the nature of dilute solutions of sodium ion in water, methanol, and tetrahydrofurana)
TRANSCRIPT

The nature of dilute solutions of sodium ion in water, methanol, andtetrahydrofuran@fa@f)Jayaraman Chandrasekhar and William L. Jorgensen Citation: J. Chem. Phys. 77, 5080 (1982); doi: 10.1063/1.443682 View online: http://dx.doi.org/10.1063/1.443682 View Table of Contents: http://jcp.aip.org/resource/1/JCPSA6/v77/i10 Published by the American Institute of Physics. Additional information on J. Chem. Phys.Journal Homepage: http://jcp.aip.org/ Journal Information: http://jcp.aip.org/about/about_the_journal Top downloads: http://jcp.aip.org/features/most_downloaded Information for Authors: http://jcp.aip.org/authors
Downloaded 08 Jun 2013 to 141.161.91.14. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions

The nature of dilute solutions of sodium ion in water, methanol, and tetrahydrofurana)
Jayaraman Chandrasekhar and William L. Jorgensen
Department of Chemistry, Purdue University, West Lafayette, Indiana 47907 (Received 22 July 1982; accepted 5 August 1982)
Monte Carlo statistical mechanics simulations have been carried out for dilute solutions of Na + in water and tetrahydrofuran (THF) at 25 ·C and 1 atm. The intermolecular interactions were described by Lennard-Jones and Coulomb terms in the TIPS format including the TIPS2 parameters for water-water interactions. In conjunction with previous simulation results for Na + in methanol, the present study provides detailed insights into the nature of ionic solvation by dipolar protic and aprotic solvents. In agreement with x-ray data, the coordination number of Na+ in water is six, identical to the value obtained for Na+ in methanol. However, the coordination number of Na+ in TIIF fluctuates between five and six. Consistently, the ion-solvent interaction is less exothermic in THF compared to the other two solvents. However, the heat of solution which is the sum of two large opposing contributions, viz., the ion-solvent energy and the solvent reorganization energy, does not differ much for the three solvents. In all three cases, the formation of the first solvation shell accounts for all the solvent structure breaking. In particular, the number of hydrogen bonds formed by water and by methanol beyond the first shell rapidly attains the value in the pure solvent. The computed radial distribution functions and bonding and dimerization energy distributions provide additional support for this analysis.
I. INTRODUCTION
It has long been recognized that protic and aprotic solvents are distinctly different in their solvating abilities. For example, the rates and equilibria for a variety of important organometallic reactions differ Significantly in these two types of solvents. 1 Even among protic solvents, water has always been held unique, both because of its anomalous properties as well as its biological importance. 2 To gain inSights into the solvation process it is desirable to examine in detail the nature of solvation of an ion by water and by typical nonaqueous protic and aprotic solvents. Until now most discussions have been based on macroscopic solvent properties like the dielectric constant or empirical parameters such as solvent polarity. 3 In recent years, theoretical procedures for studying solvation at the molecular level have matured, the most commonly employed techniques being Monte Carlo statistical mechanics and molecular dynamics calculations. 4 However, not surprisingly, pure water5,6 and aqueous solutions7- 9 have been the focus of attention. The only study of electrolytes in a nonaqueous solvent entailed Monte Carlo simulations for Na+ and CH30- in methanol reported from this laboratory. 10
In this paper, dilute solutions of Na + in three different solvents, water, methanol (a prototype nonaqueous protic solvent), and tetrahydrofuran (THF, a widely used ether), are examined via Monte Carlo statistical mechanics simulations. The intermolecular potential functions used in each case are of the same quality, yielding good results for the structure, density, and thermodynamic properties of the pure solvents. The simulations of the solutions then provide an opportunity for arriving at general conclusions regarding solvation by the three different classes of solvents. In particular, the nature and extent of structural change produced by the intro-
a) Quantum and Statistical Mechanical Studies of Liquids, Part 27,
duction of the ion into these solvents and the concomitant effects on the calculated thermodynamic properties are explored. The results are also analyzed on the basis of existing models for ionic solvation.
II. COMPUTATIONAL DETAILS
A. Simulations of dilute solutions
The Monte Carlo calculations were carried out on systems containing one sodium ion and a large number of solvent molecules (125 for water, 127 for methanol, and 127 for THF) in a cube with periodic boundary conditions. The systems do not exactly correspond to infinitely dilute solutions due to the presence of additional images of the solute. However, as in similar, previous studies, 7-10
the solvent molecules interact only with the ion in the central cube; analyses of the results demonstrate that edge effects are not problematic.
The simulations were performed in the isothermalisobaric (NPT) ensemble at 25°C and 1 atm. This choice enables the direct calculation of heats and volumes of solution which can be compared with experimental data. The previous simulations on the pure solvents were also carried out under the same conditions.5•11,12
The details of Monte Carlo calculation in the NPT ensemble have been discussed elsewhere. 13 The main point in the present case concerns the computational problem associated with dilute solutions since the number of solvent molecules is - 100 times that of the solute. Consequently, the statistics for the latter are bound to be relatively poor unless the solute is sampled preferentially. Therefore, Owicki's scheme of preferential sampling14 has been adopted here with the modifications presented in a previous study. 10 Specifically, the probability of attempting to move a solvent molecule is made proportional to 1/(r2 +C), where r is the distance between the solute and the solvent molecule and C is an adjustable parameter. The value of C was chosen to cause the nearest solvent molecules to be
