the role of neural crest during cardiac development in a mouse model of digeorge syndrome
TRANSCRIPT

Developmental Biology 251, 157–166 (2002)doi:10.1006/dbio.2002.0819
The Role of Neural Crest during CardiacDevelopment in a Mouse Modelof DiGeorge Syndrome
Lazaros Kochilas,* ,† ,1 Sandra Merscher-Gomez,‡ Min Min Lu,*Vijaya Potluri,* Jun Liao,§ Raju Kucherlapati,‡ Bernice Morrow,§and Jonathan A. Epstein**Cardiovascular Division, University of Pennsylvania, Philadelphia, Pennsylvania 19104;†A.I. duPont Hospital for Children, Wilmington, Delaware 19899; ‡Center for Genetics andGenomics, Harvard Medical School, Boston, Massachusetts 02115; and §Departmentof Molecular Genetics, Albert Einstein College of Medicine, Bronx, New York 10461
The velo-cardio-facial syndrome (VCFS)/DiGeorge syndrome (DGS) is a genetic disorder characterized by phenotypicabnormalities of the derivatives of the pharyngeal arches, including cardiac outflow tract defects. Neural crest cells play amajor role in the development of the pharyngeal arches, and defects in these cells are likely responsible for the syndrome.Most patients are hemizygous for a 1.5- to 3.0-Mb region of 22q11, that is suspected to be critical for normal pharyngeal archdevelopment. Mice hemizygous for a 1.5-Mb homologous region of chromosome 16 (Lgdel/�) exhibit conotruncal cardiacdefects similar to those seen in affected VCFS/DGS patients. To investigate the role of Lgdel genes in neural crestdevelopment, we fate mapped neural crest cells in Lgdel/� mice and we performed hemizygous neural crest-specificinactivation of Lgdel. Hemizygosity of the Lgdel region does not eliminate cardiac neural crest migration to the formingaortic arches. However, neural crest cells do not differentiate appropriately into smooth muscle in both fourth and sixthaortic arches and the affected aortic arch segments develop abnormally. Tissue-specific hemizygous inactivation of Lgdelgenes in neural crest results in normal cardiovascular development. Based on our studies, we propose that Lgdel genes arerequired for the expression of soluble signals that regulate neural crest cell differentiation. © 2002 Elsevier Science (USA)
Key Words: Tbx1; pharyngeal arches; aortic arches; smooth muscle; DiGeorge syndrome.
INTRODUCTION
The velo-cardio-facial syndrome (VCFS)/DiGeorge syn-drome (DGS) is a human genetic disorder characterized bynumerous phenotypic abnormalities, including cleft palate,cardiovascular defects, thymic hypoplasia, hypoparathy-roidism, and learning disabilities. Most patients are hemi-zygous for a 1.5- to 3.0-Mb region of 22q11 (DGCR) (Gold-muntz and Emanuel, 1997). Many of the affected tissueshave contributions of neural crest origin, and genes withinthe DGCR are widely believed to be critical for normalneural crest development (Kirby and Waldo, 1995). Micehemizygous for a 1.5-Mb homologous area on chromosome16 (Lgdel/�) that carry a targeted deletion from the Idd to
1
2094. E-mail: [email protected].
0012-1606/02 $35.00© 2002 Elsevier Science (USA)All rights reserved.
Hira loci (Lgdel) exhibit conotruncal defects and parathy-roid deficiencies (Merscher et al., 2001). Twenty-fourknown genes lie in the 1.5-Mb interval (Puech et al., 2000).Overexpression of four genes contained within a 200-kbBAC clone was able to rescue cardiovascular developmentin most Lgdel/� mice (Merscher et al., 2001). One of thefour genes, Tbx1, is expressed in the pharyngeal arches andheterozygosity of Tbx1 in mice results in cardiovasculardefects similar to those observed in VCFS/DGS (Jerome andPapaioannou, 2001; Lindsay et al., 2001; Merscher et al.,2001). Therefore, Tbx1 is considered to be primarily respon-sible for the cardiovascular defects observed in Lgdel/�mice, and may also contribute to congenital heart disease inVCFS/DGS patients.
Cardiovascular defects described in VCFS/DGS patientsand in Lgdel/� mice include those that result from abnor-
To whom correspondence should be addressed. Fax: (215) 573- mal remodeling of the symmetric pairs of aortic arch157
vessels into the mature vascular structure. This remodelingtakes place during midgestation and includes asymmetri-cally programmed persistence and regression of specificarch arteries. For instance, in vertebrates, the left fourthpharyngeal arch artery gives rise to the segment of aorticarch which extends distally from the rostral part of theoutflow tract to the site of entrance of the ductus arteriosus,while the right fourth arch artery normally reduces inrelative size and forms the brachiocephalic trunk and theproximal part of the right subclavian artery. The proximalsixth arch arteries persist bilaterally and give rise to theproximal right and left pulmonary arteries. The distal rightsixth artery regresses and eventually disappears, whereasthe distal left sixth arch becomes the ductus arteriosus.There is mounting evidence that the genetic defects inmouse models of DGS affect the development of the fourthaortic arch bilaterally, resulting in aberrant right subclavianartery or interruption of the aortic arch (Lindsay and Bal-dini, 2001). The effects of Lgdel haploinsufficiency on thedevelopment of sixth arch derivatives, including the centralportions of the branch pulmonary arteries and the ductusarteriosus, have not been described in animal models,though these segments are affected in human patients withVCFS/DGS (Momma et al., 1996).
The molecular mechanisms affecting aortic arch remod-eling are yet to be fully understood but evidence suggeststhat this process is affected by accumulating neural crest-derived cells that migrate through the pharyngeal arches.The ultimate involvement of neural crest cells in thisprocess has been suggested by the aortic arch malforma-tions resulting from the ablation of premigratory neuralcrest cells in chick embryos. These studies led to theidentification of a subgroup of neural crest cells arising atthe level of the first three somites that is required forcardiovascular development (cardiac neural crest) (Bock-man et al., 1990; Kirby, 1990; Kirby et al., 1985; Kirby andWaldo, 1990, 1995). However, it remains unclear to whatextent the human DGCR genes or the Lgdel genes on theorthologous region of mouse chromosome 16 affect neuralcrest development, and whether they modulate migration,survival, proliferation, or interaction of neural crest withthe local environment. Moreover, the mechanism by whichneural crest influences pharyngeal and aortic arch develop-ment remains poorly understood. In avian embryos, neuralcrest-derived mesenchymal cells of the anterior pharyngealarches 1 and 2, in which the aortic arch arteries disappear,differ in their phenotypic and developmental propertiesfrom those populating the posterior pharyngeal arches 3, 4,and 6, in which the aortic arch arteries persist as permanentvessels. Hence, neural crest-derived ectomesenchyme mayinfluence aortic arch artery persistence (Ciment andWeston, 1985). Also, studies in quail embryos have shownthat, although the third arch artery forms a lumen appro-priately in neural crest-ablated embryos, subsequently, itbecomes misshapen and regresses, indicating that neuralcrest is essential for the persistence of an arch artery but notfor its formation (Waldo et al., 1996).
