the p38mapk inhibitor sb203580 alleviates ultraviolet-induced phosphorylation at serine 389 but not...
TRANSCRIPT

TUb
DG*SMN
R
tdUnicspcmsmpppwTps
t
ttagmhli
O
Biochemical and Biophysical Research Communications 261, 464–471 (1999)
Article ID bbrc.1999.1023, available online at http://www.idealibrary.com on
0CA
he p38MAPK Inhibitor SB203580 Alleviatesltraviolet-Induced Phosphorylation at Serine 389ut Not Serine 15 and Activation of p53
avid Keller,* Xiaoya Zeng,* Xiaorong Li,* Mini Kapoor,† Mihail S. Iordanov,‡ Yoichi Taya,§uillermina Lozano,† Bruce Magun,‡ and Hua Lu*,1
Department of Biochemistry and Molecular Biology, ‡Department of Cell and Development Biology, Oregon Healthciences University, 3181 SW Sam Jackson Park Road, Portland, Oregon 97201; †University of Texas,.D. Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, Texas 77030; and §Biology Division,ational Cancer Center Research Institute, Tsukiji 5-1-1, Chou-ku, Tokyo, Japan
eceived June 22, 1999
irradiation differentially induce phosphorylation atdpp(3i(ptstAppeptri
Mpttsptipliwwdw
Phosphorylation of p53 at serine 389 has been showno be responsive uniquely to UV but not gamma irra-iation. This report describes identification of theV-responsive p38MAPK protein as a serine 389 ki-ase. The immunoprecipitated p38MAPK from UV-
rradiated murine embryonic testicular carcinoma F9ells phosphorylated the serine 392 residue but noterine 15 of the human p53 protein in vitro and thishosphorylation was inhibited by a p38MAPK-specifichemical inhibitor SB203580. The inhibitor also re-arkably alleviated the UV-caused induction and
erine 389 but not serine 15 phosphorylation of theurine p53 protein in vivo. Subsequently, this com-
ound suppressed transcriptional activity of p53 andartially retarded UV-induced apoptosis. Moreover,53 bound to p38 as revealed by immunoprecipitationith anti-p53 antibodies from UV-treated F9 cells.hus, these results suggest that UV-stimulated p53hosphorylation at serine 389 is mediated by thetress-responsive p38MAPK. © 1999 Academic Press
Key Words: p53; p38 MAPK; serine 389 phosphoryla-ion; UV irradiation.
In response to various genotoxic reagents, the p53umor suppressor protein inhibits tumorigenesis byriggering either programmed cell death or cell growthrrest (1). These reagents, including ultraviolet (UV) oramma (g) irradiation, activate p53 through multipleechanisms, one of which is phosphorylation (2–4). Itas been shown that phosphorylation or dephosphory-
ation of p53 is modulated in response to UV or grradiation (5–9). The two types of DNA damaging
1 Corresponding author. Fax: (503)-494-8393. E-mail: [email protected].
464006-291X/99 $30.00opyright © 1999 by Academic Pressll rights of reproduction in any form reserved.
ifferent serines of p53. g irradiation induces serine 15hosphorylation (5, 6), whereas UV stimulates phos-horylation at both the N- or C-terminal serines of p538–11). One unique site for UV is serine 389 out of the90 amino acids of the murine p53 protein (correspond-ng to serine 392 of human p53 with 393 amino acids)8, 9). Although many kinases have been reported tohosphorylate p53 in vitro or in vivo (4), only a few ofhem appear to be responsible for DNA damage respon-ive p53 phosphorylation. For instance, phosphoryla-ion at serine 15 is mediated by ATM and/or its relativeTR after g irradiation (12–15), while JNK1 was re-orted to phosphorylate the N-terminal serine 34 of53 (10, 11). UV-specific serine 389 phosphorylationnhances the sequence-specific DNA binding activity of53 in vitro (16) and is important for transcrip-ional activation in vivo (17). However, the kinase(s)esponsible for this phosphorylation has not yet beendentified.
One of the major UV responsive pathways is theKKK-MAP kinase cascade (18), in which JNK1 and
38MAPK are UV-activated. Similar to JNK1 (19),he activated p38MAPK phosphorylates some nuclearranscriptional activators (20–23), with different sub-trate preferences (24). Also, p38MAPK promotes apo-tosis and/or cell growth arrest (25, 26). This is similaro the UV-induced phenotype of p53 activation, lead-ng us to study the possible relationship between38MAPK and p53. Using immunoprecipitation fol-owed by kinase assays with anti-p38MAPK antibod-es, p38MAPK from UV-irradiated F9 or Tera-2 cellsas found to phosphorylate the serine 392 of humanild type p53 in vitro. This phosphorylation was re-uced by a pyridinyl imidazole compound SB203580,hich is a p38MAPK-specific inhibitor (27). In vivo,

this drug also abrogated phosphorylation at serine 389bisFsTor
M
StNSNT1bbipE
dpMaTwccsT
c1tpT
hcps
omifiwol
alwfPbmp
the drug and substrates used). The reaction mixtures were incubatedalt
ptupwsFCM
flmepmp(dp
R
niappUdpsinpicdwpawwr1Mldrp(Fsr
Vol. 261, No. 2, 1999 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
ut not serine 15 and thus activation of p53 by UVrradiation. Consistently, it inhibited the p53 tran-cription activity as measured by luciferase assays.inally, p38MAPK bound to p53 in vivo as demon-trated by immunoprecipitation of UV-treated cells.hus, these results suggest that p38MAPK may be onef the UV-stimulated kinases targeting the serine 389esidue of p53.
