the genomic enzymology of antibiotic resistance
TRANSCRIPT

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 1/27
GE44CH02-Wright ARI 17 July 2010 13:29
REV I E W
S
I
N
A D V A
N
C
E
The Genomic Enzymology of Antibiotic Resistance
Mariya Morar and Gerard D. Wright
M.G. DeGroote Institute for Infectious Disease Research and the Department of Biochemistry and Biomedical Sciences, McMaster University, Hamilton, Ontario,L8N 3Z5, Canada; email: [email protected]
Annu. Rev. Genet. 2010. 44:25–51
The Annual Review of Genetics is online atgenet.annualreviews.org
This article’s doi:10.1146/annurev-genet-102209-163517
Copyright c 2010 by Annual Reviews.All rights reserved
0066-4197/10/1201-0025$20.00
Key Words
antibiotic resistome, proto-resistance gene, antibiotic resistance
mechanism, enzyme structure-function.
Abstract
The need for new antibiotic therapies is acute and growing in large partbecause of the emergence of drug-resistant pathogens. A vast number
of resistance determinants are, however, found in nonpathogenic mi-
croorganisms. The resistance totality in the global microbiota is the
antibiotic resistome and includes not only established resistance genes
but alsogenes that have the potential to evolve into resistance elements.
We term these proto-resistance genes and hypothesize that they share
common ancestry with other functional units known as housekeeping
genes. Genomic enzymology is the study of protein structure–function
in light of genetic context and evolution of protein superfamilies. This
concept is highly applicable to study of antibiotic resistance evolution
from proto-resistance elements. In this review, we summarize some of
the genomic enzymology evidence for resistance enzymes pointing to
common ancestry with genes of other metabolic functions. Genomic
enzymology plays a key role in understanding the origins of antibiotic
resistance and aids in designing strategies for diagnosis and prevention
thereof.
25

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 2/27
GE44CH02-Wright ARI 17 July 2010 13:29
Antibiotic resistome:
the totality of antibiotic resistanceelements in microbialpopulations across theglobe
INTRODUCTION
The Problem of Antibiotic Resistanceand the Concept of the Resistome
Antibiotics are the cornerstone drugs of mod-
ern medicine. Ever since their discovery in
the first half of the twentieth century, clini-
cians have exploited them not only to treat in-
fections, but also to allow numerous medical
procedures by prophylactically preventing in-
fectious disease. Many modern therapies from
cancer chemotherapy to organ transplantation
would simply not be conceivable without an-
tibiotics.They truly are wonderdrugs that have
changed the practice of medicine.
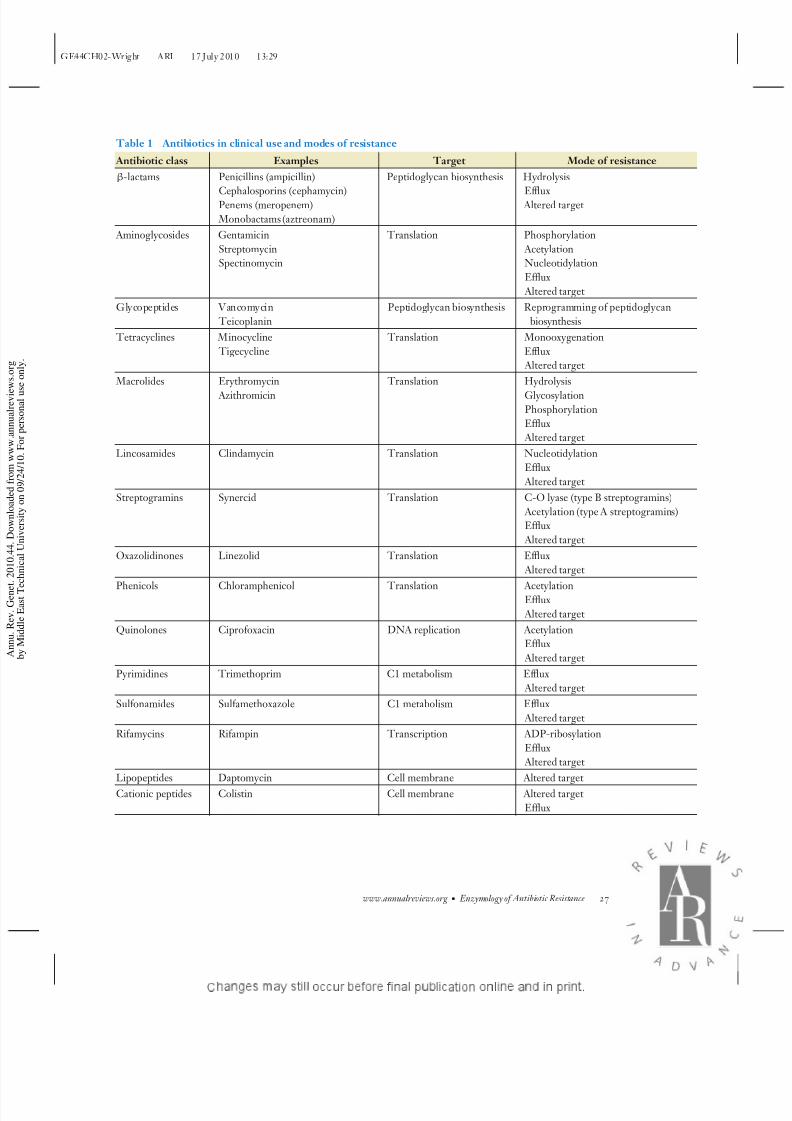
There are several classes of antibiotics avail-
able to clinicians (Table 1). These compoundstarget vital microbial biochemistry at vulner-
able metabolic and physiologic hubs such as
translation, DNA replication, and cell wall
biosynthesis. Antibiotics can arrest cell growth
(bacteriostatic) or kill the cells outright (bac-
tericidal) depending on their mode of action
and the biochemical and genetic idiosyncrasies
of individual species of pathogens. With a few
exceptions, all the members of our current ar-
senal of antibiotic drugs were discovered dur-
ing a brief 20-year period of tremendous pro-
ductivity in drug discovery following the first
clinical use of penicillin in the mid-1940s. Themajority of these antibiotics were discovered
by screening soil bacteria, mostly of the acti-
nomycete family, for the production of bioac-
tive molecules with microbial growth inhibition
activity. It is this small set of chemical scaf-
folds that has formed the basis of success of
antibiotic chemotherapy for over half a cen-
tury (45). At least for the foreseeable future,
these same scaffolds will be required to main-
tain our continued reliance on antibiotics in
medicine.
Soon after the first use of antibiotics in
medicine, resistant organisms were seen to arise
during therapy (1). The subsequent decades
have seen a cycle of emergence of resistant mi-
crobes, followed by the subsequent develop-
ment of new antibiotics. These have included
brand new classes of drugs as well as medicinal
chemical elaboration of established classes to
avoid resistance. This cyclical approach has
proven highly successful, and we are now em-ploying fourth, fifth, and sixth generation an-
tibiotics in many cases. The continued evolu-
tion andselection ofnew resistancegenes, along
with the emergence of multi-drug resistant or-
ganisms, are driving the need for new drugs
(16).
Paradoxically, despite a growing unmetclin-
ical need for new antibiotics, there has been a
general retreat over the past decades in new an-
tibiotic drug discovery by the pharmaceutical
industry. One reason is that it has proven to
be difficult to identify novel antibiotic chemi-
cal scaffolds using the screening methods thatwere so profitable in the past. At the same time,
newer target-based, high-throughput screen-
ing approaches that have been very successful
in other areas of drug discovery have so far
failed to produce in thearea of antibiotics (108).
There is therefore a chemical scaffold gap that
needs to be bridged to spur new directions in
antibiotic discovery. Compounding these chal-
lenges is the problem of resistance that contin-
ues to grow unabated.
Antibiotic resistance has evolved along with
the deployment of these cytotoxic chemicals.
The first resistance genes were no doubt asso-ciated with antibiotic-producing bacteria in the
environment and have evolved over the millen-
nia (6). Presently, the evolution and selection
of resistance is happening on a larger scale and
is being accelerated as we produce and use an-
tibiotics on the ton scale in human health and
agriculture (7, 61).
Not surprisingly, the focus of resistance re-
search has been principally on pathogenic mi-
crobes in healthcare settings. However, this is
an incomplete representation of the pool of re-
sistance elements that impact antibiotics on a
global scale. We recently proposed the con-cept of the antibiotic resistome to describe
the totality of resistance in microbial popu-
lations across the globe (36, 149). The resis-
tome includes not only those elements that
26 Morar ·Wright
8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 3/27
GE44CH02-Wright ARI 17 July 2010 13:29
Table 1 Antibiotics in clinical use and modes of resistance
Antibiotic class Examples Target Mode of resistance
β-lactams Penicillins (ampicillin) Peptidoglycan biosynthesis Hydrolysis
Cephalosporins (cephamycin) EffluxPenems (meropenem) Altered target
Monobactams (aztreonam)
Aminoglycosides Gentamicin Translation Phosphorylation
Streptomycin Acetylation
Spectinomycin Nucleotidylation
Efflux
Altered target
Glycopeptides Vancomycin
Teicoplanin
Peptidoglycan biosynthesis Reprogramming of peptidoglycan
biosynthesis
Tetracyclines Minocycline Translation Monooxygenation
Tigecycline Efflux
Altered target
Macrolides Erythromycin Translation Hydrolysis
Azithromicin GlycosylationPhosphorylation
Efflux
Altered target
Lincosamides Clindamycin Translation Nucleotidylation
Efflux
Altered target
Streptogramins Synercid Translation C-O lyase (type B streptogramins)
Acetylation (type A streptogramins)
Efflux
Altered target
Oxazolidinones Linezolid Translation Efflux
Altered target
Phenicols Chloramphenicol Translation AcetylationEfflux
Altered target
Quinolones Ciprofoxacin DNA replication Acetylation
Efflux
Altered target
Pyrimidines Trimethoprim C1 metabolism Efflux
Altered target
Sulfonamides Sulfamethoxazole C1 metabolism Efflux
Altered target
Rifamycins Rifampin Transcription ADP-ribosylation
Efflux
Altered target
Lipopeptides Daptomycin Cell membrane Altered target
Cationic peptides Colistin Cell membrane Altered target
Efflux
www.annualreviews.org • Enzymology of Antibiotic Resistance 27

