the receptor binding site of feline leukemia virus surface
TRANSCRIPT

JOURNAL OF VIROLOGY,0022-538X/98/$04.0010
Apr. 1998, p. 3268–3277 Vol. 72, No. 4
Copyright © 1998, American Society for Microbiology
The Receptor Binding Site of Feline Leukemia Virus SurfaceGlycoprotein Is Distinct from the Site Involved in
Virus NeutralizationI. K. RAMSEY,* N. SPIBEY, AND O. JARRETT
Department of Veterinary Pathology, University of Glasgow, Bearsden, Glasgow G61 1QH, United Kingdom
Received 23 July 1997/Accepted 19 December 1997
The external surface glycoprotein (SU) of feline leukemia virus (FeLV) contains sites which define the viralsubgroup and induce virus-neutralizing antibodies. The subgroup phenotypic determinants have been locatedto a small variable region, VR1, towards the amino terminus of SU. The sites which function as neutralizingepitopes in vivo are unknown. Recombinant SU proteins were produced by using baculoviruses that containedsequences encoding the SUs of FeLV subgroup A (FeLV-A), FeLV-C, and two chimeric FeLVs (FeLV-215 andFeLV-VC) in which the VR1 domain of FeLV-A had been replaced by the corresponding regions of FeLV-Cisolates. The recombinant glycoproteins, designated Bgp70-A, -C, -215, and -VC, respectively, were similar totheir wild-type counterparts in several immunoblots and inhibited infection of susceptible cell lines in asubgroup-specific manner. Thus, Bgp70-A interfered with infection by FeLV-A, whereas Bgp70-C, -VC, and-215 did not. Conversely, Bgp70-C, -VC, and -215 blocked infection with FeLV-C, while Bgp70-A had no effect.These results indicate that the site on SU which binds to the FeLV cell surface receptor was preserved in therecombinant glycoproteins. It was also found that the recombinant proteins were able to bind naturallyoccurring neutralizing antibodies. Bgp70-A, -VC, and -215 interfered with the action of anti-FeLV-A neutral-izing antibodies, whereas Bgp70-C did not. Furthermore, Bgp70-C interfered with the action of anti-FeLV-Cneutralizing antibodies, while the other proteins did not. These results indicate that the neutralizing epitope(s)of FeLV SU lies outside the subgroup-determining VR1 domain.
Feline leukemia virus (FeLV) is a major cause of degener-ative and malignant diseases in domestic cats. The envelope ofFeLV, which is a typical type C retrovirus, is studded with atransmembrane protein (TM) which anchors an external sur-face glycoprotein (SU). The FeLV SU (gp70) is the target ofvirus-neutralizing antibodies and the site of the initial virus-host cell interaction (14, 19, 35, 37, 39).
Three subgroups of FeLV (A, B, and C) have been definedon the basis of interference with superinfection (39). FeLVsubgroup A (FeLV-A) is found in all cases of FeLV infection(13). It is antigenically monotypic (36) and is believed to beresponsible for interhost transmission. FeLV-B is found inapproximately 33% of FeLV-positive cats that are otherwisehealthy (13). Available sequence data and experimental evi-dence suggest that FeLV-B arises by recombination with en-dogenous FeLV (22, 24, 26, 41). FeLV-C isolates are rare andoccur only in association with FeLV-A or with FeLV-A andFeLV-B (13). FeLV-C isolates are uniquely associated with thedevelopment of pure erythrocyte aplasia, which is one of themost acute degenerative retroviral diseases known (20, 25).FeLV-C is thought to arise by mutation from FeLV-A (22),although the prototypic FeLV-C strain FeLV-C/Sarma con-tains additional sequences, probably derived from endogenousFeLV. Rigby (32) demonstrated by site-directed mutagenesisthat the determinants of the superinfection interference phe-notype of FeLV-C were located within variable region 1 (VR1)(Fig. 1). The disease-causing and infectivity phenotypes are,however, not completely associated with this small region, andother sequence differences, possibly near VR5, may play a rolein the generation of these phenotypes (33).
The existence of three FeLV subgroups with different hostranges has traditionally been taken as evidence of the presenceof three cellular receptors, one for each subgroup (4, 11, 30, 33,39). Recently, however, some doubt has been cast on thisassumption (28). The evidence for three subgroups was origi-nally augmented by neutralization data (39). Subsequently,others found that a degree of cross-neutralization occurredbetween the subgroups, with several isolates of FeLV-C beingindistinguishable from FeLV-A in neutralization assays (36).However, FeLV-C/Sarma can be distinguished from FeLV-A,although not from an isolate of FeLV-B (FeLV-B/ST), by thistechnique. These results suggested that the neutralizationepitope(s) of FeLV SU may be distinct from the subgroup-determining regions.
This paper describes the production of recombinant surfaceglycoproteins of FeLV by using the baculovirus expressionvector system (18). In vitro experiments with these proteinsdemonstrated that they were able to block virus replication andinhibit the activity of neutralizing antibodies. Using chimericFeLV-A/C proteins, we demonstrate that the neutralizingepitopes of FeLV are distinct from the subgroup-determiningregions.
MATERIALS AND METHODS
Virus strains, plasmid vectors, and cell lines. The FeLV strains used wereFeLV-A/Glasgow 1 (15), FeLV-B/Snyder-Theilen (ST) (39), FeLV-C/Sarma(39), and FeLV-C/FA27C (25). The proviral clones of FeLV-A and FeLV-C,called pFGA and pFSC, respectively, and proviral clones of chimeric A/C FeLVscreated by site-directed mutagenesis, called pVC and pV215, were obtained fromM. Rigby. Both of these chimeric viruses have an FeLV-A backbone but containa small region near the 59 end of an FeLV-C specific sequence which confersmany of the properties of FeLV-C (33). The sequences of the two FeLV-Cisolates used in the production of these chimeras are very similar in this small 59region, differing by only one amino acid residue (isoleucine for methionine inpV215).
Autographa californica multiple nuclear polyhedrosis virus (AcMNPV) was
* Corresponding author. Mailing address: Department of ClinicalVeterinary Medicine, University of Cambridge, Cambridge CB3 0ES,United Kingdom. Phone: (44)-1223-337621. Fax: (44)-1223-330848.
3268
on January 31, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

obtained from N. Stow (Institute of Virology, Glasgow, United Kingdom). ViralDNA was obtained as described elsewhere (18). The baculovirus transfer vectorpAcLacZ1 was provided by R. Possee (NERC Institute of Virology, Oxford,United Kingdom). This vector contains 59 and 39 polyhedrin flanking sequences,between which is inserted a BamHI cloning site and polyhedrin promoter as wellas the lacZ gene under the control of the baculovirus p10 promoter. Recombi-nant viruses produced by using this vector express b-galactosidase. The vectorwas modified by the insertion of a multiple cloning site containing unique PstI,SmaI and XbaI recognition sequences into the BamHI cloning site of the originalvector. The original BamHI cloning site was also preserved, and the resultingvector was called pAcLacMCS.
The QN10S feline cell line, used for the assay of FeLV, was maintained asdescribed elsewhere (12, 27). Infections of QN10S cells with FeLV were per-formed by using 12-well tissue culture trays containing 22-mm-diameter wells.The cells were subcultured the previous day and seeded at a density of 4 3 104
per well. The virus was inoculated into 1 ml of cell medium containing 4 mg ofPolybrene per ml. The cells were incubated at 37°C for 2 h with occasionalagitation, and the inoculum was then replaced with fresh medium. Foci oftransformed cells were counted at 6 days postinfection.
