the catalytic subunit of camp-dependent protein kinase ... et al-pka.pdf · the catalytic subunit...
TRANSCRIPT

The catalytic subunit of cAMP-dependent protein kinase:prototype for an extended network of communication
Christopher M. Smitha,*, Elzbieta Radzio-Andzelmb, Madhusudanb,Pearl Akamineb, Susan S. Taylorb
aSan Diego Supercomputer Center, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0505,USA
bHoward Hughes Medical Institute, Department of Chemistry and Biochemistry, University of California, San Diego,
9500 Gilman Drive, La Jolla, CA 92093-0654, USA
Abstract
The protein kinase catalytic core in essence comprises an extended network of interactions that linkdistal parts of the molecule to the active site where they facilitate phosphoryl transfer from ATP toprotein substrate. This review de®nes key sequence and structural elements, describes what is currentlyknown about the molecular interactions, and how they are involved in catalysis. # 1999 ElsevierScience Ltd. All rights reserved.
1. Introduction
Protein phosphorylation is probably the most important mechanism of regulation in theeukaryotic cell. While allosteric regulation is important in all cells and allows many proteins tobe sensitive to their immediate environment, protein phosphorylation allows cells to beresponsive to their external environment. The signal transduction pathways that allow a cell torespond to external stimuli, whether it is a hormone, a growth factor, a photon or osmoticshock, are all regulated at critical switch points by protein phosphorylation. The enzymes thatcatalyze this transfer of a phosphate from ATP to a protein or peptide substrate, the proteinkinases, constitute a large family of enzymes. The family is comprised of two main subfamilies
Progress in Biophysics & Molecular Biology 71 (1999) 313±341
0079-6107/99/$ - see front matter # 1999 Elsevier Science Ltd. All rights reserved.PII: S0079-6107(98)00059-5
* Corresponding author. Fax: +1-619-534-5113.E-mail address: [email protected] (C.M. Smith)

Ð those that transfer phosphate to serine and threonine and those that transfer phosphate totyrosine (Hanks and Hunter, 1995). The entire family, however, has evolved from a commonorigin. Based on analysis of the yeast genome, the human genome alone is predicted to encodefor over 2000 unique protein kinases each turned on and o� in response to speci®c signals(Hunter and Plowman, 1997). This makes the protein kinases one of the largest gene families.When one considers as well the protein phosphatases and the myriad of regulatory and adapterproteins that modulate protein kinase function and location, as well as the fact that over onethird of the proteins in cells are the phosphoproteins, it is clear that a signi®cant portion of theeukaryotic cellular machinery is involved, either directly of indirectly, with proteinphosphorylation. Westheimer's elegant 1987 review of phosphates and their importance innature, while acknowledging their major role in nucleic acid structure and function and inbioenergetics, ignored this equally major role that phosphates play in the regulation of cellularprocesses (Westheimer, 1987).Protein phosphorylation was ®rst recognized as a mechanism for regulation of protein
function in the 1950s by the pioneering work of Fischer and Krebs who showed that glycogenphosphorylase was regulated by the covalent attachment of a phosphate moiety (Fischer andKrebs, 1955). This ®nding coincided with the discovery of glycogen phosphorylase kinase, the®rst protein kinase to be rigorously characterized (Krebs et al., 1959). The identi®cation ofglycogen phosphorylase kinase kinase, later renamed cAMP-dependent protein kinase (cAPK)and its activation by cAMP (Walsh et al., 1968), led to two additional and fundamentalconcepts. First was the idea of kinase cascades and the power of ampli®cation. We nowrecognize that there are many more elaborate cascades that lead from a single event at theplasma membrane to the eventual mediation of gene expression in the nucleus. From theidenti®cation of cAPK and its activation by cAMP, also came the recognition of the `secondmessenger' concept (Sutherland and Wosilait, 1955). Binding of a molecule at the surface of acell can lead to the generation of a second molecule within the cell that mediates the biologicalresponse. The three most prominent intracellular protein kinase second messengers, cAMP,Ca++ and phospholipids, all mediate their primary responses by contributing to the activationof speci®c protein kinases. Extracellular mediators such as growth factors mediate theirresponse by binding directly to a molecule having an intracellular kinase domain which isactivated upon ligand binding to the extracellular domain (Ullrich and Schlessinger, 1990).
2. cAMP-dependent protein kinase
cAMP-dependent protein kinase is not only one of the ®rst protein kinases to be discovered(Walsh et al., 1968), it remains as one of the simplest. Its simplicity derives primarily from itsdissociative mechanism of activation (Taylor et al., 1990; Francis and Corbin, 1994). It iscomprised of two subunit types, regulatory (R) and catalytic (C). In the absence of cAMP theenzyme exists as an inactive tetrameric holoenzyme complex, R2C2. The two main subfamiliesof cAPK are determined by the R subunit. The type I subunits are not autophosphorylated byC and have an absolute requirement for MgATP to form a tight complex with the C subunit.The type II R subunits, in contrast, are autophosphorylated and do not require MgATP toform a holoenzyme complex. Depending on the cell type, cAMP can be generated by the
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341314

activation of adenylyl cyclase in response to a variety of stimuli. In all cases, the initialreceptor belongs to the 7-transmembrane spanning serpentine family of G protein coupledreceptors (Luttrell et al., 1997). The primary receptors for cAMP in eukaryotic cells are the Rsubunits of cAPK. The cooperative binding of cAMP to the holoenzyme causes a ®ve order ofmagnitude decrease in the a�nity of R for C (Granot et al., 1980) and under physiologicalconditions is thought to lead to the dissociation of the holoenzyme into an R2 (cAMP)4 dimerand two active C subunits (Gill and Garren, 1969; Brostrom et al., 1970; Tao et al., 1970). It isthis unleashed C subunit that is the active form of the enzyme.The catalytic subunit has many known protein substrates (Zetterqvist et al., 1976), and the
speci®c targets again will vary depending on the cell type and on the particular proteins thatare expressed in that cell at any given time. In addition to phosphorylating known cytoplasmicproteins such as glycogen phosphorylase kinase (Yeaman et al., 1977), glycogen synthase(Parker et al., 1981), pyruvate kinase (Hjelmquist et al., 1974), phosphofructokinase II (Murrayet al., 1984), protein phosphatase inhibitor I (Cohen et al., 1977) and all proteins in the liverthat synergistically contribute to the mobilization of stored glycogen, the C subunit also leadsto increased expression of genes that are regulated in response to cAMP such as the gene forPEPCK, a gluconeogenic enzyme (Sutherland et al., 1996). These genes are typically precededby a cAMP response element (CRE) (Montminy, 1997). A cAMP response element bindingprotein (CREB) binds to this CRE and is activated in response to phosphorylation of Ser112.This phosphorylation can be mediated by the C subunit. Once phosphorylated, CREB becomesa docking site for a transcriptional activator, the CREB binding protein, CBP, which leads tothe assembly of the transcriptional complex and subsequent activation of gene expression(Goldman et al., 1997).All substrates, as well as the known physiological inhibitors, of the C subunit have the
general consensus recognition site summarized in Fig. 1. Important determinants are argininesat the P-6, P-3 and P-2 positions with most substrates having either a P-6 and P-3 Arg or a P-3and a P-2 Arg (Zetterqvist et al., 1976). A large hydrophobic residue is preferred at the P+1site while there are few constraints at the P-1 site (Kemp et al., 1975). This consensus site
Fig. 1. Substrates and Inhibitors of cAPK. The general features of the minimum consensus site, P-3 through P+1,that occupies the active site cleft, are highlighted in purple. PKI and the RI subunit share this consensus site,although unlike the RII subunits and substrates, they are pseudosubstrates. The P-site hydroxyl group is missing.The region required for high a�nity is highlighted in teal and phosphorylated residues (Serine) in blue. The essential
residues are indicated by the red diamonds. The respective binding constants for the inhibitor and hexapeptides(Kemp et al., 1977; Whitehouse and Walsh, 1983), and Kapp (for the C subunit under physiological conditions(Ho�man, 1980)) for the inhibitor domain of RIa and RIIa subunits are also listed.
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341 315

peptide is su�cient to convey low a�nity binding in the mM range. The regulatory subunits arecompetitive inhibitors of protein substrates and have a substrate-like motif that resembles theconsensus site peptide and binds to the active site cleft.In addition to the R subunits, there is another class of physiological inhibitors of C. These
are the heat stable protein kinase inhibitors (PKI's). Like the R subunits, the PKI's bind withhigh a�nity (<1 nM) to free C. Like the R subunits, they also share an inhibitor site thatresembles a cAPK substrate. As seen in Fig. 1, PKI and the RI subunits have apseudosubstrate inhibitor site, while the RII subunits have a true phosphorylation site. WhilePKI was discovered almost simultaneously with cAPK because it copuri®ed as a contaminate(Walsh et al., 1990), the true physiological role of PKI is still unknown. It is expressed in a cellcycle-dependent manner (Wen et al., 1995), is relatively unstructured when it is free in solution(Hauer et al., 1999) and is multifunctional. The high a�nity inhibitor site in PKI is locatednear the amino-terminus as indicated in Fig. 1, and the requirements for high a�nity bindingwere elegantly mapped by Walsh and Glass (Cheng et al., 1986; Scott et al., 1986). However, inaddition to its C subunit inhibitor site, PKI contains near its carboxyl-terminus a nuclearexport signal (NES). Thus, when C is bound to PKI, the complex is rapidly exported from thenucleus (Wen et al., 1995). This NES, ®rst discovered in PKI and HIV-rev, is now known tobe present in many proteins and contributes actively to the translocation of proteins betweenthe cytoplasm and the nucleus.
3. Structure of the catalytic subunit
To understand the molecular basis for the function of any protein, it is essential to have ahigh resolution structure. The solution of the crystal structure of the C-subunit (Knighton etal., 1991a, 1991b), the ®rst member of this enzyme family to be solved, was facilitated by twothings. First was the binding of PKI (5±24) which was cocrystallized with the C subunit.Second was the expression of the C subunit in E. coli (Slice and Taylor, 1989). Furthermore,the enzyme is readily expressed in a fully phosphorylated form in E. coli (Yonemoto et al.,1993a), and this led to the isolation of large quantities of very pure and active enzyme.
3.1. Overall architecture
The C subunit is a bilobal enzyme with two major subdomains that are conservedthroughout the protein kinase family (Knighton et al., 1991a, 1991b). In Fig. 2(a) the structureis correlated with the sequence motifs as de®ned by Hanks et al. (1988). The structure beginswith a myristylation motif that, in most structures of the recombinant unmyristylated enzyme,is disordered. This is followed by a nonconserved long A-helix which spans both lobes. Thehighly conserved core begins with the small domain that is dominated by a ®ve strandedantiparallel b-sheet. The single conserved helix in the small domain is the C-helix. This domainconstitutes the nucleotide binding domain. A single linker strand joins the two lobes. Althoughthe larger carboxy terminal domain is dominated by helices, a single small b-sheet lies at theactive site cleft, and this region contains many of the conserved residues that are essential forcatalysis. The C-terminal tail (residues 300±350) wraps as a mostly extended chain over the
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341316

