temporal stability of magic-number metal clusters: beyond the shell closing model
TRANSCRIPT

Nanoscale
PAPER
Dow
nloa
ded
by P
enns
ylva
nia
Stat
e U
nive
rsity
on
04 M
arch
201
3Pu
blis
hed
on 1
4 Ja
nuar
y 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3N
R33
705G
View Article OnlineView Journal | View Issue
Department of Chemistry, The University of
† Electronic supplementary informa10.1039/c3nr33705g
‡ Present address: Department of ChePittsburgh, PA.
Cite this: Nanoscale, 2013, 5, 2036
Received 19th November 2012Accepted 11th January 2013
DOI: 10.1039/c3nr33705g
www.rsc.org/nanoscale
2036 | Nanoscale, 2013, 5, 2036–204
Temporal stability of magic-number metal clusters:beyond the shell closing model†
Anil Desireddy, Santosh Kumar,‡ Jingshu Guo, Michael D. Bolan, Wendell P. Griffithand Terry P. Bigioni*
The anomalous stability of magic-number metal clusters has been associated with closed geometric and
electronic shells and the opening of HOMO–LUMO gaps. Despite this enhanced stability, magic-number
clusters are known to decay and react in the condensed phase to form other products. Improving our
understanding of their decay mechanisms and developing strategies to control or eliminate cluster
instability is a priority, to develop a more complete theory of their stability, to avoid studying mixtures of
clusters produced by the decay of purified materials, and to enable technology development. Silver
clusters are sufficiently reactive to facilitate the study of the ambient temporal stability of magic-number
metal clusters and to begin to understand their decay mechanisms. Here, the solution phase stability of a
series of silver:glutathione (Ag:SG) clusters was studied as a function of size, pH and chemical
environment. Cluster stability was found to be a non-monotonic function of size. Electrophoretic
separations showed that the dominant mechanism involved the redistribution of mass toward smaller
sizes, where the products were almost exclusively previously known cluster sizes. Optical absorption
spectra showed that the smaller clusters evolved toward the two most stable cluster sizes. The net surface
charge was found to play an important role in cluster stabilization although charge screening had no
effect on stability, contrary to DLVO theory. The decay mechanism was found to involve the loss of Ag+
ions and silver glutathionates. Clusters could be stabilized by the addition of Ag+ ions and destabilized by
either the addition of glutathione or the removal of Ag+ ions. Clusters were also found to be most stable
in near neutral pH, where they had a net negative surface charge. These results provide new mechanistic
insights into the control of post-synthesis stability and chemical decay of magic-number metal clusters,
which could be used to develop design principles for synthesizing specific cluster species.
Introduction
Magic-number clusters address the fundamental question ofwhy nanoscale objects should be stable.1–3 Their enhancedstability has been attributed to closed electronic and geometricshells, leading to the emergence of discrete size distributionscomposed of only certain sizes of metal clusters.4–13 Totalstructure determination of four members of the Au:thiolate(Au:SR) family has provided the rst opportunities to test theseshell models in detail in the condensed phase, and have thus farvalidated theory.14–18
While closed-shell congurations have generally beenaccepted as important attributes for stability, studies havefocused on cluster formation. The temporal stability of thesesystems must also be considered in order to broaden our
Toledo, Toledo, OH 43606, USA
tion (ESI) available. See DOI:
mistry, Carnegie Mellon University,
4
understanding of the general question of enhanced stability.Further, the control of cluster stability and decay will be of crit-ical importance for the development of design principles forsynthesizing specic cluster species19–21 as well as useful mate-rials and applications. For example, while most applicationswould require stability, biomedical applications might require acontrolled decay of the metal clusters.22,23 Despite these needs,the long-term chemical stability and reactivity of already-formedmagic-number clusters have yet to be systematically explored.
Although etching studies have been done,20,24–26 little atten-tion has been paid to the ambient time-evolution of metal clus-ters in the solution phase.20,25 This is not surprising, however,since gold is generally the metal of choice due to its less reactivenature. For other less stable materials systems, however, it isimperative that great care be taken to avoid studyingmixtures ofclusters produced by the chemical evolution of putatively puresamples. This is acutely important when an impurity species candominate the measurement of the species of interest, as can bethe case with uorescence, for example. Here, we take advantageof the greater reactivity of silver to examine the ambient stabilityof metal magic-number clusters. Using this advantage, the
This journal is ª The Royal Society of Chemistry 2013

Paper Nanoscale
Dow
nloa
ded
by P
enns
ylva
nia
Stat
e U
nive
rsity
on
04 M
arch
201
3Pu
blis
hed
on 1
4 Ja
nuar
y 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3N
R33
705G
View Article Online
detailed mechanisms of metal cluster evolution and decay cannow begin to be uncovered.
The use of small silver:glutathione (Ag:SG) clusters12 hasallowed cluster evolution to be studied on practical timescalesand with easily measured effects. The sizes of the smaller Ag:SGclusters are similar to those of the well-known Au:SG clusterseries,12,27 although the larger Ag:SG clusters have no sizeanalogue. While the masses and formulae of the Au:SG clustersare well known,6–8 only the formula for Ag32(SG)19 (band 6 in theAg:SG series) has been identied thus far.27 Regardless of theiridentities, however, the chemical evolution of this series ofAg:SG clusters can still provide valuable insights into thegeneral mechanisms of the chemical evolution and decay ofmetal clusters.