The F422 (29) and FL74 (17) cell lines were used as sources of purified FeLVfor immunological assays. Large quantities of the cells were grown in rollerflasks, and the viral particles from the culture fluid were concentrated by ultra-filtration followed by centrifugation in a 20 to 50% sucrose gradient. The prep-arations of purified virus were lysed with 1% Triton X-100 on ice for 30 min,diluted with an equal volume of Tris-buffered saline (TBS), and stored at 220°Cuntil used.
The Sf9 cell line was used for the production of recombinant baculoviruses andproteins. The cells were maintained without CO2 supplementation at 28°C inTC100 medium (Gibco) with 2 mM glutamine, 100 IU of penicillin per ml, 100mg of streptomycin per ml, and 10% fetal calf serum. For the production ofrecombinant proteins, the cells were maintained in a serum-free insect cellmedium (SF900 II; Gibco) after infection with the recombinant virus.
Sera and antibodies. Rabbit 709 serum was provided by G. Reid. The serumwas produced by immunization of a rabbit with FeLV glycoproteins purified bylentil lectin affinity chromatography. The serum reacts well with native FeLV SUin Western blots.
Immune cat sera P11 and P12 were derived from cats that had recovered fromnatural contact infection with FeLV-A/Glasgow 1 in the experiments ofMadewell and Jarrett (21). P11 serum had an FeLV-neutralizing titer of 128against FeLV-A and reacted specifically with FeLV-A SU, but not with FeLV-B/ST SU or FeLV-C/Sarma SU, in enzyme-linked immunosorbent assays(ELISAs). P12 serum is similar to P11 except that its FeLV-A-neutralizing titer
is slightly higher (256). Cat serum 31 was derived from an experimental infectionwith biologically cloned FeLV-C/Sarma. The neutralizing-antibody titers of thisserum were 16 against FeLV-A/Glasgow-1, 16 against FeLV-B/ST, and 1,024against FeLV-C/Sarma.
The monoclonal antibody 3-17 was donated by K. Weijer (European Veteri-nary Laboratory). This antibody neutralizes all FeLV subgroups in vitro (45).One of us (N.S.) demonstrated that the antibody recognizes a region coveringamino acid residues 204 to 208 of FeLV-A SU, which has been shown to be aneutralizing epitope present in all FeLV subgroups (6). Peptides of this regioninduce virus-neutralizing antibodies in rabbits but not in cats (23, 46). Theantibody was conjugated to alkaline phosphatase (AP) to give antibody 3-17AP.
Immunostaining. Polyacrylamide gel electrophoresis was performed underreducing conditions on 7.5% or 5 to 20% gradient gels. The proteins wereelectroblotted onto nylon or nitrocellulose membranes which were then incu-bated overnight in a blocking solution (TBS plus 3% skim milk powder plus 10%goat serum) and then washed several times in phosphate-buffered saline (PBS)with 0.05% Tween 20 (PBS-Tween). The membrane was incubated for 2 h withfresh blocking solution (to which the serum or monoclonal antibody, 0.2%[vol/vol] 0.5 M EDTA, and 0.5% [vol/vol] Tween 20 had been added). If a secondantibody was to be used, then the membrane was briefly washed in PBS-Tweenbefore being incubated with a similarly prepared solution for a further hour. Allsuch antibodies were conjugated with AP. The membrane was then washedseveral times with PBS-Tween and was finally washed in AP buffer (200 mMNaCl, 10 mM MgCl2, 100 mM Tris [pH 9.5]). The blots were developed byincubation with a substrate solution containing 1.65 mg of bromochloroin-dolylphosphate (BCIP) and 3.3 mg of nitroblue tetrazolium (NBT) in 10 ml of APbuffer.
ELISA for FeLV SU. An ELISA was used for the rapid detection of recom-binant SU as described elsewhere (27). Briefly, a microtiter plate was coated witha 1:1,000 dilution of rabbit 709 serum. The plate was then washed twice with TBSplus 0.05% Tween 20 (TBS-Tween) and blocked for 1 h with blocking solution.The antigens to be tested were diluted with 1% Empigen in TBS, added to theplate, and left for 3 h. A further two washes were followed by the addition of a1:1,000 dilution of 3-17AP in blocking solution. The plate was then washedthoroughly with TBS-Tween before addition of 50 ml of a 1-mg/ml solution of4-nitrophenyl phosphate in AP buffer to each well and incubation for approxi-mately 4 h. The reaction was stopped with 50 ml of 0.4 M NaOH, and the resultwas read at 405 nm.
Production of recombinant baculoviruses. All DNA manipulations were per-formed as described elsewhere (38). The oligonucleotide primers CCCGGATCCCTGCAGGACCAACCACC and AGGAGGTAATACCCGGTAGTGGTAGTGGTAGTGACTGGGCCCGCG were used to generate PCR fragments of the
FIG. 1. Variable regions of FeLV and recombinant surface glycoproteins. This diagram shows the predicted peptide sequences of the four recombinant proteinsproduced during this study. Above the peptide sequences is a single line representing the FeLV-A/Glasgow-1 sequence and showing the positions of the variable regions(VR) of FeLV SU (as defined by Rigby [32]) as shaded areas. The unshaded box represents the neutralizing epitope that is recognized by the monoclonal antibodiesC11D8 (6, 8) and 3-17 (40a, 47). This epitope is common to all FeLV sequences used in this diagram. The numbers refer to the predicted amino acid residues of theenvelope polyprotein precursor of FeLV-A/Glasgow-1 (41). Note that the C-terminal end of the transmembrane protein (the anchoring domain) is absent in therecombinant proteins. L, leader peptide.
VOL. 72, 1998 FeLV RECEPTOR BINDING AND VIRUS NEUTRALIZATION SITES 3269
on January 31, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

FIG. 2. Analysis of recombinant FeLV SUs. To analyze the four recombinantproteins and compare their immunoreactivities with those of their wild-typecounterparts, several sodium dodecyl sulfate–7.5% polyacrylamide gels were run,and immunoblotting was performed. (a) Nitrocellulose membrane probed with a1:600 dilution of 3-17AP. (b) Similar membrane probed with rabbit 709 anti-gp70serum diluted 1:500. The membrane was incubated with a goat anti-rabbit im-munoglobulin G–AP conjugate (Bio-Rad). (c) SPF rabbit serum used as a con-trol. (d) Similar membrane probed with a 1:100 dilution of the immune cat serumP11. A goat anti-cat immunoglobulin G–AP conjugate was used to detect bind-ing. (e) SPF cat serum used as a control. Control protein preparations of wild-type-infected and uninfected supernatants (C61 and C5L respectively), as well astwo FeLV-positive controls, were included in all blots. Lanes: 1, FL74; 2, F422;3, C6L; 4, C5L; 5, 6, 7, and 8, recombinant FeLV SUs Bgp70-215, -VC, -C, and-A, respectively; 9, molecular mass markers.
3270 RAMSEY ET AL. J. VIROL.
on January 31, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from
env genes of the proviral clones pFGA, pV215, and pVC. A similar fragment wasamplified from pFSC by using a different 39 primer (AGGAGGTAGTACCCGGTAGTGGTAGTGGTAGTGACTGGGCCCGCG). These fragments includ-ed all of the SU and the TM sequences as far as the ApaI site in FeLV-A env(nucleotide 1838) (41), which code for the first 147 amino acids at the aminoterminus, but did not include the anchoring transmembrane region. The frag-ments had six histidine codons at their carboxy termini that were intended toallow the use of a nickel-nitrilotriacetic acid adsorbent to affinity purify theproteins from the supernatants of infected cells (8). Following BamHI/SmaIdigestion, the fragments were all cloned directly into a preparation of BamHI/SmaI-digested pAcLacMCS which had been purified by agarose gel electro-phoresis followed by electroelution. The recombinant plasmids were purified ona cesium chloride gradient. To distinguish between the recombinant transfervectors, each was digested with EcoRI, HindIII, AccI, ClaI, ApaI, BglII, andKpnI. The VR1 regions of the transfer vectors and parent plasmids were alsodirectly sequenced (Sequenase; Stratagene). The results of these experimentswere in accordance with predictions based on available sequence data. Figure 1illustrates the predicted peptide sequences of the recombinant proteins that wereproduced from these cloned PCR fragments.
