transient striatal glt-1 blockade increases eaac1 expression, glutamate reuptake, and decreases...
TRANSCRIPT

Experimental Neurology 234 (2012) 428–436
Contents lists available at SciVerse ScienceDirect
Experimental Neurology
j ourna l homepage: www.e lsev ie r .com/ locate /yexnr
Transient striatal GLT-1 blockade increases EAAC1 expression, glutamate reuptake,and decreases tyrosine hydroxylase phosphorylation at ser19
Michael F. Salvatore ⁎, Richard W. Davis, Jennifer C. Arnold, Tanya ChotibutDepartment of Pharmacology, Toxicology, & Neuroscience, Louisiana State University Health Sciences Center-Shreveport, Shreveport, Louisiana 71130, USA
Abbreviations: UCPH-101, 2-Amino-5,6,7,8-tetrah(naphthalene-1-yl)-5-oxo-4H-chromene-3-carbonitrile⁎ Corresponding author at: Department of Pharmacol
ence, Louisiana State University Health Sciences Center,Highway, Shreveport, Louisiana 71130, USA. Fax: +17810(Lab).
E-mail address: [email protected] (M.F. Salvatore)
0014-4886/$ – see front matter © 2012 Elsevier Inc. Alldoi:10.1016/j.expneurol.2012.01.012
a b s t r a c t
a r t i c l e i n f oArticle history:Received 9 October 2011Revised 24 December 2011Accepted 10 January 2012Available online 17 January 2012
Keywords:EAAC1/EAAT3GLAST/EAAT1GLT-1/EAAT2GlutamateExcitotoxicityParkinson's diseaseStriatumNigrostriatalGlutathioneTyrosine hydroxylasePhosphorylation
Three glutamate transporters, GLT-1, GLAST, and EAAC1, are expressed in striatum. GLT-1 and, to a lesser ex-tent, GLAST are thought to play a primary role in glutamate reuptake and mitigate excitoxicity. Progressivetyrosine hydroxylase (TH) loss seen in Parkinson's disease (PD) is associated with increased extracellular glu-tamate. Glutamate receptor antagonists reduce nigrostriatal loss in PD models. These observations suggestthat excess synaptic glutamate contributes to nigrostriatal neuron loss seen in PD. Decreased GLT-1 expres-sion occurs in neurodegenerative disease and PD models, suggesting decreased GLT-1-mediated glutamatereuptake contributes to excitotoxicity. To determine how transient GLT-1 blockade affects glutamate reup-take dynamics and a Ca2+-dependent process in nigrostriatal terminals, ser19 phosphorylation of TH, theGLT-1 inhibitor dihydrokainic acid (DHK) was delivered unilaterally to striatum in vivo and glutamate reup-take was quantified ex vivo in crude synaptosomes 3 h later. Ca2+-influx is associated with excitotoxic con-ditions. The phosphorylation of TH at ser19 is Ca2+-dependent, and a change resulting from GLT-1 blockademay signify the potential for excitotoxicity to nigrostriatal neurons. Synaptosomes from DHK infused stria-tum had a 43% increase in glutamate reuptake in conjunction with decreased ser19 TH phosphorylation.Using a novel GLAST inhibitor and DHK, we determined that the GLAST-mediated component of increasedglutamate reuptake increased 3-fold with no change in GLAST or GLT-1 protein expression. However, GLT-1 blockade increased EAAC1 protein expression ~20%. Taken together, these results suggest that GLT-1 block-ade produces a transient increase in GLAST-mediated reuptake and EAAC1 expression coupled with reducedser19 TH phosphorylation. These responses could represent an endogenous defense against excitotoxicity tothe nigrostriatal pathway.
© 2012 Elsevier Inc. All rights reserved.
Introduction
In neurodegenerative diseases, impaired glutamate regulation inthe synapse may contribute to neuronal loss (Behrens et al., 2002;Sheldon and Robinson, 2007). Glutamatergic-signaling is critical forbasal ganglia function, (Greenamyre, 2001). However, in PD animalmodels, increased extracellular glutamate levels in striatum dooccur (Lindefors and Ungerstedt, 1990; Meredith et al., 2009;Meshul et al., 1999; Robinson et al., 2003) and glutamate receptor an-tagonists mitigate experimentally-induced loss of nigrostriatal neu-rons (Armentero et al., 2006; Masilamoni et al., 2011; Vernon et al.,2007). Thus, glutamate excitotoxicity, which may originate from
ydro-4-(4-methoxyphenyl)-7-.ogy, Toxicology, and Neurosci-School of Medicine, 1501 Kings318 675 7857, +1 318 675
.
rights reserved.
increased extracellular glutamate, could play some role in nigrostria-tal dopamine (DA) neuron loss.
High affinity Na+-dependent glutamate transport diminishes theconstant potential for excitotoxicity in the CNS (Danbolt, 2001; DelRio et al., 2008; Rosenberg and Aizenman, 1989; Robinson, 1998;Robinson et al., 1991; Rothstein et al., 1996; Velasco et al., 1996). Theglutamate transporters primarily expressed by glia, GLT-1 and GLAST,play a major role for glutamate removal from the synapse (Danbolt,2001; Rosenberg and Aizenman, 1989; Rothstein et al., 1996). EAAC1,largely expressed on neurons, could play a comparatively more minorrole in glutamate reuptake (Danbolt, 2001; Fumagalli et al., 2008;Rothstein et al., 1996; Robinson, 1998). However, EAAC1 also transportscysteine, in addition to glutamate (Zerangue and Kavanaugh, 1996) andcontributes significantly to total glutamate reuptake in the CNS in somestudies (DeSilva et al., 2009; Nieoullon et al., 2006). EAAC1 expressionlevels may be less than GLT-1 or GLAST (Danbolt et al., 2011;Robinson, 1998), which may explain why in some cases it plays aminor role in glutamate reuptake. However, EAAC1 may increase itsfunction in response to excitotoxic conditions (Nieoullon et al., 2006;Ross et al., 2011a, 2011b). Furthermore, EAAC1 expression may be crit-ical for maintaining the anti-oxidant glutathione (Aoyama et al., 2006)

429M.F. Salvatore et al. / Experimental Neurology 234 (2012) 428–436
and nigrostriatal neuron viability during aging (Berman et al., 2011) viacysteine transport.
The initial response to increased synaptic glutamate may re-veal the mechanisms by which the potential for excitotoxicity isdiminished. In the same vein, a lack or eventual failure of plastic-ity could precipitate Ca2+-mediated events in the nigrostriatalneuron that precede excitotoxic cell death. For example, if GLT-1is the major glutamate transporter, a compromise in its expres-sion or function could accelerate neuron loss when synaptic gluta-mate levels are increased (Rothstein et al., 1996, 2005; Selkirk etal., 2005; Wang et al., 1998). GLT-1 loss occurs in neurodegenera-tive diseases, (Behrens et al., 2002; Dabir et al., 2006; Schallier etal., 2011), including ALS and Alzheimer's disease patients(Sheldon and Robinson, 2007). Furthermore, decreased expressionof GLT-1 has been reported at the severe stage of nigrostriatalpathway loss in PD models (Dervan et al., 2004; Chung et al.,2008). Consequently, diminished GLT-1 function could initiateexcitotoxic conditions that exacerbate nigrostriatal DA loss.
Enhanced GLAST or EAAC1 function may be critical for preventingglutamate excitotoxicity. Aging, which is associated with PD, bringson reduced glutamate reuptake in striatum and is associated with de-creased GLAST plasma membrane expression (Nickell et al., 2005;2007). GLT-1 blockade for at least 24 h may not produce neurotoxic-ity (Selkirk et al., 2005), and this protection could be conferred, inpart, by GLAST, which increases its activity in response to increasedglutamate (Duan et al., 1999). GLAST may protect DA neurons tomaintain normal locomotor activity (Karlsson et al., 2008), and re-duced GLAST expression in PD models may hasten excitotoxic dam-age to nigrostriatal neurons (Chung et al., 2008). EAAC1 expressionmay also increase in response to increased glutamatergic tone (Rosset al., 2011a; 2011b). EAAC1 function is also affected during loss ofnigrostriatal neurons in PD models (Aoyama et al., 2008; Chung etal., 2008).
