the symmetry of energy levels parity - unibas.chtulej/spectroscopy_related_aspects/lecture32... ·...
TRANSCRIPT

Asymetric tops
continuation

For asymetric top molecules, CBA III ≠≠ and the classical energy for a rigidrotor is given
C
c
B
b
A
a
IJ
IJ
IJE
222
222
++=
C
c
B
b
A
a
IJ
IJ
IJH
2ˆ
2ˆ
2
ˆˆ222
++=
This results in in the rigid asymetric rotor Hamiltonian:
The asymmetric to Hamiltonian is solved using a symmetric top basis set,changing slightly the form of the terms in the Hamiltonian:
C
B
A
IC
IB
IA
2
2
2
2
2
2
h
h
h
=
=
=
2222 ˆˆˆˆcba JCJBJAH ++=h

])ˆ()ˆ)[(4
(ˆ)2
()ˆ)(2
(
)ˆˆ)(2
(ˆ)ˆˆ)(2
(ˆ
2222
222222
−+ −−
++
−++
=
−−
++++
=
JJBAJBACJBA
JJBAJCJJBAH
c
bacbah
The following symmetric top matrix elements are used to calculate energy levels:
21
22
21
22
222
22
)]2)(1)(1)([()ˆ(2
)]2)(1)(1)([()ˆ(2
ˆ)1(ˆ
+−−++−+=−
++−−++−=+
=
+=
+
−
KJKJKJKJJKJJK
KJKJKJKJJKJJK
JKJKJJK
JJJKJJK
c
h
h
h
h
Where J+ is a lowering operator and J- is rising operator (molecular frame). With the symmetric top basis functions the asymmetric top Hamiltonian has matrix elements with ΔK=0 and ΔK=+/-2
The Hamiltonian can be solved for particular J. Labelling of the energy levelsis carried out by considering the correlation diagram that connects the energylevels of a prolate top with those of an oblate top.

Prolate-oblate correlation diagram (labeling asymetric top levels)

Asymetric top energy levels

The energy levels of prolate and oblate symmetric tops are
)1()(
)1()(2
2
++−=
++−=
JBJKBCE
JBJKBAE
co
ap
For a given value of J the Ka levels increase in energy as Ka increases for a prolate top, while the Kc levels decrese in energy as Kc increases for an oblate top
The levels are labeled byJKaKc , where J is a good quantum number, but Ka and Kc are labels for the asymmetric top.
Ka and Kc are good quantum numbers only in the prolate or oblate symmetric top limits.
Ka+Kc= J or J+1

τ
= Ka-Kc τ =+J..0..-J
Convention:τ label (K-like) is introduced and it defines 2J+1 different possible values for every J
The degree of asymmetry can be quantified by an asymmetry parameter („ Ray‘s asymmetry parameter „ ) κ,
which runs from -1 for a prolate top to +1 for an oblate top:
CACAB
−−−
=2κ
A
B
CNotation of the asymetric top labels Ka=Kp, Kc=Ko ---- K-1 and K+1

Selection rules of asymmetric top
Generally, an arbitrary molecule has three dipole moment components μ=(μa, μb, μc). Each nonvanishing dipole moment makes a certain setof transitions possible, and leads to a set of selection rules.
J-----ΔJ=0, +/-1M----ΔM=0, +/-1
a
b
cμ
μa
μbμc
A-type:
ma, mb=mc=0 DKa=0 (+/-2,+/-4,...) DKc=+/-1, +/-3,...B-type
mb, ma=mc=0 DKa=+/-1, (+/-3, +/-5) DKc=+/-1, +/-3
C-type mc, ma=mb=0 DKa=+/-1(+/-3, +/-5) DKc=0, (+/-2, +/-4)

000
101
111
110c-type
b-type
a-type
a-,b-,c-type transitions
For molecules of low symmetry, all three types could occur together

Structure determination
The moments of inertia are related to bond lengths and bond angles.Generally, the interest is in Be rotational constant extrapolated tothe bottom of the potential well.
Be
B0
B1
B2
r0
r1
r2
re
...)21()
21(
12 2
2
++++−=
=
vvBB
rB
eee
v
γα
ννμ
ν
h
An average over a differentvibrational functions
Knowing B0 and B1 one can determineBe and α0
Diatomic potential energy curve and vibrational levels

