toxicologic pathology - faculty.mercer.edufaculty.mercer.edu/zalups_rk/pdf manuscript files/toxicol...
TRANSCRIPT

http://tpx.sagepub.com/
Toxicologic Pathology
http://tpx.sagepub.com/content/26/1/92The online version of this article can be found at:
DOI: 10.1177/019262339802600111
1998 26: 92Toxicol PatholGary L. Diamond and Rudolfs K. Zalups
Understanding Renal Toxicity of Heavy Metals
Published by:
http://www.sagepublications.com
On behalf of:
Society of Toxicologic Pathology
can be found at:Toxicologic PathologyAdditional services and information for
http://tpx.sagepub.com/cgi/alertsEmail Alerts:
http://tpx.sagepub.com/subscriptionsSubscriptions:
http://www.sagepub.com/journalsReprints.navReprints:
http://www.sagepub.com/journalsPermissions.navPermissions:
http://tpx.sagepub.com/content/26/1/92.refs.htmlCitations:
at MERCER UNIV on September 29, 2010tpx.sagepub.comDownloaded from

TOXICOLOGIC PATHOLOGY, VOl. 26, no. 1 , pp. 92-103, 1998 Copyright 0 1998 by the Society of Toxicologic Pathologists
Understanding Renal Toxicity of Heavy Metals*
GARY L. DIAMOND' AND RUDOLFS K. ZALUPS* 'Syraciise Research Corporation, 6225 Rioiriitig Ridge Road, North Syracuse, New York 13212-2510, arid
2Division of Basic Medical Science. Mercer University School of Medicine, Macon, Georgia 31207
ABSTRAC~
The mechanisms by which metals induce renal injury are, in general, poorly understood. Characteristic features of metal nephro- toxicity are lesions that tend fo predominate in specific regions of the nephron within specific cell types. This suggests that certain regions of the nephron are selectively sensitive to specific metals. Regional variability in sensitivity could result from the localization of molecular targets in certain cell populations andlor the localization of transport and binding ligands that deliver metals to targets within the nephron. Significant progress has been made in identifying various extracellular, membrane, and intracellular ligands that are important in the expression of the nephrotoxicity of metals. As an example, mercuric chloride induces n nephropathy that, at the lowest effective doses, is restricted primarily to the S , segment of the proximal tubule, with involvement of the S , and S , segments at higher doses. This spccificity appears to bc derived, at least in part, from the distribution of enzymes and transport proteins important for the uptake of mercury into proximal tubule cells: apical y-glutamyltranspeptidase and the basolateral organic anion travsport system. Regional distributions of transport mechanisms for binding proteins appear to be important in the expression of nephrotoxicity of metals. Thcsc and other new research developments are reviewed.
Albumin; cadmium; cysteine; glutathione; kidney; mercury; metallothionein; nephrotoxicity; organic anion; p-amino- Keyr.ords. hippurate; transport; uranium
INTRODUCTION Characteristic features of metal nephrotoxicity are le-
sions that tend to predominate in specific regions of the proximal tubule and in the glomerulus. This suggests that these regions of the nephron may be selectively sensitive to specific metals. Regional variability in sensitivity to metals could result from the localization of transport and binding ligands that deliver metals to targets within the nephron andor the localization of molecular targets in certain cell populations. Although significant progress has been made in identifying various extracellular, mem- brane, and intracellular ligands that are important in the expression of the nephrotoxicity of metals, the mecha- nisms by which metals induce renal injury remain poorly understood. This is due, at least in part, to the complexity of the ligand interactions that dominate the disposition of metals in living organisms. For the most part, metals af- fect molecular processes through binding interactions with molecular targets, generally nucleophiles such as proteins and nucleic acids. These interactions may alter the normal function of the target molecule by displacing essential elements from the molecule, altering its struc- ture or changing its rate of metabolism or its interactions with other important ligands. While it is possible to study in great detail the binding interactions of metals and the consequences of binding on the function of specific bind- ing ligands, it is extremely difficult to determine which of these interactions contribute to the biochemical changes that give rise to toxicity. Thus, elucidating mech- anisms of toxicity requires an understanding of the patho- physiology that underlies toxicity, within which, specific
* Address correspondence to: Gary L. Diamond, Syracusc Rcscnrch Corporation, 6225 Running Ridge Road, North Syracuse. NY 13212- 2510.
92
molecular interactions can be placed in appropriate con- text.
GENERAL FEATURES OF METAL NEPHROTOXICITY The pathophysiology of acute uranium nephrotoxicity
has been extensively examined and reviewed (26) and exemplifies some general features of metal nephrotoxic- ity. Historically, intense interest in the toxicology of ura- nium originated with the Manhattan Project and the atom- ic energy program that required the processing and han- dling of large amounts of hexavalent uranium compounds (U6+) and that presented significant occupational health challenges (46). As new techniques for studying kidney morphology and function developed, research on uranium nephrotoxicology continued with the focus of using hex- avalent uranium compounds (e.g., uranyl acetate, uranyl fluoride, and uranyl nitrate) to explore mechanisms of acute renal failure (33). The results of these studies de- scribe the sequence of events that can occur in response to an acutely toxic dose of uranium and, although not entirely applicable to other metals, illustrate important kinetic and functional characteristics of proximal tubular and glomerular injury that are relevant to other metals.
Functional and morphologic changes that follow a sin- gle parented dose of uranyl nitrate, 6-15 mg U kg body weight (BW), in rats are summarized in Figs. 1 and 2. A qualitatively similar sequence of changes has been estab- lished for dogs and rabbits exposed to uranyl acetate (56, 91) and, in less detail, for dogs and rats exposed to uranyl fluoride (26, 63, 64, 74). Principal features of this se- quence can be divided into glomerular effects, including perfusion and filtration defects and morphological abnor- malities, and tubular effects, including transport and per- meability defects and necrosis. In general, a discussion of mechanisms of metal nephrotoxicity can be similarly
Ol92-6233/98$3.00+$0.00
at MERCER UNIV on September 29, 2010tpx.sagepub.comDownloaded from

Vol. 26, No. 1 , 1998 RENAL TOXICITY OF HEAVY METALS 93
DISTAL CONVOLUTED
Macula Densa TUBULE c __-- !t -- ROXIMAL TUBULE Pars Convoluta
_---- - -
CORTICAL -- - - _ _ SEGMENT
- - - --_ -
THIN L I M B O F H E N L E
THICK ASCENDING LIMB
FIG. 1.-Primary sites of uranium-induced injury in the rat (stippled areas). Necrosis of the pars recfa segment of the proximal tubule, and to a lesser extent, distal segments of the nephron are prominent features. Ultrastructural abnormalities irrth lomerular endothelium and epithe- lium have been documented at dose 1 vels exceeding 6 mg Ukg body weight.
approached as a discussion mechanisms.
1 f glomerular and tubular
MECHANIShlS OF GLOMERULAR TOXICITY At sufficiently high dose levels, most heavy metals will
depress the glomerular filtration rate (GFR). The Starling equation for capillary filtration, which describes the ma- jor determinants of GFR, provides a conceptual model with which to explore potential mechanisms:
J , = K,[(P,, - PI) - (Tgc - T I ) ]
K , = LpAj
J, = single nephron GFR (nl min-I) K, = glomerular ultrafiltration coefficient
P,, = glomerular capillary hydraulic pressure (mmHg);
T~~ = glomerular capillary fluid oncotic
(nl min-I mmHg-I);
PI = tubular fluid hydraulic pressure (mmHg);
pressure (mmHg); nf = tubular fluid oncotic pressure (mmHg); Lp = hydraulic conductivity of the glomerular
A , = filtration surface area (cm2). filter (nl min-I cm-2 mmHg);
Net filtration pressure is determined by the balance be-
tween hydraulic (P,, - PI) and oncotic (ng, - TJ pressure gradients along the glomerular capillary. Filtration pro- ceeds along the length of the capillary as long as net hydraulic pressure exceeds net oncotic pressure. If at some point along the glomerular capillary net oncotic pressure increases sufficiently to equal net hydraulic pres- sure, then a condition of filtration pressure equilibrium is established and filtration ceases at that point.
A consistently reported feature of uranium-induced acute renal failure in dogs, rabbits and rats is a decrease in renal outer cortical blood flow and glomerular perfu- sion (34, 35, 77, 90, 91). Decreased glomerular perfusion could lower GFR by decreasing glomerular capillary hy- draulic pressure (P,,) or by shifting the point of filtration pressure equilibrium toward the afferent end of the glo- merular capillary, thereby decreasing the length of the capillary that is available for filtration. In at least one study, GFR was observed to decrease in the absence of significant changes in renal plasma flow in rats treated with uranyl nitrate (12). Therefore, decreased renal plas- ma flow does not appear to be required for decreasing GFR, and other factors must be involved.
A rise in tubular hydraulic pressure ( P J , as a result of impaired tubular solute and fluid reabsorption or tubular blockage, also may contribute to decreased GFR. Al- though elevated tubular hydraulic pressure has been re- ported to occur in rats exposed to 6 mg U k g BW par- enteral uranyl nitrate (72) and in isolated perfused dog kidneys exposed in vitro to uranyl nitrate (76), it is not a consistent finding (34, 90); thus, it cannot be the sole mechanism responsible for decreasing GFR.