Given that increased glutamatergic tone occurs in PD models, weexamined the response of glutamate reuptake dynamics in responseto GLT-1 blockade. We infused the GLT-1 inhibitor, dihydrokainicacid (DHK), into the striatum, as previously shown to increase extra-cellular glutamate (Massieu et al., 1995; Pintor et al., 2004). To exam-ine the impact of GLT-1 blockade on nigrostriatal neurons, wequantified TH phosphorylation at ser19. The glutamatergic agonistNMDA increases ser19 TH phosphorylation (Lindgren et al., 2000). In-creased ser19 TH phosphorylation from depolarizing stimuli in vivo(Haycock and Haycock, 1991), also depends upon Ca2+-influx(Salvatore et al., 2001). Excess Ca2+-influx, mediated by glutamatereceptors (Lau and Tymianski, 2010), is also a prerequisite for excito-toxicity, as mediated in part by calpain overactivation (Del Rio et al.,2008; Grant et al., 2009; Geddes and Saatman, 2010). Thus, a changein ser19 TH phosphorylation is hypothesized herein to indicate the ex-tent to which a Ca2+-dependent process, specifically in the nigros-triatal neuron, is affected following GLT-1 blockade.
Methods
Animals
Male Sprague Dawley rats of ages 3–4 months were used inthis study and were obtained from Charles River (Wilmington,MA). As the process of aging can impact glutamate reuptake dy-namics (Nickell et al., 2007), the ages of all experimental subjectswere less than 6 months old. Protocols for animal use were ap-proved by the Institutional Animal Care and Use Committee, asdeclared by the Uniform Requirements for manuscripts submittedto Biomedical journals (http://www.icmje.org). Animals werehoused according to approved guidelines with food and wateravailable ad libitum.
Infusion protocol
Rats were anesthetized with 40 mg/kg Nembutal i.p. (pentobarbi-tal Lundbeck Inc, Deerfield, IL) and 0.1 cm3 injection of 3:3:1 keta-mine, xylazine, and acepromazine. They were placed in a stereotaxicframe to target the striatum at coordinates ML +/−2.5, AP +1.0,DV −5.0 relative to Bregma according to Paxinos and Watson. Dihy-drokainic acid (DHK) (Tocris Bioscience, Ellisville, Missouri, cat#0111) was prepared in Krebs bicarbonate buffer (118 mM NaCl,4.7 mM KCl, 1.2 mM KH2PO4, 25 mM NaHCO3, 1.0 mM Na2EDTA,1.7 mM CaCl2, 10 mM glucose, 100 μM parglyline, 100 μM ascorbicacid). A volume of 4 μL was injected unilaterally into striatum andan equal volume of vehicle was injected on the contralateral side.The syringe was left in place for 10 min before removal. After 3 h, stri-atum was dissected from a chilled rodent brain matrix in 1 mm coro-nal slices.
Synaptosome preparation
Striatal tissue was homogenized in 5 mL of 0.32 M sucrose solu-tion using 10 up and down strokes of a Teflon/glass homogenizingwand (Glas-Col, Terre Haute, IN) then spun at 1000×g for 10 min.The resulting pellet was stored as the P1 fraction, from which theanalysis of total and phosphorylated TH was conducted. The superna-tant was spun further at 17,500×g for 30 min yielding the P2 fraction.The supernatant was aspirated and resuspended in 1 mL of Kreb'sbuffer. Protein concentration was determined using a BCA color-metric assay (Thermo Scientific, Rockford, IL). This protocol hasbeen used to determine the reuptake of other neurotransmitters en-dogenous to striatum (Salvatore et al., 2003).
It is well-known that mitochondria can transport glutamate(Azarias et al., 2011; Satrustegui et al., 2007) and GLT-1 can co-compartmentalize with metabolic enzymes of the mitochondria(Genda et al., 2011). Because we did not isolate mitochondria in thiscrude synaptosome preparation, we compared the conditions of ouruptake protocol (as presented in the next section) to those measuringglutamate uptake in mitochondria preparations (Dolinska andAlbrecht, 1997; Ziemińska et al., 2004). In these studies, between 12and 15 nmol glutamate/mg protein was determined for uptake,using assay conditions of 300–500 μg of mitochondrial protein, 0.2or 2 mM total glutamate, and uptake time period of 3 to 5 min.Given that our control level reuptake is 300 pmole/mg protein/minunder assay conditions using 10- to 17-fold less protein, 40- to 400-fold less [glutamate], and 2- to 3.3-fold less reuptake time, adjustingour assay conditions to the mitochondria protocols would yield 240to 6700 nmol of glutamate reuptake. Thus, ~15 nmol of reuptake inthe mitochondrial preparation could represent 0.2 to 5% of total up-take in our preparation that could be attributed to mitochondria, as-suming a linear relationship among the variables of protein,glutamate concentration, and uptake time allowed.
Uptake protocol
Synaptosomes contain glial components (Henn et al., 1976) and~70% of the levels of glial fibrillary acid protein recovered in purifiedglial plasmalemmal vesicles (Suchak et al., 2003) and thus are ade-quate for assessment of glutamate-reuptake. Synaptosomes were dis-tributed in test tubes at equal protein quantity to prepare forglutamate reuptake, with an aliquot saved for later determination ofthe protein quantities of GLT-1, GLAST, and EAAC1. The determinationof glutamate reuptake in the crude synaptosomes harvested from thevehicle- and DHK-infused striatum was conducted simultaneouslyand included assessments of reuptake in the presence of 300 μMDHK to determine the GLT-1-mediated glutamate reuptake compo-nent and a novel GLAST inhibitor UCPH-101 (2-Amino-5,6,7,8-tetra-hydro-4-(4-methoxyphenyl)-7-naphthalen-1-yl)-5-oxo-4H-chromene-

430 M.F. Salvatore et al. / Experimental Neurology 234 (2012) 428–436
3-carbonitrile) at 30 μM (Ascent Scientific, Princeton, NJ, cat# Asc-309)(Jensen et al., 2009) to determine the GLAST-mediated glutamate reup-take component. We note that in our striatal preparation UCPH-101 atconcentration of 10 μM was found to inhibit glutamate reuptake asmuch the 30 μM concentration. Each determination (no inhibitor pre-sent, DHK present, or UCPH-101 present) was done in triplicate foreach experiment.
Synaptosomes were used in a quantity 30 μg of protein in a 200 μLfinal volume for glutamate reuptake. In 100 μL, the combination of thesynaptosome prep to constitute 30 μg synaptosomal protein, oxygen-ated Kreb's buffer, and inhibitor (if indicated) was prepared at 4 °C.The synaptosomes were then warmed to 35 °C for 5 min, then100 μL of 10 μM 14C(U)-L-glutamic acid (Perkin-Elmer, specific activ-ity 260 mCi/mmol, cat# NEC290E050UC) was added to the synapto-some preparations (giving a 5 μM final [glutamate]), allowed toincubate for reuptake, and terminated after 90 s with an excess vol-ume of ice-cold Kreb's buffer and re-immersing the tubes in the ice-bath. The reuptake time was chosen to be as close as technicallyand practically possible to the reuptake time of glutamate observedin vivo, which occurs within 10 s (Nickell et al., 2005; Wassum etal., 2008). Synaptosomes were washed extensively to remove excesslabeled-glutamate with equal-osmolarity PBS buffer through a Bran-del M24-TI (Gaithersburg, MD) cell harvester through Brandel GF/Cfilter paper pretreated with a 2% polyethylenimine solution to reducenon-specific binding of label. The filter paper containing the rinsedsynaptosomes were transferred into scintillation vials containing5 mL of biodegradable scintillation cocktail (Research Products Inter-national, Mount Prospect, IL) and counted with a Beckman CoulterLS6500 scintillation counter (Brea, CA).
Determination of glutamate reuptake
To determine the quantity of glutamate reuptake, the percent ofglutamate (as the label) recovered in the synaptosomes against thetotal amount of glutamate (as the label) in the reuptake experimentwas first determined. This percentage of reuptake averaged 1.50±0.28% (mean±SEM, n=7 experiments) using 5 μM glutamate for re-uptake studies. In pilot experiments, the percent reuptake was notsignificantly different using 1 or 2 μM glutamate (n=5 experiments),and we also found glutamate reuptake was DHK- (≥200 μM) andUCPH-sensitive (30 μM), using 1 or 2 μMglutamate in the synaptosomereuptake protocol. The pmole of glutamate was then determined basedupon this percent of label recovery in the synaptosomes andnormalizedto mg protein per minute.
To determine the contributions of GLT-1 or GLAST toward gluta-mate reuptake, the reuptake of glutamate in the presence of the re-spective glutamate transporter inhibitor was subtracted from thetotal reuptake. This difference was assumed to be the component ofglutamate reuptake mediated by the targeted transporter. The reup-take value obtained with the inhibitor on-board (which is not pre-sented, but subtracted from the total, as stated) represents reuptakeby the non-targeted transporter plus that by EAAC1 (for which nospecific inhibitor currently exists). From our analysis, the percent oftotal glutamate reuptake in vehicle-infused (control) striatum was~50% for the GLT-1-mediated component and ~10% for the GLAST-mediated component.