Structure determination in polyatomic molecules
1. 3N-6 (or 5) vibrational modes and similar number of α‘s for eachA,B,C
∑
∑
∑
−
=
−
=
−
=
+==
+==
+==
63
1
63
1
63
1
)2/(
)2/(
)2/(
N
iii
Ciev
N
iii
Biev
N
iii
Aiev
dvCC
dvBB
dvAA
α
α
α
di ----is the degeneracy of the ith mode
2. More than three structural parameters need to be determined from at most three moments of inertia (planar - only 2)One makes use of data from isotopic molecules
e.g., H2 CO, H213CO, H2 C18O
and additional set of independent moments of inertia can be derived and available to determine structural parameters

Coupling of angular momenta in diatomic molecules

The theory developed so far requires quite substantial modification When the species under study possesses electronic angular momenta
The total angular momentum J can be described:
J=R+L+S+(lvib )+(Inuc )R ----Nuclear rotational angular momentumL ---- Electronic angular momentum S --- Spin angular momentumlvib --Vibrational angular momentumInuc - Spin nuclear angular momentum
These additional angular momenta will cause changes to the rotational Hamiltonian
Hrot = BR2 = B(J-L-S)2
lvib, I nuc are neglected initially

The rotational energy levels can be affected by:
intramolecular interactions involving angular momenta:
e.g., -----LS-spin orbit, RL rotational-electronic,...
-----nuclear hyperfine interactions I-S nuclear-electron spin dipolar interaction SI Fermi contact interactionIL Nuclear spin-electron orbital hyperfine couplingNuclear electrostatic quadrupole interaction
interactions with external fields B, E:----Zeeman & Stark interactions
L S
R I
L S
R I
B(E)

Construction of effective Hamiltonians
The interpretation of the microwave spectra involves understanding how the different sorts of angular momenta interact with each otherto produce the energy levels.
The couplig cases can be represented by vector diagrams
The effective Hamiltonian contains terms describing the variousangular momenta, the magnetic and electric interaction within the molecule, and interactions with external fields.
The effective Hamiltonian is designed so it operates within one electronic and one vibrational level. Final choice of H is made with terms which are likely to have an observable effect on the spectrum.
The set of basis functions is chosen in such way that it most nearly yields a diagonal matrix of Hamiltonian.
for diatomic and linear molecule one coose a basis set corresponding to HUND Cases