Fig. 2. Correlation of conserved sequences motifs with the structure of the catalytic subunit. The structure of theternary complex of C, PKI (5±24) and ATP is shown in panel a. The structure and sequence (panel b) of the Csubunit are color coordinated according to the eleven distinct sequence motifs identi®ed by Hanks et al. (1988). The
structure coordinates are from Zheng et al. (1993c) (PDB accession No.: 1ATP). Secondary structural elements: a-helices, b-sheets and loops and turns, are depicted as cylinders, planar arrows and noodle lines, respectively. Thephosphorylated Ser338 and Thr197 are indicated as white spheres, and the ATP substrate is depicted, as a ball and
stick structure, in the active site cleft.
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341 317

surface of both lobes and again is not conserved throughout the protein kinase family althoughit is shared by all members of the cAPK, PKC and PKG families (Hanks et al., 1988). Fig.
2(b) is color-coded according to the subdomains (I±XI) de®ned by Hanks et al. (1988).
As seen in Figs. 2 and 3, the active site lies at the cleft between the two lobes with the
adenine ring of ATP deeply buried at the base of the cleft. The peptide docks to the surface ofthe large lobe where the hydroxyl group of the P site Ser or Thr is poised for a direct in-line
transfer of the g-phosphate of ATP (Ho et al., 1988). The catalytic subunit of cAPK is theonly protein kinase that has been crystallized in its active form in the presence and absence ofboth substrates and inhibitors. These various structures have revealed the conformational
¯exibility that is an essential part of catalysis. However, before talking about catalysis,¯exibility and peptide recognition, we ®rst need to describe the active site more rigorously. This
review will focus in particular on the conserved motifs in the core and on our understanding ofthe molecular basis for the importance of each motif. It is based on numerous structures of the
Fig 2 (continued)
Fig. 3. Active site cleft of the catalytic subunit of cAPK is characterized by conserved loops that converge at the site
of phosphoryl transfer. The top panel (a) is a stereo view of the ternary complex, rC:PKI (5±24): MnATP. Theglycine-rich loop (residues 48±57) is in red, the linker (residues 120±127) in yellow and the catalytic loop (residues166±171) and the magnesium positioning loop (residues 184±187) in white. The side chains of His87, phosphoThr197, Arg280 and Glu208 are also indicated. The convergence of these three loops at the active site is shown in
the bottom panel (b).
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341318
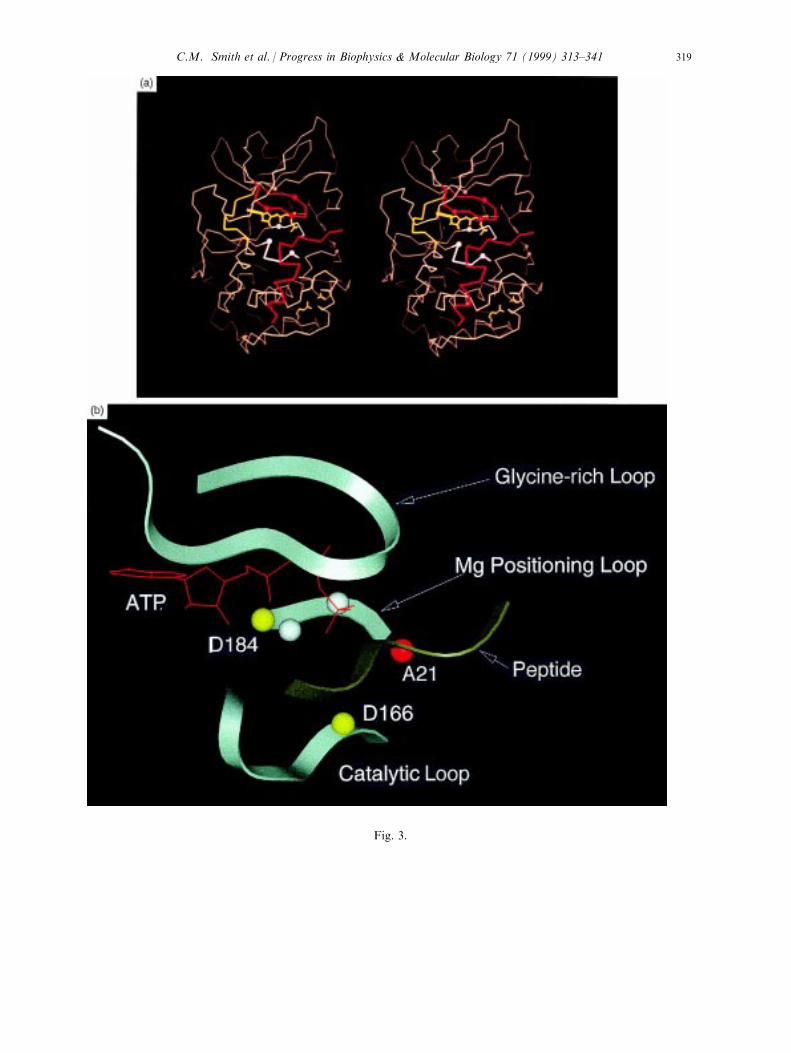
Fig. 3.
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341 319
C-subunit and on comparison with many other protein kinases that have been crystallized inthe last 4±5 years.
3.2. Conserved core
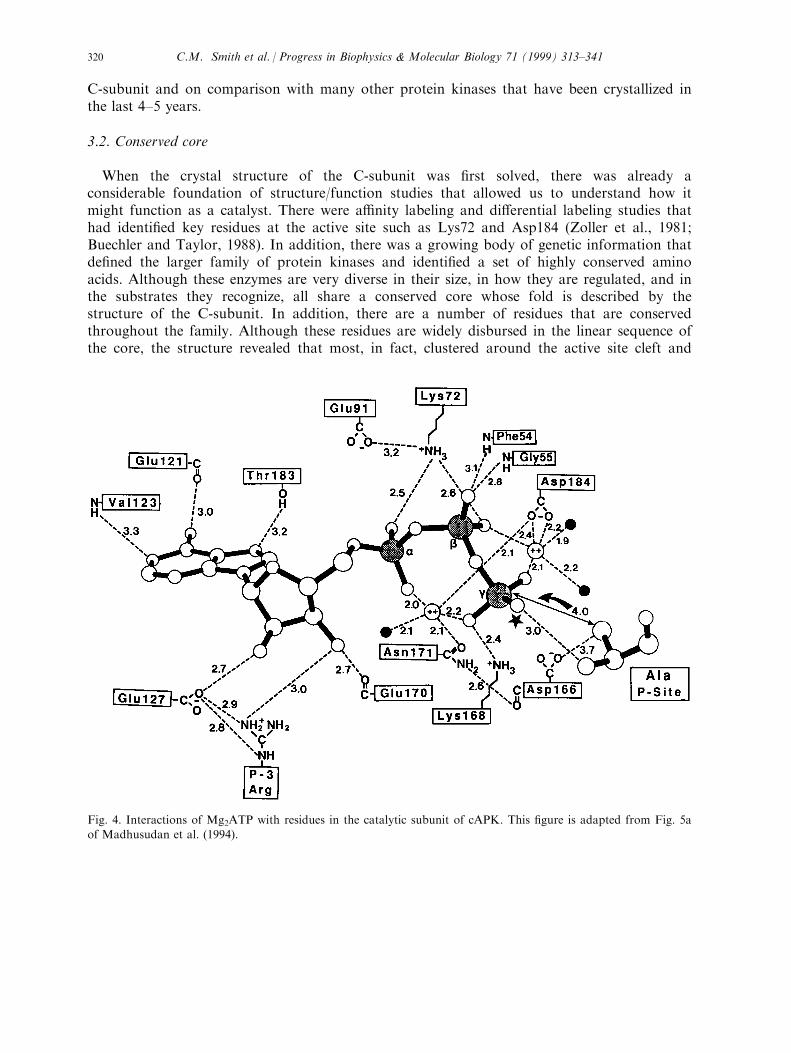
When the crystal structure of the C-subunit was ®rst solved, there was already aconsiderable foundation of structure/function studies that allowed us to understand how itmight function as a catalyst. There were a�nity labeling and di�erential labeling studies thathad identi®ed key residues at the active site such as Lys72 and Asp184 (Zoller et al., 1981;Buechler and Taylor, 1988). In addition, there was a growing body of genetic information thatde®ned the larger family of protein kinases and identi®ed a set of highly conserved aminoacids. Although these enzymes are very diverse in their size, in how they are regulated, and inthe substrates they recognize, all share a conserved core whose fold is described by thestructure of the C-subunit. In addition, there are a number of residues that are conservedthroughout the family. Although these residues are widely disbursed in the linear sequence ofthe core, the structure revealed that most, in fact, clustered around the active site cleft and
Fig. 4. Interactions of Mg2ATP with residues in the catalytic subunit of cAPK. This ®gure is adapted from Fig. 5a
of Madhusudan et al. (1994).
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341320

appeared to be important for ATP binding and catalysis. The general convergence of active siteresidues around ATP is shown in Fig. 3(b) while Fig. 4 identi®es most of the importantresidues that interact directly with ATP. As summarized in Fig. 3, most of these residues arelocalized speci®cally in three loops that converge at the active site cleft where the ATP isbound. The most dynamic of these loops is contributed by the small lobe while the other twolie in the large lobe. All of this information made interpretation of the structure feasible. Weshall de®ne these various motifs beginning ®rst with the small lobe.The function of the small lobe, shown in Fig. 5(a), is to bind nucleotide, leaving the g-
phosphate poised for transfer to a peptide or protein substrate. It is dominated by a ®ve-
Fig. 5. The small domain and its interactions with Mg2ATP. The small domain and the linker segments are shownin the upper panel (a) as a ribbon diagram with the glycine-rich loop (residues 48±57) and the C-Helix (residues 84±
98) highlighted in white. The a carbons of the highly conserved residues (Gly50, Gly52, Gly55, Lys72 and Glu91)are indicated as spheres. Relative to the ATP substrate, the position of the glycine-rich loop is shown in the lowerpanel (b) in both its open (Zheng et al., 1993b; white) and closed (Zheng et al., 1993a, 1993c; multicolor)conformations. The hydrogen bonds between backbone amides and the b- and g-phosphates are also indicated as
dashed lines.
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341 321

stranded antiparallel b-sheet with a single conserved helix. The position of the small loberelative to the large lobe leads to the opening and closing of the active site cleft and is anessential feature of the catalytic process.The ®rst two b strands, linked by a glycine-rich loop, constitute a nucleotide positioning
motif (NPM), residues 48±57. Gly50, 52 and 55 are highly conserved and are hallmark of theprotein kinase family. The NPM spans the entire length of the wedge shaped nucleotidebinding pocket and forms the ceiling of this pocket with the nucleotide ®tting snugly againstthis motif. The highly conserved Val57 lies above the ribose ring while three backbone amidesat the tip of the loop position the phosphates of ATP. The glycines at the tip of the loop areparticularly important for positioning the phosphates of ATP. The backbone amide of Ser53binds to one of the g-phosphate oxygen's while the backbone amides of Phe54 and Gly55position one of the b phosphate oxygens (Fig. 5(b)). Gly50 lies above the ribose ring. Gly52 ishighly conserved and is also the most important for catalysis (Hemmer et al., 1997; Grant etal., 1998). The presumed reason for its importance is that the hydrogen bonding of the Ser53amide to the g-phosphate as shown in Fig. 5(a), is thought to be a critical step for forming thetransition state intermediate prior to phosphoryl transfer (Bossemeyer, 1994; Zheng et al.,1993a). The tip of this loop is the most mobile part of the molecule and serves as a sensor forwhat is occupying the active site cleft ( Narayana et al., 1997a). While more than a dozendi�erent structures of the C subunit have been solved, the tip of the loop has only beenobserved in a very stable form, based on temperature factors, in the ternary complex whereATP or an ATP analog and PKI (5±24) are both bound with high a�nity (Zheng et al., 1993a;Narayana et al., 1997a).b strand 3 contains another conserved residue, Lys72. The importance of this Lys was ®rst
recognized when it was shown to be modi®ed by the ATP analog, ¯uorosulfonyl benzoyladenosine (Zoller et al., 1981). The structure revealed that Lys72 binds to the a and bphosphates of ATP. b strand 3 is followed by a small B-helix. This is the only element ofsecondary structure in the core of the C-subunit that is not shared by the overall protein kinasefamily. The long C-helix (Fig. 6) that follows is, however, conserved and houses the thirdessential motif in the small lobe, Glu91. Like the nucleotide positioning motif, this helix spansthe entire length of the wedge-shaped ATP binding pocket. In the active form of the C-subunitthis Glu is directed towards Lys72. It does not interact directly with the nucleotide but ratherpositions Lys72. Through multiple interactions the C-helix actually positions the entiremolecule for catalysis.Helical switches appear to be important modulators of protein kinase function and this C-
helix switch is critical for every protein kinase. Like railroad switches connecting di�erenttracks and determining completely di�erent fates, the twist of this helix determines thefunctional mode of the enzyme. Its capability to serve as such a global switch is due to themultiple contacts that are made by this helix to all parts of the molecule. A closer examinationof the speci®c residues in the turn of the helix that includes Glu91 is su�cient to demonstratethis global communication (Fig. 6(b)). Lys92, for example, pairs with the a-carboxylate ofPhe350. This carboxyl terminal Phe is buried in the small lobe as is the ion pair between Lys92and Phe350. The next residue in the turn, Arg93, forms part of the deep hydrophobic pocketwhere Trp30 at the end of the A-helix is docked. Replacement of Trp30 with either Tyr or Alaintroduces signi®cant instability (Herberg et al., 1998). The preceding turn of this helix
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341322

