tema 13. sequence comparison. concept of homology. sequence alignment. comparison strategies. blast,...
Post on 18-Dec-2015
220 views
TRANSCRIPT

Tema 13. Sequence comparison. Concept of homology. Sequence alignment. Comparison
strategies. BLAST, PSI-Blast. Multiple alignment, profiles. Families of proteins.
Functional prediction based on sequence.
Gabriel Pons, Departament de Ciències Fisiològiques II, Campus deCiències de la salut. Bellvitge. Universitat de Barcelona

Sequence comparison

Goals
• To take advantage from functional or structural information identifiyng homologies between sequences
• Differences between Homology and identity
• Two sequences are homologous when:– They have the same evolutive origin– They have similar function and structure

• Homologous sequences - sequences that share a commonevolutionary ancestry• Similar sequences - sequences that have a high percentage ofaligned residues with similar physicochemical properties(e.g., size, hydrophobicity, charge)
IMPORTANT:• Sequence homology:• An inference about a common ancestral relationship, drawn whentwo sequences share a high enough degree of sequence similarity• Homology is qualitative• Sequence similarity:• The direct result of observation from a sequence alignment• Similarity is quantitative; can be described using percentages

More definitions
• Orthologs: sequences which exactely correspond to the same function/structure in different species
• Paralogs: sequences produced by gene duplications in the same organism. Usually, it involves change in function, but keeping functional relationship many times.
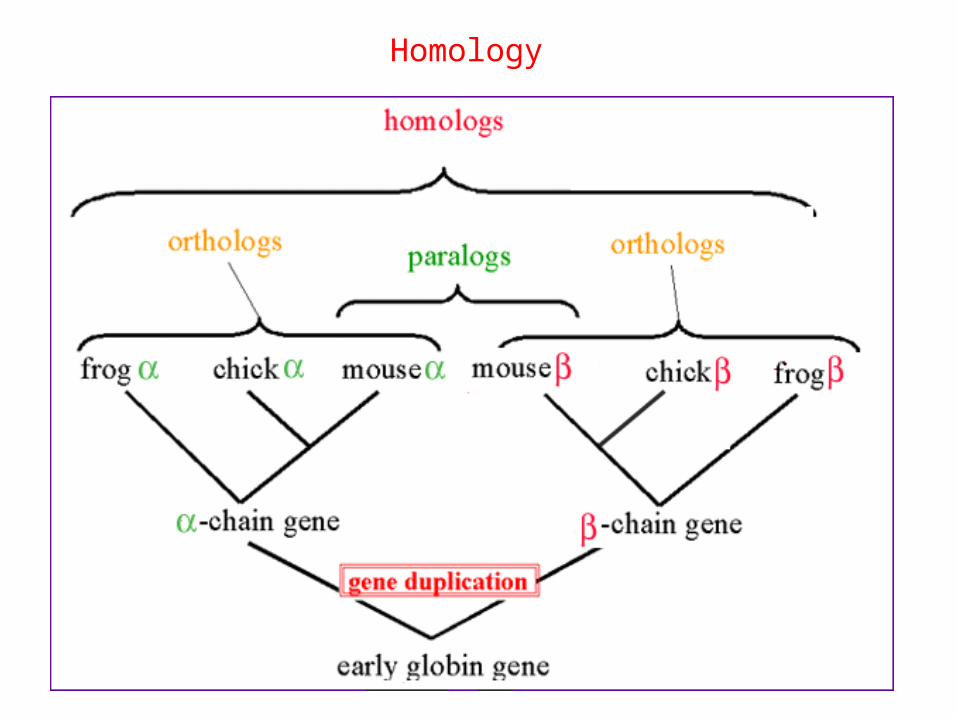
Homology

Homology and prediction
• Very divergent protein sequences may suport similar structures
• Similar protein structures will probably have related or similar functions