ATERIALS AND METHODS
Reagents and buffers. The pyridinyl imidazole compoundB203580 was purchased from CalBiochem, Inc (CA). Western blot-ing lysis buffer is composed of 50 mM Tris/HCl (pH 8.0), 0.5%P-40, 5 mM EDTA, 2 mM DTT, 150 mM NaCl and 0.2 mM PMSF.NNTE is composed of 50 mM Tris/HCl (pH 7.4), 5 mM EDTA, 1%P-40, 500 mM NaCl and 5% sucrose. RIPA is comprised of 50 mMris/HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 0.1% SDS, and% (w/v) sodium deoxycholate. Immunoprecipitation-kinase lysisuffer is 100 mM Hepes-KOH (pH 7.4), 2 mM EGTA, 50 mM-glycerophosphate, 10% glycerol, and 1% Triton X-100. LiCl buffer
s 500 mM LiCl, 100 mM Tris/HCl (pH 7.6), and 0.1% Triton X-100.38MAPK assay buffer is 10 mM Mops (pH 7.2), 25 mM MgCl2, 2 mMGTA, 0.1% Triton X-100, and 1 mCi [g-32P]ATP.
Plasmids and antibodies. pGST-Elk1 and pGSTcJun were asescribed (28). The pBP100-Luc plasmid containing a luciferase re-orter gene driven by two copies of p53RE motif derived from theDM2 promoter was as described (29). Polyclonal rabbit anti-p38
nd anti-JNK1 antibodies were purchased from Santa Cruz (CA).he polyclonal anti-p38 antibody (#12850) for immunoprecipitationas provided by Gary Johnson (30). Antibodies aS389 or aS15 spe-
ifically against the phosphorylated serine 389- or serine 15-ontaining p53 C- or N-terminal peptides were generated as de-cribed (5, 8, 9). The anti-p53 antibody Pab421, Pab248, and anti-ag antibody Pab419 were as described (31).
Cell lines and cell culture. Human teratoma tera-2 cells wereultured in Dulbecco’s modified Eagle medium supplemented with0% fetal bovine serum, 50 units/ml penicillin, and 0.1 mg/ml strep-omycin at 37°C in a 5% CO2 atmosphere. DMEM without sodiumyruvate was used for murine testicular carcinoma F9 cells. F9 andera-2 cells contain wild type p53.
Preparation of F9 or Tera-2 nuclear extracts and purification ofuman p53 and mouse alternate splice p53 proteins. F9 or Tera-2ell nuclear extracts were prepared and purification of human53 and mouse alternate splice p53 proteins were purified as de-cribed (32).
UV irradiation and chemical treatment. UV irradiation (20 J/m2)f cells was carried out using the 254 nm source of a UV transillu-inator (UVP, Inc, Upland, CA), as described (9, 30). Cells were
ncubated with different concentrations of SB203580 as indicated ingure legends for two h prior to UV irradiation. Postirradiation, cellsere harvested either at different time points as indicated in figuresr 8 h afterward for immunoprecipitation-Western blot, kinase oruciferase analyses as described below.
Immunoprecipitation-kinase assay. Immunoprecipitation-kinasessays were performed using described methods (30, 32). Briefly, cellysates containing approximately 500 mg of proteins were incubatedith 3 ml of polyclonal rabbit anti-38 antibodies at 4°C degree for 1 h,
ollowed by incubating with 30 ml of protein A-agarose (50% slurry,harmacia) for 1 h. Agarose beads were then washed with lysisuffer once, LiCl buffer once, and kinase assay buffer once. Reactionixtures were in 20 ml assay buffer including the substrates in the
resence or absence of SB203580 (see figure legends for amounts of
465
t 30°C degree for 30 min and stopped by addition of protein sampleoading buffer. Phosphorylated protein signals were detected by au-oradiography after analysis on an SDS-gel.
Immunoprecipitation-Western blot. Western blot analysis waserformed as described (9). Proteins from either immunoprecipita-ion (500 mg) or straight cell lysates (100 mg) of the UV-treated orntreated cells were separated by SDS-polyacrylamide gel electro-horesis and transferred to nitrocellulose membrane. The membraneas immunoprobed with Pab421, aS389 and aS15, respectively as
hown in Fig. 1, or with polyclonal anti-p38 or anti-p53 antibodies forig. 5. Signals were detected by the ECL reagents (Santa Cruz Inc,A). The relative quantification of p53 levels was conducted usingodel GS-700 Imaging Densitometer (BioRad).
Transient transfection and luciferase assay. F9 cells were trans-ected with a pCMV-b-galactoside reporter plasmid (0.5 mg) and auciferase reporter plasmid (1 mg) driven by two copies of the p53RE
otif derived from the MDM2 promoter (33) or by the SV-40nhance/promoter (Promega), using GenePORTER (GTS, CA). 40 hosttransfection, cells were incubated with SB203580 (10 mM, 20M) or an equal volume of DMSO (used for dissolving this com-ound) as a control. 2 h afterward, the cells were irradiated with UV20 J/m2) and harvested 7 h after irradiation for luciferase assays asescribed (34). Luciferase activity was statistically calculated andlotted in graph using CA-Cricket Graph III.