Brief review of the separate angular momenta in diatomic molecules
L
Λ
Orbital angular momentum L
Component of L along the internuclear axis is only defined: Λ.The different values of IΛI correspond to different electronic states
Electron spin S The spins of the individual electrons form a resultant S, and anassociated magnetic moment με.
. If orbital angular momentum is Present, the spin-orbit interaction will orient the spin S so that the magnetic moment due to S also lies along the nuclear axis. The component along the axis is Σ.
L
Λ
S
Σ
Coupling of L and S to the internuclear axis
Resultant component of total electronic angular momentum:
Ω=Λ+Σ
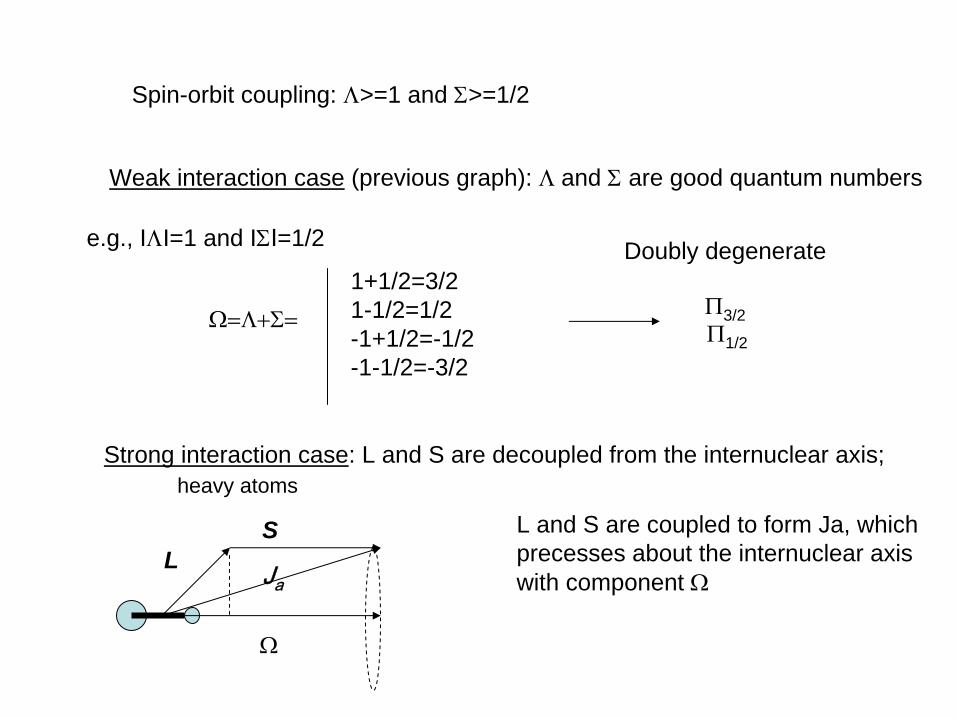
Spin-orbit coupling: Λ>=1 and Σ>=1/2
Weak interaction case (previous graph): Λ and Σ
are good quantum numbers
e.g., IΛI=1 and IΣI=1/2
Ω=Λ+Σ=
1+1/2=3/21-1/2=1/2-1+1/2=-1/2-1-1/2=-3/2
Π3/2Π1/2
Doubly degenerate
Strong interaction case: L and S are decoupled from the internuclear axis;
L Ja
S
Ω
L and S are coupled to form Ja, which precesses about the internuclear axis with component Ω
heavy atoms
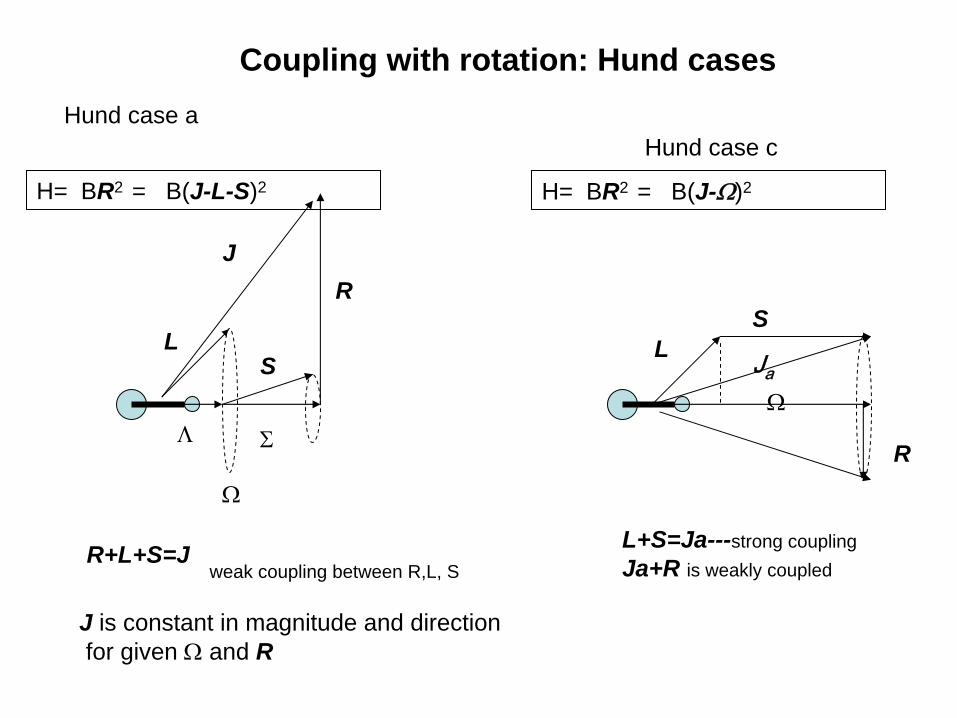
Coupling with rotation: Hund cases
L
Λ
S
Σ
JR
Ω
L Ja
S
Ω
R
Hund case aHund case c
R+L+S=J
J is constant in magnitude and directionfor given Ω
and R
weak coupling between R,L, SL+S=Ja---strong couplingJa+R is weakly coupled
H= BR2 = B(J-L-S)2 H= BR2 = B(J-Ω)2