3D STRUCTURE VERSUS SEQUENCESequence alignment between human myoglobin, and globins from hemoglobin
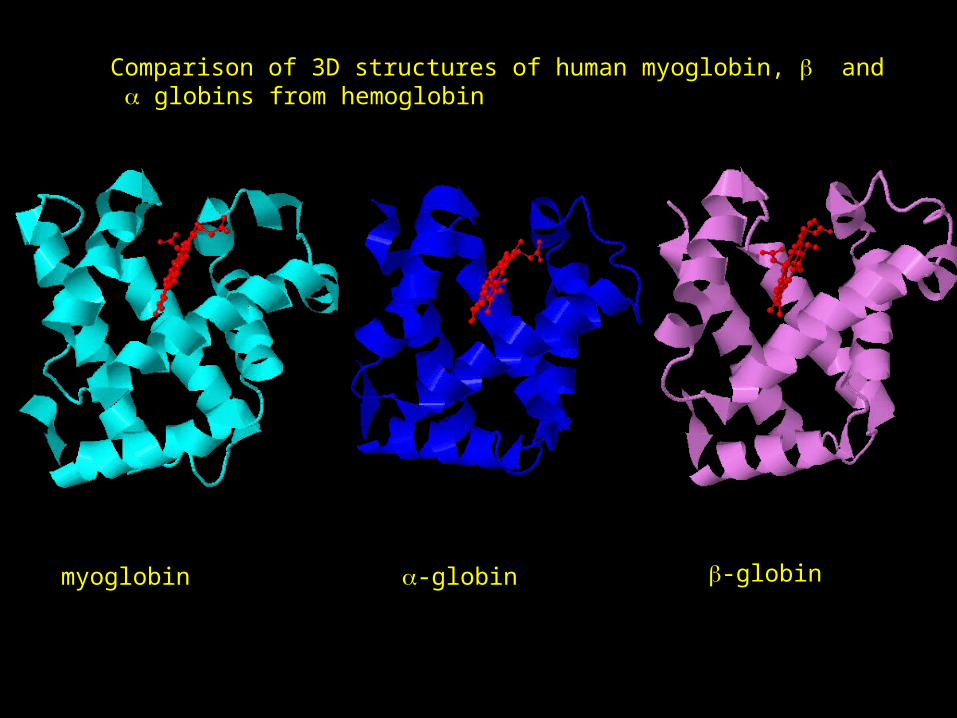
myoglobin -globin -globin
Comparison of 3D structures of human myoglobin, and globins from hemoglobin
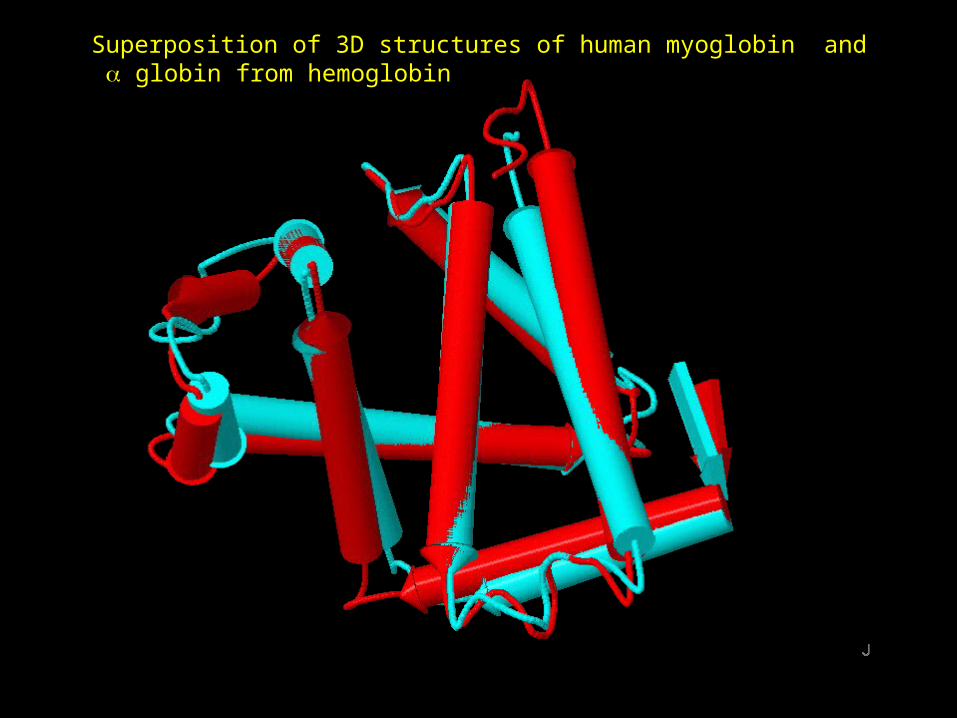
Superposition of 3D structures of human myoglobin and globin from hemoglobin

Homology and prediction
• Sequence comparison is the simplest method in order to identify the presence of homology between sequences.
• Identity > 30% in proteins involves homology (>65% nucleic)
• Identity > 80-90% usual in orthologs from close species
• Identity 10-30%. If there is homology may be not detectable (“twilight zone”)

No me gusta la bioinformaticaTeme usted la ionosfera optica
Nomegusta-labioin-forma--ticaTeme-ustedla-ionosfer-aoptica
64% identity? But…
I don´t like bioinformaticsDo you fear optical ionospher?

