trace analysis — challenges and problemsmencer/pdf_docs/chm342_paper1.pdf · trace analysis —...
TRANSCRIPT

271
1040-8347/02/$.50© 2002 by CRC Press LLC
Critical Reviews in Analytical Chemistry, 32(4):271–300 (2002)
Trace Analysis — Challenges and Problems
Jacek NamiesnikDepartment of Analytical Chemistry, Chemical Faculty, Gdansk Univ. of Technology, 11/12 G.Narutowicz Str., 80-952 Gdansk, Poland. tel.: (058) 374-10-10, fax: (058) 347-26-94. e-mail:[email protected]
ABSTRACT: Analytical procedures and techniques employed to determine trace componentsare objects of growing interest to analysts. The determination of those components in samples ofdifferent matrix composition is becoming ever more common. Hence, attention should be paidto problems involved in this type of activity.
The article presents specific problems connected with trace analysis. The problems andsolutions proposed are documented with data from the literature.
KEY WORDS: trace analysis, sources of error, prevention, wall memory effect, cross-contami-nation.
I. INTRODUCTION
Even a cursory perusal of any analyticaljournal must lead one to the conclusion thattrace and ultra-trace analysis is a domain ofchemical analysis that is gaining in impor-tance recently. This conclusion is corrobo-rated not only by the feelings and opinionsof analysts. By the current definition of theterm ‘trace component’ proposed by theIUPAC, the limit from which we can talkabout trace analysis is the concentration of100 ppm (100 µg/g). Naturally, this limit ispurely conventional and is not a constant. Asrecently as 30 years ago ‘trace analysis’ wasunderstood to denote activities aiming to de-termine components at a concentration levelone order of magnitude higher (i.e., below1000 ppm, or 0.1%).
Even today the determination of compo-nents at a concentration level of 100 ppm,even in samples with complex matrices, doesnot pose major problems and is done rou-tinely in many laboratories. This is mainlydue to the rapid development of instrumen-
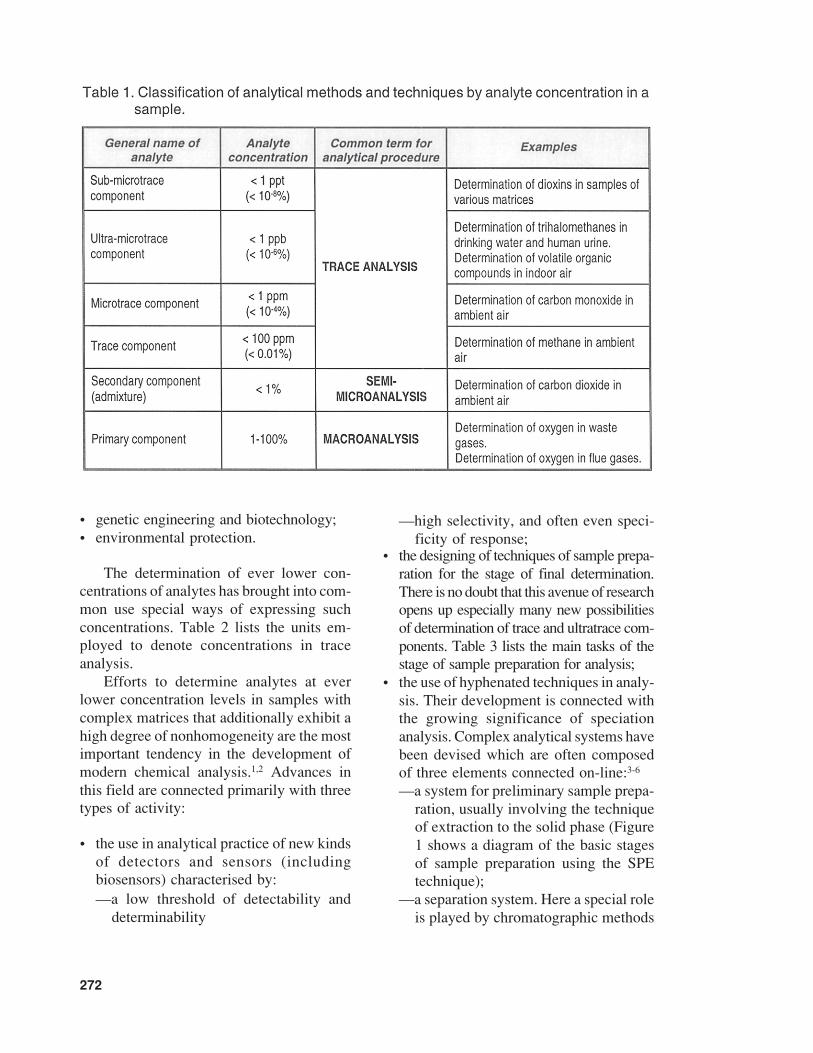
tation, or the science of the construction anduse of monitoring and measuring devices.Hence, one may expect the definition of theterm ‘trace component’ to change again soon.Table 1 presents a classification of analyti-cal methods and techniques by analyte con-centration in the sample examined.
II. AREA OF APPLICATION OFTRACE ANALYSIS
It is possible to distinguish three areas ofscience and technology that spur the devel-opment of analytical methods and techniquesemployed in the determination of low andvery low analyte contents in samples of vari-ous kinds. They are
• technologies of the production of high-purity materials; to date, the purity of thecleanest man-made material is denoted by11 N, which means that the sum total ofall impurities it contains does not exceed10–9%, or 10 ppt;
272
• genetic engineering and biotechnology;• environmental protection.
The determination of ever lower con-centrations of analytes has brought into com-mon use special ways of expressing suchconcentrations. Table 2 lists the units em-ployed to denote concentrations in traceanalysis.
Efforts to determine analytes at everlower concentration levels in samples withcomplex matrices that additionally exhibit ahigh degree of nonhomogeneity are the mostimportant tendency in the development ofmodern chemical analysis.1,2 Advances inthis field are connected primarily with threetypes of activity:
• the use in analytical practice of new kindsof detectors and sensors (includingbiosensors) characterised by:—a low threshold of detectability and
determinability
—high selectivity, and often even speci-ficity of response;
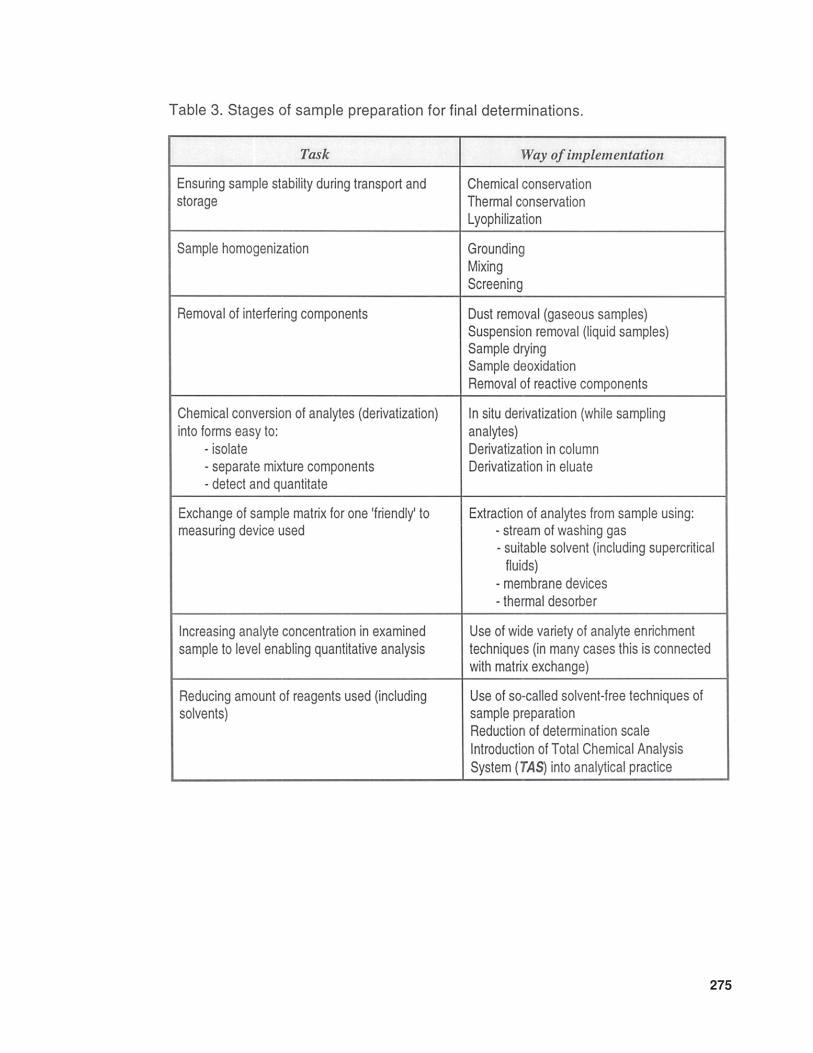
• the designing of techniques of sample prepa-ration for the stage of final determination.There is no doubt that this avenue of researchopens up especially many new possibilitiesof determination of trace and ultratrace com-ponents. Table 3 lists the main tasks of thestage of sample preparation for analysis;
• the use of hyphenated techniques in analy-sis. Their development is connected withthe growing significance of speciationanalysis. Complex analytical systems havebeen devised which are often composedof three elements connected on-line:3-6
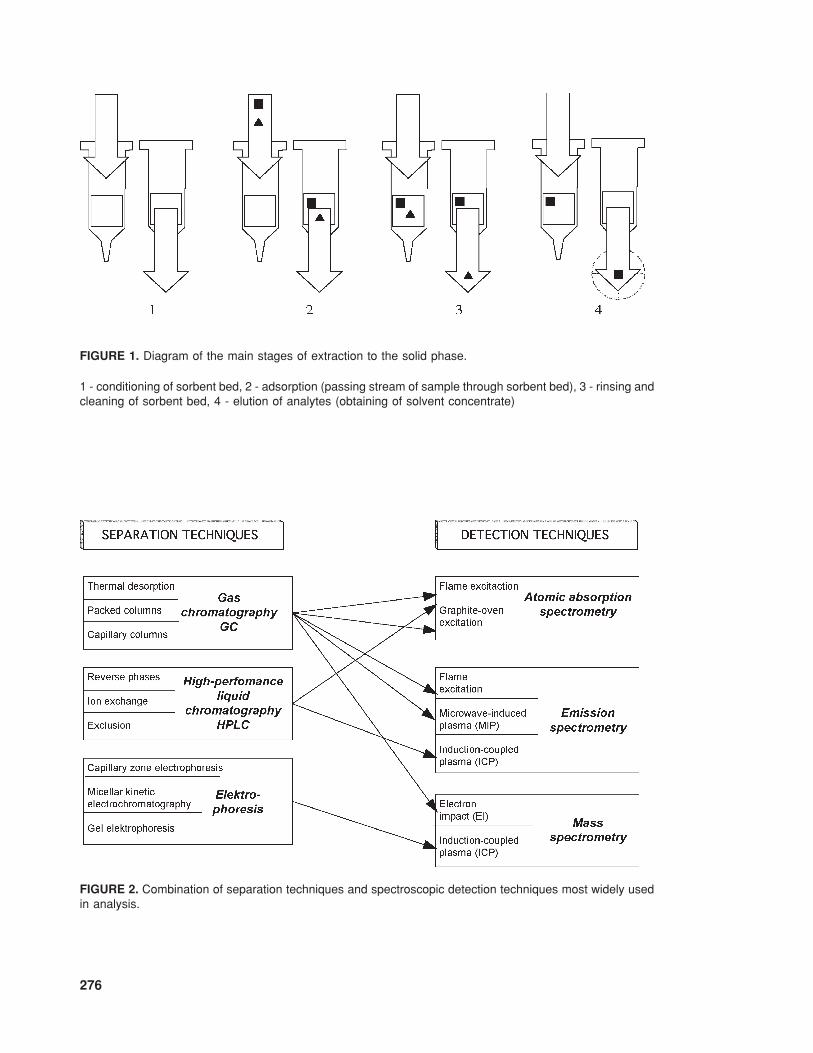
—a system for preliminary sample prepa-ration, usually involving the techniqueof extraction to the solid phase (Figure1 shows a diagram of the basic stagesof sample preparation using the SPEtechnique);
—a separation system. Here a special roleis played by chromatographic methods

