the 1.8 angstrom crystal structure of an mmp8- barbiturate inhibitor
TRANSCRIPT

The 1.8 Angstrom Crystal Structure of an MMP8- Barbiturate Inhibitor Complex
Reveals a Previously Unobserved Mechanism for Collagenase Substrate
Recognition
Hans Brandstetter1*, Frank Grams3, Dagmar Glitz2, Anja Lang1, Robert Huber1, Wolfram
Bode1, Hans-Willi Krell2 and Richard A. Engh1,2
1 Department of Structural Research, Max-Planck-Institute for Biochemistry,
D-82152 Martinsried, Germany
2 Pharmaceutical Research, Roche Diagnostics GmbH, D-82372 Penzberg, Germany
3 Molecular Design, Pharmaceuticals Division, F. Hoffmann La Roche Ltd., CH-4070 Basel,
Switzerland
* To whom correspondence should be addressed:
Email: [email protected]
Phone: ++49 89 8578-2828
Fax: ++49 89 8578-3516
Running Title: Structural Basis of Collagenase Substrate Specificity
We wish to dedicate this work to Prof. H. Tschesche on the occasion of his 65th birthday.
Structural Basis of Collagenase Substrate Recognition
1
Copyright 2001 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on January 22, 2001 as Manuscript M007475200 by guest on M
arch 28, 2018http://w
ww
.jbc.org/D
ownloaded from
by guest on M
arch 28, 2018http://w
ww
.jbc.org/D
ownloaded from
by guest on M
arch 28, 2018http://w
ww
.jbc.org/D
ownloaded from

SUMMARY
The individual zinc endoproteinases of the tissue degrading matrix metalloproteinase (MMP)
family share a common catalytic architecture but are differentiated with respect to substrate
specificity, localization, and activation. Variation in domain structure and more subtle structural
differences control their characteristic specificity profiles for substrates from among four distinct
classes (1). Exploitation of these differences may be decisive for the design of anticancer or other
drugs, which should be highly selective for their particular MMP targets. Based on the 1.8 Å
crystal structure of human neutrophil collagenase (MMP-8) in complex with an active site
directed inhibitor (RO200-1770), we identify and describe new structural determinants for
substrate and inhibitor recognition in addition to the primary substrate recognition sites. RO200-
1770 induces a major rearrangement at a position relevant to substrate recognition near the
MMP-8 active site (Ala206-Asn218). In stromelysin (MMP-3), competing stabilizing
interactions at the analogous segment hinder a similar rearrangement, consistent with kinetic
profiling of several MMPs. Despite the apparent dissimilarity of the inhibitors, the central 2-
hydroxy-pyrimidine-4,6-dione (barbiturate) ring of the inhibitor RO200-1770 mimics the
interactions of the hydroxamate-derived inhibitor batimastat (2) for binding to MMP-8. The two
additional phenyl and piperidyl ring substituents of the inhibitor bind into the S1’ and S2’ pockets
of MMP-8, respectively. The crystal lattice contains a hydrogen bond between the Oγ group of
Ser209 and Nδ1 of His207 of a symmetry related molecule; this interaction suggests a model for
recognition of hydroxyprolines present in physiological substrates. We also identify a
collagenase-characteristic cis-peptide bond, Asn188-Tyr189, on a loop essential for
collagenolytic activity. The sequence conservation pattern at this position marks this cis-peptide
bond as a determinant for triple-helical collagen recognition and processing.
Structural Basis of Collagenase Substrate Recognition
2
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

KEYWORDS
Crystal structure, matrix metalloproteinase, collagen recognition exosite, cis-peptide bond,
protein-inhibitor interaction, hydroxy-proline recognition
INTRODUCTION
The matrix metallo proteinases (MMPs), one of the five families that form the metzincin group of
zinc proteinases (3), function to degrade the extracellular matrix (ECM) during embryonic
development, reproduction, and tissue remodeling (1) but are disregulated in arthritis, cancer, and
other diseases. The “minimal” MMPs matrilysin and endometase (MMP-7 and MMP-26,
respectively), have a Zn2+ and Ca2+ binding catalytic domain , and an N-terminal pro-domain.
All other known MMPs possess additionally a hemopexin-like domain near the C-terminus
(CTD), and further domain insertions differentiate MMP subfamilies. Gelatinases A and B
(MMP-2 and MMP-9) possess three fibronectin type II-like repeats inserted at a loop in the
catalytic domain; these form an independent folding domain adjacent to the catalytic domain.
Membrane-type MMP’s (MT-MMPs) possess an anchoring transmembrane helix C-terminal to
the hemopexin-like domain (4). Hierarchical regulation of MMP activity occurs on many levels,
including gene expression control (1,5), proteolytic activation of MMP zymogens (6), inhibition
by endogenous tissue inhibitors of metalloproteinases (TIMPs) (7), and both positive and
negative proteolytic feedback loops (8,9). Crystal structures of several MMPs have been
determined (for a review, see e.g. (10)), revealing overall domain structures, catalytic
mechanisms, and many aspects of MMP regulation mechanisms; these include collagenase 1
(MMP-1) (11,12) and collagenase 2 (MMP-8). Structures of the latter are represented by two
forms of the catalytic domain, resulting from activation cleavage alternately at two cleavage sites,
leaving either Met80 (13,14) or Phe79 as the N-terminal residue (15). The latter form is
“superactivated”, as Phe79 forms a salt bridge with Asp232 and thereby prevents the N-terminal
sequence from transient or other interference with the active site. The result is a three-fold
increase in activity compared to activation cleavage at Met80 (16).
As their early nomenclature implies, collagenases I, -II, –III (17) (MMP-1, MMP-8, and
Structural Basis of Collagenase Substrate Recognition
3
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

MMP-13, respectively) degrade mainly fibrillar collagens (18-20), although the structural origin
of this specificity is not well understood (4). Disruption of MMP tissue remodeling function
causes a variety of disorders, including cancer (tumor growth, invasion and metastasis),
rheumatoid- and osteoarthritis, and a variety of diseases involving neovascularization. The
resulting clinical need has fostered an enormous interest in the development of inhibitors against
MMP’s. As part of these efforts, crystal structures of MMPs with a variety of synthetic inhibitors
have been determined. For MMP-8, complexes reported include peptide-mimetics, hydroxamic
acid derivatives, (2,14,21,22), phosphinamides and sulfodiimines (23,24), thiadiazole (25), and
malonic acid-derivatives (26,27).
Here we describe the 1.8 Å crystal structure of MMP-8 inhibited by a barbituric acid
derivative. Conformational rearrangements accompanying the inhibitor binding lead to a new
and highly ordered crystal packing arrangement. The high resolution structural data enables a
thorough analysis of determinants of MMP selectivity towards both low molecular weight
substances as well as substrate classes. A previously unreported cis-peptide bond
(Asn188—Tyr189) could be unambiguously identified. The conservation patterns of the
sequence at the cis-peptide bond position supports the hypothesis that this cis-peptide plays a
critical role in substrate recognition mechanisms specific to the collagenases I and II (MMP-1
and MMP-8).
Structural Basis of Collagenase Substrate Recognition
4
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