¿DNA or protein?
• Both give information about homología
• Protein: Exists functional equivalence between aminoacids
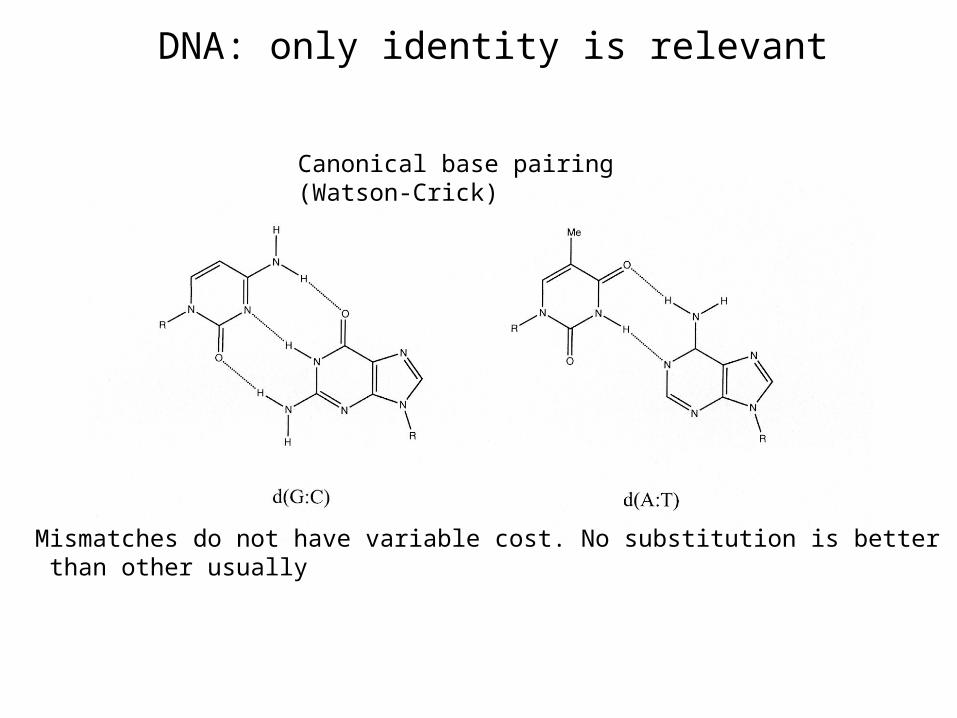
DNA: only identity is relevant
Mismatches do not have variable cost. No substitution is better than other usually
Canonical base pairing (Watson-Crick)
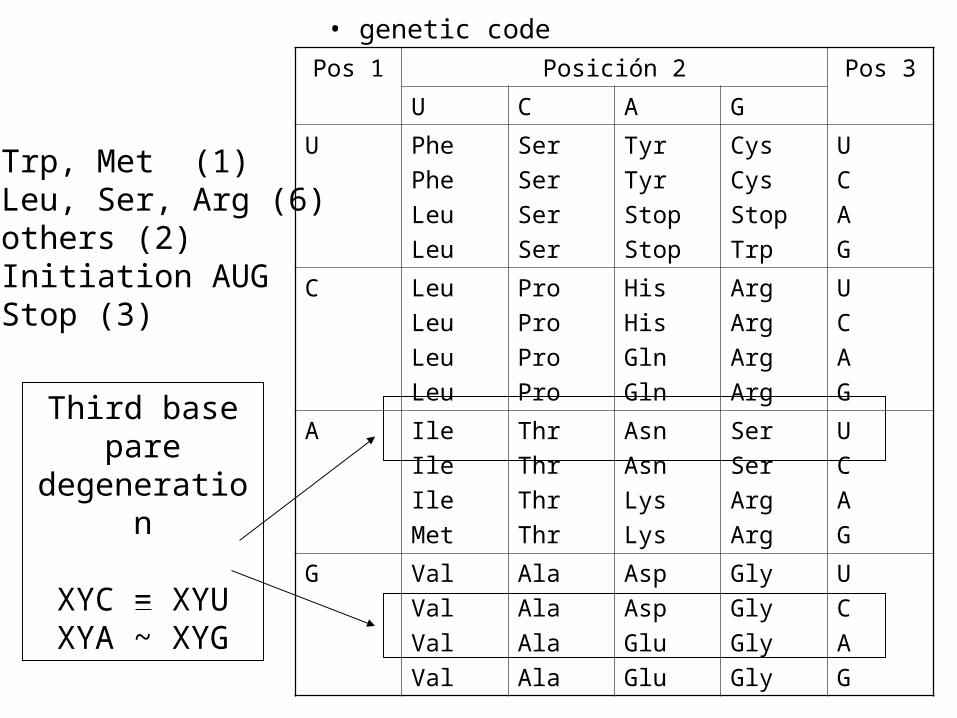
• genetic code
Pos 1 Posición 2 Pos 3
U C A G
U Phe
Phe
Leu
Leu
Ser
Ser
Ser
Ser
Tyr
Tyr
Stop
Stop
Cys
Cys
Stop
Trp
U
C
A
G
C Leu
Leu
Leu
Leu
Pro
Pro
Pro
Pro
His
His
Gln
Gln
Arg
Arg
Arg
Arg
U
C
A
G
A Ile
Ile
Ile
Met
Thr
Thr
Thr
Thr
Asn
Asn
Lys
Lys
Ser
Ser
Arg
Arg
U
C
A
G
G Val
Val
Val
Val
Ala
Ala
Ala
Ala
Asp
Asp
Glu
Glu
Gly
Gly
Gly
Gly
U
C
A
G
• Trp, Met (1)• Leu, Ser, Arg (6)• others (2)• Initiation AUG• Stop (3)
Third base pare degeneration
XYC = XYUXYA ~ XYG

