the role of paladin in endothelial cell signaling and
TRANSCRIPT

ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2016
Digital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 1225
The Role of Paladin in EndothelialCell Signaling and Angiogenesis
ANJA NITZSCHE
ISSN 1651-6206ISBN 978-91-554-9578-7urn:nbn:se:uu:diva-281708

Dissertation presented at Uppsala University to be publicly examined in Fåhraeussalen,Rudbeck laboratory (C5), Dag Hammarskjölds väg 20, Uppsala, Thursday, 9 June 2016 at09:15 for the degree of Doctor of Philosophy (Faculty of Medicine). The examination willbe conducted in English. Faculty examiner: Professor Frank-Dietmar Böhmer (Institute ofMolecular Cell Biology, Jena University Hospital, Jena, Germany).
AbstractNitzsche, A. 2016. The Role of Paladin in Endothelial Cell Signaling and Angiogenesis.Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine1225. 47 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-554-9578-7.
Angiogenesis, the formation of new blood vessels from a pre-existing vasculature, is crucialduring development and for many diseases including cancer. Despite tremendous progressin the understanding of the angiogenic process, many aspects are still not fully elucidated.Several attempts have been made to identify novel genes involved in endothelial cell biologyand angiogenesis. Here we focused on Pald1, a recently identified, vascular-enriched geneencoding paladin. Our in vitro studies indicate that paladin is a lipid phosphatase catalyzingdephosphorylation of phosphatidylinositol phosphates, a process essential for endocytosis andintracellular vesicle trafficking.
We confirmed paladin’s vascular expression pattern and revealed a shift from a broadendothelial cell expression during development to an arterial mural cell-restricted expression inseveral vascular beds in adult mice. Paladin expression in the lung, however, was not restrictedto the vasculature, but was also observed in pneumocytes and myofibroblasts. Lungs of female,but not male, Pald1 null mice displayed an obstructive lung phenotype with increased alveolarair sacs that were already apparent early in the alveolarization process. Only endothelial cells,but not other main lung cell types, were affected by loss of paladin. Endothelial cell number wasreduced in 4-week old mice, possibly due to increased endothelial turnover in Pald1 deficientlungs.
Vascular defects were also found in the retina. Loss of paladin led to reduced retinal vascularoutgrowth accompanied by a hyperdense and hypersprouting vascular front. Downstreamsignaling of the major angiogenic driver, vascular endothelial growth factor receptor 2(VEGFR2) was sustained in Pald1 null mice, and VEGFR2 degradation was impaired.Furthermore, paladin inhibited endothelial cell junction stability and loss of paladin led toreduced vascular permeability.
Whether the differences in VEGFR2 signaling and adherens junction stability are connectedremains to be fully explored. The newly identified lipid phosphatase activity of paladin and itsspecific effects on VEGFR2 signaling and adherens junction stability indicate that paladin maybe controlling the endocytic pathway.
Keywords: Pald1, endothelium, lung, vascular permeability, phosphatase, angiogenesis
Anja Nitzsche, Department of Immunology, Genetics and Pathology, Rudbecklaboratoriet,Uppsala University, SE-751 85 Uppsala, Sweden.
© Anja Nitzsche 2016
ISSN 1651-6206ISBN 978-91-554-9578-7urn:nbn:se:uu:diva-281708 (http://urn.kb.se/resolve?urn=urn:nbn:se:uu:diva-281708)

To my family
It always seems impossible until it’s done.Nelson Mandela

Cover: Postnatal day 5 retina stained for endothelial cells (isolectin B4, green) and adherens junctions (VE-cadherin, red).

List of Papers
This thesis is based on the following papers, which are referred to in the text by their Roman numerals.
I Wallgard E, Nitzsche A, Larsson J, Guo X, Dieterich LC,
Dimberg A, Olofsson T, Pontén FC, Mäkinen T, Kalen M, Hellström M (2012). Paladin (X99384) is expressed in the vasculature and shifts from endothelial to vascular smooth muscle cells during mouse development. Developmental Dy-namics, 241(4):770-786.
II Egaña I, Nitzsche A, Kaito H, Becker L, Garrett L, Niaudet C, Liu W, Vanlandewijck M, Larsson J, Hrabe de Angelis M, Fuchs H, Gailus-Durner V, Vernaleken A, Klopstock T, Hölter SM, Wurst W, Rask-Andersen H, German Mouse Clinic Con-sortium, Yildirim AÖ, Hellström M: Female mice lacking Pald1 exhibit endothelial cell apoptosis and emphysema. Manuscript
III Nitzsche A, Testini C, Ekvärn E, Larsson J, Bentley K,
Philippides A, Roche FP, Egaña I, Smith R, Hellberg C, Ballmer-Hofer K, Hellström M: Paladin (Pald1) regulates en-dothelial sprouting, VE-cadherin junction stability and vascu-lar permeability. Manuscript
Reprint of paper I was made with permission from publisher.

Additional publication
The author also contributed to the following paper not included in this thesis:
Kalen M, Heikura T, Karvinen H, Nitzsche A, Weber H, Esser N, Yla-Herttuala S, Hellström M (2011). Gamma-secretase inhibitor treatment promotes VEGF-A-driven blood vessel growth and vascular leakage but disrupts neovascular perfusion. PLoS One 6:e18709.

Contents
Introduction ..................................................................................................... 9 The vascular system ................................................................................... 9
Blood vessel formation ........................................................................ 10 In vivo models of developmental angiogenesis ................................... 11 Vascular endothelial growth factors and their receptors ...................... 11 Molecular mechanisms of sprouting angiogenesis .............................. 13 Endothelial cell adherens junctions and vascular permeability ........... 15 Endosomal trafficking ......................................................................... 18
Lung physiology and disease ................................................................... 20 Lung development ............................................................................... 20 Alveolar cell types ............................................................................... 21 Chronic obstructive pulmonary disease and emphysema .................... 22
Phosphatases............................................................................................. 23 Protein Tyrosine Phosphatases ............................................................ 23 Pseudophosphatases ............................................................................. 26
Paladin ...................................................................................................... 27
Present investigations .................................................................................... 29 Paper I ...................................................................................................... 29 Paper II ..................................................................................................... 30 Paper III .................................................................................................... 32
Concluding remarks and future perspectives ................................................ 34
Acknowledgments......................................................................................... 36
References ..................................................................................................... 38

Abbreviations
Arf ADP-ribosylation factor BASC Bronchioalveolar stem cell COPD Chronic obstructive pulmonary disease Csk C-terminal Src tyrosine protein kinase DASC Distal airway stem cell DEP1 density enhanced phosphatase-1 Dll4 Delta-like ligand 4 DUSP Dual-specificity phosphatase E# Embryonic day ECM Extracellular matrix EEA1 Early endosome antigen 1 Erk Extracellular regulated kinase FAK Focal adhesion kinase GAP GTPase-activating protein GEF Guanine nucleotide exchange factor HIF Hypoxia-inducible factor LMW-PTP Low molecular weight protein tyrosine phosphatase MMP Matrix metalloproteinase MTM myotubularin N-cadherin Neuronal cadherin P# Postnatal day PAK p21-activated kinase PIP phosphatidylinositol phosphate PLC Phospholipase C PTEN phosphatase and tensin homolog deleted on chromo-
some 10 PTP Protein tyrosine phosphatase ROS Reactive oxygen species RTK Receptor tyrosine kinase TNFα tumor necrosis factor TSAd T-cell specific adaptor VE-cadherin Vascular endothelial cadherin VEGF Vascular endothelial growth factor VEGFR Vascular endothelial growth factor receptor VE-PTP Vascular endothelial protein tyrosine phosphatase

9
Introduction
The vascular system The complex vertebrate body depends on a functional, highly branched vas-cular system, comprising both blood and lymphatic vasculatures. The blood vessel network ensures supply of oxygen and nutrients to the tissues and removal of waste products1. Oxygenated blood from the lung is transported via the heart and, through arteries and arterioles, to the periphery where gas exchange is facilitated by thin capillaries (diameter < 10 µm). Through post-capillary venules and veins deoxygenated blood is transported back to the heart and enters the pulmonary circulation to be replenished with oxygen (Figure 1)2. The lymphatic vascular system ensures the drainage of excess interstitial fluids back into the systemic circulation1.
Figure 1. The vascular system. Blood vessels are composed of endothelial cells. Pericytes sparsely cover capillaries, whereas arteries and vein are surrounded by smooth muscle cells. Peripheral arteries transport oxygenated blood to the periphery for gas exchange in the capil-laries and veins transport the deoxygenated blood back to the heart, where it enters the lung circulation. Reprinted with permission3.
Blood vessels are composed of endothelial cells, which in capillaries are sparsely covered by pericytes and surrounded by a laminin-rich basement membrane4. Pericytes tightly interact with endothelial cells and contribute to vessel stabilization, maturation and remodeling5. Arteries and veins, howev-

10
er, are surrounded by a different type of mural cells, termed vascular smooth muscle cells. Smooth muscle cells form thick concentric layers around arter-ies, which are important to withstand the high shear stress and pulsatile blood flow. In contrast, veins are exposed to low shear stress and are covered by fewer smooth muscle cells1.
Different vascular beds show unique structural and functional properties to meet the needs of their specific tissues, which results in significant heter-ogeneity among endothelial cells2. For example, endothelial cells of glomer-ular capillaries in the kidney are highly fenestrated for filtering blood6, while endothelial cells of the blood-brain barrier are almost completely impermea-ble7. The blood-brain barrier is further stabilized by the tight interaction with pericytes8.
Blood vessel formation Blood vessel formation occurs by two distinct mechanisms: the de novo for-mation called vasculogenesis and the subsequent expansion of the vascular plexus termed angiogenesis. In mice, by embryonic day (E) 6.5-7 precursor cells cluster to form blood islands in the yolk sac. Cells at the periphery of blood islands, called angioblasts, differentiate into endothelial cells, while inner blood island cells develop into hematopoietic precursors9. In contrast to the side-by-side differentiation of hematopoietic and endothelial cells in the yolk sac, intraembryonic endothelial cells differentiate from solitary an-gioblasts that are unaccompanied by hematopoietic precursors9,10. At around E8.0 the first intraembryonic endothelial cells can be detected, which will give rise to the dorsal aorta. The lateral vascular network is established be-tween E8.2 and E8.511. Intra- and extraembryonic primitive vascular plexi anastomose at around E8.5 and a connection to the developing heart tube is made9,12. First irregular contraction of cardiac myocytes can be observed at E8.25, but unidirectional blood flow is only established at E9.2513. The sub-sequent entry of erythrocytes into the circulation creates shear stress that is essential for vascular remodeling14.
Expansion of the primary plexus occurs via intussusception (splitting of a mother vessel to form two daughter vessels) and sprouting angiogenesis, which is the most prominent process of blood vessel formation during devel-opment15. The molecular mechanisms of sprouting angiogenesis are dis-cussed in more detail below.
In adults, angiogenesis is a rare event, only normally occurring during wound healing, tissue remodeling and in the female reproductive system15. Looping, described as blood vessel translocation due to biomechanical forc-es, has recently been identified as a third process of neovascularization dur-ing wound healing16. The importance of post-developmental angiogenesis is highlighted by conditions arising from insufficient blood supply leading to ischemic heart disease, neurodegeneration or stroke15. However, angiogene-

11
sis is re-activated in many pathological conditions, for example, every solid tumor exceeding 1-2 mm3 in size needs to induce angiogenesis for sufficient supply of oxygen and nutrients17. Abnormal angiogenesis is also a central process in blinding eye diseases18,19.
In vivo models of developmental angiogenesis Much of our current understanding of angiogenesis is derived from two model systems: the zebrafish embryo and the postnatal mouse retina. Both models are particularly good for angiogenesis research because of the easy accessibility of the vasculature, the wide availability of tools to visualize the vasculature, the ability to modulate gene expression or protein-protein inter-actions, and their tightly regulated, highly organized vascular development. This structured, reliable pattern of neo-vascularization facilitates detection of any irregularities after experimental intervention20-22. However, caution must be taken when attempting to generalize, as all vascular beds are distinct.
The intraretinal vasculature in the mouse eye develops postnatally. Within the first week after birth the vasculature expands radially from the optic nerve and reaches the periphery of the retina (Figure 2). From postnatal day (P) 7 onwards, the vasculature grows to the deep layer and expands to the periphery until P12; finally the intermediate layer is formed by P15. At P21 the retinal vasculature is fully mature and several interconnecting capillaries between the layers have been established20,21. Interestingly, only the superfi-cial layer will develop into a hierarchical vasculature composed of veins, arteries and capillaries, the deeper layers will only form a capillary net-work21.
Figure 2. Postnatal development of retinal vasculature. Vascular outgrowth begins from the optic nerve in the center of the retina and progresses towards the hypoxic periphery. By post-natal day 8 the vasculature has reached the rim of the retina. Reprinted with permission20.
Vascular endothelial growth factors and their receptors Angiogenesis is regulated by a fine balance of pro- and anti-angiogenic sig-nals with the relative strength of each determining the outcome. The major pro-angiogenic factors are members of the vascular endothelial growth factor (VEGF) family. There are five mammalian VEGF isoforms (VEGF-A – D,

