the key role of the lectin pathway enzyme masp-3 in the ... · ii abstract the key role of the...
TRANSCRIPT

The key role of the Lectin Pathway enzyme
MASP-3 in the innate immune protection against
Neisseria meningitidis
Thesis submitted for the degree of
Doctor of Philosophy
at the University of Leicester
By
Saleh Alshamrani
Department of Infection, Immunity and Inflammation
University of Leicester
2016

I
Statement of originality
This accompanying thesis submitted for the degree of PHD entitled (The
key role of the Lectin Pathway enzyme MASP-3 in the innate immune
protection against Neisseria meningitidis) is based on work conducted by
the author at the University of Leicester mainly during the period between
January 2012 and January 2015
All the work recorded in this thesis is original unless otherwise
acknowledged in the text or by references.
None of the work has been submitted for another degree in this or any other
University
Signed: Date:

II
Abstract
The key role of the Lectin Pathway enzyme MASP-3 in the innate
immune protection against Neisseria meningitidis
Neisseria meningitidis infections pose a worldwide threat to human health being a
major cause of morbidity and mortality. The bacterium can often be found to live as a
commensal organism in the upper respiratory-tract. However, under disease promoting
circumstances it may cause invasive infections such as bacterial meningitis with a
mortality rate of up to 10% in patients with sepsis. The complement system plays a vital
role in immune protection from Neisseria meningitidis infections and ongoing research
in our laboratories has recently observed that the serum of mice deficient in the lectin
pathway of complement effector enzyme MASP-2 had a higher bactericidal activity
towards Neisseria meningitidis as compared to MASP-2 sufficient serum. This work
also revealed a key role of the lectin pathway components MBL and MASP-3 in driving
serum bacteriolytic activity against Neisseria meningitidis and has identified a novel
link between MASP-3 and the alternative pathway of complement activation. The work
described in this thesis highlights the critical role that MASP-3 plays in the innate
immune response to this pathogen using in vitro models of serum bactericidal activity
and in vivo mouse models of Neisseria meningitidis infection. The failure of MASP-3
deficient non immune serum to lyse Neisseria meningitidis serotype A and serotype B
was restored by adding a recombinant enzymatically active MASP-3 fragment to this
serum while the therapeutic systemic injection of recombinant murine MASP-3
zymogen convincingly restored the defective alternative pathway functional activity and
with that repaired the high susceptibility of MASP-1/3 deficient mice to Neisseria
meningitidis infections. In line with the essential role that the alternative pathway plays
in driving the innate immune response against Neisseria meningitidis, the early results
of my study showed the therapeutic utility of enhancing the alternative pathway
functional activity through the addition of recombinant murine properdin to WT mice
sera and significantly increased the lytic activity against Neisseria meningitidis.

III
Acknowledgement
My greatest thanks go to my creator for his blessing and help throughout my life.
All glory and praise is due to Allah
I would like to express my deepest gratitude to my supervisor, Professor Wilhelm
Schwaeble, who has guided me during this project. His encouragement and support
have had a deep impact on my finishing this project successfully. He has always been
very patient with me and never hesitated to answer all my queries.
I would like to give my sincere thanks to my co supervisor Professor Peter Andrew for
his scientific help and advice. Many thanks go out to Dr Mohammad Y. Ali and Dr
Nicholas Lynch for their expert advice during this journey.
Next, it is a pleasure to offer particular thanks to Dr Sarah Glen for her continuous help
and advice during training in the Neisseria lab. Also, I am very thankful to all the staff
and members from lab 231; I was lucky enough to have great friends there and I have
enjoyed working with you.
Finally, I would like to express my deepest gratitude for the constant support,
remarkable patience, encouragement and love I received from my wife, Sawsan, and my
son, Abdulmalik. No words would be enough to thank my parents and siblings for
everything they have given and did for me throughout my life and especially during this
period.

IV
List of contents
Table of contents
CHAPTER 1: INTRODUCTION .................................................................................. 1
1.1 THE IMMUNE SYSTEM ............................................................................................ 1
1.2 THE COMPLEMENT SYSTEM ................................................................................... 3
1.2.1 Classical Pathway ........................................................................................................... 5
1.2.2 Alternative Pathway ...................................................................................................... 6
1.2.2.1 Complement components of alternative pathway ................................. 8
1.2.2.1.1 Factor B ............................................................................................. 8
1.2.2.1.2 Factor D ............................................................................................ 9
1.2.2.1.3 Properdin ........................................................................................... 9
1.2.3 Lectin Pathway .............................................................................................................. 10
1.2.3.1 Lectin pathway components ............................................................... 11
1.2.3.1.1 Mannan Binding Lectin .................................................................. 11
1.2.3.1.2 Ficolin ............................................................................................. 12
1.2.3.1.3 Collectin .......................................................................................... 13
1.2.3.1.4 Mannan Binding Lectin associated serine proteases ...................... 14
1.2.4 Membrane Attack Complex ...................................................................................... 17
1.2.5 Functions of the Complement System ................................................................... 18
1.2.6 Regulators of Complement System ......................................................................... 20
1.2.6.1 Fluid Phase Regulators ....................................................................... 20
1.2.6.2 Membrane-Bound Regulators: ............................................................ 21

V
1.2.7 Complement deficiency .............................................................................................. 24
1.2.7.1 Defects of the Classical Pathway ........................................................ 24
1.2.7.2 Defects of the Alternative Pathway .................................................... 25
1.2.7.3 Defects of the Lectin Pathway ............................................................ 25
1.2.7.4 Defects of terminal complement components .................................... 26
1.2.7.5 Defects of complement regulatory components ................................. 26
1.3 NEISSERIA MENINGITIDIS ....................................................................................... 28
1.3.1 The Virulence Factors of Neisseria meningitidis ................................................ 30
1.3.1.1 Pili and Pilus Subunits ........................................................................ 30
1.3.1.2 Outer Membrane Proteins ................................................................... 31
1.3.1.3 Capsule and Lipo-oligosaccharide (LOS) ........................................... 32
1.3.2 Colonization and invasion by Neisseria meningitidis ....................................... 35
1.3.3 The Immune System and Neisseria meningitidis ................................................ 37
1.3.4 The Complement System and Neisseria meningitidis ....................................... 38
1.4 THESIS AIMS ........................................................................................................ 44
CHAPTER 2: MATERIALS AND METHODS ........................................................ 45
2.1 MATERIALS .......................................................................................................... 45
2.1.1 Chemicals and materials............................................................................................. 45
2.1.2 Antibodies/proteins ...................................................................................................... 47
2.1.3 Media and buffers ........................................................................................................ 49
2.2 METHODS ............................................................................................................ 51
2.2.1 In vitro experiments ..................................................................................................... 51
2.2.1.1 Preparation of mouse and human serum ............................................. 51

VI
2.2.1.2 Preparation of Neisseria meningitidis for Enzyme Linked
Immunosorbent Assay (ELISA) ..................................................................... 51
2.2.1.3 C1q deposition assays ......................................................................... 52
2.2.1.4 MBL-A, MBL-C, Ficolin-A and CL-11 binding assays ..................... 53
2.2.1.5 C3 deposition assays ........................................................................... 54
2.2.1.6 Alternative pathway mediated C3 deposition assays .......................... 55
2.2.1.7 Lectin pathway mediated C4 deposition assays ................................. 56
2.2.1.8 Serum Bactericidal Assay (SBA) ....................................................... 57
2.2.1.9 Preparation of Neisseria meningitidis for FACS analysis .................. 57
2.2.1.10 FACS analysis for detect C3 deposition on Neisseria meningitdis .... 58
2.2.2 In vivo experiments ..................................................................................................... 59
2.2.2.1 Genotyping of fB and MASP-1/3 deficient mice ............................... 59
2.2.2.1.1 Isolation of genomic DNA from mouse ear snips .......................... 59
2.2.2.1.2 Polymerase Chain Reaction (PCR) ................................................. 59
2.2.2.2 Preparation of Neisseria meningitidis passage ................................... 61
2.2.2.3 Virulence testing of passaged stocks of Neisseria meningitidis ......... 62
2.2.2.4 Infection of mice with Neisseria meningitidis .................................... 63
2.2.2.5 Determination of Blood bacterial burden ........................................... 63
2.2.2.6 MASP-3 reconstitution experiment .................................................... 64
2.2.3 Statistical analysis ........................................................................................................ 65
CHAPTER 3: IN VITRO STUDY ............................................................................... 66
3.1 RESULTS: ............................................................................................................. 66
3.1.1 Complement pathway specific Enzyme Linked Immune Sorbent Assays
(ELISAs) ....................................................................................................................................... 66

VII
3.1.1.1 Binding of the classical pathway molecule C1q to Neisseria
meningitidis ......................................................................................................... 67
3.1.1.2 Binding of the lectin pathway recognition component MBL to
Neisseria meningitidis ......................................................................................... 68
3.1.1.3 Binding of the lectin pathway Collectin-11 to Neisseria meningitidis 69
3.1.1.4 Binding of the lectin pathway ficolin-A to Neisseria meningitidis .... 70
3.1.2 C3 deposition assays ................................................................................................... 71
3.1.3 C4 deposition assays ................................................................................................... 78
3.1.4 Serum Bactericidal Assays ........................................................................................ 82
3.1.5 Effect of recombinant properdin on complement mediated killing of
Neisseria meningitidis ............................................................................................................... 97
3.1.5.1 Recombinant properdin has the ability to enhance C3 deposition on
the surface of Neisseria meningitidis .................................................................. 97
3.1.5.2 Recombinant properdin has the ability to enhance the killing of
Neisseria meningitidis ....................................................................................... 100
3.2 DISCUSSION ....................................................................................................... 107
3.2.1 Binding of Neisseria meningitidis to different complement recognition
molecules .................................................................................................................................... 108
3.2.2 Activation of the complement system on the surface of Neisseria
meningitidis requires a close cooperation between the lectin and alternative
pathway ....................................................................................................................................... 111
3.2.3 Recombinant properdin enhances the serum bacteriolytic activity against
Neisseria meningitidis ............................................................................................................. 118

VIII
CHAPTER 4: IN VIVO STUDY ................................................................................ 121
4.1 RESULTS: ........................................................................................................... 121
4.1.1 Genotyping of factor B deficient mice and MASP-1/3 deficient mice ....... 121
4.1.2 The optimal infective dose ...................................................................................... 125
4.1.3 Survival of factor B deficient mice and factor B sufficient mice following
experimental Neisseria meningitidis infection ................................................................. 126
4.1.3.1 The viable bacterial load of Neisseria meningitidis in the blood of
infected mice ................................................................................................... 128
4.1.4 Survival of MASP-1/3 deficient mice and MASP-1/3 sufficient mice
following experimental Neisseria meningitidis infection ............................................. 130
4.1.4.1 The viable bacterial load of Neisseria meningitidis in the blood of
infected mice ..................................................................................................... 132
4.1.5 Effect of full length recombinant MASP-3 on reconstituting the absence of
the alternative pathway functional activity in MASP-1/3 deficient mice ................ 134
4.1.6 Effect of full length recombinant MASP-3 administration on mortality in a
mouse model of Neisseria meningitidis infection ........................................................... 143
4.1.6.1 The viable bacterial load of Neisseria meningitidis in the blood of
infected mice ..................................................................................................... 147
4.2 DISCUSSION ....................................................................................................... 149
4.2.1 Mice deficient in the alternative pathway functional activity show
dramatically higher susceptibility to Neisseria meningitidis infection ..................... 149
4.2.2 The therapeutic application of recombinant full-length MASP-3
dramatically improves the survival of MASP-1/3 deficient mice from Neisseria
meningitidis infection .............................................................................................................. 153

IX
CHAPTER 5: CONCLUSION AND FUTURE WORK ......................................... 158
5.1 CONCLUSION ..................................................................................................... 158
5.1.1 Binding of complement system recognition molecules to Neisseria
meningitidis ................................................................................................................................ 160
5.1.2 Activation of the complement system on the surface of Neisseria
meningitidis requires a close cooperation between the lectin and alternative
pathways ...................................................................................................................................... 162
5.1.3 Recombinant properdin enhances the serum bacteriolytic activity against
Neisseria meningitidis ............................................................................................................. 166
5.1.4 Mice deficient in the alternative pathway functional activity show
dramatically higher susceptibility to Neisseria meningitidis infection ..................... 167
5.1.5 The therapeutic application of recombinant full-length MASP-3
dramatically improves the survival of MASP-1/3 deficient mice from Neisseria
meningitidis infection .............................................................................................................. 169
5.2 FUTURE WORK ................................................................................................... 172
5.2.1 Assess the therapeutic benefit of recombinant MASP-3 in fighting other
microbial infection ................................................................................................................... 172
5.2.2 Assess the ability of recombinant properdin in restoring the killing of
properdin-deficient sera .......................................................................................................... 172
CHAPTER 6: BIBLIOGRAPHY .............................................................................. 173

X
List of tables
Table 2.1 The severity scores of disease with clinical signs of infected mice ............... 63
Table 3.1 Statistically significant differences between serum bactericidal assay of
different mouse sera (C1q-/-
, WT and HIS) against Neisseria meningitidis serogroup A
strain Z2491 ................................................................................................................... 83
Table 3.2 Statistically significant differences between serum bactericidal assay of
different mouse sera (C1q-/-
, WT and HIS) against Neisseria meningitidis serogroup B
strain MC58 . .................................................................................................................. 84
Table 3.3 Statistically significant differences between the serum bactericidal assay of
different mouse sera (FB-/-
, MASP-1/3-/-
, WT and HIS) against Neisseria meningitidis
serogroup A strain Z2491 . ............................................................................................. 86
Table 3.4 Statistically significant differences between the serum bactericidal assay of
different mouse sera (FB-/-, MASP-1/3-/-, WT and HIS) against Neisseria meningitidis
serogroup B strain MC58 ................................................................................................ 87
Table 3.5 Statistically significant differences between serum bactericidal assay of
different mouse sera (MASP-2-/-
, MBL-/-
, WT and HIS) against Neisseria meningitidis
serogroup A strain Z2491 ............................................................................................... 89
Table 3.6 Statistically significant differences between serum bactericidal assay of
different mouse sera (MASP-2-/-, MBL-/-
, WT and HIS) against Neisseria meningitidis
serogroup B strain MC58. ............................................................................................... 90
Table 3.7 Statistically significant differences between serum bactericidal assay of
different mouse sera (MASP-1/3-/-
+ truncated rMASP-3 (10µg/ml), MASP-1/3-/-
, WT
and HIS) against Neisseria meningitidis serogroup A strain Z2491. ............................. 91

XI
Table 3.8 Statistically significant differences between serum bactericidal assay of
different mouse serum (MASP-1/3-/-
+ truncated rMASP-3 (10µg/ml), MASP-1/3-/-
, WT
and HIS) against Neisseria meningitidis serogroup B strain MC58 ............................... 93
Table 3.9 Statistically significant differences between serum bactericidal assay of
different human sera (3MC + truncated rMASP-3 (6µg/ml), 3MC, MBL-/-
, Immune,
NHS and HIS) against Neisseria meningitidis serogroup A strain Z2491 ..................... 95
Table 3.10 Statistically significant differences between serum bactericidal assay of
different human sera (3MC + truncated rMASP-3 (6µg/ml), 3MC, MBL-/-
, Immune,
NHS and HIS) against Neisseria meningitidis serogroup B strain MC58 ...................... 96
Table 3.11 Statistically significant differences between serum bactericidal assay of
mouse serum with or without recombinant murine properdin against Neisseria
meningitidis serogroup A strain Z2491 ........................................................................ 101
Table 3.12 Statistically significant differences between serum bactericidal assay of
mouse serum with or without recombinant murine properdin against Neisseria
meningitidis serogroup B strain MC58 ......................................................................... 102
Table 3.13 Statistically significant differences between serum bactericidal assay of
human serum with or without recombinant human properdin against Neisseria
meningitidis serogroup A strain Z2491 ........................................................................ 104
Table 3.14 Statistically significant differences between serum bactericidal assay of
human serum with or without recombinant human properdin against Neisseria
meningitidis serogroup B strain MC58 ......................................................................... 106
Table 4.1 The design of the MASP-3 reconstitution experiment .............................. 135
Table 4.2 Statistically significant differences for the C3 deposition assay of different
mouse sera (MASP-1/3-/-
+ full-length rMASP-3 (40µg/mouse), MASP-1/3-/-
+ full
length rMASP-3 (20µg/mouse), MASP-1/3-/-, WT and HIS) ...................................... 137

XII
Table 4.3 Statistically significant difference assessed between serum bactericidal
assay of different mouse serum (MASP-1/3-/-
+ full length rMASP-3 (treated with two
doses of 20µg/mouse each given 96 and 24 hours), MASP-1/3-/-
+ full length rMASP-3
(20µg/mouse given 96 hours prior to bleeding), MASP-1/3-/-
(untreated), WT and HIS)
against Neisseria meningitidis serogroup A strain Z2491 ............................................ 140
Table 4.4 Statistically significant difference assessed between serum bactericidal
assay of different mouse serum (MASP-1/3-/-
+ full length rMASP-3 (treated with two
doses of 20µg/mouse each given 96 and 24 hours), MASP-1/3-/-
+ full length rMASP-3
(20µg/mouse given 96 hours prior to bleeding), MASP-1/3-/-
(untreated), WT and HIS)
against Neisseria meningitidis serogroup B strain MC58 ............................................ 142

XIII
List of figures
Figure 1.1 Complement system activation pathways. ..................................................... 4
Figure 1.2 Structure of the C1 Complex of the classical pathway. .................................. 5
Figure 1.3 The domain structure and oligomerization of MBL. .................................. 11
Figure 1.4 The domain structure and oligomerization of ficolins ................................ 13
Figure 1.5 MASP and Map domain organization ......................................................... 15
Figure 1.6 MASPs activation. ....................................................................................... 16
Figure 1.7 Membrane attack complex pathway ........................................................... 17
Figure 1.8 Regulation of complement activation. ......................................................... 23
Figure 1.9 Neisseria meningitidis cell membrane ........................................................ 30
Figure 1.10 Different stages in the pathogenesis of N. meningitidis. .......................... 36
Figure 3.1 C1q binding on the surface of different Neisseria meningitidis strains. .... 67
Figure 3.2 MBL-A binding on the surface of different Neisseria meningitidis
strains. ............................................................................................................................. 68
Figure 3.3 MBL-C binding on the surface of different Neisseria meningitidis strains 69
Figure 3.4 CL-11 binding to the surface of different Neisseria meningitidis strains.. . 70
Figure 3.6 C3 deposition assay on the surface of different Neisseria meningitidis
strains under specific condition (high serum dilution in BBS buffer) allows the
activation through classical and lectin pathways ............................................................ 72
Figure 3.7 C3 deposition assay on the surface of different Neisseria meningitidis
strains under alternative pathway permissive conditions (high serum concentration in
EGTA buffer) .................................................................................................................. 73

XIV
Figure 3.8 C3 deposition assay on the surface of different Neisseria meningitidis
strains under specific condition (high serum dilution in BBS buffer) using MASP-2+/+
and MASP-2-/-
serum ...................................................................................................... 74
Figure 3.9 C3 deposition assay on the surface of Neisseria meningitidis strain A
serogroup Z2491 under alternative pathway permissive condition (high serum
concentration in BBS buffer) using sera of MASP-2+/+
and MASP-2-/-
mice................. 75
Figure 3.10 C3 deposition a assay on the surface of Neisseria meningitidis strain B
serogroup MC85 under alternative pathway permissive condition (high serum
concentration in BBS buffer) using sera of MASP-2+/+
and MASP-2-/-
mice................. 76
Figure 3.11 C3 deposition assay on the surface of Neisseria meningitidis strain A
serogroup Z2491 under alternative pathway permissive conditions .............................. 77
Figure 3.12 C3 deposition assay on the surface of Neisseria meningitidis strain B
serogroup MC58 under alternative pathway permissive conditions ............................... 78
Figure 3.13 C4 deposition assay on the surface of different Neisseria meningitidis
strains under lectin pathway specific condition (high serum dilution in MBL binding
buffer) ............................................................................................................................. 79
Figure 3.14 C4 deposition assay on the surface of Neisseria meningitidis strain A
serogroup Z2491 under specific condition (high serum dilution in BBS buffer) ........... 80
Figure 3.15 C4 deposition assay on the surface of Neisseria meningitidis strain B
serogroup MC58 under specific condition (high serum dilution in BBS buffer) ........... 81
Figure 3.16 Bactericidal activity of different mouse sera (C1q-/-
, WT and HIS)
against Neisseria meningitidis serogroup A strain Z2491. ............................................. 83
Figure 3.17 Bactericidal activity of different mouse sera (C1q-/- , WT and HIS)
towards Neisseria meningitidis serogroup B strain MC58.. ........................................... 84

XV
Figure 3.18 Bactericidal activity of different mouse sera (FB-/-
, MASP-1/3-/-
, WT and
HIS) against Neisseria meningitidis serogroup A strain Z2491. .................................... 85
Figure 3.19 Bactericidal activity of different mouse sera (FB-/-
, MASP-1/3-/-
, WT and
HIS) against Neisseria meningitidis serogroup B strain MC58.. .................................... 86
Figure 3.20 Bactericidal activity of different mouse sera (MASP-2-/-
, MBL-/-
, WT and
HIS) against Neisseria meningitidis serogroup A strain Z249.. ..................................... 88
Figure 3.21 Bactericidal activity of different mouse sera (MASP-2-/-
, MBL-/-
, WT and
HIS) against Neisseria meningitidis serogroup B strain MC58 ...................................... 89
Figure 3.22 Bactericidal activity of different mouse sera (MASP-1/3-/-
+ truncated
rMASP-3 (10µg/ml), MASP-1/3-/-
, WT and HIS) against Neisseria meningitidis
serogroup A strain Z2491. .............................................................................................. 91
Figure 3.23 Bactericidal activity of different mouse sera (MASP-1/3-/-
+ truncated
rMASP-3 (10µg/ml), MASP-1/3-/-
, WT and HIS) against Neisseria meningitidis
serogroup B strain MC58. ............................................................................................... 92
Figure 3.24 Bactericidal activity of different human sera (3MC + truncated rMASP-3
(6µg/ml), 3MC, MBL-/-, Immune, NHS and HIS) against Neisseria meningitidis
serogroup A strain Z2491 ............................................................................................... 94
Figure 3.25 Bactericidal activity of different human sera (3MC + truncated rMASP-3
(6µg/ml), 3MC, MBL-/-
, Immune, NHS and HIS) against Neisseria meningitidis
serogroup B strain MC58. ............................................................................................... 95
Figure 3.26 C3 deposition assay on the surface of Neisseria meningitidis strain A
serogroup Z2491 under alternative pathway permissive conditions (high serum
concentration in EGTA buffer) ....................................................................................... 98

XVI
Figure 3.27 C3 deposition assay on the surface of Neisseria meningitidis strain B
serogroup MC58 under alternative pathway permissive conditions (high serum
concentration in EGTA buffer). ...................................................................................... 99
Figure 3.28 FACS analysis of C3 deposition on the surface of Neisseria meningitidis
serogroup B strain MC58 using human and mouse serum with or without recombinant
properdin. ...................................................................................................................... 100
Figure 3.29 Bactericidal activity of mouse serum with or without recombinant murine
properdin against Neisseria meningitidis serogroup A strain Z2491.. ......................... 101
Figure 3.30 Bactericidal activity of mouse serum with or without recombinant murine
properdin against Neisseria meningitidis serogroup B strain MC58. ........................... 102
Figure 3.31 Bactericidal activity of human serum with or without recombinant human
properdin against Neisseria meningitidis serogroup A strain Z2491. .......................... 103
Figure 3.32 Bactericidal activity of human serum with or without recombinant human
properdin against Neisseria meningitidis serogroup B strain MC58 ............................ 105
Figure 3.33 Lectin pathway effector arms. ................................................................ 117
Figure 4.1 Genotyping results for the FB targeted mouse line showed the amplified
products ......................................................................................................................... 123
Figure 4.2 Genotyping results for MASP1/3 mice showed the amplified products . 124
Figure 4.3 Survival of C57BL/6 wild-type mice following an i.p. injection with
different doses of N.meningitidis serogroup B strain MC58. ....................................... 125
Figure 4.4 Survival of wild type (on C57/BL6 background) and Factor B deficient
mice (on C57/BL6 background) following an i.p. injection with a low dose (1×105) of
Neisseria meningitidis serogroup B strain MC58. ........................................................ 127

XVII
Figure 4.5 Average illness score of wild type and Factor B deficient mice following an
i.p. injection with a low dose (1×105) of Neisseria meningitidis serogroup B strain
MC58. ........................................................................................................................... 128
Figure 4.6 Bacterial load in the blood of Factor B deficient mice and wild-type mice
given an i.p. injection of a low dose (1x105
CFU/mouse) of Neisseria meningitidis
serogroup B strain MC58 .............................................................................................. 129
Figure 4.7 Survival of WT and MASP-1/3 deficient mice (both on C57/BL6
background) following an i.p. injection with a low dose (1×105) of Neisseria
meningitidis serogroup B strain MC58.. ....................................................................... 131
Figure 4.8 Average illness score of wild type and MASP-1/3 deficient mice following
an i.p. injection with a low dose (1×105) of Neisseria meningitidis serogroup
B strain MC58 ............................................................................................................... 132
Figure 4.9 Bacterial load in blood of MASP-1/3 deficient mice and wild-type mice
given an i.p. injection of a low dose (1x105 CFU/mouse) of Neisseria meningitidis
serogroup B strain MC58. ............................................................................................. 133
Figure 4.10 C3 deposition of MASP-1/3 deficient mice treated with full length
recombinant murine MASP-3. ...................................................................................... 136
Figure 4.11 Serum Bactericidal Activity of the different mouse sera (MASP-1/3-/-
+
full length rMASP-3 (treated with two doses of 20µg/mouse each given 96 and 24
hours), MASP-1/3-/-
+ full length rMASP-3 (20µg/mouse given 96 hours prior to
bleeding), MASP-1/3-/-
(untreated) and WT mouse blood and heat-inactivated WT
mouse blood, i.e. HIS) against Neisseria meningitidis serogroup A strain Z2491. ...... 139

XVIII
Figure 4.12 Serum Bactericidal Activity of the different mouse sera (MASP-1/3-/-
+
full length rMASP-3 (treated with two doses of 20µg/mouse each given 96 and 24
hours), MASP-1/3-/-
+ full length rMASP-3 (20µg/mouse given 96 hours prior to
bleeding), MASP-1/3-/-
(untreated) and WT mouse blood and heat-inactivated WT
mouse blood, i.e. HIS) against Neisseria meningitidis serogroup B strain MC58 ....... 141
Figure 4.13 Survival of wild type mice, MASP-3 reconstituted MASP-1/3 deficient
mice (receiving 2 doses of full length recombinant MASP-3 (20µg/mouse at time points
96 hours and time point 24 hours prior to infection) and non-treated MASP-1/3 deficient
mice (all on C57/BL6 background) were infected by i.p. injections with a low dose
(1×105) of Neisseria meningitidis serogroup B strain MC58. ...................................... 145
Figure 4.14 Average illness score for wild type, treated MASP-1/3 deficient mice and
non-treated MASP-1/3 deficient mice following an i.p. injection with a low dose
(1×105) of Neisseria meningitidis serogroup B strain MC58 ....................................... 146
Figure 4.15 Bacterial load in blood of wild-type mice, treated MASP-1/3 deficient
mice and non-treated MASP-1/3 deficient mice given an i.p. injection of a low dose
(1x105 CFU/mouse) of Neisseria meningitidis serogroup B strain MC58 .................. 148

I
Abbreviations
Α Alpha
Β Beta
AP Alkaline phosphate
AP Alternative pathway
BBS Barbital buffer saline
BHI Brain heart infusion
Bp Base pair
BSA Bovine serum albumin
CCP Complement control protein
CED Carbohydrate recognition domain
CF Catalytic fragment
CFS Cerebrospinal fluid
CNL Capsule null locus
CP Classical Pathway
CPS Capsule synthesis
CRD Carbohydrate recognition domain
CrgA Contact-regulated gene A
II
DIC Disseminated intravascular coagulation
DNA Deoxyribonucleic acid
dNTPs Deoxynucleotides
EDTA Ethylenediaminetetra acetic acid
EGF Epidermal growth factor
EGTA Ethylene glycol tetraacetic acid
ELISA Enzyme Linked Immune Sorbent Assay
ELISA Enzyme Linked Immunosorbent Assay
FACS Fluorescent activated cell sorter
FB Factor B
FBG Fibrinogen-like carbohydrate recognition domain
FCS Foetal calf serum
FCS Foetal calf serum
Fd Factor D
G Grams
gDNA Genomic deoxyribonucleic acid
GNA Genome derived neisserial antigen
HAE Hereditary angioedema
HIS Heated inactivated serum
III
Ig Immunoglobulin
Kb Kilobase
kDa Kilodalton
KDO 2-keto-3-deoxyoctulosonic acid
KO Knockout
LNnt Lacto-N-neotetraose
LOS Lipo-oligosaccharide
MASP MBL- associated serine proteases
MBL Mannan Binding Lectin
MIDS Metal ion dependant adhesion site
Min Minutes
NHS Normal human serum
NK Natural killer cell
NspA Niesserial surface protein A
OD Optical density
OMPs Outer membrane proteins
Opa Outer membrane protein A
Opc Outer membrane protein C
PAMPS Pathogen associated molecular patterns

IV
PBS Phosphate buffered saline
PCR Polymerase chain reaction
PEA Phosphoethanolamine groups
PorA Meningococcal porin A
PorB Meningococcal porin B
rMASP-3 Recombinant MASP-3
RT Room temperature
SBA Serum bactericidal assay
SCRs Short consensus repeats
Sec Seconds
Tfp Type IV pili
TSRs Thrombospondin structural homology repeats
vWA Van Willebrand factor A
WT Wild-type
µ Micro

Chapter 1: Introduction
1
Chapter 1: Introduction
1.1 The Immune System
Throughout our life we are exposed to various microbial organisms as well molecular
species such as toxins and allergic substances that may impact on the normal
physiological function and integrity of our body. This exposure leads to the activation
of different defence mechanisms that protect our body by eliminating or neutralizing the
effects of these molecules and invading organisms. Together, these defence mechanisms
compose the immune system, which consists of a large number of proteins, cells, tissues
and organs that have evolved to protect the human body (Chaplin, 2010). The immune
system comprises two main systems, namely the innate immune system and the
adaptive immune system. The innate immune system, which is phylogenetically older
than the latter, provides the first line of defence against invading organism and toxic
substances (Medzhitov, 2007). It is characterized by the absence of memory function
but has the ability to mount a rapid response against invading microbes, resulting in
isolation and destruction of invading organisms (Borghesi & Milcarek, 2007). This
system works at many levels of anatomical and physiological barriers, for example, the
skin and mucosal membranes, by the production of protective chemicals such as
stomach acidity and proteins that recognise structural proteins and other molecules on
the surface of microbes known as pathogen associated molecular patterns (PAMPS).
Following recognition, the organism is then able to eliminate the microbe by a process
known as phagocytosis utilising various cells such as macrophages (Goldsby et al.,
2003; Hoffman et al., 1999; Medzhitov, 2007). The adaptive immune system, referred

Chapter 1: Introduction
2
to as the antigen-specific immune response, is more complex compared to the innate
immune system. It starts with the binding of antigens to immune cells, followed by a
series of complex steps which lead to the destruction of the invading microbe. Unlike
innate immunity, adaptive immunity has memory cells, which are established after the
primary exposure to the microbe and provide a more rapid response on secondary and
further exposures (Reid 1983). The adaptive immune system is divided into two parts,
cell-mediated immunity and humoral immunity. Humoral immunity develops when B-
lymphocytes produce antibodies after exposure to antigens while cell-mediated
immunity involves activation of immune cells (such as natural killer cells (NK) and
antigen specific cytotoxic T-lymphocytes) and the release of cytokines (Holmskov, et
al., 2003; Medzhitov, 2007).
The complement system is also an important component of the immune system. Its
largely thought of as being a part of the innate immune system but it also helps to link
innate immunity with adaptive immunity by its interaction with antibodies (Ricklin &
Lambris, 2007).

Chapter 1: Introduction
3
1.2 The Complement System
The complement system is a vital part of the immune system and plays a crucial role in
fighting against invading microbes (Trouw & Daha, 2011). The complement system
was firstly described as heat labile components found in the serum or plasma which had
the ability to destroy bacteria by complementing the ability of antibodies. Paul Ehrlich
later coined this term (complement) for these components (Holmskov, et al., 2003;
Medzhitov, 2007; Fujita et al., 2004).
The complement system consists of more than 35 proteins present in both plasma and
on the surface of cells, which interact with each other to form a network to give the
body protection against microbes. Most components are present in their inactive
proenzyme form (i.e. zymogen), which will be converted and cleaved to their active
enzymatic form during a series of sequential steps of complement activation (Sarma &
Ward, 2011; Walport, 2001). The complement system can be activated via three
different pathways, named the classical pathway, the alternative pathway and the lectin
pathway (Figure 1.1). While the activation of the classical pathway depends on the
presence of antibodies and commences once the multi-molecular C1 complex binds to
an antigen-antibody complex, the other two pathways, the alternative and the lectin
pathway, are antibody-independent, representing the linkage between innate immunity
and adaptive immunity. The activation of the lectin pathway depends on the association
of the carbohydrate recognition molecules with the key enzyme of the lectin pathway, a
serine protease called Mannan binding lectin serine protease-2 (MASP-2). The
alternative pathway forms an amplification loop of activation that supports the functions
of the classical and lectin pathways by amplifying the activation of the most abundant
third complement component called C3. C3 activation, a key step during complement

Chapter 1: Introduction
4
activation, contributes to the formation of the bactericidal membrane attack complex.
Membrane attack complexes (MAC) are initiated once the fifth complement component
C5 is cleaved via a C5 convertase to C5a and C5b. C5b is the initial component of
forming the terminal C5b-9 complement complex that leads to bacterial lysis (Stover et
al., 1999; Köhl, 2001; Schneider et al., 2006). The complement system has many
functions, such as opsonisation, lysis of microbes and cells and antigen antibody
complex clearance and it is controlled by proteins found in the serum (fluid phase
regulators) or on the surface of cells (surface bound regulators) (Unsworth et al., 2011).
Figure 1.1 Complement system activation pathways (Figure courtesy of Professor W
Schwaeble, University of Leicester, UK).

Chapter 1: Introduction
5
1.2.1 Classical Pathway
The activation of the classical pathway begins when the multimolecular C1 complex
binds to immune complexes in the presence of calcium (Gál et al., 2009). The
multimolecular C1 complex consists of the recognition subcomponent C1q (composed
of hexamers of the heterotrimeric C1q chains C1q-A, C1q-B and C1q-C) and a
heterotetramer of the classical pathway specific serine proteases C1r and C1s,
C1s:C1r:C1r:C1s complex (Wallis et al., 2010). A single C1q molecule consists of six
similar subunits that bind to collagenous stalks, ending in globular head domains
(Figure 1.2). Three homologous genes (A, B, and C), which are located in one gene
locus, encode the three homologous polypeptide chains that form each subunit of the
C1q (Arlaud et al., 2002).
Figure 1.2 Structure of the C1 Complex of the classical pathway showing the homodimers of
C1r and C1s with the C1q molecule (Pflieger et al., 2010).

Chapter 1: Introduction
6
The activation of the classical pathway starts once C1q binds indirectly to the Fc region
of the immunoglobulins IgG and IgM or directly to the surface of bacteria. This binding
leads to an auto-activation of C1r which cleaves and activates C1s. C1s in turn cleaves
C4 into two portions: a small portion C4a, which dissociates in the fluid phase as an
anaphylatoxin, and a large portion, C4b, which is bound to C2 on the surface of the
microbe (Wallis et al. 2007). C2 then cleaves into a small particle C2b released into the
fluid phase and a large particle C2a by the action of C1s. The large particle C2a will
stay attached to C4b to form the classical pathway C3 convertase (C4b2a) which works
on the most abundant component in the plasma, C3, and converts it to C3a which is
released as an anaphylatoxin and C3b that opsonises the pathogen and helps in
phagocytosis. C3b will bind to C3 convertase, leading to the formation of C5 convertase
that cleaves C5 to C5a that is released as an anaphylatoxin and C5b that binds to the
surface of the pathogen and initiates the formation of the membrane attack complex
leading to lysis of the pathogen (Arlaud et al., 2002; Schwaeble et al., 2011; Vorup-
Jensen et al., 2000).
1.2.2 Alternative Pathway
Alternative pathway activation takes place when the C3, which is abundantly available
in blood, is spontaneously hydrolysed to form C3(H2O) that binds to Factor B (FB) in
the presence of Mg+2, leading to the creation of a zymogen complex C3(H2O)B. This
binding of C3(H2O) to FactorB (FB) allows complexed FB to be cleaved by factor D
(fD) into its two activation products: Ba and Bb. While the fragment Ba is released from

Chapter 1: Introduction
7
the complex, the other fragment Bb will stay attached to the complex, forming the
alternative pathway C3 convertase C3(H2O)Bb. This C3 convertase cleaves C3 into C3a
and C3b, which binds to the pathogen surface. Factor B will bind to the newly generated
C3b to form a new C3 convertase C3bBb after cleavage of factor B by Factor D
(Thurman & Holers, 2006). Properdin, which is a positive regulatory component of the
alternative pathway, promotes alternative pathway activation by stabilizing the C3
convertase C3bBb and prevent the decay of this convertase complex as an antagonist to
the regulatory plasma component factor H. The action of properdin can increase the
half-life of the AP C3 and C5 convertase complexes by 5-10 fold. Properdin directly
blocks the downregulatory activity of factor H, which binds to C3b bound in the C3bBb
or C3bB complexes and decays these complexes and also acts as a cofactor in the factor
I –mediated conversion of C3b to iC3b, the inactive form of C3b, which subsequently
inactivates the alternative pathway C3 convertase (Schwaeble and Reid, 1999).
Recently, it was postulated that properdin can bind like a recognition molecule to
pathogen surfaces to act as receptor for C3b and initiate the formation of C3bBb
complexes and target AP activation to the surface of pathogens (Hourcade, 2006).
Another way to activate the alternative pathway amplification loop would be by the
provision of C3b from either the classical pathway or the lectin pathway which after the
binding to factor B initiated the formation of the alternative pathway C3 convertase
complex resulting in a pronounced amplification of complement activation (Schwaeble
and Reid, 1999). Host cells protect themselves from overshooting complement
activation by expressing a large number of complement regulatory components, such as
complement receptors CR-1, CR-3 or membrane bound regulators, such as MCP or

Chapter 1: Introduction
8
DAF, or fluid-phase regulators like factor H that interacts with host membrane
structures and increases its affinity towards C3b by the binding of its carboxy-terminal
domains to autologous cell surfaces (Wallis et al., 2007).
As described before for the classical pathway, the alternative pathway C3 convertase
(C3bBb) switches its substrate specificity from cleaving C3 to cleaving C5 if several
molecules of C3b bind in close proximity to C3 convertase, forming the AP C5
convertase (C3bBb(C3b)n). This C5 convertase cleaves C5 into C5a and C5b which
initiates the terminal activation cascade leading to the formation of the membrane attack
complex (Farries et al., 1988).
1.2.2.1 Complement components of alternative pathway
1.2.2.1.1 Factor B
Alternative pathway factor B (FB) is a proenzyme composed of two fragments Ba and
Bb. Bb fragment comprises van Willebrand factor A (vWA) domain that binds C3b to
FB via a metal ion dependant adhesion site (MIDS) in the presence of magnesium
Mg+2 and C-terminal serine protease (SP) domain. Ba fragment comprises three N-
terminal complement control protein (CCP) domains linked to fragment Ba via a 45
residue long linker (Pryzdial and Isenman, 1987).

Chapter 1: Introduction
9
1.2.2.1.2 Factor D
Factor D (FD) is a small serine protease consisting of a single serine protease with a
plasma concentration of around 2µg/ml. Factor D is a critical component of the
alternative pathway and is produced in different tissues, mainly in adipose tissue
(Barnum et al., 1984; Stanton et al., 2011; Volanakis and Narayana, 1996). The main
form of factor D is mature factor D. However, the rest of factor D, about 1%, is
profactor D that is converted to mature factor D after its biosynthesis (Lesavre and
Muller-Eberhard, 1978; Yamauchi et al., 1994).
1.2.2.1.3 Properdin
Properdin is a glycoprotein known as a positive regulator of complement activation and
found in its soluble form in the blood (Pillemer et al., 1991). Its normal plasma
concentration is 5-15µg/ml (Schwaeble and Reid, 1999). It is composed of a monomer
formed of six homologous structural units called thrombospondin structural homology
repeats (TSRs) and an N terminal domain. This monomer is linked together head to tail
to form different forms of properdin, which is dimer, trimer and tetramer (Perdikoulis
et al., 2001; Schwaeble and Reid, 1999; Smith et al., 1984). The functional activity of
the different forms is variable as functionality increases by increasing the size of the
polymers. Therefore, the tetramer has ten times the activity of the dimer (Pangbum,
1989). Properdin has the ability to bind to soluble C3b and cell bound C3b. However,
the affinity of properdin binding to cell bound C3b is greater than that for soluble C3b.
Moreover, properdin binds to C3b and the C3bBb complex with a greater affinity to cell
bound C3 convertase rather than to cell bound C3b (Farries et al., 1989). Properdin has
an important role in alternative pathway activation, as lack of properdin in serum leads

Chapter 1: Introduction
10
to decreased ability of serum to activate the alternative pathway. However, adding
properdin can restore the activation of the alternative pathway (Schwaeble and Reid,
1999). Furthermore, it has been found that properdin can bind to the alternative pathway
activator and initiate the activation of the alternative pathway. Moreover, it has been
claimed that properdin can bind directly to the bacterial surface (Neisseria gonorrhoeae
and Escherichia coli) and enhance C3 deposition on the bacterial surface after its
addition to properdin deficient serum (Spitzer et al., 2007).
1.2.3 Lectin Pathway
Lectin pathway activation takes place when one or more of the lectin pathway
components (Mannan-binding lectin (MBL), ficolin and CL-11) bind to the pathogen-
associated molecular patterns (PAMPs), such as polysaccharides/carbohydrates and
acetylated sugars, on the pathogens (Schwaeble et al., 2011). As a result of this binding,
mannan-binding lectin-associated serine proteases 1, 2, 3 (MASPs) and Map19, which
is a non-enzymatic truncated product of MASP-2, become active.
Activation of MASP-2 leads to the cleavage of C4 into two portions: the large one, C4b,
which remains attached to the cell surface and the small portion, C4a, which is released
as an anaphylatoxin. C2 is also cleaved by active MASP-2 into two portions: the large
portion C2a and the small portion C2b. The large portion of the cleaved C2 (C2a) binds
to the large portion of the cleaved C4 (C4b) on the pathogen to form the lectin pathway

Chapter 1: Introduction
11
C3 convertase (C4b2a) on the surface of the pathogen (Fujita, 2002). As seen in the
classical and alternative pathways, the lectin pathway C3 convertase cleaves C3, which
forms C5 convertase (C4b2a(C3b)n) that then initiates the formation of the membrane
attack complex, as described earlier for the classical and alternative pathways
(Schwaeble et al., 2002; Thiel et al., 2000).
1.2.3.1 Lectin pathway components
The activation of the lectin pathway is more complex than the classical and alternative
pathways because of the interaction between the lectin pathway components (Sorenson
et al., 2005). These components are multimeric carbohydrate recognition
subcomponents (Mannan Binding Lectin (MBL) and ficolin) and Mannan Binding
Lectin associated serine proteases (MASP-1, MASP-2, MASP-3 and MAP19) (Stover et
al., 1999; Takahshi et al., 1999).
1.2.3.1.1 Mannan Binding Lectin
Mannan Binding Lectin (MBL) is a member of collectin proteins produced mainly from
the liver. It circulates in serum as a large oligomeric complex (trimers, tetramers and
hexamers) (Brouwer et al., 2008; MacMullen et al., 2006). It is composed of multimers
of three identical polypeptide chains (homotrimers). Each polypeptide chain is
comprised of a short N-terminal cysteine-rich domain (collagen-like domain) that links
MASPs with the MBL neck region and a globular head part that contains the
carbohydrate recognition domain (CRD) (Figure 1.3). Mannose and N-acetyl-
glucosamine (GIcNAc) show affinity for MBL while other carbohydrates show no

Chapter 1: Introduction
12
affinity for MBL. In addition to this, IgM can bind to MBL and activate the lectin
pathway directly (McMullen et al., 2006). Recent reports by Ogden et al. (2001) and
Jack et al. (2005) claimed that MBL works as a complement independent opsonin.
Moreover, Jack et al. (2001) and Jack et al. (2005) show that MBL works as a
dependent opsonin by enhancing the uptake and killing of Neisseria meningitidis
bacteria by human phagocytes. While humans have one MBL gene, rodents have two
genes, MBL-A and MBL-C (Sastry et al., 1995).
Figure 1.3 The domain structure and oligomerization of MBL (Garred et al., 2009).
1.2.3.1.2 Ficolin
Ficolins are carbohydrate recognition molecules composed of three identical
polypeptide chains. Each polypeptide chain consists of a short N-terminal cysteine-rich
domain with many cysteine residues, a collagen-like domain, a neck region and a
fibrinogen-like carbohydrate recognition domain (FBG) (Figure 1.4). Most ficolins are
present in serum and activate the lectin pathway by binding to N-acetyl glucosamine

Chapter 1: Introduction
13
sugars and lipoteichoic acids of gram positive bacteria (Garred et al., 2009; Matsushita
et al., 2001; Endo et al., 2005; Lynch et al., 2004).
Ficolins in humans have three different forms which are L-ficolin, M-ficolin, and H-
ficolin. Unlike L-ficolin and H-ficolin, which are found in serum and activate the lectin
pathway, M-ficolin is found on the surface of leukocytes (neutrophils and monocytes)
and activates the lectin pathway by forming a complex with MASP-1 and MASP-2 (Liu
et al., 2005; Matsushita et al., 2000; Matsushita and Fujita, 2001). Mice have two
different forms of ficolin: ficolin-A which is found in serum and looks like human
ficolin-L and ficolin-B which is found in bone marrow cells. Although ficolin-B has a
similar structure to ficolin-A, it has no ability to activate the lectin pathway because it
does not bind to MASP-2 (Endo et al., 2005; Runza et al., 2008).
Figure 1.4 The domain structure and oligomerization of ficolins (Garred et al., 2009).
1.2.3.1.3 Collectin
Collectin 11 (CL11) is a member of the collectin family produced in many organs,
especially the kidney, adrenal gland and liver, with a plasma concentration of about 2.1

Chapter 1: Introduction
14
µg/ml. It is composed of an N-terminal domain, collagen-like domain, neck domain and
a carbohydrate recognition domain (CRD). Collectin 11 can interact with MASP-1 and
MASP-3 in the plasma. It can also bind to D-mannose and L-fucose terminal
saccharides on the surface of microbes and acts as a recognition molecule of the lectin
pathway (Hansen et al., 2010).
1.2.3.1.4 Mannan Binding Lectin associated serine proteases
Mannan Binding Lectin associated serine proteases are members of the serine protease
family and are homologous to the serine proteases C1r and C1s of the classical pathway.
In mammals, three forms of MASPs are found: MASP-1, MASP-2 and MASP-3. In
addition to the MASPs, MAP19 is a truncated product generated from the MASP-2
gene and Map44 is a truncated product generated from the MASP-1 gene, which are
non-enzymatic products and act as inhibitors of lectin pathway activation (Schwaebleet
al., 2002; Stover et al., 1999; Wallis, 2007).
MASP-1, MASP-3 and Map19 are generated from the MASP-1 gene on chromosome 3
in humans. MASP-2 and Map19 are generated from the MASP-2 gene on chromosome
1 in humans and 4 in mice (Dahl et al., 2001; Sorenson et al., 2005; Stover et al., 1999).
MASP-2 is produced only by the liver, MASP-1 is also mainly produced by the liver
and MASP-3 is produced by the liver, spleen, lung and other tissues. All MASPs share
the same domain organization which is N-terminal CUB 1, an epidermal growth factor
(EGF)-like domain, CUB 2 domain, two complement control protein domains (CCP1
and CCP2), also known as short consensus repeats (SCRs), and a chymotrypsin-like
serine protease domain (Figure 1.5). Although MASP-1 and MASP-3 have the same N-

Chapter 1: Introduction
15
terminal domain they have a different serine protease domain (Lynch et al., 2005;
Sorenson et al., 2005; Thiel, 2007).
Figure 1.5 MASP and Map domain organization as described by Yongqing et al. (2012).
Binding between MASPs, MBL and ficolin occur by the binding of CUB1 and the EGF-
like domain from MASPs and the collagen-like domain of MBL and ficolin (Wallis et
al., 2004). The enzymatically active form of MASPs occurs when there is cleavage
between CCP-2 and the serine protease domain which leads to the formation of a heavy
chain (N-terminal domains) and light chain (serine protease domain) liked together by a
disulfide bridge (Matsushita and Fujita, 1995).
Lectin pathway activation occurs only by MASP-2 which cleaves C4 and C2 that bound
to C4b to form the lectin pathway C3 convertase and neither MASP-1 nor MASP-3 can
restore the activation of the lectin pathway (Matsushita et al., 2000; Rossi et al., 2001;

Chapter 1: Introduction
16
Vorup-Jensen et al., 2000). Therefore, murine MASP-2 deficiency leads to the loss of
lectin pathway activation (Schwaeble et al., 2011). Takahshi et al. (2010) reported that
MASP-1 plays a role in activation of the alternative pathway by converting factor D to
an enzymatically active form (Figure 1.6). A recent study of Iwaki et al. (2011)
reported an important role of MASP-3 in the activation of the alternative pathway,
showing that a complex of recombinant MASP-3 and recombinant MBL has the ability
to activate the alternative pathway by cleaving C3b bound factor B on the surface of
bacteria.
Figure 1.6 MASPs activation results in the formation of a heavy and a light chain held together
through a disulfide bond (Fujita, 2002).