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 4/27
GE44CH02-Wright ARI 17 July 2010 13:29
Genomic
enzymology: study of protein structure andfunction taking intoaccount geneticcontext and evolutionwithin a proteinsuperfamily
Proto-resistanceelement: a gene thatmay have any functionbut has potential toevolve into a resistanceelement
are acute problems in the clinic but also takes
into account the vast reservoir of resistance
genes in nonpathogenic organisms (23). This
idea is important to fully understand the evo-lution of resistance, the maintenance of resis-
tance genes within microbial populations, and
the spread of these genes between species and
genera.
Resistance can arise as the result of altered
molecular targets, efflux of antibiotics from
within the cell, blockade of antibiotic entry into
the cell, and chemical modification or destruc-
tion of the compounds. In many cases, a sin-
gle antibiotic or class is impacted by more than
one, or even all, of these mechanisms (e.g., see
Table 1). Furthermore, these mechanisms can
be collected within a single organism resultingin combinatorial resistance. The range of re-
sistance mechanisms is common to pathogenic
and nonpathogenic bacteria and suggests a
complex natural history of evolution and selec-
tion. The evolution of enzymes that modify an-
tibiotic targets and the antibiotics themselves
are of particular interest. Unlike many efflux
systems, which tend to have been selected to
purge a wide variety of toxic organic molecules
from the cell (91), or single site mutations in
targets, which may confer resistance with neu-
tral evolutionary impact, enzymes likely have
evolved in specific response to antibiotics (148).Theseare particularlynoteworthy for theirsub-
strate specificity (target or antibiotic).
The wellspring of the resistome is the col-
lectionof genes that have thepotential to evolve
into resistance elements. We have termed these
proto-resistance genes as they are analogous to
the proto-oncogenes of the cancer field (98).
Proto-resistance genes are found in numerous
bacterial chromosomes and are often annotated
as putative antibiotic resistance elements during
genome sequencing. We hypothesize that it is
these proto-resistance genes, which may have
other functions in the cell and have little or noantibiotic resistance capacity, that are the ulti-
mate source of the highly efficient and robust
resistance elements whose function is to detox-
ify antibiotics (Figure 1a).
Genomic Enzymology as an Approach to Understanding Resistance Evolution
Gerlt & Babbit(50) have suggested theterm ge-nomic enzymology as a strategy to understand
the evolution of protein function within en-
zyme superfamilies. This concept is highly ap-
plicable to the study of the evolution of antibi-
otic resistance from proto-resistance enzymes
of the resistome (Figure 1b). Protein structure
and function studies clearly show that many
antibiotic resistance enzymes are members of
several enzyme superfamilies, providing clues
for the nature of ancestral proto-resistance
elements. Furthermore, bacterial genome se-
quences reveal numerous chromosomal genes
annotated as possible resistance elements, evenin the absence of the need for self protection of
antibiotic producers. These genes may encode
bona fide resistance proteins or could be proto-
resistance elements. Expression and character-
ization of the structure and function of these
proteins provides vital information for the un-
derstanding of resistance evolution and insight
into leveraging this knowledge to overcome re-
sistance in the clinic. In this review, we describe
the genomic enzymology of several classes of
resistance enzymes with a goalof understanding
how resistance evolves and what can be learned
from these studies that can have applicability
in drug discovery and medicine. We are care-
ful to note that in most cases the generally low
level of sequence similarity between resistance
enzymes and structurally similar enzymes can
often only suggest at common ancestry, but
lacks the robustness to unequivocally demon-
strate evolutionary relatedness. We therefore
have generally resisted comment on phylogeny
except where the evidence is clear.
ANTIBIOTIC HYDROLASES
AND LYASES
β-Lactamases
β-Lactam antibiotics include the penicillins,
cephalosporins, penems, and monobactams
28 Morar ·Wright

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 5/27
GE44CH02-Wright ARI 17 July 2010 13:29
Ancestralprotoresistance
protein
Housekeepingprotein
Resistanceprotein
a
b
Identify homologsand protoresistance
genes
Characterizeenzyme
Vmax
0.5Vmax
K m
V
[S]
Determine3D structure
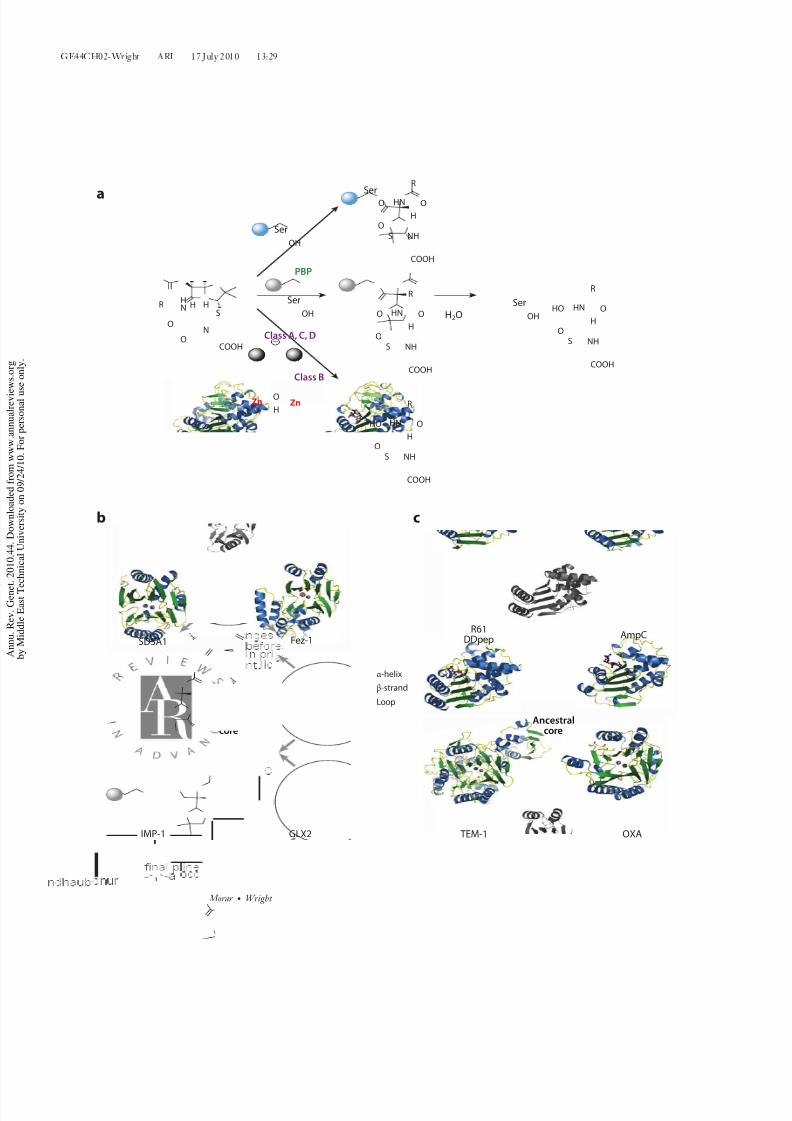
Figure 1
Key concepts in antibiotic genomic enzymology. (a) Representation of the concept of proto-resistancedepicting the ability of the same ancestral core to acquire a different function, housekeeping versusresistance. (b) The genomic enzymology strategy integrates enzyme structure, function, and genetic context.
that are mainstay drugs for the treatment of
bacterial infection. These antibiotics act by co-
valentbinding through an active siteSer residue
to so-called penicillin binding proteins (PBPs),
which are required for assembly and tailoring
of the peptidoglycan component of the bacte-
rial cell wall (92).
Resistance to β-lactam antibiotics is princi-pally the result of the expression of hydrolytic
enzymes, β-lactamases, that inactivate the
antibiotics via lactam ring opening hydrolysis
(Figure 2a). Two general chemical mecha-
nisms are recognized that accomplish this reac-
tion. The first is via canonical Ser nucleophile
attack at the lactam carbonyl carbon, resulting
in a covalent enzyme intermediate that mimics
the inactivation step of PBPs. The complex
then undergoes hydrolysis comparable to Ser
protease/esterasechemistry.The secondmech-
anism is by Zn2+-dependent activation of a
hydrolytic water molecule, here in mechanisticanalogy to metallo-peptidases. β-Lactamases
have been categorized based on protein se-
quence analysis [Ambler classification (3)] and
by function [Bush-Jacoby notation (21)]. The
Ambler notation separates β-lactamases into
www.annualreviews.org • Enzymology of Antibiotic Resistance 29
8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 6/27
GE44CH02-Wright ARI 17 July 2010 13:29
N
S
HN
COOH
R
O
O
HH
PBP
NHS
HN
COOH
R
O
OH
O
Ser
NHS
HN
COOH
R
O
OH
O
NHS
HN
COOH
R
O
OH
HOSer
OH
NHS
HN
COOH
R
O
OH
HO
O
HZn
Ser
OH
Ser
OH
Class A, C, D
Class B
a
b c
Fez-1
IMP-1
Ancestralcore
Ancestralcore
GLX2
SDSA1AmpC
OXA
R61DDpep
TEM-1
H2O
Zn
α-helix
β-strand
Loop
30 Morar ·Wright

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 7/27
GE44CH02-Wright ARI 17 July 2010 13:29
4 groups: A, B, C, and D. Class B enzymes are
Zn2+-dependent metallo enzymes, and class A,
C, D are active-site Ser enzymes. Phylogenetic
analysis reveals that the class B metallo-enzymes are distinct from the class A, C, and D
Serproteins. Theformer consistof twodiscrete
structural groups (B1–2 and B3 subclasses)
(Figure 2b) (54, 55). On the other hand,
the class A and D enzymes are homologues
and have diversified from a common shared
ancestor with the class C enzymes (52, 53).
In 1986, comparison of the three-dimensional
(3D) structure of class A enzymes with
PBPs revealed conservation of 3D structure
(Figure 2c ) and of biochemical mechanism (74,
92). Thedetermination of several 3D structures
of PBPs and class A, C, and D β-lactamasesin the past 25 years has served to solidify the
relationship between cell wall–metabolizing
enzymes and drug-resistance β-lactamases.β-Lactamases therefore share structure and
function with enzymes associated with cell wall
metabolism and general peptidase activities.
Phylogenetic evidence suggests that the class
A Ser β-lactamases emerged 2.4 billion years
ago from a common ancestor of PBPs (53).
The genes encoding these enzymes and the
related class D lactamases are widely circulated
in bacteria through the aegis of mobile genetic
elements such as plasmids. Class C enzymes,however, are chromosomally encoded (al-
though in the past decades, they have begun
to be associated with mobile genetic elements,
no doubt a recent event linked to antibiotic
selection) and diverged from the class A and
D enzymes approximately 1.8 billion years ago
(based on their association with Gram negative
proteobacteria) (53). Consistent with the
ancient divergence of β-lactamases from PBPs,
the former have no peptidoglycan biosynthetic
activity. Recent evolution has contributed
to the broadening of the antibiotic substrate
specificity of β-lactamases, and the numberof distinct enzymes with unique function
continues to grow and is approaching 1,000
entries (www.lahey.org/studies).
Streptogramin B Lyase
Thepublicationof the3D structuresof thebac-
terial ribosomal subunits in complex with an-
tibiotic inhibitors of translation over the past
decade has transformed our understanding of
antibiotic action and resistance. The large ribo-
somal subunit includes twomajorsitesof antibi-
otic binding: the peptidyl transfer center (theactive site of the ribosome) and the peptide exit
tunnel where nascent proteins transit from the
peptidyl transfer center and emerge from the
ribosomal complex. Streptogramin antibiotics
are composed of two structurally distinct com-
ponents, type A peptide-polyketide hybrids that
bind to the peptidyl transfer center and type B
cyclic depsipeptides that block the peptide exit
tunnel. These antibiotic are produced simulta-
neously by the same organism resulting in syn-
ergistic antibiotic activity (100).
Type B streptogramins are inactivated by
cleavage of the lactone link by an enzyme, vir-giniamycin B lyase (Vgb) (2). The structure-
function of this enzymehas been thoroughlyin-
vestigated showing that the ring opening takes
place via an elimination reaction, assisted by a
hexa-coordinated Mg2+ ion and a catalytic His
base (8, 78, 87, 99). Vgb has a seven-blade β-
propeller fold, similar to that of a muconate-
lactonizing enzyme (MLE) (Figure 3a). MLE
catalyzes a similar reaction, that of cyclization
←−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−
Figure 2
Genomic enzymology of β-lactamases. (a) Enzymatic reaction of a β-lactam hydrolysis catalyzed by PBPs versus class A, C, D serineβ-lactamases versus class B metallo β-lactamases, summarizing the similarities and differences in this transformation. Spheres representthe remainder of the enzyme. (b) Conservation in structure between class B1–2 (IMP-1 complexed with two Zn2+ ions; pdb id: 1DDK),B3β-lactamases (Fez-1 complexed with two Zn2+ ions; pdb id: 1L9Y), glyoxalase II (complexed with Zn2+ and Fe3+ ions; pdb id:1XM8), and alkylsulfatase (complexed with two Zn2+ ions; pdb id: 2CFZ). Their emergence from a common ancestor is represented by the common structural core. Structures are shown in ribbon diagram and color coded by secondary structure. The structural core is inblack and white. (c ) Conservation in structure between class A (TEM1 complexed with transition state analog; pdb id: 1M40), C (AmpC;pdb id: 1IEL), D β-lactamases (OXA complexed with Meropenem; pdb id: 1H8Y), and PBP DD-peptidase R61 (pdb id: 3PTE).
www.annualreviews.org • Enzymology of Antibiotic Resistance 31

