theproteasomestressreguloniscontrolledbyapairofnac ...role for 26s proteasome levels in controlling...
TRANSCRIPT

The Proteasome Stress Regulon Is Controlled by a Pair of NACTranscription Factors in Arabidopsis
Nicholas P. Gladman,a Richard S. Marshall,a,b Kwang-Hee Lee,a and Richard D. Vierstraa,b,1
a Department of Genetics, University of Wisconsin, Madison, Wisconsin 53706bDepartment of Biology, Washington University in St. Louis, St. Louis, Missouri 63130
ORCID ID: 0000-0003-0210-3516 (R.D.V.)
Proteotoxic stress, which is generated by the accumulation of unfolded or aberrant proteins due to environmental or cellularperturbations, can be mitigated by several mechanisms, including activation of the unfolded protein response and coordinatedincreases in protein chaperones and activities that direct proteolysis, such as the 26S proteasome. Using RNA-seq analysescombined with chemical inhibitors or mutants that induce proteotoxic stress by impairing 26S proteasome capacity, we definedthe transcriptional network that responds to this stress in Arabidopsis thaliana. This network includes genes encoding core andassembly factors needed to build the complete 26S particle, alternative proteasome capping factors, enzymes involved in proteinubiquitylation/deubiquitylation and cellular detoxification, protein chaperones, autophagy components, and various transcriptionalregulators. Many loci in this proteasome-stress regulon contain a consensus cis-element upstream of the transcription start site,which was previously identified as a binding site for the NAM/ATAF1/CUC2 78 (NAC78) transcription factor. Double mutantsdisrupting NAC78 and its closest relative NAC53 are compromised in the activation of this regulon and notably are stronglyhypersensitive to the proteasome inhibitors MG132 and bortezomib. Given that NAC53 and NAC78 homo- and heterodimerize, wepropose that they work as a pair in activating the expression of numerous factors that help plants survive proteotoxic stress andthus play a central regulatory role in maintaining protein homeostasis.
INTRODUCTION
All cellular organisms require mechanisms to dampen the accu-mulation of aberrant proteins caused by transcription/translationerrors, misfolding, cleavage, chemical modification, and environ-mental conditions such as heat that perturb tertiary/quaternarystructures. If allowed to hyperaccumulate, this detritus can nega-tively impact a host of intracellular activities through an imbalance inprotein homeostasis and excessive protein aggregation (Morimoto,2008; Hipp et al., 2014). The resulting proteotoxic stress has strongphysiological consequences, including genome instability, arrest ofthe cell cycle, inhibition of translation through a reduction in ribo-somes, lost membrane integrity, inhibition of metabolism, and ac-celerated senescence, as well as amino acid starvation if proteinrecyclingbecomesseverely compromised. In fact, theaccumulationof misfolded, aggregation prone proteins is the hallmark of a broadrange of human diseases (Cuanalo-Contreras et al., 2013).
Toalleviate this stress, plants, fungi, andanimals elicit a numberof cytoprotective responses, including expression of proteinchaperones and activation of the unfolded protein responsedesigned to promote refolding and sequester protein aggregates,maintenance of chromatin integrity through SUMOylation, andupregulation of several proteolytic pathways that remove theseaberrant polypeptides before they become cytotoxic (Hetz, 2012;Howell, 2013; Kim et al., 2013; Amm et al., 2014; Seifert et al.,
2015). Excessive protein aggregation and the accumulation ofdefective protein complexes (e.g., ribosomes and proteasomes)and organelles (e.g., chloroplasts andmitochondria) often engageautophagy (Li and Vierstra, 2012; Khaminets et al., 2016;Marshallet al., 2015). These damaged structures are encapsulated intocytoplasmic vesicles and delivered to the vacuole/lysosome forbreakdown, in many cases using ubiquitylation as a signal.Arguably, the most important protease upregulated during
proteotoxic stress is the 26S proteasome (Amm et al., 2014;Hanssum et al., 2014; Livnat-Levanon et al., 2014). This 2.5-MD,ATP-dependent proteolytic machine works in tandemwith ubiquitin(Ub) to direct the selective breakdown of aberrant polypeptidesand normal short-lived proteins in the cytoplasm and nucleus.Targets are first covalently modified with multiple Ubs; the re-sulting ubiquitylated species dock with the 26S proteasome,which degrades the modified protein concomitant with release ofthe Ub moieties for reuse.The 26S proteasome consists of two subcomplexes: the 20S
core protease (CP) and the 19S regulatory particle (RP) (Finley,2009; Bhattacharyya et al., 2014). The CP consists of fourstacked heteroheptameric rings of distinct a- and b-subunits inan a1-7/b1-7/b1-7/a1-7 configuration, which houses in a centralchamber the proteolytic active sites provided by the PBA (b1),PBB (b2), andPBE (b5) subunits. TheRPhas18ormore subunits; itcaps one or both ends of the CP barrel and contains receptors forubiquitylated targets and activities that remove the Ub moietiesand unfold and translocate the target polypeptides into the CPlumen before breakdown. In addition, a host of accessory factorsassociate substoichiometrically, including dedicated chaperonesthat promote the sequential assembly of theCPandRPcomplexesand final construction of the 26S particle, alternative capping
1Address correspondence to [email protected] author responsible for distribution of materials integral to the findingspresented in this article in accordance with the policy described in theInstructions for Authors (www.plantcell.org) is: Richard D. Vierstra([email protected]).www.plantcell.org/cgi/doi/10.1105/tpc.15.01022
The Plant Cell, Vol. 28: 1279–1296, June 2016, www.plantcell.org ã 2016 American Society of Plant Biologists. All rights reserved.

factors (PA200 and CDC48), shuttle proteins that help deliverubiquitylated substrates, and ubiquitin-protein ligases (or E3s)and deubiquitylating enzymes (Finley, 2009; Hanssum et al., 2014).
Given its role(s) inmaintainingprotein homeostasis, the levels ofthe 26S proteasome are highly regulated to meet demand, whichis achieved by the coordinated expression of all core subunitsand most, if not all, accessory factors. In yeast (Saccharomycescerevisiae), this regulon is driven by the C2H2-type zinc-fingertranscription factor Rpn4, which binds to the PACE (proteasomeassociated control element) cis-element found upstream of mostproteasome subunit genes (Mannhaupt et al., 1999; Xie andVarshavsky, 2001; Shirozu et al., 2015). Rpn4 responds to pro-teotoxic stress by itself being a target of proteasomal breakdown.When proteolytic demand is low, Rpn4 is rapidly degraded (t1/2;2.5 min), thus attenuating expression of 26S proteasome genes(Xie andVarshavsky, 2001;Dohmenet al., 2007).However, as 26Sproteasome capacity is challenged by proteotoxic stress, Rpn4 isstabilized, thus allowing its levels to rise and upregulate particlesynthesis.
Although orthologs of Rpn4 are not obvious outside of yeasts,a similar proteasome stress regulon (PSR) exists in many othereukaryotes, includingmammalian cells,Drosophilamelanogaster,and Arabidopsis thaliana (Meiners et al., 2003; Yang et al., 2004;Lundgren et al., 2005; Kurepa et al., 2008; Book et al., 2009). Inmice and humans, the Nrf1 (Nuclear Factor Erythroid-derived2-Related Factor 1) transcription factor, unrelated to Rpn4, wasrecently shown tobeacentral effector (Radhakrishnanetal., 2010,2014; Sha andGoldberg, 2014). Nrf1 is bound to the endoplasmicreticulum (ER) and is constitutively degradedby the ER-associatedprotein degradation pathway, and like Drpn4 yeast cells, nrf1-nullcells aremore sensitive toproteasome inhibitors.Uponproteotoxicstress,Nrf1 isproteolytically released fromtheERand isnow free toenter the nucleus and activate the PSR.
How the PSR is controlled by proteotoxic stress remainsunclear in plants. One candidate regulator in Arabidopsis isNAC78, a member of the NO APICALMERISTEM/ARABIDOPSISTRANSCRIPTION ACTIVATION FACTOR1/CUP-SHAPEDCOTYLEDONS2 (NAC) familyof transcriptional regulators.NAC78was first implicated by overexpression studies showing that itpositively regulates the expression of core 26S proteasome subunitgenes and that its putative DNA binding site is present within many,but not all, associated promoters (Yabuta et al., 2011; Nguyen et al.,2013). Whereas NAC78-OX plants are smaller than the wild type,nac78mutant plants are larger, which is consistent with a reportedrole for 26S proteasome levels in controlling cell size (Kurepa et al.,2009; Sonoda et al., 2009).
To more broadly define the plant PSR, we combined RNA-seqanalysis with both chemical inhibitors and proteasome mutantsthat induce proteotoxic stress. Gene coexpression together withprotein interactome network analyses revealed a complex net-work of PSR genes/proteins enriched in protein chaperones,autophagy components, and detoxification enzymes that pre-sumably help plants cope with proteotoxic stress, in addition tothose encoding the 26S proteasome and its assembly/cappingcomponents. Network and promoter-interaction studies thenidentified the NAC78 paralog NAC53 as a key effector that, to-gether with NAC78, likely functions as homo- and heterodimers.Importantly, seedlings simultaneously lackingNAC53andNAC78
poorly activate the PSR during proteotoxic stress, and theirgrowth is strongly hypersensitive to proteasome inhibitors. Assuch, we propose that these two NAC proteins are central reg-ulators of an extended PSR in Arabidopsis by providing sufficient26S proteasomes and other protein homeostatic factors to mit-igate proteotoxic stress.
RESULTS
26S Proteasome Subunits Are Upregulated duringProteotoxic Stress
To better understand how 26S proteasome gene expression isupregulated by proteotoxic stress, we examined the transcriptlevels for representative Arabidopsis subunits either upon short-(3 h) or long-term (24 h) exposure of seedlings to the proteasomeinhibitorMG132 (Yang et al., 2004), or inmutant backgrounds thatcompromise 26S proteasome assembly (rpn10-1 and rpn12a-1)and elicit seedling phenotypes consistent with impaired capacity(Smalle et al., 2002, 2003). The mutant alleles were fortuitouslygenerated by exon-trap mutagenesis; they are viable but havestunted growth and show pleiotropic defects in hormone sig-naling. The rpn12a-1 allele dampens expression of the full-lengthRPN12a transcript (see Figure 2A) but is relatively mild pheno-typically (Smalle et al., 2002). The rpn10-1 allele expressesa truncation of RPN10, the main Ub receptor in the 26S complex,and has much stronger developmental consequences (Smalleet al., 2003). The translated polypeptide includes the N-terminalvon Willebrand Factor-A domain that links RPN10 to the rest ofthe RP but is missing the C-terminal region containing the threeUb-interacting motifs that bind Ub, the autophagy adaptor ATG8,and cargo receptors bearing Ub-like domains, respectively(Farmer et al., 2010; Fatimababy et al., 2010;Marshall et al., 2015).Previous studies demonstrated that both long-term exposure
toMG132and the rpn10-1mutationelevate thesteadystate levelsof Ub conjugates and increase the abundance of several coresubunits of the 26S proteasome (Smalle et al., 2003; Yang et al.,2004; Kurepa et al., 2008). We extended these results here byimmunoblotting crude extracts from MG132-treated wild-typeand untreated homozygous rpn10-1 and rpn12a-1 seedlings withantibodies against Ub, the CP subunit PBA1(b1), the RP subunitsRPN1,RPT2, andRPN12a, and theCP regulator PA200 (knownasBlm10 in yeast) (Figures 1A and 1B). In particular, accumulation ofthe unprocessed form of PBA1 increased strongly upon MG132treatment, consistent with the need for active proteasomes togenerate themature, truncatedb1polypeptide (Finley,2009;Booket al., 2010). The rpn12a-1 allele had only a marginal effect on theabundance of Ub conjugates and the PBA1, RPN1, and RPT2subunits, but its action was obvious based on its strong effecton PA200 levels (Figure 1A). Why the effects on CP/RP subunitlevels weremild for the rpn12a-1 allele was unclear, but it couldreflect the relatively modest phenotype of the mutant (Smalleet al., 2002) and/or the possibility that genetically compro-mised 26S proteasomes generated by the rpn12a-1 allelemoreeffectively induce autophagic turnover of the complex as op-posed to the rpn10-1 mutation, which blocks such turnover(Marshall et al., 2015).
1280 The Plant Cell