12
placental growth factor), which normally act as homodimers. Among the three VEGF receptors (VEGFR1 – 3), homodimers as well as VEGFR1/2 and VEGFR2/3 heterodimers can be found23. VEGF-A-VEGFR2 signaling is the primary regulatory event in angiogenesis, but recent data also highlights the importance of VEGF-C-VEGFR3 signaling in angiogenesis24-28.
VEGF-A expression depends significantly upon tissue oxygenation status where, under hypoxic conditions (i.e. low oxygen tension), VEGF-A is up-regulated. Oxygen sensitive expression is mediated by the hypoxia-inducible factor (HIF), a dimer of HIFα and HIF1β. In the presence of O2, dioxygenas-es catalyze prolyl and asparaginyl hydroxylation of HIFα, which marks HIFα for proteasomal degradation and inhibits its binding to transcriptional co-activators, respectively29. Under hypoxic conditions however, HIFα can ac-cumulate to form an active HIF transcription factor, which induces expres-sion of many angiogenic factors apart from VEGF-A, such as erythropoietin, endothelial nitric oxide synthase and VEGFR129.
VEGFRs are classical receptor tyrosine kinases (RTKs) with an extracel-lular domain comprised of seven immunoglobulin-like repeats, a transmem-brane domain and an intracellular tail containing a split tyrosine kinase do-main (Figure 3). VEGF binding leads to receptor dimerization inducing au-tophosphorylation of VEGFR23,30. The three major tyrosine phosphorylation sites on the cytoplasmic tail of VEGFR2 outside the kinase domain are Y949, Y1173 and Y1212 in mice31. All of these phosphorylation sites pro-vide binding sites for a wide range of adaptors and signaling molecules lead-ing to distinct downstream signaling events. By mutating specific tyrosine residues to phenylalanine, thereby preventing site-specific phosphorylation, the function of individual VEGFR2 phosphorylation sites has been studied. Mutation of Y1173 in the mouse led to early embryonic lethality due to fail-ure in vasculogenesis and hematopoiesis, which closely matched the pheno-type seen in the Vegfr2 full knockout32,33. Phosphorylation of Y1173 trans-mits important proliferation, survival and migration signals (see Figure 3 for a detailed overview)23. In contrast to Y1173, mice carrying mutations in the other two major phosphorylation sites, Y949 and Y1212, are viable and fer-tile32,34. Phosphorylation of Y949 provides a binding site for the T-cell spe-cific adaptor (TSAd), triggering activation of src kinase and inducing vascu-lar leakage (see also below)31,34,35. Phosphorylation of Y1212 was suggested to be important for endothelial cell migration23.
Importantly, the amplitude, duration and specificity of VEGFR2 downstream signaling is also determined by the subcellular localization of VEGFR236. Activated VEGFR2 is endocytosed into clathrin-coated vesicles and trans-ported to early endosomes and either to late endosomes and lysosomes, or to recycling endosomes and back to the plasma membrane36,37. Strikingly, full activation of extracellular regulated kinase 1/2 (Erk1/2) as well as Akt re-quires internalization of VEGFR2, while phospholipase C (PLC can be activated from the cell membrane38-41. VEGFR2 endocytosis is regulated by

13
interaction with co-receptors, such as Neuropilin 1, or Ephrin-B2, a trans-membrane ligand for Eph receptors38,41. Consequently, the spatial and tem-poral regulation of VEGFR2 endocytosis and endosomal trafficking is tight-ly controlled during blood vessel formation and maturation38,39.
Another layer of complexity is added by the endothelial cell apical-basal polarity leading to differential distribution of VEGFRs on the apical and the basal cell membrane. In brain and retinal microvasculature it was shown that VEGF-A administrated from the apical side has cytoprotective functions, while VEGF-A from the tissue side leads to vascular permeability42.
Figure 3. Schematic outline of VEGFR2 phosphorylation sites and downstream signaling. VEGFR2 is composed of seven Ig-like domains (circles), one transmembrane domain and a cytoplasmic tail with a split tyrosine kinase domain (boxes). VEGFR2 phosphorylation sites of human VEGFR2 are depicted: human Y951 corresponds to Y949 in mice, Y1175 to Y1173 and Y1214 to Y1212. Binding partners and major downstream signaling effects (i.e. prolifera-tion, migration, survival and permeability) are indicated. Reprinted with permission23.
Molecular mechanisms of sprouting angiogenesis Angiogenic sprouting is the process by which a new sprout buds from an existing blood vessel to grow towards areas of incipient hypoxia, ultimately extending the existing vasculature and alleviating oxygen/nutrient depriva-tion. Angiogenic sprouting is driven mainly by VEGF-A. In the postnatal retina, VEGF-A is secreted by astrocytes in the avascular periphery43.

14
VEGF-A is presented as a gradient, which is established by retaining VEGF-A on the extracellular matrix (ECM) via binding to heparan sulfate proteo-glycans or by VEGF-A sequestration by the soluble form of high-affinity VEGFR11,24. Upon VEGF-A stimulation, a fraction of endothelial cells ac-quire a motile phenotype and lose their apical-basal polarity to form the tip cell of a new growing vessel sprout. Neighboring cells will instead become stalk cells following the tip cell. The tip cell is believed to sense guidance cues, such as UNC5B/netrin-1, Slit/Robo4 and Sema3E/PlexinD1, by using long filopodia extensions to lead the new sprout into hypoxic areas with high VEGF-A levels15,24,43. For the tip cell to invade the surrounding tissue, base-ment membrane is broken down and rearranged by matrix-degrading en-zymes, e.g. matrix metalloproteinases (MMPs). Importantly, breakdown of basement membrane and the surrounding extracellular matrix can result in release of matrix-bound VEGF-A and therefore influences the availability of VEGF-A1,24,44. Extracellular matrix components themselves have been shown to modulate endothelial cell behavior. For example, exposure to inter-stitial collagen type I induces endothelial cell morphogenesis, whereas basement membrane laminins trigger vessel stabilization45. Mechanical ten-sion transmitted via the extracellular matrix is an additional guidance cue facilitating coordinated vascular morphogenesis over a sizeable distance45,46.
One important signaling event in discriminating between tip and stalk cell is lateral inhibition via delta-like ligand 4 (Dll4)-Notch signaling47. Ligand Dll4 and its receptor Notch are membrane-bound molecules and binding can only occur between two adjacent cells. In response to VEGF-A, endothelial cells upregulate Dll448. The Dll4-mediated Notch activation on opposing cells leads to suppression of VEGFR2 and VEGFR3 expression and thus renders these cells less responsive to VEGF stimulation24,48. Additionally, secretion of high affinity soluble VEGFR1 is induced by Notch1, limiting the availability of VEGF-A and thus attenuating VEGFR2 signaling in Notch1 activated cells48-50. Consequently, endothelial cells encountering high concentrations of VEGF will inhibit neighboring cells, which are less VEGF responsive as a result of lateral inhibition.
Recent findings indicate a more complex interplay between Notch com-ponents and VEGFRs than outlined by the described VEGFR–Dll4–Notch signaling circuit50. Transcriptional profiling of endothelial cells from Dll4+/- early postnatal retinas demonstrated an upregulation of 411 genes and a downregulation of 158 genes in those heterozygous cells compared to wild type controls, thus emphasizing the complexity of Notch signaling51.
It was shown that the tip-stalk cell specification is highly dynamic and endothelial cells constantly compete for tip cell positioning, which requires constant changes in cell-cell adhesiveness52,53. Interrupting endothelial ad-herens junctions postnatally results in intensive filopodia formation in the developing retina54 and similarly in the zebrafish embryo55 highlighting the

15
importance of adherens cell-cell junctions in sprouting angiogenesis. Inter-estingly, stabilizing endothelial adherens junctions by permanent linkage to the actin cytoskeleton did not result in any gross abnormalities during devel-opmental angiogenesis56.
Sprout formation is only the first step in creating a new blood vessel. Subse-quent fusion of sprouts with each other or with a nearby vessel, termed anas-tomosis, and lumen formation are necessary to establish functional vessels. Basement membrane deposition and recruitment of pericytes lead to further maturation and stabilization of newly formed blood vessels1,24. Finally, re-modeling of the vascular network, including extensive vessel pruning and regression, is necessary to ensure a fully functional vasculature57.
Endothelial cell adherens junctions and vascular permeability To fulfill their physiological functions, blood vessels must have the ability to be tight and permeable at the same time. Endothelial cell junctions are criti-cally involved in maintaining cell monolayer integrity within the vessel wall, in order to prevent uncontrolled vascular leakage and withstand mechanical forces (i.e. blood pressure), while at the same time allowing transmission of leukocytes and small solutes across the endothelium in a selective, controlled manner58. Two major types of cell junctions control the endothelial mono-layer integrity: tight junctions and adherens junctions59. In contrast to epithe-lial cells, tight junctions and adherens junctions are intermingled in endothe-lial cells and lack a sharp spatial separation59. Adherens junctions are formed by homophilic interactions of cadherins. Endothelial cells express neuronal (N)-cadherin and the endothelial specific vascular endothelial (VE)-cadherin60-62. While N-cadherin has a more dispersed distribution and was suggested to be important for adhesion between endothelial and non-endothelial cells, such as pericytes, VE-cadherin forms a zipper-like struc-ture between endothelial cells of blood vessels60,63.
VE-cadherin belongs to the classical cadherin family and is a calcium-dependent transmembrane protein with five extracellular cadherin repeats (EC1-EC5) and a cytoplasmic tail61,64. VE-cadherin dimers can form homo-philic interaction in cis (on the same cell) or trans (between adjacent cells)65. The cytoplasmic tail of VE-cadherin shares high sequence similarity to other classical cadherins and thus provides binding sites for typical cadherin inter-action partners, called catenins (Figure 4)60. -catenin, plakoglobin (also termed -catenin) and -catenin are critical for anchoring VE-cadherin to the cytoskeleton and thereby allowing cytoskeletal forces to transmit between and within cells66,67. Plakoglobin and -catenin are structurally very similar; they are both composed of twelve armadillo repeats (named after the Dro-sophila homologue armadillo) and bind to the membrane-distal part of the VE-cadherin cytoplasmic tail65,68. However, despite their similarity, plako-

16
globin and -catenin possess different functions. Plakoglobin not only bridg-es between VE-cadherin and the actin cytoskeleton, but also mediates bind-ing to intermediate filaments via desmoplakin65. It has been shown in endo-thelial cell cultures that the ratio between plakoglobin and -catenin that is bound to VE-cadherin increases during the maturation of endothelial cell junctions69. Plakoglobin/-catenin association is just one example of the many junctional changes occurring during the maturation process from es-tablishing new cell-cell contacts to the formation of a tight, selectively per-meable endothelial monolayer60. Interestingly, even in mature adherens junc-tions there is a constant dynamic equilibrium of assembly and disassembly of adhesion proteins as shown for E-cadherin junctions in epithelial cells70.
Figure 4. VE-cadherin homodimer structure. VE-cadherin is composed of an ectodomain comprising five cadherin repeats (EC1-5) stabilized by Ca2+ ions, a single transmembrane (TM) domain and a cytoplasmic tails. The cytoplasmic tail provides binding sites for p120 catenin, plakoglobin and -catenin, the latter link VE-cadherin to the actin cytoskeleton via -catenin. Reprinted with permission58.
VE-cadherin has been found to be involved in a plethora of cellular func-tions, mediating contact inhibition of cell growth, cell polarity, lumen for-mation and vascular remodeling. VE-cadherin can be seen as a “signaling hub” in endothelial cells and associates with several kinases, phosphatases and small GTPases. VE-cadherin influences, and is influenced by, signaling through VEGFR2, Angiopoietin-Tie, TGF-Smad, FGFR, PI3K-Akt and Notch. Furthermore, VE-cadherin regulates transcription in endothelial cells, mainly by acting as a trap for transcriptionally active binding partners, such as -catenin, plakoglobin and p120, another armadillo protein58,60,71.
Adherens junctions are critical for restraining the permeability of cell sheets and, indeed, permeability control was the first function described for VE-cadherin. The addition of an activating N-terminal VE-cadherin-specific antibody led to the disruption of an endothelial cell monolayer and conse-quently increased permeability61. Since these early experiments two main concepts regarding how the endothelial barrier can be weakened have emerged: either by (1) dissociation of the VE-cadherin-catenin complex or by (2) endocytosis of the VE-cadherin-catenin complex (Figure 5)60. Both mechanisms seem to be controlled by the phosphorylation status of VE-cadherin and its associated catenins. Interestingly, leukocytes, inflammatory