5080 J, Chern, Phys. 77(101, 15 Nov. 1982 021-9606/82/225080-10$02.10 © 1982 American Institute of Physics
Downloaded 08 Jun 2013 to 141.161.91.14. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions
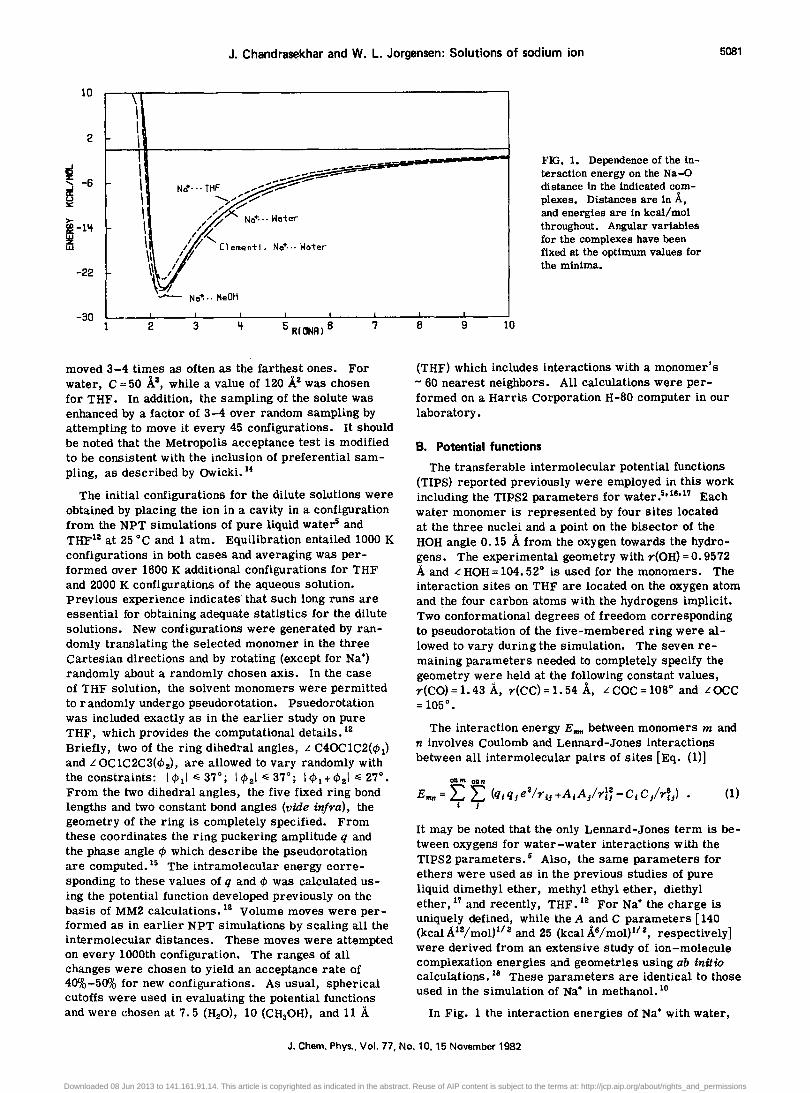
J. Chandrasekhar and W. L. Jorgensen: Solutions of sodium ion 5081
10
2
5 I :E:
" -6 I a! u \ :.::
>- \ ;-1'+ \ \ UJ
No"·· Ho-ter
No+··· Ho-ter
-22
No~·· MeOH
-30 2 3
moved 3-4 times as often as the farthest ones. For water, C =50 Aa, while a value of 120 Aa was chosen
7
for THF. In addition, the sampling of the solute was enhanced by a factor of 3-4 over random sampling by attempting to move it every 45 configurations. It should be noted that the Metropolis acceptance test is mOdified to be consistent with the inclusion of preferential sampling, as described by Owicki.14
The initial configurations for the dilute solutions were obtained by plaCing the ion in a cavity in a configuration from the NPT simulations of pure liquid waterS and THFla at 25 ° C and 1 atm. Equilibration entailed 1000 K configurations in both cases and averaging was performed over 1600 K additional configurations for THF and 2000 K configurations of the aqueous solution. Previous experience indicates that such long runs are essential for obtaining adequate statistics for the dilute solutions. New configurations were generated by randomly translating the selected monomer in the three Cartesian directions and by rotating (except for Na+) randomly about a randomly chosen axis. In the case of THF solution, the solvent monomers were permitted to randomly undergo pseudorotation. Psuedorotation was included exactly as in the earlier study on pure THF, which provides the computational details. 1a
Briefly, two of the ring dihedral angles, L C40C1C2(<1>I) and L OC 1C2C3(<1>a), are allowed to vary randomly with the constraints: 1<1>11 <f 37°; l<1>al <f 37°; 1 <1>1 + <1>a l <f 27°. From the two dihedral angles, the five fixed ring bond lengths and two constant bond angles (vide infra), the geometry of the ring is completely specified. From these coordinates the ring puckering amplitude q and the phase angle <1> which describe the pseudorotation are computed. 15 The intramolecular energy corresponding to these values of q and <1> was calculated using the potential function developed previously on the basis of MM2 calculations. 1a Volume moves were performed as in earlier NPT simulations by scaling all the intermolecular distances. These moves were attempted on every 1000th configuration. The ranges of all changes were chosen to yield an acceptance rate of 40%-50% for new configurations. As usual, spherical cutoffs were used in evaluating the potential functions and were chosen at 7.5 (HaO), 10 (CH30H), and 11 A
B 9 10
FIG. 1. Dependence of the interaction energy on the Na-O distance in the indicated complexes. Distances are in 'A. and energies are in kcal/mol throughout. Angular variables for the complexes have been fixed at the optimum values for the minima.
(THF) which includes interactions with a monomer's - 60 nearest neighbors. All calculations were performed on a Harris Corporation H-BO computer in our laboratory.
B. Potential functions
The transferable intermolecular potential functions (TIPS) reported previously were employed in this work including the TIPS2 parameters for water. 5.18.17 Each water monomer is represented by four sites located at the three nuclei and a point on the bisector of the HOH angle O. 15 A from the oxygen towards the hydrogens. The experimental geometry with r(OH) = O. 9572 A and L HOH = 104. 52° is used for the monomers. The interaction sites on THF are located on the oxygen atom and the four carbon atoms with the hydrogens implicit. Two conformational degrees of freedom corresponding to pseudorotation of the five-membered ring were allowed to vary durin g the simulation. The seven remaining parameters needed to completely specify the geometry were held at the following constant values, r(CO) =1.43 A, r(CC):::1.54 A, LCOC = 108° and LOCC ::: 105°.
The interaction energy E"," between monomers m and n involves Coulomb and Lennard-Jones interactions between all intermolecular pairs of sites [Eq. (1)]
Ollm onn
Emn=L L (qlqJea/riJ+AIA/r~~-CIC/r1J) (1) j J
It may be noted that the only Lennard-Jones term is between oxygens for water-water interactions with the TIPS2 parameters. 5 Also, the same parameters for ethers were used as in the previous studies of pure liquid dimethyl ether, methyl ethyl ether, diethyl ether,l? and recently, THF.12 For Na+ the charge is uniquely defined, while the A and C parameters [140 (kcal A12/mol) 11 2 and 25 (kcaIA6/mol)l/2, respectively] were derived from an extensive study of ion-molecule complexation energies and geometries using ab initio calculations. 18 These parameters are identical to those used in the simulation of Na+ in methanol. 10
In Fig. 1 the interaction energies of Na+ with water,
J. Chern. Phys., Vol. 77, No. 10, 15 November 1982
Downloaded 08 Jun 2013 to 141.161.91.14. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions

5082 J. Chandrasekhar. and W. L. Jorgensen: Solutions of sodium ion
methanol, and THF as a function of Na-O distance are compared. The angular variables are fixed at the optimum values for the minima (C 2v symmetry for Na+ .•. OH2 and Na+ ..• THF complexes; C s symmetry for Na+··· CH30H). The potential functions are quite similar with minima occurring near 2.25 A. The minimum energy for the water and methanol complexes is nearly the same, - 25 and - 26 kcal/mol, respectively, and that for the THF complex is slightly higher, - 23 kcal/mol. These energies are consistent with our ab
initio 6-31G* findings18 and are clearly sensitive to the charges chosen at the oxygen atoms of the bases. Experimentally, lithium is known to have a slightly greater affinity for dimethyl ether than for alcohols and water, though no more closely related data are available. 19 Some additional support for the reasonableness of the potential functions may be found in Fig. 1 which also shows the Na+ ... water potential curve obtained from the analytical function of Kistenmacher, Popkie, and Clementi. 2o This function was derived from HartreeFock level calculations and was used in a simulation of Na+ in water by Mezei and Beveridge. 7 The similarity of this potential curve to the one derived from the simple TIPS functions is striking.