J
L
S
Λ
For Λ=0 (also for Λ=/0; SO coupling weak)
R is coupled to L to form a resultant N, N is coupled to the S to form the total J
NR+L=NN+S=J
Hund‘s case b
Remarks: The Hund‘s coupling cases correspond to particular coordinate transformations.
case a: transformation of both the electron position and spin coordinates froma space-fixed axis system to the molecule-fixed rotating axes; with a case a basis set one specifies the values of J,Ω,Λ,Σ,I and the spatial components MJ and M I
case b:transformation of electron positions only;with this case basis set one specifies J,N, S, I, MJ, MI
H= BR2 = B(N-L)2

Survey of the results obtained for diatomic molecules arranged in the increasing order of complexity
1Σ
- ---CS,---Λ=Σ=I=0
Stark splitting
J=1
J=0
MJ =0
MJ =+/-1
MJ =0
The Hamiltonian determining the rotational levels contains only terms describing thenuclear rotation and centrifugal distorsion.
The Stark perturbations produced by an electric field do enable the electricdipole moment to be determined

1Π a1Π Excited state of CS [L=1, S=0] (radiofrequency/optical double resonance study)
H= BR2 = B(J-L)2 = B(J2-2JL+L2)=B(J2-2Jz Lz -2Jx Lx -2Jy Ly +Lz
2+Lx2+Ly
2)
=B(J(J+1)-Λ2)+B(Lx2+Ly
2)-“B(Jx Lx +Jy Ly )
In the absence of JL terms, energy levels are given by BJ(J+1) and each would possess the two-fold Λ
degeneracy
In terms of Hund case a - Ω=Λ=1 and in the lowest rotational level is J=1
Jz Lz and Lz2 can be replaced by
Λ2; Lx2,L y2 are independant of J
and they do not affect relative spacing of the rotational levels.
The last term represents coupling between the rotational and electronic motions, having the effect of mixing the 1Π
state with other electronic states of different Λ
Λ-degeneracy is removed; the splitting of the components can be observed and it inreases with the increase of J : Λ-DOUBLING
J3
2
1
parity-+
+-
-/+

1Δ O2 , SO, SeO, NF
Ω=Λ=2, S=0, Hund case a, the lowest rotational level starts with J=2
H= BR2 = B(J-L)2 = B(J2-2JL+L2)=B(J2-2Jz Lz -2Jx Lx -2Jy Ly +Lz
2+Lx2+Ly
2)
=B(J(J+1)-Λ2)+B(Lx2+Ly
2)-“B(Jx Lx +Jy Ly )
Jz Lz and Lz2 can be replaced by
Λ2; Lx2,L y2 are independant of J
and they do not affect relative spacing of the rotational levels.
In Δ
states, the Λ
splitting is usually to small to be observed
J4
3
2
parity-+
+-
-/+
Hrot =B0 J(J+1)

N3
2
0
J
5/2 +3/2 +
3/2 -½ -
½ +
2Σ CN, SiN and CO+
It conforms to Hund‘s case b coupling, L=0 S=1/2---
H= BR2 = B(N-L)2 = B(N2)
=BN(N+1)
Each N level is split into a doublet by the spin-rotation interaction γNS ; J
J=N+/-1/2

3Σ O2 , SO, SeO
Effective Hamiltonian:
H=BN2+2/3 λ(3S2z -S2)+γNS
Spin-spin interaction energies
J=N+1 E=-2λN/[3(2N+3)]J=N E=2/3 λJ=N-1 E=-2λ(N+1)/[3(2N-1)]
Spin-rotation interaction energies
J=N+1 E=-2γNJ=N E=-γJ=N-1 E=-γ(N+1)

2Π Majority of radicals have 2Π ground electronic state
Generally, Hund‘s case a or b is possible, usually the species show intermediate case.
In case a spin-orbit coupling split energy pattern on two stacks ½ and 3/2; these are fine structure states. The sign of spin-orbit coupling constant will determine energy ordering (normal ½ and 3/2 or inverted 3/2 and ½ order) In case b N quantum number is good. J remains good in both cases. Each level retains an extra two-fold degeneracy, irrespective of whether the coupling is close to a (Ω-doubling) or b type (Λ
doubling)

The symmetry of energy levels Parity
Diatomic or linear molecules

Parity concept
+/-, e/f, g/u, s/a
They are related to the symmetry operator which commutes withthe molecular Hamiltonian:
Types of parity:
[H, O]=0
Hψ+/- =E+/- ψ+/-Oψ+/- =E+/- ψ+/-
Since the molecule has certain symmetry, the effect of the associated symmetry operator can be used to label wavefunctions and their energy levels.
Both, the symmetry operator and the part of the total Hamiltonian underconsideration hast obe specified carefully !!!!