The recombinant pAcLacMCS transfer vectors were cotransfected by calciumphosphate precipitation with wild-type baculovirus DNA into Sf9 insect cells(18). Once numerous polyhedrin crystals could be seen, the medium was har-vested and centrifuged at 3,500 rpm for 10 min, and the supernatant fluid wasstored at 4°C. To confirm the success of the cotransfection, the cells were fixedwith 2% formaldehyde and 0.2% glutaraldehyde in PBS for 10 min at 4°C. Theywere then stained for b-galactosidase activity with 50 mM potassium ferricya-nide–50 mM potassium ferrocyanide–20 mM MgCl2–0.4 mg of X-Gal (5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside) per ml in PBS.
The recombinant viruses were purified from the wild-type viruses by a plaquepurification technique as described elsewhere (18, 27). To achieve completepurification, four or five rounds of purification were required. Once purifiedstocks were obtained, large volumes of high-titer stocks were generated byinfecting Sf9 cells in early logarithmic growth phase with 0.1 PFU/cell. Therecombinant viruses were termed AcAHS, AcCHS, AcVCHS, and Ac215HS.Restriction enzyme digests (HindIII and BglII) of the FeLV VR1 regions ob-tained as PCR fragments from the recombinant viruses were compared withdigests of fragments obtained from the original proviral clones.
Production and purification of recombinant proteins. Samples of Sf9 cells,previously infected with the recombinant viruses, and their culture media were
analyzed by immunoblotting with 3-17AP. Most of the recombinant proteinswere released into the supernatant. The optimum time at which to harvest thesupernatant was determined by detection of SU by ELISA in aliquots of mediumtaken from cells infected with several different doses of the recombinant AcCHSvirus. The results indicated that 1 to 3 PFU/cell produced optimum protein levelsat 67 to 76 h postinfection.
To prepare large quantities of each protein, 10 flasks (225 cm2) were seededwith Sf9 cells at a density of 2 3 107 cells per flask. The next day the cells wereincubated for 2 h with an inoculum containing 2 PFU of one of the recombinantbaculoviruses per cell. The cells were then washed once with SF900 II serum-freemedium and were maintained in SF900 II medium for 67 h. After this period, thesupernatant was harvested, clarified by centrifugation at 5,000 rpm, and thenconcentrated by ultrafiltration with a cellulose triacetate filter with a nominalmolecular weight cutoff of 20,000 (Sartorius) in a model 8200 stirred cell (Ami-con). The concentrated supernatant was then purified by lentil lectin affinitychromatography. A mixture of 0.5 M methyl-a-glucopyranoside together with 0.5M methyl-a-mannopyranoside in TBS was used to elute the recombinant pro-tein. The resulting solution was dialyzed extensively against TBS at 4°C. The totalquantity of protein in each preparation was assayed by using Bradford’s reagent(38). The proteins were stored in aliquots at 270°C until used. The recombinantSU proteins produced were designated Bgp70-A, -C, -VC, and -215 (Fig. 1).
A control protein solution was prepared from an uninfected Sf9 culture. Theculture medium was harvested, concentrated, and purified as described above toform a preparation designated C5L. Similarly, a control solution was preparedfrom a culture infected with wild-type AcMNPV (2 PFU/cell). The supernatantwas concentrated and purified to form a preparation designated C6L. A recom-binant feline immunodeficiency virus (FIV) SU protein, Bgp100, was also pre-pared for use as a control preparation (27).
Infection interference assay. Infection interference assays were performed toinvestigate the ability of the recombinant proteins to block FeLV infection ofsusceptible cells. The ability of detergent-disrupted FeLV particles to interferewith infection of susceptible cells had previously been demonstrated (data notshown). In preliminary experiments it was found that optimum interference bythe recombinant FeLV-A surface glycoprotein, Bgp70-A, with FeLV-A infectionwas achieved when the protein was present both during and after the virusadsorption phase of infection. Mixtures of virus and recombinant protein weremade by adding various aliquots of the recombinant (or control) proteins to 1-mlaliquots of fresh medium containing approximately 80 focus-forming units(FFU) of FeLV-A, FeLV-B, or FeLV-C. The culture fluid was removed fromQN10S cells seeded the day previously as described above and was replaced with0.5-ml aliquots of the virus-protein mixtures. Each assay was performed induplicate. The cells were incubated for 2 h, after which time the mixtures wereremoved and replaced with 0.5-ml aliquots of culture medium to which had beenadded recombinant protein alone. The cells were incubated for a further 2 days,and another 1-ml aliquot of fresh medium was added. The cells were maintainedfor a further 4 days, and the foci on each plate were counted.
Neutralization inhibition assay. The neutralization inhibition assay measuredthe ability of recombinant proteins to block the action of virus-neutralizingantibodies. Aliquots of 50 ml of dilutions of recombinant proteins and immunecat serum were mixed in a checkerboard fashion in a round-bottom microtitertray. FeLV was diluted with medium until a titer of 2.4 3 103 FFU/ml wasobtained. From this diluted stock, further volumes of 50 ml containing 120 FFUwere added to the reaction mixtures. The reaction mixtures were then incubatedfor 2 h at 37°C in a humidified incubator. Three samples of 25 ml were taken fromeach reaction mixture and were used to infect three wells of QN10S cells. Thereaction inocula were incubated with the cells for a further 90 min. The overlyingmedium was then replaced with fresh medium, and the cells were incubated fora further 6 days, when the foci were counted microscopically. The mean numberof foci was calculated from these data.
As these assays were potentially susceptible to a wide range of nonspecific orspurious reactions, a number of controls were included. To control for directinteraction of the virus and the antigen, some reactions were performed withoutserum. To demonstrate that the immune cat serum contained virus-neutralizingantibodies, other reactions were performed without recombinant protein. Toestablish that the recombinant proteins were not acting with nonspecific com-ponents of the immune cat serum to enhance viral entry, some reactions wereperformed with specific-pathogen-free (SPF) cat serum. To control for nonspe-cific cytotoxicity of the antigen, some wells had only the proteins added. Non-specific inhibition of neutralizing activity by baculovirus or insect cell proteinswas controlled by inclusion of reactions with a mixture of the two negativecontrols (C5L and C6L) in place of the recombinant protein. When one com-ponent of the reaction was omitted, fresh medium was used in its place. Inaddition, each assay was performed at least twice on separate days to reduce thelikelihood of spurious reactions.
