thesis section: brain edema
DESCRIPTION
Thesis section: brain edemahttp://yassermetwally.comhttp://yassermetwally.netTRANSCRIPT

Central Nervous SystemEdema
Essay
In Neuropsychiatry
Submitted for partial fulfillment of Master Degree
By
Mina Ibrahim Adly IbrahimM.B.B.CH
Supervisors of
Prof. Mohammed Yasser MetwallyProfessor of Neuropsychiatry
Faculty of Medicine-Ain Shams Universitywww.yassermetwally.com
Prof. Naglaa Mohamed ElkhayatProfessor of Neuropsychiatry
Faculty of Medicine-Ain Shams University
Dr. Ali Soliman Ali ShalashLecturer of Neuropsychiatry
Faculty of Medicine-Ain Shams University
Faculty of MedicineAin Shams University
2011

1
CCoonntteennttssSubject page
1. Acknowledgment………………………………………………2
2. List of abbreviations……………………………………………3
3. List of figures…………………………………………………..6
4. List of tables…………………………………………………....8
5. Introduction and aim of the work……………………………....9
6. Chapter (1): Pathogenesis of cerebral edema…………………15
7. Chapter (2): Chemical Mediators Involved in The Pathogenesis
Of Brain Edema…………………………………37
8. Chapter (3): Diagnosing cerebral edema……………………...53
9. Chapter (4): Cerebral Edema in Neurological Diseases………69
10.Chapter (5): Treatment of Cerebral Edema…………………...79
11. Chapter (6): Spinal Cord Edema In Injury and Repair……...101
12. Summary…………………………………………....………115
13. Discussion……..……………………………………………120
14. References………..…………………………………………123
15. Arabic summary……...…………………………………………

2
AAcckknnoowwlleeddggmmeenntt
Thanks to merciful lord for all the countless gifts you have
offered me, and thanks to my family for their love and support.
It is a great pleasure to acknowledge my deepest thanks and
gratitude to Prof. Mohammed Yasser Metwally, Professor of
Neuropsychiatry, Faculty of Medicine-Ain Shams University, for
suggesting the topic of this essay, and his kind supervision. It is a great
honour to work under his supervision.
I would like to express my deepest thanks and sincere appreciation
to Prof. Naglaa Mohamed Elkhayat, Professor of Neuropsychiatry,
Faculty of Medicine-Ain Shams University, for her encouragement,
creative and comprehensive advice until this work came to existence.
I would like to express my extreme sincere gratitude and
appreciation to Dr. Ali Soliman Ali Shalash, Lecturer of
Neuropsychiatry, Faculty of Medicine-Ain Shams University, for his
kind endless help, generous advice and support during the study.
Mina Ibrahim Adly
2011

3
LLiisstt ooff aabbbbrreevviiaattiioonnss
ADC: Apparent diffusion coefficient.
AMP& ADP: Adenosine monophosphate& Adenosine diphosphate.
Ang: Angiopoietin.
AQP: Aquaporins.
ATP: Adenosine triphosphate.
BBB: Blood–brain barrier.
BDNF: Brain derived neurotrophic factor.
BK: Bradykinin.
BSCB: Blood-spinal cord barrier.
Cav-1: Caveolin-1.
CBF: Cerebral blood flow.
CPP: Cerebral perfusion pressure.
CSF: Cerebrospinal fluid.
CT: Computed tomography.
Da: Dalton unit.
DPTA: Diethylenetriaminepentaacetic Acid.
DWI: Diffusion-weighted imaging.
EBA: Evans blue albumin.
ECS: Extracellular space.
FLAIR: Fluid-attenuated inversion recovery.
G: gram.
GCS: Glasgow coma scale.
HRP: Horseradish peroxidase.

4
HS: Hypertonic saline.
I 125: Iodine 125.
ICH: Intracranial hemorrhage.
ICP: Intracranial pressure.
ICUs: Intensive care units.
IGF-1: Insulin like growth factor 1.
IL: Interleukins.
JAM: Junctional adhesion molecule.
MAP: Mean arterial pressure.
MCA: Middle cerebral artery.
Meq/L: Milliequevalent per litre.
MIP: Macrophage inflammatory proteins.
MmHg: Millimetrs of mercury.
Mmol/L: Millimoles per litre.
MMPs: Matrix metalloproteinases.
MOsm/L: Milliosmoles per litre.
MRI: Magnetic resonance imaging.
mRNA: messenger Ribonucleic acid.
MS: Multiple sclerosis.
MT1-MMP: Membrane-type Matrix metalloproteinases.
Nm: Nanometre.
Nor-BNI: Nor-binaltrophimine.
NOS: Nitric oxide synthase.
PGs: Prostaglandins.
PWI: perfusion-weighted imaging.

5
SAH: Subarachnoid hemorrhage.
SCI: Spinal cord injury.
TBI: Traumatic brain injury.
TIMPs: Tissue inhibitors of metalloproteinases.
TNF-: Tumor necrosis factor alpha.
VEGF: Vascular endothelial growth factors.
ZO: zonula occludens.

6
LLiisstt ooff ffiigguurreess
Figure Page
Figure 1: Gross image demonstrating edema in human brain compared
with a normal one...………………………………..…….18
Figure 2: White matter from an area of edema…………………....…19
Figure 3: Illustrated picture of blood brain barrier…………………..20
Figure 4: An axial CT scan with glioblastoma multiforme…….……21
Figure 5: The cold injury site…………………..……………………23
Figure 6: Endothelial phosphorylated Cav-1………………………...25
Figure 7: expression of caveolins and tight junction proteins during
BBB breakdown…..……………………………….………29
Figure 8: Axial CT scans with whole right hemisphere infarction…..32
Figure 9: An axial MR image of a 4 year old with hydrocephalus….34
Figure 10: Pathways for water entry into and exit from brain……….42
Figure 11: Temporal expression of growth factor proteins is shown
during the period of BBB breakdown in the cold injury
mode……………………………………………………..51
Figure 12: Cerebral herniation syndromes..…………………………55
Figure 13: CT scan of global brain edema...…………………………60
Figure 14: CT scan showing brain edema caused by a tumor……….61
Figure 15: An area which represents an infarct………………….…..61
Figure 16: Intracranial hemorrhage depicted by MRI……………….63
Figure 17: Periventricular FLAIR hyperintensity due to hydrocephalic
edema………………………………………………….…….63

7
Figure 18: MRI showing central pontine myelinolysis…...................63
Figure 19: The cytotoxic component of acute cerebral ischemia is
demonstrated by ADC hypointensity, whereas T2 weighted
sequences may be unrevealing …….………………………..65
Figure 20: MRI of status epilepticus reveals evidence of cytotoxic
edema..............................................................................…...65
Figure 21: Disruption of the BBB associated with a glioma….…….66
Figure 22: Mass effect from infarction and midline shift.
Hemicraniectomy performed with herniation through the
skull defect…………………………………………….…100

8
LLiisstt ooff ttaabblleess
Table Page
Table 1: Vasoactive agents that increase the blood–brain barrier
permeability……………………..……………………….39
Table 2: Summary of the clinical subtypes of herniation
syndromes…………………………………………….…56
Table 3: Summary of experimental studies comparing different
formulations of hypertonic saline with mannitol 20%….…90
Table 4: Theoretical potential complications of using hypertonic saline
solutions………………..………………………………….93
Table 5: Treatment Strategies in Spinal Cord Injury…………..…...109

9
IInnttrroodduuccttiioonnSurprising as it may sound cerebral edema is a fairly common
pathophysiological entity which is encountered in many clinical
conditions. Many of these conditions present as medical emergencies.
By definition cerebral edema is the excess accumulation of water in
the intra-and/or extracellular spaces of the brain (Kempski, 2001).
To explain the consequences of cerebral edema in the simplest
terminology, it is best to take the help of Monro-Kelie hypothesis,
which says that; the total bulk of three elements inside the skull i.e.
brain, cerebral spinal fluid and blood is at all times constant. Since
skull is like a rigid box which cannot be stretched, if there is excessive
water, the volume of brain as well as blood inside the skull is
compressed. Further increase in the intracranial pressure (ICP)
eventually causes a reduction in cerebral blood flow throughout the
brain which can correspondingly cause extensive cerebral infarction. If
these changes continue further, it leads to the disastrous condition of
brain herniation, which is the fore runner of irreversible brain damage
and death (Rosenberg, 2000).
Despite the classification of edema into distinct forms as:
vasogenic, cytotoxic, hydrocephalic and osmotic, it is recognized that
in most clinical situations there is a combination of different types of
edema depending on the time course of the disease. For example, early
cerebral ischemia is associated with cellular swelling and cytotoxic
edema; however, once the capillary endothelium is damaged there is

10
BBB breakdown and vasogenic edema results. While in traumatic
brain injury both vasogenic and cytotoxic edema coexist (Marmarou
et al, 2006).
Vasogenic cerebral edema refers to the influx of fluid and solutes
into the brain through an incompetent blood brain barrier. This is the
most common type of brain edema and results from increased
permeability of the capillary endothelial cells; the white matter is
primarily affected. Breakdown in the BBB allows movement of
proteins from the intravascular space through the capillary wall into
the extracellular space. This type of edema is seen in: trauma, tumor,
abscess, hemorrhage, infarction, acute MS plaques, and cerebral
contusion (Metwally, 2009).
Cellular (cytotoxic) cerebral edema refers to a cellular swelling. It is
seen in conditions like head injury, severe hypothermia,
encephalopathy, pseudotumor cerebri and hypoxia. It results from the
swelling of brain cells, most likely due to the release of toxic factors
from neutrophils and bacteria within minutes after an insult. Cytotoxic
edema affects predominantly the gray matter (Liang et al, 2007).
Interstitial edema is seen in hydrocephalus when outflow of CSF is
obstructed and intraventricular pressure increases. The result is
movement of sodium and water across the ventricular wall into the
paraventricular space. Interstitial cerebral edema occurring during

11
meningitis is due to obstruction of normal CSF pathways (Abbott,
2004).
Osmotic cerebral edema occurs when plasma is diluted by
hyponatremia, syndrome of inappropriate antidiuretic hormone
secretion, hemodialysis, or rapid reduction of blood glucose in
hyperosmolar hyperglycemic state, the brain osmolality will then
exceed the serum osmolality creating an abnormal pressure gradient
down which water will flow into the brain causing edema (Nag, 2003)
a.
Pathophysiology of cerebral edema at cellular level is complex.
Damaged cells swell, injured blood vessels leak and blocked
absorption pathways force fluid to enter brain tissues. Cellular and
blood vessel damage follows activation of an injury cascade which
begins with glutamate release into the extracellular space. Calcium
and sodium entry channels are opened by glutamate stimulation.
Membrane ATPase pumps extrude one calcium ion exchange for 3
sodium ions. Sodium builds up within the cell creating an osmotic
gradient and increasing cell volume by entry of water (Marmarou,
2007).
It appears that injury in the spinal cord induce blood-spinal cord
barrier (BSCB) disruption. The BSCB breakdown involves cascade of
events involving several neurochemicals like: serotonin,
prostaglandins, neuropeptides and amino acids (Sharma, 2004).
Serial neuroimaging by CT scans and magnetic resonance imaging
can be particularly useful in confirming intracranial compartmental

12
and midline shifts, herniation syndromes, ischemic brain injury, and
exacerbation of cerebral edema (sulcal effacement and obliteration of
basal cisterns), and can provide valuable insights into the type of
edema present (focal or global, involvement of gray or white matter).
CT scan provides an excellent tool for determination of abnormalities
in brain water content. CT is an excellent method for following the
resolution of brain edema following therapeutic intervention. MRI
appears to be more sensitive than CT at detecting development of
cerebral edema (Kuroiwa et al, 2007).
Management of cerebral edema involves using a systematic and
algorithmic approach, from general measures to specific therapeutic
interventions, and decopressive surgery. The general measures
include: elevation of head end of bed 15-30 degrees to promote
cerebral venous drainage, fluid restriction, hypothermia, and
correction of factors increasing ICP e.g. hypercarbia, hypoxia,
hyperthermia, acidosis, hypotension and hypovolaemia (Ng et al,
2004).
Specific therapeutic interventions include: 1. osmotherapy:
mannitol, the most popular osmotic agent (Toung et al, 2007).
2. Diuretics: the osmotic effect can be prolonged by the use of loop
diuretics after the osmotic agent infusion (Thenuwara et al, 2002).
3. Corticosteroids: they lower intracranial pressure primarily in
vasogenic edema because of their effect on the blood vessel (Sinha et
al, 2004).

13
4. Controlled hyperventilation: is helpful in reducing the raised ICP
which falls within minutes of onset of hyperventilation (Mayer &
Rincon, 2005).
Cerebral edema, irrespective of the underlying origin of brain
injury, is a significant cause of morbidity and death, and though there
has been good progress in understanding pathophysiological
mechanisms associated with cerebral edema more effective treatment
is required and is still awaited (Rabinstein, 2006).

14
Aim of the work The aim of this review is to discuss different types and
etiologies of brain edema and to overview recent management of the
various chemical mediators involved in the pathogenesis of cerebral
edema.

15
CChhaapptteerr ((11))::PPaatthhooggeenneessiiss
OOff CCeerreebbrraall EEddeemmaa

16
PPaatthhooggeenneessiiss OOff CCeerreebbrraall EEddeemmaa
IInnttrroodduuccttiioonn::Brain edema is defined as an increase in brain volume resulting
from a localized or diffuse abnormal accumulation of fluid within the
brain parenchyma (Johnston & Teo, 2000). This definition excludes
volumetric enlargement due to cerebral engorgement which results
from an increase in blood volume on the basis of either vasodilatation
due to hypercapnia or impairment of venous flow secondary to
obstruction of the cerebral veins and venous sinuses (Nag, 2003) b.
Initially, the changes in brain volume are compensated by a
decrease in cerebrospinal fluid (CSF) and blood volume. In large
hemispheric lesions, progressive swelling exceeds these compensatory
mechanisms and an increase in the intracranial pressure (ICP) results
in herniations of cerebral tissue leading to death (Wolburg et al,
2008).
Hence the significance of brain edema, which continues to be a
major cause of mortality after diverse types of brain pathologies such
as major cerebral infarcts, hemorrhages, trauma, infections and
tumors. The lack of effective treatment for brain edema remains a
stimulus for continued interest and research into the pathogenesis of
this condition (Marmarou, 2007).

17
GGeenneerraall ccoonnssiiddeerraattiioonnss::The realization that brain edema is associated with either extra- or
intra-cellular accumulation of abnormal fluid led to its classification
into vasogenic and cytotoxic edema. Vasogenic edema is associated
with dysfunction of the blood–brain barrier (BBB) which allows
increased passage of plasma proteins and water into the extracellular
compartment, while cytotoxic edema results from abnormal water
uptake by injured brain cells. Other types of edema described include
hydrocephalic or interstitial edema and osmotic or hypostatic edema
(Czosnyka et al, 2004).

18
AAeettiiooppaatthhooggeenneessiiss ooff vvaarriioouuss ttyyppeess ooffcceerreebbrraall eeddeemmaa::
11.. VVaassooggeenniicc eeddeemmaa::Brain diseases such as hemorrhage, infections, seizures, trauma,
tumors, radiation injury and hypertensive encephalopathy are
associated with BBB breakdown to plasma proteins leading to
vasogenic edema. Vasogenic edema also occurs in the later stages of
brain infarction. Vasogenic edema may be localized or diffuse
depending on the underlying pathology. The overlying gyri become
more flattened, and the sulci are narrowed (Figure 1). When diffuse
edema is present the ventricles are slit-like (Hemphill et al, 2001).
Figure 1: 1b. Gross image demonstrating edema in human braincompared with a normal one (figure 1 a) (Hemphill et al, 2001).
Breakdown of the BBB to plasma proteins can be demonstrated by
immunohistochemistry using antibodies to whole serum proteins,

19
albumin, fibrinogen or fibronectin in human autopsy brain tissue or
brains of experimental animals (Kimelburg, 2004).
The white matter is more edema-prone since it has unattached
parallel bands of fibers with an intervening loose extracellular space
(ECS). The grey matter has a higher cell density with many inter-
cellular connections which reduce the number of direct linear
pathways making the grey matter ECS much less subject to swelling.
Light microscopy in acute edema shows vacuolation and pallor of the
white matter (Figure 2a & b) (Ballabh et al, 2004).
Figure 2: (figure 2a) Light microscopic appearance of normal white matterstained with hematoxylin–eosin and Luxol fast blue. (Figure 2b) White matter
from an area of edema adjacent to a meningioma (not shown) shows myelin pallorand an increased number of astrocytes (arrowheads) (Ballabh et al, 2004).
In long standing cases of edema there is fragmentation of the
myelin sheaths which are phagocytosed by macrophages resulting in
myelin pallor. An astrocytic response is present in the areas of edema.
mRNA levels are maximal on days 4–5 and they remain elevated up to
day 14 post-injury. Spatial mRNA expression follows the pattern of
post-injury edema being present in the cortex adjacent to the lesion,

20
and the ipsilateral and contralateral callosal radiations (Hawkins,
2008).
TThhee bblloooodd––bbrraaiinn bbaarrrriieerr ((BBBBBB))::It is well known that cerebral vessels differ from non-neural vessels
and have a structural, biochemical and physiological barrier, which
limits the passage of various substances including plasma proteins
from blood into brain (Nag, 2003) b.
Cellular components of the BBB include endothelium, pericytes
and the perivascular astrocytic processes, which together with their
associated neurons form the ‘‘neurovascular unit’’. The best studied
cell type is cerebral endothelium which has two distinctive structural
features that limit their permeability to plasma proteins (figure 3).
These cells have fewer caveolae or plasmalemmal vesicles than non-
neural vessels and circumferential tight junctions are present along the
interendothelial spaces. Breakdown of the BBB is assessed by tracers.
Gadolinium DPTA is the most commonly used tracer in human
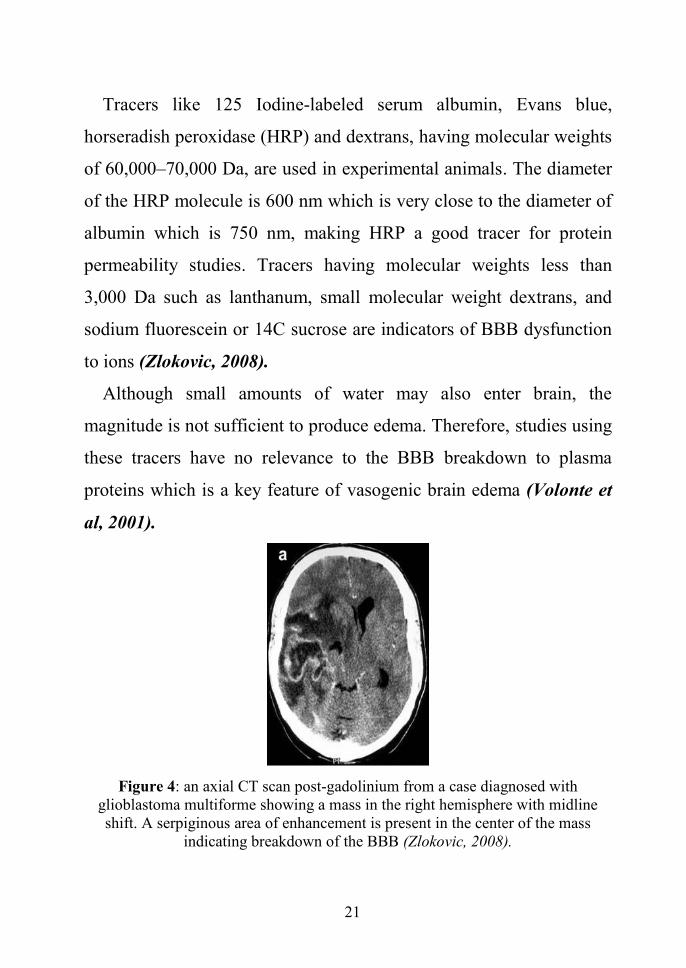
studies (Figure 4).
Figure 3: illustrated picture of blood brain barrier (Nag, 2003) b.
21
Tracers like 125 Iodine-labeled serum albumin, Evans blue,
horseradish peroxidase (HRP) and dextrans, having molecular weights
of 60,000–70,000 Da, are used in experimental animals. The diameter
of the HRP molecule is 600 nm which is very close to the diameter of
albumin which is 750 nm, making HRP a good tracer for protein
permeability studies. Tracers having molecular weights less than
3,000 Da such as lanthanum, small molecular weight dextrans, and
sodium fluorescein or 14C sucrose are indicators of BBB dysfunction
to ions (Zlokovic, 2008).
Although small amounts of water may also enter brain, the
magnitude is not sufficient to produce edema. Therefore, studies using
these tracers have no relevance to the BBB breakdown to plasma
proteins which is a key feature of vasogenic brain edema (Volonte et
al, 2001).
Figure 4: an axial CT scan post-gadolinium from a case diagnosed withglioblastoma multiforme showing a mass in the right hemisphere with midlineshift. A serpiginous area of enhancement is present in the center of the mass
indicating breakdown of the BBB (Zlokovic, 2008).