Chapter 1: Introduction
17
1.2.4 Membrane Attack Complex
Any of the complement pathway C3 convertases (classical pathway and lectin pathway
C3 convertase (C4b2a) or alternative pathway C3 convertase (C3bBb)) facilitate the
formation of the membrane attack complex by cleaving C3 into a small fragment C3a
and a large fragment C3b. Accumulation of C3b leads to the formation of C5
convertase, C4b2a(C3b)n for the classical and lectin pathways, or C3bBb(C3b)n for the
alternative pathway which cleaves C5 into C5b and C5a. The larger cleavage product
C5b binds to the surface of the pathogen, leading to the formation of a C5b-8 complex.
This results in C9 polymerisation that forms pores in the lipid bilayer of the pathogen
cell membrane which, in turn, disturbs the balance of salts and metabolites inside the
pathogen (Figure 1.7) (Podack et al., 1982).
Figure 1.7 Membrane attack complex pathway (Fujita, 2002).

Chapter 1: Introduction
18
1.2.5 Functions of the Complement System
The complement system plays several roles in the normal functioning of the immune
system, especially in the process of protection against pathogens, and links the non-
specific immune system with the specific immune system. Also, waste products (e.g.
apoptotic cells and debris) are removed by the complement system (Markiewski &
Lambris, 2007). In addition, the opsonisation of bacteria, which plays an important role
in the recognition of the presence of bacterial infection, is controlled by the complement
system through C3b, C4b, iC3b and iC4b which can attach to the surface of pathogens.
Phagocytosis can be enhanced by attracting leucocytes via C3b which uses complement
receptors 1 and 3 (CR1 and CR3) to bind pathogens. Moreover, phagocytic cells are
able to bind to MBL and L-ficolin to establish the process of phagocytosis (Matsushita,
2010).
Inflammatory cells (i.e. leucocytes such as mast cells, phagocytes and neutrophils) are
attracted and activated at the site of infection by C3a and C5a through establishing a
strong pro-inflammatory reaction. The C3a and C5a complement components are called
anaphylatoxins and they are produced during the activation of the complement system
due to the cleaved products of C3 and C5. C3a and C5a are able to attract leucocytes to
inflammatory sites as a consequence of raising the vascular permeability of endothelial
cells. Subsequently, pathogens can be recognised and excluded (Markiewski &
Lambris, 2007).

Chapter 1: Introduction
19
Moreover, C5a was shown to enhance the activation and production of some
chemokines (e.g. tumour necrosis factor (TNF)) and interleukins,e.g. IL-1 and IL-8
(Markiewski & Lambris, 2007). In addition, using a mouse model, it was shown that
C5a and C3a have many functions in releasing macrophage inflammatory protein-2 and
monocyte chemo-attractant protein-1 by exciting endothelial cells (Laudes et al., 2002).
Furthermore, the complement system has an important role in direct bacterial lysis
mediated by the MAC complex, particularly those that have thin cell walls such as
gram-negative bacteria (Wu et al., 2009).
In addition to MAC complex-mediated killing, the complement system has another
crucial role in clearing immune complexes, necrotic cells and apoptotic cells.
Phagocytic cells like macrophages have complement receptors CR3 and CR4 on their
surfaces, which mainly control this clearance. Phagocytosis happens after CR3 and CR4
identify the apoptotic cells that bind to C1q and create immune complexes (Taylor et
al., 2000). In addition, because RBCs have CR1 receptors that link to the C3b
opsonized C1 complex, immune complexes can be found in the bloodstream associated
with RBC’s. Ultimately, the spleen and liver remove RBC’s with attached immune
complexes via macrophages and the reticulo-endothelial system (Frank, 2010).
Finally, the specific immune response by B cells is activated by the complement system.
B cells have CR1 and CR2 that attach to iC3b, C3dg and C3d and leads to the activation
of B cells (Molina et al., 1994). As a result of this molecular linkage, antibody
production can be enhanced because of the lowering of the threshold required for
activation of B cells (Chen et al., 2000).

Chapter 1: Introduction
20
1.2.6 Regulators of Complement System
Initiating and controlling the activation of the complement system needs to be done
cautiously as un-regulated, activation of the complement system can lead to potentially
dangerous outcomes such as host tissue damage and inflammatory diseases (Trouw et
al., 2008). In practice, soluble regulators (fluid phase) and membrane-bound regulators
are both used to control the activation of the complement system (Figure 1.8)
(Kirschfink & Mollnes, 2003).
1.2.6.1 Fluid Phase Regulators
Regulation of the activation of classical pathway (CP) and lectin pathway (LP) occurs
via a regulatory protein called C1 inhibitor (C1-INH). C1 inhibitor can be attached to
C1 complexes to produce (C1-INH-C1r2-C1s2-C1-INH) in the classical pathway and
can be attached to LP complexes to produce (MASP-1-C1-INH and MASP-2-C1-INH)
in the lectin pathway (Ehrnthaller et al., 2011). However, C1-INH does not bind to
MASP-3 (Yongqing et al., 2012).
In addition to C1 inhibitor (C1-INH) the C4-binding protein, C4bp, is another crucial
regulator of the complement system. It works by reducing C4b-bound C2 to block C3
convertase which is required for the production of CP and LP. Moreover, by enhancing
the production of I-factor, which plays a role as a cofactor in factor I-mediated
conversion of C4b to C4dg and C3b to iC3b and C3dg, is another function of the C4-
binding protein C4bp (Jurianz et al., 1999).

Chapter 1: Introduction
21
Furthermore, control of the AP C3 convertase and C3 convertase pathways by Factor H
(which is one of the most copious fluid phase mediatory components (150 mg/L)) plays
a vital role in preventing unwanted complement system activation (Ehrnthaller et al.,
2011).
Moreover, clusterin and S-protein mediate the terminal pathway. They block pore
formation within the cell membrane through attaching to the C5b-7 complex, which
reduces the lytic activity of the MAC complex. Finally, the inflammatory role of the
anaphylatoxins within the complement system, C3a and C5a, can be blocked by
carboxypeptidase N (Ehrnthaller et al., 2011).
1.2.6.2 Membrane-Bound Regulators:
Basically, the main complement mediators that are bound to the membrane are CD35
(complement receptor 1 (CR1), CD46 (membrane cofactor protein, (MCP), CD55
(decay accelerating factor (DAF) and CD59 (protectin) (Ehrnthaller et al., 2011).
Complement receptor 1 (CR1), which can be found on RBCs (erythrocytes) and WBCs
(leukocytes) retards the activity of C3 and C5 convertase. Also, CR1 can work as a
cofactor for factor I in C3b and C4b cleavage.

Chapter 1: Introduction
22
In addition, membrane factor protein (MCP) can attach to C3b and support factor I in its
deactivation. Moreover, decay accelerating factor (DAF) can attach to C2a in the CP
and LP and Bb in the AP to retard the formation of C3 and C5 convertase (Frank, 2010).
Finally, protectin (CD59) can stop production of the MAC complex by interacting with
C8 and C9 and prevent them from attaching to the C5b-7 complex (Ehrnthaller et al.,
2011).

Chapter 1: Introduction
23
Figure 1.8 Regulation of complement activation by membrane bound and fluid phase
regulators as described by Mollnes et al. (2002).

Chapter 1: Introduction
24
1.2.7 Complement deficiency
The complement system plays a crucial role in fighting microbial infection and clearing
the body of immune complexes and apoptotic cells (Langer et al., 2010). This role is
preformed via a network of proteins which either initiate complement pathway
activation or regulate its activation. Therefore, deficiency in any component of the
complement system or in complement regulators may lead to changes in body
homeostasis and health, resulting in increased susceptibility to pathogens or the
induction of autoimmune disease (Mayilyan, 2012).
1.2.7.1 Defects of the Classical Pathway
Immune complex/autoimmune disease is often associated with the classical complement
pathway deficiency. Deficiency of the classical pathway C1q component leads to
increases in the risk of Systemic Lupus Erythematosus (SLE). This indicates that C1q
plays an important role in the clearance of immune complexes and apoptotic cells
(Leffler et al., 2014). Deficiency of C1s, C1r, and C4 is also associated with SLE
disease but is lower than with C1q deficiency. Classical pathway deficiency is
associated with recurrent bacterial infection (Brown et al., 2002). C2 deficiency is
associated with increased susceptibility to S.pneumoniae in children (Jonsson et al.,
2005). Even though, the deficiency of C3 is uncommon, it leads to autoimmune disease
and increases the risk of getting recurrent severe infection caused by encapsulated
bacteria like Haemophilus influenza and pneumococci (Ross & Densen, 1984; Singh
and Rai, 2009). In addition to this, defects in innate immunity and adaptive immunity

Chapter 1: Introduction
25
have been reported in a C3 deficient patient due to the functional impairment of the
function of B cells, T cells, and dendritic cells (Botto et al., 2009).
1.2.7.2 Defects of the Alternative Pathway
Alternative pathway malfunction is most often associated with deficiency of factor D,
factor B and properdin. Alternative pathway factor D deficiency is associated with
increased severity of meningococcal infection. Factor B deficiency also leads to
recurrent meningococcal and pneumococcal infection (Slade et al., 2013; Sprong et al.,
2006). Properdin deficiency, which is the most common deficiency of alternative
pathway, increases the risk of meningococcal disease and pneumonia (Schejbel et al.,
2009; Fijien et al., 1999). In addition, properdin deficient individuals are up to 250
times more likely to get meningococcal infection compared to normal individuals.
Furthermore, the rate of morbidity and mortality in meningococcal infection increases
due to properdin deficiency, illustrating the crucial role of properdin against this
infection (Fijien et al., 1999).
1.2.7.3 Defects of the Lectin Pathway
Lectin pathway MBL deficiency is most commonly caused by polymorphism in the
MBL gene. Even though 10% of the population has been found to be MBL deficient,
this population remains healthy and shows no increasing susceptibility to bacterial
infection and morbidity compared to normal people. However, under certain conditions,
their susceptibility to bacterial infection increases, especially when it is associated with

Chapter 1: Introduction
26
immunocompromised conditions like HIV infection (Dahl et al., 2004; Peterslund et al.,
2001; Sorensen et al., 2005; Super et al., 1989). In addition to this, MBL deficiency is
associated with impairment of opsonisation and it has been found that MBL deficiency
increases the risk of rheumatoid arthritis, atherosclerosis and arterial thrombosis
(Jacobsen et al., 2001; Ohlenschlaeger et al., 2004). L-ficolin deficiency is associated
with recurrent respiratory infection and allergies in children (Atkinson et al., 2004).
Autoimmune disease and recurrent severe infections have been found to be associated
with MASP-2 deficiency, which is a rare condition (Stengaard-Pedersen et al., 2003).
1.2.7.4 Defects of terminal complement components
Terminal complement components (C5, C6, C7, C8 and C9) play an important role in
the lysis of bacteria. Deficiency of one or more of terminal complement components
impairs this function and increases the susceptibility to recurrent meningitis (Figuera
and Densen, 1991). C7 and C9 deficiency are associated with an increased risk of
meningococcal infection. However, the risk of meningococcal infection increases 1000
fold in C9 deficient individuals and 1400 fold in C7 deficient individuals compared to
the general population (Nagata et al., 1989).
1.2.7.5 Defects of complement regulatory components
C1 inhibitor is important not only in complement regulation but also plays a role in
regulating blood clotting pathways. Therefore, deficiency of C1 inhibitor is associated
with hereditary angioedema (HAE), increased vascular permeability to plasma and

Chapter 1: Introduction
27
excessive lymphoproliferation in autoimmune disease and lymphoma. In
lymphoproliferation, the concentration of C1 inhibitor is normally high while the
concentration of C1q is often low (Cancian, 2014; Cugno et al., 2009; Markovic et al.,
2000). Deficiency of the alternative pathway regulators, Factor H and Factor I, is
associated with immune associated diseases such as SLE and atypical haemolytic
uraemic syndrome (Reis et al., 2006; Thurman and Holers, 2006). It is also associated
with increased susceptibility to infection because of continuous alternative pathway
activation that depletes levels of factor B and C3 (Reis et al., 2006).

Chapter 1: Introduction
28
1.3 Neisseria meningitidis
There are twelve species of Neisseria genus that have been isolated from humans. These
species of Neisseria can be divided into two groups according to their colony
characteristics and morphology. The species in the first group grow as non-pigmented
and translucent colonies, such as Neisseria meningitidis, Neisseria gonorrhoeae, and
Neisseria lactamica. The species in the second group grow as opaque and yellow
pigment colonies, such as Neisseria muocsa, Neisseria subflava and Neisseria sicca.
Among these, only two species are considered to be pathogenic: Neisseria meningitidis
and Neisseria gonorrhoeae (Barrett and Sneath, 1994; Knapp, 1988).
Neisseria meningitidis is the main cause of bacterial meningitis throughout the world,
significantly contributing to increased mortality (Emonts et al., 2003). While its
diagnosis, vaccination and treatment have improved considerably in recent years,
infection is still spreading worldwide, with a mortality rate of up to 10% (Connolly and
Noah, 1999).
Neisseria meningitidis is a gram negative, bean-shaped diplococcus, which attacks the
upper respiratory tract as commensals in 10-40% of population, may cause systemic
diseases. Meningococcal disease is spread between the hosts primarily through an
aerosol transmission. Although some individuals infected with Neisseria meningitidis
suffer severe disease, other individuals have it as commensals. However, it is not clear
yet why some individuals develop the severe disease (Yazdankhah et al., 2004). Due to

Chapter 1: Introduction
29
the gradual loss of passive immunity children under five years old can be at risk of
meningococcal disease (Edwards and Baker, 1981).
Neisseria meningitidis is enclosed by three layers: outer membrane lipids, outer
membrane proteins (OMPs) and lipo-oligoccharides (LOS), which are covered by a
polysaccharide capsule on the pathogenic Neisseria meningitidis (Figure 1.9) (van
Deuren et al., 2000). Neisseria meningitidis can be identified according to the
polysaccharide capsule into 13 serogroups (A, B, C, E-29, H, I, K, L, M, W-135, X, Y
and Z) (Choudhury et al., 2008). However, five strains (A, B, C, Y and W-135) are
responsible for most diseases (Spinosa et al., 2007). While the serogroups B and C are
most common in industrialized countries, the serogroups A and C are most common in
the third world (Takahashi et al., 2008). Each year, serogroup A causes an outbreak of
meningococcal disease in Sub-Saharan Africa, with an incidence rate of up to
1,000/100,000 (Moore, 1992; Riedo et al., 1995). Northern China was the source of an
outbreak of meningococcal disease caused by serogroup A, which spread throughout the
world (Wang et al., 1992). Serogroup Y caused the most cases of meningococcal
disease in the United States (Racoosin et al., 1998). Northwestern Europe is affected by
meningococcal disease caused by serogroup B, with an attack rate of up to 1 to
50/100,000. In the UK, more than 2000 people are affected each year, with 10%
mortality rate. People who recover from meningococcal disease may suffer permanent
damage, like deafness and mental retardation (Baraff et al., 1993).

Chapter 1: Introduction
30
Figure 1.9 Neisseria meningitidis cell membrane (Rosenstein et al., 2001).
1.3.1 The Virulence Factors of Neisseria meningitidis
1.3.1.1 Pili and Pilus Subunits
Type IV pili (Tfp) and pilus subunits are very important factors that play a significant
role in the interaction of Neisseria meningitidis with the host cells, resulting in a
productive infection (Ieva et al., 2005). The type IV pili subunit is made up of
thousands of main pilus subunits, PilE proteins, and a small number of pilus-associated
proteins, for example PilV and PilC, which contain two proteins PilC1 and PilC2 that
are very important in the formation of pili. During the initiation of interaction between
the epithelial mucosa and Neisseria meningitides, the expression of PilC1 will be
upregulated by control of the sequence 150-bp that is located upstream of PilC1 known
as CREN. Contact-regulated protein A (CrgA) protein, as a regulatory protein, works
when the Neisseria meningitidis starts adhesion to the host epithelial cells (Tettelin et
al., 2000). PilT is an important component of pilus that induces intimate attachment

Chapter 1: Introduction
31
once Neisseria has attached to the host cell by promoting pilus retraction, which is
required for twitching motility (Pujol et al., 1999).
1.3.1.2 Outer Membrane Proteins
Outer membrane proteins (OMPs) are other important virulence factors of Neisseria
meningitidis that facilitate the Neisseria’s adhesion to and invasion of the host cells.
These proteins are divided into five classes (class 1, 2, 3, 4 and 5) depending on the size
of proteins on sodium dodecyl sulphate polyacrylamide gels (SDS- polyacrylamide gel).
However, recently, these classes of protein have been reordered according to the
producer genes into two groups, (1) ProA, which includes proteins classes 1, 4, and 5
and (2) ProB, which includes protein classes 2 and 3 (Tsai et al., 1981; Hitchcock,
1989).
Meningococcal porins (ProA and ProB) are the most frequent types of proteins found in
the meningococcal membrane. The molecular size of these proteins varies across
different strains of meningococcal. These proteins are found in trimer form and act to
allow the small hydrophilic nutrients to enter the cell (Tommassen et al., 1990; Frasch
et al., 1985). In addition, they are used to classify meningococcal disease serologically
because of their abundance in the outer membrane. While ProA, known as class 1 porin,
has the ability to enhance the generation of B cells and the interaction between T cells
and B cells, the two forms of ProB, ProB2 known as class 2 porin and ProB3 known as
class 3 porin, have the ability to activate T cells and dendritic cells. Additionally, ProB
acts to differentiate T cells into Th2 cells which facilitate the production of antibodies
(Mackinnon et al., 1999). Meningococcal opacity proteins (Ops) comprise two proteins

Chapter 1: Introduction
32
Opa and Opc, with Opa being more abundant compared to Opc (De Vries et al., 1998).
CEACAM and heparin sulphate proteoglycan receptors that are found on the host cells
are bound to the membrane opacity proteins A (Opa), initiating the adhesion (Albiger et
al., 2003).
1.3.1.3 Capsule and Lipo-oligosaccharide (LOS)
Capsule and lipo-oligosaccharide (LOS), non-protein factors, play a crucial role in
Neisseria meningitidis infection, Neisseria protection from complement-mediated
killing and Neisseria protection from the phagocytosis by the immune cells (Takahashi
et al., 2008). Because the lipid A portion of the lipo-oligosaccharide, which mediates
the endotoxic shock, leads to the activation of macrophages and produces various
cytokines and chemokines, lipo-oligosaccharide is considered the most important
virulence factor (Albiger et al., 2003).
Neisseria meningitidis serogroups differ in their polysaccharides capsules, for example,
serogroups B, C, Y, and W-135 have polysaccharides capsules comprising mainly sialic
acid attached to glucose or galactose while the serogroup A polysaccharide capsule
comprises N-acetyl mannosamine-1-phosphate (Swartley et al., 1997). Serogroups B
and C have a similar structure of their polysaccharides capsules and they have the
ability to change their lipo-oligosaccharides to variable degrees (Mandrell et al., 1991).
Neisseria meningitidis capsule synthesis gen (cps) has five different regions. Region A
has the gene responsible for making the polysaccharides capsule. Different serogroups

Chapter 1: Introduction
33
have different genes, for example, serogroups B, C, Y, W-135 have the siaD gene which
is required to make a capsule containing sialic acid, whereas serogroup A has the myn
gene which is required to make the serogroup A capsule (Edwards et al., 1994). The
gene on region B facilitates lipid modifications (Frosch & Müller, 1993). Region C has
the Ctr gene that is responsible for polysaccharide transport and is also found conserved
in most isolated Neisseria meningitidis. It is also used as a target to clinically detect
Neisseria (Guiver and Borrow, 2001). Region D contains a gene that is responsible for
making lipopolysaccharides. Region E contains the tex gene the function of which is not
yet clear (Claus et al., 2002).
However, it has been claimed that some Neisseria meningitidis strains can be present in
the human nasopharyngeal transmission system without a gene responsible for
synthesising the capsule and replace it with the capsule null locus (cnl) sequence that
has been found at least in four different groups of Neisseria meningitidis (Claus et al.,
2002). Uncapsulated meningococcal strains show increased ability to adhere to mucosal
epithelium. Moreover, uncapsulated meningococcal strains are more susceptible to be
killed by human serum compared to capsulated meningococcal strains because of the
presence of the capsule provides a protection against human serum (Dolan-Livengood et
al., 2003; Vogel et al., 1996).
Lipooligosaccharide (LOS) is one of the Neisseria meningitidis virulence factors which
provides bacterial protection from serum and plays a role in the colonization and
invasion of host cells. Endotoxic shock is associated with the Lipid A portion of LOS. It

Chapter 1: Introduction
34
leads to the activation of macrophages which produce many of cytokines and
chemokines involved in inflammation (Albiger et al., 2003; Rosenstein et al., 2001).
Additionally, the levels of endotoxin are associated with the severity of meningococcal
disease (Brandtzaeg et al., 1992). LOS is characterized by a lack of O-antigen and
having short chains of polysaccharide comprising two to five sugars linked to the inner
core of Neisseria meningitidis through two 2-keto-3-deoxyoctulosonic acid (KDO)
molecules (Gamian et al., 1992). Some studies have shown that changing the Neisseria
meningitidis LOS by phosphoethanolamine groups (PEA) and O-acetyl groups increases
Neisseria meningitidis adhesion to epithelial or endothelial cells (Takahashi et al.,
2008). LOS has been used to serologically divide meningococcal meningitis into 12
immunotypes (L1-L12),dependingon thegroupofαandβchainsadded to the inner
core region (Mandrell and Zollinger, 1977). However, some Neisseria menigitidis have
more than one immunotype-specific epitope and are classified as the L3, 7, 9 or L2, 4
immunotypes (Tsai et al., 1983). The L3, 7, 9 meningococcal immunotype is the most
common cause of meningococcal disease (Griffiss et al., 2000).
Recent studies have claimed that in spite of the expression of PilC and Pili, Neisseria
meningitidis that lacks LOS is not able to adhere to epithelial cells and significantly
decreases the competence of DNA transformation (Albiger et al., 2003). The ability of
LOS in complement activation has been studied, showing that Neisseria menigitidis are
able to activate the complement system effectively even when they are LOS mutants
(Sprong et al., 2004).

Chapter 1: Introduction
35
1.3.2 Colonization and invasion by Neisseria meningitidis
Neisseria meningitidis is a human pathogen transmitted by aerosol and other infected
secretions. The human nasopharyngeal compartment is the normal habitat for Neisseria
menigitidis. It can colonize the mucosal surface of the upper respiratory track without
causing any symptoms in a phenomenon called carriage. However, 10% of the
population are asymptomatic carriers (Broome, 1986). The susceptibility of the host and
concomitant viral infection particularly when found in an epidemic area and the
meningococcal virulence factors all play a role in the development of meningococcal
disease (Anderson et al., 1998). The rates of neisserial carriage increase by up to 40% in
closedandsemiclosedpopulationslikeuniversities(Ala’Aldeenet al., 2000; Caugant
et al., 1992).
Meningococcal colonization starts on the surface of mucosal epithelium and
intraepithelially. Subsequently, it penetrates the blood stream as result of endocytosis
caused by phagocytosis. This penetration causes various clinical symptoms, ranging
from sepsis and mild meningococcal infection to meningococcal septicemia and
meningitis (Figure 1.10) (Van Deuren et al., 2000). If the level of bacteria in the blood
is low the bacteria will be cleared out spontaneously. However, if the bacteria is not
cleared out and the level of bacteria is high (could be up to 106
colony forming units per
ml), this leads to vascular and tissue damage (Brandtzaeg et al., 1995). Endotoxins are
released during the bacteremic stage of the disease in the form of vesicular outer
membrane structures (composed of LOS, OMPs, lipids and polysaccharides capsule),
which promotes the production of inflammatory factors in the blood. This inflammatory
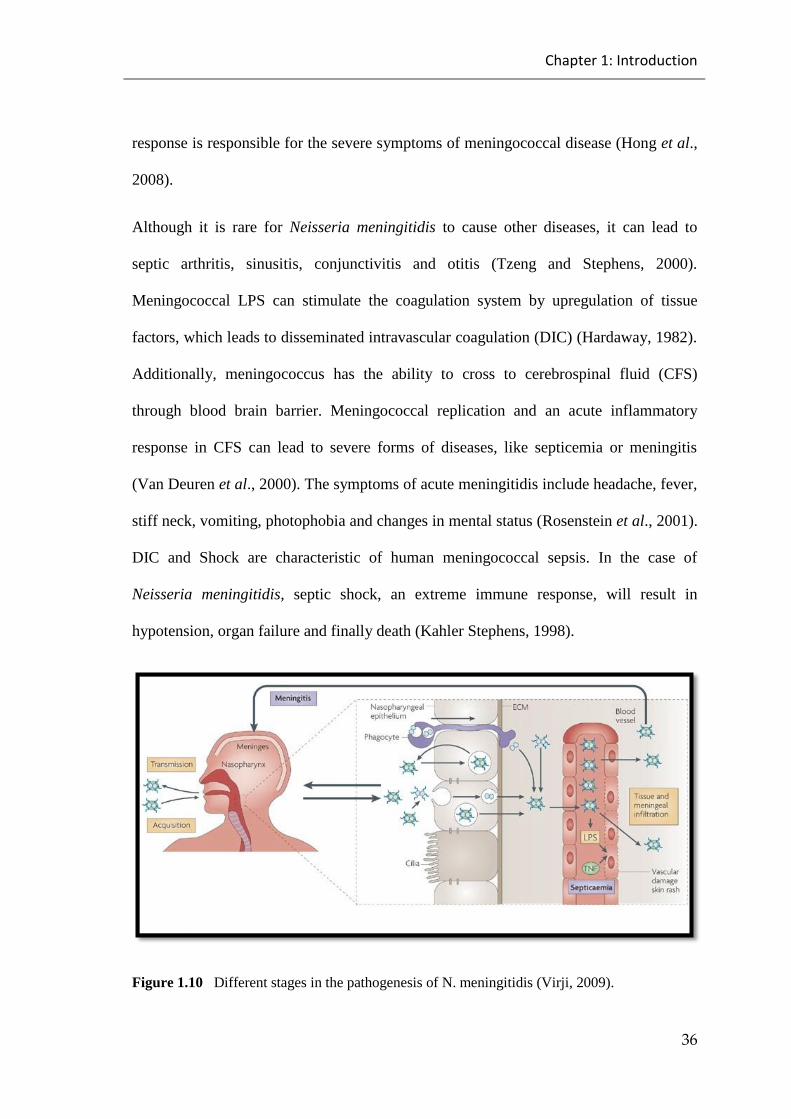
Chapter 1: Introduction
36
response is responsible for the severe symptoms of meningococcal disease (Hong et al.,
2008).
Although it is rare for Neisseria meningitidis to cause other diseases, it can lead to
septic arthritis, sinusitis, conjunctivitis and otitis (Tzeng and Stephens, 2000).
Meningococcal LPS can stimulate the coagulation system by upregulation of tissue
factors, which leads to disseminated intravascular coagulation (DIC) (Hardaway, 1982).
Additionally, meningococcus has the ability to cross to cerebrospinal fluid (CFS)
through blood brain barrier. Meningococcal replication and an acute inflammatory
response in CFS can lead to severe forms of diseases, like septicemia or meningitis
(Van Deuren et al., 2000). The symptoms of acute meningitidis include headache, fever,
stiff neck, vomiting, photophobia and changes in mental status (Rosenstein et al., 2001).
DIC and Shock are characteristic of human meningococcal sepsis. In the case of
Neisseria meningitidis, septic shock, an extreme immune response, will result in
hypotension, organ failure and finally death (Kahler Stephens, 1998).
Figure 1.10 Different stages in the pathogenesis of N. meningitidis (Virji, 2009).

Chapter 1: Introduction
37
1.3.3 The Immune System and Neisseria meningitidis
Neisseria meningitidis infection stimulates the innate immune system to clear the
Neisseria meningitidis. Short febrile flu can be observed because of low levels of
bacteremia, which usually clear out spontaneously. The induction of specific adaptive
immunity, in the form of antibodies, is more effective in the development of resistance
to Neisseria meningitidis. However, because it is a delayed response, the innate immune
response plays a crucial role in fighting against Neisseria meningitidis infection (van
Deuren et al., 2000).
Some factors, such as the complement system, cationic antimicrobial peptides (CAMPs) and
other components of the inflammatory response, work to limit the growth of Neisseria
meningitidis in the host. A potent inflammatory response which emerges following a Neisseria
meningitidis invasion is responsible for the symptoms of meningococcal disease (Hong et al.,
2008).
The host produces different antimicrobial substances after invasion from pathogens.
One of them is cationic antimicrobial peptide (CAMP). Cationic antimicrobial peptide
(CAMP) is one of the important components of innate immunity produced by
phagocytic cells and expressed by epithelial cells after pathogen invasion (Hancock,
2001). Neisseria meningitidis shows a high resistance to cationic antimicrobial peptide
through different mechanisms, such as a modification of lipid A. The Neisseria
meningitidis capsule also shows resistance to the cationic antimicrobial peptide
(Takahashi et al., 2008).
Another way of controlling Neisseria meningitidis infection has been shown by human
dendritic cells through their ability to phagocytose the pathogens (Kolb-Mäurer et al.,
2001).

Chapter 1: Introduction
38
1.3.4 The Complement System and Neisseria meningitidis
The complement system is a vital part of the innate immune system and plays a crucial
role in fighting against invading microbes (Trouw & Daha, 2011). It consists of more
than 35 proteins present in both plasma and on the surface of cells which interact with
each other to form a network of proteins that protect against microbes. The complement
system is activated by three different pathways which are the classical pathway, the
alternative pathway and the lectin pathway. This activation leads to the cleavage of C3
which is a central component of complement activation, to C3b, which binds to the
microbe and opsonises it for phagocytosis by immune cells. Moreover, C3 activation
leads to the formation of the bactericidal membrane attack complex, which can lead to
the lysis of the microbe. The membrane attack complex is initiated once the C5
molecule is cleaved by C5 convertase to C5a and C5b which are the initial components
of forming the terminal C5b-9 complement complex that leads to bacterial lysis (Kohl,
2001; Schneider et al., 2006; Stover et al., 1999; Walport, 2001). The complement
system plays a crucial role in fighting meningococcal infection so deficiency in any
complement component will increase the susceptibility to meningococcal infection
(Rosa et al., 2004; Rossi et al., 2001).
The meningococcal polysaccharide capsule is a very important structure that protects
the Neisseria meningitidis from complement mediated killing. Therefore, the absence of
the meningococcal capsule increases the susceptibility of the meningococcal to lysis by
the complement system (Schneider et al., 2006; Geoffroy et al., 2003). A study
conducted by Vogel et al. (1996) has used a modified meningococcal serogroup B strain
in which they inactivated the gene responsible for capsule expression found in the wild-

Chapter 1: Introduction
39
type strain and then compared the survival in human serum. The study showed that the
wild-type was less susceptible to killing than is more than the non-capsulated strain due
to the absence of capsule. However, they found no difference in the C3 deposition on
the surface of both strains which indicates that the absence of capsule does not affect the
cleavage of C3 on both strain surfaces (Vogel et al., 1996).
Meningococcal lipopolysaccharide is another important structure and is composed of
two chains linked to the lipid A portion of the outer membrane and provides protection
against complement mediated killing (Brandtzaeg et al., 1995). Meningococcal
serogroup B and C Strains have increased sialylation of LOS by addition of sialic acid
to Lacto-N-neotetraose (LNnt) to increase the serum resistance and avoid
meningococcal lysis by the complement system (Estabrook et al., 1997). Additionally,
serum resistance of Neisseria meningitidis can be enhanced through binding of
meningococcal Opc to vitronectin which is a very important regulator of terminal
complement activation (Viriji et al., 2008).
The classical pathway is a complement pathway that initiates binding to natural IgM
antibodies during meningococcal infection and is a potent activator of complement.
Following meningococcal colonization both specific and cross reactive antibodies may
be generated (Finne et al., 1987; Hoff and Hoiby, 1978; Liu et al., 2008). A previous
study has shown that the C4 binding (C4b) protein of the fluid-phase complement
proteins of classical and lectin pathways can bind to Neisseria meningitides and this
requires meningococcal porin A. However, the role of the C4bpin both meningococcal

Chapter 1: Introduction
40
binding and evasion of the immune system is not yet clear as it is seen in only non-
physiological sodium conditions (Jarva et al., 2005).
The alternative pathway factor H is a regulatory protein that works as a cofactor for
factor I mediating the cleavage of C3b to iC3b which is an enzymatically inactive decay
fragment (Pangburn et al., 2000). It has been suggested that Neisseria meningitidis can
bind to factor H via specific factor H binding protein (fHbp), also called genome
derived neisserial antigen (GNA), limiting complement activation on the surface of
Neisseria meningitidis (Pangburn et al., 2000; Schneider et al., 2006). Recent studies
have shown that factor H binds to Neisseria meningitidis via the neisserial surface
protein A (NspA) which has the ability to provide serum resistance to Neisseria
meningitidis even without presence of factor H binding protein (Lewis et al., 2010). In
addition, it has been reported that the sialylation of meningococcal serogroup B and C
increases the affinity of factor H to factor B or Bb bound C3b, leading to a decay of the
formed C3 convertase complex which results in the inhibition of complement activation
via the alternative pathway (Van Deuren et al., 2000).
The alternative pathway factor D is a serine protease that plays an important role in the
initiation and amplification of C3 convertase of the alternative pathway (Choy and
Spiegelman, 1996; Matsumoto et al., 1997). Sprong et al. (2006) reported that a defect
in the factor D allele could lead to factor D deficiency, which was associated with
increased invasive meningococcal disease (Sprong et al., 2006).

Chapter 1: Introduction
41
The alternative pathway includes properdin which is a serum glycoprotein that acts as a
positive regulator of alternative pathway activation (Nolan et al., 1992). Deficiency of
components in the alternative pathway have been associated with increased
susceptibility to severe meningococcal infection and thus an increased rate of mortality
compared to normal individuals (Braconier et al., 1983; Densen et al., 1987; Morgan
and Walport, 1991; Fijien et al., 1995; Spath et al., 1999). Agarwal et al (2010)
conducted a study in which they showed that native properdin could not bind to
meningococcus directly. However, unfractionated properdin also has the ability to bind
directly to meningococcus after incubation in properdin depleted serum and enhances
the level of C3 deposition on the surface of the meningococci (Agarwal et al., 2010).
Additionally, the importance of properdin has been reported in an in vivo model of
polymicrobial septic peritonitis which showed that the survival of properdin deficient
mice was decreased compared to their wild-type littermates (Stover et al., 2008).
Garred et al. (1993) studied the relationship between the MBL/lectin pathway and
meningococcal disease and found that there was no relationship between meningococcal
disease (caused by serogroup B and C) and low concentrations of lectin pathway MBL
(Garred et al., 1993). On the other hand, the Department of Paediatrics, Imperial
College School of Medicine at St Mary’s UK, conducted two independent studies,
showing that the susceptibility to meningococcal disease increased in children with
genetic variants of MBL (Hibberd et al., 1999). In addition to that, MBL deficiency in
early childhood increases the risk of meningococcal disease (Eisen et al., 2003; Faber et
al., 2007; Tully et al., 2006).

Chapter 1: Introduction
42
Although MBL binding varies in different serogroups of meningococcal, it has been
reported that LOS expression on meningococci is responsible for the major binding of
MBL and the meningococcal serogroups B and C show high activation of MBL even at
low concentrations (Jack et al., 2001; Van Emmerik et al., 1994; Kuipers et al., 2003).
It has also been shown that the binding affinity of MBL to Neisseria meningitidis
increased with low sialylation and decreased with high sialylation (Jack et al., 2001). In
a recent study it was reported that MBL binds to the meningococcal outer membrane
proteins Opa and ProB (Estabrook et al., 2004). This binding is not calcium dependent,
is not inhibited with mannose and is sensitive to 0.5M NaCl. Bjerre et al. (2002) carried
out a study using a whole blood model to study complement activation by
meningococcal strains and showed that meningococcal strains activate the complement
system through both the alternative and lectin pathways.
In summary, Neisseria meningitidis is the main causes of bacterial meningitis
throughout the world, and causes a significant rate of mortality (Emonts et al., 2003).
While there has been considerable progress in its diagnosis, vaccination and treatment
in recent years, the rates of Neisseria meningitidis infection are still increasing
worldwide, with a mortality rate of up to 10% (Connolly and Noah, 1999).
The complement system is a vital part of the immune system (activated by three
different pathways, which are the classic pathway, the alternative pathway and the lectin
pathway) and plays a crucial role in fighting microbial infection (Trouw and Daha 2011;
Schwaeble et al., 2002). Previous studies have highlighted the important role of the

Chapter 1: Introduction
43
complement system in fighting Neisseria meningitidis infection so this study has
focused on delineating the roles of defined complement component deficiencies in
increasing the susceptibility to meningococcal infection in mice and therefore to further
define the complement pathways that are involved in providing protection against
infection and disease progression (Jarva et al., 2005; Rosa et al., 2004; Rossi et al.,
2001)

Chapter 1: Introduction
44
1.4 Thesis Aims
Previous work in our laboratories has recently observed that serum of mice deficient of
the lectin pathway of complement effector enzyme MASP-2 has a higher bactericidal
activity towards Neisseria meningitidis as compared to MASP-2 sufficient serum. It
also showed that MASP-2 deficient mice were significantly protected against
meningococcal infection compared to MASP-2 sufficient mice. These findings
suggested that lectin pathway specific enzyme MASP-1 and/or MASP-3 has ability to
drive the alternative pathway mediated complement activation to fight N.meningitidis
infections. Therefore, this work aimed to achieve the following goals:
To assess the role of different complement pathways in innate immune response
against Neisseria meningitidis using in vitro assays
To assess the role of the alternative complement pathway in meningococcal
infection in experimental models of infection in a transgenic mouse strain with a
total deficiency of alternative pathway functional activity
To investigate the therapeutic benefits of recombinant MASP-3 (a lectin
pathway specific serine protease) in a murine model of meningococcal infection
To determine the potential benefits of recombinant properdin towards Neisseria
meningitidis using in vitro assays

Chapter 2: Materials and methods
45
Chapter 2: Materials and methods
2.1 Materials
2.1.1 Chemicals and materials
37% Formaldehyde solution Sigma-Aldrich
Agar-Agar Lab M
Barbital Sigma-Aldrich
Bovine serum albumin (BSA) Sigma-Aldrich
Brain heart infusion (BHI) medium Oxoid
Calcium chloride Sigma-Aldrich
Defibrinated Horse blood Oxoid
Ethylene glycol tetraacetic acid (EGTA) Sigma-Aldrich
Foetal calf serum Harlan
Heparin Sigma-Aldrich
Iron Dextran Sigma-Aldrich

Chapter 2: Materials and methods
46
Magnesium chloride Sigma-Aldrich
Mannan Sigma-Aldrich
N-acetyl BSA Promega
Phosphate Buffered Saline (PBS) Oxoid
Sigma Fast p-Nitrophenyl Phosphate tablet Sigma-Aldrich
Sodium chloride Fisher Scientific
Tris-HCl Sigma-Aldrich
Tween 20 Sigma-Aldrich
Zymosan Sigma-Aldrich

Chapter 2: Materials and methods
47
2.1.2 Antibodies/proteins
FITC-conjugated rabbit anti-human
C3c
Dako
Goat anti-human C1q polyclonal
antibody
Atlantic Antibodies Scarborough (USA)
Chicken anti-human C4c-alkaline
phosphatise
Immunsystem AB
Donkey anti-goat IgG (whole
molecule) alkaline phosphatase
antibody
Sigma-Aldrich
Goat anti-rabbit IgG (whole molecule)
Alkaline phosphatase antibody
Sigma-Aldrich
Rat anti-mouse MBL-A monoclonal
antibody
Hycult
Rat anti-mouse MBL-C monoclonal
antibody
Hycult
Rabbit anti-human C3c polyclonal
antibody
Dako
Rabbit anti-mouse Ficolin A Prof. T. Fujita, Department of Immunology,
Fukushima Medical University School of
Medicine, Fukushima, Japan
Rabbit anti-mouse IgG (whole
molecule) Alkaline phosphatase
antibody
Sigma-Aldrich
Chapter 2: Materials and methods
48
Recombinant murine MASP-3 Dr Sadam Yassin
Department of infection, Immunity &
inflammation, University of Leicester (UK)
Recombinant murine properdin Dr Youssif Mohammed Ali
Department of infection, Immunity &
inflammation, University of Leicester (UK)
Rat anti-mouse CL-11 antibody
Dr. Soren Hansen
Department of Cancer and Inflammation
Research,
University of Southern Denmark (Denmark)

Chapter 2: Materials and methods
49
2.1.3 Media and buffers
Levinthal‟ssupplement
400ml of Brain heart infusion (BHI) was mixed
with 200 ml defibrinated horse blood and heated
at 45°C for 40 minutes. Then mixtures was
cooled for 15 minutes at room temperatures and
centrifugedat5350xgfor25minutesat4◦C.
Then the supernatant of mixture was aliquoted
into 40 mland kept at -20◦C.
Coating buffer 15 mM Na2CO3
35 mM NaHCO3
pH 9.6
Tris buffer saline (TBS) 10 mMTris-HCL
140 mMNaCl
pH 7.4
BSA-TBS blocking buffer TBS with 1% (w/v) BSA
pH 7.4
Washing buffer TBS with 0.05% tween-20
5mM CaCl2
pH 7.4
Barbital buffer saline (BBS) 4 mM barbital
145 mM NaCl
1 mM MgCl2
2 mM CaCl2
pH7.4

Chapter 2: Materials and methods
50
MBL binding buffer 20 mM Tris-HCl
10 mM CaCl2
1 M NaCl
pH 7.4
Ethylene glycol tetraacetic acid
(EGTA) buffer
4 mM barbital
145 mM NaCl
1 mM MgCl2
10 mM EGTA
pH7.4

Chapter 2: Materials and methods
51
2.2 Methods
2.2.1 In vitro experiments
2.2.1.1 Preparation of mouse and human serum
Sera were used in the immune biochemical test was extracted from mouse (by cardiac
Puncture) and healthy human (by vein puncture). For some assays sera was taken from
mice and humans who have a deficient on their complement components. Blood
samples were taken and kept on ice for 5 hours to avoid complement activation and
coagulation factors. Blood was then centrifuged in a cooled microcentrifuge at 14,000
rpm for 7 minutes. Serum was aliquated as 100µl into labelled Eppendorf tubes and
stored at -80°C.
2.2.1.2 Preparation of Neisseria meningitidis for Enzyme Linked Immunosorbent
Assay (ELISA)
Tow strains of Neisseria meningitidis were used (these tow strains are the same strains
that used in the previous study in our laboratories) which are serotype B (BMC-58) and
serotype A (Z2491). Neisseria meningitidis were grown overnight on brain heat
infusionmedium(BHI)supplementedwith5%Levanthal’s (to enrich the medium with
iron which is an important element for the Neisseria meningitidis growth) at 37⁰C and
5% CO2. Then the medium containing bacteria were centrifuged at 4000 ×g for 10
minutes. The pellet was then washed three times with phosphate buffered saline (PBS),
and 10ml of 0.5 % formalin was added to resuspend the pellet and then incubated for 60