RESULTS
Recombinant FeLV surface glycoproteins are similar totheir wild-type counterparts. Four baculovirus transfer vectorswere produced, containing fragments of FeLV env coding forSU and part of TM of FeLV-A/Glasgow 1, FeLV-C/Sarma,
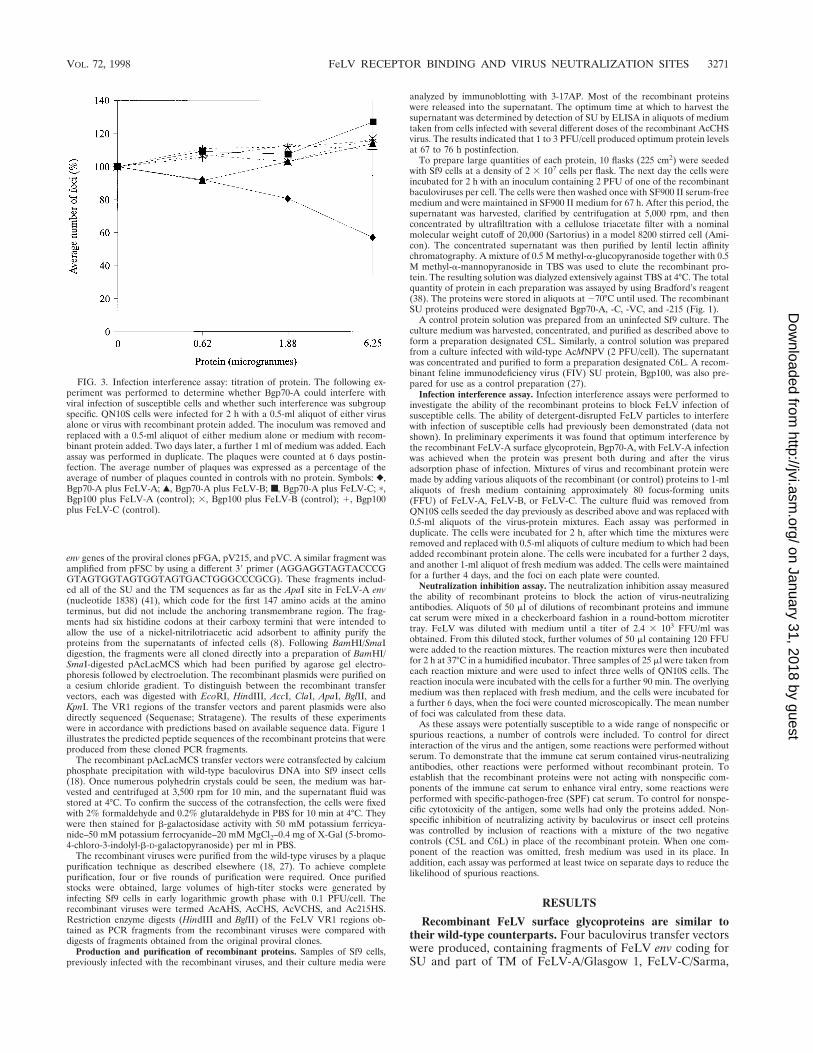
FIG. 3. Infection interference assay: titration of protein. The following ex-periment was performed to determine whether Bgp70-A could interfere withviral infection of susceptible cells and whether such interference was subgroupspecific. QN10S cells were infected for 2 h with a 0.5-ml aliquot of either virusalone or virus with recombinant protein added. The inoculum was removed andreplaced with a 0.5-ml aliquot of either medium alone or medium with recom-binant protein added. Two days later, a further 1 ml of medium was added. Eachassay was performed in duplicate. The plaques were counted at 6 days postin-fection. The average number of plaques was expressed as a percentage of theaverage of number of plaques counted in controls with no protein. Symbols: },Bgp70-A plus FeLV-A; Œ, Bgp70-A plus FeLV-B; ■, Bgp70-A plus FeLV-C; p,Bgp100 plus FeLV-A (control); 3, Bgp100 plus FeLV-B (control); 1, Bgp100plus FeLV-C (control).
VOL. 72, 1998 FeLV RECEPTOR BINDING AND VIRUS NEUTRALIZATION SITES 3271
on January 31, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

and two VR1 chimeric viruses, FeLV-A/C-FZ215 and FeLV-A/C-Sarma (32). By using electroblotting and subsequent im-munostaining with a monoclonal antibody (3-17AP), it wasdemonstrated that infection of Sf9 cells with the recombinantbaculoviruses resulted in the expression of the truncated envgenes. The four recombinant proteins (Bgp70-A, -C, -215, and-VC) were found to be exported into the culture supernatant ofthe infected Sf9 cells. The proteins were concentrated by ul-trafiltration and purified by lentil lectin affinity chromatogra-phy. The eluates from the lentil lectin columns all contained atleast one major impurity, a species of about 65 kDa which wasprobably the major baculovirus envelope glycoprotein, gp64(44). The major product in the eluate which reacted with the3-17AP monoclonal antibody was slightly larger than the SUfrom F422. The difference in size was probably partly due tothe presence of the nonanchoring portion of TM. Deglycosy-lation of the recombinant protein with PNGase F (OxfordGlycosystems) demonstrated that the proteins were glycosy-lated but not to the same extent as their native counterparts(27).
Western blotting was performed to analyze the binding offive sera to the recombinant proteins. The results of theseanalyses are shown in Fig. 2. The monoclonal antibody 3-17AP,the rabbit anti-gp70 serum, and the immune cat serum, but notthe SPF cat serum or the normal rabbit serum, recognized allof the recombinant proteins. The pattern of binding to therecombinant proteins observed with the rabbit anti-gp70 serumwas similar to that observed with the monoclonal antibody3-17AP. In contrast, the pattern of binding with the recovered-cat serum was different. Both the 709 rabbit serum and 3-17APbound to a 60-kDa species as well as to the major 75-kDaspecies. The 147-amino-acid-residue fragment of TM that wasincluded in the recombinant constructs was calculated to havea mass of 15 kDa. This suggests that the 60-kDa band repre-sented cleavage of the SU and TM portions of the proteins.The P11 serum did not bind to the smaller species and boundonly to a component of the broad 75-kDa band. Similar dif-ferences were also seen with the native FeLV positive controls,confirming a species-dependent difference in immunologicalreactivity to both native and recombinant FeLV SU. The re-sults obtained from the immunostained blots demonstratedthat the baculovirus expression system had produced Bgp70sthat were immunologically similar to the native proteins.
Recombinant FeLV surface glycoproteins interfere with vi-rus infection according to the subgroup identity of their VR1sequences. The capacity of Bgp70-A to interfere with the in-fection of susceptible cells by all three FeLV subgroups wasinvestigated. The recombinant FIV SU Bgp100 was used as acontrol in this experiment. The results are shown in Fig. 3. Areduction in the number of foci was observed when Bgp70-Awas included in the culture fluid during and after FeLV-Ainfection of QN10S cells, indicating that Bgp70-A interferedwith FeLV-A infection. The recombinant FIV SU protein,Bgp100, did not interfere with FeLV-A, FeLV-B, or FeLV-Cinfections. Bgp70-A did not interfere with FeLV-B or FeLV-C
FIG. 4. Infection interference assay: inhibition of FeLV-A and FeLV-C. (a)The following experiment was performed to investigate the ability of the recom-binant proteins to interfere with the infection of susceptible cells by FeLV-A/Glasgow-1. QN10S cells, seeded in a 12-well tray the previous day, were incu-bated with a 0.5-ml viral inoculum containing a diluted recombinant protein for2 h. After this period, the inoculum was removed and replaced with 0.5 ml of
fresh medium containing only the protein. After 2 days, a further 1 ml of freshmedium was added, and the cells were incubated for a further 5 days. Each assaywas performed in duplicate. The plaques were counted at 7 days postinfection.The average number of plaques was expressed as a percentage of the average ofnumber of plaques counted in controls with no protein. (b) The experiment wasrepeated with different batches of protein. (c) Another experiment was per-formed to investigate the ability of the same recombinant proteins to interferewith the infection of susceptible cells by FeLV-C/FA27C. Symbols: �, Bgp70-A;■, Bgp70-C; À, Bgp70-215; E, Bgp70-VC; 3, C5L (a) or C5L and C6L (b and c)(control); ç, Bgp100 (control).
3272 RAMSEY ET AL. J. VIROL.
on January 31, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from
infections, indicating that the interference observed betweenBgp70-A and FeLV-A was subgroup specific. In the controlreactions a slight increase in the number of foci was observedwith increasing volume of recombinant protein added. The risewas constant for the various controls and was not dependenton the FeLV subgroup used. The slight decrease in serumprotein concentration, caused by adding the recombinant pro-tein solution to a maximum volume of 50 ml to the original0.5-ml volume of viral inoculum, might be responsible. Alter-natively, a nonspecific interaction between FeLV and baculo-virus proteins may account for this observation.