22
Permeability properties of cerebral endothelium are not uniform in
all brain vessels. In rodents, aside from regions outside the BBB, a
significant number of normal cerebral vessels are permeable to HRP.
Thus, the demonstration of increased permeability in these areas
cannot be ascribed to pathology. Also, freeze fracture studies show
that there is variation in the number of interconnected strands that
make up tight junctions in the different types of brain vessels, with
cortical vessels having junctions of the highest complexity, while
junctions of the postcapillary venules are least complex. The latter
would explain why increased permeability of the postcapillary venules
occurs in inflammation (Nag, 2007).
TThhee ccoolldd iinnjjuurryy mmooddeell::This model was developed by Klatzo to study the pathophysiology
of vasogenic edema and has been used extensively in studies. A
unilateral focal cortical freeze lesion is produced by placing the tip of
a cold probe cooled with liquid nitrogen on the dura for 45 seconds.
There are variations in the method of producing the cold lesion which
makes it difficult to compare the results obtained from different
laboratories (Klatzo, 1958 coated from Sukriti Nag, et al, 2009).
The ensuing edema was initially studied using exogenous tracers
such as Evans blue and HRP. BBB breakdown to HRP was present at
12 h, which was the earliest time point studied and the BBB was
restored on day 6 post-injury. Similar results were obtained using
immunohistochemistry to demonstrate endogenous serum protein
23
extravasation using an antibody to serum proteins, fibrinogen or
fibronectin (Lossinsky & Shivers, 2004).
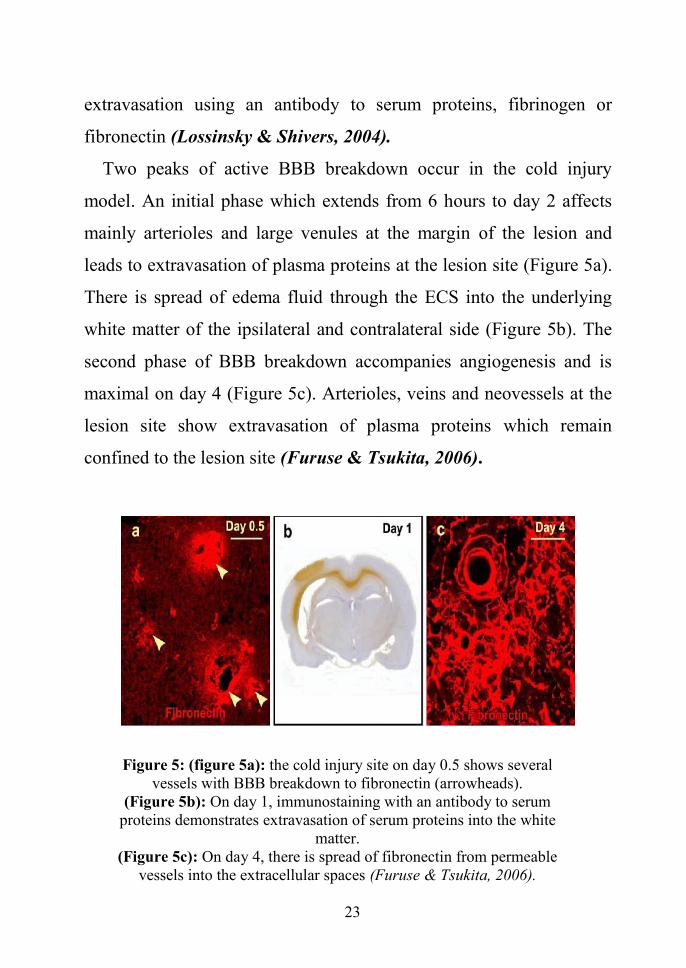
Two peaks of active BBB breakdown occur in the cold injury
model. An initial phase which extends from 6 hours to day 2 affects
mainly arterioles and large venules at the margin of the lesion and
leads to extravasation of plasma proteins at the lesion site (Figure 5a).
There is spread of edema fluid through the ECS into the underlying
white matter of the ipsilateral and contralateral side (Figure 5b). The
second phase of BBB breakdown accompanies angiogenesis and is
maximal on day 4 (Figure 5c). Arterioles, veins and neovessels at the
lesion site show extravasation of plasma proteins which remain
confined to the lesion site (Furuse & Tsukita, 2006).
Figure 5: (figure 5a): the cold injury site on day 0.5 shows severalvessels with BBB breakdown to fibronectin (arrowheads).
(Figure 5b): On day 1, immunostaining with an antibody to serumproteins demonstrates extravasation of serum proteins into the white
matter.(Figure 5c): On day 4, there is spread of fibronectin from permeable
vessels into the extracellular spaces (Furuse & Tsukita, 2006).

24
BBBBBB bbrreeaakkddoowwnn iinn vvaassooggeenniicc eeddeemmaa::Ultrastructural studies demonstrate an increase in the number of
endothelial caveolae only in the vessels with BBB breakdown to HRP
within minutes after the onset of pathological states such as
hypertension, spinal cord injury, seizures, experimental autoimmune
encephalomyelitis, excitotoxic brain damage, brain trauma, and BBB
breakdown- induced by bradykinin, histamine, and leukotriene C4
(Nag, 2002).
These findings suggest that enhanced caveolae (figure 6) are the
major route by which early passage of plasma proteins occurs in brain
diseases associated with vasogenic edema. Caveolae allow protein
passage across endothelium via fluid-phase transcytosis and
transendothelial channels. These enhanced caveolae represent the
response of viable endothelial cells to injury since both caveolar
changes and BBB breakdown are reversed 10 minutes after the onset
of acute hypertension induced by a single bolus of a pressor agent. No
alterations in tight junctions were noted in the studies mentioned
above (Parton & Simons, 2007).
Convincing demonstration of tight junction breakdown has only
been reported following the intracarotid administration of
hyperosmotic agents using the tracer lanthanum, which is a marker of
ionic permeability. Thus, junctional breakdown to proteins occurs late
in the course of brain injury probably during end-stage disease and
precedes endothelial cell breakdown. Research in the last decade has
led to the isolation of novel proteins in both caveolae and tight

25
junctions and studies are underway to define their role in brain injury
(Minshall & Malik, 2006).
Figure 6: a vein with BBB breakdown to fibronectin showsendothelial phosphorylated Cav-1 (PY14Cav-1) (Parton &
Simons, 2007).
CCaavveeoolliinn--11 ((CCaavv--11))::The specific marker and major component of caveolae is Cav-1, an
integral membrane protein, which belongs to a multigene family of
caveolin-related proteins that show similarities in structure but differ
in properties and distribution (Virgintino et al, 2002).
Of the two major isoforms of Cav-1 only the -isoform is
predominant in the brain. Cav-2 has a similar distribution as Cav-1
and non-neural endothelial cells express both Cav-1 and -2. Cav-1 has
been localized in human and murine cerebral endothelial cells. The
properties of Cav-1 are the subject of many reviews (Boyd et al,
2003).
Brain injury is associated with increased expression of Cav-1. Time
course studies in the rat cortical cold injury model demonstrate a

26
threefold increase in Cav-1 expression at the lesion site on day 0.5
post-injury. At the cellular level, a marked increase in endothelial
Cav-1 protein is present in vessels showing BBB breakdown to
fibronectin (Rizzo et al, 2003).
Further studies demonstrate that the endothelial Cav-1 in vessels
with BBB breakdown is phosphorylated. It is well established that
dilated vascular segments show enhanced permeability and leak
protein. Phosphorylation of Cav-1 is known to be an essential step for
formation of caveolae (figure 6). Thus, phosphorylation of Cav-1 is
essential for transcytosis of proteins across cerebral endothelium
leading to BBB breakdown and brain edema following brain injury
(Minshall et al, 2003).
In summary, caveolae and Cav-1 have a significant role in early
BBB breakdown; hence, they could be potential therapeutic targets in
the control of early brain edema (Williams & Lisanti, 2004).
TTiigghhtt jjuunnccttiioonn pprrootteeiinnss::Tight junctions are localized at cholesterol-enriched regions along
the plasma membrane associated with Cav-1. Tight junctions are
formed of three integral transmembrane proteins: occludin, the
claudin, and junctional adhesion molecule (JAM) families of proteins
(Forster, 2008).
The extracellular loops of these proteins originate from neighboring
cells to form the paracellular barrier of the tight junction, which

27
selectively excludes most blood borne substances from entering brain.
Several accessory cytoplasmic proteins have also been isolated which
are necessary for structural support at the tight junctions. They include
zonula occludens (ZO)-1 to -3, and cingulin (Nusrat et al, 2000).
Occludin, the first tight junction protein to be identified is an
approximately 60-kDa tetraspan membrane protein with two
extracellular loops. High expression of occludin in brain endothelial
cells as compared to nonneural endothelia provides an explanation for
the different properties of both these endothelia (Song et al, 2007).
Claudins are 18- to 27-kDa tetraspan proteins with two extracellular
loops, and they do not show any sequence similarity to occludin. The
claudin family consists of 24 members in humans and exhibits distinct
expression patterns in tissue. Claudins may be the major
transmembrane proteins of tight junctions as occludin knockout mice
are still capable of forming interendothelial tight junctions while
claudin knockout mice are nonviable (Nitta et al, 2003).
The JAMs belong to the immunoglobulin superfamily. JAM-A, the
first member of the family to be isolated has been implicated in a
variety of physiologic and pathologic processes involving cellular
adhesion including tight junction assembly and leukocyte
transmigration (Turksen & Troy, 2004).
Occludin, claudins-3, -5 and -12, JAM-A and ZO-1 proteins have
been localized in normal cerebral endothelium. Decreased expression
of the tight junction proteins in vessels with BBB breakdown in the
cold injury model follows a specific sequence with transient decreases

28
in expression of JAM-A on day 0.5 only, of claudin-5 on day 2 only
while occludin expression is attenuated from day 2 onwards and
persists up to day 6 (figure 7) (Plumb et al, 2002).
RReessoolluuttiioonn ooff eeddeemmaa::Much of our information about the resolution of vasogenic edema is
derived from the earlier studies of the cortical cold injury model.
During the period of BBB breakdown to plasma proteins there is
progressive increase in I 125-labeled albumin, paralleled by an increase
in water content (Van Itallie & Anderson, 2006).
Disappearance of serum proteins from the ECS coincides with the
return of water content to normal values. Resolution of edema occurs
immediately after closure of the BBB to proteins (figure 7). These
studies support previous observations that caveolae and Cav-1
changes precede significant tight junction changes during early BBB
breakdown (Xi et al, 2002).
Reduction of CSF pressure accelerates the clearance of edema fluid
into the ventricle. Recent evidence suggests that aquaporin 4 channels
located in the ependyma and astrocytic foot processes (digesting
serum proteins), have an important role in the clearance of the
interstitial water (Turksen & Troy, 2004).

29
(Figure 7) Expression of caveolins and junction proteins duringBBB breakdown:
Days post-lesion
0.5 2 4 6BBB break down
Caveolin-1 and PY14 Caveolin-1
Junctional adhesion molecule-A
Claudin-5
Occludin
Basal Increased Decreased
Figure 7: expression of caveolins and tight junction proteins during BBBbreakdown in the cold injury model. Increased expression of both caveolin-1 and
phosphorylated caveolin-1 (PY14 Caveolin-1) was observed. Decreasedexpression of junctional adhesion molecule-A was observed on day 0.5 only and
of claudin-5 on day 2 only, while decreased expression of occludin was present onday 2 and persisted throughout the period of observation (Vorbrodt, 2003).
Other mechanisms for clearance of edema fluid include passage of
extravasated proteins via the abluminal plasma membrane of
endothelial cells back into blood. Edema fluid can also pass across the
glia limitans externa into the CSF in the subarachnoid space and enter
the arachnoid granulations for clearance into the superior sagittal
venous sinus (Papadopoulos et al, 2004).

30
Quantitative studies of the relative involvement of the various
routes indicate that the clearance of edema by bulk flow into the CSF
is restricted to the early phase of edema. Clearance by brain
vasculature is small compared to that of CSF (Stummer, 2007).
22.. CCyyttoottooxxiicc EEddeemmaa::The most commonly encountered cytotoxic edema occurs in
cerebral ischemia, which may be focal due to vascular occlusion, or
global due to transient or permanent reduction in brain blood flow.
Other causes include traumatic brain injury, infections, and metabolic
disorders including kidney and liver failure (Vaquero & Butterworth,
2007).
Intoxications such as exposure to methionine sulfoxime, cuprizone,
and isoniazid are associated with cytotoxic edema and swelling of
astrocytes. Triethyl tin and hexachlorophene intoxications cause
accumulation of water in intramyelinic clefts and produce striking
white matter edema, while axonal swelling is a hallmark of exposure
to hydrogen cyanide. Since toxins are not involved in many cases of
cytotoxic edema some prefer the term ‘‘cellular edema’’ rather than
cytotoxic edema (Ranjan et al, 2005).
Experimental models used to study cytotoxic edema include the
focal and global ischemia models and the water intoxication model. In
cytotoxic edema astrocytes, neurons and dendrites undergo swelling
with a concomitant reduction of the brain ECS. This cellular swelling

31
does not constitute edema which implies a volumetric increase of
brain tissue (Lo et al, 2003).
Astrocytes are more prone to pathological swelling than neurons
because they are involved in clearance of potassium and glutamate,
which cause osmotic overload that in turn promotes water inflow.
Astrocytes outnumber neurons 20:1 in humans and astrocytes can
swell up to five times their normal size, therefore glial swelling is the
main finding in this type of edema (Rosenblum, 2007).
Cytotoxic edema is best studied in focal ischemia models where an
interruption of energy supply due to decrease in blood flow below a
threshold of 10 ml/100 g leads to failure of the ATP-dependent Na
pumps. This results in intracellular Na accumulation, with shift of
water from the extracellular to the intracellular compartment to
maintain osmotic equilibrium. This can occur within seconds. The Na
is accompanied by influx of Cl¯, H¯ and HCO3¯ ions (Unterberg et al,
2004).
These changes are reversible. However, ischemia of less than 6
minutes results in irreversible brain damage forming the ‘‘ischemic
core’’. This infracted tissue is surrounded by a region referred to as
the ‘‘penumbra’’ where the blood flow is greater than 20 ml/100 g per
min. Neurons and astrocytes in the penumbra undergo cytotoxic
edema. If hypoxic conditions persist, death of these neurons and glia
results in release of water into the ECS (Liang et al, 2007).
Damage to endothelium leads to vasogenic edema which can be
demonstrated by computed tomography in human brain by 24–48
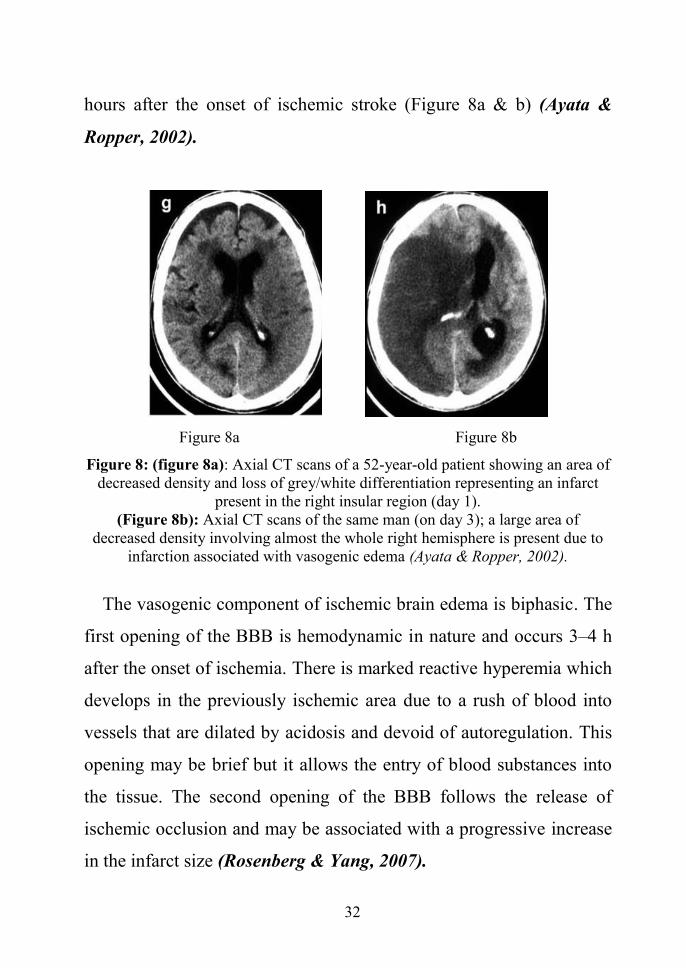
32
hours after the onset of ischemic stroke (Figure 8a & b) (Ayata &
Ropper, 2002).
Figure 8a Figure 8b
Figure 8: (figure 8a): Axial CT scans of a 52-year-old patient showing an area ofdecreased density and loss of grey/white differentiation representing an infarct
present in the right insular region (day 1).(Figure 8b): Axial CT scans of the same man (on day 3); a large area of
decreased density involving almost the whole right hemisphere is present due toinfarction associated with vasogenic edema (Ayata & Ropper, 2002).
The vasogenic component of ischemic brain edema is biphasic. The
first opening of the BBB is hemodynamic in nature and occurs 3–4 h
after the onset of ischemia. There is marked reactive hyperemia which
develops in the previously ischemic area due to a rush of blood into
vessels that are dilated by acidosis and devoid of autoregulation. This
opening may be brief but it allows the entry of blood substances into
the tissue. The second opening of the BBB follows the release of
ischemic occlusion and may be associated with a progressive increase
in the infarct size (Rosenberg & Yang, 2007).

33
Exudation of protein into the infarct area combined with an increase
in osmolarity due to breakdown of cell membranes results in an
increase in local tissue pressure. This leads to depression of regional
blood flow below the critical thresholds for viability in penumbral
regions and to further extension of the territory which undergoes
irreversible tissue damage. Elimination routes for excess water may be
the same as those in vasogenic edema (Kuroiwa et al, 2007).
33.. HHyyddrroocceepphhaalliicc oorr iinntteerrssttiittiiaall eeddeemmaa::This is best characterized in noncommunicating hydrocephalus
where there is obstruction to flow of CSF within the ventricular
system or communicating hydrocephalus where the obstruction is
distal to the ventricles and results in decreased absorption of CSF into
the subarachnoid space. In hydrocephalus, a rise in the intraventricular
pressure causes CSF to migrate through the ependyma into the
periventricular white matter, thus, increasing the extracellular fluid
volume (figure 9). The edema fluid consists of Na and water and has
the same composition as CSF (Johnston & Teo, 2000).
The white matter in the periventricular regions is spongy and on
microscopy there is widespread separation of glial cells and axons.
Astrocytic swelling is present followed by gradual atrophy and loss of
astrocytes (Abbott, 2004).
In chronic hydrocephalus, increase in the hydrostatic pressure
within the white matter results in destruction of myelin and axons and

34
this is associated with a microglial response. The end result is thinning
of the corpus callosum and compression of the periventricular white
matter. Other changes reported are destruction of the ependyma which
may be focal or widespread, distortion of cerebral vessels in the
periventricular region with collapse of capillaries and occasionally
there is injury of neurons in the adjacent cortex (Czosnyka et al,
2004).
Figure 9: An axial MR image of a 4 year old with hydrocephalus involving thelateral and third ventricles due to a posterior fossa tumor (not shown). The flair
sequence highlights the transependymal edema (Johnston & Teo, 2000).
In normal pressure hydrocephalus where normal intraventricular
pressure is recorded, ependymal damage with backflow of CSF is
postulated to produce edema. Functional manifestations in these cases
are minor unless changes are advanced when dementia and gait
disorder become prominent (Ball & Clarke, 2006).

35
44.. OOssmmoottiicc eeddeemmaa::
In this type of edema an osmotic gradient is present between plasma
and the extracellular fluid and the BBB is intact, otherwise an osmotic
gradient could not be maintained. Edema may occur with a number of
hypo-osmolar conditions including: improper administration of
intravenous fluids leading to acute dilutional hyponatremia,
inappropriate antidiuretic hormone secretion, excessive hemodialysis
of uremic patients and diabetic ketoacidosis (Kimelburg, 2004).
There is a decrease of serum osmolality due to reduction of serum
Na and when serum Na is less than 120 mmol/L, water enters the
brain and distributes evenly within the ECSs of the grey and white
matter. Astrocytic swelling may be present. The spread of edema
occurs by bulk flow along the normal interstitial fluid pathways.
Following a 10% or greater reduction of plasma osmolarity, there is a
pronounced increase in interstitial fluid volume flow, and extracellular
markers are cleared into the CSF at an increased rate (Katayama &
Katayama, 2003).
The formation of osmotic edema can lead to a significant increase
in the rate of CSF formation without any contribution of the choroid
plexuses. Since osmotic edema is vented rapidly, the increase in brain
volume tends to be modest. Experimentally, this type of edema is
induced following intraperitoneal infusion of distilled water. The BBB
is not affected and cytotoxic mechanisms are not involved. Osmotic
brain edema can also occur when the plasma osmolarity is normal but

36
tissue osmolarity is high in the core of the lesion as in brain
hemorrhage, infarcts or contusions (Nag, 2003) a.

37
CChhaapptteerr ((22)):: CChheemmiiccaallMMeeddiiaattoorrss IInnvvoollvveedd iinn
tthhee PPaatthhooggeenneessiiss ooffBBrraaiinn EEddeemmaa

38
CChheemmiiccaall MMeeddiiaattoorrss IInnvvoollvveeddiinn TThhee PPaatthhooggeenneessiiss OOff BBrraaiinn
EEddeemmaa
IInnttrroodduuccttiioonn::Brain edema continues to be a major cause of mortality after
diverse types of brain pathologies such as major cerebral infarcts,
hemorrhages, trauma, infections and tumors. The classification of
edema into vasogenic, cytotoxic, hydrocephalic and osmotic has
stood the test of time although it is recognized that in most clinical
situations there is a combination of different types of edema during
the course of the disease (Schilling & Wahl, 1999).
It is well established that vaso-active agents can increase BBB
permeability and promote vasogenic brain edema (Table 1)
(Yamamoto et al, 2001).
Basic information about the types of edema is provided for better
understanding of the expression pattern of some of the newer
molecules implicated in the pathogenesis of brain edema. These
molecules include the aquaporins (AQP), matrix metalloproteinases
(MMPs) and growth factors such as vascular endothelial growth
factors (VEGF) A and B and the angiopoietins. The potential of
these agents in the treatment of edema is the subject of many
reviews (Dolman et al, 2005).

39
Table 1: Vasoactive agents that increase blood–brain barrierpermeability:
Arachidonic acid
Bradykinin
Complement-derived polypeptide C3a-desArg Glutamate
Histamine
Interleukins: IL-1a, IL-1b, IL-2 Leukotrienes
Macrophage inflammatory proteins MIP-1, MIP-2
Nitric oxide
Oxygen-derived free radicals
Phospholipase A2, platelet activating factor,
prostaglandins
Purine nucleotides: ATP, ADP, AMP
Thrombin
Serotonin
(Yamamoto et al, 2001).

40
AAqquuaappoorriinnss aanndd bbrraaiinn eeddeemmaa::Aquaporins (AQP) are a growing family of molecular water-
channel proteins that assemble in membranes as tetramers. Each
monomer is 30 kDa and has six membrane-spanning domains
surrounding a water pore that allows bidirectional passage of water
(Badaut et al, 2001).
At least 13 AQPs have been found in mammals and more than
300 in lower organisms. Expression of AQP 1, AQP3, AQP4,
AQP5, AQP8 and AQP9 has been reported in rodent brain. Only
AQP1 and AQP4 are reported to have a role in human brain edema
and will be discussed (Oshio et al, 2005)..
AAqquuaappoorriinn11 ((AAQQPP11))::Localization of AQP1 in the apical membrane of the choroid
plexus epithelium suggests that it may have a role in CSF secretion.
This could be supported by the finding that AQP1 is upregulated in
choroid plexus tumors, which are associated with increased CSF
production. AQP1 is also expressed in tumor cells and peritumoral
astrocytes in high grade gliomas (Longatti et al, 2006).
Although AQP1 is present in endothelia of non-neural vessels, it
is not observed in normal brain capillary endothelial cells. Brain
capillary endothelial cells cultured in the absence of astrocytes and
those in brain tumors that are not surrounded by astrocytic end-feet
do express AQP1, suggesting that astrocytic end-feet may signal

41
adjacent endothelial cells to switch off AQP1 expression (Verkman,
2005).
AQP1-null mice show a 25% reduction in the rate of CSF
secretion, reduced osmotic permeability of the choroid plexus
epithelium and decreased ICP. These findings support the role of
AQP1 in facilitating CSF secretion into the cerebral ventricles by the
choroid plexuses and suggest that AQP1 inhibitors may be useful in
the treatment of hydrocephalus and benign intracranial hypertension,
both of which are associated with increased CSF formation or
accumulation (Tait et al, 2008).
AAqquuaappoorriinn44 ((AAQQPP44))::AQP4, the principal AQP in mammalian brain, is expressed in
glia at the borders between major water compartments and the brain
parenchyma (figure 10). AQP4 is expressed in the basolateral
membrane of the ependymal cells lining the cerebral ventricles and
subependymal astrocytes which are located at the ventricular CSF
fluid– brain interface (Furman et al, 2003).
Expression of AQP4 in astrocytic foot processes brings it in close
proximity to intracerebral vessels, and thus, the blood–brain
interface. Water molecules moving from the blood pass through the
luminal endothelial membranes by diffusion and across the
astrocytic foot processes through the AQP4 channels. AQP4 is also
expressed in the dense astrocytic processes that form the glia
limitans which is at the subarachnoid– CSF fluid interface (Rash et
al, 2004).