III. RESULTS AND DISCUSSION
A. Thermodynamics
A thermodynamic quantity of primary interest for a dilute solution is the energy for the process of transferring the solute from the ideal gas phase into the solvent. The energy of solution tlEsol can be computed from the simulation results for the dilute solution and the pure solvent as in Eq. (2)
(2)
The total energy of the solution consists of the solutesolvent (E5:<) and solvent-solvent (Ess) contributions, while Ets is the energy of the pure solvent and tlEss is the solvent reorganization energy. In the case of the THF solution, an extra term representing the change in intramolecular energy on going from the pure solvent to the solution is added to Eq. (2). The volume of solution, tl v.'Oh which is the difference between the volume of the dilute solution V and that of the pure solvent V* is another property which is experimentally measurable. From tlE.ol and tl Vaol the enthalpy of solution of Na+ in the various solvents can be calculated via Eq. (3)
c.Hsol = tlEsol + P tl Vsol - R T , (3)
where the last term represents the PV contribution for the solute in the ideal gas. At 1 atm, c.H.ol '" .:::.E.ol - RT.
In Table I, the calculated quantities are compared for Na+ in water, methanol, and THF. The figures for methanol and THF correspond to systems containing one solute and 127 solvent molecules in the solution and 127 solvent monomers in the pure solvent. The results for pure water and the aqueous solution correspond to the presence of 125 solvent molecules. The reported energies include cutoff corrections for the LennardJones part of the interactions. Error bars (± 2cr) for the computed quantities were estimated from separate
TABLE I. Thermodynamic results for Na+ in water, methanol, and THF at 25°C and 1 atm. ~
Property Na+ in waterb Na+ in methanolC Na+ in THFc
Esx -201± 2 -206±2 -163±2 Ess -1188±4 -791 ±7 - 806±4 E~s -1254±3 -895±6 -848±4 1::.. Ess 66±7 105 ± 12 42 ±8 1::.. Esol -135 ±7 -101± 12 -121 ±8 V 3677 ± 22 9076 ± 51 18255±65 V* 3751 ± 30 9093 ± 67 18 264± 66 C. Vaol -74±52 -17±118 -9± 131 C. Vaol (expt) _l1d - 28 ± act c. Haol -136±7 -102 ±12 -122 ±8 C. Haol (expt) _1060.3" -111±101 -110±1<Y'
aE and H in kcal/mol; V in J\3. Subscripts ss and sx indicate the solvent-solvent and solvent-solute contributions. Superscript * indicates the pure solvent.
bCalculated results are total quantities for the ion and 125 solvent molecules.
cCalculated results are total quantities for the ion and 127 solvent molecules.
dF. Kawaizumi and R. Zana, J. Phys. Chern. 78, 627 (1974). "Reference 22. f From Refs. 22 and 24. Error bars estimated. gEstimated from Refs. 22 and 24 (see the text).
averages over blocks of 50 K configurations. These values are dependent on the size of the blocks; analyses of results for long NPT simulations of pure water indicate the computed uncertainties in Table I may be too low by as much as a factor of 2.21
The calculated enthalpy of solution of Na+ in water ( - 136 ± 7 kcal/ mol) is in reasonable agreement with the experimental value of - 106 ± 3 kcal/mol. 22 Previous calculations have obtained values considerably lower than the experimental one. 7,s(m) For example, Mezei and Beveridge obtained a value of -180 kcal/mol for tlEaol of Na+ in water. 7 Since the ion-molecule potential function they employed is similar to the one used in the present study (Fig. 1), the discrepancy must be due to their use of MCY -CI potential for water. 23 In fact, the MCY -CI potential under constant volume conditions yields excessively exothermic heats of solution for methanol and ethanol in water as well. 8UO,8(h) The TIPS2 function performs conSistently better in these cases. 9 Indeed, the present result represents the most accurate estimate of the heat of solution of Na+ in water obtained from a computer simulation thus far. The result is particularly notable in view of potential problems due to three-body effects and the boundary conditions. Apparently these factors can be accommodated in an average sense through a suitable choice of empirical potential functions.
The computed heat of solution of Na+ in methanol is in even better agreement with experiment (Table I). As discussed in the earlier study, the experimental value for tlH~OI was obtained by combining the heat of transfer of Na+ from methanol to water (+ 4.8 kcal/mol)24 with the tlH ~ol for Na+ in water. Similar data are not available for THF solutions; however, the heats of transfer of Na+ from water to a variety of dipolar aprotic sol-
J. Chem. Phys., Vol. 77, No. 10,15 November 1982
Downloaded 08 Jun 2013 to 141.161.91.14. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions

J. Chandrasekhar and W. L. Jorgensen: Solutions of sodium ion 5083
TABLE II. Single-ion enthalpies of transfer from water to solvents (kcal/mol) for Na" a
Solvent
Methanol Dimethyl sulfoxide Dimethylformamide Propylene carbonate Sulfolane
-4.8 -7.2 -5.9 -3.3 -4.05
aAll values from Ref. 22.
Solvent dielectric constant at 25·C
31.5 46.7 36.7 65.1 41. 3
vents with significantly different dielectric constants vary only over a range of ± 10 kcal/mol (Table 11).24 Although the dielectric constant of THF (7) is somewhat lower than that of any of the solvents listed in Table II, it may be assumed that the heat of solution of Na+ in THF is similar to that in the other dipolar aprotic solvents, leading to an experimental estimate of -110± 10 kcal/mol shown in Table 1. The computed result (- 122 ± 8 kcal/mol) is in good agreement with this value.
The individual components of the heats of solution show an interesting variation for the three systems (Table I). The solute-solvent interaction energy EBJ< is much more negative for methanol and water than for THF, The computed trend is reasonable because: (i) the optimum ion-molecule interaction energy with the present potential functions follows the order methanol >water > THF (Fig. 1), and (ii) the coordination number of Na+ in THF is lower than that in methanol and water (vide infra).