To extend these observations, another infection interferenceassay was performed with a larger range of proteins. QN10Scells were infected with FeLV-A inocula to which had beenadded various concentrations of one of the recombinant pro-teins, Bgp70-A, Bgp70-C, Bgp70-215, or Bgp100. The C5Lpreparation was included as an additional control. The resultsare shown in Fig. 4a. The specific interference with FeLV-Ainfection by Bgp70-A observed in Fig. 3 was again demon-strated. Neither Bgp100 nor C5L interfered with the infectionof susceptible cells. Significantly, Bgp70-C and Bgp70-215failed to inhibit the infection of the cells by FeLV-A. Thisresult confirmed the subgroup-specific nature of the infectioninterference caused by the recombinant proteins. The infectionof the cells was completely inhibited at the highest dose ofBgp70-A. The assay was repeated with different batches ofprotein with similar results (Fig. 4b). A mixture of equal pro-portions of the control preparations, C5L and C6L, was alsoused in this assay, and a small increase in the number of fociproduced by the control reactions was seen again.
To determine if FeLV-C infection could be inhibited by therecombinant chimeric FeLV-A/C protein, Bgp70-215, an in-fection interference assay was performed with FeLV-C/FA27Cas the infecting virus and the same range of recombinant pro-
teins used previously. In this assay an attempt was made tokeep the volume of the protein inoculum constant by the ad-dition of appropriate volumes of PBS. The results are pre-sented in Fig. 4c. Inhibition of focus formation was observed inthose wells that contained either Bgp70-C or the FeLV-A/Cchimeric protein, Bgp70-215. This result extended previousobservations on the ability of Bgp70-A to interfere withFeLV-A infection by demonstrating that Bgp70-C interferedspecifically with FeLV-C. The ability of Bgp70-215 to interferewith FeLV-C, but not FeLV-A, demonstrates the functionalsimilarity between the A/C subgroup-determining VR1s of therecombinant proteins and native FeLVs (33). Interestingly, theslight increase in the number of foci with increasing volumes ofcontrol protein preparations identified previously was not ob-served in this experiment. It may be relevant that decreases inprotein concentration increase absorption of FeLV (34).
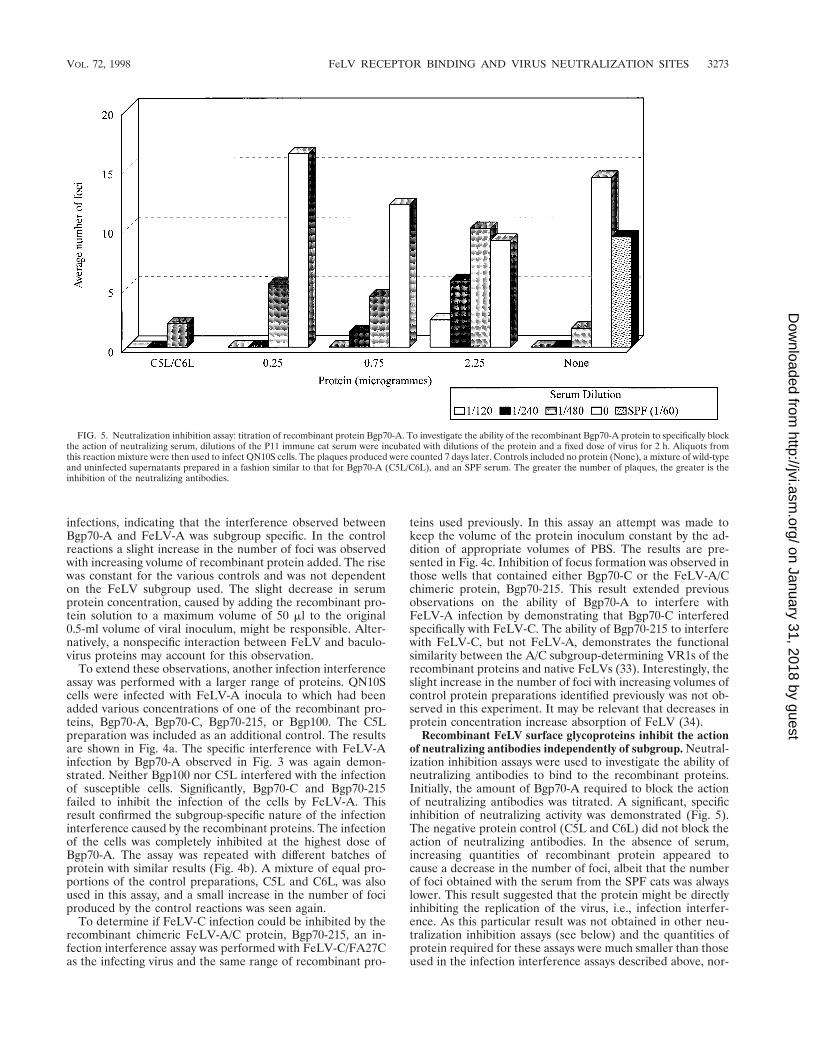
Recombinant FeLV surface glycoproteins inhibit the actionof neutralizing antibodies independently of subgroup. Neutral-ization inhibition assays were used to investigate the ability ofneutralizing antibodies to bind to the recombinant proteins.Initially, the amount of Bgp70-A required to block the actionof neutralizing antibodies was titrated. A significant, specificinhibition of neutralizing activity was demonstrated (Fig. 5).The negative protein control (C5L and C6L) did not block theaction of neutralizing antibodies. In the absence of serum,increasing quantities of recombinant protein appeared tocause a decrease in the number of foci, albeit that the numberof foci obtained with the serum from the SPF cats was alwayslower. This result suggested that the protein might be directlyinhibiting the replication of the virus, i.e., infection interfer-ence. As this particular result was not obtained in other neu-tralization inhibition assays (see below) and the quantities ofprotein required for these assays were much smaller than thoseused in the infection interference assays described above, nor-
FIG. 5. Neutralization inhibition assay: titration of recombinant protein Bgp70-A. To investigate the ability of the recombinant Bgp70-A protein to specifically blockthe action of neutralizing serum, dilutions of the P11 immune cat serum were incubated with dilutions of the protein and a fixed dose of virus for 2 h. Aliquots fromthis reaction mixture were then used to infect QN10S cells. The plaques produced were counted 7 days later. Controls included no protein (None), a mixture of wild-typeand uninfected supernatants prepared in a fashion similar to that for Bgp70-A (C5L/C6L), and an SPF serum. The greater the number of plaques, the greater is theinhibition of the neutralizing antibodies.