A complete quantitative description of the Starling forces in the glomerulus is possible in the Munich-Wistar rat, which possesses glomeruli that are located on the surface of the kidney and can be micropunctured for mea- surement of glomerular hydrostatic and oncotic pressures (14). In this animal model, a 50% decrease in single nephron filtration rate was observed during the first 6 hr after injection of 15 mg U k g BW uranyl nitrate, con- current with significant elevation of net glomerular filtra- tion pressure and in the absence of a change in renal plasma flow (12). This study demonstrates the importance of changes in the ultrafiltration coefficient (K,) in urani- um-induced depression of GFR.
Abnormalities in the ultrastructure of the glomerulus occur concurrently with the decrease in GFR. In rats, di- ameter and density of glomerular endothelial cell fenes- trae decrease 7 hr after a parented dose of 9-15 mg U k g BW uranyl nitrate (6). Such changes may give rise to a decrease in filtration surface area (A,) and, thereby, a decrease in ultrafiltration coefficient. Endothelial changes are followed by distortions of the glomerular vis- ceral epithelium (6, 43), suggesting that the endothelium, rather than the epithelium, may be the primary target of uranium-induced depression of GFR. In rabbits, concur- rent endothelial and epithelial changes in the glomerulus have been reported (56). Epithelial changes include spreading and flattening of the visceral epithelial cells (podocytes) and loss of podocyte processes, leading to decreased epithelial coverage of the glomerular endothe- lium.
at MERCER UNIV on September 29, 2010tpx.sagepub.comDownloaded from

94 DIAMOND AND ZALUPS ToxIcoLoG~c PATHOLOGY
TIMECOURSE FOR FUNCTONAL AND MORPHOLCGIC CHANGES AFTER PARENTERAL URANYL URANIUM IN THE RAT
I 1 1 1
1 CcnlW BLtoD FLO\Y
I GfR. GLO.IERULMI KI
1 NI. REABSWPlIOi.~OSL41u
I P l PERMEABILITY 1x)RhtAL GFR. UFR.IOSMIU +
FIG. 2.Time-course for functional and rnorphologic changes after parenteral uranium in the rat. Abbreviations: BB, brush border; DT, distal tubule; GFX, glomerular filtration rate; K,, glornerular filtration coefficient; OSM, osmolarity; PCT; proximal convoluted tubule (pars conlalofa); PST, proximal straight tubule (pars recta); F’T, proximal tubule; TEA’, tetraethyl ammonium; UFX, urine flow rate.
Both uranium and mercury can induce albuminuria in animals (27, 68, 116). The albuminuria may reflect im- paired reabsorption of protein, as it occurs in association with cellular and tubular necrosis; however, increased fil- tration of albumin also may be a contributing factor. Ef- facement of glomerular epithelial cells would be expected to alter the sieving properties of the glomerulus and may contribute to glomerular proteinuria; however, definitive evidence for increased filtration of albumin in animals treated with uranium has not been established. In rabbits, clearance of neutral dextrans having molecular weights of 70,000-90,000 daltons is not affected by a parenteral dose of 0.2 mgkg BW uranyl acetate, suggesting that mechanisms other than a change in the size selectivity of the glomerular filter contribute to proteinuria (36). A rig- orous examination of the effect of uranium compounds on the charge selectivity of the glomerulus has not been reported.
Our understanding of mercury-induced albuminuria is no more complete than that for uranium; that is, it is unclear to what extent albuminuria is of glomerular or tubular origin. Evidence for mercury-induced glomerular albuminuria is indirect. In rabbits, rats, and mice, multi- ple exposures to inorganic mercury induce the production of antibodies against the glomerular basement membrane, deposition of immune complexes in the mesangium and glomerular basement membrane, and glomerular nephritis (1 1, 29-31, 48, 83, 86, 96). In general, immune-complex glomerulonephritis has been associated with a disruption of the charge and/or size-selective sieving properties of
the glomerulus and increased filtration of albumin and other plasma proteins (2, 13, 99); however, this has not been rigorously explored in animals treated with mercury.
MECHANIShlS OF TUBULAR TOXICITY In general, nephrotoxic heavy metals characteristically
produce lesions in the proximal tubule. These lesions of- ten show a degree of dose-response heterogeneity across the length of the proximal tubule. For example, chronic exposure to cadmium chloride or acute injections of po- tassium dichromate characteristically produce tubular ne- crosis that initiates at the lowest effective dose levels in the pars convoIirta of the proximal tubule and then pro- gresses to more distal regions of the tubule. By contrast, mercuric chloride (1 19), uranyl compounds (43), and platinum compounds (28) produce a necrosis that origi- nates at the lowest effective dose levels in the pars recta of the proximal tubule. Uranyl compounds exemplify the extreme of this dose-response heterogeneity. In rats, mor- phological changes in the pars recta segments of the proximal tubule, evident within 6 hr after a 6-mg U k g dose of uranyl nitrate, include focal loss of periodic acid- Schiff (PAS)-positive staining and loss of brush border from the proximal tubule (34, 43). The injury progresses in severity for the first 5 days, culminating in near com- plete necrosis of the pars recta segment of the proximal tubule with the pars convolm segment remaining rela- tively free from injury (43). Distortion, focal necrosis, and casts in the thick ascending limb of the loop of Hen- le, distal tubule and collecting tubule follow proximal
at MERCER UNIV on September 29, 2010tpx.sagepub.comDownloaded from

Vol. 26, No. 1, 1998 RENAL TOXICITY OF HEAVY METALS 95
tubule injury (27, 43). Regeneration of the injury with squamous epithelium begins within 1 wk after dosing, during the time of peak severity of injury; however, atropic tubules, casts and interstitial fibrosis have been reported 56 days after 6 mg U k g BW uranyl nitrate, suggesting a chronic aspect of the injury (44). After low- er doses of uranyl fluoride, 0.7-1.3 mg U/kg.BW, resto- ration of the normal morphology of the tubule is com- pleted within 35 days after dosing (27).
The mechanisms by which uranium and other metals produce localized damage to specific regions of the prox- imal tubule are not understood and remain one of the most significant gaps in our understanding of metal neph- rotoxicology. Regional sensitivity of the proximal tubule appears to be preserved in some iii vitro preparations of kidney slices. Ruegg et a1 (85) examined histological changes that occurred when renal cortical slices of rat kidney were incubated with mercuric chloride or potas- sium dichromate. Consistent with the in vivo response with these 2 chemicals, the pars recta region was more sensitive to mercuric chloride while the pars coizvolicta region was more sensitive to potassium dichromate. Thus, the regional differences in sensitivity seem to be a characteristic of the renal tubular epithelium and are not the result of hemodynamic or neurogenic factors.
An understanding of the mechanism for dose-response heterogeneity in the proximal tubule will come with iden- tification of the various extracellular, membrane, and in- tracellular ligands that are important in the expression of the nephrotoxicity. Significant recent progress has been made on this problem as it relates to the nephrotoxicol- ogy of inorganic mercuric mercury (1 19, 120). What fol- lows is a summary of our current understanding of this subject, and; where relevant, other heavy metals are dis- cussed.
Relatioriship between Sites of Merc~ry Accunit~lation in the Kidney and Toxicity
Acute doses of inorganic mercuric mercury (Hgz+) in- duce cellular necrosis in the proximal tubule. The injury is most prominent in the pars recta (S2 and S3 segments) at low doses, with involvement of the pars coiivolitta of the proximal tubule and distal segments of the nephron at higher doses (73, 102, 119). The location of the injury along the nephron gives rise to a characteristic histolog- ical presentation typified by cellular necrosis that is most severe in the inner cortex and outer stripe of the outer medulla (OSOM). Functional correlates of mercury-in- duced cellular degeneration include loss of enzymes in the luminal brush-border membrane of the proximal tu- bule that can be detected as an increase in the excretion of brush-border enzymes in urine (e.g., alkaline phospha- tase, y-glutamyltranspeptidase [yGT]). With more ad- vanced cellular and tubular necrosis, intracellular en- zymes (e.g., lactate dehydrogenase, N-P-D-ghIcosamini- dase) are excreted in urine and impaired reabsorption of water and solutes in the proximal tubule gives rise to diuresis, glucosuria, amino aciduria, and proteinuria (73, 115, 116).
The pattern of histological changes in the renal cortex and outer medulla agrees well with the observed pattern
of accumulation of inorganic mercury that occurs pre- dominantly in the same regions of the kidney (9, 94, 123). Audioradiographic and histochemical techniques have further localized the sites of inorganic mercury ac- cumulation to the proximal tubule (82, 94). These obser- vations suggest that injury results from a direct interac- tion between mercury and the proximal tubule. Analysis of the location of mercury along the proximal tubule has been attempted to define further the relationship between the site of accumulation and the site of injury. In the BALBk mouse, after administration of a nephrotoxic dose of mercuric chloride, and in the Sprague-Dawley rat, after administration of either a nontoxic or nephrotoxic dose of mercuric chloride, mercury was localized with autometallographic techniques to the S1, S2, and S3 seg- ments with more abundant deposits in the pars rectn, S2, and S3 segments (47, 49, 103). Unilateral nephrectomy and subsequent compensatory renal growth, which in- creases the severity of mercury-induced injury to the pars recta of the proximal tubule and increases accumulation of mercury in the OSOM in the rat, also increase the abundance of autometallographically detected mercury in the S2 and S3 segments of the proximal tubule (103).
These observations suggest that injury to the pars rec- ta segment of the proximal tubule may be directly related to the association of mercury with the pars recta. In the rabbit, after administration of a nontoxic dose of mercury, the mercury contents (per unit length) of microdissected S 1 and S2 segments of proximal tubule were similar and. approximately twice that of the S3 segment (107). Thus, although the pattern of injury along the proximal tubule is consistent with the overall pattern of abundant of ac- cumulation of mercury in the pars recta, mercury is not exclusively accumulated in this segment of the proximal tubule. Variables other than the total accumulation of mercury must contribute to the pronounced vulnerability of the pars recta segment of the proximal tubule to mer- cury.