42
Figure 10: Pathways for water entry into and exit from brain are shown. TheAQP4- dependent water movement across the blood–brain barrier, through
ependymal and arachnoid barriers is shown (Furman et al, 2003).
Two AQP4 splice variants are expressed in brain, termed M1 and
M23, which can form homo- and hetero-tetramers, respectively. The
location of AQP 4 at the brain–fluid interfaces suggests that it is
important for brain water balance and may play a key role in brain
edema. AQP4 overexpression in human astrocytomas correlates with
the presence of brain edema on magnetic resonance imaging
(Silberstein et al, 2004).
However, decrease in AQP4 protein expression is associated with
early stages of edema in rodents subjected to permanent focal brain
ischemia and hypoxia-ischemia. In traumatic brain injury AQP4
mRNA is decreased in the area of edema adjacent to a cortical

43
contusion. AQP4-null mice provide strong evidence for AQP4
involvement in cerebral water balance in the various types of edema
(Warth et al, 2007).
Vasogenic edema:
Data derived from AQP4-null mice suggest that AQP4 is involved
in the clearance of extracellular fluid from the brain parenchyma in
vasogenic edema (Meng et al, 2004).
A number of models in which vasogenic edema is the
predominant form of edema, including the cortical cold injury,
tumor implantation and brain abscess models, demonstrate that the
AQP4-null mice have a significantly greater increase in brain water
content and ICP than the wild-type mice suggesting that brain water
elimination is defective after AQP4 deletion (Papadopoulos &
Verkman, 2007).
Melanoma cells implanted into the striatum of wild-type and
AQP4-null mice produce peritumoral edema and comparable sized
tumors in both groups after a week. However, the AQP4- null mice
have a higher ICP and water content. This suggests that in vasogenic
edema, excess water enters the brain ECS independently of AQP4,
but exits the brain primarily through AQP4 channels into the CSF
and via astrocytic foot processes into blood (Papadopoulos &
Verkman, 2007).

44
Cytotoxic edema:
Swelling of astrocytic foot processes is a major finding in
cytotoxic edema and since AQP4 channels are located in the
astrocytic foot processes, it was hypothesized that they may have a
role in formation of cell swelling. This was found to be the case
since water intoxicated AQP4-null mice show a significant reduction
in astrocytic foot process swelling, a decrease in brain water content
and a profound improvement in their survival (Saadoun et al, 2002).
Since water intoxication is of limited clinical significance, AQP4-
null mice were subjected to ischemic stroke and bacterial meningitis.
In both models AQP4-null mice showed decreased cerebral edema
and improved outcome and survival. These studies imply that AQP4
has a significant role in water transport and development of cellular
edema following cerebral ischemia (Zador et al, 2007).
Hydrocephalic edema:
Obstructive hydrocephalus produced by injecting kaolin in the
cistern magna of AQP4-null mice show accelerated ventricular
enlargement compared with wild-type mice.
Reduced water permeability of the ependymal layer,
subependymal astrocytes, astrocytic foot processes and glia limitans
produced by AQP4 deletion reduces the elimination rate of CSF
across these routes. Thus, AQP4 induction could be evaluated as a
nonsurgical treatment for hydrocephalus (Bloch et al, 2006).
In summary, AQP4 has opposing roles in the pathogenesis of
vasogenic and hydrocephalic edema when compared to cytotoxic

45
edema. Therefore, AQP4 activators or upregulators have the
potential to facilitate the clearance of vasogenic and hydrocephalic
edema, while AQP4 inhibitors have the potential to protect the brain
in cytotoxic edema. This is an area of ongoing research since none
of the AQP4 activators or inhibitors investigated thus far are suitable
for development for clinical use (Sun et al, 2003).
MMaattrriixx mmeettaalllloopprrootteeiinnaasseess ((MMMMPPss))::The MMPs are zinc- and calcium-dependent endopeptidases
which are known to cleave most components of the extracellular
matrix including fibronectin, proteoglycans and type IV collagen.
Activation of MMPs involves cleavage of the secreted proenzyme,
while inhibition involves a group of four endogenous tissue
inhibitors of metalloproteinases (TIMPs). The balance between
production, activation, and inhibition prevents excessive proteolysis
or inhibition (Asahi et al, 2001).
Type IV collagenases are members of the larger MMP gene
family of proteolytic enzymes that have the ability of destroying the
basal lamina of vessels and thereby play a role in the development of
many pathological processes including vasogenic edema in multiple
sclerosis and bacterial meningitis and ischemic stroke (Chang et al,
2003).
MMPs are found in all of the elements of the neurovascular unit,
but different MMPs have a predilection for certain cell types.

46
Endothelial cells express mainly MMP-9; pericytes express MMP-3
and -9, while astrocytic end-feet express MMP-2 and its activator,
membrane-type MMP (MT1-MMP) (Rosenberg, 2002).
Normally MMP-2 is expressed at low levels but is markedly
upregulated in many brain diseases. In human ischemic stroke,
active MMP-2 is increased on days 2–5 compared with active MMP-
9 which is elevated up to months after the ischemic episode.
Molecular studies in experimental permanent and temporary
ischemia have shown that MMPs contribute to disruption of the
BBB leading to vasogenic cerebral edema (Yang et al, 2007).
Middle cerebral artery occlusion in rats for 90 min with
reperfusion causes biphasic opening of the BBB in the piriform
cortex with a transient, reversible opening at 3 h which correlates
with a transient increase in expression of MMP-2. This is associated
with a decrease in claudin-5 and occludin expression in cerebral
vessels. By 24 h the tight junction proteins are no longer observed in
lesion vessels, an alteration that is reversed by treatment with the
MMP inhibitor, BB-1101. The later BBB opening between 24 and
48 h is associated with a marked increase of MMP-9 which is
released in the extracellular matrix where it degrades multiple
proteins, and produces more extensive blood vessel damage
(Rosenberg & Yang, 2007).
The role of MMPs in BBB breakdown is further supported by the
observation that treatment with MMP inhibitors or MMP
neutralizing antibodies decreases infarct size and prevents BBB

47
breakdown after focal ischemic stroke. The MMP inhibitors used so
far restore early integrity of the BBB in rodent ischemia models.
Since these inhibitors block MMPs involved in angiogenesis and
neurogenesis as well, they slow recovery. Therefore, the challenge is
to identify agents that will protect the BBB and block vasogenic
edema without interfering with recovery (Candelario-Jalil et al,
2008).
GGrroowwtthh ffaaccttoorrss aanndd bbrraaiinn eeddeemmaa:: VVaassccuullaarr eennddootthheelliiaall ggrroowwtthh ffaaccttoorr--AA ((VVEEGGFF--AA))::
VEGF, the first member of the six member VEGF family to be
discovered is now designated as VEGF-A. Initial reports described
the potent hyperpermeability effect of VEGF-A on the
microvasculature of tumors hence its designation ‘vascular
permeability factor’. VEGF-A has a significant role in vascular
permeability and angiogenesis during embryonic vasculogenesis and
in physiological and pathological angiogenesis (Adams & Alitalo,
2007).
There is agreement that vascular endothelial growth factor
receptor- 2 (VEGFR-2), which is present on endothelial cells, is the
major mediator of the mitogenic, angiogenic and permeability-
enhancing effects of VEGF-A.
The permeability inducing properties of VEGF-A have also been
demonstrated in the brain; Intracortical injections of VEGF-A

48
produces BBB breakdown at the injection site. Normal adult cortex
shows basal expression of VEGF-A mRNA and protein, while high
expression of VEGF-A mRNA and protein is present in normal
choroid plexus epithelial cells and ependymal cells (Ferrara et al,
2003).
Although several studies reported VEGF-A gene up regulation in
cerebral ischemia models, increased expression was related to
angiogenesis and not to BBB breakdown. In non-neural vessels,
VEGF-A is reported to cause vascular hyperpermeability by opening
of interendothelial junctions and induction of fenestrae in
endothelium (Marti et al, 2000).
A single ultrastructural study reported interendothelial gaps and
segmental fenestrae-like narrowings in brain vessels permeable to
endogenous albumin following a single intracortical injection of
VEGF-A. VEGF-A can also increase permeability by inducing
changes in expression of tight junction proteins. Reduced occludin
expression occurs in retinal and brain endothelial cells exposed to
VEGF-A (Machein & Plate, 2000).
VVaassccuullaarr eennddootthheelliiaall ggrroowwtthh ffaaccttoorr--BB ((VVEEGGFF--BB))::This member of the VEGF family displays strong homology to
VEGF-A. Mice embryos (day 14) and adults show high expression
of VEGF-B mRNA in most organs with very high levels in the heart
and the nervous system. Moderate down regulation of VEGF-B
occurs prior to birth and VEGF-B is the only member of the VEGF

49
family that is expressed at detectable levels in the adult CNS (Nag et
al, 2005).
Constitutive expression of VEGF-B protein is present in the
endothelium of all cerebral vessels including those of the choroid
plexuses. Thus, VEGF-B has a role in maintenance of the BBB in
steady states and VEGF-B may be protective against BBB
breakdown and edema formation (Nag et al, 2002).
AAnnggiiooppooiieettiinn ((AAnngg)) ffaammiillyy::Four members of this family have been isolated thus far and
designated Ang1–4, Ang1 and 2 are best characterized. Endothelial
Ang1 is expressed widely in normal adult tissues, consistent with it
playing a constitutive stabilization role by maintaining normal
endothelial cell to cell and cell to matrix interactions. Studies of the
rodent brain show constitutive expression of Ang1 protein in
endothelium of all cerebral cortical vessels and only weak
expression of Ang2 (Raab & Plate, 2007).
Functional studies indicate that Ang1 and Ang2 have reciprocal
effects in many systems. Ang1 has an antiapoptotic effect on
endothelial cells, while Ang2 is reported to promote apoptosis.
Presence of Ang1 is associated with smaller gaps in the endothelium
of postcapillary venules during inflammation. Ang1 is reported to
stabilize interendothelial junctions. This demonstrates that Ang1 is a
potent antileakage factor (Otrock et al, 2007).

50
Time course of growth factor expression post-
injury:The cold injury model was used to study the temporal alterations
in expression of growth factors and their relation to BBB breakdown
(figure 11). In the early phase post-injury up to day 2, there is
increased expression of VEGF-A protein, VEGFR-2 protein and a
sevenfold increase in Ang2 mRNA. During this period, vessels with
BBB breakdown show endothelial immunoreactivity for VEGF-A
and Ang2 but not for VEGF-B or Ang1 (Reiss, 2005).
On days 4 and 6 post-injury, there is progressive increase in Ang1
and VEGF-B mRNA and protein and decrease in Ang2 and VEGF-
A mRNA coinciding with maturation of neovessels and restoration
of the BBB (Roviezzo et al, 2005).
Increased expression of growth factors has been reported in
gliomas. VEGF-A is overexpressed up to 50-fold in the peri-necrotic
tumor cells in glioblastomas, Increased expression of the
angiopoietins has also been reported in glioblastomas. High
expression of Ang1 has been reported in areas of high vascular
density in all stages of glioblastoma progression while high
expression of Ang2 has been reported in endothelial cells in
glioblastomas. In these studies a strong association is made between
these growth factors and tumor angiogenesis (Roy et al, 2006).

51
Figure 11: Expression of growth factors during BBBbreakdown:
Days post-lesion0.5 2 4 6
BBB breakdown
VEGF-A
VEGF-B
VEGFR-2
Ang1
Ang2
Protein ExpressionBasal Increased Decreased
Figure 11: Temporal expression of growth factor proteins and their receptors isshown during the period of BBB breakdown in the cold injury model. Protein
expression was determined by immunohistochemistry and/orimmunofluorescence (Reiss, 2005).
There is the potential of using growth factors to treat early and
massive edema associated with large hemispheric lesions which are
lethal due to the effects of early edema. Potential candidates include
inhibitors of VEGF-A or administration of Ang1 or VEGF-B (Zadeh
& Guha, 2003).

52
Inhibitors of VEGF-A or recombinant Ang1 have been tried in
rodent models of ischemia. Pretreatment of rodents with VEGF-A
receptor protein, which inactivates endogenous VEGF-A or
recombinant Ang1 attenuates BBB breakdown and edema associated
with cerebral infarcts (Zhang, 2002).
The long-term effects of administering these agents on
angiogenesis and repair were not studied in these models. This must
be assessed before these agents can be used for the treatment of
brain edema (Yla-Herttuala et al, 2007).

53
CChhaapptteerr ((33)):: DDiiaaggnnoossiinnggcceerreebbrraall eeddeemmaa

54
DDiiaaggnnoossiinngg cceerreebbrraall eeddeemmaa
Introduction:Brain edema is a life-threatening complication following several
kinds of neurological and non-neurological conditions. Neurological
conditions include: ischemic stroke and intracerebral hemorrhage,
brain tumors meningitis, encephalitis of all etiologies and other brain
traumatic and metabolic insults (Rosenberg, 1999).
Non-neurological conditions include: diabetic ketoacidosis, lactic
acidotic coma, hypertensive encephalopathy, fulminant viral hepatitis,
hepatic encephalopathy, Reye’s syndrome systemic poisoning (carbon
monoxide and lead), hyponatraemia, opioid drug abuse and
dependence, bites of certain reptiles and marine animals, and high
altitude cerebral edema (Glasr et al, 2001).
Most cases of brain injury that result in elevated intracranial
pressure (ICP) begin as focal cerebral edema. Consistent with the
Monroe–Kellie doctrine as it applies to intracranial vault physiology,
the consequences of cerebral edema can be lethal and include cerebral
ischemia from compromised cerebral blood flow and intracranial
compartmental shifts due to ICP gradients, resulting in compression of
vital brain structures (herniation syndromes; Table 2) (Harukuni et al,
2002).
Prompt recognition of these clinical syndromes and institution of
targeted therapies constitutes the basis of cerebral resuscitation. It is
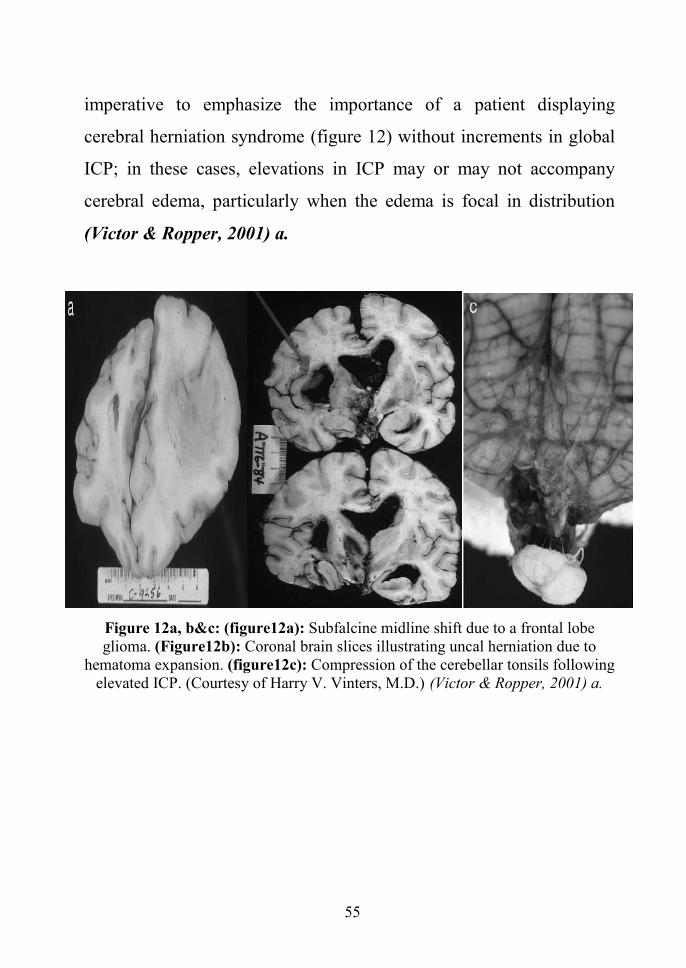
55
imperative to emphasize the importance of a patient displaying
cerebral herniation syndrome (figure 12) without increments in global
ICP; in these cases, elevations in ICP may or may not accompany
cerebral edema, particularly when the edema is focal in distribution
(Victor & Ropper, 2001) a.
Figure 12a, b&c: (figure12a): Subfalcine midline shift due to a frontal lobeglioma. (Figure12b): Coronal brain slices illustrating uncal herniation due to
hematoma expansion. (figure12c): Compression of the cerebellar tonsils followingelevated ICP. (Courtesy of Harry V. Vinters, M.D.) (Victor & Ropper, 2001) a.

56
Table 2: Summary of the clinical subtypes of herniationsyndromes:
HerniationSyndrome
Clinical Manifestations
subfalcianor cingulate
usually diagnosed using neuroimaging; cingulategyrus herniates under the falx cerebrii (usuallyanteriorly); may cause compression of ipsilateralanterior cerebral artery, resulting in contralaterallower extremity paresis
centraltentorial
downward displacement of one or both cerebralhemispheres, resulting in compression ofdiencephalon and midbrain through tentorial notch;typically due to centrally located masses; impairedconsciousness and eye movements; elevated ICP;bilateral flexor or extensor posturing
lateraltranstentorial(uncal)
most commonly observed clinically; usually due tolaterally located (hemispheric) masses (tumors andhematomas); herniation of the mesial temporal lobe,uncus, and hippocampal gyrus through the tentorialincisura; compression of oculomotor nerve,midbrain, and posterior cerebral artery; depressedlevel of consciousness; ipsilateral papillary dilationand contralateral hemiparesis; decerebrate posturing;central neurogenic hyperventilation; elevated ICP
tonsillarherniation of cerebellar tonsils through foramenmagnum, leading to medullary compression; mostfrequently due to masses in the posterior fossa;precipitous changes in blood pressure and heart rate,small pupils, ataxic breathing, disturbance ofconjugate gaze and quadriparesis
external due to penetrating injuries to the skull, loss of CSFand brain tissue; ICP may not be elevated due todural opening
(Harukuni et al, 2002)

57
CClliinniiccaall FFeeaattuurreess::A high index of suspicion is very important. The features of cerebral
edema add on to and often complicate the clinical features of the
primary underlying condition. Cerebral edema alone will not produce
obvious clinical neurological abnormalities until elevation of ICP
occurs. Symptoms of elevation of intracranial pressure are headache,
vomiting, papilledema, abnormal eye movements, neck pain or
stiffness, cognitive decline, seizures, hemiparesis, dysphasia, other
focal neurologic deficits, and depression of consciousness (Rosenberg,
2000).
The headache associated with an increased intracranial pressure,
especially when resulting from mass lesions, is mainly due to
compression or distortion of the dura mater and of the pain-sensitive
intracranial blood vessels. It is often paroxysmal, at first worse on
waking or after recumbency, throbbing in character, corresponding
with the arterial pressure wave. Exertion, coughing, sneezing,
vomiting, straining, or sudden changes in posture accentuate it. Such
headache is often frontal or occipital or both (Pollay, 1996).
The vomiting that accompanies increased intracranial pressure often
occurs in the mornings when the headache is at its height, it is more
common in children than in adults. It is generally attributed to
compression or ischemia of the vomiting center in the medulla
oblongata (Hemphil et al, 2001).

58
Similarly, the bradycardia, which is also common, results from
dysfunction in the cardiac centre but, in some patients with
infratentorial lesions, tachycardia eventually develops. Papilledema
develops more rapidly with mass lesions in the posterior fossa because
of their especial tendency to cause sudden obstructive hydrocephalus.
Obstruction of CSF flow in the subarachnoid space and impaired
absorption both appear to be important factors in patients with tumors
(Schilling, 1999).
Breathing control is often impaired. Slow and deep respiratory
movements often accompany a sudden rise in intracranial pressure
sufficient to impair consciousness. Later, breathing may become
irregular, Cheyne–Stokes respiration, and periods of apnea then
alternate with phases during which breathing waxes and wanes in
amplitude. Central neurogenic hyperventilation, or so-called ataxic
breathing, is less common effects of brainstem compression or
distortion but, in terminal coma, breathing is often rapid or shallow.
These abnormalities of respiratory rate and rhythm may be due to
compression or distortion of the brainstem (Victor & Ropper, 2001) b.