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 8/27
GE44CH02-Wright ARI 17 July 2010 13:29
Ancestralcore
MLE Vgb
a
b
N
NHO
OH
HN
O
NO
O
ONH
N
O
CH3
N(CH3)2
ON
O
O
N
NHO
OH
HN
O
NO
O–
ONH
N
O
CH3
N(CH3)2
ON
O
OVgb, Mg2+
OO
MLE
CO2–
–O2C
CO2–
β-strand
Loop
Figure 3
Genomic enzymology of streptogramin B lyase (Vgb). (a) Conservation instructure between Vgb (complexed with quinupristin; pdb id: 2Z2P) and amuconate-lactonizing enzyme (MLE; pdb id: 1JOF). Presentation and colorcoding as in Figure 2. (b) Reaction catalyzed by MLE versus Vgb, withsimilarities in the transformation highlighted in green for the housekeepingprotein and in purple for the resistance protein.
APH: aminoglycosidephosphotransferase
rather than ring opening, and also makes use of
a His base (Figure 3b) (70). Analogous to the
macrolide glycosyltransferases discussed below,
Vgb binds its antibiotic substrate through very
few hydrogen bonds, and substrate recognition
is dominated by van der Waal interactions.
ANTIBIOTIC KINASES
Aminoglycoside Phosphotransferases
Aminoglycoside antibiotics are cationic small
molecules composed of an aminocyclitol ring
linked to various amino sugars. They include
such clinically important antibiotics as to-
bramycin, gentamicin, streptomycin, and
spectinomycin. Aminoglycosides bind to the
16S rRNA of the small ribosomal subunitand interfere with proper codon-anticodon
pairing, which results in mistranslation of
proteins (the exception is spectinomycin,
which blocks protein synthesis but does not
cause mistranslation) (25, 35). These abnormal
proteins are cytotoxic and result in cell death.
Binding of the antibiotics to the 16S rRNA
requires both charge-charge interactions and
close steric alignment. It is therefore not
surprising that bacteria have evolved a series
of enzymes that alter aminoglycoside charge
and structure as a means of efficient resistance.
Interestingly, three unrelated mechanismsare known: O-phosphotransferases (APH), N -
acetyltransferases, and O-adenylyltransferases.
Structure and function studies on each of these
classes have revealed that they share traits with
well-known families of metabolic enzymes.
APHs and ePKs share a common ancestor.
Over two dozen APHs have been identified,
with protein sequence identity ranging from
20% to 40% (127, 151). These enzymes are
classified based on regiospecificity of phospho-
rylation of the same aminoglycoside substrate,as well as substrate specificity and promiscuity.
Current knowledge of the structure-function
for several APH family members and phy-
logenetic analyses place them in the atypical
kinase superfamily with eukaryotic protein ki-
nase (ePK) fold, with the closest relatives being
small molecule kinases: choline kinase(CK) and
5-methylthioribose kinase (MTRK) (71, 125)
(Figure 4a,b).
To date, four crystallographic structures of
APHs have been reported (48, 64, 106, 156).
The structure of APH(3)-IIIa was the first to
reveal conservation of the ePK fold for theseenzymes and suggest a common ancestor (64).
In fact, all four structures have the core ePK
fold, and moreover, the primary structure for
theATP-binding, HXDXXXXN,knownas the
Brenner motif, is conserved as well (18).
32 Morar ·Wright

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 9/27
GE44CH02-Wright ARI 17 July 2010 13:29
APH(9)-Ia
APH(3’)-IIIa
CAPK
CK
APH(2”)-IIa
MTRK
a
b
CPT SK
AK
c
d
Ancestral
coreAncestral
core
O
O
O
O
NH2 NH2
NH2
NH2
NH2
NH2
NH2
NH2
HOHO
HO
HO OH
OH
ATP ADP
ATP ADP
O
O
O
O
HO
HO
HO OH
OH
N+
HO
N+
NH
CHCl2 CHCl2
NO2NO2
OH
O
OH
NH
OH
O
OP
O
O––O
HO
OH
OH
OHO
OH
OH
OHO
OP
O
O––O
OP
O
O––O
OP
O
O––O
APH(3'), Mg2+
CK, Mg2+
ATP ADP
SK, Mg2+
ATP ADP
CPT, Mg2+
α-helix
β-strand
Loop
H2N H2N
Figure 4
Antibiotic phosphotransferases. (a) Conservation in structure between aminoglycoside phosphotransferases APH(2 )-IIa (pdb id:3HAV), APH(3)-IIIa (pdb id: 2B0Q), APH(9)-Ia (pdb id: 3I0O), choline kinase (CK pdb id: 2CKP), 5-methylthioribose kinase(MTRK; pdb id: 2OLC), and cAMP-dependent protein kinase (CAPK; pdb id: 1ATP). Presentation and color coding as in Figure 2.
APH(2
)-IIa is shown in complex with AMPPCP, CAPK in complex with ATP, and the remaining four are with ADP. (b) Reactioncatalyzed by CK versus APH(3)-IIIa, with similarities in the transformation highlighted in green for the housekeeping protein and inpurple for the resistance protein. (c ) Conservation in structure between chloramphenicol phosphotransferase (CPT complexed withATP; pdb id: 1QHX), adenylate kinase (AK complexed with ADP; pdb id: 1M7G), and shikimate kinase (SK complexed withAMPPNP; pdb id: 3BAF). Presentation and color coding as in Figure 2. (d ) Reaction catalyzed by SK versus CPT, with similarities inthe transformation highlighted in green for the housekeeping protein and in purple for the resistance protein.
www.annualreviews.org • Enzymology of Antibiotic Resistance 33

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 10/27
GE44CH02-Wright ARI 17 July 2010 13:29
Of the four APHs with known structure,
APH(9)-Ia is thought to be the closest link to
ePKssofar.LikeePKs,thedomainsinAPH(9)-
Ia undergo conformational changes upon sub-strate binding, which are unseen in other
APHs (48). In ePKs conformational changes are
needed for activation, whereas APHs thus far
have been thought to be constitutively in the
“on” conformation. The conformational flex-
ibility of APH(9)-Ia could either be a vestige
of ePK relation or perhaps could serve some
as yet unknown regulatory function. The ge-
ometry of nucleotide binding in APH(9)-Ia is
also more ePK-like. Furthermore, like CK and
MTRK, the APH(9)-Ia substrate binding site is
quite rigid and small in contrast with APH(3)
isozymes and APH(2
)-IIa (80, 90, 110). Thelatter have a large flexible active site pocket
capable of accommodating multiple substrates
(46, 47).
The similarity of APHs with ePKs extends
beyond structural components; these enzymes
alsoshare mechanistic aspects. Enzymatic stud-
ies have shown that like ePKs, APHs operate
via a direct phosphate transfer mechanism from
ATP to antibiotic substrate, with the Asp from
the Brenner motif responsible for activation of
the attacking hydroxyl ontoγ-phosphate of the
ATP (96, 97, 132, 139). However, for the most
thoroughly studied antibiotic kinase, APH(3)-IIIa, the transfer takes place via a dissociative
transition state (138), and this transition state
was also suggested for MTRK (80), whereas
for a well-studied ePK:cAMP-dependent pro-
tein kinase the transition is associative (157).
Mechanistic connection between APHs and
ePK is further strengthened by studies showing
that APHs can phosphorylate ePK peptide and
protein substrates, and are inhibited by ePKs
inhibitors (14, 30, 31).
Macrolide PhosphotransferasesMacrolide antibiotics include the first gen-
eration natural product drug erythromycin
as well as the semisynthetic derivatives clar-
ithromycin and azithromycin.These antibiotics
block translation by binding to the peptide exit
tunnel of the large ribosomal subunit (140).
Macrolide resistance in the clinic is most often
associated with enzyme-catalyzed methylationof the23S rRNA by Ermmethyltransferases;an
action that simultaneously confers resistance to
the structurally unrelated lincosamide and type
B streptogramins. Recently, however,genesen-
coding macrolide kinases (MPHs) have been
found to be associated with emerging resistance
(112). These enzymes inactivate macrolides
by phosphorylation of the 2-OH in a GTP-
dependent manner (128). The MPHs charac-
terized so far come in two isoforms that are dif-
ferentiated based on substrate specificity. No
detailed structure-function analysis has been
published for these enzymes. However, Sawaiand coworkers (137) have noted a presence
of well-conserved C-terminal regions between
MPHs and APHs, one of them being the
Brenner motif. Mutagenesis of the Asp in this
motif confirmed it being essential for enzyme
activity, leading the authors to suggest that like
APHs and ePKs, this residue acts as a general
base activating the 2-OH. Further characteri-
zation of this enzyme will show whether MPHs
like APHs are a result of divergent evolution
from the vast ePK superfamily.
Chloramphenicol Phosphotransferase
Like macrolides, chloramphenicol also binds to
the largesubunit of the ribosome. In the crystal
structure of chloramphenicol, with a large sub-
unit of the bacterium Haloarcula marismortui ,
theantibiotic overlaps with the macrolidebind-
ing site (57), whereas in the Deinococcus radiodu-
rans structure, it binds at a different site adja-
cent to the peptidyltransfer center (126). These
distinct binding sites are nonetheless consis-
tent with biochemical and resistance mutation
data that suggest that both sites may be relevant
to chloramphenicol action. The antibiotic hasbeenassociated with significant toxicity, includ-
ing rare but irreversible aplastic anemia, but
it still sees limited clinical use throughout the
world.
34 Morar ·Wright