To elucidate the chemical evolution of Ag:SG clusters,mixtures of different cluster sizes were separated by poly-acrylamide gel electrophoresis (PAGE).5–8,12 Once separated, thestability of each size of cluster was studied independently. Clus-ters were aged in aqueous solution under ambient conditions,since storing indry powder formor in thepolyacrylamidegelwerepreviously found to provide some level of protection.12 Usingthese methods, the general characteristics of cluster evolutionwere rst determined, followed by the details of the decaymechanisms as well as methods of slowing or halting them.
Experimental
Ag:SG clusters were synthesized as reported elsewhere.12 Briey,aqueous silver nitrate (5 � 10�3 M) was combined with gluta-thione (2 � 10�2 M) to form silver thiolate.28 This was
Fig. 1 Separation of Ag:SG clusters. (Top) Different sizes of Ag:SG clusters were selowest index. (Middle) Gel bands were separated from one another by cutting the ggels from which they were extracted, due to aging of the clusters.
This journal is ª The Royal Society of Chemistry 2013
immediately reduced with 0.2 M sodium borohydride to formAg:SG clusters. The reaction product was puried by repeatedprecipitation and washing. PAGE was then used to separate theraw product into bands of the component magic-numberedAg:SG clusters, each with widely varying colors and abun-dances,12 as shown in Fig. 1. PAGE experiments were run usinghomemade polyacrylamide gels with 30% density and withoutsurfactants.6
For some experiments, these bands were excised from the geland then the clusters in each were extracted to form solutions ofthe puried components, as shown in Fig. 1. Extraction ofclusters was done by rst crushing the gel, soaking in water for2–4 hours, and then centrifuging to remove pieces of gel fromthe resultant colored solutions. Solutions were ltered througha 0.22 mm syringe lter to remove the remaining gel fragmentsand then concentrated with a 3 kDa cutoff lter. Sample ageswere reported with respect to the time that water was rst addedto the crushed gel.
For aging experiments with Ag:SG cluster mixtures, solutionswere prepared identically by splitting one larger solution intomultiple equal aliquots. Each aliquot was then aged underdifferent conditions.
To compare aging times, all samples were initially frozen togreatly impede or stop aging. Each sample was then thawed atdifferent time points, using small sample volumes to facilitaterapid warming, and then stored under ambient conditions onthe bench top. All samples were loaded and run in the same gelonce the last sample was thawed.
To determine the effects of pH on aging, the pH of eachsolution was adjusted with an appropriate buffer for that pH
parated into differently colored bands using PAGE. The smallest clusters have theel. (Bottom) Aqueous solutions of purified clusters have different colors than the
Nanoscale, 2013, 5, 2036–2044 | 2037

Nanoscale Paper
Dow
nloa
ded
by P
enns
ylva
nia
Stat
e U
nive
rsity
on
04 M
arch
201
3Pu
blis
hed
on 1
4 Ja
nuar
y 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3N
R33
705G
View Article Online
range. Buffers were selected such that there was overlapwhenever possible, to ensure that chemical effects were notmistaken for pH effects.
Solutions of the different puried Ag:SG cluster sizes couldnot be prepared with identical concentrations, however, due tothe wide variation in abundance of each cluster species as wellas their unknown molar masses and molar absorptivities. Inthis case, only the relative concentrations were measured overtime, as were the nal mass distributions.
Optical absorption spectra were measured for extractedclusters as well as for clusters that remained in the gel. Apertureswere used to measure the absorption of individual bands in thegel, in order to minimize interference from neighboring bands.Although aging in the gel is very slow, spectra were measuredimmediately aer excising the bands to minimize aging.
Results and discussion
Although the existence of magic-numbered Ag:SG clustersimplies enhanced stability, solutions of the clusters are notindenitely stable.12 For example, changes in the color of Ag:SGcluster solutions indicated that chemical changes were occur-ring over the course of days, as can be seen by comparing themiddle and lower panels of Fig. 1. Drying the clusters appearedto stop the evolution, however, while refrigeration slowed downthe changes.
To gain a basic understanding of how Ag:SG clusters changein time, solutions of unseparated particles were aged fordifferent periods of time and then were separated together onthe same gel, as shown in Fig. 2. The changes over time werereadily apparent. In general, the smallest clusters were observedto be most stable and the largest were least stable. This isconsistent with the smallest clusters having the largest HOMO–LUMO gaps, which would make them most resistant to chem-ical reactions. Notably, the clusters in bands 2 and 6 wereamong the most stable bands and have the largest reportedHOMO–LUMO gaps.12
The trends in stability were in general non-monotonic,however. For example, the intensities of bands 13, 15, and 16 in
Fig. 2 Time evolution of Ag:SG clusters in water. Aqueous solutions were agedfor different times, as labeled, and run together on a PAGE gel. Band indices arealso labeled.
2038 | Nanoscale, 2013, 5, 2036–2044
Fig. 2 appear to rapidly decay, while bands 12 and 14 were stillvisible aer 1week. Further, somebandswere alwaysmore intensethanothers, e.g.bands2, 6, 9, 12, and14, suggesting thatmore thanjust size determines the stability of these metal clusters.