17
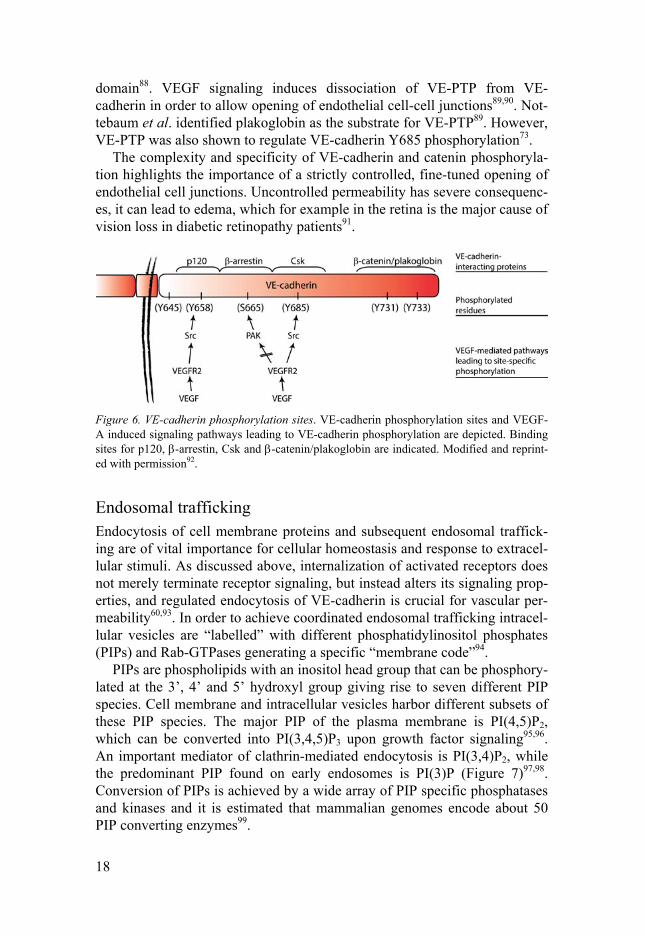
cytokines and VEGF, all target different phosphorylation sites60. Y731 in the cytoplasmic tail of VE-cadherin, for example, is pivotal for leukocyte trans-migration, whereas VEGF-mediated opening of junctions involves phos-phorylation of Y685 and Y658 (Figure 6)72-75.
Figure 5. Opening of endothelial cell junctions. Upon leukocyte attachment or receptor-mediated signaling (a) endothelial cell adherens junctions are likely to open either by disso-ciation of the VE-cadherin-catenin complex (b) or by endocytosis of the entire complex (c). Modified and reprinted with permission60.
Major regulators of VEGF-induced VE-cadherin phosphorylation are the src family kinases60,71. Interestingly, it was shown that activated src can only be found in veins and co-localizes with phosphorylated VE-cadherin, and loss of VE-cadherin phosphorylation correlates with sites of leakage in post-capillary venules and veins75. Src-induced VE-cadherin Y685 phosphoryla-tion also provides a binding site for C-terminal src tyrosine protein kinase (Csk), which is a potent inhibitor of src establishing a negative feedback loop for src activity76,77.
It has been suggested that VEGF-induced phosphorylation of VE-cadherin leads to dissociation of p120 and -catenin from VE-cadherin trig-gering clathrin-dependent VE-cadherin endocytosis and thereby junctional disintegration78-81. Besides tyrosine phosphorylation, VEGF treatment induc-es phosphorylation of S665 in the cytoplasmic tail of VE-cadherin by activa-tion of src, Vav2, Rac and p21-activated kinase (PAK), which leads to -arrestin2-dependent endocytosis82,83. Interestingly, S665 is highly conserved across mammalian VE-cadherin, but is absent in N- or E-cadherin82. Addi-tional to src family kinases, focal adhesion kinases (FAK) regulate VEGF-induced permeability. VEGF signaling leads to the activation and recruit-ment of FAK, which phosphorylates -catenin, again leading to the dissocia-tion of -catenin from VE-cadherin and a breakdown of endothelial adherens junctions84.
In addition to kinases, several phosphatases have been found at adherens junctions. For example, density enhanced phosphatase-1 (DEP-1/CD148) controls vascular permeability by regulating src activity and VE-cadherin phosphorylation75,85. The only known endothelial cell-specific phosphatase is vascular endothelial protein tyrosine phosphatase (VE-PTP)86,87. VE-PTP is a receptor-type phosphatase and binds to VE-Cadherin via its extracellular
18
domain88. VEGF signaling induces dissociation of VE-PTP from VE-cadherin in order to allow opening of endothelial cell-cell junctions89,90. Not-tebaum et al. identified plakoglobin as the substrate for VE-PTP89. However, VE-PTP was also shown to regulate VE-cadherin Y685 phosphorylation73.
The complexity and specificity of VE-cadherin and catenin phosphoryla-tion highlights the importance of a strictly controlled, fine-tuned opening of endothelial cell junctions. Uncontrolled permeability has severe consequenc-es, it can lead to edema, which for example in the retina is the major cause of vision loss in diabetic retinopathy patients91.
Figure 6. VE-cadherin phosphorylation sites. VE-cadherin phosphorylation sites and VEGF-A induced signaling pathways leading to VE-cadherin phosphorylation are depicted. Binding sites for p120, -arrestin, Csk and -catenin/plakoglobin are indicated. Modified and reprint-ed with permission92.
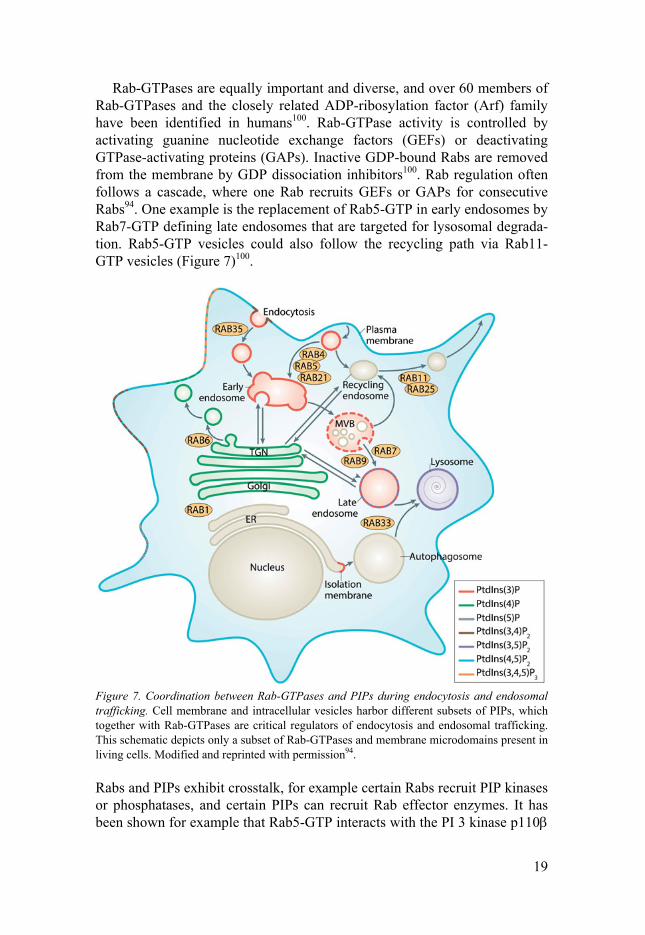
Endosomal trafficking Endocytosis of cell membrane proteins and subsequent endosomal traffick-ing are of vital importance for cellular homeostasis and response to extracel-lular stimuli. As discussed above, internalization of activated receptors does not merely terminate receptor signaling, but instead alters its signaling prop-erties, and regulated endocytosis of VE-cadherin is crucial for vascular per-meability60,93. In order to achieve coordinated endosomal trafficking intracel-lular vesicles are “labelled” with different phosphatidylinositol phosphates (PIPs) and Rab-GTPases generating a specific “membrane code”94.
PIPs are phospholipids with an inositol head group that can be phosphory-lated at the 3’, 4’ and 5’ hydroxyl group giving rise to seven different PIP species. Cell membrane and intracellular vesicles harbor different subsets of these PIP species. The major PIP of the plasma membrane is PI(4,5)P2, which can be converted into PI(3,4,5)P3 upon growth factor signaling95,96. An important mediator of clathrin-mediated endocytosis is PI(3,4)P2, while the predominant PIP found on early endosomes is PI(3)P (Figure 7)97,98. Conversion of PIPs is achieved by a wide array of PIP specific phosphatases and kinases and it is estimated that mammalian genomes encode about 50 PIP converting enzymes99.
19
Rab-GTPases are equally important and diverse, and over 60 members of Rab-GTPases and the closely related ADP-ribosylation factor (Arf) family have been identified in humans100. Rab-GTPase activity is controlled by activating guanine nucleotide exchange factors (GEFs) or deactivating GTPase-activating proteins (GAPs). Inactive GDP-bound Rabs are removed from the membrane by GDP dissociation inhibitors100. Rab regulation often follows a cascade, where one Rab recruits GEFs or GAPs for consecutive Rabs94. One example is the replacement of Rab5-GTP in early endosomes by Rab7-GTP defining late endosomes that are targeted for lysosomal degrada-tion. Rab5-GTP vesicles could also follow the recycling path via Rab11-GTP vesicles (Figure 7)100.
Figure 7. Coordination between Rab-GTPases and PIPs during endocytosis and endosomal trafficking. Cell membrane and intracellular vesicles harbor different subsets of PIPs, which together with Rab-GTPases are critical regulators of endocytosis and endosomal trafficking. This schematic depicts only a subset of Rab-GTPases and membrane microdomains present in living cells. Modified and reprinted with permission94.
Rabs and PIPs exhibit crosstalk, for example certain Rabs recruit PIP kinases or phosphatases, and certain PIPs can recruit Rab effector enzymes. It has been shown for example that Rab5-GTP interacts with the PI 3 kinase p110

20
subunit, which converts PI(4,5)P2 into PI(3,4,5)P3, and subsequently recruits PI 5-phosphatase and PI 4-phosphatase on endocytosed vesicles converting PI(3,4,5)P3 to PI(3)P101. Important for regulated endosomal trafficking is the concept of coincidence detection, i.e. a signal is only transmitted if both, specific PIPs and specific Rabs, are encountered at the same time. For ex-ample, the early endosome antigen 1 (EEA1) is only recruited to early endo-somes when both PI(3)P and Rab5-GTP are present102. Therefore, Rabs and PIPs together convey the necessary specificity and compartmentalization needed for directed endocytosis and endosomal trafficking94.
Lung physiology and disease The lung is the central organ for replenishing blood with oxygen for distribu-tion to the periphery. The lung is a highly branched system comprised of two main compartments: the air conducting compartment with the trachea branching into bronchi and bronchioles, and the distal compartment consist-ing of hundreds of millions bronchiolar terminal air sacs, called alveoli, where the gas exchange between the air and the blood takes place103. Alveoli are covered by a dense capillary network, separated only by a thin basement membrane. This air-blood barrier created by endothelium, basement mem-brane and epithelium is only 0.1 µm thick in humans2. The direct interaction between the lung and the outside environment necessitates the presence of efficient host defense mechanisms in the lung protecting the organism from inhaled pathogens and particulates. Specialized ciliated cells and secretory, non-ciliated cells along the trachea and bronchi ensure clearing of the in-haled pathogens and particulates from the lung2,104.
Lung development In mice specification of the lung endoderm is initiated around E9.0 and marked by the expression of transcription factor Nkx2.1. At E9.5 lung de-velopment commences with the formation of two lung buds from the anterior foregut endoderm, which branches and forms a tree-like network during the subsequent pseudoglandular stage (E12.5 - E16.5)105. Proximodistal differen-tiation in the canalicular stage (E16.5 – E17.5) leads to differentiation of proximal stalk and distal tip cells, the latter forming clusters of epithelial sacs during the subsequent saccular stage (E18.5 – P5). During the final al-veolarization stage maturation and secondary septation of these primitive alveolar sacs into alveoli takes place. In mice alveolarization occurs entirely postnatally, which is in contrasts to humans, where alveolarization starts in utero at around 36 weeks. However, in both cases, alveolarization is a long process and is only completed at around 3 weeks after birth in mice and 15 years in humans (Figure 8)106,107.