contains His87 which under some conditions makes direct contact with the essential phosphateon Thr197 in the large lobe (Cox and Taylor, 1995).The correct orientation of this helix is essential, although how it is positioned appears to be
unique for every kinase. The surface of the helix that faces the b-sheet and the active site ismore-or-less conserved throughout the family and in all cases, when the kinase is in an active
Fig. 6. The C-helix interacts with many parts of the molecule and contributes directly to ATP binding at the activesite. The C-helix is shown in white, while PKI (5±24) is in red. Panel (a) depicts the relative location of the C-helixin the catalytic subunit. A close up view (panel (b)) indicates many of the speci®c interactions that are made byresidues in the C-helix. Panel (c) is a perpendicular view of the image depicted in panel (d).
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341 323

conformation, this helix must be positioned such that Glu91 is oriented towards Lys72 in bstrand 3. The opposite surface of the helix is variable and is positioned di�erently in eachkinase. As indicated above, when the C subunit is in its active conformation, this surface iscorrectly positioned by both the carboxyl-terminal tail and by the amino terminal A-helix Ðtwo regions that lie outside the conserved core. In general, this positioning of the C-helix byresidues that lie outside the core appears to be true for most protein kinases. In some cases, itis the segment that lies either directly amino-terminal or carboxy-terminal to the core that isimportant while in other cases a separate regulatory subunit is required. In cdk2, for example,it is cyclin binding that correctly orients this helix (De Bondt et al., 1993; Je�rey et al., 1995).In MAP kinase or ERK2 the carboxy terminal tail, speci®cally Phe327/Phe329, ®lls this surface(Taylor and Radzio-Andzelm, 1994; Zhang et al., 1994) and the precise orientation of this partof the tail is dependent on phosphorylation of Thr183 and Tyr185 in the activation loop(Canagarajah et al., 1997; Khokhlatchev et al., 1998). In the insulin receptor kinase it is theamino-terminal segment preceding the core that complements the outer surface of the C-helix(Hubbard et al., 1994; Hubbard, 1997). In src it is the proline-rich segment that links the SH2domain to the core (Xu et al., 1997). In all of these cases, where structures of active andinactive conformations are available, the orientation of the C-helix is altered as a consequenceof activation.
The loop between the C-helix and b strand 4 spans the surface of the large lobe and isfunctionally part of the large lobe. b strands 4 and 5 complete the sheet and then fuse with thelinker strand (residues 120±127) that joins the two lobes. The adenine ring butts up against thislinker strand making two hydrogen bonds. The N6 nitrogen hydrogen bonds to the backbonecarbonyl of Glu121 while the N1 nitrogen in the adenine ring hydrogen bonds to the backboneamide of Val123. The base ®ts snugly into this pocket. There are no water molecules, and thethermostability of the enzyme is enhanced signi®cantly when this pocket is ®lled (Herberg etal., 1998). Glu127 at the end of the linker is not invariant, but plays two roles in cAPK. Itbinds to the 2 ' and 3 ' hydroxyls of the ATP ribose and also binds to the P-3 Arg of thepeptide. It also contributes to the binding of a highly ordered water molecule that links theactive site directly to Tyr330 in the nonconserved carboxy terminal tail (Narayana et al.,1997b; Shaltiel et al., 1998).
The large lobe is mostly helical with the exception of a small b-sheet comprised of 4 strandsthat line the active site cleft (Fig. 7(b)). The two remaining loops at the active site arecontributed by this segment. b strands 6 and 7 are joined by a conserved loop referred to asthe catalytic loop because it contains several conserved residues that contribute directly tocatalysis (Fig. 7(a)). Asp166 hydrogen bonds to the P site hydroxyl moiety of the substratepeptide (Madhusudan et al., 1994). It is positioned to serve as a catalytic base, although theactual rate enhancement provided by this residue appears to be small (Zhou and Adams,1997). Lys168 is conserved in Ser/Thr protein kinases and interacts directly with one of the g-phosphate oxygens of ATP. Asn171, also conserved, bridges the loop and binds to the
Fig. 7. The b-sheet in the large lobe provides most of the residues that contribute to phosphoryl transfer. Panel (a)on the left highlights residues in the catalytic loop while panel (b) highlights residues in the Mg++ positioning loop.The b-sheet at the active site cleft is summarized in the panel (c).
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341324
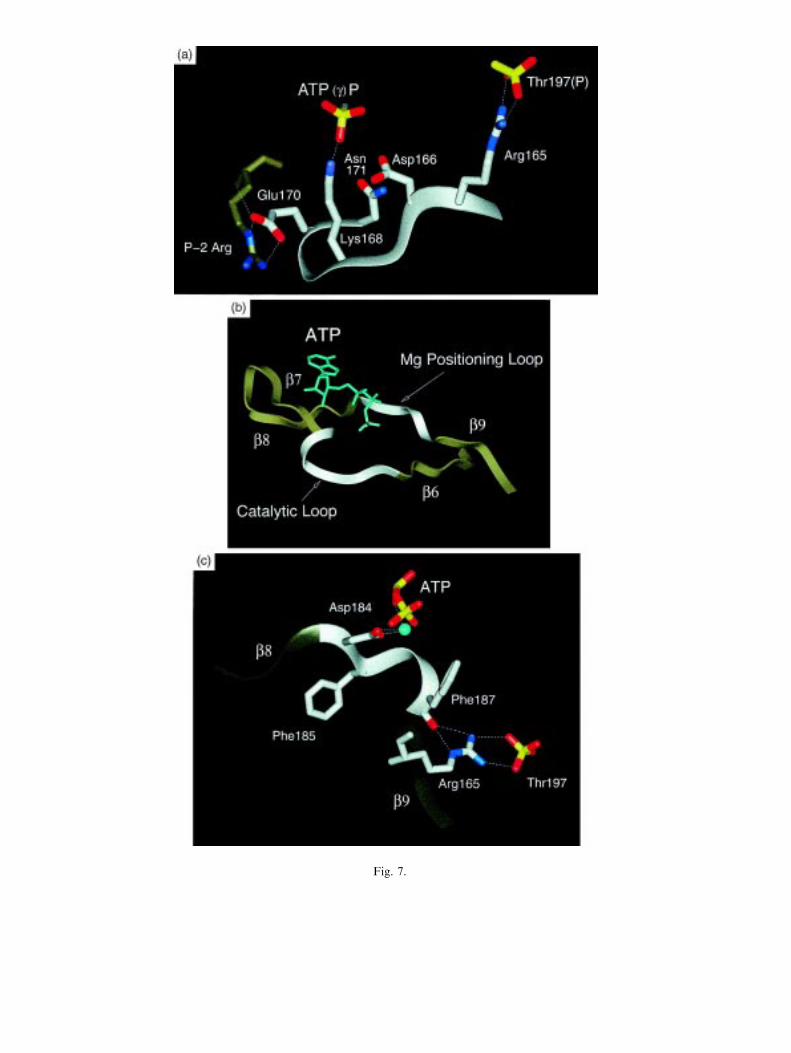
Fig. 7.
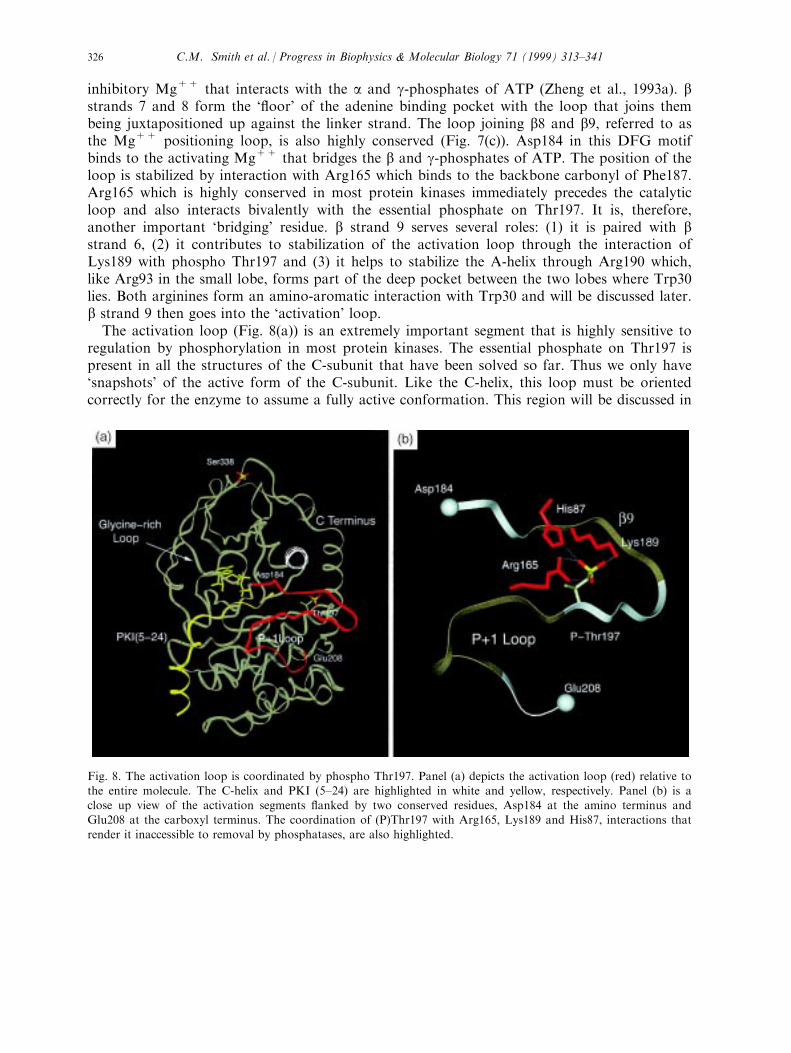
inhibitory Mg++ that interacts with the a and g-phosphates of ATP (Zheng et al., 1993a). bstrands 7 and 8 form the `¯oor' of the adenine binding pocket with the loop that joins thembeing juxtapositioned up against the linker strand. The loop joining b8 and b9, referred to asthe Mg++ positioning loop, is also highly conserved (Fig. 7(c)). Asp184 in this DFG motifbinds to the activating Mg++ that bridges the b and g-phosphates of ATP. The position of theloop is stabilized by interaction with Arg165 which binds to the backbone carbonyl of Phe187.Arg165 which is highly conserved in most protein kinases immediately precedes the catalyticloop and also interacts bivalently with the essential phosphate on Thr197. It is, therefore,another important `bridging' residue. b strand 9 serves several roles: (1) it is paired with bstrand 6, (2) it contributes to stabilization of the activation loop through the interaction ofLys189 with phospho Thr197 and (3) it helps to stabilize the A-helix through Arg190 which,like Arg93 in the small lobe, forms part of the deep pocket between the two lobes where Trp30lies. Both arginines form an amino-aromatic interaction with Trp30 and will be discussed later.b strand 9 then goes into the `activation' loop.The activation loop (Fig. 8(a)) is an extremely important segment that is highly sensitive to
regulation by phosphorylation in most protein kinases. The essential phosphate on Thr197 ispresent in all the structures of the C-subunit that have been solved so far. Thus we only have`snapshots' of the active form of the C-subunit. Like the C-helix, this loop must be orientedcorrectly for the enzyme to assume a fully active conformation. This region will be discussed in