ESULTS
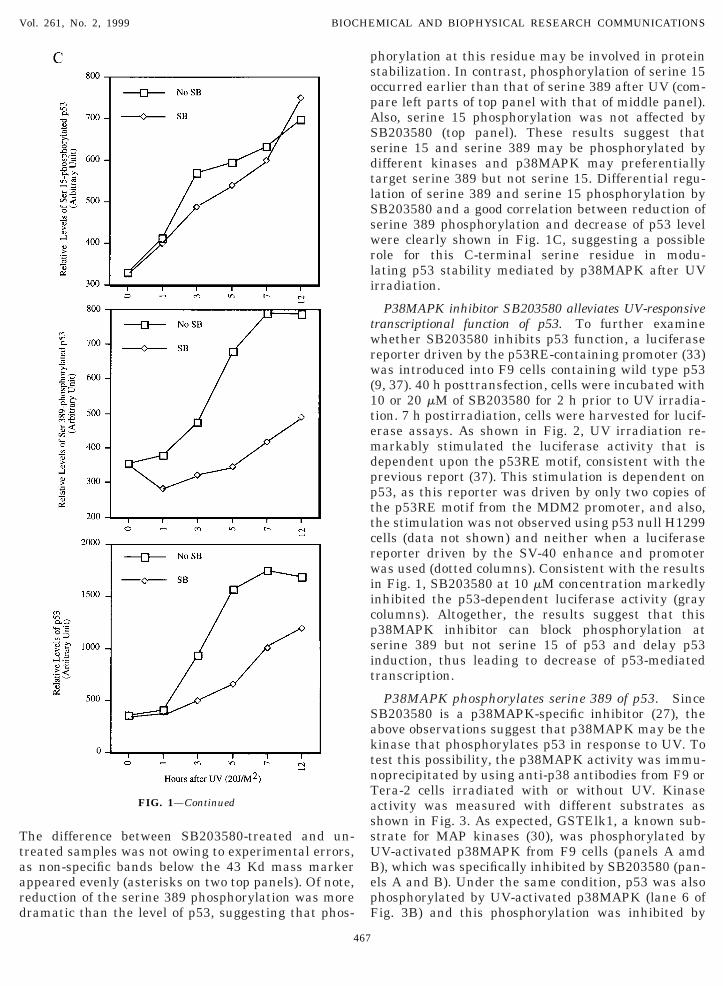
p53 induction and phosphorylation at serine 389 butot serine 15 were repressed by a p38MAPK-specificnhibitor SB203580. Because phosphorylation of p53t serine 389 is uniquely responsive to UV (8, 9) and38MAPK is activated by UV (24), we tested whether53 is a functional downstream component for thisV-responsive MAP kinase. A group of pyridinyl imi-azole compounds was shown to specifically inhibit38MAPK-mediated phosphorylation of several sub-trates with various IC50 values (20, 24, 27, 35). Thisnhibition further impairs the MKKK-p38MAPK sig-aling in response to UV irradiation (36). Thus, one38MAPK-specific inhibitor SB203580 was employedn our study. Murine testicular carcinoma F9 cells werehosen for this test because p53 response to UV irra-iation has been characterized in these cells (37). Cellsere treated with 1 and 10 mM of SB203580 for two hrior to irradiation with UV (20 J/m2). 8 h postirradi-tion, cells were harvested for Western blot analysisith anti-p53 antibody Pab421 and aS389. Consistentith our previous reports (8, 9), p53 level was induced
emarkably after UV irradiation (middle panel of Fig.A) and so was serine 389 phosphorylation (top panel).arkedly, SB203580 reduced induction of either p53
evel or phosphorylation at serine 389 in a dose-ependent manner (lanes 4–6 of top two panels). Thiseduction was specific to p53, as equal amounts of the38MAPK protein were detected in all three lanesbottom panel). p53 level was quantified in a graph ofig. 1B, which clearly indicated that the p38MAPK-pecific inhibitor at the concentration of 10 mM canetard UV-stimulated p53 induction, suggesting that

pw
adWa
epswrms
(wmlsaraa
Vol. 261, No. 2, 1999 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
38MAPK may mediate serine 389 phosphorylation asell as p53 activation.We also tested whether serine 15 phosphorylation is
lso affected by SB203580. F9 cells were treated asescribed above, but harvested at different time points.estern blot analysis was conducted with anti-p53
ntibodies Pab421, aS389 and aS15, respectively. As
FIG. 1. p38MAPK-specific inhibitor SB203580 retards UV-stimuA) 100 mg of F9 cell lysates after treatment with SB203580 and UVere detected by Western blot with aS389, Pab421 and ap38 as inolecular mass markers in Kd (same in all the following figures). The
evels of p53 were quantified in an arbitrary unit and plotted as a grerines 389 and 15 after UV irradiation in the presence or absencefter UV exposure were directly loaded onto a 10% SDS-gel. Protespectively, as indicated. Asterisks indicate non-specific bands crosss described in Materials and Methods. The relative levels of p53 anrbitrary unit and plotted as graphs in this panel.