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 11/27
GE44CH02-Wright ARI 17 July 2010 13:29
CPT:
chloramphenicolphosphotransferase
AAC: aminoglycosideacetyltransferase
CPT is a member of nucleoside monophos-
phate kinase family. The producer of chlo-
ramphenicol, Streptomyces venezuelae, uses a 3-
O-phosphotransferase (CPT) to modify theC3-hydroxyl of the antibiotic and render it in-
active. Although this microorganism utilizes
the same inactivation strategy for chloram-
phenicol as described for aminoglycosides and
macrolides, the similarity does not extend to
structure. X-ray crystallographic characteriza-
tion of CPT revealed that this enzyme belongs
to the monophosphate kinase family, which is
unrelated to ePKs. The closest structural rel-
atives of CPT are shikimate kinase (SK) and
adenylate kinase (AK) (Figure 4c ) (39, 65, 79).
Like SK and AK, CPT possesses a signature
Gly-rich P-loop, which forms an oxy-anionhole and accommodates the phosphate tail of
ATP. A second conserved motif, the B-motif,
contains an Asp to act as the catalytic base
for C3-OH activation. CPT is proposed to act
via direct phosphate transfer and an associative
transition state (65). The same type of mech-
anism was proposed for SK (Figure 4d ) (58).
Therefore, although thefold andpositioning of
the catalytic residues is different for CPT and
the monophosphate kinase family from APHs
and ePKs, the fundamental mechanistic fea-
tures are retained.
ANTIBIOTICACETYLTRANSFERASES
Aminoglycoside AcetyltransferasesAre Members of the GNAT Superfamily
Aminoglycoside inactivation via aminoglyco-
side acetyltransferase (AAC) enzymes was the
second bacterial resistance mechanism discov-
ered after penicillinases (107). Since then ex-
tensive structure-function characterization of
these enzymes has been ongoing. AACs belongto the GCN5 N-acetyl transferase (GNAT) su-
perfamily. This superfamily of enzymes utilizes
acetyl-CoA as a cofactor and contains an im-
pressive number of approximately 10,000mem-
bers (146). Of these, AACs were the first to
be identified (33, 130) and were subsequently
shown to have conserved sequence motifs with
eukaryotic transcription factor GCN5, giving
the superfamily its name (130).
Structure and function of AACs. AACs are
divided into four groups based on the re-
giospecificity of the aminoglycoside modifica-
tion and designated AAC(1), (3), (2), and (6).
Structures are available for plasmid-encoded
AAC(3)-Ia, chromosomally encoded AAC(2)-
Ic, and several AAC(6) isozymes (143, 147,
153). The structures show conservation of the
GNAT core, despite very little identity or sim-
ilarity in the primary sequence (Figure 5a)
(142). One sequence motif R/QxxGxG/A is in-
variant in these proteins and is responsible forrecognition of 3-P ADP moiety of the CoA
cofactor (105, 147). Structure of the AAC(6)-
Ii dimer is similar with AAC(3)-Ia and even
more so with eukaryoticHpa2-encoded histone
acetyltransferase (20, 153), while being distinct
from AAC(2)-Ic.
AACs have been extensively investigated
biochemically with most reports available for
AAC(6)-Ii and -Iy and AAC(2)-Ic groups.
The acetylation reaction follows a random [for
AAC(2)-Ic] or sequential [AAC(6)-I] mecha-
nism, with acetyl-CoA binding first followed by
aminoglycoside, andin eithercase, thepresenceof both substrate and cosubstrate is required
for acyl transfer, with rate-limiting product
release (37, 60, 89, 118). Further investigation
of molecular mechanism via mutagenesis of
potential catalytic residues revealed that muta-
tions ofmostactivesiteresidues donot yield in-
active enzymes. These observations are consis-
tent with the enzyme’s primary function being
binding and positioningof the substrates,rather
than specific catalytic activation via direct par-
ticipationof amino acidfunction groupsin roles
such as a general acid/base (38, 59). Substrate
specificity investigations have shown a remark-ably broad range of aminoglycosides acetylated
by both AAC(6) isozymes and AAC(2)-Ic (29,
60,117, 150). Moreover AAC(6) isozymes were
shown to acetylate eukaryotic histone proteins
and human histone H3 peptide (144, 153).
www.annualreviews.org • Enzymology of Antibiotic Resistance 35

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 12/27
GE44CH02-Wright ARI 17 July 2010 13:29
CrAT
E2pCD CAT
a
c d
b
VatDLpxA
XAT
Ancestralcore
Ancestralcore
Ancestralcore
O
O
O
O
NH2
HOHO
HO
HO OH
OH
O
O
O
O
NH
HOHO
HO
HO OH
OH
OHPA2
AAC(6')
SCoA
O
HSCoA
SCoA
O
HSCoA
NH
O
NH
CHCl2 CHCl2
NO2 NO2
OH
O
OH
CAT
SCoA
O
HSCoA
NH
OH
O
OO
O
O–OH
O
O–O
N+
N+ SCoA
O
HSCoA
CrAT
O
AAC(2’)-Ic
AAC(6’)-IiHpa2
NH2
Lys Lys
NH2
NH2
NH2
NH2
NH2NH2
H2N H2N
α-helix
β-strand
Loop
36 Morar ·Wright

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 13/27
GE44CH02-Wright ARI 17 July 2010 13:29
CAT:
chloramphenicolacetyltransferase
Divergent evolution of GNAT superfamily.
Despite a low amino acid sequence iden-
tity, the GNAT superfamily members known
to date share the core tertiary structure andfollow similar mechanisms of acetyl transfer
(Figure 5c ). In catalysis, the positioning of the
substrates appears to be most important, rather
than the presence of specific functional moi-
eties. AACs do not share the most conserved
quaternary structure among themselves. Some
function less discriminatively and are capable
of acetylating substrates other than aminogly-
cosides. Owing to this loose conservation of
tertiary and quaternary structure, as well as a
very broad substrate specificity, the true physi-
ologic function of AACs, especially those chro-
mosomally encoded, remains to be confirmed,placing these enzymes at the interface of bona
fide and proto-resistance elements in the resis-
tome (143, 150). For these same reasons, AACs
are thought the oldest members of the GNAT
family from which other members, such as eu-
karyotic histone acetyltransferases and fluoro-
quinolone acetyltransferases (see below), can
evolve toward novel function (144).
Fluoroquinolone acetyltransferases. Flu-
oroquinolones such as ciprofloxacin are
synthetic antibiotics that target DNA gy-rase and topoisomerases necessary for DNA
synthesis. Resistance is usually the result of
point mutations in target genes (e.g., gyrA,
parC ) or via efflux. By virtue of their synthetic
origins, it was considered unlikely that there
would be enzyme-catalyzed inactivation modes
of resistance. However, an AAC(6) variant,
AAC(6)-Ib-cr, containing two amino acid mu-
tations, has evolved to confer resistance fluoro-quinolone antibiotics with an available nitrogen
on a piperazine heterocycle (122). Structure-
function characterization of this enzyme led
to the mechanistic proposal for the enzyme’s
ability to acetylate both aminoglycosides and
fluoroquinolones. However, the mechanistic
details remain to be worked out (94, 145).
These findings further underscore the sig-
nificance of broad specificity for AACs and
our lack of understanding of their physiologic
function. The adaptability of these enzymes is
remarkable as only small numbers of changes
arerequiredfor theevolution of a newfunction.
AACs, CATs, and XATs are a result of
convergent evolution. Two variants of chlo-
ramphenicol acetyltransferases (CATs) have
been reported to date. Classic CATIII activity
was first identified in 1967 encoded on a plas-
mid (136). Biochemical characterization of this
enzyme yielded a mechanism consistent with
randomorder of substratebinding andthe pres-
ence of both a chloramphenicol substrate and
the acetyl-CoA cofactor for direct acetylation
(76, 77). Although the mechanism is reminis-cent of that for AACs, site-directed mutagen-
esis points to an important difference. Unlike
AACs, whose primary purpose in catalysis is
proposed to be binding and orientation of sub-
strates, CATs have an essential histidineresidue
←−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−
Figure 5
Antibiotic acetyltransferases. (a) Conservation in structure between aminoglycoside acetyltransferases AAC(6)-Ii (pdb id: 1B87),AAC(2)-Ic (CK pdb id: 1M4D), and Hpa2 histone acetyltransferase (Hpa2; pdb id: 1QSO). Presentation and color coding as inFigure 2. AAC(6)-Ii and AAC(2)-Ic are both shown in complex with Coenzyme A (CoA). (b) Conservation in structure betweenaminoglycoside acetyltransferases:chloramphenicol acetyltransferase (CAT complexed with chloramphenicol; pdb id: 3CLA), carnitine
acetyltransferase (CrAT; pdb id: 2H3P), and dihydrolipoyl transacetylase (E2pCD; pdb id: 1EAB). Presentation and color coding as inFigure 2. CrAT and E2pCD are shown in complex with acetyl-CoA and CoA, respectively. (c ) Reactions catalyzed by Hpa2 and CrATversus AAC(6) and CAT, with similarities in the transformations highlighted in green for the housekeeping proteins and in purple forthe resistance proteins. Spheres represent the remainder of the enzyme. (d ) Conservation in structure between xenobioticacetyltransferases chloramphenicol acetyltransferase (XAT complexed with chloramphenicol and desulfo-CoA; pdb id: 2XAT),streptogramin group A acetyltransferase (VatD complexed with acetyl-CoA; pdb id: 1KK4), and UDP N -acetylglucosamineO-acyltransferase (LpxA; pdb id: 1LXA). Presentation and color coding as in Figure 2.
www.annualreviews.org • Enzymology of Antibiotic Resistance 37