273

274
(mainly GC and HPLC). They are usu-ally employed to separate mixtures intoindividual components;
—a suitable detection system. For ex-ample, Figure 2 presents combina-tions of separation techniques andspectroscopic detection systems thatare most widely used in analyticalpractice.6
III. MAIN SOURCES OF ERROR INTRACE ANALYSIS
Obviously enough, the determination ofcomponents that occur in a sample at verylow concentrations presents a completely dif-ferent challenge to analysts. The various op-erations performed on the sample during itspreparation for the stage of final determina-tions can be a source of many errors cru-cially affecting the final result of the analy-sis.
The chief sources of error are changesin the composition of the sample understudy brought about by a variety of fac-tors. In Table 4 three principal kinds oferror distorting the result of analysis arepresented.
The bias of the final determination resultcaused by the errors listed (Table 4) may bedue to various processes occurring in thesample and due to the influence of the exter-nal environment on the sample. By way ofexample, Figure 3 presents diagrammaticallythe sources of error that can distort the resultof determination of a trace component in aliquid sample. The intensity of the processesand phenomena shown in the figure can varydepending on:
• the temperature of the sample and itschanges,
• the presence and intensity of solar radia-tion, and
• the presence of living organisms in the sample.
The contact of analytes present in bothgas and liquid mixtures with the walls ofvessels, tubing and appliances crucially af-fects the concentration levels of trace andultratrace components. It is the result ofadsorption and desorption of analytes onthe surface of a solid. These phenomenadistorting the sample composition are de-scribed collectively as ‘the wall memoryeffect’ and ‘cross-contamination’. Table 5shows diagrammatically the magnitude ofpolar organic analyte-wall surface interac-tion.
What gives rise to many misunderstand-ings is the use in analysis of water of suitablepurity. Table 6 lists data concerning the ef-ficiency of some water-purifying methods,while Table 7 gives specific electrical con-ductance values for water with various de-grees of purity.
Obtaining ultrapure water, which is thebasic reagent in laboratories performing traceanalysis, is usually a multistage process. Inthe installation for the production of water ofthis level of purity the following processesare employed:
• preliminary filtering to remove most sus-pended matter;
• activated-carbon filtering to remove freechlorine (to a sub-1-ppm level), small par-ticles of suspended matter (1 to 5 µm) andmost organic matter (to a level below1 ppm of carbon);
• preliminary chemical processing, con-sisting in the neutralization of water withacid or alkalies, as required, and in thetransformation of dissolved CO2 into car-bonates and bicarbonates;
• ionic exchange, during which ions presentin water, including those introduced in theprocessing, are exchanged for hydrogenand hydroxyl ions on the cation and anionexchangers;
• membrane filtration — microfiltration,ultrafiltration, reverse osmosis;
275
276
FIGURE 1. Diagram of the main stages of extraction to the solid phase.
1 - conditioning of sorbent bed, 2 - adsorption (passing stream of sample through sorbent bed), 3 - rinsing andcleaning of sorbent bed, 4 - elution of analytes (obtaining of solvent concentrate)
FIGURE 2. Combination of separation techniques and spectroscopic detection techniques most widely usedin analysis.

277
FIGURE 3. Diagrammatic presentation of factors that can affect concentrationlevels of trace components in a liquid sample.
1 - contact of sample with laboratory air; 2 - residues of components of dishwashingmixtures; 3 - distilled water; 4 - reagents and solvents employed; 5 - contact withanalyst; 6 - vaporization of the most volatile components; 7-8 - processes ofadsorption-desorption (wall memory effect); 9 - adsorption of analytes on suspen-sion; 10 - precipitation of sediment; 11 - leaching of components from vessel; 12-13 - permeation of solution components through vessel walls; 14 - reaction ofanalyte with vessel material; 15 - chemical reactions among solution components.

278
279
• water degassing to remove oxygen andcarbon dioxide, and to reduce the contentof volatile organic compounds to a levelbelow 1 ppb;
• UV sterilization, which ensures a re-duction of bacteria in water to a levelbelow 1 bacterium/100 cm3 of water,and the oxygenation of the remainingpart of organic matter to a level below 1ppb;
• water ozonation to further reduce the con-tent of organic matter.
For years the amount of organic matterin water (usually expressed by the total con-tent of organic carbon) has been consideredthe chief measure of quality of high-puritywater.
Analytic work in the field of traceanalysis should be carried out not onlywith the use of highest-purity reagents,but also in rooms conforming to the ‘cleanroom’ standards. Such a room is a spaceseparated from ambient air and only ac-cessible through a sluice. A stream of aircleaned by a system of filters is pumpedinto the room under a slight positive gaugepressure. The excess is removed through alattice floor. The HEPA filter that is partof the system stops dust particles of morethan 0.3 µm with an efficiency of up to
99.99%. With the air cleaned in this way,controlled humidity and temperature, andsuitable working rules of the personnel,the risk of sample contamination is re-duced to a minimum in such a laboratory.There has appeared a first mention of theso-called ultraclean chemical laboratory(UCCL) in the literature. In its case the airquality must meet even more stringent cri-teria. Unfortunately, a significant limita-tion to ‘clean rooms’ is the very high costof such an investment. That is why it ismuch more common for laboratories toutilize much cheaper ‘clean boxes’ with alaminar flow of an inert gas. They can beused successfully in sample processing.The introduction of the robotization andautomation of sample processing into ana-lytical practice may be a solution to manyproblems connected with the cleanness ofthe laboratory environment.
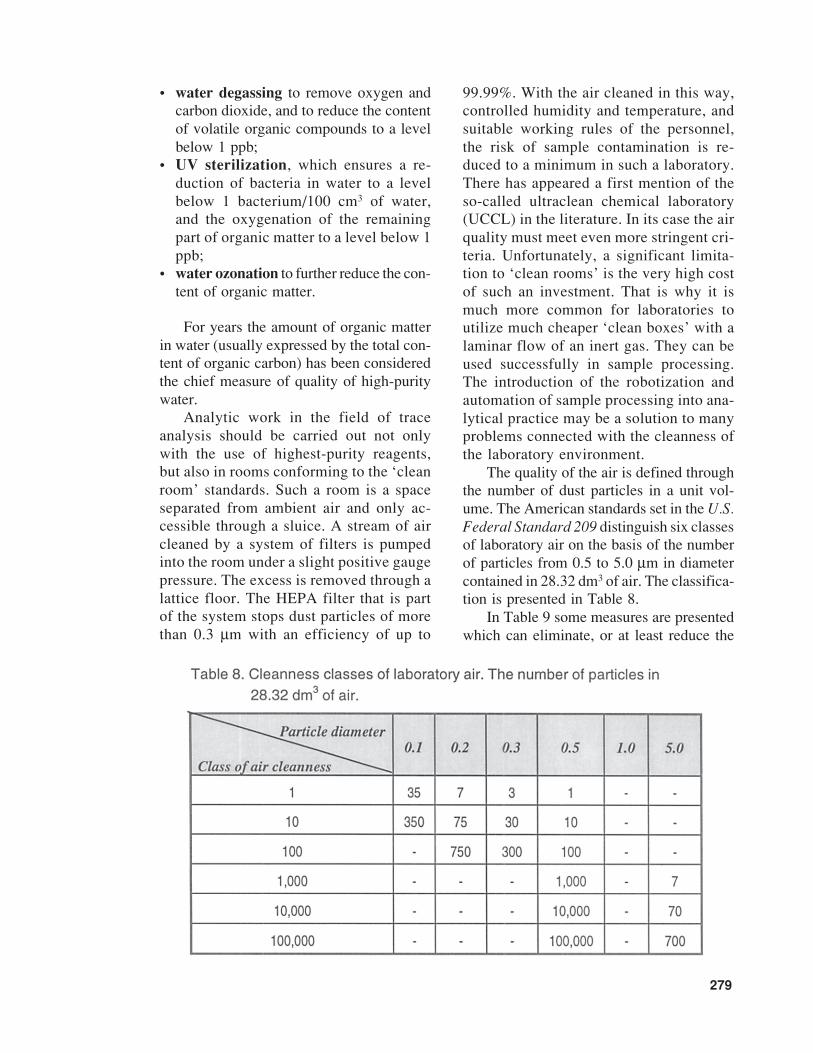
The quality of the air is defined throughthe number of dust particles in a unit vol-ume. The American standards set in the U.S.Federal Standard 209 distinguish six classesof laboratory air on the basis of the numberof particles from 0.5 to 5.0 µm in diametercontained in 28.32 dm3 of air. The classifica-tion is presented in Table 8.
In Table 9 some measures are presentedwhich can eliminate, or at least reduce the
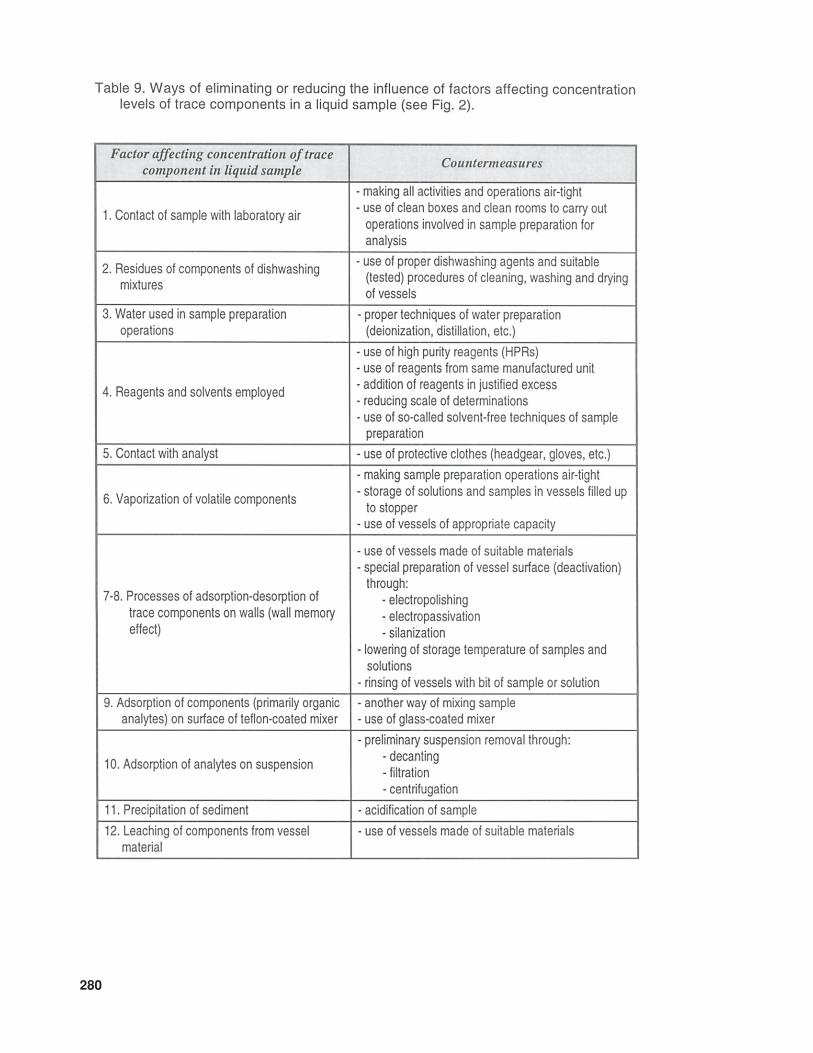
280