+/- ------total parity
The symmetry operation and the total Hamiltonian including electronic, vibrational and rotational parts are used (not nuclear spin).
The symmetry operator E* inverts all of the coordinates of the particles (nuclei and electrons) in the laboratory frame with the origin at the center of the mass:
E* ψ(Xi,Yi,Zi)=ψ(-Xi,-Yi,-Zi) = +/- ψ(Xi,Yi,Zi).
All relative positions of the particle are the same before and after inversionand the energy levels are unchanged by application of this operator.
Relevance: nuclear physics, atomic physicsleft and right hand coordinate system is equivalentsymmetry brakes in β-decay process
E* operator is used to divide all rovibronic states into two groups
E*ψ
= E*(ψel ψvib ψrot )=+/- ψ
The effects have to be determined individually

Electronic part
Evaluating the effect of inversion on ψel one has to consider that the electronic wave is define in the molecular frame (BO approximation) butinversion operation is prepared in laboratory frame.
It has been shown that E* operation in the laboratory frame is equivalent to the symmetry operation of reflection σv in the molecular frame
Hougen(1970)The calculation of rotational energy levels and rotational line intensities in diatomic molecules, US Gov. Print. Office
NBS Monograph 115, Washington, D.C., 1970
xi,yi, zi ------------ xi, -yi, zireflection is arbitrary chosen to be in the xz plane of the molecule
Z
X
Y
θ
φ
z
Diatomic molecule ccordinate system

In the next step, the detailed form of the spin and orbital parts of the electronic wavefunction has to be considered. According to Hougen:
σv IS,Σ>=(-1)S-ΣIS, -Σ>
σv IΛ>=+/-(-1)ΛI -Λ>
The effect of σv on a IΛ=0> orbital part of the electronic wavefunction is
σv I Λ=0> = +/- IΛ=0>σv IΣ+/- > = +/- I Σ+/- >
+/- are superscripts on the term symbols Σ+ and Σ- and indicate the effects ofthe σv symmetry operator on only the orbital part of the electronic wavefunction.
For Λ>0 +/- superscript is not used since the levels always occur as a +/- pair because of the twofold orbital degeneracy.

Vibrational part
The effect of E* symmetry operator on the vibrational part of the diatomic wavefunction gives + because inversion of all particle leaves r unchanged. The vibrational wavefunction is a function of the magnitude of the internuclear separation.
E*ψvib (r)=+ψvib (r)

Rotational part
For evaluation of the effect of E* on the rotational wavefunction, the properties of the rotational wave functions has to be considered.
For diatomic molecule the E* operation replaces θ
by π−θ
and φ
by φ+π in
ψrot
(θ,φ). In
Σ+ states ψrot =YJM
E* YJM =(-1)J YJM
In general,
ψrot =IΩJM>, E*IΩJM>=(-1)J-ΩIΩJM>

The effect of the σv operator on the total wavefunction:
σv (ψel ψvib ψrot ) = σv (InΛSΣ> Iv> IΩJM>)
=(-1)J-2Σ+S+σ (In,-Λ,S,-Σ> Iv> I-ΩJM>)
σ=0 for all states except Σ- sates for which σ=1
Since the σv operation changes the signs of Λ, Σ,
and Ω, the parity eigenfunctions are linear combinations of the basis functions:
2s+1Λ Ω+/−
=I2s+1ΛΩ
>+/-(-1)J-2Σ+s+σI2s+1Λ−Ω
>
σv
I2s+1ΛΩ+/-
>=+/-I2s+1Λ
Ω+/−
)

Selection rules for one photon electric dipole transition
∫ τμψψ dif * and E*μ = −μ
+ - transitions are only allowed for one photon electric dipole transition
Parity conservation rule
Parity can be violated!!!See Quack, M.(ETH) publications
The selection rules on total parity are derived by requiring a totally symmetric integrand for the transition moment integral

Examples: parity of Σ states

Examples: parity of Π
states

e/f -rotational parity
The total parity alternates always with J because of the phase factor
E*IΩJM>= (-1)J-Ω
IΩJM>
For convinience this is factorized out by defining e and f parity
For integer J:
E*ψ=+(-1)Jψ−−−−
eE*ψ=-(-1)Jψ−−−−− f
For half-integer J:
E*ψ=+(-1)J-1/2ψ−−−−
eE*ψ=-(-1)J-1/2ψ−−−−− f
e,f parity is a rotationalless parity describing the total parity with the rotational part removed.In the molecular frame, E* operator is replaced by σv