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 14/27
GE44CH02-Wright ARI 17 July 2010 13:29
XAT: xenobiotic
acetyltransferaseANT: aminoglycosidenucleotidyltransferase
acting as a general base(85, 103). Furthermore,
CAT structure reveals a completely different
fold, quaternary organization, and substrate
binding mode from that of AACs (83, 84).CATs show high specificity for their substrate,
although one variant is able to bind and acety-
late the steroidal antibiotic, fusidic acid (102).
CATs share structure-function similarity
with carnitine acetyltransferase (68, 152) and
dihydrolipoyl transacetylase (93), the latter two
also being CoA-dependent acetyltransferases
(Figure 5b,c ). Remarkably, CAT and dihy-
drolipoyl transacetylase are both trimers with
the active site located at the interface, and their
monomer organization is similar with the two
domain organization of carnitine acetyltrans-
ferase. Furthermore, the catalytic histidine ispresent at the same position for all (67).
There exists a second variant of enzyme ca-
pable of chloramphenicol acetylation, termed
CATB, that belongs to xenobiotic acetyltrans-
ferase (XAT) class (104). XAT class includes an
enzyme, VatD, responsible for group A strep-
togramin acetylation (17, 121). Structures of
both CATB and VatD have been determined
(10, 73, 135) and show an unusual left-handed
parallel β-helix (LβH) fold (119). Other en-
zymes with LβH fold are also CoA-dependent
acetyltransferases, the exception beingcarbonic
anhydrase (Figure 5d ) (75, 119). Similaritiesbetween VatD and CATB extend to primary
structure, with the conservation of key catalytic
residues. Although theLβH fold is unrelated to
the classic CAT, XATs also form trimers with
the active site located at the monomer interface
and an active site histidine proposed to serve as
catalytic base (10).
In summary, nature has adapted a least
three different protein folds to catalyze CoA-
dependent acetyl transfers, and antibiotic
acetyltransferases span all three.
ANTIBIOTIC NUCLEOTIDYLYLTRANSFERASES
Divergent Evolution of Aminoglycoside and Lincosamide NTs
Aminoglycosides can be inactivated by an
ATP-dependent adenylylation of various hy-
droxyl moieties. Enzymes responsible for this
modification, aminoglycoside nucleotidyltrans-
ferase (ANTs), are classified based on their
substrate and regiospecificity (88, 148). Of these, ANT(2 )-Ia and ANT(4)-I have been
thoroughly investigated biochemically, and al-
though these enzymes show no relatedness in
primary sequence, they follow a similar mech-
anism. The adenylyl group is transferred di-
rectly from ATPto theantibiotic via an ordered
Bi-Bi mechanism and an associative transition
state (26, 49, 51, 141). The 3D structure of
ANT(4)-I further supports this mechanism and
points to a glutamate residue as a potential cat-
alytic base responsible for substrate activation
(109, 123). The ANT(4)-I structure also re-
veals thesimilarity of this enzyme fold with thatof DNA polymerase β (Figure 6a) (63, 124).
Another class of antibiotics inactivated by
adenylylation is the lincosamides. Clindamycin
is the lincosamide antibiotic that is most of-
ten used clinically. It binds to the peptide exit
tunnel of the bacterial ribosome in the same
region as the macrolide antibiotics, consistent
−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−→
Figure 6
Antibiotic adenylyltransferases and glycosyltransferases. (a) Conservation in structure between aminoglycoside adenylyltransferase[ANT(4)-I in complex with AMPCPP; pdb id: 1KNY], lincosamide adenylyltransferase (LinB in complex with AMPCPP; pdb id:
3JZ0), DNA polymeraseβ (PolB in complex with dUMPNPP; pdb id: 2FMS) and polyA polymerase (PAP in complex with 3
-dATP;pdb id: 1Q78). Presentation and color coding as in Figure 2. (b) Reactions catalyzed by PAP versus ANT(4)-I and LinB, withsimilarities in the transformations highlighted in green for the housekeeping protein and in purple for the resistance proteins.(c ) Conservation in structure between macrolide glycosyltransferases OleI (ternary complex with UDP and oleandomycin; pdb id:2IYA) and OleD (ternary complex with UDP and erythromycin; pdb id: 2IYF), triterpene UDP-glucosyltransferase (UGT71G1 incomplex with UDP; pdb id: 2ACV), and vancomycin aglycone UDP-glucosyltransferase (GtfB; pdb id: 1IIR). Presentation and colorcoding as in Figure 2. (d ) Reactions catalyzed by GtfB versus OleD, with similarities in the transformations highlighted in green forthe housekeeping protein and in purple for the resistance protein.
38 Morar ·Wright

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 15/27
GE44CH02-Wright ARI 17 July 2010 13:29
ANT(4’)-I
LinB
PAP
PolB
a b
c d
Ancestralcore
Ancestralcore
HN Cl
ON
OO
OHHO
N
N N
N
NH2
O P
O
O–
LinB
ORO
OHHO
N
N N
N
NH2
O P
O
O–
ATP PPi
PAP
ORO
OHO
N
N N
N
NH2
O P
O
O–
O
OHHO
N
N N
N
NH2
O P
O
O–
ATP PPi
ATP PPi
ANT4'
O
O
O
O
NH2
NH2
NH2
NH2
HO
HO
HO
HO
OH
OH
O
O
O
O
NH2
NH2
NH2
NH2
OHO
H2N
HO
HO OH
OH
O
OHHO
N
N N
N
NH2
O P
O
–O
O
SOH
HO
HO
HN Cl
ON
O
SOH
HO
O
O
O
O
OHHO
O
OOH
O
OHO
NOH
O
O
O
O
OHHO
O
OOH
O
OO
NOHUDP-Glc
OleD
UDP O
OH
HO OH
HO
NHO
HN
O
NHO
HN
OHNH
O HO
O
OH
OH
HO
OCl
O
NH2
O
O
NH
Cl
HO
O
HN
HO
NH
O
HNO
NHO
HN
OHNHO HO
O
OHOH
HO
OCl
ONH2
O
O
NH
Cl
HO
O
HN
O
UDP-Glc
GtfB
UDP
O
HO
HOHO
HO
Utg71G1
OleI OleD
GtfB
H2N
α-helix
β-strand
Loop
www.annualreviews.org • Enzymology of Antibiotic Resistance 39

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 16/27
GE44CH02-Wright ARI 17 July 2010 13:29
MGT: macrolide
glycosyltransferase
with the cross resistance afforded by Erm 23S
RNA methyltransferases (140). The responsi-
ble clindamycin adenylating enzymes are en-
coded by lin genes (40, 82) and are dividedinto two major groups, LinA and LinB, based
on substrate and regiospecificity. Analogous to
ANT(2 )-Ia and ANT(4)-I, LinA and LinB
show no homology in sequence. Structure-
function characterization of LinB revealed that
similar to ANTs this enzymefollows an ordered
Bi-Bi mechanism with direct AMP transfer
(Figure 6b) (98). Moreover, it has the same
3D fold as ANT(4)-I, DNA polβ, and polyA
polymerase (PAP) (Figure 6a) (34, 123, 124).
Conservation is alsoobservedfor positioning of
ATP, as well as location and positioning of the
catalytic glutamate and Mg2+
ions responsibleforstabilizationof thetransition state. LinB and
ANT(4) are thereforea resultof divergentevo-
lution from family X polymerases (4), as these
polymerases not only share the structural simi-
larities with antibiotic NTsbut alsofunctionvia
a direct associative mechanism like antibiotic
NTs (11, 133). ANT(2 )-Ia and LinA variants
show low sequence homology with each other
and none with family X polymerases, ANT(4 )-
I, or LinB (111). Structural characterization of
these enzymes will aid in further understanding
the relation of these resistance genes with the
rest of the resistome.
OTHER ANTIBIOTIC GROUPTRANSFERASES
Macrolide GlycosyltransferasesAre Related to the GT-1 Family
Macrolide glycosyltransferase (MGT) encodes
an enzyme that inactivates macrolides such as
erythromycin using UDP-glucose as cofactor.
This activity has been detected in environmen-
tal organisms such as members of the genus
Streptomycetes , both in antibiotic producers andnonproducers (66, 81). MGT inactivates the
antibioticsby glycosylation of the 2-OHofa6-
deoxyhexose moiety; however, variants of this
enzyme have different substrate specificities
(113, 116). This is the same regiospecificity of
the MPH resistance kinases and reflects the
importance of this hydroxyl group in binding
of macrolide antibiotics to the large subunitof the ribosome as revealed by the X-ray
structures, where a hydrogen bond between
the 2-OH and A2058 (E. coli numbering) is
evident (140). The MGT catalyzed reaction
proceeds via an ordered mechanism, with
both substrate and cofactor required for direct
transfer, and antibiotic binding first followed
by UDP-glucose (114–116). Structures of two
MGTs, OleI and OleD, revealed that these
enzymes belong to glycosyltransferase-1 family
(GT-1) (15, 27) and share similarity with the
GtfB enzyme, which is responsible for gly-
cosylation of vancomycin aglycone (101) andflavonoid/triterpene GT (Figure 6c ) (129). As
proposed for GtfB, the OleI/D-substrate com-
plexes showed primarily hydrophobic interac-
tions between the enzymes and the substrate.
Another unifying factor for these enzymes is
the mechanistic postulate, which involves the
activation of hydroxyl moiety via a general
base, followed by direct transfer (Figure 6d ).
Fosfomycin Inactivation EnzymesBelong to Vicinal Oxygen
Chelate Superfamily One mechanism of fosfomycin inactivation is
via epoxide ring opening using one of three re-
sistancegenes: FosA, FosB, and FosX (5, 24,41,
42). These enzymes utilize different substrates:
FosA uses glutathione, FosB uses L-cysteine,
FosX uses water. Each enzyme shows a differ-
ent metal dependency: Mn2+/K +, Mg2+, and
Mn2+ for FosA, ForB, and FosX, respectively
(13, 24, 43, 134). Despite these differences the
three enzymes show 30% to 35% protein se-
quence identity andshare thesame fold belong-
ing to the vicinal oxygen chelate (VOC) family
of metallo-enzymes (43).FosA and FosX structure-functions have
been characterized more extensively, and these
not only share a common fold but also metal
and substrate binding sites (43, 44, 120). FosX
40 Morar ·Wright