Tissue preparation for immunoblotting
After completing glutamate reuptake experiments, the remainingquantity of striatal synaptosomes was processed for analysis of totalprotein levels of GLT-1, GLAST, and EAAC1. In each case, synaptosomepellets were sonicated in a 1% sodium dodecyl sulfate solution (pH~8) using a Branson Sonifier 150 (Danbury, CT). Protein concentra-tion was determined using the bichinchoninic acid colometric assay.After electrophoresis proteins were transferred for 500 Vh in a Tris/
glycine/methanol buffer onto nitrocellulose membranes (Bio-RadLaboratories, Hercules, CA).
The nitrocellulose membrane was stained with Ponceau S to re-veal relative protein staining in each sample lane. These lanes werescanned and quantified by Image J to normalize protein in each sam-ple. This relative total level then served as a normalizing value to de-termine the quantity of each transporter assayed. To continueprocessing, the membranes were placed in PVP buffer (1% polyvinyl-pyrrolidone and 0.05% Tween 20) for a minimum of 2 h to reducenonspecific antibody binding. The membrane was soaked in primaryantibody for 1–3 h. Specific primary antibodies were as follows;GLT-1 (Santa Cruz, Santa Cruz, CA, cat#15317), GLAST (Novus Biolog-icals, Littleton, CO, cat# NB100-1869), EAAC1 (Alpha Diagnostic Int.,San Antonio, TX, cat# EAAC11-A), TH (total, Millipore, Temecula, CA,cat# AB152: phospho-ser19, Phosphosolutions, Aurora, CO,cat#p1580-19: phospho-ser31 21st Century, (Salvatore et al., 2009).Nominal protein loads for linear detection of each transporter were20–30 μg total protein for GLT-1 and GLAST, ~50 μg for EAAC1, 10 μgfor total TH, and ~10 ng TH for ser19 and ~5 ng for ser31). After prima-ry treatment, blots were exposed to secondary antibody (swine anti-rabbit IgG) for signal enhancement, followed by 1 h incubation with[125I] protein A (PerkinElmer, Waltham, MA).
Statistics
A two-tailed Student's paired t-test was conducted on the totalglutamate reuptake studies, comparing glutamate reuptake in theDHK-infused striatum against its inherently matched vehicle-infused striatum for each test subject (n=7). The same statisticalanalysis was conducted for the assessments of the total glutamatetransporter protein for GLT-1, GLAST, and EAAC1 and assessmentof TH phosphorylation. Statistical significance is defined atpb0.05. We used a one-tailed t-test to determine the GLT-1- andGLAST-mediated glutamate reuptake components to determinesignificance of their respective involvement in total glutamate re-uptake, given that there was a significance increase in total gluta-mate reuptake.
Results
Glutamate reuptake
The concentration of glutamate released in striatum, after a phys-iological stressor (tail-pinch), ranges from 3 to 5 μM (Wassum et al.,2008), and averages ~10 μM following potassium-evoked release(Nickell et al., 2005). Thus, we used a concentration of glutamate(100% as 14C-labeled) for reuptake studies that was in similar range(5 μM). Three hours after vehicle- or DHK-infusion in dorsal striatum,synaptosomes were prepared simultaneously from the DHK-treatedand vehicle-treated side striatum to conduct glutamate reuptake(90 s uptake time). Compared with vehicle-treated striatum, therewas a significant increase (~43%) in striatal glutamate reuptake inthe DHK-treated striatum (Fig. 1). Mean reuptake in synaptosomesprepared from vehicle-infused side was 406±71 pmole glutamate/mg protein/min versus 580±67 pmole glutamate/mg protein/minin the DHK-infused striatum.
There was a significant increase in the non-GLT-1-mediatedcomponent of glutamate reuptake following GLT-1 blockade,which was determined by glutamate reuptake in the presence of300 μM DHK in the synaptosomes. The non-GLT-1-mediated com-ponent was about 50% of the total reuptake (~200 pmole/mg protein/min) in the vehicle-infused striatum (Table 1) and this reuptake com-ponent significantly increased (data not shown), suggesting that en-hanced glutamate reuptake following GLT-1 blockade in vivo ismediated, in part, by either GLAST- or EAAC1-mediated glutamatereuptake.
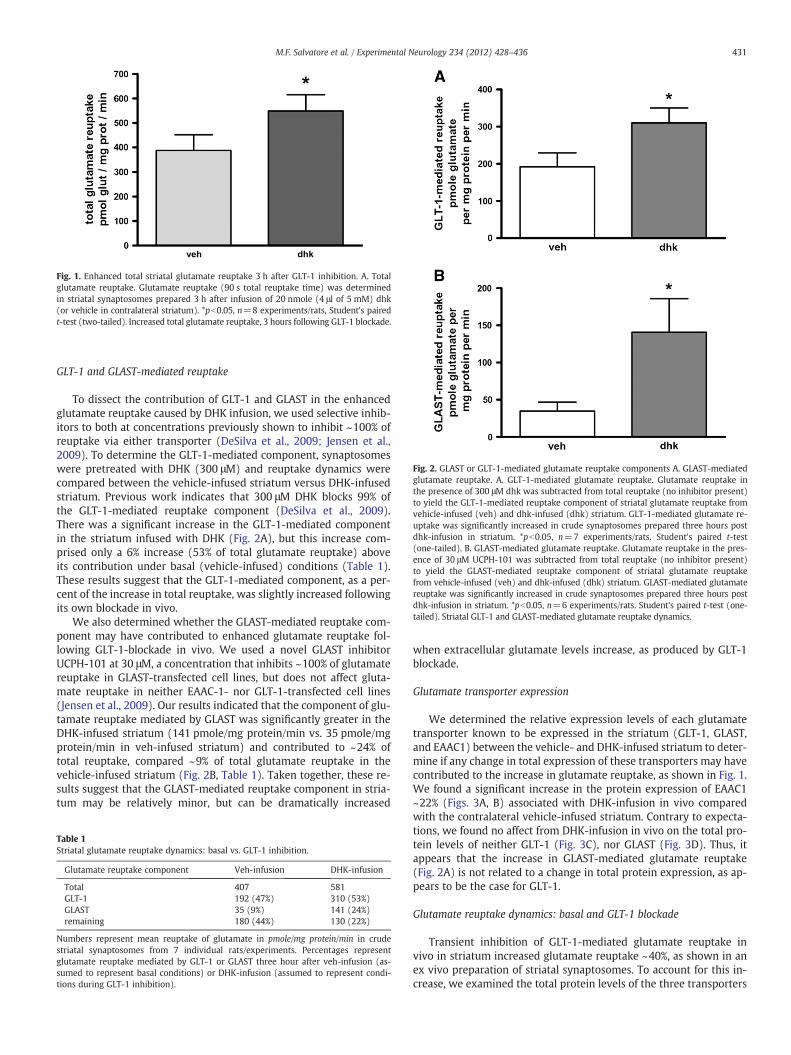
Fig. 2. GLAST or GLT-1-mediated glutamate reuptake components A. GLAST-mediatedglutamate reuptake. A. GLT-1-mediated glutamate reuptake. Glutamate reuptake inthe presence of 300 μM dhk was subtracted from total reuptake (no inhibitor present)to yield the GLT-1-mediated reuptake component of striatal glutamate reuptake fromvehicle-infused (veh) and dhk-infused (dhk) striatum. GLT-1-mediated glutamate re-uptake was significantly increased in crude synaptosomes prepared three hours postdhk-infusion in striatum. *pb0.05, n=7 experiments/rats. Student's paired t-test(one-tailed). B. GLAST-mediated glutamate reuptake. Glutamate reuptake in the pres-ence of 30 μM UCPH-101 was subtracted from total reuptake (no inhibitor present)to yield the GLAST-mediated reuptake component of striatal glutamate reuptakefrom vehicle-infused (veh) and dhk-infused (dhk) striatum. GLAST-mediated glutamatereuptake was significantly increased in crude synaptosomes prepared three hours postdhk-infusion in striatum. *pb0.05, n=6 experiments/rats. Student's paired t-test (one-tailed). Striatal GLT-1 and GLAST-mediated glutamate reuptake dynamics.
Fig. 1. Enhanced total striatal glutamate reuptake 3 h after GLT-1 inhibition. A. Totalglutamate reuptake. Glutamate reuptake (90 s total reuptake time) was determinedin striatal synaptosomes prepared 3 h after infusion of 20 nmole (4 μl of 5 mM) dhk(or vehicle in contralateral striatum). *pb0.05, n=8 experiments/rats, Student's pairedt-test (two-tailed). Increased total glutamate reuptake, 3 hours following GLT-1 blockade.