21
The lung mesoderm develops in parallel to the lung endoderm and signal-ing from the mesoderm is crucial for the developing endoderm106. For exam-ple, FGF10 producing cells around the distal tips are essential for branching morphogenesis during the pseudoglandular stage108,109. During the canalicu-lar stage capillaries start to underlie the epithelium and subsequently influ-ence the differentiation of alveolar epithelial cells103. Alveolar septation de-pends on alveolar myofibroblasts, which segregate around the primitive ter-minal alveolar sacs prior to the saccular and alveolarization stage110,111.
Figure 8. Lung development in the mouse. This schematic depicts the different stages of mouse lung development from the formation of lung buds during the embryonic stage, fol-lowed by branching morphogenesis in the pseudoglandular stage and proximodistal differen-tiation in the canalicular stage to the formation of bronchiolar terminal air sacs in the saccular stage and the final alveolarization stage. Modified and reprinted with permission107.
Alveolar cell types Two distinct epithelial cell populations, type I and type II pneumocytes, comprise the majority of the alveolar surface area (Figure 9). During the saccular stage of lung development, bipotential progenitor cells in distal tips of canalicular tubules give rise to both types of pneumocytes112,113. Type I pneumocytes are squamous-shaped and cover 95% of the alveolar surface area114. Together with the underlying, closely interacting capillaries they form the primary site for gas exchange115. In contrast, type II pneumocytes are cuboidal and reside at the “corners” where type I pneumocytes converge116. Type II pneumocytes constitute the primary source of surfac-tant, a mixture of extracellular proteins and lipids. Pulmonary surfactant is important for host defense and for reducing the alveolar surface tension cre-ated at the air-liquid interface117. Interestingly, the turnover of alveolar cells is infrequent in healthy lungs118. Only type II, but not type I pneumocytes, proliferate, instead type I pneumocytes are replenished by the transdifferen-tiation from type II pneumocytes113,119. Whether only a specialized subset of type II pneumocytes or all type II pneumocytes display these stem cell prop-erties is still under debate116. The self-renewal and differentiation potential of alveolar cells is being studied intensively and different progenitor cells have been identified112. Bronchioalveolar stem cells (BASC), distal airway stem

22
cells (DASCs) as well as secretory Clara cells might be important adult stem cell populations for lung regeneration after lung injuries112,120-122.
Figure 9. Alveoli structure. The majority of the alveoli surface is formed by type I and type II pneumocytes, which are in close contact to the underlying capillary network. Fibroblasts and pericytes cover the epithelial and endothelial cells. Ciliated cells, Clara cells and BASCs can be found in terminal bronchioles and at the bronchioalveolar duct junction. Modified and reprinted with permission104.
Chronic obstructive pulmonary disease and emphysema Long-term exposure to air-borne substances can cause many diseases of the lung, for example cigarette smoking is the single highest risk factor for de-veloping chronic obstructive pulmonary disease (COPD)123. COPD is the fourth leading cause of death and will probably become the third leading cause by 2020, due to changed smoking habits in countries like China123. COPD is an umbrella term for a number of different diseases but is generally defined by airflow obstruction associated with chronic pulmonary inflamma-tory response. COPD is typically a progressive disease which cannot be fully reversed123. If the pulmonary inflammation is established, for example as a result of smoking, COPD persists and progresses even following cessation of smoking124. In the UK, 10% of men and 11% of women between 16-65 years show moderate COPD123,125. The prevalence for COPD increases with age 123. Emphysema, the destruction of the alveolar wall, is often seen in patients with COPD125.
How COPD or emphysema develop is not yet fully understood, but much attention has been drawn to immune cells (macrophages, neutrophils and T-cells) as being important mediators in disease progression124. Alveolar mac-rophages secrete elastolytic enzymes (such as MMPs) that destroy the lung parenchyma. Neutrophils contribute to the destruction by secreting serine proteases and MMPs. This destruction will eventually result in emphysema126. In healthy individuals these proteolytic enzymes are counter-acted by endogenous anti-proteases and individuals with a deficiency in α1 antitrypsin, the endogenous inhibitor of neutrophil elastase, are predisposed to develop COPD (1-2% of COPD patients)123.

23
While it is generally agreed that emphysematous lungs show increased numbers of apoptotic cells, it is less clear which cells predominantly undergo apoptosis127. A study by Segura-Valdez et al. suggest endothelial cells as the main apoptotic cells in smokers with emphysema, also further supported by a mouse model of smoke-induced emphysema128. Interestingly, apoptosis specifically induced in lung endothelial cells in mice led to the destruction of alveoli and was associated with other typical emphysema characteristics, i.e. influx of macrophages and increased oxidative stress129. Increased endotheli-al and non-endothelial cell apoptosis could be directly linked to impaired VEGF-VEGFR2 signaling seen in emphysematous lungs130-132. It was also suggested that the increased number of apoptotic cells could be due to the inability of macrophages to efficiently clear apoptotic cells in the absence of VEGF133. VEGF also contributes to vascular remodeling, which is often seen in COPD patients134. Therefore, balancing VEGF and its receptors seems to be critical in COPD and emphysema progression. However, these diseases are very complex and many other factors contribute to disease development and progression, such as vessel wall thickening by increased ECM deposi-tion for example134.
Phosphatases Phosphorylation is the most common post-translational modification of pro-teins altering their charge and consequently the conformation and activity of many signaling molecules135-137. As discussed above, phosphorylation and dephosphorylation of, for example, VEGFR2 or VE-cadherin are critical mechanisms in endothelial biology. The human genome encodes about 518 kinases138 and about 245 phosphatases (retrieved from hupho.uniroma2.it139) to ensure a stringent regulation of phosphorylation events. Tyrosine, serine and threonine residues serve as acceptors for phosphate groups in most pro-teins140. Additionally, several non-proteinaceous substrates have been identi-fied, among them cytosolic and lipid inositol phosphates141,142, glycans143 and mRNA144,145.
Protein phosphatases can be divided into two main groups determined primarily by their dephosphorylation specificity: protein serine/threonine phosphatases (~27% of all protein phosphatases) and protein tyrosine phos-phatases (PTPs)146,147.
Protein Tyrosine Phosphatases The first step in the dephosphorylation reaction catalyzed by phosphatases is the nucleophilic attack on the phosphoester group of the substrate and the formation of a phosphoryl enzyme intermediate148. In the majority of PTPs, a highly conserved cysteine residue serves as nucleophile, but there are also

24
aspartate and histidine based phosphatases144. Cysteine-based PTPs can be grouped into three classes144. Class I PTPs comprise the majority of phos-phatases, including classical and dual-specific PTPs (DUSPs/VH1-like), the latter are able to dephosphorylate both tyrosine and serine/threonine resi-dues147. Class II and III comprise low molecular weight (LMW)-PTPs and cell division cycle (CDC) 25s, respectively (Figure 10)149.
Figure 10. The extended PTPome by Alonso and Pulido. PTPs are classified according to the nucleophilic amino acid used for phosphoester hydrolysis (cysteine, aspartate or histidine). Cysteine-based phosphatases are further subdivided in classes and subclasses based on their structural similarities. Alonso and Pulido propose four new subclasses (III-VI). The number of members per group is indicated by the number to the right (inactive pseudophosphatases are included). Reprinted with permission144.
The majority of cysteine-based PTPs share ten highly conserved signature motifs150. Signature motif 9 ([I/V]HCxxGxxR[S/T]G) describes the active site and contains the invariant, nucleophilic cysteine residue148,150. However, some phosphatases diverge from this active site signature motif and a less stringent consensus sequence has recently been proposed by Alonso and Pulido, expanding the classification to include four new class I subclasses (III-VI, Figure 10)144. Importantly, all active new members of this proposed “extended PTPome” display substrate specificities to phosphatidylinositol phosphates144. Lipid phosphatase activity has also been described for mem-bers of the classical PTPs and DUSPs151. Indeed, more and more PTPs emerge as lipid phosphatases151. The tumor suppressor PTEN (phosphatase and tensin homolog deleted on chromosome 10) is probably the best known example, hydrolyzing both phosphatidylinositol-3,4,5-triphosphate and phosphotyrosine residues152.

25
Figure 11. Dephosphorylation reaction and oxidation of PTPs. Catalysis of dephosphoryla-tion by classical PTPs is exemplified by PTP1B. The phosphoryl group of the substrate is transferred to the nucleophilic thiolate of active site cysteine. Subsequently, the cysteinyl-phosphate intermediate is hydrolyzed and the phosphate released. The enzyme can be ren-dered inactive by oxidation and the formation of sulphenic acid. This inactive form can be reduced again to the active form via a cyclic sulphenamide intermediate. Reprinted with permission148.
Essential for PTP activity is the active site cysteinyl thiolate ion that attacks the phosphoester group of the phosphotyrosines substrate (Figure 11)148,153-
155. This enzyme-substrate intermediate is stabilized by the invariant arginine in the active site, without which enzyme activity is almost completely lost156. The removal of the tyrosyl substrate from the enzyme-substrate intermediate is facilitated by an aspartic acid residue present in another highly conserved motif, the WPD loop (motif 8)148,150. The aspartic acid initially protonates the tyrosyl leaving group and subsequently acts as base to catalyze phosphate release148,153. Interestingly, not all phosphatases contain the WPD loop but instead catalyze phosphate removal via alternative aspartic or glutamic resi-dues157,158.
Mutation of the active site cysteine to serine completely abrogates phos-phatase activity, however the structure of the protein is not altered and sub-strate binding is retained, therefore these phosphatase-dead mutants are often used as “substrate trapping” mutants153,156. In contrast, substrate affinity is greatly reduced when mutating the invariant arginine to methionine (Km for PTP1B mutant increased by 10-fold)156.

26
Active site cysteine thiolate is highly susceptible to inactivating oxida-tion. However, the histidine residue juxtaposed to the cysteine quickly re-verses this oxidation, and its absence severely reduces enzyme activity (Fig-ure 11)148,159. This sensitivity to oxidation makes phosphatases a potential target for intracellularly produced reactive oxygen species (ROS) and other oxidants, thereby regulating cell signaling events160,161. It has been shown that ROS production increased VEGFR2-mediated signaling, most likely due to oxidation and consequently inactivation of VEGFR2-associated phospha-tases, DEP1 and PTP1B162,163. Localized ROS production is essential for a specific, balanced regulation of cell signaling. The extracellular superoxide dismutase, for example, is localized to caveolae/lipid rafts, which leads to a restricted formation of H2O2 and confined inactivation of DEP1 and PTP1B in endothelial cells163.
Pseudophosphatases In recent years, the existence of catalytically inactive pseudokinases and pseudophosphatases has been increasingly appreciated. To date, it is estimat-ed that ~10% of the human kinome consists of pseudokinases, and ~8% of the human phosphatome is estimated to be pseudophosphatases164. It is be-lieved that pseudokinases and pseudophosphatases play essential roles in many signaling pathways by serving as co-factors or dimerization partners, compete for substrate binding, act as spatial anchors by trapping substrates to specific subcellular locations, or serve as a scaffolding protein for several different signaling molecules164.
In order to experimentally prove lack of catalytic activity Kharitidi et al. postulated the introduction of “backmutations”, i.e. changing amino acids divergent from the consensus sequence to the corresponding conserved ami-no acids. Consequently, backmutations should restore phosphatase activity. This method is a powerful tool to prevent misinterpretation due to non-optimized assay conditions or lack of appropriate substrates165.
Pseudophosphatases may be important in human diseases, for example inactive myotubularin-related phosphatase 11 (MTMR11) has been linked to breast cancer and acute myeloid leukemia166,167. Interestingly, six out of the 15 MTM/MTMR family members encode catalytically inactive proteins, which, however, form dimers with active MTMRs and thereby modulate their activity and subcellular location148,164.