While most of the bands were observed to decay, the inten-sity of some bands increased. This indicated that mass wasredistributed among the different cluster sizes. For example,bands 2 and 12 appeared to deepen in intensity over time. Also,a new green band appeared by the end of the time series,between the positions of bands 13 and 14, but did not appear tobe part of the initial mass distribution.
Gas phase studies1–3 found that the most stable clusters werethe most abundant in the mass distributions. Further, sincethere are more opportunities for unstable clusters to decay insolution than in the gas phase, abundance and stability oughtto be closely related. Interestingly, the decay of band 13 was notconsistent with these expectations since its abundance was thehighest of any cluster size yet its decay rate was among thefastest (see Fig. 2). Again, this suggests that the nature of clusterstability is more complex than simply electronic shell closings.
It is important to note here that the band 13 cluster isdistinct from the Ag44L30 cluster.29,30 Although the clusters aresimilar in size and color,12 their spectra are quite different. Infact, none of the spectra of Ag:SG clusters resembled that of theAg44L30 cluster. This suggests that the nature of the ligands, inthis case alkyl versus phenyl, plays a role in selecting the corestructure.
To better understand the decay mechanisms at play, themixture of Ag:SG clusters was separated such that each puriedcomponent could be studied independently. The spectra of themost abundant clusters were recorded at different time pointsover the course of three days, as shown in Fig. 3. The spectra ofband 2 and band 6 clusters appeared to simply decay in time,while the spectra of band 9 and band 13 clusters appeared toevolve in shape over time.
Absorption spectra were also recorded for bands 1–14 aer 3days of aging. Interestingly, the spectra evolved into a smallsubset of forms, as can be seen in Fig. 3(e). For example, solu-tions from bands 1–4 all shared the spectral form of band 2clusters, and solutions from bands 5–10 all resembled thespectrum of the band 6 cluster. This suggests that the differentcluster sizes evolved into these two most stable cluster sizes,consistent with the electronic shell-closing model. Such aconversion of one particle size into another is well known inetching reactions, which normally involve the addition of excessligands (see Fig. S2†).24 Larger clusters, from bands 11–14, donot share a spectral form, however.
The rate of change of the spectra increased monotonicallywith increasing size, as shown in Fig. 3(f). This is consistent withthe previous observation that smaller clusters tended to bemorestable than larger clusters. As expected, band 2 was the moststable in time, followed by band 6 (the Ag32(SG)19 cluster).27
Although changes in spectra becamemore rapid with increasingparticle size, it is difficult to precisely quantify the decay kineticssince aging necessarily produces mixtures of clusters.
To gain a qualitative understanding of the decay mecha-nism, a series of separated and extracted cluster bands was aged
This journal is ª The Royal Society of Chemistry 2013

Fig. 3 (a–d) Time series of Ag:SG cluster absorption spectra. Spectra of smaller clusters appear to simply decay in intensity over time. In the cases of larger clusters,spectral evolution occurred. (e) Spectra after 3 days of aging, grouped according to their similar attributes. The smallest clusters (red) resembled band 2 and theintermediate sizes (blue) resembled band 6, which is known to be Ag32(SG)19. Larger clusters (black) did not share a common spectral form. Note: spectra are scaled forclarity, appearing in order from bottom to top at 490 nm. (f) The decay of spectral intensity of (a–d) over time, evaluated at the following wavelengths: (a) 402 nm, (b)490 nm, (c) 560 nm, (d) 530 nm.
Paper Nanoscale
Dow
nloa
ded
by P
enns
ylva
nia
Stat
e U
nive
rsity
on
04 M
arch
201
3Pu
blis
hed
on 1
4 Ja
nuar
y 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3N
R33
705G
View Article Online
in solution for 3 days before being analyzed by PAGE, as shownin Fig. 4. Resolving the products into their components revealedthat the aging process produced only previously known clusters,with the exception of the new green clusters near band 13. Theaged solutions consisted of the original clusters with a signi-cant fraction of the mass converted into lower mass clusters,suggesting that fragmentation was the main decay mechanism.
Fig. 4 Products of aging individually purified clusters from different bands. Clusteraged fractions were separated into the resultant components using PAGE. The dashetypically contained smaller clusters but also contained particles that were too large
This journal is ª The Royal Society of Chemistry 2013
This is in some ways similar to previous observations of clusteretching to produce smaller particles.20,24–26
A minor component of the nal mass distributions includedparticles that were too large to enter the resolving gel, indicatingthat a small amount of aggregation or Ostwald ripening hadoccurred. In addition, a signicant amount of mass was lostfrom the clusters as polymeric silver glutathionates, which
s from each band were initially separated into a pure fraction and then aged. Thed lines indicate the approximate positions of the original bands. Mass distributionsto enter the resolving gel. All samples were aged for 3 days.
Nanoscale, 2013, 5, 2036–2044 | 2039

Nanoscale Paper
Dow
nloa
ded
by P
enns
ylva
nia
Stat
e U
nive
rsity
on
04 M
arch
201
3Pu
blis
hed
on 1
4 Ja
nuar
y 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3N
R33
705G
View Article Online
absorb light around 350 nm and thus are not visible in the gel(see ESI†).28
It is important to point out that although the spectra of theaged band 6 solutions (shown in Fig. 3(b)) continue to look verymuch like the spectrum of the band 6 cluster, it is clear fromFig. 4 that the solution also contained a large amount of theband 2 cluster aer 3 days of aging. The lack of prominentfeatures in the spectrum of the band 2 clusters makes themessentially impossible to identify within these mixtures byabsorption spectroscopy alone. Bands 2 and 6 appear to havehigher abundances when compared to other clusters, however,consistent with accumulation of mass at these sizes due to theirenhanced stability.