Fig. 8. The activation loop is coordinated by phospho Thr197. Panel (a) depicts the activation loop (red) relative tothe entire molecule. The C-helix and PKI (5±24) are highlighted in white and yellow, respectively. Panel (b) is aclose up view of the activation segments ¯anked by two conserved residues, Asp184 at the amino terminus andGlu208 at the carboxyl terminus. The coordination of (P)Thr197 with Arg165, Lys189 and His87, interactions that
render it inaccessible to removal by phosphatases, are also highlighted.
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341326
more detail later in Section 3.4. The activation loop is followed by the P+1 loop which in
cAPK provides a hydrophobic docking surface for the P+1 residue in the consensus sitepeptide. This loop, discussed in more detail in Section 6, also contributes to recognition of theP, P-2, P-3 and the P-6 sites and can thus be thought of as the peptide positioning loop.
The remainder of the large lobe consisting of the F, G, H and I helices together with the Dand E helices, form the solid core of the large domain. Residue 300 begins the nonconservedcarboxyl terminal tail. This core has been studied less extensively than the active site regions;
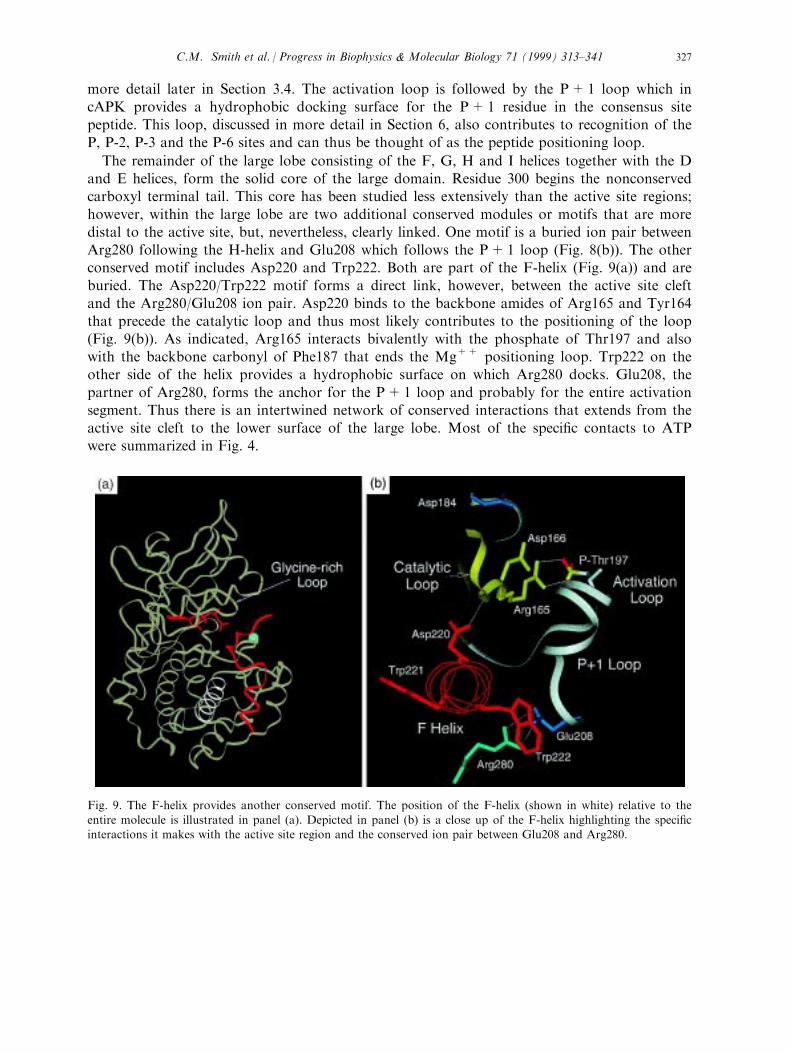
however, within the large lobe are two additional conserved modules or motifs that are moredistal to the active site, but, nevertheless, clearly linked. One motif is a buried ion pair betweenArg280 following the H-helix and Glu208 which follows the P+1 loop (Fig. 8(b)). The otherconserved motif includes Asp220 and Trp222. Both are part of the F-helix (Fig. 9(a)) and areburied. The Asp220/Trp222 motif forms a direct link, however, between the active site cleft
and the Arg280/Glu208 ion pair. Asp220 binds to the backbone amides of Arg165 and Tyr164that precede the catalytic loop and thus most likely contributes to the positioning of the loop(Fig. 9(b)). As indicated, Arg165 interacts bivalently with the phosphate of Thr197 and alsowith the backbone carbonyl of Phe187 that ends the Mg++ positioning loop. Trp222 on theother side of the helix provides a hydrophobic surface on which Arg280 docks. Glu208, the
partner of Arg280, forms the anchor for the P+1 loop and probably for the entire activationsegment. Thus there is an intertwined network of conserved interactions that extends from theactive site cleft to the lower surface of the large lobe. Most of the speci®c contacts to ATPwere summarized in Fig. 4.
Fig. 9. The F-helix provides another conserved motif. The position of the F-helix (shown in white) relative to the
entire molecule is illustrated in panel (a). Depicted in panel (b) is a close up of the F-helix highlighting the speci®cinteractions it makes with the active site region and the conserved ion pair between Glu208 and Arg280.
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341 327
3.3. Heads and tails
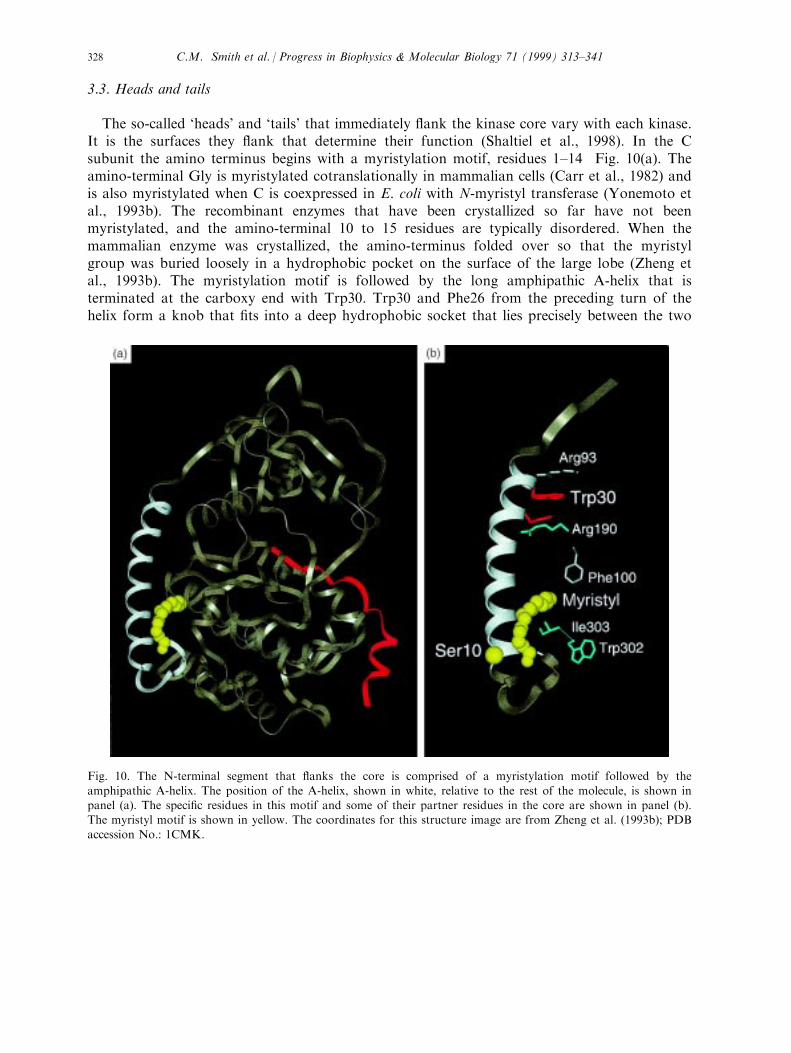
The so-called `heads' and `tails' that immediately ¯ank the kinase core vary with each kinase.It is the surfaces they ¯ank that determine their function (Shaltiel et al., 1998). In the Csubunit the amino terminus begins with a myristylation motif, residues 1±14 Fig. 10(a). Theamino-terminal Gly is myristylated cotranslationally in mammalian cells (Carr et al., 1982) andis also myristylated when C is coexpressed in E. coli with N-myristyl transferase (Yonemoto etal., 1993b). The recombinant enzymes that have been crystallized so far have not beenmyristylated, and the amino-terminal 10 to 15 residues are typically disordered. When themammalian enzyme was crystallized, the amino-terminus folded over so that the myristylgroup was buried loosely in a hydrophobic pocket on the surface of the large lobe (Zheng etal., 1993b). The myristylation motif is followed by the long amphipathic A-helix that isterminated at the carboxy end with Trp30. Trp30 and Phe26 from the preceding turn of thehelix form a knob that ®ts into a deep hydrophobic socket that lies precisely between the two
Fig. 10. The N-terminal segment that ¯anks the core is comprised of a myristylation motif followed by the
amphipathic A-helix. The position of the A-helix, shown in white, relative to the rest of the molecule, is shown inpanel (a). The speci®c residues in this motif and some of their partner residues in the core are shown in panel (b).The myristyl motif is shown in yellow. The coordinates for this structure image are from Zheng et al. (1993b); PDB
accession No.: 1CMK.
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341328
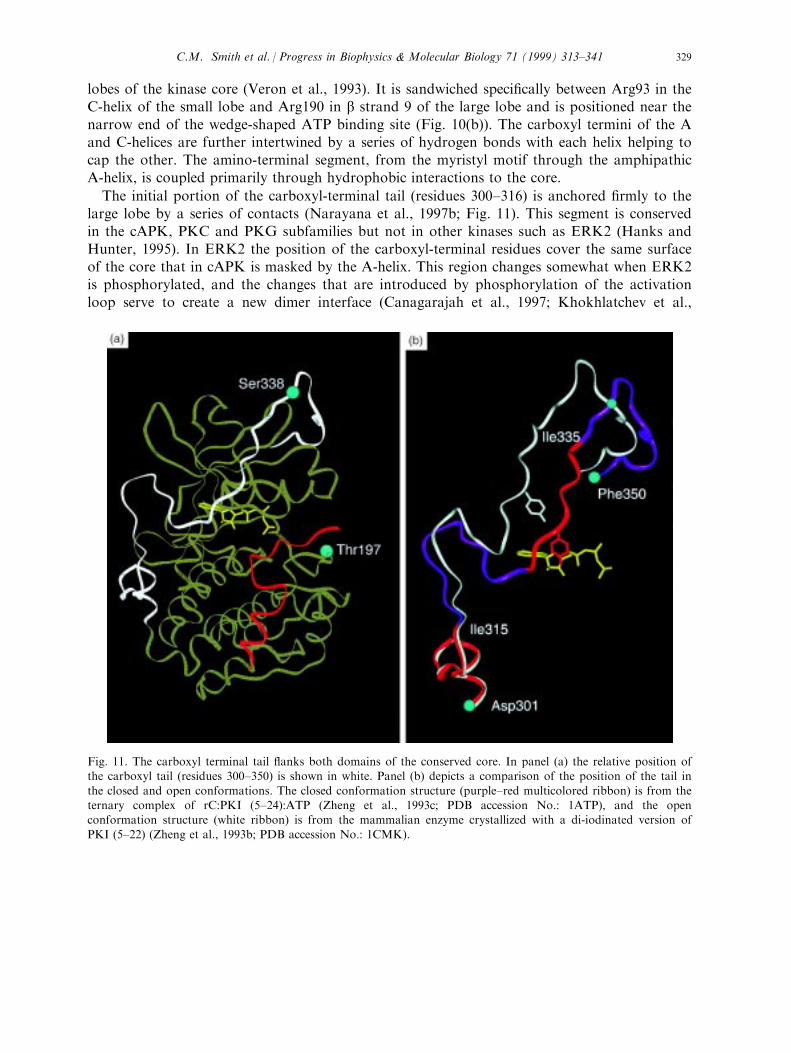
lobes of the kinase core (Veron et al., 1993). It is sandwiched speci®cally between Arg93 in theC-helix of the small lobe and Arg190 in b strand 9 of the large lobe and is positioned near thenarrow end of the wedge-shaped ATP binding site (Fig. 10(b)). The carboxyl termini of the Aand C-helices are further intertwined by a series of hydrogen bonds with each helix helping tocap the other. The amino-terminal segment, from the myristyl motif through the amphipathicA-helix, is coupled primarily through hydrophobic interactions to the core.
The initial portion of the carboxyl-terminal tail (residues 300±316) is anchored ®rmly to thelarge lobe by a series of contacts (Narayana et al., 1997b; Fig. 11). This segment is conservedin the cAPK, PKC and PKG subfamilies but not in other kinases such as ERK2 (Hanks andHunter, 1995). In ERK2 the position of the carboxyl-terminal residues cover the same surfaceof the core that in cAPK is masked by the A-helix. This region changes somewhat when ERK2is phosphorylated, and the changes that are introduced by phosphorylation of the activationloop serve to create a new dimer interface (Canagarajah et al., 1997; Khokhlatchev et al.,
Fig. 11. The carboxyl terminal tail ¯anks both domains of the conserved core. In panel (a) the relative position ofthe carboxyl tail (residues 300±350) is shown in white. Panel (b) depicts a comparison of the position of the tail in
the closed and open conformations. The closed conformation structure (purple±red multicolored ribbon) is from theternary complex of rC:PKI (5±24):ATP (Zheng et al., 1993c; PDB accession No.: 1ATP), and the openconformation structure (white ribbon) is from the mammalian enzyme crystallized with a di-iodinated version of
PKI (5±22) (Zheng et al., 1993b; PDB accession No.: 1CMK).
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341 329