59
IInnvveessttiiggaattiioonnss::A. Computed Tomography (CT):
CT technology may noninvasively illustrate the volumetric changes
and alterations in parenchymal density resulting from cerebral edema.
Expansion of brain tissue due to most forms of edema may be detected
on CT, although diffuse processes like fulminant hepatic failure may
be more difficult to discern. Diffuse swelling may be recognized by a
decrease in ventricular size with compression or obliteration of the
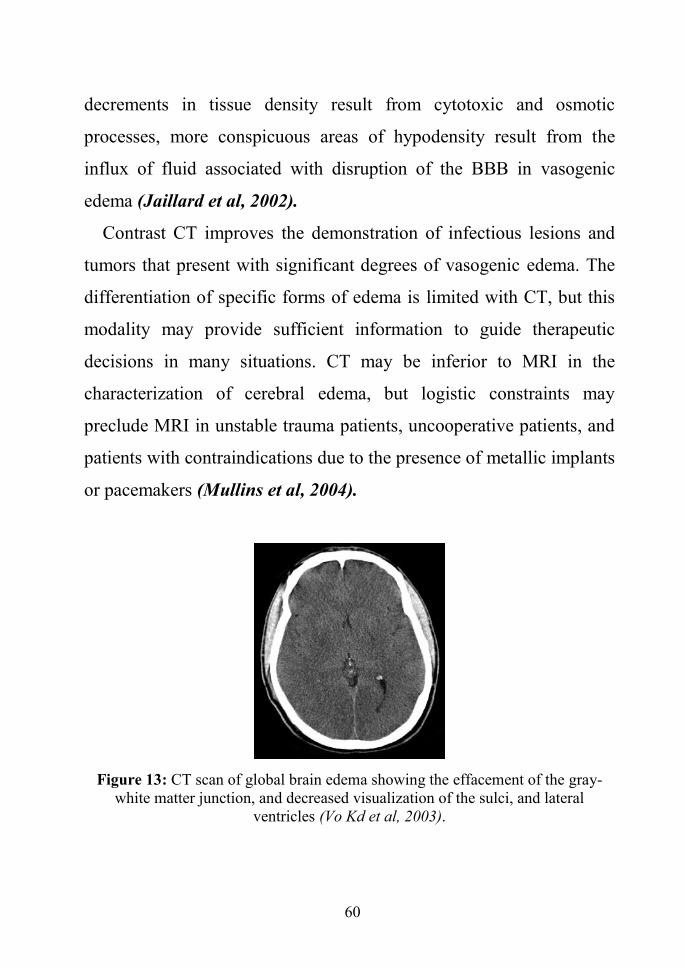
cisterns and cerebral sulci (figure 13) (Vo Kd et al, 2003).
Cellular swelling associated with cytotoxic and ischemic edema can
manifest as subtle enlargement of tissue with obscuration of normal
anatomic features, such as the differentiation between gray matter and
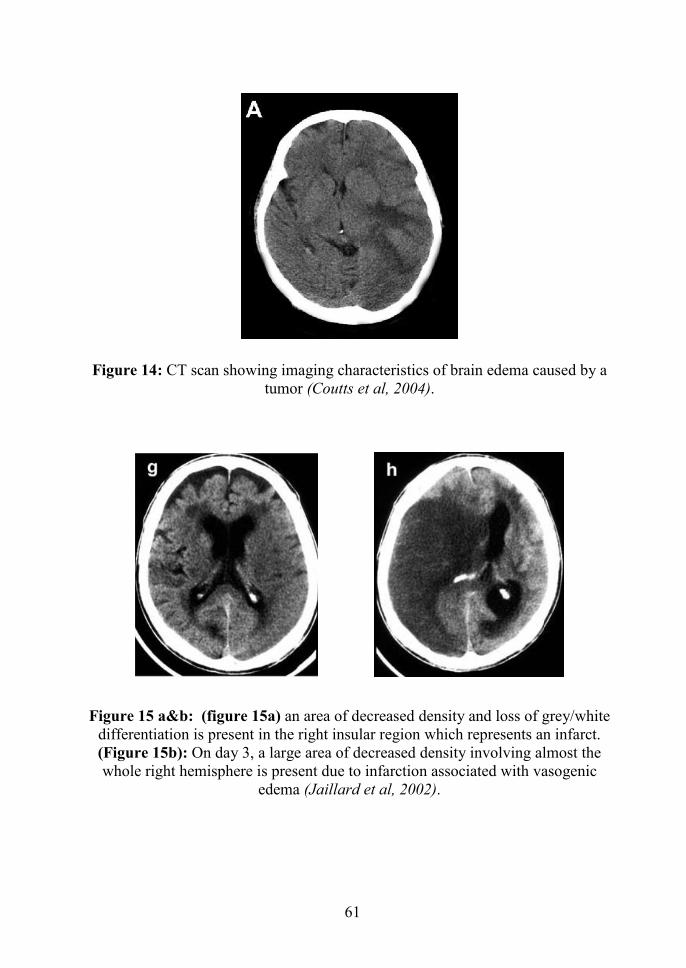
white matter tracts (figure 14). Vasogenic edema may also cause tissue
expansion, although the associated density changes may be more
prominent (Coutts et al, 2004).
In contrast, hydrocephalic edema may be suspected in cases in
which ventricular expansion has occurred. Extensive volumetric
changes and the associated pressure differentials resulting in herniation
may be noted on CT as shifts in the location of various anatomic
landmarks (Rother, 2001).
The increased water content associated with edema causes the
density of brain parenchyma to decrease on CT (figure 15). The
attenuation effects of other tissue contents complicate precise
correlation of water content with density on CT. Although slight
60
decrements in tissue density result from cytotoxic and osmotic
processes, more conspicuous areas of hypodensity result from the
influx of fluid associated with disruption of the BBB in vasogenic
edema (Jaillard et al, 2002).
Contrast CT improves the demonstration of infectious lesions and
tumors that present with significant degrees of vasogenic edema. The
differentiation of specific forms of edema is limited with CT, but this
modality may provide sufficient information to guide therapeutic
decisions in many situations. CT may be inferior to MRI in the
characterization of cerebral edema, but logistic constraints may
preclude MRI in unstable trauma patients, uncooperative patients, and
patients with contraindications due to the presence of metallic implants
or pacemakers (Mullins et al, 2004).
Figure 13: CT scan of global brain edema showing the effacement of the gray-white matter junction, and decreased visualization of the sulci, and lateral
ventricles (Vo Kd et al, 2003).
61
Figure 14: CT scan showing imaging characteristics of brain edema caused by atumor (Coutts et al, 2004).
Figure 15 a&b: (figure 15a) an area of decreased density and loss of grey/whitedifferentiation is present in the right insular region which represents an infarct.(Figure 15b): On day 3, a large area of decreased density involving almost thewhole right hemisphere is present due to infarction associated with vasogenic
edema (Jaillard et al, 2002).

62
B. Magnetic Resonance Imaging (MRI):Volumetric enlargement of brain tissue due to edema is readily
apparent on MRI and the use of gadolinium, an MRI contrast agent,
enhances regions of altered BBB. Differences in water content may be
detected on MRI by variations in the magnetic field generated
primarily by hydrogen ions. T2-weighted sequences and fluid-
attenuated inversion recovery (FLAIR) images reveal hyperintensity in
regions of increased water content (figure 16). FLAIR images
eliminate the bright signal from CSF spaces and are therefore helpful
in characterizing periventricular findings such as hydrocephalic edema
(figure 17) (Cosnard et al, 2000).
These conventional MRI sequences are more sensitive in the
detection of lesions corresponding to hypodensities on CT. MRI is also
superior in the characterization of structures in the posterior fossa
(figure 18). Recent advances in MRI technology make it possible to
specifically discern the type of edema based on signal characteristics
of a sampled tissue volume (Weber et al, 2000).
This discriminatory capability resulted from the development of
diffusion imaging techniques. The use of strong magnetic field
gradients increases the sensitivity of the MR signal to the random,
translational motion of water protons within a given volume element
(Scarabino et al, 2004).

63
Figure 16: Intracranial hemorrhage depicted by MRI. T2-weighted sequenceshowing hyperintensity associated with vasogenic edema in the right frontal lobe
(Cosnard et al, 2000).
Figure 17: Periventricular FLAIR hyperintensity due to hydrocephalic edema(Cosnard et al, 2000).
Figure 18 a&b: Central pontine myelinolysis illustrated as (a) T2- weightedhyperintensity and (b) T1-weighted hypointensity in the pons (Weber et al, 2000).

64
Cytotoxic edema and cellular swelling produce a net decrease in the
diffusion of water molecules due to the restriction of movement,
imposed by intracellular structures such as membranes and
macromolecules, and diminished diffusion within the extracellular
space due to shrinkage and tortuosity (figure 19). In contrast, the
accumulation of water within the extracellular space as the result of
vasogenic edema allows for increased diffusion (Scott et al, 2006).
Diffusion-weighted imaging (DWI) sequences yield maps of the
brain, with regions of restricted diffusion appearing bright or
hyperintense. The cytotoxic component of ischemic edema has been
demonstrated on DWI within minutes of ischemia onset (Simon et al,
2004).
Apparent diffusion coefficient (ADC) maps may be generated from a
series of DWI images acquired with varying magnetic field gradients.
ADC elevations, resulting from vasogenic edema, appear hyperintense
on ADC maps, whereas decreases in ADC due to cytotoxic edema
appear hypointense (figure 20). These maps may be sampled to
measure the ADC of a given voxel for multiple purposes, such as
differentiating tumor from tumor associated edema (Yamasaki et al,
2005).
The development of perfusion-weighted imaging (PWI) with MR
technology provided parametric maps of several hemodynamic
variables, including cerebral blood volume. Elevations in cerebral
blood volume associated with cerebral edema are detectable by this
technique. Simultaneous acquisition of multiple MRI sequences

65
enables the clinician to distinguish various forms of cerebral edema.
T2-weighted sequences and FLAIR images permit sensitive detection
of local increases in water content (Bastin et al, 2002).
Figure 19 a, b&c: the cytotoxic component of acute cerebral ischemia isdemonstrated by ADC hypointensity (a). The ischemic region appears
hyperintense on DWI (b), whereas T2 weighted sequences may be unrevealing atthis early stage (c) (Scott et al, 2006).
Figure 20 a, b&c: MRI of status epilepticus reveals evidence of cytotoxic edemawithin cortical structures, illustrated by (a) T2-weighted and (b) DWI
hyperintensity, with (c) mild hypointensity on ADC maps(Yamasaki et al, 2005)
66
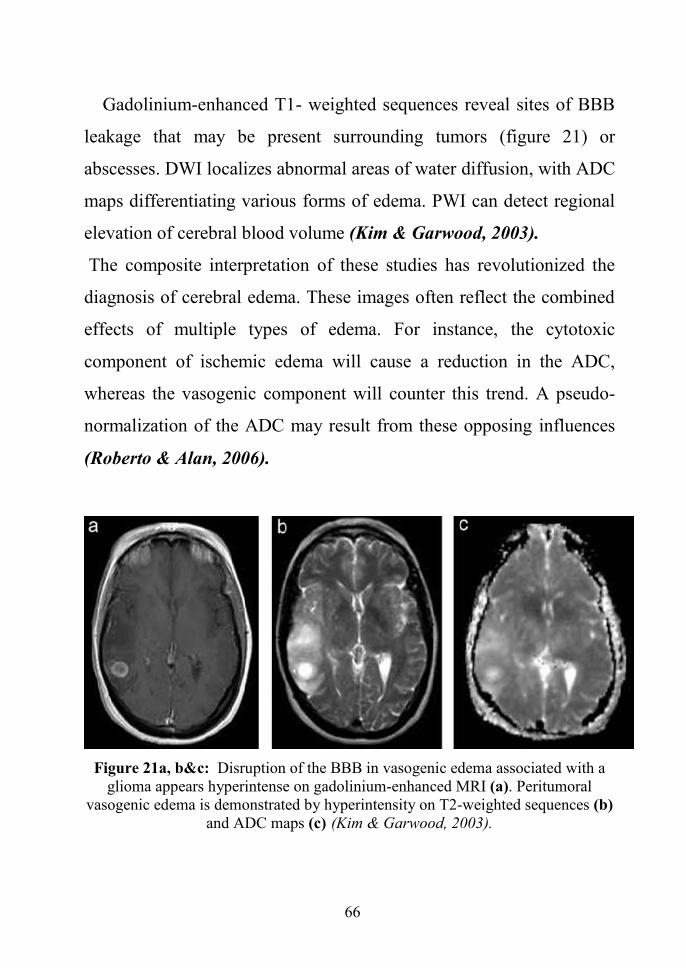
Gadolinium-enhanced T1- weighted sequences reveal sites of BBB
leakage that may be present surrounding tumors (figure 21) or
abscesses. DWI localizes abnormal areas of water diffusion, with ADC
maps differentiating various forms of edema. PWI can detect regional
elevation of cerebral blood volume (Kim & Garwood, 2003).
The composite interpretation of these studies has revolutionized the
diagnosis of cerebral edema. These images often reflect the combined
effects of multiple types of edema. For instance, the cytotoxic
component of ischemic edema will cause a reduction in the ADC,
whereas the vasogenic component will counter this trend. A pseudo-
normalization of the ADC may result from these opposing influences
(Roberto & Alan, 2006).
Figure 21a, b&c: Disruption of the BBB in vasogenic edema associated with aglioma appears hyperintense on gadolinium-enhanced MRI (a). Peritumoral
vasogenic edema is demonstrated by hyperintensity on T2-weighted sequences (b)and ADC maps (c) (Kim & Garwood, 2003).

67
Serial imaging with this noninvasive modality also allows for the
temporal characterization of edema evolution. The relative
contributions of cytotoxic and vasogenic edema with respect to the
ADC during acute ischemic stroke and TBI have been investigated in
this manner. The main limitations of this technology logistically relate
to cost, availability, contraindications, and its restricted use in
critically ill individuals (Doerfler et al, 2002).
C. Intracranial pressure monitoring:ICP monitoring is an important tool to monitor cases where cerebral
edema is present or anticipated and is routinely done in all neurology
and neurosurgery ICUs. Unfortunately, the direct measurements of
ICP and aggressive measures to counteract high pressures have not
yielded uniformly beneficial results, and after two decades of
popularity the routine use of ICP monitoring remains controversial
(Bullock et al, 1996).
The problem may be partly a matter of the timing of monitoring and
the proper selection of patients for aggressive treatment of raised ICP.
Only if the ICP measurements are to be used as a guide to medical
therapy and the timing of surgical decompression is the insertion of a
monitor justified (Ayata & Ropper, 2002).
Monitoring of ICP is helpful in patients in whom neurological status
is difficult to ascertain serially, particularly in the setting of
pharmacological sedation and neuromuscular paralysis. The Brain
Trauma Foundation guidelines recommend ICP monitoring in patients

68
with TBI, a GCS score of less than 9, and abnormal CT scans, or in
patients with a GCS score less than 9 and normal CT scans in the
presence of two or more of the following: age greater than 40 years,
unilateral or bilateral motor posturing, or systolic blood pressure
greater than 90 mmHg (Suarez, 2001).
No such guidelines exist for ICP monitoring in other brain injury
paradigms (ischemic stroke, ICH, cerebral neoplasm), and decisions
made for ICP monitoring in this setting are frequently based on the
clinical neurological status of the patient and data from neuroimaging
studies. Whether ICP monitoring adds much to the management of
patients of stroke is still open to question, clinical signs and imaging
data on shift of brain tissue are probably more useful (Xi, et al 2006).

69
CChhaapptteerr ((44)):: CCeerreebbrraallEEddeemmaa iinn NNeeuurroollooggiiccaall
DDiisseeaasseess

70
CCeerreebbrraall EEddeemmaa iinn NNeeuurroollooggiiccaallDDiisseeaasseess
IInnttrroodduuccttiioonn::Cerebral edema is associated with a wide spectrum of clinical
disorders. Edema can either result from regional abnormalities
related to primary disease of the central nervous system or be a
component of the remote effects of systemic toxic–metabolic
derangements. In either scenario, cerebral edema may be a life
threatening complication that deserves immediate medical attention
(Banasiak et al, 2004).
Several challenges surround the management of cerebral edema,
because the clinical presentation is extremely variable. This
variability reflects the temporal evolution of a diverse combination
of edema types because most forms of cerebral edema have the
capacity to generate other types. The specific clinical
manifestations are difficult to categorize by type and are better
described by precipitating etiology. In other words, it is essential to
outline the prominent forms of edema that are present in a given
clinical scenario. The location of edema fluid determines
symptomatology. Focal neurologic deficits result from isolated
regions of involvement, whereas diffuse edema produces
generalized symptoms such as lethargy (Amiry-Moghaddam &
Ottersen, 2003).

71
11.. CCeerreebbrroovvaassccuullaarr DDiisseeaassee::Cerebral ischemia frequently causes cerebral edema. Tissue
hypoxia that results from ischemic conditions triggers a cascade of
events that leads to cellular injury. The onset of ischemic edema
initially manifests as glial swelling occurring as early as 5 min
following interruption of the energy supply. This cytotoxic phase of
edema occurs when the BBB remains intact, although continued
ischemia leads to infarction and the development of vasogenic
edema after 48–96 hours (Latour et al, 2004).
Clinical symptoms are initially representative of neuronal
dysfunction within the ischemic territory, although the spread of
edema may elicit further neurological deficits in patients with large
hemispheric infarction. This clinical syndrome involves increasing
lethargy, asymmetrical pupillary examination, and abnormal
breathing. The mechanism of neurologic deterioration appears to
involve pressure on brain stem structures due to the mass effect of
infarcted and edematous tissue. Elevation of ICP may be
generalized or display focal gradients that precipitate herniation
syndromes. Herniation may lead to compression and infarction of
other vascular territories, in turn initiating a new cycle of infarction
and edema (Hawkins & Davis, 2005).
Intracerebral hemorrhage presents with focal neurologic deficits,
headache, nausea, vomiting, and evidence of mass effect. The
edema associated with intracerebral hemorrhage is predominantly
vasogenic, climaxing 48–72 hours following the initial event.

72
Secondary ischemia with a component of cytotoxic edema may
result from impaired diffusion in the extracellular space of the
perihemorrhage region. Other forms of hemorrhage, including
hemorrhagic transformation of ischemic territories and
subarachnoid hemorrhage may be associated with edema that
results from the noxious effects of blood degradation products
(Wang X & Lo, 2003).
22.. TTrraauummaattiicc BBrraaiinn IInnjjuurryy (TTBBII))::Raised ICP attributed to cerebral edema is the most frequent
cause of death in TBI. Focal or diffuse cerebral edema of mixed
types may develop following TBI. Following contusion of the
brain, the damaged BBB permits the extravasation of fluid into the
interstitial space. Areas of contusion or infarction may release or
induce chemical mediators that can spread to other regions. These
factors activated during tissue damage are powerful mediators of
extravasation and vasodilation (Marcella et al 2007).
TBI is associated with a biphasic pathophysiologic response
heralded by a brief period of vasogenic edema immediately
following injury, followed after 45–60 minutes by the development
of cytotoxic edema. Vasogenic edema may be detected by
neuroimaging modalities within 24–48 hours and reach maximal
severity between Days 4 and 8. Autoregulatory dysfunction is a
common sequela of TBI that may promote the formation of
hydrostatic edema in regions where the BBB remains intact. Recent

73
efforts have also demonstrated a prominent role of cytotoxic edema
in head-injured patients. Tissue hypoxia with ischemic edema
formation and neurotoxic injury due to ionic disruption contribute
to this cytotoxic component. In addition, osmotic edema may result
from hyponatremia, and hydrocephalic edema may complicate the
acute phase of TBI when subarachnoid hemorrhage or infections
predominate. Diffuse axonal injury may produce focal edema in
white matter tracts experiencing shear-strain forces during
acceleration/deceleration of the head (Stanley & Swierzewski,
2011).
33.. IInnffeeccttiioonnss::A combination of vasogenic and cytotoxic edema arises from
many infectious processes within the central nervous system. Other
forms of edema may also occur in infections, including
hydrocephalic edema secondary to CSF obstruction and osmotic
edema due to SIADH. Numerous infectious agents have direct toxic
effects generating vasogenic edema through alteration of the BBB
and cytotoxic edema from endotoxin-mediated cellular injury.
Bacterial wall products stimulate the release of various endothelial
factors, resulting in excessive vascular permeability (Simon &
Beckman, 2002).
Cerebral edema is a critical determinant of morbidity and
mortality in pediatric meningitis. Abscess formation or focal
invasion of the brain results in an isolated site of infection

74
surrounded by a perimeter of edema encroaching on the
neighboring parenchyma. This ring of vasogenic and cytotoxic
edema may produce more symptoms than the actual focus of
infection. Similar regions of focal or diffuse edema may
accompany encephalitis, particularly viral infections such as herpes
simplex encephalitis (Nathan & Scheld, 2000).
44.. CCeerreebbrraall VVeennoouuss SSiinnuuss TThhrroommbboossiiss::A major life-threatening consequence of cerebral venous sinus
thrombosis is cerebral edema. Two different kinds of cerebral
edema can develop. The first, cytotoxic edema is caused by
ischemia, which damages the energy-dependent cellular membrane
pumps, leading to intracellular swelling. The second type,
vasogenic edema, is caused by a disruption in the blood–brain
barrier and leakage of blood plasma into the interstitial space
(Masuhr et al, 2004).
The clinical manifestations of cerebral venous thrombosis are
highly variable. Individuals may be asymptomatic, and others may
suffer a progressive neurologic deterioration with headaches,
seizures, focal neurologic deficits, and severe obtundation leading
to death (Lemke & Hacein-Bey, 2005).

75
55.. NNeeooppllaassttiicc DDiisseeaassee::The detrimental effects of cerebral edema considerably influence
the morbidity and mortality associated with brain tumors. Tumor-
associated edema continues to be a formidable challenge,
producing symptoms such as headache and focal neurologic deficits
and, considerably altering the clinical outcome (partial resection,
chemotherapeutic agents and radiation have also been shown to
encourage the formation of edema). The predominant form of
tumor-associated edema is vasogenic, although cytotoxic edema
may occur through secondary mechanisms, such as tumor
compression of the local microcirculation or tissue shifts with
herniation. Individuals with hydrocephalus can also develop
hydrocephalic edema because of ventricular outflow obstruction
(Pouyssegur et al, 2006).
66.. SSeeiizzuurreess::Prolonged seizure activity may lead to neuronal energy depletion
with eventual failure of the Na+/K+ ATPase pump and concomitant
development of cytotoxic or ischemic edema. Unlike ischemia
produced by occlusion of a cerebral artery, a more heterogeneous
cellular population is affected. The reactive hyperemic response
driven by excessive metabolic demands increases the hydrostatic
forces across a BBB already damaged by the vasogenic component
of ischemic edema. The disruption of normal ionic gradients,
extracellular accumulation of excitotoxic factors, and lactic acidosis

76
further exacerbate vasogenic edema. Consequently, cessation of
seizure activity usually results in the complete resolution of
cerebral edema (Vespa et al, 2003).
77.. MMuullttiippllee SScclleerroossiiss::One of the crucial stages in the evolution of a multiple sclerosis
lesion is considered to be the disruption of the blood brain barrier,
leading to edema in the CNS by accumulation of plasma fluids.
This process is believed to be initiated by autoreactive CD4+
lymphocytes which migrate into the CNS and start an inflammatory
response. Although BBB breakdown imaged as focal enhancement
in T1- weighted MRI after gadolinium DTPA injection is the gold
standard of lesion detection during the course of the disease, the
deposition of contrast agent in the CNS has been shown to correlate
with clinical disability (Vos et al, 2005).
88.. HHyyddrroocceepphhaalluuss::Isolated hydrocephalic edema may result from acute obstructive
hydrocephalus with impairment of CSF drainage. Transependymal
pressure gradients result in edema within periventricular white
matter tracts. The rapid disappearance of myelin lipids under
pressure causes the periventricular white matter to decrease in
volume. The clinical manifestations may be minor, unless
progression to chronic hydrocephalus becomes apparent with

77
symptoms including dementia and gait abnormalities (Abbott,
2004).
99.. HHyyppeerrtteennssiivvee EEnncceepphhaallooppaatthhyy::This potentially reversible condition presents with rapidly
progressive neurological signs, headache, seizures, altered mental
status, and visual disturbances. The pathogenesis of edema
formation is controversial but is thought to involve elevated
hydrostatic forces due to excessive blood pressure, with lesser
degrees of involvement attributed to vasogenic edema and
secondary ischemic components. The rate of blood pressure
elevation is a critical factor, because hypertensive encephalopathy
usually develops during acute exacerbations of hypertension. Early
recognition and treatment of hypertensive encephalopathy may
reverse cerebral edema, preventing permanent damage to the BBB,
and ischemia, although severe cases may be fatal (Johnston et al,
2005).
1100.. HHyyppeerrtthheerrmmiiaa::The pathophysiology of this rare cause of cerebral edema is poorly
understood. Although the fatal consequences of heat stroke have been
recognized since ancient times, the underlying mechanisms await
clarification. Scant pathologic material suggests a combination of
cytotoxic and vasogenic components, secondary to an increase in
BBB permeability due to the release of multiple chemical factors and

78
direct cytotoxic damage. Age and physiologic state of the individual
appear to be important determinants of clinical outcome in
hyperthermic injury (Bruno et al, 2004).