Using this general understanding of the products of clusterdecay, the question of the mechanism can now be addressed. Aspart of this effort, aging experiments were carried out undervarious conditions.
The canonical concerns with silver clusters are for theirprotection from oxidation and from light. Experiments thatexcluded light and oxygen from the cluster solutions had littleto no effect on cluster decay, however (Fig. S1†). As none of thesereduced the observed decay, an entirely different mechanismmust be operative.
The relative stability of the clusters in the gel versus insolution suggests that interparticle collisions might play animportant role in the decay mechanism. Further, the excellentsolid phase stability suggests that collisions in solutionpresumably facilitate the rearrangement and loss of material.DLVO theory31 states that surface charges can be used to effec-tively impede collisions by providing interparticle repulsions,thereby stabilizing the particles. Further, the strength of theinteractions can be modulated by using an electrolyte to screenthe surface charge interactions.31
To test the role of surface charges, different salts were addedto screen the clusters before being aged. Surprisingly, saltscreening was not observed to accelerate cluster decay, as
Fig. 5 The effect of salts on decay. (A) Freshly prepared (not aged, no salt), (B) nosalt, (C) 30 mM NaNO3, (D) 30 mM NaCl, and (E) 30 mM AgNO3. Samples wereaged for 2 days.
2040 | Nanoscale, 2013, 5, 2036–2044
shown in Fig. 5. Cluster solutions containing NaNO3 did notdecay any faster than solutions without salt, even at saltconcentrations as high as 300 mM. Curiously, samples withNaCl added faired slightly better than samples without, despitethe ability of Cl� ions to precipitate Ag+ ions to form solid AgCl.
Most dramatic, however, was the effect of AgNO3 on thestability of the clusters. Samples containing 30 mM or moreAgNO3 suffered very little decomposition over 4 days of moni-toring. Since NaNO3 had no effect on cluster stability it is clearthat the NO3
� ions played no role. This implicates the Ag+ ions ina chemical, rather than electrostatic, cluster stabilization role.
If cluster decay involves the loss of Ag+ ions, then one wouldexpect that the addition of AgNO3 would raise the chemicalpotential of the Ag+ solution to inhibit further Ag+ ion loss andtherefore inhibit cluster decay. Indeed, increasing the AgNO3
concentration improved the long-term stability of the clusters.The best results were for solution concentrations between 30and 300 mM AgNO3. In separate experiments, Ag+ ions wereprecipitated from aged Ag:SG cluster solutions using Cl� ions.Elemental analysis identied AgCl as the precipitate (see ESI†),directly conrming Ag+ ions as a decay product.
Since screening the surface charges had no noticeable effecton the stability of the clusters, pH was used to instead directlymodify the surface charge and the strength of cluster–clusterrepulsions. Identical solutions of Ag:SG clusters were preparedand then the pH was adjusted using a variety of buffers.Samples were aged for three days and then analyzed usingPAGE, as shown in Fig. 6.
A complex pattern of stability emerged as a function of pH.Clusters were on averagemost stable from pH 5 to 8 and at pH 2,however they were least stable at pH 3 and 4. Comparisons ofdifferent buffers at the same pH ensured that major differencesin stability were dependent solely on pH, with only minorchemical effects arising from the different buffers. This patternof stability can therefore be understood by considering only thecharge state on the glutathione ligands as a function of pH.
Glutathione is a tripeptide that consists of a glutamate, acysteine, and a glycine, each of which is ionizable such that acomplex set of charge states is possible.32 It is the complexity ofglutathione, however, that allows insights about the role ofcluster surface charge to be gained. The glutamate possessesone amine and one carboxylate group while the glycine has onecarboxylate, the charge state of each being controlled by the pH.Note that the thiol group on the cysteine is not considered hereas it is bound to the metal core. While any combination ofcharges on these functional groups is in principle possible,32
the most probable ionization states of the free ligand can bepredicted from the acid dissociation constants: pK1 ¼ 2.12(Glu–COOH), pK2 ¼ 3.59 (Gly–COOH), pK3 ¼ 8.75 (Glu–NH3
+).33
All three groups would be protonated at the lowest pH andsequentially depronated with an increase in pH. The glutamiccarboxylate would be deprotonated rst, since it is stabilized bythe proximal ammonium, followed by the glycyl carboxylate andnally the glutamic amine.