Chapter 2: Materials and methods
52
minutes at room temperature. The pellet were centrifuged and washed three times with
PBS after the incubation. A loopful of the pellet was grown overnight on BHI agar
supplementedwith5%Levanthal’sat37⁰C and 5% CO2 to ensure the formalin fixation
by the absence of growth. Finally the pellet was resuspended in coating buffer (15 mM
Na2CO3, 35 mM NaHCO3, 0.02% sodium azide, pH 9.6) for later use after adjusting the
optical density (OD) of the resuspension to be at 0.6 (OD550= 0.6).
2.2.1.3 C1q deposition assays
Microtitre ELISA plates were coated with 100μl of different fixed Neisseria
meningitidis strains (OD550=0.6). As positive controls ELISA plate wells were coated
with1μg/mlbovineserumalbuminutes(BSA).Alsoas negative controls ELISA plate
wells received 100μlofthecoatingbuffer. Then the plates were incubated overnight in
a 4°C fridge. Next day, ELISA plates were incubated at RT for 2hours. Plates were
washed three times by washing buffer (TBS with 0.05% tween-20 and 5mM CaCl2)
before adding 100 μl of rabbit anti-BSA (2 μg/ml) to positive control wells and
incubated for 90 minutes at room temperature. Two fold serial dilutions of mouse serum
were prepared in barbital buffered saline (BBS; 4 mM barbital, 145 mM NaCl, 2 mM
CaCl2, 1 mM MgCl2, pH 7.4) starting from 1/80 (high dilution). Plates were washed
three times by washing buffer (TBS with 0.05% tween-20 and 5mM CaCl2) before
adding the 100μl of serum dilutions in duplicates into corresponding wells and
incubating at 37°C for 1hour. Washing buffer was used to repeat the washing followed
by the addition of 100μl of goat anti-humanC1q (2μg/ml). Plateswere incubated at
37°Cfor90minutesbeforerepeatingthewashingagain.100μlofsecondaryantibodies
(anti-goat IgG-alkaline phosphatase conjugate diluted 1/10000) was added to the wells

Chapter 2: Materials and methods
53
and incubated again at RT for 90 minutes. Plates were washed again, followed by the
addition of 100μl substrate solution (Fast pNPP tablet (p-Nitrophenyl phosphate Tablet)
sets, Sigma). Hydrolysis of substrates was monitored using BioRad microtitre ELISA
plate reader by measuring the absorption at 405nm.
2.2.1.4 MBL-A, MBL-C, Ficolin-A and CL-11 binding assays
Microtitre ELISA plates were coated with 100μl of different fixed Neisseria
meningitidis strains (OD550=0.6). As positive controls ELISA plate wells were coated
by1μg/mlmannaninMBL-A and MBL-Cassays,10μg/mlZymosaninCL-11 assays,
and10μg/mlN-acetylated BSA in ficolin-A assays. Also as negative controls ELISA
plate Wells received 100μl of TBS buffer (10 mM Tris, 140 mM NaCl, pH 7.4)
containing 1% of bovine serum albuminutes (BSA). Then the plates were incubated
overnightina4°Cfridge.Nextday,ELISAplateswereblockedby250μlofTBSbuffer
containing 1% BSA and incubated at room temperature (RT) for 2hours. Two fold serial
dilutions of mouse serum were prepared in MBL binding buffer (20 mM Tris-HCl, 10
mM CaCl2, 1 M NaCl, pH 7.4) starting from 1/20 (low dilution). Plates were washed
three times with washing buffer (TBS with 0.05% tween-20 and 5mM CaCl2) before
added100μlofserumdilutionsinduplicatesintocorrespondingwellsandincubatedat
37°C for 1hour. Washing buffer was used to repeat the washing, followed by the
additionof100μlofprimaryantibodiesratanti-mouse MBL-A (Hycult; 1mg/ml stock
solution), rat anti-mouse MBL-C (Hycult; 1mg/ml stock solution), rabbit anti-mouse
Ficolin A (Prof. T. Fujita, Department of Immunology, Fukushima Medical University
School of Medicine, Fukushima, Japan: 0.7 mg/ml stock solution) antibodies diluted
1/1000, rat anti-mouse CL-11 (Dr. Soren Hansen, Department of Cancer and Inflammation

Chapter 2: Materials and methods
54
Research, University of Southern Denmark: 2.04mg/ml stock solution) antibodies diluted
1/500. Plates were incubated at RT for 90 minutes before repeating the washing again.
100μl of secondary antibody (goat anti mouse IgG-alkaline phosphatase conjugate or
goat anti rabbit IgG-alkaline phosphatase conjugate diluted 1/10000) dilutions were
added to wells and incubated again at RT for 90 minutes. Plates were washed again,
followed by the addition of 100μl substrates solution (Fast pNPP tablet sets, Sigma).
Hydrolysis of substrates was monitored using BioRad microtitre ELISA plate reader by
measuring the absorption at 405nm.
2.2.1.5 C3 deposition assays
Microtitre ELISA plates were coated with 100μl of different fixed Neisseria
meningitidis strains (OD550=0.6). As positive controls ELISA plate wells were coated
with10μg/mlZymosan(Sigma).AlsoasnegativecontrolsELISAplateWellsreceived
100μlofTBSbuffer(10mMTris,140mMNaCl,pH7.4) containing 1% BSA. Then
the plates were incubated overnight in a 4°C fridge. Next day, ELISA plates were
blockedby250μlof TBS buffer (10 mM Tris, 140 mM NaCl, pH 7.4) containing of 1%
BSA and incubated at RT for 2hours. Two fold serial dilutions of mouse serum were
prepared in barbital buffered saline (BBS; 4 mM barbital, 145 mM NaCl, 2 mM CaCl2,
1 mM MgCl2, pH 7.4) starting from 1/80 (high dilution). Plates were washed three
times by washing buffer (TBS with 0.05% tween-20 and 5mM CaCl2) before addition
of the100μlofserumdilutionsinduplicatesintocorrespondingwellsandincubatingat
37°C for 1hour. Washing buffer was used to repeat the washing followed by the
addition of 100μl of rabbit anti-human C3c antibodies (Dako) diluted 1/5000. Plates
were incubated at 37°C for 90minutes before repeating thewashing again. 100μl of

Chapter 2: Materials and methods
55
secondary antibodies (goat anti rabbit IgG-alkaline phosphatase conjugate (Sigma)
diluted 1/10000) was added to the wells and incubated again at RT for 90 minutes.
Plates were washedagain, followedby theadditionof100μl substrates solution (Fast
pNPP tablet sets, Sigma). Hydrolysis of substrates was monitored using BioRad
microtitre ELISA plate reader by measuring the absorption at 405nm.
2.2.1.6 Alternative pathway mediated C3 deposition assays
Microtitre ELISA plates were coated with 100μl of different fixed Neisseria
meningitidis strains (OD550=0.6). As positive controls ELISA plate wells were coated
with10μg/mlZymosan(Sigma).AlsoasnegativecontrolsELISAplate Wells received
100μlofTBS buffer (10 mM Tris, 140 mM NaCl, pH 7.4) containing 1% BSA. Then
the plates were incubated overnight in a 4°C fridge. Next day, ELISA plates were
blockedby250μlofTBS buffer (10 mM Tris, 140 mM NaCl, pH 7.4) containing 1%
BSA and incubated at RT for 2hours. Two fold serial dilutions of mouse serum were
prepared in Ethylene glycol tetra acetic acid (EGTA) buffered (4 mM barbital, 145 mM
NaCl, 10 mM EGTA, 1 mM MgCl2, pH 7.4) starting from ½ (low dilution). Plates were
washed three times by washing buffer (TBS with 0.05% tween-20) before adding the
100μlofserumdilutionsinduplicatesintocorrespondingwellsandincubatingat37°C
for 1hour. Washing buffer was used to repeat the washing followed by the addition of
100μlofrabbitanti-human C3c antibodies (Dako) diluted 1/5000. Plates were incubated
at 37°C for 90 minutes before repeating the washing again. 100μl of secondary
antibodies (goat anti rabbit IgG-alkaline phosphatase conjugate (Sigma) diluted
1/10000) was added to the wells and incubated again at RT for 90 minutes. Plates were
washedagain,followedbytheadditionof100μlsubstratessolution(FastpNPPtablet

Chapter 2: Materials and methods
56
sets, Sigma). Hydrolysis of substrates was monitored using BioRad microtitre ELISA
plate reader by measuring the absorption at 405nm.
2.2.1.7 Lectin pathway mediated C4 deposition assays
Microtitre ELISA plates were coated with 100μl of different fixed Neisseria
meningitidis strains (OD550=0.6). As positive controls ELISA plate wells were coated
with10μg/mlZymosan(Sigma).Alsoasnegativecontrols ELISA plate Wells received
100μlofTBSbuffer(10mMTris,140mMNaCl,pH7.4) containing 1% BSA. Then
the plate incubated overnight in a 4°C fridge. Next day, ELISA plates were blocked by
250μl ofTBS buffer (10 mM Tris, 140 mM NaCl, pH 7.4) containing 1% BSA and
incubated at RT for 2hours. Two fold serial dilutions of mouse serum were prepared in
MBL binding buffer (20 mM Tris-HCl, 10 mM CaCl2, 1 M NaCl, pH 7.4) starting from
1/80 (high dilution). Plates were washed three times by washing buffer (TBS with
0.05% tween-20 and 5mM CaCl2) before adding the 100μl of serum dilutions in
duplicates into corresponding wells and incubating at 37°C for 1hour. Washing buffer
wasusedtorepeatthewashingfollowedbytheadditionof100μlof1μg/mlhumanC4
diluted in BBS. Plates were incubated at 37°C for 90 minutes before repeating the
washing again. 100μl of chicken anti-human C4c antibody alkaline phosphatase
conjugate diluted 1/10000 was added to the wells and incubated again at RT for 90
minutes. Plates were washed again, followed by the addition of 100μl substrates
solution (Fast pNPP tablet sets, Sigma). Hydrolysis of substrates was monitored using
BioRad microtitre ELISA plate reader by measuring the absorption at 405nm.

Chapter 2: Materials and methods
57
2.2.1.8 Serum Bactericidal Assay (SBA)
Serum bactericidal assay (after modification on Estabrook et al (1997) was used to
assess the sensitivity of Neisseria meningitidis toward different serum samples.
Neisseria meningitidis wasgrownin10mlBHIsupplementedwith5%Levanthal’sand
incubated 3 hours at 37⁰C in 5% CO2. After this time, a sample of the growth was taken
and the optical density at 600nm was measured (which corresponds to 109 CFU/ml) to
check the count. Then aliquots of 500µl were made of the growth and kept at -80⁰C.
Viable counts were done to confirm the bacterial count. An aliquot was resuspended in
phosphate buffered saline (PBS) to get the desired count of bacteria. A suspension of a
known concentration of Neisseria meningitidis (105) was mixed with a desired
concentration of serum samples on Barbital buffer saline and incubated at 37oC, with
shaking at 120rpm. Samples of reaction were taken at 0, 30, 60, 90, and 120 min, and
serial dilutions in phosphate buffered saline (PBS) have prepared and plated onto Brain
heart infusion (BHI) agar with5%Levanthal’ssupplement and incubated overnight at
37oC in 5% CO2.
2.2.1.9 Preparation of Neisseria meningitidis for FACS analysis
Neisseria meningitidis Strain B serogroup MC58 grew overnight on brain heat infusion
medium (BHI) supplemented with 5% Levanthal’s at 37⁰C and 5% CO2. Then the
medium containing bacteria were centrifuged at 4000 ×g for 10 minutes. The pellet was
then washed three times with phosphate buffered saline (PBS), and 10ml of 0.5 %
formalin was added to resuspend the pellet, which was then incubated it for 60 minutes
at room temperature. Subsequently, the pellet was centrifuged and washed three times

Chapter 2: Materials and methods
58
with TBS buffer (10 mM Tris, 140 mM NaCl, pH 7.4). A loopful of the pellet was
grownovernightonBHIagarsupplementedwith5%Levanthal’sat37⁰C and 5% CO2
to ensure the formalin fixation by the absence of growth. Finally, the pellet was
resuspended in EGTA buffer (4 mM barbital, 145 mM NaCl, 10 mM EGTA, 1 mM
MgCl2, pH 7.4) for later use.
2.2.1.10 FACS analysis for detect C3 deposition on Neisseria meningitdis
Fixed Neisseria meningitidis was washed three times with TBS buffer (10 mM Tris, 140
mM NaCl, pH 7.4) before resuspended the pellets in EGTA buffer (4 mM barbital, 145
mM NaCl, 10 mM EGTA, 1 mM MgCl2, pH 7.4). 100µl of the suspend bacteria were
opsonised with 200µl of 5% normal human serum or 15% wildtype mouse serum at
37°C with or without 5µg/ml of recombinant properdin. Nonopsonised bacteria were
used as negative control. After 1 hour, bacterial samples were centrifuged and washed
three times with TBS buffer. 200µl of FITC-conjugated rabbit anti-human C3c diluted
1/5000 was added to bacteria and incubated at RT for I hour. Bacterial sample was then
centrifuged and washed three times again with TBS buffer before resuspending the
bacteria in 1ml PBS buffer. The fluorescence intensity was measured with a
FACSCalibur cell analyser (BD Biosciences).

Chapter 2: Materials and methods
59
2.2.2 In vivo experiments
2.2.2.1 Genotyping of fB and MASP-1/3 deficient mice
2.2.2.1.1 Isolation of genomic DNA from mouse ear snips
Around 4mm of mice ear snips were incubated overnight at 55°C and gently mixed in
master mix containing 250µl of Nuclei Lysis Solution, 60µl of 0.5M EDTA solution,
and10µlofProteinaseK(Qiagen).Onthenextday,1.5μlofRNasesolution(4mg/ml)
was added to the mixture and mixed by inverting the tube three times, and incubated at
37°Cfor30mins.Thesamplewascooledtoroomtemperatureforfivemins,100μlof
protein precipitation was added to the tube and it was vortexed at high speed for 20 sec.
Ice was used to cool the sample for five mins, then it was centrifuged for four mins at
13,000rpm. Supernatant was taken to a clean labelled tube, then300μlof isopropanol
was added to the supernatant and then centrifuged for five mins at 13000rpm.
Supernatant wasdiscardedand300μlof70% ethanol was added. The tube was gently
inverted three times then centrifuged for one min at 13000rpm. The tube was dried from
ethanolfor30minbyinvertingthetubeoncleanabsorbentpaper,then100μlofDNA
re-hydration solution was added and it was stored at 4°C overnight.
2.2.2.1.2 Polymerase Chain Reaction (PCR)
PCR is a powerful method to amplify a specific region of DNA that lies between known
sequences of DNA. A standard PCR reaction is composed of three main steps
(denaturation, annealing and DNA synthesis), starting with a denaturation step during
which the DNA template is denatured by heating to a high temperature, followed by a
cooling step which allows the primer to link to its target, and finally an amplification
end step by optimal temperature that allows synthesis of the DNA. These steps are
repeated 35 times using an automated thermal cycler.

Chapter 2: Materials and methods
60
PCR was done to determine the genomic DNA (gDNA) and therefore identify the fB+/+
,
fB+/-
, fB-/-
, MASP-1/3 +/+
, MASP-1/3 +/-
, and MASP-1/3 -/-
mice.
The primers used for genotyping of fB are
Primer name Primer sequence
FB_F25’- GAAGGACCTAGAAACAGCGCTCA-3’
FB_WTO_R15’- CTGATCTACCTTCTCAATCAAGTTGGTGA-3’
Neo3_F5 5’-CTGTTGTGCCCAGTCATAGCCGA-3’
The primers used for genotyping of MASP-1/3 are
Primer name Primer sequence
NeoU 5’- CAT CGC CTT CTA TCG CCT TCT TGA-3
M1U5’- CTC CCT GCC TCA GAC TGT TTG ATA-3’
Mil5’- GCT GAT GCT GAT GTT AGG ATG GTA TTC-3’
Each PCR reaction tube contains:
Genomic DNA(200ng/μl) 1μl
Reaction buffer (10x) 1.5μl
MgCl2 (2.5mM) 1.5μl
dNTP mix (10mM) 0.3μl
M2screen_F1 1.5μl
M2wto_R1 1.5μl
Neo5_R1 1.5μl
Taq-DNA polymerase 0.12μl
Nanopure distilled water 6.08μl

Chapter 2: Materials and methods
61
2.2.2.2 Preparation of Neisseria meningitidis passage
Neisseria meningitidis replication depends on the presence of iron as it an important
factor to perform some metabolic functions by acting as a cofactor for some enzymes
including enzymes required for DNA replication, oxygen metabolism and electron
transport. A previous study made it clear that the mice that were injected with iron (iron
dextran or human transferrin) before challenging them with Neisseria meningitidis had
ability to develop a lethal infection. However, the mice that not injected with iron could
not develop a lethal infection (Griffiths, 1999; Holbein, 1980; Holbein et al., 1979).
A mouse was injected with iron intraperitoneally (400mg/kg body weight) 12 hours
before the infective dose. Neisseria meningitidis was grown in 10ml BHI supplemented
with5%Levanthal’sandincubatedfor3hoursat37⁰C in 5% CO2. After this time, a
sample of the growth was taken and the optical density at 600nm was measured (which
corresponds to 109 CFU/ml) to check the count. Subsequently, aliquots of 500µl of the
growth were made and kept at -80⁰C. Viable counts were done to confirm the bacterial
count. An aliquot of it was resuspended in phosphate buffered saline (PBS) and 100µl
of resuspended bacteria (109
CFU/ML) was injected into the mouse intraperitoneally.
On the following day, the uses mouse culled by cervical dislocation once it developed
the sign of terminal illness and a blood sample was collected by cardiac puncture. The
blood sample was streaked on brain heart infusion (BHI) agar supplemented with 5%
Levanthal’sandincubatedovernightat37⁰C in 5% CO2.
A loopful of colonies from the overnight culture was picked up and resuspended in
10ml serumBHI (80% BHI supplementedwith 5% Levanthal’s and 20% foetal calf

Chapter 2: Materials and methods
62
serum (FCS)) and incubated at 37⁰C in 5% CO2 for 3 hours. After this time, a sample of
the growth was taken and the optical density at 600nm was measured (which
corresponds to 109 CFU/ml) to check the count. Afterwards, aliquots of 500µl of the
growth were made and kept at -80⁰C. Viable counts were done to confirm the bacterial
count of the stocks. For each infection experiment, an aliquot of the stock was thawed at
room temperature, spun down, and resuspended in PBS to get the desired concentration
of bacteria. The bacterial concentration was confirmed by doing serial dilution in PBS
andplatingitontobrainheart infusion(BHI)agarsupplementedwith5%Levanthal’s
and incubated overnight at 37⁰C in 5% CO2.
2.2.2.3 Virulence testing of passaged stocks of Neisseria meningitidis
Wild-type mice were used to assess the virulence of the passaged stocks of Neisseria
meningitidis. Mice were injected intraperitoneally with iron dextran (400mg/kg body
weight) 12 hours before infection dose. An aliquot of passaged stock of Neisseria
meningitidis was thawed at room temperature, centrifuged and resuspended in PBS to
obtain the required viable count. Mice were divided to three groups and injected with
required bacterial doses of 1 x105, 1 x10
6, and 1 x10
7 CFU/mouse diluted in sterile 100
µl PBS. Following infection, mice were regularly monitored every six hours for the
illness symptoms. The signs of illness were scored (table 2.1) based on the scheme
(with some modifications) of Fransen etal (2010). Once mice developed signs of
terminal illness, they were culled by cervical dislocation and a blood sample was
collected by cardiac puncture under aseptic conditions for bacterial load determination.

Chapter 2: Materials and methods
63
Table 2.1 The severity scores of disease with clinical signs of infected mice
Signs Score
Normal 0
Slightly ruffled fur 1
Ruffled fur, slow and sticky eyes 2
Ruffled fur, lethargic and eyes shut 3
Very sick and no movement after
stimulation 4
Dead 5
2.2.2.4 Infection of mice with Neisseria meningitidis
Wild-type mice and mice deficient in their alternative pathway components (fB-/-
or
MASP-1/3-/-
) have been used in the infection studies. Mice ranged in age from 12 to 14
weeks. Mice were injected intraperitoneally with iron dextran (400mg/kg body weight)
12 hours before infection dose. An aliquot of passaged stock of Neisseria meningitidis
was thawed at room temperature, centrifuged and resuspended in PBS to obtain the
required viable count 1 x105 CFU/mouse diluted in sterile 100 µl PBS. Following
infection, mice were monitored every six hours for the illness symptoms. Once mice
developed the signs of terminal illness (very sick and no movement after simulation), it
was culled immediately by cervical dislocation and a blood sample collected by cardiac
puncture following by determined of Blood bacterial burden.
2.2.2.5 Determination of Blood bacterial burden
Following the introducing of the pathogen to the mice intraperitoneally, the course of
infection was monitored by time course of bacteraemia. Every six hours of infection,

Chapter 2: Materials and methods
64
mice were placed in thermocage at 37°C for 20 minutes to worm the mice and dilate the
veins in mice tails. Mice blood was collected from the tails of mice and serial diluted in
sterile PBSandplatingonBHIagar,supplementedwith5%Levanthal’sandincubated
overnight at 37⁰C in 5% CO2 to count the bacteria.
2.2.2.6 MASP-3 reconstitution experiment
Wild-type mice and MASP-1/3-/-
mice were divided to three groups: Group A wild type
mice were injected intravenously with PBS, group B MASP-1/3-/-
mice were injected
intravenously with20μg/mouseofrecombinantmouseMASP-3 at 0 hour, and group C
MASP-1/3-/-
micewereinjectedintravenouslywith20μg/mouseofrecombinantmouse
MASP-3 at 0 hour and 72 hours. Mice were bled at 0, 24, 48, 72 and 96 hours post
reconstitution. Alternative pathway C3 deposition assay was performed. Microtitre
ELISA plates were coated with with 10μg/ml Zymosan (Sigma) and incubated
overnightina4°Cfridge.Nextday,ELISAplateswereblockedby250μlof1%bovine
serum albuminutes (BSA) in TBS buffer (10 mM Tris, 140 mM NaCl, pH 7.4) and
incubated at RT for 2hours. Two fold serial dilutions of mice sera were prepared in
Ethylene glycol tetraacetic acid (EGTA) buffered saline (4 mM barbital, 145 mM NaCl,
10 mM EGTA, 1 mM MgCl2, pH 7.4) starting at 13% concentration. Plates were
washed three times by washing buffer (TBS with 0.05% tween-20) before adding the
100μlofserumdilutionsinduplicatesintocorrespondingwellsandincubatingat37°C
for 1hour. Wells received only washing buffer were used as negative controls. Washing
bufferwasusedtorepeatthewashing,followedbytheadditionof100μlofrabbitanti-

Chapter 2: Materials and methods
65
human C3c antibodies diluted 1/5000. Plates were incubated at 37°C for 90 minutes
before repeating the washing again. 100μl of secondary antibodies (α-rabbit IgG-
alkaline phosphatase conjugate diluted 1/10000) was added to the wells and incubated
again at RT for 90 minutes. Plates were washed again, followed by the addition of
100μlsubstratessolution(FastpNPPtabletsets,Sigma).Hydrolysisofsubstrates was
measured using BioRad microtitre ELISA plate reader to read an absorbance at 405nm.
2.2.3 Statistical analysis
GraphPad prism version 6 was used to preform experimental statistical analysis
by using unpaired t-test for all experiments except the in vivo survival
experiments where Log-rank (Mantel-Cox) test was used.

Chapter 3: In vitro study
66
Chapter 3: In vitro study
The complement system is a vital part of the immune system and plays a crucial role in
fighting against invading microbes including Neisseria meningitidis. However the role
of different complement pathways in fighting Neisseria meningitidis infection needs to
be clarified which could be done by studying the binding of the complement pathways
recognition molecules to the Neisseria meningitidis and by studying the bacteriolytic
activity of different mice sera against Neisseria meningitidis.
3.1 Results:
3.1.1 Complement pathway specific Enzyme Linked Immune Sorbent
Assays (ELISAs)
The initiation of either of the three complement activation pathways on the surface of N.
meningitidis depends on the binding of complement recognition molecules to
meningococcus. The binding of various complement recognition molecules to different
serotypes of Neisseria meningitidis strains (i.e. serotype A strain Z2491 and serotype B
strain MC58) were examined. This results show that meningococcus serotype A strain
Z2491 and serotype B strain MC58 have the ability to bind to C1q, the recognition
molecule of classical pathway, and recognition molecules of the lectin pathway
including mouse MBL-A, MBL-C and CL-11. In contrast, ficolin-A a recognition
molecule of lectin pathway in the mouse showed no binding to any of the
meningococcal serotypes, i.e. serotype A strain Z2491 and serotype B strain MC58.

Chapter 3: In vitro study
67
3.1.1.1 Binding of the classical pathway molecule C1q to Neisseria meningitidis
A C1q binding assay was used to investigate the role of the classical pathway in fighting
Neisseria meningitidis infection in the absence of specific antibodies against Neisseria
meningitidis by studying the binding of C1q to the surface of different Neisseria
meningitidis strains. This work showed that C1q bound to the surface of Neisseria
meningitidis strains, indicating that the classical pathway may possibly contribute to the
formation of C3 convertases deposited on the surface of Neisseria meningitidis (See
Figure 3.1).
0 .0 0 .2 0 .4 0 .6 0 .8 1 .0 1 .2 1 .4
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
1 .2
1 .4
1 .6
1 .8
% s e r u m c o n c e n t r a t io n
OD
At
40
5n
m
B S A im m u n e co m p le x
S e ro A -Z 2 4 9 1
S e ro B -M C 5 8
N e g a tiv e c o n tro l
Figure 3.1 C1q binding on the surface of different Neisseria meningitidis strains.
Mouse serum was diluted in BBS. BSA anti-BSA coating was used as positive control.
The negative control wells received BSA blocking buffer as a coating. Binding of C1q
was detected by using Goat anti-human C1q polyclonal antibody (the data is presented
as a mean of all three independent experiments with duplicate for each +/- SEM).

Chapter 3: In vitro study
68
3.1.1.2 Binding of the lectin pathway recognition component MBL to Neisseria
meningitidis
The contribution of the lectin pathway recognition subcomponent Mannan Binding Lectin
(MBL) in fighting Neisseria meningitidis infection was examined by assessing the binding of
the murine lectin pathway recognition molecules MBL-A and MBL-C to the surface of
Neisseria meningitidis. The binding of MBL-A and MBL-C to the surface of Neisseria
meningitidis outer membrane proteins (outer membrane protein A and porin B) had been
reported previously (Estabrook et al., 2004). It has also been reported that the structure and
sialylation of lipooligoccharides (LOS) acts as a binding site for MBL (Jack et al., 1998; Jack et
al., 2001). MBL-A and MBL-C binding assays were carried to assess the relative binding
affinity of the recognition subcomponents to the bacterial surface of different N. meningitidis
serotypes. These assays showed that MBL-A and MBL-C had the ability to bind to the surface
of all tested Neisseria meningitidis strains (see Figures 3.2 and 3.3).
0 .0 0 .2 0 .4 0 .6 0 .8 1 .0 1 .2 1 .4
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
1 .2
1 .4
% s e r u m c o n c e n t r a t io n
OD
At
40
5n
m
M a n n a n , M B L -A
S e ro B -M C 5 8 , M B L -A
S e ro A -Z 2 4 9 1 , M B L -A
N e g a tiv e C o n tro l M B L - A
Figure 3.2 MBL-A binding on the surface of different Neisseria meningitidis strains. Mouse
serum was diluted in MBL binding buffer. Mannan coating was used as a positive control. The
negative control wells received BSA blocking buffer as a coating. Binding of MBL-A was
detected by using rat anti-mouse MBL-A monoclonal antibody (the data is presented as a mean
of three independent experiments with duplicate for each +/- SEM).

Chapter 3: In vitro study
69
0 .0 0 .2 0 .4 0 .6 0 .8 1 .0 1 .2 1 .4
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
1 .2
1 .4
1 .6
% S e r u m C o n c e n t r a t io n
OD
At
40
5n
m
S e ro B -M C 5 8, M B L -C
S e ro A -Z 2 4 9 1 , M B L -C
M an na n, M B L-C
N e ga tiv e C on tro l, M B L - C
Figure 3.3 MBL-C binding on the surface of different Neisseria meningitidis strains. Mouse
serum was diluted in MBL binding buffer. Mannan coating was used as a positive control. The
negative control wells received BSA blocking buffer as a coating. Binding of MBL-C was
detected by using rat anti-mouse MBL-C monoclonal antibody (the data is presented as a mean
of all three independent experiments with duplicate for each +/- SEM).
3.1.1.3 Binding of the lectin pathway Collectin-11 to Neisseria meningitidis
Collectin-11 (CL-11) is a member of the lectin pathway and has recently been identified
as a recognition molecule of the lectin pathway, which can bind to the surface of the
pathogen (1-fucose and d-mannose sugars) and activate the lectin pathway. Binding of
CL-11 to Neisseria meningitidis has been done to assess the binding of CL-11 to the
Neisseria meningitidis. Using the direct binding assay shown in Figure 3.4 it was shown
that CL-11 had the ability to bind to the surface of the two Neisseria meningitidis strains
tested (see Figure 3.4).

Chapter 3: In vitro study
70
0 .0 0 .2 0 .4 0 .6 0 .8 1 .0 1 .2 1 .4
0 .0
0 .1
0 .2
0 .3
0 .4
0 .5
0 .6
0 .7
0 .8
0 .9
Z y m o s a n
S e ro A -Z 2 4 9 1
S e ro B -M C 5 8
% s e r u m c o n c e n t r a t io n
OD
At
40
5n
m
N e g a tiv e c o n tro l
Figure 3.4 CL-11 binding to the surface of different Neisseria meningitidis strains. Mouse
serum was diluted in MBL binding buffer. Zymosan coating was used as a positive control. The
negative control wells received BSA blocking buffer as a coating. Binding of CL-11 was
detected by using Rat anti-mouse CL-11 antibody (the data is presented as a mean of all three
independent experiments with duplicate for each +/- SEM).
3.1.1.4 Binding of the lectin pathway ficolin-A to Neisseria meningitidis
Ficolin-A is one of the recognition molecules of the lectin pathway belonging to the ficolin
family that can activate the lectin pathway by binding to N-acetylglucosamine on the surface of
pathogens. In mice, there are two types of ficolins, ficolin- A and ficolin-B. While ficolin-B is
synthesised mainly in myeloid cells in the bone marrow, ficolin-A is predominantly synthesised
in the liver. Ficolin A is considered to be the only murine ficolin that can bind to the mannan
binding lectin associated serine proteases (MASPs) to activate the lectin pathway (Endo et al.,
2005; Runza et al., 2008). The binding of ficolin-A to Neisseria meningitidis was assessed to
evaluate the binding of ficolin-A the Neisseria meningitidis. The failure to detect any binding of
ficolin-A to the surface of Neisseria meningitidis strains indicates that deficiency of ficolin-A

Chapter 3: In vitro study
71
might not predispose for a higher predisposition to Neisseria meningitidis infections (see Figure
3.5).
0 .0 0 .2 0 .4 0 .6 0 .8 1 .0 1 .2 1 .4
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
1 .2
1 .4
1 .6
% s e r u m c o n c e n t r a t io n
OD
At
40
5n
m
N -A c e ty l a lb u m in
S e ro B -M C 5 8
S e ro A -Z 2 4 9 1
N e g a tiv e c o n tro l
Figure 3.5 Ficolin-A does not bind to the surface of different Neisseria meningitidis strains.
Mouse serum was diluted in MBL binding buffer. N-Acetyl albumin coating was used as a
positive control. The negative control wells received BSA blocking buffer as a coating. Binding
of the ficolin-A was detected by using rabbit anti-mouse ficolin-A antibody (the data is
presented as a mean of all three independent experiments with duplicate for each +/- SEM).
3.1.2 C3 deposition assays
C3 deposition assays were performed to assess the ability of Neisseria meningitidis to
activate complement pathways by measuring the amount of C3 that was deposited on
the surface of Neisseria meningitidis strains. In these assays, an antibody against C3c,
which is generated from the cleavage of C3 and binds to ELISA plates, was used to

Chapter 3: In vitro study
72
detect the cleavage of C3. Specific conditions were chosen to allow activation of the
classical and lectin pathways, but not the alternative pathway (as highly diluted serum
makes the alternative pathway dysfunctional). High levels of C3 deposition was
detected on the surface of the two Neisseria meningitidis strains tested. Serogroup B
strain MC-58 showed more C3 depositions compared to serogroup A strain Z2491 (see
Figure 3.6).
0 .0 0 .2 0 .4 0 .6 0 .8 1 .0 1 .2 1 .4
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
1 .2
1 .4
1 .6
1 .8
% s e r u m c o n c e n t r a t io n
OD
At
40
5n
m
Z y m o s a n
S e ro A -Z 2 4 9 1
S e ro B -M C 5 8
N e g a t iv e c o n tro l
Figure 3.6 C3 deposition assay on the surface of different Neisseria meningitidis strains under
specific condition (high serum dilution in BBS buffer) allows the activation through classical
and lectin pathways. Mouse serum was diluted on BBS binding buffer. Zymosan coating has
used as a positive control while the negative control wells received BSA blocking buffer as a
coating. The binding of the C3 cleavage product (C3c) was detected by using rabbit anti-human
C3c polyclonal antibody (the data is presented as a mean of all three independent experiments
with duplicate for each +/- SEM).
To assess the role of the alternative pathway in fighting Neisseria meningitidis, an
alternative pathway specific C3 deposition assay was designed using high serum

Chapter 3: In vitro study
73
concentrations (sera were diluted in and alternative pathway permissive EGTA buffer
that contains Mg++
(in absence of Ca++
complement can only be activated via the
alternative pathway). A high level of C3 deposition was observed on Neisseria
meningitidis serogroup B strain MC58, while a moderate level of C3 deposition was
observed on Neisseria meningitidis serogroup A strain Z2491. These results suggests
that the alternative pathway plays a role in fighting Neisseria meningitidis infection (see
Figure 3.7).
0 1 0 2 0 3 0 4 0 5 0 6 0
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
1 .2
1 .4
% s e r u m c o n c e n t r a t io n
OD
At
40
5n
m
Z y m o s a n
S e ro B -M C 5 8
S e ro A -Z 2 4 9 1
N e g a t iv e c o n tro l
Figure 3.7 C3 deposition assay on the surface of different Neisseria meningitidis strains under
alternative pathway permissive conditions (high serum concentration in EGTA buffer). Mouse
serum was diluted in EGTA buffer. Zymosan coating was used as a positive control while the
negative control wells received BSA blocking buffer as a coating. The binding of the C3
cleavage product (C3c) was detected by using rabbit anti-human C3c polyclonal antibody (the
data is presented as a mean of all three independent experiments with duplicate for each +/-
SEM).
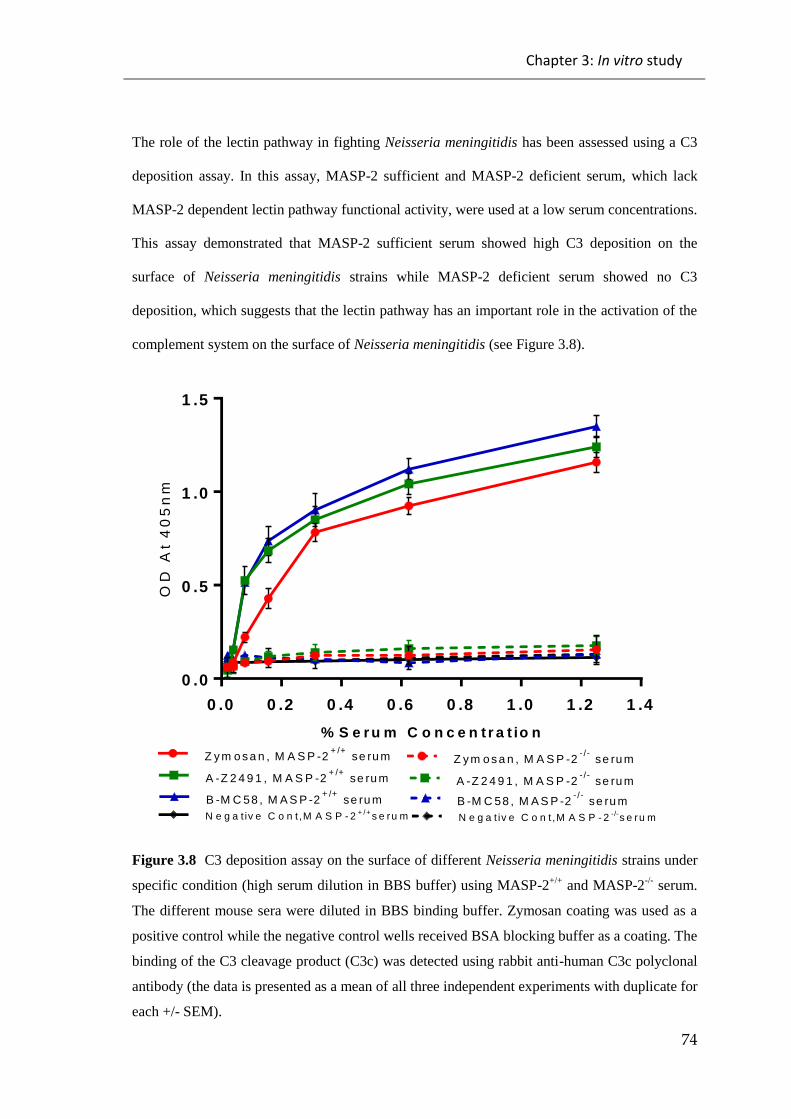
Chapter 3: In vitro study
74
The role of the lectin pathway in fighting Neisseria meningitidis has been assessed using a C3
deposition assay. In this assay, MASP-2 sufficient and MASP-2 deficient serum, which lack
MASP-2 dependent lectin pathway functional activity, were used at a low serum concentrations.
This assay demonstrated that MASP-2 sufficient serum showed high C3 deposition on the
surface of Neisseria meningitidis strains while MASP-2 deficient serum showed no C3
deposition, which suggests that the lectin pathway has an important role in the activation of the
complement system on the surface of Neisseria meningitidis (see Figure 3.8).
0 .0 0 .2 0 .4 0 .6 0 .8 1 .0 1 .2 1 .4
0 .0
0 .5
1 .0
1 .5
% S e r u m C o n c e n t r a t io n
OD
At
40
5n
m
Z y m o s a n , M A S P -2+ /+
se ru m Z y m o s a n , M A S P -2- /-
se ru m
A -Z 2 4 9 1 , M A S P -2+ /+
se ru m A -Z 2 4 9 1 , M A S P -2- /-
se ru m
B -M C 58 , M A S P -2+ /+
se ru m B -M C 58 , M A S P -2- /-
se ru m
N e g a t iv e C o n t ,M A S P - 2+ /+
s e ru m N e g a t iv e C o n t ,M A S P - 2-/-
s e ru m
Figure 3.8 C3 deposition assay on the surface of different Neisseria meningitidis strains under
specific condition (high serum dilution in BBS buffer) using MASP-2+/+
and MASP-2-/-
serum.
The different mouse sera were diluted in BBS binding buffer. Zymosan coating was used as a
positive control while the negative control wells received BSA blocking buffer as a coating. The
binding of the C3 cleavage product (C3c) was detected using rabbit anti-human C3c polyclonal
antibody (the data is presented as a mean of all three independent experiments with duplicate for
each +/- SEM).
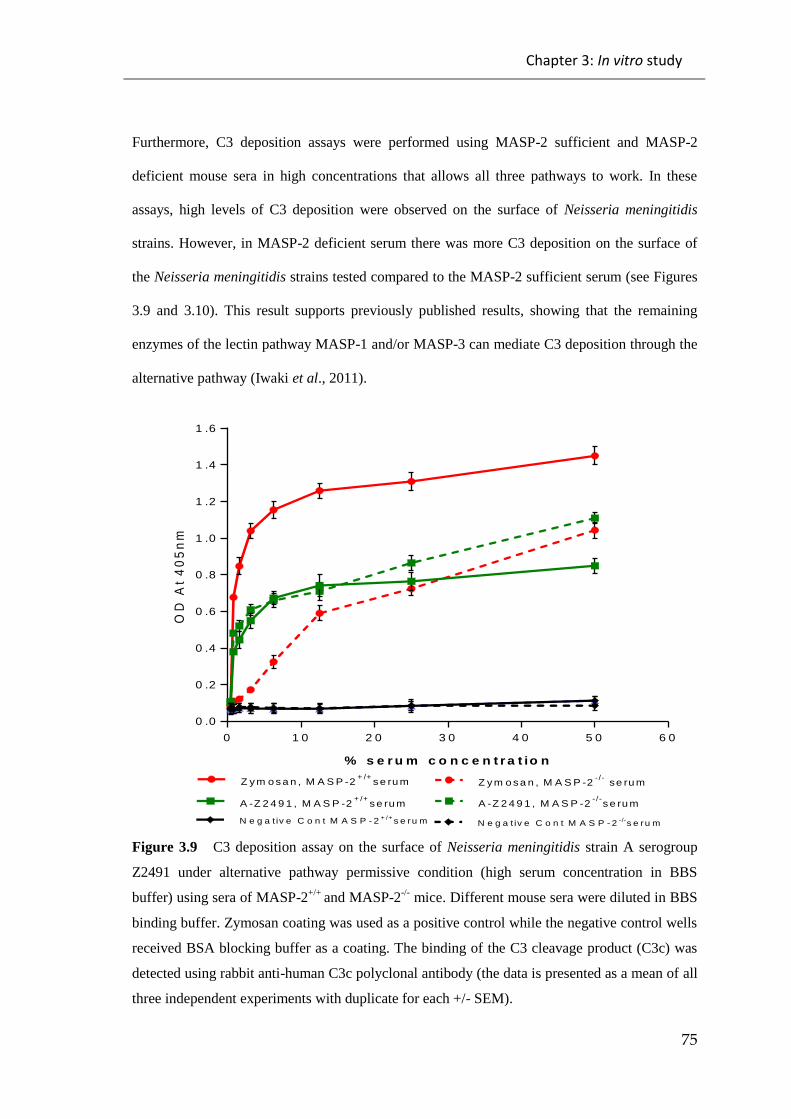
Chapter 3: In vitro study
75
Furthermore, C3 deposition assays were performed using MASP-2 sufficient and MASP-2
deficient mouse sera in high concentrations that allows all three pathways to work. In these
assays, high levels of C3 deposition were observed on the surface of Neisseria meningitidis
strains. However, in MASP-2 deficient serum there was more C3 deposition on the surface of
the Neisseria meningitidis strains tested compared to the MASP-2 sufficient serum (see Figures
3.9 and 3.10). This result supports previously published results, showing that the remaining
enzymes of the lectin pathway MASP-1 and/or MASP-3 can mediate C3 deposition through the
alternative pathway (Iwaki et al., 2011).
0 1 0 2 0 3 0 4 0 5 0 6 0
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
1 .2
1 .4
1 .6
% s e r u m c o n c e n t r a t io n
OD
At
40
5n
m
Z y m o s a n , M A S P -2+ /+
s e ru m Z y m o s a n , M A S P -2- /-
se ru m
N e g a t iv e C o n t M A S P - 2-/-
s e ru mN e g a t iv e C o n t M A S P - 2+ /+
s e ru m
A -Z 2 4 9 1 , M A S P -2+ /+
s e ru m A -Z 2 4 9 1 , M A S P -2- /-
s e ru m
Figure 3.9 C3 deposition assay on the surface of Neisseria meningitidis strain A serogroup
Z2491 under alternative pathway permissive condition (high serum concentration in BBS
buffer) using sera of MASP-2+/+
and MASP-2-/-
mice. Different mouse sera were diluted in BBS
binding buffer. Zymosan coating was used as a positive control while the negative control wells
received BSA blocking buffer as a coating. The binding of the C3 cleavage product (C3c) was
detected using rabbit anti-human C3c polyclonal antibody (the data is presented as a mean of all
three independent experiments with duplicate for each +/- SEM).
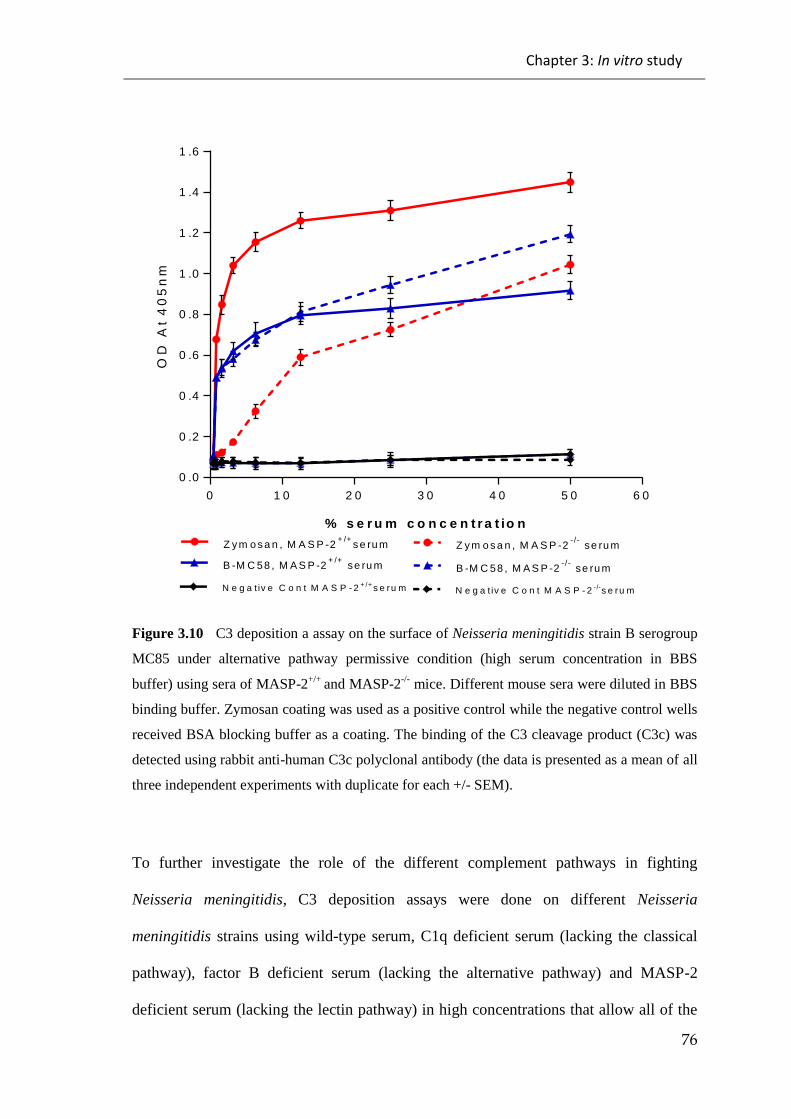
Chapter 3: In vitro study
76
0 1 0 2 0 3 0 4 0 5 0 6 0
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
1 .2
1 .4
1 .6
% s e r u m c o n c e n t r a t io n
OD
At
40
5n
m
Z y m o s a n , M A S P -2+ /+
s e ru m Z y m o s a n , M A S P -2- /-
se ru m
B -M C 58 , M A S P -2+ /+
se ru m
N e g a t iv e C o n t M A S P - 2-/-
s e ru mN e g a t iv e C o n t M A S P - 2+ /+
s e ru m
B -M C 58 , M A S P -2- /-
se ru m
Figure 3.10 C3 deposition a assay on the surface of Neisseria meningitidis strain B serogroup
MC85 under alternative pathway permissive condition (high serum concentration in BBS
buffer) using sera of MASP-2+/+
and MASP-2-/-
mice. Different mouse sera were diluted in BBS
binding buffer. Zymosan coating was used as a positive control while the negative control wells
received BSA blocking buffer as a coating. The binding of the C3 cleavage product (C3c) was
detected using rabbit anti-human C3c polyclonal antibody (the data is presented as a mean of all
three independent experiments with duplicate for each +/- SEM).
To further investigate the role of the different complement pathways in fighting
Neisseria meningitidis, C3 deposition assays were done on different Neisseria
meningitidis strains using wild-type serum, C1q deficient serum (lacking the classical
pathway), factor B deficient serum (lacking the alternative pathway) and MASP-2
deficient serum (lacking the lectin pathway) in high concentrations that allow all of the