1998). In particular, Phe329 which interfaced with the core in a manner analogous to Trp30 incAPK ¯ips out and now contributes directly to the dimer interface. In src the short carboxylterminal tail contains an inhibitory Tyr that is constitutively phosphorylated in the inactiveenzyme. In the inactive form of src this Tyr docks to its own SH2 domain thereby helping totether the enzyme in an inactive conformation (Xu et al., 1997). The next segment of thecarboxy-terminal tail (residues 315±335) serves as the `gate' that allows for entry and exit ofthe nucleotide from the active site cleft (Chestukhin et al., 1996a; Narayana et al., 1997b;Shaltiel et al., 1998). This segment shows considerable ¯exibility depending on what isoccupying the active site cleft. The highly acidic segment surrounding an important tyrosine,Tyr330, also is thought to serve as a docking mechanism for basic substrates. In addition, thisregion is the cleavage site for a highly speci®c kinase splitting membrane protease recentlyidenti®ed as meparin (Chestukhin et al., 1997, 1996b). The last segment of the tail is ®rmlyanchored to the small lobe with Phe350 deeply buried in the small lobe. As discussedpreviously, the buried a-carboxylate is anchored to Lys92 in the C-helix.
3.4. Covalent modi®cations
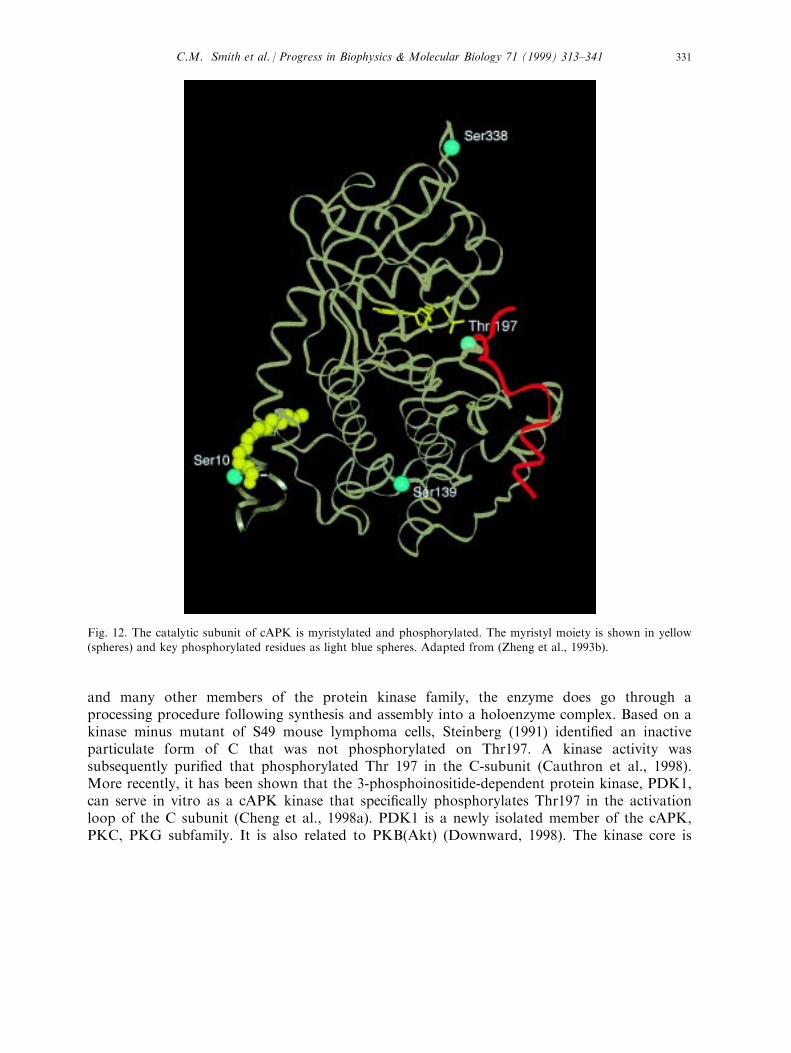
The catalytic subunit is subject to several types of covalent modi®cation (Fig. 12). Inaddition to the myristyl moiety which is added cotranslationally to the N-terminal Gly (Carr etal., 1982), the enzyme is phosphorylated. In E. coli the C-subunit is autophosphorylated atfour sites, Ser10, Ser139, Thr197 and Ser338 (Yonemoto et al., 1997). In the mammalianenzyme Thr197 and Ser338 are very stable phosphorylation sites and are very resistant toremoval by phosphatases (Shoji et al., 1979, 1983). Ser139 has not been observed as aphosphorylated residue in the mammalian enzyme while Ser 10 can be autophosphorylatedslowly in vitro (Toner-Webb et al., 1992). Whether Ser10 is phosphorylated physiologically isunknown. Based on mutational analysis, the phosphate on Ser338 is thought to contribute tostability but does not in¯uence the kinetic properties of the enzyme (Yonemoto et al., 1997).Thr197, in contrast, is essential for maximum activity and contributes to the correctcon®guration of residues at the active site cleft. When Thr197 was replaced with Asp, theenzyme was still reasonably active. When replaced with Ala, however, the Km for ATPincreased 140-fold from 10 mM to 1.4 mM and the phosphoryl transfer step was reduced bymore than two orders (Adams et al., 1995).In many other protein kinases, the conformation of the activation loop is very dynamic.
While we have yet to see the conformation in the inactive, dephosphorylated form of cAPK, inthe active form of the enzyme this loop is very stable. As was seen in Fig. 8, it provides part ofthe surface on which PKI (5±24) docks, and it does not appear to undergo any majorconformational changes as a consequence of substrate or inhibitor binding. A structure of anadenosine binary complex represented the ®rst structure of the C subunit that had no peptidebound, and there were no changes in this region (Narayana et al., 1997b). It does indeedappear to be a stable surface on which the substrate protein or inhibitor docks.The catalytic subunit is typically assembled as a fully active enzyme phosphorylated on
Ser338 and Thr197. The enzyme is then kept in an inactive state by its association withregulatory subunits. The active enzyme is then unleashed in response to cAMP. Although thephosphate on Thr197 is not thought to turnover rapidly in cAPK, unlike src and MAP kinase
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341330
and many other members of the protein kinase family, the enzyme does go through aprocessing procedure following synthesis and assembly into a holoenzyme complex. Based on akinase minus mutant of S49 mouse lymphoma cells, Steinberg (1991) identi®ed an inactiveparticulate form of C that was not phosphorylated on Thr197. A kinase activity wassubsequently puri®ed that phosphorylated Thr 197 in the C-subunit (Cauthron et al., 1998).More recently, it has been shown that the 3-phosphoinositide-dependent protein kinase, PDK1,can serve in vitro as a cAPK kinase that speci®cally phosphorylates Thr197 in the activationloop of the C subunit (Cheng et al., 1998a). PDK1 is a newly isolated member of the cAPK,PKC, PKG subfamily. It is also related to PKB(Akt) (Downward, 1998). The kinase core is
Fig. 12. The catalytic subunit of cAPK is myristylated and phosphorylated. The myristyl moiety is shown in yellow(spheres) and key phosphorylated residues as light blue spheres. Adapted from (Zheng et al., 1993b).
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341 331