A more severe injury to the renal proximal pars recta, relative to the pars convoli4ta, has been observed in rat renal cortical slices incubated in vifro with 100 phi mer- curic chloride (85), suggesting that the mechanisms re- sponsible for the pronounced sensitivity of the pars recta segment may not require glomerular filtration or other hemodynamic factors present in vivo. In contrast to the slice, when mercuric chloride (18 phi) was perfused di- rectly into isolated rabbit proximal tubular segments, tox- icity occurred in S1, S2, and S3 segments without evi- dence for selective or more severe toxicity to the S2 or S3 segments (8, 124). Three hypotheses can be derived from these observations: (a) 18 p M mercuric chloride, presented to the luminal side of the proximal tubule ep- ithelium, is above the threshold for toxicity of the Sl , S2, and S3 segments of the proximal tubule, (b) selective toxicity to the S2 and S3 derives from the delivery of mercury to the basolateral side of the proximal tubule; or (c) selective toxicity requires a level of structural or met- abolic integrity of the renal cortex and outer medulla that is preserved in the slice but not in the isolated perfused tubule.
at MERCER UNIV on September 29, 2010tpx.sagepub.comDownloaded from
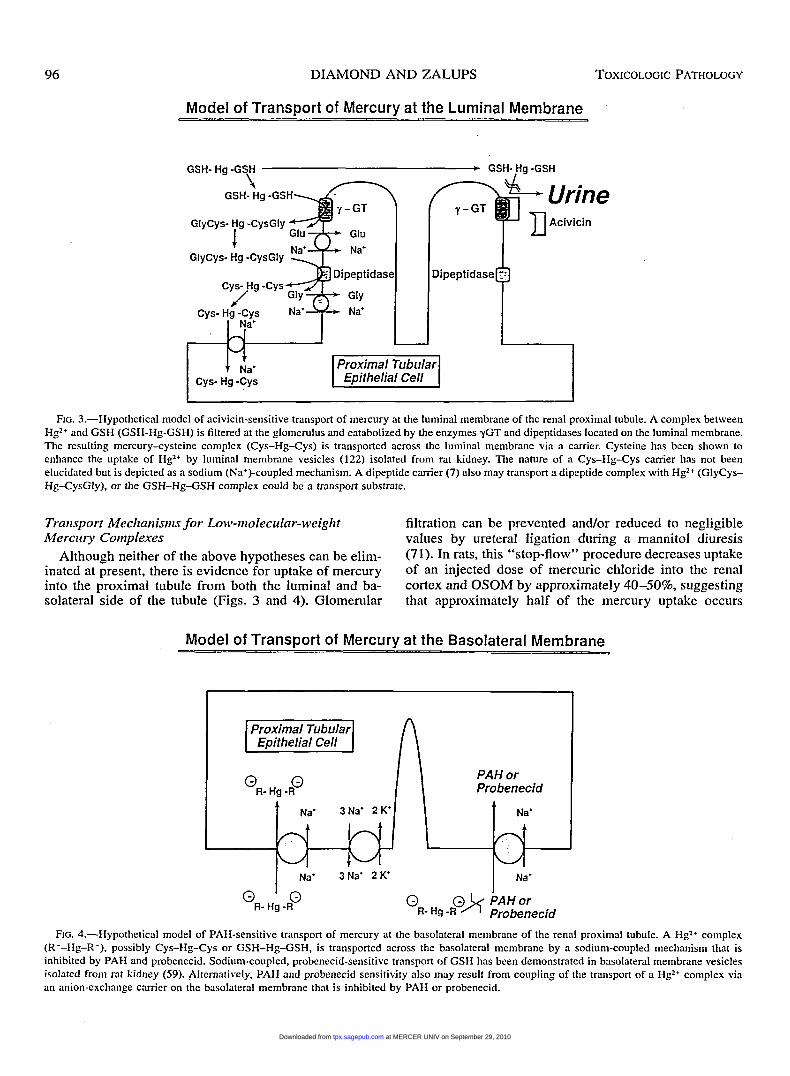
96 DIAMOND AND ZALUPS TOXICOLOGIC PATHOLOGY
Model of Transport of Mercury at the Luminal Membrane
GSH- Hg -GSH * GSH-,Hg-GSH I
CYS- Hg -CYS
FIG. 3.-Hypothetical model of acivicin-sensitive transport of mercury at the luminal membrane of the renal proximal tubule. A complex between Hg2+ and GSH (GSH-Hg-GSH) is filtered at the glomerulus and catabolized by the enzymes yGT and dipeptidases located on the luminal membrane. The resulting mercury-cysteine complex (Cys-Hg-Cys) is transported across the luminal membrane via a carrier. Cysteine has been shown to enhance the uptake of Hg2+ by luminal membrane vesicles (122) isolated from rat kidney. The nature of a Cys-Hg-Cys camer has not been elucidated but is depicted as a sodium (Na+)-coupled mechanism. A dipeptide carrier (7) also may transport a dipeptide complex with Hg2+ (GIyCys- Hg-CysGly), or the GSH-Hg-GSH complex could be a transport substrate.
Trmsport Meclimzisms for Low-niolecular-weight Mercitry Coniplexes
Although neither of the above hypotheses can be elim- inated at present, there is evidence for uptake of mercury into the proximal tubule from both the luminal and ba- solateral side of the tubule (Figs. 3 and 4). Glomerular
filtration can be prevented and/or reduced to negligible values by ureteral ligation during a mannitol diuresis (71). In rats, this ‘‘Stop-floW’’ procedure decreases uptake of an injected dose of mercuric chloride into the renal cortex and OSOM by approximately 40-50%, suggesting that approximately half of the mercury uptake occurs
Model of Transport of Mercury at the Basolateral Membrane
0 0 R- Hg -R Pro benecid I I PAHor
I Na’ I Na’ 3Na’ 2K‘ I
R- Hg -R 0 0
FIG. 4.-Hypothetical model of PAH-sensitive transport of mercury at the basolateral membrane of the renal proximal tubule. A Hgz+ complex (R--Hg-R-), possibly Cys-Hg-Cys or GSH-Hg-GSH, is transported across the basolateral membrane by a sodium-coupled mechanism that is inhibited by PAH and probenecid. Sodium-coupled, probenecid-sensitive transport of GSH has been demonstrated in basolateral membrane vesicles isolated from rat kidney (59). Altcrnatively, PAH and probenecid sensitivity also may result from coupling of the transport of a Hg2+ complex via an anion-exchange carrier on the basolateral membrane that is inhibited by PAH or probenecid.
at MERCER UNIV on September 29, 2010tpx.sagepub.comDownloaded from

Vol. 26, No. 1, 1998 RENAL TOXICITY OF HEAVY METALS 97
from the glomerular filtrate and, therefore, from the lu- minal side of the tubule, and half of the uptake occurs at the basolateral side of the tubule (123). Most of this “ba- solateral component” of mercury uptake, approximately 75-80%, can be inhibited by an intravenous injection of the organic anion p-aminohippurate (PAH) given prior to mercuric chloride during stop-flow conditions; suggesting the involvement of an organic anion transport mechanism in the basolateral uptake of mercury (123). The enhanced uptake of mercury into the OSOM that occurs in rats that have undergone unilateral nephrectomy and compensa- tory renal growth is completely abolished under stop- flow conditions and uptake is further decreased by a com- bination of stop-flow and pretreatment with PAH (106). Therefore, unilateral nephrectomy and compensatory re- nal growth appear to enhance, predominantly, a luminal uptake mechanism for mercury.
Evidence has been provided for at least 3 mechanisms for the uptake of mercury into the proximal tubule epi- thelium, although numerous other mechanisms may exist. An organic anion-sensitive mechanism, previously eluded to, appears to be involved in the uptake of mercury from the basolateral side of the rat proximal tubule (123). An acivicin-sensitive mechanism has been shown to exist in the mouse kidney (92, 93) and rat kidney (10, 25, 105). Acivicin is an inhibitor of the enzyme yGT which cleaves the y-glutamyl moiety from glutathione (GSH). The en- zyme is located in the kidney, predominantly in the pars recta segment of the proximal tubule (43 , and predom- inantly in the luminal membrane, although the enzyme also has been associated with the basolateral membrane and renal microvasculature (24, 88). In rats, after an in- jection of a nontoxic dose of mercuric chloride (0.5 pmoVkg), near complete inhibition of renal yGT activity by acivicin decreases uptake of mercury in the renal cor- tex by approximately 65%. The acivicin-induced de- crease in mercury uptake occurs almost exclusively in the renal cortex, whereas PAH inhibits uptake of mercury in both the cortex and outer medulla (105). Treatment of rats with acivicin and PAH in combination inhibits mer- cury uptake in the renal cortex by approximately 90% and in the outer medulla by approximately 50% (105). Furthermore, ureteral ligation abolishes the effect of aciv- icin on mercury uptake, suggesting that the acivicin-sen- sitive mechanisms resides predominantly, if not exclu- sively, on the luminal side of the proximal tubule. Thus, the acivicin-sensitive and PAH-sensitive mechanisms ap- pear to be distinct mechanisms and, together, account for most (possibly as much as 80%) of the mercury uptake in the proximal tubule.