Chapter 3: In vitro study
77
three complement pathways to work. The highest level of C3 deposition was shown
with the MASP-2 deficient serum which could be mediated by the alternative and the
classical pathways. C1q deficient serum showed significant amounts of C3 deposition
which could have been mediated by both the lectin and the alternative pathways. Factor
B deficient serum showed a limited amount of C3 deposition which could have been
mediated by both the classical and the lectin pathways. These results suggested that the
alternative pathway plays an role in fighting Neisseria meningitidis (see Figures 3.11
and 3.12).
0 2 0 4 0 6 0
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
1 .2
1 .4
M A S P -2- /-
s e ru m
C 1q- /-
s e ru m
W t s e ru m
FB- /-
s e ru m
N e g a tiv e c o n tro l
% s e r u m c o n c e n t r a t io n
OD
At
40
5n
m
Figure 3.11 C3 deposition assay on the surface of Neisseria meningitidis strain A serogroup
Z2491 under alternative pathway permissive conditions (high serum concentration in BBS
buffer) the presence of both Ca++
and Mg++
allows the activation through all the complement
pathways. Different mouse sera were diluted in BBS binding buffer. Zymosan coating was used
as a positive control while the negative control wells received BSA blocking buffer as a coating.
The binding of the C3 cleavage product (C3c) was detected by using rabbit anti-human C3c
polyclonal antibody (the data is presented as a mean of all three independent experiments with
duplicate for each +/- SEM).
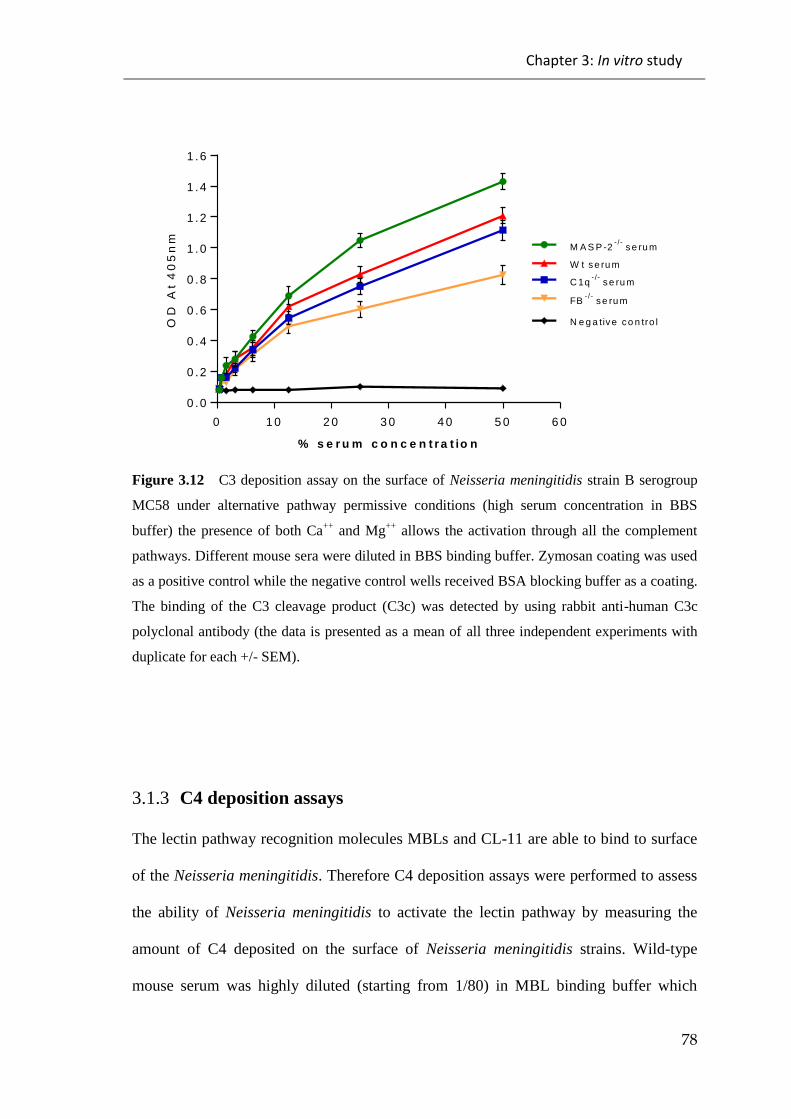
Chapter 3: In vitro study
78
0 1 0 2 0 3 0 4 0 5 0 6 0
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
1 .2
1 .4
1 .6
M A S P -2- /-
s e ru m
C 1q- /-
s e ru m
W t s e ru m
FB- /-
s e ru m
N e g a tiv e c o n tro l
% s e r u m c o n c e n t r a t io n
OD
At
40
5n
m
Figure 3.12 C3 deposition assay on the surface of Neisseria meningitidis strain B serogroup
MC58 under alternative pathway permissive conditions (high serum concentration in BBS
buffer) the presence of both Ca++
and Mg++
allows the activation through all the complement
pathways. Different mouse sera were diluted in BBS binding buffer. Zymosan coating was used
as a positive control while the negative control wells received BSA blocking buffer as a coating.
The binding of the C3 cleavage product (C3c) was detected by using rabbit anti-human C3c
polyclonal antibody (the data is presented as a mean of all three independent experiments with
duplicate for each +/- SEM).
3.1.3 C4 deposition assays
The lectin pathway recognition molecules MBLs and CL-11 are able to bind to surface
of the Neisseria meningitidis. Therefore C4 deposition assays were performed to assess
the ability of Neisseria meningitidis to activate the lectin pathway by measuring the
amount of C4 deposited on the surface of Neisseria meningitidis strains. Wild-type
mouse serum was highly diluted (starting from 1/80) in MBL binding buffer which
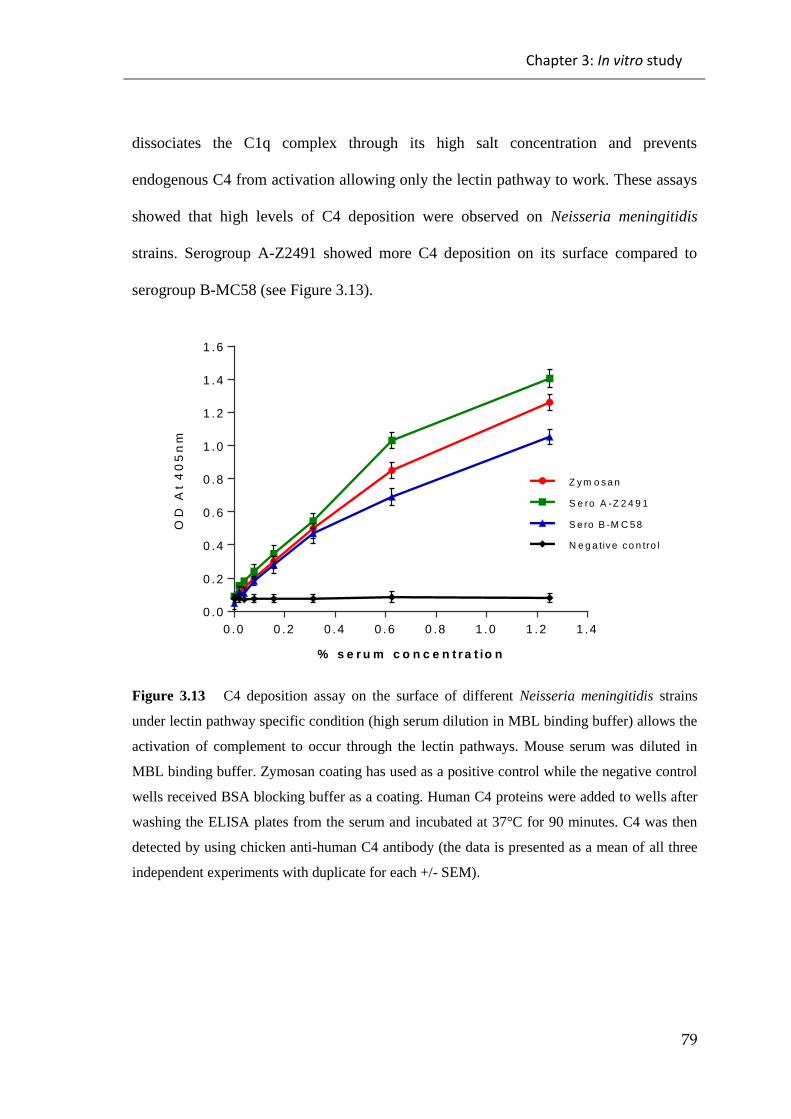
Chapter 3: In vitro study
79
dissociates the C1q complex through its high salt concentration and prevents
endogenous C4 from activation allowing only the lectin pathway to work. These assays
showed that high levels of C4 deposition were observed on Neisseria meningitidis
strains. Serogroup A-Z2491 showed more C4 deposition on its surface compared to
serogroup B-MC58 (see Figure 3.13).
0 .0 0 .2 0 .4 0 .6 0 .8 1 .0 1 .2 1 .4
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
1 .2
1 .4
1 .6
% s e r u m c o n c e n t r a t io n
OD
At
40
5n
m
Z y m o s a n
S e ro A -Z 2 4 9 1
S e ro B -M C 5 8
N e g a tiv e c o n tro l
Figure 3.13 C4 deposition assay on the surface of different Neisseria meningitidis strains
under lectin pathway specific condition (high serum dilution in MBL binding buffer) allows the
activation of complement to occur through the lectin pathways. Mouse serum was diluted in
MBL binding buffer. Zymosan coating has used as a positive control while the negative control
wells received BSA blocking buffer as a coating. Human C4 proteins were added to wells after
washing the ELISA plates from the serum and incubated at 37°C for 90 minutes. C4 was then
detected by using chicken anti-human C4 antibody (the data is presented as a mean of all three
independent experiments with duplicate for each +/- SEM).
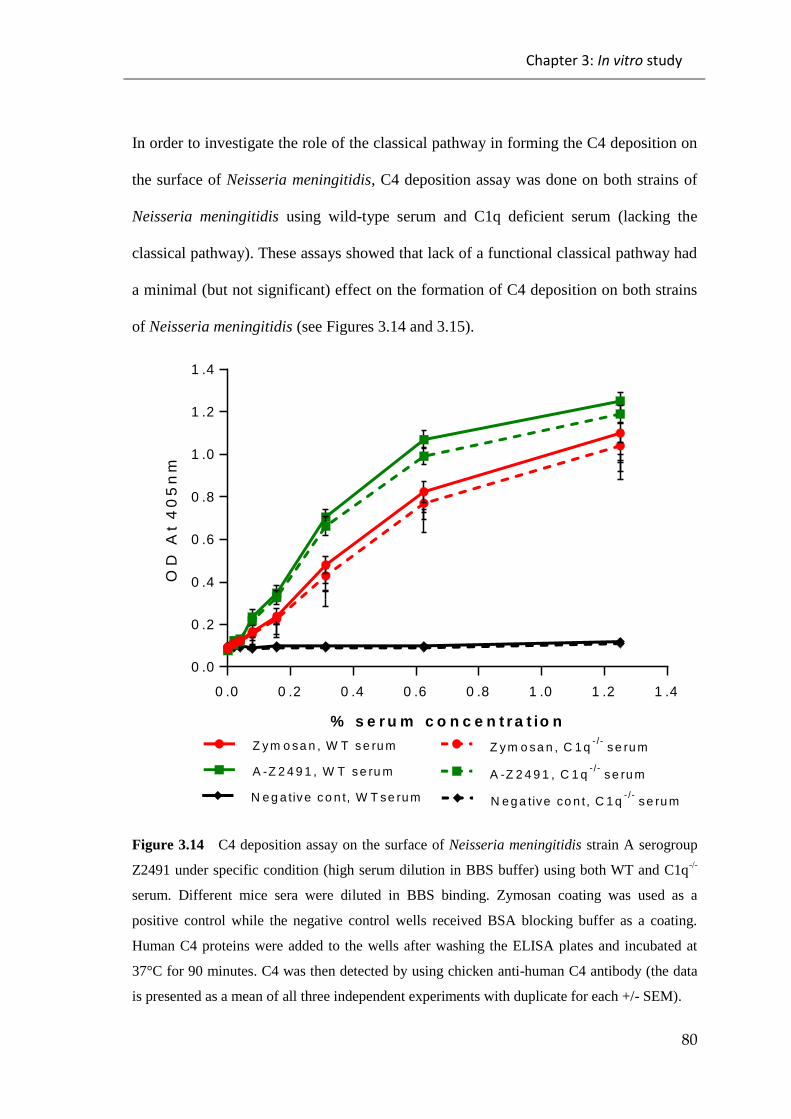
Chapter 3: In vitro study
80
In order to investigate the role of the classical pathway in forming the C4 deposition on
the surface of Neisseria meningitidis, C4 deposition assay was done on both strains of
Neisseria meningitidis using wild-type serum and C1q deficient serum (lacking the
classical pathway). These assays showed that lack of a functional classical pathway had
a minimal (but not significant) effect on the formation of C4 deposition on both strains
of Neisseria meningitidis (see Figures 3.14 and 3.15).
0 .0 0 .2 0 .4 0 .6 0 .8 1 .0 1 .2 1 .4
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
1 .2
1 .4
% s e r u m c o n c e n t r a t io n
OD
At
40
5n
m
Z y m o sa n , W T s e ru m Z y m o sa n , C 1 q- /-
s e ru m
N e g a tiv e c o n t, W T se ru m N e g a tive co n t, C 1 q- /-
s e ru m
A -Z 2 4 9 1 , C 1 q- /-
s e ru mA -Z 2 4 9 1 , W T s e ru m
Figure 3.14 C4 deposition assay on the surface of Neisseria meningitidis strain A serogroup
Z2491 under specific condition (high serum dilution in BBS buffer) using both WT and C1q-/-
serum. Different mice sera were diluted in BBS binding. Zymosan coating was used as a
positive control while the negative control wells received BSA blocking buffer as a coating.
Human C4 proteins were added to the wells after washing the ELISA plates and incubated at
37°C for 90 minutes. C4 was then detected by using chicken anti-human C4 antibody (the data
is presented as a mean of all three independent experiments with duplicate for each +/- SEM).
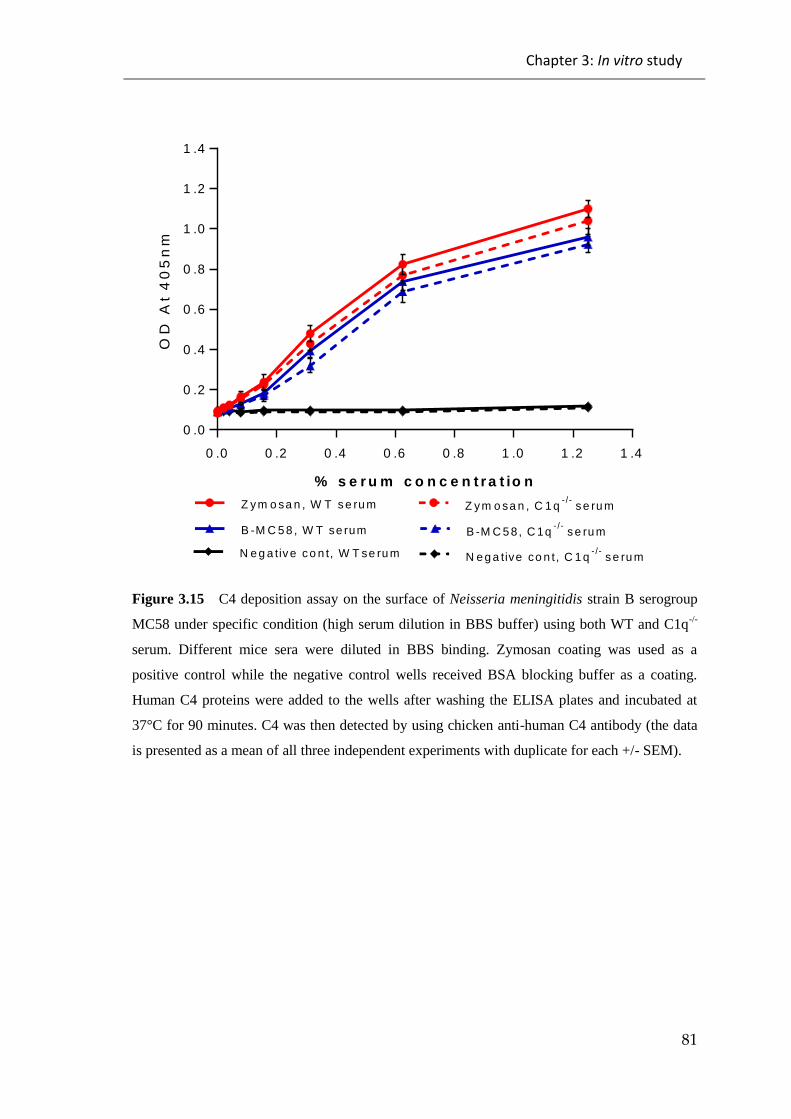
Chapter 3: In vitro study
81
0 .0 0 .2 0 .4 0 .6 0 .8 1 .0 1 .2 1 .4
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
1 .2
1 .4
% s e r u m c o n c e n t r a t io n
OD
At
40
5n
m
Z y m o sa n , W T s e ru m Z y m o sa n , C 1 q- /-
s e ru m
N e g a tiv e c o n t, W T se ru m N e g a tive co n t, C 1 q- /-
s e ru m
B -M C 5 8, C 1q- /-
s e ru mB -M C 5 8 , W T se rum
Figure 3.15 C4 deposition assay on the surface of Neisseria meningitidis strain B serogroup
MC58 under specific condition (high serum dilution in BBS buffer) using both WT and C1q-/-
serum. Different mice sera were diluted in BBS binding. Zymosan coating was used as a
positive control while the negative control wells received BSA blocking buffer as a coating.
Human C4 proteins were added to the wells after washing the ELISA plates and incubated at
37°C for 90 minutes. C4 was then detected by using chicken anti-human C4 antibody (the data
is presented as a mean of all three independent experiments with duplicate for each +/- SEM).

Chapter 3: In vitro study
82
3.1.4 Serum Bactericidal Assays
Serum bactericidal assays were done to assess the serum bacteriolytic activity of mice
sera against Neisseria meningitidis strains. In these assays, different mice sera, which
were deficient in one of the complement pathways (classical, alternative or lectin
pathways) were compared with the wild-type mice sera in terms of their ability to kill
Neisseria meningitidis strains.
C1q deficient serum (C1q-/-
), which is considered to be a classical pathway deficient
serum, was used to assess the role of the classical pathway in fighting the
meningococcus strains by comparing it to the wild-type (WT) control serum. This assay
showed no significant difference between the C1q deficient serum (classical pathway
deficient serum) and wild-type serum which suggests that the classical pathway might
plays a minimal role in fighting Neisseria meningitidis infection (see Figures 3.16 and
3.17). Additionally, it suggests that meningococcus could be killed through other
complement pathways (the alternative and lectin pathways).

Chapter 3: In vitro study
83
0 2 0 4 0 6 0 8 0 1 0 0
4 .2
4 .4
4 .6
4 .8
M i n u t e s
log
cfu
/ml
HIS
W T
C 1q- /-
Figure 3.16 Bactericidal activity of different mouse sera (C1q-/-
, WT and HIS) against
Neisseria meningitidis serogroup A strain Z2491. Bacteria and sera (30% concentration) were
incubated at 37°C with shaking. Samples were taken at time points 0, 30, 60 And 90 minutes
and plated out and the viable count was calculated. No significant difference emerged between
the C1q-/-
serum and the WT serum. However, the difference between the C1q-/-
, WT and the
heated inactivated sera (HIS) which was used as a negative control, was significant (the data is
presented as a mean of all three independent experiments with duplicate for each +/- SEM).
Table 3.1 Statistically significant differences between serum bactericidal assay of different
mouse sera (C1q-/-
, WT and HIS) against Neisseria meningitidis serogroup A strain Z2491 using
theStudent’st-test at time point 90 minutes.
Student’st-test at time point 90 minutues
Significant? P > 0.05 P value Summary
WT vs C1q-/- NO Ns (0.4385)
WT vs HIS YES * (0.0154)
C1q-/-
vs HIS YES * (0.0133)
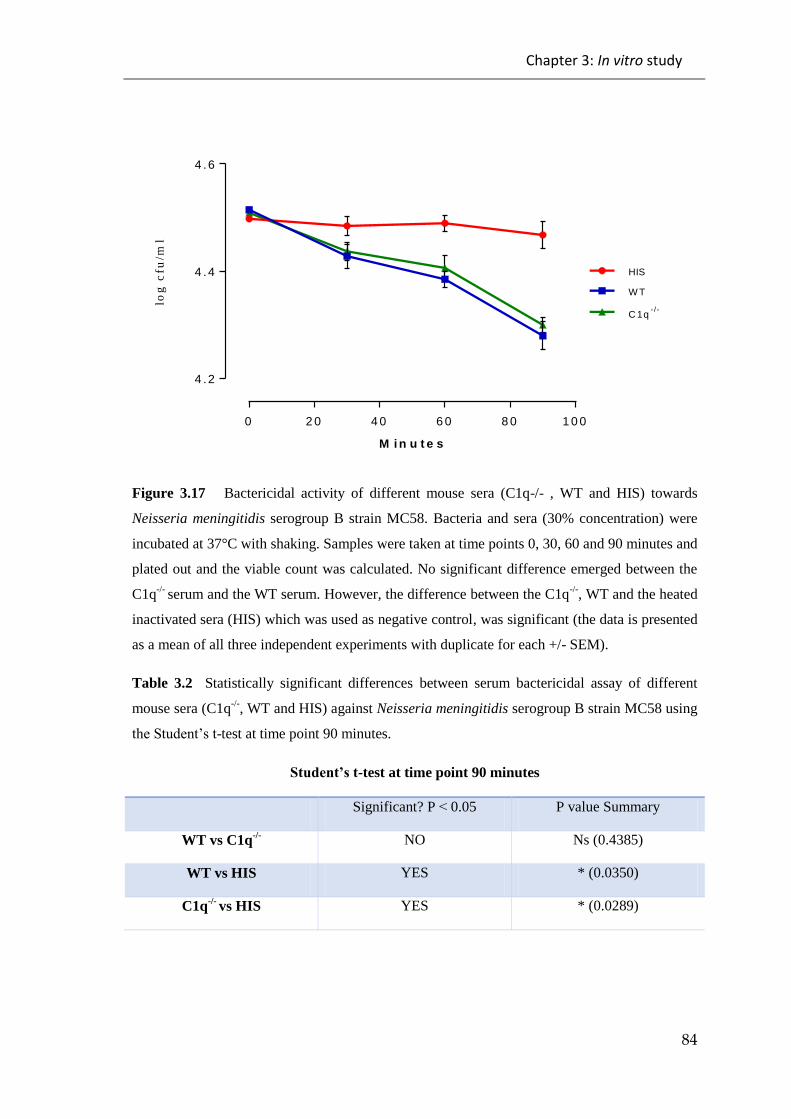
Chapter 3: In vitro study
84
0 2 0 4 0 6 0 8 0 1 0 0
4 .2
4 .4
4 .6
M i n u t e s
log
cfu
/ml
HIS
W T
C 1q- /-
Figure 3.17 Bactericidal activity of different mouse sera (C1q-/- , WT and HIS) towards
Neisseria meningitidis serogroup B strain MC58. Bacteria and sera (30% concentration) were
incubated at 37°C with shaking. Samples were taken at time points 0, 30, 60 and 90 minutes and
plated out and the viable count was calculated. No significant difference emerged between the
C1q-/-
serum and the WT serum. However, the difference between the C1q-/-
, WT and the heated
inactivated sera (HIS) which was used as negative control, was significant (the data is presented
as a mean of all three independent experiments with duplicate for each +/- SEM).
Table 3.2 Statistically significant differences between serum bactericidal assay of different
mouse sera (C1q-/-
, WT and HIS) against Neisseria meningitidis serogroup B strain MC58 using
theStudent’st-test at time point 90 minutes.
Student’st-test at time point 90 minutes
Significant? P > 0.05 P value Summary
WT vs C1q-/- NO Ns (0.4385)
WT vs HIS YES * (0.0350)
C1q-/-
vs HIS YES * (0.0289)

Chapter 3: In vitro study
85
In order to assess the role of the alternative pathway in fighting Neisseria meningitidis,
Factor B deficient (FB-/-
) serum and MASP-1/3 deficient (MASP-1/3-/-
) serum, a
previous study by Takahashi et al (2010) had showed that MASP-1/3 deficient mice are
lacking the full function of alternative pathway, were used to assess the ability of these
sera to kill Neisseria meningitidis. The assays showed that unlike the wild-type serum,
neither sera (FB-/-
and MASP-1/3-/-
) has the ability to kill meningococcus,
demonstrating the important role of the alternative pathway in fighting Neisseria
meningitidis (see Figures 3.18 and 3.19).
0 2 0 4 0 6 0 8 0 1 0 0
4 .2
4 .4
4 .6
4 .8
M i n u t e s
log
cfu
/ml HIS
W T
FB- /-
M A S P -1 /3- /-
Figure 3.18 Bactericidal activity of different mouse sera (FB-/-
, MASP-1/3-/-
, WT and HIS)
against Neisseria meningitidis serogroup A strain Z2491. Bacteria and sera (30% concentration)
were incubated at 37°C with shaking. Samples were taken at time points 0, 30, 60 And 90
minutes and plated out and the viable count was calculated. The ability of FB-/-
and MASP-1/3-/-
deficient serum was disrupted compared to the WT serum. In this assay heated inactivated
serum (HIS) was used as a negative control (the data is presented as a mean of all three
independent experiments with duplicate for each +/- SEM).
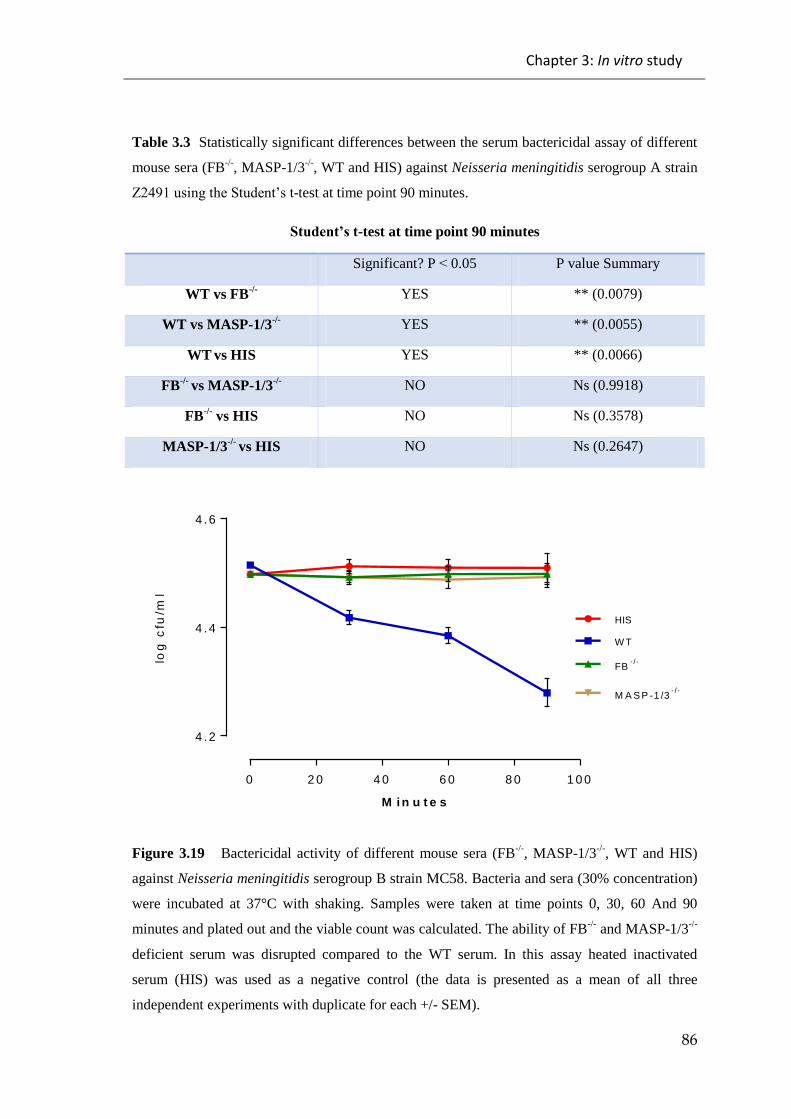
Chapter 3: In vitro study
86
Table 3.3 Statistically significant differences between the serum bactericidal assay of different
mouse sera (FB-/-
, MASP-1/3-/-
, WT and HIS) against Neisseria meningitidis serogroup A strain
Z2491usingtheStudent’st-test at time point 90 minutes.
Student’st-test at time point 90 minutes
Significant? P > 0.05 P value Summary
WT vs FB-/- YES ** (0.0079)
WT vs MASP-1/3-/- YES ** (0.0055)
WT vs HIS YES ** (0.0066)
FB-/-
vs MASP-1/3-/-
NO Ns (0.9918)
FB-/-
vs HIS NO Ns (0.3578)
MASP-1/3-/-
vs HIS NO Ns (0.2647)
0 2 0 4 0 6 0 8 0 1 0 0
4 .2
4 .4
4 .6
M i n u t e s
log
cfu
/ml
HIS
W T
FB- /-
M A S P -1 /3- /-
Figure 3.19 Bactericidal activity of different mouse sera (FB-/-
, MASP-1/3-/-
, WT and HIS)
against Neisseria meningitidis serogroup B strain MC58. Bacteria and sera (30% concentration)
were incubated at 37°C with shaking. Samples were taken at time points 0, 30, 60 And 90
minutes and plated out and the viable count was calculated. The ability of FB-/-
and MASP-1/3-/-
deficient serum was disrupted compared to the WT serum. In this assay heated inactivated
serum (HIS) was used as a negative control (the data is presented as a mean of all three
independent experiments with duplicate for each +/- SEM).

Chapter 3: In vitro study
87
Table 3.4 Statistically significant differences between the serum bactericidal assay of different
mouse sera (FB-/-, MASP-1/3-/-, WT and HIS) against Neisseria meningitidis serogroup B
strainMC58usingtheStudent’st-test at time point 90 minutes.
Student’st-test at time point 90 minutes
Significant? P > 0.05 P value Summary
WT vs FB-/- YES * (0.0218)
WT vs MASP-1/3-/- YES * (0.0230)
WT vs HIS YES * (0.0256)
FB-/-
vs MASP-1/3-/-
NO Ns (0.8531)
FB-/-
vs HIS NO Ns (0.7737)
MASP-1/3-/-
vs HIS NO Ns (0.6635)
MASP-2 deficient serum (MASP-2-/-
) and MBL deficient serum, lectin pathway
deficient sera, were used to assess the role of the lectin pathway in fighting the
meningococcus strains by comparing them to the wild-type (WT) control serum. The
assays showed that the ability of the MBL deficient serum to kill Neisseria meningitidis
was impaired compared to the wild-type serum. This result is consistent with previous
reports which showed an association between MBL deficiency and Neisseria
meningitidis infection (Faber et al., 2007; Tully et al., 2006). In addition, the assays
showed that the ability of MASP-2 deficient serum to kill Neisseria meningitidis was
significantly higher compared to the wild-type serum which suggests that the functional
activity of the alternative pathway may compensate for the absence of the functional
activity of the lectin pathway (see Figures 3.120 and 3.21). This result is in the line with
previous results, which showed that MASP-2-/-
deficient serum showed high levels of
C3 deposition on the surface of meningococcus strains when used at a high
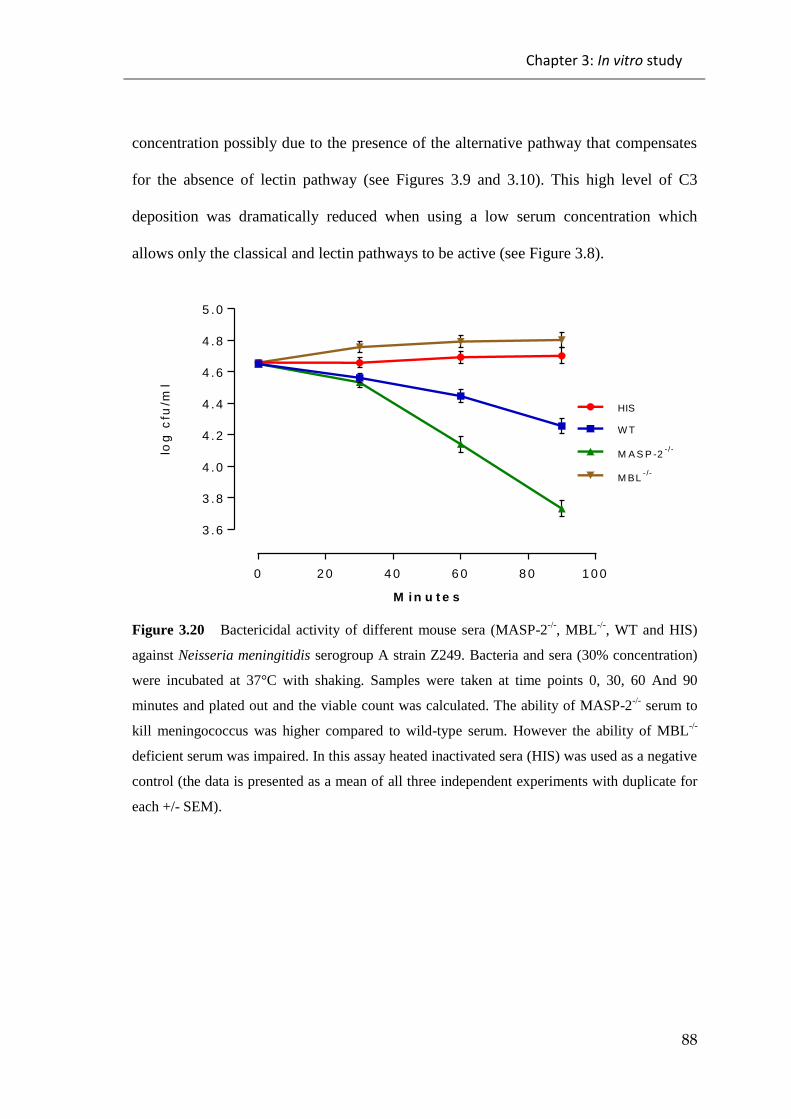
Chapter 3: In vitro study
88
concentration possibly due to the presence of the alternative pathway that compensates
for the absence of lectin pathway (see Figures 3.9 and 3.10). This high level of C3
deposition was dramatically reduced when using a low serum concentration which
allows only the classical and lectin pathways to be active (see Figure 3.8).
0 2 0 4 0 6 0 8 0 1 0 0
3 .6
3 .8
4 .0
4 .2
4 .4
4 .6
4 .8
5 .0
M i n u t e s
log
cfu
/ml
HIS
W T
M A S P -2- /-
M BL- /-
Figure 3.20 Bactericidal activity of different mouse sera (MASP-2-/-
, MBL-/-
, WT and HIS)
against Neisseria meningitidis serogroup A strain Z249. Bacteria and sera (30% concentration)
were incubated at 37°C with shaking. Samples were taken at time points 0, 30, 60 And 90
minutes and plated out and the viable count was calculated. The ability of MASP-2-/-
serum to
kill meningococcus was higher compared to wild-type serum. However the ability of MBL-/-
deficient serum was impaired. In this assay heated inactivated sera (HIS) was used as a negative
control (the data is presented as a mean of all three independent experiments with duplicate for
each +/- SEM).
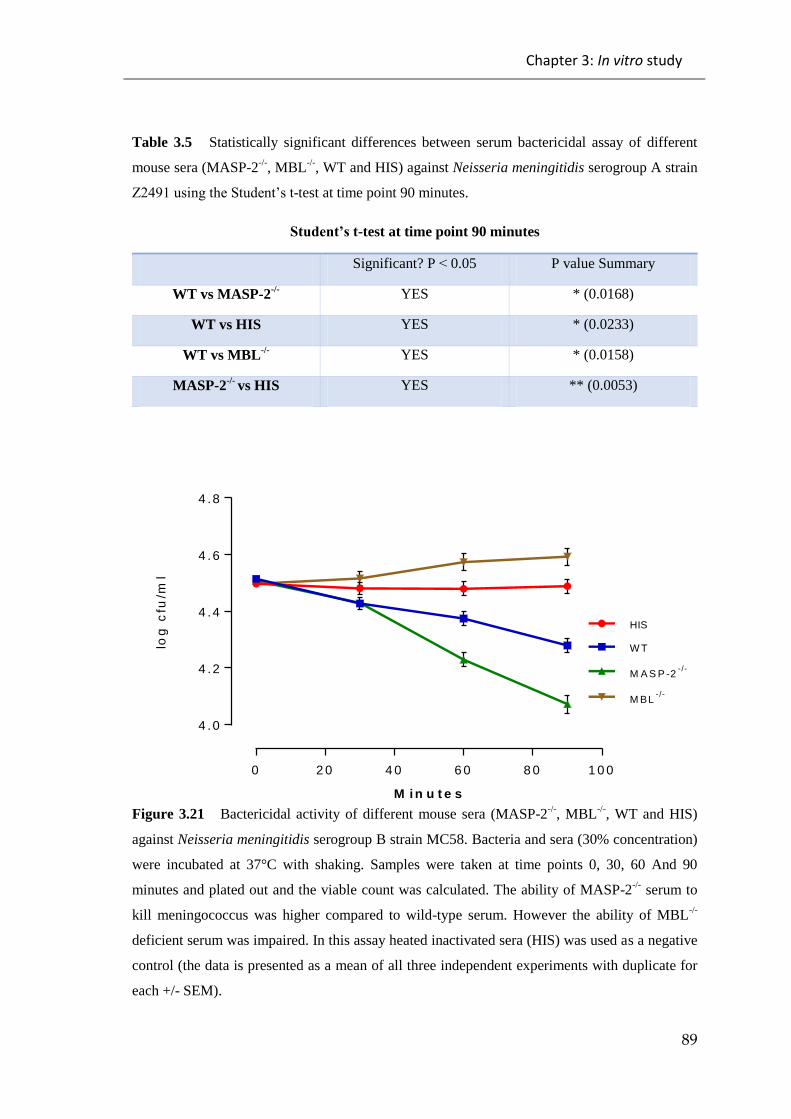
Chapter 3: In vitro study
89
Table 3.5 Statistically significant differences between serum bactericidal assay of different
mouse sera (MASP-2-/-
, MBL-/-
, WT and HIS) against Neisseria meningitidis serogroup A strain
Z2491usingtheStudent’st-test at time point 90 minutes.
Student’st-test at time point 90 minutes
Significant? P > 0.05 P value Summary
WT vs MASP-2-/- YES * (0.0168)
WT vs HIS YES * (0.0233)
WT vs MBL-/- YES * (0.0158)
MASP-2-/-
vs HIS YES ** (0.0053)
0 2 0 4 0 6 0 8 0 1 0 0
4 .0
4 .2
4 .4
4 .6
4 .8
M i n u t e s
log
cfu
/ml
HIS
W T
M A S P -2- /-
M BL- /-
Figure 3.21 Bactericidal activity of different mouse sera (MASP-2-/-
, MBL-/-
, WT and HIS)
against Neisseria meningitidis serogroup B strain MC58. Bacteria and sera (30% concentration)
were incubated at 37°C with shaking. Samples were taken at time points 0, 30, 60 And 90
minutes and plated out and the viable count was calculated. The ability of MASP-2-/-
serum to
kill meningococcus was higher compared to wild-type serum. However the ability of MBL-/-
deficient serum was impaired. In this assay heated inactivated sera (HIS) was used as a negative
control (the data is presented as a mean of all three independent experiments with duplicate for
each +/- SEM).
Chapter 3: In vitro study
90
Table 3.6 Statistically significant differences between serum bactericidal assay of different
mouse sera (MASP-2-/-, MBL-/-
, WT and HIS) against Neisseria meningitidis serogroup B
strainMC58usingtheStudent’st-test at time point 90 minutes.
Student’st-test at time point 90 minutes
Significant? P > 0.05 P value Summary
WT vs MASP-2-/- YES * (0.0371)
WT vs HIS YES * (0.0278)
WT vs MBL-/- YES * (0.0157)
MASP-2-/-
vs HIS YES ** (0.0091)
To study the role of the alternative pathway in bacteriocidal activity further, MASP-1/3
deficient serum which lacks functional activity of alternative pathway was used
(Takahshi et al., 2010), this showed no killing of Neisseria meningitidis (see Figures
3.18 and 3.19), following the addition of truncated recombinant of MASP-3 which
could restores the functional activity of the alternative pathway to kill Neisseria
meningitidis. The results from these assays showed that the ability to kill
meningococcus; and thus the activity of alternative pathway, was restored by the
addition of truncated recombinant MASP-3 (see Figures 3.22 and 3.23). This result is in
line with previous work by Iwaki et al., (2011) who suggested that MASP-3 of the
lectin pathway had the ability to induce the activity of the alternative pathway by
cleaving C4b bound factor B on the surface of pathogen, forming C3 convertase of
alternative pathway.

Chapter 3: In vitro study
91
0 2 0 4 0 6 0 8 0 1 0 0
4 .2
4 .4
4 .6
M i n u t e s
log
cfu
/ml
HIS
W T
M A S P -1 /3- /-
+
rM A S P -3 (1 0 µ g /m l)
M A S P -1 /3- /-
Figure 3.22 Bactericidal activity of different mouse sera (MASP-1/3-/-
+ truncated rMASP-3
(10µg/ml), MASP-1/3-/-
, WT and HIS) against Neisseria meningitidis serogroup A strain Z2491.
Bacteria and sera with or without truncated recombinant MASP-3 10ug/ml (30% serum
concentration) were incubated at 37°C with shaking. Samples were taken at time points 0, 30,
60 And 90 minutes and plated out and the viable count was calculated. The ability of MASP-
1/3-/-
deficient serum to kill Neisseria meningitidis; thus, the functional activity of the
alternative pathway was restored by adding 6ug/ml of truncated recombinant MASP-3 (rMASP-
3). In this assay heated inactivated serum (HIS) was used as a negative control (the data is
presented as a mean of all three independent experiments with duplicate for each +/- SEM)
Table 3.7 Statistically significant differences between serum bactericidal assay of different
mouse sera (MASP-1/3-/-
+ truncated rMASP-3 (10µg/ml), MASP-1/3-/-
, WT and HIS) against
Neisseria meningitidis serogroup A strain Z2491 using the Student’s t-test at time point 90
minutes.
Student’st-test at time point 90 minutes
Significant? P > 0.05 P value Summary
WT vs MASP-1/3-/- YES ** (0.0055)
WT vs MASP-1/3-/-
+rMASP-3 NO Ns (0.0513)
WT vs HIS YES ** (0.0091)
MASP-1/3-/-
vs MASP-1/3-/-
+ rMASP-3 YES * (0.0143)
MASP-1/3-/- vs HIS NO Ns (0.2025)
MASP-1/3-/-
+ rMASP-3 vs
HIS YES * (0.0277)

Chapter 3: In vitro study
92
0 2 0 4 0 6 0 8 0 1 0 0
4 .4
4 .6
4 .8
M i n u t e s
log
c
fu
/m
l
HIS
W T
M A S P -1 /3- /-
+
rM A S P -3 (1 0 µ g /m l)
M A S P -1 /3- /-
Figure 3.23 Bactericidal activity of different mouse sera (MASP-1/3-/-
+ truncated rMASP-3
(10µg/ml), MASP-1/3-/-
, WT and HIS) against Neisseria meningitidis serogroup B strain MC58.
Bacteria and sera with or without truncated recombinant MASP-3 10ug/ml (30% serum
concentration) were incubated at 37°C with shaking. Samples were taken at time points 0, 30,
60 And 90 minutes and plated out and the viable count was calculated. The ability of MASP-
1/3-/-
deficient serum to kill Neisseria meningitidis; thus, the functional activity of the
alternative pathway was been restored by adding 6ug/ml of truncated recombinant MASP-3
(rMASP-3). In this assay heated inactivated serum (HIS) was used as a negative control (the
data is presented as a mean of all three independent experiments with duplicate for each +/-
SEM).