closely related to PKC, PKG and cAPK; however, PDK1 as well as PKB (Akt) also contains a
plextrin homology domain that is thought to bind to phosphoinositide lipids. PDK1 was
shown to phosphorylate the activation loop of Akt (Alessi et al., 1997a, 1997b) and of p60 S6
kinase (Pullen et al., 1998). Based on sequence similarities in the activation loops of these two
enzymes and the activation loop of cAPK, its ability to phosphorylate the activation loop of
the C-subunit was tested. A dephosphorylated form of the C-subunit was isolated by
expressing the enzyme in E. coli in the presence of H89, a speci®c inhibitor of cAPK. This C-
subunit was readily phosphorylated and activated by PDK1 (Cheng et al., 1998a). PKC z(Chou et al., 1998; Le Good et al., 1998), like MAP kinase appears to be activated by the
dimeric turnover of the activation loop phosphate, whereas PKCa and PKCb more closely
resemble cAPK in that they are assembled as an active phosphorylated protein and then
activated in response to second messengers including both Ca++ and lipids (Dutil et al., 1998).
A new aspect of the processing of the C-subunit has thus been opened up by these ®ndings.
This will be particularly intriguing in light of recent suggestions that the catalytic subunit may
function in other ways that are completely independent of R subunits and cAMP (Zhong et al.,
1997).
The myristyl group is loosely anchored to the core and may play a role in phosphorylating
membrane proteins; however, as yet there is no convincing evidence to support this. Certainly
in other proteins such as MARCKS, src and recoverin, the myristyl group provides a loose
membrane anchor that is joined by patches of basic residues to create a tight membrane
anchor (Murray et al., 1997). In the case of MARCKS and recoverin, the membrane anchored
conformation is regulated by a switch mechanism. Membrane anchoring of MARCKS is
released by PKC phosphorylation while Ca++ regulates the accessibility of the myristyl group
in recoverin (McLaughlin and Aderem, 1995; Ames et al., 1997). Although the potential for
such a switch mechanism exists for the C-subunit, it has not yet been demonstrated.
A novel type of covalent modi®cation was also identi®ed recently at the amino-terminus of
the C subunit, deamidation of Asn2 (Jedrzejewski et al., 1998). This modi®cation appears to
account for the two major isoforms of the C subunit, A and B, that were identi®ed in
mammalian tissues (Van Patten et al., 1986). When Asn2 is deamidated, the amino-terminus of
the protein (residues 1±14) are disordered in the crystal structure as they are in the crystal
structures of the recombinant C-subunit that is not myristylated and is phosphorylated on
Ser10. The crystal structure of the mammalian C-subunit that is not deamidated shows the
amino-terminal mytristyl moiety folded into a hydrophobic pocket on the surface of the C-
subunit. Although the chain can be traced, the temperature factors for residues 1±10 remain
high suggesting that the acyl group may be loosely anchored. The amino-terminus thus appears
to be sensitive to a number of modi®cations that may contribute in ways that are still
unrecognizable to physiological function.
Fig. 13. Stereo views of the open and closed conformations of the catalytic subunit. The closed conformation (light
blue trace) is from the ternary complex C:PKI(5±24):ATP (PDB accession No.: 1ATP) (Zheng et al., 1993c), andthe open conformation (pink trace) is the mammalian binary complex (Zheng et al., 1993b; PDB accession No.:1CMK). The image in panel (b) is a perpendicular view of the image depicted in panel (a).
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341332
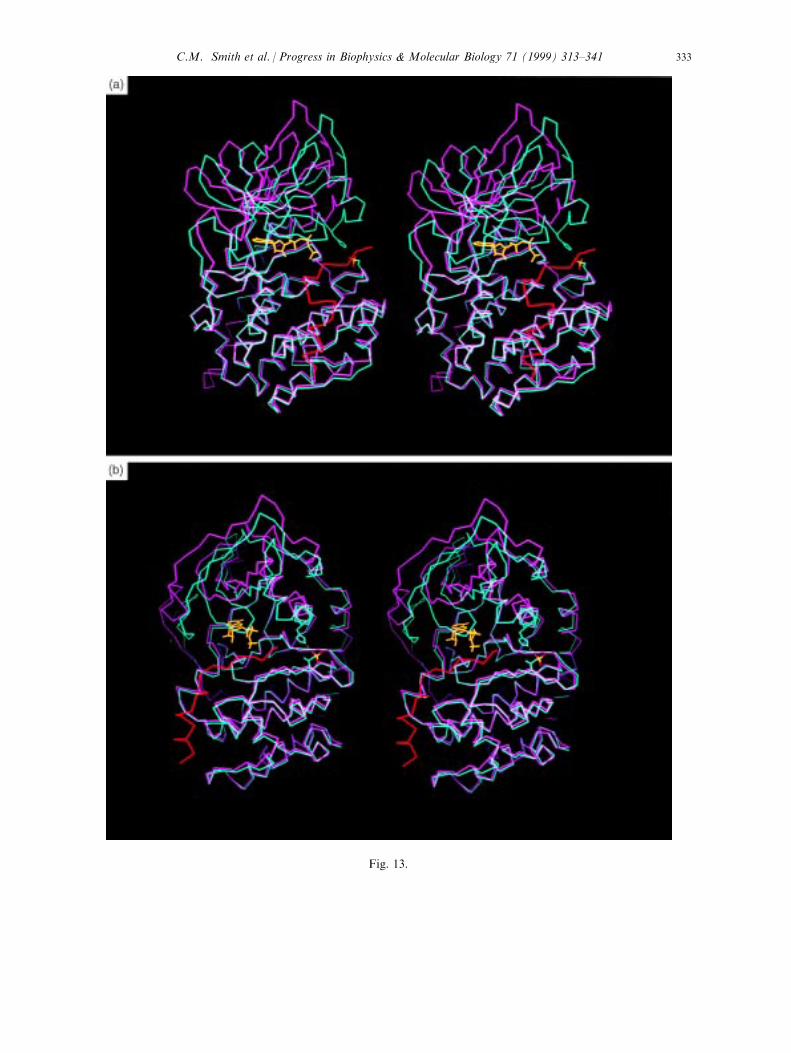
Fig ?? (continued)
Fig. 13.
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341 333

4. Conformational ¯exibility
4.1. Crystallographic evidence
The conformational ¯exibility of the active C subunit is revealed, in part, by the variouscrystal structures that have been solved. The two ternary complexes, C:PKI (5±24):MgATPand the C:PKI:MgAMPPNP, both show a tightly closed conformation (Bossemeyer et al.,1993; Zheng et al., 1993a; Fig. 13). In this conformation the small lobe is oriented so thatHis87 at the beginning of the C-helix forms an ion pair with the phosphate on Thr197 in theactivation loop. The glycine-rich loop is folded over the phosphates of ATP so that thebackbone amide of Ser53 is close enough to hydrogen bond to the g-phosphate as was shownin Fig. 5. In contrast to all of the other crystal structures of the C-subunit, the temperaturefactors for the tip of the loop are very low for this ternary complex. This structure very likelyresembles the transition state in catalysis. Although the side chain of Ser53 also interacts withthe backbone carbonyl of the P site residue in the inhibitor peptide (Bossemeyer, 1994),replacement of Ser53 with Ala does not hinder catalysis (Aimes and Taylor, 1998). This sidechain interaction is thus not essential. Replacement of Gly52 with Ala does, however, lead toimpaired catalysis, most likely because it sterically interferes with formation of the hydrogenbond between the Ser53 amide and the g-phosphate of ATP thus reinforcing the importance ofthat hydrogen bond for catalysis.While the fully closed conformation is essential for e�cient phosphoryl transfer, the
nucleotide cannot be released when the enzyme is in this state. Several di�erent more openconformations have been observed. The structure of the mammalian enzyme that is bound to adi-iodinated form of PKI (5±22) revealed a much more open conformation (Karlsson et al.,1993; Zheng et al., 1993b; Fig. 13), whereas other complexes such as the rC:adenosine binarycomplex reveal an intermediate conformation (Narayana et al., 1997b). A comparison of thesestructures has allowed us to de®ne the features that are associated with opening of the cleft aswell as the rigid body motions that comprise the enzyme (Shaltiel et al., 1998). The generaldegree of openness can be approximated by three measurements: (1) the distance betweenHis87 and Thr197, (2) the distance between Ser53 and Asp166 and (3) the distance betweenTyr330 and Glu127. In the most open conformation the C-helix has pivoted with thepositioning of the carboxyl terminal portion unchanged but with the amino terminal portionnow lifted away from the large lobe. His87 which interacts with the phosphate of Thr197 inthe closed conformation is now more than 6 AÊ away (Zheng et al., 1993b). This is the primaryelectrostatic or hydrogen bond contact between the two lobes. The Ser53 distance re¯ects thetip of the glycine loop. In the open conformation this distance is now >7.1 AÊ . This opening ofthe cleft and lifting of the glycine-rich lid is contributed by two kinds of motions Ð a lifting ofthe tip of the loop and a shearing of the small domain (Narayana et al., 1997b). The thirddistance re¯ects the movement of the carboxyl terminal tail which serves as a `gate' that allowsfor entry and exit of the nucleotide. This segment mostly stays linked to the surface of thesmall lobe. In the closed conformation Tyr330 contacts a primary structured water moleculeand the P-3 Arg and thus contributes directly to the con®guration of residues at the active sitecleft. Replacement of Tyr330 leads to a decrease in catalytic e�ciency (Chestukhin et al.,1996b). This segment is very stable with relatively low temperature factors when the nucleotide
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341334

gate is `closed'. In the open conformation this distance between Tyr330 and Asp127 is greaterthan 7 AÊ , and the temperature factors of the tail are very high, indicating a signi®cant degreeof disorder. The `malleability' of this tail, predicted ®rst on the basis of di�erential reactivity ofthe endogenous cysteines, Cys199 and Cys343 (Jimenez et al., 1982), was subsequentlycon®rmed by the susceptibility of the free subunit to cleavage at this site by the kinase speci®cmembrane protease, KSMP (Chestukhin et al., 1996b). That malleability has been con®rmeddramatically by the crystal structures. The intermediate structures reveal di�erences primarilyin the opening and ¯exibility of the glycine-rich loop.In de®ning the kinase core and the conserved active site residues, it is clear that a number of
highly conserved loops converge at the active site cleft. These include the glycine-rich loop inthe small lobe and the catalytic and magnesium positioning loops in the large lobe. Incomparing these loops in the various binary and ternary complexes observed so far, it isstriking that only the glycine-rich loop is subject to major conformational changes.
4.2. Solution evidence
While the crystal structures reveal the detailed molecular structures of the enzyme, thestructures represent an ensemble of rigid `snapshots'. Other methods are required to probe thedynamics of the enzyme in solution. Recent footprinting using Fe-EDTA free radicals as acleavage reagent revealed distinct conformations for the free enzyme and the C:PKI (5±24):ATP ternary complex (Cheng et al., 1998b). The pattern of footprinting when correlatedwith the crystal structures is consistent with the open and closed conformations andfurthermore demonstrates that the free enzyme adopts a much `looser' conformation.Surprisingly, when the e�ects of PKI(5±24) and MgATP were characterized separately, it wasfound the peptide had little e�ect on global conformation. In contrast, the footprintingobserved in the presence of MgATP was indistinguishable from the ternary complex indicatingthat the nucleotide is primarily responsible for inducing the changes that lead to eventualcatalysis (Cheng et al., 1998b).Fluorescence anisotropy provides an alternative strategy to characterize local conformational
changes. The catalytic subunit contains only two endogenous cysteines, Cys199 and Cys343.Since both can be protected against covalent modi®cation by MgATP, it is possible tointroduce single cysteines into various sites on the C subunit and then label them with a¯uorescent probe such as ¯uoresceine. The conformational ¯exibility of ®ve such C-subunits(Cys16, Cys81, Cys244, Cys327 and Cys343) have now been characterized. The ¯uorescenceanisotropy measurements reveal a range of backbone ¯exibility (Gangal et al., 1998), and thein¯uence of binding of the regulatory subunits is now being evaluated.
5. Catalysis
The early studies of Cook and Walsh demonstrated that the reaction assumes a preferredordered mechanism with ATP most likely binding ®rst (Cook et al., 1982; Whitehouse andWalsh, 1983; Yoon and Cook, 1987). The kinetic properties of the enzyme are highly sensitiveto Mg++ (Cook et al., 1982; Adams and Taylor, 1993). In trying to de®ne the individual steps
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341 335