EXPERIMENTAL PROCEDURES
Materials. MMP-1, MMP-3, and MMP-8 were kindly provided by Profs. G. Murphy
(Norwich),H. Nagase (London), and H. Tschesche, respectively; MMP-2 and MMP-9 were
obtained from Roche Molecular Biochemicals (Penzberg); MT1-MMP and MT3-MMP were
provided by Prof J. Foidart (Liege). The inhibitors RO200-1770, RO204-1924, I-COL043,
RO206-0027, and RO206-0032 were synthesized as previously described (28). The fluorogenic
substrate M-1855 (Dnp-Pro-Leu-Gly-Leu-Trp-Ala-D-Arg-NH2) was purchased from
BACHEM (Heidelberg), all other chemicals were of highest purity commercially available.
Inhibition assay. All measurements were performed at 25 0C using a buffering solution of
50 mM Tris pH 7.6, 100 mM NaCl, 10 mM CaCl2. Based on multiple measurements, all data are
precise to within 5%. Depending on activity, enzymes were used at 5-50 nM concentration range
with a substrate concentration of 2.55 µM. The enzyme was briefly pre-incubated with the
inhibitor at a resultant DMSO concentration of 1%. The reaction was started with the addition of
the substrate M-1855. Substrate was excited at 280 nm and the substrate fluorescence was
monitored at 346 nm using the FuoroMax-3 fluorometer (SPEX, HORIBA Group,
Grasbrunn/Munich)
Crystallization, data collection, and structure refinement. MMP-8 was concentrated to 8
mg/ml and then mixed with 3-fold molar excess of an aqueous solution of RO200-1770 for a
final MMP-8 concentration of 6 mg/ml. 3 µl of protein-inhibitor complex was mixed with 2 µl
precipitant solution containing 100 mM cacodylate pH 5.5-6.5, 10 mM CaCl2, 100 mM NaCl,
and 10 % PEG 6000. The hanging drop was equilibrated by vapor diffusion at room temperature
against a reservoir containing 1.0-1.5 M phosphate buffer. Data were collected on a multiwire
detector (X1000, Bruker AXS) to 1.8 Å resolution and processed using SAINT data reduction
software (29) yielding an agreement of redundant measurements of Rmerge= 9.3% over all data
and 41 % in the outer resolution shell (completeness 98% and 87%, respectively). The space
group of the crystal was determined as I222 with unit cell dimensions a= 61.02 Å b= 69.24 Å c=
Structural Basis of Collagenase Substrate Recognition
5
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

88.47 Å. The orientation and translation of the molecule within the crystallographic unit cell was
determined with Patterson search techniques (30-32) using the program AMoRe (33). Electron
density calculation, and model building proceeded using the program MAIN (34). The structure
has been refined by using the program X-PLOR (35) to a crystallographic R-value of 21.1%
(Rfree= 29.6%) with bond deviations of 0.009 Å and angle deviations of 1.70 from ideality (36).
The molecular structure was analyzed and compared using appropriate tools within the program
MAIN (34).
Structural Basis of Collagenase Substrate Recognition
6
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

RESULTS
Inhibitor conformation
The inhibitor RO200-1770 is a barbituric acid derivative, doubly substituted with a
phenyl and 4-ethanolpiperidyl rings as depicted in Figure 1. The barbiturate ring chelates the
zinc and rigidly orients the two cyclic substituents into the S1’ and S2’ substrate binding sites.
Neither substituent ring system appears strained by the protein environment, although their
relative orientations may be induced by protein binding. The phenyl moiety occupies the MMP-
8 binding site and is perfectly planar to within the 1.8 Å resolution. The electron density
observed for the piperidine ring allows an interpretation whereby two chair conformations related
by a 180 degree rotation along the C5-pN1 bond might be superimposed; either conformation
would allow favorable hydrophobic contacts in the S2’-site. Adopting an all-trans
conformation, the alcohol group points towards the solvent. SUGGESTED LOCATION OF
FIGURE 1
The relative orientations of the rings of the inhibitor may be described by considering the
ring planes and the bonds linking the substituent rings to the C5 atom of the barbiturate ring. The
C5—fC1 bond linking the phenyl ring is nearly perpendicular to the plane of the barbiturate ring
(excluding C5). This arrangement necessarily orients the plane of the phenyl ring likewise
perpendicular to the barbiturate plane. The dihedral angle C6—C5—fC1—fC2 fixes the ring
orientation with an eclipsed geometry (at 1.20, while the C4—C5—C1—fC2 dihedral is
staggered at 60.80). In contrast, the C5—pN1 bond lies nearly in the plane of the barbiturate,
extending the P2’-piperidyl ring away from the barbiturate; all dihedrals across the C5—pN1
bond have staggered orientations. This results in an arrangement where all three rings are
mutually perpendicular, as follows: the angle between (the normal vectors) of the barbiturate and
phenyl rings is 910, between the barbiturate and the piperidyl rings is 1030, and between the
phenyl and piperidyl rings is 1110. SUGGESTED LOCATION OF FIGURE 2
Structural Basis of Collagenase Substrate Recognition
7
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Protein-inhibitor interaction
The MMP-8 substrate recognition sites are shown schematically in Figure 3A.
Comparison with the binding mode of RO200-1770 as depicted in Figure 3B highlights the
inhibitor binding at the “primed” substrate recognition sites and at the Zn2+ ion. The Zn2+ is
coordinated by atoms N3 and O2 of the barbiturate ring. The Zn2+—N3 coordination has a
favorable distance of 2.09 Å and highly symmetric Zn2+-N3-C2 and Zn2+-N3-C4 angles of
1190 and 1170, respectively. Positioned where the catalytic water is expected for peptidic
substrates, a partial negative charge at the hydroxyl O2 is stabilized by the adjacent Glu198,
thereby strengthening its binding to Zn2+ (Figures 2 & 3, Table 2). The enol form of the
barbiturate is thus favored by the protein matrix over the tautomeric keto form which dominates
in solution (37). In addition, the polar H1-N1-C6=O6 atoms of the barbiturate, Figure 3B,
mimic the P1’-S1’ antiparallel main chain interactions of a substrate (Figure 3A). The amide
N1-H1 thereby is hydrogen bonded to the carbonyl of Ala161, and the ketone C6=O6 is stabilized by
the amides of Leu160 and Ala161. This latter interaction is reminiscent of the oxyanion hole
binding of serine proteinases, although here there is no evidence of oxygen anion stabilization.
The C4=O4 ketone seems unlikely to contribute to the binding energy for two reasons. First,
unfavorable geometry (Table 1) precludes a role as a third ligand in the Zn2+ chelation. More
importantly, the C4=O4 ketone would collide with the carbonyl oxygen position of Pro217 at the
“southern” rim of the active site as defined by other MMP-8 structures (2,14,26). Instead, the
inhibitor induces a reorientation at the Pro217 position at an energy cost we discuss below.
SUGGESTED LOCATION OF FIGURE 3
The pentacoordinated Zn2+ binding geometry resembles a highly distorted trigonal
bipyramidal structure with O2, Nε2197His, and Nε2207His approximately in plane with the
Zn2+ ion, with Nε2201His and N3 lying above and below the basal plane, respectively (Table 1).
Alternatively, the coordination can be described as a distorted square pyramid where O2, N3,
Nε2201His, and Nε2207His form the basal ligands (dihedral deviation from planarity 150, Table 1).
The metal ion lies outside of the basal plane but within 0.5 Å, and the fifth ligand Nε2197His
Structural Basis of Collagenase Substrate Recognition
8
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