27
Paladin The importance of angiogenesis in pathological conditions has stimulated the search for potential drugs targeting angiogenic pathways. While many key players in angiogenesis are known, the therapeutic success of anti-angiogenic therapy has been complicated by severe side effects and drug resistance15,168. Several screens have been performed to find vascular-enriched genes that could be potential drug targets for anti- or pro-angiogenic therapies. Examples are microarray studies on primary endotheli-al cells169 or specific vascular beds170 and compared to the non-endothelial compartment, as well as a reverse and chemical genetic screen employing the zebrafish embryo and an in vitro sprouting assay171. Remarkably, all of these screens identified paladin as a vascular-enriched gene. Paladin is en-coded by PALD1 (or KIAA1274 in humans and Pald1, mKIAA1274 or x99384 in mice) on chromosome 10 (q22.1 in humans) and is highly con-served across vertebrate genomes (HomoloGene, release 68, 2014). PALD1 contains 20 exons (Figure 12) and currently there are no splice variants re-ported for either human or murine paladin (Ensembl annotation release 76), which is in contrast to earlier annotations where three splice variants of hu-man paladin were predicted and referenced in paper I172.
Figure 12. Overview of paladin protein and Pald1 gene. (A) Paladin protein sequence with putative phosphatase domains: minimal signature motif CxxxxxR (white boxes, x = any ami-no acid), Interpro predictions (grey boxes), SCOP SUPERFAMILY algorithm (black box). (B) Pald1 gene contains 20 exons both in humans and mice. Modified and reprinted with permission172.
Structurally, paladin possesses several putative PTP domains (up to four depending on the recognition motif used, see Figure 12). However, none of these phosphatase domains are fully homologous to the conserved consensus active site PTP motif and they all lack the highly conserved active site histi-dine residue. Furthermore, no WPD loop can be identified in the paladin amino acid sequence. Consequently, despite a lack of experimental proof, paladin had been proposed to be a pseudophosphatase by two independent reviewers, Reiterer et al.164 and Kharitidi et.al.165. However, a recent ge-nome-wide analysis by Alonso and Pulido144 suggests that paladin could catalyze removal of phosphates from inositol rings. Indeed, phosphatases with similar amino acid sequences in the active site all catalyze dephosphor-ylation of phosphatidylinositol phosphates144. Strikingly, similar to paladin, several catalytically active lipid phosphatases lack the histidine juxtaposed to

28
the active site cysteine and display alterations in the WPD loop, e.g. PTPRQ encodes glutamic acid instead of aspartic acid, while MTMs completely lack the WPD loop144,151,173.
The subcellular localization of paladin has not yet been described, but paladin has been shown to be myristoylated174. Such a lipid modification could facilitate protein-lipid interaction, therefore, paladin might be located at the plasma membrane or at intracellular vesicles175.
Functionally, paladin has been shown to be required for normal develop-ment of intersegmental blood vessels in zebrafish171. However, the detailed role of paladin in vascular biology has not been further elucidated. In an in vitro assay paladin was found to interfere with insulin signaling by negative-ly regulating insulin receptor expression and phosphorylation, as well as phosphorylation of the downstream target Akt176. Roffers-Agarwal and col-leagues demonstrated that paladin is expressed in, and regulates the for-mation and migration of neural crest cells in chick embryos177. This function seems to be independent of the putative phosphatase activity, since a phos-phatase-dead C/S mutant did not abolish paladin’s function in the neural crest, and the phosphatase-dead mutant was also able to almost completely rescue a Pald1 knock-down177. Furthermore, Li and colleagues identified paladin as part of the innate immunity protein interaction network and found that paladin interacts with the DNA sensor DAI, Toll-like receptor 9 and interferon regulatory factor 7178. Paladin expression in mouse macrophages is upregulated after infection with the DNA virus herpes simplex virus (HPV) and knock-down of paladin in human embryonic kidney cells (HEK293) resulted in a greater HPV-induced interferon-β production. Tak-ing these finding together, Li and colleagues postulate that paladin negative-ly regulates immune responses to DNA viruses178.
From the published expression and function data, paladin seems to be en-riched, but not fully restricted to the vasculature and exerts a broad range of functions from neural crest development to innate immunity and insulin sig-naling. The aim of my thesis was to shed more light upon this complex pic-ture by (1) studying the expression pattern of paladin in vivo, (2) characteriz-ing the Pald1 knock-out mouse and (3) investigating the biochemical proper-ties of paladin.

29
Present investigations
Paper I
Paladin (X99384) is expressed in the vasculature and shifts from endothelial to vascular smooth muscle cells during mouse development
The scope of paper I was to examine the expression pattern of paladin. We made use of a newly generated genetically modified mouse model, anti-paladin antibodies and published in situ hybridization data.
To generate a Pald1 null mouse, exon 1-18 were targeted and replaced in frame with a functional LacZ expression cassette. LacZ encodes the bacterial β-galactosidase and its activity can be visualized by providing the modified sugar molecule x-gal (5-bromo-4-chloro-3-indolyl-beta-D-galactopyra-noside) as a substrate179. Thus, LacZ activity was used to map expression in Pald1LacZ/+ mice.
We found that paladin is strongly expressed during embryogenesis. At day E9.5 LacZ expression was found in the intersomitic mesenchyme and along the longitudinal axis of the embryo as well as in the hindbrain (rhom-bomere 2 and 4), in certain pharyngeal arches (which give rise to specific arteries, nerves, skeletal and muscle tissue) and the heart. At E10.5 blood vessels of the heart and the intersomitic mesenchyme were LacZ positive. The neural tube, which had been LacZ negative at E9.5, displayed LacZ expression in the roof- and floor plate at E10.5. Furthermore, LacZ was found in the cervical nerve trunk, perineural vascular plexus, blood vessels of the brain as well as the cardinal veins, and to a lesser extent in the dorsal aorta. At E14.5, virtually all vascular beds demonstrated strong LacZ expres-sion. Additionally, non-vascular staining was seen in solitary cells in the liver, heart myocardium, lung and kidney mesenchyme.
The strong vascular LacZ expression was also seen in adult tissues, how-ever in some organs expression appeared largely restricted to major blood vessels. Indeed, upon further investigation of brain and kidney, we found expression only in arteries and arterioles, but not veins and venules. More specifically, LacZ expression had almost completely shifted from endothelial cells to arterial mural cells. This shift from an initial broad endothelial ex-

30
pression to specifically arteries and arterioles was also observed in the retinal vasculature. In retinas of P5 pups, LacZ expression was strong in capillaries and veins and somewhat weaker in arteries, but in adult retinas this expres-sion pattern dramatically changed and LacZ expression was restricted to arteries and absent in capillaries and veins.
However, in other organs, such as the lung and brown adipose tissue, this shift in expression between vascular cells could not be observed, and the non-vascular staining of the lung mesenchyme was still present. Further-more, the heart seemed to express LacZ both in smooth muscle cells, capil-laries and the myocardium. Moreover, LacZ expression was confirmed in a subset of hematopoietic cells during embryogenesis and in adult mice. In humans, paladin was also found in the vasculature of brain tissue and glio-mas.
There are other vascular-specific genes that can be found both in endothe-lial and mural cells (e.g. Ephrin-B2180 or GRP124181), but the observed dy-namic shift has not been reported previously. It is not possible to estimate how common such dynamic endothelial to mural cell expression is among vascular genes, because of the lack of studies addressing temporal expres-sion patterns.
Paper II
Female mice lacking Pald1 exhibit endothelial cell apoptosis and emphysema After investigating the expression pattern of paladin in paper I, the scope of paper II was to characterize the Pald1 knock-out mice as a first step in un-derstanding paladin’s physiological function. In collaboration with the German Mouse Clinic, we performed a global phe-notypic analysis of adult Pald1-/- mice employing behavioral, physiological and biochemical tests182,183. Two major physiological defects were identified in Pald1-/- mice: impaired lung function and increased hearing sensitivity.
We demonstrated that the increased hearing sensitivity was due to an un-related single nucleotide polymorphism in Cdh23 originating from the back-ground strain 129SvEv used to create the Pald1-/- mouse. However, paladin was expressed broadly in the mouse cochlea, including the organ of Corti, which mediates the mechanotransduction of sound waves into neural action potentials. Therefore, a role of paladin in the inner ear cannot be excluded, but cannot be evaluated with the current mouse model.
Lung function was assessed in adult female mice using two different in-vasive methods measuring both steady-state and forced lung volume and

31
flow parameters. Mice lacking Pald1 demonstrated an increase in lung vol-ume and compliance as well as a decrease in lung resistance. These findings clinically resemble COPD, a condition often accompanied by emphysema125. Indeed, an emphysema-like histology was observed in 19-week old female mice. Interestingly, this emphysema-like lung appearance was present al-ready at the beginning of alveolarization at P5 and most prominent after completed alveoli septation at four weeks of age. Strikingly, increase in al-veolar size was only observed in female mice, but not in males. Sex-specific differences in susceptibility to COPD and emphysema have been described in a mouse model of smoke-induced emphysema184, and are also known from humans, where women have an earlier onset of COPD and more severe disease progression than men185.
As shown in paper I, paladin is expressed in the lung both during embry-ogenesis and in the adult. A more detailed analysis of the LacZ reporter ac-tivity revealed expression in smooth muscle cells around bigger vessels and in all main cell types of mature alveoli, i.e. endothelial cells of capillaries, type I and type II pneumocytes and myofibroblasts. A similar expression pattern was also seen in P5 lungs with the exception of type II pneumocytes that were LacZ negative. Despite the broad expression of paladin, only endo-thelial cells were affected by loss of paladin. At four weeks, emphysematous lungs displayed a reduced number of endothelial cells, probably due to in-creased apoptosis, which could not be fully compensated for by the observed increase in proliferation. Both endothelial cell apoptosis and proliferation was still increased at 19 weeks, but to a lesser extent than at 4 weeks. How-ever, the endothelial cell number was unaltered in Pald1-/- female lungs at 19 weeks.
How the loss of Pald1 in female lungs leads to emphysema is at present unclear. However, loss of VEGF signaling and endothelial apoptosis has previously been shown to trigger emphysematous changes in lungs129-131. Even though endothelial cell number was not altered in P5 lungs at the be-ginning stage of alveolarization, we have not yet assessed the turnover of endothelial cells at this stage. It remains a possibility that already at P5 endo-thelial cells undergo apoptosis more frequently in lungs lacking Pald1 than in wild type littermates, but that increased proliferation compensates for that loss at this early age. In order to gain more understanding of the underlying mechanisms leading to the emphysematous phenotype, we are currently per-forming broad analyses of the proteome and transcriptome of Pald1+/+ and Pald1-/- lungs.
In conclusion, we could show that Pald1 is required for normal lung de-velopment and function in a sex-specific manner.