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 17/27
GE44CH02-Wright ARI 17 July 2010 13:29
catalyzes direct addition of water with Mn(II)
and a base involved in activation of the nucle-
ophile(43, 44), anddirect addition hasalso been
proposed for the glutathione transferase FosA (43, 44). Furthermore, the relationshipbetween
the two enzymes is strengthened by identifica-
tion ofa FosX homolog from Mesorhizobium loti
containing both activities (43). A triple mutant
of this enzyme improved FosA catalytic effi-
ciency and abolished that of FosX, demonstrat-
ing theease of manipulation of theproperties of
these enzymes (19). Besides Fos enzymes, VOC
family includes extradiol dioxygenase, glyox-
alase I, and methylmalonyl-CoA epimerase
(Figure 7a,c ) (22, 56, 95). All are metallo-
enzymes with a requirement for a catalytic base
and with a conserved metal binding site. Inter-estingly, Fos proteins show no structural simi-
laritywith an ancient superfamilyof glutathione
transferases, catalyzing analogous reactions of
glutathione-mediated small molecule detoxifi-
cation (131), and therefore represent an exam-
ple of convergent evolution.
ADP-Ribosylation of Rifamycin Antibiotics
Rifamycin antibiotics are inactivated by ADP-
ribosylation reaction catalyzed by NAD-
dependent Arr enzymes (28). This is theonly small molecule ADP-ribosylation enzyme
known to date. Structure-function analysis of
Arr from Mycobacterium smegmatis revealed that
despite nonexistent sequence homology, Arr
is a member of the ADP-ribosyltransferase
(ART) superfamily (9). All ART enzymes cat-
alyze transfer of ADP-ribose moiety with re-
lease of nicotinamide (62, 154). The conser-
vation of Arr structure is most significant for
the NAD+ binding core to that of RNA 2-
phosphotransferase (72) and bacterial toxins:
exotoxin A domain III (86) and cholera toxin
(69) (Figure 7b,d ). Of these, Arr representsthe minimal unit necessary for NAD-binding
and catalysis (9). Besides the tertiary structure,
Arr also contains three catalytically essential
residues characteristic of the ART family. Two,
His and Tyr, are part of a conserved H-Y-
E motif, proposed to be directly involved in
NAD binding. However, the catalytic Glu of
ART family is not conserved in Arr; instead Arrutilizes an Asp found elsewhere in the struc-
ture. This enzyme, capable of modifying both
natural-product and semisynthetic rifamycins,
is an excellent example of nature exploiting ex-
istingprotein scaffoldsin evolution of antibiotic
resistance.
OPPORTUNITIES ANDOUTCOMES OF ANTIBIOTICRESISTANCE GENOMICENZYMOLOGY
Antibiotic resistance is an integral componentof the natural history of antibiotics. The most
parsimonious hypothesis on the origins of re-
sistance is that it must have first coevolved with
biosynthesis as a means of auto-immunity to
the production of toxic secondary metabolites.
This chemical strategy then could have inde-
pendently evolved de novo in neighboring or-
ganisms as a means of protection or could have
been imported via horizontal gene transfer. As
long as there was selection, the genes for resis-
tance would have been maintained and even-
tually stably integrated into the genome. The
number of resistance elements that we nowsee scattered in virtually all bacterial genomes
may reflect prior exposure during the evolu-
tion of the species. This idea is not incompati-
ble with the new hypothesis that naturally pro-
duced antibiotics are not in fact antibiotics at
all in the concentrations produced in the envi-
ronment, but rather signaling molecules (155).
Resistance elements could have evolved as
receptors or mediators of such signaling
molecules. The fact that resistance is so
widespread in the environment and that we can
readily select for resistance even to completely
synthetic antibiotics, speaks to the depth andplasticity of the resistome.
The genomic enzymology strategy offers an
integrated approach to the study of the evolu-
tion of resistance and possible solutions to the
www.annualreviews.org • Enzymology of Antibiotic Resistance 41

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 18/27
GE44CH02-Wright ARI 17 July 2010 13:29
a
c
b
d
Ancestralcore
Ancestralcore
O
P–O
OHO
FosX, Mn2+
OH
HO
P
O–
HO
O
OH
OH O2
DHBD, Fe2+O
OH–O
O
OO
R1
OH
HO
NHO
OH
OOO
O
OO
O
OHHO
N
N N
N
NH2
O P
O
O
OP
O– O
OHHO
O–
OO
R1
OH
HO
NHO
OH
OOO
O
O
NAD+
ARR
R2R2
N
NH2
O
OHO
OHHO
N
N N
N
NH2
O PO
O
ORP
O
PARPelongationO
O
OHHO
N
N N
N
NH2
O P
O
OOP
O– O
OHHO
O–O
OHO
N
N N
N
NH2
O
NAD+
N
NH2
O
–OOHP
O
O
ORP
O–O
DHBDFosA
FosXGlxI
ARR
PARPpeIII
RNA2P
H2O
α-helix
β-strand
Loop
42 Morar ·Wright

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 19/27
GE44CH02-Wright ARI 17 July 2010 13:29
challenge resistance poses for the therapeutic
use of antibiotics. Using structure and function
studies, it is now evident as discussed above that
either resistance has emerged directly from el-ements with other metabolic functions or that
they share common ancestors. This idea first
emerged with the comparison of the struc-
tures of β-lactamases and PBPs over 20 years
ago (74) and the observations of Benveniste &
Davies (12) on aminoglycoside resistance al-
most 40 years ago. The availability of micro-
bial genomes coupled with theability to express
enzymes in quantity and study both structure
and function have only strengthened the links
between the genes and proteins of antibiotic
resistance and cellular metabolism. Given the
vast numbers of microbes on the planet (>1030
)(146), the depth of metabolic enzymes that can
serve as proto-resistance elements, and the ra-
pidity of bacterial cell growth, antibiotic resis-
tance is inevitable.
While the inevitability of resistance is
reflected in the clinical experience of the
past 50 years, it is a reality that can serve
to strengthen antibiotic drug discovery and
stewardship. For example, the identification
of resistance and proto-resistance elements
in environmental organisms early in the drug
discovery process can be very valuable. First, it
can serve to tailor candidate molecules duringlead optimization to avoid or otherwise modify
particularly vulnerable features, e.g., hydroxyl
groups that can be modified by kinases.
Second, by identifying resistance elements
early, scans of bacterial genomes can inform
on the distribution and density of resistance
genes. Third, this information could be used
to prepare molecular diagnostics that can be
used to routinely survey clinically relevant
organisms for the emergence of resistanceduring therapy. Finally, by understanding the
molecular basis of resistance, strategies such as
the codevelopment of inhibitors of resistance
can be pursued. For example, the relationship
between aminoglycoside kinases and eukariotic
Ser-Thr-Tyr protein kinases led to a study
demonstrating that protein kinase inhibitors
block the action of resistance kinases (31).
Because many pharmaceutical companies have
extensive chemical libraries targeted towards
protein kinase inhibition, leads for antiresis-
tance molecules that could be coadministered
with antibiotics may be readily identified andrepurposed for infectious disease therapy.
Given the continuing emergence of multi-
drug resistant pathogens, the need for new an-
tibiotics is acute and growing. With the ad-
ventof nextgenerationgenome sequencingthat
promises to ever increase the rapidity and num-
ber of sequenced bacterial genomes along with
downward pressures on the per genome costs,
there will be a large influx of new genomic
data from pathogenic and nonpathogenic bac-
teria over the next decade. The application of
this information to infectious disease biology
and drug discovery requires robust platformsthat can link gene with function and struc-
ture. Genomic enzymology is a modern inte-
grated strategy that offers a pathway to study
antibiotic resistance and apply this knowledge
in the development and management of new
drugs.
←−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−
Figure 7
Genomic enzymology of fosfomycin inactivation enzymes and rifamycin ADP-ribosyltransferase. (a) Conservation in structure betweenFosA (complexed with fosfomycin; pdb id: 1LQP), FosX (pdb id: 1R9C), glyoxalase I (GlxI complexed with methyl-gerfelin; pdb id:
2ZA0), and biphenyl-cleaving extradiol dioxygenase (DHBD; pdb id: 1HAN). Presentation and color coding as in Figure 2.(b) Conservation in structure between Arr (complexed with rifampicin; pdb id: 2HW2), PARP (complexed with NAD+ analogue; pdbid: 1A26), exotoxin A domain III (peIII complexed with NAD+ analogue; pdb id: 1AER), and RNA 2-phosphotransferase (RNA2P;pdb id: 1WFX). Presentation and color coding as in Figure 2. (c ) Reactions catalyzed by DHBD versus FosX, with similarities in thetransformations highlighted in green for the housekeeping protein and in purple for the resistance protein. (d ) Reactions catalyzed by poly (ADP-ribose) polymerase (PARP) versus rifamycin ADP-ribosylase (ARR), with similarities in the transformations highlighted ingreen for the housekeeping protein and in purple for the resistance protein.
www.annualreviews.org • Enzymology of Antibiotic Resistance 43

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 20/27
GE44CH02-Wright ARI 17 July 2010 13:29
SUMMARY POINTS
1. The antibiotic resistome accounts for all bone fide and potential antibiotic resistance
elements found not only in the clinic but also in any other environments (e.g., soil). Inorder to fully understand various aspects of antimicrobial drug resistance, all parts of the
resistome need to be investigated.
2. Proto-resistance elements are genes in the resistome that have potential to become re-
sistance determinants. Proto-resistance genes may share common ancestry with genes of
other cellular functions.
3. Genomic enzymology can be used to study the evolution of antibiotic resistance from
proto-resistance enzymes. Genomic enzymology of several classes of antibiotic resistance
enzymes revealed their evolutionary relation with other cellular functionalities.
4. Genomic enzymology of antibiotic resistance is a powerful approach for learning the
dynamics of antibiotic resistance. This knowledge can be applied in development and
management of antibiotic therapies.
FUTURE ISSUES
1. How can we track and differentiate resistance and proto-resistance elements in microbial
communities?
2. Can resistance genes in nonpathogenic microbes be directly linked to the emergence of
resistance in pathogens? What is the role of horizontal gene transfer in this process?
3. Can we identify the metabolic roles of proto-resistance elements in the resistome?
4. What will aid leveraging genomic enzymology strategies in drug discovery?
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that
might be perceived as affecting the objectivity of this review.
ACKNOWLEDGMENTS
Research in the author’s lab on antibiotic resistance is supported by a Canada Research Chair (to
GDW)and the Canadian Institutes of Health Research and the Natural Sciences and Engineering
Research Council of Canada.
LITERATURE CITED
1. Abraham EP, Chain E. 1940. An enzyme from bacteria able to destroy penicillin. Nature 146:837
2. Allignet J, Loncle V, Mazodier P, el Solh N. 1988. Nucleotide sequence of a staphylococcal plasmid
gene, vgb, encoding a hydrolase inactivating the B components of virginiamycin-like antibiotics. Plasmid
20:271–75
3. Ambler RP. 1980. The structure of beta-lactamases. Philos. Trans. R. Soc. Lond. Ser. B 289:321–31
44 Morar ·Wright