281
Table 9 (continued)
intensity of, factors likely to affect the con-centration levels of trace components in aliquid sample. In the case of gaseous or solidsamples, there will appear not only their spe-cific sources of error, but also those (or atleast some of those) characteristic of liquidsamples.
In the literature one can find plenty ofmore detailed information about sources oferror in trace analysis. There have appearedcomprehensive overviews devoted to theseissues7-10. They deal primarily with inorganictrace analysis.
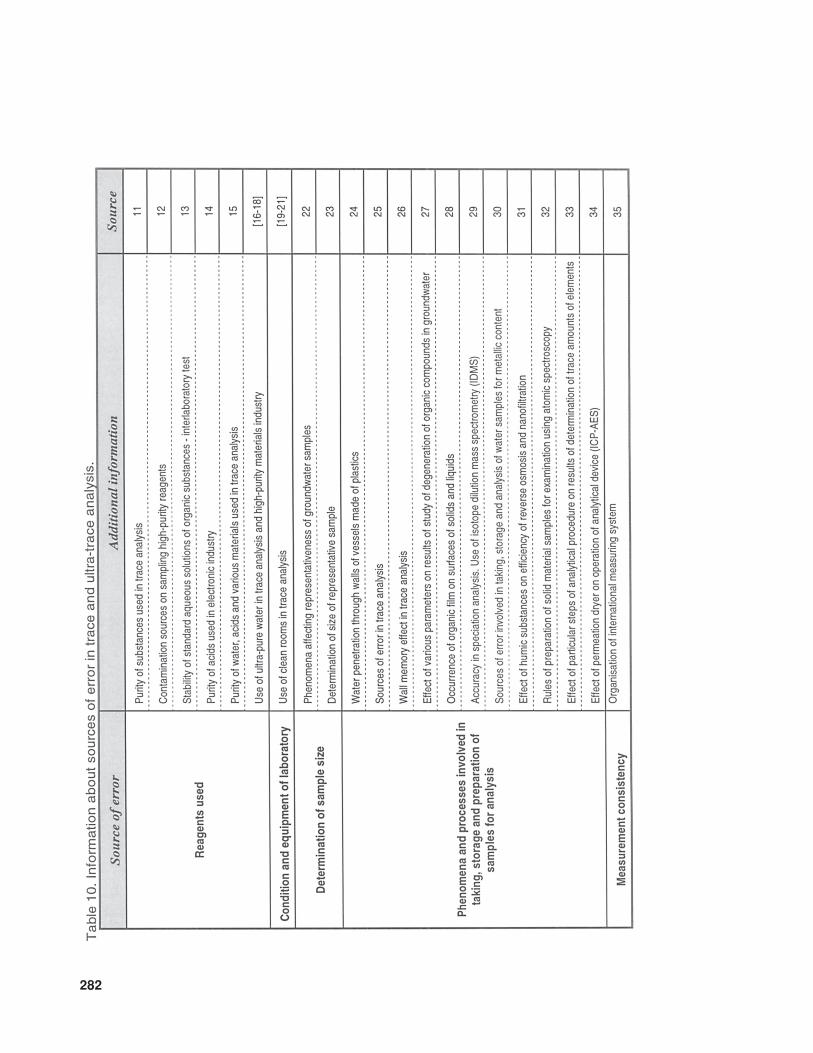
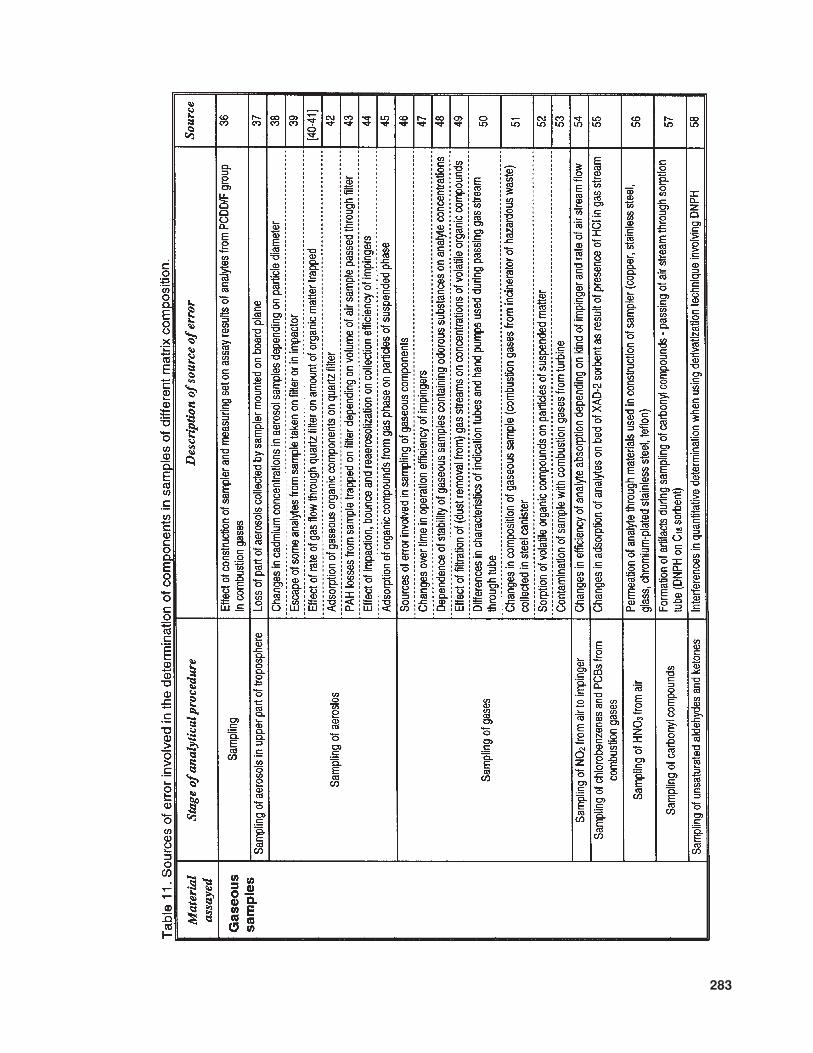
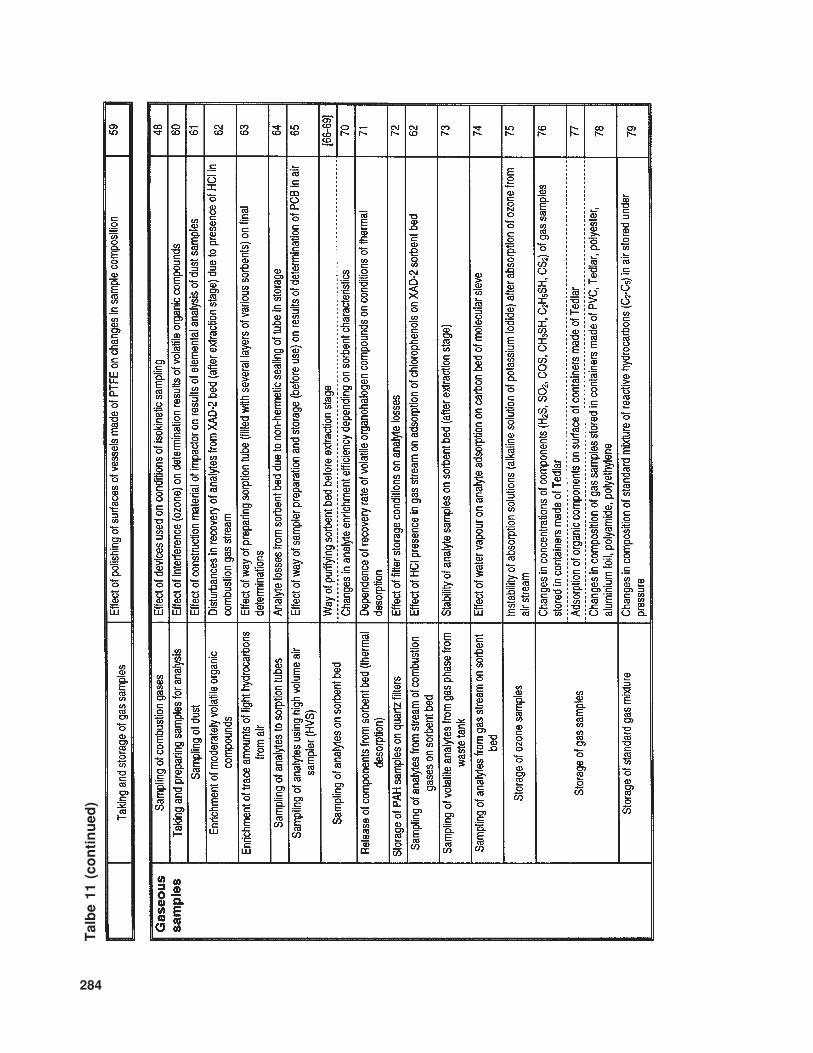
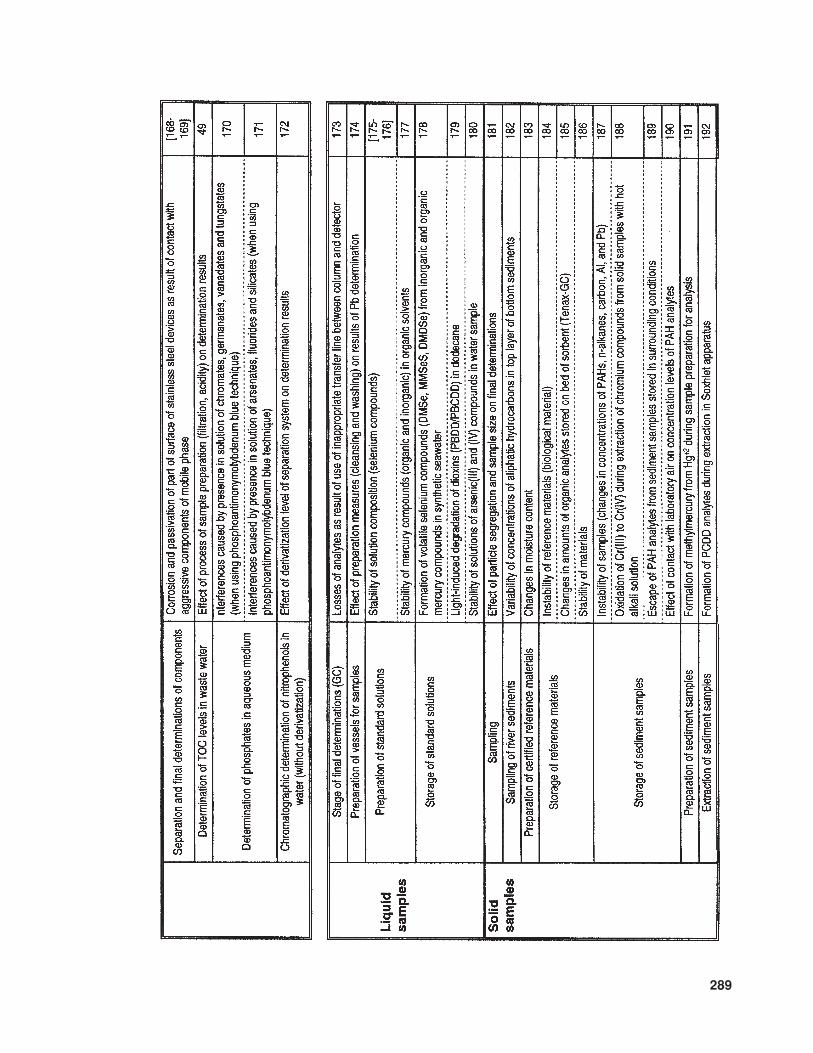
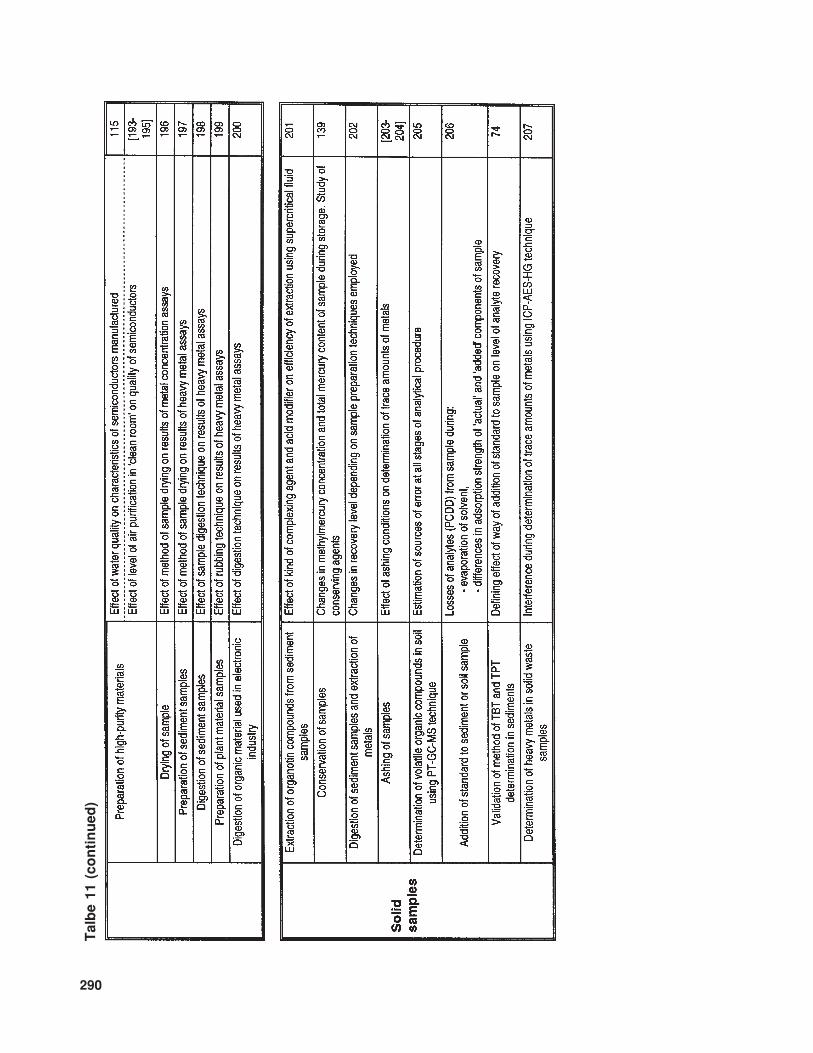
The available literature also suppliesmuch information (of various levels of de-tail) concerning problems and sources of errorinvolved in the determination of trace com-ponents in samples with complex matrices.In Table 10 the relevant information is listedthat concerns trace analysis, while Table 11provides data about specific issues related totrace analysis in three basic types of sample(gaseous, liquid, and solid).
SUMMARY
Trace analysis is an important part ofchemical analysis and is becoming an objectof interest to ever growing ranks of analysts.In solving analytical tasks in this field, moreand more complicated monitoring and mea-suring equipment is used. Unfortunately, thisdoes not always go hand in hand with thelevel of knowledge of the personnel thatoperates such devices. There have been agrowing number of cases when an analyticaldevice is treated like a typical black box intowhich it suffices to put a sample to get therequired analytical information. This ap-proach is, of course, false and the determina-tion results obtained in this way are often asource of misinformation. The author hopesthat the present article will be useful in point-ing out several problems relevant in deter-mining components occurring at low andvery low concentrations in samples with com-plex compositions.
282
283
284
Tal
be
11 (
con
tin
ued
)
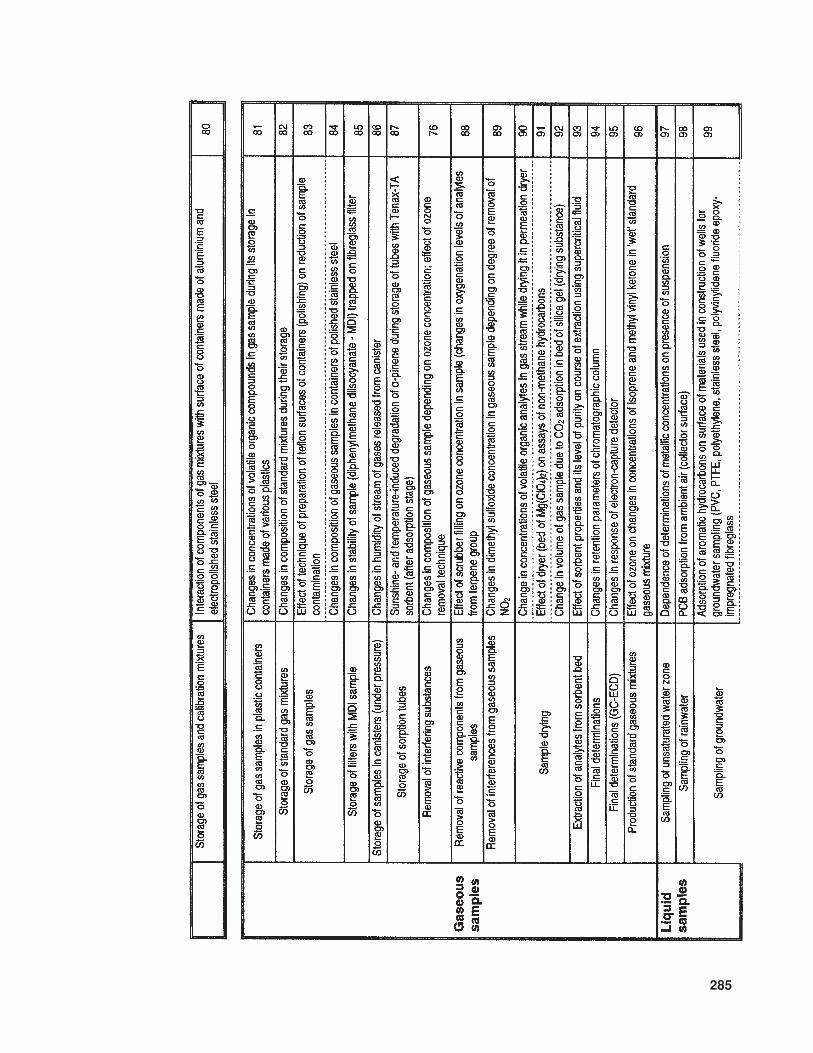
285
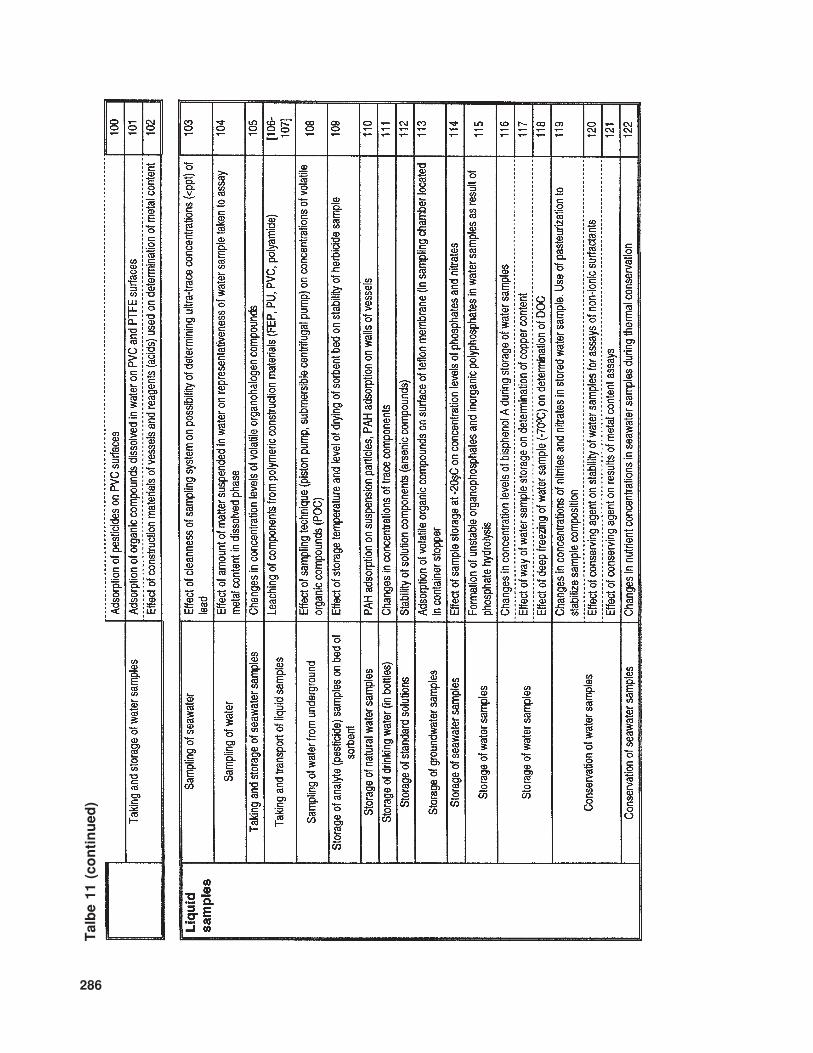
286
Tal
be
11 (
con
tin
ued
)
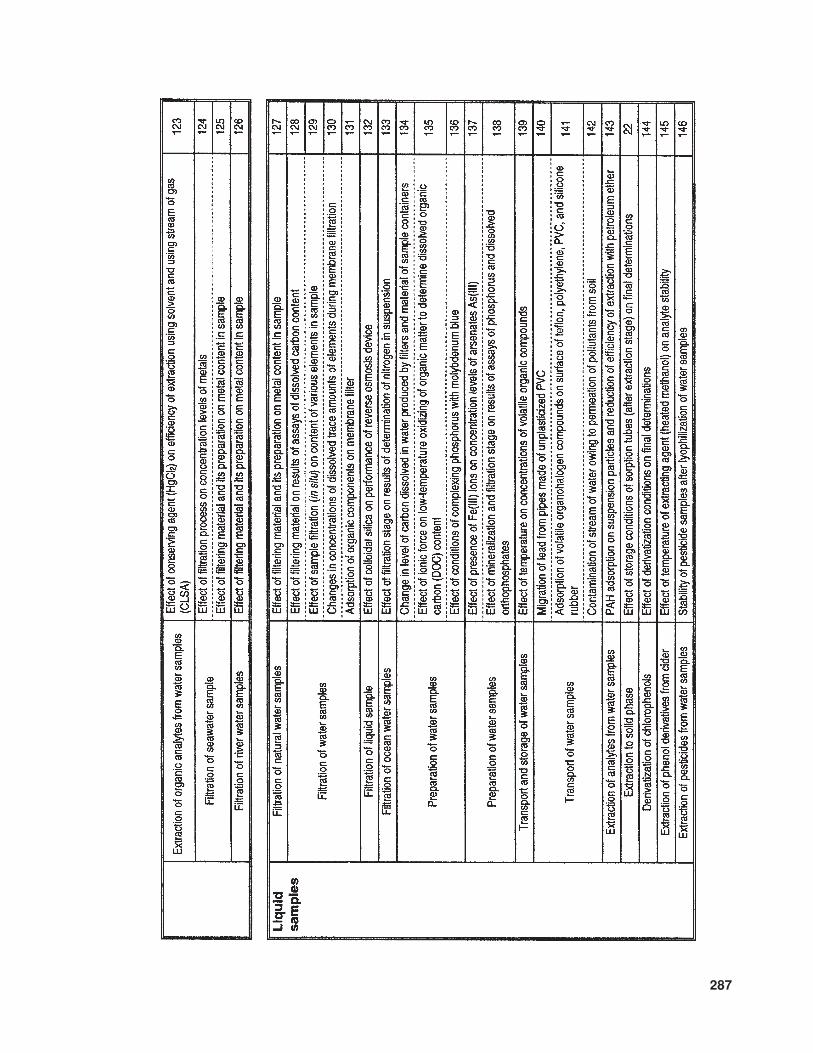
287
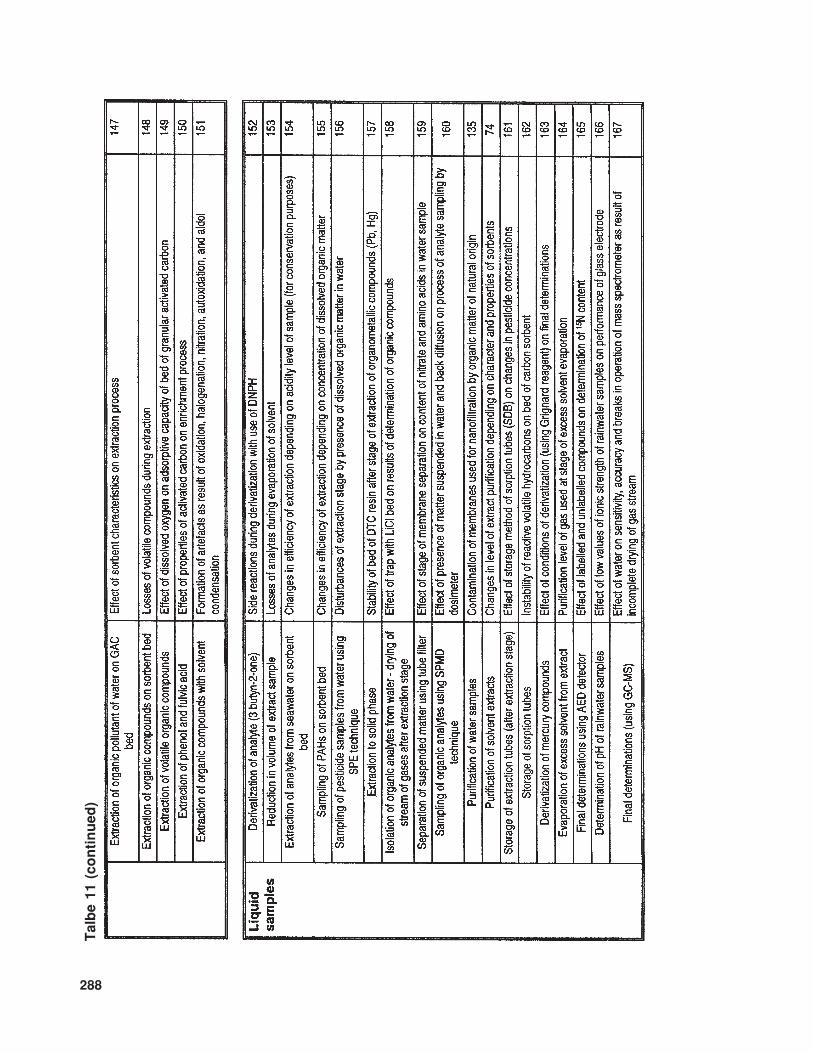
288
Tal
be
11 (
con
tin
ued
)
289
290
Tal
be
11 (
con
tin
ued
)