VOL. 72, 1998 FeLV RECEPTOR BINDING AND VIRUS NEUTRALIZATION SITES 3273
on January 31, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

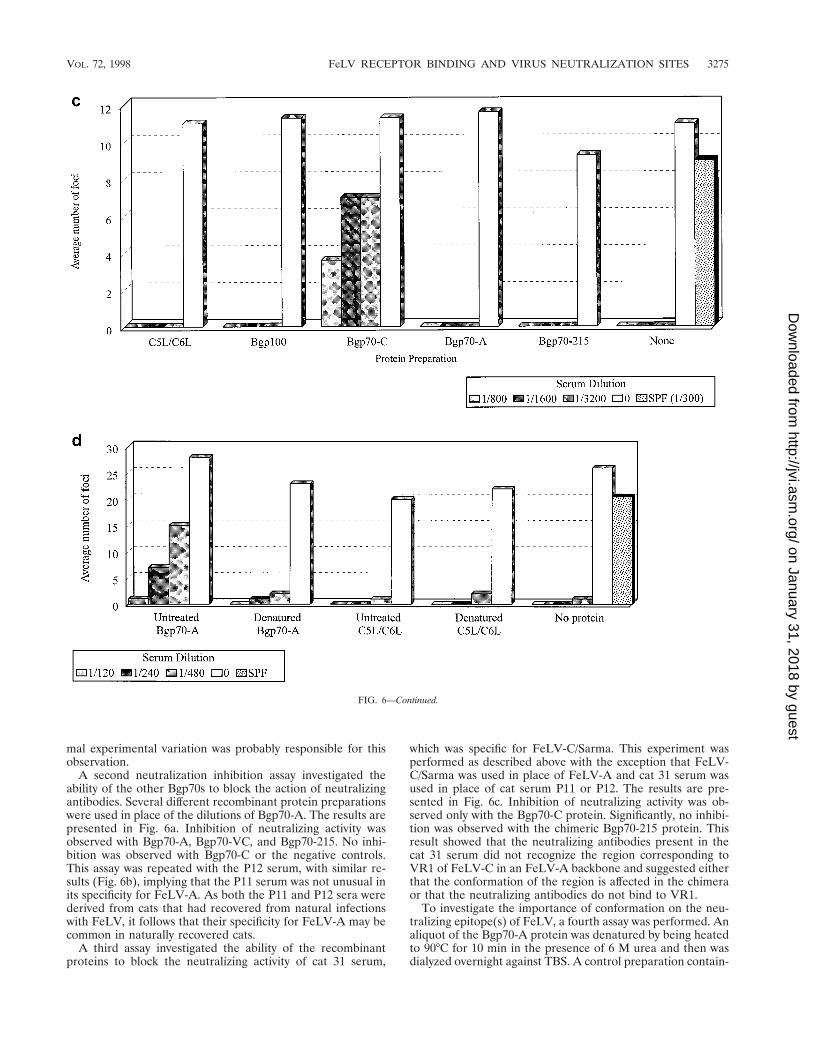
FIG. 6. Neutralization inhibition assay: reaction of recombinant proteins with neutralizing sera P11, P12, and cat 31. (a) To investigate the ability of the recombinantBgp70 proteins to specifically block the action of a neutralizing serum, dilutions of the P11 immune cat serum were incubated with 2.25 mg of the proteins and a fixeddose of virus for 2 h. Aliquots from these reaction mixtures were then used to infect QN10S cells. The plaques produced were counted 7 days later. Controls includedno protein (None), a mixture of wild-type and uninfected supernatants prepared in a fashion similar to that for Bgp70-A (C5L/C6L), and an SPF cat serum. (b) A similarexperiment was performed with P12 immune cat serum. (c) The reciprocal experiment of that for panels a and b, that is, determination of the ability of the recombinantBgp70 proteins to specifically block the action of an FeLV-C-specific neutralizing serum, was also performed. Dilutions of cat 31 serum were incubated with 20 ml ofthe proteins and a fixed dose of FeLV-C/Sarma for 2 h. Aliquots from these reaction mixtures were then used to infect QN10S cells. The plaques produced were counted7 days later. Controls included no protein (None), a mixture of wild-type and uninfected supernatants prepared in a fashion similar to that for Bgp70-C (C5L/C6L),a recombinant FIV SU protein (Bgp100), and an SPF serum. (d) Inability of a denatured preparation of recombinant Bgp70-A protein to specifically block the actionof an FeLV-A-neutralizing serum. The protein was denatured by heating to 90°C for 10 min in the presence of 6 M urea. A mixture of wild-type and uninfectedsupernatants (C5L/C6L) was treated in a fashion similar to that for the Bgp70-A preparation. Dilutions of the P11 immune cat serum were incubated with 25-ml aliquotsof the protein samples and a fixed dose of FeLV-A for 2 h. Aliquots from these reactions were then used to infect QN10S cells. The plaques produced were counted7 days later. A no-protein control was included (None). An SPF serum was also used. In all of these experiments, the greater the number of plaques, the greater isthe inhibition of the neutralizing antibodies.
3274 RAMSEY ET AL. J. VIROL.
on January 31, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from
mal experimental variation was probably responsible for thisobservation.
A second neutralization inhibition assay investigated theability of the other Bgp70s to block the action of neutralizingantibodies. Several different recombinant protein preparationswere used in place of the dilutions of Bgp70-A. The results arepresented in Fig. 6a. Inhibition of neutralizing activity wasobserved with Bgp70-A, Bgp70-VC, and Bgp70-215. No inhi-bition was observed with Bgp70-C or the negative controls.This assay was repeated with the P12 serum, with similar re-sults (Fig. 6b), implying that the P11 serum was not unusual inits specificity for FeLV-A. As both the P11 and P12 sera werederived from cats that had recovered from natural infectionswith FeLV, it follows that their specificity for FeLV-A may becommon in naturally recovered cats.
A third assay investigated the ability of the recombinantproteins to block the neutralizing activity of cat 31 serum,
which was specific for FeLV-C/Sarma. This experiment wasperformed as described above with the exception that FeLV-C/Sarma was used in place of FeLV-A and cat 31 serum wasused in place of cat serum P11 or P12. The results are pre-sented in Fig. 6c. Inhibition of neutralizing activity was ob-served only with the Bgp70-C protein. Significantly, no inhibi-tion was observed with the chimeric Bgp70-215 protein. Thisresult showed that the neutralizing antibodies present in thecat 31 serum did not recognize the region corresponding toVR1 of FeLV-C in an FeLV-A backbone and suggested eitherthat the conformation of the region is affected in the chimeraor that the neutralizing antibodies do not bind to VR1.
To investigate the importance of conformation on the neu-tralizing epitope(s) of FeLV, a fourth assay was performed. Analiquot of the Bgp70-A protein was denatured by being heatedto 90°C for 10 min in the presence of 6 M urea and then wasdialyzed overnight against TBS. A control preparation contain-
FIG. 6—Continued.
VOL. 72, 1998 FeLV RECEPTOR BINDING AND VIRUS NEUTRALIZATION SITES 3275
on January 31, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

ing equal proportions of C5L and C6L was treated in a similarfashion. Untreated controls were assayed concurrently. Theresults are presented in Fig. 6d. As in previous experiments,the untreated Bgp70-A protein inhibited neutralization whilethe control preparations did not. The denatured Bgp70-Apreparation did not inhibit neutralization, suggesting that theneutralizing epitope(s) of FeLV-A is conformational.
DISCUSSION
This paper describes the production, purification, and anal-ysis of recombinant FeLV surface glycoproteins. Other work-ers have expressed the entire FeLV env gene in the baculovirusexpression vector system and found that their product was notsecreted or processed into gp70 and p15(E) (42). Similarlyhuman immunodeficiency virus, human T-cell leukemia virustype 1, and FIV env gene products are not cleaved when ex-pressed in insect cells (1, 9, 43). However, the proteins in thisstudy were produced in the baculovirus expression system fromtruncated env genes of FeLV-A, FeLV-C/Sarma, and two chi-meric FeLV-A/C chimeras produced by Rigby (32). Truncatedenv genes were used, as it is known that retrovirus surfaceglycoproteins are secreted in quantity only if the transmem-brane portion of the TM protein is deleted and the aminoterminus of TM is left intact (5). As expected, the recombinantproteins were efficiently exported and could be detected in theculture fluid of infected cells. The proteins were purified bylentil lectin affinity chromatography and were demonstrated tobe immunologically similar to their native counterparts inELISAs and immunoblots. The purified recombinant proteinswere found to be approximately 75 kDa in size and wereaberrantly glycosylated. A minor degree of cleavage of theproteins was observed in immunoblots probed with the 3-17APor rabbit 709 antibody. It is not obvious why cat serum P11 didnot bind to the lower species seen in the other blots.