The remaining contribution to the heat of solution is the solvent reorganization energy !lEss which is computed to be positive in all three cases. However, this quantity is much smaller for THF than for methanol (42 and 105 kcal/mol, respectively). This can be readily understood on the basis of the effect of Na+ on the structure of the solvent. In order to effectively solvate the ion, the hydrogen bonding network in methanol necessarily needs to be disrupted at least in the vicinity of the ion resulting in a large reorganization energy. Since liquid THF lacks comparable structure,12 little energy is required to reorient the solvent around the ion. Surprisingly, the solvent reorganization energy in water is calculated to be only 66 kcal/mol. It is possible that this
6.0 NIHI RADIAL DISTRIBUTION FUNCTIONS
'1.5 WATER
HeOH
THF
G 3.0
1.5
0.0 I 2 8 9
FIG. 2. Computed Na-O rdf's for Na+ in water, methanol, and THF.
6.o~ ______ ~~~~R~~I~~D~IS~TR~I~BU~T~IO=N~ru~~~TI~!INS~ ______ -,
I I
'1.5
3.0
1.5
I I I I I ,- M.OH
I I I I I I I , I \ I \ J
------0.01L---~2~L-~3--~~~~5----~6----~7----LB--~9
R
FIG. 3. Computed Na-H rdf's for Na+ in water and methanol.
value is too low by - 30 kcal/mol and that it accounts for the difference between the calculated and the experimental !lH.o1 ' It should be emphasized that unlike the Es% term, the solvent reorganization energy is obtained as a difference from two simulations and is therefore subject to substantial error bars. In any event, it is clear from Table I that the heat of solution is made up of two large opposing contributions in ionic solutions. The solute-solvent energy alone does not determine the variation in the heat of solution. The limited solvent disruption in THF completely compensates for the weaker solute-solvent attraction resulting in a heat of solution similar to those for water and methanol. This accounts for the excellent solvating ability of dipolar aprotic solvents in general. Protic solvents, on the other hand, function comparably due to the more attractive solute-solvent interactiolB offsetting the greater solvent disruption.
The calculated volumes of solution are compared with available experimental data in Table I. Solvation of Na+ is accompanied by a reduction in volume in aU three solvents but no quantitative conclusions can be drawn in view of the error bars.
B. Solute-solvent structure
The computed solute-solvent radial distribution functions (rdf's) are shown in Figs. 2-4. The distribution functions show at least two well resolved peaks in all cases. The first maximum in the Na-O rdf's occurs at nearly the same distance (2.35 A) in all three solutions. This value is in good accord with the Na-O distances of
S.O tfl-C RroIAL DISTRIBUTION FUNCTIONS
I I I I
'1.5 I I - H.OH
I I -- TH!', (I
I I --- THF, (2 G 3.0 I \
I \ I \ 1.5
0.0 I 2 3 'I 5 6 7 8 9
R
FIG. 4. Computed Na-C rdf's for Na+ in methanol and THF.
J. Chern. Phys., Vol. 77, No. 10, 15 November 1982
Downloaded 08 Jun 2013 to 141.161.91.14. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions

5084 J. Chandrasekhar and W. L. Jorgensen: Solutions of sodium ion
2.38 and 2.4 A for Na+ in aqueous solutions obtained from x-ray experiments. 25.26
When comparing the rdf's for the different solutions an important point needs to be noted. By definition, the rdf gXy(R) describes the probability of occurrence of y type atoms about x type atoms as a function of x-y separation R in the liquid. This is expressed in Eq. (4):
(4)
where the numerator is the average number of y atoms in the shell between Rand R + dR, and the denominator normalizes the distribution so that gXY = 1 when (N) is the same as expected from the bulk density of y atoms py(Ny/ V). There is no ambiguity in interpreting the peaks in the rdf's as representing structure relative to the bulk. Furthermore, the higher first peak in the Na-O rdf for THF states that there is more structuring of the first shell in THF relative to bulk THF than in water or methanol. However, this results largely from the lower bulk density of oxygens in THF since the molar volumes are in the order THF > CH30H > H20. A related point is that the apparent areas of the peaks can be misinterpreted. Although the first peak in the Na-O rdf is broader and higher for THF, integration to obtain the coordination numbers requires evaluating f 47T'rpygxy(r)dr and yields nearly the same result for all three solvents.
The computed coordination number for Na+ in both water and methanol is exactly six, identical to the value obtained by Mezei and Beveridge for the aqueous solution. 7 The first peaks in the Na-O, Na-C1 and Na-C2 distributions in THF integrate to a slightly smaller value of 5.7. The figure for water is also in exact agreement with x-ray diffraction results for dilute aqueous solutions of NaBF4 • 25,26 Values of 5-6 for Na+ in methanol have been derived from mobility measurements via Stokes' law, although the interpretation of results from this method is controversial. 27.28 Similar stUdies of dipolar aprotic solvents yield a lower coordination number although no data are available for THF. 27 However, an investigation of the solvation of Na+ by THF has been carried out using NMR. 29 When THF is added to a solution of sodium tetrabutylaluminate in cyclohexane, the THF proton signals are shifted due to the complexation with sodium ion. A plot of the chemical shifts as a function of THF concentration shows a break corresponding to a THF: salt ratio of 4: 1 suggesting the formation of a four coordinated species. However, this experiment does not rule out the formation of higher coordinated clusters nOr is it immune from complications due to aggregation and ion pairing, though this has not been fully recognized. 30
The second maximum in the Na-O rdf's occur at the same distance (- 4.5 A) for water and methanol. Interestingly, the height and width of the second peak is greater for water than for methanol in spite of the smaller molar volume of the former. The second peaks in the Na-H rdf's (Fig. 3) also exhibit the same pattern. Integration shows the second peak contains only six solvent molecules in the methanolic solution, but 21-25 solvent molecules for water. The large number of water
molecules reflects the differences in the interfac-ing of the first shell of solvent molecules into the hydrogen bonded networks of the bulk (vide infra). In both the methanolic and aqueous solutions this interfacing is efficient; however, the average number of hydrogen bonds per monomer in the bulk is greater for water than for methanol, - 3.6 vs 1. 7. Similarly the number of potential sites for hydrogen bonds protruding from the first shells is greater for water. To accommodate these sites and the greater solvent-solvent hydrogen bonding necessitates a more substantial second layer for water. Thus, as found previously,10 solvent-solvent interactions dominate the structure of the second layers because there are so many of them relative to the number of ion-solvent interactions. The Na-O rdf for THF also shows a well defined second peak. The separation between the first two layers is greater in this case which is consistent with the lack of strong solvent-solvent attractions like hydrogen bonding between the first two shells and with the larger size of the monomers. Integration of the peak indicates the presence of 9-11 monomers in the second layer for THF.