Chapter 3: In vitro study
93
Table 3.8 Statistically significant differences between serum bactericidal assay of different
mouse serum (MASP-1/3-/-
+ truncated rMASP-3 (10µg/ml), MASP-1/3-/-
, WT and HIS) against
Neisseria meningitidis serogroup B strain MC58 using the Student’s t-test at time point 90
minutes.
Student’st-test at time point 90 minutes
Significant? P > 0.05 P value Summary
WT vs MASP-1/3-/- YES * (0.0112)
WT vs MASP-1/3-/-
+rMASP-3 NO Ns (0.0559)
WT vs HIS YES ** (0.0086)
MASP-1/3-/-
vs MASP-1/3-/-
+ rMASP-3 YES * (0.0349)
MASP-1/3-/- vs HIS NO Ns (0.7007)
MASP-1/3-/-
+rMASP-3 vs
HIS YES * (0.0261)
Furthermore, the serum bactericidal assay was done using HIS serum, MBL-/-
serum,
NHS, immune serum and 3MC serum with or without truncated recombinant MASP-3
6µg/ml (this serum was obtained from a patient, with MASP-1 but not MASP-3, with a
disease called Carnevale, Mingarelli, Malpuech, Michels (3MC) syndrome) to study
their bacteriolytic activity against Neisseria meningitidis. This assay showed that the
ability of MBL deficient serum to kill Neisseria meningitidis was impaired compared to
normal human serum. Additionally this assay showed that the functional activity of the
alternative pathway was restored by the addition of truncated recombinant MASP-3,
which enhanced the killing of Neisseria meningitidis (see Figure 3.24 and 3.25). This
result is consistent with previous results, which showed the importance of the
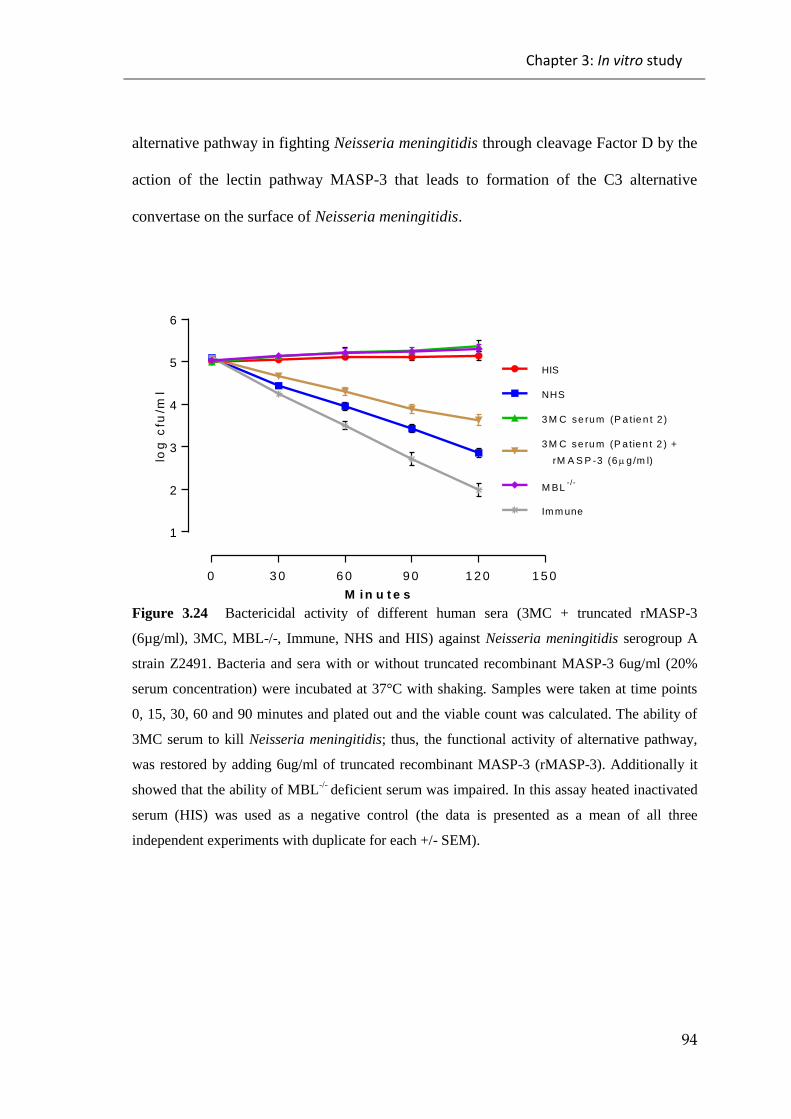
Chapter 3: In vitro study
94
alternative pathway in fighting Neisseria meningitidis through cleavage Factor D by the
action of the lectin pathway MASP-3 that leads to formation of the C3 alternative
convertase on the surface of Neisseria meningitidis.
0 3 0 6 0 9 0 1 2 0 1 5 0
1
2
3
4
5
6
M i n u t e s
log
cfu
/ml
HIS
Im m une
3 M C se rum (P a tie n t 2 )
3 M C se ru m (P a tie n t 2 ) +
rM A S P -3 (6 g/m l)
M BL- /-
NHS
Figure 3.24 Bactericidal activity of different human sera (3MC + truncated rMASP-3
(6µg/ml), 3MC, MBL-/-, Immune, NHS and HIS) against Neisseria meningitidis serogroup A
strain Z2491. Bacteria and sera with or without truncated recombinant MASP-3 6ug/ml (20%
serum concentration) were incubated at 37°C with shaking. Samples were taken at time points
0, 15, 30, 60 and 90 minutes and plated out and the viable count was calculated. The ability of
3MC serum to kill Neisseria meningitidis; thus, the functional activity of alternative pathway,
was restored by adding 6ug/ml of truncated recombinant MASP-3 (rMASP-3). Additionally it
showed that the ability of MBL-/-
deficient serum was impaired. In this assay heated inactivated
serum (HIS) was used as a negative control (the data is presented as a mean of all three
independent experiments with duplicate for each +/- SEM).
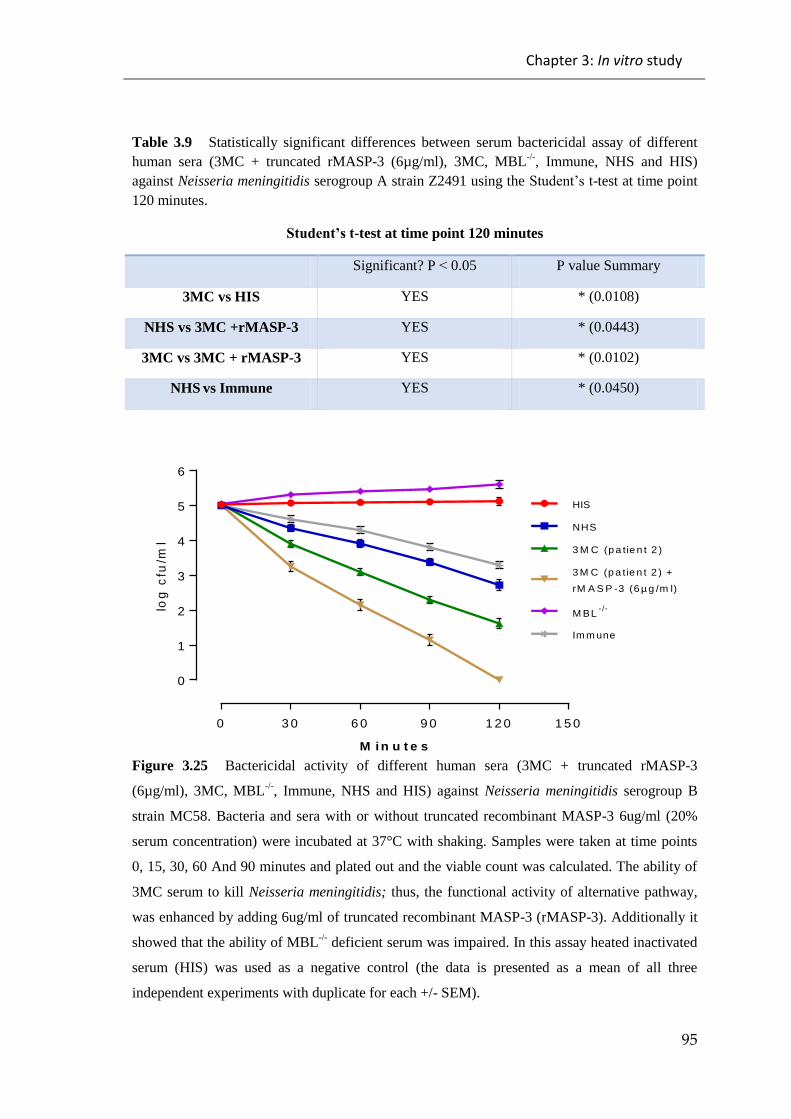
Chapter 3: In vitro study
95
Table 3.9 Statistically significant differences between serum bactericidal assay of different
human sera (3MC + truncated rMASP-3 (6µg/ml), 3MC, MBL-/-
, Immune, NHS and HIS)
against Neisseria meningitidis serogroup A strain Z2491 using the Student’st-test at time point
120 minutes.
Student’st-test at time point 120 minutes
Significant? P > 0.05 P value Summary
3MC vs HIS YES * (0.0108)
NHS vs 3MC +rMASP-3 YES * (0.0443)
3MC vs 3MC + rMASP-3 YES * (0.0102)
NHS vs Immune YES * (0.0450)
0 3 0 6 0 9 0 1 2 0 1 5 0
0
1
2
3
4
5
6
M i n u t e s
log
cfu
/ml
HIS
3 M C (p a tie n t 2 )
NHS
3 M C (p a tie n t 2 ) +
rM A S P -3 (6 µ g /m l)
M BL- /-
Im m une
Figure 3.25 Bactericidal activity of different human sera (3MC + truncated rMASP-3
(6µg/ml), 3MC, MBL-/-
, Immune, NHS and HIS) against Neisseria meningitidis serogroup B
strain MC58. Bacteria and sera with or without truncated recombinant MASP-3 6ug/ml (20%
serum concentration) were incubated at 37°C with shaking. Samples were taken at time points
0, 15, 30, 60 And 90 minutes and plated out and the viable count was calculated. The ability of
3MC serum to kill Neisseria meningitidis; thus, the functional activity of alternative pathway,
was enhanced by adding 6ug/ml of truncated recombinant MASP-3 (rMASP-3). Additionally it
showed that the ability of MBL-/-
deficient serum was impaired. In this assay heated inactivated
serum (HIS) was used as a negative control (the data is presented as a mean of all three
independent experiments with duplicate for each +/- SEM).

Chapter 3: In vitro study
96
Table 3.10 Statistically significant differences between serum bactericidal assay of different
human sera (3MC + truncated rMASP-3 (6µg/ml), 3MC, MBL-/-
, Immune, NHS and HIS)
against Neisseria meningitidis serogroup B strain MC58 using the Student’st-test at time point
120 minutes.
Student’st-test at time point 120 minutes
Significant? P > 0.05 P value Summary
Immune vs HIS YES ** (0.0078)
NHS vs Immune NO ns(0.0920)
NHS vs 3MC YES * (0.0351)
3MC vs 3MC + rMASP-3 YES ** (0.0084)
3MC vs Immune YES * (0.0121)

Chapter 3: In vitro study
97
3.1.5 Effect of recombinant properdin on complement mediated
killing of Neisseria meningitidis
Properdin is a positive regulator of the alternative pathway activation and can bind to
and stabilize C3 convertase by increasing its half-life. In addition, it has an important
role in alternative pathway activation, as properdin deficiency in serum decreases the
ability of the serum to activate the alternative pathway. However adding properdin can
restore normal activation of alternative pathway (Schwaeble and Reid, 1999). In
addition, it has been claimed that properdin can bind directly to the bacterial surface and
enhance C3 deposition on the bacterial surface (Spitzer et al., 2007).
3.1.5.1 Recombinant properdin has the ability to enhance C3 deposition on the
surface of Neisseria meningitidis
In order to assess whether recombinant properdin has the ability to enhance the
formation of C3 deposition on the surface of Neisseria meningitidis, ELISA and FACS
analysis were used to measure C3 deposition on the surface of Neisseria meningitidis
under specific conditions that block all complement pathway activation except the
alternative pathway. Both ways of measurement showed recombinant properdin could
enhance the alternative C3 deposition on the surface of Neisseria meningitidis (see
Figures 3.26, 3.27 and 3.28).

Chapter 3: In vitro study
98
0 1 0 2 0 3 0 4 0 5 0 6 0
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
1 .2
1 .4
1 .6
1 .8
% S e r u m C o n c e n t r a t io n
OD
At
40
5n
m
Z y m o s a n , w t Z y m o s a n , w t + rP ro p e rd in (5 µ g /m l)
S e ro A -Z 2 4 9 1 , w t S e ro A -Z 2 4 9 1 , w t + rP ro p e rd in (5 µ g /m l)
N e g a tiv e C o n t, w t N e g a t iv e C o n t, w t + rP ro p e rd in (5 u g /m l)
Figure 3.26 C3 deposition assay on the surface of Neisseria meningitidis strain A serogroup
Z2491 under alternative pathway permissive conditions (high serum concentration in EGTA
buffer). Mouse serum was diluted in EGTA buffer with or without recombinant properdin
(5µg/ml). Zymosan coating was used as a positive control while the negative control wells
received BSA blocking buffer as a coating. The binding of the C3 cleavage product (C3c) was
detected by using rabbit anti-human C3c polyclonal antibody (the data is presented as a mean of
all three independent experiments with duplicate for each +/- SEM).

Chapter 3: In vitro study
99
0 1 0 2 0 3 0 4 0 5 0 6 0
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
1 .2
1 .4
1 .6
1 .8
2 .0
% S e r u m C o n c e n t r a t io n
OD
At
40
5n
m
Z y m o s a n , w t Z y m o s a n , w t + rP ro p e rd in (5 µ g /m l)
S e ro B -M C 5 8 , w t S e ro B -M C 5 8 , w t + rP ro p e rd in (5 µ g /m l)
N e g a tiv e C o n t, w t N e g a t iv e C o n t, w t + rP ro p e rd in (5 u g /m l)
Figure 3.27 C3 deposition assay on the surface of Neisseria meningitidis strain B serogroup
MC58 under alternative pathway permissive conditions (high serum concentration in EGTA
buffer). Mouse serum was diluted in EGTA buffer with or without recombinant properdin
(5µg/ml). Zymosan coating was used as a positive control while the negative control wells
received BSA blocking buffer as a coating. The binding of the C3 cleavage product (C3c) was
detected by using rabbit anti-human C3c polyclonal antibody (the data is presented as a mean of
all three independent experiments with duplicate for each +/- SEM).
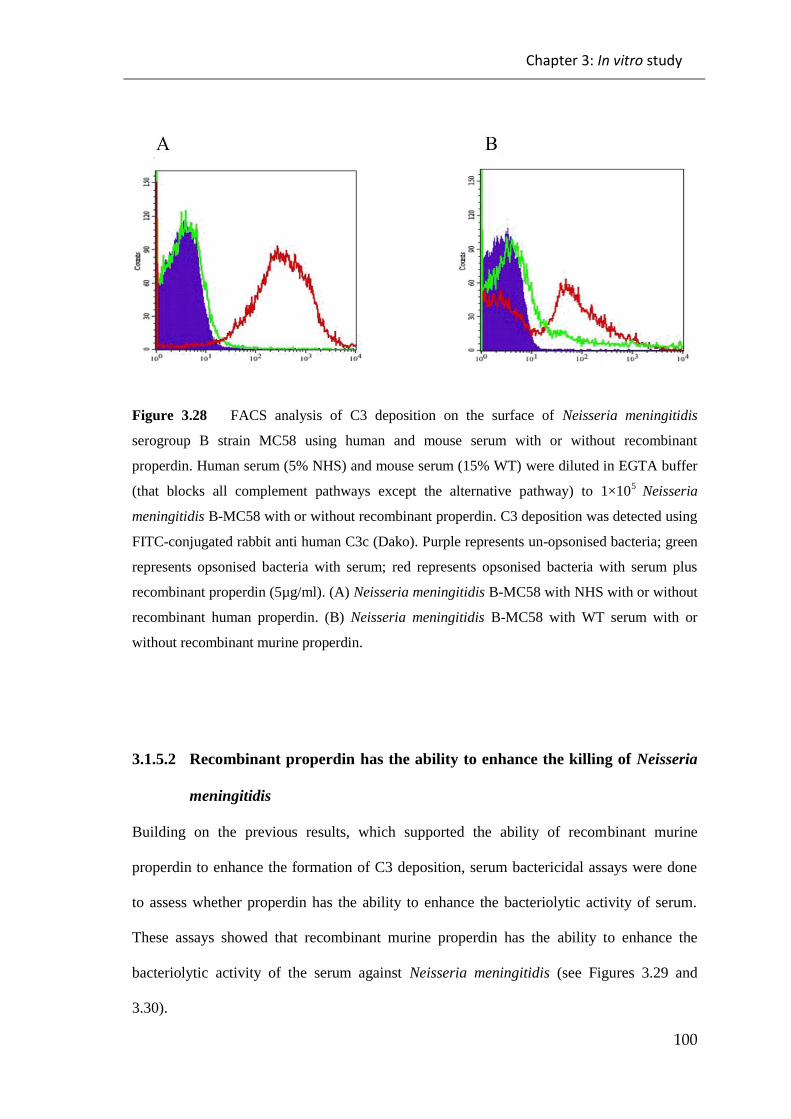
Chapter 3: In vitro study
100
Figure 3.28 FACS analysis of C3 deposition on the surface of Neisseria meningitidis
serogroup B strain MC58 using human and mouse serum with or without recombinant
properdin. Human serum (5% NHS) and mouse serum (15% WT) were diluted in EGTA buffer
(that blocks all complement pathways except the alternative pathway) to 1×105
Neisseria
meningitidis B-MC58 with or without recombinant properdin. C3 deposition was detected using
FITC-conjugated rabbit anti human C3c (Dako). Purple represents un-opsonised bacteria; green
represents opsonised bacteria with serum; red represents opsonised bacteria with serum plus
recombinant properdin (5µg/ml). (A) Neisseria meningitidis B-MC58 with NHS with or without
recombinant human properdin. (B) Neisseria meningitidis B-MC58 with WT serum with or
without recombinant murine properdin.
3.1.5.2 Recombinant properdin has the ability to enhance the killing of Neisseria
meningitidis
Building on the previous results, which supported the ability of recombinant murine
properdin to enhance the formation of C3 deposition, serum bactericidal assays were done
to assess whether properdin has the ability to enhance the bacteriolytic activity of serum.
These assays showed that recombinant murine properdin has the ability to enhance the
bacteriolytic activity of the serum against Neisseria meningitidis (see Figures 3.29 and
3.30).

Chapter 3: In vitro study
101
0 2 0 4 0 6 0 8 0 1 0 0
3 .6
3 .9
4 .2
4 .5
4 .8
M i n u t e s
log
cfu
/ml
H IS + rP ro p e rd in (5 µ g /m l)
W T
W T + rP ro p e rd in (5 µ g /m l)
HIS
Figure 3.29 Bactericidal activity of mouse serum with or without recombinant murine properdin against
Neisseria meningitidis serogroup A strain Z2491. Bacteria and serum with or without recombinant murine
properdin 5µg/ml (30% serum concentration) were incubated at 37°C with shaking. Samples were taken
at time points 0, 30, 60 and 90 minutes and plated out and the viable count was then calculated. It was
shown that the ability of recombinant murine properdin to kill Neisseria meningitidis was enhanced by
adding 5ug/ml of recombinant properdin, which indicates that properdin enhanced the functional activity
of the alternative pathway. In this assay heated inactivated serum (HIS) was used as a negative control
(the data is presented as a mean of all three independent experiments with duplicate for each +/- SEM).
Table 3.11 Statistically significant differences between serum bactericidal assay of mouse serum with or
without recombinant murine properdin against Neisseria meningitidis serogroup A strain Z2491 using the
Student’st-test at time point 90 minutes.
Student’st-test at time point 90 minutes
Significant? P > 0.05 P value Summary
WT vs WT + rProperdin YES * (0.0221)
WT vs HIS YES * (0.0230)
WT vs HIS + rProperdin YES *(0.0222)
WT + rProperdin vs HIS YES ** (0.0059)
WT + rProperdin vs HIS +
rProperdin YES **(0.0051)
HIS + rProperdin vs HIS NO Ns(0.5536)
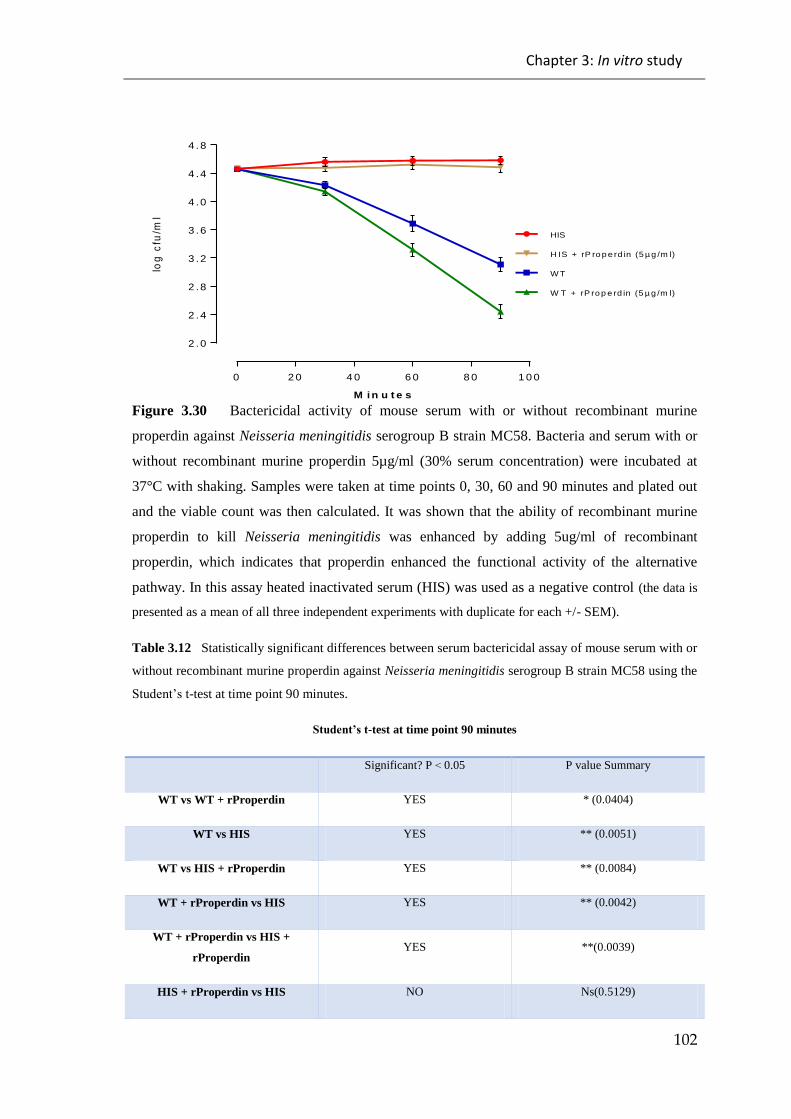
Chapter 3: In vitro study
102
0 2 0 4 0 6 0 8 0 1 0 0
2 .0
2 .4
2 .8
3 .2
3 .6
4 .0
4 .4
4 .8
M i n u t e s
log
cfu
/ml
HIS
W T
W T + rP ro p e rd in (5 µ g /m l)
H IS + rP ro p e rd in (5 µ g /m l)
Figure 3.30 Bactericidal activity of mouse serum with or without recombinant murine
properdin against Neisseria meningitidis serogroup B strain MC58. Bacteria and serum with or
without recombinant murine properdin 5µg/ml (30% serum concentration) were incubated at
37°C with shaking. Samples were taken at time points 0, 30, 60 and 90 minutes and plated out
and the viable count was then calculated. It was shown that the ability of recombinant murine
properdin to kill Neisseria meningitidis was enhanced by adding 5ug/ml of recombinant
properdin, which indicates that properdin enhanced the functional activity of the alternative
pathway. In this assay heated inactivated serum (HIS) was used as a negative control (the data is
presented as a mean of all three independent experiments with duplicate for each +/- SEM).
Table 3.12 Statistically significant differences between serum bactericidal assay of mouse serum with or
without recombinant murine properdin against Neisseria meningitidis serogroup B strain MC58 using the
Student’st-test at time point 90 minutes.
Student’st-test at time point 90 minutes
Significant? P > 0.05 P value Summary
WT vs WT + rProperdin YES * (0.0404)
WT vs HIS YES ** (0.0051)
WT vs HIS + rProperdin YES ** (0.0084)
WT + rProperdin vs HIS YES ** (0.0042)
WT + rProperdin vs HIS +
rProperdin YES **(0.0039)
HIS + rProperdin vs HIS NO Ns(0.5129)

Chapter 3: In vitro study
103
To further study the ability of recombinant properdin to enhance the killing of Neisseria
meningitidis, serum bactericidal assays were done against meningococcus using human
serum with or without recombinant human properdin. The results of these assays
showed that the ability of serum to kill Neisseria meningitidis was enhanced by adding
recombinant properdin (see Figures 3.31 and 3.32). The results support the earlier
finding that demonstrated the important role of the alternative pathway in fighting
Neisseria meningitidis.
0 2 0 4 0 6 0 8 0 1 0 0
1
2
3
4
5
6
HIS
H IS + rP ro p e rd in (5 µ g /m l)
N H S
N H S + rP ro p e rd in (5 µg /m l)
M i n u t e s
log
cfu
/ml
Figure 3.31 Bactericidal activity of human serum with or without recombinant human
properdin against Neisseria meningitidis serogroup A strain Z2491. Bacteria and serum with or
without recombinant human properdin 5µg/ml (20% serum concentration) were incubated at
37°C with shaking. Samples were taken at time points 0, 30, 60 and 90 minutes and plated out
and the viable count was then calculated. It was shown that the ability of recombinant human
properdin to kill Neisseria meningitidis was enhanced by adding 5ug/ml of recombinant
properdin, which indicates that properdin enhanced the functional activity of the alternative
pathway. In this assay heated inactivated serum (HIS) was used as a negative control (the data is
presented as a mean of all three independent experiments with duplicate for each +/- SEM).

Chapter 3: In vitro study
104
Table 3.13 Statistically significant differences between serum bactericidal assay of
human serum with or without recombinant human properdin against Neisseria
meningitidis serogroup A strain Z2491 using the Student’s t-test at time point 90
minutes.
Student’st-test at time point 90 minutes
Significant? P > 0.05 P value Summary
NHS vs NHS +
rProperdin YES * (0.0453)
NHS vs HIS YES * (0.0107)
NHS vs HIS + rProperdin YES *(0.0144)
NHS + rProperdin vs HIS YES ** (0.0050)
NHS + rProperdin vs HIS
+ rProperdin YES **(0.0063)
HIS + rProperdin vs HIS NO Ns(0.6701)

Chapter 3: In vitro study
105
0 2 0 4 0 6 0 8 0 1 0 0
2 .0
2 .5
3 .0
3 .5
4 .0
4 .5
5 .0
M i n u t e s
log
cfu
/ml
H IS + rP ro p e rd in (5 µ g /m l)
N H S + rP ro p e rd in (5 µg /m l)
NHS
HIS
Figure 3.32 Bactericidal activity of human serum with or without recombinant human
properdin against Neisseria meningitidis serogroup B strain MC58. Bacteria and serum
with or without recombinant human properdin 5µg/ml (20% serum concentration) were
incubated at 37°C with shaking. Samples were taken at time points 0, 30, 60 and 90
minutes and plated out and the viable count was then calculated. It was shown that the
ability of recombinant human properdin to kill Neisseria meningitidis was enhanced by
adding 5ug/ml of recombinant properdin, which indicates that properdin enhanced the
functional activity of the alternative pathway. In this assay heated inactivated serum
(HIS) was used as a negative control (the data is presented as a mean of all three
independent experiments with duplicate for each +/- SEM).
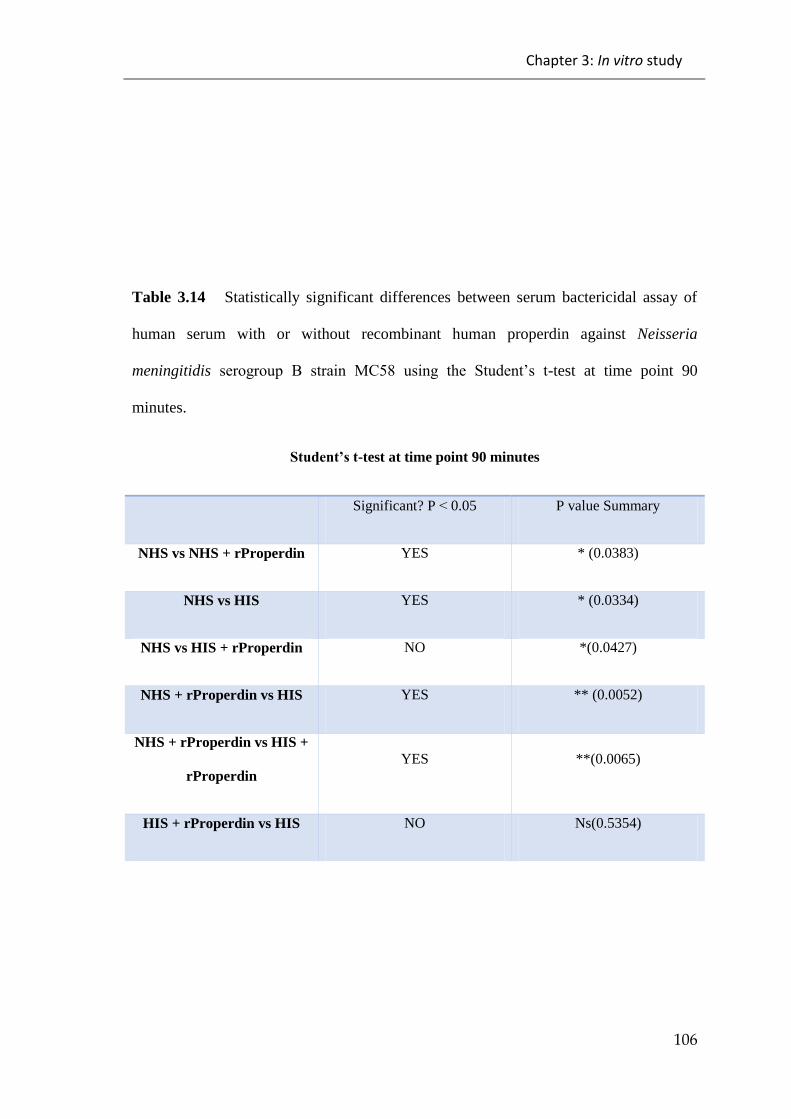
Chapter 3: In vitro study
106
Table 3.14 Statistically significant differences between serum bactericidal assay of
human serum with or without recombinant human properdin against Neisseria
meningitidis serogroup B strain MC58 using the Student’s t-test at time point 90
minutes.
Student’st-test at time point 90 minutes
Significant? P > 0.05 P value Summary
NHS vs NHS + rProperdin YES * (0.0383)
NHS vs HIS YES * (0.0334)
NHS vs HIS + rProperdin NO *(0.0427)
NHS + rProperdin vs HIS YES ** (0.0052)
NHS + rProperdin vs HIS +
rProperdin
YES **(0.0065)
HIS + rProperdin vs HIS NO Ns(0.5354)

Chapter 3: In vitro study
107
3.2 Discussion
The complement system is a vital part of the immune system and plays a crucial role in
fighting against invading microbes including Neisseria meningitidis. This important role
has been illustrated in many published reports (Finne et al., 1987; Estabrook et al.,
1997; Rossi et al., 2001; Jarva et al., 2005). Therefore deficiency in complement
components increases the susceptibility to Neisseria meningitidis infection (Trouw &
Daha, 2011; Rosa et al., 2004; Rossi et al., 2001). For example, increased susceptibility
to Neisseria meningitidis infection is associated with deficiency of terminal pathway
components (C5, C6, C7, C8, or C9) or alternative pathway components such as factor
D and properdin (Morgan and Walport, 1991).
Previous work had shown that MASP-2 deficient serum had a higher bactericidal
activity against Neisseria meningitidis than NHS which correlated with a better survival
of MASP-2 deficient mice in an Neisseria meningitidis infection study compared to
wild-type mice (Hayat 2012). Therefore, this work aimed to explain this finding by
emphasizing the crucial role of the alternative pathway in fighting Neisseria
meningitidis and to define the role of the MASP-3 lectin pathway in activating the
alternative pathway of the complement system and therefore in improving resistance to
Neisseria meningitidis. To achieve this aim, several complement specific in vitro assays
(Chapter 3) were carried out which were followed by in vivo infection experiments
(Chapter 4) in a mouse model of Neisseria meningitidis infection. The mouse model
was a unique mouse strain with a gene-targeted disruption of the murine factor B gene
or MASP-1/3 gene that blocked the activity of the alternative complement pathway
(Takahashi et al., 2010).

Chapter 3: In vitro study
108
3.2.1 Binding of Neisseria meningitidis to different complement
recognition molecules
The activation of complement pathways is dependent on initial binding of complement
recognition molecules to the surface of microbes (Carroll and Sim, 2011). Therefore,
ELISA assays were done in order to determine which murine complement recognition
molecules, C1q of the classical pathway and MBL-A, MBL-C, Cl-11 and ficolin-A of
Lectin pathway, bind to the surface of Neisseria meningitidis.
The activation of the classical pathway starts once C1q binds indirectly to the Fc region
of immunoglobulins IgG or IgM or directly to the surface of bacteria (Wallis et al.
2007). The ELISA based binding assays that were done showed that the C1q molecule
had the ability to bind to both strains of Neisseria meningitidis (see Figure 3.1). Thus, in
order to assess the importance of this binding in activating the classical pathway on the
surface of Neisseria meningitidis, a C4 deposition assay using C1q deficient serum and
wild-type serum was used. This assay showed that the absence of C1q and therefore the
classical pathway from the serum had a minimal (but not significant) effect on the
amount of the C4 deposition on the surface of Neisseria meningitidis (see Figures 3.14
and 3.15). This finding could be explained by the source of the serum that was used in
this assay. This serum was collected from mice that had not been exposed to Neisseria
meningitidis before; therefore, it did not have any specific antibodies against Neisseria
meningitidis that could drive the activation of the classical pathway on the surface of
Neisseria meningitidis. This finding is line with previous work (Agarwal et al., 2014)
that there was minimal C4 deposition on the surface of Neisseria meningitidis following
incubation of Neisseria with C1q and C4 only, as the activation of the classical pathway

Chapter 3: In vitro study
109
on the surface of Neisseria meningitidis was dependent on the presence of specific
antibodies.
MBL-A and MBL-C are mouse lectin pathway carbohydrate recognition molecules
which could initiate the activation of the lectin pathway by binding to the surface of
Neisseria meningitidis. Therefore in order to assess whether these recognition molecules
have the ability to bind to Neisseria meningitidis, ELISA based binding assays were
done. These assays showed that both murine MBL-A and MBL-C bind to the surface of
the both strains of Neisseria Meningitidis (see Figures 3.2 and 3.3). This result
supported previous work by Van Emmerik et al., (1994) using radiolabelled human
mannan binding protein which showed binding of human MBL to unencapsulated
Neisseria meningitidis, intermediate binding to Neisseria meningitidis serogroup A and
weak binding to other Neisseria meningitides strains. Additionally Jack et al. (2001)
supported this result and showed that MBL has the ability to bind to encapsulated
Neisseria meningitidis serogroup C. Another study carried out by Kuipers et al. (2003)
showed high binding of MBL to Neisseria meningitidis serogroup B and C.
Ficolins are lectin carbohydrate recognition molecules mostly present in serum that
have ability to bind to N-acetyl glucosamine sugars and lipoteichoic acids of bacteria
(Garred et al., 2009; Endo et al., 2005). While three types of ficolin exist in humans,
mice have only two types of ficolin: ficolin-A and ficolin-B. Although both ficolins
have almost similar structure, ficolin-A, is the only ficolin in mice that has the ability to
activate the lectin pathway by binding to MASP-2 (Runza et al., 2008). Thus, in order
to assess whether mouse ficolin-A can bind to Neisseria meningitidis a binding assay

Chapter 3: In vitro study
110
based on ELISA showed that ficolin-A has no ability to bind to either strains of
Neisseria meningitidis; thus, it is unable to activate the lectin pathway on the surface of
Neisseria meningitidis (see Figure 3.5).
Collectin 11 is one of the lectin recognition molecules that can bind to D-mannose and
L-fucose terminal saccharides on the surface of microbes. Collectin 11 has the ability to
activate the lectin pathway by interacting with MASP-1 and MASP-3 (Hansen et al.,
2010). Therefore, an ELISA based binding assay was used to assess the ability of
Collectin 11 to bind to the surface of Neisseria meningitidis. This assay showed that
Collectin 11 had the ability to bind to both strains of Neisseria meningitidis (see Figure
3.4).
Following the results of the binding assays of lectin pathway recognition molecules a
C4 deposition assay was used to assess the role of the independent lectin pathway in the
activation of the complement system on the surface of strains of Neisseria meningitidis.
Mouse serum was diluted in MBL binding buffer in order to dissociate the C1q complex
and inhibit the classical pathway. The result of this study showed high levels of C4
deposition on the surface of tested strains of Neisseria meningitidis (see Figure 3.13).
This result is compatible with previously published results, which reported that a
significant amount of C4 was found on the surface of Neisseria meningitidis following
incubation with whole blood. This deposition of C4 can be reduced by using
monoclonal anti-MBL antibodies. The inhibition of the amount of C4 deposited on the
surface of Neisseria meningitidis confirms that C4 deposition occurred only via the
lectin pathway (Sprong et al., 2003).

Chapter 3: In vitro study
111
3.2.2 Activation of the complement system on the surface of Neisseria
meningitidis requires a close cooperation between the lectin and
alternative pathway
The ability of Neisseria meningitidis to activate the complement system was studied
using several in vitro assays, which showed that both the strains of Neisseria
meningitidis used throughout this study had the ability to activate the complement
system mainly through the lectin and alternative pathways (see Figures 3.6 and 3.7).
This finding is line with previous reports (Jarvis, 1994; Vogel et al., 1997) that show an
ability of Neisseria meningitidis to activate the complement system as determined by
measuring the C3 deposition on the surface of Neisseria meningitidis.
In order to investigate the role of the different complement pathways in complement
activation by Neisseria meningitidis, wild-type serum and MASp-2 deficient serum
lacking the lectin pathway was used to assess the role of the lectin pathway leading to
C3 deposition on the surface of Neisseria meningitidis. This assay was carried out under
conditions (i.e. high serum dilution in Barbital buffer saline buffer) that allowed the
activation to occur only through the classical and lectin pathways. The assay showed
impairment of the ability of MASP-2 deficient serum to allow C3 deposition on the
surface of both tested strains of Neisseria meningitidis, which suggested that there is an
important role of the lectin pathway C3 deposition on the surface of Neisseria
meningitidis (see Figure 3.8) Following this result the same assay was repeated with a
high serum concentration in BBS buffer which allows functional activity of all three
complement pathways. Surprisingly, the results showed an increase in the amount of C3

Chapter 3: In vitro study
112
deposition on Neisseria meningitidis with MASP-2 deficient serum than with wild-type
serum (see Figures 3.9 and 3.10). This observation suggested that alternative pathway
may provide compensation for the absence of the lectin pathway (MASP-2) through the
remaining lectin pathway molecules MASP-1 and/or MASP-3 that could also drive the
alternative pathway and therefore form C3 deposition on the surface of Neisseria
meningitidis. This result is in line with previous work by Iwaki et al., (2011) who
suggested that MASP-3 of lectin pathway has the ability to induce the activity of the
alternative pathway on the surface of Staphylococcus aureus by cleaving Factor D and
therefore form C3 convertase via the alternative pathway.
To further analyse these results, C3 deposition assays were done on both tested strains
of Neisseria meningitidis using different mice sera (wild-type, C1q deficient, FB
deficient and MASP-2 deficient) under conditions (high serum concentration in BBS
buffer) that allow all three complement pathways to work. These assays showed
different levels of C3 deposition on the surface of both strains of Neisseria meningitidis.
The lowest level of the C3 deposition on both strains came from factor B deficient
serum presumably because it lacks a functionally active alternative pathway which
could then have contributed to both classical and lectin pathway activation. It also
suggests that the alternative pathway plays an important role in inducing C3 deposition
on the surface of the Neisseria meningitidis strains tested. On the other hand, C3
deposition on the surface of Neisseria meningitidis was minimally affected compared to
wild-type serum by the use of C1q deficient serum lacking classical pathway functional
activity suggesting that the classical pathway plays a minimal role in forming the
initiation of C3 deposition on the surface of Neisseria meningitidis. Additionally, it

Chapter 3: In vitro study
113
emphasized the important role of the other two complement pathways, alternative and
lectin pathways, which contributed to the formation of the C3 deposition on the surface
Neisseria meningitidis. The minimal role of the classical pathway could be explained by
the source of the serum, which came from mice that had not been exposed to Neisseria
meningitidis before; therefore, they did not have any specific antibodies against
Neisseria meningitidis that could drive the activation of the classical pathway on the
surface of Neisseria meningitidis. This finding is in line with previous work (Agarwal et
al., 2014) that showed minimal activation of classical pathway on the surface Neisseria
meningitidis when incubating the Neisseria with C1q and C4 only, as the activation of
the classical pathway on the surface of Neisseria meningitidis is dependent on
antibodies.
However the highest level of the C3 deposition was seen with MASP-2 deficient serum,
which lacks the action of the lectin pathway and may have enhanced activation of the
alternative pathway. The results using the MASP-2 deficient serum suggest that the
remaining lectin pathway molecules, i.e. MASP-1and/or MASP-3, could drive the
alternative pathway and therefore form C3 deposition on the surface of Neisseria
meningitidis. This result supports the previous work by Bjerre et al., (2011) who studied
the activation of the complement system on the surface of Neisseria meningitidis using
a whole blood model and he reported that the activation of the complement system on
the surface of Neisseria meningitidis was mainly dependent on the lectin and alternative
pathways (see Figures 3.11 and 3.12).

Chapter 3: In vitro study
114
To conduct further analysis, serum bactericidal assays were used to measure the lytic
activity of serum against Neisseria meningitidis. In these assays different mice sera
(wild-type, C1q deficient, factor B deficient, Masp-1/3 deficient and MASP-2 deficient)
were used under conditions that did not block any of the three complement pathways.
The results of these assays were consistent (and in agreement) with the previous results
of C3 deposition assays and demonstrated that the absences of the classical pathway, i.e.
C1q deficiency, had minimal effect on the lytic activity of C1q deficient serum due to
the presence of the other two (alternative and lectin) complement pathways (see Figures
3.16 and 3.17). However, in the absence of the lectin pathway, i.e. MASP-2 deficiency,
there was an increase in the lytic activity of the MASP-2 deficient serum (see Figures
3.20 and 3.21), which could be explained by the presence of the lectin pathway
molecules MASP-1 and/or MASP-3 that could also drive activation of the alternative
pathway (Iwaki et al., 2011). In addition to that it demonstrates that the MASP-2 lectin
pathway does not seem to play a major role in the lysis of Neisseria meningitides and
confirms previous work carried out in our lab (Hayat, 2012).
The important role of the alternative pathway was highlighted by using different
alternative pathway deficient sera (factor B deficient and MASP-1/3 deficient) which
showed that even in the presence of the other two complement pathways, classical and
lectin pathways, the lytic activity of the sera was inefficient due to the deficiency of the
alternative pathway (see Figures 3.18 and 3.19). This result is in agreement with
previous reports (Biesma et al., 2001; Hiemstra et al., 1989; Sprong et al., 2006) that
showed an association between alternative pathway deficiency and Neisseria
meningitidis infection. To further study whether the impairment of alternative pathway

Chapter 3: In vitro study
115
lytic activity was due to deficiency of MASP-3, experiments were undertaken to restore
alternative pathway by the addition of truncated recombinant MASP-3 to the MASP-1/3
deficient sera. Serum bactericidal assays using MASP-1/3 deficient serum with and
without truncated recombinant murine MASP-3 showed that the lytic activity of
alternative pathway against both strains of Neisseria meningitidis was completely
restored following the addition of truncated recombinant murine MASP-3 (see Figures
3.22 and 3.23). In addition to this, it highlighted the importance of the alternative
pathway in fighting Neisseria meningitidis infection. This result supported previous
work (Takahshi et al., 2010) which showed that MASP-1/3 deficient mice lack
alternative pathway activity. This is also consistent with Iwaki et al., (2011) who
showed that MASP-3 of lectin pathway has the ability to induce the activity of the
alternative pathway by cleaving C4b bound factor B on the surface of Staphylococcus
aureus.
To further analyse this, the bacteriolytic activity of human serum from a patient with
Carnevale, Mingarelli, Malpuech and Michels syndrome (3MC) - these patients have
MASP-1 but not MASP-3 (Rooryck et al., 2011). The mutation in MASP-3 makes it
dysfunctional. This serum showed lytic activity toward Neisseria meningitidis
serogroup B strain MC58, but not to serogroup A strain Z2491, which could be due to
the presence of antibodies against Neisseria meningitidis in the serum, which binds to
the surface and then activates the classical pathway. In addition to this, restoring the
activity of the alternative pathway by adding truncated recombinant MASP-3 enhanced
the lytic activity of the serum (see Figures 3.24 and 3.25). This finding supports the
previous results which showed that MASP-3 was crucial in driving the activity of the

Chapter 3: In vitro study
116
alternative pathway and therefore enhancing the lytic activity of the serum toward the
Neisseria meningitidis, as well as the role of alternative pathway in fighting Neisseria
meningitidis.
Additionally, other in vitro assays demonstrated the importance of the lectin pathway
molecule MBL in fighting Neisseria meningitidis. This was demonstrated when testing
the bacteriolytic activity of MBL deficient serum against Neisseria meningitidis, which
showed that the ability of mouse MBL deficient serum to kill Neisseria meningitidis
was impaired compared to mouse MBL sufficient serum (see Figures 3.20 and 3.21).
The same observation was also seen with human serum, where the ability of human
MBL deficient serum to lyse Neisseria meningitidis was impaired compared to normal
human serum (see Figures 3.24 and 3.25). These results are in agreement with previous
results (Hibberd et al., 1999) which showed the important role of MBL in fighting
Neisseria meningitidis infection.
Taken together, the results strongly suggest that both lectin pathway molecules, i.e.
MBL and MASP-3, play crucial roles in mediating the lytic activity toward Neisseria
meningitidis. This observation is in line with Iwaski et al.’s(2011)reportwhichshowed
the complex of MASP-3 and MBL was able to trigger the activation of the alternative
pathway on the surface of Staphylococcus aureus by cleaving the proenzymes
C3(H2O)B or C3bB to their enzymaticly active forms. Moreover, the findings presented
in this chapter and in previously publisheded work (Schwaeble et al., 2011)
demonstrates that the two effector arms of lectin pathway, which are:
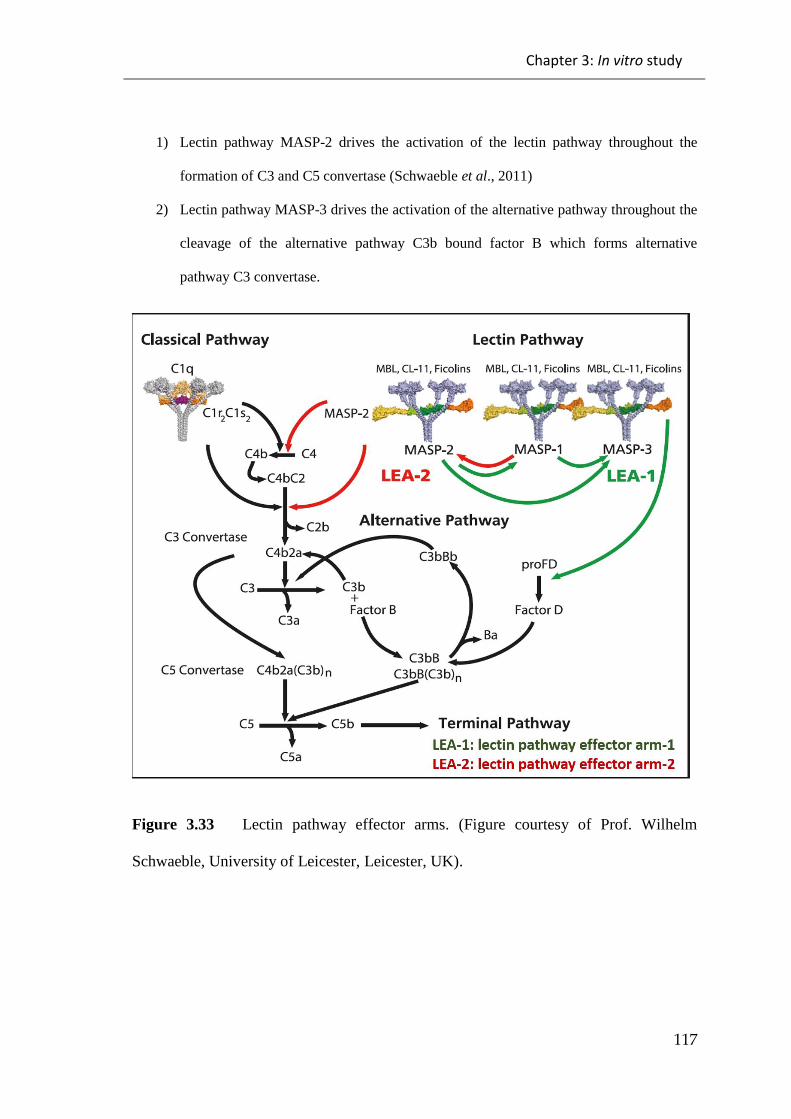
Chapter 3: In vitro study
117
1) Lectin pathway MASP-2 drives the activation of the lectin pathway throughout the
formation of C3 and C5 convertase (Schwaeble et al., 2011)
2) Lectin pathway MASP-3 drives the activation of the alternative pathway throughout the
cleavage of the alternative pathway C3b bound factor B which forms alternative
pathway C3 convertase.
Figure 3.33 Lectin pathway effector arms. (Figure courtesy of Prof. Wilhelm
Schwaeble, University of Leicester, Leicester, UK).