466
xpected (8, 9) and shown in the left part of Fig. 1B,53 level (bottom panel) as well as phosphorylation oferine 389 (middle panel) and serine 15 (top panel)ere induced gradually after UV. In line with the
esult of Fig. 1A, serine 389 phosphorylation wasarkedly reduced within the period of first seven h and
o was the p53 induction (middle and bottom panels).
ed serine 389 but not serine 15 phosphorylation and p53 induction.indicated on top were directly loaded onto a 10% SDS-gel. Proteinsted. SB denotes SB203580. Numbers on the left of the gel denote
iddle blot of panel A was scanned using a densitometer. The relativeon right. (B) Time-course of p53 induction and phosphorylation at
B203580 (10 mM). 100 mg of F9 cell lysates of different time pointss were detected by Western blot with aS15, aS389 and Pab421,acting with the antibodies. (C) All the blots of panel A were scannedts phosphorylation at serine 389 or serine 15 were quantified in an
latas
dicam
aphof Sein-red i
Ttaard
phorylation at this residue may be involved in proteinsopASsdtlSswrli
twrw(1temdppttcrwiicpsit
SaktnTassUBepF
Vol. 261, No. 2, 1999 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
he difference between SB203580-treated and un-reated samples was not owing to experimental errors,s non-specific bands below the 43 Kd mass markerppeared evenly (asterisks on two top panels). Of note,eduction of the serine 389 phosphorylation was moreramatic than the level of p53, suggesting that phos-
FIG. 1—Continued
467
tabilization. In contrast, phosphorylation of serine 15ccurred earlier than that of serine 389 after UV (com-are left parts of top panel with that of middle panel).lso, serine 15 phosphorylation was not affected byB203580 (top panel). These results suggest thaterine 15 and serine 389 may be phosphorylated byifferent kinases and p38MAPK may preferentiallyarget serine 389 but not serine 15. Differential regu-ation of serine 389 and serine 15 phosphorylation byB203580 and a good correlation between reduction oferine 389 phosphorylation and decrease of p53 levelere clearly shown in Fig. 1C, suggesting a possible
ole for this C-terminal serine residue in modu-ating p53 stability mediated by p38MAPK after UVrradiation.
P38MAPK inhibitor SB203580 alleviates UV-responsiveranscriptional function of p53. To further examinehether SB203580 inhibits p53 function, a luciferase
eporter driven by the p53RE-containing promoter (33)as introduced into F9 cells containing wild type p53
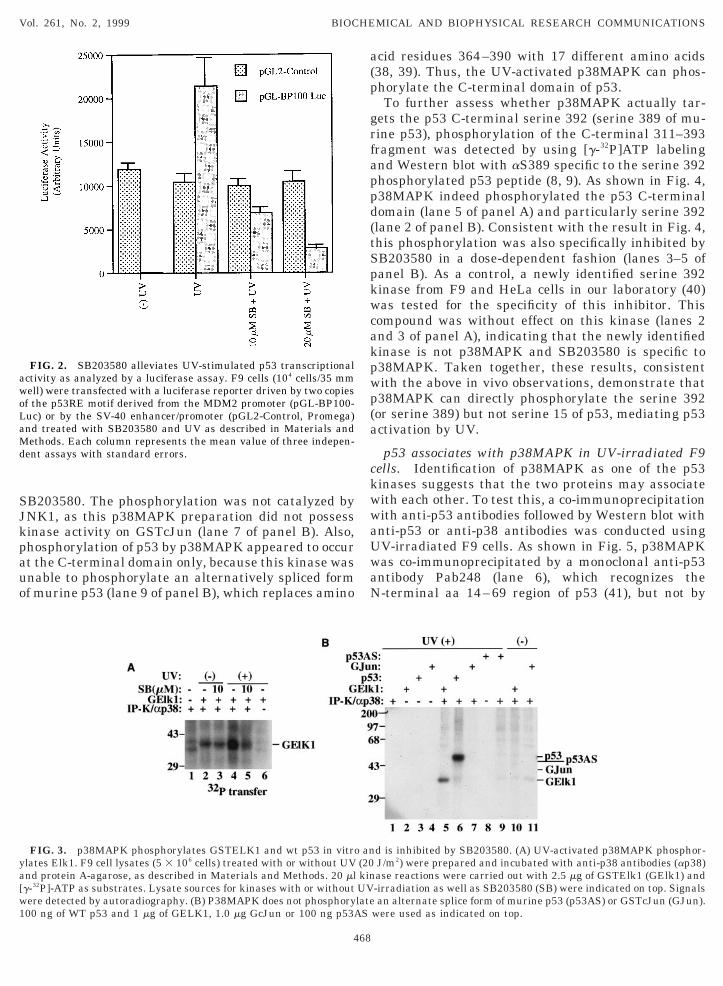
9, 37). 40 h posttransfection, cells were incubated with0 or 20 mM of SB203580 for 2 h prior to UV irradia-ion. 7 h postirradiation, cells were harvested for lucif-rase assays. As shown in Fig. 2, UV irradiation re-arkably stimulated the luciferase activity that is
ependent upon the p53RE motif, consistent with therevious report (37). This stimulation is dependent on53, as this reporter was driven by only two copies ofhe p53RE motif from the MDM2 promoter, and also,he stimulation was not observed using p53 null H1299ells (data not shown) and neither when a luciferaseeporter driven by the SV-40 enhance and promoteras used (dotted columns). Consistent with the results
n Fig. 1, SB203580 at 10 mM concentration markedlynhibited the p53-dependent luciferase activity (grayolumns). Altogether, the results suggest that this38MAPK inhibitor can block phosphorylation aterine 389 but not serine 15 of p53 and delay p53nduction, thus leading to decrease of p53-mediatedranscription.