431M.F. Salvatore et al. / Experimental Neurology 234 (2012) 428–436
GLT-1 and GLAST-mediated reuptake
To dissect the contribution of GLT-1 and GLAST in the enhancedglutamate reuptake caused by DHK infusion, we used selective inhib-itors to both at concentrations previously shown to inhibit ~100% ofreuptake via either transporter (DeSilva et al., 2009; Jensen et al.,2009). To determine the GLT-1-mediated component, synaptosomeswere pretreated with DHK (300 μM) and reuptake dynamics werecompared between the vehicle-infused striatum versus DHK-infusedstriatum. Previous work indicates that 300 μM DHK blocks 99% ofthe GLT-1-mediated reuptake component (DeSilva et al., 2009).There was a significant increase in the GLT-1-mediated componentin the striatum infused with DHK (Fig. 2A), but this increase com-prised only a 6% increase (53% of total glutamate reuptake) aboveits contribution under basal (vehicle-infused) conditions (Table 1).These results suggest that the GLT-1-mediated component, as a per-cent of the increase in total reuptake, was slightly increased followingits own blockade in vivo.
We also determined whether the GLAST-mediated reuptake com-ponent may have contributed to enhanced glutamate reuptake fol-lowing GLT-1-blockade in vivo. We used a novel GLAST inhibitorUCPH-101 at 30 μM, a concentration that inhibits ~100% of glutamatereuptake in GLAST-transfected cell lines, but does not affect gluta-mate reuptake in neither EAAC-1- nor GLT-1-transfected cell lines(Jensen et al., 2009). Our results indicated that the component of glu-tamate reuptake mediated by GLAST was significantly greater in theDHK-infused striatum (141 pmole/mg protein/min vs. 35 pmole/mgprotein/min in veh-infused striatum) and contributed to ~24% oftotal reuptake, compared ~9% of total glutamate reuptake in thevehicle-infused striatum (Fig. 2B, Table 1). Taken together, these re-sults suggest that the GLAST-mediated reuptake component in stria-tum may be relatively minor, but can be dramatically increased
Table 1Striatal glutamate reuptake dynamics: basal vs. GLT-1 inhibition.
Glutamate reuptake component Veh-infusion DHK-infusion
Total 407 581GLT-1 192 (47%) 310 (53%)GLAST 35 (9%) 141 (24%)remaining 180 (44%) 130 (22%)
Numbers represent mean reuptake of glutamate in pmole/mg protein/min in crudestriatal synaptosomes from 7 individual rats/experiments. Percentages representglutamate reuptake mediated by GLT-1 or GLAST three hour after veh-infusion (as-sumed to represent basal conditions) or DHK-infusion (assumed to represent condi-tions during GLT-1 inhibition).
when extracellular glutamate levels increase, as produced by GLT-1blockade.
Glutamate transporter expression
We determined the relative expression levels of each glutamatetransporter known to be expressed in the striatum (GLT-1, GLAST,and EAAC1) between the vehicle- and DHK-infused striatum to deter-mine if any change in total expression of these transporters may havecontributed to the increase in glutamate reuptake, as shown in Fig. 1.We found a significant increase in the protein expression of EAAC1~22% (Figs. 3A, B) associated with DHK-infusion in vivo comparedwith the contralateral vehicle-infused striatum. Contrary to expecta-tions, we found no affect from DHK-infusion in vivo on the total pro-tein levels of neither GLT-1 (Fig. 3C), nor GLAST (Fig. 3D). Thus, itappears that the increase in GLAST-mediated glutamate reuptake(Fig. 2A) is not related to a change in total protein expression, as ap-pears to be the case for GLT-1.
Glutamate reuptake dynamics: basal and GLT-1 blockade
Transient inhibition of GLT-1-mediated glutamate reuptake invivo in striatum increased glutamate reuptake ~40%, as shown in anex vivo preparation of striatal synaptosomes. To account for this in-crease, we examined the total protein levels of the three transporters
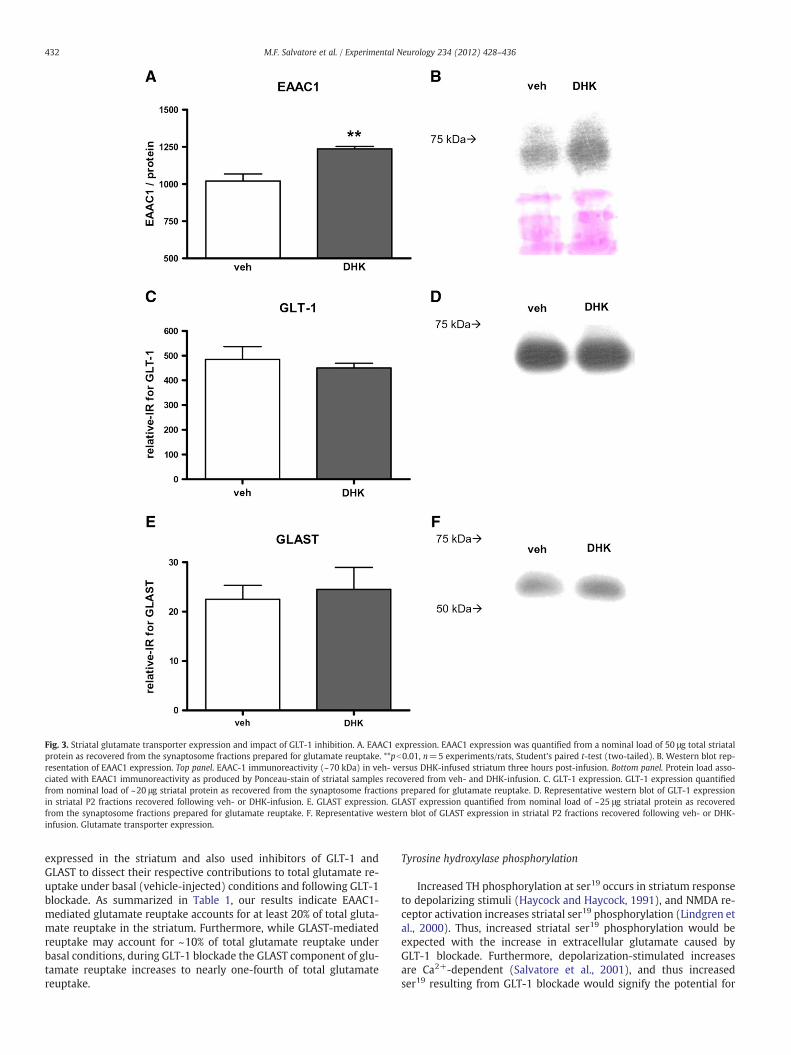
Fig. 3. Striatal glutamate transporter expression and impact of GLT-1 inhibition. A. EAAC1 expression. EAAC1 expression was quantified from a nominal load of 50 μg total striatalprotein as recovered from the synaptosome fractions prepared for glutamate reuptake. **pb0.01, n=5 experiments/rats, Student's paired t-test (two-tailed). B. Western blot rep-resentation of EAAC1 expression. Top panel. EAAC-1 immunoreactivity (~70 kDa) in veh- versus DHK-infused striatum three hours post-infusion. Bottom panel. Protein load asso-ciated with EAAC1 immunoreactivity as produced by Ponceau-stain of striatal samples recovered from veh- and DHK-infusion. C. GLT-1 expression. GLT-1 expression quantifiedfrom nominal load of ~20 μg striatal protein as recovered from the synaptosome fractions prepared for glutamate reuptake. D. Representative western blot of GLT-1 expressionin striatal P2 fractions recovered following veh- or DHK-infusion. E. GLAST expression. GLAST expression quantified from nominal load of ~25 μg striatal protein as recoveredfrom the synaptosome fractions prepared for glutamate reuptake. F. Representative western blot of GLAST expression in striatal P2 fractions recovered following veh- or DHK-infusion. Glutamate transporter expression.
432 M.F. Salvatore et al. / Experimental Neurology 234 (2012) 428–436
expressed in the striatum and also used inhibitors of GLT-1 andGLAST to dissect their respective contributions to total glutamate re-uptake under basal (vehicle-injected) conditions and following GLT-1blockade. As summarized in Table 1, our results indicate EAAC1-mediated glutamate reuptake accounts for at least 20% of total gluta-mate reuptake in the striatum. Furthermore, while GLAST-mediatedreuptake may account for ~10% of total glutamate reuptake underbasal conditions, during GLT-1 blockade the GLAST component of glu-tamate reuptake increases to nearly one-fourth of total glutamatereuptake.
