� Europhysics Letters� ���������� �����
���

����� F� Gygi� Ab initio molecular dynamics in adaptive coordinates� Physical ReviewB� �������������� �����
����� F� Gygi and G Galli� Real�space adaptive�coordinate electronic�structure cal�culations� Physical Review B� ��R�����R����� �����
����� K� Cho� T� A� Arias� and J� D� Joannopoulos� Wavelets in electronic structurecalculations� Physical Review Letters� ������������ �����
����� A� Devenyi� K� Cho� T� A� Arias� and J� D� Joannopoulos� Adaptive Riemannianmetric for all�electron calculations� Physical Review B� �������������� �����
����� K� J� Chang and M� L� Cohen� High�pressure behavior of MgO Structural andelectronic properties� Physical Review B� ������������ �����
����� A� De Vita� M� J� Gillan� J� S� Lin� M� C� Payne� I� Stich� and L� J� Clarke�Defect energetics in MgO treated by �rst�principles methods� Physical ReviewB� �������������� �����
����� J� F� Mammone� H� K� Mao� and P� M� Bell� Equations of state of CaO understatic pressure conditions� Geophysical Research Letters� ��������� �����
����� H� K� Mao and P� M� Bell� Equations of state of MgO and epsilon Fe under staticpressure conditions� Journal of Geophysical Research� ������������ �����
����� M� J� L� Sangster� G� Peckham� and D� H� Saunderson� Lattice dynamics ofmagnesium oxide� Journal of Physics C� ����������� �����
����� O� L� Anderson and P� Andreatch Jr� Pressure derivatives of elastic constantsof single�crystal MgO at ��o and ������o C� Journal of the American CeramicSociety� ���������� �����
����� A� S� Rao and R� J� Kearney� Logarithmic derivative re�ectance spectra of BaOand SrO� Physica Status Solidi B� ���������� �����
����� D� M� Roessler and W� C� Walker� Electronic spectrum and ultraviolet opticalproperties of crystalline MgO� Physical Review� ����������� �����
����� The International Union of Crystallography� International Tables for Crystal�lography� Kluwer Academic Publishers� Dordrecht� �����
����� J� P� Perdew� R� G� Parr� M� Levy� and J� L� Balduz Jr� Density�functionaltheory for fractional particle number derivative discontinuities of the energy�Physical Review Letters� ������������ �����
����� D� K� Smith and H� W� Newkirk� The crystal structure of baddeleyite �mono�clinic ZrO�� and its relation to the polymorphism of ZrO�� Acta Cryst�� ���������� �����
���

����� P� Aldebert and J� P� Traverse� Structure and ionic mobility of zirconia at hightemperature� Journal of the American Ceramic Society� �������� �����
����� V� S� Stubican� Phase equilibria and metastabilities in the systems ZrO��MgO�ZrO��CaO� and ZrO��Y�O�� In S� Somiya� N� Yamamoto� and H� Yanagida�editors� Science and technology of zirconia III� volume ��� pages ������ West�erville� Ohio� ����� The American Ceramic Society�
����� J� R� Hellmann and V� S� Stubican� The existence and stability of Ca�Zr�O��
compound in the system ZrO��CaO� Materials Research Bulletin� ���������������
����� H� J� A� Koopmans� G� M� H� van de Velde� and P� J� Gellings� Powder neutrondiraction study of perovskites CaTiO� and CaZrO�� Acta Crystallographica�C������������ �����
����� A� Dwivedi and A� N� Cormak� A computer simulation study of the defectstructure of calcia�stabilized zirconia� Philosophical Magazine A� ������� �����
����� R� M� McMeeking and A� G� Evans� Mechanics of transformation�toughening inbrittle materials� Journal of the American Ceramic Society� ���������� �����
����� R� Orlando� C� Pisani� C� Roetti� and E� Stefanovich� Ab initio Hartree�Fockstudy of tetragonal and cubic phases of zirconium dioxide� Physical Review B����������� �����
����� H� J� F� Jensen� Electronic structure of cubic and tetragonal zirconia� PhysicalReview B� ������������ �����
����� H� J� F� Jensen and J� A� Gardner� Total energy calculations for ZrO�� PhysicalReview B� ��������� �����
����� F� Zandiehnadem� R� A� Murray� and W� Y� Ching� Electronic structures ofthree phases of zirconium oxide� Physica B� ��������� �����
����� R� E� Cohen� M� J� Mehl� and L� L� Boyer� Phase transitions and elasticity inzirconia� Physica B� ������� �����
����� R� J� Ackermann� E� G� Rauh� and C� A� Alexander� The thermodynamicproperties of ZrO��g�� High Temperature Science� ��������� �����
����� C� J� Howard� R� J� Hill� and B� E� Reichert� Structures of the ZrO� poly�morphs at room temperature by high�resolution neutron powder diraction�Acta Cryst�� ���������� �����
����� H� T� Stokes� Private communication�
����� R� Pawellek� M� Fahnle� C� Elsasser� K� M Ho� and C� T� Chan� First�principlescalculation of the relaxation around a vacancy and the vacancy formation energyin bcc Li� Journal of PhysicsCondensed Matter� ����������� �����
���

����� R� Magri� S� H� Wei� and A� Zunger� Ground�state structures and the random�state energy of the Madelung lattice� Physical Review B� �������������� �����
����� C� Wolverton� A� Zunger� S� Froyen� and S��H�Wei� Point�charge electrostaticsin disordered alloys� Physical Review B� ������������ �����
����� V� Heine� Electronic structure from the point of view of the local atomic en�vironment� In H� Ehrenreich� F� Seitz� and D� Turnbull� editors� Solid StatePhysics� volume ��� pages ������ New York� ����� Academic Press�
����� D� W� Bullet� The renaissance and quantitative development of the tight�binding method� In H� Ehrenreich� F� Seitz� and D� Turnbull� editors� SolidState Physics� volume ��� pages �������� New York� ����� Academic Press�
����� R� Haydock� The recursive solution of the Schr#odinger equation� In H� Ehren�reich� F� Seitz� and D� Turnbull� editors� Solid State Physics� volume ��� pages�������� New York� ����� Academic Press�
����� M� J� Kelley� Applications of the recursion method to the electronic structurefrom an atomic point of view� In H� Ehrenreich� F� Seitz� and D� Turnbull� edi�tors� Solid State Physics� volume ��� pages �������� New York� ����� AcademicPress�
����� J� A� Majewski and P� Vogl� Quantum theory of structure Tight�bindingsystems� In F� R� de Boer and D� G� Pettifor� editors� The Structure of BinaryCompounds� volume II��� page ���� New York� ����� North Holland�
����� E� E� Lafon and C� C� Lin� Energy band structure of lithium by the tight�binding method� Physical Review� ����������� �����
����� J� C� Browne and R� D� Poshusta� Quantum mechanical integrals over Gaussianatomic orbitals� Journal of Chemical Physics� ������������ �����
����� A� T� Paxton� A� P� Sutton� and C� M� M� Nex� Structural stability of silicon intight�binding models� Journal of Physics C Solid State Physics� ��L����L���������
����� D� G� Pettifor and R� Podloucky� Microscopic theory of the structural stabilityof pd�bonded AB compounds� Physical Review Letters� ������������ �����
����� M� J� Mehl and D� A� Papaconstantopoulos� Applications of a tight�bindingtotal�energy method for transition and noble metals Elastic constants� vacan�cies� and surfaces of monoatomic metals� Physical Review B� �����������������
����� P� O� L#owdin� On the non�orthogonality problem connected with the use ofatomic wave functions in the theory of molecules and crystals� The Journal ofChemical Physics� ���������� �����
���

����� R� R� Sharma� General expressions for reducing the Slater�Koster linear com�bination of atomic orbitals integrals to the two center approximation� PhysicalReview B� ������������ �����
����� R� R� Sharma� Improved general expressions for the Slater�Koster integrals inthe two center approximation� Physical Review B� ������������ �����
����� S� Froyen and W� A� Harrison� Elementary prediction of linear combination ofatomic orbitals matrix elements� Physical Review B� ������������ �����
����� W� A� Harrison� New tight�binding parameters for covalent solids obtainedusing Louie peripheral states� Physical Review B� ������������ �����
����� W� A� Harrison and G� K� Straub� Electronic structure and properties of d�and f�shell�metal compounds� Physical Review B� ������������ �����
����� J� L� Mercer and M� Y� Chou� Tight�binding model with intra�atomic matrixelements� Physical Review B� ������������ �����
����� W� M� C� Foulkes and R� Haydock� Tight�binding models and density�functional theory� Physical Review B� �������������� �����
����� L� Goodwin� A� J� Skinner� and D� G� Pettifor� Generating transferable tight�binding parameters Application to silicon� Europhysics Letters� ��������������
����� P� O� L#owdin� Quantum theory of cohesive properties of solids� Advances inPhysics� ������� �����
����� P� W� Anderson� Localized magnetic states in metals� Physical Review� ��������� �����
����� O� F� Sankey and J� D� Dow� Theory of tetrahedral�site interstitial s� andp�bonded impurities in Si� Physical Review B� ������������ �����
����� C� Huang� J� A� Moriarty� and A� Sher� Two�electron bond�orbital model� II�Physical Review B� ������������ �����
����� R� H� Parmenter� Eect of orbital degeneracy on the Anderson model of alocalized moment in a metal� Physical Review B� ����������� �����
����� Walter A� Harrison� Coulomb interactions in semiconductors and insulators�Physical Review B� ������������ �����
����� F� Liu� Self�consistent tight�binding method� Physical Review B� �������������� �����
����� M� O� Robbins and L� M� Falicov� Electronic theory of ordering and segregationin transition�metal alloys� Physical Review B� ������������ �����
���

����� M� van Schilfgaarde� A� B� Chen� and A� Sher� Coulomb energy in pseudobinaryalloys� Physical Review Letters� ������������ �����
����� J� A� Majewski and P� Vogl� Simple model for structural properties and crystalstability of sp�bonded solids� Physical Review B� ������������ �����
����� M� Kohyama� R� Yamamoto� Y� Ebata� and M� Kinoshita� Atomic forces in theself�consistent tight�binding model� Phys� Stat� Sol� �b�� ����������� �����
����� R� Homan� An extended H#uckel theory� I� Hydrocarbons� Journal of ChemicalPhysics� ������������ �����
����� W� A� Harrison� Theory of the two�center bond� Physical Review B� ������������ �����
����� D� W� Marquardt� An algorithm for least�squares estimation of nonlinear pa�rameters� J� Soc� Ind� Appl� Math� ���������� �����
����� O� K� Andersen� Z� Pawlowska� and O� Jepsen� Illustration of the linear�mu�n�tin�orbital tight�binding representation Compact orbitals and charge densityin Si� Physical Review B� ������������ �����
����� H� Shiba� A reformulation of the coherent potential approximation and itsapplications� Progress of Theoretical Physics� �������� �����
����� M� Monkenbusch� A set of routines for e�cient and accurate computation oflattice sums of � rn�potentials� Computer Physics Communications� ���������� �����
����� P� H� Dederichs and R� Zeller� Self�consistency iterations in electronic�structurecalculations� Physical Review B� ������������ �����
����� E� Issacson and H� B� Keller� Analysis of Numerical Methods� John Wiley -Sons� New York� �����
����� E� Anderson et� al� LAPACK Users� Guide� Society for Industrial and AppliedMathematics� Philadelphia� �����
����� M� Methfessel and A� T� Paxton� High�precision sampling for Brillouin�zoneintegration in metals� Physical Review B� ������������ �����
����� A� F� Kohan and G� Ceder� Calculation of total energies in multicomponentoxides� Computational Materials Science� �� ����� in press�
����� P� Vogl� H� P� Hjalmarson� and J� D� Dow� A semi�empirical tight�bindingtheory of the electronic structure of semiconductors� J� Phys� Chem� Solids����������� �����
����� D� A� Papaconstantopoulos� Handbook of the band structure of elemental solids�Plenum Press� New York� �����
���

����� M� Rosenbauer and H� J� F� Jansen� Slater�Koster interpolation of energybands of complex crystal structures Tetragonal zirconia� Physical Review B��������������� �����
����� N� F� Mott� The energy of the superlattice in � brass� The Proceedings of thePhysical Society �London�� ���������� �����
����� J� S� Faulkner� Y� Wang� and G� M� Stocks� Electrons in extended systems�Physical Review B� �������������� �����
����� J� P� Perdew� K� Burke� and M� Ernzerhof� Generalized gradient approximationmade simple� Physical Review Letters� ������������ �����
����� D� R� Hamann� Generalized gradient theory for silica phase transitions� PhysicalReview Letters� ���������� �����
����� C� Filippi� D� J� Singh� and C� J� Umrigar� All�electron local�density andgeneralized�gradient calculations of the structural properties of semiconductors�Physical Review B� �������������� �����
����� V� I� Anisimov� J� Zaanen� and O� K� Andersen� Band theory and Mott insula�tors Hubbard U instead of Stoner I� Physical Review B� ���������� �����
����� V� I� Anisimov� F� Aryasetiawan� and A� I� Lichtenstein� First�principles cal�culations of the electronic structure and spectra of strongly correlated systemsthe LDA � U method� Journal of Physics� ��������� �����
����� T� A� Arias and J� D� Joannopoulos� Ab initio theory of dislocation interactionsfrom close�range spontaneous annihilation to the long�range continuum limit�Physical Review Letters� ���������� �����
����� J� Rath and A� J� Freeman� Generalized magnetic susceptibilities in metalsApplication of the analytic tetrahedron linear energy method� Physical ReviewB� ����������� �����
����� J� Hama and M� Watanabe� General formulae for the special points and theirweighting factors in k�space integration� J� Phys�Condens� Matter� ����������� �����
����� D� J� Chadi and M� L� Cohen� Special points in the Brillouin zone� PhysicalReview B� ����������� �����
����� D� J� Chadi� Special points for Brillouin�zone integrations� Physical Review B������������� �����
����� J� D� Pack and H� J� Monkhorst� Special points for Brillouin�zoneintegrations��a reply� Physical Review B� ������������ �����
����� H� J� Monkhorst and J� D� Pack� Special points for Brillouin�zone integrations�Physical Review B� ������������ �����
���

����� S� Froyen� Brillouin�zone integration by Fourier quadrature special points forsuperlattice and supercell calculations� Physical Review B� ������������ �����
����� M� K� Aydinol� A� F� Kohan� and G� Ceder� Ab�initio calculation of the inter�calation voltage of lithium�transition�metal oxide electrodes for rechargeablebatteries� Journal of Power Sources� ����� in press�
����� G� Ceder� M� K� Aydinol� and A� F� Kohan� Application of �rst�principlescalculations to the design of rechargeable Li�Batteries� Computational MaterialsScience� �� ����� in press�
����� M� K� Aydinol� G� Ceder� and A� F� Kohan� Eect of chemistry and structureon voltage of Li intercalation oxides� In A� Jacobson� P� Davies� T� Vanderah�and C� Torardi� editors� Solid�State Chemistry of Inorganic Materials� MaterialsResearch Society Symposium Proceedings� volume ���� Pittsburgh� ����� Mater�Res� Soc�� in press�
����� M� K� Aydinol� A� F� Kohan� G� Ceder� K� Cho� and J� Joannopoulos� Ab initiostudy of lithium intercalation in oxides and metal dichalcogenides� PhysicalReview B� ����� in press�
����� W� R� McKinnon� Insertion electrodes I Atomic and electronic structure of thehosts and their insertion compounds� In P� G� Bruce� editor� Solid state electro�chemistry� volume ��� pages �������� Cambridge� ����� Cambridge UniversityPress�
����� K� Mizushima� P� J� Jones Wiseman� and J� B� Goodenough� LixCoO� �� x� �� a new cathode material for batteries of high energy density� MaterialsResearch Bulletin� ���������� �����
����� M� M� Thackeray� L� A� de Picciotto� W� I� F David� P� G Bruce� and J� B�Goodenough� Structural re�nement of delithiated LiVO� by neutron diraction�Journal of Solid State Chemistry� ���������� �����
����� L� A� De Picciotto� M� M� Thackeray� and G� Pistoia� An electrochemicalstudy of the systems Li��xV�O� and Li��xVO� ���x���� Solid State Ionics���������������� �����
����� T� Ohzuku and A� Ueda� Solid�state redox reactions of LiCoO� �R(�m� for � voltsecondary lithium cells� Journal of the Electrochemical Society� ������������������
����� T� Ohzuku� A� Ueda� and M� Nagayama� Electrochemistry and structural chem�istry of LiNiO� �R(�m� for � volt secondary lithium cells� Journal of the Elec�trochemical Society� ������������� �����
����� W� B� Pearson� P Villars� and L� D� Calvert� Pearson�s Handbook of Crystal�lographic data for intermetallic phases� American Society for Metals� MetalsPark� Ohio� �����
���