P38MAPK phosphorylates serine 389 of p53. SinceB203580 is a p38MAPK-specific inhibitor (27), thebove observations suggest that p38MAPK may be theinase that phosphorylates p53 in response to UV. Toest this possibility, the p38MAPK activity was immu-oprecipitated by using anti-p38 antibodies from F9 orera-2 cells irradiated with or without UV. Kinasectivity was measured with different substrates ashown in Fig. 3. As expected, GSTElk1, a known sub-trate for MAP kinases (30), was phosphorylated byV-activated p38MAPK from F9 cells (panels A amd), which was specifically inhibited by SB203580 (pan-ls A and B). Under the same condition, p53 was alsohosphorylated by UV-activated p38MAPK (lane 6 ofig. 3B) and this phosphorylation was inhibited by
SJkpauo
acid residues 364–390 with 17 different amino acids(p
grfappd(tSpkwcakpwp(a
ckwwaUwaN
awoLaMd
ya[w1
Vol. 261, No. 2, 1999 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
B203580. The phosphorylation was not catalyzed byNK1, as this p38MAPK preparation did not possessinase activity on GSTcJun (lane 7 of panel B). Also,hosphorylation of p53 by p38MAPK appeared to occurt the C-terminal domain only, because this kinase wasnable to phosphorylate an alternatively spliced formf murine p53 (lane 9 of panel B), which replaces amino
FIG. 2. SB203580 alleviates UV-stimulated p53 transcriptionalctivity as analyzed by a luciferase assay. F9 cells (104 cells/35 mmell) were transfected with a luciferase reporter driven by two copiesf the p53RE motif derived from the MDM2 promoter (pGL-BP100-uc) or by the SV-40 enhancer/promoter (pGL2-Control, Promega)nd treated with SB203580 and UV as described in Materials andethods. Each column represents the mean value of three indepen-
ent assays with standard errors.
FIG. 3. p38MAPK phosphorylates GSTELK1 and wt p53 in vitrolates Elk1. F9 cell lysates (5 3 106 cells) treated with or without UVnd protein A-agarose, as described in Materials and Methods. 20 mlg-32P]-ATP as substrates. Lysate sources for kinases with or withoutere detected by autoradiography. (B) P38MAPK does not phosphory00 ng of WT p53 and 1 mg of GELK1, 1.0 mg GcJun or 100 ng p53A
468
38, 39). Thus, the UV-activated p38MAPK can phos-horylate the C-terminal domain of p53.To further assess whether p38MAPK actually tar-
ets the p53 C-terminal serine 392 (serine 389 of mu-ine p53), phosphorylation of the C-terminal 311–393ragment was detected by using [g-32P]ATP labelingnd Western blot with aS389 specific to the serine 392hosphorylated p53 peptide (8, 9). As shown in Fig. 4,38MAPK indeed phosphorylated the p53 C-terminalomain (lane 5 of panel A) and particularly serine 392lane 2 of panel B). Consistent with the result in Fig. 4,his phosphorylation was also specifically inhibited byB203580 in a dose-dependent fashion (lanes 3–5 ofanel B). As a control, a newly identified serine 392inase from F9 and HeLa cells in our laboratory (40)as tested for the specificity of this inhibitor. This
ompound was without effect on this kinase (lanes 2nd 3 of panel A), indicating that the newly identifiedinase is not p38MAPK and SB203580 is specific to38MAPK. Taken together, these results, consistentith the above in vivo observations, demonstrate that38MAPK can directly phosphorylate the serine 392or serine 389) but not serine 15 of p53, mediating p53ctivation by UV.
p53 associates with p38MAPK in UV-irradiated F9ells. Identification of p38MAPK as one of the p53inases suggests that the two proteins may associateith each other. To test this, a co-immunoprecipitationith anti-p53 antibodies followed by Western blot withnti-p53 or anti-p38 antibodies was conducted usingV-irradiated F9 cells. As shown in Fig. 5, p38MAPKas co-immunoprecipitated by a monoclonal anti-p53ntibody Pab248 (lane 6), which recognizes the-terminal aa 14–69 region of p53 (41), but not by
d is inhibited by SB203580. (A) UV-activated p38MAPK phosphor-J/m2) were prepared and incubated with anti-p38 antibodies (ap38)ase reactions were carried out with 2.5 mg of GSTElk1 (GElk1) and-irradiation as well as SB203580 (SB) were indicated on top. Signalsan alternate splice form of murine p53 (p53AS) or GSTcJun (GJun).
were used as indicated on top.
an(20kinUVlateS

P3cSpTwmtom(
D
asd(Ussppsiwpps
p1p
serine 389 of this tumor suppressor, indicating that theUmbowMibl(pa
pcbepttosf(tsoPyayib4TbnasU
tbamnaSsauti
dlPaoa
Vol. 261, No. 2, 1999 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
ab421 (lane 5), which recognizes the C-terminal aa70–380 region (41), from only the UV-irradiated F9ells (compare lanes 3 with 6). As a negative control, anV40 Tag-specific antibody Pab419 (41) was unable toull down any proteins in either cases (lanes 1 and 4).his result indicates that p38MAPK may associateith the C-terminal domain but not N-terminal do-ain of p53, as Pab421 competed with p38MAPK for
he C-terminus (lane 5), consistent with the abovebservations that p38MAPK phosphorylates the C-ter-inal serine 392 but not N-terminal serine 15 of p53
Figs. 3 and 4).