Chapter 3: In vitro study
118
3.2.3 Recombinant properdin enhances the serum bacteriolytic
activity against Neisseria meningitidis
Properdin is a glycoprotein known as positive regulator of complement activation. It
works to stabilize the C3 convertase C3bBb on the pathogen through increasing its half-
life. This function (stabilizing C3 convertase) can be done in two ways. One of them is
by binding to the C3 convertase on the surface of the pathogen. The other way is
through binding to the surface of the pathogen and then starting the formation of the
alternative pathway C3 convertase (Schwaeble and Reid, 1999). Kemper et al., (2008)
claimed that properdin could initiate alternative pathway activation by working as a
pattern recognition molecule. However, Harboe et al., (2012) studied the binding of
properdin to the pathogen surface and found that the binding between properdin and the
pathogen surface could not be found in the absence of C3 deposition indicating that this
binding does not initiate the alternative pathway. The association between properdin
deficiency and Neisseria meningitidis infection has been reported previously,
illustrating the important role of properdin in fighting Neisseria meningitidis infection
(Fijen et al., 1995).
Therefore, in order to assess the effect of properdin on the formation of C3 deposition
and thus the lytic activity of the serum, the effect of adding recombinant properdin to
serum on the formation of C3 deposition was examined by measuring the amount of the
C3 deposition on the surface of Neisseria meningitidis using both ELISA assay and
FACS analysis. The results of these assays showed an increased amount of C3
deposition on the surface of the tested Neisseria meningitidis strains following the
addition of recombinant properdin (see Figures 3.26, 3.27 and 3.28). This finding is

Chapter 3: In vitro study
119
consistent with that of Agarwal et al., (2010) who reported an increase in C3 deposition
on the surface of Neisseria meningitidis that were pre-incubated with properdin,
followed by the addition of properdin depleted serum.
To further analyse and confirm the important role of properdin in enhancing the serum
bactericidal activity against Neisseria meningitidis. It was shown that serum bactericidal
activity of human or mouse was enhanced significantly by adding recombinant
properdin compared to normal serum alone (See Figures 3.29, 3.30, 3.31 and 3.32). This
result further demonstrated the important role of the alternative pathway in fighting
Neisseria meningitidis infection.
In conclusion, the in vitro study demonstrated the important roles of the lectin and the
alternative pathways in fighting Neisseria meningitidis infection. It also highlighted the
major role of the lectin pathway complex molecule i.e. MASP-3 and MBL, in driving
the activation of the alternative pathway on the surface of Neisseria meningitidis
through the cleavage of the alternative pathway zymogen pro-factor D to Factor D
which is essential to allow C3bB complexes to be converted into the alternative
pathway C3 convertase complex C3bBb in a factor D dependent fashion. The
amplification loop that the alternative pathway provides to generate optimal activation
rates for the formation of MAC complexes appears to be critical in allowing an effective
lysis of Neisseria meningitidis. This is in line with the observation that serum
bactericidal activity against Neisseria meningitidis can be significantly increased
through the addition of highly active, high grade polymerised recombinant properdin
which in turn increases the half-live of the alternative pathway C3 and C5 convertase

Chapter 3: In vitro study
120
complexes by protecting C3b within these complexes from the decay activity of serum
factor H and other cofactors, like DAF or MCP. Thus the promotion of alternative
pathway functional activity was shown to enhance the ability of mice to fight Neisseria
meningitidesinfection.

Chapter 4: In vivo study
121
Chapter 4: In vivo study
The current in vitro study of the alternative pathway against Neisseria meningitidis
shows that a limited level of C3 deposition was observed following the use of
alternative pathway deficient serum compared to wild-type serum. Furthermore, the
serum bactericidal assay of sera from factor B deficient mice and MASP-1/3 deficient
mice showed no killing of Neisseria meningitidis compared to the wild-type serum.
These results led to do in vivo infection experiments in a mice model of Neisseria
meningitidis infection in Factor B deficient mice and MASP-1/3 deficient mice which
provide a mice model with a total defect in the alternative pathway.
4.1 Results:
4.1.1 Genotyping of factor B deficient mice and MASP-1/3 deficient
mice
All gene-targeted complement deficient mouse lines, i.e. factor B deficient mice and
MASP-1/3 deficient mice that have been used throughout this study were bred and
housed in the Central Research Facility at the University of Leicester which allows to
breed mice in a high quality pathogen free environment.
The alternative pathway Factor B molecule is a single glycoprotein that upon activation
is attached to C3b cleaved by Factor D into two fragments, the major Bb fragment that
is composed of a von Willebrand factor domain and the serine protease domain and the

Chapter 4: In vivo study
122
Ba fragment (composed of 3 CCP domains and a spacer fragment (alpha-L). While the
Bb fragment remains bound to C3b to form the alternative pathway C3 convertase
C3bBb, the Ba fragment is released and does not contribute to further complement
activation processes. (Mole et al. 1984, Williams et al. 1999). This molecule is encoded
by the complement Factor B gene which consists of 18 exons. Targeting exons 4-9
disrupted the Factor B gene resulting a complete deficiency of Factor B (Matsumoto et
al., 1997).
The results of the PCR show the amplified products which are a single band at
approximately 986bp identified as wild-type allele (wild-type mouse FB gene), a single
band at approximately 650bp identified as the gene targeted allele (factor B deficient
allele). If both bands of approximately 986bp and 650bp were amplified on DNA of our
FB line, they indicate that these mice are heterozygous containing both the WT and the
targeted FB alleles (heterozygous mice) (see Figure 4.1).
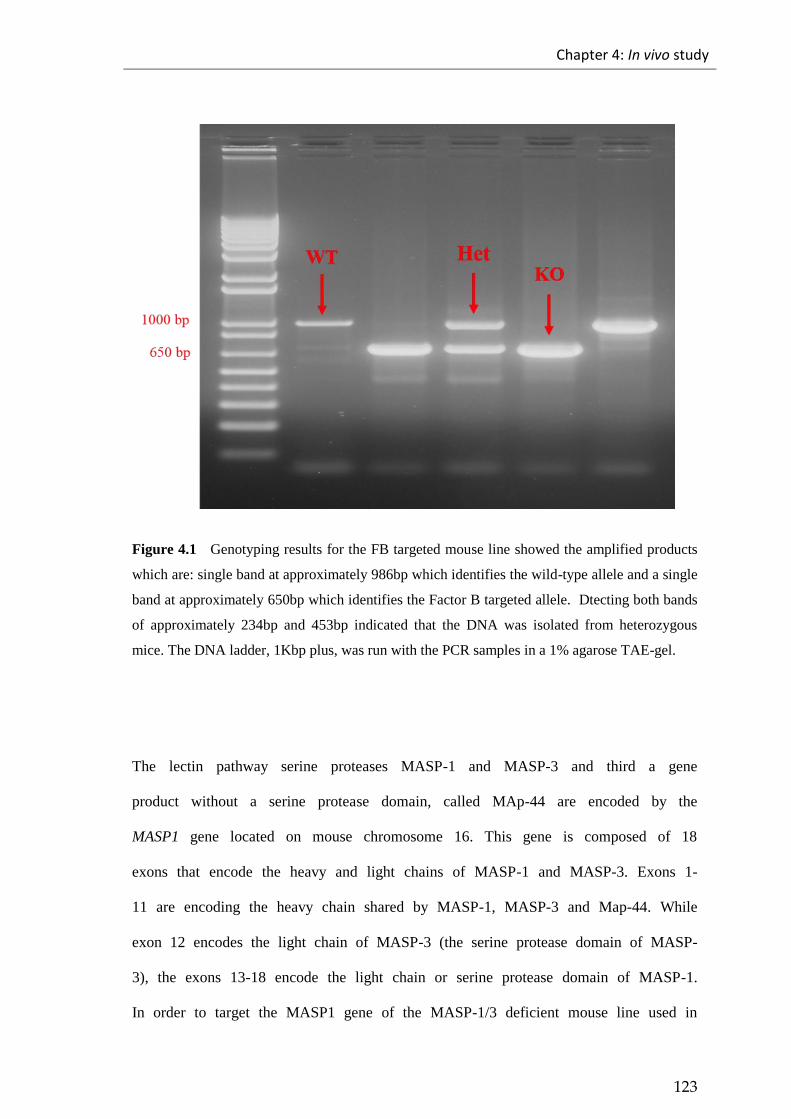
Chapter 4: In vivo study
123
Figure 4.1 Genotyping results for the FB targeted mouse line showed the amplified products
which are: single band at approximately 986bp which identifies the wild-type allele and a single
band at approximately 650bp which identifies the Factor B targeted allele. Dtecting both bands
of approximately 234bp and 453bp indicated that the DNA was isolated from heterozygous
mice. The DNA ladder, 1Kbp plus, was run with the PCR samples in a 1% agarose TAE-gel.
The lectin pathway serine proteases MASP-1 and MASP-3 and third a gene
product without a serine protease domain, called MAp-44 are encoded by the
MASP1 gene located on mouse chromosome 16. This gene is composed of 18
exons that encode the heavy and light chains of MASP-1 and MASP-3. Exons 1-
11 are encoding the heavy chain shared by MASP-1, MASP-3 and Map-44. While
exon 12 encodes the light chain of MASP-3 (the serine protease domain of MASP-
3), the exons 13-18 encode the light chain or serine protease domain of MASP-1.
In order to target the MASP1 gene of the MASP-1/3 deficient mouse line used in
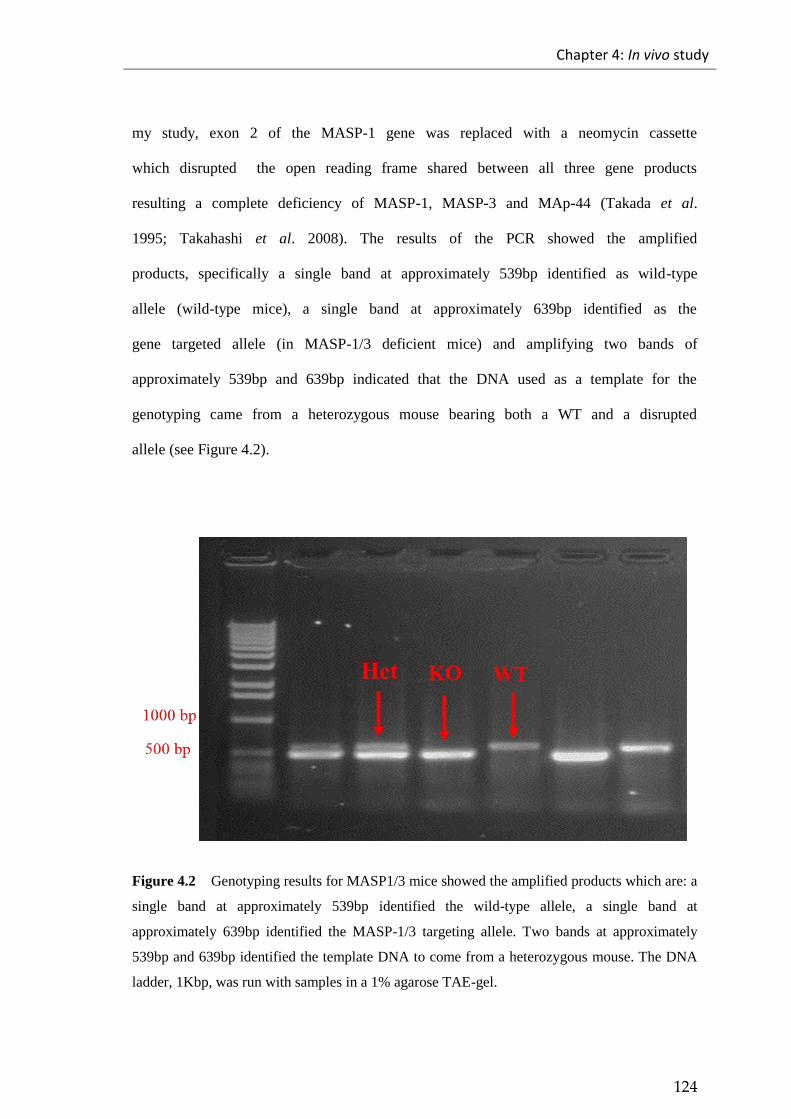
Chapter 4: In vivo study
124
my study, exon 2 of the MASP-1 gene was replaced with a neomycin cassette
which disrupted the open reading frame shared between all three gene products
resulting a complete deficiency of MASP-1, MASP-3 and MAp-44 (Takada et al.
1995; Takahashi et al. 2008). The results of the PCR showed the amplified
products, specifically a single band at approximately 539bp identified as wild-type
allele (wild-type mice), a single band at approximately 639bp identified as the
gene targeted allele (in MASP-1/3 deficient mice) and amplifying two bands of
approximately 539bp and 639bp indicated that the DNA used as a template for the
genotyping came from a heterozygous mouse bearing both a WT and a disrupted
allele (see Figure 4.2).
Figure 4.2 Genotyping results for MASP1/3 mice showed the amplified products which are: a
single band at approximately 539bp identified the wild-type allele, a single band at
approximately 639bp identified the MASP-1/3 targeting allele. Two bands at approximately
539bp and 639bp identified the template DNA to come from a heterozygous mouse. The DNA
ladder, 1Kbp, was run with samples in a 1% agarose TAE-gel.
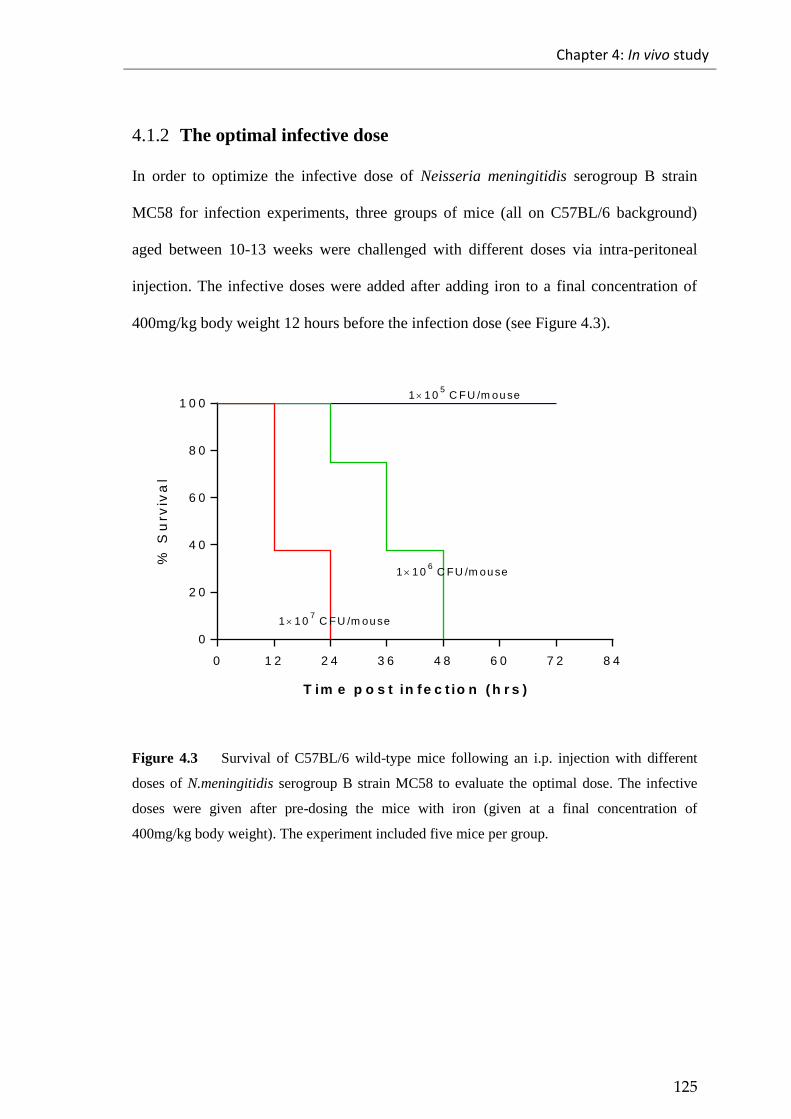
Chapter 4: In vivo study
125
4.1.2 The optimal infective dose
In order to optimize the infective dose of Neisseria meningitidis serogroup B strain
MC58 for infection experiments, three groups of mice (all on C57BL/6 background)
aged between 10-13 weeks were challenged with different doses via intra-peritoneal
injection. The infective doses were added after adding iron to a final concentration of
400mg/kg body weight 12 hours before the infection dose (see Figure 4.3).
0 1 2 2 4 3 6 4 8 6 0 7 2 8 4
0
2 0
4 0
6 0
8 0
1 0 0
T im e p o s t in fe c t io n ( h r s )
% S
urv
iva
l
1 1 05 C FU /m ou se
1 1 06 C FU /m ou se
1 1 07 C FU /m ou se
Figure 4.3 Survival of C57BL/6 wild-type mice following an i.p. injection with different
doses of N.meningitidis serogroup B strain MC58 to evaluate the optimal dose. The infective
doses were given after pre-dosing the mice with iron (given at a final concentration of
400mg/kg body weight). The experiment included five mice per group.

Chapter 4: In vivo study
126
4.1.3 Survival of factor B deficient mice and factor B sufficient mice
following experimental Neisseria meningitidis infection
The host immune response to Neisseria meningitidis infection in terms of the role of
alternative pathway was assessed using Factor B (FB) deficient mice and Factor B (FB)
sufficient mice (WT). The two groups of mice aged between 10-13 weeks, i.e. Factor B
deficient and Factor B sufficient, were challenged with a low dose (1×105 colony
forming unit (CFU)/mouse) of Neisseria meningitidis serogroup B strain MC58, via an
intra-peritoneal route, as a higher dose would kill the wild-type control mice. Mice
received iron dextran (400mg/kg body weight) 12 hours before the administration of the
infection dose. Mice were constantly monitored for the development or onset of the
signs of illness throughout the experiment.
Figures 4.4, 4.5 and 4.6 show the survival of Factor B deficient mice, illness scores and
the recovery of viable bacterial counts from blood in FB deficient compared to FB
sufficient WT mice.
At 12 hours post infection, all FB deficient mice started developing clear signs of illness
and at the time point 24 hours post infection 50% of the FB deficient mice had to be
euthanized before they progressed to a lethargic stage. At the same time points, Factor
B sufficient WT mice looked healthy with no signs of illness. The rest of the Factor B
deficient mice continued developing signs of illness, and the experiment had to be
terminated as the remaining mice approached a lethargic end stage with a mortality rate
of 100% at 48 hours post infection (see Figure 4.4), the Factor B sufficient mice showed

Chapter 4: In vivo study
127
only minor signs of illness at 36 hours post infection and later they started to recover
(see Figure 4.5). These results showed that there was a dramatically increased
susceptibility of Factor B deficient mice to Neisseria meningitidis infection compared
with the Factor B sufficient WT control mice which showed significantly lower signs of
illness in terms of disease severity.
The difference between the survival of Factor B deficient mice and Factor B sufficient
mice has been statistically analysed using the Log-rank (Mantel-Cox) test, which
showed a significant difference between the two groups of mice, emphasizing the
important role of the alternative pathway in terms of the host immune response to
Neisseria meningitidis infection.
0 1 2 2 4 3 6 4 8 6 0 7 2
0
2 0
4 0
6 0
8 0
1 0 0
T im e p o s t in fe c t io n ( h r s )
% S
urv
iva
l
W T
F B- / -
* * * * p < 0 .0 0 0 1
Figure 4.4 Survival of wild type (on C57/BL6 background) and Factor B deficient mice (on
C57/BL6 background) following an i.p. injection with a low dose (1×105) of Neisseria
meningitidis serogroup B-MC58. The infective dose was added after adding iron to a final
concentration of 400mg/kg body weight. It was shown that Factor B deficient mice were
significantly more susceptible to Neisseria meningitidis infection compared to the Factor B
sufficient mice. ****p < 0.0001 Log-rank (Mantel-Cox) test; n=10/group.

Chapter 4: In vivo study
128
612
24
36
48
72
0
1
2
3
4
5
T im e p o s t in fe c t io n ( h r s )
Av
era
ge
ill
ne
ss
sc
ore
W T
FB- / -
* * * *
* * * ** * * *
* * * *
Figure 4.5 Average illness score of wild type and Factor B deficient mice following an i.p.
injection with a low dose (1×105) of Neisseria meningitidis serogroup B-MC58. The data are
presented as mean with SEM. ****p < 0.0001, Student t test.
4.1.3.1 The viable bacterial load of Neisseria meningitidis in the blood of infected
mice
Throughout the course of the infection experiment of Factor B deficient mice and their
sex and age matched Factor B sufficient WT control mice (WT) infected with a low
dose (1×105 CFU/mouse) of Neisseria meningitidis serogroup B strain MC58, the mice
were monitored and blood was taken at constant time intervals via a tail bleed route to
assess the viable bacterial load in the blood at defined time points 12, 24, 36 and 48
hours post infection. This showed that the bacterial load in the blood of FB deficient

Chapter 4: In vivo study
129
mice was significantly higher compared to the bacterial load in the blood of Factor B
sufficient WT control mice, indicating the development of bacteraemia (see Figure 4.6).
At 48 hours post infection, the Factor B sufficient mice started to clear the bacteria from
the blood, while the survivors of the FB deficient mice group had developed higher
bacteraemia, showing signs of terminal disease; thus, they had to be euthanized.
12.
24.
36.
48.
0
1
2
3
4
5
6
T im e p o s t in f e c t io n ( h r s )
Lo
g C
FU
/ml
of
blo
od W T
F B- /-
*
*
* *
* *
Figure 4.6 Bacterial load in the blood of Factor B deficient mice and wild-type mice given an
i.p. injection of a low dose (1x105
CFU/mouse) of Neisseria meningitidis serogroup B strain
MC58 at time points 12, 24, 36 and 48 hours post infection. The data are presented as mean
with SEM. *p < 0.0156, **p < 0.0063, Student t test.

Chapter 4: In vivo study
130
4.1.4 Survival of MASP-1/3 deficient mice and MASP-1/3 sufficient
mice following experimental Neisseria meningitidis infection
The lectin pathway molecule MASP-1 was recently shown to play an important role in
maintaining alternative pathway functional activity (Takahashi et al. 2010). I therefore
assessed if MASP-1/3 deficient mice are similarly compromised in their ability to
Neisseria meningitidis infections like FB deficient mice. Thus, in order to assess this
role of MASP-1/3 deficiency in vivo, two groups of mice, MASP-1/3 deficient mice and
MASP-1/3 sufficient mice (WT), aged between 10-13 weeks (all on C57/BL6
background), were challenged with a low dose of Neisseria meningitidis serogroup B
strain MC58 (1×105
CFU/mouse) via an intra-peritoneal route after receiving iron
dextran (400mg/kg body weight) 12 hours prior to challenge. The mice were
subsequently checked, and the development of meningitis was monitored throughout the
experiment. Figures 4.7, 4.8 and 4.9 show the survival of MASP-1/3 deficient mice,
their illness scores and the recoverable viable bacterial load from blood samples taken at
different time points after infection were compared to MASP-1/3 sufficient WT mice.
The MASP-1/3 deficient mice started developing the signs of illness at 12 hours post
infection. These signs progressed, leading to the death of 40% of these mice at 24 hours
post infection. At the same time, the wild-type mice remained healthy with no sign of
illness. At 54 hours post infection, the remaining MASP-1/3 deficient mice showed
signs of terminal disease and had to be euthanized (see Figure 4.7) while the MASP-1/3
sufficient mice were have started to recover with only minor signs of illness that
developed at 36 hours post infection (see Figure 4.8). These results highlighted the
importance of the lectin pathway molecule MASP-1/3 in driving the alternative pathway
to clear the bacteria. In contrast MASP-1/3 deficiency increases the susceptibility of

Chapter 4: In vivo study
131
mice to Neisseria meningitidis infection, unlike in MASP-1/3 sufficient WT mice,
which show significantly lower signs of illness in terms of disease severity when
compared to MASP-1/3 deficient mice.
The difference between the survival of MASP-1/3 deficient mice and MASP-1/3
sufficient WT mice was statistically analysed using the Log-rank (Mantel-Cox) test,
which showed a significant difference between the two groups of mice emphasizing the
important role of MASP-1/3 in driving the alternative pathway activation in terms of
host immune response to Neisseria meningitidis infection.
0 1 2 2 4 3 6 4 8 6 0 7 2
0
2 0
4 0
6 0
8 0
1 0 0
T im e p o s t in fe c t io n ( h r s )
% S
urv
iva
l
W T
M A S P -1 /3- / -
****p < 0 .0 0 0 1
Figure 4.7 Survival of WT and MASP-1/3 deficient mice (both on C57/BL6 background)
following an i.p. injection with a low dose (1×105) of Neisseria meningitidis serogroup B-
MC58. The infective dose was added after adding iron to a final concentration of 400mg/kg
body weight. . It was shown that MASP-1/3 deficient mice were more susceptible to
Neisseria meningitidis infection compared to the MASP-1/3 sufficient mice. ****p < 0.0001
Log-rank (Mantel-Cox) test; n=10/group.
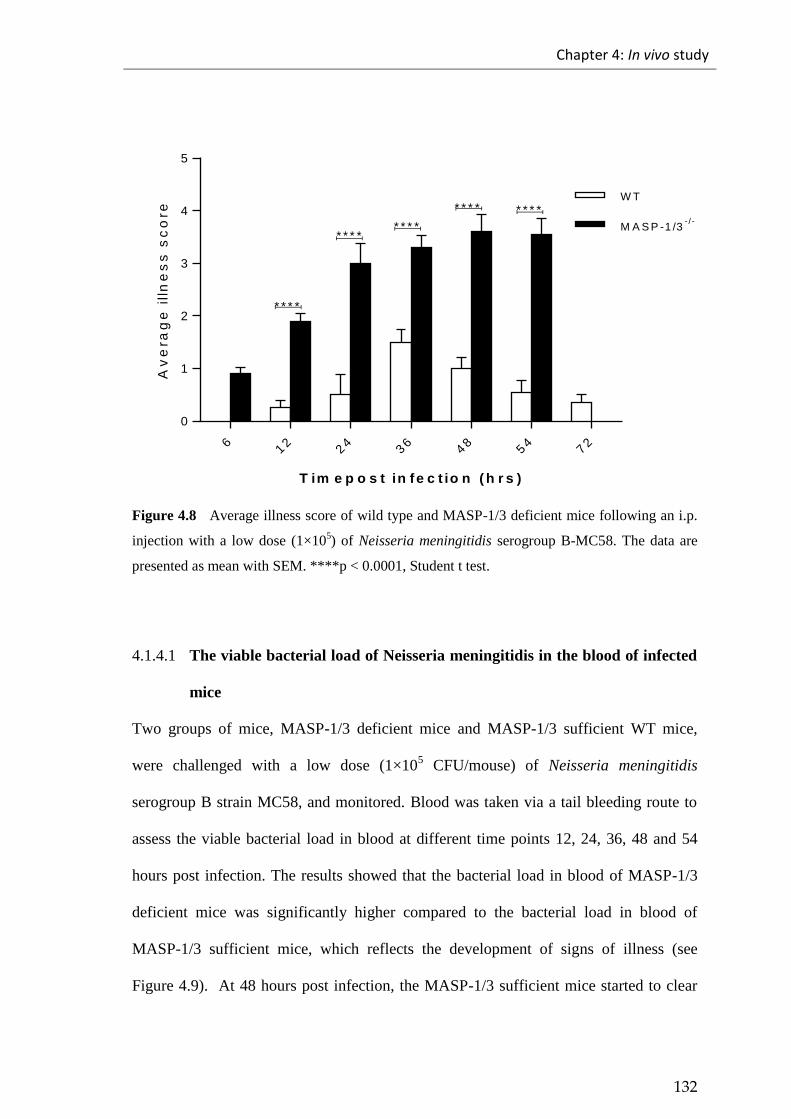
Chapter 4: In vivo study
132
612
24
36
48
54
72
0
1
2
3
4
5
T im e p o s t in fe c t io n ( h r s )
Av
era
ge
ill
ne
ss
sc
ore
W T
M A S P -1 /3- / -
* * * *
* * * ** * * *
* * * * * * * *
Figure 4.8 Average illness score of wild type and MASP-1/3 deficient mice following an i.p.
injection with a low dose (1×105) of Neisseria meningitidis serogroup B-MC58. The data are
presented as mean with SEM. ****p < 0.0001, Student t test.
4.1.4.1 The viable bacterial load of Neisseria meningitidis in the blood of infected
mice
Two groups of mice, MASP-1/3 deficient mice and MASP-1/3 sufficient WT mice,
were challenged with a low dose (1×105 CFU/mouse) of Neisseria meningitidis
serogroup B strain MC58, and monitored. Blood was taken via a tail bleeding route to
assess the viable bacterial load in blood at different time points 12, 24, 36, 48 and 54
hours post infection. The results showed that the bacterial load in blood of MASP-1/3
deficient mice was significantly higher compared to the bacterial load in blood of
MASP-1/3 sufficient mice, which reflects the development of signs of illness (see
Figure 4.9). At 48 hours post infection, the MASP-1/3 sufficient mice started to clear

Chapter 4: In vivo study
133
the bacteria from the blood while the remaining MASP-1/3 deficient mice had
developed higher bacteraemia, leading to the development of the signs of terminal
illness; thus, these animals had to be euthanized.
12.
24.
36.
48.
54
0
1
2
3
4
5
6
T im e p o s t in f e c t io n ( h r s )
Lo
g C
FU
/ml
of
blo
od
W T
M A S P - 1 /3- /-
*
*
* *
* ** *
Figure 4.9 Bacterial load in blood of MASP-1/3 deficient mice and wild-type mice given an
i.p. injection of a low dose (1x105 CFU/mouse) of Neisseria meningitidis serogroup B strain
MC58 at time points 12, 24, 36, 48 and 54 hours post infection. The data are presented as mean
with SEM. *p < 0.0161, **p < 0.0047, Student t test.

Chapter 4: In vivo study
134
4.1.5 Effect of full length recombinant MASP-3 on reconstituting the
absence of the alternative pathway functional activity in MASP-
1/3 deficient mice
In order to assess whether full length recombinant murine MASP-3 had the ability to
reconstitute the functional activity of the alternative pathway in MASP-1/3 deficient
mice and therefore the bacteriolytic activity toward the Neisseria meningitidis, MASP-
1/3 sufficient mice and MASP-1/3 deficient mice were divided into four groups. The
first group are MASP-1/3 deficient mice used as a control. The second groups are
MASP-1/3 deficient mice treated with a single dose of full-length recombinant murine
MASP-3 (20 µg/mouse) given i.v. 96 hours prior to the final bleed. The third groups
comprised the MASP-1/3 deficient mice treated twice with full length recombinant
murine MASP-3 (20 µg/mouse) given in two separate doses at 96 hours before infection
and 24 hours prior to the final bleed. The fourth groups is made up of sex and age
matched MASP-1/3 sufficient WT mice used as a reference for mice with full
alternative pathway functional activity. These mice were monitored and bled every day
until the end of the experiment (day 4).

Chapter 4: In vivo study
135
Table 4.1 The design of the MASP-3 reconstitution experiment
Mice group
Dosage
Full length recombinant
MASP-3
Days of injection
Group one MASP-1/3 deficient mice
i.v.20μg/mice Day zero
Group two MASP-1/3 deficient mice
i.v.20μg/mice
i.v.20μg/mice
Day zero
Day three
Group three MASP-1/3 deficient mice
Saline only Day zero
Group four MASP-1/3 sufficient mice (WT)
Saline only Day zero
The ability of full length recombinant murine MASP-3 to reconstitute the alternative
pathway functional activity was assessed by measuring C3 deposition of alternative
pathway. This assay showed that the functional activity of MASP-1/3 deficient mice
was restored by 35% at 24 hours post injection. The functional activity of alternative
pathway was nearly fully restored at 72 hours post injection. However, at 96 hours post
injection, the alternative pathway activity started to decline again in MASP-1/3
deficient mice as shown in the group of mice that was treated only once, while the
second group (treated twice) kept the level of the alternative pathway activity steady as
it was treated twice (see Figure 4.10).

Chapter 4: In vivo study
136
24
48
72
96
0 .0
0 .3
0 .6
0 .9
1 .2
T im e p o s t in je c t io n (d y s )
OD
At
40
5n
m
W T
M A S P -1 /3- / -
M A S P -1 /3- / -
+ rM A S P -3 (4 0 u g /m o u s e )
M A S P -1 /3- / -
+ rM A S P -3 (2 0 u g /m o u s e )
Figure 4.10 C3 deposition of MASP-1/3 deficient mice treated with full length recombinant
murine MASP-3. Mice were treated once or twice with rMASP-3 (20µg/mouse) by i.v.
injection. Mice were monitored and bled 24, 48, 72 and 96 hours post injection. The C3
deposition assay was used to assess the functional activity of the alternative pathway. Mouse
serum was diluted 13% with EGTA buffer. Zymosan was used to coat the plates. The binding of
the C3 cleavage product (C3c) was detected by using rabbit anti-human C3c polyclonal
antibody. It was shown that the full functional activity of the alternative pathway was restored
gradually by 72 hours post injection. This restored functionality started to decline in the group
that was treated once while in the group that was treated twice, it remained unchanged. (Data
presented as a mean of two independent experiments with duplicate for each +/- SEM).

Chapter 4: In vivo study
137
Table 4.2 Statistically significant differences for the C3 deposition assay of different mouse
sera (MASP-1/3-/-
+ full-length rMASP-3 (40µg/mouse), MASP-1/3-/-
+ full length rMASP-3
(20µg/mouse), MASP-1/3-/-,WTandHIS)usingtheStudent’st-test at time point 96 hours.
Student’st-test at time point 96 hours
Significant? P > 0.05 P value Summary
WT vs MASP-1/3-/-
YES ** (0.0031)
WT vs MASP-1/3-/-
+rMASP-3
(20µg/mouse) YES
* (0.0126)
WT vs MASP-1/3-/-
+rMASP-3
(40µg/mouse) YES
* (0.0276)
MASP-1/3-/-
vs MASP-1/3-/-
+ rMASP-
3 (20µg/mouse) YES
* (0.0050)
MASP-1/3-/-
vs MASP-1/3-/-
+ rMASP-
3 (40µg/mouse) YES
** (0.0015)
MASP-1/3-/-
+ rMASP-3 (20µg/mouse)
vs MASP-1/3-/-
+ rMASP-3
(40µg/mouse)
YES
* (0.0356)

Chapter 4: In vivo study
138
To study this reconstitution of a deficient AP functional activity by recombinant MASP-
3 further, serum bactericidal assays were performed to assess the effect of this
reconstitution on serum bactericidal activity (SBA) of mouse serum towards Neisseria
meningitidis after collecting blood from the mice by cardiac punctures. The results from
these assays showed that the killing of meningococcus was restored through
reconstitution of MASP-1/3 deficient mice by the full length recombinant murine
MASP-3 indicating that the activity of alternative pathway towards Neisseria
meningitidis had been restored (see Figures 4.11 and 4.12). The SBA of the mice treated
with a single dose of full length recombinant murine MASP-3 was nearly half of that
seen in mice treated with two doses of full length recombinant murine MASP-3.
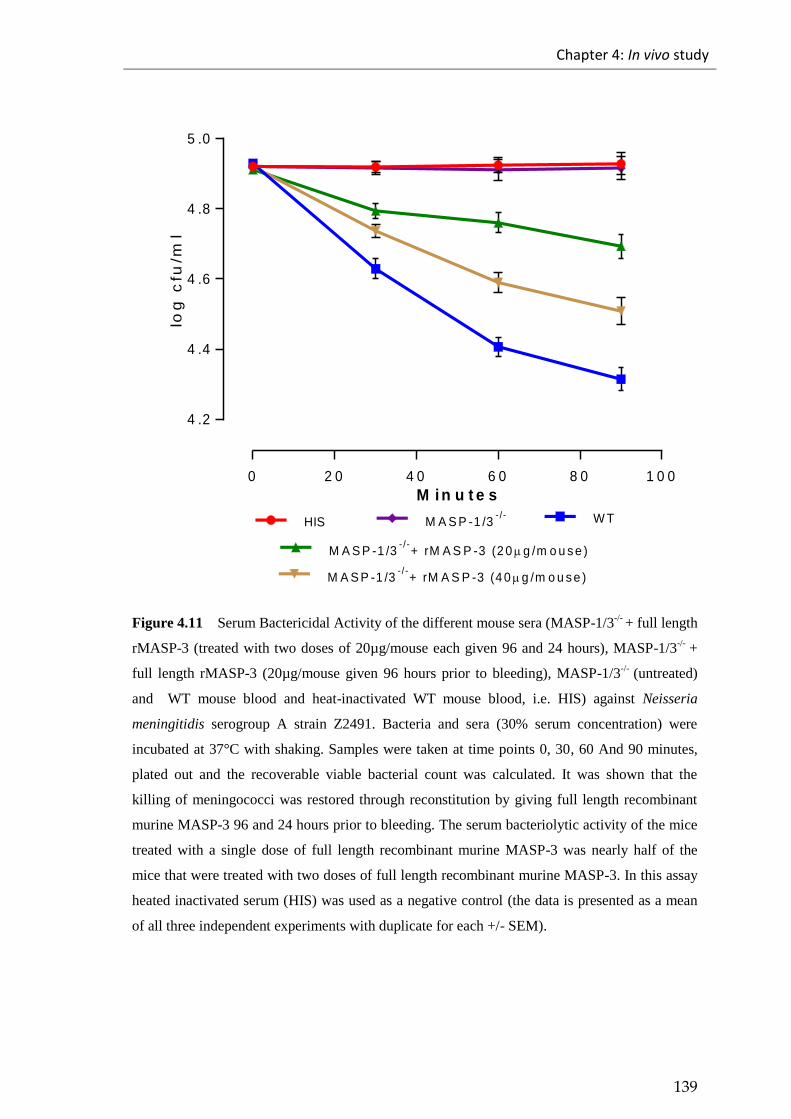
Chapter 4: In vivo study
139
0 2 0 4 0 6 0 8 0 1 0 0
4 .2
4 .4
4 .6
4 .8
5 .0
M i n u t e s
log
cfu
/ml
HIS W T
M A S P -1 /3- /-
+ rM A S P -3 (4 0 g /m o u s e )
M A S P -1 /3- /-
+ rM A S P -3 (2 0 g /m o u s e )
M A S P -1 /3- /-
Figure 4.11 Serum Bactericidal Activity of the different mouse sera (MASP-1/3-/-
+ full length
rMASP-3 (treated with two doses of 20µg/mouse each given 96 and 24 hours), MASP-1/3-/-
+
full length rMASP-3 (20µg/mouse given 96 hours prior to bleeding), MASP-1/3-/-
(untreated)
and WT mouse blood and heat-inactivated WT mouse blood, i.e. HIS) against Neisseria
meningitidis serogroup A strain Z2491. Bacteria and sera (30% serum concentration) were
incubated at 37°C with shaking. Samples were taken at time points 0, 30, 60 And 90 minutes,
plated out and the recoverable viable bacterial count was calculated. It was shown that the
killing of meningococci was restored through reconstitution by giving full length recombinant
murine MASP-3 96 and 24 hours prior to bleeding. The serum bacteriolytic activity of the mice
treated with a single dose of full length recombinant murine MASP-3 was nearly half of the
mice that were treated with two doses of full length recombinant murine MASP-3. In this assay
heated inactivated serum (HIS) was used as a negative control (the data is presented as a mean
of all three independent experiments with duplicate for each +/- SEM).

Chapter 4: In vivo study
140
Table 4.3 Statistically significant difference assessed between serum bactericidal assay of
different mouse serum (MASP-1/3-/-
+ full length rMASP-3 (treated with two doses of
20µg/mouse each given 96 and 24 hours), MASP-1/3-/-
+ full length rMASP-3 (20µg/mouse
given 96 hours prior to bleeding), MASP-1/3-/-
(untreated), WT and HIS) against Neisseria
meningitidis serogroupAstrainZ2491usingtheStudent’st-test at time point 90 min.
Student’st-test at time point 90 minutes
Significant? P > 0.05 P value Summary
WT vs MASP-1/3-/-
YES ** (0.0032)
WT vs MASP-1/3-/-
+rMASP-3
(20µg/mouse) YES
** (0.0081)
WT vs MASP-1/3-/-
+rMASP-3
(40µg/mouse) YES
* (0.0335)
MASP-1/3-/-
vsMASP-1/3 -/-
+
rMASP-3 (20µg/mouse) YES
* (0.0237)
MASP-1/3-/-
vsMASP-1/3-/-
+
rMASP-3 (40µg/mouse) YES
** (0.0080)
MASP-1/3-/-
+ rMASP-3
(20µg/mouse) vs MASP-1/3-/-
+
rMASP-3 (40µg/mouse)
YES
* (0.0379)

Chapter 4: In vivo study
141
0 2 0 4 0 6 0 8 0 1 0 0
4 .0
4 .5
M i n u t e s
log
cfu
/ml
HISW T
M A S P -1 /3- /-
+ rM A S P -3 (4 0 g /m o u s e )
M A S P -1 /3- /-
+ rM A S P -3 (2 0 g /m o u s e )
M A S P -1 /3- /-
Figure 4.12 Serum Bactericidal Activity of the different mouse sera (MASP-1/3-/-
+ full length
rMASP-3 (treated with two doses of 20µg/mouse each given 96 and 24 hours), MASP-1/3-/-
+
full length rMASP-3 (20µg/mouse given 96 hours prior to bleeding), MASP-1/3-/-
(untreated)
and WT mouse blood and heat-inactivated WT mouse blood, i.e. HIS) against Neisseria
meningitidis serogroup B strain MC58. Bacteria and sera (30% serum concentration) were
incubated at 37°C with shaking. Samples were taken at time points 0, 30, 60 And 90 minutes,
plated out and the recoverable viable bacterial count was calculated. It was shown that the
killing of meningococci was restored through reconstitution by giving full length recombinant
murine MASP-3 96 and 24 hours prior to bleeding. The serum bacteriolytic activity of the mice
treated with a single dose of full length recombinant murine MASP-3 was nearly half of the
mice that were treated with two doses of full length recombinant murine MASP-3. In this assay
heated inactivated serum (HIS) was used as a negative control (the data is presented as a mean
of all three independent experiments with duplicate for each +/- SEM).