291
REFERENCES
1. J. Namiesnik, Trends in environmentalanalytics and monitoring. Crit. Rev. Anal.Chem., 30, 221–269 (2000).
2. J. Namiesnik, Modern trends in monitoringand analysis of environmental pollutants. Pol.J. Environ. Stud., 10, 127–140 (2001).
3. U.A.Th. Brinkman, Multidimensional ap-proaches in environmental analysis. Anal.Commun. 34, 9H-14H (1997).
4. O.F.X. Donard, F.M. Martin, Hypenated tech-niques applied to environmental speciationstudies. Trends Anal. Chem., 11, 17–26 (1992).
5. Th.H.M. Noij, F.L. Schulting, Trends in ana-lytical techniques for the analysis and identi-fication of organic micropollutants in water.Water Supply, 11, 31–49 (1993).
6. R. –Lobinski, Advanced techniques forbioinorganic and environmental speciationanalysis for heavy metals. NATO ASI Ser.,26, 389–409 (1997).
7. M. Hoenig, Preparation steps in environmen-tal trace element analysis – facts and traps.Talanta, 54, 1021–1038 (2001).
8. J. Namiesnik, T. Górecki, Preparation of en-vironmental samples or the determination oftrace constituents. Pol. J. Environ. Stud., 10,77–84 (2001).
9. J. Namiesnik, T. Górecki, Sample prepara-tion for chromatographic analysis of plantmaterial. J. Planar Chromatogr., 13, 404–413 (2000).
10. G. Tölg, Problems and trends in extreme traceanalysis for the elements. Anal. Chim. Acta,283, 3–18 (1993).
11. S. Noack, Of what purity are pure substances,GIT Fachz. Lab., 40, 771–775 (1996).
12. W. Büttner, J. Dahmen, N. Harder, J.Heckenkamp, Contamination during the sam-pling of ultraclean chemicals. 37, 992–996(1993).
13. M. Gardner, Stability of trace organic substancesin water: data obtained from interlaboratory tests.Anal. Commun., 35, 373–375 (1998).
14. M. Feuerstein, G. Schlemmer, B. Kraus, Thesimultaneous GFAAS determination of
various elements at ultratrace levels inultrapure acids and photoresist stripper solu-tions. At. Spectrosc., 19, 1–5 (1998).
15. G. Knapp, Mechanised methods of sampledecomposition in trace and ultra-trace analy-sis. Analytical Proceedings, 27, 112–114(1990).
16. J. Wibowo, F. Shadman, D. Blackford, Mea-suring and removing dissolved and colloidalsilica in ultrapure water. Micro (May 1997).
17. M. K. Balazs, Ultrapure water, friend or en-emy?. Solid State Technol., 36, 75–81 (1993).
18. K. Clark, M. Retzik, D. Darbouret, Measur-ing TOC to maintain high-purity water.Ultrapure Water, 21–24 (February 1997).
19. H. Schleibinger, H. Rüden, Air filters fromHVAC systems as possible source of volatileorganic compounds (VOC) – laboratory andfield assays. Atmospheric Environment, 33,4571–4577 (1999).
20. A. Lamberty, J. R. Moody, E. van Duffel, P.de Bièvre, J. Broothaerts, Ph. Taylor, C.Lathen, The ultra-clean chemical laboratory(UCCL) at the Institute for Reference Mate-rials and Measurements (IRMM). FreseniusJ. Anal. Chem., 357, 359–363 (1997).
21. C. P. Ferrari, A. L. Moreau, C. F. Boutron,Clean conditions for the determination of ul-tra-low levels of mercury in ice and snowsamples. Fresenius J. Anal. Chem., 366, 433–437 (2000).
22. C. Aguilar, I. Ferrer, F. Borrull, R. M. Marcé,D. Barceló, Monitoring of pesticides in riverwater based on samples previously stored inpolymeric cartridges followed by on-linesolid-phase extraction-liquid chromatography-diode array detection and confirmation byatmospheric pressure chemical ionizationmass spectrometry. Anal. Chim. Acta, 386,237–248 (1999).
23. G. A. Sorial, M. T. Suidan, R. D. Vidic, R. C.Brenner, Effect of GAC Characteristics onadsorption of organic pollutants. WaterEnviron. Res., 65, 53–57 (1993).
24. H. Gerstenberger, Zur Durchlässigkeit von Wasserdurch die Wandungen von Kunststffflaschen(Water penetration through the walls of contain-ers made from plastics). GIT Fachz. Lab. 4, 279–284 (1994).