It should be possible to predict the relative net charges on theclusters for different pH values based on the above pKa values forthe free ligands, as shown in Fig. 6. Based on this model, it is
This journal is ª The Royal Society of Chemistry 2013
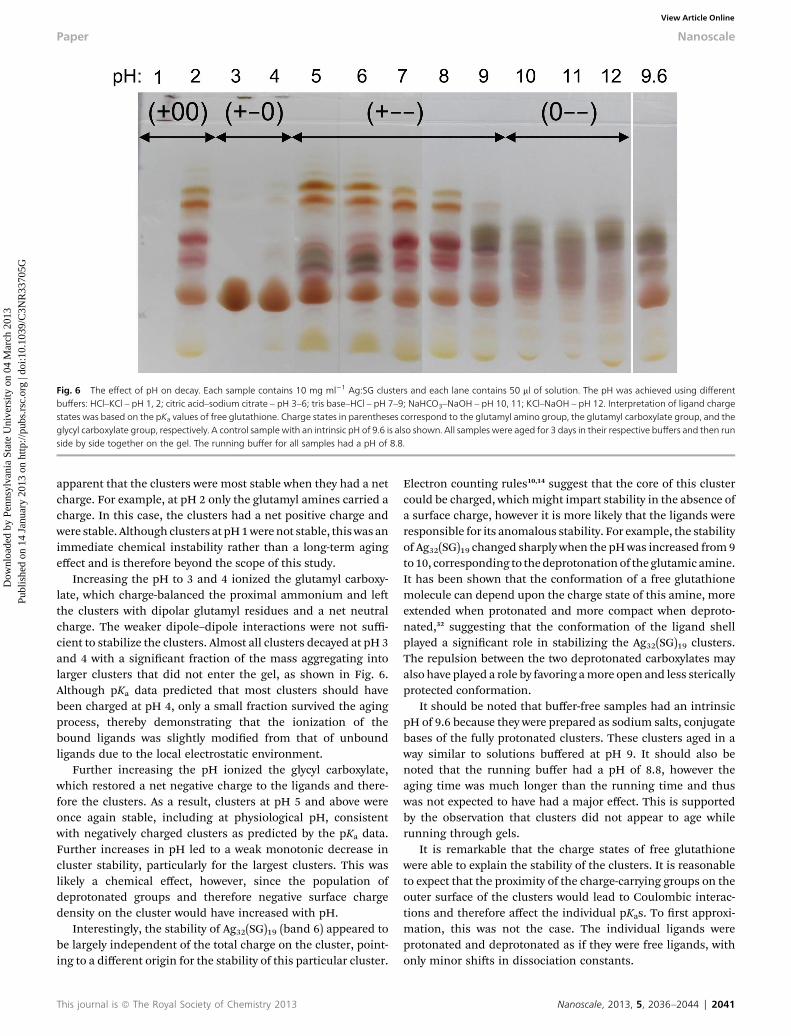
Fig. 6 The effect of pH on decay. Each sample contains 10 mg ml�1 Ag:SG clusters and each lane contains 50 ml of solution. The pH was achieved using differentbuffers: HCl–KCl – pH 1, 2; citric acid–sodium citrate – pH 3–6; tris base–HCl – pH 7–9; NaHCO3–NaOH – pH 10, 11; KCl–NaOH – pH 12. Interpretation of ligand chargestates was based on the pKa values of free glutathione. Charge states in parentheses correspond to the glutamyl amino group, the glutamyl carboxylate group, and theglycyl carboxylate group, respectively. A control sample with an intrinsic pH of 9.6 is also shown. All samples were aged for 3 days in their respective buffers and then runside by side together on the gel. The running buffer for all samples had a pH of 8.8.
Paper Nanoscale
Dow
nloa
ded
by P
enns
ylva
nia
Stat
e U
nive
rsity
on
04 M
arch
201
3Pu
blis
hed
on 1
4 Ja
nuar
y 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3N
R33
705G
View Article Online
apparent that the clusters were most stable when they had a netcharge. For example, at pH 2 only the glutamyl amines carried acharge. In this case, the clusters had a net positive charge andwere stable. Although clusters at pH1werenot stable, thiswas animmediate chemical instability rather than a long-term agingeffect and is therefore beyond the scope of this study.
Increasing the pH to 3 and 4 ionized the glutamyl carboxy-late, which charge-balanced the proximal ammonium and lethe clusters with dipolar glutamyl residues and a net neutralcharge. The weaker dipole–dipole interactions were not suffi-cient to stabilize the clusters. Almost all clusters decayed at pH 3and 4 with a signicant fraction of the mass aggregating intolarger clusters that did not enter the gel, as shown in Fig. 6.Although pKa data predicted that most clusters should havebeen charged at pH 4, only a small fraction survived the agingprocess, thereby demonstrating that the ionization of thebound ligands was slightly modied from that of unboundligands due to the local electrostatic environment.
Further increasing the pH ionized the glycyl carboxylate,which restored a net negative charge to the ligands and there-fore the clusters. As a result, clusters at pH 5 and above wereonce again stable, including at physiological pH, consistentwith negatively charged clusters as predicted by the pKa data.Further increases in pH led to a weak monotonic decrease incluster stability, particularly for the largest clusters. This waslikely a chemical effect, however, since the population ofdeprotonated groups and therefore negative surface chargedensity on the cluster would have increased with pH.
Interestingly, the stability of Ag32(SG)19 (band 6) appeared tobe largely independent of the total charge on the cluster, point-ing to a different origin for the stability of this particular cluster.