The recombinant proteins interfered with the infection ofsusceptible cells in a subgroup-specific manner. A chimericprotein containing VR1 of FeLV-C in an FeLV-A backbone(Bgp70-215) blocked the infection of FeLV-C but notFeLV-A. This result supports data demonstrating that VR1 ofFeLV determines the subgroup phenotype of FeLV-A andFeLV-C (33). The existence of three FeLV subgroups hasoften been suggested to imply that there are three distinctsubgroup-specific cell receptors for FeLV (2, 31, 33, 39). Theability of the recombinant glycoproteins to block viral replica-tion may be useful in the identification of the FeLV recep-tor(s). Preliminary studies on a putative FeLV-A receptor havebeen performed by others (7).
It has been demonstrated that the neutralizing antibodiesinduced in cats by FeLV infection are only partially subgroupspecific (36). If the FeLV-A/C phenotype is determined mostlyby VR1 (33), then it is logical to suggest that VR1 may beinvolved in the binding of neutralizing antibodies. Neutraliza-tion inhibition assays demonstrated that the recombinant pro-tein Bgp70-A, but not Bgp70-C, was able to block the action ofneutralizing antibodies to FeLV-A. However, the two FeLV-A/C chimeric proteins, Bgp70-VC and Bgp70-215, were alsoable to block the neutralizing activities of these sera. Con-versely, the chimeric proteins and Bgp70-A were unable toblock the virus-neutralizing activity of a serum derived from acat infected with FeLV-C/Sarma. These results indicated thatmost of the neutralizing activity of these sera was directed atregions other than that which determined the subgroup phe-notype. This provided direct evidence that the subgroup-de-termining and neutralizing-antibody binding sites of FeLV aredistinct (36). Indeed, the subgroup-determining VR1 region
appears to play little or no part in the binding of neutralizingantibodies.
The nature of viral epitopes involved in neutralization isknown for only some viruses, including the V3 loop of humanimmunodeficiency virus (16, 40) and a loop of influenza virushemagglutinin (47). Insofar as generalizations can be madefrom these examples, it appears that neutralizing epitopes arecomposed of contiguous amino acids, albeit often arranged ina particular conformation. Other regions of the molecule mayplay a peripheral role in binding. Our neutralization inhibitiondata suggest that a conformational epitope is present on FeLV.In the future it may be possible to identify a region or regionsthat are present in Bgp70-A, but that are altered in Bgp70-C,which may serve as binding sites for neutralizing antibodies. IfVR1 is not directly involved in the binding of neutralizingantibodies, then by comparison of sequence data (Fig. 1), itfollows that the neutralizing-antibody binding site(s) is likely tobe VR4 and/or VR5. Both regions are hydrophilic and possessmoderately high surface probabilities when examined by usingthe PLOTSTRUCTURE program, which is part of the Genet-ics Computer Group package for the VAX network (3, 10).The production of FeLV-A/C VR4 and VR5 chimeric SUproteins would allow these hypotheses to be tested. Specifi-cally, by mutation of nucleotide residues 811 and 812 (cytidineto thymidine and cytidine to guanosine, respectively) (41), aconvenient XbaI site could be introduced. Similarly, alterationof nucleotide residue 1019 (cytidine to guanosine) introducesan XhoI site. None of these mutations would result in a changeto the amino acid sequence.
All of our results agree with previously published work onthe specificity of neutralizing antibodies in relation to sub-group phenotype (35, 36). By utilizing subgroup chimeras andisolated recombinant SUs, we have confirmed that the regionsof SU responsible for subgroup specification are distinct fromthe regions that are recognized by neutralizing antibodies. Theability to produce recombinant FeLV SUs which will block theneutralizing activity of sera from cats that have recovered froma natural challenge represents a useful tool for further studiesdirected at identifying those epitopes that are recognized byvirus-neutralizing antibodies.
ACKNOWLEDGMENTS
We acknowledge the invaluable assistance of J. MacDonald, M.Heatherington, and M. Golder.
I.K.R. received a Wellcome Trust Veterinary Research TrainingScholarship during this project. N.S. was supported by Intervet Inter-national.
REFERENCES
1. Arp, J., C. M. Ford, T. J. Palker, E. E. King, and G. A. Dekaban. 1993.Expression and immunogenicity of the entire human T cell leukaemia virustype 1 envelope protein produced in a baculovirus system. J. Gen. Virol.74:211–222.
2. Brojatsch, J., B. S. Kristal, G. A. Viglianti, R. Khiroya, E. A. Hoover, and J. I.Mullins. 1992. Feline leukemia virus subgroup C phenotype evolovesthrough distinct alterations near the N terminus of the envelope surfaceglycoprotein. Proc. Natl. Acad. Sci. USA 89:8457–8461.
3. Devereux, J., P. Haeberle, and O. Smithies. 1984. A comprehensive set ofsequence analysis programs for the VAX. Nucleic Acids Res. 12:387–395.
4. Donahue, P. R., E. A. Hoover, G. A. Beltz, N. Riedel, V. Hirsch, J. Over-baugh, and J. I. Mullins. 1988. Strong sequence conservation among hori-zontally transmissible, minimally pathogenic feline leukemia viruses. J. Virol.62:722–731.
5. Einfeld, D., and E. Hunter. 1988. Oligomeric structure of a prototype ret-rovirus glycoprotein. Proc. Natl. Acad. Sci. USA 85:8688–8692.
6. Elder, J. H., J. S. McGee, M. Munson, R. A. Houghten, W. Kloetzer, J. L.Bittle, and C. K. Grant. 1987. Localization of neutralizing regions of theenvelope gene of feline leukemia virus using anti-synthetic peptide antibod-ies. J. Virol. 61:8–15.
7. Ghosh, A. K., M. H. Bachmann, E. A. Hoover, and J. I. Mullins. 1992.
3276 RAMSEY ET AL. J. VIROL.
on January 31, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

Identification of a putative receptor for subgroup A feline leukemia virus onfeline T cells. J. Virol. 66:3707–3714.
8. Hochuli, E., H. Dobeli, and A. Schacher. 1987. New metal chelate adsorbentselective for proteins and peptides containing neighbouring histidine resi-dues. J. Chromatogr. 411:177–184.
9. Hu, S., S. G. Kosowski, and K. F. Schaaf. 1987. Expression of envelopeglycoproteins of human immunodeficiency virus by an insect virus vector.J. Virol. 61:3617–3620.
10. Jameson, B. A., and H. Wolf. 1988. The antigenic index: a novel algorithm forpredicting antigenic determinants. CABIOS 4:181–186.
11. Jarrett, O. 1984. Pathogenesis of feline leukaemia virus-related diseases, p.135–154. In J. M. Goldman and O. Jarrett (ed.), Mechanisms of viral leu-kaemogenesis. Churchill-Livingstone, Edinburgh, United Kingdom.
12. Jarrett, O., and J. Ganiere. 1996. Comparative studies of the efficacy of arecombinant feline leukaemia virus vaccine. Vet. Rec. 138:7–11.
13. Jarrett, O., W. D. J. Hardy, M. C. Golder, and D. Hay. 1978. The frequencyof occurrence of feline leukemia virus subgroups in cats. Int. J. Cancer21:334–337.
14. Jarrett, O., H. M. Laird, and D. Hay. 1972. Restricted host range of a felineleukaemia virus. Nature 238:220–221.
15. Jarrett, O., H. M. Laird, and D. Hay. 1973. Determinants of the host rangeof feline leukaemia viruses. J. Gen. Virol. 20:169–175.