Chapter 4: In vivo study
142
Table 4.4 Statistically significant difference assessed between serum bactericidal assay of
different mouse serum (MASP-1/3-/-
+ full length rMASP-3 (treated with two doses of
20µg/mouse each given 96 and 24 hours), MASP-1/3-/-
+ full length rMASP-3 (20µg/mouse
given 96 hours prior to bleeding), MASP-1/3-/-
(untreated), WT and HIS) against Neisseria
meningitidis serogroupBstrainMC58usingtheStudent’st-test at time point 90 min
Student’st-test at time point 90 minutes
Significant? P > 0.05 P value Summary
WT vs MASP-1/3-/- YES ** (0.0083)
WT vs MASP-1/3-/-
+rMASP-3
(20µg/mouse) YES
** (0.0095)
WT vs MASP-1/3-/-
+rMASP-3
(40µg/mouse) YES
* (0.0356)
MASP-1/3-/-
vsMASP-1/3-/-
+
rMASP-3 (20µg/mouse) YES * (0.0128)
MASP-1/3-/-
vsMASP-1/3-/-
+
rMASP-3 (40µg/mouse) YES
** (0.0091)
MASP-1/3-/-
+ rMASP-3
(20µg/mouse) vs MASP-1/3-/-
+
rMASP-3 (40µg/mouse)
YES * (0.0475)

Chapter 4: In vivo study
143
4.1.6 Effect of full length recombinant MASP-3 administration on
mortality in a mouse model of Neisseria meningitidis infection
MASP-1, a lectin pathway specific serine protease, has previously been reported to play
a role in driving the alternative pathway activation on the surface of pathogens by
cleaving pro-Factor D (FD) to Factor D (FD) to allow the alternative pathway C3
convertase zymogen C3bB to be converted into its active form C3bBb (Takahashi et al.,
2010). My work demonstrates that it is in fact the absence of MASP-3 that leads to the
loss of AP functional activity and not the absence of MASP-1. Reconstitution
experiments of the MASP-1/3 deficient mouse line with recombinant MASP-1 failed to
restore the AP deficient phenotype (Takahashi et al., 2010). My work shows that the
administration of MASP-3 in MASP-1/3 deficient mice restores the functional activity
of the alternative pathway and also restores the resistance of reconstituted MASP-1/3
deficient mice against Neisseria meningitidis both in vitro and as you see below also in
vivo.
In order to assess the potential benefits of the therapeutic administration of full length
recombinant MASP-3 in MASP-1/3 deficient mice in fighting the Neisseria
meningitidis infections, three groups of mice aged between 11-13 weeks were
challenged with a low dose (1×105 CFU/mouse) of
Neisseria meningitidis serogroup B
strain MC58, via the intra-peritoneal route, following injection the mice with or without
full length recombinant murine MASP-3 via an intravenous route. Prior to infection, all
mice received iron (400mg/kg body weight) 12 hours prior to the infection dose. The
mouse groups were: i) group A MASP-1/3 deficient mice, ii) group B composed of

Chapter 4: In vivo study
144
MASP-1/3 deficient mice treated twice with full length recombinant murine MASP-3,
96 hours and 24 hours prior to infection, and iii) group C composed of MASP-1/3
sufficient mice (WT) used as a positive control. Following the infection dose, the mice
were checked and continuously monitored for the development of signs of illness
throughout the experiment. The MASP-1/3 deficient mice, with or without being given
full-length recombinant MASP-3, were compared in terms of survival, illness scores
and bacterial viable load of blood with the MASP-1/3 sufficient mice (see Figures 4.13,
4.14 and 4.15) .
While both groups of MASP-1/3 deficient mice developed signs of illness, the MASP-
1/3 deficient mice that were not treated with full-length recombinant MASP-3
developed more severe signs compared to the treated ones. At the same time the MASP-
1/3 sufficient mice were healthy with no sign of illness. The severity of disease in
MASP-1/3 deficient mice that were not reconstituted with MASP-3 continued to
progress, and at time point 24 hours post infection 30% of this group had to be
euthanized because of the severity of disease signs. At time point 54 hours post
infection the rest of the MASP-1/3 deficient mice progressed to terminal disease
symptoms and had to be euthanized. In comparison, at this time point (i.e. 54 hours post
infection) only 30% of the MASP-3 reconstituted MASP-1/3 deficient mice developed
signs of terminal disease and had to be euthanized. The remaining mice in this group
recovered from the infection with an overall survival of 70% in the treatment group,
compared to 0% survival in the non-treated MASP-1/3 deficient littermates. MASP-1/3
sufficient WT mice developed minor signs of illness, but all of them recovered fully and
survived. At the end of experiment i.e. 96 hours post infection, we observed no
significant difference in illness scores when comparing the MASP-3 reconstituted
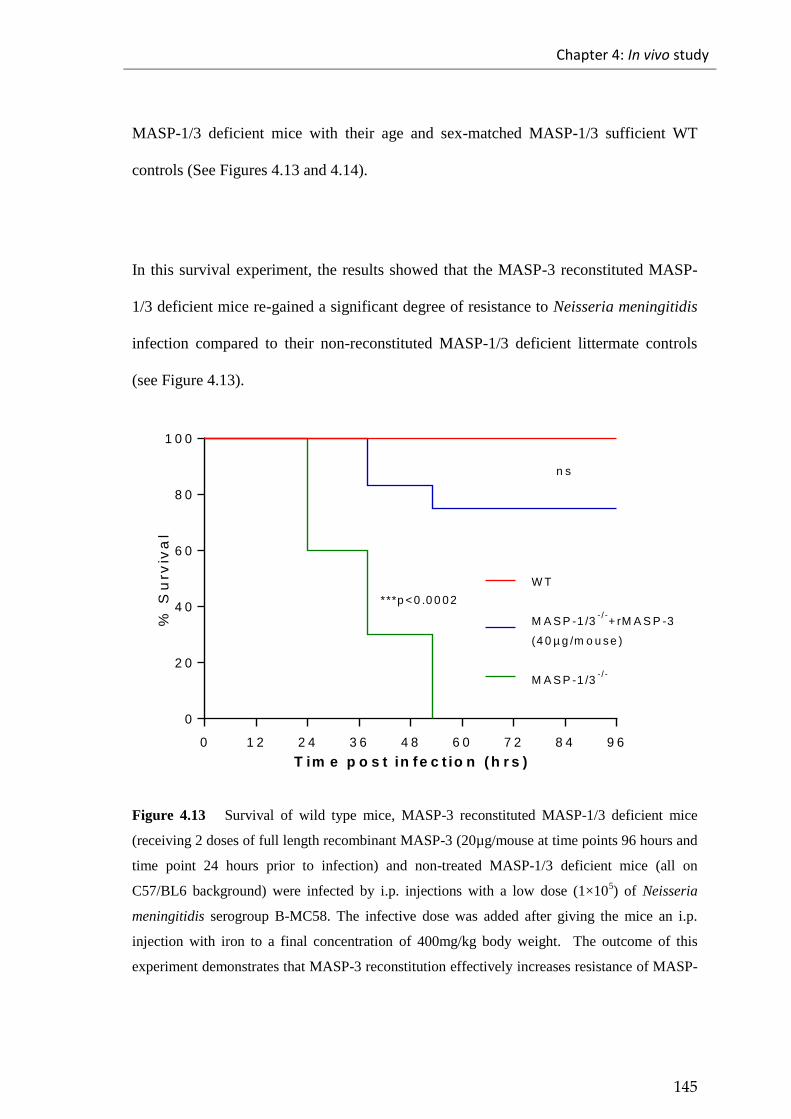
Chapter 4: In vivo study
145
MASP-1/3 deficient mice with their age and sex-matched MASP-1/3 sufficient WT
controls (See Figures 4.13 and 4.14).
In this survival experiment, the results showed that the MASP-3 reconstituted MASP-
1/3 deficient mice re-gained a significant degree of resistance to Neisseria meningitidis
infection compared to their non-reconstituted MASP-1/3 deficient littermate controls
(see Figure 4.13).
0 1 2 2 4 3 6 4 8 6 0 7 2 8 4 9 6
0
2 0
4 0
6 0
8 0
1 0 0
T im e p o s t in fe c t io n ( h r s )
% S
urv
iva
l
W T
M A S P -1 /3- / -
+ rM A S P -3
(4 0 µ g /m o u s e )
M A S P -1 /3- / -
***p <0 .0 0 0 2
n s
Figure 4.13 Survival of wild type mice, MASP-3 reconstituted MASP-1/3 deficient mice
(receiving 2 doses of full length recombinant MASP-3 (20µg/mouse at time points 96 hours and
time point 24 hours prior to infection) and non-treated MASP-1/3 deficient mice (all on
C57/BL6 background) were infected by i.p. injections with a low dose (1×105) of Neisseria
meningitidis serogroup B-MC58. The infective dose was added after giving the mice an i.p.
injection with iron to a final concentration of 400mg/kg body weight. The outcome of this
experiment demonstrates that MASP-3 reconstitution effectively increases resistance of MASP-

Chapter 4: In vivo study
146
1/3 deficient mice against Neisseria meningitidis infection compared to the non-treated MASP-
1/3 deficient mice. ***p < 0.0002 Log-rank (Mantel-Cox) test; n=10/group.
0 .0
0 .5
1 .0
1 .5
2 .0
2 .5
1 2 h o u r s p o s t in f e c t io n
Av
era
ge
ill
ne
ss
sc
ore
W T M A S P -1 /3- /-M A S P -1 /3
- /-+
r M A S P -3 (4 0 g /m o u s e )
***
***
0
1
2
3
4
3 6 h o u r s p o s t in f e c t io n
Av
era
ge
ill
ne
ss
sc
ore
W T M A S P -1 /3- /-M A S P -1 /3
- /-+
r M A S P -3 (4 0 g /m o u s e )
****
***
0
1
2
3
4
5
4 8 h o u r s p o s t in f e c t io n
Av
era
ge
ill
ne
ss
sc
ore
W T M A S P -1 /3- /-M A S P -1 /3
- /-+
r M A S P -3 (4 0 g /m o u s e )
****
****
0
1
2
3
4
5 4 h o u r s p o s t in f e c t io n
Av
era
ge
ill
ne
ss
sc
ore
W T M A S P -1 /3- /-M A S P -1 /3
- /-+
r M A S P -3 (4 0 g /m o u s e )
***
***
0 .0
0 .5
1 .0
1 .5
7 2 h o u r s p o s t in f e c t io n
Av
era
ge
ill
ne
ss
sc
ore
W T M A S P -1 /3- /-M A S P -1 /3
- /-+
r M A S P -3 (4 0 g /m o u s e )
**
a ll d e a d
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
9 6 h o u r s p o s t in f e c t io n
Av
era
ge
ill
ne
ss
sc
ore
W T M A S P -1 /3- /-M A S P -1 /3
- /-+
r M A S P -3 (4 0 g /m o u s e )
n s
a ll d e a d
Figure 4.14 Average illness score for wild type, treated MASP-1/3 deficient mice and non-
treated MASP-1/3 deficient mice following an i.p. injection with a low dose (1×105) of
Neisseria meningitidis serogroup B-MC58. The data are presented as mean with SEM. ***p <
0.0001, **p < 0.0041, Student t test.

Chapter 4: In vivo study
147
4.1.6.1 The viable bacterial load of Neisseria meningitidis in the blood of infected
mice
Three groups of mice, i.e. non-treated MASP-1/3 deficient mice, treated MASP-1/3
deficient mice and MASP-1/3 sufficient mice (WT), were challenged with a low dose
(1×105 CFU/mouse) of Neisseria meningitidis serogroup B strain MC58. The mice were
monitored and bled via a tail vein route was collected to assess the viable bacterial load
in blood at different time points 12, 24, 36, 54, 72 and 96 hours post infection. This
assessment showed that the bacterial load in blood of MASP-1/3 deficient mice was
significantly higher compared to the bacterial load in blood of treated MASP-1/3
deficient mice, which indicates that the treated mice had better ability to clear the
bacteria compared to non-treated mice. (See Figure 4.15). At 54 hours post infection,
the MASP-1/3 sufficient mice started to clear the bacteria from the blood while the rest
of the treated MASP-1/3 deficient mice started to clear the bacteria from the blood 60
hours post infection. At 96 hours post infection, the difference in the bacterial load of
blood between the treated MASP-1/3 deficient mice and MASP-1/3 sufficient mice was
not significant. However at an earlier stage, non-treated MASP-1/3 deficient mice had
developed a greater bacteremia, indicating signs of terminal illness; thus, these mice had
to be euthanized.
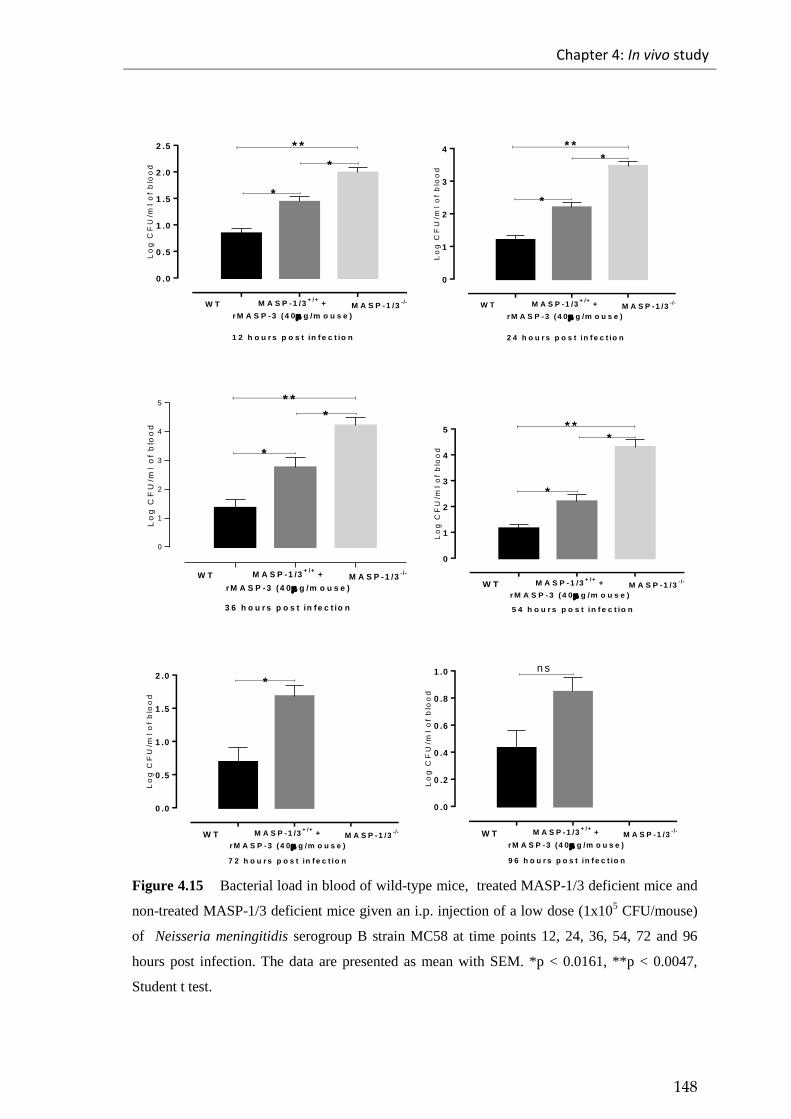
Chapter 4: In vivo study
148
0 .0
0 .5
1 .0
1 .5
2 .0
2 .5L
og
CF
U/m
l o
f b
loo
d
W T M A S P -1 /3+ /+
+
r M A S P -3 (4 0 g /m o u s e )
M A S P -1 /3- /-
* *
*
*
1 2 h o u r s p o s t in f e c t io n
0
1
2
3
4
Lo
g C
FU
/ml
of
blo
od
W T M A S P -1 /3+ /+
+
r M A S P -3 (4 0 g /m o u s e )
M A S P -1 /3- /-
* **
*
2 4 h o u r s p o s t in f e c t io n
0
1
2
3
4
5
Lo
g C
FU
/ml
of
blo
od
W T M A S P -1 /3+ /+
+
r M A S P -3 (4 0 g /m o u s e )
M A S P -1 /3- /-
* *
*
*
3 6 h o u r s p o s t in fe c t io n
0
1
2
3
4
5L
og
CF
U/m
l o
f b
loo
d
W T M A S P -1 /3+ /+
+
r M A S P -3 (4 0 g /m o u s e )
M A S P -1 /3- /-
* **
*
5 4 h o u r s p o s t in f e c t io n
0 .0
0 .5
1 .0
1 .5
2 .0
Lo
g C
FU
/ml
of
blo
od
W T M A S P -1 /3+ /+
+
r M A S P -3 (4 0 g /m o u s e )
M A S P -1 /3- /-
*
7 2 h o u r s p o s t in f e c t io n
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0
Lo
g C
FU
/ml
of
blo
od
W T M A S P -1 /3+ /+
+
r M A S P -3 (4 0 g /m o u s e )
M A S P -1 /3- /-
n s
9 6 h o u r s p o s t in f e c t io n
Figure 4.15 Bacterial load in blood of wild-type mice, treated MASP-1/3 deficient mice and
non-treated MASP-1/3 deficient mice given an i.p. injection of a low dose (1x105 CFU/mouse)
of Neisseria meningitidis serogroup B strain MC58 at time points 12, 24, 36, 54, 72 and 96
hours post infection. The data are presented as mean with SEM. *p < 0.0161, **p < 0.0047,
Student t test.

Chapter 4: In vivo study
149
4.2 Discussion
4.2.1 Mice deficient in the alternative pathway functional activity
show dramatically higher susceptibility to Neisseria meningitidis
infection
In Chapter 3, in vitro assays showed that lectin pathway deficient sera (MASP-2
deficient sera) showed greater C3 deposition than MASP-2 sufficient sera in conditions
that allow all three complement activation pathways to be active. When the assay
conditions were changed to allow only the classical or the lectin pathway to be active,
the ability of the MASP-2 deficient sera to allow C3 deposition was impaired, while
MASP-2 sufficient sera showed a higher degree of C3 deposition on the surface of
Neisseria meningitidis.
In further analyses, the bacteriolytic activity of different sera was tested against
Neisseria meningitidis, showing that the killing activity of MASP-2 deficient serum was
higher than the killing activity the MASP-2 sufficient serum. These results from in vitro
assays suggested that the alternative pathway had an important role in mediating the
complement system for killing Neisseria meningitidis.
The alternative pathway is the part of the complement system that maintains a
continuous activation of complement pathways. It is initiated when C3 is spontaneously
hydrolysed to form C3(H2O) that binds to Factor B (FB) in the presence of Mg+2
,

Chapter 4: In vivo study
150
leading to the creation of a zymogen complex C3(H2O)B. C3(H2O)B is then cleaved by
factor D (FD) into two parts: Ba and Bb. While the fragment Ba is released from the
complex, the other fragment Bb stays attached to the complex, leading to the formation
of the alternative pathway C3 convertase C3(H2O)Bb. This C3 convertase cleaves C3
into C3a and C3b, which can bind to a pathogen surface. Factor B will bind to the newly
generated C3b to form new C3 convertase C3bBb after cleaving Factor B via Factor D
(Thurman & Holers, 2006). This binding is stabilized by Properdin, which is one of the
components of the alternative pathway, and acts as positive regulator of the alternative
pathway that works to stabilize the C3 convertase C3bBb on the pathogen by increasing
its half-life 5-10 folds (Schwaeble and Reid, 1999).
The current in vitro study of the alternative pathway against Neisseria meningitidis
shows that a limited level of C3 deposition was observed following the use of factor B
deficient serum (i.e. alternative pathway deficient serum) compared to wild-type serum.
Furthermore, the serum bactericidal assay of factor B deficient serum showed no killing
of Neisseria meningitidis compared to the wild-type serum. These results also suggest
the importance of the alternative pathway in fighting Neisseria meningitidis. To further
analyse these results in vivo infection experiments were used in a mice model of
Neisseria meningitidis infection in Factor B deficient mice, mice with gene-targeted
disruption of Factor B gene, providing a mouse model with a total defect in the
alternative pathway.

Chapter 4: In vivo study
151
The infective dose for the infection experiment was a low dose (1×105
CFU/mouse) of
Neisseria meningitidis serogroup B strain MC58 administrated via an intra-peritoneal
route. The virulence of the Neisseria meningitidis infection was enhanced by
administrating iron into the murine model of infection (Holbein, 1981; Perkins-Balding
et al., 2004). Therefore in all the infection experiments mice received iron (400mg/kg
body weight) intra-peritoneally 12 hours before the infection dose.
Although the infective dose was low, there was a significant difference in the survival
of the two groups of mice (i.e. Factor B deficient mice and Factor B sufficient control
mice). The Factor B deficient mice were more susceptible to Neisseria meningitidis
infection with 100 % mortality compared to the Factor B sufficient mice that were more
resistance to Neisseria meningitidis infection with a survival rate of 100%. The illness
scores of the mice reflected this observation. While Factor B sufficient mice showed
only minor signs of illness, the Factor B deficient mice showed significantly higher
signs of illness within 24 hours post infection, leading some mice to reach a lethargic
end stage. These mice were euthanized according to the home office regulations.
Although none of the Factor B sufficient mice progressed to a lethargic end stage, all
the Factor B deficient mice showed higher signs of illness and reached the lethargic end
stage by 48 hours post infection (see Figures 4.4 and 4.5).
The presence of culturable Neisseria meningitidis in blood gave a better illustration of
disease progression during the infection experiment. Although the presence of the
bacteria in the blood started to appear in both groups at 12 hours post infection, the

Chapter 4: In vivo study
152
viable count of bacteria in the blood of Factor B deficient mice was higher than the
viable count of bacteria in the blood of Factor B sufficient mice at each time post
infection. The results also showed that Factor B sufficient mice were able to
significantly reduce and clear the bacteria from the blood, which was in contrast to the
Factor B deficient mice (see Figure 4.6). The results of this infection experiment are in
line with previous reports showing an association between an alternative pathway
deficiency and Neisseria meningitidis infection (Biesma et al., 2001; Hiemstra et al.,
1989; Sprong et al., 2006).
MASP-3 is a component of the lectin pathway of MBL associated serine proteases
discovered by Dahl et al. (2001) and is produced by the same gene that produces the
MASP-1(MASP-1 gene) located on chromosome 16. Although they have the same
heavy chain, the linker regions are different (the last 15 C-terminal residues). Unlike the
other MBL serine protease (MASP-1 and MASP-2) MASP-3 is not an auto activated
molecule and requires the action of MASP-1 or C1r to be activated (Megyeri et al.,
2014; Wijeyewickrema et al., 2013). MASP-3 was firstly thought to act as a negative
regulator of the lectin pathway by competing with the binding site of MASP-1 and
MASP-2 to MBL (Dahl et al., 2001). However, Takahashi et al (2010) showed that
MASP-1/3 deficient mice lack the full function of an alternative pathway.
Therefore in order to assess role of MASP-1/3 in fighting Neisseria meningitidis
infection, an infection experiment using MASP-1/3 deficient mice and MASP-1/3
sufficient mice was performed and showed that MASP-1/3 deficient mice were more

Chapter 4: In vivo study
153
susceptible to Neisseria meningitidis than MASP-1/3 sufficient mice. While all of the
MASP-1/3 deficient mice developed signs of illness 6 hours post infection that led to all
of the mice reaching a lethargic end stage and were euthanized by 53 hours post
infection, the MASP-1/3 sufficient mice were healthy with only minor signs of illness
and recovered by 72 hours post infection. The viable count of Neisseria meningitidis in
the blood of mice showed the ability of MASP-1/3 sufficient mice to fight and clear the
bacteria, while the MASP-1/3 deficient mice showed a significantly higher count of
bacteria in their blood. The results of this infection study suggest that MASP-1/3 has an
important role in fighting Neisseria meningitidis infection (see Figures 4.7, 4.8 and 4.9).
4.2.2 The therapeutic application of recombinant full-length MASP-3
dramatically improves the survival of MASP-1/3 deficient mice
from Neisseria meningitidis infection
Recently published work by Iwaki et al. (2011) postulated that on the surface of
Staphylococcus aureus the lectin pathway components MASP-3 and MBL may actually
allow alternative pathway activation to be initiated through the direct cleavage of
zymogen C3(H2O)B or C3bB and convert these zymogen complexes into their active
form by converting C3bB to C3Bb. In order to assess whether MASP-3 has an ability to
drive activation of the alternative pathway and therefore increase the lysis of Neisseria
meningitidis, an in vivo MASP-3 reconstitution experiment in MASP-1/3 deficient mice
was performed. These mice were divided into two groups. The first group received one
dose of full-length recombinant MASP-3 (20µg/mouse) 96 hours before terminal

Chapter 4: In vivo study
154
bleeding. The second group received two doses of full length recombinant MASP-3
(20µg/mouse) 96 and 24 hours before the terminal bleeding. The results of this
reconstitution experiment showed gradual restoration of the functional activity of the
alternative pathway. The levels of this restoration were monitored and measured by
assessing the formation of the alternative pathway C3 deposition at 24, 48, 72 and 96
hours post MASP-3 injection.
This experiment showed that while the restoration levels were nearly 35% in both
groups at 24 hours post injection, this increased to 60% at 48 hours post injection
compared to the wild-type mice serum. At 72 hours post injection alternative pathway
functional activity was nearly fully restored in both groups of mice. However, at 96
hours post injection the level of the alternative pathway function was reduced in the
group of mice that received only one dose of full-length recombinant MASP-3
compared to the second groups of mice that received two doses of full-length
recombinant MASP-3 (see Figure 4.10). My results clearly show that it is MASP-3 and
not MASP-1 (as previously claimed by Takahashi et al. 2010) that is responsible for the
loss of alternative pathway functional activity in the blood of MASP-1 and MASP-3
double deficient mice.
Serum bactericidal assays were also done to assess the cytolytic activity of the sera
taken from both groups of mice at 96 hours post injection to Neisseria meningitidis. The
results of these assays showed that both sera had restored the ability to kill the Neisseria
meningitidis, which is in line with findings by Iwaki et al. (2011). However in this

Chapter 4: In vivo study
155
experiment, it was observed that even though both sera had the ability to lyse the
Neisseria meningitidis, the sera of the group that received two doses of full length
recombinant MASP-3 showed significantly higher lysis than the group receiving only
one dose of full length recombinant MASP-3. This may be due to the instability and
subsequent breakdown of the administrated recombinant MASP-3 (see Figures 4.11 and
4.12).
Current results of the reconstitution experiment suggest that reconstitution of full-length
recombinant MASP-3 in MASP-1/3 deficient mice reduced the mortality in MASP-1/3
deficient mice in a model of Neisseria meningitidis infection. Therefore an experiment
to assess this hypothesis was performed using three groups of mice: MASP-1/3
sufficient mice, MASP-1/3 deficient mice and MASP-1/3 deficient mice that received
two doses of full length recombinant MASP-3 at 96 and 24 hours before the Neisseria
meningitidis dose. The infective dose was a relatively low dose (1×105 CFU/mouse) of
Neisseria meningitidis serogroup B strain MC58 and this was used after adding iron to a
final concentration of 400mg/kg body weight.
The result of this infection experiment showed a significant difference in the survival of
the mouse groups showing that MASP-1/3 deficient mice were more susceptible to
Neisseria meningitidis infection with 100 % mortality compared to the wild-type mice
which were more resistant to Neisseria meningitidis infection with a survival rate of
100%. Interestingly the survival rate of the treated MASP-1/3 deficient mice was 70%
compared to the non-treated MASP-1/3 deficient mice. This result shows the enhanced

Chapter 4: In vivo study
156
ability of the treated MASP-1/3 to fight Neisseria meningitidis infection. The illness
scores of the mice also reflected this observation. While MASP-1/3 sufficient mice
showed minor signs of illness and none of them progressed to the lethargic end stage,
the MASP-1/3 deficient mice show significantly higher signs of illness that lead some
of the mice to reach a lethargic end stage at 36 hours post infection and had to be
euthanized. The remaining MASP-1/3 deficient mice developed greater signs of illness
and reached the lethargic end stage by 54 hours post infection. In contrast even though
the treated MASP-1/3 deficient mice showed signs of illness that lead some of the mice
to the lethargic end stage at 54 hours post infection, the other mice recovered and
returned to a normal health status at 96 hours post infection ( see Figures 4.13 and 4.14).
In line with the survival and illness scores results, the viable bacterial count in blood
showed a significantly higher bacterial count in the blood of none treated MASP-1/3
deficient mice than the treated MASP-1/3 deficient and MASP-1/3 sufficient mice.
Although the presence of the bacteria in the blood started to appear in all the groups at
12 hours post infection, the viable count of bacteria in the blood of non-treated MASP-
1/3 deficient mice was higher than the viable count of bacteria in blood of treated
MASP-1/3 deficient mice at each time point post infection, which indicated that the
treated MASP-1/3 deficient mice were able to more significantly reduce and clear the
bacteria of the blood than the MASP-1/3 deficient mice. Interestingly, even though the
count of bacteria in the blood of treated MASP-1/3 deficient mice was significant higher
than the bacterial count in the MASP-1/3 sufficient mice at each time point post
infection, the difference between the two groups become non-significant at the end of
the experiment (see Figure 4.15).

Chapter 4: In vivo study
157
In summary, results of the survival, illness scores and viable bacterial count
demonstrated that the lectin pathway molecule MASP-3 was able to drive
the activation of the complement system (alternative pathway) which
resulted in a reduction and clearance of the bacteria and led to a dramatic
increase in the survival rate of MASP-3 reconstituted MASP-1/3 deficient
mice.

Chapter 5: Conclusion and future work
158
Chapter 5: Conclusion and future work
5.1 Conclusion
The complement system is a vital part of the immune system, composed of many
proteins that interact with each other to form a complex interacting network. This
network of proteins is activated by three different pathways designated as the classical,
the alternative and the lectin pathways. Regardless of the pathways involved, the
complement system plays several vital physiological roles especially in the process of
protection against pathogens and links the non-specific (innate) immune system with the
specific (adaptive) immune system. Also waste products such as apoptotic cells and
debris are removed by the complement system by facilitating their uptake by phagocytic
cells (Schwaeble et al., 2002; Walport, 2001).
Neisseria meningitidis is the main cause of bacterial meningitis throughout the world
and causes a significant rate of mortality. While there has been considerable progress in
its diagnosis, vaccination and treatment in recent years meningococcal disease is still a
major health threat with a mortality rate by septicaemia of up to 10%. Neisseria
meningitidis colonisation starts on the surface of the mucosal epithelium and
intraepithelially on the host nasopharynx. It is the invasion of the blood stream that
causes the well-known clinical symptoms ranging from mild meningococcal infection to

Chapter 5: Conclusion and future work
159
sepsis and to full-blown meningococcal septicemia and meningitis (Schneider et al.,
2007; Connolly and Noah, 1999).
The association between Neisseria meningitidis and the complement system has been
extensively studied and many published reports (Finne et al., 1987; Estabrook et al.,
1997; Rossi et al., 2001; Rosa et al., 2004; Jarva et al., 2005; Trouw & Daha, 2011)
indicate the important role of the complement system in fighting Neisseria meningitidis
infection. In this study, I have focussed on the crucial role of the alternative pathway in
fighting Neisseria meningitidis infection and managed to define the so far unknown role
of the lectin pathway component, serine protease MASP-3, in directing alternative
pathway functional activity to the surface of Neisseria meningitidis to enable the
complement system to lyse these pathogens, a mechanism that is vital in fighting off
infection and clearing the bacteria from the blood stream. To define the role of the lectin
pathway dependent enzyme MASP-3 in more detail, several complement-specific in
vitro assays were established and the lessons learned from these were tested in
subsequent in vivo infection studies using mouse models of Neisseria meningitidis
infection. The mouse models of infection included wild-type C57/BL6 mice as well as
gene-targeted mouse strains of the same genetic background with deficiencies of
essential alternative pathway effector enzymes [factor B gene (FB-/-
mice)] or mice with
a disrupted MASP1 gene which were deficient in the MASP1 gene products MASP-1.
MASP-3 and MAp44 (MASP-1/3-/-
mice) that have been previously reported to present
with a deficiency in alternative pathway functional activity (Takahashi et al., 2010).

Chapter 5: Conclusion and future work
160
The results showed that, the in vitro study has newly demonstrated the important role of
a new link between the lectin and the alternative pathways in fighting Neisseria
meningitidis infection. It has also highlighted the major role of the lectin pathway
complex molecules MASP-3 and MBL, in driving the activation of the alternative
pathway on the surface of Neisseria meningitidis through the cleavage of C3b bound
Factor B leading to the formation of the alternative pathway C3 convertase and
subsequently to the formation of the MAC complex and thus lysis of Neisseria
meningitidis. The findings emphasize the ability of recombinant properdin to enhance
the lytic activity of serum and this knowledge could be used to develop a therapeutic
approach to fight Neisseria meningitidis infection. The in vivo study emphasizes the
importance of the alternative pathway in fighting Neisseria meningitidis infection. Also,
it has highlighted the important role of the lectin pathway component MASP-3 in
driving the activation of the alternative pathway and fighting Neisseria meningitidis
infection.
5.1.1 Binding of complement system recognition molecules to Neisseria
meningitidis
As complement activation on the surface of Neisseria meningitidis may be dependent
on the binding of the classical pathway recognition molecule C1q and/or lectin pathway
recognition molecules such as Ficolin-A, MBL-A, MBL-C and CL-11, the binding of
C1q and other lectin pathway recognition molecules to different serotypes of Neisseria
meningitidis strains (serotype A strain Z2491 and serotype B strain MC58) was studied.

Chapter 5: Conclusion and future work
161
The results showed that Neisseria meningitidis serotype A strain Z2491 and serotype B
strain MC58 were recognised by the classical pathway recognition molecule C1q as
shown by the binding of C1q from sera of non-immune mice to the N. meningitidis
strains tested (see Figure 3.1). However, this direct binding of C1q to the tested strains
of Neisseria meningitidis did not induce classical pathway dependent complement
activation, as shown by the lack of CP sufficient serum to deposit C3 activation
products when serum of MBL-null mice or serum of non-immune MBL-deficient blood
donors was used.
Then the binding of the recognition molecules of the lectin pathway such as ficolin-A,
MBL-A, MBL-C and CL-11 to Neisseria meningitidis were assessed. The murine lectin
pathway recognition molecule ficolin-A showed no binding to either of the two
different serotypes of meningococcus strains tested, i.e. serotype A strain Z2491 and
serotype B strain MC58, while both murine MBL molecules MBL-A and MBL-C as
well as the MBL-related collectin CL-11 all showed binding to both Neisseria
meningitidis serotypes (see Figures 3.2 to 3.5).

Chapter 5: Conclusion and future work
162
5.1.2 Activation of the complement system on the surface of Neisseria
meningitidis requires a close cooperation between the lectin and
alternative pathways
The ability of Neisseria meningitidis to activate the complement system was studied
using several in vitro assays which showed that both strains of Neisseria meningitidis
used throughout this study had the ability to activate complement (see Figures 3.6 and
3.7). These results encouraged me to investigate the specific contributions of each of the
different complement pathways towards this activation. The results revealed different
levels of C3 deposition on the surface of both strains of Neisseria meningitidis
depending on which sera were used to measure the relative amount of C3 deposition on
the surface of strains tested. The lowest level of the C3 deposition on both strains was
observed when using serum from factor B deficient mice (see Figures 3.11 and 3.12).
This serum was totally deficient of alternative pathway functional activity while both
the classical and lectin pathways were unaffected. This result suggests that there is a
predominant role of the alternative pathway in driving C3 deposition and complement
activation on the surface of the strains tested. C3 deposition on the surface of Neisseria
meningitidis was either not or only minimally affected when using C1q deficient non-
immune serum (which totally lacks classical pathway functional activity) in comparison
to wild-type serum (see Figures 3.11 and 3.12). This result suggests that the classical
pathway plays a minimal (and rather insignificant) role in mediating C3 deposition on
the surface of Neisseria meningitidis. Additionally, it emphasized the important role of
the other two complement pathways, the alternative and lectin pathways, which
contributed to the majority of C3 deposition on the surface of Neisseria meningitidis in
non-immune sera. Surprisingly, the highest level of the C3 deposition was seen in sera

Chapter 5: Conclusion and future work
163
of MASP-2 deficient mice, which is deficient in its ability to form the lectin pathway
C3 convertase C4aC2b (see Figures 3.11 and 3.12). This finding again underlined the
important contribution of the alternative pathway in driving complement activation on
the surface of this pathogen. The high bactericidal activity of MASP-2 deficient serum
and the finding that MBL is required to trigger this bactericidal activity in non-immune
serum implies that a lectin pathway enzyme other than MASP-2 may support alternative
pathway function and contribute to C3 deposition and initiation of serum killing of
Neisseria meningitidis.
Serum bactericidal assays to quantify the lytic activity against Neisseria meningitidis
included both human and different mouse sera (wild-type, C1q deficient, factor B
deficient, MASP-1/3 deficient and MASP-2 deficient mouse sera) were performed.
These assays were run under conditions that did allow activation of each of the three
complement activation pathways. The results of these assays were consistent (i.e. in full
agreement) with the results obtained using the C3 deposition assays. Like the C3
deposition results, these results showed that in the serum bactericidal assays, in the
absence of classical pathway activity, i.e. in C1q deficient serum, there was no, or only
a minimal contribution of the C1q driven classical pathway towards the bacteriolytic
activity of the non-immune serum. The other two complement activation pathways, i.e.
the alternative and the lectin pathways, fully compensated for the loss of classical
pathway functional activity in non-immune serum (see Figures 3.16 and 3.17). Similar
to my observations using the C3 deposition assays, MASP-2 deficient serum showed a
significantly increased lytic activity (see Figures 3.20 and 3.21) which could be
explained by the previously postulated phenomenon that either of the two remaining

Chapter 5: Conclusion and future work
164
lectin pathway serine proteases, MASP-1 or MASP-3, could be involved in driving the
alternative pathway (Takahashi et. 2010; Iwaki et al., 2011). Notably, these findings
demonstrate that the MASP-2 dependent effector arm of the lectin pathway does not
play a major role in the lysis of Neisseria meningitidis and that the therapeutic
inhibition of MASP-2 functional activity does not increase the predisposition of patients
treated with therapeutic MASP-2 inhibitors to suffer more frequent or more severe
meningococcal infections.
The important role of the alternative pathway was highlighted when using different
alternative pathway deficient sera (Factor B deficient and MASP-1/3 deficient). In both
sera highly compromised and lacking in bactericidal activity was observed even in the
presence of the other two complement activation pathways, classical and the MASP-2
dependent lectin pathway activation route (see Figures 3.18 and 3.19).
To further analyse whether the loss of bacteriolytic activity in MASP-1 and MASP-3
deficient mouse sera or MASP-1 and MASP-3 deficient human 3MC serum was due to
the deficiency of either MASP-1 or MASP-3 or both, the sera were reconstituted with
functionally active recombinant MASP-3, a recombinant fragment composed of the
MASP-3 domains CCP-1/CCP-2 and the serine protease domain, termed rMASP-3
(catalytic fragment, cf). Bactericidal assays were performed using MASP-1/3 deficient
serum and human 3MC serum, with or without rMASP-3 (cf). The results of these
assays showed that the alternative pathway dependent lytic activity against both strains
of Neisseria meningitidis was restored by the addition of rMASP-3 (cf). In addition the

Chapter 5: Conclusion and future work
165
results highlighted the importance of the alternative pathway in fighting Neisseria
meningitidis infection (see Figures 3.22 to 3.25).
In vitro assays also demonstrated the importance of the lectin pathway recognition
molecule MBL in mediating bacteriolytic activity towards Neisseria meningitidis.
Human and murine MBL deficient sera were not able to kill Neisseria meningitidis
when tested in a serum bactericidal assay. This observation was consistent in both
human and murine MBL deficient sera (see Figures 3.20, 3.21, 3.24 and 3.25).
In context with the new essential role that this work has identified for the lectin pathway
serine protease MASP-3, the requirement of both MASP-3 and the lectin pathway
recognition subcomponent MBL implies that the MASP-3 dependent initiation of
alternative pathway mediated killing of Neisseria meningitidis is dependent on lectin
pathway activation/function that takes place on the surface of Neisseria meningitidis
since this event requires both the recognition component, MBL (that binds to the
surface of the pathogen), and the lectin pathway enzyme MASP-3 that converts pro-
Factor D to enzymatically active Factor D which in turn is required to convert the
alternative pathway C3 convertase zymogen complex C3bB into its enzymatically
active form (i.e. C3bBb). These results on the role of MBL in mediating serum
bacteriolytic activity towards Neisseria meningitidis are in full agreement with a
previously published report which implied that MBL had an important role in fighting
Neisseria meningitidis infection (Hibberd et al., 1999).

Chapter 5: Conclusion and future work
166
Taken together, the results strongly suggest that both lectin pathway molecules, i.e.
MBL and MASP-3, play crucial roles in mediating the lytic activity toward Neisseria
meningitidis.
5.1.3 Recombinant properdin enhances the serum bacteriolytic
activity against Neisseria meningitidis
Properdin is a glycoprotein known as a positive regulator of complement activation. It
works to stabilize the C3 convertase C3bBb on the pathogen through increasing its half-
life. The association between properdin deficiency and Neisseria meningitidis infection
has been reported previously, illustrating the important role of properdin in fighting
Neisseria meningitidis infection (Fijen et al., 1995).
Therefore, in order to assess the effect of properdin on the formation of C3 deposition
(and thus the lytic activity) on the surface of Neisseria meningitidis was measured
using ELISA assay and FACS analysis. The results of these assays showed increased
amounts of C3 deposition on the surface of Neisseria meningitidis strains following the
addition of recombinant properdin (see Figures 3.26, 3.27 and 3.28).
To further analyse and confirm the important role of properdin in enhancing the serum
bactericidal activity against Neisseria meningitidis it was shown that the bactericidal
activity of human or mouse serum was enhanced significantly by adding recombinant
properdin compared to normal serum alone (see Figures 3.29 to 3.32).

Chapter 5: Conclusion and future work
167
5.1.4 Mice deficient in the alternative pathway functional activity
show dramatically higher susceptibility to Neisseria meningitidis
infection
The current in vitro study of the alternative pathway against Neisseria meningitidis
shows that a limited level of C3 deposition was observed following use of Factor B
deficient serum, (alternative pathway deficient serum) compared to wild-type serum.
Further, the serum bactericidal assay of Factor B deficient serum showed no killing of
Neisseria meningitidis compared to the wild-type serum. These results also suggest the
importance of the alternative pathway in fighting Neisseria meningitidis. To further
analyse these results, an in vivo infection experiment in a mouse model of Neisseria
meningitidis infection in Factor B deficient mice (i.e. mice with a gene-targeted
disruption of Factor B gene) which provides a mouse model with a total defect in the
alternative pathway, was performed.
Although the infective dose was low, there was a significant difference in the survival
of the Factor B deficient mice and the Factor B sufficient control mice. The Factor B
deficient mice were more susceptible to Neisseria meningitidis infection, with 100%
mortality, compared to the Factor B sufficient mice which were more resistant to
Neisseria meningitidis infection with a survival rate of 100%. The illness scores of the
mice reflected this observation. While Factor B sufficient mice showed only minor
signs of illness, the Factor B deficient mice showed significantly higher signs of illness.
Additionally, the results showed that Factor B sufficient mice were able to significantly

Chapter 5: Conclusion and future work
168
reduce and clear the bacteria from the blood, which was in contrast to the Factor B
deficient mice (see Figures 4.4 to 4.6).
MASP-3 is one of the lectin pathway MBL associated serine proteases which was firstly
thought to act as a negative regulator of the lectin pathway by competing with the
binding site of MASP-1 and MASP-2 to MBL (Dahl et al., 2001). However, Takahashi
et al. (2010) showed that MASP-1/3 deficient mice are lacking in the full function of an
alternative pathway. Thus, in order to assess its role in fighting Neisseria meningitidis
infection, an infection experiment using two groups of mice; MASP-1/3 deficient mice
and MASP-1/3 sufficient mice was performed. The results from this infection study
showed that MASP-1/3 deficient mice were more susceptible to Neisseria meningitidis
than MASP-1/3 sufficient mice. Illness scores of mice showed higher signs of illness in
MASP-1/3 deficient mice compared to MASP-1/3 sufficient mice that were healthy
with only minor signs of illness. Also, the viable count of Neisseria meningitidis in the
blood of mice showed the ability of MASP-1/3 sufficient mice to fight and clear the
bacteria while the MASP-1/3 deficient mice showed a significantly higher count of
bacteria in their blood (see Figures 4.7 to 4.9).

Chapter 5: Conclusion and future work
169
5.1.5 The therapeutic application of recombinant full-length MASP-3
dramatically improves the survival of MASP-1/3 deficient mice
from Neisseria meningitidis infection
Recently published work by Iwaki et al. (2011) showed that the activation of an
alternative pathway on the surface of Staphylococcus aureus through the lectin pathway
complex between the MASP-3 and MBL which cleaves the proenzymes C3(H2O)B or
C3bB to their enzymatic active form. Therefore, in order to assess whether MASP-3 had
the ability to drive the activation of the alternative pathway and therefore increase the
lysis of Neisseria meningitidis, an in vivo MASP-3 reconstitution experiment in MASP-
1/3 deficient mice using recombinant murine full-length MASP-3 zymogen (rMASP-3
(flz)), produced by Dr. SadamYaseen in Professor Schwaeble’s laboratory were
performed. rMASP-3 (flz) (20 micrograms) was given i.v. to MASP-1/3-/-
mice and
reconstitution of alternative pathway functional activity was measured in blood taken at
24, 36, 48 and 96 hours after injection. After 24 hours alternative pathway functional
activity was restored to approximately 35% of that of wild-type mice, after 48 hours to
about 60% and after 72 hours to more than 90%. The results of this reconstitution
experiment clearly demonstrated the ability of the lectin pathway serine protease
MASP-3 to restore the severely compromised alternative pathway functional activity in
MASP-1/3-/-
mice (see Figures 4.10) and the subsequent experiment that was conducted
by infecting MASP-3 reconstituted MASP-1/3-/-
mice with Neisseria meningitidis
demonstrated the essential role of MASP-3 in the innate immune response to Neisseria
meningitidis infection.

Chapter 5: Conclusion and future work
170
The results of the reconstitution experiment demonstrated for the first time that
reconstitution of MASP-1/3-/-
mice with full length recombinant MASP-3 dramatically
reduced the mortality in the MASP-1/3 deficient mice in a model of Neisseria
meningitidis infection. Therefore this infection experiment was repeated to confirm this
findings using three groups of mice: MASP-1/3 sufficient WT mice, MASP-1/3
deficient mice and MASP-1/3 deficient mice that received two doses of full length
recombinant MASP-3 at 96 and 24 hours prior to intraperitoneal Neisseria meningitidis
infection. The results of this infection experiment showed that the survival rate of the
treated MASP-1/3 deficient mice was 70%, compared to the survival rate of non-treated
MASP-1/3 deficient mice which was 0% with 100% mortality. The wild-type mice were
resistant to the relatively low infectious dose of 1x 105 Neisseria meningitidis serogroup
B strain MC58 given i.p. leading to a survival rate of 100%. The illness scores
established for the different experimental mouse groups were fully in line with the
overall survival results. While MASP-1/3 sufficient mice showed minor signs of illness,
the MASP-1/3 deficient mice show significantly higher signs of illness. In contrast,
even though the treated MASP-1/3 deficient mice showed signs of illness that led 30%
of the mice to the lethargic end stage, the remaining treated MASP-1/3 deficient mice
recovered and returned to a normal health status. In line with the survival and illness
scores results, the viable bacterial count in blood showed a significantly higher bacterial
count in the blood of non-treated MASP-1/3 deficient mice than the treated MASP-1/3
deficient and MASP-1/3 sufficient mice. Interestingly, even though the count of bacteria
in the blood of treated MASP-1/3 deficient mice was significantly higher than the
bacterial count in the MASP-1/3 sufficient mice at each time point post-infection, the

Chapter 5: Conclusion and future work
171
difference between the two groups was non-significant at the end of the infection
experiment (see Figures 4.11 to 4.15).
In summary, results of the survival, illness scores and viable bacterial count
demonstrated that the lectin pathway molecule MASP-3 was able to drive the activation
of the complement system (alternative pathway) and resulted in the reduction and
clearance of the bacteria and led to an increase in the survival rate of treated MASP-1/3
deficient mice.

Chapter 5: Conclusion and future work
172
5.2 Future work
A number of interesting aspects of the research arose during this work, but due to time
constraints these could not be followed up.
5.2.1 Assess the therapeutic benefit of recombinant MASP-3 in
fighting other microbial infection
My work has highlighted the major role of the lectin pathway molecule i.e. MASP-3, in
driving the alternative pathway, and thus fighting the Neisseria meningitidis infection.
Therefore, it would be interesting to assess the therapeutic benefit of recombinant
MASP-3 in fighting other microbial infection.
5.2.2 Assess the ability of recombinant properdin in restoring the
killing of properdin-deficient sera
This study has shown the important role of properdin in enhancing the serum
bactericidal activity against Neisseria meningitidis. It was shown that serum bactericidal
activity of human or mouse was enhanced significantly by adding recombinant
properdin compared to normal serum alone. Therefore the ability of recombinant
properdin in restoring the killing activity of properdin-deficient sera should be tested to
assess to what extent the administration of properdin can be used as a therapy in
properdin-deficient individuals.