This journal is ª The Royal Society of Chemistry 2013
Electron counting rules10,14 suggest that the core of this clustercould be charged, whichmight impart stability in the absence ofa surface charge, however it is more likely that the ligands wereresponsible for its anomalous stability. For example, the stabilityof Ag32(SG)19 changed sharply when the pHwas increased from9to 10, corresponding to thedeprotonation of the glutamic amine.It has been shown that the conformation of a free glutathionemolecule can depend upon the charge state of this amine, moreextended when protonated and more compact when deproto-nated,32 suggesting that the conformation of the ligand shellplayed a signicant role in stabilizing the Ag32(SG)19 clusters.The repulsion between the two deprotonated carboxylates mayalso have played a role by favoring amore open and less stericallyprotected conformation.
It should be noted that buffer-free samples had an intrinsicpH of 9.6 because they were prepared as sodium salts, conjugatebases of the fully protonated clusters. These clusters aged in away similar to solutions buffered at pH 9. It should also benoted that the running buffer had a pH of 8.8, however theaging time was much longer than the running time and thuswas not expected to have had a major effect. This is supportedby the observation that clusters did not appear to age whilerunning through gels.
It is remarkable that the charge states of free glutathionewere able to explain the stability of the clusters. It is reasonableto expect that the proximity of the charge-carrying groups on theouter surface of the clusters would lead to Coulombic interac-tions and therefore affect the individual pKas. To rst approxi-mation, this was not the case. The individual ligands wereprotonated and deprotonated as if they were free ligands, withonly minor shis in dissociation constants.
Nanoscale, 2013, 5, 2036–2044 | 2041

Fig. 7 Negative-ion electrospray-ionization mass spectrum showing the mostabundant fragment species of Ag32(SG)19 clusters (A) freshly prepared and (B)after 10 days of aging, normalized to the base peaks in each spectrum. All speciesare single anions unless otherwise noted. The trap collision energy was 0.5 eV.
Nanoscale Paper
Dow
nloa
ded
by P
enns
ylva
nia
Stat
e U
nive
rsity
on
04 M
arch
201
3Pu
blis
hed
on 1
4 Ja
nuar
y 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3N
R33
705G
View Article Online
It is clear that DLVO theory failed to fully predict the stabilityof these small clusters. Although changing the pH showed thatsurface charge interactions were important (consistent withDLVO theory), screening with salts had no effect on clusterstability (inconsistent with DLVO theory). For large colloids, ahigh surface charge is necessary for stability since it preventsparticles from approaching one another close enough for vander Waals interactions to lead to irreversible aggregation. Forsmall clusters, however, van der Waals interactions are weak atdistances sufficiently large for enough salt ions to screen thesurface charges. Further, the clusters form thermodynamicallystable solutions, unlike the colloids, and therefore do not irre-versibly aggregate due to van der Waals interactions.
The above ndings provide insights into the decay mecha-nism. The primary Ag:SG cluster decay products have now beenidentied as smaller Ag:SG clusters, silver glutathionate poly-mers, and Ag+ ions. Cluster decay was not induced by light oroxygen, nor was excess glutathione involved as it was carefullyremoved from the samples before aging. Further, the decaypattern was similar over a wide range of pH (with the exceptionof pH 3 and 4), indicating that H+ was not responsible foroxidation of Ag from the core. Since the aqueous solvent is notcapable of oxidizing Ag, the only remaining possibility is for thethiolate fragments to have oxidized Ag as they le, in a unim-olecular reaction. In the case of pH 3 and 4, however, the netsurface charge was zero and the decay was more rapid than fornon-zero surface charges, indicating a more complex bimolec-ular reaction pathway involving cluster–cluster collisions.
To form smaller clusters from larger clusters, more silverthan glutathione must be lost in order to increase the surface-area-to-volume ratio for smaller clusters. This requires the lossof silver as Ag+ in addition to AgSG polymer, as observed. Toform the polymer, however, small silver glutathionate inter-mediates must rst be lost from the Ag:SG clusters.
Electrospray-ionization mass spectrometry (ESI-MS) studiescomparing the fragments found in fresh and aged solutions ofAg32(SG)19 clusters showed that the aged sample contained asignicantly higher abundance of anionic silver glutathionatespecies, as shown in Fig. 7. Although ESI-MS can involve gas-phase dissociation, the differences between the samplessuggest that the anionic silver glutathionate species played arole in the solution-phase decay mechanism. These species hadthe general form Agx(SG)y
�, where x $ y with only two excep-tions. Similar fragment species were observed for the Au25(SG)18cluster34 (see also Fig. S7†). Since AgSG� was a major product offragmentation, it is used here as an example to illustrate onepossible decay channel, corresponding to the reaction:
Agn(SG)m / Ag(n�2y)(SG)(m�y) + yAg+ + yAgSG�
where the AgSG� intermediates would then be oxidized to formthe stable polymeric solid. Analogous reactions could also bewritten for other fragment species since it is likely that manysolution phase fragmentation pathways would be possible. It isalso possible that neutral fragments species (undetected inmass spectrometry) could also have been lost, directly formingthe polymer.