All energy levels of 1Σ+ states have e parity1Σ- states have f parity
1Π states have pairs e/f
The selection rules for total parity +<->- becomes:
e-e ----for P
branch (J=J‘-J‘‘=-1)f-f -----for R branch (J=J‘-J‘‘=+1)e-f------for Q branch (J=J‘-J‘‘=0

Gerade (g)/ ungerade (u) parity
g/u are used to classify the electronic orbital part of the total wavefunction It is applied to to homonuclear diatomic molecules: C2 , N2 , O2 ....
This parity is defined using an inversion operation i applied inthe molecular frame and only the electrons are inverted throughthe center of the molecule:
ι ψel
(x,y,z)=ψ
el (-xi,-yi,-zi)= +/-ψel (xi,yi,zi)
ι ψ IΛ>=+/- IΛ>
g ug, u parity is appended as a subscript to the term symbol of a diatomic molecule:
E,g., 2Σ+g
2Πg1Δg

s/a (symmetric/antisymmetric) parity
It is used for homonuclear diatomic molecules to classify the rotational energy levels
To define the parity of the rotational levels, one uses the Pauly exclussion principle:
the total wavefunction including nuclear spin has to e symmetric or antisymmetric with respect to interchange of the two identical nuclei
P12 (ψψnuc )=+ (ψψnuc ) for bosons I=0,1,2...P12 (ψψnuc )= -(ψψnuc ) for fermions I=1/2, 3/2,...
Operator is applied in the laboratory frame
Wavefunction includes electron spin,orbital, vibrational and rotational parts

Examples:
1. H2 , F2 nuclei are fermions I=1/2
α(1)α(2)ψnuc (sym)= ( ½)1/2 [ α(1)β(2)+β(1)α(2)]
β(1)β(2)
ψnuc (antisym) = (½ )1/2 [α(1)β(2)−β(1)α(2)]
Four nuclear spin wavefunctions exists and they can be grouped in
The antimetric nuclear spin wavefunction ψnuc (antisym) is then combined witha symmteric normal wavefunction to give overall antisymmetric product
the rovibrational energy levels are symmetric s para
The antimetric nuclear spin wavefunction ψnuc (sym) is then combined witha antisymmteric normal wavefunction to give overall antisymmetric product
the rovibrational energy levels are symmetric a ortho

2. The ground state of O2 3Σ-
g
The nuclear spin of O16 is zero -----bosonposseses only symmetric nuclear spin function
The even N values have – parity(a) and the odd N values + parity (s)
The symmetric ψnuc waavefunction must be combined with the s symmetry ψ
function.The a levels of oxygen (even N) cannot exist and are therefore missing in the spectrum
The parity of O2 X3Σ-g

Nuclear spin weight and electric dipole selection rules
The s and a symmetry levels are useful in establishing the relative intensitiesof rotational lines. In general the relative nuclear spin weight (spin statistics) of ortho levels relative to para levels is given by:
[(2I+1)(I+1)]/ [(2I+1)I = (I+1)/I
The electric dipole selection rules for s and a symmetry is s-s and a-a since there is no transition between para and ortho form (nuclear spin flip during the transition is forbidden
it can be weakly observed

Application of P12
Representing E* in the molecular frame –σv
the permutatio-iversion operation can be seen as reflection in mol frame
The P12 operation is a permutation-inversion operation that switchesthe two nuclei of a homonuclear diatomic molecule.It can be applied to the wavefunction in two steps.
All of the electrons and all of the nuclei are inverted through the origin by applying the E* operation in the laboratory frame;
Only the electrons are inverted back by applying ι in the molecular frame.
All + rotational energy levels have s symmetry for g electronic states or a symmetry for u electronic states
All - rotational energy levels have a symmetry for g electronic states or a symmetry for u electronic states
P12 = σvxz i =σv
xzσvxzC2(y)=C2
(y)
Similarly, the ι operation in the molecularframe is equivalent to:
E*P12
inversion of the electrons and nuclei and switching the nuclei back leaving only the electrons inverted

s/a parity of Σ
states