forms the apex of the pyramid Table 1. (If considering O4 to be a sixth ligand, the geometry may
be described as a pentagonal pyramid with O4 as basal ligand in addition to O2, N3, Nε2201His,
and Nε2207His). SUGGESTED LOCATION OF TABLE 1
In contrast to the polar interactions of the barbiturate ring, the interactions mediated by the
phenyl and piperidyl rings are predominantly hydrophobic and involve the S1’ and S2’ pockets,
respectively. The most prominent interaction in the S1’ pocket is the ideally parallel planar
stacking of the phenyl ring and His197 at a distance of 3.6 Å (Figure 4). The conserved Leu160
contributes to ligand binding also with its side chain in the S1’ site. The phenyl ring does not by
itself fill the S1’ site, but leaves space filled by a network of three ordered water molecules. The
first of these (Sol595) is probably incompletely occupied and forms hydrogen bonds with the
inhibitor, with MMP-8, and with a second water molecule. The proximity of the inhibitor phenyl
fC4 atom to Sol595 (3.1 Å) indicates a O...H-C interaction (38). The carbonyl group of Leu193
forms a 2.9 Å hydrogen bond with Sol595 with however an unfavorable C=O193-O595 angle of
1130. The second water molecule, Sol602, is positioned deeper inside the S1’ pocket at a
hydrogen bonding distance of 2.7 Å from Sol595. Sol602 in turn is hydrogen bonded (2.8 Å) with
the third solvent molecule in the S1’ pocket, Sol592. Sol592 is in a channel bounded by Arg222
which forms a link between the three water network in S1’ and, via Sol667 (2.9 Å from Sol602),
water in the adjacent cavity. Mutation of Arg222 would connect the two cavities, opening a
“back door” to S1’ for solvent access. The guanidinium group atoms Nη2 and Nη1 of Arg222
are fixed by hydrogen bonds to the carbonyl oxygen of Ala213 (3.3 Å) and the Oη of Tyr227 (3.3
Å), respectively. Since most MMPs lack an equivalently stabilized Arg, MMP-8 has a
comparatively restricted S1’ site. SUGGESTED LOCATION OF FIGURE 4
The hydrophobic interactions of the piperidine ring are mediated by aliphatic surfaces
from Pro217-Asn218-Tyr219 at the “southern” rim of S2’ and by the main chain Gly158-
Ile159-Leu160 at the “northern” rim. The latter residue (Leu160) separates the S1’ and S2’
pockets. No ordered water molecule can be detected in the vicinity of the hydroxyl group pOH9,
although the position of this solvent exposed ethanol group is well defined by electron density,
Structural Basis of Collagenase Substrate Recognition
9
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Figure 2, and is thus presumably hydrated by disordered water.
Protein conformational changes
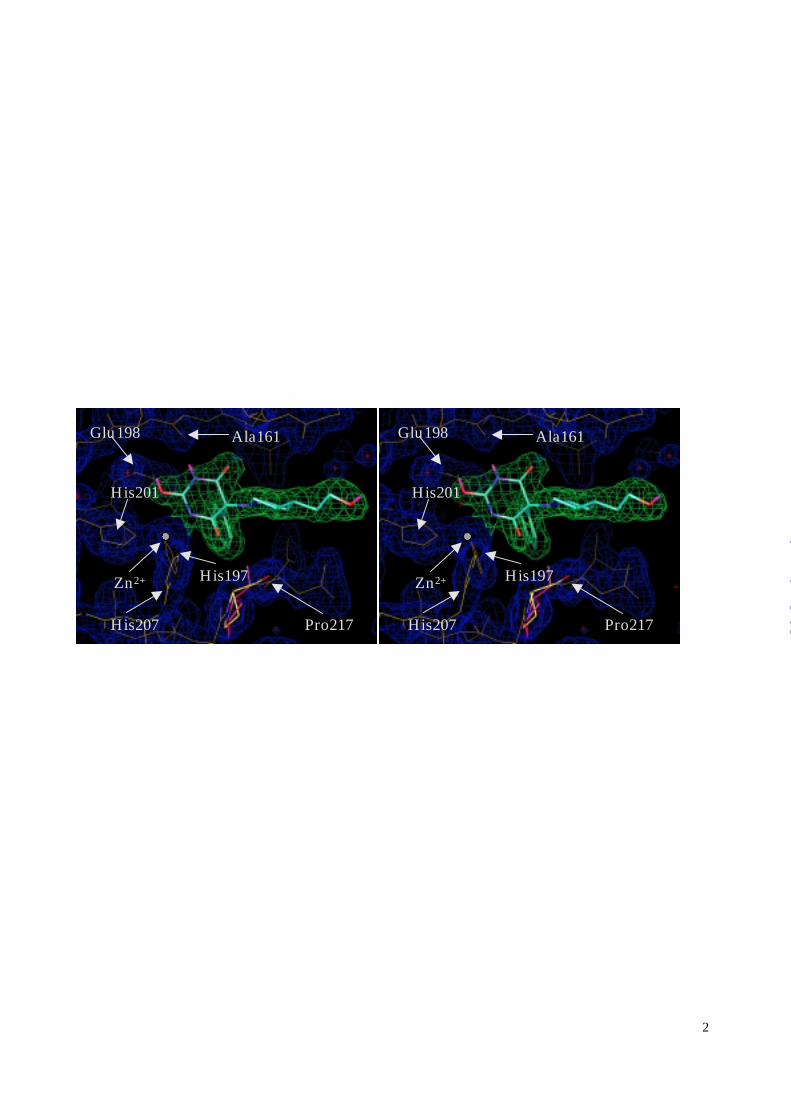
Significant differences are apparent in the protein structure compared to previously determined
MMP-8 structures (2,14,21). The catalytic Zn2+ ion of the three reference structures occupies
the same position to within 0.2 Å; it is however shifted from that average position by 0.6 Å in the
RO200-1770 complex structure. Corresponding shifts of the Zn2+ protein ligand positions are
also apparent, with the respective Nε2 and Cγ values measured as follows: His197 (0.4 Å, 0.3 Å),
His201 (0.3 Å, 0.3 Å), and His207 (0.6 Å, 0.7 Å). Consistent with this overall shift, the side
chain of the catalytic Glu198 is translated by 0.2 Å. This displacement of the catalytic Zn2+ and
its protein ligands is evidently induced by inhibitor binding, as the net effect of the optimization
of barbiturate—Zn chelation geometry and the inhibitor orienting forces arising from the other
inhibitor—protein interactions.
Of the two partial sequences harboring the Zn2+ binding histidine residues, the loop
Ala206—His207—Asn218 is more exposed to the solvent and anchored by fewer protein
contacts than the internal helix Leu191—H197EXXH201—Leu203. The conformation of this
loop is altered by several effects associated with the binding of RO200-1770. First, the greater
inherent plasticity of this loop leads to greater compensation by the Zn ligand His207 for shear
stresses induced at the catalytic site. Second, the Pro217-Asn218 peptide bond is rotated by
approximately 1000, evidently to prevent a repulsive interaction between the barbiturate C4=O4
keto group with the Pro217 carbonyl. Third, residues Ser209, Tyr216, Pro217, and Asn218 form
crystal contacts. These effects in combination lead to a translation along the entire loop from
Ala206 to Pro217, which however is relatively rigid, leaving most dihedral angles similar to those
in the reference structures. In the “north” rim of the active site, the largest change compared to
the inhibitor free MMP-8 structure is a 0.98 Å displacement and disorder of the Ile159 side
chain; the electron density shows a branched but symmetric side chain interpretable as two
equally populated rotamers which “swap” Cγ1 and Cγ2 positions.
Structural Basis of Collagenase Substrate Recognition
10
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Enzyme inhibition analysis
Utilizing the crystal structure of the MMP8-RO200-1770 complex, several follow-up
compounds were synthesized and tested against the panel of metalloenzymes shown in Table 2.
SUGGESTED LOCATION OF TABLE 2 The lead compound RO200-1770 shows broadly
nonspecific micromolar inhibition, excepting only stromelysin 1 (MMP-3) with its
approximately 10-fold weaker binding affinity to RO200-1770. To facilitate synthesis, the
piperidine of the lead compound RO200-1770 was substituted by an essentially isosteric
piperazine, RO204-1924. The almost uniform decrease in binding affinity might be rationalized
by higher desolvation penalties for piperazine binding. The theoretical clogP values calculated
for 1,4 dimethylpiperidine (1.9) and 1,4 dimethylpiperazine (0.8) support this hypothesis (39). I-
COL043 and RO206-0027 represent the results of two orthogonal approaches to optimize P1’-
S1’ and P2’-S2’ binding, respectively. For each inhibitor, an approximately 10-fold increase in
inhibition towards MMP-8, -2, -9, and –3 was accomplished, while inhibition of MT1-MMP
and MMP-1 was weakened or remained relatively unchanged . With its 4-fold weaker inhibition
of MMP-1, I-COL043 showed significantly enhanced selectivity potential against the latter
enzyme. The P1’ and P2’ optimizations of I-COL043 and RO206-0027 are combined in
RO206-0032 and the inhibition values demonstrate, to a first approximation, additivity of the
effects for MMP-8, -2, -9, and MT1-MMP. The improvement in its binding affinity to
stromelysin (MMP-3) is less distinct, while fibroblast collagenase (MMP-1) binding averages
rather than sums the effects of the precedent compounds.
Crystal Packing Effects
The MMP8-RO200-1770 complex did not crystallize as previously described (26), but
also under the previously reported crystallization conditions formed the crystal packing
arrangement described here. Thus, the inhibitor induces a conformational rearrangement that
leads to the new crystal packing. As discussed above, repulsion between the barbiturate C4=O4
and Pro217 carbonyl groups displaces the Pro217-Asn218 peptide. Its new orientation is
Structural Basis of Collagenase Substrate Recognition
11
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