In this report, we examine the effect of haploinsufficiencyof Lgdel genes on neural crest migration and gene expres-sion. Using a binary Cre-lox system to fate-map cardiacneural crest progenitors, we demonstrate that these cellsmigrate through the pharyngeal arches and reach theirappropriate targets in affected Lgdel/� mice. Molecularmarkers of migrating neural crest progenitors are expressednormally, even in the complete absence of Tbx1, butexpression of the secreted growth factors Fgf8 and Fgf10 isreduced in the pharyngeal regions of Tbx1�/� mutants.Furthermore, haploinsufficiency of the Lgdel region inneural crest cells does not reproduce the Lgdel/� cardiovas-cular phenotype. Examination of neural crest differentia-tion using an SM22�-lacZ knock-in allele revealed abnor-malities of smooth muscle differentiation in the fourth andsixth aortic arches in Lgdel/� embryos. These data suggestthat haploinsufficiency of the Lgdel region affects neuralcrest differentiation into smooth muscle through non-cell-autonomous signaling pathways.
MATERIALS AND METHODS
Animal Husbandry and Genotyping
Mice were kept on a 12-h light/dark cycle, and noon of the plugdate was considered as 0.5 d.p.c. (E0.5). P3proCre (Li et al., 2000),
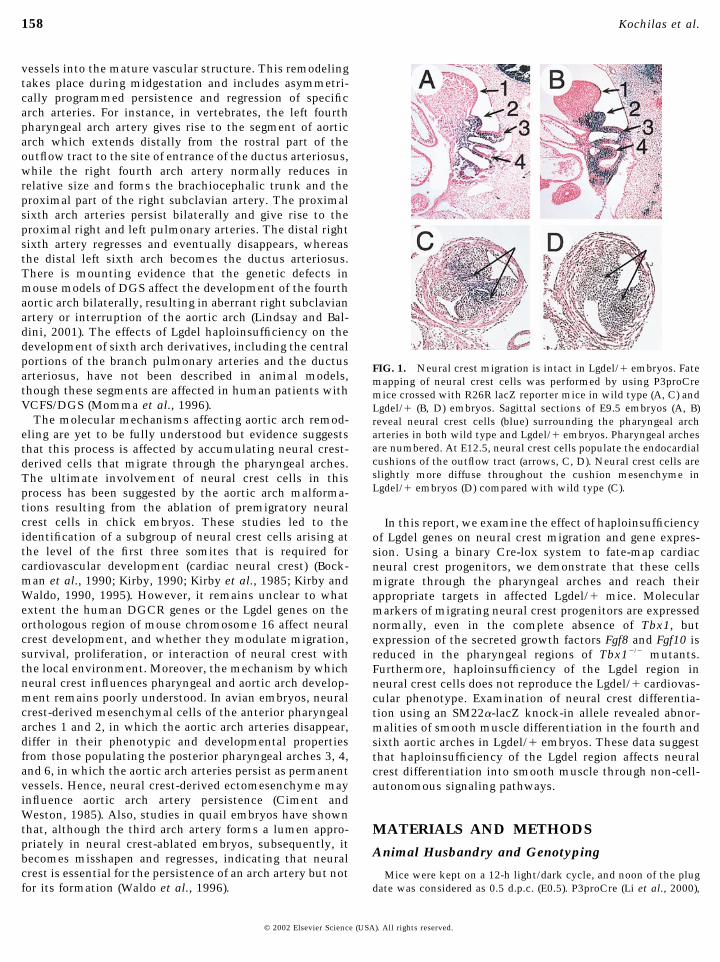
FIG. 1. Neural crest migration is intact in Lgdel/� embryos. Fatemapping of neural crest cells was performed by using P3proCremice crossed with R26R lacZ reporter mice in wild type (A, C) andLgdel/� (B, D) embryos. Sagittal sections of E9.5 embryos (A, B)reveal neural crest cells (blue) surrounding the pharyngeal archarteries in both wild type and Lgdel/� embryos. Pharyngeal archesare numbered. At E12.5, neural crest cells populate the endocardialcushions of the outflow tract (arrows, C, D). Neural crest cells areslightly more diffuse throughout the cushion mesenchyme inLgdel/� embryos (D) compared with wild type (C).
158 Kochilas et al.
© 2002 Elsevier Science (USA). All rights reserved.

FIG. 2. Downregulation of Fgf8 and Fgf10 expression in Tbx1�/� embryos. Littermates at E11.5 (A–H) were examined for expression of Fgf8(A, B) and Fgf10 (E, F). Sagittal sections through the pharyngeal region (A–H, dorsal is left, ventral is right) reveal loss of expression of Fgf8(arrows A, B) in the endoderm of the third pharyngeal pouch (3p) of Tbx1�/� (B) embryos. The third pharyngeal pouch is easily identifiablein the adjacent sections stained with hematoxylin and eosin in both wild type and mutant embryos, respectively (C, D). Fgf10 expressionwas also significantly reduced in Tbx1�/� embryos (F) within discrete domains (arrows, E, F) that involves a very discrete area of thepharyngeal mesenchymal tissue in close proximity with the developing fourth aortic arch. Arrows in (G, H) indicate in H&E adjacentsections the areas of tissue where differences in Fgf10 expression was detected.
159Neural Crest and DiGeorge Syndrome Mice
© 2002 Elsevier Science (USA). All rights reserved.

SM22�-lacZ (Zhang et al., 2001), R26R (Soriano, 1999), andLgdel/� (Merscher et al., 2001) mice have been described and weremaintained on a C57/B6 background. LgdelloxP/� mice contain the1.5-Mb Lgdel region flanked by loxP sites that disrupt the Idd andHira loci; these mice were previously referred to as Cis-Idd/Hira-KO (Merscher et al., 2001).
The following primers were used for genotyping: PGK1, 5�-GCTAAAGCGCATGCTCCAGAC-3�; Neo5F, 5�-ACCGCTAT-CAGGACATAGCGT-3�; Idd-KO1, 5�-CTGTTGTTGACACAG-CACATG-3�; WT, 5�-AACTCTACCTGTTCCTACTG-3�; CreF,5�-GGACATGTTCAGGGATCGCCAGGCG-3�; and CreR, 5�-GCATAACCAGTGAAACAGCATTGCTG-3�.
The primers were combined as follows: PGK1/Neo5F recognizesthe deleted allele (Lgdel) of Lgdel/� mice and of LgdelloxP/� miceafter removal of the Lgdel genes. The Idd-KO1/WT combinationamplifies the Idd wild type allele (WT). CreF/CreR recognizes theCre transgene. PCR conditions were 94°C for 3 min, followed by94°C for 30 s, 58°C for 45 s, 72°C for 45 s (35 cycles), and 72°C for5 min (Merscher et al., 2001).
Histology
Embryos were fixed in 4% paraformaldehyde in PBS for 1–2 h,dehydraded through an EtOH series, and maintained at –20o untilthe time of analysis. Near term, embryos (E18.5) were analyzed bydissection and by preparation of corrosion casts, as described (Li etal., 1999). �-Galactosidase activity was determined by stainingwith X-Gal solution as previously described (Chang et al., 1995).After dehydration and embedding in paraffin wax, 10-�m sectionswere stained with hematoxylin and eosin. Radioactive in situhybridization was performed as described (Epstein et al., 1996). Toassess smooth muscle differentiation, we performed immunohis-tochemistry on mouse tissue sections by using an anti-�-smoothmuscle actin monoclonal antibody (SMA, Sigma, A2547) andapplying the indirect conjugated method as previously described(Epstein et al., 2000). Digital images were collected by using a ZeissAxioplan 2 microscope and were processed with Adobe Photoshop.
RESULTS
Fate Mapping Neural Crest Derivatives in Lgdel/�Embryos
Since cardiovascular defects in Lgdel/� mice resemblethose resulting from ablation of premigratory neural crestin chick embryos, we sought to determine whether neuralcrest migration was altered in Lgdel/� embryos. In order todo so, we utilized a binary Cre-lox system that we andothers have developed to fate map neural crest precursors tothe heart and pharyngeal arch region. P3proCre mice ex-press Cre recombinase in Pax3-expressing premigratoryneural crest cells (Li et al., 2000). By crossing these trans-genic mice with R26R Cre reporter mice (Soriano, 1999)which activate expression of �-galactosidase in Cre-expressing cells, we have been able to follow descendents ofcardiac neural crest precursors as they encase the pharyn-geal arch arteries, form the aortopulmonary septation com-plex, and differentiate into smooth muscle in the aorticarch, ductus arteriosus, and great vessels (Li et al., 2000). AtE9.5, labeled neural crest cells are seen in the pharyngeal
arches surrounding the forming aortic arch arteries in wildtype embryos (Fig. 1A). In Lgdel/� embryos, a similarpattern of �-galactosidase expression is observed (Fig. 1B),indicating that neural crest migration is intact in Lgdel/�embryos. By E12.5, neural crest descendents form themesenchymal tissue that divides the truncus arteriosusinto the aorta and pulmonary artery (Fig. 1C, arrows). InLgdel/� embryos with significant vascular defects, neuralcrest descendents effectively divide the outflow tract intotwo vessels, although a more scattered and less densepattern of neural crest derivatives is observed in the endo-cardial cushions compared with wild type (Fig. 1D, arrows).Similar findings were reported by Vitelli et al. (2002) whodemonstrated that in Tbx1�/� embryos, neural crest cellsmaintain their ability to migrate but appear to lack direc-tional cues. Together these results suggest that cardiacneural crest migration and patterning are not grossly im-paired in Lgdel/� embryos, though subtle abnormalitiesexist.
Neural Crest-Related Gene Expression in Lgdel/�and Tbx1 Null Embryos
We next sought to determine whether neural crest-related gene expression was altered in Lgdel/� embryos orin embryos that were homozygous deficient for Tbx1.Heterozygous deficient Tbx1 mice display a phenotypesimilar to Lgdel/� mice (Merscher et al., 2001), whilehomozygous deficient Tbx1 mice have more severe aorticarch artery defects and might be expected to display moredramatic gene-expression alterations (Jerome and Papaioan-nou, 2001; Vitelli et al., 2002). We examined the pattern ofexpression of molecular markers and potential downstreamtargets in neural crest cells, mesenchyme, adjacentendoderm, and aortic arch endothelium.
Fibroblast growth factors (Fgfs), such as Fgf8, that aredown-regulated in other models of abnormal arch formation(Wendling et al., 2000), could mediate interactions betweenendoderm or ectoderm and migrating neural crest cells. Fgf8is expressed in the developing CNS, facial mesenchyme,and pharyngeal endoderm (Crossley and Martin, 1995;Trumpp et al., 1999). At E11.5, we noted a specific loss ofpharyngeal expression of Fgf8 in the endoderm of the thirdpharyngeal pouch in sagittal sections of Tbx1�/� embryos(Figs. 2A and 2C) compared with wild type littermates(compare arrows, Figs. 2B and 2D), while the expression inthe pharyngeal ectoderm was preserved. Fgf10, a relatedmember of the Fgf family, is likewise down-regulated ina similar region (compare arrows, Figs. 2E and 2G, and2F and 2H). In this case, the loss of signal involves notonly a very discrete area of the pharyngeal mesenchymaltissue in close proximity with the developing fourth aorticarch. In a different model of abnormal pharyngeal archdevelopment, inhibition of retinoid signaling is accompa-nied by loss of the Fgf8 expression exactly in the same areaof the third pharyngeal pouch endoderm (Wendling et al.,2000). Since Tbx1 is also expressed by pharyngeal endoderm
160 Kochilas et al.
© 2002 Elsevier Science (USA). All rights reserved.

(Jerome and Papaioannou, 2001; Lindsay et al., 2001), theseresults suggest that Tbx1 may function upstream of Fgfsignaling in this tissue.
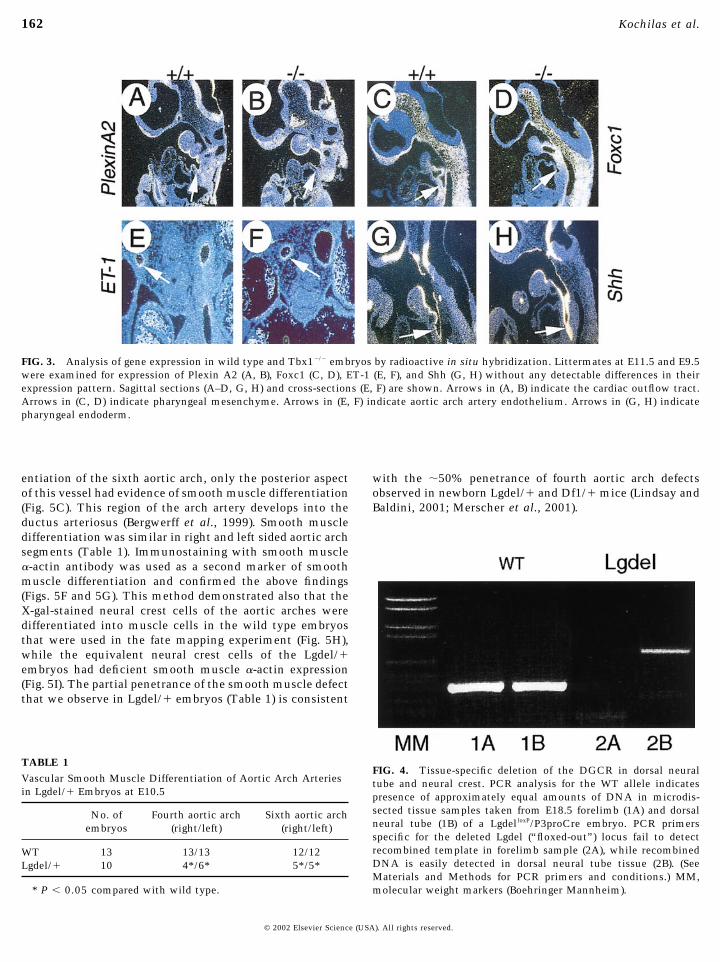
Expression of several neural crest-specific genes, includ-ing Pax3 and EdnrA (Clouthier et al., 1998; Epstein et al.,2000; Yanagisawa et al., 1998), is normal in Lgdel/� andTbx1�/� embryos at E9.5–E11.5 (data not shown). PlexinA2is also expressed by migrating neural crest cells (Brown etal., 2001) including those entering the outflow tract of theheart (Figs. 3A and 3B, arrows,) and its expression is alsounchanged. Foxc1 (Figs. 3C and 3D), Foxc2 (not shown), andSema3C (not shown) are coexpressed with Tbx1 in head andpharyngeal mesenchyme (Feiner et al., 2001; Iida et al.,1997; Winnier et al., 1999). Expression of these genes isunchanged in Lgdel/� and Tbx1�/� embryos. Endothelin 1(ET-1) is expressed by aortic arch endothelial cells. Al-though the aortic arch arteries are grossly abnormal inTbx1�/� embryos, ET-1 expression is normal at E9.5 (notshown) and E11.5 (Figs. 3E and 3F).
Sonic hedgehog (Shh) and Pitx2 are expressed by pharyn-geal endoderm, and Shh may regulate Pitx2 and Tbx1expression. Tbx1 expression was shown to be down-regulated in the pharyngeal endoderm of E10.5 Shh mutantmouse embryos, and Shh was sufficient to induce Tbx1expression (Garg et al., 2001). On the other hand, theabnormalities of the pharyngeal apparatus in Tbx1�/� mu-tants are more severe than in Shh�/�, suggesting that Tbx1may be upstream of Shh in regulating pharyngeal develop-ment (Chiang et al., 1996) and feedback regulation hasbeen implicated in other Shh-dependent systems. However,we did not identify any alterations in Shh expression inTbx1�/� embryos (Figs. 3G and 3H). Pitx2 expression isalso unchanged (not shown). These results suggest thatTbx1 functions downstream or independently of Shh andPitx2.
Hemizygous Neural Crest-Specific Inactivation ofLgdel Genes Results in Normal CardiovascularDevelopment
We sought to determine whether Lgdel genes are requiredin a cell-autonomous fashion within neural crest cells.LgdelloxP/� mice carry a modified region of chromosome 16with loxP sites flanking the Lgdel region between the Iddand Hira loci (Merscher et al., 2001). LgdelloxP/� mice werecrossed with P3proCre mice in order to delete the entireIdd-Hira interval, on one chromosome, in neural crest cells.We demonstrated Cre-mediated excision of Lgdel by per-forming PCR on microdissected tissue isolated from result-ing embryos. Cre-mediated recombination was tissue-specific, since it was evident in neural crest derived tissuewhere Pax3 is expressed, while we did not detect recombi-nation in tissue from the limb where Cre is not expressed(Fig. 4). More detailed analysis of Cre activity is compli-cated by the fact that neural crest cells do not form ahomogenous population and our dissections are contami-nated by non-Pax3 expressing cells. Hence, it is not possible
to determine the efficiency of our Cre-mediated inactiva-tion. In separate studies, we have demonstrated by coim-munohistochemistry that �90% of cells expressing Pax3 inthe dorsal neural tube display functional expression of Crerecombinase as assayed by activation of �-galactosidaseprotein expression in R26R embryos (data not shown).
We examined cardiovascular anatomy by corrosion cast-ing (Merscher et al., 2001) and gross dissection at E18.5 in49 embryos resulting from 7 litters born to LgdelloxP/� andP3proCre matings. We expected 25% (12) to inherit bothLgdelloxP and Cre, and we identified 14 such embryos.Although 50% of Lgdel/� mice display gross abnormalitiesof vascular patterning, we found no abnormalities in the 14LgdelloxP/P3proCre mice despite conserved C57/B6 geneticbackgrounds. The absence of cardiovascular anomalies inmice, harboring Lgdel haploinsufficient cells suggests anon-cell-autonomous role for Tbx1 and other genes in theLgdel region in neural crest cells during vascular remodel-ing.
Deficient Smooth Muscle Differentiation of NeuralCrest Cells in Lgdel/� Mice
Although neural crest migration and gene expressionappeared normal in Lgdel/� mice, and the Lgdel genes didnot seem to be required within neural crest cells, wewondered whether neural crest differentiation was affectedin Lgdel/� mice. Studies in chick (Kirby and Waldo, 1995)and mice (Li et al., 2000) have demonstrated that cardiacneural crest cells can differentiate into smooth muscle cellspopulating the aortic arch arteries. SM22� is an earlymarker of cardiac and smooth muscle differentiation. Weutilized a recombinant mouse in which lacZ had beeninserted into the SM22� locus as a sensitive assay for earlysmooth muscle differentiation (Zhang et al., 2001). AtE10.5, we were able to detect evidence of smooth muscledifferentiation in the third aortic arches of both wild typeand Lgdel/� embryos (Fig. 5). However, in the fourth aorticarch, smooth muscle differentiation was completely absentin about half of Lgdel/� embryos in contrast to wild typelittermates, which all displayed significant evidence of�-galactosidase activity (Figs. 5A–5E). Table 1 summarizesthe number of right and left aortic arch segments that hadevidence of �-galactosidase expression in fourth and sixtharch arteries. Moreover, even in those cases recorded inTable 1 where some smooth muscle differentiation waspresent, it was reduced in most Lgdel/� embryos whencompared with wild type.
Smooth muscle differentiation was also notably deficientin the sixth aortic arches of Lgdel/� embryos. Half of theLgdel/� embryos examined were devoid of smooth muscledifferentiation in the sixth aortic arch, while an additional40% demonstrated only minimal evidence of smoothmuscle differentiation (Fig. 5, Table 1). In contrast, 92% ofthe wild type littermates displayed significant smoothmuscle differentiation in the sixth arches (Figs. 5A and 5D,Table 1). In Lgdel/� embryos with partial muscular differ-
161Neural Crest and DiGeorge Syndrome Mice
© 2002 Elsevier Science (USA). All rights reserved.
entiation of the sixth aortic arch, only the posterior aspectof this vessel had evidence of smooth muscle differentiation(Fig. 5C). This region of the arch artery develops into theductus arteriosus (Bergwerff et al., 1999). Smooth muscledifferentiation was similar in right and left sided aortic archsegments (Table 1). Immunostaining with smooth muscle�-actin antibody was used as a second marker of smoothmuscle differentiation and confirmed the above findings(Figs. 5F and 5G). This method demonstrated also that theX-gal-stained neural crest cells of the aortic arches weredifferentiated into muscle cells in the wild type embryosthat were used in the fate mapping experiment (Fig. 5H),while the equivalent neural crest cells of the Lgdel/�embryos had deficient smooth muscle �-actin expression(Fig. 5I). The partial penetrance of the smooth muscle defectthat we observe in Lgdel/� embryos (Table 1) is consistent
with the �50% penetrance of fourth aortic arch defectsobserved in newborn Lgdel/� and Df1/� mice (Lindsay andBaldini, 2001; Merscher et al., 2001).
FIG. 3. Analysis of gene expression in wild type and Tbx1�/� embryos by radioactive in situ hybridization. Littermates at E11.5 and E9.5were examined for expression of Plexin A2 (A, B), Foxc1 (C, D), ET-1 (E, F), and Shh (G, H) without any detectable differences in theirexpression pattern. Sagittal sections (A–D, G, H) and cross-sections (E, F) are shown. Arrows in (A, B) indicate the cardiac outflow tract.Arrows in (C, D) indicate pharyngeal mesenchyme. Arrows in (E, F) indicate aortic arch artery endothelium. Arrows in (G, H) indicatepharyngeal endoderm.
FIG. 4. Tissue-specific deletion of the DGCR in dorsal neuraltube and neural crest. PCR analysis for the WT allele indicatespresence of approximately equal amounts of DNA in microdis-sected tissue samples taken from E18.5 forelimb (1A) and dorsalneural tube (1B) of a LgdelloxP/P3proCre embryo. PCR primersspecific for the deleted Lgdel (“floxed-out”) locus fail to detectrecombined template in forelimb sample (2A), while recombinedDNA is easily detected in dorsal neural tube tissue (2B). (SeeMaterials and Methods for PCR primers and conditions.) MM,molecular weight markers (Boehringer Mannheim).
TABLE 1Vascular Smooth Muscle Differentiation of Aortic Arch Arteriesin Lgdel/� Embryos at E10.5
No. ofembryos
Fourth aortic arch(right/left)
Sixth aortic arch(right/left)
WT 13 13/13 12/12Lgdel/� 10 4*/6* 5*/5*
* P � 0.05 compared with wild type.
162 Kochilas et al.
© 2002 Elsevier Science (USA). All rights reserved.

DISCUSSION
The cardiovascular and pharyngeal pouch defects seen inVCFS/DGS have been attributed to neural crest defects andresemble abnormalities produced in chick embryos by neu-ral crest ablation. Recently, animal models of VCFS/DGShave been created (Df1/�, Lgdel/�) using gene targetingapproaches to recapitulate the chromosomal defects foundin human patients (Lindsay et al., 1999; Merscher et al.,2001). In this report, we provide evidence that neural crestmigration remains intact in a mouse model of VCFS/DGS
(Lgdel/�) and that Lgdel haploinsufficiency produced in atissue-restricted fashion within neural crest cells does notproduce cardiovascular defects. However, neural crest dif-ferentiation into smooth muscle cells populating the aorticarches is deficient in the DiGeorge mouse model (Lgdel/�).These data suggest that a gene(s) in the Lgdel region affectsneural crest differentiation in the aortic arches through anon-cell-autonomous process. More specifically for Tbx1,which is considered the main candidate among the Lgdelgenes to be responsible for the cardiovascular anomalies inthe DiGeorge syndrome, the lack of expression in the
FIG. 5. Deficient smooth muscle differentiation in the aortic arch arteries of Lgdel/� embryos. SM22�-lacZ knockin mice were used todetect smooth muscle differentiation in wild type (A, D) and Lgdel/� (B, C, E) embryos at E10.5. The aortic arch arteries are numbered. Thethird aortic arch arteries are stained blue indicating smooth muscle differentiation in both wild type (A, D) and Lgdel/� embryo (B, C, E),while smooth muscle differentiation was severely deficient in the fourth and sixth aortic arch arteries of Lgdel/� embryos (B, C, E). Theblack arrow in (B) points to a hypoplastic fourth aortic arch artery that is devoid of smooth muscle. In (C), there is minimal evidence ofsmooth muscle differentiation in the dorsal segment of the sixth aortic arch artery. Coronal sections (D, E) confirm �-galactosidase activityin the third, fourth, and sixth aortic arch arteries bilaterally in wild type embryos, while no evidence of smooth muscle differentiation ispresent in the fourth arch artery of a Lgdel/� embryo (E). The sixth arch artery is not visible in this section. Double staining of embryosusing X-gal to identify Sm22� (blue) and anti-�-smooth muscle actin antibody to detect smooth muscle �-actin (dark brown) (F, G) confirmsthe deficient smooth muscle differentiation in the fourth aortic arch of Lgdel/� embryos (G) compared with wild type (F). A segment of theaortic arch from wild type (H) and Lgdel/� (I) in P3ProCreR26R background is shown at higher magnification. In these embryos, neural crestcells are labeled blue with X-gal. Neural crest cells that are differentiated into muscle cells are stained also dark brown with anti-�-smoothmuscle actin antibody. Note that the neural crest cells in the wild type embryo display uniformally expression of smooth muscle �-actin(brown arrows, H), while in the Lgdel/� embryo, there is a paucity of smooth muscle differentiation (dark brown and blue arrows, I).
163Neural Crest and DiGeorge Syndrome Mice
© 2002 Elsevier Science (USA). All rights reserved.

affected neural crest cells and the lack of effect of itshaploinsufficiency in the neural crest population on thecardiovascular development establish its nonautonomousrole in this setting. Since Tbx1 is expressed by the coremesenchyme and endoderm of the pharyngeal arches adja-cent to migrating neural crest (Garg et al., 2001), we proposea model in which Tbx1 regulates the expression of asecreted growth factor that signals to postmigratory neuralcrest cells to induce smooth muscle differentiation. Re-duced gene dosage of Tbx1 results in decreased or absentsecretion of the putative differentiation signal, and hencereduced and delayed smooth muscle differentiation.
Previous studies in chicken embryos have suggested thatthe absence of mature neural crest derivatives populatingaortic arch arteries results in regression of these arterialsegments (Waldo et al., 1996), and we propose that a similarmechanism results in regression of aortic arch arteries inLgdel/� mice. Fgfs, such as Fgf8 or Fgf10, that are down-regulated in Tbx1 null embryos and other models of abnor-mal arch formation, are possible secreted growth factorsthat could mediate interactions between endoderm or ecto-derm and migrating neural crest cells. However, furtherexperiments will be required to implicate Fgfs directly inthis pathway.
The aortic arch arteries develop in a craniocaudal se-quence forming a set of bilaterally symmetric arterialchannels. The first two pair, the most rostral of the pharyn-geal arch arteries, participate in the formation of cranialvessels and largely disappear by the time the most caudalvessels have fully differentiated. The third arch arteries giverise to the proximal common carotid arteries and moredistal cranial branches. The fourth aortic arches undergoextensive remodeling and form the distal segment of theaortic arch on the left (the “preductal” portion of thedescending aorta) and the branchiocephalic artery andproximal part of the right subclavian artery on the right.The fifth arch arteries are transient structures in highervertebrates and do not contribute to the mature vascula-ture. The sixth aortic arches undergo extensive remodelingand form the left ductus arteriosus and the proximal parts ofthe pulmonary arteries bilaterally (Edwards, 1948, 1977).The cardiovascular phenotype in the human VCFS/DGSand related mouse models (Lgdel/�, Df1/� and Tbx1�/�)includes abnormalities of structures derived from thefourth and sixth aortic arches The extensive analysis of theVCFS/DGS mouse models by our laboratory and others hasconfirmed the presence of fourth aortic arch defects de-tected as early as E10.5. We were unable to detect aorticarch defects in Lgdel/� embryos prior to that time point, inagreement with results of others studying a distinct butsimilar mouse model (Df1/�) (Lindsay and Baldini, 2001).Importantly, we were able to detect the presence of anarterial tube in the region of the fourth aortic arch at E10.5,despite the fact that smooth muscle differentiation wasdeficient or absent. Hence, we attribute subsequent vascu-lar defects to inappropriate regression of aortic arch seg-ments rather than to a lack of formation. We hypothesize
that regression is secondary to defective smooth muscledifferentiation, though we cannot rule out the possibilitythat these two defects are mechanistically unrelated.
Our results concerning the development of the sixthaortic arches in Lgdel/� mice are different than thosepreviously reported by Lindsay et al. These authors, study-ing Df1/� embryos, reported normal expression of smoothmuscle actin and normal development of sixth aortic archsegments (Lindsay and Baldini, 2001). Using Lgdel/� mice,we noted a significant deficiency of Sm22�-lacZ-expressingcells in the sixth aortic arch regions. We believe that thisobservation is significant for several reasons. Most impor-tantly, abnormalities of the central portion of the branchpulmonary arteries or absence of the ductus arteriosusoccur in human VCFS/DGS (Momma et al., 1996). Lgdel/�embryos do not have obvious sixth arch artery defectspossibly because the sixth arch arteries are less sensitive toabnormal Tbx1 dosage and the defect is not severe as in thefourth arch artery or the recovery from arterial growth thatwas described by Lindsay and Baldini (2001) is more suc-cessful in this case. Tbx1�/� embryos, though, display suchabnormalities of the sixth arch derivatives such as smallbranch pulmonary arteries (Jerome and Papaioannou, 2001),and interestingly enough, transgenics overexpressing Tbx1have related defects like absent main pulmonary artery(Merscher et al., 2001). These observations suggest thatsixth aortic arch defects contribute to human and mouseVCFS/DGS and may be related to abnormal levels of Tbx1signaling. Finally, cardiac neural crest cells (NCCs) mixextensively within the circumpharyngeal region beforecompleting migration into the pharyngeal arches. The sixtharch receives NCCs originating from the somite 1 to somite3 level, and therefore largely shares its cellular compositionwith that of the fourth arch, in contrast to the first twoarches that contain cranial neural crest cells (Kuratani andKirby, 1991). It is therefore not surprising that anomaliescoexist in the formation of the fourth and sixth aorticarches and their derivatives.
These data suggest the existence of an inductive interac-tion between pharyngeal tissues and postmigratory cardiacneural crest cells. There is precedent for such interactionswhich may occur at many stages of cardiac neural crestmigration and differentiation. Neural crest cells possessspecific positional information acquired prior to pharyngealarch formation but its implementation to form the muscu-loskeletal elements and vessels characteristic of each archis dependent on signals from both ectodermal and endoder-mal tissues in the developing pharyngeal region. Premigra-tory neural crest does not differentiate into cartilage, but ifcultured with cranial ectoderm or pharyngeal endodermdoes (Graveson and Armstrong, 1987), and outgrowth of thepharyngeal arches requires epithelial/mesenchymal inter-actions (Richman and Tickle, 1989). In Lgdel/� mice,abnormal inductive signals do not appear to affect neuralcrest migration, patterning, or early gene expression, atleast in terms of the molecular markers of migratory neuralcrest that we were able to assess. The first defects in neural
164 Kochilas et al.
© 2002 Elsevier Science (USA). All rights reserved.

crest gene expression that we were able to detect related tosmooth muscle differentiation. However, other mousemodels support the importance of signaling pathways andtissue–tissue interactions at alternate developmentalstages. Examples of such pathways include the semaphorinand endothelin signaling cascades (Brown et al., 2001;Clouthier et al., 1998; Feiner et al., 2001; Yanagisawa et al.,1998). Our results demonstrating normal endothelin 1 andSema3C expression in Tbx1-deficient embryos suggest thatTbx1 is functioning downstream of these factors or in analternate pathway.
In conclusion, our studies demonstrate that hemizygosityof the Lgdel region, homologous to the human DGCR, doesnot eliminate cardiac neural crest migration. However,neural crest cells in Lgdel/� embryos are defective in theirability to differentiate into smooth muscle. This defectinvolves cells of both fourth and sixth aortic arches. Theseabnormalities are associated with inappropriate regressionof aortic arch segments, thus accounting for the anomaliesobserved in Lgdel/� mice and humans with VCFS/DGS.Results of our neural crest-specific hemizygous deletion ofLgdel lead us to postulate a model in which Tbx1 and otherLgdel genes on mouse chromosome 16 display a haploin-sufficient phenotype because of a function outside of neuralcrest. Future studies will focus on the mechanisms bywhich Tbx1-expressing cells signal to postmigratory cardiacneural crest to trigger smooth muscle differentiation.
ACKNOWLEDGMENTS
We thank Dr. Eric Meyers for kindly providing the mouse Fgf8and Fgf10 cDNA clones. This work was supported by grants fromthe NIH (HL62974, HL61475 to J.A.E., HL67448 to L.K., HD34980to R.K and B.E.M.), the AHA (to J.A.E and B.E.M.), the WW SmithCharitable Trust (to J.A.E.), the Children’s Heart Foundation (toL.K.), and the Nemours Foundation (to L.K.).
Note added in proof. While this manuscript was in press, severalarticles appeared relevant to the role of Fgf8 in aortic arch remod-eling and Tbx1 function (Vitelli et al., Abu-Issa et al., Frank et al.,Development, 129(19), 2002).
REFERENCES
Bergwerff, M., DeRuiter, M. C., and Gittenberger-de Groot, A. C.(1999). Comparative anatomy and ontogeny of the ductus arte-riosus, a vascular outsider. Anat. Embryol. (Berl.) 200, 559–571.
Bockman, D. E., Redmond, M. E., and Kirby, M. L. (1990). Altereddevelopment of pharyngeal arch vessels after neural crest abla-tion. Ann. N. Y. Acad. Sci. 588, 296–304.
Brown, C. B., Feiner, L., Lu, M. M., Li, J., Ma, X., Webber, A. L., Jia,L., Raper, J. A., and Epstein, J. A. (2001). PlexinA2 and sema-phorin signaling during cardiac neural crest development. Devel-opment 128, 3071–3080.
Chang, M. W., Barr, E., Seltzer, J., Jiang, Y. Q., Nabel, G. J., Nabel,E. G., Parmacek, M. S., and Leiden, J. M. (1995). Cytostatic genetherapy for vascular proliferative disorders with a constitutively
active form of the retinoblastoma gene product. Science 267,518–522.
Chiang, C., Litingtung, Y., Lee, E., Young, K. E., Corden, J. L.,Westphal, H., and Beachy, P. A. (1996). Cyclopia and defectiveaxial patterning in mice lacking Sonic hedgehog gene function.Nature 383, 407–413.
Ciment, G., and Weston, J. A. (1985). Segregation of developmentalabilities in neural-crest-derived cells: Identification of partiallyrestricted intermediate cell types in the pharyngeal arches ofavian embryos. Dev. Biol. 111, 73–83.
Clouthier, D. E., Hosoda, K., Richardson, J. A., Williams, S. C.,Yanagisawa, H., Kuwaki, T., Kumada, M., Hammer, R. E., andYanagisawa, M. (1998). Cranial and cardiac neural crest defects inendothelin-A receptor-deficient mice. Development 125, 813–824.
Crossley, P. H., and Martin, G. R. (1995). The mouse Fgf8 geneencodes a family of polypeptides and is expressed in regions thatdirect outgrowth and patterning in the developing embryo.Development 121, 439–451.
Edwards, J. E. (1948). Anomalies of the derivatives of the aortic archsystem. Med. Clin. North Am. 32, 925–949.
Edwards, J. E. (1977). Anomalies of the aortic arch system. BirthDefects Orig. Artic Ser. 13, 47–63.
Epstein, J. A., Li, J., Lang, D., Chen, F., Brown, C. B., Jin, F., Lu,M. M., Thomas, M., Liu, E., Wessels, A., and Lo, C. W. (2000).Migration of cardiac neural crest cells in Splotch embryos.Development 127, 1869–1878.
Epstein, J. A., Shapiro, D. N., Cheng, J., Lam, P. Y., and Maas, R. L.(1996). Pax3 modulates expression of the c-Met receptor duringlimb muscle development. Proc. Natl. Acad. Sci. USA 93,4213–4218.
Feiner, L., Webber, A. L., Brown, C. B., Lu, M. M., Jia, L., Feinstein,P., Mombaerts, P., Epstein, J. A., and Raper, J. A. (2001). Targeteddisruption of semaphorin 3C leads to persistent truncus arterio-sus and aortic arch interruption. Development 128, 3061–3070.
Garg, V., Yamagishi, C., Hu, T., Kathiriya, I. S., Yamagishi, H., andSrivastava, D. (2001). Tbx1, a DiGeorge syndrome candidategene, is regulated by sonic hedgehog during pharyngeal archdevelopment. Dev. Biol. 235, 62–73.
Goldmuntz, E., and Emanuel, B. S. (1997). Genetic disorders ofcardiac morphogenesis. The DiGeorge and velocardiofacial syn-dromes. Circ. Res. 80, 437–443.
Graveson, A. C., and Armstrong, J. B. (1987). Differentiation ofcartilage from cranial neural crest in the axolotl (Ambystomamexicanum). Differentiation 35, 16–20.
Iida, K., Koseki, H., Kakinuma, H., Kato, N., Mizutani-Koseki, Y.,Ohuchi, H., Yoshioka, H., Noji, S., Kawamura, K., Kataoka, Y.,Ueno, F., Taniguchi, M., Yoshida, N., Sugiyama, T., and Miura,N. (1997). Essential roles of the winged helix transcription factorMFH-1 in aortic arch patterning and skeletogenesis. Develop-ment 124, 4627–4638.
Jerome, L. A., and Papaioannou, V. E. (2001). DiGeorge syndromephenotype in mice mutant for the T-box gene, Tbx1. Nat. Genet.27, 286–291.
Kirby, M. L. (1990). Alteration of cardiogenesis after neural crestablation. Ann. N. Y. Acad. Sci. 588, 289–295.
Kirby, M. L., Turnage, K. L., 3rd, and Hays, B. M. (1985). Charac-terization of conotruncal malformations following ablation of“cardiac” neural crest. Anat. Rec. 213, 87–93.
Kirby, M. L., and Waldo, K. L. (1990). Role of neural crest incongenital heart disease. Circulation 82, 332–340.
165Neural Crest and DiGeorge Syndrome Mice
© 2002 Elsevier Science (USA). All rights reserved.

Kirby, M. L., and Waldo, K. L. (1995). Neural crest and cardiovas-cular patterning. Circ. Res. 77, 211–215.
Kuratani, S. C., and Kirby, M. L. (1991). Initial migration anddistribution of the cardiac neural crest in the avian embryo: anintroduction to the concept of the circumpharyngeal crest. Am. J.Anat. 191, 215–227.
Li, J., Chen, F., and Epstein, J. A. (2000). Neural crest expression ofCre recombinase directed by the proximal Pax3 promoter intransgenic mice. Genesis 26, 162–164.
Li, J., Liu, K. C., Jin, F., Lu, M. M., and Epstein, J. A. (1999).Transgenic rescue of congenital heart disease and spina bifida inSplotch mice. Development 126, 2495–2503.
Lindsay, E. A., and Baldini, A. (2001). Recovery from arterial growthdelay reduces penetrance of cardiovascular defects in mice de-leted for the DiGeorge syndrome region. Hum. Mol. Genet. 10,997–1002.
Lindsay, E. A., Botta, A., Jurecic, V., Carattini-Rivera, S., Cheah,Y. C., Rosenblatt, H. M., Bradley, A., and Baldini, A. (1999).Congenital heart disease in mice deficient for the DiGeorgesyndrome region. Nature 401, 379–383.
Lindsay, E. A., Vitelli, F., Su, H., Morishima, M., Huynh, T.,Pramparo, T., Jurecic, V., Ogunrinu, G., Sutherland, H. F.,Scambler, P. J., Bradley, A., and Baldini, A. (2001). Tbx1 haplo-insufficieny in the DiGeorge syndrome region causes aortic archdefects in mice. Nature 410, 97–101.
Merscher, S., Funke, B., Epstein, J. A., Heyer, J., Puech, A., Lu,M. M., Xavier, R. J., Demay, M. B., Russell, R. G., Factor, S.,Tokooya, K., Jore, B. S., Lopez, M., Pandita, R. K., Lia, M.,Carrion, D., Xu, H., Schorle, H., Kobler, J. B., Scambler, P.,Wynshaw-Boris, A., Skoultchi, A. I., Morrow, B. E., and Kucher-lapati, R. (2001). TBX1 is responsible for cardiovascular defects invelo-cardio- facial/DiGeorge syndrome. Cell 104, 619–629.
Momma, K., Kondo, C., and Matsuoka, R. (1996). Tetralogy ofFallot with pulmonary atresia associated with chromosome22q11 deletion. J. Am. Coll. Cardiol. 27, 198–202.
Puech, A., Saint-Jore, B., Merscher, S., Russell, R. G., Cherif, D.,Sirotkin, H., Xu, H., Factor, S., Kucherlapati, R., and Skoultchi,A. I. (2000). Normal cardiovascular development in mice defi-cient for 16 genes in 550 kb of the velocardiofacial/DiGeorgesyndrome region. Proc. Natl. Acad. Sci. USA 97, 10090–10095.
Richman, J. M., and Tickle, C. (1989). Epithelia are interchangeablebetween facial primordia of chick embryos and morphogenesis iscontrolled by the mesenchyme. Dev. Biol. 136, 201–210.
Soriano, P. (1999). Generalized lacZ expression with the ROSA26Cre reporter strain. Nat. Genet. 21, 70–71.
Trumpp, A., Depew, M. J., Rubenstein, J. L., Bishop, J. M., andMartin, G. R. (1999). Cre-mediated gene inactivation demon-strates that FGF8 is required for cell survival and patterning ofthe first branchial arch. Genes Dev. 13, 3136–3148.
Vitelli, F., Morishima, M., Taddei, I., Lindsay, E. A., and Baldini, A.(2002). Tbx1 mutation causes multiple cardiovascular defectsand disrupts neural crest and cranial nerve migratory pathways.Hum. Mol. Genet. 11, 915–922.
Waldo, K. L., Kumiski, D., and Kirby, M. L. (1996). Cardiac neuralcrest is essential for the persistence rather than the formation ofan arch artery. Dev. Dyn. 205, 281–292.
Wendling, O., Dennefeld, C., Chambon, P., and Mark, M. (2000).Retinoid signaling is essential for patterning the endoderm of thethird and fourth pharyngeal arches. Development 127, 1553–1562.
Winnier, G. E., Kume, T., Deng, K., Rogers, R., Bundy, J., Raines,C., Walter, M. A., Hogan, B. L., and Conway, S. J. (1999). Roles forthe winged helix transcription factors MF1 and MFH1 in cardio-vascular development revealed by nonallelic noncomplementa-tion of null alleles. Dev. Biol. 213, 418–431.
Yanagisawa, H., Hammer, R. E., Richardson, J. A., Williams, S. C.,Clouthier, D. E., and Yanagisawa, M. (1998). Role of Endothelin-1/Endothelin-A receptor-mediated signaling pathway in the aor-tic arch patterning in mice. J. Clin. Invest. 102, 22–33.
Zhang, J. C., Kim, S., Helmke, B. P., Yu, W. W., Du, K. L., Lu,M. M., Strobeck, M., Yu, Q., and Parmacek, M. S. (2001).Analysis of SM22alpha-deficient mice reveals unanticipated in-sights into smooth muscle cell differentiation and function. Mol.Cell. Biol. 21, 1336–1344.
Received for publication May 31, 2002Revised August 15, 2002
Accepted August 15, 2002Published online September 30, 2002
166 Kochilas et al.
© 2002 Elsevier Science (USA). All rights reserved.