ISCUSSION
This report documents identification of p38MAPK aspotential p53 upstream player that phosphorylates
erine 389 in a cellular response to UV-caused DNAamage. Supporting this are several lines of evidence:1) A p38MAPK-specific inhibitor SB203580 inhibitedV-responsive phosphorylation at serine 389 but not
erine 15; (2) this compound delayed p53 induction andubsequently inhibited p53 transcription activity andartially impeded apoptosis (data not shown); (3)38MAPK phosphorylated p53 at serine 389 but noterine 15 in vitro and this phosphorylation was specif-cally inhibited by SB203580; (4) p38MAPK associatedith p53 as shown by immunoprecipitation with anti-53 antibodies. Thus, the UV-responsive MAP kinaserobably mediates p53 activation by phosphorylatingerine 389.It is interesting that another MAP kinase partici-
ates in the UV-p53 signaling, in addition to JNK1 (10,1). Unlike JNK1, which phosphorylates serine 34 of53 (10), p38MAPK specifically targets the C-terminal
FIG. 4. p38MAPK phosphorylates the C-terminal serine 392 ofhe human p53. (A) Phosphorylation of the p53 C-terminal domainy P38MAPK is inhibited by SB203580. A similar kinase assay to thebove ones was carried out, but the p53 C-terminal 311–393 frag-ent (100 ng) was used as a substrate. Besides p38MAPK, anotherewly identified kinase temporarily named 392K (100 ng, Keller etl., unpublished data) was also included as a control. The amount ofB203580 used is indicated on top. (B) P38MAPK phosphorylateserine 392, which is also sensitive to SB203580. The same kinasessay as that in panel A was performed except non-labeled ATP wassed. The phosphorylated human p53 C-terminal peptides were de-ected by Western blot with aS389. The amounts of SB203580 arendicated on top.
469
V-p53 signaling is through multiple mechanisms andore complicated than g-p53 signaling which can be
locked by simply deleting the ATM gene (42). The rolef JNK1 appears to be more than phosphorylation, as itas recently reported to bind to and degrade p53 in anDM2-independent fashion when this kinase is in an
nactive (dephosphorylated) form (43). Upon activationy MEKK1 or UV irradiation, JNK1 inversely stabi-izes and activates p53 probably by phosphorylating it11). It would be reasonable to see whether p38MAPKlays a similar role in regulating the stability andctivity of this transcriptional activator.Is p38MAPK the only kinase responsible for phos-
horylating serine 389 of the murine p53 protein inellular response to UV irradiation? The answer woulde no. First, in this study, we found that SB203580ven at higher concentration (20 mM) could not com-letely abolish serine 389 phosphorylation, indicatinghe existence of other potential UV-responsive kinasesargeting this site. In accordance with this possibility,ur lab has recently identified and purified anothererine 392 kinase complex devoid of p38MAPK or CKIIrom F9 or HeLa cells, as analyzed by Western blot40). Also, phosphorylation of p53 by this newly iden-ified kinase was not inhibited by the p38MAPK-pecific inhibitor (Fig. 4A). Hence, there are more thanne kinase targeting serine 389 in response to UV.38MAPK may present an early response by phosphor-lating p53. It is also possible that p38MAPK mayctivate other kinases that in turn directly phosphor-late p53. There are at least four isoforms of p38MAPKdentified thus far (18), among which p38an and p38b2ut not p38g and p38d are sensitive to SB203580 (44,5). Thus, p38an and/or p38b2 may phosphorylate p53.his phosphorylation may be specific to serine 389,ecause this kinase did not phosphorylate the alter-ate spliced form of p53 (Fig. 3B), excluding serine 315nd the N-terminal serines, and phosphorylation aterines 371, 376 and 378 of p53 were not responsive toV (7; our unpublished data).
FIG. 5. p38MAPK interacts with p53 in vivo following UV irra-iation. Lysates (500 mg proteins) from F9 cells either exposed to UVight (10 J/m2) or not were incubated with 2 mg of Pab419, Pab421 orab248 antibodies for immunoprecipitation, as described in Materi-ls and Methods. After transferred to nitrocellulose membrane, p53r p38 was detected by Western blot with polyclonal anti-p53 ornti-p38 antibodies, as indicated at the bottom.