“Equivalent aminoacids”
• Hydrophobics– Ala (A), Val (V), Met (M), Leu (L), Ile (I), Phe (F), Trp (W), Tyr (Y)
• Small– Gly (G), Ala (A), Ser (S)
• Polar– Ser (S), Thr (T), Asn (N), Gln (Q), Tyr (Y) – En la superficie de la proteína polares y cargados son equivalentes
• With charge– Asp (D), Glu (E) / Lys (K), Arg (R)
• Difficult to be substituted– Gly (G), Pro (P), Cys (C), His (H)
• BE CAREFULL: aminoacids do not always perform the same function in proteins
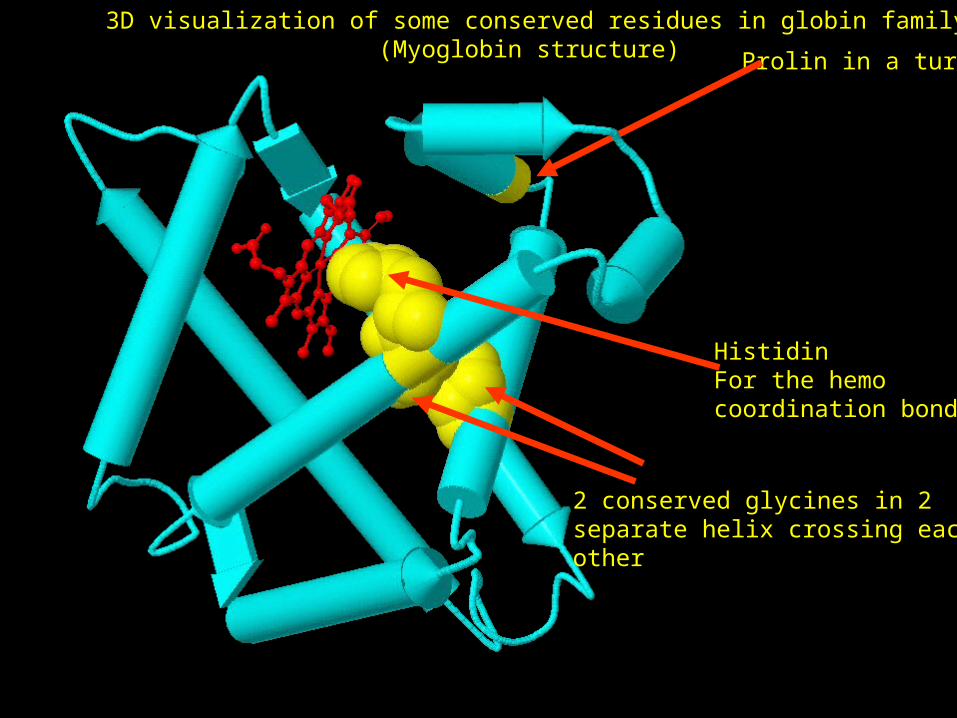
HistidinFor the hemo coordination bonds
Prolin in a turn
2 conserved glycines in 2 separate helix crossing each other
3D visualization of some conserved residues in globin family (Myoglobin structure)