32
Paper III
Paladin (Pald1) regulates endothelial sprouting, VE-cadherin junction stability and vascular permeability
In paper III we addressed the biochemical properties of paladin. As de-scribed above, amino acid sequence analysis proposed a function as a protein phosphatase. To test this, we performed in vitro phosphatase assays and ex-amined paladin’s ability to dephosphorylate phosphotyrosine peptides. Un-der the conditions tested we could not observe any protein phosphatase ac-tivity, suggesting that paladin could be a catalytically-inactive pseudophos-phatase. However, the introduction of backmutations in order to restore phosphatase activity did not lead to detectable enzyme activity under the conditions tested, therefore not fulfilling the requirements for defining pseu-dophosphatases. Instead, preliminary results demonstrated that paladin is an active lipid phosphatase dephosphorylating several PIPs. Detailed substrate kinetics remain to be determined.
As reported in paper I, paladin expression is lost in endothelial cells of a mature retinal vascular plexus. We sought to investigate if paladin expres-sion could be connected to decreasing VEGF levels seen in the mature vas-culature. Indeed, we found that paladin expression is downstream of VEGF signaling in vitro and in retinas, suggesting VEGF as an important determi-nant for paladin expression in endothelial cells.
Based on the expression of paladin in endothelial tip cells, the link be-tween VEGF and paladin, and the indication that paladin acts as a negative regulator of tyrosine (insulin receptor) and threonine/serine kinases (Akt) we analyzed early postnatal retinas as a model for sprouting angiogenesis. Pald1-/- retinas showed hypersprouting and a hyperdense vascular front as well as a general delay in maturation. In search for the underlying mecha-nism for hypersprouting, we assessed alterations in the Dll4-Notch-VEGFR2 signaling circuit by quantitative PCR, but we could not detect any significant differences in Pald1-/- retinas compared to wildtype littermates. We further investigated whether VEGF-mediated signaling is altered in the absence of paladin. Indeed, we found that in total heart lysates VEGFR2 downstream signaling was prolonged (Erk1/2) or increased (Akt), which could be due to altered internalization and degradation kinetics of VEGFR2 and consequent-ly sustained signaling from the plasma membrane and early endosomal com-partments. Direct assessment of phosphorylation states of VEGFR2, Erk1/2 or Akt in the retinas proved to be difficult, therefore it remains to be eluci-dated whether altered VEGFR2 signaling could provide an explanation for the observed retinal phenotype.
Interestingly, we observed a reduction of VE-cadherin mRNA levels in early postnatal retinas. Further investigation of the VE-cadherin complex in Pald1-/- total heart lysates and isolated lung endothelial cells revealed chang-

33
es in plakoglobin and -catenin-VE-cadherin association that were con-sistent with a more mature junctional phenotype. In line with these findings, overexpression of paladin led to a more activated junction morphology. Tak-en together, these findings indicate a role for paladin in VE-cadherin turno-ver. Paladin interaction with the VE-cadherin/VEGFR2 complex depended on stimulation with VEGF and/or pan-phosphatase inhibitor peroxyvanadate. Such treatment leads to VEGFR2 and VE-cadherin phosphorylation and subsequent endocytosis, suggesting that paladin interacts with internalized, but not cell membrane-bound VE-cadherin or VEGFR2.
Next, we addressed whether altered endothelial cell biology in Pald1-/- mice could affect the opening of endothelial cell junctions by employing the microsphere extravasation assay in the trachea. Strikingly, lack of paladin led to reduced vascular leakage in response to VEGF stimulation, which could be due to the altered VE-cadherin junction morphology or an altered response to VEGF signaling.
In conclusion, we demonstrated that paladin is likely a lipid phosphatase that is important in developmental angiogenesis, regulates VEGFR2 signal-ing and VE-cadherin junction stability. These changes in endothelial cell biology might explain the defects in VEGF-induced vascular permeability.

34
Concluding remarks and future perspectives
Many new players in angiogenesis are being discovered as we learn more about endothelial cell biology in general and the angiogenic sprouting pro-cess in particular. Remarkably, when the core-cluster of vascular-enriched genes was published in 2008, 32 out of 58 genes were still of unknown func-tion170. Paladin was one of those novel genes with very little information on its biochemical properties or biological functions.
Our thorough expression analysis confirmed the vascular expression pat-tern of paladin and revealed a remarkable shift in expression from a broad endothelial cell expression during mouse development to a specific arterial smooth muscle cell expression in the central nervous system and other vas-cular beds in adult mice. The constitutive Pald1 knock-out led to endothelial specific defects, both in the the developing vasculature in the retina as well as lung, heart and trachea. The lack of paladin resulted in altered VEGFR2 signaling, reduced VEGFR2 degradation, increased junctional stability and reduced vascular leakage. In line with this observation, overexpression of paladin led to a more activated endothelial cell junction morphology. To-gether with the newly identified phosphatase activity of paladin towards PIPs, a picture evolves where paladin may play a role in the regulation of endocytosis and endosomal trafficking. Preliminary findings on the subcellu-lar localization of paladin suggest association with intracellular vesicles. By further investigating the substrate specificities and the subcellular localiza-tion of paladin, we hope to be able to pinpoint the step in endosomal traffick-ing where paladin might play a role. Consequently, we propose a model by which paladin modifies the composition of the cell membrane and/or intra-cellular vesicles thus modulating endosomal trafficking of VEGFR2, VE-cadherin and probably other transmembrane and signaling molecules.
Interestingly, endocytosis followed by transcytosis is also important in the lung, shown for the elastase inhibitor 1-antitrypsin. Circulating 1-antitrypsin needs to be transported from blood vessels, across the endothelial cells towards the epithelium186. If, however, endocytosis and transcytosis is impaired in Pald1-/- mice, the natural balance between proteases and anti-proteases will be shifted towards proteases, which in turn leads to alveolar wall destruction. Imbalanced vesicle trafficking due to the lack of paladin could induce stress and consequently apoptosis in endothelial cells. It re-mains to be elucidated whether the increased endothelial cell turnover is the

35
cause for the lack of proper alveolarization or the consequence of alterations in other cell types.
This thesis has focused on the role of paladin in endothelial cells, but in adult mice paladin is also strongly expressed in arterial smooth muscle cells. Therefore, it would be interesting to also investigate the role of paladin in arterial smooth muscle cells and to understand what induces arterial mural cell expression.

36
Acknowledgments
This work was carried out at the Department of Immunology, Genetics and Pathology at Uppsala University, supported by the Swedish Cancer founda-tion, Beijer foundation, Åke Wiberg’s foundation, Magnus Bergwall’s foun-dation and Lion's cancer foundation. I would like to express my sincere gratitude to our co-workers for the fruitful collaborations and everyone who supported and helped me during my PhD studies. In particular I thank:
My supervisor Mats Hellström, for taking me on as a PhD student, for your advice, enthusiasm and support throughout my PhD studies. The paladin project gave me the freedom to explore many different aspects of cell biolo-gy and I am very grateful for that opportunity!
My co-supervisor Lena Claesson-Welsh, for your advice and support and welcoming me in your group. Thank you for making the lab such a nice place to work in!
My opponent Frank-D. Böhmer, for coming to Uppsala and taking your time to scrutinize my thesis. I very much appreciate your effort!
I am very grateful to all past and present members of Mats’ group: Elisabet for welcoming me into the group and introducing me to the secrets of LacZ staining and retina work. Isabel, it was a pleasure working with you, you surely lightened up the lab with your positive attitude! Hiroshi for your con-tributions to the paladin project, it is very nice working with you. Chiara for fruitful discussions about paladin, science and life in general! I am grateful for your support and friendship! Jimmy, I always enjoyed working with you! Thank you for your support, checking up on me once in a while and the many fun moments inside and outside the lab! I also thank my student Evelyn, it was a great experience working with you!
I would like to express my gratitude to all former and present members of Lena’s group for being wonderful colleagues, always supportive and open for scientific discussions! There is not enough space to mention everyone, but I will never forget the Fikas and all the fun lab events with you! In par-ticular, I would like to thank Ross, for your help with virtually everything – from bike repair and ImageJ to difficult injections! Xiujuan, you were a great lab bench neighbor and an endless source of knowledge! You are such

37
a kind and helpful person! Mimmi, thank you very much for all your help with the animal experiments! Charlotte – it was great to have you! Sina, you were so nice to me when I first came to the lab and made it so much easier for me to settle in! Thank you for all the good times! Simone, thank you for your help in the lab and for the good times inside and now outside the lab! Eric and Miguel - travelling with you was a blast! Emma G, thank you for your support and all the fun times at various non-work related occasions! Thank you for proof reading this thesis! Frank, I very much ap-preciate your help and support, the laughter, but also being there in difficult times! Putting up with me during the writing of this thesis and taking the time to proof read it! I truly value our friendship!
A big thank you also to all past and present members of the VascBio community at Rudbeck! I consider myself very lucky to have had the oppor-tunity to work with you! You are all such a great source of knowledge, end-lessly helpful and create such an enjoyable working environment!
I thank all the wonderful people at IGP and Rudbeck that made my time here a great experience! Including Christina Magnusson for being always there for us PhD students! My old teaching crew: Vasil, Lucy and Diego – teaching with you was a lot of fun! And including all the other PhD stu-dents and colleagues: Verónica, Emma Y, Ram, Tanja, Vicky, Doroteya, Antonia and Manoj, Leonor, Marco, Colin, Bong, Maike, Yang, Ines, Lei, Hua, Maria, Luuk, Sara, Marta, Viktor and many more for all the help, discussions and fun times at beer clubs, Alternative Journal Clubs, Norreda weekends and various after works! Mohammad, thank you for your friendship and the good times back in Flogsta! A big thank you also to Elise for your support, great discussions and making me dance!
During the past eight years that I have spent in Uppsala many amazing peo-ple have crossed my path! I especially thank Franzi, Anja, Kasia, Marlen, and Susanna – for all the great parties and hiking trips, an unforgettable trip to St. Petersburg and a wonderful time in Paris, thank you for your support and friendship throughout the years, you made me feel at home!
Not to forget my friends in Germany, especially Jana and Jule! Jana, we have known each other since forever – thank you for your friendship and that I can always count on you! Jule – I am so happy to call you my friend, thank you for all the good times, the amazing trips together and that I can always rely on you!
Last, but not least, a big thank you to my beloved family! Ich bin euch sehr dankbar für eure ununterbrochene Unterstützung und Liebe! Ihr habt mir immer alles ermöglicht und mich in meinen Entscheidungen ermutigt! Vie-len Dank für alles!

38
References
1. Adams, R.H. & Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 8, 464-478 (2007).
2. Aird, W.C. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res 100, 174-190 (2007).
3. Barkefors, I. Directing angiogenesis: cellular responses to gradients in vitro. Acta Universitatis Upsaliensis. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 643, 52 (2011).
4. Jakobsson, L. & Claesson-Welsh, L. Vascular basement membrane components in angiogenesis--an act of balance. ScientificWorldJournal 8, 1246-1249 (2008).
5. Gaengel, K., Genove, G., Armulik, A. & Betsholtz, C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol 29, 630-638 (2009).
6. Rostgaard, J. & Qvortrup, K. Electron microscopic demonstrations of filamentous molecular sieve plugs in capillary fenestrae. Microvasc Res 53, 1-13 (1997).
7. Wolburg, H., Noell, S., Mack, A., Wolburg-Buchholz, K. & Fallier-Becker, P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res 335, 75-96 (2009).
8. Armulik, A., et al. Pericytes regulate the blood-brain barrier. Nature 468, 557-561 (2010).
9. Patel-Hett, S. & D'Amore, P.A. Signal transduction in vasculogenesis and developmental angiogenesis. Int J Dev Biol 55, 353-363 (2011).
10. Udan, R.S., Culver, J.C. & Dickinson, M.E. Understanding vascular development. Wiley Interdiscip Rev Dev Biol 2, 327-346 (2013).
11. Drake, C.J. & Fleming, P.A. Vasculogenesis in the day 6.5 to 9.5 mouse embryo. Blood 95, 1671-1679 (2000).
12. Risau, W. & Flamme, I. Vasculogenesis. Annu Rev Cell Dev Biol 11, 73-91 (1995).
13. Nishii, K. & Shibata, Y. Mode and determination of the initial contraction stage in the mouse embryo heart. Anat Embryol (Berl) 211, 95-100 (2006).
14. Lucitti, J.L., et al. Vascular remodeling of the mouse yolk sac requires hemodynamic force. Development 134, 3317-3326 (2007).
15. Carmeliet, P. & Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 473, 298-307 (2011).
16. Kilarski, W.W., Samolov, B., Petersson, L., Kvanta, A. & Gerwins, P. Biomechanical regulation of blood vessel growth during tissue vascularization. Nat Med 15, 657-664 (2009).
17. Folkman, J. Tumor angiogenesis: therapeutic implications. N Engl J Med 285, 1182-1186 (1971).
18. Congdon, N., et al. Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol 122, 477-485 (2004).