stabilized by hydrogen bonds to the alcohol of Ser209 of a neighboring molecule in the crystal.
This serine alcohol also forms a hydrogen bond (2.7 Å) with the Nδ1 of the zinc ligand His207,
reminiscent of the charge relay system of serine-proteases. This interaction thus bridges Pro217
and His207 from one MMP-8 molecule with Ser209 of the neighboring enzyme. To create this
hydrogen bond, the Ser209 side-chain adopts a different χ1 rotamer different from earlier MMP-
8 structures. A crystallographic two-fold axis is located adjacent to Tyr216 and Pro217. The
side-chain of Asn218 is reoriented compared to typical MMP-8 structures (where an
intermolecular hydrogen to a symmetry related Thr129 exists) and forms a hydrogen bond with a
symmetry-related Tyr216 Oη. None of the crystal contacts interfere with expected peptidic
binding sites. A symmetry related Gln133, however, forms a hydrophobic contact at a depression
bounded by Ile159 and Ser151, which could serve as an alternative S2/S3 binding site. There is
no direct contact of the inhibitor with a symmetry-related protein molecule.
Secondary substrate recognition site – A collagenase-type I characteristic cis-peptide bond
SUGGESTED LOCATION OF FIGURE 5 The recognition and processing of natural
collagen substrates is known to involve the C-terminal hemopexin-like domain in addition to the
active site (4,40,41). The relative domain arrangement of the catalytic and C-terminal domains,
as seen for MMP-1 (11) and MMP-2 (42), shows the importance of the primed substrate
recognition sites, since these are located at the interface of the two collagenase domains.
Intriguingly, Asn188—Tyr189, located at the corridor connecting the catalytic and the C-
terminal domain, adopts a cis-peptide bond, Figure 5. Although not yet recognized, this cis-
peptide bond is not unique to the present crystal form; re-inspection confirmed its presence also
in the alternative crystal form (26). This cis-peptide bond is located on the solvent-exposed loop
preceding the catalytic α-helix Leu191H197E198XXH201—Leu203. The only restraint
apparent for this structural framework is a stabilizing hydrogen bond between carbonyl oxygen of
Thr181 with the amide of Tyr189. Sequence comparison of this segment with related MMP’s
reveals a subdivision within the MMP family. Only collagenases 1 and 2 (MMP-1 and MMP-8)
Structural Basis of Collagenase Substrate Recognition
12
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

lack a glycine at position 188, a feature otherwise absolutely conserved, including non-human
species as well. SUGGESTED LOCATION OF FIGURE 6. We therefore predict that the
Glu188—Tyr189 peptide bond of MMP-1 also adopts a cis-conformation. As exemplified by
the crystal structure of stromelysin 1 (MMP-3) (23,43), Gly188 exhibits dihedral angles (φ,ψ) =
(1500,-1650) which correspond to a conformation allowed only for glycine. Therefore, glycine is
conserved at position 188 presumably to stabilize the local fold; conservation of a nonglycine
residue (MMP-1, MMP-8) suggests a function related to the cis-peptide bond.
Structural Basis of Collagenase Substrate Recognition
13
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

DISCUSSION
Inhibitor conformation and its interaction with the protein
Although identified as potent collagenase inhibitor by an independent screening program,
the barbiturate-based inhibitor family exhibits striking similarities with well characterized classes
of inhibitors, namely hydroxamic and malonic acid based compounds (2,26). Figure 3C
illustrates that the Zn2+ chelation geometry of the hydroxamate, exemplified by batimastat, is
mirrored by the barbiturate with its N3 nitrogen substituting for the keto group of the
hydroxamate. Additionally, the interaction of the barbiturate N1-H1 and O6 with the protein
backbone Ala160-Ala161 parallels that of batimastat (Figure 3). A subtle difference is found at
the O6 interaction of the barbiturate ring, since the additional amide interaction with Ala161
could stabilize a greater negative charge on O6.
These findings present opportunities with challenges. The structural similarity of both
inhibitor classes for example enables the application of knowledge of optimization criteria for one
class to the other. On the other hand, the similarity might also indicate a limitation in finding
specific metalloproteinase inhibitors: The presence of a similar metal chelation topology in
independently identified and structurally unrelated lead compounds indicates that the Zn2+
binding follows a rather universal recognition motif which dominates the binding characteristics.
Consequently, many if not most potent active site directed Zn2+ protease inhibitor will exploit
such a universal binding motif and are likely to exhibit a low specificity profile, at least prior to
optimization.
The barbituric acid carries no net charge
The charge assignment of the Zn2+-chelating barbiturate ring aids an understanding of
the binding interaction. For the crystallization experiment, the pH was adjusted to 6.0, see
Experimental Procedures. This information is, however, insufficient to allow for a reliable
prediction of the protonation state of the barbiturate ring. Firstly, its pKa varies dramatically with
the presence of ring substituents: While the pKa of unsubstituted barbituric acid is about 4, its
5,5-diethyl substituted analog (”barbital”) has a pKa of around 8 (37). Secondly, the surrounding
Structural Basis of Collagenase Substrate Recognition
14
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

protein will also strongly affect the protonation of the barbiturate.
To address this issue, we inspected each polar group of the inhibitor for possible hydrogen
bonding partners. The 2.0 Å distance of the catalytic Zn2+ to N3 excludes its protonation, and
the O2H2 hydroxyl group is necessary to avoid repulsion of the carboxylate of Glu198. N1 and
O6 are involved in main chain hydrogen bonds. Consequently, their protonation appears well
defined as depicted in Figure 1. O4 is the only polar group without apparent attractive
interactions with the protein. However, the reorientation of the Pro217 carbonyl described above
would seemingly not occur if O4 is protonated as an alcohol. These arguments summarize the
case for the formula depicted in Fig. 1 which carries no net charge. Tunneling of the proton H2
(Figure 1) which bridges the carboxylate group of Glu198 however transfers a partial negative
charge to O2 and by resonance also to N3 (Figure 1). (A second line of investigation using
conformational correlation analysis of the 1.8 Å resolution structure presented here with
barbiturate derivatives deposited in the Cambridge small molecule database was not conclusive.)
S1’ and S2’ interaction, and enzyme inhibition profiles
Compared to MMP-8, human fibroblast collagenase (MMP-1) has a more restricted S1’
site with its Arg instead of Leu at position 193 . Its guanidinium group approximately occupies
the three S1’ solvent sites of MMP-8, namely Sol595, Sol602, and Sol592. Conversely, three
solvent molecules are found near Thr222 in MMP-1 where in MMP-8 the guanidinium group of
Arg222 is found. It appears, therefore, possible to enlarge the MMP-1 S1’ subsite to an MMP-8
size by swapping its Arg side-chain and solvent molecules. While such a swapped conformation
has been confirmed (12) for MMP-1, the rehydration is likely to create a considerable kinetic
barrier. Consequently, MMP-1 is expected to bind large P1’ residues with a kon kinetic rate
considerably lower than for MMP-8. The S1’ site of TACE appears rather too large to properly
accommodate the large P1’ residue of ICOL 043 and RO206-0032, Table 2. A unique feature of
the TACE active site (44) is the occurrence of Ala at the equivalent position of the strictly
conserved Tyr219 (MMP-8 numbering) of MMPs. This renders the TACE S1’ site both larger
Structural Basis of Collagenase Substrate Recognition
15
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