• DNA sequence diverges quicker than protein– Mutation or recombination may alter DNA but must
mantain function/structure
• Protein sequence comparison permits finding and localize very distant homologous proteins

Sequence alignment
• Measure the degree of similarity/identity and thus the existence of homology requires un “alignment”
Strong identity/similarity:
AWTRRATVHDGLMEDEFAAAWTRRATVHDGLCEDEFAA
Weak identity/similarity:
AWTKLATAVVVFEGLCEDEWGGAWTRRAT---VHDGLMEDEFAA

Alignments
• “pairwise”– 2 sequences
• Multiple– More than 2 sequences
• Global– Whole sequence is considered
• Local– Only similar regions are aligned

StrategiesDepends of the goal
• Sequence comparison– Goal: establish homology, identify equivalent
aminoacuds • global, ”pairwise”/multiple
• Search in data bases– Goal: Identify homologous proteins in a big
group of sequences• Local, “pairwise”

Automatic Alignment
• Requires – Objective method to compare aminoacids or bases in
order to “score” the alignment (comparison matrix)– Algoritm to find the best alignment with the maximal
score
• Quick and easy to reproduce
• Do not permit, in general, introduce additional information

Matrix types
• Identity
• Physico-chemical properties
• Genetics (codon substitution)
• Evolution

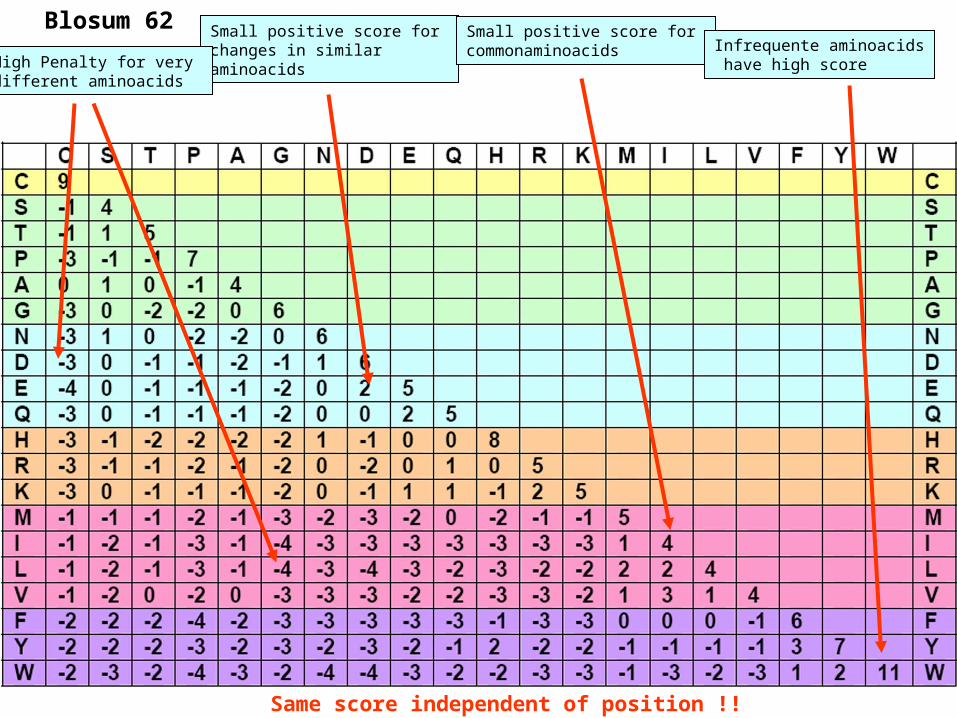
Blosum 62 Small positive score for changes in similaraminoacids
Small positive score for commonaminoacids Infrequente aminoacids
have high scoreHigh Penalty for very different aminoacids
Same score independent of position !!