The first peak in the Na-C1 rdf in the THF solution occurs at the same position as the Na-C peak in the methanolic solution (3.4 A). A well defined first peak is also observed in the Na-C2 rdf of the THF solution with a maximum at 4.6 A. The relative positions of the first peaks for the rdf's with sodium clearly demonstrate that the oxygens of the solvent molecules in the first shell are oriented towards the ion in all three solutions. Interestingly, this alignment persists in the second solvent layer as seen from the similar variation in the second peak pOSitions. Although this result could be interpreted as evidence for the long range influence of the ion, solvent-solvent interactions also lead to the same orientation. Efficient interfaCing of the first shell molecules with the bulk through hydrogen bonding would force the same alignment for second shell monomers. In the case of THF, the preference of oxygen atoms to stay near C1 atoms due to Coulombic attraction is also consistent with this order.
Stereoplots of random configurations of the aqueous and THF solutions are provided in Figs. 5 and 6 for a detailed view of the solvation shells around the ion. For clarity only the first two solvent shells are shown in each case. Just as in the methanolic solution,10 the first shell of the aqueous solution consists of six solvent molecules in a roughly octahedral arrangement about the ion. Although the oxygen atom in each water monomer is closest to the ion, the dipole axes of the monomers are not always aligned along the O-Na direction. This orientation permits waters in the first shell to act both as hydrogen bond donors and acceptors with second shell molecules. The second shell contains a large number of molecules which are clearly seen to be engaged in hydrogen bonding among themselves as well as with the first shell water molecules. The six THF molecules in the first shell are seen to be coordinated unsymmetrically to the ion in Fig. 6. Five of the solvent molecules are closer to the ion in an approximately square pyramidal arrangement with the sixth THF
J. Chem. Phys., Vol. 77, No. 10, 15 November 1982
Downloaded 08 Jun 2013 to 141.161.91.14. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions

J. Chandrasekhar and W. L. Jorgensen: Solutions of sodium ion 5085
~A ~ \r---------(/l H 0
I
;-----'1 I il ~.
I~T__---_{/
FIG. 5. Stereoplots showing the first and second solvation shells from one configuration in the simulation of Na+ in water.
molecule along the vacant position in the octahedron at a longer distance from the ion. The rings in the basal plane are seen to be staggered, suggesting solvent-solvent repulsions in the first shell are important. A few monomers in the second shell are found to be oriented with their oxygens nearest the ion, while a few others seem to be orienting to benefit from the attractive O-Cl interactions with molecules in the first shell.
To check whether the calculated coordination numbers remain static or rather represent averages of distributions, the distributions for neighbors within the range of the first peaks in the Na-O rdf's were constructed. The analysis was performed using configurations saved at intervals of 2.5 K during the simulations. It is found that sodium ion is always six coordinated in water as it is in methanol; no significant occurrence of other coordination numbers is found during the simulations. On the other hand, the coordination number of Na+ in THF was found to fluctuate between five (30%) and six (70%). The exchange of solvent molecules in and out of the first shell is apparent from the nonzero minimum between the first and second maxima in the Na-O rdf. Such exchange is virtually absent in the aqueous and methanolic solutions for runs of the present length.
C. Solute-solvent energetics
The total bonding energy distributions for the solute are shown in Fig. 7. The unimodal curves for water and methanol cover 40 kcaI/mol ranges and are consistent with continuous distributions over one basic environment for the ion. The distribution for the methanoHc solution is somewhat more attractive in accordance with the calculated average solute-solvent interaction energy (Table I). On the other hand, the bonding energy distribution for Na+ in THF is far less attractive. The plateau and structure near the maximum of the distribution indicate the presence of at least two distinctly different environments for the ion in this solution, reinforcing the results obtained from the preceding analysis
of coordination numbers.
The energy pair distributions for the ion-solvent interactions are illustrated in Fig. 8. The low energy bands represent the interaction of the ion with the solvent molecules in the first shell. The spikes near 0 kcaI/mol arise from the interactions between the ion and the many distant solvent molecules in the bulk. For the aqueous solution only these two features are present, a broad peak between - 25 and - 12 kcallmol and another one between -10 and 8 kcaI/mol. The first peak integrates to six solvent molecules as expected. The interaction of the ion with the second solvent shell is immersed in the second peak. Interestingly, quite a few water molecules interact repulsively with the sodium ion, contributing to the width of the spike. Integration of this feature from + 2 and + 8 kcaI/mol yields seven solvent monomers probably belonging to the second solvent shell. This reflects that solvent-solvent hydrogen bonding is the dominant interaction beyond the first shell. This is also apparent in the computed energy pair distribution for the methanolic solution. The first peak again integrates to six molecules. The attractive ion-molecule interactions with the second solvation shell are clearly discernible as a shoulder near - 6 kcaI/mol. Monomers having repulsive interactions with the ion are also present but are fewer than in the aqueous solution. Since the hydrogen bonding network is less extensive in methanol compared to water, fewer molecules are forced into unfavorable orientations with respect to the ion.
The first peak in the energy pair distribution for the THF solution is sharp but tails off gradually from -16 to - 5 kcaI/mol. The spike near 0 kcal/mol is particularly narrow and encompasses all solvent molecules other than those in the first shell. In view of the large ion-molecule distances involved for the second shell the interactions are not more attractive than - 6 kcal/ mol (cf. Figs. 1 and 2). Additionally, since the solvent-
\\-- ------ ---(/1 I cmq I
I ~ I
i J1r ep: I
)-~------" I -------------~
1
• qj\(I I
I i ifep I , t---.~--. . / \ Ie" ___ _
FIG. 6. stereoplots showing the first and second solvation shells from one configuration in the simulation of Na+ in THF.
J. Chern. Phys., Vol. 77, No. 10, 15 November 1982
Downloaded 08 Jun 2013 to 141.161.91.14. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions

5086 J. Chandrasekhar and W. L. Jorgensen: Solutions of sodium ion
B~NOING ENERGY DISTRIBUTI~NS .08 r---------~~~~~~~~~~------------~
.06 z .:l .... t- MeOH u
~ .04 ........... / /- ,..-" .... , .... ,
I "-, THF
FIG. 7. Computed solutesolvent bonding energy distributions for Na+ in water, methanol, and THF. The ordinate gives the mole fraction of solute with the bonding energy shown on the abscissa. Units for the ordinate are mole fraction per kcal/mol.