2042 | Nanoscale, 2013, 5, 2036–2044
Based on the above chemistry, the addition of excess gluta-thione ligands ought to facilitate the removal of Ag and therebyaccelerate decay by pushing the equilibrium to the right.Experiments indeed showed that glutathione had a strongetching effect. This was evident by an almost complete loss ofoptical density (see Fig. S2†) and the production of colorless Agthiolates,28 which were identied by the appearance of a peak at350 nm in the absorption spectrum (see Fig. S3†). This under-scores the need to carefully clean the cluster solutions of excessligands. The addition of glutathione leads to the conversion ofAg+ ions to thiolates, such that:
Agn(SG)m + (x � y)GSH / Ag(n�x)(SG)(m�y)
+ xAgSG + ½(x � y)H2
It is worth noting that these etching results for Ag:SG clusterssuggest that Au+ ion loss may be required for Au:SR clusteretching, especially considering that gold glutathionate stapleloss would be metal poor.
It is natural to seek strategies for stabilizing these silverclusters in order to realize their potential in applications. Whilethe insights gained herein could be used to design new stabi-lization strategies, there are also established strategies used forsilver materials that could be evaluated for use with silverclusters. Examples include benzotriazole, an anticorrosiveagent commonly used in dishwashing detergents, and sodiumcitrate and ascorbic acid, both of which are used asantioxidants.
Samples of silver clusters were prepared with these additives,along with AgNO3, and aged for 3 days, as shown in Fig. 8. Themost common additives, benzotriazole and sodium citrate, didnot succeed in stabilizing the clusters. Ascorbic acid and AgNO3
did succeed, however. Although the total concentration of theclusters decreased over time, there was relatively little decaywhen compared to the control solution without additives. Inparticular, the largest clusters that were normally lost in just afew hours survived aer 3 days of aging.
This journal is ª The Royal Society of Chemistry 2013

Fig. 8 The effect of additives on decay. (A) Freshly prepared (not aged, noadditives), (B) no additives, (C) 10 mM sodium citrate, (D) 10 mM ascorbic acid, (E)10 mM benzotriazole, and (F) 30 mM AgNO3. Sample F initially contained 600 mgof Ag:SG clusters. The remaining samples initially contained 800 mg of Ag:SGclusters. All samples were aged for 3 days.
Paper Nanoscale
Dow
nloa
ded
by P
enns
ylva
nia
Stat
e U
nive
rsity
on
04 M
arch
201
3Pu
blis
hed
on 1
4 Ja
nuar
y 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3N
R33
705G
View Article Online
While sodium citrate was not signicantly different than thecontrol samples, ascorbic acid had a signicant stabilizingeffect. Both of these results can be understood based solely onelectrostatic effects, due to the pH of the solutions, rather thantheir chemical antioxidant properties. Sodium citrate is a weakbase and the pH of the cluster solution was 8.8 and corre-spondedmost closely to the pH 9 and 10 solutions. The ascorbicacid cluster solution had a pH of 4.5 and stability correspondedto the pH 5 solution.
Although benzotriazole is commonly used to protect silvermetal, it acted to increase the rate of decay of the silver clusters.In contrast, the addition of AgNO3 clearly suppressed decay.Both of these effects can be understood based on chemicaleffects due to Ag+ ions in solution. Benzotriazole is known toscavenge and precipitate Ag+ ions. Since the loss of Ag+ ionsfrom the clusters is an important component of the decaymechanism, removal of Ag+ ions from solution accelerateddecay. Since the addition of AgNO3 increased the Ag+ ionconcentration, the cluster decay rate decreased.
It is worth noting that these new mechanistic insights intothe stability and decay of cluster compounds could also be usedto develop rational design principles for synthesizing speciccluster species. New synthetic strategies could be designedbased on the properties of the target clusters. For example, theband 2 clusters appear to be most favored under conditionswhere little Ag+ is available while band 6 (Ag32(SG)19) could bethe most favorable product under conditions wherein theligands have a net neutral charge. Further, promoting the decayof clusters to produce Ag+ could create new opportunities forbiomedical applications such as antibiotic technologies.22,23
Conclusions
The stability of metal cluster compounds in the solution phasedepends on more factors than simply electronic and geometricshell closings. The ambient stability of Ag magic number
This journal is ª The Royal Society of Chemistry 2013
clusters was found to be a function of size, where the smallestclusters were most stable, although this dependence was ingeneral non-monotonic. The chemical and electrostatic envi-ronments were found to have strong effects on the stability ofthe clusters.
Clusters were observed to interconvert primarily betweenknown sizes, generally from larger to smaller. This involved theloss of Ag+ ions and silver glutathionates. Screening the surfacecharges carried by the Ag:SG clusters was not found to beeffective, however modifying the surface charges by changingthe pH had a direct effect on stability. Clusters with a netsurface charge were most stable while those without were theleast stable. The Ag32(SG)19 cluster was a notable exception,however, as it did not require surface charge to remain stablesuggesting that ligand conformation played an important rolein protecting the cluster from mass loss.
These results provide mechanistic insights into controllingthe temporal stability of metal cluster solutions, providingguidelines for suppressing or accelerating their decay asneeded. For example, clusters could be stabilized by the addi-tion of Ag+ ions while they could be destabilized by either theremoval of Ag+ ions or the addition of glutathione. Clusters werealso found to be most stable near neutral pH, where they had anegative surface charge. This knowledge is also important if oneis to avoid unknowingly studying mixtures of clusters, which isacutely important when dealing with metals less stable thangold. It is expected that these insights could be applied towardthe rational design of new synthetic strategies, including thosethat target the synthesis of specic and pure cluster species.
Acknowledgements
The authors thank Prof Robert Whetten for useful discussionsand gratefully acknowledge the College of Natural Sciences andMathematics Instrumentation Center at the University ofToledo.