It is possible that the PAH-sensitive and acivicin-sen- sitive mechanisms involve some common transport sub- strates. Hg2+ readily forms highly stable S-Hg complexes with thiols such as cysteine and GSH (38, Sl), and such complexes would be expected to form in plasma given that the concentrations of reduced cysteine and GSH in plasma are approximately 10 phi (60). Furthermore, the catabolism of GSH to its constituent amino acids by yGT and dipeptidases in the brush-border membrane of the proximal tubule would be expected to give rise to Hg- cysteine in the tubular lumen. Studies of basolateral
membrane vesicles isolated from rat kidney cortex and primary cultures of rat proximal tubular cells have re- vealed PAH- andor probenecid-sensitive transport mech- anisms for GSH, S-GSH conjugates, and S-cysteine con- jugates (57-59). Thus, it is conceivable that Hg-cysteine and Hg-GSH complexes may be substrates for a PAH- sensitive transport mechanism in the proximal tubule, al- though evidence for this is currently lacking. Both GSH and cysteine, when administered with mercuric chloride, increase the uptake of mercury in the renal cortex and outer medulla (108, 109). Coadministration of cysteine increases the severity of nephrotoxicity of mercuric chlo- ride (110). Studies of the transport of Hg-cysteine and Hg-GSH complexes have been attempted with prepara- tions of luminal and basolateral membrane vesicles iso- lated from rat kidney; however, rapid and extensive bind- ing of mercury to the membranes severely limits the quantification of mercury transport fluxes in this prepa- ration. Therefore, an effect of cysteine or GSH on the amount of mercury taken up by the membranes could represent an effect on binding and/or transport because binding could occur before, during, or after transport has occurred. Cysteine in a 3- or 10-fold excess with HgZ+ increased mercury uptake by the luminal membrane ves- icles, relative to that which occurred with Hg2+ alone (121, 122). GSH, on the other hand, decreased mercury uptake in both the luminal and basolateral membrane ves- icles. The effect of cysteine on mercury uptake may re- flect a direct or indirect coupling of Hg2+ and cysteine fluxes into or across the luminal membrane vesicle or, possibly, a ligand exchange between cysteine and sulfhy- dryls at the membrane surface. It also is conceivable that a dipeptide carrier on the luminal membrane (7) may transport a dipeptide complex with Hg2+ (GlyCys-Hg- Cys-Gly). Further study is needed to sort out these pos- sible mechanisms.
Transport of Mercury-Protein Coinplexes A third possible mechanism of uptake mercury in the
proximal tubule is the endocytosis of Hgz+ bound to fil- tered serum albumin. Mercuric mercury in plasma is bound extensively to albumin (17), the most abundant (approximately 1 mi) sulfhydryl in plasma (15,5 1). The filtration fraction of albumin. in the rat is approximately 0.0004 (79, 80); therefore, a small fraction of the Hg- albumin in plasma would be expected to enter the lumen of the early proximal tubule in the glomerular filtrate. Crude estimates of the relative magnitudes of filtered Hg- albumin and filtered low molecular weight mercury com- plexes (Hg-LMW) can be made by assuming the follow- ing: 50% of the mercury in blood is in the plasma frac- tion (104), of which 98% is bound to albumin (17) and the remaining 2% consists of Hg-LMW (e.g., Hg-GSH), a GFR of 1 ml-min-’ per kidney in the rat, and a filtration fraction of 0.0003 for Hg-albumin complex and 1.0 for Hg-LMW. The above assumptions yield a plasma clear- ance of Hg-albumin due to glomerular filtration that is approximately 2% that of filtered Hg-LMW. Filtered al- bumin is extracted from the tubular fluid in the proximal tubule through the process of endocytosis and transferred to lysosomes for catabolism (67). A similar mechanism
at MERCER UNIV on September 29, 2010tpx.sagepub.comDownloaded from

98 DIAMOND AND ZALUPS TOXICOLOGIC PATHOLOGY
could result in the endocytosis of filtered Hg-albumin. Proteinuric rats given an injection of a nontoxic dose of mercuric chloride excrete a substantially larger fraction of urinary mercury as Hg-albumin compared to normal nonproteinuric rats and have a larger fraction of total cel- lular mercury associated with the lysosomal fraction (68, 70). These observations suggest that Hg-albumi‘n may be taken up from the lumen of the proximal tubule, possibly by endocytosis. Although it has been reported that al- bumin may increase the severity of toxicity of mercuric chloride in the isolated perfused rabbit proximal tubule (124), more recent studies suggest that albumin substan- tially decreases the uptake and toxicity of mercury in this preparation (R. K. Zalups, unpublished observations). Mercury can induce proteinuria in animals and, in rats, mercuric chloride-induced proteinuria is associated with increased excretion of Hg-albumin in urine (68). The al- buminuria may reflect impaired reabsorption of protein, as it occurs in association with cellular and tubular ne- crosis (1 15); however, increased filtration of albumin also may contribute to the albuminuria. If the endocytosis of filtered Hg-albumin is a significant pathway for entry of mercury into epithelial cells of the proximal tubule, then glomerular proteinuria induced by mercury may further exacerbate toxicity by increasing the delivery of Hg-al- bumin to the lumen of the proximal tubule.
Complexes between Hg2+ and filtered proteins other than albumin also may be taken up by the proximal tu- bule epithelium. Rats administered Hg-metallothionein complex (Hg-Mt) accumulate more mercury in the renal cortex and outer medulla and excrete more mercury in urine than rats administered mercuric chloride, suggesting that the complex is more available for uptake in the prox- imal tubule than HgZ+ bound to other endogenous ligands (1 13). Metallothionein is a 6,000-7,000-dalton, sulfhy- dryl-rich protein that forms highly stable complexes with Hg2+, Cd2+, and other divalent, thiolphilic metals (53, 54). The mechanism for the effect of metallothionein on mercury uptake has not been determined. Cd-metallothi- onein complex (Cd-Mt) is filtered at the glomerulus and undergoes reabsorption (32, 37), presumably in the prox- imal tubule where other low-molecular-weight proteins are known to be reabsorbed (67, 80). A similar mecha- nism may explain the increased uptake of mercury when Hg-metallothionein is administered to rats. Isolated per- fused rabbit proximal tubule segments do not take up mercury when presented with Hg-Mt complex in the tu- bule lumen or on the basolateral side of the tubule, which is unfortunate because this preparation would be other- wise ideal for examining mechanisms of uptake of Hg- Mt (114). Possibly, the kinetics of endocytosis are too slow to be accommodated in this preparation, or the prep- aration lacks some necessary property for endocytosis of low-molecular-weight proteins. It also is possible that mercury impairs or inhibits endocytosis. In the rat, mer- curic chloride has been shown to inhibit lysosomal ca- tabolism of the low-molecular-weight protein, lysotyme (69). Examination of the transport and metabolism of oth- er low-molecular-weight proteins (e.g., lysozyme, azp- globulin, retinol binding protein) in the isolated perfused
tubule preparation would provide greater insight into these problems.
The extent to which metallothionein plays a role in the mercury nephropathy remains unresolved; however, its role in the pathogenesis of cadmium nephropathy has been much more thoroughly explored. Cd-Mt is highly toxic to the proximal tubule. Rats fed 1 mM cadmium chloride in drinking water ad libititin develop nephropa- thy after a 40-50-wk exposure (55) when the amount of cadmium in renal tissue exceeds 2 pmol Cd/g wet weight (-10 pmol Cd/g dry weight); however, a single injection of 2-10 pmol Cdkg BW, as Cd-Mt (21, 9 3 , produces nephropathy in rats within 4-12 hr when the amount of cadmium in renal tissue is only 0.1-0.5 pmol Cd/g wet weight (-0.5-2.5 pmol Cd/g dry weight). The mecha- nism by which Cd-Mt increases the toxicity of cadmium is not completely understood. Cd-Mt itself may be toxic, or the rapid extraction of Cd-Mt from the circulation by the kidney (89), endocytosis of Cd-Mt in the proximal tubule, and its subsequent degradation in lysosomes (16) may serve as a mechanism for delivering large amounts of Cd2+ into proximal tubular cells where it can bind to and impair the function of critical proteins. Thus, it has been suggested that the development of cadmium ne- phropathy occurs when some critical amount of “free cadmium’’ (i.e., cadmium that is not bound to metallo- thionein) occurs in proximal tubule cells (78). Hg-Mt has not been shown to be more toxic than mercuric chloride; however, when mercury is administered as Hg-Mt, the segmental distribution of injury is shifted along the prox- imal tubule. The pars convolirta and early pars recta ( S 1 and S2) segments of the proximal tubule appear to be more vulnerable to Hg-Mt than is the late pars recta (S3) segment (1 8), whereas the opposite pattern of vulnera- bility is characteristic of mercuric chloride (73, 102, 119). It is tempting to speculate that this change in the location of the injury may reflect endocytosis of Hg-Mt in the pars coitvolirta segments of the proximal tubule. In sup- port of this, the nephropathy induced by cadmium chlo- ride or Cd-Mt is characteristically more severe in the pars convolirta than in the pars recta segments of the proximal tubule; however, an injected dose of cadmium chloride together with cysteine induces an acute injury predominantly in the pars recta segment (75). As previ- ously noted, coadministration of cysteine with mercuric chloride increases the uptake of mercury in the renal cor- tex and OSOM where the pars recta segments of the proximal tubule are located (1 10).