When mRNA abundance was then examined by RT-qPCRanalysis of seedlings, levels of the CP subunit PBA1 and theRP subunits RPN5a, RPN10, and RPN12a were found to riserapidly uponshort treatmentswithMG132andwere constitutivelyupregulated in the rpn10-1and rpn12a-1backgrounds (Figure2A).(Throughout this study, we used transcripts from the ACTIN2[ACT2] and/or TYPE-2A SERINE/THREONINE PROTEINPHOSPHATASE [PP2A] genes as controls given their relativeimmunity to proteotoxic stress [Supplemental Figure 1].) As withpreviousstudies (Galloisetal., 2009;Leeetal.,2011), this increasedtranscript abundance could also be demonstrated with transgenicplants expressing the GUS reporter under the control of various 26Sproteasome gene promoters. Upregulation upon MG132 treat-mentwasvisualizedcolorimetrically by staining the seedlingswithX-Gluc and quantitatively by 4-methylumbelliferyl-b-D-glucuro-nide (MUG)-based fluorescence activity assays of crude seedlingextracts (Figures2Band2C). Interestingly, comparisonsofseveralgene pairs that encode individual proteasome subunits revealedthat often only one responds to MG132, implying that the manypairs havesubfunctionalizedwithone locusmainly responsible forincreasing subunit mRNA levels during proteotoxic stress (e.g.,RPN3a, RPT1a, RPT2a, and RPT4b; Figures 2B and 2C).
RNA-Seq Analysis of the ProteasomeStress-Induced Regulon
Tomore fully identify the suite of genes that are upregulatedwhenproteasome capacity is compromised, we performed RNA-seqanalysis with our cohort of wild-type seedlings treated withMG132 (3 and 24 h) and untreated rpn10-1 and rpn12a-1 seed-lings. These transcriptome studies identified a large collection ofmRNAs whose abundance was significantly affected as com-pared with untreated wild-type seedlings based on negative bi-nomial normalization using edgeR differential expression analysis(P value < 0.01, false discovery rate [FDR] < 0.05; SupplementalData Sets 1 and 2). By merging the data sets, we identified119genes (includingRPN10 andRPN12agiven their upregulationin two of the three conditions) that were coordinately upregulatedunder all three conditions (3-h treatment with MG132 and in therpn10-1 and rpn12a-1 backgrounds), which we designated asmembers of the PSR (Figure 3A; Supplemental Data Set 3). RNA-seq analysis after a 24-h exposure to MG132 identified an addi-tional set of 865 upregulated genes; these loci were not included inthe final PSR as their long-term induction might be more indirectlyrelated to the prolonged effects of the inhibitor and the downstreamresponses to severe proteotoxic stress (Supplemental Data Set 4).Gene Ontology analyses via DAVID (database for annotation,
visualization, and integrated discovery; https://david.ncifcrf.gov)of the 119 PSR upregulated genes detected a significant func-tional enrichment for the 26S proteasome, Ub-related events, andgeneral stress response processes and identified several tran-scription factors thatmight transcriptionally activate thePSR.Thisenrichment for 26S proteasome genes was especially strong inthe more robustly affected group with a$2-fold increase (44% oftotal; Figure 3B). Of the 53 genes encoding core 26S proteasomesubunits (Book et al., 2010; Russell et al., 2013), 48 (91%) weresignificantly upregulated (Figures 3B and 3C). We note that therank order of the upregulated and downregulated genes in theMG132and rpn10-1/rpn12a-1data setsbasedon their strengthofchange differed markedly (Figure 3B); this deviation could reflecttwo distinct subtypes of proteotoxic stress, one elicited rapidlyand strongly by the inhibitor and the other being a more subtlechronic stress elicited by the mutations. We also detected33 genes whose expression was downregulated by all threeconditions (Figures 3A and 3B; Supplemental Data Set 3); therewas no significant functional enrichment in this group, thusleaving their collective action(s) unclear.From analysis of genes known or predicted to be associated
with the Arabidopsis 26S proteasome (Finley, 2009; Book et al.,2010), a strikingly coordinated upregulation was evident for mostloci. This was apparent for both core CP and RP subunits as wellas acknowledged accessory factors (PA200 and ECM29) andchaperones (HSM3, NAS6, PBAC1, PBAC2, and UMP1a) that arepresumably needed during proteotoxic stress to assist in particleregulation/assembly (Figure 3C). The proteasome binding proteinPROTEASOME REGULATOR1, recently identified as a positiveregulator of auxin signaling through activation of the 26Sproteasome(Yang et al., 2016), was also upregulated, as was the possible plant-specific chaperone PAP1. The only exception for subunits encodedby a single gene was the locus encoding the Ub receptor RPN13,whose expression was relatively immune to MG132 and was
Figure 1. Both theProteasome InhibitorMG132 andProteasomeMutantsIncrease the Accumulation of Proteasome Components and Ub Con-jugates in Arabidopsis.
Wild-type seedlings treatedwith 100 mMMG132 or untreated rpn10-1 andrpn12a-1 seedlings were grown for 5 d. Total extracts were probed byimmunoblotting with the indicated antibodies, using anti-histone H3 an-tibodies to verify near equal protein loading.(A) Levels of individual subunits of the proteasome. The open and closedarrowheads identify the unprocessed and processed forms of the b1subunit PBA1.(B) Levels of Ub conjugates. Closed arrowheads locate free Ub and pol-y-Ub chains assembled with varying numbers of Ub monomers. Bracketindicates high molecular mass Ub conjugates.
Transcriptional Regulation of Proteotoxic Stress 1281

unchanged by the mutations. In agreement with the RT-qPCR andproteasome promoter:GUS reporter studies above, we found thatmultiple gene pairs encoding individual subunits had at least onelocus that was PSR regulated with the second locus sometimes
nonresponsive. Examples include thePAA2,PBB2,RPT1a,RPT6b,RPN3a, and RPN9b loci, which were highly sensitive to proteotoxicstress, whereas their paralogs PAA1, PBB1, RPT1b, RPT6a, RPN3,and RPN9a were comparatively silent (Figure 3C).
Figure 2. Expression of Arabidopsis Proteasome Genes Is Upregulated in Response to Proteasome Stress.
(A) Expression of 26S proteasome subunit genes following proteasome inhibition with MG132 or in rpn10-1 or rpn12a-1 mutant backgrounds thatcompromise assembly. Total RNA from 5-d-old seedlings, either untreated or treated for 3 and 24 h with 100 mMMG132, was subjected to RT-qPCR. Theexpressionvalueswerecalculatedusing theACT2 transcriptasa referenceandnormalized to those fromuntreatedwild-typeseedlings.Eachbar representsthe average of at least three biological replicates (6SD).(B) Effects of MG132 on the expression of proteasome promoter:GUS transgenes. Transgenic wild-type seedlings expressing the fusions were grown for3 d, treated for 1 d with 100 mM MG132, and then incubated overnight with the X-Gluc substrate.(C) Quantitative measure of proteasome promoter:GUS expression following MG132 treatment. Ten-day-old seedlings were incubated overnight with orwithout 100mMMG132andhomogenized, and theGUSactivity in the resulting cell extractswasassayedusing theMUGsubstrate. Eachbar represents theanalysis of at least 30 independent T1 lines, each assayed in triplicate (6SD). The data in (B) and (C) for the RPT2a and RPT2b promoter:GUS fusions werereported previously and are included here for comparison (Lee et al., 2011).
1282 The Plant Cell

When the analysis was extended to nonproteasome tran-scripts, additional loci potentially relevant to the PSR were detected(Supplemental Data Set 3). Included were mRNAs encodingmembers of the NAC, WRKY, and heat shock protein transcriptionfactor (HSF) families that often have prominent roles in various plantstress responses (Rushton et al., 2010; Nakashima et al., 2012;Scharf et al., 2012), other Ub/proteasome pathway componentsincluding Ub, several Ub E3 ligases (AIP2, At1g71020, At4g27050,and At5g27920), deubiquitylating enzymes (UBP6, UBP7, andUCH3), extraproteasomal Ub binding proteins (DSK2a and DDI1),and components of the autophagy system (Supplemental Figure 2).Notable autophagy factors included several members of the ATG8family required for autophagic vesicle dynamics and cargo deliveryand the autophagy receptor NBR1, which is devoted to the re-cruitment of ubiquitylated cargo (Li and Vierstra, 2012; Zhou et al.,2013). Strong transcriptional upregulation of NBR1, especially byMG132, implied a role for autophagy in removing Ub conjugatesthat become stabilized when proteasome capacity is limited.
The MG132-Induced PSR Network
To better visualize the MG132-induced PSR, we entered the336 MG132-induced genes that were significantly upregulated
$2-fold after a 3-h treatment (Supplemental Data Set 1) into theSearch Tool for the Retrieval of Interacting Genes/Proteins(STRING) coexpression and interactome database (Jensen et al.,2009). Upon visualization in Cytoscape v3.0 (Shannon et al.,2003), an extensive collage of networks involving 275 output lociemerged that clustered into three main groups (Figure 4A). Asexpected, one tight cluster included loci encoding 26S protea-some subunits and accessory factors/proteasome chaperones. Asecond cluster encompassed a large collection of general heatshock protein (HSP) chaperones (several HSP70 isoforms,HSA32, HSP81, and HSP101, and their dedicated transcriptionalregulator HSF8A) that presumably direct protein refolding and/orminimize aggregation of insoluble proteins awaiting proteaso-mal/autophagic turnover (Lin et al., 2014). A third more diffusecluster was enriched in the stress-related NAC, WRKY, and HSFtranscription factors that might help activate the PSR andtransferase activities (e.g., GSTs and O-methyltransferases) thatcould mitigate proteotoxic stress (Livnat-Levanon et al., 2014).The third cluster also contained several UDP-glycosyl trans-ferases and the main mitochondrial alternative oxidase, AOX1a,which were shown previously to be upregulated upon mito-chondrial perturbation (De Clercq et al., 2013). Many of these lociwere also seen in clusters generated with the 24-h MG132
Figure 3. Characterization of the PSR in Arabidopsis.
(A) Venn diagrams of transcripts that were differentially expressed significantly (P value < 0.01, FDR < 0.05) under three proteasome stress conditions: thewild type after a 3-h treatmentwith 100mMMG132, and the rpn10-1and rpn12a-1genetic backgrounds. The shared 119upregulated (includingRPN10 andRPN12a) and 33 downregulated genes determined by edgeR analysis comprise the PSR.(B) A heat map of the PSR for the three treatments ranked by the fold change in expression obtained with the MG132-treated wild-type seedlings. Thebrackets indicate genes with more or less than a 2-fold increase (log2(fold change) = 1) in expression compared with the wild type. Categories of enrichedgene classes are indicated. TFs, transcription factors. See Supplemental Data Set 3 for the full list.(C) Expression change heatmaps of 26S proteasome subunit and assembly factor genes after proteasome stress comparedwith untreatedwild type. ESTvalues obtained from The Arabidopsis Information Resource (TAIR v10) are listed.
Transcriptional Regulation of Proteotoxic Stress 1283