292
25. B. Griepink., Improving the quality of envi-ronmental trace analysis. Fresenius Z. Anal.Chem., 317, 210–212 (1984).
26. P. Bachhausen, N. Buchholz, H. Hartkamp,Wandeffekte in der Gasspurenanalyse.Fresenius Z. Anal. Chem. 322, 336–342(1985).
27. S. K. Papiernik, A raview of in situ measure-ment of organic compound transformation ingroundwater. Pest. Manag. Sci., 57, 325–332(2001).
28. N. L. Law, M. L. Diamond, The role of or-ganic films and the effect on hydrophobicorganic compounds in urban areas: an hy-pothesis. Chemosphere, 36, 2607–2620(1998).
29. H. M. “Skip” Kingston, D. Huo, Y. Lu, S.Chalk, Accuracy in species analysis: speci-ated isotope dilution mass spectrometry(SIDMS) exemplified by the evaluation ofchromium species. Spectrochimica Acta B,53, 299–309 (1998).
30. G. Benoit, K. S. Hunter, T. F. Rozan, Sourcesof trace metal contamination artifacts duringcollection, handling, and analysis of freshwa-ters. Anal. Chem., 69, 1006–1011 (1997).
31. A. E. Childress, M. Elimelech, Effect of solu-tion chemistry in the surface charge of poly-meric reverse osmosis and nanofiltration mem-branes. J. Membrane Sci., 119, 253–268(1996).
32. M. Hoenig, A.-M. de Kersabiec, Samplepreparation steps for analysis by atomic spec-troscopy methods: present status. Spectrochim.Acta B, 51, 1297–1307 (1996).
33. J. R. W. Woitiez, J. E. Sloof, Sampling andsample preparation. In: Determination of traceelements (Ed.: Z. B. Alfassi), VCH Weinheim(1994) Chapter 3, 59–107.
34. J. Yang, T. S. Conver, J. A. Koropchak, D. A.Leighty, Use of a multi-tube Nafion® mem-brane dryer for desolvation with thermospraysample introduction to inductively coupledplasma-atomic emission spectrometry.Spectrochim. Acta B, 51, 1491–1503 (1996).
35. B. King, Development of an internationalchemical measurement system plenary lec-ture. Analyst, 118, 587–591 (1993).
36. W. Funcke, H. Linnemann, Sampling of poly-chlorinated dibenzofurans (PCDF) andibenzo(p)dioxins (PCDD) in emissions fromcombustion facilities using an adsorptionmethod. Chemosphere, 24, 1563–1572(1992).
37. V. L. Foltescu, E. Selin, Corrections for par-ticle losses and sizing errors during aircraftaerosol sampling using a rosemount inlet andthe PMS LAS-X. Atmospheric Environment,29, 449–453 (1995).
38. K. G. Heumann, Determination of inorganicand organic traces in the clean room compart-ment of Antarctica. Anal. Chim. Acta, 283,230–245 (1993).
39. M. Chih Chang, C. Sioutas, S. Kim, H GongJr., W. S. Linn, Reduction of nitrate lossesfrom filter and impactor samplers by meansof concentration enrichment. AtmosphericEnvironment, 34, 825–98 (2000).
40. S. R. McDow, J. J. Huntzicker, Vapor ad-sorption artifact in the sampling of organicsaerosol: face velocity effects. AtmosphericEnvironment, 24A, 2563–2571 (1990).
41. C. E. Miller, R. Lewis, The effect of samplingrate on the efficiency of the warren springsampler in the determination of ambient con-centrations of airborne particles. Technicalnote. Atmospheric Environment, 24A, 2267–2270 (1990).
42. Th. W. Kirschstetter, C. E. Corrigan, T.Novakov, Laboratory and field investigationof the adsorption of gaseous organic com-pounds onto quartz filters. Atmospheric Envi-ronment, 35, 1663–1671 (2001).
43. U. Knecht, H.-J. Woitowitz, PAH-losses fromglass fiber filters under the conditions of dif-ferent air volume sampling: results of fieldevaluations in occupational atmospheres.Fresenius Z. Anal. Chem., 331, 8–13, (1988).
44. S. A. Grinshpun, K. Willeke, V. Ulevicius, A.Juozaitis, S. Terzeiva, J. Donnely, G. N.Stelma, K. P. Brenner, Effect of impaction,bounce and reaerosolization on the collectionefficiency of impingers. Aerosol. Sci. Technol.,26, 326–342 (1997).
45. E. D. Winegar, L. H. Keith, Sampling andanalysis of airborne pollutants. 191–208,Lewis Publishers (1993).