Cellitlar ~echaiiisiiis of Mercirry Toxicity
The disposition of mercury in cells is dominated by its high affinity for the thiolate anion and, therefore, poten- tial molecuIar targets for mercury will include nonprotein and protein thiols. Hg2+ binds to metallothionein and ad- ministration of mercuric chloride to rats increases the concentration of metallothionein in the renal cortex and OSOM (1 11). Pretreatment of rats with zinc, which in- duces the synthesis of metallothionein affords protection against a subsequent acute nephrotoxic dose of mercuric chloride (1 12). Thus, metallothionein seems to serve as
at MERCER UNIV on September 29, 2010tpx.sagepub.comDownloaded from

Vol. 26, No. 1, 1998 RENAL TOXICITY OF HEAVY METALS 99
a buffer for Hg2+ ions, presumably preventing its inter- action with other critical sulfhydryls in the cell.
Hg2+ also complexes avidly with GSH. Treatment of rats with nontoxic to moderately nephrotoxic doses of mercuric chloride increases the GSH concentration in the renal cortex and OSOM (118). Renal proximal tubular epithelial cells, but not distal tubular cells, frtshly isolat- ed from rats treated with a nontoxic dose of mercuric chloride, show an increase in the activity of y-glutamyl- cysteine synthetase, which catalyzes the rate-limiting step in the synthesis of GSH (62). Therefore, the increase in GSH induced by mercuric chloride appears to derive from an induction of the synthesis of GSH. In rats, de- pletion of GSH from the kidney by treatment with diethyl maleate (DEM) and buthionine sulfoximine (BSO), prior to a moderately toxic parented dose of mercuric chloride (4 mgkg, -15 pmoVkg), decreases the uptake of mer- cury in the renal cortex and increases the severity of nephrotoxicity (10). Thus, GSH, in addition to metallo- thionein, may serve as an important intracellular buffer for Hg2+.
The mechanism by which GSH depletion decreases mercury uptake in the kidney is not completely under- stood. The concentration of GSH in the kidney does not appear to be the sole determining factor. Zalups and Lash (121) pretreated rats with either DEM or BSO to produce similar levels of depletion of GSH (60% depletion) and similar GSH concentrations in the renal cortex or outer medulla and then administered a nontoxic dose of mer- curic chloride (0.5 pmolkg). Mercury uptake in the cor- tex and OSOM was significantly decreased by the DEM; however, BSO significantly decreased mercury uptake only in the OSOM and not in the renal cortex. Diethyl maleate depletes GSH in tissues by forming disulfide conjugates with GSH, whereas BSO depletes GSH by inhibiting the y-glutamylcysteine synthetase step in the synthesis pathway for GSH. Therefore, it appears that depletion of GSH resulting from inhibition of the syn- thesis of GSH has less of an effect on mercury uptake in the renal cortex than does conjugation of GSH with DEM. By contrast, depletion of GSH by inhibiting GSH synthesis or conjugating GSH both decrease mercury up- take in the renal OSOM.
In contrast to the induction of GSH that occurs with nontoxic or moderately toxic doses, depletion of GSH from the kidney occurs in animals treated with severely toxic doses of mercuric chloride (1, 3, 39-42, 61, 118, 125). Depletion of GSH would not only reduce cellular Hg2+ buffering capacity but would expose the cell to ox- idative damage, as GSH is an important free-radical scav- enger. Mercury-induced depletion of renal GSH has been associated with increased free-radical-mediated oxidation of reduced porphryins (100, 101). Depletion of cellular GSH in the kidney appears to occur in association with a general collapse of antioxidant mechanisms in the cell, including depletion of renal ascorbic acid and vitamin E levels; and decreased activities of superoxide dismutase, catalase, GSH peroxidase, and GSSG reductase in renal cortex (22, 40, 42). All of these effects would imply se- vere oxidative stress in the renal cortex subsequent to a severely nephrotoxic dose of mercuric chloride.
The ATPases are a critical class of sulfhydryls in cells (84). In isolated, reconstituted preparations of renal med- ullary (Na+-K+)-ATPase, Hg2+ binds to the ATPase, in- hibits its activity and ATPase-driven transport (4, 5, 50). Binding of Hg2+ to the ATPase is reversible and occurs at a site distinct from the oubain binding site (5) and appears to affect the interaction between the 01 and p subunits of the ATPase protein complex (50). The bind- ing site is on the cytosolic domain of the ATPase (4). The (Na+-K+)-ATPase is the energy-requiring step in the development of the electrochemical gradients that drive solute and water transport in the proximal tubule. Inhi- bition of the (Na+-K+)-ATPase would be expected not only to impair solute and water reabsorption in the prox- imal tubule but would also impair the transport of sub- strates for energy metabolism and synthesis in the kidney (e.g., amino acids, citrate, fatty acids, glucose, lactate) (23). The above observations refer specifically to the plasma membrane (Na+-K+)-ATPase; however, other ATPases, including the (Ca2+-Mg2+)-ATPase and mito- chondrial ATPases also may be targets of Hg2+, in which case, more diverse effects on cellular function might be anticipated.
Mercuric chloride depletes cellular ATP in the proxi- mal tubule. In isolated microdissected segments of rat proximal tubule, incubation with 1 p h i mercuric chloride depletes ATP only from the S2 segment and not from the S1 or S3 segments of the proximal tubule, thick ascend- ing limb, or distal tubule (52). This pattern is consistent with greater in vivo sensitivity of the pars recta segment of the proximal tubule to injury. The mechanism for the ATP depletion derives, at least in part, from a direct im- pairment of mitochondrial respiration. Zit vitro incubation with mercuric chloride decreases nystatin-stimulated ox- ygen consumption in suspensions of isolated rabbit prox- imal tubules. This affect occurs prior to the release of intracellular enzymes (i-e., lactate dehydrogenase), sug- gesting that the effect of mercury on mitochondrial func- tion precedes and may contribute to irreversible cellular injury (1 17). Mercuric chloride has been shown to induce a variety of changes in function of mitochondria isolated from renal cortex. These include increases in state 4 ox- ygen consumption (i.e., uncouples respiration) and de- creases in state 3 (substrate-stimulated) oxygen consump- tion (97, 98); increases in calcium efflux and decreases in mitochondrial membrane potential (19, 20); and de- pletion of mitochondrial GSH, increased mitochondrial hydrogen peroxide production, and lipid peroxidation (65, 66). The above effects suggest that mercury, at suf- ficient doses, will induce a general collapse of oxidative metabolism and generation of ATP to support transport and synthesis.
A variety of critical cell functions depend on the reg- ulation of the intracellular Ca2+ concentration. In primary cultures of renal tubular cells from rabbit cortex, incu- bation with mercuric chloride increases intracellular Ca2+ concentration (87). Exposures to 2.5-10 phI mercuric chloride resulted in a 2-10-fold increase in Ca2+ concen- tration, whereas, exposure to 25-100 p h l mercuric chlo- ride induced a biphasic change in Ca2+ concentration. The initial phase consists of a rapid rise in Ca2+ that is in-
at MERCER UNIV on September 29, 2010tpx.sagepub.comDownloaded from

100 DIAMOND AND ZALUPS TOXICOLOGIC PATHOLOGY
dependent of extracellular Ca2+ concentration and pre- sumably reflects release of Ca2+ from intracellular stores. This is followed by a decrease and subsequent gradual increase in intracellular Ca2+ that is dependent on extra- cellular Ca2+, which may indicate a change in perme- ability of the plasma membrane to Ca2+. These results indicate that mercuric chloride can perturb intracellular Ca2+ concentrations by at least 2 different mechanisms.
SUhlhlARY AND CONCLUSIONS The picture that is emerging from these studies is that
injury to the pars recta segment of the renal proximal tubule results from a direct effect of mercury accumula- tion in this region of the nephron. The mechanisms by which mercury enters cells of the renal proximal tubule remain obscure; however, .it appears that pathways from both the luminal and basolateral side of the tubule are active. One pathway appears to be dependent on activity of the enzyme yGT; a second pathway appears to involve an organic anion transport mechanism. A common trans- port substrate for both mechanisms may be a Hg-cysteine complex. The exact mechanisms underlying the cellular degeneration associated with the accumulation of mer- cury in the pars recta segment of the proximal tubule have not been elucidated. Early biochemical changes in- duced by mercury include an induction of GSH and me- tallothionein that may be a direct effect of mercury on their metabolism or may represent a more general re- sponse to oxidative stress induced by mercury, possibly through impairment of membrane ATPase and/or mito- chondrial function. At some point, or level of mercury in the cell, a multifocal collapse of cell metabolism occurs, including oxidative metabolism, cellular free-radical de- fense mechanisms, and membrane transport and perme- ability, initiating the terminal phases of cellular toxicity leading to cell degeneration and death. It is at this point that the effects of mercury on the tubular epithelium be- come evident histologically and the commonly applied urinary biomarkers reveal injury and functional impair- ment of the kidney.
While continued progress is being made in understand- ing the mechanism of toxicity of mercury and other met- als, our current understanding remains frustratingly un- satisfactory. Mercury is illustrative in that we probably know more about the mechanisms of nephrotoxicity of inorganic mercuric mercury than we do of any other met- al; yet many challenging problems remain. The exact mechanisms by which Hg2+, or its complexes, cross renal tubular cell membranes remain one of the more important unresolved problems. The same applies to all of the ne- phrotoxic heavy metals. The induction of GSH and me- tallothionein by mercury is intriguing; however, its pre- cise role in the development of toxicity and the mecha- nism for the induction itself remain to be elucidated. The rise in intracellular Ca2+ that accompanies low-level ex- posures of renal tubular cells to mercury also is intriguing in view of the extreme importance of Ca2+ in signal trans- duction in the control of cellular metabolism. A mecha- nistic explanation for the effect of mercury on intracel- lular Ca2+ is not at hand. The mechanisms by which mer- cury disrupts mitochondria1 functions needs to be eluci-
dated if we are to understand the pathways that lead to catastrophic collapse of oxidative metabolism that occurs in mercury nephrotoxicity. These intriguing problems, for the most part, can be generalized to most of the nephro- toxic heavy metals. Further research applying the latest techniques for studying membrane transport and cellular metabolism should greatly improve our understanding of the mechanisms of nephrotoxicity of metals and of the pathophysiology of renal injury.