treatment data set, especially for the 26S proteasome and relatedfactors (Supplemental Figure 3). Additions to the 24-h MG132network included a pronounced autophagy cluster and a largecluster enriched in protein kinases that could extend the PSRsignaling network given the roles of some in stress defenseand developmental signal transduction cascades (e.g., MEK1,MAPKKK1, RLP24, HLECRK, CRCK1, and CRCK11).
Under the assumption that many of the 336 loci are tran-scriptionally regulated by a common set of transcription factors,we searched their promoter regions for shared cis-regulatoryDNAelements using the motif-based sequence analysis tool MEME(Multiple Expectation Maximization for Motif Elicitation; Baileyet al., 2006). Highly enriched were the Proteasome-Related cis-Element (PRCE) with a TGGGC core sequence, which was pre-viously implicated by Nguyen et al. (2013) in the regulation ofproteasome genes, and the mitochondrial dysfunction motif(MDM) associated with oxidative stress responses (De Clercqet al., 2013) (Figures4Band4C).Bothmotifswereprominent in the26S proteasome gene cluster, with few promoters (14 of 48)predicted to be devoid of either motif (Figure 4D). When placedwithin the region upstream of the translation start site for genesencoding core 26S proteasome subunits, a wide dispersion of thePRCE and MDM motifs was seen, with some having these ele-ments close to the transcription start site and others placed>1000 bp away (Supplemental Figure 4). Whereas single MDMelements were common, we often detected multiple PRCE-related sequences in tandem, sometimes on opposite DNAstrands, suggesting that PRCE is acted upon by DNA bindingproteins that work palindromically. We also detected other po-tentially relevant cis-elements associated with the PSR at lowerfrequencies (Supplemental Figure 5). Included were the SORLIP2andT-boxmotifs that are involved in light-induced developmentalresponses (Chan et al., 2001; Hudson and Quail, 2003).
The NAC53 and NAC78 Transcription Factors AreAssociated with the PSR
To help identify the transcriptional regulators that might co-ordinate the PSR, we screened a yeast one-hybrid (Y1H) libraryof ;1700 known or predicted DNA binding proteins from Arabi-dopsis by an automated luciferase-based expression system(Gaudinier et al., 2011), usingboth thePA200orRPN12apromoterregions as bait (1000- and 253-bp fragments upstream of thetranslation start codon, respectively). The activity of both pro-moters is strongly upregulated by MG132 exposure, and ac-cordingly they contain multiple segments related to theconsensus PRCE sequence. (It should be noted that the RPN12apromoter is predicted to be short given that only 253 bp separatesits translation start site from the coding region immediately up-stream.) Among the list of 15 interactors that activated both Y1HassayswasNAC53(At3g10500,alsoknownasNTL4;SupplementalTable 1), a 555-residue NAC protein that we also discovered asa locus significantly upregulated by brief MG132 exposure andpositioned by Cytoscape as a central hub within the PSR de-toxification cluster (Figure 4A; Supplemental Figure 2). Uponscanning theArabidopsisgenome for relatedproteins,we identifiedNAC78 (At5g04410, also known as NTL11/RPX1), a 584-residueNACprotein that is its closest relative (;72%amino acid sequence
identity) among the ;109 NAC-type transcriptional regulators(Olsenet al., 2005a;Nakashimaetal., 2012;Wuetal., 2012).BindingofNAC53andNAC78 tobothpromoterswasconfirmedbydirectedY1H assays using 500 bp of the PA200 promoter and 253 bp of theRPN12a promoter as baits.Intriguingly, several previous studies implicated NAC53 and
NAC78 in the PSR and/or in binding PRCE/MDM-type sequences.NAC78 was first linked by overexpression screens that searchedfor factors that regulate either plant size or seedling responsesto light stress and was subsequently shown by transcriptomestudies to impact proteasome gene expression (Morishita et al.,2009;Yabutaet al., 2011;Nguyenetal., 2013).Nguyenet al. (2013)further connected NAC78 to the PSR by reporting that it recog-nizes the same PRCE element identified here as common withinthe regulon (Figure 3C). A further connection to the PSR wasprovided by DNA binding studies showing that both NAC53 andNAC78 might also recognize MDM elements found here to becommon within PSR loci (De Clercq et al., 2013). Finally, NAC53was previously linked to heat stress, reactive oxygen species(ROS) production, and senescence, all of which are related toproteotoxic stress through cursory phenotypic analyses ofNAC53 mutant and overexpression lines (Lee et al., 2012, 2014).To provide an evolutionary connection between NAC53 and
NAC78, we examined the two loci phylogenetically in the contextof the entire NAC family. Bayesian analyses clustered NAC53 andNAC78 togetherwheneither the109 full-lengthNACproteinswerecompared or when just the DNA-binding NAM domains wereanalyzed, indicating that this pair forms a unique subclade withinthe NAC family (Figure 5A; Supplemental Figures 6 and 7). In fact,strong amino acid sequence identity was seen throughout theNAM domain (;93%), implying that they bind the same DNAsequence (Supplemental Figure 8). Close sequence relatives(;50% sequence identity) were also detected in other plantspecies, including variousmonocots and dicots and the seedlessplants Selaginella moelendorffii and Physcomitrella patens, butnot in the alga Chlamydomonas reinhardtii, implying that the pairhas a conserved function in land plants. Relatives of both NAC53and NAC78 could be found in Arabidopsis lyrata that clusteredapart from likely orthologs in other dicots, suggesting that NAC53and NAC78 originated from a duplication that occurred recentlywithin the Brassicaceae lineage.The C-terminal ends of NAC53 and NAC78 are predicted to
contain a 24-amino acid membrane-spanning sequence that isalso found within 11 other Arabidopsis NAC proteins, which arecollectively designated as NTLs (NACs with transmembrane-likeregion; Kim et al., 2007; Supplemental Figure 6). Like the NAMdomain, this putative membrane-spanning region is highly con-served between NAC53 and NAC78 (75% sequence identity),even when aligning their sequences next to that of their closestNTL relative NAC13 (30% full polypeptide sequence identify and65% NAM domain sequence identity; Supplemental Figure 8).Even within the NTL subfamily, NAC53/NAC78 stand apartphylogenetically, suggesting that they have distinct function(s)(Figure 5A; Supplemental Figures 6 and 7). The role(s) of themembrane-spanning domain is not yet clear; for at least oneArabidopsis NTL, it has been proposed to facilitate binding of theNACprotein tocytosolicmembranes,withproteolytic release thenpermitting nuclear import (Kim et al., 2006; Kim et al., 2007).
1284 The Plant Cell
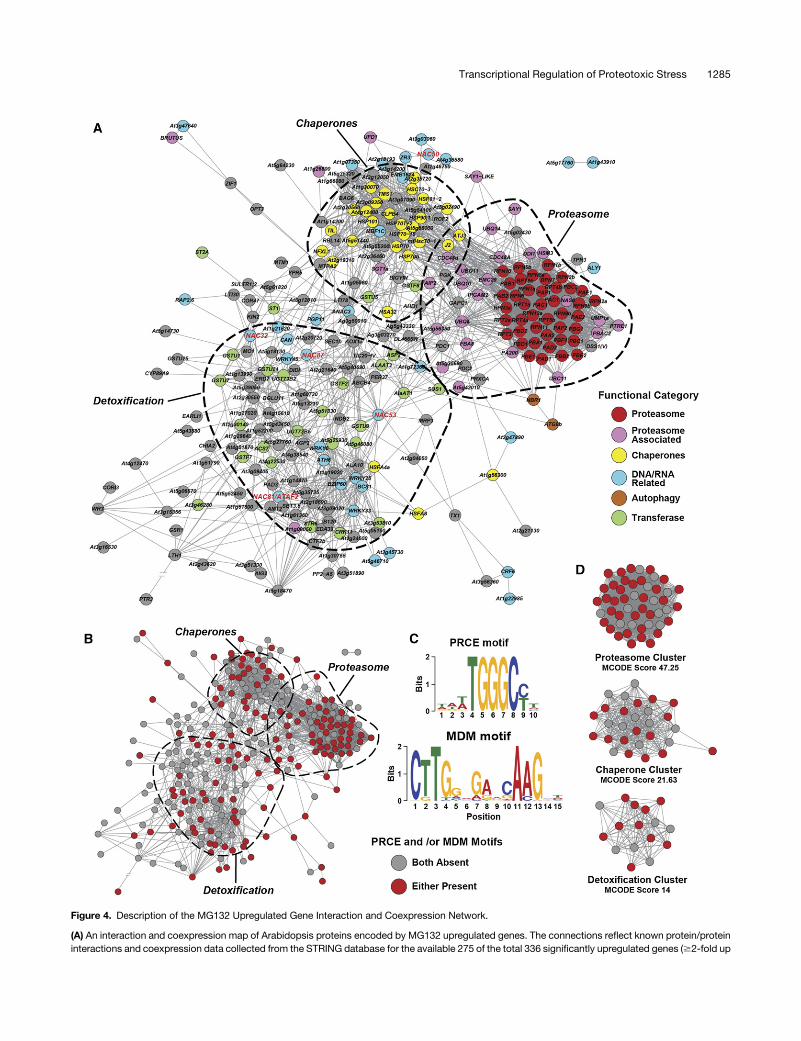
Figure 4. Description of the MG132 Upregulated Gene Interaction and Coexpression Network.
(A) An interaction and coexpression map of Arabidopsis proteins encoded by MG132 upregulated genes. The connections reflect known protein/proteininteractions and coexpression data collected from the STRING database for the available 275 of the total 336 significantly upregulated genes ($2-fold up
Transcriptional Regulation of Proteotoxic Stress 1285

NAC53 and NAC78 Regulate PSR Gene Expression
Given the aforementioned connections of NAC53/NAC78 toproteasome gene regulation and their likely ability to bind PRCE-type motifs (Yabuta et al., 2011; De Clercq et al., 2013; Nguyenet al., 2013; this report), we hypothesized that NAC53 and NAC78work in concert as homo- and heterodimers to upregulate thePSR. As a first validation, we tested whether NAC53 and NAC78interact, as is common for paralogs within the NAC family
(Nakashima et al., 2012; De Clercq et al., 2013). By yeast two-hybrid (Y2H) assays, we detected binding of NAC53 and NAC78to themselves and to each other, using the Gal4/LacZ pDESTsystem, thus generating colonies that grew in the absence of Hisandwere resistant to 50mM3-amino-1,2,4-triazole (3-AT) (Figure5B). This assembly of homo- and heterodimers was then con-firmed in planta by bimolecular fluorescence complementation(BiFC)with the split YFPsystem transiently expressed inNicotianabenthamiana leaf epidermal cells.Onlywhen theNAC53orNAC78
Figure 4. (continued).
compared with untreated wild type) after a 3-h exposure to 100 mM MG132. Classifications of major functional groups in the network are highlighted.Members of the NAC transcription factor family are in red. Portions of the map enclosed by the dashed lines are statistically enriched for genes with thedenoted functional categories (DAVID, P value < 0.01). If available, specific gene names were used instead of the TAIR locus identifier. See SupplementalData Set 1 for the full list.(B)Members of the network that contain the consensus PRCE or MDM cis-motifs within their promoter regions (motif-containing gene nodes are in red).(C) Sequence descriptions of the PRCE and MDM cis-motifs as determined by MEME.(D) Interactomemaps focusing on the statistically significant central hubspresent in the proteasome, chaperone, and detoxification clusters as determinedby MCODE analysis and colored based on the presence of PRCE and/or MDM sequences.
Figure 5. NAC53 and NAC78 Are Closely Related and Physically Interact.
(A)ABayesian phylogenetic tree of all 109NACproteins in Arabidopsis rooted to aP. patensNACprotein (gi_168025227). Nodes highlighted in red indicatetheNTL subclasswith apredicted transmembrane-spanningmotif. The same treewith the names included for eachprotein is in Supplemental Figure 6, anda text file of the alignment used is presented as Supplemental Data Set 5.(B) Y2H assays showing that NAC53 and NAC78 homo- and heterodimerize. The full-length proteins were expressed as N-terminal fusions with either theGAL4 activating (AD) or binding (BD) domains. Shown are cells grown on selectivemedium lacking Leu andTrp, or lacking Leu, Trp, andHis, and containing50 mM 3-AT.(C)BiFC analysis showing that NAC53 andNAC78 homo- and heterodimerize in planta and partially localize to the nucleus.N. benthamiana leaf epidermalcellswere coinfiltratedwithplasmids expressing theN-andC-terminal fragments ofYFP (nYFPandcYFP, respectively) fused to theN terminusofNAC53orNAC78. Shown are reconstituted BiFC signals, as detected by confocal fluorescence microscopy of leaf epidermal cells 36 h after infiltration, along withDAPI staining of nuclei (white arrowheads) and a bright-field (BF) image of the cells. Bar = 10 mm.(D) Y2H assays testing interactions between NAC53 and NAC78 and other NAC proteins within the PSR. The assays were conducted as in (B) with theselective medium lacking Leu, Trp, and His and containing 25 mM 3-AT.
1286 The Plant Cell