and less hydrophobic than in MMPs by almost completely removing the barrier to the S3’ site.
Consequently, the hydrophobic P1’ residue of ICOL 043 and RO206-0032 is not optimally
anchored in the TACE S1’ pocket. Further, incomplete dehydration of the voluminous site is
likely to disrupt the solvent structure within the TACE S1’ site (44) without the energy
compensation of a good fit.
Considering MMP-3, the southern rim of the active site, and in particular Pro221 (Pro217
in MMP-8), is rigidified by His224 (Ala220 in MMP-8) which hydrogen bonds via its Nδ1 and
Nε2 atoms to the backbone carbonyl groups of Leu222 and Thr215 (Asn218 and Pro211 in
MMP-8). The MMP-3 active site is thus incompatible with binding the barbiturate ring, as
reflected by the overall lower binding constants. His224 is unique to MMP-3. The observed
progressive increase of binding affinity with enlarging P1’ or P2’ residues is likely due to
generally increasing hydrophobic surface areas.
Optimization of binding affinity and selectivity.
Excepting C4=O4, all polar groups of the barbiturate bind within the protein matrix and should,
therefore, remain invariant in optimization strategies. The C4=O4 keto group, however,
presumably weakens the binding because of repulsive interactions with the Pro217 carbonyl
group, as described in the paragraph “Protein conformational changes”. Consequently, we
identify this group as a major variable in optimizing the Zn2+-chelating moiety. In fact,
modeling studies together with small molecule crystallographic database analyses indicate that
analogous five-membered ring systems might be an appropriate substitute Zn2+-binding group,
provided that the Zn2+ chelating properties are maintained. For example, deletion of the C4=O4
ketone and retention of the position of the chelating group N3-C2-OH2 will allow the new five-
membered ring to relax to a chemically reasonable geometry. With appropriate restraints for the
relaxation, the result will still possess favorable hydrogen bonding interactions between the
HN1-C6=O6 segment and the protein. To compensate for concomitant displacements of the
phenyl ring, it would be necessary to add a spacer atom to restore unstrained occupancies of both
Structural Basis of Collagenase Substrate Recognition
16
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

the S1’ and S2’ pockets.
Additional optimization approaches are suggested by kinetic analyses of earlier X-ray
structures of MMP’s with peptidic, hydroxamic, and malonic acid based inhibitors (2,26,27,45),
whereby significant improvement in binding affinity is achieved by better filling the respective
binding pockets (see (46) for a comprehensive review of these approaches). Expansion of the P1’
residue with an appropriate heterocyclic ring should supply both the necessary flexibility to
optimally fill the curved S1’ pocket and the hydrophilicity to adequately replace the binding sites
of the three water molecules found in the S1’ pocket. In summary, the major contribution to
specificity can be attributed to the P1’-S1’ interaction, while both substituents similarly
contribute to MMP-8 binding, Table 2.
Protein conformational changes
As described in the Results section, the most striking structural changes induced by inhibitor
binding occur near the catalytic Zn2+ with a major contribution from repulsive interactions
between the C4=O4 ketone and the Pro217 carbonyl group. For the collagenases, considerable
flexibility near the active site environment appears physiologically necessary for triple helical
peptide processing (47). The flexibility observed in the present structure suggests that the loop
Ala206—Asn218 will provide much of the flexibility necessary for collagen substrate recognition
(48), along with the additional plasticity seen at the catalytic Zn2+ and its ligating residues. The
generally weaker inhibition constants indicate that stromelysin 1 (MMP-3) does not possess the
necessary plasticity in this segment for barbiturate binding.
The intermolecular crystal contact Ser209—His207 might provide an unexpected
opportunity for synthetic drug design. In particular, this interaction offers the welcome
possibility to deviate from peptide-like binding patterns at a highly ordered position in the active
site. Intriguingly, since hydroxyprolines and other hydroxylated amino acids are present in the
physiological substrates of the extracellular matrix, we speculate that this crystal contact may
mimic a hydroxylated substrate interaction. An alternative possibility which also would exploit
Structural Basis of Collagenase Substrate Recognition
17
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

the common His-Ser motif would be an interaction with His201 Nδ1
Type-I collagen recognition exosite
Independent investigations by others on rat MMP-8 have shown the 188-loop to be required for
collagenase activity. The single site directed mutation to N209K, corresponding to N188K in
human MMP-8, disrupts collagenolytic activity 1. In addition, hybrid molecule studies involving
stromelysin 1 (N-terminal) and collagenase 1 (C-terminal) underscore the importance of this
loop for collagenolytic activity. The segment R181WTNNFREY189 of collagenase 1 is critical
for triple-helicase activity (49). In addition to collagenase 1, 2, and 3, the two gelatinases MMP-
2 and MMP-9 have tyrosine at position 189, both of which are preceded by a large insertion of
three fibronectin-II domains, Figure 6. These domains also are known to be critical for substrate
recognition (4,48,50). Therefore, the 188 exosite serves as a collagen substrate recognition site in
both collagenases (MMP-1,8,13) and gelatinases (MMP-2,9). This proposed substrate
recognition site is the position of the cis peptide bond described here for MMP-8 and predicted
for MMP-1. As such, it distinguishes collagenases 1 and 2 (MMP-1,8) from the other MMPs
known to cleave collagen which have a glycine at this position, including MMP-13 (17), MMP-
14 (51), and MMP-18 (52). Thus, collagenase 3 (MMP-13) (12,53), MMP-14 (54), and
presumably MMP-18 have a different backbone conformation in this loop segment. This
structural relationship is reflected by the biochemical properties of the respective enzymes.
MMP-13 is distinct from MMP-1 and –8, as it preferentially hydrolyzes type II collagen, while
the enzyme was 5 or 6 times less efficient at cleaving type I or III collagen (17). Similarly,
MMP-14 is 5-7 times less efficient at hydrolyzing type I collagen than MMP-1 while its
gelatinolytic activity is 8 times higher than that of MMP-1 (51).
To further investigate the role of the 189 exosite for macromolecular substrate
recognition, we docked a collagen triple helix to a full-length collagenase (MMP-1). In addition
to optimizing overall contact areas of the substrate-enzyme complex, we were guided by the
following localized interactions: (a) the contact of the collagen helix with the primary substrate
Structural Basis of Collagenase Substrate Recognition
18
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

recognition sites, including the catalytic Zn2+, (b) the contact of the collagen helix with the 189
exosite, and (c) the interaction of the collagen hydroxyproline with His207. We used published
data for modeling the structures of the isolated components (11,55,56). The most reliable and
powerful conclusion from these modeling studies is the orientation of the extended collagen
peptide relative to the enzyme. Earlier models postulated that the triple helix makes major
contacts with the first “blade” of the propeller-like hemopexin domain (48). We conclude
however that the triple helix will not lie in the MMP active site oriented along the shortest route
to the hemopexin-like domain, which would bring it into contact with its first blade. Instead, we
propose that the substrate runs through the 188 exosite, leading to major contacts to blade 2 of the
C-terminal collagenase domain, consistent with the chimera mutant studies by Nagase and
coworkers (49). The extended contact of the collagen substrate with the catalytic domain is
consistent with a collagenolytic activity of the catalytic domain alone, as described for MMP-1
(57). On the other hand, the conservation of the substrate exosite within MMP-1 and MMP-8
would suggest that the catalytic domain of MMP-8 should also exhibit a collagenolytic activity
that however has not been observed (48). A second important consequence of these modeling
studies is that at least one of the collagen strands must be bent or arched by approximately 20o;
an perfectly straight, rod-like collagen model binding to the active site of MMP-8/1 remains
approximately 6.5 Å distant from the collagen exosite (Tyr189-Asn190). This bending may be
part of the unwinding mechanism of the collagen triple helix necessary for its proteolysis (48).
The occurrence of a presumably functional cis-peptide bond at the type-I collagen-exosite of
collagenase 1 and 2 poses the question of its precise mechanism in collagenolysis. The function
of the 188 loop should be structurally linked to either (A) the amino acid 188 side chain or (B) the
backbone conformation due to the cis-peptide bond. Since the residue at the “non-glycine”
position 188 of MMP-1 and MMP-8 (Figure 6) shows conserved similarity but not identity, a
contribution of option (B) seems likely. A detailed understanding of the 188-exosite’s
mechanism in collagen processing, and in particular the role of the amino at 188, awaits further
experiments.
Structural Basis of Collagenase Substrate Recognition
19
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