Chapter 6: Bibliography
173
Chapter 6: Bibliography
Agarwal, S., Ferreira, V. P., Cortes, C., Pangburn, M. K., Rice, P. A., and Ram,
S., 2010. An evaluation of the role of properdin in alternative pathway
activation on Neisseria meningitidis and Neisseria gonorrhoeae. J. Immunol.,
185, 507-516.
Ala'Aldeen, D. A., Neal, K. R., Ait-Tahar, K., Nguyen-Van-Tam, J. S., English,
A., Falla, T. J., Hawkey, P. M., and Slack, R. C., 2000. Dynamics of
meningococcal long-term carriage among university students and their
implications for mass vaccination. J. Clin. Microbiol., 38, 2311–2316.
Albiger, B., Johansson, L., and Jonsson., A.-B., 2003. Lipooligosaccharide-deficient
Neisseria meningitidis shows altered pilus-associated characteristics. Infect. Immun., 71,
155-162.
Anderson, M. S., Glode, M. P., and Smith, A. L., 1998. Meningococcal
diseases. In R. D. Feigin and J. D. Cherry (ed.), Textbook of pediatric
infectious diseases. W. B. Saunders Co., Philadelphia, Pa., 1143-1156.
Arlaud, G.J., Gaboriaud, C., Thielens N.M., Budayova-Spano, M., Rossi V.,
and Fontecilla-Camps, J.C., 2002. Structural biology of the C1 complex of
complement unveils the mechanisms of its activation and proteolytic activity.
Mol. Immunol., 39, 383–394.
Barnum, S.R., Niemann, M.A., Kearney, J.F., Volanakis, J.E., 1984.
Quantitation of complement factor D in human serum by a solid-phase
radioimmunoassay.Journal of Immunological Methods.67, 303-309.
Barrett, S.J. and Sneath, P.H.A., 1994. A numerical phenotypic taxonomic
study of the genus Neisseria. Microbiol., 140, 2867-2891.

Chapter 6: Bibliography
174
Bjerre, A., Brusletto B., Mollnes T.E., et al., 2002. Complement activation
induced by purified Neisseria meningitidis lipopolysaccharide (LPS), outer
membrane vesicles, whole bacteria and an LPS free mutant. J. Infect. Dis., 15,
220-228.
Botto, M., Kirschfink, M., Macor, P., Pickering, M.C., Wurzner, R., Tedesco,
F., 2009. Complement in human diseases: Lessons from complement
deficiencies. Molecular Immunology.46, 2774-2783.
Braconier, J. H., Sjo¨holm, A. G., and So¨derstro¨m, C., 1983. Fulminant
meningococcal infections in a family with inherited deficiency of properdin.
Scand. J. Infect. Dis., 15, 339–345.
Brandtzaeg, P., Halstensen A., Kierulf P., Espevik T., Waage A., 1992.
Molecular mechanisms in the compartmentalized inflammatory response
presenting as meningococcal meningitis or septic shock. Microbiol.
Path., 13 (6), 423-431.
Brandtzaeg, P., Ovstebo, R., and Kierulf, P., 1995. Bacteremia and
compartmentalization of LPS in meningococcal disease. Prog. Clin. Biol. Res.,
392, 219–33.
Broome, C. V., 1986. The carrier state, Neisseria meningitidis. J. Antimicrob.
Chemo., 18, 25–34.
Brouwer N., Dolman, K. M., van Houdt, M., Sta, M., Roos, D., and Kuijpers, T.W.,
2008. Mannose-Binding Lectin (MBL) facilitates opsonophagocytosis of yeasts but not
of bacteria despite MBL binding. J. Immunol., 180, 4124-4132.
Brown, J.S., Hussell, T., Gilliland, S.M., Holden, D.W., Paton, J.C.,
Ehrenstein, M.R., Walport, M.J., and Botto, M., 2002. The classical pathway is
the dominant complement pathway required for innate immunity to
Streptococcus pneumoniae infection in mice. Proc. Natl. Acad. Sci. USA.,
99(26), 16969-16974.

Chapter 6: Bibliography
175
Burke, J. M., Ganley-Leal, L. M. Khatri, K. and Wetzler. L. M., 2007.
Neisseria meningitidis PorB, a TLR2 ligand, induces an antigen-specific
eosinophil recall response, potential adjuvant for helminth vaccines?. J.
Immunol., 179, 3222-3230.
Caugant, D. A., Høiby, E. A., Rosenqvist, E., Frøholm, L. O., and Selander, R.
K., 1992. Transmission of Neisseria meningitidis among asymptomatic military
recruits and antibody analysis. Epidemiol. Infect.,109, 241–253.
Choudhury B., C. M. Kahler, A. Datta, D. S. Stephens and R.l W. Carlson The
structure of the L9 immunotype lipooligosaccharide from Neisseria meningitidis NMA
Z2491, (2008) Carbohy. Res. 343: 2971-2980.
Choy, L. N., and Spiegelman, B. M., 1996. Regulation of alternative pathway
activation and C3a production by adipose cells. Obes. Res., 4, 521-532.
Connolly M. and N. Noah. Is group C meningococcal disease increasing in Europe? A
report of surveillance of meningococcal infection in Europe 1993–6, (1999) Epidemiol.
and Inf. 122: 41-49.
Cooper, N. R., 1985. The classical complement pathway, activation and regulation of
the first complement component. Adv Immunol., 37, 151-216.
Cugno, M., Zanichelli, A., Foieni, F., Caccia, S., Cicardi, M., 2009. C1-
inhibitor deficiency and angioedema: molecular mechanisms and clinical
progress. Trends in Molecular Medicine.15, 69-78.
Dahl, M., Tybjaerg-Hansen, A., Schnohr, P. and Nordestgaard, B.G., 2004. A
population-based study of morbidity and mortality in mannose-binding lectin
deficiency. J. Exp. Med., 199(10), 1391-1399.
Dahl, M.R., Thiel, S. Matsushita, M., Fujita, T., Willis, A.C., Christensen, T., Vorup-
Jensen, T., and Jensenius, J.C., 2001. Masp-3 and its association with distinct
complexes of the mannan-binding lectin complement activation pathway. Immunity., 15,
127–133.

Chapter 6: Bibliography
176
Defranco, A.L., Locksley, R.M., and Robertson, M., 2007. IMMUNITY, The immune
response in Infectious and Inflammatory disease. Oxford University Press.
Densen, P., Weiler, J. M., Griffiss, J. M., and Hoffmann, L. G., 1987. Familial
properdin deficiency and fatal meningococcemia. correction of the bactericidal
defect by vaccination. N. Engl. J. Med. 316, 922–926.
Edwards U., A. Müller, S. Hammerschmidt, R. Gerardy-Schahn and M. Frosch.
Molecular analysis of the biosynthesis pathway of the alpha-2, 8 polysialic acid capsule
by Neisseria meningitidis serogroup B, (1994). Mol. Microbiol. 14, 141-149.
Edwards, M. S., and Baker, C. J., 1981. Complications and sequelae of
meningococcal infections in children. J. Pediatr. 99,540-545.
EHRNTHALLER, C., IGNATIUS, A., GEBHARD, F. and HUBER-LANG, M., 2011.
New insights of an old defense system: structure, function, and clinical relevance of the
complement system. Molecular medicine (Cambridge, Mass.), 17(3-4), pp. 317.
Eisen, D. P., and Minchinton, R. M., 2003. Impact of mannose-binding lectin
on susceptibility to infectious diseases, Clin. Infect. Dis., 37:1496-1505.
Emonts M., J.A. Hazelzet, R. de Groot and P.W. Hermans. Host genetic determinants of
Neisseria meningitidis infections, (2003) Lancet Infect Dis 3:565–577.
Endo, Y., Nakazawa, N., Liu, Y., Iwaki, D., Takahashi, M., Fujita, T., Nakata, M., and
Matsushita, M., 2005. Carbohydrate-binding specificities of mouse ficolin A, a splicing
variant of ficolin A and ficolin B and their complex formation with MASP-2 and sMAP.
Immunogenetics., 57, 837-844.
Estabrook, M. M., Jack, D. L., Klein, N. J., and Jarvis, G. A., 2004. Mannose-
binding lectin binds to two major outer membrane proteins, opacity protein and
porin, of Neisseria meningitidis. J. Immunol., 172, 3784-3792.

Chapter 6: Bibliography
177
Estabrook, M.M., Griffiss J.M., and Jarvis G.A., 1997. Sialylation of Neisseria
meningitidis lipooligosaccharide inhibits serum bactericidal activity by
masking lacto-N-neotetraose. Infect. Immun., 65,4436–4444.
Faber, J., Schuessler, T., Finn, A., Murdoch, C., Zenz, W., Habermehl, P.,
Meyer, C. U., Zabel, B. U., Schmitt, H., Zepp, F., and Knuf, M., 2007. Age-
dependent association of human mannose-binding lectin mutations with
susceptibility to invasive meningococcal disease in childhood. Pediatr. Infect.
Dis. J. 26:243-246.
Farries, T.C. & Atkinson, J.P., 1989.Biosynthesis of properdin.Journal of
Immunology (Baltimore, Md.: 1950).142, 842-847.
Farries, T.C., Lachmann, P. J. and Harrison, R. A., 1988. Analysis of the interaction
between properdin and factor B, components of the alternative-pathway C3 convertase
of complement. Biochem J., 253, 667-675.
Figueroa, J.E. & Densen, P., 1991. Infectious diseases associated with
complement deficiencies. Clinical Microbiology Reviews.4, 359-395.
Fijen C.A., B.H. Derkx, E.J. Kuijper, M. Mannens, S.R. Poort, M. Peters, M.R. Daha
and J. Dankert, Fulminant meningococcal septic shock in a boy with combined inherited
properdin and protein C deficiency, (1995) Clin. Exp. Immunol. 102:290–296.
Fijen, C.A., Kuijper, E.J., te Bulte, M.T., Daha, M.R., Dankert, J., 1999.
Assessment of complement deficiency in patients with meningococcal disease
in The Netherlands.Clinical Infectious Diseases : An Official Publication of the
Infectious Diseases Society of America. 28, 98-105.
Finne, J., Bitter-Suermann, D., Goridis, C., and Finne, U., 1987. An IgG
monoclonal antibody to group B meningococci cross-reacts with
developmentally regulated polysialic acid units of glycoproteins in neural and
extraneural tissues. J. Immunol., 138, 4402-4407.

Chapter 6: Bibliography
178
FRANK, M.M., 2010. Complement disorders and hereditary angioedema. The Journal
of allergy and clinical immunology, 125(2 Suppl 2), pp. S262-S271.
Frasch, C.E., Zollinger, W.D., and Poolman, J.T., 1985. Serogype antigens of
Neisseria meningitidis and a proposed scheme from designation of serotypes.
Rev. Infect. Dis., 7 (4), 504-510.
Frosch M. and A. Müller. Phospholipid substitution of capsular polysaccharides and
mechanisms of capsule formation in Neisseria meningitidis, (1993). Mol. Microbiol. 8,
483-493.
FUJITA, T., 2002. Evolution of the lectin-complement pathway and its role in
innate immunity. Nature reviews.Immunology, 2(5), pp. 346-353.
FUJITA, T., MATSUSHITA, M. and ENDO, Y., 2004. The lectin-complement
pathway--its role in innate immunity and evolution. Immunological reviews,
198(1), pp. 185-202.
GÁL, P., DOBÓ, J., ZÁVODSZKY, P. and SIM, R.B.M., 2009. Early
complement proteases: C1r, C1s and MASPs. A structural insight into
activation and functions. Molecular immunology, 46(14), pp. 2745-2752.
Garred, P., Honoré, C., Ma, Y.J., Rørvig, S., Cowland, J., Borregaard, N., and
Hummelshøj, T., 2009. The genetics of ficolins. J. Innate. Immun., 2: 3-16
Garred, P., Michaelsen, T. E., Bjune, G., Thiel, S., and Svejgaard, A., 1993. A
low serum concentration of mannan-binding protein is not associated with
serogroup B or C meningococcal disease. Scand. J. Immunol., 37, 468-470.
Geoffroy, M.C., Floquet, S., Metais, A., Nassif, X., and Pelicic, V., Large-scale
analysis of the meningococcus genome by gene disruption, resistance to
complement-mediated lysis. Gen. Res., 2003; 13, 391–98.
Goldsby, R. A., Kindt, T. J., Osborne, B. A., and Kuby, J., 2003. Immunology. 5th ed. W.
H. Freeman and Co., New York, NY.

Chapter 6: Bibliography
179
Griffiths,E., 1999. Iron in biological system, In Iron and infection: molecular,
physiological and clinical aspects, Bullen, J.j. and Griffiths,E. (ed.), 2nd
ed., vol. 1. John
Wiley & Sons, Ltd., West Sussex, United Kingdom. Pp.1-26.
Griffiss J. M. Mechanisms of host immunity, (1995) K. Cartwright (ed.),Meningococcal
disease. John Wiley & Sons, Chichester, England. 35-70.
Griffiss, J., Brandt, B. L., Saunders, N. B., and Zollinger, W., 2000. Structural
relationships and sialylation among meningococcal L1, L8, and L3,7
lipooligosaccharide serotypes. J. Biol. Chem., 275, 9716-9724.
Guiver M. and R. Borrow. PCR diagnosis. In Meningococcal Disease Methods and
Protocols, (2001) N. J. Hum. Press. 23-39.
Hancock R.E. Cationic peptides: effectors in innate immunity and novel antimicrobials,
(2001) Lancet Infect Dis. 1:156–164.
Hansen S., Selman, L., Palaniyar, N., Ziegler, K., Brandt, J., Kliem, A.,
Jonasson, M., Skjoedt, M., Nielsen, O., Hartshorn, K., Jørgensen, T. J. D.,
Skjødt, K., and Holmskov, U., 2010. Collectin 11 (CL-11, CL-K1) is a MASP-
1/3–Associated plasma collectin with microbial-binding activity. J. Immunol.,
185, 6096-6104.
Harboe M, et al. (2012) The role of properdin in zymosan- and Escherichia coliinduced
complement activation. J Immunol 189(5):2606–2613
Hardaway, R. M., 1982. Pathology and pathophysiology of disseminated
intravascular coagulation. In R. A. Cowley, and B. F. Trump (ed.),
Pathophysiology of shock, anoxia and ischaemia. The Williams & Wilkins Co.,
Baltimore, Md., 186-197.
Hayat, A., 2012. The molecular interactions between two activation pathways of
complement are essential for a protective innate immune response to Neisseria
meningitidis infection. Leicester: University of Leicester.

Chapter 6: Bibliography
180
Hermaszewski R. A. and A. D. B. Webster.Primary hypogammaglobulinemia: a survey
of clinical manifestations and complications, (1993) Q. J. Med. 81:31-42.
Hibberd, M.L., Sumiya, M., Summerfield, J.A., Booy, R., and Levin, M., 1999.
Association of variants of the gene for mannose-binding lectin with susceptibility to
meningococcal disease. Lancet., 353, 1049–1053.
Hiemstra, P.S., Langeler, E., Compier, B., Keepers, Y., Leijh, P.C., van den Barselaar,
M.T., Overbosch, D., Daha, M.R., 1989. Complete and partial deficiencies of
complement factor D in a Dutch family. J. Clin. Invest. 84, 1957–1961.
Hitchcock, P.J.,1989. Unified nomenclature for pathogenic Neisseria species.
Clin. Microbiol. Rev.,2 , 64-65.
Hoff, G.E., and Høiby, N., 1978. Cross-reactions between
Neisseriameningitidis and twenty-seven other bacterial species. Acta. Pathol.
Microbiol. Scand., 86, 87–92.
Holbein, B. E., 1980. Iron-controlled infection with Neisseria meningitidis in
mice. Infect. Immun., 29,886-891.
Holbein, B. E., 1981. Enhancement of Neisseria meningitidis infection in mice
by addition of iron bound to transferrin. Infect. Immun., 34,120-125.
Holbein, B. E., Jericho, K. W. and Likes, G. C., 1979. Neisseria meningitidis
infection in mice, influence of iron, variations in virulence among strains, and
pathology. Infect. Immun., 24,545-551.
Holmskov, U., Thiel, S. and Jensenius, J. C., 2003. Collectins and ficolins,
humoral lectins of the innate immune defense. Annu. Rev. Immunol. 21, 547–
578.
Hong, S., Mogensen, T. H., Kilian, M., Jonsson, A.-B., and Paludan, S. R., 2008.
Important role for Toll-Like Receptor 9 in host defense against meningococcal sepsis.
Infect. Immun., 76, 5421-5428.

Chapter 6: Bibliography
181
Hourcade, D.E., 2006. The role of properdin in the assembly of the alternative
pathway C3 convertases of complement.The Journal of Biological
Chemistry.281, 2128-2132.
Ieva R., C. Alaimo, I. Delany, G. Spohn, R. Rappuoli and V. Scarlato. CrgA is an
inducible LysR-type regulator of Neisseria meningitidis, acting both as a repressor and
as an activator of gene transcription, (2005) J. Bacteriol. 187:3421-3430.
Iwaki, D., Kanno, K., Takahashi, M., Endo, Y., Matsushita, M., and Fujita, T.,
2011. The role of mannose-binding lectin-associated serine protease-3 in
activation of the alternative complement pathway. J. Immunol., 187, 3751-
3758.
Jack, D. L., Dodds, A. W., Anwar, N., Ison, C. A., Law, A., Frosch, M.,
Turner, M. W., and Klein, N. J., 1998. Activation of complement by mannose-
binding lectin on isogenic mutants of Neisseria meningitidis serogroup B. J.
Immunol., 160, 1346-1353.
Jack, D.L., Jarvis, G.A., Booth, C.L., Turner M.W., and Klein, N.J., 2001.
Mannose-binding lectin accelerates complement activation and increases serum
killing of Neisseria meningitidis serogroup C. J. Infect. Dis., 184, 836–845.
Jack, D.L., Lee, M.E., Turner, M.W., Klein, N.J., and Read, R.C., 2005.
Mannose-binding lectin enhances phagocytosis and killing of Neisseria
meningitidis by human macrophages. J. Leukoc. Biol., 77, 328-336.
Jarva, H., Ram, S., Vogel, U., Blom, A. M., and Meri, S., 2005. Binding of the
Complement Inhibitor C4bp to Serogroup B Neisseria meningitidis. J. Immunol., 174,
6299-6307.
Jonsson A.B., G. Nyberg and S. Normark. Jonsson Kahler C. M. and D. S. Stephens.
Genetic basis for biosynthesis, structure, and function of meningococcal
lipooligosaccharide (endotoxin), (1998) Crit. Rev. Microbiol. 24: 281-334.

Chapter 6: Bibliography
182
Jonsson, G., Truedsson, L., Sturfelt, G., Oxelius, V.A., Braconier, J.H. and
Sjoholm, A.G., 2005. Hereditary C2 deficiency in Sweden, frequent occurrence
of invasive infection, atherosclerosis, and rheumatic disease. Med., 84(1), 23-
34.
JURIANZ, K., ZIEGLER, S., GARCIA-SCHÜLER, H., KRAUS, S.,
BOHANA-KASHTAN, O., FISHELSON, Z. and KIRSCHFINK, M., 1999.
Complement resistance of tumor cells: basal and induced mechanisms.
Molecular immunology, 36(13), pp. 929-939.
Kahler, C. M., and Stephens, D. S., 1998. Genetic basis for biosynthesis, structure, and
function of meningococcal lipooligosaccharide (endotoxin). Crit. Rev. Microbiol., 24,
281-334.
Kamberi P., Sobel, R. A., Clemons, K. V., Stevens, D. A., Pappagianis, D., and
Williams, P. L., 2003. A murine model of coccidioidal meningitis. J. Infect. Dis., 187,
453-460.
Kemper C, Mitchell LM, Zhang L, Hourcade DE (2008) The complement protein
properdin Proc Natl Acad Sci USA 105(26):9023–9028.
KIRSCHFINK, M. and MOLLNES, T.E., 2003. Modern complement analysis. Clinical
and diagnostic laboratory immunology, 10(6), pp. 982-989.
Knapp, J. S., 1988. Historical perspectives and identification of Neisseria and
related species. Clin. Microbiol. Rev., 1, 415–431.
Kolb-Maurer A., Unkmeir, A., Kammerer, U., Hubner, C., Leimbach, T., Stade, A.,
Kampgen, E., Frosch, M., & Dietrich, G., 2001. Interaction of Neisseria meningitidis
with human dendritic cells. Infect. Immun., 69, 6912–6922.
Kuipers, S., Aerts, P.C., and Van Dijk, D.H., 2003. Differential
microorganism-induced mannose-binding lectin activation. FEMS Immunol.
Med. Microbiol., 36, 33–39.

Chapter 6: Bibliography
183
Langer, H.F., Chung, K.J., Orlova, V.V., Choi, E.Y., Kaul, S., Kruhlak, M.J.,
Alatsatianos, M., DeAngelis, R.A., Roche, P.A., Magotti, P., Li, X.,
Economopoulou, M., Rafail, S., Lambris, J.D., Chavakis, T., 2010.
Complement-mediated inhibition of neovascularization reveals a point of
convergence between innate immunity and angiogenesis. Blood.116, 4395-
4403.
LAUDES, I.J., CHU, J.C., HUBER-LANG, M., GUO, R., RIEDEMANN,
N.C., SARMA, J.V., MAHDI, F., MURPHY, H.S., SPEYER, C., LU, K.T.,
LAMBRIS, J.D., ZETOUNE, F.S. and WARD, P.A., 2002. Expression and
function of C5a receptor in mouse microvascular endothelial cells. Journal of
immunology (Baltimore, Md.: 1950), 169(10), pp. 5962.
Leffler, J., Bengtsson, A.A., Blom, A.M., 2014. The complement system in
systemic lupus erythematosus: an update. Annals of the Rheumatic
Diseases.73, 1601-1606.
Lesavre, P.H. & Muller-Eberhard, H.J., 1978.Mechanism of action of factor D
of the alternative complement pathway.The Journal of Experimental
Medicine.148, 1498-1509.
Lewis L.A., Ngampasutadol J., Wallace R., Reid J.E.A., Vogel U., and Ram,
S., 2010. The meningococcal vaccine candidate neisserial surface protein A
(NspA) binds to factor H and enhances meningococcal resistance to
complement. PLoS Path., 6(7) e1001027.
Liu, Yu., Endo, Y., Iwaki, D., Nakata, M., Matsushita, M., Wada, I., Inoue, K.,
Munakata, M., and Fujita, T., 2005. Human M-Ficolin is a secretory protein that
activates the lectin complement pathway. J. Immunol., 175, 1350-1356.
Lynch, N.J., Khan, S.U., Stover, C.M., Sandrini, S.M., Marston, D., Presanis,
J.S. and Schwaeble, W.J., 2005. Composition of the lectin pathway of

Chapter 6: Bibliography
184
complement in Gallus gallus, absence of mannan-binding lectin-associated
serine protease-1 in birds. J. Immunol immunol., 174(8), 4998-5006.
Lynch, N.J., Roscher, S., Hartung, T., Morath, S., Matsushita, M., Maennel,
D.N., Kuraya, M., Fujita, T., and Schwaeble, W.J., 2004. L-ficolin specifically
binds to lipoteichoic acid, a cell wall constituent of gram-positive bacteria, and
activates the lectin pathway of complement. J. Immunol.,172, 1198-1202.
Mackinnon, F.G, Ho, Y., Blake, M.S., Michon, F., Chandraker, A., Sayegh,
M.H., and Wetzler, L.M., 1999. The role of B/T costimulatory signals in the
immunopotentiating activity of neisserial porin. J. Infect. Dis., 180,755–761.
Mandrell R.E., J.J. Kim, C.M. John, B.W. Gibson, J.V. Sugai, M.A. Apicella, J.M.
Griffiss and R. Yamasaki. Endogenous sialylation of the lipooligosaccharides of
Neisseria meningitidis, (1991) J. Bacteriol. 173(9):2823–2832.
Mandrell, R. E., and Zollinger, W. D., 1977. Lipopolysaccharide serotyping of
Neisseria meningitidis by hemagglutination inhibition. Infect. Immun., 16,471-
475.
MARKIEWSKI, M.M. and LAMBRIS, J.D., 2007. The Role of Complement
in Inflammatory Diseases From Behind the Scenes into the Spotlight. The
American Journal of Pathology, 171(3), pp. 715-727.
Markovic, S.N., Inwards, D.J., Frigas, E.A., Phyliky, R.P., 2000.Acquired C1
esterase inhibitor deficiency.Annals of Internal Medicine.132, 144-150.
Matsumoto, M., Fukuda, W., Circolo, A., Goellner, J., Strauss-Schoenberger, J.
Wang, X., Fujita, S., Hidvegi, T., Chaplin, D. D., and Colten, H. R., 1997.
Abrogation of the alternative complement pathway by targeted deletion of
murine factor B. Proc. Natl. Acad. Sci. USA 94, 8720-8725.
MATSUSHITA, M., 2010. Ficolins: complement-activating lectins involved in innate
immunity. Journal of innate immunity, 2(1), pp. 24-32.

Chapter 6: Bibliography
185
Matsushita, M., and Fujita, T., 1995. Cleavage of the third component of
complement (C3) by mannose-binding protein-associated serine protease
(MASP) with subsequent complement activation. Immunobiol. 194, 443-48.
Matsushita, M., Endo, Y. and Fujita, T., 2000. Cutting edge, complement-activating
complex of ficolin and mannose-binding lectin-associated serine protease. J. Immunol.,
164, 2281-2284.
Matsushita, M., Endo, Y., Hamasaki, N., and Fujita, T., 2001. Activation of the lectin
complement pathway by ficolins. Int. Immunopharmacol. 1, 359–363.
Mayilyan, K.R., 2012. Complement genetics, deficiencies, and disease
associations. Protein & Cell.3, 487-496.
McMullen, M.E., Hart, M.L., Walsh, M.C., Buras, J., Takahashi, K., and Stahl, G.L.,
2006. Mannose-binding lectin binds IgM to activate the lectin complement pathway in
vitro and in vivo. Immunobiol., 211, 759–766.
Medzhitov, R., 2007. Recognition of microorganisms and activation of the immune
response. Nature , 449, 819-826.
MOLINA, H., KINOSHITA, T., WEBSTER, C.B. and HOLERS, V.M., 1994.
Analysis of C3b/C3d binding sites and factor I cofactor regions within mouse
complement receptors 1 and 2. Journal of immunology (Baltimore, Md.: 1950),
153(2), pp. 789.
Moore, P. S., 1992. Meningococcal meningitis in sub-Saharan Africa, a model
for the epidemic process. Clin. Infect. Dis. 14,515-525.
Morgan, B.P., and Walport, M.J., 1991. Complement deficiency and diseases. Immunol.
Today., 12,301−306.

Chapter 6: Bibliography
186
Nagata, M., Hara, T., Aoki, T., Mizuno, Y., Akeda, H., Inaba, S., Tsumoto, K.,
Ueda, K., 1989. Inherited deficiency of ninth component of complement: an
increased risk of meningococcal meningitis.The Journal of Pediatrics.114,
260-264.
Nolan, K. F., Kaluz, S., Higgins, J. M., Goundis, D., and Reid, K. B., 1992.
Characterization of the human properdin gene. Biochem. J., 287, 291–297.
Ogden, C. A., deCathelineau, A., Hoffmann, P. R., Bratton, D., Ghebrehiwet,
B., Fadok, V. A., and Henson, P. M., 2001. C1q and mannose-binding lectin
engagement of cell surface calreticulin and CD91 initiates macropinocytosis
and uptake of apoptotic cells. J. Exp. Med., 194, 781-796.
Pangburn, M. K., Pangburn, K. L., Koistinen, V., Meri, S., Sharma, A. K.,
2000. Molecular mechanisms of target recognition in an innate immune
system, interactions among factor H, C3b, and target in the alternative pathway
of human complement. J. Immunol., 164, 4742-4751.
Pangburn, M.K., 1989. Analysis of the natural polymeric forms of human
properdin and their functions in complement activation.Journal of Immunology
(Baltimore, Md.: 1950).142, 202-207.
Perdikoulis, M.V., Kishore, U., Reid, K.B., 2001. Expression and
characterisation of the thrombospondin type I repeats of human properdin.
Biochimica Et Biophysica Acta. 1548, 265-277.
Perkins-Balding, D., Ratliff-Griffin, M., and Stojiljkovic, I., 2004. Iron Transport
Systems in Neisseria meningitidis. Microbiol. Mol. Biol. Rev., 68, 154-171.
Pflieger D., Przypylski C., Gonnet F., Le Caer J.P., Lunardi T., Arlaud G.J.,
and Daniel R., 2010. Analysis of Human C1q by Combined Bottom-up and
Top-down Mass Spectrometry: detailes mapping of post-translational
modifications and insights into the C1R/C1S binding sites. Mol. Cell.
Proteomics., 9 (4), 593-610.

Chapter 6: Bibliography
187
PILLEMER, L., BLUM, L., LEPOW, I.H., ROSS, O.A., TODD, E.W.,
WARDLAW, A.C., 1954. The properdin system and immunity. I.
Demonstration and isolation of a new serum protein, properdin, and its role in
immune phenomena. Science (New York, N.Y.). 120, 279-285.
PODACK, E., MULLER-EBERHARD, H., HORST, H. and HOPPE, W., 1982.
Membrane attach complex of complement (MAC): three-dimensional analysis of MAC-
phospholipid vesicle recombinants. The Journal of Immunology, 128(5), pp. 2353.
Pryzdial, E.L. & Isenman, D.E., 1987. Alternative complement pathway
activation fragment Ba binds to C3b. Evidence that formation of the factor B-
C3b complex involves two discrete points of contact. The Journal of Biological
Chemistry.262, 1519-1525.
Pujol, C., Eugene, E., Marceau, M., and Nassif. X., 1999. The meningococcal
PilT protein is required for induction of intimate attachment to epithelial cells
following pilus-mediated adhesion. Proc. Natl. Acad. Sci. U. S. A., 96, 4017-
4022.
Racoosin J.A., Whitney C.G., Conover C.,and Diaz P.S., 1998. Serogroup Y
meningococcal disease in Chicago, 1991-1997. JAMA., 280,2094-2098.
Reid, K. B., 1983. Proteins involved in the activation and control of the two pathways
of human complement. Biochem Soc Trans, 11, pp. 1-12.
RICKLIN, D. and LAMBRIS, J.D., 2007. Complement-targeted therapeutics.
Nature biotechnology, 25(11), pp. 1265-1275.
Riedo, F. X., Plikaytis, B. D., and Broome, C. V., 1995. Epidemiology and
prevention of meningococcal disease. Pediatr. Infect. Dis. J. 14,643-657.
Rooryck, C., Diaz-Font, A., Osborn, D.P.S., Chabchoub, E., et al., 2011.
Mutations in the lectin complement pathway genes COLEC11 and MASP1
cause 3MC syndrome. Nat. Genet., 43: 197-203.

Chapter 6: Bibliography
188
Roos A., L. H. Bouwman, D.J. van Gijlswijk-Janssen, M.C. Faber-Krol, G.L. Stahl,
M.R. Daha. Human IgA activates the complement system via the mannan-binding lectin
pathway, (2001) J. Immunol. 167: 2861–2868.
Rosenstein, N.E., Perkins, B.A., Stephens, D.S., Popovic, T., and Hughes, J.M., 2001.
Meningococcal disease. N. Engl. J. Med., 344, 1378-1388.
Ross, S.C., and Densen, P., 1984. Complement deficiency states and infection,
epidemiology, pathogenesis and consequences of Neisserial and other
infections in an immune deficiency. Med., 63 (5), 243-73.
Rossi, V., Cseh, S., Ball, I., Thielens, N.M., Jensenius, J.C., and Arlaud, G.J., 2001.
Substrate specificities of recombinant mannan-binding lectin-associated serine
proteases-1 and -2, J. Biol. Chem., 276, 40880-40887.
SARMA, J.V. and WARD, P.A., 2011. The complement system. Cell and
tissue research, 343(1), pp. 227-235.
Sastry, R., Wang, J.S., Brown, D.C., Ezekowitz, R.A., Tauber, A.I., and Sastry, K.N.,
1995. Characterization of murine mannose-binding protein genes Mbl1 and Mbl2
reveals features common to other collectin genes. Mamm. Genome., 6 , 103–110.
Schejbel, L., Rosenfeldt, V., Marquart, H., Valerius, N.H., Garred, P., 2009.
Properdin deficiency associated with recurrent otitis media and pneumonia, and
identification of male carrier with Klinefelter syndrome. Clinical Immunology
(Orlando, Fla.).131, 456-462.
Schelenz S., R. Malhotra, R.B. Sim, U. Holmskov and G.J. Bancroft. Binding of host
collectins to the pathogenic yeast Cryptococcus neoformans: human surfactant protein
D acts as an agglutinin for acapsular yeast cells, (1995) Infect. Immun. 63:3360–3366.
Schneider M. C., R. M. Exley, H. Chan, I. Feavers, Y. H. Kang, R. B. Sim and C. M.
Tang. Functional significance of factor H binding to Neisseria meningitidis, (2006) J.
Immunol. 176:7566-7575.

Chapter 6: Bibliography
189
Schwaeble, W.J. & Reid, K.B., 1999. Does properdin crosslink the cellular and
the humoral immune response? Immunology Today.20, 17-21.
Schwaeble, W.J., Dahl, M. R., Thiel, S., Stover C.M., and Jensenius, J. C., 2002. The
mannan-binding lectin-associated serine proteases (MASPs) and MAp19, four
components of the lectin pathway activation complex encoded by two genes.
Immunobiol., 205, 455-466.
SCHWAEBLE, W.J., LYNCH, N.J., CLARK, J.E., MARBER, M., SAMANI,
N.J., ALI, Y.M., DUDLER, T., PARENT, B., LHOTTA, K., WALLIS, R.,
FARRAR, C.A., SACKS, S., LEE, H., ZHANG, M., IWAKI, D.,
TAKAHASHI, M., FUJITA, T., TEDFORD, C.E. and STOVER, C.M., 2011.
Targeting of mannan-binding lectin-associated serine protease-2 confers
protection from myocardial and gastrointestinal ischemia/reperfusion injury.
Proceedings of the National Academy of Sciences of the United States of
America, 108(18), pp. 7523-7528.
Singh, D.K. & Rai, R., 2009.Recurrent meningitis secondary to isolated C3
deficiency.Indian Journal of Pediatrics.76, 95-96.
Slade, C., Bosco, J., Unglik, G., Bleasel, K., Nagel, M., Winship, I.,
2013.Deficiency in complement factor B.The New England Journal of
Medicine.369, 1667-1669.
Smith, C.A., Pangburn, M.K., Vogel, C.W., Muller-Eberhard, H.J.,
1984.Molecular architecture of human properdin, a positive regulator of the
alternative pathway of complement.The Journal of Biological Chemistry.259,
4582-4588.
Sorensen, R., Thiel S., and Jensenius, J.C., 2005. Mannan-binding-lectin-associated
serine proteases, characteristics and disease associations. Springer Semin.
Immunopathol., 27, 299–319.

Chapter 6: Bibliography
190
Spath, P.J., Sjoholm, A.G., Fredrikson, G.N., Misiano, G., Scherz, R., Schaad, U.B.,
Uhring-Lambert, B., Hauptmann, G., Westberg, J., Uhlen, M., Wadelius, C., and
Truedsson, L., 1999. Properdin deficiency in a large Swiss family, identification of a
stop codon in the properdin gene, and association of meningococcal disease with lack of
the IgG2 allotype marker G2m (n). Clin. Exp. Immunol., 118, 278–284.
Spinosa M. R., C. Progida, Tala, A., Cogli, L., Alifano, P., and Bucci, C. The Neisseria
meningitidis Capsule Is Important for Intracellular Survival in Human Cells, (2007)
Infect. Immun. 75: 3594–3603.
Sprong, T., Møller, A.-S. W., Bjerre, A., Wedege, E.,
Kierulf, P., van der Meer, J. W.
M., Brandtzaeg, P., van Deuren, M.,
and Mollnes, T. E., 2004. Complement activation
and complement-dependent inflammation by Neisseria meningitidis are independent of
lipopolysaccharide. Infect. Immun., 72(6), 3344–3349.
Sprong, T., Roos, D., Weemaes, C., Neeleman, C., Geesing, C.L., Mollnes,
T.E., van Deuren, M., 2006. Deficient alternative complement pathway
activation due to factor D deficiency by 2 novel mutations in the complement
factor D gene in a family with meningococcal infections. Blood.107, 4865-
4870.
Stanton, C.M., Yates, J.R., den Hollander, A.I., Seddon, J.M., Swaroop, A.,
Stambolian, D., Fauser, S., Hoyng, C., Yu, Y., Atsuhiro, K., Branham, K.,
Othman, M., Chen, W., Kortvely, E., Chalmers, K., Hayward, C., Moore, A.T.,
Dhillon, B., Ueffing, M., Wright, A.F., 2011. Complement factor D in age-
related macular degeneration. Investigative Ophthalmology & Visual
Science.52, 8828-8834.
Stengaard-Pedersen, K., Thiel, S., Gadjeva, M., Moller-Kristensen, M.,
Sorensen, R., Jensen, L.T., Sjoholm, A.G., Fugger, L., Jensenius, J.C.,
2003.Inherited deficiency of mannan-binding lectin-associated serine protease
2.The New England Journal of Medicine.349, 554-560.

Chapter 6: Bibliography
191
Stover C. M., Thiel, S., Thelen, M., Lynch, N. J., Vorup-Jensen, T., Jensenius, J. C., and
Schwaeble, W.J., 1999. Two constituents of the initiation complex of the mannan-
binding lectin activation pathway of complement are encoded by a single structural
gene. J. Immunol., 162, 3481-3490.
Stover, C.M., Luckett, J.C., Echtenacher, B., Dupont, A., Figgitt, S.E., Brown,
J., Mannel, D.N. and Schwaeble, W.J., 2008. Properdin plays a protective role
in polymicrobial septic peritonitis. J. Immunol., 180(5), 3313-3318.
Swartley J. S., A. A. Marfin, S. Edupuganti, L.-J. Liu, P. Cieslak, B. Perkins, J. D.
Wenger and D. S. Stephens. Capsule switching of Neisseria meningitidis, (1997) Proc.
Natl. Acad. Sci. U S A. 94(1): 271–276.
Taha M. K., P. C. Morand, Y. Pereira, E. Eugene, D. Giorgini, M. Larribe and X.
Nassif. Pilus-mediated adhesion of Neisseria meningitidis: the essential role of cell
contact-dependent transcriptional upregulation of the PilC1 protein, (1998) Mol.
Microbiol. 28:1153-1163.
Takahashi H., R. W. Carlson, A. Muszynski, B. Choudhury, K.S. Kim, D.S. Stephens
and H. Watanabe. Modification of Lipooligosaccharide with Phosphoethanolamine by
LptA in Neisseria meningitidis Enhances Meningococcal Adhesion to Human
Endothelial and Epithelial Cells, (2008) Infect. Immun. 76: 5777-5789.
Takahashi, M., Endo, Y., Fujita, T., and Matsushita, M., 1999. A truncated form of
mannose-binding lectin-associated serine protease (MASP)-2 expressed by alternative
polyadenylation is a component of the lectin complement pathway. Int. Immunol., 11,
859-863.
Takahashi, M., Ishida, Y., Iwaki, D., Kanno, K., Suzuki, T., Endo, Y., Homma,
Y. and Fujita, T., 2010. Essential role of mannose-binding lectin-associated
serine protease-1 in activation of the complement factor D. J. Exp. Med.,
207:29-37.

Chapter 6: Bibliography
192
TAYLOR, P.R., CARUGATI, A., FADOK, V.A., COOK, H.T., ANDREWS,
M., CARROLL, M.C., SAVILL, J.S., HENSON, P.M., BOTTO, M. and
WALPORT, M.J., 2000. A hierarchical role for classical pathway complement
proteins in the clearance of apoptotic cells in vivo. The Journal of experimental
medicine, 192(3), pp. 359-366.
Tettelin H., N. J. Saunders, J. Heidelberg, A. C. Jeffries, K. E. Nelson, J. A. Eisen, K.
A. Ketchum, D. W. Hood, J. F. Peden, R. J. Dodson, W. C. Nelson, M. L. Gwinn, R.
DeBoy, J. D. Peterson, E. K. Hickey, D. H. Haft, S. L. Salzberg, O. White, R. D.
Fleischmann, B. A. Dougherty, T. Mason, A. Ciecko, D. S. Parksey, E. Blair, H.
Cittone, E. B. Clark, M. D. Cotton, T. R. Utterback, H. Khouri, H. Qin, J. Vamathevan,
J. Gill, V. Scarlato, V. Masignani, M. Pizza, G. Grandi, L. Sun, H. O. Smith, C. M.
Fraser, E. R. Moxon, R. Rappuoli and J. C. Venter. Complete genome sequence of
Neisseria meningitidis serogroup B strain MC58, (2000) Science 287:1809-1815.
Thiel S., 2007. Complement activating soluble pattern recognition molecules with
collagen-like regions, mannan-binding lectin, ficolins and associated proteins. Mol.
Immunol., 44, 3875-3888.
Thiel, S., Petersen, S. V., Vorup-Jensen, T., Matsushita, M., Fujita, T., Stover, C.,
Schwaeble, W. J., and Jensenius, J. C., 2000. Interaction of C1q and mannan-binding
lectin (MBL) with C1r, C1s, MBL-associated serine proteases 1 and 2, and the MBL-
associated protein Map19. J. Immunol., 165, 878-887.
THURMAN, J.M. and HOLERS, V.M., 2006. The central role of the
alternative complement pathway in human disease. Journal of immunology
(Baltimore, Md.: 1950), 176(3), pp. 1305.
Tommassen, J., Vermeij, P., Struyve, M., Benz, R., and Poolman, J.T., 1990.
Isolation of Neisseria meningitidis mutants deficient in class 1 (PorA) and class
3 (PorB) outer membrane proteins. Infect. Immun., 5, 1355-1359.
Trouw, L.A. and Daha, M.R., 2011. Role of complement in innate immunity and host
defense. Immunology letter., 138(1), pp. 35-37.

Chapter 6: Bibliography
193
TROUW, L.A., BLOM, A.M. and GASQUE, P., 2008. Role of complement and
complement regulators in the removal of apoptotic cells. Molecular immunology, 45(5),
pp. 1199.
Tsai, C. M., Frasch, C. E., and Mocca, L. F., 1981. Five structural classes of
major outer membrane proteins in Neisseria meningitidis., J. Bacteriol., 146,
69-78.
Tsai, C.M., Boykins, R., and Frasch, C.E., 1983. Heterogeneity and variation
among Neisseria meningitidis lipopolysaccharides. J. Bacteriol., 155, 498-504.
Tully, J., Viner, R. M., Coen, P. G., Stuart, J. M., Zambon, M., Peckham, C.,
Booth, C., Klein, N., Kaczmarski, E., and Booy, R., 2006. Risk and protective
factors for meningococcal disease in adolescents: matched cohort
study. B.M.J., 332:445-450.
Tzeng, Y. L., and Stephens, D. S., 2000. Epidemiology and pathogenesis of
Neisseria meningitidis. Microb. Infect., 2, 687–700.
UNSWORTH, D.J., VIRGO, P.F. and LOCK, R.J., 2011. Immunoglobulin E
deficiency: a forgotten clue pointing to possible immunodeficiency? Annals of
Clinical Biochemistry, 48(Pt 5), pp. 459-461.
Van Deuren M., P. Brandtzaeg and
J. W. M. van der Meer. Update on Meningococcal
Disease with Emphasis on Pathogenesis and Clinical Management, (2000) Clin.
Microbiol. Rev. 13:144-166.
Van Emmerik, L.C., Kuijper, E.J., Fijen, C.A.P, Dankert, J., and Thiel, S., 1994.
Binding of mannan-binding protein to various pathogens of meningitis. Clin. Exp.
Immunol., 97, 411–416.

Chapter 6: Bibliography
194
Virji, M., and Griffiths, N.J., 2008. Binding of Opc to vitronectin contributes to
increased serum resistance of Neisseria meningitidis isolates. In, van Alphen L,
van der Ley P, van den Dobbelsteen G, editors. Rotterdam, The Netherlands,
166–167.
Vogel, U., Weinberger, A., Frank, R., Müller, A., Köhl, J., Atkinson, J.P., and
Frosch, M., 1997. Complement factor C3 deposition and serum resistance in
isogenic capsule and lipooligosaccharide sialic acid mutants of serogroup B
Neisseria meningitidis. Infect. Immun., 65 (10), 4022-4029.
Volanakis, J.E. & Narayana, S.V., 1996. Complement factor D, a novel serine
protease. Protein Science : A Publication of the Protein Society. 5, 553-564.
Vorup-Jensen, T., Petersen, S.V., Hansen, A.G., Poulsen, K., Schwaeble, W. J., Sim,
R.B., Reid, K.B., Davis, S.J., Thiel, S., and Jensenius, J.C., 2000. Distinct Pathways of
Mannan-Binding Lectin (MBL) - and C1-Complex Autoactivation Revealed by
Reconstitution of MBL with Recombinant MBL-Associated Serine Protease-2. J.
Immunol., 165, 2093–2100.
Wallis, R., Dodds, A. W., Mitchell, D. A., Sim, R. B., Reid, K. B. and Schwaeble,
W.J., 2007. Molecular interaction between MASP-2, C4, and C2 and their activation
fragments leading to complement activation via the lectin pathway. J Biol Chem., 282,
pp. 7844-7851.
Wallis, R., Mitchell, D.A., Schmid, R., Schwaeble, W.J., Keeble, A.H., 2010. Paths
reunited: Initiation of the classical and lectin pathways of complement activation.
Immunobiology. 215, 1-11.
Wallis, R., Shaw, J.M., Uitdehaag, J., Chen, C.B., Torgersen, D., and Drickamer, K.,
2004. Localization of the serine protease-binding sites in the collagen-like domain of
mannose-binding protein, indirect effects of naturally occurring mutations on protease
binding and activation. J. Biol. Chem., 279(14), 14065-14073.
Walport, M. J., 2001. Complement. First of two parts. N. Engl. J. Med., 344, 1058–
1066.

Chapter 6: Bibliography
195
Wang, J.F., Caugant, D.A., Li, X., Hu, X., Poolman, J.T., Crowe, B.A., and
Achtman, M., 1992. Clonal and antigenic analysis of serogroup A Neisseria
meningitidis with particular reference to epidemiological features of epidemic
meningitis in the People's Republic of China. Infect Immun. 60, 5267-5282.
WU, G., HU, W., SHAHSAFAEI, A., SONG, W., DOBARRO, M., SUKHOVA, G.K.,
BRONSON, R.R., SHI, G., ROTHER, R.P., HALPERIN, J.A. and QIN, X., 2009.
Complement regulator CD59 protects against atherosclerosis by restricting the
formation of complement membrane attack complex. Circulation research, 104(4), pp.
550-558.
Yamauchi, Y., Stevens, J.W., Macon, K.J., Volanakis, J.E., 1994. Recombinant
and native zymogen forms of human complement factor D. Journal of
Immunology (Baltimore, Md.: 1950). 152, 3645-3653.
Yazdankhah S. P., P. Kriz, G. Tzanakaki, J. Kremastinou, J. Kalmusova, M. Musilek, T.
Alvestad, K. A. Jolley, D. J. Wilson, N. D. McCarthy, D. A. Caugant and M. C.
Maiden. Distribution of serogroups and genotypes among disease-associated and carried
isolates of Neisseria meningitidis from the Czech Republic, Greece, and Norway,
(2004) J. Clin. Microbiol. 42: 5146–5153.