coding regions were simultaneously expressed as fusions to ei-ther the nYFP or cYFP fragments was fluorescence reconstituted(Figure 5C; Supplemental Figures 9A and 9C). Fluorescence waseasily detected in the cytoplasm and nucleus, suggesting that thetwo proteins interact in each compartment.
It had been reported that when a GFP fusion of full-lengthNAC78 is transiently expressed in onion (Allium cepa) epidermalcells, it mainly localizes to the cytosol but becomes concentratedin the nucleus when expressed without the membrane-spanningregion (Morishita et al., 2009). To examine whether proteotoxicstress could recapitulate this redistribution, we attempted togenerate transgenic plants that stably express GFP-NAC53 andGFP-NAC78 with or without this transmembrane domain. Un-fortunately, no fluorescent signals were detected, in agreementwith a previous failure with another NTL, NAC13 (De Clercq et al.,2013). As an alternative, we examined the BiFC signal from theNAC53/NAC78 pair in N. benthamiana cells simultaneouslytreated withMG132. Unfortunately, under our conditions, MG132treatment neither impacted the strength nor the distribution of theBiFC signals (Supplemental Figure 9B).
As the next step in connecting at least one of this NAC pairto thePSR,we examined transgenic lines expressingNAC78 fromthe b-estradiol-inducible XVE promoter, which was provided bythe TRANSPLANTA collection (Coego et al., 2014). In our hands,NAC78mRNA levels rose;60-fold after an overnight b-estradiolexposure. As shown in Figure 6, pEST:NAC78 seedlings, but notwild-type seedlings, treated with b-estradiol also strongly in-creased the mRNA abundance for several representative 26Sproteasome subunits, as first observed by Nguyen et al. (2013).Importantly, this upregulation extended to other members of thePSR outside of the core 26S particle, including genes encodingthe proteasome accessory factor PA200, the NAS6 assemblychaperone, the UPS component UFD1, and the HSP transcrip-tional regulator HSF8A. Two other PSR genes encoding theproteasome accessory factor PA200 and the glutathioneS-transferase GSTU25 also displayed increases close to signifi-cance, consistent with NAC78 more broadly controlling the PSR.
Assuming that the NAC53/NAC78 proteins work together, wegenerated double mutants impacting the pair to observe anyadverse effects on PSR regulation. Potentially useful alleles wereidentified in the SALK collection of T-DNA insertion mutants thateither disrupted the coding region downstream from theN-terminal NAM domain (nac53-1), within the NAM domain(nac78-1), just upstreamof that for theC-terminal transmembranesequence (nac53-2), or toward the end of the coding region(nac78-2; Figure 7A). Genomic sequencing mapped the insertionsites to 735 and 372 nucleotides downstream of the translationalstart site for the nac53-1 and nac78-1 mutations and 87 and21 nucleotides upstream of the translational stop site for thenac53-2 and nac78-2 mutations, respectively.
Transcript analyses by RT-PCR failed to detect the corre-sponding full-length mRNAs in homozygous nac53-1 andnac78-1 seedlings (Figure 7B). For nac53-1, we also could notdetect transcripts downstream of the insertion site, whereas fornac78-1wecoulddetect a transcript of slightly greater length thanexpected. Collectively, the RT-PCR data imply that nac53-1 andnac78-1 represent null alleles. For homozygous nac53-2 andnac78-2 seedlings, we detected low levels of near-full length
transcripts originating upstream of the insertion sites; thesemRNAs included the NAM domain coding sequence. For thenac78-2 mutant, only a small portion of the proposed C-terminalmembrane-spanning region domain was eliminated, whereasnearly the entire membrane-spanning region was missing for thenac53-2 mutant (Supplemental Figure 8), suggesting that theyrepresent weaker alleles under the assumption that the fulltransmembrane domain is not essential (at least for NAC53).When grown at 24°C under a normal 16-h-light/8-h-dark
photoperiod on agar medium or soil, all the single and doublemutant combinations germinated well, generated phenotypicallynormal rosettes, flowered at the same time, and were equallyfertile as the wild type, indicating that NAC53 and NAC78 aloneor in combination are not necessary for Arabidopsis growth,development, and fecundity under nonstressed conditions. Atleast under our growth conditions, the mutant seedlings were notlarger than the wild type, as had been reported previously for aT-DNA insertion line disruptingNAC78by itself (Nguyen et al., 2013).Importantly, RT-qPCR analyses of PSR-related genes revealed
that NAC53 and NAC78 combine to activate the PSR. Whereasthe nac53-1 mutation had little effect on expression of repre-sentative PSR genes after an overnight MG132 treatment, a mildbutstatistically significantdampening (Pvalue<0.01)wasseen forthe nac78-1 line (Figure 8A). Moreover, when the double nac53-1nac78-1 mutant was tested, a substantially attenuated PSR re-sponse was observed. Compromised loci included genes en-coding subunits of the 26S particle (PBA1, RPN5a, RPN10, andRPN12a), the accessory factors PA200 and CDC48, the NAS6chaperone, the UPS factor UFD1, the glutathione S-transferaseGSTU25, and the heat shock transcription factor HSF8A (P value <0.01). For some loci, the nac53-1 nac78-1 combination almostcompletely ablated the transcriptional upregulation. A smaller butsignificant drop in PSR gene expression was also seen for thenac53-2 nac78-2 doublemutant, consistent with themilder effectof these mutations on the NAC53/NAC78 mRNAs (Figure 8A).The diminished PSR in turn reduced the steady state levels of
some of the corresponding proteins. Whereas MG132 treatmentincreased the seedling abundance of the 26S accessory proteinPA200, the RP subunit RPN12a, and the unprocessed form of theCP subunit PBA1 in wild-type seedlings, this increase was sub-stantially weaker in the nac53-1 nac78-1 background (Figure 8B).However, this drop did not appear to translate into a substantialstabilization of Ub conjugates, as the pool of immunodetectablespecies was only mildly increased in the nac53-1 nac78-1 plantscomparedwith thewild type (Supplemental Figure 10). Consistentwith the reduced strength of the nac53-2 and nac78-2 alleles onPSR gene expression, the doublemutants had little impact on theRPN12aandPBA1protein levels andpossiblyonly amildeffect onPA200 (Figure 8B). As the polypeptides derived from the nac53-2nac78-2 alleles should be missing all or part of the presumedtransmembrane domain, respectively, membrane associationmight not be essential for NAC53/NAC78 function.
Plants Missing NAC53 and NAC78 Are Hypersensitive toProteotoxic Stress
Phenotypic analysis of homozygous nac53-1 nac78-1 plantsin turn revealed that both transcription factors together help
Transcriptional Regulation of Proteotoxic Stress 1287

Arabidopsis survive proteotoxic stress. Whereas wild-type plantscould tolerate long-term exposure to sublethal doses of MG132beginning at germination, growth of the doublemutant plantswassubstantially arrested (Figures 9A and 9B). In fact, developmentof nac53-1 nac78-1 seeds stalled soon after germination, andfor many, the radicals failed to exit the seed coat after rupture.Quantification of the response also detected a significant growthinhibition (P value < 0.01) for the nac78-1 singlemutant and for theweaker nac53-2 nac78-2 double mutant compared with the wildtype (Figure 9B).
Given that the smaller plants observed for the nac53-1 nac78-1line upon MG132 exposure might have arisen from defective ordelayed germination, we also germinated and grew the seedlingsfor 6 d in the absence of MG132 and then placed them oninhibitor-containing medium and measured seedling freshweight 6dafterwards. Even in this situation, growthof thenac53-1nac78-1 plants was significantly impaired compared with the wildtype (Figure 9C). AsMG132might impact other proteases besidesthose within the CP, we also tested a second 26S proteasomeinhibitor with potentially greater specificity and potency: borte-zomib (Kisselev et al., 2012). Here, growth of the nac53-1 nac78-1plants was severely inhibited at submicromolar concentrations,with amild but significant effect also seen for thenac53-2 nac78-2plants (Figures 9D and 9E).
NAC53 and NAC78 Work Together and with Other Membersof the NAC Family
While our studies indicated that NAC53 and NAC78 work together,possiblyasheterodimersbasedonourY2Hassays, toactivate thePSR, they could also have temporally distinct functions within theresponse (e.g., early versus late) given thatNAC53 but notNAC78transcripts were increased rapidly by MG132 treatment. To testthese possibilities, we compared the time course for PSR acti-vation by MG132 in the single mutant nac53-1 and nac78-1backgrounds byRT-qPCR analysis of representative PSR loci. Asshown in Supplemental Figure 11, the induction time courseswere identical (but slightly diminished) for genes encoding the CPand RP subunits, CDC48, the RP chaperone NAS6, andGSTU25,with the nac53-1 nac78-1 double mutant lacking a significantresponse for all loci tested but GSTU25. Together, the data implythat NAC53 and NAC78 work within the same time frame.Themild attenuation of the PSR for the nac53-1 nac78-1 plants
in response to MG132 as seen with GSTU25 and other loci (e.g.,WRKY25) (Figure 8A; Supplemental Figure 11) suggested thatother transcription factors also contribute to the PSR. Particularlynotable were several other members of the NAC family that weretranscriptionally upregulated by the inhibitor, including NAC1, 13,32, 44, 50, 55, 81/ATAF2, 82, and 87 (Supplemental Figure 2). To
Figure 6. NAC78 Overexpression Induces the Expression of Some Arabidopsis PSR Genes.
Upregulation of proteasome subunit and other PSR genes in seedlings expressing NAC78 from an estradiol-inducible promoter. Total RNA from 6-d-oldwild-type and pEST:NAC78 seedlings incubated for 24 h with or without 10 mM b-estradiol were subjected to RT-qPCR. The expression values werecalculated using ACT2 (black) and PP2A (gray) transcripts as references and normalized to those obtained from untreated wild-type seedlings. Barsrepresent the average of at least three biological replicates (6SD), each measured in triplicate. Asterisks indicate significant differences between pEST:NAC78 and the wild type based on Student’s t test (P value < 0.05). Dashed lines indicate the average values for untreated wild-type seedlings.
1288 The Plant Cell