REFERENCES 1. Addya A, Chakravanti K, Basu A, Santa hl, Haldar S , and Chat-
terjee GC (1984). Effects of mercuric chloride on several scav- enging enzymes in rat kidney and influence of vitamin E supple- mentation. A C I ~ Vitaniinol. Erzzyrzol. 6: 103-107.
2. Adler SG, Wang H, Ward HJ, Cohen AH, and Border WA (1983). Electrical charge. Its role in the pathogenesis and prevention of experimental membranous nephropathy in the rabbit. J. Clirz. In - \vest. 71: 487-499.
3. Alco MD, Taum ML, Olson JR, Nickerson PA, and Kostyniak PJ (1987). Cellular uptake and response of primary cultures of rabbit renal proximal tubule cells exposed to mercuric chloride and methylmercury chloride. In: 6 z Vitro To.xicology: Approaches to
Validntiorz, AM Goldbcrg (ed). Mary Ann Liebert, New York, pp.
4. Anner BM and Moosmayer M (1992). Mercury inhibits Na-K ATPase primarily at the cytosolic side. Anz. J. PIzysiol. 262: F843- F848.
5. Anner BM, Moosmayer M, and Imesch E (1992). Mercury blocks Na-K-ATPase by a ligand-dependent reversible mechanism. Am. J. Plzysiol. 262: F830-F836.
6. Avasti PS, Evan AP, and Hay D (1980). Glomerular endothelial cells in uranyl nitrate-induced acute renal failure in rats. J. Clin. Invest. 65: 121-127.
7. Barfuss DW, Ganapathy V, and Leibach FH (1988). Evidence for active dipeptide transport in isolated proximal straight tubules. Am. J. Pliysiol. 255: F177-FI81.
8. Barfuss DW, Robinson MK, and Zalups RK (1990). Inorganic mercury transport in the proximal tubule of the rabbit. J. Arm Soc. Nephrol. 1: 910-917.
9. Bergstrand A, Friberg L, Mendel L, and Odeblad E (1959). The localization of subcutaneously administered radioactive mercury in the rat kidney. J. Ultrastrirct. Res. 3: 238-239.
10. Bemdt WO, Baggett JMcC, Blacker A, and Houser M (1985). Renal glutathione and mercury uptake by kidney. Fiuzdatrz. Appl. Toxicol. 5: 832-839.
1 1 . Bigazzi PE (1992). Lessons from animal models: The scope of mercury-induced autoimmunity. Cliri. 1rtzrriiuzoI. Irtzniirrzopnthol.
12. Blantz RC (1977). The mechanisms of acute renal failure after uranyl nitrate. J. Clirz. Invest. 55: 621-635.
13. Border WA, Ward HJ, Kami ES, and Cohen AH (1982). Induction of membranous nephropathy in rabbits by administration of an exogenous cationic antigen. J. Clitz. Invest. 69: 451-461.
14. Brenner BM, Troy JL, and Daugharty Thl (1971). The dynamics of glomerular filtration in the rat. J. Chi. /rivest. 50: 1776-1789.
15. Brown JR and Shockley P (1982). Serum albumin: Structure and characterization of its ligand binding sites. In: Lipid-Protein In- teractions, Vol. 1 , PC Jost and OH Griffith (cds). Wiley, New York, pp. 25-68.
16. Cain K and Holt DE (1983). Studies of cadmium-thionein induced nephropathy: Time course of cadmium-thionein uptake and deg- radation. Clzerm-Biol. Interact. 43: 223-237.
17. Cember H, Gallagher P, and Faulkner A (1968). Distribution of mercury among blood fractions and serum proteinx Am. Ind. Hyg.
18. Chan Hhl. Satoh hl, zlllups RK. and Cherian hlG (1992). Exog-
21 1-226.
65: 81-84.
ASSOC. J. 29: 233-237.
at MERCER UNIV on September 29, 2010tpx.sagepub.comDownloaded from

Vol. 26, No. 1, 1998 RENAL TOXICITY OF HEAVY METALS 101
enous metallothionein and renal toxicity of cadmium and mercury in rats. To.ricology 76: 15-26.
19. Chavez E and Holguin JA (1988). Mitochondria1 calcium release induced by Hg2+. J. Biol. Chem. 283: 3581-3587.
20. Chavez E. Zazueta C, Osornio A, Holguin JA, and hliranda ME (1991). Protective behavior of captopril on Hg++-induced toxicity on kidney mitochondria. In vivo and in vitro experi.ments. J. Phar- ntacol. Exp. Ther. 256: 385-390.
21. Cherian MG, Goyer RA, and DeLaquemere-Rechardson L (1976). Cadmium-metallothionein induced nephropathy. To.xicol. Appl. PIzannacol. 38: 399-408.
22. Chung A-S, Maines hlD, and Reynolds WA (1982). Inhibition of the enzymes of glutathione metabolism by mercuric chloride in the rat kidney: Reversal by selenium. Bioclzenz. Phanriacol. 3 1:
23. Cohen JJ and Kamm DE (1981). Renal metabolism: Relation to renal function. In: The Kidney, BM Brenncr and FC Rector (eds). WB Saunders, Philadelphia, pp. 144-248.
24. Dass PD, Misra RP, and Welbourne TC (1981). Presence of y- glutamyltransferase in the renal microvascular compartment. Can. J. Biochenz. 59: 383-386.
25. de Ceaumz J, Pyan JP, Morel G, and Brondeau MT (1994). Role of extracellular glutathione and y-glutamyltranspeptidase in the disposition and kidney toxicity of mercury. J. Appf . Toxicof. 1 4
26. Diamond GL (1989). Biological consequences of exposure to sol- uble forms of natural uranium. Radiat. Protect. Dosini. 26: 23- 33.
27. Diamond GL, Morrow PE, Panner BJ, Gelein Rhf, and Baggs RB (1989). Reversible uranyl fluoride nephrotoxicity in the Long Evans rat. Fzmd. Appl. To.ricol. 13: 65-78.
28. Dobyan DC, Levi J, Jacobs C, Kosek J, and Weiner hlW (1980). Mechanism of cis-platinum nephrotoxicity. 11. Morphologic ob- servations. J. Phannacol. Exp. Ther. 213: 551-562.
29. Druet P, Druet E, Potdevin E and Sapin C (1978). Immune type glomerulonephritis induced by HgCI, in the Brown-Norway rat. Ann. bnrtiunol. (Irtst. Pasteur) 129C: 777-792.
30. Enestrom S and Hultman P (1984). Immune-mediated glomerular- nephritis induced by mercuric chloride in mice. Experienfia 40: 1234-1240.
31. Enestrom S and Hultman P (1992). Dose-response studies in mu- rine mercury-induced autoimmunity and immune-complex dis- ease. To.rico1. Appl. PIzannncol. 113: 199-208.
32. Felley-Bosco E and Diezi J (1987). Fate of cadmium in rat renal tubules: A microinjection study. Toxicol. Appl. Plznnnacol. 9 1 : 204-21 1 .
33. Flamenbaum W (1973). Pathophysiology of acute renal failure. Arch. Intern. Meed. 131: 91 1-928.
34. Flamenbaum I , Huddleston ML, McNeil JS, and Hamburger lU (1974). Uranyl nitrite-induced acute renal failure in the rat: Mi- cropuncture and hemodynamic studies. Kidney Int. 6: 408-418.
35. Flamenbaum W, McNeil JS, Kotchen TA, and Saladino AJ (1972). Experimental acute renal failure induced by uranyl nitrate in the dog. Circ. Res. 31: 682-697.
36. Foulkes EC (1971). Glomerular filtration and renal plasma flow in uranium poisoned rabbits. Toxicol. Appl. Plzannacol. 20: 380- 385.
37. Foulkes EC (1978). Renal tubular transport of Cd-metallothi- onein. To-ricol. Appl. Phannacol. 45: 505-5 12.
38. Fuhr BJ and Rabenstein DL (1973). Nuclear magnetic-resonance studies of solution chemistry of metal-complexes. 9. Binding of cadmium, zinc, lead and mercury by glutathione. J. Ant. Clzem.
39. Fukino H, Hirai M, Hsueh YM, Moriyasu S . and Yamane Y (1986). Mechanism of protection by zinc against mercuric chloride toxicity in rats: Effects of zinc and mercury on glutathione me- tabolism. J. To.rico1. Environ. Health 19: 75-89.
40. Fukino H, Hirai hl, Hsueh Yhl, and Yamane Y (1984). Effect of zinc pretreatment on mercuric chloride-induced lipid peroxidation in the rat kidney. To.xico1. Appl, PIznnnacol. 73: 395-401.
3093-3100.
201-206.
SOC.' 95: 6944-6950.
41. Girardi C and Elias MM (1991). Effectiveness of N-acetylcystcinc in protecting against mercuric chloride-induced nephrotoxicity. To,ricology 67: 155-164.