ACKNOWLEDGEMENTS
We are grateful to Drs. Angelika Esswein, Valeria Livi, Ernesto Menta, and Gerd Zimmermann
for synthesizing the compounds RO204-1924, I-COL-043, RO206-0027, and RO206-0032.
We thank Dr. Christopher M. Overall for communicating site directed mutagenesis results prior
to publication.
Structural Basis of Collagenase Substrate Recognition
20
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

REFERENCES
1. Nagase, H., and Woessner, J. F. J. (1999) J. Biol. Chem. 274(31), 21491-21494
2. Grams, F., Reinemer, P., Powers, J. C., Kleine, T., Pieper, M., Tschesche, H., Huber, R.,
and Bode, W. (1995) Eur. J. Biochem. 228(3), 830-841
3. Stöcker, W., Grams, F., Baumann, U., Reinemer, P., Gomis-Rüth, F. X., McKay, D. B.,
and Bode, W. (1995) Protein Sci. 4, 823-840
4. Murphy, G., and Knäuper, V. (1997) Matrix Biology 15, 511-518
5. Westmays, J. A., Strissel, K. J., Sadow, P. M., and Fini, M. E. (1995) Proc. Natl. Acad.
Sci. USA 92(15), 6768-6772
6. van Wart, H., and Birkedal-Hansen, H. (1990) Proc. Natl. Acad. Sci. USA 87, 5578-
5582
7. Gomez, D. E., Alonso, D. F., Yoshiji, H., and Thorgeirsson, U. P. (1997) Eur. J. Cell Biol.
74(2), 111-122
8. Patterson, B. C., and Sang, Q. X. A. (1997) J. Biol. Chem. 272(46), 28823-28825
9. Werb, Z. (1997) Cell 91, 439-442
10. Bode, W., Fernandez-Catalan, C., Tschesche, H., Grams, F., Nagase, H., and Maskos, K.
(1999) Cell. & Mol. Life Sciences 55(4), 639-652
11. Li, J., Brick, P., O’Hare, M. C., Skarzynski, T., Lloyd, L. F., Curry, V. A., Clark, I. M.,
Bigg, H. F., Hazleman, B. L., Cawston, T. E., al., e., and Blow, D. M. (1995) Structure
3(6), 541-549
12. Lovejoy, B., Welch, A. R., Carr, S., Luong, C., Broka, C., Hendricks, R. T., Campbell, J.
A., Walker, K. A. M., Martin, R., Van Wart, H., and Browner, M. F. (1999) Nature Struct.
Bio. 6(3), 217-221
13. Stams, T., Spurlino, J. C., Smith, D. L., Wahl, R. C., Ho, T. F., Qoronfleh, M. W., Banks,
T. M., and Rubin, B. (1994) Nature Structural Biology 1(2), 119-123
14. Bode, W., Reinemer, P., Huber, R., Kleine, T., Schnierer, S., and Tschesche, H. (1994)
EMBO J. 13(6), 1263-1269
Structural Basis of Collagenase Substrate Recognition
21
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

15. Reinemer, P., Grams, F., Huber, R., Kleine, T., Schnierer, S., Piper, M., Tschesche, H.,
and Bode, W. (1994) FEBS Letters 338(2), 227-233
16. Knäuper, V., Wilhelm, S. M., Seperack, P. K., DeClerck, Y. A., Langley, K. E., Osthues,
A., and Tschesche, H. (1993) Biochem. J. 295, 581-586
17. Knäuper, V., Lopezotin, C., Smith, B., Knight, G., and Murphy, G. (1996) J. Biol. Chem.
271(3), 1544-1550
18. Netzel-Arnett, S., Fields, G. B., Birkedal-Hansen, H., and Van Wart, H. E. (1991) J.
Biol. Chem. 266(11), 6747-6755
19. Ottl, J., Battistuta, R., Pieper, M., Tschesche, H., Bode, W., Kuhn, K., and Moroder, L.
(1996) FEBS Letters 398(1), 31-36
20. Knäuper, V., Cowell, S., Smith, B., Lopezotin, C., Oshea, M., Morris, H., Zardi, L., and
Murphy, G. (1997) J. Biol. Chem. 272(12), 7608-7616
21. Betz, M., Huxley, P., Davies, S. J., Mushtaq, Y., Pieper, M., Tschesche, H., Bode, W., and
Gomis-Rüth, F. X. (1997) Eur. J. Biochem. 247(1), 356-363
22. Dhanaraj, V., Williams, M. G., Ye, Q. Z., Molina, F., Johnson, L. L., Ortwine, D. F.,
Pavlovsky, A., Rubin, J. R., Skeean, R. W., White, A. D., Hubmlet, C., Hupe, D. J., and
Blundell, T. L. (1999) Croatica Chem. Acta 72(2-3), 575-591
23. Chen, L. Y., Tydel, T. J., Gu, F., Dunaway, C. M., Pikul, S., Dunham, K. M., and Barnett,
B. L. (1999) J. Mol. Biol. 293(3), 545-557
24. Browner, M. F., Smith, W. W., and Castelhano, A. L. (1995) Biochemistry 34(20), 6602-
6610
25. Finzel, B. C., Baldwin, E. T., Bryant, G. L., Hess, G. F., Wilks, J. W., Trepod, C. M.,
Mott, J. E., Marshall, V. P., Petzold, G. L., Poorman, R. A., Osullivan, T. J., Schostarez,
H. J., and Mitchell, M. A. (1998) Protein Science 7(10), 2118-2126
26. Brandstetter, H., Engh, R. A., von Roedern, E. G., Moroder, L., Huber, R., Bode, W., and
Grams, F. (1998) Protein Science 7(6), 1303-1309
27. von Roedern, E., Brandstetter, H., Engh, R. A., Bode, W., Grams, F., and Moroder, L.
Structural Basis of Collagenase Substrate Recognition
22
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