test whether they also heterodimerize with NAC53/NAC78, wesubjected each to Y2H assays using NAC53 and NAC78 as prey.Interestingly, NAC13 and NAC81, but not the others, generatedpositive Y2H signals, suggesting that they also contribute to PSRactivation in combination with NAC53/NAC78 (Figure 5D).Whereas NAC13 bound to both NAC53 and NAC78, NAC81 onlyshowed an interaction with NAC53 under these conditions.NAC13 is especially intriguing given its ability to recognize MDMcis-elements common within the PSR and its membership in thetransmembrane-containing NTL-NAC subfamily like NAC53/NAC78.
DISCUSSION
Given the importance of protein quality control to maintaininghealthy cellular functions and the likelihood that plants routinelyexperience proteotoxic stress (e.g., ER-associated protein deg-radation; Howell 2013), we considered it likely that plants havestrong protective responses. Examples include various
environmental insults such as heat, cold, salt, drought, excesslight, photooxidative stress, and ROS production that perturbprotein folding, carbon and nitrogen starvation that limits syn-thesis of new polypeptides, pathogen invasion that often co-incides with the massive translation of pathogen-associatedpolypeptides that might express improperly in the host, and ex-posure to toxins made by the pathogen, such as the proteasomeinhibitors epoxomicin and syringolin A that block host proteinturnover (Yanget al., 2004;Groll et al., 2008).OurRNA-seqstudieson Arabidopsis seedlings induced to experience proteotoxicstress through impairment of 26S proteasome capacity (MG132inhibition and the rpn10-1 and rpn12a-1 mutants) revealed anintricate network of PSR genes/proteins that presumably repre-sent parts of these protective mechanisms. Many of the associ-ated activities are also important for protein homeostasis in yeastand animals, indicating that they reflect conserved protein ho-meostatic processes among eukaryotes (Morimoto, 2008; Hippet al., 2014).A prominent PSR cluster is the 26S proteasome itself and its
associated assembly/regulatory factors that are needed to builda functional holoenzyme, demonstrating that increasing 26Sproteasome capacity is one crucial protectivemechanism. In fact,given the plethora of core subunits and assembly chaperonesrequired to construct 26Sproteasomes, a remarkable coordinationofexpressionhasevolved toensure faithful productionof completeparticles. As shown by mutational studies affecting individualsubunits, substoichiometric accumulation of only one subunit issufficient to stall construction of the entire complex and induce off-product accumulation of assembly intermediates (Smalle et al.,2002, 2003; Book et al., 2009; Lee et al., 2011). The importance of26S proteasomes to protein homeostasis is also demonstrated bythe fact that nearly all core subunits are encoded by at least onegene that is strongly sensitive to proteotoxic stress. The presenceof a nonresponsive paralog likely reflects subfunctionalization ofthe pair, which might be important for maintaining proteasomelevels in specific spatio-temporal contexts.A second PSR cluster includes a collection of protein chap-
erones that help protein folding andminimize protein aggregation,along with several HSF transcription factors that promote theirexpressionduringstress.Their inclusionwasanticipatedgiven thecentral roles of chaperones in protein quality control and main-taining protein homeostasis (Morimoto, 2008; Hipp et al., 2014). Athird PSR cluster is enriched in a collection of enzymes importantfor cellular detoxification, such as glutathione S-transferases, O-methyltransferases, and AOX1a, the last of which is responsiblefor alternative respiration in mitochondria. Presumably, theseproteinshelp repair damagedproteinsand reduceoxidativestressand ROS. Several autophagy components are also members ofthe PSR, including ATG8 and the Ub receptor NBR1 (Kraft et al.,2010), thusconnecting autophagy toplant protein homeostasis. Arole for NBR1 is in agreement with studies on nbr1mutants; theyaccumulate substantial amounts of high molecular mass Ubconjugates during heat stress, which possibly represent ubiq-uitylatedprotein aggregates awaiting autophagic clearance (Zhouet al., 2013). Interestingly, additional autophagy genes appearedin the network created upon long-term MG132 exposure(Supplemental Figure 3), suggesting that autophagy representsa last line of defense during proteotoxic stress. It should also be
Figure 7. Description of Mutations Impacting NAC53 and NAC78.
(A) Diagrams of the NAC53 and NAC78 transcribed regions. Boxes rep-resent coding regions (colored) andpredicted untranslated regions (white).The blue andorangeboxes identify theDNAbindingNAMdomains and thepredicted membrane-spanning regions, respectively. Lines represent in-trons. The positions of the T-DNA insertions are indicated by the red tri-angles; their exact locations in the amino acid sequences are indicated inSupplemental Figure 8.(B)RT-PCRanalysis of theNAC53 andNAC78 transcripts in the single anddoublemutants. Total RNA isolated fromwild-type or homozygousmutantplants was subjected to RT-PCR using the primer pairs indicated in (A).RT-PCRwith primers specific forACT2was included to confirm analysis ofequal amounts of cDNA.
Transcriptional Regulation of Proteotoxic Stress 1289

emphasized that we discovered a number of genes within theArabidopsis PSRwhose functions remain to be discovered. Oncetheir purposes become clear, it is likely that additional proteinhomeostatic mechanisms will be revealed.
Combined with previous studies (Yabuta et al., 2011; Nguyenet al., 2013), our network and promoter interaction studiesidentified NAC53 and NAC78 as key regulators of the PSR.Support here includes: (1) Y1H binding of NAC53 to the PA200 andRPN12apromoters that strongly respond toproteotoxic stress, (2)the ability of NAC53 and NAC78 to homo- and heterodimerize, (3)b-estradiol-induced transcription of an assortment of PSR genesin plants harboring the pEST:NAC78 transgene, and (4) strongtranscriptional attenuation of representative PSR genes in plantsmissing both NAC53 and NAC78. The ability to dimerize is con-sistent with other NACs that require dimerization to bind a pair ofpalindromically oriented DNA elements (Olsen et al., 2005b).Connections of NAC53 and/or NAC78 to heat, intense light,drought stress, ROS production, and senescence have also been
reported based on the analysis of mutants and overexpressionlines (Morishita et al., 2009; Yabuta et al., 2011; Lee et al., 2012,2014). It is conceivable that these phenotypes are indirectlymanifested by direct participation of the pair in proteotoxic stressprotection and regulation of 26S proteasome synthesis.As expected for plants with a dampened PSR, we found that
nac53 nac78 double mutants are highly sensitive to proteasomeinhibitors. Seedling growth was completely blocked with 50 mMMG132 and 0.5 mM bortezomib for the null nac53-1 nac78-1plants, with partial inhibition seen for plants harboring the weakernac53-2 nac78-2 alleles. A similarly modest growth inhibition wasseen for the nac78-1 single mutant but not for the single nac53-1mutant, suggesting that the NAC78 protein ismore critical to PSRactivation. Although the proteotoxic stress conditions studiedhere activated expression of NAC53 more robustly than NAC78,we consider it likely that they work simultaneously within the PSRgiven the similar temporal induction seen for the nac53-1 andnac78-1 single mutants. Regardless of their functions, we note
Figure 8. Loss of NAC53 and NAC78 Compromises Activation of the Proteasome Stress Regulon.
(A)RT-qPCR analysis of representative PSRmRNAs during proteasome stress. Total RNAwas extracted from6-d-oldwild-type and nacmutant seedlingsafter a 24-h incubationwith orwithout 100mMMG132. Transcript abundancewasdetermined viaRT-qPCRusing theACT2 (black) andPP2A (gray)mRNAsas references and normalized to those obtained from untreatedwild-type seedlings. Bars represent the average of at least three biological replicates (6SD),each measured in triplicate. Asterisks indicate significant differences between the nac51-1 nac78-1 seedlings and the wild type (+MG132) based onStudent’s t test (P value <0.05). Dashed lines indicate the average values for untreated wild-type seedlings.(B) Increased levels of several 26S proteasome subunits during proteasome stress depend on NAC53 and NAC78. Seedlings were treated with or withoutMG132 as in (A), and the resulting crude extractswere immunoblottedwith the indicated antibodies. HistoneH3was included to confirm near equal proteinloading. Open and closed arrowheads identify the unprocessed and processed forms of PBA1, respectively.
1290 The Plant Cell

that nac53 nac78 double null mutants are phenotypically normalunder nonstress conditions and do not hyperaccumulate Ub con-jugates upon MG132 treatment (Supplemental Figure 10). Theseobservations imply that other transcription factors are responsiblefor the basal synthesis of 26S proteasomes and other protein ho-meostasis regulators and that during proteotoxic stress, additionalrecycling pathways are engaged to eliminate the excess Ub con-jugates that accumulate if 26S proteasome capacity is insufficient(e.g., autophagy). As an aside, we note that Arabidopsis growth ismore sensitive to bortezomib thanMG132 and thusmight representa better agent to block 26S proteasome activity in planta.
Weconsider it likely thatNAC53andNAC78recognize thesamecis-element given their strong amino acid sequence similaritywithin theNAMDNAbindingmotif.Unfortunately, thenatureof theelement is unclear. Prior studies identified the PRCE element withthe consensus core TGGGC sequence as the preferred NAC78binding site (Yabuta et al., 2010; Nguyen et al., 2013), whereasmore recent interaction studies with NAC53 and NAC78 reportedthat both prefers MDM-type motifs (Lee et al., 2012; De Clercq
et al., 2013). Understanding this discrepancy will certainly requiremore in-depth binding studies using NAC53 and NAC78 aloneand in combination. Both cis-motifs were common in many of thePSR loci and are present upstream of most 26S proteasomegenes. Given the close proximity of PRCE and MDM sequenceswithin proteasome subunit promoters, it is conceivable that theNAC53/NAC78 dimers bind both motifs simultaneously and/orwork with other transcription factors that bind.How NAC53 and NAC78 activate the PSR during proteotoxic
stress is unclear. In yeast, their functional counterpart Rpn4participates in a simple negative feedback circuit whereby Rpn4becomes stabilized during proteotoxic stress as proteasomecapacity becomes overloaded. In mice and humans, its potentialcounterpart is Nrf1, a transmembrane-containing basic leucinezipper transcription factor that resides on theERmembrane undernonstressed conditions. Upon proteotoxic stress, the DNAbinding region of Nrf1 is proteolytically released from themembrane-spanning segment, which then permits its nuclearimport to drive 26S proteasome gene expression. Given that
Figure 9. Plants Lacking Both NAC53 and NAC78 Are Hypersensitive to Proteasome Inhibitors.
(A) Double homozygous nac53-1 nac78-1 plants are hypersensitive to MG132. Ten-day-old seedlings of the indicated genotypes were germinated andgrown on MS medium plus sucrose and containing either DMSO (control) or 30 or 50 mM MG132.(B) Quantification of fresh weight for seedlings shown in (A).(C) Growth inhibition of 6-d-old nac53-1 nac78-1 seedlings first germinated on MG132-free medium and then transferred to medium containing 50 mMMG132 2 d after germination.(D) Double homozygous nac53-1 nac78-1 plants are strongly hypersensitive to bortezomib. Seedlings were germinated and grown for 7 d on variousconcentrations of bortezomib.(E) Fresh weight of 7-d-old wild-type, nac53-1 nac78-1, and nac53-2 nac78-2 seedlings grown on 1 mM bortezomib (Btz).Asterisks in panels (B), (C), and (E) indicate a P value < 0.01 based on one-way ANOVA.
Transcriptional Regulation of Proteotoxic Stress 1291