Rat versus mouse protein
Rat versus bacterialprotein
BLOSUM90PAM30
BLOSUM45PAM240
BLOSUM80PAM120
BLOSUM62PAM180
Choice of a Matrix!
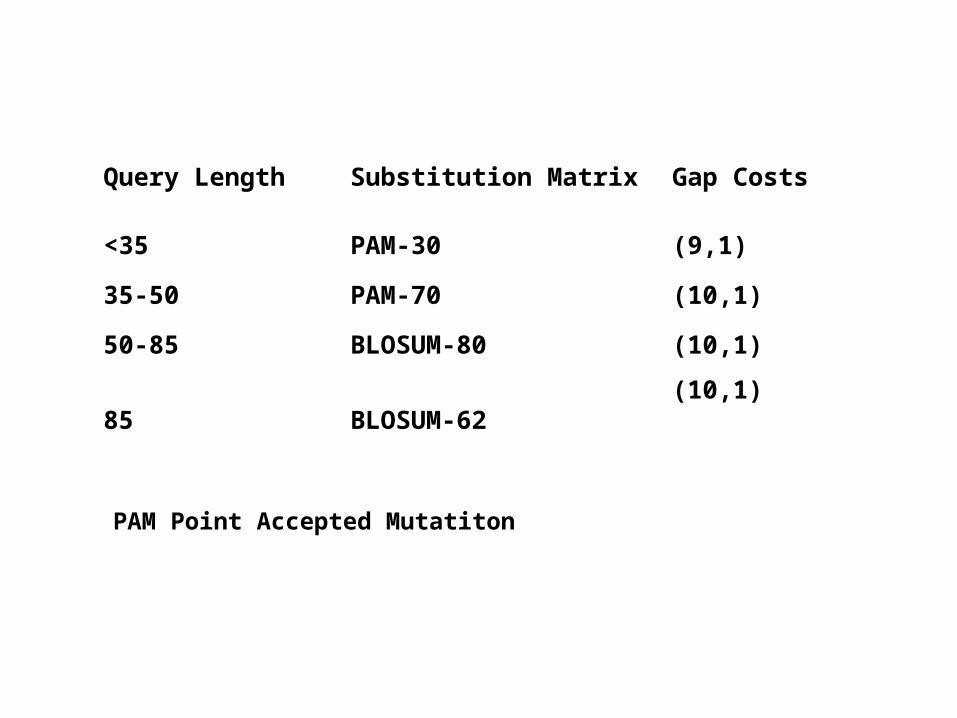
Query Length Substitution Matrix Gap Costs
<35 PAM-30 (9,1)
35-50 PAM-70 (10,1)
50-85 BLOSUM-80 (10,1)
85 BLOSUM-62(10,1)
PAM Point Accepted Mutatiton
Gaps (inserciones/delecciones)
• Normalmente localizados en loops
AWTKLATAVVVFEGLCEDEWGAWTKLATAVVVFEGLCEDEWGGGAWTRRAT---AWTRRAT---VHDGLMEDEFAAVHDGLMEDEFAA

Global versus local alignment
• Global alignment– Finds best possible alignment across entire length of 2
sequences– Aligned sequences assumed to be generally similar over entire
length• Local alignment
– Finds local regions with highest similarity between 2 sequences– Aligns these without regard for rest of sequence– Sequences are not assumed to be similar over entire length

Comparación de secuencias contra bases de datos
Secuencia incógnitaATTVG...LMN
Base de datos De secuencias
AGLM...WTKRTCGGLMN..HICGWRKCPGL...
Requiere algoritmos de comparación muy rápidos