(1998) J. Med. Chem. 41(16), 3041-3047
28. Bosies, E., Esswein, A., Grams, F., and Krell, H. W. (1996), Boehringer Mannheim, USA
29. Howard, A. J., Gilliland, G. L., Finzel, B. C., Poulos, T. L., Ohlendorf, D. H., and
Salemme, F. R. (1987) J. Appl. Cryst. 20(5), 383-387
30. Huber, R. (1965) Acta Cryst. 19, 353-356
31. Hoppe, W. (1957) Acta Cryst. 10, 750-751
32. Rossmann, M. G., and Blow, D. M. (1962) Acta Cryst. 15, 24-31
33. Navaza, J. (1994) Acta Cryst. A50, 157-163
34. Turk, D. (1992) in Chemistry, Technische Universität München, München
35. Brunger, A. T. (1992) X-PLOR version 3.1. A system for X-ray crystallography and
NMR, Yale Univeristy Press, New Haven, CT
36. Engh, R. A., and Huber, R. (1991) Acta Cryst. A47, 392-400
37. Budavari, S., O’Neil, M. J., Smith, A., and Heckelman, P. E. (eds) (1989) The Merck
Index, 11 Ed., Merck & Co., Inc., Rahway, N.J., USA
38. Wahl, M. C., and Sundaralingam, M. (1997) TIBS 22(3), 97-102
39. Ghose, A. K., Viswanadhan, V. N., and Wendoloski, J. J. (1998) J. Phys. Chem. 102(21),
3762-3772
40. Allan, J. A., Hembry, R. M., Angal, S., Reynolds, J. J., and Murphy, G. (1991) J. Cell Sci.
99, 789-795
41. Schnierer, S., Kleine, T., Gote, T., Hillemann, A., Knäuper, V., and Tschesche, H. (1993)
Biochem. Biophys. Res. Commun. 191, 319-326
42. Morgunova, E., Tuuttila, A., Bergmann, U., Isupov, M., Lindqvist, Y., Schneider, G., and
Tryggvason, K. (1999) Science 284, 1667-1670
43. Becker, J. W., Marcy, A. I., Rokosz, L. L., Axel, M. G., Burbaum, J. J., Fitzgerald, P. M.,
Cameron, P. M., Esser, C. K., Hagmann, W. K., Hermes, J. D., and al., e. (1995) Prot.
Science 4(10), 1966-1976
44. Maskos, K., Fernandez Catalan, C., Huber, R., Bourenkov, G. P., Bartunik, H., Ellestad,
Structural Basis of Collagenase Substrate Recognition
23
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

G. A., Reddy, P., Wolfson, M. F., Rauch, C. T., Castner, B. J., Davis, R., Clarke, H. R. G.,
Petersen, M., Fitzner, J. N., Cerretti, D. P., March, C. J., Paxton, R. J., Black, R. A., and
Bode, W. (1998) PNAS USA 95(7), 3408-3412
45. von Roedern, E., Grams, F., Brandstetter, H., and Moroder, L. (1998) J. Med. Chem.
41(3), 339-345
46. Grams, F., Brandstetter, H., Engh, R. A., Glitz, D., Krell, H.-W., Livi, V., Menta, E.,
Moroder, L., Müller, J. C. D., Roedern, E. G. v., and Zimmermann. (2000) in Matrix
Metalloproteinases-Inihibitors in Cancer Therapy (Clendeninn, N. J., and Appelt, K., eds)
Vol. in press
47. Ottl, J., and Moroder, L. (1999) J. Am. Chem. Soc. 121(4), 653-661
48. Ottl, J., Gabriel, D., Murphy, G., Knauper, V., Tominaga, Y., Nagase, H., Kroger, M.,
Tschesche, H., Bode, W., and Moroder, L. (2000) Chemistry & Biology 7(2), 119-132
49. Chung, L., Shimokawa, K., Dinakarpandian, D., Grams, F., G.B., F., and Nagase, H.
(2000) J. Biol. Chem. 275(38), 29610-29617
50. Banyai, L., Tordai, H., and Patthy, L. (1994) Biochem. J. 298, 403-407
51. Ohuchi, E., Imai, K., Fujii, Y., Sato, H., Seiki, M., and Okada, Y. (1997) J. Biol. Chem.
272(4), 2446-2451
52. Stolow, M. A., Bauzon, D. D., Li, J., Sedgwick, T., Liang, V. C., Sang, Q. A., and Shi, Y.
B. (1996) Mol. Biol. Cell 7(10), 1471-1483
53. Botos, I., Meyer, E., Swanson, S. M., Lemaitre, V., Eeckhout, Y., and Meyer, E. F. (1999)
J. Mol. Biol. 292(4), 837-844
54. Fernandez-Catalan, C., Bode, W., Huber, R., Turk, D., Calvete, J. J., Lichte, A.,
Tschesche, H., and Maskos, K. (1998) EMBO J. 17(17), 5238-5248
55. Li, M. H., Fan, P., Brodsky, B., and Baum, J. (1993) Biochemistry 32(29), 7377-7387
56. Kramer, R. Z., Vitagliano, L., Bella, J., Berisio, R., Mazzarella, L., Brodsky, B., Zagari,
A., and Berman, H. M. (1998) J. Mol. Biol. 280(4), 623-638
57. Lauer-Fields, J. L., Tuzinski, K. A., Shimokawa, K., Nagase, H., and Fields, G. B. (2000)
Structural Basis of Collagenase Substrate Recognition
24
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

J. Biol. Chem. 275(18), 13282-13290
Structural Basis of Collagenase Substrate Recognition
25
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

FOOTNOTES
1 Personal communication by Dr. Christopher M. Overall
Structural Basis of Collagenase Substrate Recognition
26
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