Tyrosine hydroxylase phosphorylation
Increased TH phosphorylation at ser19 occurs in striatum responseto depolarizing stimuli (Haycock and Haycock, 1991), and NMDA re-ceptor activation increases striatal ser19 phosphorylation (Lindgren etal., 2000). Thus, increased striatal ser19 phosphorylation would beexpected with the increase in extracellular glutamate caused byGLT-1 blockade. Furthermore, depolarization-stimulated increasesare Ca2+-dependent (Salvatore et al., 2001), and thus increasedser19 resulting from GLT-1 blockade would signify the potential for
433M.F. Salvatore et al. / Experimental Neurology 234 (2012) 428–436
excitotoxicity given the influx of Ca2+ necessary to increase ser19
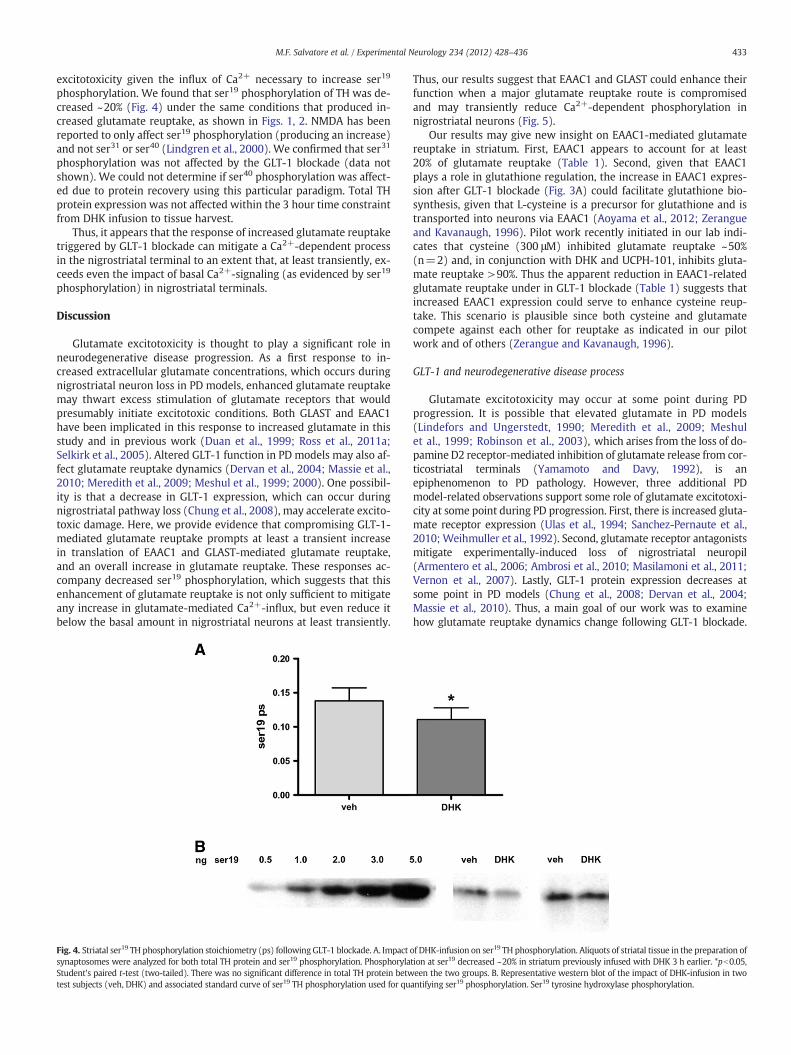
phosphorylation. We found that ser19 phosphorylation of TH was de-creased ~20% (Fig. 4) under the same conditions that produced in-creased glutamate reuptake, as shown in Figs. 1, 2. NMDA has beenreported to only affect ser19 phosphorylation (producing an increase)and not ser31 or ser40 (Lindgren et al., 2000). We confirmed that ser31
phosphorylation was not affected by the GLT-1 blockade (data notshown). We could not determine if ser40 phosphorylation was affect-ed due to protein recovery using this particular paradigm. Total THprotein expression was not affected within the 3 hour time constraintfrom DHK infusion to tissue harvest.
Thus, it appears that the response of increased glutamate reuptaketriggered by GLT-1 blockade can mitigate a Ca2+-dependent processin the nigrostriatal terminal to an extent that, at least transiently, ex-ceeds even the impact of basal Ca2+-signaling (as evidenced by ser19
phosphorylation) in nigrostriatal terminals.
Discussion
Glutamate excitotoxicity is thought to play a significant role inneurodegenerative disease progression. As a first response to in-creased extracellular glutamate concentrations, which occurs duringnigrostriatal neuron loss in PD models, enhanced glutamate reuptakemay thwart excess stimulation of glutamate receptors that wouldpresumably initiate excitotoxic conditions. Both GLAST and EAAC1have been implicated in this response to increased glutamate in thisstudy and in previous work (Duan et al., 1999; Ross et al., 2011a;Selkirk et al., 2005). Altered GLT-1 function in PD models may also af-fect glutamate reuptake dynamics (Dervan et al., 2004; Massie et al.,2010; Meredith et al., 2009; Meshul et al., 1999; 2000). One possibil-ity is that a decrease in GLT-1 expression, which can occur duringnigrostriatal pathway loss (Chung et al., 2008), may accelerate excito-toxic damage. Here, we provide evidence that compromising GLT-1-mediated glutamate reuptake prompts at least a transient increasein translation of EAAC1 and GLAST-mediated glutamate reuptake,and an overall increase in glutamate reuptake. These responses ac-company decreased ser19 phosphorylation, which suggests that thisenhancement of glutamate reuptake is not only sufficient to mitigateany increase in glutamate-mediated Ca2+-influx, but even reduce itbelow the basal amount in nigrostriatal neurons at least transiently.
Fig. 4. Striatal ser19 TH phosphorylation stoichiometry (ps) followingGLT-1 blockade. A. Impactsynaptosomes were analyzed for both total TH protein and ser19 phosphorylation. PhosphorylaStudent's paired t-test (two-tailed). There was no significant difference in total TH protein betwtest subjects (veh, DHK) and associated standard curve of ser19 TH phosphorylation used for qu
Thus, our results suggest that EAAC1 and GLAST could enhance theirfunction when a major glutamate reuptake route is compromisedand may transiently reduce Ca2+-dependent phosphorylation innigrostriatal neurons (Fig. 5).
Our results may give new insight on EAAC1-mediated glutamatereuptake in striatum. First, EAAC1 appears to account for at least20% of glutamate reuptake (Table 1). Second, given that EAAC1plays a role in glutathione regulation, the increase in EAAC1 expres-sion after GLT-1 blockade (Fig. 3A) could facilitate glutathione bio-synthesis, given that L-cysteine is a precursor for glutathione and istransported into neurons via EAAC1 (Aoyama et al., 2012; Zerangueand Kavanaugh, 1996). Pilot work recently initiated in our lab indi-cates that cysteine (300 μM) inhibited glutamate reuptake ~50%(n=2) and, in conjunction with DHK and UCPH-101, inhibits gluta-mate reuptake >90%. Thus the apparent reduction in EAAC1-relatedglutamate reuptake under in GLT-1 blockade (Table 1) suggests thatincreased EAAC1 expression could serve to enhance cysteine reup-take. This scenario is plausible since both cysteine and glutamatecompete against each other for reuptake as indicated in our pilotwork and of others (Zerangue and Kavanaugh, 1996).
GLT-1 and neurodegenerative disease process
Glutamate excitotoxicity may occur at some point during PDprogression. It is possible that elevated glutamate in PD models(Lindefors and Ungerstedt, 1990; Meredith et al., 2009; Meshulet al., 1999; Robinson et al., 2003), which arises from the loss of do-pamine D2 receptor-mediated inhibition of glutamate release from cor-ticostriatal terminals (Yamamoto and Davy, 1992), is anepiphenomenon to PD pathology. However, three additional PDmodel-related observations support some role of glutamate excitotoxi-city at some point during PD progression. First, there is increased gluta-mate receptor expression (Ulas et al., 1994; Sanchez-Pernaute et al.,2010; Weihmuller et al., 1992). Second, glutamate receptor antagonistsmitigate experimentally-induced loss of nigrostriatal neuropil(Armentero et al., 2006; Ambrosi et al., 2010; Masilamoni et al., 2011;Vernon et al., 2007). Lastly, GLT-1 protein expression decreases atsome point in PD models (Chung et al., 2008; Dervan et al., 2004;Massie et al., 2010). Thus, a main goal of our work was to examinehow glutamate reuptake dynamics change following GLT-1 blockade.
of DHK-infusion on ser19 TH phosphorylation. Aliquots of striatal tissue in the preparation oftion at ser19 decreased ~20% in striatum previously infused with DHK 3 h earlier. *pb0.05,een the two groups. B. Representative western blot of the impact of DHK-infusion in twoantifying ser19 phosphorylation. Ser19 tyrosine hydroxylase phosphorylation.