42. Gstraunthaler C, Pfallcr \V, and Kotankp P (1983). Glutathione depletion and in vifro lipid peroxidation in mercury or maleate- induced acute renal failure. Biochenz. P han1zacol. 3 2 2969-2972.
43. Haley DP (1982). Morphologic changes in uranyl nitrate-induced acute renal failure in saline- and water-drinking rats. Lab. Invest. 46: 196-208.
44. Haley DP, Bulger RE, and Dobyan DC (1982). The long-term effect of uranyl nitrate on the structure and function of the rat kidney. Virchonvs Arch. (Cell Pathol.) 4: 18 1-192.
45. Heinle H and Wendel A (1977). The activities of the key enzymes of the y-glutamyl cycle in microdissected segments of the rat ne- phron. FEBS k f f . 73: 220-224.
46. Hodge HC (1973). A history of uranium poisoning. In: Uraniicriz. Phttoniunz, Transplictonic Elements, HC Hodge, JN Stannnrd, and JB Hursh (eds). Springer-Verlag. New York, pp. 5-68.
47. Hultman P and Enestrom S (1986). Localization of mercury in the kidney during experimental acute renal tubular necrosis studied by the cytochemical silver amplification method. Br. J. Erp. Pathol.
48. Hultman P and Enestrom S (1992). Dose-response studies in mu- rine mercury-induced autoimmunity and immune-complex dis- ease. Toxicol. Appl. PIzarrnocol. 1 13: 199-208.
49. Hultman P, Enestrom S, and von Schenck H (1985). Renal han- dling of inorganic mercury in mice. The early excretion phase following a single intravenous injection of mercuric chloride stud- ies by the silver amplification method. Virchows Arch. (Cell Path-
50. Imesch E, hloosmayer M, and Anner BM (1992). hfercury weak- ens membrane anchoring of Na-K ATPase. Am. J. Physiol. 262:
51. Jocelyn PC (1972). Biocliemistry of the SH Grofrp. Academic Press, London.
52. Jung KY, Uchida S , and Edou H (1989). Nephrotoxicity assess- ment by measuring cellular ATP content. I. Substrate specificities in the maintenance of ATP content in isolated rat nephron seg- ments. Toxicol. Appl. Plzanimcol. 100: 369-382.
53. Kagi JHR and Nordberg G F (1979). hfetallothiorzeirz. Beerkhauser Verlag, Basel.
54. Kagi JHR and Vallee BL (1961). Metallothionein: A cadmium and zinc-containing protein from equine cortex. 11. Physiochemical properties. J. Biol. Chenz. 236: 2435-2442.
55. Kajikawa K, Nakanishi I, and Kudoa K (1981). Morphological changes in the kidney and bone of rats in chronic cadmium poi- soning. Exp. hfol. Pathol. 34: 9-24
56. Kobayashi S, Nagase M, Honda N, and Hishida A (1984). Glo- merular alterations in uranyl acetate-induced acute renal failure in rabbits. Kidney Int. 26: 808-815
57. Lash LH and Anders MW (1989). Uptake of nephrotoxic S-con- jugates by isolated proximal tubule cells. J. Phonnacol. Erp. Ther.
58. Lash LH and Jones DP (1983). Transport of glutathione by renal glutathione by renal basal-lateral membrane vesicles. Bioclieni. Bioplijs. Cornrniaz. 1 12: 55-60.
59. Lash-LH and Jones DP (1984). Renal glutathione transport: Char- acteristics of the sodium-dependent system in the basal-lateral membrane. J. Biol. Chem. 259: 14508-145 14.
60. Lash LH and Jones DP (1985). Distribution of oxidized and re- duced forms of glutathione and cysteine in rat plasma. Arch. Bio- chenz. Biophys. 240: 583-592.
61. Lash LH and Zalups RK (1992). Mercuric chloride-induced cy- totoxicity and compensatory hypertrophy in rat kidney proximal tubular cells. J. Phannacol. Exp. T k r . 261: 819-829.
62. Lash LH and Zalups RK (1996). Alterations in renal cellular glu- tathione metabolism after in riro administration of a subtoxic dose of mercuric chloride. J. Biocliem. Toxicol. 11: 1-9.
63. Leach LJ, Gelein Rhl, Panner BJ. Yuile CL, Cox CC, Balys hfhl, and Rolchigo Phl (1984). The acute toxicity of the hydrolysis
67: 493-503.
01.) 49: 209-224.
F837-F842.
248: 531-537.
at MERCER UNIV on September 29, 2010tpx.sagepub.comDownloaded from

102 DIAMOND AND ZALUPS TOXICOLOGIC PATHOLOGY
products of uranium hexanuoride when inhaled by the rat and guinea pig. U.S. Department of Energy Report WSUB/81-9039/ 3.
63. Leach LJ. Yuile CL, Hodge HC, Sylvester GE, and Wilson HB (1973). A five-year inhalation study with natural uranium dioxide (UO,) dust. 11. Postexposure retention and biologic effects in the monkey, dog and rat. Health Plrys. 25: 239-258.
65. Lund B-0, hliller DIM, and Woods JS (1991). Mercuiy-induced H,0, production and lipid peroxidation in vifro in rat kidney mi- tochondria. Biochetn. Pliannacol. 42(suppl.): S I8 I-S 187.
66. Lund, B-0, hliller DM, and Woods JS (1993). Studies in Hg(I1)- induced H,O, formation and oxidative stress in vivo and in vifro in rat kidney mitochondria. Biochem. Pharrnocol. 45: 2017-2024.
67. hlaack T (1992). Renal handling of proteins and polypeptides. In: Handbook of Physiology, Secfion 8, Renal Physiology, Vol. 2. EE Windhager (ed). Oxford University Press, New York, pp. 2039- 2082.
68. Madsen Khl (1980). Mercury accumulation in kidney lysosomes of proteinuric rats. Kidney hit. 18: 445-453.
69. Madsen Khl and Christensen EL (1978). Effects of mcrcury on lysosomal protein digestion in the kidney proximal tubule. Lab. Itwesf. 38: 165-174.
70. hladsen KM and Hansen JC (1980). Subcellular distribution of mercury in the rat kidney cortex after exposure to mercuric chlo- ride. Toxicol. Appl. Pharnzacol. 54: 443-453.
71. hlalvin RL and WIde hlS (1973). Stop-flow technique. In: Hand- book of Physiology, Secfion 8, Renal Physiology, Vol. 1, J Orloff and RW Berliner (eds). American Physiology Society, Washing- ton, DC, pp. 119-128.
72. Mason J, Olbricht C, Takabatake T, and Thurau K (1977). The early phase of experimental acute renal failure. I. Intratubule prcs- sure and obstruction. Pfliigers Arch. 370: 155-163.
73. McDowell EM, Nagle RB, Zalme RC, McNeil JS, Flamenbaum W, and Trump BF (1976). Studies of the pathophysiology of acute renal failure. I. Correlation of ultrastructure and function in the proximal tubule of the rat following administration of mercuric chloride. Virchoits Arch. (Cell Paflrol.) 22: 173-196.
74. Morrow PE, Leach L, Smith FA, Gelein RM, Scott JM, Beiter HD, Amato FJ, Picano JJ, Yule CL, and Consler TG (1981). Met- abolic fate and evaluation of injury in rats and dogs following exposure to the hydrolysis products of uranium hexanuoride. U S . Nuclear Regulatory Commission Report NUREGICR-2268.
75. Murakami M and Webb M (1981). A morphological and biochem- ical study of the effects of I-cysteine on the renal uptake and nephrotoxicity of cadmium. Br. J. Exp. Pafhol. 62: 115-130.
76. Nizet A (1981). Influenee of uranyl nitrate upon tubular reabsorp- tion and glomerular filtration in blood perfused isolated dog kid- neys. Pfliigers Arch. 391: 296-300.
77. Nomiyama K and Foulkes EC (1968). Some effects of uranyl ac- etate on proximal tubular function in rabbit kidney. Toxicol. Appl. Phanrmcol. 13: 89-98.
78. Nordberg GE Coyer R, and Nordberg hl (1975). Comparative toxicity of cadmium-metallothionein and cadmium chloride on mouse kidney. Arch. Pat ld . 99: 192-197.
79. Oken DE, Cotes SC, and hlende CW (1972). hlicropuncture study of tubular transport of albumin in rats with aminonucleoside ne- phrosis. Kidney I n f . 1: 3-1 1.
80. Pesce AJ and First hlR (1979). Proteinitria: An Integrated Revieiv. Marcel Dekkcr, New York, pp. 5-31.
81. Rabenstein DL (1989). Metal complexes of glutathione and their biological significance. In: Gliifafhiorre: C/rernical, Biochemical arid Medical Aspects. Coenzymes and Cofacfors, Vol 3, D Dol- phin, 0 Auramovbic, and R Poulson (eds). Wiley, New York, pp.
82. Rodier Phl, Kates B, and Simons R (1988). hlercury localization in mouse kidney over time: Audoradiography versus silver stain- ing. Toxicol. Appl. Plrnrtnacol. 257: 235-245.
83. Roman-Franco AA, Turiello hl. Albini B, Ossi E, hlilgrom E and Andres GA (1978). Anti-basement membrane antibodies and an-
147-1 86.
tigen-antibody complexes in rabbits injected with mercuric chlo- ride. Clin. Itnrnunol. Itiirniinopafhol. 9: 464-481.