Although phosphorylation at multiple serines of p53ofpbsmcpbdCNTtiorommnotpbsifi
A
sIvp9tr
R
10. Milne, D. M., Campbell, L. E., Campbell, D. G., and Meek, D. W.
1
1
1
1
1
1
1
1
1
2
2
22
2
2
2
2
2
2
3
3
3
3
3
3
Vol. 261, No. 2, 1999 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
ccurs after UV irradiation, how cells coordinate dif-erent kinases in regulating the stability and activity of53 needs to be further studied. One mechanism woulde that this posttranslational modification at differentites provides p53 with different properties or confor-ations such that MDM2, a cellular p53 inhibitor (46),
ould no longer recognize this target. For instance,hosphorylation at serine 15 was shown to preventinding of MDM2 to p53 (5). MDM2-mediated degra-ation of p53 (47, 48) requires both the N- and-termini of both the proteins (47–49), though their-termini directly interact with each other (50, 51).his implicates that other unidentified cellular pro-eins in addition to MDM2 may be involved in regulat-ng p53 stability probably by targeting the C-terminusf p53. Therefore, phosphorylation at the C-terminalesidues may affect targeting of p53 by those moleculesr ubiquitination of p53 (52). Although this notion re-ains to be tested, it is clear that diverse pathwaysediate stress responsive p53 activation (4). This phe-omenon is correlated well with the central importancef this tumor suppressor in monitoring cell growth sohat it is still functional when one mechanism, such ashosphorylation (53, 54), of the activating pathwaysecomes defective. Thus, molecular dissection of theophisticated UV-p53 signaling would provide muchnsight into understanding of cellular networking innely tuning p53 function.
CKNOWLEDGMENTS
We thank Philip Stork, Moshe Oren, Zhijun Luo, and Gary John-on for reagents. We thank Philip Stork for active discussion. M.ordavnov and B. Magun were supported by US Public Health Ser-ice grants (CA-39360 and ES-08456). This work was supportedartly by grants to Hua Lu from the American Cancer Society (RPG-8-191-01-CBE), NIH (R01 CA 79721), Medical Research Founda-ion of Oregon, and Oregon Division of American Cancer Society,espectively.
EFERENCES
1. Levine, A. J. (1997) Cell 88, 323–331.2. Gottlieb, T. M., and Oren, M. (1996) Biochim. Biophys. Acta
1287, 77–102.3. Ko, J. L., and Prives, C. (1996) Genes Dev. 10, 1054–1072.4. Giaccia, A. J., and Kastan, M. B. (1998) Genes Dev. 12, 2973–
2983.5. Shieh, S. Y., Ikeda, M., Taya, Y., and Prives, C. (1997) Cell 91,
325–334.6. Siliciano, J. D., Canman, C. E., Taya, Y., Sakaguchi, K., Appella,
E., and Kastan, M. B. (1997) Genes Dev. 11, 3471–3481.7. Waterman, M. J., Stavridi, E. S., Waterman, J. L., and Hala-
zonetis, T. D. (1998) Nat. Genet. 19, 175–178.8. Kapoor, M., and Lozano, G. (1998) Proc. Natl. Acad. Sci. USA 95,
2834–2837.9. Lu, H., Taya, Y., Ikeda, M., and Levine, A. J. (1998) Proc. Natl.
Acad. Sci. USA 95, 6399–6402.
470
(1995) J. Biol. Chem. 270, 5511–5518.1. Fuchs, S. Y., Adler, V., Pincus, M. R., and Ronai, Z. (1998) Proc.
Natl. Acad. Sci. USA 95, 10541–10546.2. Canman, C. E., Lim, D. S., Cimprich, K. A., Taya, Y., Tamai, K.,
Sakaguchi, K., Appella, E., Kastan, M. B., and Siliciano, J. D.(1998) Science 281, 1677–1679.
3. Banin, S., Moyal, L., Shieh, S. Y., Taya, Y., Anderson, C. W.,Chessa, L., Smorodinsky, N. I., Prives, C., Reiss, Y., Shiloh, Y.,and Ziv, Y. (1998) Science 281, 1674–1677.
4. Tibbetts, R. S., Brumbaugh, K. M., Williams, J. M., Sarkaria,J. N., Cliby, W. A., Shieh, S. Y., Taya, Y., Prives, C., and Abra-ham, R. T. (1998) Genes Dev. 13, 152–157.
5. Khanna, K. K., Keating, K. E., Kozlov, S., Scott, S., Gatei, M.,Hobson, K., Taya, Y., Gabrielli, B., Chan, D., Less-Miller, S. P.,and Lavin, M. F. (1998) Nature Genetics 20, 398–400.
6. Hupp, T. R., and Lane, D. P. (1995) J. Biol. Chem. 270, 18165–18174.
7. Hao, M., Lowy, A. M., Kapoor, M., Deffie, A., Liu, G., and Lozano,G. (1996) J. Biol. Chem. 271, 29380–29385.
8. Dhanasekaran, N., and Reddy, E. P. (1998) Oncogene 17, 1447–1455.
9. Holbrook, N. J., and Fornace, A. J., Jr. (1991) New Biol. 3,825–833.
0. Raingeaud, J., Gupta, S., Rogers, J. S., Dickens, M., Han, J.,Ulevitch, R. J., and Davis, R. J. (1995) J. Biol. Chem. 270,7420–7426.
1. Tan, Y., Rouse, J., Zhang, A., Cariati, S., Coehn, P., and Comb,M. J. (1996) EMBO J. 15, 4629–4642.
2. Wang, X., and Ron, D. (1996) Science 272, 1347–1349.3. Han, J., Jiang, Y., Li, Z., Kravchenko, V. V., and Ulevitch, R. J.