Fig. 5. Summary of the impact of GLT-1 blockade in striatum. With the blockade of GLT-1-mediated glutamate reuptake, the concentration of glutamate produced by basal releasefrom corticostriatal terminals is increased. This increase results in three significant events; 1) an increase in GLAST-mediated glutamate reuptake, 2) increased EAAC1 protein ex-pression, and 3) decreased ser19 TH phosphorylation. The decreased in ser19 phosphorylation is believed to arise, at least in part, from increased GLAST-mediated glutamate reup-take. This increase not only reduces the increase in extracellular glutamate produced from GLT-1 blockade but may also reduce normal extracellular glutamate concentration, aswould be indicated by reduced glutamate stimulation of NMDA receptors and subsequent Ca2+-influx and ser19 phosphorylation. An unresolved question is whether the increasein EAAC1 expression could also increase cysteine uptake and, as a result, promote glutathione synthesis. Proposed impact of transient GLT-1 blockade in striatum.
434 M.F. Salvatore et al. / Experimental Neurology 234 (2012) 428–436
GLT-1 blockade has been previously conducted in vivo (Bechtholt-Gompf et al., 2010; Lee et al., 2007) and the quantity of DHK infusedin striatum is similar to that used in studies that produce increased ex-tracellular glutamate (Massieu et al., 1995; Pintor et al., 2004). Thequantity of DHK infused in our study also likely has minimal immediateimpact on neuronal viability, which was 20-fold less than a quantitythat produces neuron loss after 7 days (Massieu et al., 1995). Thus, ourstudymay reveal how striatal glutamate reuptake dynamics are alteredwhenGLT-1 function is compromised and extracellular glutamate levelsincrease, as seen in PD models.
GLAST: an emerging player in responding to threat of excitotoxicity
Our results here indicate that GLAST-mediated reuptake, whileminor under basal conditions (assumed to be represented by thevehicle-infusion), can be induced when GLT-1-mediated reuptake iscompromised. Our paradigm of DHK infusion does increase extracel-lular glutamate, as previously discussed. Glutamate itself acceleratesits own reuptake and the mechanism is related to an increase in theplasma membrane expression of GLAST (Duan et al., 1999). Our con-clusion of GLAST-mediated enhancement of glutamate reuptake fol-lowing GLT-1 blockade is based upon the use of a novel GLASTinhibitor (Jensen et al., 2009). In their work, 30 μM of UCPH-101inhibited nearly 100% of glutamate reuptake in GLAST-transfectedHEK293 cell lines, but was without effect on glutamate reuptake inGLT-1- or EAAC1-transfected HEK293 cells. To our knowledge, thisis the first report of using UCPH-101 in CNS tissue. Future workwith this compound should more fully reveal how GLAST contributesto glutamate reuptake in the CNS.
GLAST plasma membrane expression was not examined in thisstudy. We would predict an enhancement of its plasma membraneexpression in this paradigm, which would be congruent with findingsof Duan et al. (1999). GLAST plasmamembrane expression does affectglutamate reuptake dynamics in vivo (Nickell et al., 2007). The age-related deficit in GLAST expression may contribute to increased extra-cellular glutamate levels reported in aging (Donzanti et al., 1993).Thus, because Parkinson's disease incidence increases with aging, adecline in GLAST-mediated reuptake, as shown in this study, mightcontribute to the excitoxicity that affects nigrostriatal neuron loss,
particularly if GLT-1 function decreases, as indicated in PD models(Chung et al., 2008).
Significance of EAAC1 upregulation: reuptake of glutamate vs. cysteine
Increased expression of EAAC1 accompanied transient GLT-1blockade. Others have also observed increased EAAC1 expressionunder potentially excitotoxic conditions (Miller et al., 1997; Ross etal., 2011a, 2011b). However, our results leave a yet to be determinedconclusion if increased EAAC1 expression contributed to increasedglutamate or cysteine reuptake following DHK-infusion, though ourpilot work (previously mentioned) suggests a cysteine sensitive inhi-bition of glutamate reuptake is operational in striatum, with andwithout GLT-1 and GLAST inhibition. This transporter is fully capableof glutamate reuptake (DeSilva et al., 2009; Fumagalli et al., 2008;Nieoullon et al., 2006; Zerangue and Kavanaugh, 1996), and our phar-macological profiling (which assumes ~100% inhibition of GLT-1 andGLAST in the reuptake studies using the crude synaptosomes) indi-cates EAAC1 could contribute between 20 and 40% of glutamate reup-take in striatum (Table 1). Conversely, increased expression of EAAC1,which is expressed in nigrostriatal neurons (Plaitakis andShashidharan, 2000), could be a mechanism by which to transportcysteine to increase glutathione synthesis, as previously suggestedin other studies (Aoyama et al., 2006, 2008; Berman et al., 2011;Himi et al., 2003), because EAAC1 is also a primary transporter of cys-teine (Aoyama et al., 2006; Zerangue and Kavanaugh, 1996), the es-sential substrate required for glutathione synthesis that has anessential role in nigrostriatal neuron viability (Garrido et al., 2011).Thus, increased EAAC1 could be a response to promote glutathionesynthesis and represent an initial defense to protect againstexcitotoxicity.
Future work should resolve whether increased EAAC1 expressionprotects nigrostriatal neurons during increased synaptic levels of glu-tamate, as seen in PD progression. Furthermore, given thatglutamate-stimulated GLAST-mediated uptake could be mitigated inaging, our findings here illustrate that striatal tissue may possess aninnate capacity to protect nigrostriatal neurons from excitotoxicityat least for some time, but this defense could be compromised overtime or in aging. Thus, increased EAAC1 expression and GLAST-

435M.F. Salvatore et al. / Experimental Neurology 234 (2012) 428–436
mediated glutamate reuptake in conjunction with the decrease of aCa2+-dependent event in nigrostriatal terminals (ser19 phosphoryla-tion), in response to GLT-1 blockade, may represent pharmacologicaltargets for improving the longevity of nigrostriatal neurons during PDprogression.
Acknowledgments
We thank Dr. Paul A. Rosenberg and colleagues for insights anddiscussion.
References
Ambrosi, G., Armentero, M.-T., Levandis, G., Bramanti, P., Nappi, G., Blandini, F., 2010.Effects of early and delayed treatment with an mGluR5 antagonist on motor im-pairment, nigrostriatal damage and neuroinflammation in a rodent model of Par-kinson's disease. Brain Res. Bull. 82, 29–38.
Aoyama, K., Suh, S.W., Hamby, A.M., Liu, J., Chan, W.Y., Chen, Y., Swanson, R.A., 2006.Neuronal glutathione deficiency and age-dependent neurodegeneration in theEAAC1 deficient mouse. Nat. Neurosci. 9, 119–126.
Aoyama, K., Matsumura, N., Watabe, M., Nakaki, T., 2008. Oxidative stress on EAAC1 isinvolved in MPTP-induced glutathione depletion and motor dysfunction. Eur. J.Neurosci. 27, 20–30.
Aoyama, K., Watabe, M., Nakaki, T., 2012. Modulation of neuronal glutathione synthesisby EAAC1 and its interacting protein GTRAP3-18. Amino Acids 42, 163–169.
Armentero, M.-T., Fancellu, R., Nappi, G., Bramanti, P., Blandini, F., 2006. Prolongedblockade of NMDA or mGluR5 glutamate receptors reduces nigrostriatal degener-ation while inducing selective metabolic changes in the basal ganglia circuitry ina rodent model of Parkinson's disease. Neurobiol. Dis. 22, 1–9.
Azarias, G., Perreten, H., Lengacher, S., Poburko, D., Demaurex, N., Magistretti, P.J., Chat-ton, J.-Y., 2011. Glutamate transport decreases mitocondrial pH and modulates ox-idative metabolism in astrocytes. J. Neurosci. 31, 3550–3559.
Bechtholt-Gompf, A.J., Walther, H.V., Adams, M.A., Carlezon, W.A., Ongur, D., Cohen,B.M., 2010. Blockade of astrocytic glutamate uptake in rats induces signs of anhe-donia and impaired spatial memory. Neuropsychopharmacology 35, 2049–2059.
Behrens, P.F., Franz, P., Woodman, B., Lindenberg, K.S., Landwehrmeyer, G.B., 2002. Im-paired glutamate transport and glutamate-glutamine cycling: downstream effectsof the Huntington mutation. Brain 125, 1908–1922.
Berman, A.E., Chan, W.Y., Brennan, A.M., Reyes, R.C., Adler, B.L., Suh, S.W., Kauppinen,T.M., Edling, Y., Swanson, R.A., 2011. N-acetylcysteine prevents loss of dopaminer-gic neurons in the EAAC1−/− mouse. Ann. Neurol. 69, 509–520.