associated with catalysis and to evaluate mutants, it was essential to ®rst characterize the rateof phosphoryl transfer more carefully. The Km's for a heptapeptide substrate and ATP areapproximately 10±20 mM; the kcat is typically about 20 sÿ1. When the e�ect of viscogens on thekcat was measured, however, it was found that the kcat was highly dependent on viscosity(Adams and Taylor, 1992). This suggested that the kcat did not re¯ect the true chemicaltransfer step but instead re¯ected the rate of product release. To con®rm this, Grant andAdams used a rapid quench assay to measure the presteady state rate constants for theformation of the phosphopeptide. The rapid burst (500 sÿ1) was predicted to correlate with thephospohoryl transfer step while the slower rate (20 sÿ1) corresponded to the kcat (Grant andAdams, 1996). Based on the analysis of a ¯uorescently labeled form of the subunit, a rapidburst was also observed that correlated well with the 500 sÿ1 seen in the rapid quench assay(Lew et al., 1997). The slower rate was thought to be comprised of the ADP o� rate plusconformational changes that are associated with the release of ADP. The reaction pathway forthe C-subunit can thus be described by the following reaction pathway:
E� ATPÿÿ*)ÿÿk2
kÿ2E� ATP� S ÿÿÿ4k3
500 sÿ1E� ADP� Pÿÿ*)ÿÿ
k4
20 sÿ1E:
This reaction pathway with a very rapid phosphoryl transfer step and a relatively slow productrelease step also demonstrates that the Km for peptide (approximately 20 mM for Kemptide:Leu±Arg±Arg±Ala±Ser±Leu±Gly) does not re¯ect the true (Adams and Taylor, 1993). The Kd
for peptide binding is actually much greater, approximately 200 mM for Kemptide.In considering the reaction pathway, it is also important to recognize that the
conformational changes described in the preceding section are all associated with the activeform of the enzyme. Traversing that reaction pathway requires conformational changes thatallow for opening and closing of the cleft between the two lobes. Furthermore, it is theopening of the cleft allowing for release of the nucleotide that is rate-limiting. The proteinkinases thus appear to be remarkably dynamic proteins. Tethering their intrinsic mobility inany way will inevitably limit their catalytic e�ciency.
6. Extended network of interactions
The C-subunit is remarkable for the extended network of interactions that link distal partsof the molecule to the active site where the primary mission of the enzyme is to transfer the g-phosphate of ATP to a protein substrate. In discussing the activation loop and the carboxyterminal tail, several speci®c residues such as Thr197 and Tyr330 have already been discussedthat demonstrate the extent of this network. Mutation of these residues that lie quite a distancefrom the phosphoryl transfer site signi®cantly reduced catalytic e�ciency not only byincreasing the Km for ATP but also by reducing k3. The peptide binding site is also remarkablenot only for the way in which di�erent parts of the molecule, relatively far apart in linearsequence come together to recognize speci®c residues, but also for the network that linkspeptide recognition to the site of phosphoryl transfer. Glu230, one of the residues associated
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341336
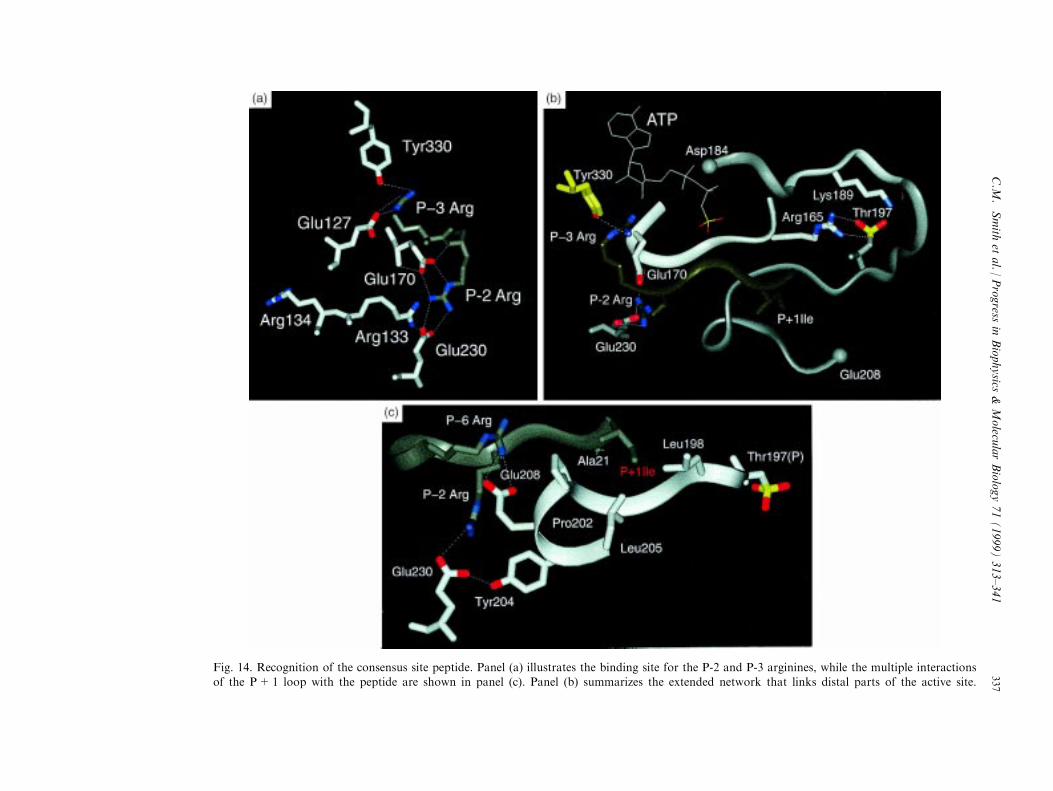
Fig. 14. Recognition of the consensus site peptide. Panel (a) illustrates the binding site for the P-2 and P-3 arginines, while the multiple interactionsof the P+1 loop with the peptide are shown in panel (c). Panel (b) summarizes the extended network that links distal parts of the active site.
C.M
.Smith
etal./
Progress
inBiophysics
&Molecu
larBiology71(1999)313±341
337

with recognition of the P-2 Arg in the peptide substrate, is another excellent example todemonstrate this networking (Grant et al., 1996).The minimum peptide recognition site can be de®ned as the region at the active site cleft
where the consensus site peptide docks. This region is su�cient to convey low a�nity bindingtypical of most substrates. As was seen in Fig. 1, the arginines at the P-3 and the P-2 positionsare key determinants for peptide recognition of the consensus peptide by the catalytic subunit.Each site for recognition of these arginines is comprised of several acidic residues assummarized in Fig. 14(a). The P-2 Arg interacts with two carboxylates, Glu230 and Glu170.Replacement of Glu230 with Gln had two consequences (Grant et al., 1996). As anticipated theKm was increased from 6.9 mM to approximately 1.4 mM. However, the k3 also was reducedfrom 500 to 20 sÿ1. While the change in the electrostatic properties are signi®cant (Tsigelny etal., 1995), an examination of this residue and its links to other parts of the molecule isrevealing. As seen in Fig. 14, the other residue that forms the P-2 recognition site is Glu170which is located in the middle of the catalytic loop. The adjacent P-3 Arg interacts directlywith Glu127 in the linker segment that joins the two lobes. Glu127 also interacts with the 2 'and 3 ' -hydroxyl of the ATP ribose. The P-3 Arg also interacts with Tyr330 in the carboxyterminal tail which coordinates a highly ordered water molecule that was discussed earlier. Thehydrophobic P+1 residue in the consensus peptide is bound to the P+1 loop that followsphospho Thr197. The speci®c hydrophobic residues that form the binding pocket are Leu198,Phe202, and Leu205. As seen in Fig. 14 (right), in addition to contributing to recognition ofthe P+1 residue, this loop communicates with many other parts of the protein. Gly200hydrogen bonds to the backbone amide of the P site residue while Thr201 interacts directlywith Lys168 and, in some cases, Asp166 in the catalytic loop where it appears to contribute tothe actual phosphoryl transfer step. Glu203 forms the site for recognition of the P-6 Arg, whileTyr204 hydrogen bonds to Glu230, a primary determinant of the P-2 Arg recognition site. Thishighly interactive network that allows the kinase to recognize peptides and inhibitors as well asATP extends in all directions.The interactiveness of these listed residues at the active site is apparent from several
mutational studies. For example, replacing Glu230 with Gln had two major consequences.First it reduced the Km for Kemptide as expected, to 6 mM, however, it also signi®cantlyreduced the rate of phosphoryl transfer to 20 sÿ1. Thus there appears to be quite an extendednetwork that contributes to catalytic e�ciency.
7. Summary
Our understanding of the protein kinase family has increased considerably since the ®rststructure was solved in 1991 (Knighton et al., 1991a, 1991b). Not only have many new proteinkinases been discovered making this one of the largest enzyme families, but structures are nowavailable for quite a few di�erent protein kinases. Several things are striking. First is theremarkable rea�rmation of the original prediction that the core would be conserved. Althoughthere are some di�erences between the protein kinases that phosphorylate Ser/Thr and thosethat phosphorylate Tyr, the di�erences are subtle. The conserved residues that we havereviewed here are more-or-less a constellation of ®xed points that can be superimposed readily
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341338

for all members of the family. Second is the incredible diversity in how each core is activatedand regulated. The mechanisms by which the core can be tethered or locked into an inactiveconformation are limitless. Finally is the dynamic nature of these enzymes. We have focusedhere on the dynamics of the active enzyme as it undergoes catalysis. Equally dynamic are themodules that ¯ank the core and the inhibitory molecules that latch onto the core.Understanding these dynamics will be a major challenge Ð one that will require a diverse setof approaches to fully understood at the molecular level.
Acknowledgements
This research was supported by the National Institutes of Health (GM19301). Other supportwas provided by the National Biomedical Computation Resource (NIH P41 RR08605-05) andthe National Partnership for Advanced Computation Infrastructure (NPACI; http://www.npaci.edu). Structure images were prepared using InsightII (Molecular Simulations, Inc.,San Diego, CA) and Molscript v2.1 (Kraulis, 1991; Avatar Software AB [http://www.avatar.se]). The authors would also like to acknowledge the assistance of DavidBaraclough in the preparation of this manuscript.
References
Adams, J.A., McGlone, M.L., Gibson, R., Taylor, S.S., 1995. Biochem. 34, 2447±2454.
Adams, J.A., Taylor, S.S., 1992. Biochem. 31, 8516±8522.
Adams, J.A., Taylor, S.S., 1993. Prot. Sci. 2, 2177±2186.
Alessi, D., James, S., Downes, C., Holmes, A., Ga�ney, P., Reese, C., Cohen, P., 1997a. Curr. Biol. 7, 261±269.
Alessi, D.R., Deak, M., Casamayor, A., Caudwell, F.B., Morrice, N., Norman, D.G., Ga�ney, P., Reese, C.B.,
MacDougall, C.N., Harbison, D., Ashworth, A., Bownes, M., 1997b. Curr. Biol. 7, 776±789.
Aimes, R., Taylor, S., 1998. Personal communication.
Ames, J.B., Ishima, R., Tanaka, T., Gordon, J.I., Stryer, L., Ikura, M., 1997. Nature 389, 198±202.
Bossemeyer, D., 1994. TIBS Rev. 19, 201±205.
Bossemeyer, D., Engh, R.A., Kinzel, V., Ponstingl, H., Huber, R., 1993. EMBO J. 12, 849±859.
Brostrom, M.A., Reimann, E.M., Walsh, D.A., Krebs, E.G., 1970. Adv. Enzyme Regul. 8, 191±203.
Buechler, J.A., Taylor, S.S., 1988. Biochem. 27, 7356±7361.
Canagarajah, B.J., Khokhlatchev, A., Cobb, M.H., Goldsmith, E.J., 1997. Cell 5, 859±869.
Carr, S.A., Biemann, K., Shoji, S., Parmalee, D.C., Titani, K., 1982. Proc. Natl. Acad. Sci. USA 79, 6128±6131.
Cauthron, R.D., Carter, K.B., Liauw, S., Steinberg, R.A., 1998. Mol. Cell. Biol. 18, 1416±1423.
Cheng, H.-C., van Patten, S.M., Smith, A.J., Walsh, D.A., 1986. Biochem. J. 231, 655±661.
Cheng, X., Ma, Y., Moore, M., Hemmings, B.A., Taylor, S.S., 1998a. Proc. Nat. Acad. Sci. USA 95, 9849±9854.
Cheng, X., Shaltiel, S., Taylor, S.S., 1998b. Biochem. 37, 14005±14013.
Chestukhin, A., Litovchick, L., Schourov, D., Cox, S., Taylor, S.S., Shaltiel, S., 1996a. J. Biol. Chem. 271, 10175±10182.
Chestukhin, A., Muradov, K., Litovchick, L., Shaltiel, S., 1996b. J. Biol. Chem. 271, 30272±30280.
Chestukhin, A., Litovchick, L., Muradov, K., Batkin, M., Shaltiel, S., 1997. J. Biol. Chem. 272, 3153±3160.
Chou, M.M., Hou, W., Johnson, J., Graham, L.K., Lee, M.H., Chen, C.-S., 1998. Curr. Biol. 8, 1069±1077.
Cohen, P., Rylatt, D.B., Nimmo, G.A., 1977. FEBS Lett. 76, 182.
Cook, P.F., Neville, M.E., Vrana, K.E., Hartl, F.T., Roskoski, R., 1982. Biochem. 21, 5794±5799.
Cox, S., Taylor, S.S., 1995. Biochem. 34, 16203±16209.
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341 339