293
46. R. Niessner, Sampling techniques for air pol-lutants. In: Environmental Analysis (Ed.: D.Barceló): Techniques, Applications and Qual-ity Assurance (Chapter 1), 3–22, Elsevier Sci.Publ., 1993.
47. X. Lin, K. Willeke, V. Ulevicius, S. A.Grinshpun, Effect of sampling time on thecollection efficiency of all-glass impingers.Am. Ind. Hyg. Assoc. J., 58, 480–488 (1997).
48. G. Leonardos, F. Sullivan, S. P. Levine, R. T.Stordeur, T. M. Harvey, D. Schuetzle, A com-parison of polymer adsorbent and bag sam-pling techniques for paint bake oven odorousemissions. J. Air Poll. Control Assoc., 30,22–29 (1980).
49. S. D. Wachasunder, Sh. Satyanarayan, Theeffect of sample preparation techniques onthe measurement of total organic carbon ofsynthetic waste. Indian J. Environmental Pro-tection, 9, 19–21 (1989).
50. W. R. Haag, Interchangeability of gas detec-tion tubes and hand pumps. AIHAJ, 62, 65–69 (2001).
51. A. R. Ghoison, J. F. Storm, R. K. M. Jayanty,R. G. Fuerst, T. J. Logan, M. R. Midgett,Evaluation of canisters for measuring emis-sions of volatile organic air pollutants fromhazardous waste incineration. J. Air Pollut.Control Assoc., 39, 1210–1217 (1989).
52. K.-U. Goss, S. J. Eisenreich, Sorption of vola-tile organic compounds to particles from acombustion source at different temperaturesand relative humidities. Atmospheric Envi-ronment, 31, 2827–2834 (1997).
53. G. Desmet, G. Dumont, D. Tielemans, R. deLathouwer, E. J. Roekens, Measurements ofatmospheric pollutants using helicopters:evaluation of the possible contamination ofthe sample air by turbine exhausts. Atmo-spheric Environ., 29, 2547–2552 (1995).
54. S. K. Goyal, Measurement of NO2 in ambientair — effect of bubbler type and samole flowrate in sodium arsenite method. Indian J. En-vironmental Protection, 19, 161–165 (1999).
55. Z. Tuba, Z. Csintalan, Z. Nagy, K. Szente, Z.Takács, Sampling of terricolous lichen andmoss species for trace element analysis, withspecial reference to bioindication of air pollu-tion., In: Environmental sampling for trace
analysis (Ed.: B. Markert), VCH Weinheim(1994) Chapter 22, 415–434.
56. F. de Santis, A. Febo, C. Perrino, Negativeinterference of teflon sampling devices in thedetermination of nitric acid and particulatenitrate. Science Total Environ., 76, 93–99(1988).
57. M. Pires, L. R. F. Carvalho, An artifact in aircarbonyls sampling using C18 DNPH-coatedcartridge. Anal. Chim. Acta, 367, 223–231(1998).
58. R. Schulte-Ladbeck, R. Lindahl, J.-O. Levin,U. Karst, Characterization of chemical inter-ferences in the determination of unsaturatedaldehydes using aromatic hydrazine reagentsand liquid chromatography. J. Environ. Monit.,3, 306–310 (2001).
59. M. Takenaka, M. Hayashi, I. Suzuki, Y.Yamada, K. Takamatsu, M. Kageyama, Evalu-ation of a mirror-polishing technique for fluo-rocarbon polymer surfaces for reduction ofcontamination from containers used inultratrace analysis. Anal. Chem., 69, 972–976(1997).
60. D. Helmig, Ozone removal techniques in thesampling of atmospheric volatile organic tracegases. Atmospheric Environment, 31, 3635–3651 (1997).
61. H. Bayer, A. von Bohlen, R. Klockenkämper,D. Klockow, Choice of a suitable material forconstruction of a battelle type impactor tominimize systematic errors in sampling ofairborne dust. Mikrochim. Acta, 119, 167–176 (1995).
62. L. K. Tan, A. J. Liem, Effect of hydrochloricacid on sampling and analysis of semivolatileorganic compounds in incineration flue gas.1. chlorophenols. Environ. Sci. Technol., 30,1053–1060 (1996).
63. A. Castellnou, N. Gonzales-Flesca, J. O.Grimalt, Refrigerated multibed adsorption insampling and analysis of atmospheric lighthydrocarbons at ppb (v/v) and sub-ppb (v/v)concentrations, J. Chromatogr. A, 778, 269–277 (1997).
64. P. J. H. D. Verkoelen, M. W. F. Nielen, Short-term sample loss and sample uptake by Tenaxadsorption tubes. J. High Res. Chromatogr.Chromatogr. Comm., 11, 291–293 (1988).

294
65. I. Basu, J. M. O’Dell, K. Arnold, R. A. Hites,A source of PCB contamination in modifiedhigh-volume samplers. Environ. Sci. Technol.,34, 527–529 (2000).
66. G. Hunt, N. Pangaro, Potential contaminationfrom the use of synthetic adsorbents in airsampling procedures. Anal. Chem., 54, 369–372 (1982).
67. N. Schmidbauer, M. Oehme, Comparison ofsolid adsorbent and stainless steel canistersampling for very low ppt-concentrations ofaromatic compounds (≥ C6) in ambient airfrom remote areas. Fresenius Z. Anal. Chem.,331, 14–19 (1988).
68. D. Helming, Artifact-free preparation, stor-age and analysis of solid adsorbent samplingcartridges used in the analysis of volatile or-ganic compounds in air. J. Chromatogr. A,732, 414–417 (1996).
69. S. A. Hubbard, G. M. Russwurm, S. G.Walburn, A method for reducing and evaluat-ing blanks in Tenax air sampling cartridges.Atmospheric Environ., 15, 905–908 (1981).
70. I. Devai, R. D. DeLaune, Trapping efficiency ofvarious solid adsorbents for sampling and quan-titative gas chromatographic analysis of carbonylsulfide. Analytical Letters, 30, 187–198 (1997).
71. U. Herber, H.-U. Meisch, Studies about thedesorption of volatile halocarbons from acti-vated carbon by application of static headspacegas chromatography. Fresenius J. Anal.Chem., 353, 219–221 (1995).
72. J. Oda, A. Yasuhara, K. Matsunaga, Y. Saito,Stability of polycyclic aromatic hydrocarbonsand their oxygenated derivatives during vari-ous storage. Jpn. J. Toxicol. Environ. Health44, 352–363 (1998).
73. C.-Y. Ma, J. T. Skeen, A. B. Dindal, Ch. K.Bayne, R. A. Jenkins, Performance evalua-tion of a thermal desorption/gas chromato-graphic/mass spectrometric method for thecharacterization of waste tank headspacesamples. Environ. Sci. Technol., 31, 853–859(1997).
74. M. Gawrys, P. Fastyn, J. Gawlowski, T.Gierczak, J. Niedzielski, Prevention of watervapour adsorption by carbon molecular sievesin sampling humid gases. J. Chromatogr. A,933, 107–116 (2001).
75. M. Hekmat, P. Fung, R. Smith, Instability ofozone samples collected in alkaline potas-sium iodide solution. Am. Ind. Hyg. Assoc. J.,53, 672 (1992).
76. Y. K. Lau, Measurement of sulphur gases inambient air. Environmental Monitoring andAssessment, 13, 69–74 (1989).
77. L. J. Mc Garvey, Ch. V. Shorten, The effectsof adsorption on the reusability of Tedlar®air sampling bags. AIHAJ, 61, 375–380(2000).
78. J. C. Polasek, J. A. Bullin, Evaluation of bagsequential sampling technique for ambient airanalysis. Environ. Sci. Technol., 12, 708–712(1978).
79. J. Mowrer, A. Lindskog, Automatic unat-tended sampling and analysis of backgroundlevels of C2 – C5 hydrocarbons. AtmosphericEnvironment, 25A, 1971–1979 (1991).
80. J. J. F. McAndrew, D. Znamensky, D. DeBord,R. Inman, Interaction of gas-phase organicmolecules with aluminium and electropolishedstainless steel tubing. Anal. Chem., 67, 3075–3078 (1995).
81. J. C. Possner, W. J. Woodfin, Sampling withgas bags I: losses of analyte with time. Appl.Ind. Hyg., 4, 163–168 (1986).
82. R. K. M. Jayanty, J. R. Albritton, Y. H. Straley,D. J. von Lehmden, Stability of PPB organiccylinder gases used for calibration and au-dits., Proc. 84th Ann. Meet. Exhib., June 16–21, 1991, Vancouver, BC, Canada, paper 91–58.2.
83. J. H. Shreffler, Comparison of nonmethaneorganic compound concentration data col-lected by two methods in Atlanta. J. Air WasteManage. Assoc., 43, 1576–1584 (1993).
84. K. D. Oliver, J. D. Pleil, W. A. McClenny,Sample integrity of trace level volatile or-ganic compounds in ambient air stored insumma® polished canisters. Atmospheric En-vironment, 20, 1403–1411 (1986).
85. W. J. Karoly, Stability studies of diphenylmethanediisocyanate (MDI) on glass fiber filters. Am.Ind. Hyg. Assoc. J. 59, 645–647 (1998).
86. W. A. McClenny, S. M. Schmidt, K. G.Kronmiller, Variation of the relative humid-ity of air released from canisters after ambi-

295
ent sampling. J. Air & Waste Manage. Assoc.,49, 64–69 (1999).
87. W. Schrader, J. Geiger, D. Klockow, E.H.Korte, Degradation of α-pinene on Tenaxduring sample storage: effects of daylight ra-diation and temperature. Environ. Sci.Technol., 35, 2717–2720 (2001).
88. J. Fick, L. Pommer, B. Andersson, C. Nilsson,Ozone removal in the sampling of parts perbillion levels of terpenoid compounds: anevaluation of different scrubber materials.Environ. Sci. Technol., 35, 1458–1462 (2001).
89. B. A. Rodrigues, L. R. M. Pitombo, A. A. Cardoso,Study on the use of oxidant scrubbers for elimi-nation of interferences due to nitrogen dioxide inanalysis of atmospheric dimethylsulfide. J. Braz.Chem. Sci., 11, 71–77 (2000).
90. J. D. Pieil, K. D. Oliver, W. A. McClenny,Enhanced performance of nafion dryers onremoving water air samples prior to gas chro-matographic analysis. J. Air Poll. ControlAssoc., 37, 244–248 (1987).
91. P. V. Doskey, The effect of treating air sampleswith magnesium perchlorate for water removalduring analysis. J. High Resol. Chromatogr.14, 724–728 (1991).
92. S. W. Klamm, G. W. Scheil, Sample volumemeasurement errors caused by CO2 adsorp-tion in desiccants. Environ. Sci. Technol., 23,1420–1422 (1989).
93. J. R. Bowyer, Supercritical fluid extraction asa means of cleaning and desorbing commonair sampling sorbents. Chemosphere, 31,2905–2918 (1995).
94. J. B. Laurens, J. M. Swinley, J. P. de Coning,Gas chromatographic analysis of trace impu-rities in chlorine trifluoride. J. Chromatogr.A, 873, 229–235 (2000).
95. J. Lasa, I. Sliwka, Stability extraction of theresponse of a GC equipped with an ECDworking in a constant current mode. Chem.Anal. (Warsaw), 46, 421–431 (2001).
96. E. Leibrock, J. Slemr, Method for measure-ment of volatile oxygenated hydrocarbons inambient air. Atmospheric Environment, 31,3329–3339 (1997).
97. S. E. Bufflap, H. E. Allen, Sediment porewater collection methods for trace metal analy-sis: a review. Wat. Res., 29, 165–177 (1995).
98. T. J. Murphy, C. W. Sweet, Contamination ofteflon surfaces by PCBs in the atmosphere.Atmospheric Environment, 28, 361–364(1994).
99. R. W. Gillham, S. F. O’Hannesin, Sorption ofaromatic hydrocarbons by materials used inconstruction of ground-water sampling wells.ASTM STP, 1053, 108–122 (1989).
100. W. C. Koskinen, A. M. Cecchi, R. H. Dowdy,K. A. Norberg, Adsorption of selected pesti-cides on a rigid PVC lysimeter. J. Environ.Qual., 28, 732–734 (1999).
101. L. V. Parker, T. A. Ranney, Effect of concen-tration on sorption of dissolved organics byPVC, PTFE and stainless steel well casings.GWMR, 139–149 (summer 1994).
102. M. Gasparon, Trace metals in water samples:minimizing contamination during samplingand storage. Environmental Geology, 36, 207–214 (1998).
103. V. Stukas, C. S. Womg, W. K. Johnson, Sub-part per trillion levels of lead and isotopicprofiles in a fjord, using an ultra-clean pump-ing system. Marine Chemistry, 68, 133–143(1999).
104. I. G. Droppo, C. Jaksot, Impact of river trans-port characteristics on contaminant samplingerror and design. Environ. Sci. Technol., 28,161–170 (1995).
105. J. Pruvost, O. Connan, Y. Marty, P. Le Corre,A sampling device for collection and analysisof volatile halocarbons in coastal and oceanicwaters. Analyst, 124, 1389–1394 (1999).
106. L. V. Parker, Th. A. Ranney, Sampling trace-level organic solutes with polymeric tubing.Part I. Static studies. GWMR, 115–124 (Fall1997).
107. L. V. Parker, Th. A. Ranney, Sampling trace-level organic solutes with polymeric tubing.Part II. Dynamic studies. GWMR, 148–155(Winter 1998).
108. L. L. Knobel, L. J. Mann, Sampling forpurgeable organic compounds using positive-displacement piston and centrifugal submers-ible pumps: a comparative study. GroundWater Monit. Remed., 142–148 (spring 1993).
109. H. Sabik, R. Jeannot, E. Sauvard, Stability ofherbicides and their degradation products ongraphitized carbon black extraction cartridges