Chung, E.K., Chen, L.W., Chan, Y.S., Yung, K.K., 2008. Downregulation of glial glutamatetransporters after dopamine denervation in the striatum of 6-hydroxydopamine-lesioned rats. J. Comp. Neurol. 511, 421–437.
Dabir, D.V., Robinson, M.B., Swanson, E., Zhang, B., Trojanowski, J.Q., Lee, V.M.-Y., For-man, M.S., 2006. Impaired glutamate transport in a mouse model of tau pathologyin astrocytes. J. Neurosci. 26, 644–654.
Danbolt, N.C., 2001. Glutamate uptake. Prog. Neurobiol. 65, 1–105.Danbolt, N.C., Holmseth, S., Dehnes, Y., Huang, Y.H., Follin-Arbelet, V., Grutle, N.J., Zhou,
Y., Plachez, C., Furness, D.N., Bergles, D.E., Lehre, K.P., 2011. Quantification and lo-calization of the EAAC1 glutamate transporter in brain. Soc. Neurosci. Abstracts,342.12.
Del Rio, P., Montiel, T., Massieu, L., 2008. Contribution of NMDA and non-NMDA recep-tors to in vivo glutamate-induced calpain activation in the rat striatum. Relation toneuronal damage. Neurochem. Res. 33, 1475–1483.
Dervan, A.G., Meshul, C.K., Beales, M., McBean, G.J., Moore, C., Totterdell, S., Snyder, A.K.,Meredith, G.E., 2004. Astroglial plasticity and glutamate function in a chronicmouse model of Parkinson's disease. Exp. Neurol. 190, 145–156.
DeSilva, T.M., Kabakov, A.Y., Goldhoff, P.E., Volpe, J.J., Rosenberg, P.A., 2009. Regulationof glutamate transport in developing rat oligodendrocytes. J. Neurosci. 29,7898–7908.
Dolinska, M., Albrecht, J., 1997. Glutamate uptake is inhibited by L-arginine in mito-chondria isolated from rat cerebrum. Neuroreport 8, 2365–2368.
Donzanti, B.A., Hite, J.F., Yamamoto, B.K., 1993. Extracellular glutamate levels increasewith age in the lateral striatum: potential involvement of presynaptic D-2 recep-tors. Synapse 13, 376–382.
Duan, S., Anderson, C.M., Stein, B.A., Swanson, R.A., 1999. Glutamate induces rapidupregulation of astrocyte glutamate transport and cell-surface expression ofGLAST. J. Neurosci. 19, 10193–10200.
Fumagalli, E., Funicello, M., Rauen, T., Gobbi, M., Mennini, T., 2008. Riluzole enhancesthe activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur. J. Pharmacol.587, 171–176.
Garrido, M., Tereshchenko, Y., Zhevtsova, Z., Taschenberger, G., Bahr, M., Kugler, S.,2011. Glutathione depletion and overproduction both initiate degeneration ofnigral dopaminergic neurons. Acta Neuropathol. 121, 475–485.
Geddes, J.W., Saatman, K.E., 2010. Targeting individual calpain isoforms for neuropro-tection. Exp. Neurol. 226, 6–7.
Genda, E.N., Jackson, J.G., Sheldon, A.L., Locke, S.F., Greco, T.M., O'Donnell, J.C., Spruce,L.A., Xiao, R., Guo, W., Putt, M., Seeholzer, S., Ischiropoulos, H., Robinson, M.B.,2011. Co-compartmentalization of the astroglial glutamate transporter, GLT-1,with glycolytic enzymes and mitochondria. J. Neurosci. 31, 18275–18288.
Grant, R.J., Sellings, L.H., Crocker, S.J., Melloni, E., Park, D.S., Clarke, P.B., 2009. Effects ofcalpain inhibition on dopaminergic markers and motor function following intras-triatal 6-hydroxydopamine administration in rats. Neuroscience 158, 558–569.
Greenamyre, J.T., 2001. Glutamatergic influences on the basal ganglia. Clin. Neurophar-macol. 24, 65–70.
Haycock, J.W., Haycock, D.A., 1991. Tyrosine hydroxylase in rat brain dopaminergicnerve terminals: multiple-site phosphorylation in vivo and in synaptosomes. J.Biol. Chem. 266, 5650–5657.
Henn, F.A., Anderson, D.J., Rustad, D.G., 1976. Glial contamination of synaptosomal frac-tions. Brain Res. 101, 341–344.
Himi, T., Ikeda, M., Yasuhara, T., Nishida, M., Morita, I., 2003. Role of neuronal glutamatetransporter in the cysteine uptake and intracellular glutathione levels in culturedcortical neurons. J. Neural Transm. 110, 1337–1348.
Jensen, A.A., Erichsen, M.N., Nielsen, C.W., Stensbol, T.B., Kehler, J., Bunch, L., 2009. Dis-covery of the first selective inhibitor of excitatory amino acid transporter subtype1. J. Med. Chem. 52, 912–915.
Karlsson, R.-M., Tanaka, K., Heilig, M., Holmes, A., 2008. Loss of glial glutamate and as-partate transporter (excitatory amino acid transporter 1) causes locomotor hyper-activity and exaggerated responses to psychotomimetics: rescue by haloperidoland metabotropic glutamate 2/3 agonist. Biol. Psychiatry 64, 810–814.
Lau, A., Tymianski, M., 2010. Glutamate receptors, neurotoxicity and neurodegenera-tion. Pflugers Arch. 460, 525–542.
Lee, Y., Gaskins, D., Anand, A., Shekar, A., 2007. Glia mechanisms in mood regulation: anovel model of mood disorders. Psychopharmacology 191, 55–65.
Lindefors, N., Ungerstedt, U., 1990. Bilateral regulation of glutamate tissue and extra-cellular levels in caudate-putamen by midbrain dopamine neurons. Neurosci.Lett. 115, 248–252.
Lindgren, N., Xu, Z.Q.D., Lindskog, M., Herrera-Marschitz, M., Goiny, M., Haycock, J.W.,Goldstein, M., Hökfelt, T., Fisone, G., 2000. Regulation of tyrosine hydroxylase activ-ity and phosphorylation at ser19 and ser40 via activation of glutamate NMDA recep-tors in rat striatum. J. Neurochem. 74, 2470–2477.
Masilamoni, G.J., Bogenpohl, J.W., Alagille, D., Delevich, K., Tamagnan, G., Votaw, J.R.,Wichmann, T., Smith, Y., 2011. Metabotropic glutamate receptor 5 antagonist pro-tects dopaminergic and noradrenergic neurons from degeneration in MPTP-treated monkeys. Brain 134, 2057–2073.
Massie, A., Goursaud, S., Schallier, A., Vermoesen, K., Meshul, C.K., Hermans, E.,Michotte, Y., 2010. Time-dependent changes in GLT-1 functioning in striatum ofhemi-Parkinson rats. Neurochem. Int. 57, 572–578.
Massieu, L., Tapia, R., 1997. Glutamate uptake impairment and neuronal damage inyoung and aged rats in vivo. J. Neurochem. 69, 1151–1160.
Massieu, L., Morales-Villagran, A., Tapia, R., 1995. Accumulation of extracellular gluta-mate by inhibition of its uptake is not sufficient for inducing neuronal damage:an in vivo microdialysis study. J. Neurochem. 64, 2272.
Meredith, G.E., Totterdell, S., Beales, M., Meshul, C.K., 2009. Impaired glutamate ho-meostasis and programmed cell death in a chronic MPTP mouse model of Parkin-son's disease. Exp. Neurol. 219, 334–340.
Meshul, C.K., Emre, N., Nakamura, C.M., Allen, C., Donohue, M.K., Buckman, J.F., 1999.Time-dependent changes in striatal glutamate synapses following a 6-hydroxydopamine lesion. Neuroscience 88, 1–16.
Meshul, C.K., Cogen, J.P., Cheng, H.W., Moore, C., Krentz, L., McNeill, T.H., 2000. Alter-ations in rat striatal glutamate synapses following a lesion of the cortico- and/ornigrostriatal pathway. Exp. Neurol. 165, 191–206.
Miller, H.P., Levey, A.I., Rothstein, J.D., Tzingounis, A.V., Conn, P.J., 1997. Alterations inglutamate transporter protein levels in kindling-induced epilepsy. J. Neurochem.68, 1564–1570.
Nickell, J., Pomerleau, F., Allen, J., Gerhardt, G.A., 2005. Age-related changes in the dy-namics of potassium-evoked L-glutamate release in the striatum of Fisher 344rats. J. Neural Transm. 112, 87–96.