/ _ WATER I ,/'
I , w ...J
I .:l / \ lE: I \
I \ .02 / I \ I '\ / I ,
/ ........... JI " '~,/ , o .00 L......<.L----''--__ ---' ____ --L ____ --L~ __ __L_ ____ _L_ ____ ...l..._ __ __...J
-225 -215 -205 -195 -185 -175 BONDING ENERGY
-165
solvent interactions are relatively weak in THF, the solvent monomers are not forced to adopt orientations yielding unfavorable interactions with the ion.
D. Solvent structure
The solvent-solvent rdf's were also computed in the simulations. The 0-0 rdf for the aqueous solution is compared to the result for pure TIPS2 water in Fig. 9. The structure of liquid water has been discussed at length previously so the earlier work can be consulted for details. 5,6 For the present purposes the key point from Fig. 9 is that introduction of sodium ion has negligible influence on the structure of water as measured by the 0-0 rdf. The computed O-H and H-H rdf's are also essentially identical for the solution and pure solvent. The results for the pure liquid can be found in Ref. 5. Consequently, it is apparent that the hydrogen bonding in water is not Significantly affected at low ion concentrations. The same situation was found to prevail in the methanolic solution as well. 10
Only molecules in the first solvent shell are likely to have contacts different from the rest and hence may
2.00
~ I'
1.50 I' I'
II) (\ I \ w ...J a I ~ \ ~1.00 I \ 10 E
D I z: I .50
I I
0.00 -28
HATER MeOH THF
-16 -10 -4 INTERACTION ENERGY
I
I' I' I' \1 I I \ I I \ \ \ \ \ \ \ ,
2 8
FIG. 8. Computed solute-solvent energy pair distributions for Na+ in water, methanol, and THF. The ordinate records the number of solvent molecules bound to the ion with the energy given on the abscissa. Units for the ordinate are molecules per kcal/mol.
-155 -145
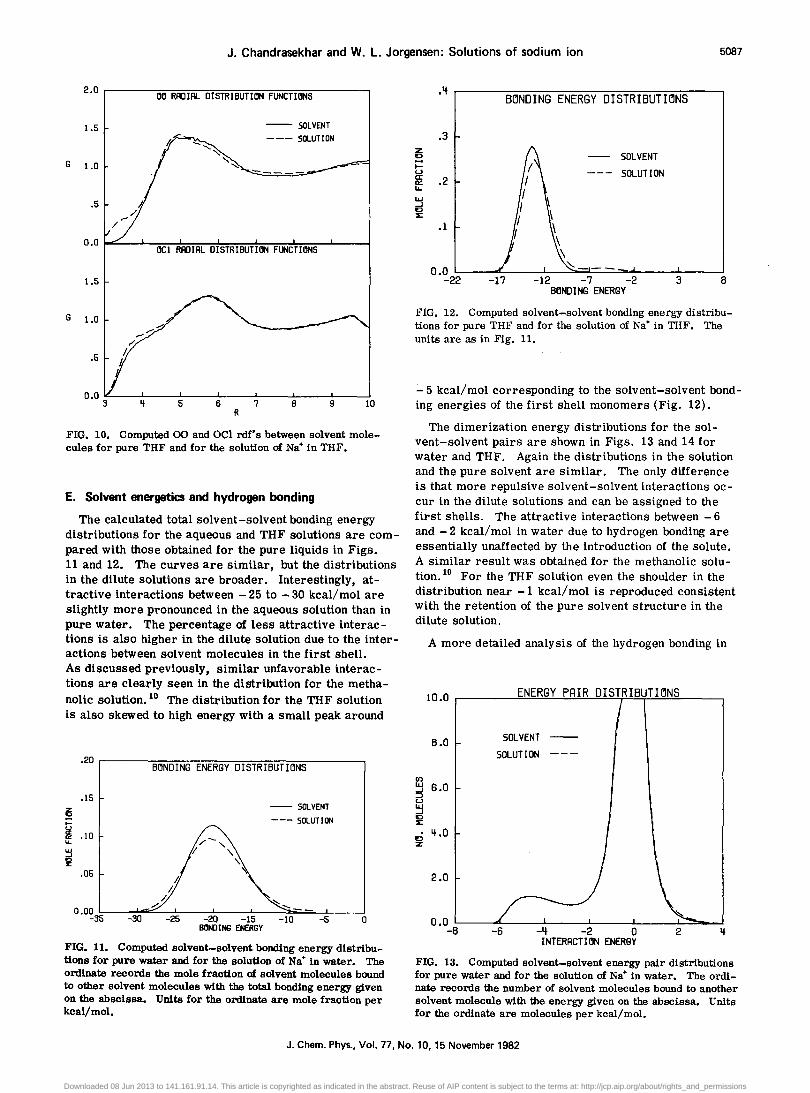
alter the solvent-solvent rdf's. However, there are only six molecules in the first shell and their contributions to these rdf's are overwhelmed by the many bulk monomers for water and methanol. Interestingly, the presence of the first solvation shell does show up as shoulders in the 0-0 and 0-C1 rdf's in the THF solution (Fig. 10). Although the contributions from the first shell are small, the features are discernible because the rdf's are otherwise nearly zero at these distances for THF. In contrast, the 0-0 rdf between 3 and 4 'A is Significant even in the absence of first shell-first shell contacts in water and methanol. The latter are thus submerged in the total rdf in these two cases.
Even for the THF solution the presence of the ion is only apparent in the additional short 0-0 and 0-C1 contacts. The remainders of these rdf's and the other rdf's (0-C2, C1-C1, C1-C2, and C2-C2) are essentially indistinguishable for the solution and pure solvent. The latter four rdf's are displayed for pure THF in the accompanying paper. 12 In addition, it should be noted that the conformational results, i. e., the phase angle and puckering amplitude distributions, for the THF monomers in the dilute solution and the pure liquid are also identical. Thus, the ion has negligible effect on the structure of the solvent beyond the first shell in all three solvents. Mezei and Beveridge arrived at a similar conclusion in their study of Na+ in water. 7
00 RADIAL DISTRIBUTION FUNCTIONS
3
-- SOLVENT
G 2 --- SOLUTION
O'--__ __L_~J~~--~--~~--~--~~~ 1 2 3 ~ 5 6 7 8
R
FIG. 9. Computed 00 rdf's between solvent molecules for pure water and for the solution of Na+ in water.
J. Chem. Phys., Vol. 77, No. 10, 15 November 1982
Downloaded 08 Jun 2013 to 141.161.91.14. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions
J. Chandrasekhar and W. L. Jorgensen: Solutions of sodium ion 5087
2.0 ~~ RADIAL DISTRIBUTION FUNCTI~NS
1.5 -- SOLVENT
--- SOLUTION
G 1.0 -----.5
0.0 ~I RADIAL OISTRIBUTI~N FUNCTI~NS
1.5
G 1.0
• 5
0.0 3 'I 5 6 7 B 9 10
R
FIG. 10. Computed 00 and OCl rdf's between solvent molecules for pure THF and for the solution of Na+ in THF.