79
CChhaapptteerr ((55)):: TTrreeaattmmeennttooff CCeerreebbrraall EEddeemmaa

80
TTrreeaattmmeenntt ooff CCeerreebbrraall EEddeemmaa
IInnttrroodduuccttiioonn::Cerebral edema is frequently encountered in clinical practice in
critically ill patients with acute brain injury from diverse origins and
is a major cause of increased morbidity and death in this subset of
patients. The consequences of cerebral edema can be lethal and
include cerebral ischemia from compromised regional or global
cerebral blood flow (CBF) and intracranial compartmental shifts due
to intracranial pressure gradients that result in compression of vital
brain structures (Rabinstein, 2004).
The overall goal of treatment of cerebral edema is to maintain
regional and global CBF to meet the metabolic requirements of the
brain and prevent secondary neuronal injury from cerebral ischemia
(Broderick et al, 1999).
Treatment of cerebral edema involves using a systematic and
algorithmic approach, from general measures (optimal head and neck
positioning for facilitating intracranial venous outflow, avoidance of
dehydration and systemic hypotension, and maintenance of
normothermia) to specific therapeutic interventions (controlled
hyperventilation, administration of corticosteroids and diuretics,
osmotherapy, and pharmacological cerebral metabolic suppression)
,and decompressive surgery (Wakai et al, 2007).

81
II.. GGeenneerraall mmeeaassuurreess ffoorr ttrreeaattiinnggCCeerreebbrraall eeddeemmaa::
Several general measures that are supported by principles of altered
cerebral physiology and clinical data from patients with brain injury
should be applied to patients with cerebral edema. The primary goal
of these measures is to optimize cerebral perfusion, oxygenation, and
venous drainage; minimize cerebral metabolic demands; and avoid
interventions that may disturb the ionic or osmolar gradient between
the brain and the vascular compartment (Ahmed & Anish, 2007).
1. Optimizing head and neck positions:Finding the optimal neutral head position in patients with cerebral
edema is essential for avoiding jugular compression and impedance of
venous outflow from the cranium, and for decreasing CSF hydrostatic
pressure. In normal uninjured patients, as well as in patients with
brain injury, head elevation decreases ICP (Ng et al, 2004).
These observations have led most clinicians to incorporate a 30°
elevation of the head in patients with poor intracranial compliance.
Head position elevation may be a significant concern in patients with
ischemic stroke, however, because it may compromise perfusion to
ischemic tissue at risk. It is also imperative to avoid the use of
restricting devices and garments around the neck (such as devices
used to secure endotracheal tubes), as these may lead to impaired
cerebral venous outflow via compression of the internal jugular veins
(Ropper et al, 2004).

82
2. Ventilation and oxygenation:Hypoxia and hypercapnia are potent cerebral vasodilator and
should be avoided in patients with cerebra edema. It is recommended
that any patients with Glasgow coma scale (GCS) scores less than or
equal to 8 and those with poor upper airway reflexes be intubated
preemptively for airway protection. This strategy is also applicable to
patients with concomitant pulmonary disease, such as aspiration
pneumonitis, pulmonary contusion, and acute respiratory distress
syndrome (Eccher & Suarez, 2004).
Avoidance of hypoxemia and maintenance of PaO2 at
approximately 100 mmHg are recommended. Careful monitoring of
clinical neurological status, ICP is recommended in mechanically
ventilated patients with cerebral edema with or without elevations in
ICP. Blunting of upper airway reflexes (coughing) with endobronchial
lidocaine before suctioning, sedation, or, rarely, pharmacological
paralysis may be necessary for avoiding increases in ICP (Schwarz et
al, 2002).
3. Seizure prophylaxis:Anticonvulsants (predominantly phenytoin) are widely used
empirically in clinical practice in patients with acute brain injury of
diverse origins, including traumatic brain injury (TBI), subarachnoid
hemorrhage (SAH), and intracranial hemorrhage (ICH), although data
supporting their use are lacking (Vespa et al, 2003).

83
Early seizures in TBI can be effectively reduced by prophylactic
administration of phenytoin for 1 or 2 weeks without a significant
increase in drug-related side effects. The use of prophylactic
anticonvulsants in ICH can be justified, as subclinical seizure activity
may cause progression of shift and worsen outcome in critically ill
patients with ICH. Yet the benefits of prophylactic use of
anticonvulsants in most causes leading to brain edema remain
unproven, and caution is advised in their use (Glantz et al, 2000).
4. Management of fever and hyperglycemia:Numerous experimental and clinical studies have demonstrated the
deleterious effects of fever on outcome following brain injury, which
theoretically result from increases in oxygen demand. Therefore,
normothermia is strongly recommended in patients with cerebral
edema, irrespective of underlying origin. Acetaminophen (325–650
mg orally, or rectally every 4–6 hours) is the most common, and the
safest agent used, and is recommended to avoid elevations in body
temperature (Bruno et al, 2004).
Evidence from clinical studies in patients with ischemic stroke,
subarachnoid hemorrhage, and TBI suggests a strong correlation
between hyperglycemia and worse clinical outcomes. Hyperglycemia
can exacerbate brain injury and cerebral edema. Significantly
improved outcome has been reported in general ICU patients with
good glycemic control; although larger studies focused on specific
brain injury paradigms are forthcoming. Nevertheless, current
evidence suggests that rigorous glycemic control may be beneficial in

84
all patients with brain injury and cerebral edema (Parsons et al,
2002).
5. Blood pressure management:The ideal blood pressure will depend on the underlying cause of the
brain edema. In trauma and stroke patients, blood pressure should be
supported to maintain adequate perfusion, avoiding sudden rises and
very high levels of hypertension. Keeping cerebral perfusion pressure
above 60–70 mm Hg is generally recommended after traumatic brain
injury (Johnston et al, 2005).
6. Nutritional support and fluid management:Prompt maintenance of nutritional support is imperative in all
patients with acute brain injury. Unless contraindicated, the enteral
route of nutrition is preferred. Special attention should be given to the
osmotic content of formulations Low serum osmolality must be
avoided in all patients with brain swelling since it will exacerbate
cytotoxic edema. This objective can be achieved by strictly limiting
the intake of hypotonic fluids. In fact, there is clear evidence that free
water should be avoided in patients with head injuries and brain
edema (Leira et al, 2004).
In patients with pronounced, prolonged serum hyperosmolality, the
disorder must be corrected slowly to prevent rebound cellular
swelling. Fluid balance should be maintained neutral. Negative fluid
balance has been reported to be independently associated with adverse
outcomes in patients with severe brain trauma. Avoiding negative

85
cumulative fluid balance is essential to limit the risk of renal failure in
patients receiving mannitol (Powers et al, 2001).
IIII.. SSppeecciiffiicc mmeeaassuurreess ffoorr mmaannaaggiinnggCCeerreebbrraall eeddeemmaa::
1. CCoonnttrroolllleedd hhyyppeerrvveennttiillaattiioonn::Based on principles of altered cerebral pathophysiology associated
with brain injury, controlled hyperventilation remains the most
efficacious therapeutic intervention for cerebral edema, particularly
when the edema is associated with elevations in ICP (Carmona et al,
2000).
A decrease in PaCO2 by 10 mmHg produces proportional
decreases in regional CBF, resulting in rapid ICP reduction. The
vasoconstrictive effect of respiratory alkalosis on cerebral arterioles
has been shown to last for 10 to 20 hours, beyond which vascular
dilation may result in exacerbation of cerebral edema and rebound
elevations in ICP (Mayer & Rincon, 2005).
Overaggressive hyperventilation may actually result in cerebral
ischemia. Therefore, the common clinical practice is to lower and
maintain PaCO2 by 10 mmHg to a target level of approximately 30–
35 mmHg for 4 to 6 hours, although identifying the correct strategy
for achieving this goal is unclear in terms of adjusting tidal volumes
and respiratory rate (Marion et al, 2002).

86
It should be noted that controlled hyperventilation is to be used as a
rescue or resuscitative measure for a short duration until more
definitive therapies are instituted and maintained. Caution is advised
when reversing hyperventilation gradually over 6 to 24 hours, to
avoid cerebral hyperemia and rebound elevations in ICP secondary to
effects of reequilibration (Diringer, 2002).
22.. OOssmmootthheerraappyy uussee::Historical perspective:
The earliest description of the use of osmotic agents dates back to
1919, Weed and McKibben observed that intravenous administration
of a concentrated salt solution resulted in an inability to withdraw
CSF from the lumbar cistern due to a collapse of the thecal sac. This
observation was followed by a set of experiments in an animal model
in which they demonstrated (under direct visualization via a
craniotomy) egress of the brain away from the cranial vault with
intravenous infusion of hypertonic saline solutions and herniation of
brain tissue with administration of hypotonic fluids (Weed et al, 1919,
coated from Ahmed & Anish, 2007).
This set of observations has formed the basis for osmotherapy.
Concentrated urea was the first agent to be used clinically as an
osmotic agent. Its use was short-lived and is of historic interest only
because of several untoward side effects (nausea, vomiting, diarrhea,
and coagulopathy). The interest in elevating plasma oncotic pressure
as a strategy to ameliorate cerebral edema with the use of
concentrated human plasma proteins, which appeared briefly in 1940,

87
was short-lived due to several concerns, including cost, short half-life,
cardiopulmonary effects, and allergic reactions. Glycerol was possibly
the second osmotic agent to be used clinically and is still used
(Alejandro & Rabinstein, 2006).
Mannitol, an alcohol derivative of simple sugar mannose, was
introduced in 1960 and has since remained the major osmotic agent of
choice in clinical practice. Its long duration of action and relative
stability in solution has enhanced its use over the years (Dennis,
2003).
Renewed interest in hypertonic saline solutions reappeared in the
1980s, in these studies; cerebral effects of these solutions were
investigated in well-controlled experimental studies in animal models
of acute brain injury. These studies continue to provide evidence for
the potential use of these solutions in the clinical domains (Harukuni
et al, 2002).
Therapeutic basis and goal of osmotherapy:
Put simply, the fundamental goal of osmotherapy is to create an
osmotic gradient to cause egress of water from the brain extracellular
(and possibly intracellular) compartment into the vasculature, thereby
decreasing intracranial volume. A serum osmolality in the range of
300 to 320 mOsm/L has traditionally been recommended for patients
with acute brain injury who demonstrate poor intracranial
compliance; however, values greater than 320 mOsm/L can be
attained with caution, without apparent untoward side effects
(Korenkov et al, 2000).

88
An ideal osmotic agent is one that produces a favorable osmotic
gradient, is inert and nontoxic, is excluded from an intact BBB, and
has minimal systemic side effects. Mannitol has remained the major
osmotic agent of choice in clinical practice. Its long duration of action
(4–6 hours) and relative stability in solution have enhanced its use
over the years (Battison et al, 2005).
The extraosmotic properties of mannitol have been studied
extensively and may provide additional beneficial effects in brain
injury, including decreases in blood viscosity, resulting in increases in
CBF and CPP, free radical Scavenging and inhibition of apoptosis
(Qureshi et al, 2000).
Like mannitol, hypertonic saline also possesses unique
extraosmotic properties, including modulation of CSF production and
resorption, accentuation of tissue oxygen delivery, and modulation of
inflammatory and neurohumoral responses (arginine-vasopressin and
atrial natriuretic peptide) following brain injury that may act together
to ameliorate cerebral edema (Bhardwaj et al, 2004).
Comparison between mannitol and hypertonic saline:
Few studies have made direct comparisons between mannitol and
hypertonic saline (table 3). In a prospective, randomized comparison
of 2.5 ml/kg of either 20% mannitol (1400 mOsm/kg) or 7.5%
hypertonic saline (2560 mOsm/ kg) in patients undergoing elective
supratentorial procedures, ICP and intraoperative clinical assessment
of brain swelling were similar in both treatment groups (Toung et al,
2005).

89
In a prospective, randomized trial of hypertonic saline with
hydroxyethyl starch, hypertonic saline was shown to be more
effective than equiosmolar doses of mannitol in lowering elevated
ICP and augmenting CPP in patients with ischemic stroke (Mirski et
al, 2000).
Likewise, intravenous bolus injection of 10% hypertonic saline was
shown to be effective in lowering ICP in patients with ischemic stroke
who failed to show such a response to conventional doses of
mannitol. More recently, in a small prospective study, isovolemic
intravenous infusion of 7.5% hypertonic saline was more effective in
the control of ICP following TBI, compared with mannitol treatment
(Vialet et al, 2003).
In summary, the literature supports the use of hypertonic saline as a
therapy to decrease ICP in patients following TBI and stroke and to
optimize intravascular fluid status in patients with SAH-induced
vasospasm. However, no definite conclusions can be drawn at present
because the studies involved a wide range of saline concentrations,
and equiosmolar solutions were not consistently used. Further
carefully designed studies comparing the 2 agents are needed before
superiority of one of them can be firmly postulated (Ware et al,
2005).

90
Table 3: Summary of experimental studies comparing differentformulations of hypertonic saline (HS) with mannitol 20% (M) (Toung et
al, 2005).Study: Experimental
Model:HS Formulation &Mode of Infusion:
Results:
Gemmaet al, 1997
(50/Electiveneurosurgery)
7.5% NaClBolus
No differences in CSF pressure
Schwarzet al, 1998
(9/Ischemicinfarction withraised ICP)
75 g/L NaCl plus 60 g/Lhydroxyethylstarch (2570 mOsm/L)Serial boluses
HS lowered ICP moreeffectively M increasedCPP more effectively.
Vialet etal, 2003
(20/TBI withcoma and raisedICP)
7.5% NaClSerial boluses
HS had lower rate offailure to drop ICP.
Battisonet al,
2005
(9/TBI) 7.5% NaCl plus 6%dextran-70*Two boluses of HS and M
HS produced greater andlonger ICP reductions.
Mirski etal, 2000
Focal cryogeniclesion in rats
11 mOsm/kg NaCl*Bolus
Greater and longer ICPreduction with HS.Similar brain watercontent.
Tuong etal, 2005
Temporary MCAocclusion (2 h) inrats
7.5%NaCl/acetateContinuous
HS attenuated maximaledema in bothhemispheres less robustlythan M
Tuong etal, 2002
Permanent MCAocclusion in rats
5% And 7.5% NaCl/acetateContinuous
HS (both concentrations)reduced lung and brainwater content moreeffectively than M.
Zornowet al,
1990
Focal cryogeniclesion in rabbits
3.2% NaClBolus
Similar ICP reduction.Similar MAP response.
Freshmanet al, 1993
ICP elevation byepidural ballooninflation in sheep
7.5% NaClBolus
Similar ICP reduction.Similar brain watercontent.
CPP, cerebral perfusion pressure; MAP, mean arterial pressure; MCA, middle cerebralartery.*Equiosmolar doses of mannitol 20% (osmolarity 1160 mOsm/L) and HS were used

91
Treatment protocol for osmotherapy:
The conventional osmotic agent mannitol, when administered at a
dose of 0.25 to 1.5 g/kg by intravenous bolus injection, usually lowers
ICP, with maximal effects observed 20 to 40 minutes following its
administration. Repeated dosing of mannitol may be instituted every 6
hours and should be guided by serum osmolality to a recommended
target value of approximately 320 mOsm/L; higher values result in
renal tubular damage (Alejandro & Rabinstein, 2006).
A variety of formulations of hypertonic saline solutions (2, 3, 7.5,
10, and 23%) are used in clinical practice for the treatment of cerebral
edema with or without elevations in ICP. Hypertonic saline solutions
of 2, 3, or 7.5% contain equal amounts of sodium chloride and
sodium acetate (50:50) to avoid hyperchloremic acidosis. Potassium
supplementation (20–40 meq/L) is added to the solution as needed
(Ahmed & Anish, 2007).
Continuous intravenous infusions are begun through a central
venous catheter at a variable rate to achieve euvolemia or slight
hypervolemia (1–2 ml/ kg/hr). A 250-ml bolus of hypertonic saline
can be administered cautiously in select patients if more aggressive
and rapid resuscitation is warranted. Normovolemic fluid status is
maintained, guided by central venous pressure (Battison et al, 2005).
The goal in using hypertonic saline is to increase serum sodium
concentration to a range of 145 to 155 mEq/L (serum osmolality
approximately 300–320 mOsm/L), but higher levels can be targeted
cautiously. This level of serum sodium is maintained for 48 to 72

92
hours until patients demonstrate clinical improvement or there is a
lack of response despite achieving the serum sodium target (Toung et
al, 2002).
During withdrawal of therapy, special caution is emphasized due to
the possibility of rebound hyponatremia leading to exacerbation of
cerebral edema. Serum sodium and potassium are monitored every 4
to 6 hours, during both institution and withdrawal of therapy. Chest
radiographs are obtained to find evidence of pulmonary edema from
congestive heart failure, especially in elderly patients (Mirski et al,
2000).
Intravenous bolus injections (30 ml) of 23.4% hypertonic saline
have been used in cases of intracranial hypertension refractory to
conventional ICP-lowering therapies; repeated injections of 30 ml
boluses of 23.4% saline may be given if needed to lower ICP.
Administration of this osmotic load, to lower ICP and maintain CPP,
may allow extra time for other diagnostic or therapeutic interventions
(such as decompressive surgery) in critically ill patients (Diringer et
al, 2004).
Potential complications of osmotherapy:
Safety concerns with mannitol include hypotension, hemolysis,
hyperkalemia, renal insufficiency, and pulmonary edema. Clinical
experiences suggest that the side-effect profile of hypertonic saline is
superior to mannitol, but some theoretical complications that are

93
possible with hypertonic saline therapy are notable (Table 4) (Dennis,
2003).
Table 4: Theoretical potential complications of using hypertonicsaline solutions:
1. CNS changes (encephalopathy, lethargy, seizures, coma)central pontine myelinolysis.
2. Congestive heart failure, pulmonary edema.3. Electrolyte derangements (hypokalemia, hypomagnesemia,
hypocalcemia).4. Cardiac arrhythmias.5. Metabolic academia (hyperchloremic with use of chloride
solutions).6. Potentiation of non tamponaded bleeding.7. Subdural hematomas that result from shearing of bridging veins
due to hyperosmolar contracture of brain.8. Hemolysis with rapid infusions.9. Phlebitis with infusion via peripheral route.10.Coagulopathy (elevated prothrombin and partial thromboplastin
time, platelet dysfunction).11.Rebound hyponatremia leading to cerebral edema with rapid
withdrawal.Modified from Bhardwaj and Ulatowski, 1999 and Shell et al. (Dennis, 2003).
Myelinolysis, the most serious complication of hypertonic saline
therapy, typically occurs when rapid corrections in serum sodium
arise from a chronic hyponatremic state to a normonatremic or
hypernatremic state. Experimental studies suggest that for myelin
injury to occur, the degree of rapid change in serum sodium is much
greater from a normonatremic to a hypernatremic state (change of

94
approximately 40 mEq/L), but further study with neuroimaging
techniques is required (Takefuji et al, 2007).
3.. LLoooopp ddiiuurreettiiccss::
The use of loop diuretics (commonly furosemide) for the treatment
of cerebral edema, particularly when used alone, remains
controversial. Combining furosemide with mannitol produces a
profound diuresis; however, the efficacy and optimum duration of this
treatment remain unknown (Steiner et al, 2001).
If loop diuretics are used, rigorous attention to systemic hydration
status is advised, as the risk of serious volume depletion is substantial
and cerebral perfusion may be compromised. A common strategy
used to raise serum sodium rapidly is to administer an intravenous
bolus of furosemide (10 to 20 mg) to enhance free water excretion
and to replace it with a 250-ml intravenous bolus of 2 or 3%
hypertonic saline (Thenuwara et al, 2002).
Acetazolamide, a carbonic anhydrase inhibitor that acts as a weak
diuretic and modulates CSF production, does not have a role in
cerebral edema that results from acute brain injuries; however, it is
frequently used in outpatient practice, particularly for the treatment of
cerebral edema associated with pseudo tumor cerebrii (Eccher &
Suarez, 2004).
4. CCoorrttiiccoosstteerrooiidd aaddmmiinniissttrraattiioonn::
The main indication for the use of steroids is for the treatment of
vasogenic edema associated with brain tumors or accompanying brain

95
irradiation and surgical manipulation. Although the precise
mechanisms of the beneficial effects of steroids in this paradigm are
unknown, steroids decrease tight-junction permeability and, in turn,
stabilize the disrupted BBB (Rabinstein, 2006).
Glucocorticoids, especially dexamethasone, are the preferred
steroidal agents, due to their low mineralocorticoid activity; the usual
initial dose is 10 mg intravenously or by mouth, followed by 4 mg
every 6 hours. This is equivalent to 20 times the normal physiologic
production of cortisol (Papadopoulos et al, 2004).
Responses are often prompt and remarkable, sometimes dramatic,
but some tumors are less responsive. Higher doses, up to 96 mg per
day, may be used with chances of success in more refractory cases.
After several days of use, steroids should be tapered gradually to
avoid potentially serious complications from recurrent edema and
adrenal suppression (Kaal & Vecht, 2004).
The therapeutic role of steroids in TBI and stroke has been studied
extensively. In TBI, steroids failed to control elevations in ICP or to
show any benefit in outcome, and they may even be harmful. In
stroke, steroids have failed to show any substantial benefit despite
some success in animal models. Given the deleterious side effects of
steroid use (peptic ulcers, hyperglycemia, impairment of wound
healing, psychosis, and immunosuppression), until further studies are
published, caution is advised in the use of steroids for cerebral edema
unless absolutely indicated (Roberts et al, 2004).

96
Glucocorticoids are also useful to treat brain edema in cases of
bacterial meningitis. Edema in these patients develops as part of the
inflammatory reaction triggered by the lysis of bacterial cell walls
induced by antibiotics. Inflammation is mediated through the
increased production of cytokines and chemokines by microglia,
astrocytes, and macrophages. Interleukin-1 (IL-1) and tumor necrosis
factor (TNF) increase vascular permeability both directly and
indirectly by increasing leukocyte adherence to the endothelium
(Sinha et al, 2004).
Apart from previously mentioned mechanisms, glucocorticoids
exert a depressant effect on both the synthesis and translation of IL-1
and TNF mRNA. The timing of glucocorticoid use may be critical as
the maximal reduction in the production of these inflammatory
cytokines occurs only if therapy is started prior to the release of the
bacterial cell wall components (Slivka & Murphy, 2001).
5. PPhhaarrmmaaccoollooggiiccaall ccoommaa::
Barbiturates were introduced since the 1960s, and have gained
acceptance for the treatment of cerebral edema associated with
intractable elevations in ICP. Barbiturates lower ICP, principally via a
reduction in cerebral metabolic activity, resulting in a coupled
reduction in CBF and CBV (Mayer & Rincon, 2005).
Yet their use in clinical practice is not without controversy. In
patients with TBI, barbiturates are effective in reducing ICP, but have
failed to show evidence of improvement in clinical outcome.