Nickell, J., Salvatore, M.F., Pomerleau, F., Apparsundaram, S., Gerhardt, G.A., 2007. Re-duced plasma membrane surface expression of GLAST mediates decreased gluta-mate regulation in the aged striatum. Neurobiol. Aging 28, 1737–1748.
Nieoullon, A., Canolle, B., Masmejan, F., Guillet, B., Pisano, P., Lortet, S., 2006. The neu-ronal excitatory amino acid transporter EAAC1/EAAT3: does it represent a majoractor at the brain excitatory synapse? J. Neurochem. 98, 1007–1018.
Pintor, A., Galluzzo, M., Grieco, R., Pezzola, A., Reggio, R., Popoli, P., 2004. Adenosine A2A receptor antagonists prevent the increase in striatal glutamate levels inducedby glutamate uptake inhibitors. J. Neurochem. 89, 152–156.
Plaitakis, A., Shashidharan, P., 2000. Glutamate transport and metabolism in dopami-nergic neurons of substantia nigra: implication for the pathogenesis of Parkinson'sdisease. J. Neurol. 247, II/25–II/35.
Robinson, M.B., 1998. The family of sodium-dependent glutamate transporters: a focuson the GLT-1/EAAT2 subtype. Neurochem. Int. 33, 479–491.
Robinson, M.B., Hunter-Ensor, M., Sinor, J., 1991. Pharmacologically distinct sodium-dependent L-[3H]glutamate transport processes in rat brain. Brain Res. 544, 192–202.
Robinson, S., Freeman, P., Moore, C., Touchon, J.C., Krentz, L., Meshul, C.K., 2003. Acuteand subchronic MPTP administration differentially affects striatal glutamate syn-aptic function. Exp. Neurol. 180, 73–86.
Rosenberg, P.A., Aizenman, E., 1989. Hundred-fold increase in neuronal vulnerability toglutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci. Lett.103, 162–168.
Ross, J.R., Ramakrishnan, H., Porter, B.E., Robinson, M.B., 2011a. Group I mGluR-regulated translation of the neuronal glutamate transporter, excitatory aminoacid carrier 1. J. Neurochem. 117, 812–823.
Ross, J.R., Porter, B.E., Buckley, P.T., Eberwine, J.H., Robinson, M.B., 2011b. mRNA for theEAAC1 subtype of glutamate transporter is present in neuronal dendrites in vitroand dramatically increases in vivo after a seizure. Neurochem. Int. 58, 366–375.

436 M.F. Salvatore et al. / Experimental Neurology 234 (2012) 428–436
Rothstein, J.D., Dykes-Hoberg, M., Pardo, C.A., Bristol, L.A., Jin, L., Kunci, R.W., Kanai, Y.,Hediger, M.A., Wang, Y., Schielke, J.P., Welty, D.F., 1996. Knockout of glutamatetransporters reveals a major role for astroglial transport in excitotoxicity and clear-ance of glutamate. Neuron 16, 675–686.
Rothstein, J.D., Patel, S., Regan, M.R., Haenggeli, C., Huang, Y.H., Bergles, D.E., Jin, L.,Hoberg, M.D., Vidensky, S., Chung, D.S., Toan, S.V., Bruijn, L.I., Su, Z., Gupta, P., Fisher,P.B., 2005. Beta-lactam antibiotics offer neuroprotection by increasing glutamatetransporter expression. Nature 433, 73–77.
Salvatore, M.F., Waymire, J.C., Haycock, J.W., 2001. Depolarization-stimulated catechol-amine biosynthesis: involvement of protein kinases and tyrosine hydroxylasephosphorylation sites in situ. J. Neurochem. 79, 349–360.
Salvatore, M.F., Apparsundaram, S., Gerhardt, G.A., 2003. Decreased plasma membraneexpression of striatal dopamine transporter in aging. Neurobiol. Aging 24,1147–1154.
Salvatore, M.F., Pruett, B.S., Spann, S.L., Dempsey, C., 2009. Aging reveals a role fornigral tyrosine hydroxylase ser31 phosphorylation in locomotor activity genera-tion. PLoS One 5, e8466.
Sanchez-Pernaute, R., Wang, J.-Q., Kuruppu, D., Cao, L., Tueckmantel, W., Kozikowski,A., Isacson, O., Brownell, A.-L., 2010. Enhanced binding of metabotropic glutamatereceptor type 5 (mGluR5) PET tracers in the brain of parkinsonian primates. Neu-roImage 42, 248–251.
Satrustegui, J., Contreras, L., Ramos, M., Marmol, P., del Arco, A., Saheki, T., Pardo, B.,2007. Role of aralar, the mitochondrial transporter of aspartate-glutamate, inbrain N-acetylaspartate formation and Ca(2+) signaling in neuronal mitochondria.J. Neurosci. Res. 85, 3359–3366.
Schallier, A., Smolders, I., Van Dam, D., Loyens, E., De Deyn, P.P., Michotte, A., Michotte,Y., Massie, A., 2011. Region- and age-specific changes in glutamate transport in theAßPP23 mouse model for Alzheimer's disease. J. Alzheimer's Dis. 24, 287–300.
Selkirk, J.V., Nottebaum, L.M., Vana, A.M., Verge, G.M., MacKay, K.B., Stiefel, T.H., Naeve,G.S., Pomeroy, J.E., Petroski, R.E., Moyer, J., Dunlop, J., Foster, A.C., 2005. Role of theGLT-1 subtype of glutamate transporter in glutamate homeostasis: the GLT-1-preferring inhibitor WAY-855 produces marginal neurotoxicity in the rat hippo-campus. Eur. J. Neurosci. 21, 3217–3228.
Sheldon,A.L., Robinson,M.B., 2007. The role of glutamate transporters in neurodegenerativediseases and potential opportunities for intervention. Neurochem. Int. 51, 333–355.
Suchak, S.K., Baloyianni, N.V., Perkinton, M.S., Williams, R.J., Meldrum, B.S., Rattray, M.,2003. The ‘glial’ glutamate transporter, EAAT2 (Glt-1) accounts for high affinityglutamate uptake into adult rodent nerve endings. J. Neurochem. 84, 522–532.
Ulas, J., Weihmuller, F.B., Brunner, L.C., Joyce, J.N., Marshall, J.F., Cotman, C.W., 1994. Se-lective increase of NMDA-sensitive glutamate binding in the striatum of Parkin-son's disease, Alzheimer's disease, and mixed Parkinson's disease/Alzheimer'sdisease patients: an autoradiographic study. J. Neurosci. 14, 6317–6324.
Velasco, I., Tapia, R., Massieu, L., 1996. Inhibition of glutamate uptake induces progres-sive accumulation of extracellular glutamate and neuronal damage in rat corticalcultures. J. Neurosci. Res. 44, 551–561.
Vernon, A.C., Zbarsky, V., Datla, K.P., Croucher, M.J., Dexter, D.T., 2007. Subtype selectiveantagonism of substantia nigra pars compacta Group I metabotropic glutamate re-ceptors protects the nigrostriatal system against 6-hydroxydopamine toxicity invivo. J. Neurochem. 103, 1075–1091.
Wang, G.J., Chung, H.J., Schnuer, J., Lea, E., Robinson, M.B., Potthoff, W.K., Aizenman, E.,Rosenberg, P.A., 1998. Dihydrokainate-sensitive neuronal glutamate transport isrequired for protection of rat cortical neurons in culture against synaptically re-leased glutamate. Eur. J. Neurosci. 10, 2523–2531.
Wassum, K.M., Tolosa, V.M., Wang, J., Walker, E., Monbouquette, H.G., Maidment, N.T.,2008. Silicon wafer-based platinum microelectrode array biosensor for near real-time measurement of glutamate in vivo. Sens. Basel Sens. 8, 5023–5036.
Weihmuller, F.B., Ulas, J., Nguyen, L., Cotman, C.W., Marshall, J.F., 1992. Elevated NMDAreceptors in parkinsonian striatum. Neuroreport 3, 977–980.
Yamamoto, B.K., Davy, S., 1992. Dopaminergic modulation of glutamate release in stri-atum as measured by microdialysis. J. Neurochem. 58, 1736–1742.
Zerangue, N., Kavanaugh, M.P., 1996. Interaction of L-cysteine with a human excitatoryamino acid transporter. J. Physiol. 493, 419–423.
Ziemińska, E., Hilgier, W., Waagepetersen, H.S., Hertz, L., Sonnewald, U., Schousboe, A.,Albrecht, J., 2004. Analysis of glutamine accumulation in rat brain mitochondria inthe presence of a glutamine uptake inhibitor, histidine, reveals glutamine poolswith a distinct access to deamidation. Neurochem. Res. 29, 2121–2123.