16. Javaherian, K., A. J. Langlois, C. McDanal, K. L. Ross, L. I. Eckler, C. L.Jellis, A. T. Profy, J. R. Rusche, D. P. Bolognesi, S. D. Putney, and T. J.Matthews. 1989. Principal neutralizing domain of the human immunodefi-ciency virus type 1 envelope glycoprotein. Proc. Natl. Acad. Sci. USA 86:6768–6772.
17. Kawakami, T. G., G. H. Theilen, D. L. Dungworth, R. J. Munn, and S. G.Beall. 1967. C-type viral particles in plasma of cats with feline leukemia.Science 158:1049.
18. King, L. A., and R. D. Possee. 1992. The baculovirus expression system. Alaboratory guide. Chapman & Hall, London, United Kingdom.
19. Lutz, H., N. Pedersen, J. Higgins, U. Hubscher, F. A. Troy, and G. H.Theilen. 1980. Humoral immune reactivity to feline leukemia virus andassociated antigens in cats naturally infected with feline leukemia virus.Cancer Res. 40:3642–3651.
20. Mackey, L., W. Jarrett, O. Jarrett, and H. M. Laird. 1975. Anemia associ-ated with feline leukemia virus infection in cats. J. Natl. Cancer Inst. 54:209–217.
21. Madewell, B. R., and O. Jarrett. 1983. Recovery of feline leukaemia virusfrom non-viraemic cats. Vet. Rec. 112:339–342.
22. Neil, J. C., R. Fulton, M. Rigby, and M. Stewart. 1991. Evolution of patho-genic and oncogenic variants of feline leukaemia virus. Curr. Top. Microbiol.Immunol. 171:67–93.
23. Nick, S., J. Klaws, K. Friebel, C. Birr, G. Hunsmann, and H. Bayer. 1990.Virus neutralising and enhancing epitopes characterised by synthetic oli-gopeptides derived from the feline leukaemia virus glycoprotein sequence.J. Gen. Virol. 71:77–83.
24. Nunberg, J. H., M. E. Williams, and M. A. Innis. 1984. Nucleotide sequencesof the envelope genes of two isolates of feline leukemia virus subgroup B. J.Virol. 49:629–632.
25. Onions, D., O. Jarrett, N. Testa, F. Frassoni, and S. Toth. 1982. Selectiveeffect of feline leukaemia virus on early erythroid precursors. Nature 296:156–158.
26. Overbaugh, J., N. Riedel, E. A. Hoover, and J. I. Mullins. 1988. Transductionof endogenous envelope genes by feline leukaemia virus in vitro. Nature332:731–734.
27. Ramsey, I. K. 1993. Recombinant surface glycoproteins of feline leukaemiavirus. Ph.D. thesis. University of Glasgow, Glasgow, United Kingdom.
28. Reinhart, T. A., A. K. Ghosh, E. A. Hoover, and J. I. Mullins. 1993. Distinctsuperinfection interference properties yet similar receptor utilization by cy-topathic and noncytopathic feline leukemia viruses. J. Virol. 67:5153–5162.
29. Rickard, D. G., J. E. Post, F. deNoronha, and L. M. Barry. 1969. A trans-missible virus-induced lymphocytic leukemia of the cat. J. Natl. Cancer Inst.42:987–1014.
30. Riedel, N., E. A. Hoover, R. E. Dornsife, and J. I. Mullins. 1988. Pathogenicand host range determinants of the feline aplastic anemia retrovirus. Proc.Natl. Acad. Sci. USA 85:2758–2762.
31. Riedel, N., E. A. Hoover, P. W. Gasper, M. O. Nicolson, and J. I. Mullins.1986. Molecular analysis and pathogenesis of the feline aplastic anemiaretrovirus, FeLV-C-Sarma. J. Virol. 60:242–250.
32. Rigby, M. A. 1989. Molecular basis of the subgroup determinants of felineleukaemia virus. Ph.D. thesis. University of Glasgow, Glasgow, United King-dom.
33. Rigby, M. A., J. L. Rojko, M. A. Stewart, G. J. Kociba, C. M. Cheney, L. J.Rezanka, L. E. Mathes, J. Hartke, O. Jarrett, and J. C. Neil. 1992. Partialdissociation of subgroup C phenotype and in vivo behaviour in feline leu-kaemia viruses with chimeric envelope genes. J. Gen. Virol. 73:2839–2847.
34. Russell, P. H. 1977. Neutralising antibodies in feline leukaemia virus infec-tions. Ph.D. thesis. University of Glasgow, Glasgow, United Kingdom.
35. Russell, P. H., and O. Jarrett. 1978. The occurrence of feline leukaemia virusneutralizing antibodies in cats. Int. J. Cancer 22:351–357.
36. Russell, P. H., and O. Jarrett. 1978. The specificity of neutralizing antibodiesto feline leukaemia viruses. Int. J. Cancer 21:768–778.
37. Salerno, R. A., V. M. Larson, A. H. Phelps, and M. R. Hilleman. 1979.Infection and immunization of cats with the Kawakami-Theilen strain offeline leukemia virus. Proc. Soc. Exp. Biol. Med. 160:18–23.
38. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: alaboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, ColdSpring Harbor, N.Y.
39. Sarma, P. S., and T. Log. 1973. Subgroup classification of feline leukemiaand sarcoma viruses by viral interference and neutralization tests. Virology54:160–169.
40. Skinner, M. A., R. Ting, A. Langlois, K. J. Weinhold, H. K. Lyerly, K.Javaherian, and T. J. Matthews. 1988. Characteristics of a neutralizingmonoclonal antibody to the HIV envelope protein. AIDS Res. Hum. Ret-roviruses 4:187–197.
40a.Spibey, N. Unpublished data.41. Stewart, M. A., M. Warnock, A. Wheeler, N. Wilkie, J. I. Mullins, D. E.
Onions, and J. C. Neil. 1986. Nucleotide sequences of a feline leukemia virussubgroup A envelope gene and long terminal repeat and evidence for therecombinational origin of subgroup B viruses. J. Virol. 58:825–834.
42. Thomsen, D. R., A. L. Meyer, and L. E. Post. 1992. Expression of felineleukaemia virus gp85 and gag proteins and assembly into virus like particlesusing the baculovirus expression vector system. J. Gen. Virol. 73:1819–1824.
43. Verschoor, E. J., A. L. W. van Vliet, H. F. Egberink, W. Hesselink, M. C.Horizinek, and A. de Ronde. 1993. Expression of feline immunodeficiencyvirus gag and env precursor proteins in Spodoptera frugeriperda cells and theiruse in immunodiagnosis. J. Clin. Microbiol. 31:2350–2355.
44. Volkman, L. E. 1986. The 64K envelope protein of budded Autographacalifornica nuclear polyhedrosis virus. Curr. Top. Microbiol. Immunol. 131:103–118.
45. Weijer, K., G. C. M. Fons, O. Jarrett, H. Lutz, and A. D. M. E. Osterhaus.1986. Post-exposure treatment with monoclonal antibodies in a retrovirussystem: failure to protect cats against feline leukemia virus infection withvirus neutralizing monoclonal antibodies. Int. J. Cancer 38:81–87.
46. Weijer, K., A. Pfauth, R. van Herwijnen, O. Jarrett, R. H. Meloen, C. Tomee,and A. D. M. E. Osterhaus. 1993. Induction of feline leukaemia virus-neutralizing antibodies by immunisation with synthetic peptides derivedfrom the FeLV env gene. Vaccine 11:946–956.
47. Wiley, D. C., I. A. Wilson, and J. J. Skehel. 1981. Structural identification ofthe antibody-binding sites of Hong-Kong influenza haemagglutinin and theirinvolvement in antigenic variation. Nature 289:373–378.
VOL. 72, 1998 FeLV RECEPTOR BINDING AND VIRUS NEUTRALIZATION SITES 3277
on January 31, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from