39
19. Klein, B.E. Overview of epidemiologic studies of diabetic retinopathy. Ophthalmic Epidemiol 14, 179-183 (2007).
20. Stahl, A., et al. The mouse retina as an angiogenesis model. Invest Ophthalmol Vis Sci 51, 2813-2826 (2010).
21. Uemura, A., Kusuhara, S., Katsuta, H. & Nishikawa, S. Angiogenesis in the mouse retina: a model system for experimental manipulation. Exp Cell Res 312, 676-683 (2006).
22. Ellertsdottir, E., et al. Vascular morphogenesis in the zebrafish embryo. Dev Biol 341, 56-65 (2010).
23. Koch, S., Tugues, S., Li, X., Gualandi, L. & Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem J 437, 169-183 (2011).
24. Eilken, H.M. & Adams, R.H. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol 22, 617-625 (2010).
25. Benedito, R., et al. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature 484, 110-114 (2012).
26. Tammela, T., et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 454, 656-660 (2008).
27. Tammela, T., et al. VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat Cell Biol 13, 1202-1213 (2011).
28. Zarkada, G., Heinolainen, K., Makinen, T., Kubota, Y. & Alitalo, K. VEGFR3 does not sustain retinal angiogenesis without VEGFR2. Proc Natl Acad Sci U S A 112, 761-766 (2015).
29. Fong, G.H. Regulation of angiogenesis by oxygen sensing mechanisms. J Mol Med (Berl) 87, 549-560 (2009).
30. Dougher, M. & Terman, B.I. Autophosphorylation of KDR in the kinase domain is required for maximal VEGF-stimulated kinase activity and receptor internalization. Oncogene 18, 1619-1627 (1999).
31. Matsumoto, T., et al. VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. Embo J 24, 2342-2353 (2005).
32. Sakurai, Y., Ohgimoto, K., Kataoka, Y., Yoshida, N. & Shibuya, M. Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc Natl Acad Sci U S A 102, 1076-1081 (2005).
33. Shalaby, F., et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376, 62-66 (1995).
34. Li, X., et al. VEGFR2 pY949 signalling regulates adherens junction integrity and metastatic spread. Nat Commun 7, 11017 (2016).
35. Sun, Z., et al. VEGFR2 induces c-Src signaling and vascular permeability in vivo via the adaptor protein TSAd. J Exp Med 209, 1363-1377 (2012).
36. Gaengel, K. & Betsholtz, C. Endocytosis regulates VEGF signalling during angiogenesis. Nat Cell Biol 15, 233-235 (2013).
37. Horowitz, A. & Seerapu, H.R. Regulation of VEGF signaling by membrane traffic. Cell Signal 24, 1810-1820 (2012).
38. Koch, S., et al. NRP1 presented in trans to the endothelium arrests VEGFR2 endocytosis, preventing angiogenic signaling and tumor initiation. Dev Cell 28, 633-646 (2014).
39. Nakayama, M., et al. Spatial regulation of VEGF receptor endocytosis in angiogenesis. Nat Cell Biol 15, 249-260 (2013).
40. Wang, Y., et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483-486 (2010).
41. Sawamiphak, S., et al. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature 465, 487-491 (2010).

40
42. Hudson, N., et al. Differential apicobasal VEGF signaling at vascular blood-neural barriers. Dev Cell 30, 541-552 (2014).
43. Gerhardt, H., et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 161, 1163-1177 (2003).
44. van Hinsbergh, V.W. & Koolwijk, P. Endothelial sprouting and angiogenesis: matrix metalloproteinases in the lead. Cardiovasc Res 78, 203-212 (2008).
45. Davis, G.E. & Senger, D.R. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res 97, 1093-1107 (2005).
46. Korff, T. & Augustin, H.G. Tensional forces in fibrillar extracellular matrices control directional capillary sprouting. J Cell Sci 112 ( Pt 19), 3249-3258 (1999).
47. Hellstrom, M., et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445, 776-780 (2007).
48. Suchting, S., et al. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci U S A 104, 3225-3230 (2007).
49. Funahashi, Y., et al. Notch regulates the angiogenic response via induction of VEGFR-1. J Angiogenes Res 2, 3 (2010).
50. Siekmann, A.F., Affolter, M. & Belting, H.G. The tip cell concept 10 years after: new players tune in for a common theme. Exp Cell Res 319, 1255-1263 (2013).
51. del Toro, R., et al. Identification and functional analysis of endothelial tip cell-enriched genes. Blood 116, 4025-4033 (2010).
52. Jakobsson, L., et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol 12, 943-953 (2010).
53. Bentley, K., et al. The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nat Cell Biol 16, 309-321 (2014).
54. Gaengel, K., et al. The sphingosine-1-phosphate receptor S1PR1 restricts sprouting angiogenesis by regulating the interplay between VE-cadherin and VEGFR2. Dev Cell 23, 587-599 (2012).
55. Montero-Balaguer, M., et al. Stable vascular connections and remodeling require full expression of VE-cadherin in zebrafish embryos. PLoS One 4, e5772 (2009).
56. Dartsch, N., Schulte, D., Hagerling, R., Kiefer, F. & Vestweber, D. Fusing VE-cadherin to alpha-catenin impairs fetal liver hematopoiesis and lymph but not blood vessel formation. Mol Cell Biol 34, 1634-1648 (2014).
57. Korn, C. & Augustin, H.G. Mechanisms of Vessel Pruning and Regression. Dev Cell 34, 5-17 (2015).
58. Lagendijk, A.K. & Hogan, B.M. VE-cadherin in vascular development: a coordinator of cell signaling and tissue morphogenesis. Curr Top Dev Biol 112, 325-352 (2015).
59. Dejana, E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol 5, 261-270 (2004).
60. Dejana, E. & Vestweber, D. The role of VE-cadherin in vascular morphogenesis and permeability control. Prog Mol Biol Transl Sci 116, 119-144 (2013).
61. Lampugnani, M.G., et al. A novel endothelial-specific membrane protein is a marker of cell-cell contacts. J Cell Biol 118, 1511-1522 (1992).
62. Suzuki, S., Sano, K. & Tanihara, H. Diversity of the cadherin family: evidence for eight new cadherins in nervous tissue. Cell Regul 2, 261-270 (1991).

41
63. Navarro, P., Ruco, L. & Dejana, E. Differential localization of VE- and N-cadherins in human endothelial cells: VE-cadherin competes with N-cadherin for junctional localization. J Cell Biol 140, 1475-1484 (1998).
64. Tanihara, H., et al. Characterization of cadherin-4 and cadherin-5 reveals new aspects of cadherins. J Cell Sci 107 ( Pt 6), 1697-1704 (1994).
65. Vincent, P.A., Xiao, K., Buckley, K.M. & Kowalczyk, A.P. VE-cadherin: adhesion at arm's length. Am J Physiol Cell Physiol 286, C987-997 (2004).
66. Navarro, P., et al. Catenin-dependent and -independent functions of vascular endothelial cadherin. J Biol Chem 270, 30965-30972 (1995).
67. Sauteur, L., et al. Cdh5/VE-cadherin Promotes Endothelial Cell Interface Elongation via Cortical Actin Polymerization during Angiogenic Sprouting. Cell Rep 9, 504-513 (2014).
68. Peifer, M., McCrea, P.D., Green, K.J., Wieschaus, E. & Gumbiner, B.M. The vertebrate adhesive junction proteins beta-catenin and plakoglobin and the Drosophila segment polarity gene armadillo form a multigene family with similar properties. J Cell Biol 118, 681-691 (1992).
69. Lampugnani, M.G., et al. The molecular organization of endothelial cell to cell junctions: differential association of plakoglobin, beta-catenin, and alpha-catenin with vascular endothelial cadherin (VE-cadherin). J Cell Biol 129, 203-217 (1995).
70. Hong, S., Troyanovsky, R.B. & Troyanovsky, S.M. Spontaneous assembly and active disassembly balance adherens junction homeostasis. Proc Natl Acad Sci U S A 107, 3528-3533 (2010).
71. Kuppers, V., Vockel, M., Nottebaum, A.F. & Vestweber, D. Phosphatases and kinases as regulators of the endothelial barrier function. Cell Tissue Res 355, 577-586 (2014).
72. Wallez, Y., et al. Src kinase phosphorylates vascular endothelial-cadherin in response to vascular endothelial growth factor: identification of tyrosine 685 as the unique target site. Oncogene 26, 1067-1077 (2007).
73. Wessel, F., et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat Immunol 15, 223-230 (2014).
74. Turowski, P., et al. Phosphorylation of vascular endothelial cadherin controls lymphocyte emigration. J Cell Sci 121, 29-37 (2008).
75. Orsenigo, F., et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun 3, 1208 (2012).
76. Baumeister, U., et al. Association of Csk to VE-cadherin and inhibition of cell proliferation. Embo J 24, 1686-1695 (2005).
77. Nada, S., Okada, M., MacAuley, A., Cooper, J.A. & Nakagawa, H. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature 351, 69-72 (1991).
78. Kourtidis, A., Ngok, S.P. & Anastasiadis, P.Z. p120 catenin: an essential regulator of cadherin stability, adhesion-induced signaling, and cancer progression. Prog Mol Biol Transl Sci 116, 409-432 (2013).
79. Monaghan-Benson, E. & Burridge, K. The regulation of vascular endothelial growth factor-induced microvascular permeability requires Rac and reactive oxygen species. J Biol Chem 284, 25602-25611 (2009).
80. Chiasson, C.M., Wittich, K.B., Vincent, P.A., Faundez, V. & Kowalczyk, A.P. p120-catenin inhibits VE-cadherin internalization through a Rho-independent mechanism. Mol Biol Cell 20, 1970-1980 (2009).

42
81. Xiao, K., et al. p120-Catenin regulates clathrin-dependent endocytosis of VE-cadherin. Mol Biol Cell 16, 5141-5151 (2005).
82. Gavard, J. & Gutkind, J.S. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol 8, 1223-1234 (2006).
83. Gavard, J., Patel, V. & Gutkind, J.S. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev Cell 14, 25-36 (2008).
84. Chen, X.L., et al. VEGF-induced vascular permeability is mediated by FAK. Dev Cell 22, 146-157 (2012).
85. Spring, K., et al. Tyrosine phosphorylation of DEP-1/CD148 as a mechanism controlling Src kinase activation, endothelial cell permeability, invasion, and capillary formation. Blood 120, 2745-2756 (2012).
86. Baumer, S., et al. Vascular endothelial cell-specific phosphotyrosine phosphatase (VE-PTP) activity is required for blood vessel development. Blood 107, 4754-4762 (2006).
87. Dominguez, M.G., et al. Vascular endothelial tyrosine phosphatase (VE-PTP)-null mice undergo vasculogenesis but die embryonically because of defects in angiogenesis. Proc Natl Acad Sci U S A 104, 3243-3248 (2007).
88. Nawroth, R., et al. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. Embo J 21, 4885-4895 (2002).
89. Nottebaum, A.F., et al. VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J Exp Med 205, 2929-2945 (2008).
90. Broermann, A., et al. Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. J Exp Med 208, 2393-2401 (2011).
91. Schlingemann, R.O. & van Hinsbergh, V.W. Role of vascular permeability factor/vascular endothelial growth factor in eye disease. Br J Ophthalmol 81, 501-512 (1997).
92. Dejana, E., Orsenigo, F. & Lampugnani, M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci 121, 2115-2122 (2008).
93. Zhang, X. & Simons, M. Receptor tyrosine kinases endocytosis in endothelium: biology and signaling. Arterioscler Thromb Vasc Biol 34, 1831-1837 (2014).
94. Jean, S. & Kiger, A.A. Coordination between RAB GTPase and phosphoinositide regulation and functions. Nat Rev Mol Cell Biol 13, 463-470 (2012).
95. Watt, S.A., Kular, G., Fleming, I.N., Downes, C.P. & Lucocq, J.M. Subcellular localization of phosphatidylinositol 4,5-bisphosphate using the pleckstrin homology domain of phospholipase C delta1. Biochem J 363, 657-666 (2002).
96. Vanhaesebroeck, B., Guillermet-Guibert, J., Graupera, M. & Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol 11, 329-341 (2010).
97. Posor, Y., et al. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 499, 233-237 (2013).
98. Gillooly, D.J., et al. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. Embo J 19, 4577-4588 (2000).
99. Sasaki, T., et al. Mammalian phosphoinositide kinases and phosphatases. Prog Lipid Res 48, 307-343 (2009).