97
Evidence is limited for the utility of barbiturate treatment in cerebral
diseases that include space-occupying lesions such as tumor and ICH
(Schwab et al, 1997).
When used in the acute setting, pentobarbital, a barbiturate with an
intermediate physiological half-life (approximately 20 hours) is the
preferred agent rather than phenobarbital. The recommended regimen
entails a loading intravenous bolus dose of pentobarbital (3–10
mg/kg), followed by a continuous intravenous infusion (0.5–3.0
mg/Kg/hr, serum levels of 3 mg/dL) (Alejandro & Rabinstein, 2006).
Several adverse effects of barbiturates that limit their clinical use
are to be noted, including sustained lowering of systemic blood
pressure and CPP, cardiodepression, immunosuppression, and
systemic hypothermia. Perhaps the most important limitation with
barbiturate coma treatment is the inability to track subtle changes in a
patient’s clinical neurological status, which necessitates frequent
serial neuroimaging (Ropper et al, 2004).
6. HHyyppootthheerrmmiiaa::
Induced hypothermia has generated enormous interest as a potential
neuroprotective intervention in patients with acute brain insults.
Sound experimental data provide a solid foundation to the clinical
evaluation of hypothermia to treat acute brain ischemia and traumatic
injury (Krieger et al, 2001).
Different cooling methods are currently available, including
external (ice packs, iced gastric lavage, water or air circulating

98
blankets, cooling vest) and endovascular means. The superiority of
endovascular cooling is probable but still under evaluation. Target
core temperature is usually 32–34°C, measured with thermistors
placed inside the urinary bladder (Clifton et al, 2001).
Shivering must be prevented using deep sedation and
neuromuscular paralysis when necessary; the combination of oral
buspirone and intravenous meperidine. Hypothermia is usually
maintained for 12–72 hours, followed by a period of controlled
rewarming over 12–24 hours (Gadkary et al, 2002).
Induction of hypothermia is associated with several potential
complications. The most frequent and dangerous are sepsis
(particularly from pneumonia), cardiac arrhythmias and hemodynamic
instability (often seen during rewarming), coagulopathy (especially
thrombocytopenia), and electrolyte disturbances (potassium,
magnesium, calcium, phosphate) (Holzer et al, 2005).

99
IIIIII.. SSuurrggiiccaall iinntteerrvveennttiioonnss::In patients with ICP elevation, cerebrospinal fluid drainage is a fast
and highly effective treatment measure. This assertion holds true even
in the absence of hydrocephalus. Unfortunately, external ventricular
drainage carries a substantial risk of ventriculitis, even under the best
care. Controlled lumbar drainage may be a safe alternative, though its
use should be accompanied by extreme caution (Buschmann et al,
2007).
A comprehensive and updated discussion on the value of
hemicraniectomy to treat ischemic brain edema associated with
massive hemispheric strokes has been recently published. While it is
clear that hemicraniectomy can be lifesaving, its beneficial impact on
the long-term functional outcome of survivors remains unproven. An
example of this surgical intervention is presented in (Figure 22)
(Coplin et al, 2001).
In patients with critical intracranial hypertension after head trauma
who fail to respond to all other therapeutic measures, craniectomy
with duraplasty may be a valuable alternative. Hemicraniectomy may
be preferable in patients with focal lesions, such as hemorrhagic
contusions. Good long-term functional outcomes have been reported
in 25–56% of young patients after this surgery (Bullock, 2006).
Although the optimal timing and indications for this intervention
are not well established, the expeditious decision by an experienced
neurosurgeon to proceed with holocraniectomy in a young patient
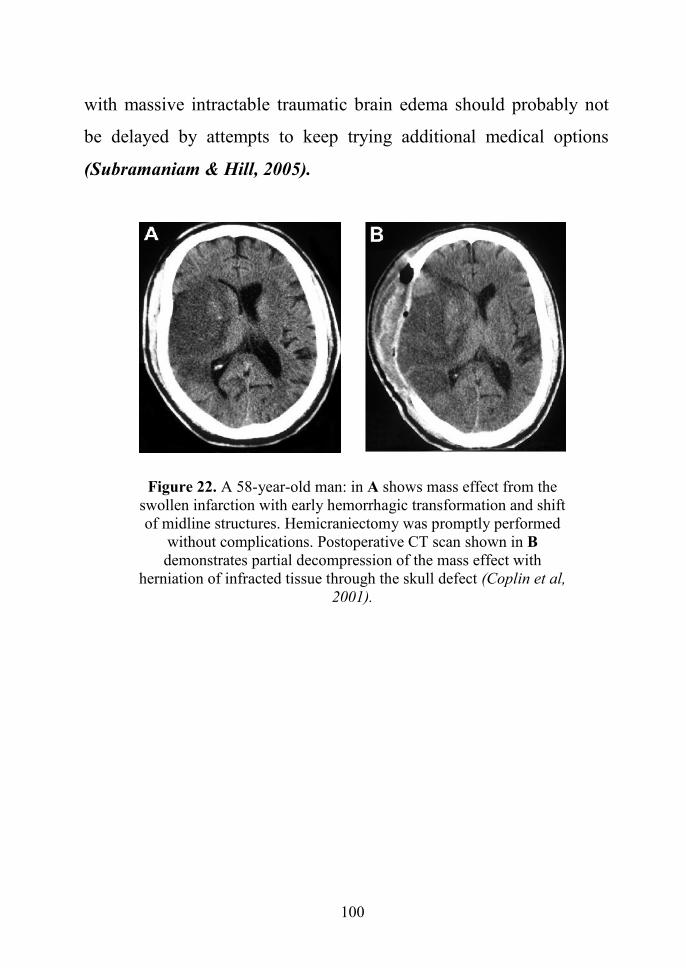
100
with massive intractable traumatic brain edema should probably not
be delayed by attempts to keep trying additional medical options
(Subramaniam & Hill, 2005).
Figure 22. A 58-year-old man: in A shows mass effect from theswollen infarction with early hemorrhagic transformation and shiftof midline structures. Hemicraniectomy was promptly performed
without complications. Postoperative CT scan shown in Bdemonstrates partial decompression of the mass effect with
herniation of infracted tissue through the skull defect (Coplin et al,2001).

101
CChhaapptteerr ((66)):: SSppiinnaall CCoorrddEEddeemmaa IInn
IInnjjuurryy aanndd RReeppaaiirr

102
SSppiinnaall CCoorrdd EEddeemmaa IInn IInnjjuurryy aannddRReeppaaiirr
IInnttrroodduuccttiioonn::The blood-spinal cord barrier (BSCB) regulates the fluid
microenvironment of the spinal cord within a narrow limit. The
details of structural and functional properties of the BSCB in normal
and pathological conditions are not well known in all details
(Leskovar et al, 2000).
Traumatic insults to the spinal cord disrupt the functional integrity
of the BSCB and results into an increased transport of several
substances from the vascular compartment to the spinal cord cellular
microenvironment. Breakdown of the BSCB thus appears to play
important roles in cell and tissue reaction as well as regeneration and
repair processes (Popovich et al, 1997).
An increased understanding of BSCB in spinal cord injury (SCI)
is important for the development of suitable therapeutic strategies to
minimize cell and tissue destruction and to enhance regeneration and
functional recovery (Sharma, 2004).
There are reasons to believe that the characteristics of the BSCB
are similar to that of the blood-brain barrier (BBB). The spinal cord
endothelial cells are connected with tight junctions and do not
exhibit vesicular transport. The spinal endothelial cells are
surrounded by a thick basement membrane like the BBB. However,

103
a minor difference in astrocytes-microvessel interactions is seen in
the superficial spinal cord microvessels. The large superficial vessels
of the spinal cord contain enough deposits of glycogen, not normally
seen in the brain microvessels. The functional significance of
glycogen deposits in relation with the barrier properties is not well
understood (James et al, 1997).
Interestingly, impairment of local circulation in the spinal cord
induces much less cell damage compared to the brain. A less marked
regional difference in the spinal cord microcirculation and/or
metabolism compared to the brain could be the main reason behind
this phenomenon (Stålberg et al, 1998).
In traumatic brain injuries, breakdown of the BBB results in
abnormal leakage of proteins leading to vasogenic edema formation
and brain pathology. Edematous swelling of brain in a closed
cranium compresses vital centers resulting in instant death.
However, in the spinal cord, the vertebral canal provides some space
to accommodate edematous expansion of the spinal cord up to some
extent (Mendelow et al, 2000).

104
EEppiiddeemmiioollooggyy ooff SSppiinnaall CCoorrdd IInnjjuurryy::In the United States of America, about 30 to 50 cases per million
populations are recorded per year that is quite comparable to Europe
and other continents. The common cause of SCI is due to motor
vehicle accidents followed by fall, penetrating injuries like gun shot,
knife wounds or sports injuries (Schwab & Bartholdi, 1996).
Majority of cases show injury to the cervical spinal cord or
thoracolumbar junctions. The victims of SCI are generally young
men of 20 to 30 years of age while only 20 to 30 % of cases involve
women (Holmes, 1915, coated from Sharma, 2005).
Quadriplegia followed by paraplegia is the main symptoms of
SCI. Complete injuries without any signs of voluntary motor or
sensory perception below the level of the lesion are seen in about
50% cases of the SCI victims. The other causes of paralysis
involving the spinal cord are multiple sclerosis, ischemia and
tumors. Currently, no suitable therapeutic strategies are effective in
improving the quality of life of SCI patients. Thus, exploration of
new pharmacological avenues with possibility of regeneration of the
damaged spinal cord axons is urgently needed. Knowledge on the
structure and function of the BSCB and the spinal cord
microenvironment in SCI is thus crucial for the development of
novel pharmacological tools to minimize cell and tissue injuries as
well as to enhance recovery (Sharma, 2000).

105
PPaatthhoopphhyyssiioollooggyy ooff SSppiinnaall CCoorrdd IInnjjuurryy::Pathophysiology of SCI is complex and includes several
immediate and late cell and tissue reactions. The progression and
persistence of these pathological changes mainly depends on the
severity of the primary lesion. Depending on the magnitude and
severity of the initial impact, microhaemorrhages and leakage of
erythrocytes are present in the perivascular space across
microvessels, arterioles, veins and venules as well as in the spinal
cord neuropil within 3 minutes (Sharma, 2005).
Damage to neuropil, swollen astrocytes, ruptured cell membranes
and basal lamina are frequent within 6 to 10 minutes after SCI.
Swollen endothelial cells with electron dense cytoplasm exhibiting
large numbers of vesicles (60 to 70 nm diameter) without widening
of the tight junctions are common at this time. In some microvessels,
the perivascular spaces contain proteinaceous fluid. The endothelial
balloons are evident in some microvessels 4 to 6 hours after injury
(Mautes & Noble, 2000).
A detailed account of BSCB permeability following spinal cord
transection and contusion is previously described by Noble and co-
workers. Extravasation of exogenous horseradish peroxidase (HRP)
is seen in 0.5 to 2.0 cm proximal and distal segments of the cord to
the transection site. The segment located 1 cm away from the lesion
site showed extravasation of HRP between 30 minutes and 3 hours
on day 1. A less pronounced increase in BSCB disruption is seen in

106
the proximal segment compared to the distal segment. The
permeability to HRP is restored within 14 days after injury (Noble,
1978, coated from Sharma, 2005).
Vesicular transport rather than widening of the tight junctions is
responsible for HRP extravasation in the transection and contusion
injuries. These observations suggest that the mechanisms of leakage
across the BSCB are similar in nature irrespective of the types of
injury (Sharma, 2004).
At the ultrastructural level, lanthanum tracer was mainly confined
within the lumen of the endothelial of normal rats. SCI resulted in
the occurrence of lanthanum filled vesicles within the endothelial
cell cytoplasm. Marked increase in the endothelial cell membrane
permeability to lanthanum is seen in several vascular profiles that
appear to be very specific. In some microvascular profiles, the
lanthanum is present in the basal lamina. However, the tight junction
remained intact to lanthanum in SCI. These observations suggest
that increased endothelial cell membrane permeability seems be one
additional way of vascular leakage (Sharma, 2000).
Spinal cord edema formation:After SCI, edema formation is apparent as early as 30 seconds and
becomes prominent within 2 to 5 minutes that could last up to 15
days. The labeled Evans blue albumin (EBA) spreads up to one
segment from the injury site. Traumatic injuries resulting in
permanent paraplegia increase tissue water content above and below
the lesion site. Adjacent spinal cord segments also exhibit leakage of

107
albumin and dextran as well as tissue damage. On the other hand,
transient paraplegia is not associated with extravasation of albumin
or dextran and/or increase in spinal cord water content (Sharma,
2003).
Edema, as measured by water content is seen as early as 5 minutes
after impact injury that persisted up to 15 days. The edema
formation is most prominent in the gray matter. On the other hand,
using specific gravity gradient column, about 127 % increase in
edema and volume swelling was observed near the impact site in the
gray matter compared to only 24 % increase in white matter after 1
hour injury. The regression of edema is evident after 9 days. This
indicates that progression, persistence and resolution of edema are
crucial for cell and tissue injury following SCI (Li & Tator, 1998).
Local microhaemorrhage and tissue necrosis near the lesion site
also influence increase in the water content. Increased tissue water
content in the adjacent non-traumatised segment, thus represents true
edema formation. Tissue pressure gradients develop within 1 or 2
hours after primary injury between the lesioned site and the remote
areas in both rostral and caudal directions. The tissue pressure
gradients influence spread of edema fluid across the spinal cord
(Sharma, 2002).
Profound edema development is seen within 30 min after SCI near
the lesion site that is progressive with time. Interestingly, the caudal
segments exhibited more pronounced edema development compared
to the rostral segments indicating that release of neurochemicals and

108
BSCB breakdown following SCI influences edema formation
(Mautes et al, 2000).
TTrreeaattmmeenntt SSttrraatteeggiieess iinn SSppiinnaall CCoorrddIInnjjuurryy::
There are reasons to believe that BSCB could be an important target
for the drugs used to treat SCI induced cell injury and sensory-motor
recovery. However, the current pharmacological strategies are not
well focused on the changes in the BSCB function after trauma in
relation to cord pathology or the functional outcome (Sharma,
2003).
The altered spinal cord microenvironment appears to be one of the
key factors in neuroprotection or sensory-motor recovery following
SCI. It is quite likely that drugs or therapeutic agents that offer
neuroprotection are able to minimize the BSCB disturbances. The
potential of these therapeutic agents in the treatment of SCI is the
subject of many researches. The main treatment strategies in spinal
cord injury can be summarized in (table 5) (Hagg & Oudega, 1998).

109
Table. 5: Treatment Strategies in Spinal Cord Injury:
1) Neuroprotective approach:directed against interrupting the cascade ofsecondary injury processes.limiting tissue damage.arrest or reverse sensory/motor function impairment.
2) Rehabilitating approach:directed against consequences of spinal cord injurystabilization of current status with traumatraining of reflexes and residual circuitsfor optimal living conditions
3) Regenerative approach:directed towards enhancement of axonal regenerationpurely experimental at this stageno experience in human spinal cord injury
(Sharma, 2003)
PPhhaarrmmaaccoollooggyy ooff tthhee BBSSCCBB iinn ssppiinnaall ccoorrdd
iinnjjuurryy::The pharmacological strategies in SCI are used to influence the
process of secondary injury cascade to limit tissue damage and to
improve sensory-motor function. Another pharmacological aspect in
SCI is to enhance axonal regeneration. This can either be achieved
using neurotrophic factors or blocking regeneration inhibiting
factors. There are many therapeutic aspects that can be used, and
will be summarized:

110
1) Neurotrophic factors:
Neurotrophic factors and their receptors are present in the
developing and adult spinal cord. The neurotrophin receptors
influence neuronal survival by modulation of neurotransmitters,
neuropeptides as well as their release in the spinal cord. The
receptors for both neurotrophins and cytokines are located on
neurons, glial cells, inflammatory cells, meninges, and blood vessels
in scar tissue. There are evidences that neurotrophins effect signaling
of cytokines (Oudega & Hagg, 1999).
Brain derived neurotrophic factor (BDNF) and insulin like growth
factor 1 (IGF-1) are members of neurotrophins family and induce
neuroprotection during ischemia and trauma. Exogenous supplement
of growth factors induces neuroprotection either by neutralizing the
influence of neurodestructive agents or by enhancing the influence
of neuroprotective substances. Pretreatment with BDNF or IGF-1
markedly attenuated the occurrence of gross visual swelling after
injury without influencing microhaemorrhages (Ruitenberg et al,
2003).
Attenuation of the BSCB permeability with neurotrophins
indicates their involvement in the secondary injury mechanisms
following trauma. A reduction in BSCB permeability reduces
leakage of plasma proteins and thus able to prevent vasogenic edema
formation (Lu & Waite, 1999).

111
2) Tumor necrosis factor alpha (TNF-) antiserum:
In the CNS, tumor necrosis factor alpha (TNF-) is a cytotoxic
cytokine that is upregulated within 1 to 6 hours following traumatic,
ischemic or hypoxic insults. Intrathecal administration of TNF-
antiserum attenuates nitric oxide (NO) production and induces
neuroprotection by neutralizing the effects of endogenous TNF-
(Lee et al, 2000).
3) Nitric oxide synthase antiserum:
Treatment with nitric oxide synthase (NOS) antiserum resulted in
a decrease in peptide or protein extravasation across the BSCB
following trauma. This indicates that NOS activation increases NO
production that disrupts the BSCB through intracellular signal
transduction. To further establish the therapeutic values of the NOS
antiserum, studies using its application at longer time intervals
following SCI on the BSCB breakdown and cell injury are needed
(Hooper et al, 2000).
4) Antioxidant compounds:
Microhaemorrhages and extravasation of blood components
caused by SCI is one of the important sources of oxidative stress and
generation of free radicals that disrupt myelin sheaths and induce
cell damage, hemoglobin is an important source of iron to catalyze
oxygen radicals and lipid peroxidation (Calbrese et al, 2000).
Treatment with one potent chain-breaking antioxidant compound
H-290/51 attenuated trauma induced BSCB disruption to Evans blue

112
albumin (EBA) and radioiodine tracers. These observations suggest
that lipid peroxidation and generation of free radicals contributes to
the BSCB breakdown in SCI (Mustafa et al, 1995).
A significant reduction in water content and mild perivascular
edema, swelling of nerve cells and myelin vesiculation at the
ultrastructural level in the drug treated group supports this idea
(Tong et al, 1998).
5) Prostaglandins:
The precursor of prostaglandins (PGs) arachidonic acid and its
metabolite are involved in the secondary injury processes.
Pretreatment with indomethacin, a potent inhibitor of
cyclooxygenase enzyme, significantly attenuated edema formation
and cell damage. These results support a role of PGs in the
endothelial cell membrane permeability. Whether the effects of PGs
on BSCB permeability are mediated by specific PG receptors, are
still unclear (Leskovar et al, 2000).
6) Bradykinin (BK):
Blockade of BK2 receptor antagonist slightly but significantly
reduced the breakdown of the BSCB to EBA, radioiodine and
lanthanum tracers. Edema formation and cell injury in the drug
treated traumatized cord are considerably reduced. These
observations demonstrate that bradykinin is involved in the
breakdown of the BSCB permeability probably through BK2
receptors (Bogar et al, 1999).

113
7) Opioid Peptides:
Opioid and non opioid neuropeptides, together with monoamines
and amino acids play integral roles in the neurotransmission in the
spinal cord. Intrathecal or systemic administration of selective -
opioid antagonist nor-binaltrophimine (nor-BNI) enhances
neurological recovery after spinal cord trauma suggests an
involvement of - opioid receptors in SCI (Tang et al, 2000).
The natural ligand of the - opioid receptors, dynorphin that is
well known to participate in the pathophysiology of SCI supports
this idea. Treatment with dynorphin A (1–17) antiserum improves
the neurological outcome after SCI, At 5 h the gross swellings of the
spinal cord, BSCB disruption and edema formation are significantly
reduced. Trauma induced cell injury; myelin vesiculation and
membrane disruption are also reduced by dynorphin antiserum
(Hauser et al, 2001).
8) Adrenergic receptor blockers:
On the basis of norepinephrine accumulation in the traumatized
cord, role of catecholamines in SCI was suggested by Osterholm and
Mathews. However, inhibition of catecholamines synthesis with -
methyltyrosine; or blockade of - adrenergic receptor with clonidine
yielded controversial results (Faden & Salzman, 1992).
Some trials which examined the influence of potent - and -
adrenergic receptor antagonists, phenoxybenzamine and propranolol,
respectively on edema formation and BSCB disruption in SCI

114
yielded that: pretreatment with - or - adrenergic receptor blockers
did not attenuate BSCB permeability and edema formation. Thus,
further studies using adrenoceptor agonists are needed to clarify the
involvement of catecholamines in SCI (Winkler et al, 1998).

115
SSuummmmaarryy
The concept of cerebral edema has been recognized for more
than 2000 years, yet an understanding of the complex physiology of
this condition has evolved only within the past 30 years.
Hippocrates noted that removal of the overlying skull bones
allowed the injured brain to swell outward, thus minimizing
compression of normal tissue trapped within the cranial vault.
The Monro–Kellie doctrine later recapitulated this concept,
affirming that when ‘‘water or other matter is effused or secreted
from the blood vessels ... a quantity of blood equal in bulk to the
effused matter, will be pressed out of the cranium.’’
This indiscriminate concept of brain swelling was cited in a
diverse range of clinical settings until 1967, when Igor Klatzo
defined the modern classification of edema based on
pathophysiology. Cerebral edema, according to Klatzo, was defined
as ‘‘an abnormal accumulation of fluid associated with volumetric
enlargement of the brain.’’
This entity was divided into vasogenic edema, characterized by
derangement of the blood–brain barrier (BBB), and cytotoxic
edema, related to intracellular swelling in the absence of changes at
the BBB. Klatzo emphasized that these two forms usually
coexisted.

116
In 1975, Robert Fishman added interstitial edema as a distinct
entity by describing the transependymal flow of cerebrospinal fluid
(CSF) into the periventricular white matter in individuals with
acute obstructive hydrocephalus; this form was later termed
hydrocephalic edema.
This classification is highly simplistic, given that it pertains to
complex pathophysiological and molecular mechanisms, but is
valuable as a simple therapeutic guide for treatment of cerebral
edema. Most brain insults involve a combination of these
fundamental subtypes of edema, although one can predominate
depending on the type and duration of injury.
Cytotoxic edema results from swelling of the cellular elements
(neurons, glia, and endothelial cells) because of substrate and
energy failure, and affects both gray and white matter. This edema
subtype is conventionally encountered in: cerebral ischemia,
traumatic brain injury, infections, and metabolic disorders
including kidney and liver failure.
Vasogenic edema that results from breakdown of the BBB due to
increased vascular permeability, as commonly encountered in:
hemorrhage, later stages of brain infarction, TBI, infections,
seizures, trauma, tumors, radiation injury and hypertensive
encephalopathy, predominantly affects white matter.
This edema subtype is responsive to both steroid administration
(notably edema associated with neoplasms) and osmotherapy.
Other causes of vasogenic edema include tissue hypoxia and water

117
intoxication that may be responsive to osmotherapy but resistant to
steroid administration.
Interstitial edema, a consequence of impaired absorption of CSF,
leads to increases in transependymal CSF flow, resulting in acute
hydrocephalus. This edema subtype is also not responsive to steroid
administration, and its response to osmotherapy is debatable.
In osmotic edema there is an osmotic gradient which is present
between plasma and the extracellular fluid. Edema may occur with
a number of hypo-osmolar conditions including: improper
administration of intravenous fluids leading to acute dilutional
hyponatremia, inappropriate antidiuretic hormone secretion,
excessive hemodialysis of uremic patients and diabetic
ketoacidosis.
Basic information about the types of edema is provided for better
understanding of the expression pattern of some of the newer
molecules implicated in the pathogenesis of brain edema. These
molecules include the aquaporins (AQP), matrix metalloproteinases
(MMPs) and growth factors such as vascular endothelial growth
factors (VEGF) A and B and the angiopoietins. The potential of
these agents in the treatment of edema is the subject of many
reviews.
Blood-spinal cord barrier (BSCB) plays an important role in the
regulation of the fluid microenvironment of the spinal cord. Trauma
to the spinal cord impairs the BSCB permeability to proteins
leading to vasogenic edema formation. Several endogenous

118
neurochemical mediators and growth factors contribute to trauma
induced BSCB disruption.
Studies carried out suggest that those drugs and neurotrophic
factors capable to attenuate the BSCB dysfunction following
trauma are neuroprotective in nature. Whereas, agents that do not
exert any influence on the BSCB disruption failed to reduce cell
injury. These observations are in line with the idea that BSCB
disruption plays an important role in the pathophysiology of spinal
cord injuries.
Neuroimaging by CT scans and magnetic resonance imaging can
be particularly useful in confirming intracranial compartmental and
midline shifts, herniation syndromes, ischemic brain injury, and
exacerbation of cerebral edema (sulcal effacement and obliteration
of basal cisterns).
The consequences of cerebral edema can be lethal and include
cerebral ischemia from compromised regional or global cerebral
blood flow (CBF) and intracranial compartmental shifts due to
intracranial pressure gradients that result in compression of vital
brain structures. The overall goal of medical management of
cerebral edema is to maintain regional and global CBF to meet the
metabolic requirements of the brain and prevent secondary
neuronal injury from cerebral ischemia.
Medical management of cerebral edema involves using a
systematic and algorithmic approach, from general measures
(optimal head and neck positioning for facilitating intracranial

119
venous outflow, avoidance of dehydration and systemic
hypotension, and maintenance of normothermia) to specific
therapeutic interventions (controlled hyperventilation,
administration of corticosteroids and diuretics, osmotherapy, and
pharmacological cerebral metabolic suppression).