296
used for large volumes of surface water.Analusis, 28, 835–842 (2000).
110. H. Zawadzka, J. Zerbe, D. Baratkiewicz, Stratypolicyklicznychweglowodorów aromatycznych(PWA) podczas przechowywania próbek wódnaturalnych., Losses of polycyclic aromatic hy-drocarbons (PAH) during storage of natural wa-ter samples. Chem. Anal., 25, 469–471(1980).
111. R. B. Thurman, S. J. Sterry, How safe isbottled water? Water, 7–10 (March/April1998).
112. M. A. Palacios, M. Gómez, C. Cámara, M. A.López, Stability studies of arsenate, mono-methylarsonate, dimethylarsinate, arsenobe-taine and arsenocholine in deionozed water,urine and clean-up dry residuefrom urinesamples and determination by liquid chroma-tography with microwave-assisted oxidation-hydride generation atomic absorption spec-trometric detection. Anal. Chim. Acta, 340,209–220 (1997).
113. D. A. Kovacs, D. H. Kampbell, Improvedmethod for the storage of groundwater samplescontaining volatile organic analytes. Arch.Environ. Contam. Toxicol., 36, 242–247(1999).
114. L. A. Clementson, S. E. Wayte. The effect offrozen storage of open-ocean seawater sampleson the concentration of dissolved phosphateand nitrate. Wat. Res., 26, 1171–1176 (1992).
115. F. H. Denison, Ph. M. Haygarth, W. A. House,A. W. Bristow, The measurement of dissolvedphosphorus compounds: evidence for hydroly-sis during storage and implications for ana-lytical definitons in environmental analysis.Int. J. Environ. Anal. Chem., 69, 111–123(1998).
116. D. A. Markham, D. A. McNett, J. H. Birk, G.M. Klecka, M. J. Bartels, Ch. A. Staples,Quantitative determination of bisphenol-A inriver water by cool on-column injection-gaschromatography-mass spectrometry. Int. J.Environ. Anal. Chem., 69, 83–98 (1998).
117. S. L. Simpson, S. C. Apte, G. E. Batley,Sample storage artifacts, affecting the mea-surement of dissolved copper in sulfidic wa-ters. Anal. Chem., 70, 4204–4205 (1998).
118. K. Hiraki, M. Kimura, H. Hattori, Y.Nakaguchi, H. Tsubota, Determination of
dissolved organic carbon in Northwest Pa-cific Ocean. The application of ultra low longtime storage. Kiuki Daigaku Rikogakubu, 32,19–24 (1996).
119. L. M. Avalone, D. W. Toohey, W. H. Brune,R. J. Salawitch, A. E. Dessler, J. G. Ander-son, Balloon-borne in situ measurements ofchlorine oxide and ozone: implications forheterogenous chemistry and mid-latitudeozone loss. Geophys. Res. Lett., 20, 1795–1798 (1993), Acc. to Chem. Abstr. Vol. 120,249657s (1994).
120. A. Szymanski, Z. Zwit, Z. –Lukaszewski, Stud-ies of preservation of water samples for thedetermination of non-ionic surfactants. Anal.Chim. Acta, 311, 31–36 (1995).
121. J. T. Creed, Th. D. Martin, M. Sivaganesan,Preservation of trace metals in water samples.Jour. AWWA, 104–114 (February 1995).
122. A. Aminot, R. Kérouel, Assessment of heattreatment for nutrient preservation in seawa-ter samples. Anal. Chim. Acta, 351, 299–309(1997).
123. W. T. Foreman, S. D. Zaugg, L. M. Faires, M.G. Werner, Th. J. Lelker, P. F. Rogerson,Analytical interferences of mercuric chloridepreservative in environmental water samples:determination of organic compounds isolatedby continuous liquid-liquid extraction orclosed–loop stripping. Environ. Sci. Technol.,26, 1307–1312 (1992).
124. M. Gardner, S. Comber, Sample filtration asa source of error in the determination of tracemetals in marine waters. Analyst, 122, 1029–1032 (1997).
125. M. Gardner, S. Comber, Sample determina-tion of trace metals in marine waters. Analyst,122, 1029–1032, (1997).
126. A. J. Horovitz, Some thoughts on problemsassociated with various sampling media usedfor environmental monitoring. Analyst, 122,1193–1200 (1997).
127. A. J. Horovitz, Problems associated with us-ing filtration to define dissolvent trace ele-ment concentrations in natural water samples.Environ. Sci. Technol. 30, 954–963 (1996).
128. B. Norrman, Filtration of water samples forDOC studies. Marine Chemistry, 41, 239–242 (1993).

297
129. F. Ödman, Th. Ruth, Ch. Pontér, Validationof a field diltration technique for character-ization of suspended particulate matter fromfreshwater. Part I. Major elements. AppliedGeochemistry, 14, 301–317 (1999).
130. A. J. Horovitz, K. R. Lum, J. R. Garbardino,G. E. M. Hall, C. Lemieux, Ch. R. Demas,The effect of membrane filtration on dissolvedtrace element concentrations. Water, Air andSoil Pollution, 90, 281–294 (1996).
131. D. O. Cooney, Adsorption of organic soluteson membrane filters during aqueous phasefiltration. I. Basic rate and equilibrium stud-ies using toluidine blue. Separation Scienceand Technology, 27, 2001–2019 (1992).
132. X. Zhu, M. Elimelech, Colloidal fouling ofreverse osmosis membranes: measurementsand fouling mechanisms. Environ. Sci.Technol., 31, 3654–3662 (1997).
133. M. Pujo-Pay, P. Raimbault, P. Conan, Sous-estimation des teneurs en azote particulaireen milieu océanique par l’utilisation des filtresGF/F. C. R. Acad. Sci. Paris, 324, 401–407(1997).
134. S. C. Yoro, Ch. Panagiotopoulos, R. Semperé,Dissolved organic carbon contamination in-duced by filters and storage bottles. Wat. Res.,33, 1956–1959 (1999).
135. Y. R. Hunter, J. S. Kuwabara, Ionic strengthand DOC determinations from various fresh-water sources to the San Francisco Bay. Bull.Environ. Contam. Toxicol., 52, 311–318(1994).
136. A. Sjösten S. Blomqvist, Influence of phos-phate concentration and reaction temperaturewhen using the molybdenium blue methodfor determination of phosphate in water. Wat.Res. 31, 1818–1823 (1997).
137. M. Borho, P. Wilderer, A reliable method forpreservation and determination of arsenate(III)concentrations in ground water and waterworks samples. J. Water SRT – Aqua, 46,138–143 (1997).
138. E. Turtola, Peroxodisulphate digestion andfiltration as sources of inaccuracy in determi-nations of total phosphorus and dissolved or-thophosphate phosphorus in water samplescontaining suspended soil particles. BorealEnvironment Research, 1, 17–26 (1996).
139. M. P. Maskarinec, L. H. Johnson, S. K.Holladay, R. L. Moody, Ch. K. Bayne, R. A.Jenkins, Stability of volatile organic com-pounds in environmental water samples dur-ing transport and storage. Env. Sci. Technol.,24, 1665–1670 (1990).
140. M. H. Al.-Malack, Migration of lead fromunplasticized polyvinyl chloride pipes. Jour-nal of Hazardous Materials, B 82, 263–274(2001).
141. M. J. Barcelona, J. A. Helfirch, E. E. Garske,Sampling tubing effects on groundwatersamples. Anal. Chem., 57, 460–464 (1985).
142. R. Hopman, T. J. J. van den Hoven, Perme-ation of organic chemicals through plasticwater pipes. J. Water SRT – Aqua, 41, 158–162 (1992).
143. J. Zerbe, D. Baratkiewicz, H. Gramowska, J.·Zminkowska, Efektywnosc144. ekstrakcjipolicyklicznych wecglowodorów aroma-tycznych z wód o du·zej zawartosci zawiesin.Chem. Anal., 29, 455–459 (1984).
144. M. Lyytikäinen, J. Pellinen, Some issues con-cerning the gas chromatographic determina-tion of chlorinated phenolics in water. Toxi-cological and Environmental Chemistry, 63,185-197 (1997).
145. M. Palma, Z. Piñeiro, C. G. Barroso, Stabilityof phenolic compounds during extraction withsuperheated solvents. J. Chromatogr. A , 921,169–174 (2001).
146. I. Ferrer, D. Barceló, Determination and sta-bility of pesticides in freeze-dried watersamples by automated on-line solid-phase ex-traction followed by liquid chromatographywith diode-array detection. J. Chromatogr. A,737, 93–99 (1996).
147. G. A. Sorial, M. T. Suidan, R. D. Vidic, R. C.Brenner, Effect of GAC characteristics onadsorption of organic pollutants. WaterEnviron. Res., 65, 53–57 (1993).
148. B. Maichin, P. Kettish, G. Knapp, Investiga-tion of microwave assisted drying of samplesand evaporation of aqueous solutions in traceelement analysis. Fresenius J. Anal. Chem.,366, 26–29 (2000).
149. P. J. Cerminara, G. A. Sorial, S. P. Papadimas,M. T. Suidan, M. A. Moteleb, Th. F. Speth,Effect of influent oxygen concentration on