NAC53 andNAC78, as part of the Arabidopsis NTL subfamily, arealso predicted to have a transmembrane domain, and that otherswithin this subfamily havebeen reported touseproteolytic releasefrom cytoplasmic stores to regulate their transcriptional activity(Kim et al., 2006; Kim et al., 2007), it is plausible that a shuttlemechanism similar to that of Nrf1 exists. However, this proteolyticstep and membrane release, if they occur, might not be essentialas the truncated nac53-2 protein described here (Figure 8A), andan engineered truncation ofNAC78byNguyen et al. (2013) did notappear to constitutively activate the PSR even in the absence ofMG132. A similar lack of effect of the C-terminal truncation wasseen for another NAC-NTL, NAC13, in its ability to activatetranscription (DeClercqet al., 2013), thusquestioning the role(s) ofthe transmembrane domain in NTL-NAC action.
Interestingly, thePSR is populatedwith transcripts for a numberof other NAC transcription factors besides NAC53, includingNAC1, 13, 32, 44, 50, 55, 81/ATAF2, 82, and 87, that are robustlyupregulated upon MG132 exposure and/or in the rpn10-1 andrpn12a-1 backgrounds. These factors could represent additionaltranscriptional regulators that assist in the immediate activationof the PSR or reflect part of a transcriptional cascade workingdownstreamthathelpsactivate the full suiteofPSR loci. Insupportof the former, Y2H analyses of these PSR-associated NAC pro-teins identified NAC13 and NAC81 as NAC53/NAC78 bindingpartners that coulddirectly participate inPSRactivation.NAC13 isparticularly intriguing as it, like NAC53 andNAC78, is amember ofthe NTL subclade and has been implicated in the transcriptionalresponse to mitochondrial dysfunction and binding to MDM se-quences (De Clercq et al., 2013). Notably, a number of the genetargets of the mitochondrial dysfunction response are shared withthe PSR (e.g., encoding UDP-glycosyl transferases and AOX1a),which combined with the prevalence of MDM sequences in PSRgenes, suggests that the two stress responses share a number ofprotective functions (e.g., oxidative stress defense). A similar overlapbetween proteotoxic stress and mitochondrial dysfunction has alsobeenobservedinmammals(Livnat-Levanonetal.,2014).Takingtheseresults together, an attractive hypothesis is that the PSR loci con-taining bothPRCEandMDMelements are activatedby heterodimersbearing NAC13 in combination with either NAC53 or NAC78.
We also note that a subpopulation of PSR genes is likelyimmune or only weakly responsive to NAC53 and NAC78 regu-lation (e.g.,WRKY25), indicating that other transcription factors areresponsible for their activationunderproteotoxicstress.Oneormoreof these factorsmight be foundwithin thePSR (e.g.,WRKY6, 25, 33,and 45; Supplemental Figure 2) or present in the list of DNA bindingproteins identified as common in our Y1H screen with the PA200and RPN12a promoters (Supplemental Table 1 and SupplementalFigure5).Consequently, it ispossible that thePSR iscontrolledbyanadditional suite of transcriptional regulators beyond NAC53/NAC78that generate a multilayered system to fully activate the regulonduring the various iterations of proteotoxic stress.
METHODS
Plant Materials and Growth Conditions
The T-DNA insertion mutants for NAC53 (nac53-1, SALK_009578C;nac53-2, SALK_018311C) andNAC78 (nac78-1, SALK_025098; nac78-2,
SALK_040812C) in the Arabidopsis thaliana ecotype Col-0 (Alonso andStepanova, 2003) were obtained from the ABRC at Ohio State University(https://abrc.osu.edu/). The rpn10-1 and rpn12a-1 exon-trap lines in theC24 background were generated as previously described (Smalle et al.,2002, 2003). TheNAC78 overexpression line (Col-0 background) driven bythe b-estradiol-inducible XVE promoter (NAC78 #2138 and #2319) wasprovided by the TRANSPLANTA resource (Coego et al., 2014).
The proteasome promoter:GUS transgenic lines were generated byPCRamplification of the 59upstream region for representative proteasomesubunit loci, starting at the end of the upstream coding region (or 2 kb) andterminating at the transcriptional start site. The PCR products wereintroduced upstream of the full GUS coding region present in the pCAM-BIA3301 or pMDC163 vectors. The chimeric genes were transformed intoArabidopsis (Col-0 ecotype) by thefloral dipmethodusing theAgrobacteriumtumefaciens strain GV3101 (Lee et al., 2011). Basta-resistant seedlings werescreened forGUSactivity byhistochemical stainingwith the substrateX-Gluc(5-bromo-4-chloro-3-indolyl-b-glucuronic acid; Sigma-Aldrich). For quanti-tative studies, total extracts from 10-d-old seedlings were assayed for GUSactivity using the fluorescence-based MUG assay (Sigma-Aldrich; Lee et al.,2011). At least 30 independent transformants were examined for each con-struction toavoidartifactsgeneratedbythesiteof the transgene insertion.Forexamination of GUS staining patterns, 6-d-old seedlings were incubatedovernight in X-Gluc following a 12-hexposure to 100mMMG132dissolved inDMSO, using an equivalent volume of DMSO as the control.
Unless otherwise noted, seeds were surface sterilized, stratified inthe dark at 4°C for 2 d, and then germinated on 0.7% agar containinghalf-strength Murashige and Skoog (MS) medium (Caisson Labs), 1%sucrose, and 0.5%MES (pH5.7), with or without various concentrations ofMG132 [N-(benzyloxycarbonyl)-leucinyl-leucinyl-leucinal; SelleckChem]orbortezomib [(1R)-3-methyl-1-(((2S)-3-phenyl-2-(pyrazin-2-carbonylamino)propanoyl)amino)butyl)boronic acid; SelleckChem]. For the RNA-seqstudies, the seedlings were grown for 5 d in 12-well liquid cultures platesunder continuous fluorescent white light at 22°Cprior toRNA isolation. Forstudies on thephenotypic effects ofMG132or bortezomib, stratified seedswere plated on the samemediumwith the addition of 0.7% agar. After 6 to10 d growth under a 16-h-light/8-h-dark photoperiod at 22°C, the plantswere weighed individually or in batches of two to five seedlings to de-termine fresh weight. One-way ANOVA was used to determine statisticalsignificance among the various genetic backgrounds and treatments.
RT-qPCR and RNA-Seq Analyses
Following various treatments, liquid-grown seedlings were pressed dry,frozen in liquid nitrogen, and pulverized. Total RNA was extracted withQIAzol lysis reagent (Qiagen), treated with DNaseI (Promega), and thenconverted to cDNA using Superscript III reverse transcriptase (LifeTechnologies). RT-qPCR amplifications on the cDNA populations wereperformed with a Roche LightCycler 480 using either Roche LightCycler480 SYBR Green Master Mix or MidSci Bullseye SYBR Green Master Mix.Appropriate priming sites for each locus were identified using Primer3Plus(http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi). SeeSupplemental Table 2 for the list of primers. Primer efficiencies were ex-perimentally determined to be between 1.90 and 2.10, based on assay ofa standard dilution series (Marshall et al., 2015). The relative abundance ofeach transcript was determined by the comparative threshold cyclemethod (Pfaffl, 2001) using theACT2 andPP2A reference genes as internalcontrols. All data were normalized to untreated wild type.
mRNA enrichment and library generation for RNA-seq studies wereperformedusing theTruSeqRNA library samplepreparationkit v2 (Illumina)with help from the University of Wisconsin-Madison Gene ExpressionCenter (http://www.biotech.wisc.edu/services/gec). Multiplexed sequenc-ing was performed on the Illumina HiSeq 2500 platform using 100-bpsingle-ended reads. At least two biological replicates were analyzed for
1292 The Plant Cell

each condition with each amplification yielding between 10 and 25 millionraw sequencing reads. FASTQ files were quality checked with theTrimmomatic package (Bolger et al., 2014). Between 70 and 80% of readswere retained in all biological replicates. Trimmed reads were alignedby TopHat-Bowtie 2 to the TAIR 10 genome using the Ensemble2013 Arabidopsis.gtf file annotations (Langmead and Salzberg, 2012).Read counts were generated through HTSeq (Anders et al., 2015),quantified via edgeR (Robinson et al., 2010), and then normalized to thewild-type Col-0 untreated control. Only those transcripts that meta FDR # 0.05 cutoff were included in the analyses.
All available gene coexpression and protein interaction data werecollected from the STRING database (Jensen et al., 2009) and mappedusing the organic layout option from Cytoscape 3.0.1 (Shannon et al.,2003). Gene Ontology and functional term enrichments were determinedusingDAVID (Huangetal., 2009).Highly interconnectedgenenodeclusterswere identifiedusing theMCODEplug-in forCytoscape (Bader andHogue,2003).Heatmapsof significantly expressedgeneswere generatedbyJavaTreeView v1.1643 (Saldanha, 2004).
Immunoblot Analysis
Immunoblot analyses were conducted with 6-d-old seedlings homoge-nized directly into SDS-PAGE sample buffer. Following SDS-PAGE, pro-teins were transferred onto Millipore Immobilon-P or Immobilon-FLmembranes and probed with antibodies against PA200 (Book et al.,2010), Ub (van Nocker et al., 1996), RPN1, RPN5, RPN10, RPN12a, andRPT2a (Smalle et al., 2002; Yang et al., 2004), and histone H3 (AbCam;AB1791).
Phylogenetic Comparisons of NAC Protein Sequences
Amino acid sequences were aligned by MEGA v6.0 under the MUSCLEdefault settings (Tamura et al., 2013). Alignments are provided inSupplemental Data Sets 5 and 6. The locations of the possible NAM andmembrane spanning regions were predicted by PFAM v28.0 (http://pfam.xfam.org).Phylogenetic analysesof thepredicted full-lengthNACproteinsor only their NAM DNA binding regions from Arabidopsis and other plantspecies available in Phytozome (www.phytozome.jgi.doe.gov/) or NCBI(www.ncbi.nlm.nih.gov/genbank) were performed using MrBayes 3.2(Ronquist et al., 2012) and the mixed amino acid model (aamodelpr =mixed) until convergence (average SD of split frequencies) reached below0.05 or plateaued. The trees were rooted with the Physcomitrella patensNAC protein gi_168025227 as the outgroup and visualized using FigTreev1.4.2. Nucleotide sequences related to the consensus PRCE and MDMcis-motifs (DeClercq et al., 2013; Nguyen et al., 2013) were detected in theregion upstream of the translation start site using the MEME sequenceenrichment identification algorithm with a 7th-order background modelcalculated from all Arabidopsis intergenic sequences (Bailey et al., 2006).
BiFC, Y1H, and Y2H Analyses
For BiFC, the full-length NAC53 or NAC78 coding sequences in thepDONR221 plasmid were introduced into either the pSITE-N-EYFP-C1 orpSITE-C-EYFP-C1 vectors (ARBC stock numbers CD3-1648 [nYFP] orCD3-1649 [cYFP], respectively), and transformed into the Agrobacteriumtumefaciens strain GV3101 (Li et al., 2014). Overnight cultures were dilutedto OD600 of 0.5 in resuspension buffer (10 mMMgCl2, 10mMMES, pH 5.7,and 100 mM acetosyringone) and syringe-infiltrated into 4- to 6-week-oldNicotiana benthamiana leaves. Fluorescence within the infiltrated regionswas visualized after 36 h using a Zeiss 510 Meta confocal laser scanningmicroscope. For MG132 treatments and 49,6-diamidino-2-phenylindole(DAPI) staining, the resuspension buffer without acetosyringone andcontaining 100mMMG132 and/or 1mMDAPI was infiltrated into the sameleaf regions 24 or 1.5 h prior to visualization, respectively.
Y2H assays were performed using the ProQuest Two-Hybrid System(Life Technologies). The indicatedNAC geneswere amplified by PCR fromArabidopsis cDNA generated as described above and recombined intopDONR221 via the Gateway BP clonase II reaction. These fragments werethen recombined in-frame to either the GAL4 activation domain or GAL4binding domain coding sequences in the pDEST22 or pDEST32 vectors(Life Technologies). Constructs were verified by sequencing, and pairwisecombinations of genes in pDEST22 and pDEST32 (or the empty vectors ascontrols) were cotransformed into the Saccharomyces cerevisiae strainMaV203 (Vidal et al., 1996). Y2H assays were performed by dilutingovernight yeast cultures with either YPD medium lacking Leu and Trp orlacking Leu, Trp, and His and containing the indicated concentrations of3-AT, and then growing the yeast for 2 d at 30°C on these media fornonselective and selective growth, respectively.
For anonbiasedY1Hscreen, 1000- and253-bp fragments of thePA200and RPN12a upstream regions, respectively, were cloned into a pLacZplasmid and screened individually against 1700 Arabidopsis transcriptionfactors cloned into the MaV203 yeast strain (Gaudinier et al., 2011). Forconfirmatory Y1H screens, lacZ activation was quantified using the Millerb-galactosidase assay and the substrate 5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside aspreviously described (ZhangandBremer, 1996).At least three biological replicates were averaged for each measurement.
Accession Numbers
Sequence data from this article can be found in the Arabidopsis GenomeInitiative or GenBank/EMBL databases under the following accession num-bers:ACT2 (At3g18780),CDC48a (At3g09840),GSTU25 (At1g17180),HSF8A(At1g67970), NAC1 (At1g01010), NAC13 (At1g32870), NAC32 (At1g77450),NAC44 (At3g01600), NAC50 (At3g10480), NAC53 (At3g10500), NAC55(At3g15500), NAC78 (At5g04410), NAC81 (At5g08790), NAC82 (At5g09330),NAC87 (At5g18270), NAS6 (At2g03430), PA200, (At3g13330), PBA1(At4g31300), PP2A (At1g13320), RPN1a (At2g20580), RPN1b (At4g28470),RPN3a (At1g20200), RPN3b (At1g75990), RPN5a (At5g09900), RPN5b(At5g64760), RPN10 (At4g38630), RPN12a (At1g64520), RPT1a At1g53750),RPT1b (At1g53780), RPT2a (At4g29040), RPT2b (At2g20140), RPT4a(At5g43010), RPT4b (At1g45000), UFD1 (At2g21270), and WRKY25(At2g30250). TheRNA-seq data are available in theNCBIGeneExpressionOmnibus under accession number GSE81668.
Supplemental Data
Supplemental Figure 1. RNA-Seq Comparison of Proteotoxic Stresson the Expression of Several Transcripts Commonly Used as Stand-ards for RT-qPCR Studies.
Supplemental Figure 2. Expression Heat Maps of RepresentativePSR Genes Outside of Those Encoding 26S Proteasome Subunits.
Supplemental Figure 3. Description of the 24-h MG132 UpregulatedGene Interaction and Coexpression Network.
Supplemental Figure 4. PRCE and MDM Sequences Are Located inthe Upstream DNA Region of 26S Proteasome Subunits.
Supplemental Figure 5. Occurrence of AGRIS-Annotated DNABinding Motifs in the Upstream Region of 26S Proteasome and OtherPSR Genes.
Supplemental Figure 6. Phylogenetic Tree of the 109 ArabidopsisNAC Transcription Factors Using the Full Amino Acid Sequences forComparison.
Supplemental Figure 7. Phylogenetic Tree of the 109 ArabidopsisNAC Transcription Factors Using just the NAM Domain Sequences forComparison.
Transcriptional Regulation of Proteotoxic Stress 1293