120
DDiissccuussssiioonn::
Hence the significance of brain edema, which continues to be a
major cause of mortality after diverse types of brain pathologies, the
lack of effective treatment, remains a stimulus for continued interest
and research into the pathogenesis of this condition (Kempski,
2001).
Though there has been good progress in understanding of
pathophysiological mechanisms associated with cerebral edema
more effective treatment is required and is still awaited (Marmarou
et al, 2006).
Certainly, the “ideal” agent for the treatment of cerebral edema-
one that would selectively mobilize and / or prevent the formation of
edema fluid with a rapid onset and prolonged duration of action, and
with minimal side effects, remains to be discovered (Abbott, 2004).
The treatment of cerebral edema remains largely empirical.
Options are relatively limited, and the mechanisms of action of most
of the therapeutic agents and interventions currently used are not
fully elucidated (Ahmed & Anish, 2007).
Research in the last decade has led to an appreciation of the
complexity of brain edema pathogenesis and to the awareness that
many molecules are involved acting simultaneously or at different
stages during the edema process (Johnston & Teo, 2000)

121
This suggests that effective treatment of brain edema cannot be
achieved by a single agent, but will require the administration of a
‘‘magic bullet’’ containing a variety of agents released at different
times during the course of edema in order to be successful
(Alejandro & Rabinstein, 2006)
Although protocols and algorithms exist to treat brain edema
associated with specific neurologic entities, these are not based on
rigorous scientific data (Kimelburg, 2004).
Current uncertainties and deficiencies must be resolved by
continuing research, fueled by growing understanding of the
pathophysiological processes responsible for the formation of the
different forms of brain edema (Nag, 2003) b.
Probably in the days to come we can look forward to newer
agents specifically acting on the various chemical mediators
involved in the pathogenesis of cerebral edema (Kuroiwa et al,
2007).
Traumatic insults to the spinal cord disrupt the functional
integrity of the blood-spinal cord barrier (BSCB) and results into an
increased transport of several substances from the vascular
compartment to the spinal cord cellular microenvironment.
Transport of macromolecules like proteins from the vascular
compartment to the spinal cord microenvironment induces vasogenic
edema (Sharma, 2003).
New pharmacotherapeutic agents and compounds that reduce
trauma induced alterations in the BSCB and cell injury may

122
strengthen the effects of endogenous neuroprotective agents and
minimize the adverse influence of endogenous neurodestructive
elements. Thus, drugs, compounds or agents that are capable to
minimize trauma induced BSCB breakdown could be the promising
therapeutic agents for the treatment of SCI in the future (Sharma,
2005).

123
RReeffeerreenncceess::
Abbott A: Evidence for bulk flow of brain interstitial
fluid: significance for physiology and pathology. Neurochem Int
(2004) 45, 545 – 52.
Adams RH & Alitalo K: Molecular regulation of
angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol (2007)
8:464– 478. doi:10.1038/nrm2183.
Ahmed R & Anish B: Medical management of cerebral
edema. Neurosurg Focus (2007), 22 (5):E12.
Alejandro A. & Rabinstein: Treatment of Cerebral
Edema. The Neurologist (2006); 12: 59–73.
Amiry-Moghaddam M, Ottersen OP. The molecular
basis of water transport in the brain. Nat Rev Neurosci (2003); 4: 991–
1001.
Asahi M, Wang X, Mori T, et al: Effects of matrix
metalloproteinase-9 gene knock-out on the proteolysis of blood–
brain barrier and white matter components after cerebral ischemia. J
Neurosci (2001) 21:7724–7732.
Ayata C & Ropper AH: Ischaemic brain oedema. J
Clin Neurosci (2002) 9:113–124. doi:10.1054/jocn.2001.1031.
Badaut J, Hirt L, Granziera C, et al: Astrocyte-
specific expression of aquaporin-9 in mouse brain is increased after

124
transient focal cerebral ischemia. J Cereb Blood Flow Metab (2001)
21:477–482. doi:10.1097/00004647- 200105000-00001.
Ball AK & Clarke CE: Idiopathic intracranial
hypertension. Lancet Neurol (2006), 5, 433-42.
Ballabh P, Braun A, Nedergaard M: The blood brain
barrier: an overview. Structure regulation and clinical implication.
Neurobiol Dis (2004),16, 1-13.
Banasiak KJ, Burenkova O, Haddad GG. Activation
of voltage sensitive sodium channels during oxygen deprivation
leads to apoptotic neuronal death. Neuroscience (2004); 126: 31–44.
Bastin ME, Sinha S, Whittle IR, et al.: Measurements
of water diffusion and T1 values in peritumoural oedematous brain.
Neuroreport (2002); 13:1335–40.
Battison C, Andrews PJ, Graham C, et al:
Randomized, controlled trial on the effect of a 20% mannitol solution
and a 7.5% saline/6% dextran solution on increased intracranial
pressure after brain injury. Crit Care Med (2005) 33:196–202.
Berger S, Schurer L, Hartl R, et al.: Reduction of
post-traumatic intracranial hypertension by hypertonic/hyperoncotic
saline/dextran and hypertonic mannitol. Neurosurgery. (1995); 37:98
–107.
Bhardwaj A & Ulatowski JA: Hypertonic saline
solutions in brain injury. Curr Opin Crit Care (2004) 10:126–131.
Bloch O, Auguste KI, Manley GT, et al: Accelerated
progression of kaolin-induced hydrocephalus in aquaporin-4-

125
deficient mice. J Cereb Blood Flow Metab (2006) 26:1527–1537.
doi:10.1038/sj.jcbfm.9600306.
Bogar LJ, Bartula LL, Parkman HP, et al: Enhanced
bradykinin-stimulated prostaglandin release in the acutely inflamed
guinea pig gallbladder is due to new synthesis of cyclooxygenase 1
and prostacyclin synthase. J Surg Res (1999); 84: 71-76.
Boyd NL, Park H, Yi H, et al: Chronic shear induces
caveolae formation and alters ERK and Akt responses in endothelial
cells. Am J Physiol Heart Circ Physiol (2003) 285:H1113–H1122.
Broderick JP, Adams HP Jr, Barsan W, et al:
Guidelines for the management of spontaneous intracerebral
hemorrhage. A statement for healthcare professionals from a special
group of the Stroke Council, American Heart Association. (1999)
Stroke 30:905–915.
Bruno A, Williams LS, Kent TA: How important is
hyperglycemia during acute brain infarction? Neurologist (2004) 10:
195–200.
Bullock R, Chesnut RM, Clifton G, et al: Guidelines
for the management of severe head injury: Brain Trauma Foundation.
Eur J Emerg Med (1996) 3:109–127.
Bullock M, Chestnut R, Ghajar J, et al.: Guidelines
for the Surgical Management of Traumatic Brain Injury. Surgical
management of traumatic parenchymal lesions. Neurosurgery.
(2006); 58(3 suppl):S25-S46.

126
Buschmann U, Yonekawa Y, Fortunati M, et al:
Decompressive hemicraniectomy in patients with subarachnoid
hemorrhage and intractable intracranial hypertension. Acta Neurochir
(Wien). (2007); 149: 59-65.
Calbrese V, Bates TE, Giuffrida Stella AM.: NO
synthase and NOdependent signal pathways in brain aging and
neurodegenerative disorders: The role of oxidant/antioxidant balance.
Neurochem Res (2000); 25: 1315-341.
Candelario-Jalil E, Yang Y, Rosenberg GA: Diverse
roles of matrix metalloproteinases and tissue inhibitors of
metalloproteinases in neuroinflammation and cerebral ischemia.
Neuroscience.(2008).158:983 994.doi:10.1016/j.neuroscience06.
025
Carmona Suazo JA, Maas AI, van den Brink WA, et
al.: CO2 reactivity and brain oxygen pressure monitoring in severe
head injury. Crit Care Med. (2000); 28:3268 –3274.
Chang DI, Hosomi N, Lucero J, et al: Activation
systems for latent matrix metalloproteinase-2 are upregulated
immediately after focal cerebral ischemia. J Cereb Blood Flow
Metab (2003) 23:1408–1419.
Clifton GL, Miller ER, Choi SC, et al: Lack of effect
of induction of hypothermia after acute brain injury. N Eng J Med
(2001) 344:556–563.
Coplin WM, Cullen NK, Policherla PN, et al.: Safety
and feasibility of craniectomy with duraplasty as the initial surgical

127
intervention for severe traumatic brain injury. J Trauma. (2001);
50:1050 –1059.
Cosnard G, Duprez T, Grandin C, et al.: Diffusion
and perfusion-weighted MR imaging during the hyperacute phase of
stroke. J Radiol (2000) 81(8):858–869.
Coutts SB, Lev MH, Eliasziw M, et al.: ASPECTS on
CTA source images versus unenhanced CT: added value in
predicting final infarct extent and clinical outcome. Stroke (2004);
35:2472–6.
Czosnyka M, Czosnyka & Z Momjian S, et al:
Cerebrospinal fluid dynamics. Physiolog Measurement (2004) 25,
R51-76.
Demediuk P, Saunders RD, Anderson DK, et al.:
Early membrane lipid changes in laminectomized and traumatized
cat spinal cord. Neurochem Pathol (1989); 7: 79-89.
Dennis MS: Outcome after brain hemorrhage.
Cerebrovasc Dis (2003), 16, 9-13.
Diringer M.: Hyperventilation in head injury: what
have we learned in 43 years? Crit Care Med. (2002); 30:2142–2143.
Diringer MN & Zazulia AR: Osmotic therapy: fact or
fiction. Neurocrit Care(2004) 1:219–234.
Doerfler A, Engelhorn T, Heiland S, et al: Perfusion-
and diffusion-weighted magnetic resonance imaging for monitoring
decompressive craniectomy in animals with experimental
hemispheric stroke. J Neurosurg. (2002); 96:933-940.

128
Dolman D, Drndarski S, Abbott NJ, et al: Induction
of aquaporin 1 but not aquaporin 4 messenger RNA in rat primary
brain microvessel endothelial cells in culture. J Neurochem (2005)
93:825–833. doi:10.1111/j.1471-4159.2005.03111.x.
Eccher M & Suarez JI: Cerebral edema and
intracranial pressure. Monitoring and intracranial dynamics, in
Suarez JI (ed): Critical Care Neurology and Neurosurgery. Totowa,
NJ: Humana Press, (2004), pp 47–100.
Faden AI & Salzman S.: Pharmacological strategies in
CNS trauma. Trends Pharmacol Sci (1992); 13: 29-35.
Ferrara N, Gerber HP, LeCouter J: The biology of
VEGF and its receptors. Nat Med (2003) 9:669–676.
doi:10.1038/nm0603-669.
Forster C: Tight junctions and the modulation of barrier
function in disease. Histochem Cell Biol (2008) 130:55–70. doi:
10.1007/s00418-008-0424-9.
Freshman SP, Battistella FD, Matteucci M, et al.:
Hypertonic saline (7.5%) versus mannitol: a comparison for
treatment of acute head injuries. J Trauma. (1993); 35:344 –348.
Furman CS, Gorelick-Feldman DA, Davidson KG, et
al: Aquaporin-4 square array assembly: opposing actions of M1 and
M23 isoforms. Proc Natl Acad Sci USA (2003) 100:13609–13614.
doi:10.1073/pnas.2235843100.

129
Furuse M & Tsukita S: Claudins in occluding
junctions of humans and flies. Trends Cell Biol: (2006) 16:181–188.
doi:10.1016/ j.tcb.2006.02.006.
Gadkary CS, Alderson P, Signorini DF: Therapeutic
hypothermia for head injury. Cochrane Database Syst Rev (2002) 1:
CD001048.
Gemma M, Cozzi S, Tommasino C, et al.: 7.5%
Hypertonic saline versus 20% mannitol during elective neurosurgical
supratentorial procedures. J Neurosurg Anesthesiol. (1997); 9:329 –
334.
Glantz MJ, Cole BF, Forsyth PA, et al: Practice
parameter: anticonvulsant prophylaxis in patients with newly
diagnosed brain tumors. Report of the Quality Standards
Subcommittee of the American Academy of Neurology (2000).
Neurology 54:1886–1893.
Glasr N, Barnett P, McCaslin D et al.: Risk factors for
cerebral edema in children with diabetic ketoacidosis. N Engl J Med
(2001); 344:264 9.
Hagg T & Oudega M.: Neurotrophic factors and CNS
regeneration. In: Stålberg E, Sharma HS, Olsson Y Eds, Spinal Cord
Monitoring. Basic Principles, Regeneration, Pathophysiology and
Clinical Aspects, Springer Wien New York (1998); 129-55.
Harukuni I, Kirsch J, Bhardwaj A: Cerebral
resuscitation: role of osmotherapy. J Anesth (2002) 16:229–237.

130
Hauser KF, Knapp PE, Turbek CS.: Structure-
activity analysis of dynorphin A toxicity in spinal cord neurons:
Intrinsic neurotoxicity of dynorphin A and its carboxyl-terminal,
nonopioid metabolites. Exp Neurol (2001); 168: 78-87.
Hawkins BT & Davis TP. The blood-brain barrier /
neuro - vascular unit in health and disease. Pharmacol Rev (2005);
57: 173–85.
Hawkins BT & Egleton RD: Pathophysiology of the
blood– brain barrier: animal models and methods. Curr Top Dev Biol
(2008) 80:277–309. doi:10.1016/S0070-2153(07)80007-X.
Hemphill JC, Beal MF, Gress DR.: Critical care in
neurology. In : Braunwald E, Fauci AS, Kasper DL, Hauser SL,
Longo DL, Jameson JL, editors. Harrison’s Principles of Internal
Medicine. 15th ed. New York: Mc Graw Hill, (2001):2491-8.
Holzer M, Bernard SA, Hachimi-Idrissi S, et al:
Hypothermia for neuroprotection after cardiac arrest: systematic
review and individual patient data metaanalysis. Crit Care Med
(2005) 33:414–418.
Hooper DC, Scott GS, Zborek A, et al.: Uric acid, a
peroxynitrite scavenger, inhibits CNS inflammation, and tissue
damage in a mouse model of multiple sclerosis. FASEB J (2000); 14:
691-98.
Jaillard A, Hommel M, Baird AE, et al.: Significance
of early CT signs in acute stroke: a CT scandiffusion MRI study.
Cerebrovasc Dis (2002); 13: 47 56.

131
James HE, Marshall LF, Reulen H-J, et al.: Brain
Edema X, Springer Wien, (1997) New York.
Johnston AJ, Steiner LA, Coles JP, et al. Effect of
cerebral perfusion pressure augmentation on regional oxygenation
and metabolism after head injury. Crit Care Med. (2005); 33:189 –
195.
Johnston IH & Teo C: Disorders of CSF
hydrodynamics. Cilds nerv syst (2000), 16, 776-99.
Kaal EC & Vecht CJ.: The management of brain
edema in brain tumors. Curr Opin Oncol. (2004); 16:593– 600.
Katayama Y & Kawamata T: Edema fluid
accumulation within necrotic brain tissue as a cause of the mass
effect of cerebral contusion in head trauma patients. Acta Neurochir
Suppl (Wien) (2003) 86:323–327.
Kempski O, von AU, Schurer L, et al: Intravenous
glutamate enhances edema formation after a freezing lesion. Adv
Neurol (1990) 52:219–223.
Kempski O: cerebral edema. Semin Nephrol (2001)
21:303–307. [PubMed: 11320499].
Kim DS & Garwood M: High field magnetic resonance
techniques for brain research. Curr Opin Neurobiol (2003) 13:612–
619.
Kimelburg HK: Water homeostasis in the brain: basic
concepts. Neuroscience (2004), 129, 851-60.

132
Korenkov AI, Pahnke J, Frei K, et al: Treatment with
nimodipine or mannitol reduces programmed cell death and infarct
size following focal cerebral ischemia. Neurosurg Rev (2000)
23:145–150.
Krieger DW, De Georgia MA, Abou-Chebl A, et al:
Cooling for acute ischemic brain damage (Cool Aid): an open pilot
study of induced hypothermia in acute ischemic stroke. Stroke (2001)
32:1847–1854.
Kuroiwa T, Miyasaka N, Fengyo Z, et al:
Experimental ischemic brain edema: morphological and magnetic
resonance imaging findings. Neurosurg Focus (2007) 22:E11.
doi:10.3171/foc.2007.22. 5.12.
Latour LL, Kang DW, Ezzeddine MA, et al:
Early blood-brain barrier disruption in human focal brain
ischemia. Ann Neurol (2004); 56: 468–77.
Lee YB, Yune TY, Bail SY, et al.: Role of tumor
necrosis factor-a in neural and glial apoptosis after spinal cord injury.
Exp Neurol (2000); 166: 190-95.
Leira R, Davalos A, Silva Y, et al.: Early neurologic
deterioration in intracerebral hemorrhage: predictors and associated
factors. Neurology. (2004); 63:461– 467.
Lemke DM & Hacein-Bey L. Cerebral venous sinus
thrombosis. J Neurosci Nurs. (2005); 37:258 –264.
Leskovar A, Moriarty LJ, Turek JJ, et al: Tha
macrophage in acute neural injury: Changes in cell number over time

133
and levels of cytokine prosuction in mammalian central and
peripheral nervous systems. J Exp Biol (2000); 203: 1783-795.
Li S & Tator Ch.: Spinal cord blood flow and evoked
potentials as outcome measures for experimental spinal cord injury.
In: Stålberg E, Sharma HS, Olsson Y, Eds., Spinal Cord Monitoring.
Basic Principles, Regeneration, Pathophysiology and Clinical
Aspects, Springer, Wien, New York (1998); pp 365 392.
Liang D, Bhatta S, Gerzanich V, et al: Cytotoxic
edema: mechanisms of pathological cell swelling. Neurosurg Focus
(2007) 22:E2. doi:10.3171/foc.22.5.3.
Lo EH, Dalkara T, Moskowitz MA: Mechanisms,
challenges and opportunities in stroke. Nat Rev Neurosci (2003)
4:399–415. doi:10.1038/nrn1106.
Longatti P, Basaldella L, Orvieto E, et al:
Aquaporin(s) expression in choroid plexus tumours. Pediatr
Neurosurg (2006) 42:228–233. doi:10.1159/000092359.
Lossinsky AS & Shivers RR: Structural pathways for
macromolecular and cellular transport across the blood–brain barrier
during inflammatory conditions. Histol Histopathol (2004) 19:535–
564 (Review).
Lu J & Waite P.: Advances in spinal cord regeneration.
Spine (1999); 24: 926-30.
Machein MR & Plate KH: VEGF in brain tumors. J
Neurooncol (2000) 50:109–120. doi:10.1023/A:1006416003964.

134
Marcella A. Madera, MD; Raj K. et al: Traumatic
brain injury. http://www.merckmanuals.com (2007).
Marion DW, Puccio A, Wisniewski SR, et al.: Effect
of hyperventilation on extracellular concentrations of glutamate,
lactate, pyruvate, and local cerebral blood flow in patients with
severe traumatic brain injury. Crit Care Med. (2002); 30:2619 –
2625.
Marmarou A, Signoretti S, Aygok G, et al: Traumatic
brain edema in diffuse and focal injury: cellular or vasogenic? Acta
Neurochir Suppl(2006);96:24–29. [PubMed: 16671417].
Marmarou A: A Review of progress in understanding
the pathophysiology and treatment of brain edema. Neurosurg Focus
(2007) 22:E1.
Marti HJ, Bernaudin M, Bellail A, et al: Hypoxia-
induced vascular endothelial growth factor expression precedes
neovascularization after cerebral ischemia. Am J Pathol (2000)
156:965–976.
Masuhr F, Mehraein S, Einhaupl K.: Cerebral venous
and sinus thrombosis. J Neurol. (2004); 251:11–23.
Mautes AE, Bergeron M, Sharp FR, et al.; Sustained
induction of heme oxygenase-1 in the traumatized spinal cord. Exp
Neurol (2000); 166: 254-65.
Mautes AE & Noble LJ.: Co-induction of HSP70 and
heme oxygenase-1 in macrophages and glia after spinal cord
contusion in the rat. Brain Res (2000); 883: 233-37.

135
Mayer SA & Rincon F: Treatment of intracerebral
hemorrhage. Lancet Neurol (2005), 4, 662-72.
Mendelow AD, Baethmann A, Czernicki Z, et al.:
Brain Edema XI, Springer Wien (2000), New York.
Meng S, Qiao M, Lin L, et al: Correspondence of
AQP4 expression and hypoxicischaemic brain oedema monitored by
magnetic resonance imaging in the immature and juvenile rat. Eur J
Neurosci (2004) 19:2261–2269. doi:10.1111/j.0953-
816X.2004.03315.x.
Milhorat TH, Clark RG, Hammock MK:
Experimental hydrocephalus. 2. Gross pathological findings in acute
and subacute obstructive hydrocephalus in the dog and monkey. J
Neurosurg (1970) 32:390–399.
Minshall RD, Sessa WC, Stan RV, et al: Caveolin
regulation of endothelial function. Am J Physiol Lung Cell Mol
Physiol (2003) 285:L1179–L1183.
Minshall RD & Malik AB: Transport across the
endothelium: regulation of endothelial permeability. Handb Exp
Pharmacol (2006) 107–144.
Mirski AM, Denchev ID, Schnitzer SM, et al.:
Comparison between hypertonic saline and mannitol in the reduction
of elevated intracranial pressure in a rodent model of acute cerebral
injury. J Neurosurg Anesthesiol. (2000); 12:334 –344.