(1997) Nature 386, 296–299.4. Derijard, B., Raingeaud, J., Barrett, T., Wu, I. H., Han, J.,
Ulevitch, R. J., and Davis, R. J. (1995) Science 267, 682–685.5. Xia, Z., Dichens, M., Raingeaud, J., Davis, R. J., and Greenberg,
M. E. (1995) Science 270, 1326–1331.6. Lavoie, J. N., L’Allemain, G., Brunet, A., Muller, R., and Poys-
segur, J. (1996) J. Biol. Chem. 271, 20608–20616.7. Lee, J. C., Laydon, J. T., McDonnell, P. C., Gallagher, T. F.,
Kumar, S., Gree, D., McNutty, D., Blumenthal, M. J., Heys, J. R.,Landvatter, S. W., Strickler, J. E., McLaoughlin, M. M., Sie-mens, I. R., Fisher, S. M., Livi, G. P., White, J. R., Adams, J. C.,and Young, P. R. (1994) Nature 371, 739–746.
8. Iordanov, M. S., Pribnow, D., Magun, J. L., Dinh, T., Pearson,J. A., Chen, S. L., and Magun, B. E. (1997) Mol. Cell. Biol. 17,3373–3381.
9. Rothe, J., Dobbelstein, M., Freedman, D. A., Shenk, T., andLevine, A. J. (1998) EMBO J. 17, 554–564.
0. Iordanov, M. S., Pribnow, D., Magun, J. L., Dinh, T., Pearson,J. A., and Magun, B. E. (1998) J. Biol. Chem. 273, 15794–15803.
1. Zeng, X. Y., Chen, L. H., Jost, C. A., Maya, R., Keller, D., Wang,X., Kaelin, W. C., Jr., Oren, M., Chen, J. D., and Lu, H. (1999)Mol. Cell. Biol. 19, 3257–3266.
2. Lu, H., Fisher, R., Bailey, P., and Levine, A. J. (1997) Mol. Cell.Biol. 17, 5923–5934.
3. Wu, X., Bayle, J. H., Olson, D., and Levine, A. J. (1993) Genes &Dev. 7, 1126–1132.
4. Zeng, X. Y., Xiaorong, Li, Miller, A., Wu, C., Kwok, R., Goodman,R., and Lu, H. (1999b). submitted.
5. Kramer, R. M., Roberts, E. F., Um, S. L., Borsch-Haulold, A. G.,Watson, S. P., Fisher, M. J., and Jakubowski, J. A. (1996) J. Biol.Chem. 271, 27723–27729.

36. Assefa, Z., Vantieghem, A., Declercq, W., Vandenabeele, P., Van-
33
3
4
4
4
4
4
45. Goedert, M., Cuenda, A., Craxton, M., Jackes, R., and Cohen, P.
4
4
4
4
5
5
55
5
Vol. 261, No. 2, 1999 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
denheede, J. R., Merlevede, W., Witte, P. D., and Agostinis, P.(1999) J. Biol. Chem. 274, 8788–8796.
7. Lutzker, S., and Levine, A. J. (1996) Nature Med. 2, 804–810.8. Han, K. A., and Kulesz-Martin, M. F. (1992) Nucleic Acids Res.
20, 1979–1981.9. Bayle, J. H., Elenbaas, B., and Levine, A. J. (1995) Proc. Natl.
Acad. Sci. USA 92, 5729–5733.0. Keller, D., Zeng, X. Y., Kapoor, M., Taya, Y., Lozano, G., and Lu,
H. (1999) Unpublished study.1. Harlow, E., Crawford, L., Pim, D. C., and Williamson, N. M.
(1981) J. Virol. 39, 861–869.2. Kasten, M. B., Zhan, Q., El-Diary, W., Carrier, F., Kacks, T.,
Walsh, W. V., Plunkett, B. S., Vogelsteins, B., and Fornace, A. J.(1992) Cell 71, 587–597.
3. Fuchs, S. Y., Adler, V., Buschmann, T., Yin, Z., Wu, X. W., Jones,S. N., and Ronai, Z. (1999) Genes Dev. 12, 2658–2663.
4. Cuenda, A., Rouse, J. R., Doza, Y. N., Meier, R., Cohen, P.,Gallagher, T. F., Young, P. R., and Lee, J. C. (1995) FEBS Lett.364, 229–231.
471
(1997) EMBO J. 16, 3563–3571.6. Momand, J., Zambetti, G. P., Olson, D. C., George, D., and
Levine, A. J. (1992) Cell 69, 1237–1245.7. Haupt, Y., Maya, R., Kazaz, A., and Oren, M. (1997) Nature 387,
296–299.8. Kubbutat, M. H. G., Jones, S. N., and Vousden, K. H. (1997)
Nature 387, 299–303.9. Kubbutat, M. H., Ludwig, R. L., Ashcroft, M., and Vousden, K. H.
(1998) Mol. Cell Biol. 18, 5690–5698.0. Chen, J., Marechal, V., and Levine, A. J. (1993) Mol. Cell Biol.
13, 4107–4114.1. Cho, Y., Gorina, S., Jeffrey, P. D., and Pavletich, N. P. (1994)
Science 265, 346–355.2. Honda, R., Tanaka, H., and Yasuda, H. (1997) FEBS Lett. 420, 25–27.3. Ashcroft, M., Kubbutat, M. H., and Vousden, K. H. (1999) Mol.
Cell Biol. 19, 1751–1758.4. Blattner, C., Tobiasch, E., Litfen, M., Rahmsdorf, H. J., and
Herrlich, P. (1999) Oncogene 18, 1723–1732.