Alignments
• “pairwise”– 2 sequences
• Multiple– More than 2 sequences
• Global– Whole sequence is considered
• Local– Only similar regions are aligned
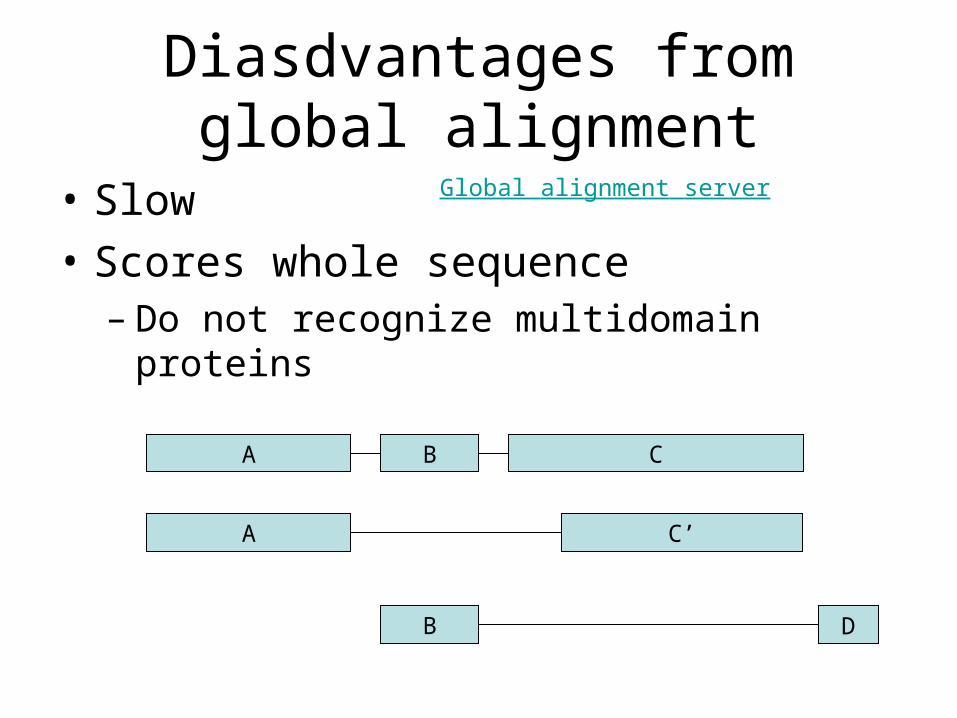
Diasdvantages from global alignment
• Slow
• Scores whole sequence– Do not recognize multidomain proteins
A B C
A C’
B D
Global alignment server
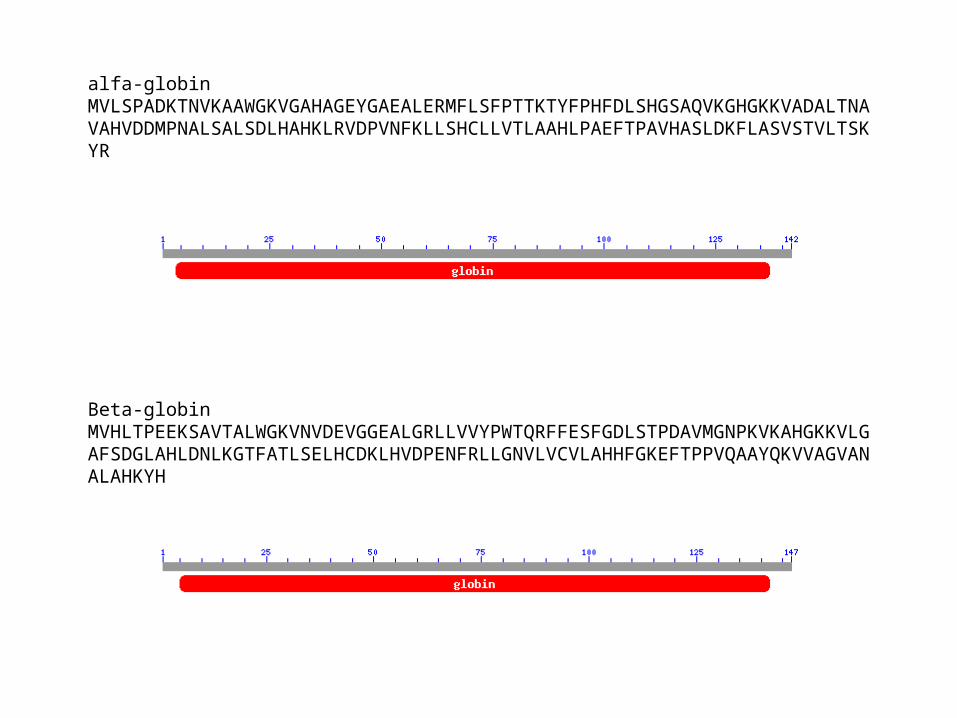
alfa-globinMVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYR
Beta-globinMVHLTPEEKSAVTALWGKVNVDEVGGEALGRLLVVYPWTQRFFESFGDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVANALAHKYH

Alfa-actininMNQIEPGVQYNYVYDEDEYMIQEEEWDRDLLLDPAWEKQQRKTFTAWCNSHLRKAGTQIENIEEDFRNGLKLMLLLEVISGERLPKPDRGKMRFHKIANVNKALDYIASKGVKLVSIGAEEIVDGNVKMTLGMIWTIILRFAIQDISVEETSAKEGLLLWCQRKTAPYRNVNIQNFHTSWKDGLGLCALIHRHRPDLIDYSKLNKDDPIGNINLAMEIAEKHLDIPKMLDAEDIVNTPKPDERAIMTYVSCFYHAFAGAEQAETAANRICKVLAVNQENERLMEEYERLASELLEWIRRTIPWLENRTPEKTMQAMQKKLEDFRDYRRKHKPPKVQEKCQLEINFNTLQTKLRISNRPAFMPSEGKMVSDIAGAWQRLEQAEKGYEEWLLNEIRRLERLEHLAEKFRQKASTHETWAYGKEQILLQKDYESASLTEVRALLRKHEAFESDLAAHQDRVEQIAAIAQELNELDYHDAVNVNDRCQKICDQWDRLGTLTQKRREALERMEKLLETIDQLHLEFAKRAAPFNNWMEGAMEDLQDMFIVHSIEEIQSLITAHEQFKATLPEADGERQSIMAIQNEVEKVIQSYNIRISSSNPYSTVTMDELRTKWDKVKQLVPIRDQSLQEELARQHANERLRRQFAAQANAIGPWIQNKMEEIARSSIQITGALEDQMNQLKQYEHNIINYKNNIDKLEGDHQLIQEALVFDNKHTNYTMEHIRVGWELLLTTIARTINEVETQILTRDAKGITQEQMNEFRASFNHFDRRKNGLMDHEDFRACLISMGYDLGEAEFARIMTLVDPNGQGTVTFQSFIDFMTRETADTDTAEQVIASFRILASDKPYILAEELRRELPPDQAQYCIKRMPAYSGPGSVPGALDYAAFSSALYGESDL
CalmodulinMADQLTEEQIAEFKEAFSLFDKDGDGTITTKELGTVMRSLGQNPTEAELQDMINEVDADGNGTIDFPEFLTMMARKMKDTDSEEEIREAFRVFDKDGNGYISAAELRHVMTNLGEKLTDEEVDEMIREADIDGDGQVNYEEFVQMMTAK

Alineamiento local
• 10 – 100x más rápidos
• Reconocen dominios individuales
• No proporcionan necesariamente el mejor alineamiento!
• BLAST, FASTA

Basic Local Alignment Search ToolBlast NCBI



E value (Expect)
• E value:• Expect: This setting specifies the statistical significance threshold for reporting
matches against database sequences. The default value (10) means that 10 such matches are expected to be found merely by chance, according to the stochastic model of Karlin and Altschul (1990). If the statistical significance ascribed to a match is greater than the EXPECT threshold, the match will not be reported. Lower EXPECT thresholds are more stringent, leading to fewer chance matches being reported.
E = K.m.n.e-.S
• Warning:
• E → Falsos negativos
Score
Normalization factors
Number of letters in query
Number of letters in data baseScore

E parameter (More)• Expect
For example, an E value of 1 assigned to a hit can be interpreted as meaning that in a database of the current size one might expect to see 1 match with a similar score simply by chance. This means that the lower the E-value, or the closer it is to "0" the more "significant" the match is. However, keep in mind that searches with short sequences, can be virtually indentical and have relatively high EValue. This is because the calculation of the E-value also takes into account the length of the Query sequence. This is because shorter sequences have a high probability of occuring in the database purely by chance.

Exercice
• Find mouse orthologous. Data
• Find closest human paralogous
• Find highest significant homolog in drosophila