298
the GAC adsorption of VOCs in the presenceof BOM. Wat. Res., 29, 409–419 (1995).
150. E. Diamadopoulos, P. Samaras, G. P.Sakellaropoulos, The effect of activated car-bon properties on the adsorption of toxic sub-stances. Wat. Sci. Tech., 25, 153–160 (1992).
151. P. H. Chen, W. A. Van Ausdale, W. S. Keeran,D. F. Roberts, GC/MS identification of arti-facts formed during sample preparation usingUSEPA methods 625 and 8270. Chemosphere,26, 1743–1749 (1992).
152. M. Vogel, W. Pötter, U. Karst, Characteriza-tion of a chemical artifact in the liquid chro-matographic determination of 3–butyn-2–oneusing the 2, 4–dinitrophenylhydrazine method.J. Chromatogr. A, 886, 303–307 (2000).
153. W. D. Bowers, M. L. Parsons, R. E. Clement,F. W. Karasek., Component loss during evapo-ration-reconstruction of organic environmen-tal samples for gas chromatographic analysis.J. Chromatogr., 207, 203–211 (1981.
154. D. E. Slauenwhite, P. J. Wangersky, Extractonof marine organic matter on XAD-2: Effectof sample acidification and development ofan in situ preacidification technique. Mar.Chem., 54, 107–117 (1996).
155. J. Kukkonen, J. F. McCarthy, A. Oikari, Ef-fects of XAD-8 fractions of dissolved organiccarbon on the sorption and bioavailability oforganic micropollutants. Arch. Environ.Contam. Toxicol., 19, 551–557 (1990).
156. W. E. Johnson, N. J. Fendinger, J. R. Plimmer,Solid-phase extraction of pesticides fromwater: possible interferences from dissolvedorganic material. Anal. Chem., 63, 1510–1513(1991).
157. M. Johansson, H. Emteborg, B. Glad, F.Reinholdsson, D. C. Baxter, Preliminary ap-praisal of a novel sampling and storage tech-nique for the speciation analysis of lead andmercury in seawater. Fresenius J. Anal. Chem.351, 461–466 (1995).
158. B. Kolb, G. Zwick, M. Auer, A water trap forstatic cryo-headspace gas chromatography. J.High Resol. Chromatogr., 19, 37–42 (1996).
159. F. Jüttner, B. Meon, O. Köster, Quasi in situseparation of particulate matter from lakewaterby hollow-fibre filters to overcome errors
caused by short turnover times of dissolvedcompounds. Wat. Res., 31, 1637–1642 (1997).
160. H. R. Rogers, Influence of suspended solidsand back diffusion on organic contaminantuptake by semi-permeable membranes(SPDMs). Chemosphere, 35, 1651–1657(1997).
161. C. Molina, P. Grasso, E. Benfenati, D. Barceló,Determination and stability of phenmediphan,ethofumesate and fenamiphos in ground wa-ter samples using automated solid phase ex-traction cartridges followed by liquid chro-matography high flow pneumatically assistedelectrospray mass spectrometry. Int. J.Environ. Anal. Chem, 65, 69–82 (1996).
162. K. Dettmer, Th. Knobloch, W. Engewald,Stability of reactive low boiling hydrocar-bons on carbon based adsorbents typicallyused for adsorptive enrichment and thermaldesorption. Fresenius J. Anal. Chem., 366,70–78 (2000).
163. H. Emteborg, J. Snell, J. Qian, W. Frech,Sources of systematic errors in mercury spe-ciation using Grignard reagents and capillarygas chromatography coupled to atomic spec-trometry. Chemosphere, 39, 1137–1152(1999).
164. T. J. Nestrick, L. L. Lamparski, Purification ofcylinder gases in solvent evaporation for traceanalysis. Anal. Chem. 53, 122–124 (1981).
165. N. A. Stevens, M. F. Borgerding, Optimiza-tion of 15N detection with an Atomic Emis-sion Detector. Anal. Chem., 71, 1083–1086(1999).
166. T. Ozeki, Y. Tsubosaka, S. Nalayama, N.Ogawa, T. Kimoto, Study of errors in deter-mination of hydrogen ion concentrations inrainwater samples using glass electrodemethod. Anal. Sci., 14, 749–756 (1998).
167. Ch. Weston, D. Speis, R. P. Albert, A com-parison of the performance characteristics ofsorbent column packing materials used forpurgeable organic analysis. Technical infor-mation, Environmental Testing and Certifica-tion Corp., Edison, New Jersey, 1991.
168. K. E. Collins, C. H. Collins, C. A. Bertran,Stainless steel surfaces in LC systems, part I– corrosion and erosion. LCGC, 18, 600–608(2000).

299
169. K. E. Collins, C. H. Collins, C. A. Bertran,Stainless steel surfaces in LC systems, part II— passivation and practical recommendations.LCGC, 18, 688–692 (2000).
170. S. Blomqvist, S. Westin, Interference fromchromate, germanate, tungastate and vadnadatewhen determining phosphate in aqueous solu-tion by the phosphoantimonylomolybdenumblue method. Anal. Chim. Acta, 3580, 245–254 (1998).
171. S. Blomqvist, K. Hjellström, A. Sjösten, Inter-ference from arsenate, fluoride and silicatewhen determining phosphate in water by thephosphoantimonylomolybdenum blue method.Intern. J. Environ. Anal. Chem, 54, 31–43(1993).
172. L. Wennrich, J. Efer, W. Engewald, Gas chro-matographic trace analysis of underivatizednitrophenols. Chromatographia, 41, 361–366(1995).
173. J. Poehlman, B. W. Pack, G. M. Hieftje, Aheated transfer line for coupling GC withplasma source spectrometry. Am. Lab., 50C-53C (Oct. 1998).
174. M. J. Lichon, Environmental monitoring prob-lems with stream sampling. Water, 1–2, 30–32 (1996).
175. I. Héninger, M. Potin-Gautier, I. de Gregori,H. Pinochet, Storage of aqueous solutions ofselenium for speciation at trace level.Fresenius J. Anal. Chem., 357, 600–610(1997).
176. R. Muñoz Olivas, P. Quevauviller, O. F. X.Donard, Long term stability of organic sele-nium species in aqueous solutions. FreseniusJ. Anal. Chem., 360, 512–519 (1998).
177. J. Snell, J. Qian, M. Johanssos, K. Smit, W.Frech, Stability and reactions of mercury spe-cies in organic solution. Analyst, 123, 905–909 (1998).
178. D. Amoroux, Ch. Pécheiran, O. F. X Donard,Formation of volatile selenium species insynthetic seawater under light and dark ex-perimental conditions. Appl. Organometal.Chem., 14, 236–244 (2000).
179. W. Chatkittikunwong, C. S. Creaser, Stabil-ity of bromo- and bromochloro-dibenzo-p-dioxins under laboratory and environmental
conditions. Chemosphere, 29, 547–557(1994).
180. G. E. M. Hall, J. C. Pelchat, G. Gauthier,Stability of inorganic arsenic(III) andarsenic(V) in water samples. J. Anal. At.Spectrom., 14, 205–213 (1999).
181. F. P. L. Lamé, P. R. Defize, Sampling ofcontaminated soil: sampling error in relationto sample size and segregation. Environ. Sci.Technol., 27, 2035–2044 (1993).
182. R. Djingova, I. Kuleff, On the sampling ofvascular plants for monitoring of heavy metalpollution. In: Environmental Sampling forTrace Analysis (Ed.: B. Markert), VCHWeinheim (1994) Chapter 21, 395–414.
183. R. S. Daniels, A. L. Fraser, Ph. Quevauviller,The variability if dry-mass correction for cer-tified reference materials. J. Environ. Anal.Chem., 63, 167–177 (1996).
184. U. Faure, P. J. Wagstaffe, Stability of refer-ence materials. Fresenius J. Anal. Chem., 345,124–126 (1993).
185. S. Vandendriessche, B. Griepink, J. C. Th.Hollander, J. W. J. Gielen, F. G. G. M.Langelaan, K. J. Saunders, R. H. Brown, Cer-tification of a reference material for aromatichydrocarbons in Tenax samplers. Analyst, 116,437–441 (1991).
186. D. van Dijk, V. J. G. Houba, Homogeneityand stability of materials distributed withinthe wageningen evaluating programmes foranalytical laboratories. Commun. Soil Sci.Plant. Anal., 31, 1745–1756 (2000).
187. P. V. Doskey, R. W. Talbot, Sediment chro-nologies of atmospheric deposition in a pre-cipitation-dominated seepage lake. Limnol.Oceanogr., 45, 895–904 (2000).
188. D. Huo, H. M. “Kip” Kingston, Correction ofspecies transformations in the analysis ofCr(VI) in solid environmental samples usingspeciated isotops dilution mass spectrometry.Anal. Chem., 72, 5047–5054 (2000).
189. S. B. Hawthorne, C. B. Grabanski, Vaporiza-tion of polycyclic aromatic hydrocarbons(PAHs) from sediments at ambient conditions.Environ. Sci. Technol., 34, 4348–4353 (2000).
190. I. T. Cousins, H. Kreibich, L. E. Hudson, W.A. Lead, K. C. Jones, PAHs in soils: contem-

300
porary UK data and evidence for potentialcontamination problems caused by exposureof samples to laboratory air. Sci. Total.Environ., 203, 141–156 (1997).
191. H. Hintelmann, Comparison of different ex-traction techniques used for methylmercuryanalysis with respect to accidental formationof methylmercury during sample preparation.Chemosphere, 39, 1093–1105 (1999).
192. L.-O. Kjeller, Ch. Rappe, Soxhlet extractionparameters’ influence on the recovery of poly-chlorinated dibenzo-p-dioxins and dibenzofurans(PCDD/F) from a sediment and artifact forma-tion of PCDD. Toxicol. Environ. Chem., 67,481–490 (1998).
193. K. Takada, T. Ashino, K. Wagatsuma, Devel-opment of a nitrogen atmosphere pretreat-ment chamber for the determination of traceamounts of carbon in high-purity iron by theinfrared absorption method after combustionin an electric resistance furnace. AnalyticalSciences, 13, 867–871 (1997).
194. S. J. Lue, C. Huang, Applications of ion chro-matography in the semiconductor industry.II. Determination of basic airborne contami-nants in a clean room. J. Chromatogr. A, 850,283–287 (1999).
195. P. Sun, M. Adams, L. Shive, S. Pirooz, Mo-lecular and ionic contamination monitoringfor cleanroom air and water surfaces. SPIE,3215, 118–127 (1997).
196. P. A. Tanner, L. S. Leong, The effects ofdifferent drying methods for marine sedimentupon moisture content and metal determina-tion. Marine Pollution Bulletin, 31, 325–329(1995).
197. F. Bordas, A. C. M. Bourg, A critical evalu-ation of sample pretreatment for storage ofcontaminated sediments to be investigated forthe potential mobility of their heavy metalload. Water, Air and Soil Pollution 103, 137–149, (1998).
198. M. Siaka, C. M. Owens, G. F. Birch, Evalu-ation of some digestion methods for the de-termination of heavy metals in sediment
samples by flame-AAS. Analytical Letters,31, 703–718 (1998).
199. G. Stringari, I. Pancheri, F. Möller, O. Failla,Influence of two grinding methods on theuncertainty of determinations of heavy met-als in atomic absorption spectrometry/elec-trothermal atomization of plant samples.Accred. Qual. Assur., 3, 122–126 (1998).
200. M. Takenaka, S. Kozuka, M. Hayashi, H.Endo, Determination of ultratrace amounts ofmetallic and chloride ion impurities in or-ganic materials for microelectronics devicesafter a microwave digestion method. Analyst,122, 129–132 (1997)
201. Y. Cai, M. Abalos, J. M. Bayona, Effects ofcomplexing agents and acid modifiers on thesupercritical fluid extraction of native phe-nyl- and butyl-tins from sediment. Appl.Organometal. Chem., 12, 577–584 (1998).
202. I. L. Marr, P. Kluge, L. Main, V. Margerin, C.Lescop, Digests or extracts? Some interestingbut conflicting results for the widely differingpolluted sediment samples. Mikrochim. Acta,119, 219–232 (1995).
203. L. Jorhem, Dry ashing, sources of error andperformance evaluation in AAS. MikrochimActa, 119, 211–218 (1995).
204. I. Novozamsky, H. J. van der Lee, V. J. G.Houba, Sample digestion procedures for traceelement determination. Mikrochim. Acta 119,183–189 (1995).
205. B. A. Schumacher, S. E. Ward, Quantitationreference compounds and VOC recoveriesfrom soils by purge-and-trap GC/MS. Environ.Sci. Technol., 31, 2287–2291 (1997).
206. L.-O. Kjeller, Addition of internal standardsto particulate sample matrices for routine traceanalyses of semivolatile organic compounds:a source of systematical and random errors.Fresenius J. Anal. Chem., 361, 791–796(1998).
207. C. Sarzanini, E. Mentasti, Removal of instru-mental interferences in evaluating solid wasteinteraction with soil. Ann. Chim., 85 407–418(1995).