E. Solvent energetics and hydrogen bonding
The calculated total solvent-solvent bonding energy distributions for the aqueous and THF solutions are compared with those obtained for the pure liquids in Figs. 11 and 12. The curves are Similar, but the distributions in the dilute solutions are broader. Interestingly, attractive interactions between - 25 to - 30 kcal/mol are slightly more pronounced in the aqueous solution than in pure water. The percentage of less attractive interactions is also higher in the dilute solution due to the interactions between solvent molecules in the first shell. As discussed previously, similar unfavorable interactions are clearly seen in the distribution for the methanolic solution. 10 The distribution for the THF solution is also skewed to high energy with a small peak around
.20 BONDING ENERGY DISTRIBUTIONS
.15
~ 0-
-- SOLVENT
--- SOLUTION !i .10 '" "-W ..J
"" z: .05
0.00 -35 -30 -25 -20 -15 -10 -5 o
BONDING ENERGY
FIG. 11. Computed solvent-solvent bonding energy distributions for pure water and for the solution of Na+ in water. The ordinate records the mole fraction of solvent molecules bound to other solvent molecules with the total bonding energy given on the abscissa. Units for the ordinate are mole fraction per kcal/mol.
z D
~ U a: 0:: lL.
lLI ...J D ::I:
.4
.3
.2
.1
B~NDING ENERGY DISTRIBUTI~NS
SOLVENT SOLUTION
0.0 ~ ___ L-__ ~~~~~~A-__ ~_~
-22 -17 -12 -7 -2 3 8 BONDING ENERGY
FIG. 12. Computed solvent-solvent bonding energy distributions for pure THF and for the solution of Na+ in THF. The units are as in Fig. 11 •
- 5 kcal/mol corresponding to the solvent-solvent bonding energies of the first shell monomers (Fig. 12).
The dimerization energy distributions for the solvent-solvent pairs are shown in Figs. 13 and 14 for water and THF. Again the distributions in the solution and the pure solvent are similar. The only difference is that more repulsive solvent-solvent interactions occur in the dilute solutions and can be assigned to the first shells. The attractive interactions between - 6 and - 2 kcal/mol in water due to hydrogen bonding are essentially unaffected by the introduction of the solute. A similar result was obtained for the methanolic solution. 10 For the THF solution even the shoulder in the distribution near -1 kcal/mol is reproduced consistent with the retention of the pure solvent structure in the dilute solution.
A more detailed analysis of the hydrogen bonding in
10.0 ENERGY PAIR DISTRIBUTI~NS
B.O SOLVENT SOLUTION
rn lLI S.O ...J :;) U lLI ...J D ::I:
.0 4.0 z
2.0
0.0 -8 -S -4 -2 a 4
INTERACTION ENERGY
FIG. 13. Computed solvent-solvent energy pair distributions for pure water and for the solution of Na+ in water. The ordinate records the number of solvent molecules bound to another solvent molecule with the energy given on the abscissa. Units for the ordinate are molecules per kcal/mol.
J. Chern. Phys., Vol. 77, No. 10, 15 November 1982
Downloaded 08 Jun 2013 to 141.161.91.14. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions

5088 J. Chandrasekhar and W. L. Jorgensen: Solutions of sodium ion
the aqueous solution was carried out using the configurations saved during the simulation. The usual energetic definition of a hydrogen bond corresponding to the position of the minimum in solvent-solvent energy pair distribution was used. Thus, any pair of water molecules with an interaction below - 2. 25 kcal/mol is considered to be hydrogen bonded. The average number of hydrogen bonds per monomer was computed as a function of distance from the sodium ion for 1 A thick shells between 1. 5 and 7. 5 A from the ion. The results are compared with a similar analysis performed on the methanolic solution with an energetic criterion of - 2.375 kcal/mol for hydrogen bonding in Fig. 15. Horizontal lines at 3.6 and 1. 7 hydrogen bonds correspond to the average value in pure water and methanol, respectively. In the aqueous solution, the solvent molecules in the first solvation shell with Na-Q distances in the range 2-3.5 A participate in a significantly fewer number of hydrogen bonds, - 2. 4. The second shell monomers at distances of 3.5-5. 5 A form practically the same number of hydrogen bonds as the bulk solvent molecules. There is no region in the solution apart from the first solvation shell in which the solvent structure is Significantly broken. In fact, the number of hydrogen bonds per water molecule seems to be higher around 6 A from the ion, before the value reaches that of the pure solvent. A very similar result is found in the methanolic solution. The first shell monomers form fewer hydrogen bonds, the second shell actually more hydrogen bonds than the bulk, and the value smoothly approaches the pure solvent limit at distances farther away from the ion.
Figure 15 provides no evidence for the presence of the interfacial B region proposed by Frank and Wen for aqueous electrolyte solutions. 31,32 The first solvation shell (A region) smoothly blends into the bulk without an interfacial region. The existence of negative B viscosity coefficients is often cited as evidence for structure broken regions in aqueous electrolyte solutions. How-
25~~E~N~E~RG~Y_PwA~I~R~D~I~ST~R~IB~U~T£IO~N~S __ ~
I
20 SOLVENT --
SOLUTION ---
15
10
5
I I I I I I I I I I I I I I L ~6~----~~----~-~2----~0~--~2----~q
INTERACTION ENERGY
FIG. 14. Computed solvent-solvent energy pair distributions for pure THF and the solution of Na+ in THF. Units are as in Fig. 13.
5.00.---------------------------~
en 3.75
~ I
;:c:
~2.50
! 1.25
WATER
HeOH
O.OO+-----~----r_----._--~r_--~
2 4 6 8 10 12 R
FIG. 15. The average number of hydrogen bonds for each solvent molecule as a function of distance from the ion in the solutions of Na+ in water and methanol. The horizontal lines at 3. 6 and 1. 7 correspond to the average numbers of hydrogen bonds calculated for monomers in pure liquid water and methanol, respectively.
ever, the B viscosity coefficient is positive for Na+ in water33 as well as in methanol. 27 The present result is also in complete accord with the conclusion of Mezei and Beveridge who found no disruption of water by Na+ beyond the A region on the basis of quasicomponent distribution function analyses. 7
IV. CONCLUSION
Two key conclusions are drawn from the present computations. First, the heat of solution of a monoatomic cation in a basic solvent is composed of two large contributions of opposite sign. The ion-molecule interaction energy is tempered by a positive solvent reorganization energy. As a result, the heats of solution of Na+ in solvents with considerably different polarity and dielectric constants are nearly of the same magnitude.