84. Rothstein A (1970). Sulfhydryl groups in membrane structure and function. Ciirr. Top. dfetnhr. Tramp. 1: 135-176.
85. Ruegg CE, Gandolfi AJ. Nagle RB, and Brendel K (1987). Dif- ferential patterns of injury to the proximal tubule of renal cortical slices following in tvifro exposure to mercuric chloride, potassium dichromate, or hypoxie conditions. To.xicol. Appl. Plramiacol. 90:
86. Sapin C, Druet E, and Druet P (1977). Induction of anti-glomer- ular basement membrane antibodies in the Brown-Norway rat by mercuric chloride. Clin. Exp. Irnnnmol. 28: 173-179.
87. Smith MW, Ambudkar IS, Phelps PC, Regec AL, and Trump BF (1987). HgC1,-induced changes in cytosolic Ca2+ of cultured rabbit renal tubular cells. Biochirn. Bioplzys. Acfa 931: 130-142.
88. Spater HW, Poruchynsky MS, Quintana N, Inoue M, and Novikoff AB (1982). Immunocytochemical localization of y-glutamyltrans- ferase in rat kidney with protein A-horseradish peroxidase. Proc. Nafl. Acad. Sci. USA 79: 3547-3550.
89. Squibb KS, Riddlington JW, Carmichael NG, and Folwer BA (1979). Early cellular effects of circulating cadmium-thionein on proximal tubules. Environ. Health Perspecf. 28: 287-296.
90. Stein JH, Gottschall J, Osgood W, and Fenis TF (1975). Patho- physiology of a nephrotoxic model of acute renal failure. Kidney
91. Sudo hl, Honda N, Hishida A, and Nagase hl (1977). Renal he- modynamics in uranyl acetate-induced actute renal failure in of rabbits. Kidney bit. 11: 35-43.
92. Tanaka T, Naganuma A, and Imura N (1990). Role of y-gluta- myltranspeptidase in renal uptake and toxicity of inorganic mer- cury in mice. Toxicology 60: 187-198.
93. Tanaka-Kagawa T, Naganuma A, and Imura N (1993). Tubular secretion and reabsorption of mercury compounds in mouse kid- ney. J. Phnnnncol. Exp. Ther. 264: 776-782.
93. Taugner R, \Vinkel K, and Iravani J (1966). Zur lokalization der sublimatanreicherung in der ratenniere. Virchotts Arch. Pafliol. Anaf. Physiol. 340: 369-383.
95. Webb M and Etienne T (1977). Studies on the toxicity and me- tabolism of Cd-thionein. Biochetn. Phannacol. 26: 25-30.
96. Weening JJ, Hoedemaefer PJ, and Bakker WW (1981). Imuno- regulation and anti-nuclear antibodies in mercury-induced glom- erulopathy in the rat. Clin. Exp. Imnirrtol . 45: 64-71.
97. Weinberg JM, Harding PG, and Humes HD (1982). Mitochondria1 bioenergetics during the initiation of mercuric chloride-induced renal injury. I. Direct effects of in vifro mercuric chloride on renal cortical mitochondria1 function. J. Biol. Cltern. 257: 60-67.
98. Weinberg JM, Harding PG, and Humes HD (1982). Mitochondria1 bioenergetics during the initiation of mercuric chloride-induced renal injury. 11. Functional alterations of renal cortical mitochon- dria isolated after mercuric chloride treatment. J. B i d . Cliern. 257:
99. Wilson CD and Dixon FJ (1986). The renal response to immu- nological injury. In: The Kidney, Bhl Brenner and FC Rector (eds). WB Saunders, Philadelphia, pp. 800-889.
100. Woods JS, Calas CA, and Aicher LD (1990). Stimulation of por- phyrinogen oxidation by mercuric ion. 11. Promotion of oxidation from the interaction of mercuric ion by glutathione, and mito- chondrial-gencratcd hydrogen peroxide. Afol. Plrannacol. 38:
101. Woods JS, Calas CA, Aicher LD, Robinson BH, and Mailer C (1990). Stimulation of porphyrinogen oxidation by mercuric ion. I. Evidence of free radical formation in the presence of thiols and hydrogen peroxide. Mol. Phnnnncol. 38: 253-260.
102. Zalme RC, hlcDowell Ehl, Nagle RB, hlcNeil JS, Flamenbaum WV, and Trump BF (1976). Studies of the pathophysiology of acute renal failure. 11. A histochemical study of the proximal tubule of the rat following administration of mercuric chloride. Virchoivs Arch. (Cell Pnfliol.) 22: 197-216.
103. Zalups RK (1991). Autometallographic localization of inorganic
261-271.
I t l f . 6: 27-41.
68-74.
261-266.
at MERCER UNIV on September 29, 2010tpx.sagepub.comDownloaded from

Vol. 26, No. 1, 1998 RENAL TOXICITY OF HEAVY METALS 103
cury in isolated perfused proximal tubules exposed to mercury- metallothionein. J. Toxicol. Environ. Health 44: 101-1 13.
115. ZalUPS RK, COX c, and Diamond GL (1988). Histological and urinalysis assessment of nephrotoxicity induced by mercuric chlo- ride in normal and uninephrectomized rats. In: Biological dfoni- toring of Toxic Mernls, TW Clarkson, L Friberg, GF Nordberg, and PR Sager (eds). Plenum, New York, pp. 531-545.
116. Zalups RK and Diamond GL (1987). Mercuric chlondc-induced nephrotoxicity in the rat following unilateral nephrectomy and compensatory renal growth. Virclzo%vs Arch. B 53: 336-346.
117. Zalups RK, Knutson KL, and Schnellman RC (1993). In vifro analysis of the accumulation and toxicity of inorganic mercury in segments of the proximal tubule isolated from the rabbit kidney. To.ricol. Appl. Phamzacol. 116: 221-227.
118. Zalups RK and Lash LH (1990). Effects of uninephrectomy and mercuric chloride on renal glutathione homeostasis. J. Phamiacol. fip. Tlzer. 254: 962-970.
119. n l u p s RK and Lash LH (1994). Advances in understanding the renal transport and toxicity of mercury. J. Toxicol. Ind. Health 42:
mercury in the kidneys of rats: Effect of unilateral nephrectomy and compensatory renal growth. Exp. Mol. Pathol. 54: 10-21.
104. Zalups RK (1993). Early aspects of the intrarenal distribution of mercury after the intravenous administration of mercuric chloride. Toxicology 79: 215-228.
105. .Zalups RK (1995). Organic anion transport and action of y-glu- tamyl transpeptidase in kidney linked mechanistically to renal tu- bular uptake of inorganic mercury. To.ricol. Appl. Plbn?acol. 132:
106. ZalUpS RK (1997). EnhancedrenaloutermedullarY UPtakeofmer- cury associated with unilateral nephrectomy: Implication of a lu- minal mechanism. J. To.ricol. Emiron. Healrh 50: 173-194.
107. Zalups RK and Barfuss D (1990). Accumulation of inorganic mer- cury along the renal proximal tubule of the rabbit. Toxicol. Appl. Pkannacol. 106: 245-253.
108. Zalups RK and Barfuss D (1995). Accumulation and handling of inorganic mercury in the kidney after coadministration with glu- tathione. J. To.ricol. Environ. Health 44: 385-399.
289-298.
109. Zalups RK and Barfuss D (1995). Renal disposition of mercury in rats after intravenous injection of inorganic mercury and cy- steine. J. Toxicol. Environ. Health 44: 401-413.
110. Zalups RK and Barfuss D (1996). Nephrotoxicity of inorganic mercury co-administered with L-cysteine. To.ricology 109: 15-29.
111. Zalups RK and Cherian MG (1992). Renal metallothionein me- tabolism after a reduction of renal mass. I. Effect of unilateral nephrectomy and compensatory renal growth on basal and metal- induced renal metallothionein metabolism. To.ricology 73: 8 1- 102.
112. Zalups RK and Cherian MG (1992). Renal metallothionein me- tabolism after a reduction of renal mass. 11. Effect of zinc pre- treatment on the renal toxicity and intrarenal accumulation of in- organic mercury. Toxicology 71: 103-1 17.
113. Zalups RK, Cherian MG, and Barfuss DW (1995). Mercury-me- tallothionein and the renal accumulation and handling of mercury. Toxciology 83: 61-78.
114. Zalups RK, Cherian MG, and Barfuss DW (1995). Lack of lu- minal or basolateral uptake and transepithelial transport of mer-
1-44. 120. Zalups RK and Lash LH (1996). Interactions between glutathione
and mercury in the kidney, liver and blood. In: Toxicology of Metals, LW Chang (ed). CRC Lewis Publishers, New York, pp. 145-1 63.
121. Zalups RF and Lash LH (1997). Depletion of glutathione in the kidney and the renal disposition of administered inorganic mer- cury. Drug. hfetab. Disp 25: 516-523.
122. Zalups RK and Lash LH (1997). Binding of mercury in renal brush-border and basolateral membrane-vesicles. Implication of a cysteine conjugate of mercury involved in the luminal uptake of inorganic mercury in the kidney. Biochem. Phannacol. 53: 1889- 1900.
123. Zalups RK and Minor KH (1995). Luminal and basolateral rnech- anisms involved in the renal tubular uptake of inorganic mercury. J. Toxicol. Environ. Healrh 46: 73-100.
124. Zalups RK, Robinson MK, and Barfuss DW (1991). Factors af- fecting inorganic mercury transport and toxicity in the isolated perfused tubule. J. h i . Soc. Neplzrol. 2: 866-878.
125. Zalups RK and Veltman JC (1988). Renal glutathione homeostasis in compensatory renal growth. Life Sci 42: 2171-2176.
at MERCER UNIV on September 29, 2010tpx.sagepub.comDownloaded from