Supplemental Figure 8. Amino Acid Sequence Alignment of NTLProteins NAC53, NAC78, and NAC13.
Supplemental Figure 9. Bimolecular Fluorescence Complementationof NAC53 and NAC78 with or without MG132 Treatment.
Supplemental Figure 10. Ub Conjugate Levels in nac53-1 and nac78-1Mutants with or without MG132 Treatment.
Supplemental Figure 11. The PSR in Single nac53 and nac78Mutants Has the Same Temporal Response to MG132 as the WildType.
Supplemental Table 1. List of Y1H Prey in Common When Using theArabidopsis PA200 and RPN12a Promoters as Bait.
Supplemental Table 2. List of Oligonucleotide Primers Used forRT-qPCR Analysis.
Supplemental Data Set 1. List of Genes Whose Expression WasSignificantly Affected by 3-h Exposure to MG132.
Supplemental Data Set 2. List of Genes Whose Expression WasSignificantly Upregulated in the rpn10-1 and rpn12a-1 Mutant Back-grounds.
Supplemental Data Set 3. List of Genes within the PSR.
Supplemental Data Set 4. List of Genes Whose Expression WasSignificantly Affected by Long-Term Exposure to MG132.
Supplemental Data Set 5. Text File of the Full NAC Protein AlignmentCorresponding to the Phylogenetic Analysis in Supplemental Figure 6.
Supplemental Data Set 6. Text File of the NAM Domain AlignmentCorresponding to the Phylogenetic Analysis in Supplemental Figure 7.
ACKNOWLEDGMENTS
We thank Jose L. Pruneda-Paz for help with the Y1H screens, DavidC. Gemperline and Joseph M. Walker for helpful discussions, and LucasM. Slivicke for technical assistance. This work and N.P.G. were supportedby a grant from the U.S. Department of Energy Office of Science, Office ofBasic Energy Sciences, Chemical Sciences, Geosciences, and Bioscien-ces Division (DE-FG02-88ER13968). N.P.G. was also funded by aNationalInstitutes of Health Training fellowship provided to the University ofWisconsin-Madison Department of Genetics.
AUTHOR CONTRIBUTIONS
N.P.G.andR.D.V.designed the research.N.P.G.performedmost research,analyzed data, and performed all the computational analyses. R.S.M.assisted with the Y2H assays, RT-PCR, and RT-qPCR analyses. K.-H.L.generated the transgenic lines expressing the promoter:GUS fusions andanalyzed their responses. N.P.G. and R.D.V. wrote the article.
Received December 18, 2015; revised April 19, 2016; accepted May 11,2016; published May 18, 2016.
REFERENCES
Alonso, J.M., and Stepanova, A.N. (2003). T-DNA mutagenesis inArabidopsis. Methods Mol. Biol. 236: 177–188.
Amm, I., Sommer, T., and Wolf, D.H. (2014). Protein quality controland elimination of protein waste: the role of the ubiquitin-proteasomesystem. Biochim. Biophys. Acta 1843: 182–196.
Anders, S., Pyl, P.T., and Huber, W. (2015). HTSeq: a Pythonframework to work with high-throughput sequencing data. Bio-informatics 31: 166–169.
Bader, G.D., and Hogue, C.W. (2003). An automated method forfinding molecular complexes in large protein interaction networks.BMC Bioinformatics 4: 2.
Bailey, T.L., Williams, N., Misleh, C., and Li, W.W. (2006). MEME:discovering and analyzing DNA and protein sequence motifs. Nu-cleic Acids Res. 43: W369–W373.
Bhattacharyya, S., Yu, H., Mim, C., and Matouschek, A. (2014).Regulated protein turnover: snapshots of the proteasome in action.Nat. Rev. Mol. Cell Biol. 15: 122–133.
Bolger, A.M., Lohse, M., and Usadel, B. (2014). Trimmomatic:a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120.
Book, A.J., Gladman, N.P., Lee, S.S., Scalf, M., Smith, L.M., andVierstra, R.D. (2010). Affinity purification of the Arabidopsis 26Sproteasome reveals a diverse array of plant proteolytic complexes.J. Biol. Chem. 285: 25554–25569.
Book, A.J., Smalle, J., Lee, K.H., Yang, P., Walker, J.M., Casper, S.,Holmes, J.H., Russo, L.A., Buzzinotti, Z.W., Jenik, P.D., andVierstra, R.D. (2009). The RPN5 subunit of the 26S proteasome isessential for gametogenesis, sporophyte development, and com-plex assembly in Arabidopsis. Plant Cell 21: 460–478.
Chan, C.S., Guo, L., and Shih, M.C. (2001). Promoter analysis of thenuclear gene encoding the chloroplast glyceraldehyde-3-phosphatedehydrogenase B subunit of Arabidopsis thaliana. Plant Mol. Biol. 46:131–141.
Coego, A., Brizuela, E., Castillejo, P., Ruíz, S., Koncz, C., del Pozo,J.C., Piñeiro, M., Jarillo, J.A., Paz-Ares, J., and León, J.;TRANSPLANTA Consortium (2014). The TRANSPLANTA collec-tion of Arabidopsis lines: a resource for functional analysis oftranscription factors based on their conditional overexpression.Plant J. 77: 944–953.
Cuanalo-Contreras, K., Mukherjee, A., and Soto, C. (2013). Role ofprotein misfolding and proteostasis deficiency in protein misfoldingdiseases and aging. Int. J. Cell Biol. 2013: 638083.
De Clercq, I., et al. (2013). The membrane-bound NAC transcriptionfactor ANAC013 functions in mitochondrial retrograde regulation ofthe oxidative stress response in Arabidopsis. Plant Cell 25: 3472–3490.
Dohmen, R.J., Willers, I., and Marques, A.J. (2007). Biting the handthat feeds: Rpn4-dependent feedback regulation of proteasomefunction. Biochim. Biophys. Acta 1773: 1599–1604.
Farmer, L.M., Book, A.J., Lee, K.H., Lin, Y.L., Fu, H., and Vierstra,R.D. (2010). The RAD23 family provides an essential connectionbetween the 26S proteasome and ubiquitylated proteins in Arabi-dopsis. Plant Cell 22: 124–142.
Fatimababy, A.S., Lin, Y.L., Usharani, R., Radjacommare, R.,Wang, H.T., Tsai, H.L., Lee, Y., and Fu, H. (2010). Cross-speciesdivergence of the major recognition pathways of ubiquitylatedsubstrates for ubiquitin/26S proteasome-mediated proteolysis.FEBS J. 277: 796–816.
Finley, D. (2009). Recognition and processing of ubiquitin-proteinconjugates by the proteasome. Annu. Rev. Biochem. 78: 477–513.
Gallois, J.L., Guyon-Debast, A., Lécureuil, A., Vezon, D., Carpentier, V.,Bonhomme, S., and Guerche, P. (2009). The Arabidopsis proteasomeRPT5 subunits are essential for gametophyte development and showaccession-dependent redundancy. Plant Cell 21: 442–459.
Gaudinier, A., et al. (2011). Enhanced Y1H assays for Arabidopsis.Nat. Methods 8: 1053–1055.
Groll, M., Schellenberg, B., Bachmann, A.S., Archer, C.R., Huber,R., Powell, T.K., Lindow, S., Kaiser, M., and Dudler, R. (2008). A
1294 The Plant Cell