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 21/27
GE44CH02-Wright ARI 17 July 2010 13:29
4. Aravind L, Koonin EV. 1999. DNA polymerase beta-like nucleotidyltransferase superfamily: identifica-
tion of three new families, classification and evolutionary history. Nucleic Acids Res. 27:1609–185. Arca P, Rico M, Brana AF, Villar CJ, Hardisson C, Suarez JE. 1988. Formation of an adduct between
fosfomycin and glutathione: a new mechanism of antibiotic resistance in bacteria. Antimicrob. Agents
Chemother. 32:1552–566. Baltz RH. 2005. Antibiotic discovery from actinomycetes: Will a renaisssance follow the decline and
fall? SIM News 55:186–967. Bancroft EA. 2007. Antimicrobial resistance: it’s not just for hospitals. JAMA 298:1803–48. Bateman KP, Yang K, Thibault P, White RL, Vining LC. 1996. Inactivation of Etamycin by a novel
elimination mechanism in Streptomyces lividans. J. Am. Chem. Soc. 118:5335–389. Baysarowich J, Koteva K, Hughes DW, Ejim L, Griffiths E, et al. 2008. Rifamycin antibiotic resistance
by ADP-ribosylation: Structure and diversity of Arr. Proc. Natl. Acad. Sci. USA 105:4886–9110. Beaman TW, SugantinoM, RoderickSL. 1998. Structureof the hexapeptide xenobiotic acetyltransferase
from Pseudomonas aeruginosa. Biochemistry 37:6689–9611. Beard WA, Wilson SH. 2006. Structure and mechanism of DNA polymerase Beta. Chem. Rev. 106:361–
8212. Benveniste R, Davies J. 1973. Aminoglycoside antibiotic-inactivating enzymes in actinomycetes similar
to those present in clinical isolates of antibiotic-resistant bacteria. Proc. Natl. Acad. Sci. USA 70:2276–80
13. Bernat BA, Laughlin LT, Armstrong RN. 1999. Elucidation of a monovalent cation dependenceand characterization of the divalent cation binding site of the fosfomycin resistance protein (FosA).
Biochemistry 38:7462–6914. Boehr DD, Draker KA, Koteva K, Bains M, Hancock RE, Wright GD. 2003. Broad-spectrum peptide
inhibitors of aminoglycoside antibiotic resistance enzymes. Chem. Biol. 10:189–9615. BolamDN, Roberts S, Proctor MR,Turkenburg JP,Dodson EJ,et al. 2007. Thecrystal structureof two
macrolide glycosyltransferases provides a blueprint for host cell antibiotic immunity. Proc. Natl. Acad.
Sci. USA 104:5336–4116. Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, et al. 2009. Bad bugs, no drugs: no
ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48:1–1217. Bozdogan B, Leclercq R. 1999. Effects of genes encoding resistance to streptogramins A and B on the
activity of quinupristin-dalfopristin against Enterococcus faecium. Antimicrob. Agents Chemother. 43:2720–
25
18. Brenner S. 1987. Phosphotransferase sequence homology. Nature 329:21
19. Brown DW, Schaab MR, Birmingham WR, Armstrong RN. 2009. Evolution of the antibiotic resistanceprotein, FosA, is linked to a catalytically promiscuous progenitor. Biochemistry 48:1847–49
20. Burk DL, Ghuman N, Wybenga-Groot LE, Berghuis AM. 2003. X-ray structure of the AAC(6 )-Ii
antibiotic resistance enzyme at 1.8 A resolution; examination of oligomeric arrangements in GNAT
superfamily members. Protein Sci. 12:426–3721. Bush K, Jacoby GA. 2010. Updated functional classification of beta-lactamases. Antimicrob. Agents
Chemother. 54:969–7622. Cameron AD, Olin B, Ridderstrom M, Mannervik B, Jones TA. 1997. Crystal structure of human
glyoxalase I–evidence for gene duplication and 3D domain swapping. EMBO J. 16:3386–9523. Canton R. 2009. Antibiotic resistance genes fromthe environment:a perspective through newly identified
antibiotic resistance mechanisms in the clinical setting. Clin. Microbiol. Infect. 15(Suppl. 1):20–2524. Cao M, Bernat BA, WangZ, ArmstrongRN, Helmann JD. 2001. FosB, a cysteine-dependent fosfomycin
resistance protein under the control of sigma(W), an extracytoplasmic-function sigma factor in Bacillus
subtilis . J. Bacteriol. 183:2380–83
25. Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. 2000.Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics.
Nature 407:340–4826. Chen-Goodspeed M, Vanhooke JL, Holden HM, Raushel FM. 1999. Kinetic mechanism of kanamycin
nucleotidyltransferase from Staphylococcus aureus . Bioorg. Chem. 27:395–40827. Coutinho PM, Deleury E, Davies GJ, Henrissat B. 2003. An evolving hierarchical family classification
for glycosyltransferases. J. Mol. Biol. 328:307–17
www.annualreviews.org • Enzymology of Antibiotic Resistance 45

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 22/27
GE44CH02-Wright ARI 17 July 2010 13:29
28. Dabbs ER, Yazawa K, Mikami Y, Miyaji M, Morisaki N, et al. 1995. Ribosylation by mycobacterial
strains as a new mechanism of rifampin inactivation. Antimicrob. Agents Chemother. 39:1007–929. Daigle DM, Hughes DW, Wright GD. 1999. Prodigious substrate specificity of AAC(6)-APH(2 ), an
aminoglycoside antibiotic resistance determinant in enterococci and staphylococci. Chem. Biol. 6:99–110
30. Daigle DM, McKay GA, Thompson PR, Wright GD. 1999. Aminoglycoside antibiotic phosphotrans-ferases are also serine protein kinases. Chem. Biol. 6:11–18
31. Daigle DM, McKay GA, Wright GD. 1997. Inhibition of aminoglycoside antibiotic resistance enzymes
by protein kinase inhibitors. J. Biol. Chem. 272:24755–58
32. Deleted in proof.33. Davies J, Wright GD.1997. Bacterialresistance to aminoglycoside antibiotics. Trends Microbiol.5:234–40
34. Davies JF 2nd, Almassy RJ, Hostomska Z, Ferre RA, Hostomsky Z. 1994. 2.3 A crystal structure of the
catalytic domain of DNA polymerase beta. Cell 76:1123–33
35. Davis BD. 1987. Mechanism of action of aminoglycosides. Microbiol. Rev. 51:341–5036. D’Costa VM, McGrann KM, Hughes DW, Wright GD. 2006. Samplingthe antibiotic resistome. Science
311:374–7737. Draker KA, Northrop DB, Wright GD. 2003. Kinetic mechanism of the GCN5-related chromoso-
mal aminoglycoside acetyltransferase AAC(6)-Ii from Enterococcus faecium: evidence of dimer subunit
cooperativity. Biochemistry 42:6565–74
38. Draker KA, Wright GD. 2004. Molecular mechanism of the enterococcal aminoglycoside 6 -N-acetyltransferase’: role of GNAT-conserved residues in the chemistry of antibiotic inactivation.
Biochemistry 43:446–5439. Dreusicke D, Karplus PA, Schulz GE. 1988. Refined structure of porcine cytosolic adenylate kinase at
2.1 A resolution. J. Mol. Biol. 199:359–7140. Dutta GN, Devriese LA. 1982. Resistance to macrolide, lincosamide and streptogramin antibiotics and
degradation of lincosamide antibiotics in streptococci from bovine mastitis. J. Antimicrob. Chemother.
10:403–8
41. Etienne J, Gerbaud G, Courvalin P, Fleurette J. 1989. Plasmid-mediated resistance to fosfomycin in
Staphylococcus epidermidis. FEMS Microbiol. Lett. 52:133–37
42. Fillgrove KL, Pakhomova S, Newcomer ME, Armstrong RN. 2003. Mechanistic diversity of fosfomycin
resistance in pathogenic microorganisms. J. Am. Chem. Soc. 125:15730–31
43. Fillgrove KL, Pakhomova S, Newcomer ME, Armstrong RN. 2003. Mechanistic diversity of fosfomycin
resistance in pathogenic microorganisms. J. Am. Chem. Soc. 125:15730–31
44. Fillgrove KL, Pakhomova S, Schaab MR, Newcomer ME, Armstrong RN. 2007. Structure and mech-anism of the genomically encoded fosfomycin resistance protein, FosX, from Listeria monocytogenes.
Biochemistry 46:8110–20
45. Fischbach MA, Walsh CT. 2009. Antibiotics for emerging pathogens. Science 325:1089–9346. Fong DH, Berghuis AM. 2002. Substrate promiscuity of an aminoglycoside antibiotic resistance enzyme
via target mimicry. EMBO J. 21:2323–3147. Fong DH, Berghuis AM. 2009. Structural basis of APH(3)-IIIa-mediated resistance to N1-substituted
aminoglycoside antibiotics. Antimicrob. Agents Chemother. 53:3049–5548. Fong DH, Lemke CT, Hwang J, Xiong B, Berghuis AM. 2010. Structure of the antibiotic resistance
factor spectinomycin phosphotransferase from Legionella pneumophila. J. Biol. Chem. 285:9545–5549. Gates CA, Northrop DB. 1988. Alternative substrate and inhibition kinetics of aminoglycoside nu-
cleotidyltransferase 2-I in support of a Theorell-Chance kinetic mechanism. Biochemistry 27:3826–3350. Gerlt JA, Babbitt PC. 2001. Divergent evolution of enzymatic function: mechanistically diverse super-
families and functionally distinct suprafamilies. Annu. Rev. Biochem. 70:209–46
51. Gerratana B, Frey PA, Cleland WW. 2001. Characterization of the transition-state structure of thereaction of kanamycinnucleotidyltransferaseby heavy-atom kinetic isotope effects. Biochemistry 40:2972–
7752. Hall BG, Barlow M. 2003. Structure-based phylogenies of the serine beta-lactamases. J. Mol. Evol.
57:255–6053. Hall BG, Barlow M. 2004. Evolution of the serine beta-lactamases: past, present and future. Drug Resist.
Updates 7:111–23
46 Morar ·Wright