References
1 T. P. Martin, T. Bergman, H. Gohlich and T. Lange, J. Phys.Chem., 1991, 95, 6421–6429.
2 W. A. deHeer, Rev. Mod. Phys., 1993, 65, 611–676.3 T. P. Martin, Phys. Rep., 1996, 273, 199–241.4 R. L. Whetten, J. T. Khoury, M. M. Alvarez, S. Murthy,I. Vezmar, Z. L. Wang, P. W. Stephens, C. L. Cleveland,W. D. Luedtke and U. Landman, Adv. Mater., 1996, 8, 428–433.
5 T. G. Schaaff, G. Knight, M. N. Shagullin, R. F. Borkmanand R. L. Whetten, J. Phys. Chem. B, 1998, 102, 10643–10646.
6 T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 2000, 104,2630–2641.
7 Y. Negishi, Y. Takasugi, S. Sato, H. Yao, K. Kimura andT. Tsukuda, J. Am. Chem. Soc., 2004, 126, 6518–6519.
8 Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc.,2005, 127, 5261–5270.
9 Y. Negishi, N. K. Chaki, Y. Shichibu, R. L. Whetten andT. Tsukuda, J. Am. Chem. Soc., 2007, 129, 11322–11323.
Nanoscale, 2013, 5, 2036–2044 | 2043

Nanoscale Paper
Dow
nloa
ded
by P
enns
ylva
nia
Stat
e U
nive
rsity
on
04 M
arch
201
3Pu
blis
hed
on 1
4 Ja
nuar
y 20
13 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C3N
R33
705G
View Article Online
10 J. Akola, M. Walter, R. L. Whetten, H. Hakkinen andH. Gronbeck, J. Am. Chem. Soc., 2008, 130, 3756–3757.
11 M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky,G. Calero, C. J. Ackerson, R. L. Whetten, H. Gronbeck andH. Hakkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105,9157–9162.
12 S. Kumar, M. D. Bolan and T. P. Bigioni, J. Am. Chem. Soc.,2010, 132, 13141–13143.
13 M. A. Tofanelli and C. J. Ackerson, J. Am. Chem. Soc., 2012,134, 16937–16940.
14 P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell andR. D. Kornberg, Science, 2007, 318, 430–433.
15 M. W. Heaven, A. Dass, P. S. White, K. M. Holt andR. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755.
16 M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin,J. Am. Chem. Soc., 2008, 130, 5883–5885.
17 H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin,J. Am. Chem. Soc., 2010, 132, 8280–8281.
18 C. Zeng, H. Qian, T. Li, G. Li, N. L. Rosi, B. Yoon,R. N. Barnett, R. L. Whetten, U. Landman and R. Jin,Angew. Chem., Int. Ed., 2012, 51, 13114–13118.
19 Z. Wu, E. Lanni, W. Chen, M. E. Bier, D. Ly and R. Jin, J. Am.Chem. Soc., 2009, 131, 16672–16674.
20 A. C. Dharmaratne, T. Krick and A. Dass, J. Am. Chem. Soc.,2009, 131, 13604–13605.
21 N. Cathcart and V. Kitaev, J. Phys. Chem. C, 2010, 114, 16010–16017.
2044 | Nanoscale, 2013, 5, 2036–2044
22 I. Chopra, J. Antimicrob. Chemother., 2007, 59, 587–590.23 V. K. Sharma, R. A. Yngard and Y. Lin, Adv. Colloid Interface
Sci., 2009, 145, 83–96.24 T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 1999, 103,
9394–9396.25 J. P. Wilcoxon and P. Provencio, J. Phys. Chem. B, 2003, 107,
12949–12957.26 Y. Shichibu, Y. Negishi, H. Tsunoyama, M. Kanehara,
T. Teranishi and T. Tsukuda, Small, 2007, 3, 835–839.27 J. Guo, S. Kumar, M. Bolan, A. Desireddy, T. P. Bigioni and
W. P. Griffith, Anal. Chem., 2012, 84, 5304–5308.28 I. G. Dance, K. J. Fisher, R. M. Herath Banda and
M. L. Scudder, Inorg. Chem., 1991, 30, 183–187.29 O. M. Bakr, V. Amendola, C. M. Aikens, W. Wenseleers, R. Li,
L. Dal Negro, G. C. Schatz and F. Stellacci, Angew. Chem., Int.Ed., 2009, 48, 5921–5926.
30 K. M. Harkness, Y. Tang, A. Dass, J. Pan, N. Kothalawala,V. J. Reddy, D. E. Cliffel, B. Demeler, F. Stellacci,O. M. Bakr and J. A. McLean, Nanoscale, 2012, 4, 4269–4274.
31 J. Israelachvili Intermolecular and Surface Forces, AcademicPress, New York, 1992.
32 O. Lampela, A. H. Juffer and A. Rauk, J. Phys. Chem. A, 2003,107, 9208–9220.
33 The Merck Index, ed. S. Budavari, Merck & Co., Inc., Rahway,NJ, 11th edn, 1989
34 L. A. Angel, L. T. Majors, A. C. Dharmaratne and A. Dass, ACSNano, 2010, 4, 4691–4700.
This journal is ª The Royal Society of Chemistry 2013