FIGURE LEGENDS
Figure 1: Schematic representation of the active site directed inhibitors.
RO200-1770: 2-hydroxy-5-phenyl-5-N-(4-(2-hydroxyethyl)-piperidyl)-4,6-
pyrimidinedione
RO204-1924: 2-hydroxy-5-phenyl-5-N-(4-(2-hydroxyethyl)-piperazyl)-4,6-
pyrimidinedione
I-COL043: 2-hydroxy-5-n-octyl-5-N-(4-(2-hydroxyethyl)-piperazyl)-4,6-
pyrimidinedione
RO206-0027: 2-hydroxy-5-phenyl-5-N-(4-paranitrophenyl-piperazyl)-4,6-
pyrimidinedione
RO206-0032: 2-hydroxy-5-n-octyl-5-N-(4-paranitrophenyl-piperazyl)-4,6-
pyrimidinedione
Figure 2: The RO200-1770 inhibitor bound to the active site of MMP-8 (yellow). The 2Fo-Fc
electron density map is contoured 1.0 σ over the mean. Pro217 (red) from a reference
structure was superimposed to the protein model (yellow) to illustrate the conflict of its
carbonyl oxygen with barbiturate binding. The figure was prepared by using the program
MAIN (34).
Figure 3:
A. Schematic representation of the active site determinants for substrate recognition
B. Schematic representation of the interaction of the 2-hydroxy-pyrimidinedione with
the MMP-8 active site.
C. Schematic representation of the Zn2+-interaction geometry of a hydroxamic acid
based inhibitor (batimastat) (2).
Figure 4: Details of the histidine-phenyl stacking interaction.
Figure 5: Cis-peptide bond between Asn188 and Tyr189. The cis-peptide bond is highlighted in
red. The figure was prepared by using the program MAIN (34).
Structural Basis of Collagenase Substrate Recognition
27
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Figure 6: Local sequence comparison of related MMP’s around the collagen recognition exosite.
All sequences are human unless otherwise noted. The complete list of known MMP
sequences shows the same pattern that only MMP-1 or MMP-8 have nonglycine
residues at position 188. Sequences were derived from Swissprot.
Structural Basis of Collagenase Substrate Recognition
28
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Table 1: Zn2+ coordination geometry: Bonds, angles, and dihedral angles
Zn2+ — N3Bar 2.09 ÅZn2+ — O2Bar 2.93 ÅZn2+ — O4Bar 3.15 ÅZn2+ Nε2197 2.03 ÅZn2+ Nε2201 1.95 ÅZn2+ Nε2207 2.00 Å
Zn2+ O2Bar C2Bar o78.2 Zn2+ O4Bar C4Bar o70.4
Trigonal bipyramidal description of the Zn2+ coordinationZn2+ Nε2197 Nε2207 O2Bar o-4.8
O2Bar Zn2+ Nε2197 o101.5 Nε2197 Zn2+ Nε2207 o119.7 Nε2207 Zn2+ O2Bar o137.8
Nε2201 Zn2+ N3Bar o125.6
Square pyramidal description of the Zn2+ coordinationNε2207 O2Bar N3Bar Nε2201 o15.2
Nε2197 Zn2+ Nε2207 o119.7 Nε2197 Zn2+ N3Bar o95.5 Nε2197 Zn2+ O2Bar o101.5 Nε2197 Zn2+ Nε2201 o100.3
Pentagonal pyramidal description of the Zn2+ coordinationNε2207 O2Bar O4Bar Nε2201 o20.0
Structural Basis of Collagenase Substrate Recognition
29
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Nε2197 Zn2+ O4Bar o88.0
Structural Basis of Collagenase Substrate Recognition
30
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from
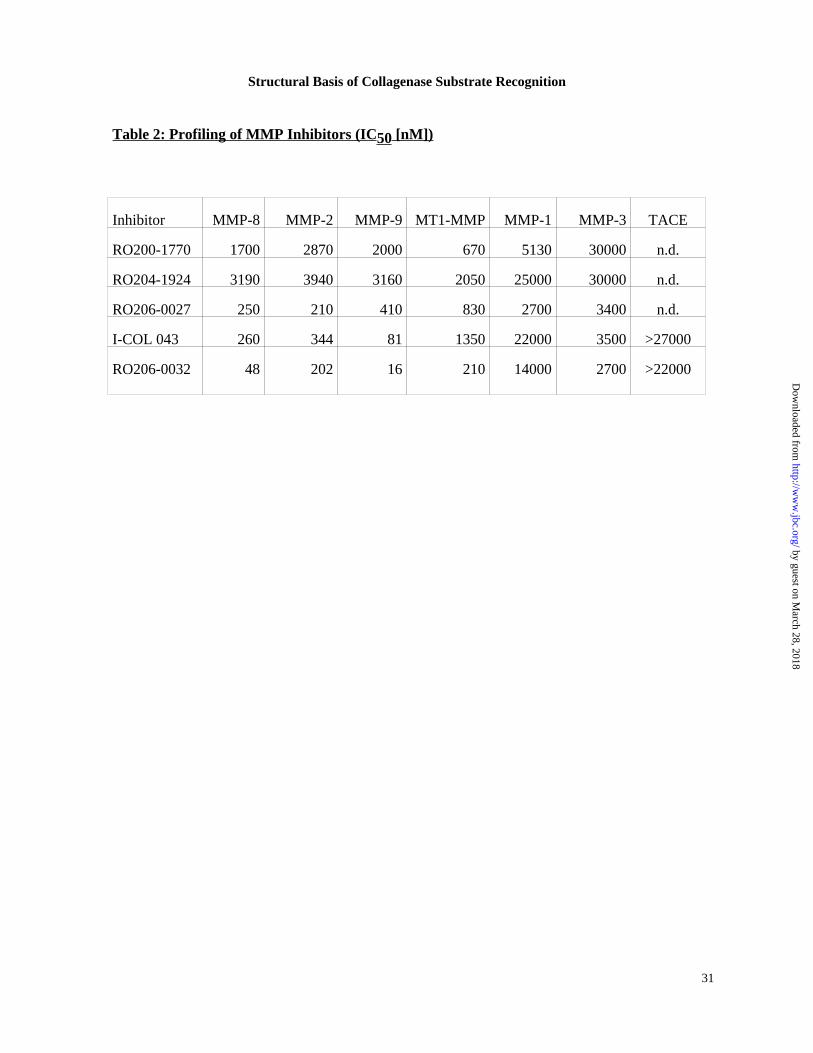
Table 2: Profiling of MMP Inhibitors (IC 50 [nM])
Inhibitor MMP-8 MMP-2 MMP-9 MT1-MMP MMP-1 MMP-3 TACE
RO200-1770 1700 2870 2000 670 5130 30000 n.d.
RO204-1924 3190 3940 3160 2050 25000 30000 n.d.
RO206-0027 250 210 410 830 2700 3400 n.d.
I-COL 043 260 344 81 1350 22000 3500 >27000
RO206-0032 48 202 16 210 14000 2700 >22000
Structural Basis of Collagenase Substrate Recognition
31
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

1
��
��
��
��
��
��
�
��
�
�
��
�
��
��
��
��
��
��
�
��
�
�
���
�
��
��
��
��
��
��
�
��
�
�
���
�
�������
�� ���
�� �� �
��
��
��
��
��
��
�
��
I&�
I&�
I&�
I&� I&
�
I&�
S1�
S&�
S&�S&
�
S&�
S&�
S&�
S&�
S2+�
�
�� ����
��
��
��
��
��
��
�
��
�
�
��
�
�� ���
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from
2
His197 Zn2+Zn2+
His207His207
His201 His201
His197
Glu198Ala161 Ala161Glu198
Pro217 Pro217
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

3
N
O
OO
O
N
O
N
N
O
O
N
O
N
N
O
NN
O
N
O
O
Ala163
P2-S2
Ala161Ala160
Tyr219
Asn218Pro217
Zn2+
Glu198
P1-S1
P1‘-S1‘
P2‘-S2‘
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

4
OO
O
N
O
N
N
O
O
N
O
N
Ala161Ala160
Tyr219
Asn218Pro217
Zn2+
Glu198
N1O2
O4
O6
H1
P1‘
P2‘
N3
H
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

5
OO
O
N
O
N
N
O
O
N
O
N
Ala161Ala160
Tyr219
Asn218Pro217
Zn2+
Glu198
HO N
H
OR
P1‘
P2‘
O
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from


7
Tyr189Asn188
Thr181
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

8
Sequence Alignment at Secondary Collagen Recognition Site
Coll-1 (MMP-1) WT NNFR EYNL
Coll-1 (bovine) WT SNFQ DYNL
Coll-1 (porcine) WT KNFR DYNL
Coll-1 (rabbit) WT KDFR NYNL
Coll-2 (MMP-8) WT NTSA NYNL
Coll-2 (rat) WT QDSK NYNL
Coll-2 (mouse) WT QDSK NYNL
Coll-3 (MMP-13) WT SSSK GYNL
Coll-4 (MMP-18) WT EGTYR GVNL
Gel A (MMP-2) WT (Fn-II)3 GYSL
Gel B (MMP-9) WS (Fn-II)3 GYSL
StrL-1 (MMP-3) WT KDTT GTNL
StrL-2 (MMP-10) WT EDAS GTNL
StrL-3 (MMP-11) WT THSG GYNL
MatrL (MMP-7) WT DGSS GINF
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Hans-Willi Krell and Richard A. EnghHans Brandstetter, Frank Grams, Dagmar Glitz, Anja Lang, Robert Huber, Wolfram Bode,
reveals a previously unobserved mechanism for collagenase substrate recognitionThe 1.8 angstrom crystal structure of an MMP8- barbiturate inhibitor complex
published online January 22, 2001J. Biol. Chem.
10.1074/jbc.M007475200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on March 28, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Additions and Corrections
Vol. 276 (2001) 17405–17412
The 1.8-Å crystal structure of a matrix metalloproteinase8-barbiturate inhibitor complex reveals a previouslyunobserved mechanism for collagenase substraterecognition.
Hans Brandstetter, Frank Grams, Dagmar Glitz, Anja Lang,Robert Huber, Wolfram Bode, Hans-Willi Krell, and Richard A.Engh
The Protein Data Bank accession number was omitted fromthis paper. The PDB entry code is 1JJ9.
Vol. 276 (2001) 9297–9302
The cytosolic O-acetylserine(thiol)lyase gene is regu-lated by heavy metals and can function in cadmiumtolerance.
Jose R. Domínguez-Solís, Gloria Gutierrez-Alcala, Jose M.Vega, Luis C. Romero, and Cecilia Gotor
Jose M. Vega was inadvertently omitted from the list ofauthors. The correct list is shown above.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 276, No. 33, Issue of August 17, p. 31474, 2001Printed in U.S.A.
We suggest that subscribers photocopy these corrections and insert the photocopies at the appropriateplaces where the article to be corrected originally appeared. Authors are urged to introduce thesecorrections into any reprints they distribute. Secondary (abstract) services are urged to carry notice ofthese corrections as prominently as they carried the original abstracts.
31474