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 23/27

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 24/27
GE44CH02-Wright ARI 17 July 2010 13:29
79. Krell T, Coggins JR, Lapthorn AJ. 1998. The three-dimensional structure of shikimate kinase. J. Mol.
Biol. 278:983–97
80. Ku SY, Yip P, Cornell KA, Riscoe MK, Behr JB, et al. 2007. Structures of 5-methylthioribose kinase
reveal substrate specificity and unusual mode of nucleotide binding. J. Biol. Chem. 282:22195–206
81. Kuo MS, Chirby DG,Argoudelis AD,Cialdella JI, CoatsJH, Marshall VP. 1989. Microbialglycosylation
of erythromycin A. Antimicrob. Agents Chemother. 33:2089–91
82. Leclercq R, Carlier C, Duval J, Courvalin P. 1985. Plasmid-mediated resistance to lincomycin by inac-
tivation in Staphylococcus haemolyticus . Antimicrob. Agents Chemother. 28:421–24
83. Leslie AG. 1990. Refined crystal structure of type III chloramphenicol acetyltransferase at 1.75 A reso-
lution. J. Mol. Biol. 213:167–86
84. Leslie AG, Moody PC, Shaw WV. 1988. Structure of chloramphenicol acetyltransferase at 1.75-A res-
olution. Proc. Natl. Acad. Sci. USA 85:4133–37
85. Lewendon A, Murray IA, Kleanthous C, Cullis PM, Shaw WV. 1988. Substitutions in the active site of
chloramphenicol acetyltransferase: role of a conserved aspartate. Biochemistry 27:7385–90
86. Li M, Dyda F, Benhar I, Pastan I, Davies DR. 1996. Crystal structure of the catalytic domain of Pseu-
domonas exotoxin A complexed with a nicotinamide adenine dinucleotide analog: implications for the
activation process and for ADP ribosylation. Proc. Natl. Acad. Sci. USA 93:6902–6
87. Lipka M, Filipek R, Bochtler M. 2008. Crystal structure and mechanism of the Staphylococcus cohniivirginiamycin B lyase (Vgb). Biochemistry 47:4257–65
88. Magnet S, Blanchard JS. 2005. Molecular insights into aminoglycoside action and resistance. Chem. Rev.
105:477–98
89. Magnet S, Lambert T, Courvalin P, Blanchard JS. 2001. Kinetic and mutagenic characterization of the
chromosomally encoded Salmonella enterica AAC(6)-Iy aminoglycoside N-acetyltransferase. Biochem-
istry 40:3700–9
90. Malito E, Sekulic N, Too WC, Konrad M, Lavie A. 2006. Elucidation of human choline kinase crystal
structures in complex with the products ADP or phosphocholine. J. Mol. Biol. 364:136–51
91. Martinez JL, Sanchez MB, Martınez-Solano L, Hernandez A, Garmendia L, et al. 2009. Functional role
of bacterial multidrug efflux pumps in microbial natural ecosystems. FEMS Microbiol. Rev. 33:430–49
92. Massova I, Mobashery S. 1998. Kinship and diversification of bacterial penicillin-binding proteins and
beta-lactamases. Antimicrob. Agents Chemother. 42:1–17
93. Mattevi A, Obmolova G, Kalk KH, Teplyakov A, Hol WG. 1993. Crystallographic analysis of substrate
binding and catalysis in dihydrolipoyl transacetylase (E2p). Biochemistry 32:3887–901
94. Maurice F, Broutin I, Podglajen I, Benas P, Collatz E, Dardel F. 2008. Enzyme structural plasticity and
the emergence of broad-spectrum antibiotic resistance. EMBO Rep. 9:344–49
95. McCarthy AA, Baker HM, Shewry SC, Patchett ML, Baker EN. 2001. Crystal structure of
methylmalonyl-coenzyme A epimerase from P. shermanii: a novel enzymatic function on an ancient
metal binding scaffold. Structure 9:637–46
96. McKay GA, Wright GD. 1995. Kinetic mechanism of aminoglycoside phosphotransferase type IIIa.
Evidence for a Theorell-Chance mechanism. J. Biol. Chem. 270:24686–92
97. McKay GA, Wright GD. 1996. Catalytic mechanism of enterococcal kanamycin kinase (APH(3)-IIIa):
viscosity, thio, and solvent isotope effects support a Theorell-Chance mechanism. Biochemistry 35:8680–
85
98. Morar M, Bhullar K, Hughes DW, Junop M, Wright GD. 2009. Structure and mechanism of the
lincosamide antibiotic adenylyltransferase LinB. Structure 17:1649–59
99. Mukhtar TA, Koteva KP, Hughes DW, Wright GD. 2001. Vgb from Staphylococcus aureus inactivatesstreptogramin B antibiotics by an elimination mechanism not hydrolysis. Biochemistry 40:8877–86
100. Mukhtar TA,WrightGD. 2005. Streptogramins, oxazolidinones, andother inhibitors ofbacterialprotein
synthesis. Chem. Rev. 105:529–42
101. Mulichak AM, Losey HC, Walsh CT, Garavito RM. 2001. Structure of the UDP-glucosyltransferase
GtfB that modifies the heptapeptide aglycone in the biosynthesis of vancomycin group antibiotics.
Structure 9:547–57
48 Morar ·Wright

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 25/27
GE44CH02-Wright ARI 17 July 2010 13:29
102. Murray IA, Cann PA, Day PJ, Derrick JP, Sutcliffe MJ, et al. 1995. Steroid recognition by chloram-
phenicol acetyltransferase: engineering and structural analysis of a high affinity fusidic acid binding site.
J. Mol. Biol. 254:993–1005
103. Murray IA, Hawkins AR, Keyte JW, Shaw WV. 1988. Nucleotide sequence analysis and overexpression
of the gene encoding a type III chloramphenicol acetyltransferase. Biochem. J. 252:173–79
104. Murray IA, Shaw WV. 1997. O-Acetyltransferases for chloramphenicol and other natural products.
Antimicrob. Agents Chemother. 41:1–6
105. Neuwald AF, Landsman D. 1997. GCN5-related histone N-acetyltransferases belong to a diverse su-
perfamily that includes the yeast SPT10 protein. Trends Biochem. Sci. 22:154–55
106. Nurizzo D, Shewry SC, Perlin MH, Brown SA, Dholakia JN, et al. 2003. The crystal structure of
aminoglycoside-3-phosphotransferase-IIa, an enzyme responsible for antibiotic resistance. J. Mol. Biol.
327:491–506
107. Okamoto S, Suzuki Y. 1965. Chloramphenicol-, dihydrostreptomycin-, and kanamycin-inactivating en-
zymes from multiple drug-resistant Escherichia coli carrying episome ‘R’. Nature 208:1301–3
108. PayneDJ,GwynnMN,HolmesDJ,PomplianoDL.2007.Drugsforbadbugs:confrontingthechallenges
of antibacterial discovery. Nat. Rev. Drug Discov. 6:29–40
109. Pedersen LC, Benning MM, Holden HM. 1995. Structural investigation of the antibiotic and ATP-
binding sites in kanamycin nucleotidyltransferase. Biochemsitry 34:13305–11
110. Peisach D, Gee P, Kent C, Xu Z. 2003. The crystal structure of choline kinase reveals a eukaryotic
protein kinase fold. Structure 11:703–13
111. PetinakiE, Guerin-FaubleeV, Pichereau V, Villers C, AchardA, et al.2008.Lincomycinresistance gene
lnu(D) in Streptococcus uberis. Antimicrob. Agents Chemother. 52:626–30
112. Phuc Nguyen MC, Woerther PL, Bouvet M, Andremont A, Leclercq R, Canu A. 2009. Escherichia coli
as reservoir for macrolide resistance genes. Emerg. Infect. Dis. 15:1648–50
113. Quiros LM, Aguirrezabalaga I, Olano C, Mendez C, Salas JA. 1998. Two glycosyltransferases and a
glycosidase are involvedin oleandomycin modification during its biosynthesisby Streptomyces antibioticus .
Mol. Microbiol. 28:1177–85
114. Quiros LM, Carbajo RJ, Brana AF, Salas JA. 2000. Glycosylation of macrolide antibiotics. Purifica-
tion and kinetic studies of a macrolide glycosyltransferase from Streptomyces antibioticus . J. Biol. Chem.
275:11713–20
115. Quiros LM, Carbajo RJ, Salas JA. 2000. Inversion of the anomeric configuration of the transferred sugar
during inactivation of the macrolide antibiotic oleandomycin catalyzed by a macrolide glycosyltrans-ferase. FEBS Lett. 476:186–89
116. Quiros LM,SalasJA. 1995. Biosynthesisof themacrolide oleandomycin by Streptomyces antibioticus. Pu-
rification and kinetic characterization of an oleandomycin glucosyltransferase. J. Biol. Chem. 270:18234–
39
117. Radika K, Northrop DB. 1984. Substrate specificities and structure-activity relationships for acylation
of antibiotics catalyzed by kanamycin acetyltransferase. Biochemistry 23:5118–22
118. Radika K, Northrop DB. 1984. The kinetic mechanism of kanamycin acetyltransferase derived from the
use of alternative antibiotics and coenzymes. J. Biol. Chem. 259:12543–46
119. Raetz CR, Roderick SL. 1995. A left-handed parallel beta helix in the structure of UDP-N-
acetylglucosamine acyltransferase. Science 270:997–1000
120. Rife CL, Pharris RE, Newcomer ME, Armstrong RN. 2002. Crystal structure of a genomically encoded
fosfomycin resistance protein (FosA) at 1.19 A resolution by MAD phasing off the L-III edge of Tl(+).
J. Am. Chem. Soc. 124:11001–3
121. Roberts MC, Sutcliffe J, Courvalin P, Jensen LB, Rood J, Seppala H. 1999. Nomenclature for macrolideand macrolide-lincosamide-streptogramin B resistance determinants. Antimicrob. Agents Chemother.
43:2823–30
122. Robicsek A, Strahilevitz J, Jacoby GA, Macielag M, Abbanat D, et al. 2006. Fluoroquinolone-modifying
enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat. Med. 12:83–88
123. Sakon J, Liao HH, Kanikula AM, Benning MM, Rayment I, Holden HM. 1993. Molecular structure of
kanamycin nucleotidyltransferase determined to 3.0-A resolution. Biochemistry 32:11977–84
www.annualreviews.org • Enzymology of Antibiotic Resistance 49

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 26/27

8/8/2019 The Genomic Enzymology of Antibiotic Resistance
http://slidepdf.com/reader/full/the-genomic-enzymology-of-antibiotic-resistance 27/27
GE44CH02-Wright ARI 17 July 2010 13:29
149. Wright GD. 2007. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat. Rev.
Microbiol. 5:175–86
150. Wright GD, Ladak P. 1997. Overexpression and characterization of the chromosomal aminoglycoside
6-N-acetyltransferase from Enterococcus faecium. Antimicrob. Agents Chemother. 41:956–60
151. Wright GD, Thompson PR. 1999. Aminoglycoside phosphotransferases: proteins, structure, and mech-
anism. Front. Biosci. 4:D9–21
152. Wu D, Govindasamy L, Lian W, Gu Y, Kukar T, et al. 2003. Structure of human carnitine acetyltrans-
ferase. Molecular basis for fatty acyl transfer. J. Biol. Chem. 278:13159–65
153. Wybenga-Groot LE, Draker K, Wright GD, Berghuis AM. 1999. Crystal structure of an aminoglyco-
side 6-N-acetyltransferase: defining the GCN5-related N-acetyltransferase superfamily fold. Structure
7:497–507
154. Yates SP, Jørgensen R, Andersen GR, Merrill AR. 2006. Stealth and mimicry by deadly bacterial toxins.
Trends. Biochem. Sci. 31:123–33
155. Yim G, Wang HH, Davies J. 2007. Antibiotics as signaling molecules. Philos. Trans. R. Soc. Lond. Ser. B
362:1195–200
156. YoungPG, WalanjR, Lakshmi V,Byrnes LJ,MetcalfP, etal. 2009.The crystal structures ofsubstrate and
nucleotide complexes of Enterococcus faecium aminoglycoside-2-phosphotransferase-IIa [APH(2)-IIa]
provide insights into substrate selectivity in the APH(2) subfamily. J. Bacteriol. 191:4133–43
157. Zheng J, Knighton DR, ten Eyck LF, Karlsson R, Xuong N, et al. 1993. Crystal structure of the catalytic
subunit of cAMP-dependent protein kinase complexed with MgATP and peptide inhibitor. Biochemistry
32:2154–61
www.annualreviews.org • Enzymology of Antibiotic Resistance 51