De Bondt, H.L., Rosenblatt, J., Jancarik, J., Jones, H.D., Morgan, D.O., Kim, S.H., 1993. Nature 363, 595±602.
Downward, J., 1998. Curr. Opin. Cell. Biol. 10, 262±267.
Dutil, E.M., Toker, A., Newton, A., 1998. Curr. Biol. 8, 1366±1375.
Fischer, E.H., Krebs, E.G., 1955. J. Biol. Chem. 216, 121±132.
Francis, S., Corbin, J., 1994. Ann. Rev. Physiol. 56, 237±272.
Gangal, M., Cox, S., Lew, J., Cli�ord, T., Garrod, S., Aschbaher, M., Taylor, S.S., Johnson, D.A., 1998. Biochem.37, 13728±13735.
Gill, G.N., Garren, L.D., 1969. Proc. Natl. Acad. Sci. USA 63, 512±519.
Goldman, P.S., Tran, V.K., Goodman, R.H., 1997. Recent Prog. Hormone Res. 52, 103±120.
Granot, J., Mildvan, A.S., Hiyama, K., Kondo, H., Kaiser, E.T., 1980. J. Biol. Chem. 255, 4569±4573.
Grant, B.D., Adams, J.A., 1996. Biochem. 35, 1533±1539.
Grant, B.D., Tsigelny, I., Adams, J.A., Taylor, S.S., 1996. Prot. Sci. 5, 1316±1324.
Grant, B.D., Hemmer, W., Tsigelny, I., Adams, J.A., Taylor, S.S., 1998. Biochem. 37, 7708±7715.
Hanks, S.K., Hunter, T., 1995. FASEB J. 9, 576±596.
Hanks, S.K., Quinn, A.M., Hunter, T., 1988. Science 241, 42±52.
Hauer, J.A., Barthe, P., Taylor, S.S., Parello, J., Padilla, A., 1999. Pro. Sci., in press.
Hemmer, W., McGlone, M.L., Taylor, S.S., 1997. J. Biol. Chem. 272, 16946±16954.
Herberg, F.W., Doyle, M., Cox, S., Taylor, S.S., 1998. Biochem., submitted for publication.
Hjelmquist, G., Anderson, J., Edlund, B., Engstrom, L., 1974. Biochem. Biophys. Res. Comm. 61, 559±563.
Ho�man, F., 1980. J. Biol. Chem. 255, 1559±1564.
Ho, M.-f., Bramson, H.N., Hansen, D.E., Knowles, J.R., Kaiser, E.T., 1988. J. Am. Chem. Soc. 110, 2680±2681.
Hubbard, S.R., 1997. EMBO J. 16, 5572±5581.
Hubbard, S.R., Wei, L., Ellis, L., Hendrickson, W.A., 1994. Nature 372, 746±754.
Hunter, T., Plowman, G.D., 1997. Trends Biochem. Sci. 22, 18±22.
Jedrzejewski, P.T., Girod, A., Tholey, A., Keonig, N., Thiller, S., Kinzel, B., Bossemeyer, D., 1998. Prot. Sci. 7,457±469.
Je�rey, P.D., Russo, A., Polyak, K., Gibbs, E., Hurwitz, J., Massague, J., Pavletich, N.P., 1995. Nature 376, 313±
320.
Jimenez, J.S., Kupfer, A., Gani, V., Shaltiel, S., 1982. Biochem. 21, 1623±1630.
Karlsson, R., Zheng, J., Xuong, N.-h., Taylor, S.S., Sowadski, J.M., 1993. Acta Crystal. D49, 381±388.
Kemp, B.E., Bylund, D.B., Huang, T.-S., Krebs, E.G., 1975. Proc. Natl. Acad. Sci. USA 72, 3448.
Kemp, B.F., Graves, D.J., Benjamini, E., Krebs, E.G., 1977. J. Biol. Chem. 252, 4888±4894.
Knighton, D.R., Zheng, J., Ten Eyck, L.F., Ashford, V.A., Xuong, N.-h., Taylor, S.S., Sowadski, J.M., 1991a.Science 253, 407±414.
Knighton, D.R., Zheng, J., Ten Eyck, L.F., Xuong, N.-h., Taylor, S.S., Sowadski, J.M., 1991b. Science 253, 414±420.
Khokhlatchev, A.V., Canagarajah, B., Wilsbacher, J., Robinson, M., Atkinson, M., Goldsmith, E., Cobb, M.H.,1998. Cell 93, 605±615.
Kraulis, P.J., 1991. J. Appl. Crystal. 24, 946±950.
Krebs, E.G., Graves, D.J., Fischer, E.H., 1959. J. Biol. Chem. 234, 2867±2873.
Lew, J., Taylor, S.S., Adams, J.A., 1997. Biochem. 36, 6717±6724.
Luttrell, L.M., van Biesen, T., Hawes, B.E., Koch, W.J., Krueger, K.M., Touhara, K., Lefkowitz, R.J., 1997. Adv.Second Messenger Phosphoprotein Res. 31, 263±277.
Le Good, J.A., Ziegler, W.H., Parekh, D.B., Alessi, D.R., Cohen, P., Parker, P.J., 1998. Science 281, 2042±2045.
Madhusudan, K.R., Trafny, E.A., Xuong, N.-h., Adams, J.A., Ten Eyck, L.F., Taylor, S.S., Sowadski, J.M., 1994.
Prot. Sci. 3, 176±187.
McLaughlin, S., Aderem, A., 1995. Trends Biochem. Sci. 20, 272±276.
Montminy, M., 1997. Annu. Rev. Biochem. 66, 807±822.
Murray, K.J., El-Maghrabi, M.R., Kountz, P.D., Lukas, T.J., Soderling, T.R., Pilkis, S.J., 1984. J. Biol. Chem. 259,7673±7681.
Murray, D., Ben-Tal, N., Honig, B., McLaughlin, S., 1997. Structure 5, 985±989.
Narayana, N., Cox, S., Shaltiel, S., Taylor, S.S., Xuong, N., 1997a. Biochem. 36, 4438±4448.
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341340

Narayana, N., Cox, S., Xuong, N.-h., TenEyck, L.F., Taylor, S.S., 1997b. Structure 5, 921±935.Parker, P.J., Aitken, A., Bilham, T., Embi, N., Cohen, P., 1981. FEBS Lett. 123, 332±335.
Pullen, N., Dennes, P.B., Andjelkovic, M., Dufner, A., Kozma, S.C., Hemmings, B.A., Thomas, G., 1998. Science279, 707±710.
Scott, J.D., Glaccum, M.B., Fischer, E.H., Krebs, E.G., 1986. Proc. Natl. Acad. Sci. USA 83, 1613±1616.
Shaltiel, S., Cox, S., Taylor, S.S., 1998. Proc. Natl. Acad. Sci. USA 95, 484±491.Shoji, S., Titani, K., Demaille, J.G., Fischer, E.H., 1979. J. Biol. Chem. 254, 6211±6214.Shoji, S., Ericsson, L.H., Walsh, D.A., Fischer, E.H., Titani, K., 1983. Biochem. 22, 3702±3709.
Slice, L.W., Taylor, S.S., 1989. J. Biol. Chem. 264, 20940±20946.Steinberg, R.A., 1991. Mol. Cell. Biol. 11, 705±712.Sutherland, C., O'Brien, R.M., Granner, D.K., 1996. Phil. Trans. R. Soc. London. Series B: Biol. Sci. 351, 191±199.
Sutherland, E.W., Wosilait, W.D., 1955. Nature 175, 169±170.Tao, M., Salas, M.L., Lipmann, F., 1970. Proc. Natl. Acad. Sci. USA 67, 408±414.Taylor, S.S., Buechler, J.A., Yonemoto, W., 1990. Annu. Rev. Biochem. 59, 971±1005.Taylor, S.S., Radzio-Andzelm, E., 1994. Structure 2, 345±355.
Toner-Webb, J., van Patten, S.M., Walsh, D.A., Taylor, S.S., 1992. J. Biol. Chem. 267, 25174±25180.Tsigelny, I., Grant, B.D., Taylor, S.S., Ten Eyck, L.F., 1995. Biopolymers 39, 353±365.Ullrich, A., Schlessinger, J., 1990. Cell 61, 203±212.
Van Patten, S.M., Fletcher, W.H., Walsh, D.A., 1986. J. Biol. Chem. 261, 5514±5523.Veron, M., Radzio-Andzelm, E., Tsigelny, I., Ten Eyck, L.F., Taylor, S.S., 1993. Proc. Nat. Acad. Sci. USA 90,
10618±10622.
Walsh, D.A., Perkins, J.P., Krebs, E.G., 1968. J. Biol. Chem. 243, 3763±3765.Walsh, D.A., Angelos, K.L., Van Patten, S.M., Glass, D.B., Garetto, L.P., 1990. In: Kemp, B.E. (Ed.), Peptides
and Protein Phosphorylation CRC Press, Boca Raton, pp. 43±84.
Wen, W., Taylor, S.S., Meinkoth, J.L., 1995. J. Biol. Chem. 270, 2041±2046.Westheimer, F.H., 1987. Science 235, 1173±1178.Whitehouse, S., Walsh, D.A., 1983. J. Biol. Chem. 258, 3682±3692.Xu, W., Harrison, S.C., Eck, M.J., 1997. Nature 385, 595±599.
Yeaman, S.J., Cohen, P., Watson, D.C., Dixon, G.H., 1977. Biochem. J. 162, 411.Yonemoto, W., Garrod, S.M., Bell, S.M., Taylor, S.S., 1993a. J. Biol. Chem. 268, 18626±18632.Yonemoto, W., McGlone, M.L., Taylor, S.S., 1993b. J. Biol. Chem. 268, 2348±2352.
Yonemoto, W., McGlone, M.L., Taylor, S.S., 1997. Prot. Eng. 10, 915±925.Yoon, M.-Y., Cook, P.F., 1987. Biochem. 26, 4118±4125.Zetterqvist, O., Ragnarsson, U., Humble, E., Berglund, L., Engstrom, L.T., 1976. Biochem. Biophys. Res. Comm.
70, 696±703.Zhang, F., Strand, A., Robbins, D., Cobb, M.H., Goldsmith, E.J., 1994. Nature 367, 704±711.Zheng, J., Knighton, D.R., Ten Eyck, L.F., Karlsson, R., Xuong, N.-h., Taylor, S.S., Sowadski, J.M., 1993a.
Biochem. 32, 2154±2161.
Zheng, J., Knighton, D.R., Xuong, N.-h., Taylor, S.S., Sowadski, J.M., Ten Eyck, L.F., 1993b. Prot. Sci. 2, 1559±1573.
Zheng, J., Trafny, E.A., Knighton, D.R., Xuong, N.-h., Taylor, S.S., Ten Eyck, L.F., Sowadski, J.M., 1993c. Acta
Cryst. D49, 362±365.Zhong, H., SuYang, H., Erdjument-Bromage, H., Tempst, P., Ghosh, S., 1997. Cell 89, 413±424.Zhou, J., Adams, J., 1997. Biochem. 36, 2977±2984.
Zoller, M.J., Nelson, N.C., Taylor, S.S., 1981. J. Biol. Chem. 256, 10837±10842.
C.M. Smith et al. / Progress in Biophysics & Molecular Biology 71 (1999) 313±341 341