136
Muizelaar JP, Marmarou A, Ward JD, et al: Adverse
effects of prolonged hyperventilation in patients with severe head
injury: a randomized clinical trial. J Neurosurg (1991) 75:731–739.
Mullins ME, Lev MH, Bove P, et al.: Comparison of
image quality between conventional and low-dose nonenhanced head
CT. AJNR Am J Neuroradiol (2004); 25:533–8.
Mustafa A, Sharma HS, Olsson Y, et al.: Vascular
permeability to growth hormone in the rat central nervous system
after focal spinal cord injury. Influence of a new antioxidant
compound H-290/51 and age. Neurosci Res (1995); 23: 185-94.
Nag S: The blood–brain barrier and cerebral
angiogenesis: lessons from the cold-injury model. Trends Mol Med
(2002) 8:38– 44. doi:10.1016/S1471-4914(01)02221-3.
Nag S, Eskandarian MR, Davis J, et al: Differential
expression of vascular endothelial growth factor-A (VEGF-A) and
VEGF-B after brain injury. J Neuropathol Exp Neurol (2002)
61:778–788.
Nag S: Pathophysiology of blood–brain barrier
breakdown. Methods Mol Med (2003) a 89:97–119.
Nag S: Blood–brain barrier permeability using tracers
and immunohistochemistry. Methods Mol Med (2003) b 89:133–144.
Nag S, Papneja T, Venugopalan R, et al: Increased
angiopoietin2 expression is associated with endothelial apoptosis and
blood–brain barrier breakdown. Lab Invest (2005) 85:1189– 1198.
doi:10.1038/labinvest.3700325.

137
Nag S: Structure and pathology of the blood–brain
barrier. In: Lathja A (ed) Handbook of neurochemistry and molecular
neurobiology. Springer, New York, (2007) pp 58–78.
Nathan BR & Scheld WM: New advances in the
pathogenesis and pathophysiology of bacterial meningitis. Curr
Infect Dis Rep (2000); 2:332-6.
Ng I, Lim J, Wong HB: Effects of head posture on
cerebral hemodynamics: its influences on intracranial pressure,
cerebral perfusion pressure, and cerebral oxygenation. Neurosurgery
(2004)54:593–597.
Nitta T, Hata M, Gotoh S, et al: Size-selective
loosening of the blood–brain barrier in claudin-5-deficient mice. J
Cell Biol (2003) 161:653–660. doi:10.1083/jcb.200302070.
Noble LJ & Wrathall JR.: Blood-spinal cord barrier
disruption proximal to a spinal cord transection in the rat: time
course and pathways associated with protein leakage. Exp Neurol
(1988); 99: 567-78.
Nusrat A, Parkos CA, Verkade P, et al: Tight
junctions are membrane microdomains. J Cell Sci (2000) 113(Pt
10):1771–1781.
Olsson Y, Sharma HS, Pettersson CÅV.: Effects of
pchlorophenylalanine on microvascular permeability changes in
spinal cord trauma. An experimental study in the rat using
131Isodium and lanthanum tracers. Acta Neuropathol (Berl) (1990);
79: 595-603.

138
Olsson Y, Sharma HS, Pettersson Å, et al:
Endogenous Release of Neurochemicals may increase Vascular
Permeability, induce Edema and Influence on Cell Changes in
Trauma to the Spinal Cord. Progress in Brain Research (1992); 91:
197-203.
Oshio K, Watanabe H, Song Y, et al: Reduced
cerebrospinal fluid production and intracranial pressure in mice
lacking choroid plexus water channel aquaporin- 1. FASEB (2005) J
19:76–78.
Otrock ZK, Makarem JA, Shamseddine AI: Vascular
endothelial growth factor family of ligands and receptors. Blood
Cells Mol Dis (2007) 38:258–268. doi:10.1016/j.bcmd.12.003
(Review).
Oudega M & Hagg T.: Neurotrophins promote
regeneration of sensory axons in the adult rat spinal cord. Brain Res
(1999); 818: 431-38.
PapadopoulosMC, Saadoun S, Binder DK, et al:
Molecular mechanisms of brain tumor edema. Journal Neuroscience
(2004) 129:1011–1020. doi:10.1016/j. neuroscience.2004. 05.044.
Papadopoulos MC & Verkman AS: Aquaporin-4 and
brain edema. Pediatr Nephrol (2007) 22:778–784.
doi:10.1007/s00467-006- 0411-0.
Papadopoulos MC & Verkman AS: Potential utility of
aquaporin modulators for therapy of brain disorders. Prog Brain Res
(2008) 170:589–601. doi:10.1016/S0079-6123(08)00446-9.

139
Parsons MW, Barber PA, Desmond PM, et al: Acute
hyperglycemia adversely affects stroke outcome: a magnetic
resonance and spectroscopy study. Ann Neurol (2002) 52:20–28.
Parton RG & Simons K: The multiple faces of
caveolae. Nat Rev Mol Cell Biol (2007) 8:185–194.
doi:10.1038/nrm2122.
Plumb J, McQuaid S, Mirakhur M, et al: Abnormal
endothelial tight junctions in active lesions and normal-appearing
white matter in multiple sclerosis. Brain Pathol (2002) 12:154–169.
Pollay M.: Blood-Brain Barrier, Cerebral Edema. In:
Wilkins RH, Rengachary SS, editors. Neurosurgery. 2nd ed. New
York: Mc Graw Hill Book Co., (1996); 335-44.
Popovich PG, Wei P, Stokes BT.: Cellular
inflammatory response after spinal cord injury in Sprague-Dawley
and Lewis rats. J Comp Neurol (1997); 377: 443-64.
Pouyssegur J, Dayan F, Mazure NM. Hypoxia
signalling in cancer and approaches to enforce tumour regression.
Nature (2006); 441: 437–43.
Powers WJ, Zazulia AR, Videen TO, et al.:
Autoregulation of cerebral blood flow surrounding acute (6 to 22
hours) intracerebral hemorrhage. Neurology. (2001); 57:18 –24.
Qureshi AI, Wilson DA, Traystman RJ.: Treatment of
elevated intracranial pressure in experimental intracerebral
hemorrhage: comparison between mannitol and hypertonic saline.
Neurosurgery. (1999); 44:1055–1063.

140
Qureshi AI & Suarez JI: Use of hypertonic saline
solutions in treatment of cerebral edema and intracranial
hypertension. Crit Care Med (2000) 28:3301–3313.
Raab S & Plate KH: Different networks, common
growth factors: shared growth factors and receptors of the vascular
and the nervous system. Acta Neuropathol (2007) 113:607–626. doi:
10.1007/s00401-007-0228-3.
Rabinstein AA: Found comatose, in Rabinstein AA,
Wijdicks EFM (eds): Tough Calls in Acute Neurology. Philadelphia:
Elsevier, (2004), pp 3–18.
Rabinstein AA: Treatment of brain edema. Neurologist
(2006)12: 59–73.
Ranjan P, Mishra AM & Kale R et al: Cytotoxic
edema is responsible for raised intracranial pressure in fulminant
hepatic failure: in vivo demonstration using DW-MRI in human
subjects. Metab Brain Dis (2005), 20, 181-92.
Rash JE, Davidson KG, Yasumura T, et al: Freeze
fracture and immunogold analysis of aquaporin-4 (AQP4) square
arrays, with models of AQP4 lattice assembly. Neuroscience (2004)
129:915–934. doi:10.1016/j.neuroscience, (2004).06.076.
Reiss Y, Machein MR, Plate KH: The role of
angiopoietins during angiogenesis in gliomas. Brain Pathol (2005)
15:311– 317.
Rizzo V, Morton C, DePaola N, et al: Recruitment of
endothelial caveolae into mechanotransduction pathways by flow

141
conditioning in vitro. Am J Physiol Heart Circ Physiol (2003)
285:H1720–H1729.
Roberto Cabeza & Alan Kingstone, Editors,
Handbook of Functional Neuroimaging of Cognition, Second
Edition, MIT Press, (2006), ISBN 0-262-03344-5.
Roberts I, Yates D, Sandercock P, et al: Effect of
intravenous corticosteroids on death within 14 days in 10008 adults
with clinically significant head injury (MRC CRASH trial):
randomised placebo controlled trial. Lancet (2004) 364:1321–1328.
Ropper AH, Gress DR, Diringer MN, et al:
Neurological and Neurosurgical Intensive Care. Philadelphia:
Lippincott, Williams & Wilkins (2004), pp 26–51.
Rosenberg GA.: Ischemic brain edema. Prog
cardiovasc Dis (1999); 42(3):209-16.
Rosenberg GA: Brain edema and disorders of
cerebrospinal fluid circulation. In: Bradley WG, Daroff RB, Ferichel
GM, Marsden CD, editors. Neurology in clinical practice. Vol 2.3rd
ed. Boston: Butterworth Heinmann(2000); 2:1545-59.
Rosenberg GA: Matrix metalloproteinases in
neuroinflammation. Glia, (2002), 39:279–291.
doi:10.1002/glia.10108.
Rosenberg GA & Yang Y: Vasogenic edema due to
tight junction disruption by matrix metalloproteinases in cerebral
ischemia. Neurosurg Focus (2007) 22:E4.
doi:10.3171/foc.2007.22.5.5.

142
Rosenblum WI: Cytotoxic edema: monitoring its
magnitude and contribution to brain swelling. J Neuropathol Exp
Neurol (2007) 66:771–778. doi:10.1097/nen.0b013e3181461965.
Rother J.: CT and MRI in the diagnosis of acute stroke
and their role in thrombolysis. Thromb Res, (2001); 103(Suppl 1):S1
25–33.
Roviezzo F, Tsigkos S, Kotanidou A, et al:
Angiopoietin-2 causes inflammation in vivo by promoting vascular
leakage. J Pharmacol Exp Ther (2005) 314:738–744.
doi:10.1124/jpet.105.086553.
Roy H, Bhardwaj S, Yla-Herttuala S: Biology of
vascular endothelial growth factors. FEBS Lett (2006) 580:2879–
2887. doi:10.1016/j.febslet.2006.03.087.
Saadoun S, Papadopoulos MC, Davies DC, et al:
Aquaporin-4 expression is increased in edematous human brain
tumors. J Neurol Neurosurg Psychiatry (2002) 72:262– 265.
doi:10.1136/jnnp.72.2.262.
Scarabino T, Nemore F, Giannatempo GM, et al.: 3.0
T imaging. Riv Neuroradiol (2004) 17:765–776.
Schell RM, Applegate RL II, Cole DJ: Salt, starch,
and water on the brain. J Neurosurg Anesthesiol (1996) 8:178–182.
Schilling L & Wahl M.: Mediators of cerebral edema.
Adv Exp Med Biol (1999); 474:123-41.

143
Schwab ME & Bartholdi D.: Degeneration and
regeneration of axons in the lesioned spinal cord. Phys Rev (1996);
76: 319-70.
Schwab S, Spranger M, Schwarz S, et al: Barbiturate
coma in acute hemispheric stroke: useful or obsolete? Neurology
(1997) 48: 1608–1613.
Schwarz S, Schwab S, Bertram M, et al.: Effects of
hypertonic saline hydroxyethyl starch solution and mannitol in
patients with increased intracranial pressure after stroke. Stroke.
(1998); 29:1550 –1555.
Schwarz S, Georgiadis D, Aschoff A, et al: Effects of
body position on intracranial pressure and cerebral perfusion in
patients with large hemispheric stroke. Stroke (2002) 33:497–501.
Scott A. Huettel, Allen W. Song, Gregory McCarthy:
Functional Magnetic Resonance Imaging, Sinauer Associates,
(2004), ISBN 0-87893-288-7.
Sharma HS, Olsson Y, Dey P K.: Early accumulation
of serotonin in rat spinal cord subjected to traumatic injury. Relation
to edema and blood flow changes. Neuroscience (1990); 36: 725-30.
Sharma HS.: Neurobiology of the CNS injury and
repair: new roles of amino acids, growth factors and neuropeptides.
Amino Acids (2002); 23: 217-19.
Sharma HS: Neurotrophic factors attenuate
microvascular permeability disturbances and axonal injury following

144
trauma to the rat spinal cord. Acta Neurochir (Wien) (2003); Suppl
86: 383- 88.
Sharma HS: Pathophysiology of the blood-spinal cord
barrier in traumatic injury. In: Sharma HS and Westman J Eds, The
Blood- Spinal Cord and Brain Barriers in Health and Disease.
Elsevier Academic Press, San Diego (2004); 437-518.
Sharma HS: Pathophysiology of Blood-Spinal Cord
Barrier in Traumatic Injury and Repair. Current Pharmaceutical
Design, (2005), 11, 1353-1389.
Silberstein C, Bouley R, Huang Y, et al: Membrane
organization and function of M1 and M23 isoforms of aquaporin-4 in
epithelial cells. Am J Physiol Renal Physiol (2004) 287:F501–F511.
doi:10.1152/ ajprenal.00439.2003.
Simma B, Burger R, Falk M, et al: A prospective,
randomized, and controlled study of fluid management in children
with severe head injury: lactated Ringer’s solution versus hypertonic
saline. Crit Care Med (1998) 26:1265–1270.
Simon RP & Beckman JS: Why pus is bad for the
brain. Neurology (2002); 58:167-8.
Simon J, Czechowsky D, Hill M, et al.: Fluid-
attenuated inversion recovery preparation: Not an improvement over
conventional diffusion-weighted imaging at 3 T in acute ischemic
stroke. AJNR (2004) 25:1653–1658.

145
Sinha S, Bastin ME, Wardlaw JM, et al: Effects of
dexamethasone on peritumoral oedematous brain: a DT-MRI study. J
Neurol Neurosurg Psychiatry (2004)75:1632 –1635.
Slivka AP & Murphy EJ: High-dose
methylprednisolone treatment in experimental focal cerebral
ischemia. Exp Neurol (2001) 167: 166–172.
Song L, Ge S, Pachter JS: Caveolin-1 regulates
expression of junction-associated proteins in brain microvascular
endothelial cells. Blood (2007) 109:1515–1523. doi:10.1182/blood-
2006-07-034009.
Stålberg E, Sharma HS, Olsson Y.: Spinal Cord
Monitoring. Basic Principles, Regeneration, Pathophysiology and
Clinical Aspects, Springer, Wien, New York (1998); pp 1-527.
Stanley J. & Swierzewski: Traumatic Brain Injury
(TBI) Prognosis. Http://www. HealthCommunities.com (2011).
Steiner T, Ringleb P, Hacke W.: Treatment options for
large hemispheric stroke. Neurology. (2001); 57(5 suppl 2):S61–S68.
Stummer W: Mechanisms of tumor-related brain
edema. Neurosurg Focus (2007) 22:E8. doi:10.3171/foc.2007.22.5.9.
Suarez JI. Treatment of acute brain edema. Rev Neurol
(2001); 32(3):275-81.
Subramaniam S & Hill MD.: Massive cerebral
infarction. Neurologist. (2005); 11:150 –160.

146
Sukriti Nag, Janet L. & Manias, et al: Pathology and
new players in the pathogenesis of brain edema. Acta Neuropathol
(2009), 118:197–217.
Sun MC, Honey CR, Berk C, et al: Regulation of
aquaporin-4 in a traumatic brain injury model in rats. J Neurosurg
(2003) 98:565–569.
Sypert GW: Stabilization and management of cervical
injuries. In: Pitt L H and Wagner F C Eds, Craniospinal Trauma,
New York, Thieme (1990); pp 363-70.
Tait MJ, Saadoun S, Bell BA, et al: Water movements
in the brain: role of aquaporins. Trends Neurosci (2008) 31:37–43.
doi:10.1016/j.tins.2007.11.003.
Takefuji S, Murase T & Sugimura Y: role of
microglia in the pathogenesis of osmotic- induced demyelination.
Exp Neurol (2007), 204, 88-92.
Tang Q, Lynch RM, Porreca F, et al: Dynorphin A
elicits an increase in intracellular calcium in cultured neurons via a
nonopioid, non-NMDA mechanism. J Neurophysiol (2000); 83:
2610- 615
Thenuwara K, Todd MM, Brian JE Jr: Effect of
mannitol and furosemide on plasma osmolality and brain water.
Anesthesiology (2002) 96:416–421
Toung TJ, Hurn PD, Traystman RJ, et al.: Global
brain water increases after experimental focal cerebral ischemia:
effect of hypertonic saline. Crit Care Med. (2002); 30:644–649.

147
Toung TJ, Chang Y, Lin J, et al.: Increases in lung
and brain water following experimental stroke: effect of mannitol
and hypertonic saline. Crit Care Med. (2005); 33:203–208.
Toung TJ, Chen CH, Lin C, et al: Osmotherapy with
hypertonic saline attenuates water content in brain and extracerebral
organs. Crit Care Med (2007)35:526–531.
Turksen K & Troy TC: Barriers built on claudins. J
Cell Sci (2004) 117:2435–2447. doi:10.1242/jcs.01235.
Unterberg AW, Stover J, Kress B, et al: Edema and
brain trauma. Neuroscience (2004) 129:1021–1029. doi:10.1016/
j.neuroscience.2004.06.046.
Van Itallie CM & Anderson JM: Claudins and
epithelial paracellular transport. Annu Rev Physiol (2006) 68:403–
429. doi:10.1146/annurev.physiol.68.040104.131404.
Vaquero J & Butterworth RF: Mechanisms of brain
edema in acute liver failure and impact of novel therapeutic
interventions. Neurol Res (2007) 29:683–690.
doi:10.1179/016164107X240099
Verkman AS: More than just water channels:
unexpected cellular roles of aquaporins. J Cell Sci (2005) 118:3225–
3232. doi:10.1242/jcs.02519.
Vespa PM, O’Phelan K, Shah M, et al: Acute seizures
after intracerebral hemorrhage: a factor in progressive midline shift
and outcome. Neurology (2003) 60:1441–1446.

148
Vialet R, Albanese J, Thomachot L, et al.: Isovolume
hypertonic solutes (sodium chloride or mannitol) in the treatment of
refractory posttraumatic intracranial hypertension: 2 mL/kg 7.5%
saline is more effective than 2 mL/kg 20% mannitol. Crit Care Med.
(2003); 31:1683–1687.
Victor M & Ropper AH: Disturbances of
cerebrospinal fluid and its circulation. Principles of Neurology. 7th
ed. New York: Mc Graw Hill, (2001) a; 655-75.
Victor M & Ropper AH: Cerebrovascular diseases.
Principles of Neurology. 7th ed. New York: Mc Graw Hill, (2001) b;
821-924.
Virgintino D, Robertson D, Errede M, et al:
Expression of caveolin-1 in human brain microvessels. Neuroscience
(2002) 115:145–152. doi:10.1016/ S0306-4522(02)00374-3.
VO KD, Lin W, Lee JM.: Evidence-based
neuroimaging in acute ischemic stroke. Neuroimaging Clin N Am
(2003); 13:167–83.
Volonte D, Galbiati F, Pestell RG, et al: Cellular stress
induces the tyrosine phosphorylation of caveolin-1 (Tyr14) via
activation of p38 mitogen-activated protein kinase and c-Src kinase.
J Biol Chem (2001) 276:8094–8103. doi:10.1074/ jbc.M009245200.
Vorbrodt AW & Dobrogowska DH: Molecular
anatomy of intercellular junctions in brain endothelial and epithelial
barriers: electron microscopist’s view. Brain Res Brain Res Rev
(2003) 42:221–242. doi:10.1016/S0165-0173(03)00177-2.

149
Vos CM, Geurts JJ, Montagne L, et al. Blood–brain
barrier alterations in both focal and diffuse abnormalities on
postmortem MRI in multiple sclerosis. Neurobiol Dis (2005);
20(3):953–60.
Wakai A, Roberts I, Schierhout G: Mannitol for acute
traumatic brain injury. Cochrane Database Syst Rev 1, (2007)
CD001049.
Wang X & Lo EH. Triggers and mediators of
hemorrhagic transformation in cerebral ischemia. Mol Neurobiol
(2003); 28: 229–44.
Ware ML, Nemani VM, Meeker M, et al: Effects of
23.4% sodium chloride solution in reducing intracranial pressure in
patients with traumatic brain injury: a preliminary study.
Neurosurgery (2005) 57:727–736.
Warth A, Simon P, Capper D, et al: Expression
pattern of the water channel aquaporin-4 in human gliomas is
associated with blood–brain barrier disturbance but not with patient
survival. J Neurosci Res (2007) 85:1336–1346.
doi:10.1002/jnr.21224.
Weed LH & McKibben PS: Experimental alteration of
brain bulk. Am J Physiol (1919) 48:531–555.
Williams TM & Lisanti MP: The caveolin genes: from
cell biology to medicine. Ann Med (2004) 36:584–595. doi:10.1080/
07853890410018899.

150
Winkler T, Sharma HS, Stålberg E, et al.: Spinal cord
bioelectrical activity, edema and cell injury following a focal trauma
to the spinal cord. An experimental study using pharmacological and
morphological approach. In: Stålberg E, Sharma HS, Olsson Y Eds,
Spinal Cord Monitoring. Basic Principles, Regeneration,
Pathophysiology and Clinical Aspects, Springer Wien New York
(1998); pp. 283-363.
Wolburg H, Noell S, Mack A, et al: Brain endothelial
cells and the glio-vascular complex. Cell Tissue Res (2008) 335:75–
96. doi:10.1007/s00441- 008-0658-9.
Xi G, Keep RF, Hoff JT: Pathophysiology of brain
edema formation. Neurosurg Clin N Am (2002) 13:371–383.
doi:10.1016/ S1042-3680(02)00007-4.
Xi G, Keep R, Hoff J.: Mechanisms of brain injury
after intracerebral haemorrhage. Lancet Neurol. (2006); 5:53–63.
Yamamoto N, Yoneda K, Asai K, et al: Alterations in
the expression of the AQP family in cultured rat astrocytes during
hypoxia and reoxygenation. Brain Res Mol Brain Res (2001) 90:26–
38. doi:10.1016/S0169- 328X(01)00064-X.
Yasser metwally: Vasogenic edema.
Http://yassermetwally.com (2009) Vasogenic edema.
Yla-Herttuala S, Rissanen TT, Vajanto I, et al:
Vascular endothelial growth factors: biology and current status of
clinical applications in cardiovascular medicine. J Am Coll Cardiol
(2007) 49:1015–1026. doi:10.1016/j.jacc.2006.09.053.

151
Zadeh G & Guha A: Neoangiogenesis in human
astrocytomas: expression and functional role of angiopoietins and
their cognate receptors. Front Biosci (2003) 8:e128–e137.
doi:10.2741/964.
Zador Z, Bloch O, Yao X, et al: Aquaporins: role in
cerebral edema and brain water balance. Prog Brain Res (2007)
161:185–194. doi:10.1016/S0079-6123(06)61012-1.
Zhang ZG, Zhang L, Croll SD, et al: Angiopoietin-1
reduces cerebral blood vessel leakage and ischemic lesion volume
after focal cerebral embolic ischemia in mice. Neuroscience (2002)
113:683–687. doi:10.1016/S0306-4522(02)00175-6.
Zlokovic BV: The blood–brain barrier in health and
chronic neurodegenerative disorders. Neuron (2008) 57:178–201.
doi:10.1016/j.neuron.2008.01.003.