plant pathogen virulence factor inhibits the eukaryotic proteasomeby a novel mechanism. Nature 452: 755–758.
Hanssum, A., Zhong, Z., Rousseau, A., Krzyzosiak, A., Sigurdardottir, A.,and Bertolotti, A. (2014). An inducible chaperone adapts proteasomeassembly to stress. Mol. Cell 55: 566–577.
Hetz, C. (2012). The unfolded protein response: controlling cell fatedecisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13:89–102.
Hipp, M.S., Park, S.H., and Hartl, F.U. (2014). Proteostasis impair-ment in protein-misfolding and -aggregation diseases. Trends CellBiol. 24: 506–514.
Howell, S.H. (2013). Endoplasmic reticulum stress responses inplants. Annu. Rev. Plant Biol. 64: 477–499.
Huang, W., Sherman, B.T., and Lempicki, R.A. (2009). Systematicand integrative analysis of large gene lists using DAVID bio-informatics resources. Nat. Protoc. 4: 44–57.
Hudson, M.E., and Quail, P.H. (2003). Identification of promotermotifs involved in the network of phytochrome A-regulated geneexpression by combined analysis of genomic sequence and mi-croarray data. Plant Physiol. 133: 1605–1616.
Jensen, L.J., Kuhn, M., Stark, M., Chaffron, S., Creevey, C., Muller,J., Doerks, T., Julien, P., Roth, A., Simonovic, M., Bork, P., andvon Mering, C. (2009). STRING 8, a global view on proteins andtheir functional interactions in 630 organisms. Nucleic Acids Res.37: D412–D416.
Khaminets, A., Behl, C., and Dikic, I. (2016). Ubiquitin-dependentand independent signals in selective autophagy. Trends Cell Biol.26: 6–16.
Kim, S.Y., Kim, S.G., Kim, Y.S., Seo, P.J., Bae, M., Yoon, H.K., andPark, C.M. (2007). Exploring membrane-associated NAC tran-scription factors in Arabidopsis: implications for membrane biologyin genome regulation. Nucleic Acids Res. 35: 203–213.
Kim, Y.E., Hipp, M.S., Bracher, A., Hayer-Hartl, M., and Hartl, F.U.(2013). Molecular chaperone functions in protein folding and pro-teostasis. Annu. Rev. Biochem. 82: 323–355.
Kim, Y.S., Kim, S.G., Park, J.E., Park, H.Y., Lim, M.H., Chua, N.H.,and Park, C.M. (2006). A membrane-bound NAC transcriptionfactor regulates cell division in Arabidopsis. Plant Cell 18: 3132–3144.
Kisselev, A.F., van der Linden, W.A., and Overkleeft, H.S. (2012).Proteasome inhibitors: an expanding army attacking a unique tar-get. Chem. Biol. 19: 99–115.
Kraft, C., Peter, M., and Hofmann, K. (2010). Selective autophagy:ubiquitin-mediated recognition and beyond. Nat. Cell Biol. 12: 836–841.
Kurepa, J., Toh-e, A., and Smalle, J.A. (2008). 26S proteasomeregulatory particle mutants have increased oxidative stress toler-ance. Plant J. 53: 102–114.
Kurepa, J., Wang, S., Li, Y., Zaitlin, D., Pierce, A.J., and Smalle,J.A. (2009). Loss of 26S proteasome function leads to increased cellsize and decreased cell number in Arabidopsis shoot organs. PlantPhysiol. 150: 178–189.
Langmead, B., and Salzberg, S.L. (2012). Fast gapped-read align-ment with Bowtie 2. Nat. Methods 9: 357–359.
Lee, K.H., Minami, A., Marshall, R.S., Book, A.J., Farmer, L.M.,Walker, J.M., and Vierstra, R.D. (2011). The RPT2 subunit of the26S proteasome directs complex assembly, histone dynamics, andgametophyte and sporophyte development in Arabidopsis. PlantCell 23: 4298–4317.
Lee, S., Seo, P.J., Lee, H.-J., and Park, C.-M. (2012). A NAC tran-scription factor NTL4 promotes reactive oxygen species productionduring drought-induced leaf senescence in Arabidopsis. Plant J. 70:831–844.
Lee, S., Lee, H.-J., Huh, S.U., Paek, K.-H., Ha, J.-H., and Park, C.-M.(2014). The Arabidopsis NAC transcription factor NTL4 participates ina positive feedback loop that induces programmed cell death under heatstress conditions. Plant Sci. 227: 76–83.
Li, F., and Vierstra, R.D. (2012). Autophagy: a multifaceted in-tracellular system for bulk and selective recycling. Trends Plant Sci.17: 526–537.
Li, F., Chung, T., and Vierstra, R.D. (2014). AUTOPHAGY-RELATED11plays a critical role in general autophagy- and senescence-induced mi-tophagy in Arabidopsis. Plant Cell 26: 788–807.
Lin, M.Y., Chai, K.H., Ko, S.S., Kuang, L.Y., Lur, H.S., and Charng,Y.Y. (2014). A positive feedback loop between HEAT SHOCKPROTEIN101 and HEAT STRESS-ASSOCIATED 32-KD PROTEINmodulates long-term acquired thermotolerance illustrating diverseheat stress responses in rice varieties. Plant Physiol. 164: 2045–2053.
Livnat-Levanon, N., Kevei, É., Kleifeld, O., Krutauz, D., Segref, A.,Rinaldi, T., Erpapazoglou, Z., Cohen, M., Reis, N., Hoppe, T., andGlickman, M.H. (2014). Reversible 26S proteasome disassemblyupon mitochondrial stress. Cell Reports 7: 1371–1380.
Lundgren, J., Masson, P., Mirzaei, Z., and Young, P. (2005). Iden-tification and characterization of a Drosophila proteasome regula-tory network. Mol. Cell. Biol. 25: 4662–4675.
Mannhaupt, G., Schnall, R., Karpov, V., Vetter, I., and Feldmann, H.(1999). Rpn4p acts as a transcription factor by binding to PACE,a nonamer box found upstream of 26S proteasomal and othergenes in yeast. FEBS Lett. 450: 27–34.
Marshall, R.S., Li, F., Gemperline, D.C., Book, A.J., and Vierstra,R.D. (2015). Autophagic degradation of the 26S proteasome ismediated by the dual ATG8/ubiquitin receptor RPN10 in Arabi-dopsis. Mol. Cell 58: 1053–1066.
Meiners, S., Heyken, D., Weller, A., Ludwig, A., Stangl, K., Kloetzel,P.M., and Krüger, E. (2003). Inhibition of proteasome activity in-duces concerted expression of proteasome genes and de novoformation of mammalian proteasomes. J. Biol. Chem. 278: 21517–21525.
Morimoto, R.I. (2008). Proteotoxic stress and inducible chaperonenetworks in neurodegenerative disease and aging. Genes Dev. 22:1427–1438.
Morishita, T., Kojima, Y., Maruta, T., Nishizawa-Yokoi, A., Yabuta,Y., and Shigeoka, S. (2009). Arabidopsis NAC transcription factor,ANAC078, regulates flavonoid biosynthesis under high-light. PlantCell Physiol. 50: 2210–2222.
Nakashima, K., Takasaki, H., Mizoi, J., Shinozaki, K., andYamaguchi-Shinozaki, K. (2012). NAC transcription factors inplant abiotic stress responses. Biochim. Biophys. Acta 1819: 97–103.
Nguyen, H.M., Schippers, J.H., Gõni-Ramos, O., Christoph, M.P.,Dortay, H., van der Hoorn, R.A., and Mueller-Roeber, B. (2013).An upstream regulator of the 26S proteasome modulates organ sizein Arabidopsis thaliana. Plant J. 74: 25–36.
Olsen, A.N., Ernst, H.A., Leggio, L.L., and Skriver, K. (2005a). NACtranscription factors: structurally distinct, functionally diverse.Trends Plant Sci. 10: 79–87.
Olsen, A.N., Ernst, H.A., Leggio, L.L., and Skriver, K. (2005b). DNA-binding specificity and molecular functions of NAC transcriptionfactors. Plant Sci. 169: 785–797.
Pfaffl, M.W. (2001). A new mathematical model for relative quantifi-cation in real-time RT-PCR. Nucleic Acids Res. 29: e45.
Radhakrishnan, S.K., den Besten, W., and Deshaies, R.J. (2014).p97-dependent retrotranslocation and proteolytic processing gov-ern formation of active Nrf1 upon proteasome inhibition. eLife 3:e01856.
Transcriptional Regulation of Proteotoxic Stress 1295

Radhakrishnan, S.K., Lee, C.S., Young, P., Beskow, A., Chan, J.Y.,and Deshaies, R.J. (2010). Transcription factor Nrf1 mediates theproteasome recovery pathway after proteasome inhibition in mammaliancells. Mol. Cell 38: 17–28.
Robinson, M.D., McCarthy, D.J., and Smyth, G.K. (2010). edgeR:a Bioconductor package for differential expression analysis ofdigital gene expression data. Bioinformatics 26: 139–140.
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D.L., Darling, A.,Höhna, S., Larget, B., Liu, L., Suchard, M.A., and Huelsenbeck, J.P.(2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and modelchoice across a large model space. Syst. Biol. 61: 539–542.
Rushton, P.J., Somssich, I.E., Ringler, P., and Shen, Q.J. (2010).WRKY transcription factors. Trends Plant Sci. 15: 247–258.
Russell, J.D., Scalf, M., Book, A.J., Ladror, D.T., Vierstra, R.D.,Smith, L.M., and Coon, J.J. (2013). Characterization and quantifi-cation of intact 26S proteasome proteins by real-time measurementof intrinsic fluorescence prior to top-down mass spectrometry.PLoS One 8: e58157.
Saldanha, A.J. (2004). Java Treeview: extensible visualization of mi-croarray data. Bioinformatics 20: 3246–3248.
Scharf, K.D., Berberich, T., Ebersberger, I., and Nover, L. (2012).The plant heat stress transcription factor (Hsf) family: structure,function and evolution. Biochim. Biophys. Acta 1819: 104–119.
Seifert, A., Schofield, P., Barton, G.J., and Hay, R.T. (2015). Pro-teotoxic stress reprograms the chromatin landscape of SUMOmodification. Sci. Signal. 8: rs7.
Sha, Z., and Goldberg, A.L. (2014). Proteasome-mediated process-ing of Nrf1 is essential for coordinate induction of all proteasomesubunits and p97. Curr. Biol. 24: 1573–1583.
Shannon, P., Markiel, A., Ozier, O., Baliga, N.S., Wang, J.T.,Ramage, D., Amin, N., Schwikowski, B., and Ideker, T. (2003).Cytoscape: a software environment for integrated models of bio-molecular interaction networks. Genome Res. 13: 2498–2504.
Shirozu, R., Yashiroda, H., and Murata, S. (2015). Identification ofminimum Rpn4-responsive elements in genes related to protea-some functions. FEBS Lett. 589: 933–940.
Smalle, J., Kurepa, J., Yang, P., Babiychuk, E., Kushnir, S., Durski, A.,and Vierstra, R.D. (2002). Cytokinin growth responses in Arabidopsisinvolve the 26S proteasome subunit RPN12. Plant Cell 14: 17–32.
Smalle, J., Kurepa, J., Yang, P., Emborg, T.J., Babiychuk, E.,Kushnir, S., and Vierstra, R.D. (2003). The pleiotropic role of the26S proteasome subunit RPN10 in Arabidopsis growth and de-velopment supports a substrate-specific function in abscisic acidsignaling. Plant Cell 15: 965–980.
Sonoda, Y., Sako, K., Maki, Y., Yamazaki, N., Yamamoto, H., Ikeda,A., and Yamaguchi, J. (2009). Regulation of leaf organ size by theArabidopsis RPT2a 19S proteasome subunit. Plant J. 60: 68–78.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S.(2013). MEGA6: molecular evolutionary genetics analysis version6.0. Mol. Biol. Evol. 30: 2725–2729.
van Nocker, S., Sadis, S., Rubin, D.M., Glickman, M., Fu, H., Coux, O.,Wefes, I., Finley, D., and Vierstra, R.D. (1996). The multiubiquitin-chain-binding protein Mcb1 is a component of the 26S proteasome inSaccharomyces cerevisiae and plays a nonessential, substrate-specificrole in protein turnover. Mol. Cell. Biol. 16: 6020–6028.
Vidal, M., Brachmann, R.K., Fattaey, A., Harlow, E., and Boeke,J.D. (1996). Reverse two-hybrid and one-hybrid systems to detectdissociation of protein-protein and DNA-protein interactions. Proc.Natl. Acad. Sci. USA 93: 10315–10320.
Wu, A., et al. (2012). JUNGBRUNNEN1, a reactive oxygen species-responsive NAC transcription factor, regulates longevity in Arabi-dopsis. Plant Cell 24: 482–506.
Xie, Y., and Varshavsky, A. (2001). RPN4 is a ligand, substrate, andtranscriptional regulator of the 26S proteasome: a negative feed-back circuit. Proc. Natl. Acad. Sci. USA 98: 3056–3061.
Yabuta, Y., Morishita, T., Kojima, Y., Maruta, T., Nishizawa-Yokoi,A., and Shigeoka, S. (2010). Identification of recognition sequenceof ANAC078 protein by the cyclic amplification and selection oftargets technique. Plant Signal. Behav. 5: 695–697.
Yabuta, Y., Osada, R., Morishita, T., Nishizawa-Yokoi, A., Tamoi,M., Maruta, T., and Shigeoka, S. (2011). Involvement of Arabi-dopsis NAC transcription factor in the regulation of 20S and 26Sproteasomes. Plant Sci. 181: 421–427.
Yang, B.-J., Han, X.-X., Yin, L.-L., Xing, M.-Q., Xu, Z.-H., and Xue,H.-W. (2016). Arabidopsis PROTEASOME REGULATOR1 is re-quired for auxin-mediated suppression of proteasome activity andregulates auxin signaling. Nat. Commun. 7: 11388.
Yang, P., Fu, H., Walker, J., Papa, C.M., Smalle, J., Ju, Y.M., andVierstra, R.D. (2004). Purification of the Arabidopsis 26S protea-some: biochemical and molecular analyses revealed the presenceof multiple isoforms. J. Biol. Chem. 279: 6401–6413.
Zhang, X., and Bremer, H. (1996). Effects of Fis on ribosome syn-thesis and activity and on rRNA promoter activities in Escherichiacoli. J. Mol. Biol. 259: 27–40.
Zhou, J., Wang, J., Cheng, Y., Chi, Y.J., Fan, B., Yu, J.Q., and Chen,Z. (2013). NBR1-mediated selective autophagy targets insolubleubiquitinated protein aggregates in plant stress responses. PLoSGenet. 9: e1003196.
1296 The Plant Cell

DOI 10.1105/tpc.15.01022; originally published online May 18, 2016; 2016;28;1279-1296Plant Cell
Nicholas P. Gladman, Richard S. Marshall, Kwang-Hee Lee and Richard D. VierstraArabidopsis
The Proteasome Stress Regulon Is Controlled by a Pair of NAC Transcription Factors in
This information is current as of February 26, 2020
Supplemental Data /content/suppl/2016/05/18/tpc.15.01022.DC1.html
References /content/28/6/1279.full.html#ref-list-1
This article cites 80 articles, 23 of which can be accessed free at:
Permissions https://www.copyright.com/ccc/openurl.do?sid=pd_hw1532298X&issn=1532298X&WT.mc_id=pd_hw1532298X
eTOCs http://www.plantcell.org/cgi/alerts/ctmain
Sign up for eTOCs at:
CiteTrack Alerts http://www.plantcell.org/cgi/alerts/ctmain
Sign up for CiteTrack Alerts at:
Subscription Information http://www.aspb.org/publications/subscriptions.cfm
is available at:Plant Physiology and The Plant CellSubscription Information for
ADVANCING THE SCIENCE OF PLANT BIOLOGY © American Society of Plant Biologists