43
100. Li, G. & Marlin, M.C. Rab family of GTPases. Methods Mol Biol 1298, 1-15 (2015).
101. Shin, H.W., et al. An enzymatic cascade of Rab5 effectors regulates phosphoinositide turnover in the endocytic pathway. J Cell Biol 170, 607-618 (2005).
102. Simonsen, A., et al. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature 394, 494-498 (1998).
103. Hislop, A.A. Airway and blood vessel interaction during lung development. J Anat 201, 325-334 (2002).
104. Rock, J.R. & Hogan, B.L. Epithelial progenitor cells in lung development, maintenance, repair, and disease. Annu Rev Cell Dev Biol 27, 493-512 (2011).
105. Metzger, R.J., Klein, O.D., Martin, G.R. & Krasnow, M.A. The branching programme of mouse lung development. Nature 453, 745-750 (2008).
106. Herriges, M. & Morrisey, E.E. Lung development: orchestrating the generation and regeneration of a complex organ. Development 141, 502-513 (2014).
107. Snoeck, H.W. Modeling human lung development and disease using pluripotent stem cells. Development 142, 13-16 (2015).
108. Ohuchi, H., et al. FGF10 acts as a major ligand for FGF receptor 2 IIIb in mouse multi-organ development. Biochem Biophys Res Commun 277, 643-649 (2000).
109. Sekine, K., et al. Fgf10 is essential for limb and lung formation. Nat Genet 21, 138-141 (1999).
110. Bostrom, H., et al. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 85, 863-873 (1996).
111. Lindahl, P., et al. Alveogenesis failure in PDGF-A-deficient mice is coupled to lack of distal spreading of alveolar smooth muscle cell progenitors during lung development. Development 124, 3943-3953 (1997).
112. Hogan, B.L., et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 15, 123-138 (2014).
113. Desai, T.J., Brownfield, D.G. & Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 507, 190-194 (2014).
114. Crapo, J.D., Barry, B.E., Gehr, P., Bachofen, M. & Weibel, E.R. Cell number and cell characteristics of the normal human lung. Am Rev Respir Dis 126, 332-337 (1982).
115. Williams, M.C. Alveolar type I cells: molecular phenotype and development. Annu Rev Physiol 65, 669-695 (2003).
116. Logan, C.Y. & Desai, T.J. Keeping it together: Pulmonary alveoli are maintained by a hierarchy of cellular programs. Bioessays 37, 1028-1037 (2015).
117. Whitsett, J.A., Wert, S.E. & Weaver, T.E. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu Rev Med 61, 105-119 (2010).
118. Uhal, B.D. Cell cycle kinetics in the alveolar epithelium. Am J Physiol 272, L1031-1045 (1997).
119. Barkauskas, C.E., et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest 123, 3025-3036 (2013).
120. Kumar, P.A., et al. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 147, 525-538 (2011).
121. Giangreco, A., Reynolds, S.D. & Stripp, B.R. Terminal bronchioles harbor a unique airway stem cell population that localizes to the bronchoalveolar duct junction. Am J Pathol 161, 173-182 (2002).

44
122. Kim, C.F., et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121, 823-835 (2005).
123. Devereux, G. ABC of chronic obstructive pulmonary disease. Definition, epidemiology, and risk factors. Bmj 332, 1142-1144 (2006).
124. Vlahos, R. & Bozinovski, S. Recent advances in pre-clinical mouse models of COPD. Clin Sci (Lond) 126, 253-265 (2014).
125. Vestbo, J., et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 187, 347-365 (2013).
126. Shapiro, S.D. Proteinases in chronic obstructive pulmonary disease. Biochem Soc Trans 30, 98-102 (2002).
127. Morissette, M.C., Parent, J. & Milot, J. Alveolar epithelial and endothelial cell apoptosis in emphysema: what we know and what we need to know. Int J Chron Obstruct Pulmon Dis 4, 19-31 (2009).
128. Segura-Valdez, L., et al. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest 117, 684-694 (2000).
129. Giordano, R.J., et al. Targeted induction of lung endothelial cell apoptosis causes emphysema-like changes in the mouse. J Biol Chem 283, 29447-29460 (2008).
130. Kasahara, Y., et al. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med 163, 737-744 (2001).
131. Kasahara, Y., et al. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest 106, 1311-1319 (2000).
132. Tang, K., Rossiter, H.B., Wagner, P.D. & Breen, E.C. Lung-targeted VEGF inactivation leads to an emphysema phenotype in mice. J Appl Physiol (1985) 97, 1559-1566; discussion 1549 (2004).
133. Kearns, M.T., et al. Vascular endothelial growth factor enhances macrophage clearance of apoptotic cells. Am J Physiol Lung Cell Mol Physiol 302, L711-718 (2012).
134. Alagappan, V.K., de Boer, W.I., Misra, V.K., Mooi, W.J. & Sharma, H.S. Angiogenesis and vascular remodeling in chronic airway diseases. Cell Biochem Biophys 67, 219-234 (2013).
135. Johnson, L.N. & Barford, D. The effects of phosphorylation on the structure and function of proteins. Annu Rev Biophys Biomol Struct 22, 199-232 (1993).
136. Johnson, L.N. & O'Reilly, M. Control by phosphorylation. Curr Opin Struct Biol 6, 762-769 (1996).
137. Khoury, G.A., Baliban, R.C. & Floudas, C.A. Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci Rep 1(2011).
138. Manning, G., Whyte, D.B., Martinez, R., Hunter, T. & Sudarsanam, S. The protein kinase complement of the human genome. Science 298, 1912-1934 (2002).
139. Liberti, S., et al. HuPho: the human phosphatase portal. Febs J 280, 379-387 (2013).
140. Ciesla, J., Fraczyk, T. & Rode, W. Phosphorylation of basic amino acid residues in proteins: important but easily missed. Acta Biochim Pol 58, 137-148 (2011).
141. Bridges, D. & Saltiel, A.R. Phosphoinositides: Key modulators of energy metabolism. Biochim Biophys Acta 1851, 857-866 (2015).

45
142. Barker, C.J. & Berggren, P.O. New horizons in cellular regulation by inositol polyphosphates: insights from the pancreatic beta-cell. Pharmacol Rev 65, 641-669 (2013).
143. Muthana, S.M., Campbell, C.T. & Gildersleeve, J.C. Modifications of glycans: biological significance and therapeutic opportunities. ACS Chem Biol 7, 31-43 (2012).
144. Alonso, A. & Pulido, R. The extended human PTPome: a growing tyrosine phosphatase family. Febs J (2015).
145. Deshpande, T., Takagi, T., Hao, L., Buratowski, S. & Charbonneau, H. Human PIR1 of the protein-tyrosine phosphatase superfamily has RNA 5'-triphosphatase and diphosphatase activities. J Biol Chem 274, 16590-16594 (1999).
146. Shi, Y. Serine/threonine phosphatases: mechanism through structure. Cell 139, 468-484 (2009).
147. Alonso, A., et al. Protein tyrosine phosphatases in the human genome. Cell 117, 699-711 (2004).
148. Tonks, N.K. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol 7, 833-846 (2006).
149. Tonks, N.K. Protein tyrosine phosphatases--from housekeeping enzymes to master regulators of signal transduction. Febs J 280, 346-378 (2013).
150. Andersen, J.N., et al. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol Cell Biol 21, 7117-7136 (2001).
151. Pulido, R., Stoker, A.W. & Hendriks, W.J. PTPs emerge as PIPs: protein tyrosine phosphatases with lipid-phosphatase activities in human disease. Hum Mol Genet 22, R66-76 (2013).
152. Leslie, N.R., Maccario, H., Spinelli, L. & Davidson, L. The significance of PTEN's protein phosphatase activity. Adv Enzyme Regul 49, 190-196 (2009).
153. Zhang, Z.Y. Mechanistic studies on protein tyrosine phosphatases. Prog Nucleic Acid Res Mol Biol 73, 171-220 (2003).
154. Jia, Z., Barford, D., Flint, A.J. & Tonks, N.K. Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science 268, 1754-1758 (1995).
155. Pannifer, A.D., Flint, A.J., Tonks, N.K. & Barford, D. Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by x-ray crystallography. J Biol Chem 273, 10454-10462 (1998).
156. Flint, A.J., Tiganis, T., Barford, D. & Tonks, N.K. Development of "substrate-trapping" mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci U S A 94, 1680-1685 (1997).
157. Begley, M.J. & Dixon, J.E. The structure and regulation of myotubularin phosphatases. Curr Opin Struct Biol 15, 614-620 (2005).
158. Arantes, G.M. The catalytic acid in the dephosphorylation of the Cdk2-pTpY/CycA protein complex by Cdc25B phosphatase. J Phys Chem B 112, 15244-15247 (2008).
159. Zhang, Z.Y. & Dixon, J.E. Active site labeling of the Yersinia protein tyrosine phosphatase: the determination of the pKa of the active site cysteine and the function of the conserved histidine 402. Biochemistry 32, 9340-9345 (1993).
160. Ostman, A., Frijhoff, J., Sandin, A. & Bohmer, F.D. Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem 150, 345-356 (2011).
161. Chiarugi, P., et al. Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol 161, 933-944 (2003).

46
162. Colavitti, R., et al. Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. J Biol Chem 277, 3101-3108 (2002).
163. Oshikawa, J., et al. Extracellular SOD-derived H2O2 promotes VEGF signaling in caveolae/lipid rafts and post-ischemic angiogenesis in mice. PLoS One 5, e10189 (2010).
164. Reiterer, V., Eyers, P.A. & Farhan, H. Day of the dead: pseudokinases and pseudophosphatases in physiology and disease. Trends in cell biology 24, 489-505 (2014).
165. Kharitidi, D., Manteghi, S. & Pause, A. Pseudophosphatases: methods of analysis and physiological functions. Methods 65, 207-218 (2014).
166. Kabir, N.N., Ronnstrand, L. & Kazi, J.U. Deregulation of protein phosphatase expression in acute myeloid leukemia. Med Oncol 30, 517 (2013).
167. Lucci, M.A., et al. Expression profile of tyrosine phosphatases in HER2 breast cancer cells and tumors. Cell Oncol 32, 361-372 (2010).
168. Ferrara, N. & Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat Rev Drug Discov (2016).
169. Bhasin, M., et al. Bioinformatic identification and characterization of human endothelial cell-restricted genes. BMC Genomics 11, 342 (2010).
170. Wallgard, E., et al. Identification of a core set of 58 gene transcripts with broad and specific expression in the microvasculature. Arterioscler Thromb Vasc Biol 28, 1469-1476 (2008).
171. Kalen, M., et al. Combination of reverse and chemical genetic screens reveals angiogenesis inhibitors and targets. Chem Biol 16, 432-441 (2009).
172. Wallgard, E., et al. Paladin (X99384) is expressed in the vasculature and shifts from endothelial to vascular smooth muscle cells during mouse development. Dev Dyn 241, 770-786 (2012).
173. Oganesian, A., et al. Protein tyrosine phosphatase RQ is a phosphatidylinositol phosphatase that can regulate cell survival and proliferation. Proc Natl Acad Sci U S A 100, 7563-7568 (2003).
174. Suzuki, T., et al. Strategy for comprehensive identification of human N-myristoylated proteins using an insect cell-free protein synthesis system. Proteomics 10, 1780-1793 (2010).
175. Resh, M.D. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim Biophys Acta 1451, 1-16 (1999).
176. Huang, S.M., Hancock, M.K., Pitman, J.L., Orth, A.P. & Gekakis, N. Negative regulators of insulin signaling revealed in a genome-wide functional screen. PLoS One 4, e6871 (2009).
177. Roffers-Agarwal, J., Hutt, K.J. & Gammill, L.S. Paladin is an antiphosphatase that regulates neural crest cell formation and migration. Dev Biol 371, 180-190 (2012).
178. Li, S., Wang, L., Berman, M., Kong, Y.Y. & Dorf, M.E. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 35, 426-440 (2011).
179. Burn, S.F. Detection of beta-galactosidase activity: X-gal staining. Methods Mol Biol 886, 241-250 (2012).
180. Gale, N.W., et al. Ephrin-B2 selectively marks arterial vessels and neovascularization sites in the adult, with expression in both endothelial and smooth-muscle cells. Dev Biol 230, 151-160 (2001).
181. Kuhnert, F., et al. Essential regulation of CNS angiogenesis by the orphan G protein-coupled receptor GPR124. Science 330, 985-989 (2010).

47
182. Fuchs, H., et al. Mouse phenotyping. Methods 53, 120-135 (2011). 183. Gailus-Durner, V., et al. Introducing the German Mouse Clinic: open access
platform for standardized phenotyping. Nat Methods 2, 403-404 (2005). 184. March, T.H., et al. Modulators of cigarette smoke-induced pulmonary
emphysema in A/J mice. Toxicol Sci 92, 545-559 (2006). 185. Hardin, M., et al. Sex-specific features of emphysema among current and
former smokers with COPD. Eur Respir J 47, 104-112 (2016). 186. Lockett, A.D., et al. Active trafficking of alpha 1 antitrypsin across the lung
endothelium. PLoS One 9, e93979 (2014).

Acta Universitatis UpsaliensisDigital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 1225
Editor: The Dean of the Faculty of Medicine
A doctoral dissertation from the Faculty of Medicine, UppsalaUniversity, is usually a summary of a number of papers. A fewcopies of the complete dissertation are kept at major Swedishresearch libraries, while the summary alone is distributedinternationally through the series Digital ComprehensiveSummaries of Uppsala Dissertations from the Faculty ofMedicine. (Prior to January, 2005, the series was publishedunder the title “Comprehensive Summaries of UppsalaDissertations from the Faculty of Medicine”.)
Distribution: publications.uu.seurn:nbn:se:uu:diva-281708
ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2016