Secondly, the solvent reorganization is almost entirely associated with the formation of the first solvation shell around the ion. In the aqueous and methanolic solutions the hydrogen bonded networks dominate the liquid structure beyond the first shell and they are virtually unaffected by the presence of the ion. In the THF solution, sodium ion is well buried inside the first shell of solvent molecules and due to the size of the monomers the molecules beyond the first shell are too far from the ion to be significantly influenced by it.
ACKNOWLEDGMENTS
Gratitude is expressed to the National Science Foundation (CHE80-20466) for support of this work. The authors are also grateful to Dr. Phillip Cheeseman for use of his stereo plotting program.
lJ. F. Garst, in Solute-Solvent Interactions, edited by J. F. Coetzee and C. D. Ritchie (Marcel Dekker, New York, 1969), p. 539.
2 Water: A Comprehensive Treatise, edited by F. Franks (Plenum, New York, 1973), Vol. 3.
J. Chern. Phys., Vol. 77, No. 10,15 November 1982
Downloaded 08 Jun 2013 to 141.161.91.14. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions

J. Chandrasekhar and W. L. Jorgensen: Solutions of sodium ion 5089
:Ie. Reichardt, in Organic Liquids, edited by A. D. Buckingham, E. Lippert, and S. Bratos (WUey-Interscience, New York, 1978), p. 269.
'Computer Modeling of Matter, edited by P. G. Lykos (American Chemical Society, Washington, D. C., 1978).
5W. L. Jorgensen, J. Chern. Phys. 77, 4156 (1982). 6For a review, see, D. L. Beveridge, M. Mezei, P. K.
Mehrotra, F. T. Marchese, G. Ravi-Shanker, T. Vasu, and S. Swaminathan, Adv. Chern. (in press).
7M• Mezei and D. L. Beveridge, J. Chern. Phys. 74, 6902 (1981).
8(a) S. Swaminathan, S. W. Harrison, and D. L. Beveridge, J. Am. Chern. Soc. 100, 5705 (1978); (b) P. K. Mehrotra and D. L. Beveridge, ibid. 102, 4287 (1980); (c) J. C. Owicki and H. A. Scheraga, ibid. 99, 7413 (1977); (d) D. C. Rapaport and H. A. Scheraga, J. Phys. Chern. 86, 873 (1982); (e) G. Bolis and E. Clementi, Chern. Phys. Lett. 82, 147 (1981); (f) G. Bolis, .G. Corongiu, and E. Clementi, ibid. 86, 299 (1982); (g) G. Alagona and A. Tani, ibid. 87, 337 (1982); (h) K. Nakanishi, S. Okazaki, K. Ikari, and H. Touhara, ibid. 84, 428 (1981); (i) S. Okazaki, K. Nakanishi, H. Touhara, and N. Watanabe, J. Chern. Phys. 74, 5863 (1981); (j) K. Heinzinger and P. C. Vogel, ibid. 31, 463 (1976); (k) P. C. Vogel and K. Heinzinger, ibid. 31, 476 (1976); (1) G. Palinkas, W. O. Riede, and K. Heinzinger, ibid. 32, 1137 (1977); (m) M. R. Mruzik, F. F. Abraham, D. E. Schreiber, and C. M. Pound, ibid. 64, 481 (1976).
Bw. L. Jorgensen and J. D. Madura, J. Am. Chern. Soc. (submitted for publication).
tOW. L. Jorgensen, B. Bigot, and J. Chandrasekhar, J. Am. Chern. Soc. (in press).
l1W. L. Jorgensen, J. Am. Chern. Soc. 103, 341 (1981); W. L. Jorgensen and M. Ibrahim, ibid. 104, 373 (1982).
t2J. Chandrasekhar and W. L. Jorgensen, J. Chern. Phys. 77, 5073 (1982).
t:Jw. L. Jorgensen and M. Ibrahim, J. Am. Chern. Soc. 103, 3976 (1981); W. L. Jorgensen, ibid. 103, 4721 (1981).
14J • C. Owicki, ACS Symp. Ser. 86, 159 (1978).
15n. Cremer and J. A. Pople, J. Am. Chern. Soc. 97, 1354 (1975).
IBw. L. Jorgensen, J. Am. Chern. Soc. 103, 335 (1981). t1W. L. Jorgensen and M. Ibrahim, J. Am. Chern. Soc. 103,
3976 (1981). t8S. F. Smith, J. Chandrasekhar, and W. L. Jorgensen, J.
Phys. Chern. 86, 3308 (1982). t9R. H. Staley and J. L. Beauchamp, J. Am. Chern. Soc. 97,
5920 (1975). 2~. Kistenmacher, H. Popkie, and E. Clementi, J. Chern.
Phys. 59, 5842 (1973). 2tW. L. Jorgensen, Chern. Phys. Lett. (in press). 22J. E. Desnoyers and C. Jolicoeur, in Modern Aspects of
Electrochemistry, edited by J. O'M, Bockris and B. E. Conway (Plenum, New York, 1969), Vol. 5, p. 1; D. R. Rosseinsky, Chern. Rev. 65, 467 (1965).
230. Matsuoka, E. Clementi, and M. Yoshimine, J. Chern. Phys. 64, 1351 (1976).
2'G. Choux and R. L. Benoit, J. Am. Chern. Soc. 91, 6221 (1969).
25G. W. Neilson and J. E. Enderby, Annu. Rep. Prog. Chern. Sec. C 76, 185 (1979).
26A. I. Ryss and I. V. Radchenko, J. struct. Chern. 5, 489 (1964).
27J. Padova, in Water and Aqueous Solutions, edited by R. A. Horne (Wiley-Interscience, New York, 1973), p. 109.
2~. Ulich, Trans. Faraday Soc. 23, 388 (1927). 29E. Schaschel and M. C. Day, J. Am. Chern. Soc. 90, 503
(1968). 3OW. F. Edgell, in Infrared and Raman Spectroscopy Part A,
edited by E. G. Brame, Jr. and J. Grasselli (Dekker, New York, 1976), Chap. 4, p. 279.
3tH. S. Frank and W. -Yo Wen, Discuss. Faraday Soc. 24, 133 (1957).
32R. W. Gurney, Ionic Processes in Solution (McGraw-Hill, New York, 1953).
33F. J. MUlero, in Water and Aqueous Solutions, edited by R. A. Horne (Wiley-Interscience, New York, 1973), p. 519.
J. Chern. Phys., Vol. 77, No. 10,15 November 1982
Downloaded 08 Jun 2013 to 141.161.91.14. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions