university of balamand st. georges hospital complex
TRANSCRIPT

John J. Haddad, PhD
University of Balamand
ST. Georges Hospital Complex
Faculty of Health Sciences
Department of Medical Laboratory Science
A Report of Academic and Professional Liaison
Authored and Contributed for and by:
Dr. John J. Haddad
Biomedical Research Scientist and Educating Professor – 2009 Page | 1

John J. Haddad, PhD
Introduction
• General Background of Dr. John Haddad (1993 – 1998):
Dr. John Haddad’s academic career goes back to 1993, when he was awarded a full ‘Research Assistant’ scholarship in Biological Sciences at the American University of Beirut (AUB), Beirut, Lebanon. The above-mentioned award is granted to individuals who have had achieved a high-profile status during the years leading to Bachelor of Science in Biological Sciences (B.Sc. [Pre-medical Biology]). Dr. Haddad, then Mr. John Haddad, was awarded a 2-year fellowship that eventually led to attaining the degree of Masters of Science (M.Sc. [Neuroimmunology]). The award was determined for only 9 graduate students chosen from an applicant pool of about 350 undergraduate individuals, and was directly supervised and approved by the Dean of the Faculty of Arts & Sciences at AUB.
During an extensive 2-year program, Dr. Haddad envisioned the creation of an independent research project, under the supervision of highly qualified faculty members, Dr. Salim A. Kanaan and Professor Bared Safieh-Garabedian, then at the department of Biology (AUB). The project was executed with close collaboration with an eminent professor at the departments of Physiology and Human Morphology, Faculty of Medicine (AUB), Professor Nayef E. Saadé, an internationally recognized scholar who co-sponsored Dr. Haddad’s project. Professor Suhayl J. Jabbur, a prominent physiologist at AUB, also supervised the aformentioned project. The details of this project are outlined below.
1. The theory proposed by Dr. Haddad involved the understanding of pain (hyperalgesia) mechanisms in mammals, including humans, in response to noxious stimulation. Dr. Haddad designed and conducted experiments to study the aforementioned theory, basically in applications involving laboratory animals (rats and mice – which are very close by their mammalian nature [Biologically and Physiologically] to human beings), using various protocols he has established for pain measurement and perception.
2. While establishing the aforementioned theories, Dr. Haddad has developed an
extensive experience in work involving the handling of laboratory animals and more specifically for testing both thermal and mechanical hyperalgesia by using different protocols for pain tests. In addition, he has working experience in performing enzyme-linked immunosorbent assays (ELISA immunoassays) for the measurement of cytokines and growth factors in tissues and blood, in addition to molecular cell biology techniques and methodologies. He was also involved in animal surgical procedures such as thymectomy, sympathectomy, cordotomy, vagotomy and brain electrolytic lesions. Because studying pain mechanisms demanded it, Dr. Haddad also developed experience in monitoring machines such as the Coulter counter and the Scintillation counter for assaying radioactivity in samples for physiological purposes. Due to the nature of the program that Dr. Haddad has conceived and performed, his major area of interest was focused in studying
Biomedical Research Scientist and Educating Professor – 2009 Page | 2

John J. Haddad, PhD
neuroimmunology, nociception, neurogenic inflammation and the mechanisms of inflammatory hyperalgesia.
3. The so far performed research by Dr. Haddad has had potential clinical
implications involved in understanding and treating inflammatory pain. He well examined the mechanisms of inflammatory pain, a condition that is induced by the accumulation of relatively small molecules peptides, termed cytokines, which play a critical role in exacerbating the pain condition by causing lesions of infection and inflammation in peripheral and central body tissues. Dr. Haddad’s program project a number of theories to discuss the mechanisms of how we perceive pain due to noxious stimulation, and how the brain, with special communication with the spinal cord, processes and controls the perception, analysis and alleviation of the pain mechanism. Dr. Haddad significant research at AUB was culminated by:
• Concluding that during peripheral injury (local) or central injury
(systemic), the pain process is exacerbated by exogenous or endogenous agents, thereby requiring the application of potentially counteractive analgesic steroidal and non-steroidal anti-inflammatory drugs (NSAIDs), which alleviate and/or control pain transduction, perception and analysis via multifaceted mechanisms (molecular and physiological).
• Publishing the aforementioned observations in high profile journals,
reviewed by experts in the field of Biological Sciences and Neuroimmunology (Safieh-Garabedian et al., Brain Research 717: 179-183, 1996; Kanaan et al., Pain 66: 373-379, 1996; Safieh-Garabedian et al., British Journal of Pharmacology 121: 1619-1626, 1997; Barbour et al., Vaccine 16: 1650-1655, 1998; Kreydiyyeh et al., Life Sciences 63: 1913-1919, 1998).
• General Background of Dr. John Haddad (1998 – 2001):
After the conclusion of his Masters at AUB, Dr. Haddad continued developing research
projects at the same institution from 1996-1998, culminating as indicated above in further peer-reviewed publications, numbering 7 in total, including presentations at experimental meetings for the Society for Neuroscience (SFN) in the USA.
Because of the encompassed high caliber of Dr. Haddad’s biomedical research at AUB,
he was selected to receive a very prestigious award, bestowed upon him by the Georges John Livanos Trust in London (England, UK). In 1998, Dr. Haddad was awarded a full scholarship to enroll in the Ph.D. program at the Center for Human Development (Oxygen Sensing Division) at the Tayside Institute of Maternal and Child Health, Faculty of Medicine, Ninewells Hospital and Medical School, University of Dundee, Dundee, Scotland, UK. Briefly, the aforementioned Trust is concerned with biomedical and clinical research that may help understand the basis for respiratory distress syndrome in premature infants and in adults due either to lung immaturity
Biomedical Research Scientist and Educating Professor – 2009 Page | 3

John J. Haddad, PhD
or lung inflammation/infection caused by a pathogenic microorganism, such as a virulent bacterium and/or an infectious virus.
Dr. Haddad spent the 3-year period in Scotland investigating patterns of lung
inflammation associated with oxygen therapy in premature births, a condition clinically termed RDS or ARDS (respiratory distress syndrome or acute respiratory distress syndrome). This syndrome is a debilitating condition from which millions of susceptible infants below the age of 5 in the USA, UK and across the globe, suffer. His accumulated expertise while training at AUB led Dr. Haddad to provide direct leadership and technological knowledge to biomedical and clinical research groups at Ninewells Hospital, which were focusing on understanding the differential regulation of target DNA genes involved in regulation RDS/ARDS. Dr. Haddad adamantly believes that the genetic factor is crucial for lung development and adaptability to hostile environments, therefore, the perpetuation of distress-related syndromes, such as RDS, must require the involvement of specific genes the functions of which are coordinated by complex mechanisms. In this regard, he designed experimentations and methodology techniques to investigate how oxygen potentially regulates target genes, such as hypoxia-inducible factor 1α (HIF-1α) and nuclear factor-κB (NF-κB), involved in lung physiology and pathophysiology.
Dr. Haddad paid exceptional attention to the theory that states that under low oxygen
environment (hypoxia) and/or high oxygen environment (hyperoxia) – from which preterm infants drastically suffer – the lung responds as to ensure survival of its tissues in hostile conditions. The condition could most often be overwhelming and, in that particular case, certain tissues in the lung and further afield, such as the brain and liver, begin to gradually loose their cellular activities. In other words, body tissues start disintegrating and dying. The diagnostic treatment proposed by Dr. Haddad then incorporated, but was not confined to, the adoption of a clinical strategy of exposing the immature lung to low, yet effective, oxygen ventilation, and of monitoring the genetic and molecular processes predisposing the lung to oxidative stress, such as in RDS.
The high impact of Dr. Haddad’s biomedical and clinical research on lung development
and oxidative stress was widely welcome by peers, professors, physicians and scientists both in the USA and UK. This unprecedented type of research was manifested in (citing few examples):
1. Invitations to lecture on the adoption of potential therapeutic strategies in
Lebanon, UK, Switzerland and, most importantly, USA. Dr. Haddad subsequently received invitations from prominent academic intuitions, thereafter listed:
• Departments of Human Morphology and Physiology, Faculty of
Medicine, and Department of Biology, Faculty of Arts and Sciences, American University of Beirut, Beirut, Lebanon. “Oxygen-sensing mechanisms in the developing lung: Regulation of gene transcription.” Invitation was received in 1999.
Biomedical Research Scientist and Educating Professor – 2009 Page | 4

John J. Haddad, PhD
• Tayside Institute of Maternal and Child Health, Faculty of Medicine, University of Dundee, Dundee, Scotland, UK. “Redox regulation of HIF-1α and NF-κB in the alveolar epithelium.” Invitation was received in 2000.
• Departments of Human Morphology and Physiology, Faculty of
Medicine, and Department of Biology, Faculty of Arts and Sciences, American University of Beirut, Beirut, Lebanon. “Redox regulation of oxygen-sensitive transcription factors.” Invitation was received in 2000.
• Department of Medicine, National Jewish Medical and Research
Center, global leader in lung, allergic and immune diseases, Denver, Colorado, USA. “The role of reactive oxygen species in inducing a pro-inflammatory signal: Therapeutic implications.” Invitation was received in 2001.
• Developmental Biology Program, Children’s Hospital Los Angeles
Research Institute, Smith Research Tower, University of Southern California School of Medicine (UCLA), Los Angeles, California, USA. “Redox regulation of pro-inflammatory cytokines in the developing lung.” Invitation was received in 2001.
• Division of Molecular Cardiovascular Biology, The Children’s
Hospital Research Foundation, Children’s Hospital Medical Center, Cincinnati, Ohio, USA. “Oxygen sensing and redox mechanisms in the perinatal airway epithelium.” Invitation was received in 2001.
• Boehringer Ingelheim International GmbH, Transatlantic Airway
Conference (17th TAC annual meeting), Lucerne, Switzerland. “Redox and oxidant-mediated regulation of apoptosis signaling pathways.” Invitation was received in 2002.
• Division of Pulmonary, Critical Care and Occupational Medicine,
Department of Internal Medicine, Health Sciences Center, School of Medicine, Saint Louis University, Saint Louis, Missouri, USA. “Antioxidant/prooxidant mechanisms in the regulation of redox-sensitive transcription factors, apoptosis signaling cofactors and inflammation.” Invitation was received in 2002.
• Programs in Pharmacology and Toxicology, Department of
Pharmaceutical Sciences, College of Pharmacy, Health Sciences Center, University of Oklahoma, Oklahoma City, Oklahoma, USA. “Immunopharmacologic antioxidant/prooxidant mechanisms in the regulation of redox-sensitive transcription factors, apoptosis
Biomedical Research Scientist and Educating Professor – 2009 Page | 5

John J. Haddad, PhD
signaling cofactors and inflammation.” Invitation was received in 2002.
• Center for Environmental Health Sciences, Department of Pharmaceutical Sciences, University of Montana, Missoula, Montana, USA. “Antioxidant/prooxidant mechanisms in the regulation of redox-sensitive transcription factors, apoptosis signaling cofactors and inflammation.” Invitation was received in 2002.
• Spinal Cord and Brain Injury Research Center, Sanders Brown Center
in Aging, University of Kentucky, Lexington, Kentucky, USA. “Antioxidant/prooxidant mechanisms in the regulation of redox-sensitive transcription factors, apoptosis signaling cofactors and inflammation.” Invitation was received in 2003.
2. Publishing outstanding research of high caliber in peer-reviewed specialized
journals, accredited nationally and worldwide. The list is thereafter mentioned in brevity below (please see updated c.v. for further details):
• Haddad and Land, The American Journal of Physiology: Lung,
Cellular and Molecular Physiology 278: L492-L503, 2000.
This journal is one of the official publication of the accredited and well established American Physiological Society (APS), member of the Federation of the American Societies for Experimental Biology (FASEB) (Please also note the letter of support from the executive director of APS, Dr. Martin Frank, who recognized the national interest and scope of Dr. Haddad’s research and reputation). APS is an accredited and recognized academic institution.
This paper discussed that oxygen-linked genetic regulation in the
perinatal lung is responsive to dynamic developmental changes in antioxidant capacity.
• Haddad and Land, Biochemical and Biophysical Research
Communications 271: 257-267, 2000. • Haddad et al., The Journal of Biological Chemistry 275: 21130-
21139, 2000.
This journal is the official publication of the accredited and well-established American Society for Biochemistry and Molecular Biology (ASBMB), member of FASEB. The high impact factor of this journal indicates that the ability of Dr. Haddad to publish his research in JBC would certainly help disseminate cutting edge biomedical research knowledge to the national scientific community in the USA. ASBMB is an accredited and recognized academic institution.
Biomedical Research Scientist and Educating Professor – 2009 Page | 6

John J. Haddad, PhD
This paper highlighted the fact that antioxidants play a key
regulatory role in determining genetic responsiveness to oxidant/antioxidant disequilibria in normal lung development and pathophysiological conditions.
• Haddad et al., Biochemical and Biophysical Research
Communications 274: 500-505, 2000. • Haddad, Cytokines, Cellular and Molecular Therapy 6: 177-187,
2000. • Haddad et al., Cytokine 13: 138-147, 2001.
• Haddad et al., Biochemical and Biophysical Research
Communications 281: 311-316, 2001.
This paper discussed the essential role of clinical oxygenation during the transition from placental-based respiration to pulmonary-based respiration.
• Haddad et al., The Journal of Pharmacology and Experimental
Therapeutics 296: 996-1005, 2001.
This journal is the official publication of the accredited and well-established American Society for Pharmacology and Experimental Therapeutics (ASPET), member of FASEB. The high impact factor of this journal indicates that the ability of Dr. Haddad to publish his research in JPET would certainly help disseminate cutting edge biomedical research knowledge to the national scientific community in the USA. ASPET is an accredited and recognized academic institution.
This paper discussed the therapeutic immunopharmacological potential of antioxidants in the treatment of inflammation in the lung that is caused by cytokines.
• Haddad et al., Biochemical and Biophysical Research
Communications 281: 987-992, 2001.
This paper discussed the role of genes in regulating ion transport and fluid accumulation, such as in asthmatic children, in the lung.
• Haddad et al., Biochemical Journal 355: 29-38, 2001. • Baines et al., Journal of Physiology 532: 105-113, 2001.
Biomedical Research Scientist and Educating Professor – 2009 Page | 7

John J. Haddad, PhD
This paper discussed an extension of the role of oxygenes in regulating ion transport and fluid accumulation, such as in asthmatic children, in the lung.
• Haddad et al., British Journal of Pharmacology 133: 49-60, 2001. • Haddad et al., Biochemical and Biophysical Research
Communications 285: 267-272, 2001.
• Haddad and Land, Federation of European Biochemical Societies Letters 505: 269-274, 2001.
• Haddad and Land, The American Journal of Respiratory Cell and
Molecular Biology 26: 114-126, 2002.
This journal is the official publication of the accredited and well-established American Thoracic Society (ATS). The high impact factor of this journal indicates that the ability of Dr. Haddad to publish his research in AJRCMB would certainly help disseminate cutting edge biomedical research knowledge to the national scientific community in the USA. ATS is an accredited and recognized academic institution.
This paper discussed the therapeutic potential of amiloride, a drug which blocks sodium channels in the lung, in the treatment of inflammation in the lung that is caused by cytokines.
• Haddad et al., The Journal of Pharmacology and Experimental Therapeutics 300: 559-566, 2002.
This journal is the official publication of the accredited and well-
established American Society for Pharmacology and Experimental Therapeutics (ASPET), member of FASEB. The high impact factor of this journal indicates that the ability of Dr. Haddad to publish his research in JPET would certainly help disseminate cutting edge biomedical research knowledge to the national scientific community in the USA. ASPET is an accredited and recognized academic institution.
This paper discussed the therapeutic immunopharmacological potential of phosphodiesterase blockers in the treatment of inflammation in the lung that is caused by cytokines.
• Haddad et al., The Journal of Pharmacology and Experimental
Therapeutics 300: 567-576, 2002.
This journal is the official publication of the accredited and well-established American Society for Pharmacology and Experimental
Biomedical Research Scientist and Educating Professor – 2009 Page | 8

John J. Haddad, PhD
Therapeutics (ASPET), member of FASEB. The high impact factor of this journal indicates that the ability of Dr. Haddad to publish his research in JPET would certainly help disseminate cutting edge biomedical research knowledge to the national scientific community in the USA. ASPET is an accredited and recognized academic institution.
This paper discussed the therapeutic immunopharmacological potential of phosphodiesterase blockers in the treatment of inflammation in the lung that is caused by cytokines from the perspective of gene regulation.
• Haddad et al., Cellular Signalling 14: 211-218, 2002. • Haddad and Land, British Journal of Pharmacology 135: 520-536,
2002. This paper discussed the role MAPK enzymes in the regulation
cytokine-mediated inflammation/infection.
• Haddad, Biochemical Pharmacology 63: 305-320, 2002.
This paper extended the discussion on the role MAPK enzymes in the regulation cytokine-mediated inflammation/infection.
• Haddad and Fahlman, Biochemical and Biophysical Research
Communications 291: 1045-1051, 2002. • Haddad and Land, Antioxidants and Redox Signaling 4: 179-193.
• Safieh-Garabedian et al., Neuropharmacology 42: 864-872, 2002.
• Haddad, Biochemical and Biophysical Research Communications
293: 252-257, 2002. • Haddad, Cytokine 17: 301-310, 2002. • Haddad, European Cytokine Network 13: 250-260, 2002.
The complete list of publications could be found at the official website for MedLine (www.pubmed.gov).
In recapitulation, during the aforementioned period Dr. Haddad developed and
helped disseminate scientific knowledgeable expertise in molecular cell biology, while investigating the differential regulation of oxygen-sensitive transcription factors in the developing perinatal lung and the alveolar epithelium, with particular emphasis on redox
Biomedical Research Scientist and Educating Professor – 2009 Page | 9

John J. Haddad, PhD
variations and cytokine-dependent pathways associated with the respiratory distress (RDS) in pre-term infants.
The molecular physiology techniques invested included ELISA, EMSA, in situ
hybridization, histochemistry and immunofluorescence, Western/Northern/Southern blotting, RT-PCR, RNase protection assay, transfection/infection and plasmid genetic vector assays, DNA laddering, nick-end labeling and terminal transferase immunoreactivity and fluorescent microscopy, apoptosis-related techniques, and electrophysiology (bioelectric patch clamp Ussing chamber experiments) – all of which represent state-of-the-art techniques mastered and, in many occasions, developed by Dr. Haddad.
• General Background of Dr. John Haddad (2001 – Present):
Because of the outstanding biomedical research performed by Dr. Haddad, he was
awarded a prestigious postdoctoral fellowship funded by the National Institutes of Health (NIH) at the University of California, School of Medicine, San Francisco (UCSF).
Dr. Haddad immediately embarked, by leading a group of several scientists at
UCSF, on conceiving and elaborating biomedical research aimed at understanding oxidative stress, stroke (heart attack), anesthesiology medicine and brain injury.
Dr. Haddad’s conceived research was subdivided into sections, each of which theorized a
possible perspective in tackling the potential complications and clinical problems affiliated with oxidative stress and brain injury. These sections – Research Parts I-III – are thereafter briefly, but comprehensively, introduced.
♣
Biomedical Research Scientist and Educating Professor – 2009 Page | 10

John J. Haddad, PhD
Research Part I (National Institutes of Health): The goal of the proposed research is to demonstrate the role of intracellular Ca2+ ([Ca2+]i)
in the injury defense mechanism of neurons deprived of oxygen. This view of Ca2+ is contrary to the prevailing dogma that elevated [Ca2+]i is always deleterious in the context of hypoxic or ischemia stress. Previous work with hypoxia-tolerant neurons from freshwater turtles and neonatal rats has shown that an elevation in [Ca2+]i of 100-200 nM increases survival by suppressing glutamate excitotoxicity. In the work proposed here, it will be shown that the survival of hypoxia-sensitive neurons is increased when [Ca2+]i pre- and post-ischemia is moderately elevated within a “neuroprotective window.” It was also demonstrated that the neuroprotective effects of anesthetics occurs because the elevated [Ca2+]i produced by the anesthetic suppresses glutamate excitotoxicity. Finally, it’s shown that this window for Ca2+ protection closes with advancing age, making senile neurons more susceptible to Ca2+-induced death. Thus, these studies will investigate the mechanisms by which [Ca2+]i plays a more complex age and concentration-dependent role in protecting or injuring neurons than has been previously appreciated. To examine these roles for Ca2+, the following studies will be carried out:
Aim 1. To determine the role of Ca2+ as a neuroprotective signaling mechanism in
hypoxia-tolerant neurons.
It was proposed that hypoxia-tolerant neurons from turtle cerebrocortex and from neonatal rat hippocampus use moderately elevated [Ca2+]i as a signal for suppressing glutamate excitotoxicity during oxygen deprivation. It will be shown that:
1. Elevation of [Ca2+]i by 100-200 nM from baseline is essential for the long-
term survival of turtle and neonatal rat neurons exposed to hypoxia. Dr. Haddad next will test whether hippocampal neurons from mature rats are protected by similar elevations of [Ca2+]i before and following hypoxic/ischemic insults (oxygen and glucose deprivation in vitro).
2. Suppression of N-methyl-D-aspartate receptors (NMDARs) by Ca2+-sensitive
phosphatases is a key mechanism by which elevated [Ca2+]i protects hypoxia-tolerant neurons.
Aim 2. To determine the relationship between increased [Ca2+]i produced by
volatile anesthetics and their neuroprotective qualities. It’s proposed that the elevation in [Ca2+]i produced by a volatile anesthetic (isoflurane)
prevents post-ischemic cell death in organotypic hippocampal slice cultures. It will be shown that:
1. The 30-50 nM increase in [Ca2+]i produced by clinically relevant
concentrations of isoflurane (1-2% vapor volume) is necessary for neuronal protection.
Biomedical Research Scientist and Educating Professor – 2009 Page | 11

John J. Haddad, PhD
2. The mechanism of isoflurane’s protection is suppression of NMDA receptor activity and excitotoxicity, caused by an elevation of [Ca2+]i into a neuroprotective range.
Aim 3. To determine whether altered Ca2+ homeostasis in aged neurons makes
them more vulnerable to ischemia.
Intracellular [Ca2+]i homeostasis may be impaired in aged/senile neurons, and elevated [Ca2+]i may contribute to cell loss in the aged brain. It will be shown that:
1. Aging (from neonatal to 2 year-old rats) alters [Ca2+]i homeostasis in
hippocampal neurons during and following in vitro ischemia. 2. Anesthetics, because they elevate [Ca2+]i and alter [Ca2+]i homeostasis during
or post ischemia, negatively influence the survival of senile neurons. What is the biomedical and clinical significance of the pioneering work of Dr.
Haddad? Analysis of how this could be projected into the national scientific interest of the United States.
Background: Brain damage from asphyxia or ischemia is an important cause of
human mortality and neurological impairment, conditions for which no adequate therapy exists. While many details remain unclear, a consensus view is that increases in [Ca2+]i cause both acute and delayed or programmed cell death. From this view have emerged a variety of therapies directed at preventing one of the many sources of [Ca2+]i increase, including glutamate receptors, voltage-gated Ca2+ channels, and intracellular stores. None of these has produced a breakthrough therapeutic victory over brain ischemia. Dr. Haddad proposes that part of this frustration is the result of failure to understand the complex, multifaceted role that [Ca2+]i homeostasis plays in ischemic or post-ischemic neurons. He will present arguments and preliminary data suggesting that elevations in [Ca2+]i serve a protective as well as an injurious role, depending on timing and concentration, and that elevations in [Ca2+]i are normally used by ischemic or post-ischemic neurons as a signal for a protective suppression of neuronal excitability and survival. Therapies that merely block certain avenues of [Ca2+]i increase may inadvertently deprive the neurons of a signaling modality needed to initiate protective events. More effective therapies might emerge from this alternative view.
The dual role of Ca2+ as a mediator of cell death and a neuroprotective signaling
mechanism: A window for neuroprotection? The vast majority of information on the role of Ca2+ in hypoxic/ischemic brain injury is related to the role that massive increases in [Ca2+]i play in acute neuronal necrosis. Ca2+ influx, particularly via the N-methyl-D-aspartate receptor (NMDAR), is specifically linked to neural injury. Delayed or apoptotic cell death is also very important in ischemic brain injury, but in apoptosis the role of fluctuations in [Ca2+]i is much less clear. Following ischemia, [Ca2+]i may recover towards previous baseline or even fall below baseline for a period of time before increasing again as a terminal part of the apoptosis cascade. Delayed elevation in [Ca2+]i may play a role in the activation of key mediators of apoptosis such as endonucleases. However, apoptosis of neurons (caused for example by growth factor
Biomedical Research Scientist and Educating Professor – 2009 Page | 12

John J. Haddad, PhD
deprivation) can be reduced by increasing [Ca2+]i by activating voltage-gated Ca2+ channels, inhibiting Ca2+ sequestration and even by low concentrations of NMDA.
These observations suggest that therapies directed strictly at preventing [Ca2+]I
increases throughout the peri-ischemic period might backfire. Indeed, toxicity from NMDAR antagonists has been a frustrating problem in pre-clinical and clinical trials, a problem perhaps related to too little Ca2+ at critical times. A variety of evidence suggests that the survival of neurons may depend on a level of [Ca2+]i that is state and time dependent: (1) a low [Ca2+]i where neurons are at risk for apoptosis if not supported by growth factors; (2) an intermediate level where some elevation in [Ca2+]i is protective and takes the place of other support; and (3) grossly elevated [Ca2+]i that is cytotoxic. Dr. Haddad suggests that survival of neurons might be enhanced if we knew more about the relationship between [Ca2+]i and cell survival at different times in the peri-ischemic period. The proposed effects of different windows of [Ca2+]i on survival are shown in Fig. 1.
Fig. 1. The Ca2+ windows concept: Hypothetical
relationship between [Ca2+]i and fate of anoxic neurons. In typical mammalian neurons, anoxia causes a rapid increase of [Ca2+]i to toxic levels, which may result in acute, or after some recovery and rebound, delayed cell death. In anoxia tolerant neurons, [Ca2+]i rises modestly, to levels causing a protective arrest of excitability. In mammalian neurons treated with an NMDA antagonist, the initial catastrophic rise in [Ca2+]i is avoided, but the “protective arrest” zone is never entered, and ultimately Ca2+ starvation and apoptosis occur.
Intr
acel
lula
r [C
a2+]
Time during anoxia
Normal
Low
Protective
ToxicNecrosis
Survival
Apoptosis
typical mammalian neuron
Anoxia tolerant neuron
mammalian neuronwith NMDA antagonist
What remains unknown is what [Ca2+]i is optimal before and after ischemia and if cell survival can be influenced by keeping [Ca2+]i within a protective zone during these periods. Increasing [Ca2+]i before or after ischemia to promote neuronal survival may seem paradoxical, but the concentrations that inhibit apoptosis (180-240 nM in sensory neurons for example), are much lower than those associated with ischemic Ca2+ overload. Interestingly, [Ca2+]i in anoxia-tolerant turtle neurons increases from 100 to about 200 nM during several hours to several weeks of anoxia.
Hypoxia-tolerant neurons may use Ca2+ as a neuroprotective signal. Hypoxia-tolerant
neurons are valuable models to test the Ca2+ window theory, because these neurons appear to use elevated [Ca2+]i as a signaling mechanism to survive anoxia. The best-studied hypoxia-tolerant neurons are CA1 neurons from the neonatal rat hippocampus and cortical neurons from freshwater turtles. The laboratory where Dr. Haddad has been working in since 2001 has been at the forefront of understanding their survival mechanisms.
Turtles are the most anoxia-tolerant vertebrates, surviving 4-5 months of anoxia during
winter dormancy. During anoxia an increase in [Ca2+]i occurs in turtle cortical neurons that causes a substantial decrease in whole cell ionic conductance that is probably critical for reducing energy consumption by over 90%. The increase in [Ca2+]i during hypoxia in both turtle
Biomedical Research Scientist and Educating Professor – 2009 Page | 13

John J. Haddad, PhD
and neonatal mammal neurons is correlated with a suppression of NMDARs. Elevated [Ca2+]i suppresses NMDARs in these cells through several mechanisms, including dephosphorylation and depolymerization of actin. Some of the controls, which regulate NMDARs in both turtle at neonatal rat neurons during hypoxia, are shown in Fig. 2, but in large measure these controls have not been elucidated in detail.
The NMDA receptor is phosphorylated by PKC, PKA, and tyrosine kinases. The
proposed studies will contribute to understanding the significance of phosphorylation control of NMDARs during and following ischemia. During hypoxia in rat brain slices, NR2A/2B subunits are dephosphorylated consistent with receptor suppression, but conversely PKC may be activated in post-ischemic states in intact animals potentially contributing to injury.
Therefore, studies examining the role of NMDAR phosphorylation in anoxia-
tolerant neurons will help clarify the significance of these processes in injury and disease and help guide new approaches to neuroprotection.
(ActiveNMDARs)
(InactiveNMDARs)
P P
Ca2+ Ca2+
Actin Microfilaments
NR1 or NR2subunits
Ca2+ Ca 2+ -Calmodulin
ActivatedPhosphatases
Protein Kinases
RegulatoryProtein
α-Actinin 2
Calcium-Calmodulin
Ca2+ Ca2+
Ca2+ Ca2+Ca 2+ Ca2+
glutamateWith O 2
Anoxia
Fig. 2. Some of the known mechanisms for suppression of NMDARs in anoxia tolerant neurons. With O2 (top), polymerized actin, maintained in this state by low [Ca2+]i, enhances NMDA receptor activity via a regulatory protein. Several residues on the cytosolic loops are phosphorylated. In turtle neurons during anoxia, increased [Ca2+]i causes dephosphorylation of these sites via calmodulin and phosphatases 1, 2A and 2B. In neonatal CA1 neurons, elevated [Ca2+]i depolymerizes actin and thereby reduces NMDAR activity.
Role of [Ca2+]i in neuroprotection with anesthetics: Clinical significance. General
anesthetics are neuroprotective in various in vivo and in vitro models of cerebral ischemia, but the degree of protection and the mechanisms producing it are unclear and continue to be debated. The prevailing view is that suppression of brain metabolic rate by anesthetics is important for protection, although depression of metabolic rate by itself does not tightly predict benefit. More recently, it has been proposed that volatile anesthetics (VA’s) reduce glutamate excitotoxicity. VA’s such as isoflurane reduce glutamate accumulation, glutamate receptor mediated Ca2+ influx, and the activity of NMDARs. The mechanisms for these actions are have not been clarified.
Biomedical Research Scientist and Educating Professor – 2009 Page | 14

John J. Haddad, PhD
Dr. Haddad and colleagues have found that volatile anesthetics increase [Ca2+]i by 30-70 nM. Although he originally proposed that this increase in [Ca2+]i is a key factor in producing suppression of synaptic transmission (i.e. the anesthetic state), Dr. Haddad believes the anesthetic might also have the effect of reducing glutamate release and glutamate toxicity in the context of hypoxia or ischemia (for example by suppressing the NMDA receptor by the calcium-dependent mechanisms in Fig. 2). This idea remains unstudied. VA’s are therefore useful tools to test the hypothesis that elevations in [Ca2+]i have neuroprotective effects.
Whether volatile anesthetics prevent delayed post-ischemic neuron death is an
important unanswered question. In a recent report, isoflurane was found to provide only short-term protection; when isoflurane treated animals were examined two weeks after focal cerebral ischemia the volume of brain infarction was the same as controls. In contrast, it was found in preliminary studies that isoflurane is highly effective at preventing delayed cell death in an organotypic slice culture model of ischemia. Because this benefit does not occur in intact animals, our model offers the ability to understand the intrinsic neuroprotective qualities of anesthetics, and will provide a rationale for discovering what occurs in intact animals to limit the protection.
The significance of these studies is that they contribute to the understanding of the
basis and limitations for volatile anesthetic neuroprotection in clinical care. Also, understanding how anesthetics produce neuroprotection will, therefore, identify new targets for neuroprotection.
Aging and Ca2+ dishomeostasis. Understanding and preventing the functional
decline of the aging brain has huge significance for the American society. One prominent theory explaining the functional decline and death of senile neurons is that with aging, neurons lose the ability to tightly regulate [Ca2+]i and that the Ca2+ dishomeostasis suppresses normal excitability and accelerates cell loss. Some studies have indeed indicated that [Ca2+]i rises gradually with aging, but other studies report the opposite, illustrating that even basic information on aged neurons such as [Ca2+]i levels remains controversial. The best evidence suggests that [Ca2+]i rises with aging, and that this rise is associated with a decline of pumps and channels responsible for keeping internal Ca2+ low.
Several different mechanisms, including a progressive decline in the ability of oxidatively
damaged calmodulin to activate the plasma membrane Ca2+-ATPase, have been proposed to account for this elevation in [Ca2+]i. Elevated [Ca2+]i appears to suppress synaptic function and may explain decreased brain function in the aged. It was found that the relative synaptic depression in hippocampal slices from 2-year old rats could be reversed by loading CA1 neurons with a Ca2+ chelator (EGTA-AM), which restored [Ca2+]i to youthful levels. An increased [Ca2+]i in aged neurons has important implications for protecting the brain with the strategies discussed above. For example, an age-related decrement in Ca2+ homeostasis may explain why the aged brain is more sensitive to ischemia. Surprisingly this has not been studied directly.
In addition, because volatile anesthetics increase [Ca2+]i in brain neurons, a reasonable
question to ask is whether VA’s could be toxic to aged neurons under certain circumstances.
Biomedical Research Scientist and Educating Professor – 2009 Page | 15

John J. Haddad, PhD
Further, one may ask whether the increased frequency of postoperative cognitive deficits in the elderly are related to anesthetics increasing [Ca2+]I in senile neurons to toxic concentrations. Fig. 3 illustrates the concept that [Ca2+]i in aged neurons might be made to exceed the survivable threshold by the presence of a [Ca2+]i-elevating anesthetic.
Intr
acel
lula
r [C
a2+]
Time
Normal
Low
Suppressive
ToxicSenile neuron
Young neuron
Anestheticapplication
Survival
Death
Anesthetics that cause the largest increases in [Ca2+]i may produce the greatest problems with Ca2+ homeostasis in aged/senile neurons. Relatively low concentrations of nitrous oxide increase [Ca2+]i in hippocampal neurons compared to volatile anesthetics. Nitrous oxide (N2O) also impairs post-hypoxia recovery of synaptic function in hippocampal slices. Selective neural necrosis after N2O administration has been observed in cingulate cortex and striatum of rats. It is possible that this toxicity is due to altered Ca2+ homeostasis that impairs the ability of neurons to clear Ca2+ from the cytosol in the usual manner, a problem exacerbated by hypoxia or ischemia. The situation may be further complicated by old age, when normal Ca2+ homeostasis mechanisms are diminished. Exploring the cellular effects of anesthetics on aged neurons is critical to understanding the basis of increased cognitive deficits following anesthesia in the elderly in the United States.
In summary, disregulation of [Ca2+]i clearly plays a key role in the pathogenesis of
brain hypoxia/ischemia, but its role may be considerably more complex than merely transducing injury in a linear fashion. Strong circumstantial evidence shows that Ca2+ can play a protective role, possibly by regulating ion channels that govern excitability and excitotoxicity. Hypoxia-tolerant neurons appear to use Ca2+ as such a signal, but whether these same rules apply to hypoxia-sensitive neurons is not known. The timing of such signaling may be crucial, since Ca2+ disregulation may be context-specific, with [Ca2+]i playing different roles before, during, and after ischemic stress, but again major gaps remain in our knowledge. The changing role of [Ca2+]i during development and during senility also is little known, and its study may suggest new ways to protect the aging brain from additional cell loss. Finally, insights provided by proposed studies will identify mechanisms by which anesthetics protect neurons and ways in which the neuroprotective efficacy of anesthetic management could be maximized.
The main findings thus far suggest that moderate increases in [Ca2+]i serve as a
neuroprotective signal that triggers endogenous neuroprotective pathways in both mammalian and reptilian neurons, and may also explain the neuroprotective properties of
Fig. 3. Hypothetical relationship between [Ca2+]i and neuron viability in young and senile neurons exposed to a volatile anesthetic. In young neurons, the anesthetic causes [Ca2+]i to rise into the zone associated with protective suppression of excitation. In neurons from senile animals, the basal [Ca2+]I is already borderline for normal function, and in some cases in the suppressive range. Exposure to an anesthetic then increases [Ca2+]i into the toxic range.
Biomedical Research Scientist and Educating Professor – 2009 Page | 16
John J. Haddad, PhD
some anesthetics. This view is contrary to the prevailing view that elevation in [Ca2+]i in the context of hypoxic stress is always deleterious.
Survival of anoxia tolerant turtle neurons may involve Ca2+ as a protective signal.
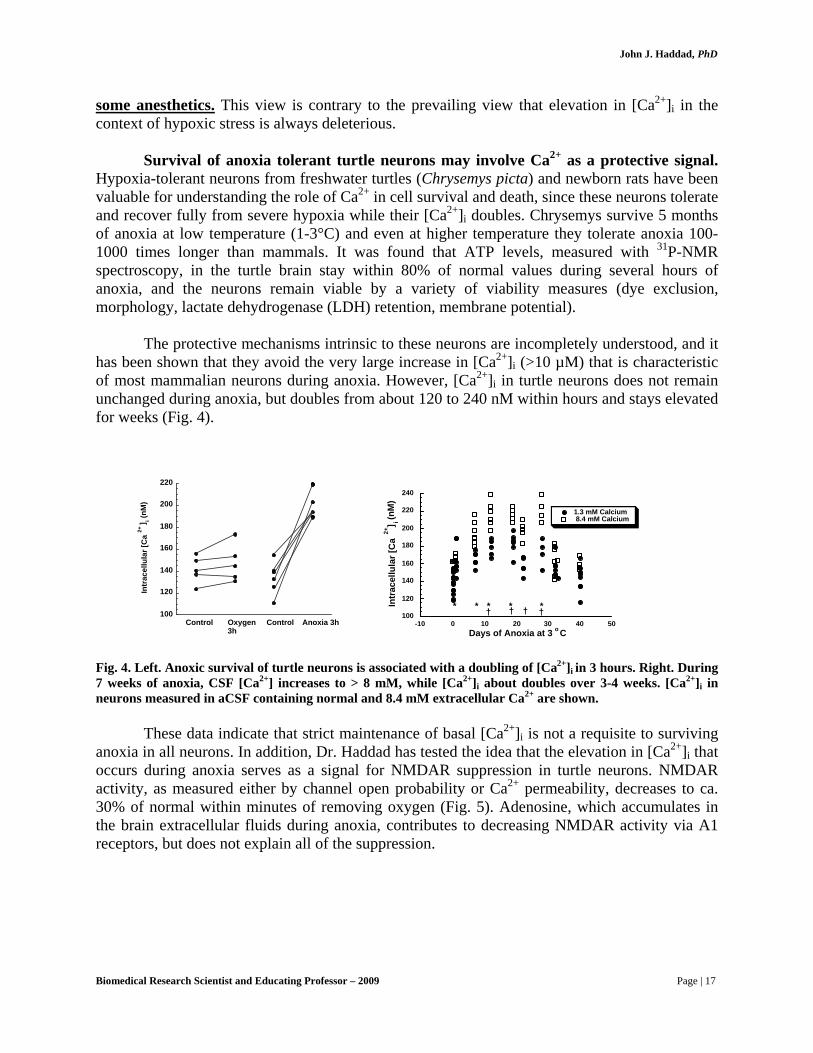
Hypoxia-tolerant neurons from freshwater turtles (Chrysemys picta) and newborn rats have been valuable for understanding the role of Ca2+ in cell survival and death, since these neurons tolerate and recover fully from severe hypoxia while their [Ca2+]i doubles. Chrysemys survive 5 months of anoxia at low temperature (1-3°C) and even at higher temperature they tolerate anoxia 100-1000 times longer than mammals. It was found that ATP levels, measured with 31P-NMR spectroscopy, in the turtle brain stay within 80% of normal values during several hours of anoxia, and the neurons remain viable by a variety of viability measures (dye exclusion, morphology, lactate dehydrogenase (LDH) retention, membrane potential).
The protective mechanisms intrinsic to these neurons are incompletely understood, and it
has been shown that they avoid the very large increase in [Ca2+]i (>10 µM) that is characteristic of most mammalian neurons during anoxia. However, [Ca2+]i in turtle neurons does not remain unchanged during anoxia, but doubles from about 120 to 240 nM within hours and stays elevated for weeks (Fig. 4).
100
120
140
160
180
200
220
Intr
acel
lula
r [C
a2+
] i (nM
)
Control Oxygen3h
Control Anoxia 3h100
120
140
160
180
200
220
240
-10 0 10 20 30 40 50
1.3 mM Calcium 8.4 mM Calcium
Intr
acel
lula
r [C
a2+
] i (nM
)
Days of Anoxia at 3 o C
* * * * *† † † †
Fig. 4. Left. Anoxic survival of turtle neurons is associated with a doubling of [Ca2+]i in 3 hours. Right. During 7 weeks of anoxia, CSF [Ca2+] increases to > 8 mM, while [Ca2+]i about doubles over 3-4 weeks. [Ca2+]i in neurons measured in aCSF containing normal and 8.4 mM extracellular Ca2+ are shown.
These data indicate that strict maintenance of basal [Ca2+]i is not a requisite to surviving anoxia in all neurons. In addition, Dr. Haddad has tested the idea that the elevation in [Ca2+]i that occurs during anoxia serves as a signal for NMDAR suppression in turtle neurons. NMDAR activity, as measured either by channel open probability or Ca2+ permeability, decreases to ca. 30% of normal within minutes of removing oxygen (Fig. 5). Adenosine, which accumulates in the brain extracellular fluids during anoxia, contributes to decreasing NMDAR activity via A1 receptors, but does not explain all of the suppression.
Biomedical Research Scientist and Educating Professor – 2009 Page | 17

John J. Haddad, PhD
Control (normoxia)
Anoxia* **
**P o p e n
.02
.06
.08
.10
0
A
0 15 30 45 60 75 900.5
1
1.5
2
2.5
3
0 100 200 300 400 500
ntra
cellu
larC
alci
um
Time (sec)
NMDA NMDA
95% oxygen Anoxia
B
Fig. 5. Decrease in NMDAR activity in anoxic turtle neurons. (A) Decrease in mean NMDAR open probability (± SEM) in cell-attached patches during 90 min of anoxia. Patch pipettes contained 10 µM NMDA. No change in receptor current amplitude was seen during anoxia. * Significant decrease compared to normoxic control. (B) Example of decrease in NMDAR activity in isolated neurons during anoxia (perfusate switch from 95% O2/5% CO2 to 95% N2/5% CO2). NMDA (100 µM) was perfusion applied for 10 sec periods (horizontal bars), with immediate washout.
Involvement of Ca2+ and phosphorylation in the control of turtle NMDARs during
anoxia. The suppression of NMDAR activity in turtle neurons during anoxia is linearly related to the increase in basal [Ca2+]i (Fig. 6), but whether this is a cause-and-effect relationship is not yet known. The Ca2+ binding protein calmodulin is involved in transducing the elevated signal into receptor suppression because the calmodulin inhibitor calmidazolium reduces the capacity of anoxia to suppress NMDAR function (Fig. 6 right).
0
50
100
150
200
250
300
100 120 140 160 180 200 220
NormoxiaAnoxia
NM
DA
rece
ptor
act
ivity
(NM
DA
C
a2+, n
M)
Basal [Ca 2+ ]i (nM)
Δ
0
0.2
0.4
0.6
0.8
1
1.2
Control Calmidazolium
NormoxiaAnoxia
Rel
ativ
e N
MD
AR
Act
ivity
7
7
78
p=0.04
Fig. 6. Evidence that [Ca2+]i plays a role in regulating the suppression of NMDAR function during anoxia. LEFT. Increase in [Ca2+]i correlates with decrease in NMDAR activity in oxic and anoxic neurons. RIGHT. The calmodulin inhibitor calmidazolium (1 µM) substantially reduces the suppression of NMDARs by anoxia.
One possible link between a rise in [Ca2+]i and receptor suppression is the activation of
Ca2+-dependent phosphatases (Fig. 2). Phosphatase activation during anoxia probably plays a role, since several phosphatase inhibitors prevent NMDAR suppression (Fig. 7).
Biomedical Research Scientist and Educating Professor – 2009 Page | 18

John J. Haddad, PhD
0
. 0 4
. 0 8
. 1 2
. 1 6
1 5 3 0 4 5 6 0 9 0 1 0 50 7 5
A nox i a + Ca l y c u l i n A
A nox i a* * *
* *
Po p
T im e ( m i n)
AB
0.8
1
1.2
1.4
1.6
1.8
0 100 200 300 400 500 600 700 800
Intr
acel
lula
r Cal
cium
(345
/380
fluo
resc
ence
)
95% O2 Anoxia + 120 nM okadaic acid
NMDA NMDA
Time (sec)
Fig.7. Role of phosphatases in NMDAR suppression. A. Inhibition of phosphatases with the non-specific inhibitor okadaic acid (100 nM) prevents NMDAR silencing in a dissociated neuron (compare with Fig. 5 B). B. Decreases in NMDAR open probability during anoxia are prevented with calyculin (1 µM), an inhibitor of phosphatase 1/2A. N=5-7 each group, * denotes significant difference from control, Dunnett’s test, p<0.05.
The above data suggest that changes in NMDAR phosphorylation state are one
factor in anoxia-induced receptor suppression. With the use of antibodies to phosphotyrosine residues on NMDARs (PKA sites), preliminary data obtained has shown that 3 h anoxia is associated with de-phosphorylation of NMDARs (Fig. 8). These data provide a rationale for efforts to determine the pathways involved in NMDAR phosphorylation control during anoxia.
Fig. 8. Western blots of phosphorylated NMDA R1 subunits (PKA sites) in turtle brain homogenates. Note reduction in staining in homogenates obtained from turtles after 3 h of anoxia in vivo. Abundance of NR1 subunits does not change during 3 h anoxia.
It was reported that the suppression of NMDARs is critical to the survival of anoxic turtle
neurons. When stimulating cAMP with forskolin prevents receptor suppression, the neurons succumb to anoxia (Fig. 9). (NMDA suppression in anoxia is also critical for the survival of neonatal mammalian neurons during hypoxia, as shown below).
Biomedical Research Scientist and Educating Professor – 2009 Page | 19

John J. Haddad, PhD
Fig. 9. Anoxia prevents NMDA neurotoxicity (200 µM NMDA for 5 min exposure, 6 h recovery prior to fixation) in turtle cortical neurons. In the anoxia + NMDA group, neurons were anoxic for 2 h before NMDA exposure. Increasing NMDAR activity with forskolin increased NMDA toxicity. Exposing forskolin treated slices with MK-801 (10 µM), a non-competitive NMDAR antagonist, reveals that survival during anoxia is dependent on inactivation of NMDARs. N=6-8 for each group, * denotes significant difference from control, Dunnett’s test, p<0.05.
0
20
40
60
80
100
NormoxiaAnoxia
% S
urvi
ving
Neu
rons
Control NMDA Anoxia+ NMDA
Anoxia +Forskolin
Anoxia +Forskolin+ MK-801
6
6
7
5
5
*
*
Neonatal CA1 neurons also use Ca2+ as a protective signal during anoxia. To
determine whether similar endogenous neuroprotective mechanisms exist in mammals, Dr. Haddad has examined neurons from neonatal rats, which tolerate anoxia 10 times longer than mature rats, and have found that survival of anoxia in hippocampal neurons appears related to small increases in [Ca2+]i that serve to suppress NMDAR function.
During anoxia, neonatal CA1 neurons are protected from experiencing the large increases
in [Ca2+]i that characterize mature neurons. In part this is due to less accumulation of glutamate, but there is also compelling evidence that NMDA receptors are suppressed as well. Although neonatal (P2-7) neurons are more hypoxia-tolerant, glutamate or NMDA cause greater increase in [Ca2+]i than do P30 neurons, indicating that P2-7 neurons have higher permeability of NMDA receptors. Thus to be hypoxia-tolerant, these receptors must be suppressed by hypoxia.
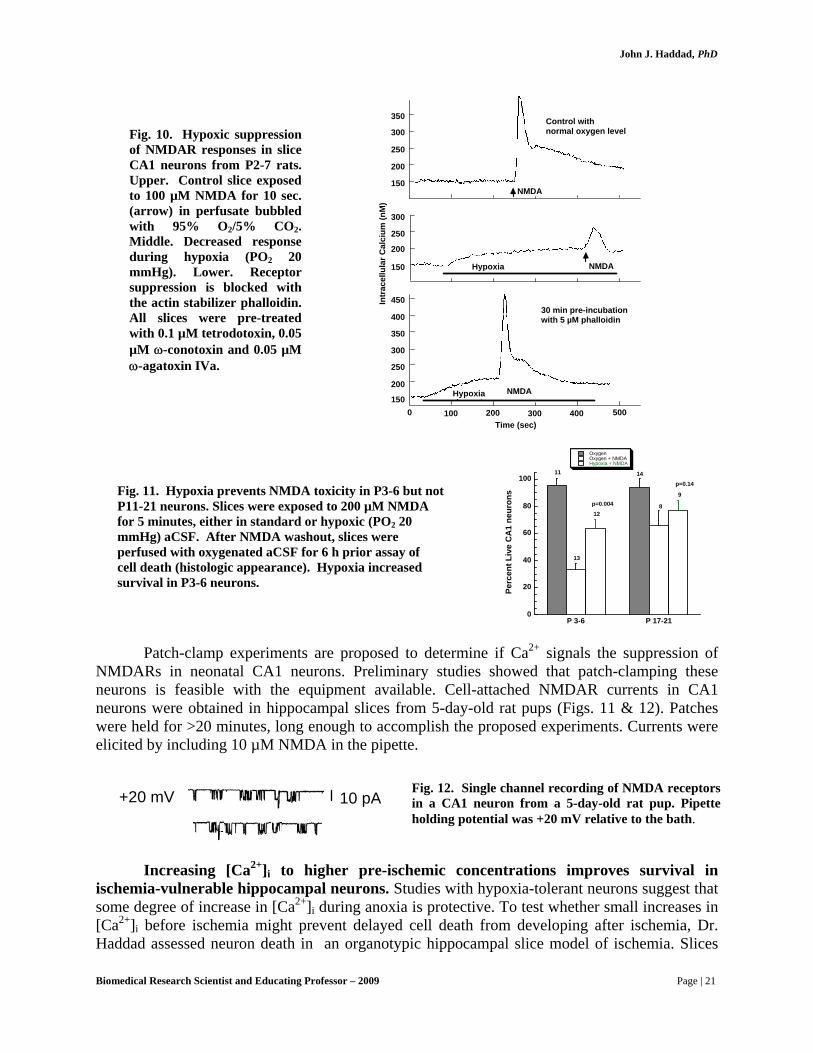
Evidence for this suppression is shown in Fig. 9. In Fig. 10, this suppression is shown to
have survival value. How this occurs is of obvious developmental and neuroprotective interest, particularly in view of the fact that the subtypes of NMDA receptor expressed in neonates (relative abundance of NR2B compared to NR1 and NR2A or 2C) are probably more Ca2+ permeable, creating the potential for greater Ca2+ dishomeostasis during ischemia in the immature brain.
Biomedical Research Scientist and Educating Professor – 2009 Page | 20
John J. Haddad, PhD
150
200
250
350
150
200
250
300
Hypoxia NMDA
NMDA
300
150
200
250
300
350
400
450
Hypoxia NMDA
30 min pre-incubationwith 5 µM phalloidin
Time (sec)
Control withnormal oxygen level
0
Intr
acel
lula
r Cal
cium
(nM
)
100 200 300 400 500
Fig. 10. Hypoxic suppression of NMDAR responses in slice CA1 neurons from P2-7 rats. Upper. Control slice exposed to 100 µM NMDA for 10 sec. (arrow) in perfusate bubbled with 95% O2/5% CO2. Middle. Decreased response during hypoxia (PO2 20 mmHg). Lower. Receptor suppression is blocked with the actin stabilizer phalloidin. All slices were pre-treated with 0.1 µM tetrodotoxin, 0.05 µM ω-conotoxin and 0.05 µM ω-agatoxin IVa.
0
20
40
60
80
100
P 3-6 P 17-21
OxygenOxygen + NMDAHypoxia + NMDA
Perc
ent L
ive
CA
1 ne
uron
s
11
13
12
14
8
9
p=0.14
p=0.004
Fig. 11. Hypoxia prevents NMDA toxicity in P3-6 but not P11-21 neurons. Slices were exposed to 200 µM NMDA for 5 minutes, either in standard or hypoxic (PO2 20 mmHg) aCSF. After NMDA washout, slices were perfused with oxygenated aCSF for 6 h prior assay of cell death (histologic appearance). Hypoxia increased survival in P3-6 neurons.
Patch-clamp experiments are proposed to determine if Ca2+ signals the suppression of NMDARs in neonatal CA1 neurons. Preliminary studies showed that patch-clamping these neurons is feasible with the equipment available. Cell-attached NMDAR currents in CA1 neurons were obtained in hippocampal slices from 5-day-old rat pups (Figs. 11 & 12). Patches were held for >20 minutes, long enough to accomplish the proposed experiments. Currents were elicited by including 10 µM NMDA in the pipette.
Fig. 12. Single channel recording of NMDA receptors in a CA1 neuron from a 5-day-old rat pup. Pipette holding potential was +20 mV relative to the bath.
10 pA+20 mV
Increasing [Ca2+]i to higher pre-ischemic concentrations improves survival in ischemia-vulnerable hippocampal neurons. Studies with hypoxia-tolerant neurons suggest that some degree of increase in [Ca2+]i during anoxia is protective. To test whether small increases in [Ca2+]i before ischemia might prevent delayed cell death from developing after ischemia, Dr. Haddad assessed neuron death in an organotypic hippocampal slice model of ischemia. Slices
Biomedical Research Scientist and Educating Professor – 2009 Page | 21

John J. Haddad, PhD
for culture were prepared from 7-10 day old Sprague-Dawley rats. It was found that increasing [Ca2+]i with the Ca2+ ionophore A23187 (10-1000 nM concentrations ) prevented delayed post ischemic cell death (Fig. 13). 10-1000 nM concentrations of A23187 increase [Ca2+]i by 50-150 nM, while 1 nM has no effect. The temporal pattern of [Ca2+]i changes during and after the in vitro ischemia remains to be studied.
-40-20
0
2040
6080
100
0 2 3 7
0 nM10 nM
100 nM1000 nM
% C
ell D
eath
Time post ischemia (days)
n=5 each data point*= p<0.05 compared to control *
*
Effects of a Ca 2+ ionophore(A-23187) on post-ischemic survival CA 1 region
*
-20
0
20
40
60
80
0 2 3 7
A23187: CA3
0 nM10 nM100 nM1000 nM
% c
ell d
eath
Time post ischemia (days)
* *
*
n=5 each data point* = p<0.05 compared to control
Fig. 13. The calcium ionophore A23187 prevented delayed cell death in hippocampal neurons subjected to 45 min in vitro ischemia (combined oxygen/glucose deprivation at 37 °C).
Volatile anesthetics prevent both acute and delayed cell loss in the hippocampus. Dr.
Haddad has found that clinical concentrations of anesthetics increase [Ca2+]i in CA1 neurons in hippocampal slices by 20-100 nM (mean 30 nM). Therefore, whether these changes might be sufficient to suppress Ca2+-influx pathways, (NMDARs), similar to that occurring in hypoxia-tolerant neurons was subsequently tested. It was shown that during anoxia, isoflurane decreases glutamate release, limits the increase of [Ca2+]i and increases survival, selectively reducing delayed cell death in CA1 but not dentate neurons.
Two experimental models were developed by Dr. Haddad to study these proposed
mechanisms for VA neuroprotection: 1) An “acute” hippocampal slice model designed to examine early cell death (within 6-8 hours of in vitro ischemia); and 2) Hippocampal slices maintained in culture for up to a month, to enable measurement of delayed cell death for up to 2 weeks after in vitro ischemia.
Acute slices: Isoflurane prevents acute cell death in hippocampal slices exposed to 20
min of anoxia/glucose deprivation, similar to protection afforded by a barbiturate and mild hypothermia. Protection seems related to inhibition of glutamate receptors, since isoflurane attenuates toxicity from exogenous glutamate, but the specific role of the NMDA receptor has not been studied (Fig. 14).
Biomedical Research Scientist and Educating Professor – 2009 Page | 22

John J. Haddad, PhD
0
20
40
60
80
100
% re
mai
ning
CA
1 ne
uron
s
12
10 10
*
No glutamate1 mM glutamate
30
Sham treatment (control)10 µM MK-801Isoflurane 2%
Fig 14. LEFT. Transverse sections of rat hippocampal slices stained with hematoxylin/eosin showing neuroprotection with several anesthetics and comparable protection with hypothermia. RIGHT. Glutamate toxicity is attenuated by isoflurane. Slices were incubated in glutamate (1 mM) for 20 minutes, with or without the non-competitive NMDA antagonist MK-801 (10 µM), or 2% isoflurane. * Denotes p<0.01 compared to no glutamate group.
Cultured slices: An organotypic slice culture model was used to show that isoflurane
prevents delayed cell death for up to a week (see Fig. 15). The mechanism for this protection will be determined in the proposed studies.
-20
0
20
40
60
80
100
0 1 2 3 4 5 6 7
CA1 region
Ischemia (n=15)Control (n=3)Isoflurane (n=13)MK-801 (n=5)
% C
ell D
eath
Days post ischemia
*
* *
-40
-20
0
20
40
60
80
0 1 2 3 4 5 6 7
Dentate gyrus
Ischemia (n=12)Control (n=3)Isoflurane (n=13)MK-801 (n=5)
% C
ell D
eath
Days post ischemia
***
Fig. 15. Isoflurane (1%) prevents delayed cell death in organotypic hippocampal slice cultures for at least 7 days after 45 min combined O2/glucose deprivation (simulated ischemia). At left, propidium iodide nuclear staining showing dead neurons in different cell body regions. At right, percentage neuron death in CA1 and dentate for controls, isoflurane/ischemia and 10 µM MK-801/ischemia groups. Isoflurane is as protective as the non-competitive NMDA antagonist MK-801.
Biomedical Research Scientist and Educating Professor – 2009 Page | 23

John J. Haddad, PhD
In collaboration, Dr. Haddad developed methods to characterize the nature of the cell death pathways. Fig. 16 shows evidence for increased caspase-3 activity in rat brain 3 days after a hypoxic/ischemic insult. These methods will be used to characterize cell death in the organotypic slice culture model.
Fig. 16. Demonstration of caspase-3 immunoreactivity in transverse cryostat sections of rat pup cortex .
Aging and nitrous oxide anesthesia produce significant perturbations in Ca2+
homeostasis. Dr. Haddad found that aging and anesthesia significantly alter [Ca2+]i homeostasis in CA1 neurons. In the case of aging, [Ca2+]i is significantly higher during both basal conditions and during anoxia in two year old neurons (Fig. 17). Nitrous oxide, the most commonly used vapor anesthetic in the USA and the world, also significantly changes [Ca2+]i homeostasis, with even sub-anesthetic levels having effects larger than other vapor anesthetics (Fig. 17, right). These observations provide the rationale for asking the question of whether nitrous oxide has deleterious effects in aged neurons, and whether such injury is worse when hypoxia/ischemia is involved.
0
100
200
300
400
500
600
700
800
Basal P30 Anoxia P30 Basal P800 Anoxia P800
Intr
acel
lula
r [C
a2+
] i (nM
)
Treatment Group
n=10
n=8
n=3
n=3
Fig. 17. Basal [Ca2+]i and [Ca2+]i after anoxia are greater in 2 year old (P800) than juvenile (P30) CA1 neurons isolated from hippocampal slices. At right, nitrous oxide reversibly increases [Ca2+]i in a P20 hippocampal CA1 neuron.
In summary, the biological and clinical significance of the aforementioned research, which could bear and accrue enormous national interest in biological sciences and medicine, is summarized below:
Biomedical Research Scientist and Educating Professor – 2009 Page | 24
John J. Haddad, PhD
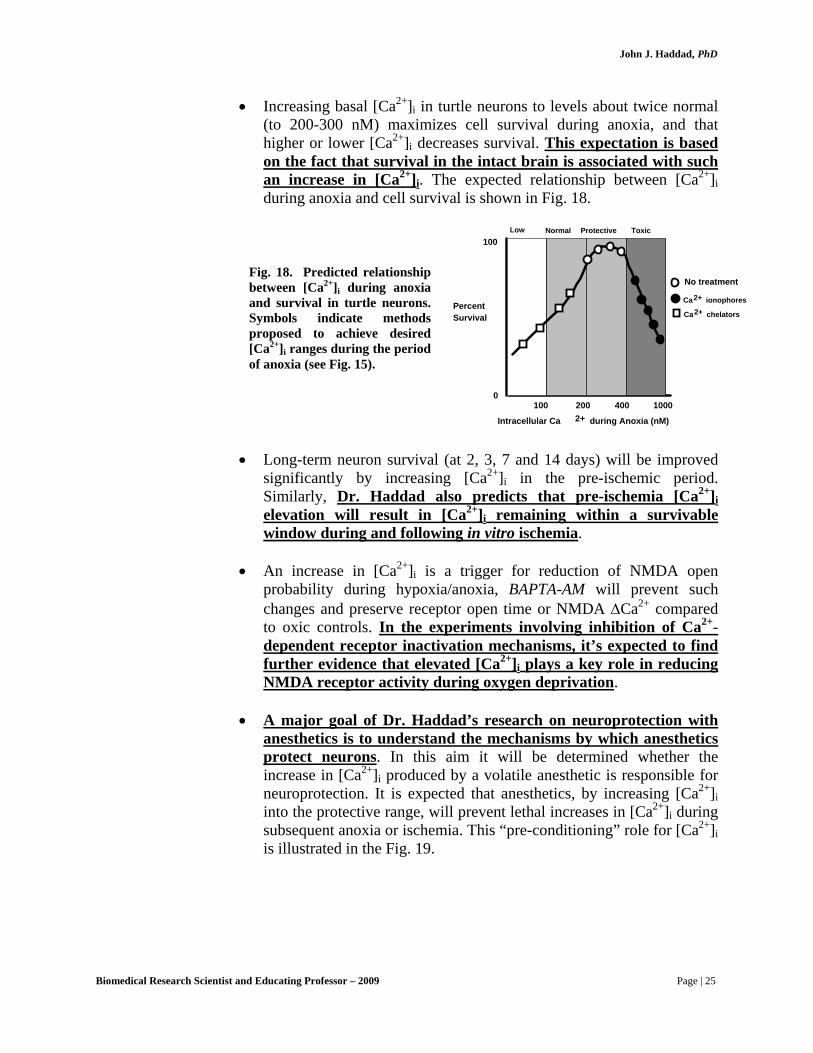
• Increasing basal [Ca2+]i in turtle neurons to levels about twice normal (to 200-300 nM) maximizes cell survival during anoxia, and that higher or lower [Ca2+]i decreases survival. This expectation is based on the fact that survival in the intact brain is associated with such an increase in [Ca2+]i. The expected relationship between [Ca2+]i during anoxia and cell survival is shown in Fig. 18.
Low Normal Protective Toxic
100 200 400 1000
Intracellular Ca 2+ during Anoxia (nM)
PercentSurvival
100
0
Ca2+ ionophores
Ca2+ chelators
No treatment
Fig. 18. Predicted relationship between [Ca2+]i during anoxia and survival in turtle neurons. Symbols indicate methods proposed to achieve desired [Ca2+]i ranges during the period of anoxia (see Fig. 15).
• Long-term neuron survival (at 2, 3, 7 and 14 days) will be improved
significantly by increasing [Ca2+]i in the pre-ischemic period. Similarly, Dr. Haddad also predicts that pre-ischemia [Ca2+]i elevation will result in [Ca2+]i remaining within a survivable window during and following in vitro ischemia.
• An increase in [Ca2+]i is a trigger for reduction of NMDA open
probability during hypoxia/anoxia, BAPTA-AM will prevent such changes and preserve receptor open time or NMDA ΔCa2+ compared to oxic controls. In the experiments involving inhibition of Ca2+-dependent receptor inactivation mechanisms, it’s expected to find further evidence that elevated [Ca2+]i plays a key role in reducing NMDA receptor activity during oxygen deprivation.
• A major goal of Dr. Haddad’s research on neuroprotection with
anesthetics is to understand the mechanisms by which anesthetics protect neurons. In this aim it will be determined whether the increase in [Ca2+]i produced by a volatile anesthetic is responsible for neuroprotection. It is expected that anesthetics, by increasing [Ca2+]i into the protective range, will prevent lethal increases in [Ca2+]i during subsequent anoxia or ischemia. This “pre-conditioning” role for [Ca2+]i is illustrated in the Fig. 19.
Biomedical Research Scientist and Educating Professor – 2009 Page | 25

John J. Haddad, PhD
Intracellular Ca 2+ during pre-conditioning
Low Normal Protective Toxic
Percentsurvivalfollowingischemia
NoTreatment
Anesthetic
Ca2+Ionophore
Fig. 19. Proposed relationship between [Ca2+]i and neuron survival in the context of normoxia, presence of a volatile anesthetic, and during anoxia/ischemia. Cell survival is expected to be influenced by [Ca2+]i, regardless of the cause of the perturbation in [Ca2+]i (buffers, alteration in [Ca2+]e, Ca2+ ionophores).
• Isoflurane protects when present before, during and after
ischemia, since it’s proposed that it triggers processes of protective value during each of these periods. In particular, however, it is believed that it will prove to be protective before ischemia, when it has a “pre-conditioning” quality, and after ischemia, when it prevents “Ca2+ starvation”. It is, therefore, expected that isoflurane will provide maximal protection against simulated ischemia in the same cell regions of the slice showing maximal sensitivity to exogenously applied NMDA, since isoflurane is, predictably, protective because it attenuates NMDA receptor activity. Based on preliminary data, it is anticipated that protection be most evident in CA1, with fewer efficacies in CA4 and dentate. This may indicate that particular subtypes of the NMDA receptor are more sensitive to influence by isoflurane. The diagnosis of cell death in the various strata depends on propidium iodide fluorescence staining in those regions. While these areas mostly consist of CA cell bodies, some other cellular elements are there and might contribute to the signals. Therefore, Dr. Haddad and colleagues are expected to perform standard histologic examination (fixation in paraformaldehyde, staining with hematoxylin/eosin) to further assess cell death. It is expected that the slices made from the 2 year-old rats will show higher basal [Ca2+]i, and higher [Ca2+]i at all time points compared to youthful controls. Cell death will be greater at all time points as well.
• Neuroprotection with isoflurane or N2O will wane with age, and
possibly be related to interference with [Ca2+]i homeostasis in aged neurons. It is possible that the dose-dependency of isoflurane neuroprotection might change with age, complicating our interpretation. Dr. Haddad thinks this is unlikely because he has previously found equal neuroprotection with 0.7% and 2.0% isoflurane, and also because the 1% dose studied is in the middle of the MAC range for this compound in young and aged rats.
Biomedical Research Scientist and Educating Professor – 2009 Page | 26

John J. Haddad, PhD
• N2O will impair [Ca2+]i homeostasis, and worsen cell death in the aged slices, but not in the young ones. That is, [Ca2+]i in N2O treated slices will be higher before, during and after ischemia in slices from older animals. The data will not directly suggest why this is the case, beyond the correlation with presumed alteration of [Ca2+]i homeostasis, because the design is primarily descriptive, not mechanistic. That is, it will describe whether nitrous oxide might be a problem for the aged brain, but will not tell us directly why. The value of this observation is that it might be a clue as to why so many elderly individuals suffer neurological (primarily cognitive) deficits following anesthesia. This information will be of value in epidemiological studies in the United States, by indicating the potential value of stratifying subjects according to whether or not they received nitrous oxide as part of their anesthetic.
The schematic in Fig. 20 summarizes how hypoxia affects key signaling events in the
brain during stroke-induced ischemia.
ig. 20. xygen sensing mechanisms during hypoxia involve the membrane-bound NADPH oxidase, the ome complex system (A) and chemoreceptors (B), such as K+ channels. This ultimately
leads to sensory recognition and activity up-regulation.
Fmitochondria-cytochr
O
Biomedical Research Scientist and Educating Professor – 2009 Page | 27

John J. Haddad, PhD
The schematic in Fig. 21 depicts the role of ischemia in mediating cell death, calcium dishomeostasis in brain injury and death.
Ischemia
Reduction of oxygen and glucose supply: Energy failure
Membrane depolarization
Glutamate release
[Ca2+]i ↑
Glutamate receptor activation
DAG ↑
PKC ↑
Protein phosphorylation Proteolysis lipolysis Reoxygenation NOS activity ↑ Mitochondrial damage
Changes in gene expression
Changes in membraneReceptor & ion channel
activities
Degradation of thecytoskeleton
Membrane degradation
Inflammatorymediators
Free radicals(ROS/RNS)
Caspase activation
(Oxidative) Damage toProteins, lipids, DNA
Membrane andVascular dysfunction
Cell death
Protein synthesis ↓
g. 21. Hypoxia (ischemia)-mediated cell death and brain injury during stroke. Fi
♣
Biomedical Research Scientist and Educating Professor – 2009 Page | 28

John J. Haddad, PhD
Research Part II (American Heart Association [AHA]): The major aim of this project is to identify which mitogen-activated protein kinase
(MAPK) signaling pathways are associated with surviving hypoxia and which are involved with brain cell death. The significance of this study is that it will help clarify major gaps in our knowledge about the balance between brain survival and death as regulated by the MAPK pathways. This work will clarify the neuroprotective potential of modulating these signaling pathways. Specifically, Dr. Haddad tested and will continue to investigate the following hypotheses:
1. MAPKJNK and MAPKp38 activation is associated with a neuro-injurious
response mediated by hypoxia. 2. MAPKERK activation is associated with a neuroprotective response mediated
by hypoxia.
3. Sustained expression of the dominant mutated genes of MAPK modules differentially regulates cell survival and death in response to hypoxia/ischemia.
Degeneration and death of neurons of the central nervous system (CNS) are
responsible, at least in part, for the clinical manifestations of stroke- and ischemia-mediated injury. A large body of evidence suggests that intracellular pathways including apoptosis cofactors constitute a major component of cell death following ischemia. We are attempting to identify the signaling mechanisms associated with surviving hypoxia and to determine which pathways regulate the fate of the cell. Fig. 1 depicts the major components of functional signaling pathways of hypoxia/ischemia whose modules and cofactors remain to be determined.
Ischemia and Ischemia-reoxygenation
Bcl-2Baxp53
Effectors(Caspases)
Inhibitors(NF-κB)
Antagonism(Anti-apoptotic)
Survival Loop Agonism
(Pro - apoptotic) Death Loop
Apoptosis Survival
G-coupled cofactors
Protein kinases
MAPK p38 pathway MAPKJNKpathway MAPKERKpathway
Bcl-xLBad
?
?
?
? ?
Fig. 1. Ischemia/hypoxia-mediated regulation of potential signaling transduction pathways relevant to determining the fate of neuronal cells.
Biomedical Research Scientist and Educating Professor – 2009 Page | 29

John J. Haddad, PhD
Amongst the signal transduction pathways that control cell fate (apoptosis vs. necrosis) are the MAPK cascades, whose components are evolutionarily highly conserved in structure and organization. Each component consists of a module of three cytoplasmic kinases: a MAP kinase kinase kinase (MAPKKK), a MAP kinase kinase (MAPKK) and a MAP kinase (MAPK). MAPKKK is a serine-threonine kinase that receives activating signals from a membrane-spanning receptor and then phosphorylates and activates its substrate, MAPKK. MAPKK is a dual-specificity kinase with the potential to phosphorylate threonine-tyrosine residues in its substrate protein, MAPK. MAPKs represent a family of serine-threonine kinases with the potential to phosphorylate other cytoplasmic proteins and to translocate to the nucleus, where they can directly regulate the activity of transcription factors controlling gene expression. MAPKs fall into three subgroups: i) extracellular signal-regulated kinase-1 (ERK1 or MAPKp44) and ERK2 or MAPKp42; ii) Jun N-terminal kinases (JNKs), which received this name because they can activate the Jun transcription factor by phosphorylation of residues near its N-terminus; and iii) MAPKp38, named because of the molecular weight (38 kDa) of the first representative of the subgroup to be discovered.
Although MAPK signaling pathways are relatively well characterized, little is
known about the critical role these modules play in regulating the response of neuronal cells to ischemia/hypoxia, which may determine cell fate. In addition, a direct relationship between the activation of these modules and the regulation of caspases, cytochrome c release and Bax/Bcl-2/p53 expression is ambiguous; the molecular mechanisms involved are not known. For instance, MAPKERK activation is associated with estrogen-mediated neuroprotection following glutamate toxicity in cortical neurons. Similarly, neuroprotection by brain-derived neurotrophic factor (BDNF) is mediated by MAPKERK and phosphatidylinositol 3-kinase in cortical neurons. Furthermore, neuroprotection mediated by glial cell line-derived neurotrophic factor (GDNF) requires a reduction of NMDA-induced calcium influx in a MAPKERK-dependent mechanism. In contrast, it was recently shown that inhibition of MAPKERK with U0126 mediates neuroprotection against oxidative stress in neuronal cells.
On the other hand, it has been reported that selective inhibition of the MAPKp38
pathway reduces brain injury and neurological deficits in cerebral focal ischemia. Moreover, it has been shown that synergistic activation of MAPKp38 and caspase-3-like proteases was involved in calyculin A-induced apoptosis in cortical neurons. In addition, selective inhibition of the MAPKp38 pathway has been proposed as a therapeutic strategy for pre-clinical evaluation.
With respect to the MAPKJNK pathway, hypoxia selectively induces SAPK/JNK-2-AP-1
in the nucleus tractus solitarii, MAPKJNK1 in pulmonary arteries, and c-Jun/AP-1 in hepatocytes, but little is known about its regulation in the brain. In contrast to these observations, hypoxia has no effect on activating MAPKJNK pathway in association with L-type voltage gated Ca2+ channels, despite the up-regulation of MAPKERK and MAPKp38. Moreover, the involvement of MAPKJNK in regulating cell fate (apoptosis) is still controversial; for instance, activation of c-Jun NH2-terminal kinase/stress-activated protein kinase (JNK/SAPK) is critical for hypoxia-induced apoptosis of human malignant melanoma. Similarly, inhibition of hypoxia/reoxygenation-induced apoptosis is achieved with an antisense oligonucleotide targeted to JNK1/SAPK1 in kidney cells. However, inhibition of c-Jun N-terminal kinase 1, but not c-Jun N-terminal kinase
Biomedical Research Scientist and Educating Professor – 2009 Page | 30

John J. Haddad, PhD
2, suppresses apoptosis induced by ischemia/reoxygenation in cardiac myocytes. In addition, sequential activation of AP-1-related transcription factors and MAPKJNK protein kinases is partly, but not critically involved, in mediating apoptotic death induced by transient hypoxia in developing brain neurons. The lack of specific MAPKJNK inhibitors has made it difficult to discern MAPKJNK involvement in regulating cell death and survival in cortical neurons. Recently, however, the compound SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase, and the strategy of incorporating antisense oligonucleotides against MAPKJNK have only emerged as an effective tool to help understand the discordant involvement of this pathway in regulating cell death.
Dr. Haddad’s experiments in the following three specific aims are defining a specific
role for MAPK modules: that of signaling pathways that regulate cell fate in response to hypoxia/ischemia in the brain. These experiments will be performed in a cortical neuron model. Trypan exclusion assay and/or propidium iodide staining determine cell viability.
Aim 1. This aim will define the role of MAPKJNK and MAPKp38 as neuro-injurious modules associated with cell injury and death mediated by hypoxia/ischemia.
1. Identification of G-coupled cofactors associated with MAPK during
hypoxia.
• Dr. Haddad is trying to identify the upstream cofactors that regulate the phosphorylation of MAPKJNK and MAPKp38. Specifically, he will target small G-coupled proteins, including RAF, PAK, CDC42 and RAC, up-regulated with the imposition of hypoxia.
• In order to address the involvement of G-proteins in regulating MAPK
signaling in hypoxia, the following approaches will be adopted:
- The protein expression of G-coupled cofactors in hypoxia will be assessed with immunoblotting using selective antibodies.
- The localization and distribution of G-coupled proteins
during hypoxia will be assessed with immunofluorescence staining using specific antibodies, which are HRP-conjugated and fluorescein-labeled.
- The interaction of G-coupled proteins with the upstream
kinases (MAPKKK and MAPKK) that regulate MAPK signaling will be addressed by co-immunoprecipitation to determine, if any, the level and degree of interaction.
Biomedical Research Scientist and Educating Professor – 2009 Page | 31

John J. Haddad, PhD
2. Characterization of upstream kinases that regulate MAPK during hypoxia.
Following identification of the small G proteins in response to hypoxia, we will identify
the downstream kinases (MAPKKK/MAPKK) that act upstream of MAPKJNK and MAPKp38.
• Identification of MAPKKK, a serine-threonine kinase that receives signals from G-coupled cofactors. MAPKKK kinase assay will be performed with MAPKK as substrate. The degree of phosphorylation of MAPKK is directly proportional to activity of upstream MAPKKK.
• Identification of MAPKK, a threonine-tyrosine kinase that receives
signals from MAPKKK. MAPKK kinase assay is performed with MAPK as substrate.
• Another approach to determine the role of MAPKKK/MAPKK in
hypoxia will be achieved by the construction of glutathione S-transferase (GST)-MAPKKKΔ, MAPKKΔ and the kinase-deficient GST-MAPKKKΔ/MAPKKΔ plasmid mutants subcloned into pGEX-KG vectors, which will be transfected into cortical neurons. Subsequently, these neurons are exposed to controlled period of hypoxia and then the activity and expression of these upstream kinases will be analyzed by co-immunoprecipitation/immunoblotting analysis.
3. Assessment of MAPK regulation during hypoxia.
After having identified which upstream modules regulate MAPK signaling in hypoxia, the focus is now turned into defining the effect of hypoxia in modulating MAPKJNK and MAPKp38 responses in cortical neurons. Preliminary experiments have shown that cortical neurons exposed to hypoxia induced a prompt phosphorylation of MAPKp38 with maximum activation at about 30-60 min in mouse and 5-10 min in rat, declining thereafter. Although the molecular basis of this species variation is unclear, it is observed that in mouse cortical neurons there is suppression of MAPKp38 with prompt hypoxia (≈ 10 min), an effect reversed (activation) with prolonged hypoxia (30-60 min), as compared with normoxic exposure (Fig. 2). On the other hand, hypoxia induced a quick activation of MAPKp38 (≈ 5-10 min) in rat cortical neurons, subsiding thereafter (Fig. 3).
Norm
oxia
Phospho-MAPKp38 10 M
in20
Min
30 M
in
120 M
in
Hypoxia
60 M
in
Figure 2
Norm
oxia
5 Min
Phospho-MAPKp38 10 M
in20
Min
30 M
in
120 M
in
Hypoxia
60 M
in
Figure 3
Fig. 2. Hypoxia activates, in a biphasic manner, the phosphorylation/activation of MAPKp38 in mouse cortical neurons. Fig. 3. Hypoxia transiently induces the phosphorylation/activation of MAPKp38 in rat cortical neurons.
Biomedical Research Scientist and Educating Professor – 2009 Page | 32

John J. Haddad, PhD
The regulation by hypoxia of MAPKJNK signaling will be investigated by assessing the phosphorylation and activation of downstream transcription factors as follows:
• Stress-activated protein kinase (SAPK), also referred to as MAPKJNK,
functions through a protein kinase cascade that transduces cellular stress signals. Activation of SAPK/MAPKJNK occurs via phosphorylation at threionine183 and tyrosine185 by the dual specificity enzyme MAPKK, particularly MAPKK4. We will determine hypoxia-mediated regulation of MAPKJNK by assessing the activity of MAPKK4 using phospho-specific anti-MAPKK4 antibody. This antibody recognizes the phosphorylation of MAPKK4 at threonine223, as will be determined by western immunoblotting.
• Dr. Haddad determines the degree of phosphorylation of MAPKJNK using a
specific anti-phospho-MAPKJNK antibody that recognizes the phosphorylated forms of threionine183 and tyrosine185 residues, in comparison with the steady, non-phosphorylated form using a non-phospho-specific antibody. In addition, the localization and distribution of MAPKJNK will be detected in situ with immunofluorescence.
• MAPKJNK regulates a transcription factor called ATF-2. ATF-2 interacts
with oncoproteins and cellular tumor suppressors and forms a downstream target for both MAPKJNK and MAPKp38. The activation of ATF-2 during hypoxia will be studied by determining the dual phosphorylation of threonine69 and threonine71, using specific antibodies by immunoblotting. Distribution of ATF-2 during hypoxia is determined by immunofluorescence staining.
• The c-Jun subunit, a component of the transcription factor activator
protein-1 (AP-1), is considered a downstream target of MAPKJNK. The transcriptional activity of c-Jun is regulated by phosphorylation at serine63 and serine73. The regulation by hypoxia of c-Jun activation will be studied using a specific phospho-c-Jun antibody by western analysis.
The regulation by hypoxia of MAPKp38 signaling will be investigated by assessing the
phosphorylation and activation of downstream transcription factors as follows:
• MAPKp38 phosphorylation is regulated by MAPKK, specifically MAPKK3 and MAPKK6. MAPKK3 and MAPKK6 are activated through phosphorylation at serine189 and threonine193 (MAPKK3), and serine207 and threonine211 (MAPKK6). Western analysis using phospho-MAPKK3/6 antibodies will be used to study the regulation of MAPKp38 by hypoxia.
• The degree of phosphorylation of MAPKp38 will be determined using a
specific anti-phospho-MAPKp38 antibody that recognizes the
Biomedical Research Scientist and Educating Professor – 2009 Page | 33

John J. Haddad, PhD
phosphorylated forms of threionine180 and tyrosine182 residues, in comparison with the steady, non-phosphorylated form using a non-phospho-specific antibody. In addition, the localization and distribution of MAPKp38 will be detected in situ with immunofluorescence.
• MAPKp38 regulates ATF-2. The activation of ATF-2 during hypoxia will
be studied by determining the dual phosphorylation of threonine69 and threonine71, using specific antibodies by immunoblotting. Distribution of ATF-2 during hypoxia is determined by immunofluorescence staining.
• Another transcription factor regulated by MAPKp38, Elk-1 is a component
of the ternary complex that binds the serum response element (SRE) and mediates gene activity in response to serum and growth factors. We will investigate the regulation of Elk-1 by hypoxia by studying its phosphorylation at a cluster of S/T motifs at its C-terminus using a specific phospho-antibody raised against serine383, which is critical for Elk-1 transcriptional activity.
• Active phosphorylated MAPKp38 also regulates a kinase cascade, which
may be followed by determining the terminal phosphorylation of heat-shock protein (Hsp27) by MAPKAP-K2. After exposure to hypoxia, cells are ruptured in lysis buffer for 30 min on ice. Cell debris is removed by centrifugation at 10,000g for 10 minutes at 4oC. The kinase activity of MAPKAPK-2, which is regulated by the phosphorylation of the upstream MAPKp38, is assayed with recombinant Hsp27 as a substrate. Assessment of Hsp27 phosphorylation by co-precipitation with MAPKAP-K2 kinase will be performed.
4. Hypoxia-dependent MAPK-mediated regulation of apoptosis signaling.
In this section, after having identified the regulation of MAPK signaling by hypoxia, Dr. Haddad shifts attention to address the key question in ‘Aim 1”: “Is the activation of MAPKJNK and MAPKp38 pathways associated with a neuro-injurious response in hypoxia?”
• Assessment of cell viability in hypoxia by Trypan blue exclusion and
propidium iodide staining. The energy charge (ATP; luciferase-lucifer measurement with a lumunometer) and MTT reduction to formazan will be also considered as independent indices of cell viability.
• Determination of nuclear matrix protein (NMP) release to the extracellular
medium in response to hypoxia by enzyme-linked immunosorbent assay (ELISA).
• Detection of DNA fragmentation using either DNA laddering in agarose
gels or TdT-mediated dUTP Nick End Labelling (TUNEL).
Biomedical Research Scientist and Educating Professor – 2009 Page | 34

John J. Haddad, PhD
• Evaluation of the translocation of membrane phospholipid phosphatidylserine (PS) from the inner leaflet to the exterior side of the cell membrane in response to hypoxia.
• Evaluation of the expression of signaling cofactors associated with
programmed cell death in response to hypoxia as baseline experiments. The main components we will investigate include pro- and anti-apoptosis cofactors. The regulation of these cofactors by hypoxia will then be investigated as follows:
- Bax, Bcl-2 and p53 protein expression will be assessed
with specific antibodies by western analysis in response to hypoxia.
- Transcriptional regulation by hypoxia of these signaling
cofactors will be studied by determining RNA expression with semi-quantitative technique RT-PCR and RNase protection assay.
- The distribution of these cofactors in hypoxic neurons
will be determined by immunocytochemistry using specific antibodies.
- Assessment of the expression and distribution of pro-
and anti-apoptotic cofactors in response to MAPKJNK and MAPKp38 activation by hypoxia. In this regard, we will identify the role of the selective inhibitors of MAPKp38 (VX-745 and SB-203580) and MAPKJNK (SP600125 and the antisense oligonucleotide 5′-CTCTCTGTAGGCCCGCTTGG-3′, which targets MAPKJNK1) in regulating the expression of Bcl-2, Bax and p53.
• The involvement of caspases and cytochrome c in MAPKJNK- and
MAPKp38-mediated injury will be investigated. To achieve this aim, the following approaches are adopted:
- The expression of caspases is studied by western analysis
using specific antibodies, thereby allowing determination of the distribution of pro-caspase forms and their cleavage into active subunits induced by hypoxia.
- The expression of caspases in response to MAPK
selective inhibitor pretreatment prior to exposure to hypoxia will be assessed as above.
Biomedical Research Scientist and Educating Professor – 2009 Page | 35

John J. Haddad, PhD
- The release of mitochondrial cytochrome c will be determined in response to hypoxia by specific antibodies using western analysis.
- The likely interaction of cytochrome c with caspases in
response to hypoxia is studied by co-immunoprecipitation to determine whether caspases and their cleavage are downstream components cytochrome c.
- Selective inhibition of the MAPKJNK and MAPKp38
pathways will allow us to determine the relationship between MAPK signaling and caspase/cytochrome c regulation during hypoxia, and whether these molecules are part of the process mediating MAPK-induced neuroinjurious response to hypoxia.
Aim 2. This aim will define the role of MAPKERK as a neuroprotective module
associated with cell survival mediated by hypoxia/ischemia. In cortical neurons: 1. Identification of G-coupled cofactors associated with MAPK regulation
during hypoxia.
Dr. Haddad will identify the upstream signaling cofactors (G-coupled proteins) that regulate the phosphorylation/activation of MAPKERK in response to hypoxia (See Aim 1).
2. Characterization of upstream kinases that regulate MAPK during hypoxia.
Following the identification of small G proteins in response to hypoxia, Dr. Haddad will
unravel the potential downstream kinases (MAPKKK and MAPKK) that act upstream of MAPKERK (See Aim 1).
3. Assessment of MAPK regulation during hypoxia. After having identified which upstream modules regulate MAPK signaling during
hypoxia, the focus is then turned into defining the effect of hypoxia on cortical neurons in modulating MAPKERK pathway.
Preliminary experiments have shown that cortical neurons exposed to hypoxia induced a
prompt phosphorylation of MAPKERK, with maximum activation between 10-30 min (Fig. 4).
Biomedical Research Scientist and Educating Professor – 2009 Page | 36

John J. Haddad, PhD
Normox
ia5 M
in
Phospho-MAPKErk 10 M
in20
Min
30 M
in
120 M
in
Hypoxia
60 M
in
Erk-1 p44Erk-2 p42
Fig. 4. Hypoxia transiently regulates the phosphorylation/activation of MAPKERK in rat cortical neurons.
The regulation by hypoxia of MAPKERK signaling will be investigated by assessing the
phosphorylation and activation of downstream transcription factors as follows:
• MAPKERK activation involves the regulation of a protein kinase cascade that sequentially activates RAF, MAPKK1/2, MAPKERK and then Elk-1 and p90RSK. The activation of RAF, particularly RAF-1, in response to hypoxia will be investigated by studying RAF-1 phosphorylation at serine259 and serine 621.
• MAPKERK phosphorylation is regulated by MAPKK, specifically
MAPKK1 and MAPKK2. MAPKK1 and MAPKK2 are activated through phosphorylation at serine189 and serine221 (MAPKK1/2). Western analysis using phospho-MAPKK1/2 antibodies will be used to study the regulation of this kinase by hypoxia.
• The degree of phosphorylation of MAPKERK will also be determined using
a specific anti-phospho-MAPKERK antibody that recognizes the phosphorylated forms of threionine202 and tyrosine204 residues. The localization of MAPKERK will be detected in situ with immunofluorescence.
• The transcription factor Elk-1 is regulated by MAPKERK. The regulation of
Elk-1 by hypoxia will be investigated by studying its phosphorylation using a specific antibody raised against serine383.
• Another downstream transcription factor regulated by MAPKERK is
p90RSK, which is phosphorylated at serine380. The regulation of p90RSK by hypoxia will be investigated using a specific anti-phospho-p90RSK antibody by western analysis.
Biomedical Research Scientist and Educating Professor – 2009 Page | 37

John J. Haddad, PhD
4. Hypoxia-dependent MAPK-mediated regulation of apoptosis signaling.
In this section, after having identified the regulation of MAPKERK signaling by hypoxia, the attention is shifted to address the key question in ‘Specific Aim # 2’: “Is the activation of MAPKERK associated with neuroprotection in hypoxia?”
• The expression and distribution of pro- and anti-apoptotic cofactors in
response to MAPKERK activation by hypoxia will be investigated. In addition, the role of the selective inhibitors of MAPKK1/2 (U0126) and MAPKERK (PD-98059) in regulating the expression of Bcl-2, Bax and p53 will be determined.
• Dr. Haddad will also investigate the involvement of caspases and
cytochrome c in MAPKERK-mediated neuroprotection.
Aim 3.
1. Over-expression of MAPKJNK and MAPKp38. The pCMV-Flag-JNK1 and pCMV-Flag-p38 plasmids are constructed.
• Transient transfection experiments in cortical neurons are performed using
the lipofectamine reagent.
• Following transfection, cells are exposed to controlled periods of hypoxia. Analysis of downstream components, including apoptosis signalling cofactors (Bax, Bcl-2, p53, caspases and cytochrome c), is performed as detailed above.
2. Over-expression of MAPKERK.
The pCMV-Flag-ERK wild and mutant plasmid is constructed using the expression vector pCMV5 and MAPKERK cDNA. The mutant plasmid thus formed would be pCMV-Flag-MAPK. The sequence of all plasmids is confirmed by automated sequencing.
• Transient transfection experiments in cortical neurons are performed.
• Following transfection, cells are exposed to controlled periods of hypoxia
and analysis of downstream components, including apoptosis signaling cofactors, is subsequently performed.
In summary, the biological and clinical significance of the aforementioned research, which could bear and accrue a national interest scope in biological sciences and medicine, is summarized below:
Biomedical Research Scientist and Educating Professor – 2009 Page | 38

John J. Haddad, PhD
• Hypoxia/ischemia leads to activation of MAPKJNK and MAPKp38 in association with up-regulating the expression of pro-apoptosis cofactors and down-regulating that of anti-apoptotic cofactors in the brain.
• Hypoxia/ischemia leads to activation of MAPKERK in association with up-
regulating the expression of anti-apoptosis cofactors and down-regulating that of pro-apoptotic cofactors, creating a balance in favor of neurons cell survival.
• Sustained expression of MAPKJNK and MAPKp38 would exacerbate
hypoxia-mediated injury; in contrast, sustained expression of MAPKERK
would enhance cell survival by counteracting the injurious effect of the hypoxic insult to the brain. The following schematic depicts the hypothetical regulatory MAPK pathways that control cell survival and death in hypoxia (Fig. 5).
Hypoxia
RAS RAC CDC42
MAPKKK1/4
MAPKK4
MAPK JNK
ATF-2 c-Jun
MAPKKK
MAPKK3/6
MAPK p38
ATF-2 Elk-1 MAPKAP -K2
RAF MAPKKK
MAPKK1/2
MAPK ERK
Elk - 1 p90RSK
Cell Death Cell Survival
Fig. 5. Hypoxia-mediated regulation of MAPK signaling pathways in the brain. • The patterns of MAPK signaling in the brain are crucial in
neuroprotection during ischemia, especially in cases closely resembling heart attack (stroke-mediated oxygen deprivation).
• In patients suffering from stroke, the level of oxygen reaching for the
brain decreases. Those particular cortical areas suffer from a severe hypoxic condition to an extent cells can no longer support effective brain functioning, due in part to differential expression of MAPKs.
• The aforementioned experiments designed and performed by Dr.
Haddad will identify why certain areas in the brain are more vulnerable to stroke-mediated ischemia, what might the potential
Biomedical Research Scientist and Educating Professor – 2009 Page | 39

John J. Haddad, PhD
responsible mechanisms (MAPK) and what strategies should be proposed as an alleviating clinical approach.
The schematic in Fig. 6 summarizes the various signaling pathways encompassing
MAPKs and their potential regulation.
ig. 6. An overview of the complex network of MAPK signaling pathways and their interactions and
The high impact of Dr. Haddad’s biomedical and clinical research on brain injury, anesthe
. Receiving invitations to lecture in prominent academic institutions and
. Publishing this pioneer work independently and/or with other colleagues in
Rec
epto
r Ty
K
Rec
epto
r Ty
K
Grb2Ras-GRF SHC Grb2 Sos-1 GTPP
GDP
RasGDP
RasGTPP
GAP
P
Ca2+/CaMR-Ras GTPase
Rho
Raf-1 A-Raf B-Raf C-Raf Phosphatide PKC DAG
Rac-1
Stress/Cytokines
RhoCdc42
DPK
MEKK3 MEKK1/2 ASK-1
TAK-1
MKK3MKK4 MKK6MKK7
p38αp38βp38β2
p38δp38γ
MBPATP-2MAPKAP2/3MSK-1
MAPKAP5
p47phox
ATF-2
MEF2CCHOP1
JNK1JNK2
c-JunMBP
Elk-1SAP-1
MEK1/2
ERK1/2MBP
MAP
MAPKAP1 p90RSK c-Myc Est1/2
p53
PKA
JIP
MKP1/2/3
p53
Pyk2
Src
Src
Pyk2Ca2+/CaM
Fbifurcations. The sign ( ) denotes inhibition as (⎯⏐); the solid arrows indicate activation (stimulation); P, phosphorylation.
tics, stroke and oxidative stress was widely praised and welcome by peers, professors, physicians and scientists in the USA, UK and further afield. This unprecedented type of research is manifested in:
1
laboratories across the USA (Please see details of the list stated above). 2
internationally renowned and USA-based publications, as detailed below (please see c.v. for complete list):
Biomedical Research Scientist and Educating Professor – 2009 Page | 40

John J. Haddad, PhD
• Haddad, Cellular Signalling 14: 799-810, 2002.
his paper discussed oxygen homeostasis in the regulation of sele
Haddad, Cytokines, Cellular and Molecular Therapy 7: 1-14, 2002.
Haddad, Biochemical and Biophysical Research Communications 296:
• Haddad, Cellular Signalling 14: 879-897, 2002.
This paper discussed antioxidant/prooxidant mechanisms in the
reg
Haddad and Fahlman, Biochemical and Biophysical Research
This paper discussed breakthroughs and views on interlekin-10 as an ant
• addad, Biochemical and Biophysical Research Communications 297:
This paper discussed breakthroughs and views on cytokines and the
• addad, Free Radical Biology & Medicine 33: 907-926, 2002.
This paper discussed breakthroughs and views on the pharmaco-gen
• addad et al., International Immunopharmacology 2: 1567-1583, 2002.
Haddad et al., Journal of Neuroimmunology 133: 1-19, 2002.
his paper discussed neuro-immune-endocrine interactions from the perspective of regulating the hypothalamic-pituitary-adrenal revolving
Tctive genes.
• •
847-856, 2002.
ulation of genetic transcription factors. Specifically, this paper is elaborating on the performance of basic biomedical research that has profound clinical impact and projections.
•Communications 297: 163-176, 2002.
i-inflammatory agent. Specifically, this paper is elaborating on the performance of basic biomedical research that profound has clinical impact and projections. H700-713, 2002.
ir receptors in physiology and pathophysiology. Specifically, this paper is elaborating on the performance of basic biomedical research that has profound clinical impact and projections. H
omics of cytokine-related pathways. Specifically, this paper is elaborating on the performance of basic biomedical research that has profound clinical impact and projections. H
•
T
Biomedical Research Scientist and Educating Professor – 2009 Page | 41

John J. Haddad, PhD
axi
•
scription genetic factors in the
regulation of injury and their influence in critical care. Specifically, this pap
•
nce of basic biomedical research that has profound clinical impact and projections.
•
anscription genetic factors in the regulation of injury and their influence in critical care. Specifically, this pap
•
ce of basic biomedical research that has profound clinical impact and projections.
s, thereby communicating the brain and the nervous system with the endocrine and exocrine body functions in stress. Specifically, this paper is elaborating on the performance of basic biomedical research that has profound clinical impact and projections.
Haddad, Critical Care 6: 481-490, 2002.
This paper discussed the role of tran
er is elaborating on the performance of basic biomedical research that has profound clinical impact and projections. Haddad, Respiratory Research 3: 26-53, 2003.
This paper is elaborating on the performa
Haddad, Critical Care 7: 47-54, 2003.
This paper discussed the role of tr
er is elaborating on the performance of basic biomedical research that has profound clinical impact and projections. Haddad, Cellular Signalling 15: 255-267, 2003.
This paper is elaborating on the performan
♣
Biomedical Research Scientist and Educating Professor – 2009 Page | 42

John J. Haddad, PhD
Research Part III (Tenovus Scotland For Medical Research *):
* This grant was awarded while Dr. Haddad was still in Scotland training for his biomedical Ph.D.
Background: Tenovus Scotland exists to foster high quality research within the health care professions in Scotland (UK) and invites applications for grants in Medicine, Dentistry, the Medical Sciences and allied professions. Preference is given to innovative, patient-related projects and particularly to preliminary "pump priming" studies, which are thought likely to lead to subsequent funding from major grant-giving bodies. Applications from junior staff and from established staff in the early stages of a new project is particularly encouraged. Support is mainly provided for the purchase of equipment for research and/or running costs although, from time to time when funds are available, applications may be invited for salary support or for research studentships. All projects must have a clear research component and fall within the general areas of clinical or medical science. (All applications are submitted to detailed peer review.)
CYTOKINE-MEDIATED ACTIVATION OF HIF-1α AND NF-κB IN ALVEOLAR EPITHELIA: SIGNALLING PATHWAYS OF APOPTOSIS IN OXIDATIVE STRES
Throughout pregnancy, the fluid-filled lung of the unborn infant develops at an O2 concentration, which is the same as that found at the summit of Mt. Everest, about 3% of air at sea level. During delivery, fluid drains from the lung and the first breath raises the O2 concentration in the newly created space 5 times, to 15%. Such sudden, sustained changes in O2 availability are known to regulate a suite of genetic switches which determine the survival of cells according to the severity of the shift in O2 tension experienced. In cases where this ability to “sense” and adapt to changes in O2 tension is improperly developed (in premature babies receiving O2 therapy, for example), consequent lung damage is severely debilitating and frequently life threatening. An understanding of the pathways which transmit changes in O2 availability towards cellular survival responses are therefore of major importance to perinatal medicine. This study aims to determine the role of a candidate family of O2 sensitive molecular signals, called “cytokines”, in relaying signals about the O2 environment towards the genetic switches which control whether or not a cell will survive, grow or die during the stress period. If it can be demonstrated that O2, its poisonous metabolites, and cytokines co-operate in sending vital messages, which regulate lung cell survival, then a foundation is laid for developing therapeutic strategies for the treatment of infant respiratory distress conditions.
In pre-term infants, sustained therapy with high-inspired oxygen tensions (e.g. for respiratory distress syndrome) causes epithelial damage, septal fibrosis and a scattered occlusion of distal airways characteristic of broncho-pulmonary dysphasia (BPD). The severity of damage incurred follows the degree of septal occlusion as lobules that are fully blocked experience lower sustained Po2's than those that remain patent. As O2/redox-responsive transcription factors may direct the expression of genes involved in promoting either cellular survival or apoptosis, it is increasingly apparent that defined O2-linked pathways could mediate lung damage caused during chronic O2 therapy, in part. Thus, the global objective of this proposal is to probe the molecular signaling pathways, which relay information regarding
Biomedical Research Scientist and Educating Professor – 2009 Page | 43

John J. Haddad, PhD
changes in oxygen availability, and the pattern of stress response mounted by the distal lung epithelium of the neonate.
Studies will utilize whole lung and distal lung epithelial cultures from perinatal rats to assess the capacity of cells to engage in growth, apoptosis or necrotic lysis during shifts in Po2 designed to mimic the transition from pre- to post-natal Po2’s and beyond. A key mediator of cell cycle changes from the onset of stress to programmed cell death (PCD) is the interleukin-1β converting enzyme (ICE/caspase 1). Since ICE cleaves pro IL-1β into the active cytokine, IL-1β, a stress induced lymphokine, it represents a candidate signal transducer involved in cell death signaling pathways implicated under oxidative conditions.
Intracellular signaling pathways, which are linked to changes in oxygen availability, can determine both function and survivorship of cells by regulating the expression of adaptive or defensive gene families in proportion to the direction and magnitude of the oxygen shift. As the perinatal lung epithelium constitutes the front line of oxygen transfer into the circulation, the change in lung luminal Po2 from fetal (23 Torr) to postnatal (100 Torr) tensions constitutes a profound, yet substantially overlooked, signaling event which we believe plays a major role in adapting the lung towards aerobic, postnatal function. As such, molecular oxygen and its charged derivatives may bear central importance to ensuring the successful transition from placental to pulmonary modes of gas exchange at birth.
Dr. Haddad’s work in this area has recently demonstrated that the epithelial lining
of the distal lung possesses a finely tuned capacity for mounting regulated genetic responses to modest shifts in oxygen tension and that reactive oxygen species (ROS) are likely involved as second messengers in this oxygen signaling pathway. Hypoxia inducible factor-1α (HIF-1α) and nuclear factor-κB (NF-κB) are genetic switches (transcription factors) whose activation states are potentiated by hypoxic and hyperoxic stresses, respectively. HIF-1α regulates the expression of a suite of genes involved in the provision of glycolytically derived ATP, vascular growth and apoptosis and as such, its activities are consistent with the co-ordination of adaptive responses to hypoxia. NF-κB was identified as a factor that regulates the expression of the κ light chains in B-lymphocytes, and increasing evidence suggests that it is profoundly responsive to oxidizing stresses, including hyperoxia. Under such conditions, its cytosolic association with an inhibitory protein (IκB) is relieved, resulting in its translocation to the nucleus and activation of hyperoxic stress-response genes, including those involved in the regulation of cell cycle events. By modulating the ROS buffering capacity of fetal distal lung epithelial cells via the glutathione biosynthetic pathway under modest shifts in oxygen tension, we have established that the reduction/oxidation potential is a key component of the signaling pathways involved in the activation of HIF-1α and NF-κB. Given the regulatory roles of these transcription factors in mediating cell survivorship in hypoxic or hyperoxic stress, the broad thrust of this proposal is to determine the molecular signaling components which link oxygen availability to the activities of these transcription factors and the subsequent potential for engaging in apoptosis in the lung.
The activation of transcription factors mediated by ROS is a potential determinant of
apoptosis (programmed cell death, PCD) in the alveolar epithelium. In early apoptosis, the level of ROS increases in parallel with depressed glutathione concentrations. This is further supported
Biomedical Research Scientist and Educating Professor – 2009 Page | 44

John J. Haddad, PhD
by observations that depletion of glutathione accelerates onset of apoptosis, and that the inhibition of its extrusion defends against apoptosis. Moreover, ROS contribution to cell death in many cell models has been attenuated, at least in part, by the antioxidant capacity of reduced glutathione (GSH). As epithelial apoptosis and necrosis found in bronchopulmonary dysplasia (BPD) and respiratory distress syndrome (RDS) are intimately linked to oxidative ROS production, and as glutathione ROS-buffering capacity is substantially depressed in the perinatal period, it would appear that the O2 shift in the lung at birth, or indeed elevations in lung lumen Po2 by O2 therapy, may evoke PCD in the perinatal lung.
A major transducer in PCD is the mammalian interleukin-1β (IL-1β)-converting enzyme
(ICE), a cysteine protease that cleaves pro-IL-1β to generate the active form of the cytokine ⎯ a peptide hormone, which regulates stress-induced responses, and a mediator of apoptotic signaling pathways. There is substantial evidence suggesting that IL-1β participates as a transducing signal in oxidative cell injury via ROS, and has stimulatory effects on regulatory transcription factors in mediating apoptosis. Furthermore, cytokines such as IL-1β have been shown to be potent inducers of oxidative stress in several cell models. This is further supported by the observations that the blockade of ICE protease activity by specific inhibitors and preventing the activity of IL-1β by either anti-IL-1β antibodies or IL-1 receptor antagonist (IL-ra) rescued cells from apoptosis induced by down-regulating superoxide dismutase, a major superoxide (O2
−) scavenger. Moreover, it has been reported that IL-1β mediates the apoptotic signal by specifically alerting neighboring, as well as distant, cells that the signaling/effector phase of apoptosis is at onset, and that the cytokine’s apoptotic autocrine and paracrine signaling mechanisms recruit immune cells to activate and accumulate at the site of injury and/or stress to trigger a suicide response. Dr. Haddad therefore suggests that cytokine-based signaling pathways likely constitute a significant component of the oxygen-sensing pathway of the perinatal lung epithelium.
To further elucidate the molecular mechanisms involved in the activation of HIF-1α and
NF-κB in alveolar epithelia which might provide clues to the signaling pathways involved in PCD, it has been reasoned that ICE and its substrate, IL-1β, could be potential regulators of the genetic response of the fetal lung in oxidative stress. In the face of the evidence that the perinatal lung redox-sensitivity is controlled at the gene level, Dr. Haddad foresees that cytokine-mediated signaling via regulatory genetic factors may constitute a major target for therapeutic intervention in the treatment of respiratory diseases.
Dr. Haddad’s broad goal is to determine how cytokines participate in dictating cell cycle responses of the perinatal lung epithelium during mild, moderate and severe forms of hypoxic or hyperoxic stress. Specific hypotheses are:
1. Form, duration and intensity of oxidative stress constitute a potential determinant of cellular survival or apoptosis to alveolar epithelial cells exposed to ascending ΔpO2 regimes. A key mediator of this signaling pathway from the onset of stress to PCD is the interleukin-1β-converting enzyme, ICE (Caspase-1).
Biomedical Research Scientist and Educating Professor – 2009 Page | 45

John J. Haddad, PhD
2. ICE-regulated IL-1β production modulates the activation profiles of HIF-1α and NF-κB, both implicated in the regulation of PCD, in response to oxygen-linked stress. In this way, ICE activation state may serve to finely tune genetic responsiveness to oxygen availability.
3. ICE activation state (and down-stream effects on HIF-1α and NF-κB
activities) is dependent upon both species and concentration of reactive oxygen species.
Living organisms, from prokaryotes to complex eukaryotes, have developed through
evolution an elaborate sequence of adaptive mechanisms to maintain oxygen (O2) homeostasis. In mammals, for instance, the development of the respiratory and cardiovascular systems allow the capture and appropriate distribution of O2 as a substrate for oxidative phosphorylation, the major biochemical reaction for the derivation of ATP as the vital biological currency necessary to maintain cell survival. The process of breathing is the initiating step of respiration, which also includes the movement of O2 from the lungs to peripheral tissues, and the process of cellular respiration, which produces ATP. The role of the lung in adult life is essentially one of gas exchange. The lung is responsible for providing a moist epithelial barrier for the transport of atmospheric O2 into the blood via a network of fine capillaries that envelope the alveolar sacs, and concomitantly remove from the body accumulating waste CO2. The cone-shaped lungs are divided into lobes, each of which is subdivided into lobules having bronchioles that serve many alveoli. Each alveolar sac is made up of simple squamous epithelium surrounded by blood capillaries, thereby allowing for efficient and rapid gas exchange across this barrier. The development of a mature lung, therefore, is crucial for survival and, within the context of an integral physiological system, tight regulation of the partial pressure of O2 (pO2) is important in the face of a continuously changing environment. Lung Maturation at Different Stages of Gestational Development
The development of the human lung begins approximately on the 26th day of gestation (4 weeks post conception). However, lung maturation continues postnatally and is not completed until late childhood (up to 8 years), although postnatal development generally consists of an increase in the number of mature alveoli. The major stages of lung development from a glandular structure to an alveolar structure capable of efficient gaseous exchange with the capillary network take place commencing the 8th week of gestation and continue to term (40 weeks) and postnatally. Therefore, the 32 weeks of gestational development are classified into stages in accordance with the visual appearance of lung tissue: i) embryonic; ii) pseudo-glandular; iii) canalicular; iv) saccular; and v) alveolar.
The embryonic stage of lung development (26 days - ≈ 6 weeks) begins when the
respiratory diverticulum, or lung bud, appears as an outgrowth from the ventral wall of the foregut. This stage is followed by the separation of the lung bud from the foregut, thus forming the trachea (windpipe) and bronchial buds, which successively enlarge at the beginning of the 5th week to form the main bronchi. The embryonic stage is marked by the formation of the lobular and segmental sections of the respiratory tree as columnar epithelial lined tubes evident by the end of the 5th or 6th week.
Biomedical Research Scientist and Educating Professor – 2009 Page | 46

John J. Haddad, PhD
The pseudo-glandular stage roughly begins at the 5th/6th week of gestation and lasts up until 16th/17th week. What marks this period is the histological appearance of the fetal lungs as an exocrine gland and the completion of the proliferation of the primitive airways. At this stage, cartilage is formed around the larger airways, and smooth muscles begin to envelop airways and blood vessels. Upon completion of this stage, acinar outlines first begin to appear as epithelial tubes continue to grow and branch. The undifferentiated columnar epithelial cells lining the tubular glandular structures are destined to evolve into the many cell types that populate the airways, including serous, goblet, ciliated, Clara and alveolar cells.
This stage comprises the period commencing on the 16th/17th week and continues to
weeks 25th-27th of gestation. The enlargement of the lumina of bronchi and terminal bronchioles characterizes the canalicular stage, in addition to the formation of capillaries at the site of the future air space and the appearance of surfactant, resembling the major developments that are absolutely crucial to extra-uterine life. During this stage, in addition, the acini subdivisions are formed, and the epithelial lining begins to differentiate into alveolar type I (ATI) and II (ATII) cells.
The saccular stage, or terminal sac stage (28th-35th week of gestation), represents the
development of terminal air sacs from alveolar ducts, refinement of gas exchange sites, decrease in the thickness of the interstitium, thinning of the epithelium and separation of the terminal air units. This stage also marks the terminal differentiation stages of alveolar ATI and ATII epithelial cells.
The final 5 weeks of fetal lung development, termed the alveolar period, encompass the
alveolar stage in which millions of alveoli are formed, with the surface area increased by thinning of the septal walls and attenuation of the cuboidal epithelium. The terminal sub-saccules are now separated by loose connective tissue and cellular maturation continues specifically with ATII cells developing a greater density of lamellar bodies.
Originally, the fetal lung develops as a fluid-filled organ; as respiration commences most
of the lung fluid is reabsorbed into blood and lymph capillaries, allowing the newborn to breathe normally. Postnatal lung development continues and the ≈ 50 million alveoli at birth, which have a surface area of 3-4 m2, represent ≈ 15-20% of the 300 million alveoli present in the adult lung (surface area ≈ 75-100 m2).
Concomitant with the development of various lung structures is the cellular
differentiation of ATI and ATII cells occurring as the alveolar epithelium matures. During the first four months of gestation the epithelial lining is more or less columnar to cuboidal. By six months ATI and ATII cells can be relatively distinguished in the more localized differentiated zones of pseudo-cuboidal cells. ATI cells are thin, flat and squamous epithelia conspicuous due to their small perinuclear body and long cytoplasmic extrusions; they are developed from the cuboidal cells that line bronchioles and cover most of the alveolar wall at later stages of development. ATI cells are characterized by having a low compliment of organelles indicating low metabolic activity, thus reflecting the quiescent natures of these cells. The morphology of ATI cells, however, is suitably convenient to provide a large surface area with a small volume, ideal therefore for rapid and efficient gas exchange.
Biomedical Research Scientist and Educating Professor – 2009 Page | 47

John J. Haddad, PhD
ATII cells are identifiable owing to their granular and cuboidal appearance, as a result of the dense packing of cytoplasmic organelles (indicating metabolically active cells), and lamellar bodies, dense layered organelles that synthesize and store pulmonary surfactants. The major function of a surfactant, which is a mixture of surfactant proteins and disaturated dipalmitoyl phosphatidylcholine, is to reduce the surface tension, thus facilitating lung expansion during inhalation/exhalation. ATII cells are small in diameter (≈ 400 μm3 in rat and ≈ 900 μm3 in human) but otherwise essential for proper gas exchange. Situated at the corners of the alveolar sacs, ATII cells represent little obstruction to gaseous diffusion and are fed by a capillary network. Intracellularly, these cells are richly endowed with cytoplasmic organelles associated with the biosynthesis of surfactant phospholipid and related proteins. In summary, ATII cells function to serve as thin, gas-permeable entities for diffusion and act as a protective barrier against water and electrolyte leakage.
The airway epithelium is not only an inert barrier but also a major participant in
signaling mechanisms during development and under pathophysiological conditions. Any damage caused to the airway epithelium can adversely affect its normal physiology and regulatory processes. The major functions of the airway epithelium include the following: i) It acts as a physiological barrier to diffusion and osmotic processes; ii) the epithelial layer has an integral metabolic function by synthesizing and degrading chemical components either endogenously produced or exogenously introduced; and iii) it possess a secretory property in that the epithelium has an inherent capacity to produce mucous, cytokines and chemokines, hormones, growth factors and enzymes. This underlies the significance of a physiologically competent epithelium where in the case of metabolic failure or noxious damage this would lead to abnormalities in the normal development and functioning of the lung.
The fetal lung develops as a fluid-filled organ and is continuously situated in an
environment that is relatively hypoxic (≤ 3 % O2), resembling the potential O2-carrying capacity of the umbilical vein. At birth the lung undergoes a dramatic change from a fluid-filled to a gas-filled organ, thereby subjecting the fetus, or the neonate, lung to a transition from a relatively hypoxic environment to that which is hyperoxic (10-15 % O2). The transition from placental to pulmonary-based respiration, therefore, constitutes a potential signaling mechanism for the continuation of lung development and maturation as the lung experiences dramatic and dynamic variations in pO2.
Although the transition from placental to pulmonary-based respiration is perceived
as normal in fully mature babies, the pre-term infants suffer tremendously as the lungs are insufficiently developed, thus incapable of sustaining normal breathing. The neonate as such can suffer from a variety of clinical illnesses with the probability of developing chronic lung diseases with the supplementation of exogenous O2. In normal breathing, however, the incomplete reduction of inhaled O2 may lead to accumulation of toxic reactive O2 species that may contribute to capillary injury and lung tissue perturbations.
All forms of aerobic life are thus faced with the threat of oxidation from molecular O2
and have developed elaborate mechanisms of antioxidant defenses to cope with this potential problem. Any deviation from homeostatic, or physiological, changes in pO2 is recognized as an exposure to oxidative stress. In particular, key developmental changes in the late gestational
Biomedical Research Scientist and Educating Professor – 2009 Page | 48

John J. Haddad, PhD
(pre-term) lung have evolved to allow production of surfactants and enzymatic and non-enzymatic antioxidants in preparation for the first breaths at birth. Moreover, the chronology of maturation of the lung antioxidant system parallels that of prenatal maturation of the surfactant system, pointing out to the developing stages fetuses undergo in preparation to be born into an O2-rich environment. Apparently, any perturbations in maintaining homeostatic mechanisms in response to changes in O2 levels are critical, thus determining cellular characteristic integrity. The clinical, biochemical and histologic responses of the lungs to such variations consequently characterize the efficiency and specificity of the antioxidant system in combating stress. In certain lung pathophysiological conditions, for instance, oxygenation of terminal airways becomes uneven, such that this temporal and spatial variance in O2 abundance essentially determines the survival of lung cells via O2-dependent activation of cell regulators and genes critical to defending their structural/functional characteristics.
Accumulating evidence in recent years has linked the pathogenesis of human
diseases to increased oxidative stress. In particular, reactive O2 species (ROS), which are partially reduced metabolites of O2, may contribute to alveolar-capillary membrane perturbations and the development of lung injury. A wealth of data has drawn attention to the significance of maintaining reducing conditions in cells and to fight against the damaging effect of ROS intermediates. Oxidants, for instance, can cause carcinogenesis, sclerosis, Alzheimer’s disease and other neurological disorders, acute lung injury and chronic lung diseases. Oxidative cell injury involves the modification of cellular macromolecules by ROS, often leading to cell death and lysis of sensitive cells, resulting in the microvascular and alveolar perturbations. Oxidative stress appears to increase lung antioxidants in some experimental models, and hypoxia and hyperoxia modulate fetal lung growth. Furthermore, there is growing evidence supporting the concept of cross talk between oxidative stress and up-regulation of a pro-inflammatory signal, through participation of cytokines. These cytokines, which are peptide hormones that participate in autocrine and paracrine signaling, are major participants in the pathophysiology of respiratory distress and have been recognized as signaling molecules responsive to dynamic variation in pO2. Cytokines, such as interleukin (IL)-1β, IL-6, IL-8, tumor necrosis factor (TNF)-α, transforming growth factor (TGF-β), granulocyte macrophage colony stimulating factor (GM-CSF) and a plethora of other inflammatory mediators play important roles not necessarily inflammatory during fetal life, initiation of labor, and in neonatal immunity and diseases. Hematopoietic growth factors, for example, regulate the maturation of progenitors in fetal and neonatal hematopoietic organs. Cytokines act as extra-hematopoietic growth factors, modulators of feto-maternal tolerance and are involved in selective apoptosis during tissue remodeling. Inter-regulation of cytokine networks is therefore critical for normal function and maturation of neonatal host defenses. Antigen specific immunity develops later in life and neonates initially depend on natural (innate) immunity. Cytokines regulate innate immunity and connect it with antigen specific adaptive immunity. This integral association between oxidative stress and a pro-inflammatory state may effect cellular reduction-oxidation (redox) potential, thereby imposing a direct role in modulating the pattern of gene expression in lung tissues and, accordingly, could be pivotal in determining cellular fate under these conditions.
As the fetus leaves a hypoxic environment and enters a relatively hyperoxic environment
during the transition from placental to pulmonary-based respiration, it is imperative that the fetus develops antioxidant mechanisms to guard against the potential harm posed by O2-derived
Biomedical Research Scientist and Educating Professor – 2009 Page | 49

John J. Haddad, PhD
species. Defense mechanisms, for example, include the reduction of ROS by antioxidant enzymes such as catalase, manganese-, copper- and zinc-containing superoxide dismutase (Mn-SOD/Cu-SOD/Zn-SOD), and glutathione peroxidase. ROS may not, however, impose a real threat to the fetus if these end products were detoxified and balanced against the amount of ROS generated. In keeping with this, glutathione (GSH), one of the major detoxifying antioxidants, has evolved as the most abundant thiol in almost all mammalian cells. GSH determines intracellular redox potential and detoxifies harmful ROS by the glutathione-peroxidase-coupled reaction (Fig. 1). Therefore, the transfer of O2 signaling across membranes to intercellular compartments may be converted into, and coupled to, a certain redox state that might be crucial in regulating the magnitude and pattern of gene expression of O2- and redox-responsive transcription factors. Such transcription factors are implicated in determining cellular responses under both physiological and pathophysiological conditions.
ig. 1. The schematic of the redox cycle shows an intimate relationship between antioxidant enzymes and
NADPH/H comes from the conversion of glucose, a reaction blocked by dehydroepiandrosterone (DHEA).
L-Glutamate + L-Cysteine
ROS (H2O2)
ATPγ-GCS
L-γ-Glutamyl-L-cysteine
Glycine/AT
BSO
PGS
Fglutathione. Glutathione (GSH) is synthesized from amino acids by the action of γ-glutamylcysteine synthetase (γ-GCS), the rate-limiting enzyme, and glutamyl synthase (GS). This reaction requires energy, is ATP-limited and is specifically inhibited at the level of γ-GCS by L-buthionine-(S,R)-sulfoximine (BSO). GSH undergoes the glutathione-peroxidase (GSH-PX) coupled reaction, thereby detoxifying reactive oxygen species (ROS) such as hydrogen peroxide (H2O2). A major source of H2O2 is the biochemical conversion of superoxide anion (O2
−.) by the action of superoxide dismutase (SOD). During this reaction, GSH is oxidized to generate GSSG, which is recycled back to GSH by the action of glutathione reductase (GSSG-RD) at the expense of reduced nicotinamide (NADPH/H+), thus forming the redox cycle. The reduction of the glutathione pathway is blocked by the action of 1,3-bis-(2-chloroethyl)-1-nitrosourea (BCNU). The major source of
+
Glutathione (2GSH)
Glutathione (GSSG)
GSSG-RDGSH-PX
ROH (H2O)
O2−.
SOD
NADPH/H+
NADP+
BCNU
DHEA
G-6-P 6-PG
Glucose
Biomedical Research Scientist and Educating Professor – 2009 Page | 50

John J. Haddad, PhD
Redox-sensitive transcription factors are therefore likely to be differentially regulated by O2 availability, bind specific DNA consensus sequences, and activate the expression of several genes, particularly those controlling adaptive homeostasis to a hostile environment. Among such factors, the hypoxia-inducible factor-1α (HIF-1α) and nuclear factor-κB (NF-κB), whose activation states are differentially regulated in oxidative stress, remains of particular importance. HIF-1α first identified in vitro as a DNA-binding activity expressed under hypoxic conditions, has its concentration and activity increased exponentially on lowering O2 tensions over physiologically relevant ranges.
The ubiquitous activation of HIF-1α is thus consistent with the significant role that this
factor
gen sensing and the underlying molecular stratagems utilized from prokaryotes to the ost co
odels for O2 sensing, certain pharmacological studies suggested that O
CytFe2+ + O2 → CytFe2+O2
CytFe3 AD(P)
plays in coordinating adaptive responses to hypoxia. NF-κB, on the other hand, first identified as a transcriptional factor that regulates antibody release in B cells, is central to the regulation and expression of stress response genes in the face of inflammatory and oxidative challenge.
Oxym mplex eukaryotes have been the focus of experimental investigations trying to find an answer to the question: “What is the identity of the O2 sensor?” The proposed molecular mechanism underlying O2 sensing in mammalian cells involved an O2 sensor that is a heme protein. Studies on erythropoietin (EPO), a glycoprotein hormone required for the proliferation and differentiation of eryhthroid cells, demonstrated that EPO production was enhanced under hypoxic conditions. Furthermore, the induction of EPO expression by transition metals, such as cobalt and nickel, supported the hypothesis that the O2 sensor for the induction of this glycoprotein is a heme protein and that these metal atoms can substitute for the iron atom within the heme moiety. Further evidence supporting the notion that the O2 sensor is a heme protein came with additional studies utilizing carbon monoxide (CO), which can non-covalently bind to ferrous (Fe2+) heme groups in hemoglobin, myoglobin, cytochromes and other heme proteins, where its ligation state is structurally identical to that of O2. It was subsequently proposed that the effect of CO on O2 sensing might occur via locking the sensor in an oxy conformation, which could involve a multi-subunit mechanism.
In addition to the aforementioned m
2 sensing might involve a microsomal mixed function oxidase. Based on these studies, it was suggested that O2 sensing for EPO involve an interaction between cytochrome P450 and cytochrome P450 reductase, thereby allowing the conversion of molecular O2 to superoxide and hydrogen peroxide radicals. Another has provided support for the central role of an oxidase in O2 sensing based on spectral evidence. It was found that b-cytochrome functions as a NAD(P)H oxidase, converting O2 to superoxide. The enzymatic complex in mammalian cells is membrane-bound and transduces the conversion of molecular O2 to ROS, according to the following equations:
CytFe2+O2 → CytFe3+ + O2
−
+ + NAD(P)H → CytFe2+ + N
Biomedical Research Scientist and Educating Professor – 2009 Page | 51

John J. Haddad, PhD
Further experimen the mitochondrion in O2 sensing
CO + 2Fe + H2O → CO2 + 2Fe2+ + 2H+
The hypothesis that there exists a specific O2 sensor(s), which relays chemical signals
intracel
he expression of genes is predominantly determined by conditions of the
ts showed a likely potential involvement of . For instance, a spectroscopic photolysis with monochromatic light has identified a CO-
binding heme protein falling within the spectrum of the mitochondrial cytochrome a3. It was consequently proposed (Dr. Haddad) that a heme protein, presumably located on the plasma membrane, has a low affinity for O2 and a relatively high affinity for CO. The same model predicted that another heme protein in the mitochondrion has a relatively higher affinity for O2 and a lower affinity for CO. The biochemical reaction, which was proposed as an alternative way of regenerating ferroheme in the O2 sensor, is given below:
3+
lularly, is therefore consistent with the notion that there is a mechanism involved in transducing dynamic changes in pO2 to the nucleus. In response to ΔpO2 there is a coordinate expression of genes needed to confer appropriate responses to hypoxia or hyperoxia. The regulation of physiological important O2-responsive genes, therefore, would dictate well-controlled responses of the cell within a challenging environment and necessarily determines the specificity of cellular adaptation.
T
microenvironment of cells. Prime examples of such regulation are found in embryonic development of all multicellular organisms. The naturally occurring regulating agents, for example, interact with specific receptors, which subsequently transduce a signal onto the nucleus for the regulation of gene expression/activation. The putative O2 sensor responds to dynamic variation in pO2 such as those occurring during the birth transition period. Upon ligand binding, this membrane-bound receptor transduces intracellular chemical/redox signals that relay messages for the regulation of gene expression, a phenomenon mainly involving the activation of transcription factors.
In order to maintain O2 homeostasis, a process that is essential for survival, pO2 delivery
to the
daptive responses to hypoxia involve regulation of gene expression by HIF-1α, whose express
magnitude of the response was inversely related to pO2.
mitochondrial electron transport chain must be tightly maintained within a narrow physiological range. However, this system may fail with subsequent induction of hypoxia, resulting in a failure to generate sufficient ATP to sustain metabolic activities, or hyperoxia that contribute to the generation of ROS, which, in excess, could be cytotoxic and often cytocidal.
Aion, stability and transcriptional activity increase exponentially on lowering pO2. HIF-1α
is the only known mammalian transcription factor expressed uniquely in response to physiologically relevant hypoxic conditions. Studies of the EPO gene led to the identification of a cis-acting hypoxia-response element (HRE) in the 3′ -flanking regions. HIF-1 was identified as a hypoxia-inducible HRE-binding activity. The HIF-1 binding site was subsequently used for purification of the HIF-1α and HIF-1β subunits by DNA affinity chromatography. Both HIF-1 subunits were basic helix-loop-helix (bHLH)-PAS proteins: HIF-1α was a novel protein; HIF-1β was identical to the aryl hydrocarbon receptor nuclear translocator (ARNT) protein. HIF-1α DNA-binding activity and HIF-1α protein expression were rapidly induced by hypoxia and the
Biomedical Research Scientist and Educating Professor – 2009 Page | 52

John J. Haddad, PhD
In hypoxia, multiple systemic responses are induced, including angiogenesis, erythropoiesis and glycolysis. HREs containing functionally essential HIF-1-binding sites were identifi
llels this to decreased O2 availability through expression and activation of glucose transporters and glycolytic enzymes. EPO is responsible for increas
own to activate transcription of genes encoding inducible nitric oxide synthase (iNOS) and heme oxygenase 1 (HO-1) ⎯ which are responsible for the synthesis of the
directly controlled by HIF-1α is given in Table 1. It is expected that any reduction of tissue oxygenation in vivo and in vitro would therefore provide a mechanistic stimulu
ed in genes encoding vascular endothelial growth factor (VEGF), glucose transporter 1 (GLUT-1), and the glycolytic enzymes aldolase A, enolase 1 (ENO-1), lactate dehydrogenase A (LDH-A) and phoshoglycerate kinase 1. HIF-1α is an important mediator for increasing the efficiency of O2 delivery through EPO and VEGF.
A well-controlled process of adaptation para
ing blood O2-carrying capacity by stimulating erythropoiesis; VEGF is a transcriptional regulator of vascularization; and glycolytic transporters and enzymes increase the efficiency of anaerobic generation of ATP.
HIF-1 has also been sh
vasoactive molecules nitric oxide (NO) and CO, respectively ⎯ and transferrin ⎯ that, like EPO, is essential for erythropoiesis. Each of these genes contains an HRE sequence of < 100 bp that includes one or more HIF-1-binding sites containing the core DNA consensus sequence 5′-RCGTG-3′.
The array of genes
s for a graded and adaptive response mediated by HIF-1α. Hypoxia signal transduction is schematized in Fig. 2.
Biomedical Research Scientist and Educating Professor – 2009 Page | 53

John J. Haddad, PhD
Table 1. Direct HIF-1 target genes. ______________________________________________________________________________ Glucose/Energy Metabolism and Cell Proliferation/Viability Adenylate Kinase 3, Aldolase A, Aldolase C, Enolase 1 (ENO-1), Glucose Transporter 1, Glucose Transporter 3, Glyceraldehyde-3-phosphate Dehydrogenase, Hexokinase 1, Hexokinase 2, Insulin-like Growth Factor 2 (IGF-2), IGF Binding Protein 1 (IGFBP-1), IGFBP-3, Lactate Dehydrogenase A, Phosphoglycerate Kinase 1, Pyruvate Kinase M, p21, Transforming Growth Factor 3 (TGF3). Erythropoiesis and Iron Metabolism Ceruloplasmin, Erythropoietin (EPO), Transferrin, Transferrin Receptor. Vascular Development/Remodeling and Vasomotor Tone Adrenergic Receptor, Adrenomedullin, Endothelin-1, Heme Oxygenase 1 (HO-1), Nitric Oxide Synthase 2, Plasminogen Activator Inhibitor 1, Vascular Endothelial Growth Factor (VEGF), VEGF Receptor FLT-1. ______________________________________________________________________________
Biomedical Research Scientist and Educating Professor – 2009 Page | 54

John J. Haddad, PhD
↓ [O2]
H2O2
CatalaseCO
DesferrioxamineThioredoxin
Δ Redox
GenisteinNaF V-Src
Δ Phosphorylation
MG-132
↑ HIF-1α protein stability
↑ HIF-1α transcriptional activity
↑ Expression of hypoxia-inducible genes Fig. 2. Hypoxia signal transduction. Reduction of cellular O2 concentration is associated with redox changes that lead to altered phosphorylation of HIF-1α, which increases its stability and transcriptional activity, resulting in the induction of downstream gene expression. Putative inducers (horizontal arrows) and inhibitors (blocked arrows) of different stages in the proposed pathway are indicated.
Biomedical Research Scientist and Educating Professor – 2009 Page | 55

John J. Haddad, PhD
An important and widely investigated transcription factor that is a major participant in signaling pathways governing cellular responses to environmental stresses is NF-κB. First identified as a factor that regulates the expression of the immunoglobulin κ light chains in B lymphocytes, NF-κB is now recognized, as Dr. Haddad indicates, as a sequence-specific transcription factor involved in the activation of an exceptionally large number of genes in response to inflammation, viral and bacterial infections, and other stressful situations requiring rapid reprogramming of gene expression such as in oxidative challenge.
In un-stimulated cells under resting conditions (inactive state), NF-κB exists as homo- or
heterodimers of members of the Rel family. The dimers of NF-κB are sequestered in the cytosol through non-covalent interactions with inhibitory proteins termed IκBs, of which the most important may be IκB-α. The translocation and activation of NF-κB in response to various stimuli, such as cytokines (IL-1 and TNF-α), microbial agents (endotoxin lipopolysaccharide; LPS), oxidative challenge (ROS) and irradiation (UV and γ-rays), are sequentially organized at the molecular level. NF-κB activation occurs through the signal-induced phosphorylation of a multisubunit upstream kinase, termed IκB kinase (IKK), by NF-κB inducing kinase (NIK). Stimulation leads to rapid phosphorylation of IκB, thereby marking it for ubiquitinylation and ultimately proteolytic degradation. This exposes the nuclear localization signal on NF-κB, thus allowing the nuclear translocation of the subunit and activation of the transcription of its target genes. The array of genes directly controlled by NF-κB is given in Table 2.
IκB-independent pathways, however, have been recently recognized as alternative factors
that regulate the activation of NF-κB, Dr. Haddad notes. As an example, direct phosphorylation of RelA (p65), the major transactivating member of the κB family, in one of two of its transactivation domains, has been shown to regulate NF-κB activation. A further mechanism was revealed for NF-κB regulation with the discovery of transcription factor-IIB/D (TF-IIB/D) and TATA-binding protein (TBP), recognized as two important regulators of NF-κB transcriptional activity. The dominant-negative form of mitogen-activated protein kinase (MAPK) (MAPKp38) expression vector abrogated the interaction of TF-IID/TBP with a co-transfected His-p65 fusion protein, and selective inhibition of MAPKp38 by SB203580 reduced TF-IID/TBP in vitro. Finally, modulating intracellular redox equilibrium constitutes such a potential mechanism that can manipulate the localization and activation of NF-κB. Hyperoxia and other stress conditions signal transduction schematized model is shown in Fig. 3.
Biomedical Research Scientist and Educating Professor – 2009 Page | 56

John J. Haddad, PhD
Table 2. Direct NF-κB target genes ______________________________________________________________________________ Cytokines/Growth Factor Interleukin (IL)-1α, IL-1β, IL-2, IL-3, IL-6, IL-8, IL-12, Tumor Necrosis Factor (TNF)-α, Lymphotoxin (LT)-α, Interferon (IFN)-β, Granulocyte Colony-Stimulating Factor (G-CSF), Macrophage Colony-Stimulating Factor (M-CSF), Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF). Cytokine Receptors IL-2 Receptor α-Chain (IL-2Rα). Stress Proteins Serum Amyloid A Protein (SAA), Complement Factors B, C3 and C4, α1-Acid Glycoprotein. Adhesion Molecules Intracellular Adhesion Molecule 1 (ICAM-1), Vascular Cell Adhesion Molecule 1 (VCAM-1), Mucosal Addressin Cell Adhesion Molecule 1 (MAdCAM-1), E-Selectin. Immunoregulatory Molecules Immunoglobulin κ Light Chain (Igκ), Major Histocompatibility complex (MHC class I and II), T-Cell Receptor (TCRα and β), β2-Microglobulin, Invariant Chain (Ii), Transporter Associated with Antigen Processing (TAP-1), Proteasome Subunit (LMP-2), Inducible Nitric Oxide Synthase (iNOS), Inhibitory κB (IκB), p53. ______________________________________________________________________________
Biomedical Research Scientist and Educating Professor – 2009 Page | 57

John J. Haddad, PhD
Environmental Stress
LPS Cytokines Hyperoxia UV
PAK/RAC
MEKK/NIK
MKK IKK IκB/NF-κBp38/RK
MAPKAP-K2Hsp27
ARE
IκB Degradation
NF-κB Translocation
Stress Gene Expression and Regulation
SB203580
Fig. 3. Hyperoxia and other stress conditions signal transduction. Environmental stress, including hyperoxia, is associated with the activation of upstream kinases that diverge at either activating the p38 mitogen-activated protein kinase pathway or the IκB/NF-κB pathway. Translocation of NF-κB into the nucleus triggers the activation of stress-response genes. Abbreviations: ARE, AU-rich element; Hsp27, heat shock protein 27; IκB, inhibitory κB; IKK, IκB kinase; MEKK, mitogen-activated protein kinase kinase kinase; MKK, mitogen-activated protein kinase kinase; NIK, NF-κB inducing kinase; RAC/PAC, small G-coupled proteins; RK, reactivity kinase.
In the last few decades since the term apoptosis was coined, a vast quantity of work has been performed in search of the cause of the phenomenon it originally alluded to. It became certain, moreover, that some cells were genetically programmed, or destined, for death during the normal development of multi-cellular organisms. Thus the general model is one of intercellular signaling molecules playing on intracellular effector systems that balance the individual cell’s progress to either life (survival) or death (apoptosis).
Apoptosis, first identified as shrinkage necrosis, was originally observed in mature
human/vertebrate tissues as stochastic loss of cells that showed distinctive histopathic morphology and induced minor inflammatory response. Simply, it was argued by Dr. Haddad and other colleagues that the key tenets of this model state that there is a universal genetic program that governs cell death at different stages of development, that a variety of stimuli can elicit or activate this program, and that even though many transduction mechanisms
Biomedical Research Scientist and Educating Professor – 2009 Page | 58

John J. Haddad, PhD
are involved, eventually apoptosis requires activation of a downstream converging, final common pathway.
At least two distinct forms of death are known by which cells were found to undergo
death: i) the well-characterized, and usually rapid, necrotic tissue damage induced by trauma and noxious stimuli, and ii) a more protracted and morphologically distinct form of cell death that was then termed apoptosis. In apoptosis, cells often shrink, dissociate from surrounding cells and undergo cytoplasmic membrane blebbing, when their chromosomes rapidly condense and aggregate around the nuclear periphery and small apoptotic bodies are formed. However those during apoptosis cellular organelles do retain their definition for a long time, the nucleus in particular displays a distinctive pattern of heterochromatization and eventual fragmentation. In many, but not all, apoptotic cells, the condensed chromosomes are acted upon by specific nucleases that cleave the DNA, thereby producing a characteristic ladder.
Necrotic cell death, on the other hand, is relatively violent, characterized by cytoplasmic
swelling, the rupturing of cell membranes, the dilatation of the mitochondrion, and the disintegration of subcellular and nuclear components. Conversely, apoptosis is characterized by an ordered series of events that take place over a longer period of time. Although necrosis may be more analogous to random acts of violence that characterize murder, apoptosis is more appropriately referred to as cellular suicide. The cell initiates apoptotic death when it senses that its environment of physical state has been vigorously compromised; this is, indeed, the ultimate self-sacrifice.
If there is no simple dichotomy in modes of cell death, perhaps there is more than
one basic genetic program for death and more than one final common pathway (Fig. 4). Perhaps some ligand-induced cell death could result from confused or inappropriate regulation of gene expression rather than from turning on pre-set genetic programs. Studies of C. elegans development, for example, have contributed significantly to the bio-molecular understanding of cell death. Their genetic analysis led to the identification of cellular genes required for programmed cell death during the development of C. elegans. The isolation and molecular characterization of C. elegans death (ced) genes demonstrated that ced-3 gene was homologous to the mammalian interleukin-1β-converting enzyme (ICE; Caspase-1). ICE was originally isolated from mammalian cells as an enzyme essential for the proper processing and biologic activation of pro-interleukin-1β, a cytokine involved in mediating cellular inflammatory processes. The expression of ced-3/ICE rapidly induced apoptosis, demonstrating that this gene encodes a cysteine protease essential for programmed cell death.
Molecular cloning isolated many of the ICE-like proteases, henceforth referred to as
caspases, Dr. Haddad notes. The term caspase is based on a common nomenclature unanimously adopted: c reflects a cysteine protease mechanism, and aspase refers to the ability of these proteases to cleave a protein following an aspartic acid residue. Many of these caspases contain a conserved sequence, QAC(R/Q)G, required for the catalytic activity of these enzymes. The activation of the caspase proteases has been linked to the aggregation of cell surface receptors, when receptor-sensitive target cells are exposed to the appropriate ligand, or when the receptor self-aggregate in response to their high cell-surface density. Therefore, a functional caspase enzyme can be generated following receptor oligomerization by autocatalysis, or by the
Biomedical Research Scientist and Educating Professor – 2009 Page | 59

John J. Haddad, PhD
action of another alerted caspase. Recent evidence suggested that caspases regulate the process of apoptosis by regulating additional cellular processes such as the progression through the well-defined cell cycle and its regulators (Fig. 4).
Agent-1
Non-caspase Proteases
Various Biochemical SequelaeDNA Lysis
Lamin BreakdownPhosphatidylserine Asymmetry
Membrane Blebbing
Agent-2
Caspase Activation
Agent-3
Altered Gene Expression
MacromolecularSynthesis Inhibitors
Morphologic Changes: Cell Death
Caspase InhibitorsClass-specific Protease Inhibitors
Fig. 4. Potential pathways to apoptosis (cell suicide), or programmed cell death.
As noted, multicellular organisms eliminate redundant, damaged, or infected cells by a stereotypic program of cell suicide. The first mammalian regulator of apoptosis emerged when bcl-2, the gene activated by chromosome translocation in human follicular lymphoma, was unexpectedly found to permit the survival of cytokine-dependent hematopoietic cells, in a quiescent state, in the absence of exogenous cytokine. Ced-9 of C. elegans and the mammalian Bcl-2 proved to be functional and structural homologues, and their survival function is opposed either by close relatives such and Bax, or by distant cousins such as the mammalian Bik, or Nbk, and nematode EGL-1. All members possess at least one of four conserved motifs known as Bcl-2 homology domains (BH-1 – BH-4). Most pro-survival members, which can inhibit apoptosis in the face of a wide variety of cytotoxic insults, contain at least BH-1 and BH-2, and those most similar in structure to Bcl-2 have all four BH domains.
Pro- and anti-apoptotic family members can heterodimerize and seemingly titrate one
another’s function, suggesting that their relative concentration may act as a rheostat for the suicide program. Bcl-2 resides on the cytoplasmic face of the mitochondrial outer membrane, endoplasmic reticulum (ER), and nuclear envelope and may register damage to these
Biomedical Research Scientist and Educating Professor – 2009 Page | 60

John J. Haddad, PhD
compartments and affect their behavior, possibly by modifying the flux of small molecules and proteins. On the other hand, Bax, for instance, is cytosolic before an apoptotic stimulus, even though it, like most other family members, bears a hydrophobic domain. Biochemical evidence suggested that the pro-survival proteins might function by directly inhibiting the activity of caspases, directly or indirectly prevent the release of cytochrome c from the mitochondrion, which, along with ATP, may facilitate structural changes in the pro-caspase domain, allowing its cleavage and activation. Bax and Bax-like proteins and the anti-survival proteins may promote apoptosis by cleaving and activating caspases, but also can initiate caspase-independent death via channel-forming activity, which could promote the mitochondrial permeability transition or puncture the mitochondrial outer membrane.
Reactive oxygen species (ROS) are generated in all aerobic cells during normal
mitochondrial respiration, are used by specialized phagocytic cells to destroy invading pathogens and are byproducts of the intracellular metabolism of toxic drugs and their environmental metabolites. Dr. Haddad observed that incubating cells, for instance, with exogenous oxidants, free radicals, or adding redox-active compounds, has been shown to trigger the apoptotic process. It has been hypothesized that the oxidation of cellular molecules could trigger general protection alert system, and these sensors in turn detect and assess the damage and subsequently activate the apoptotic machinery. A classic example of this model is p53-mediated detection of DNA damage. Alternatively, low-level oxidative stress is known to activate different protein cascades and transcription factors, such as AP-1, HIF-1α, and NF-κB. In addition, the ability of nitric oxide (NO) to rapidly react with the heme group of guanylate cyclase has been used as a message in activating downstream apoptotic cellular pathways. Recent studies, moreover, have focused on the role of hydrogen peroxide (H2O2) in regulating apoptosis in several cell models. This peroxide is an oxidant that has been shown to trigger caspase activation and subsequent apoptosis. Of note, H2O2-mediated caspase activation was dependent on the release of cytochrome c from the mitochondrion, suggesting that a key role for this peroxide in mitochondrial permeability and leakage.
The importance of dissecting the pathway from oxidative stress/redox signaling to
apoptosis is not restricted to models where cells are exposed to exogenous oxidants. Intracellular oxidant production has been detected in cells incubated with a wide range of seemingly independent apoptotic agents, and some of these changes are suggested, however, to occur sufficiently early to be intricately involved in the activation of apoptosis. A recent example is p53-mediated apoptosis, which is hypothesized to occur by the increased transcription of pro-oxidant factors, thereby leading to caspase activation and apoptosis. Interestingly, antioxidants and glutathione (GSH) precursors conferred protective effect against ROS-mediated injury and subsequent apoptosis, implicating a critical role for ROS in initiating the death machinery.
It is becoming apparent that the redox status of a cell can have complex and multi-layered
effects on apoptosis (Fig. 5). It was postulated that the mitochondrion could be the principle sensor and that the release of such mitochondrial factors as cytochrome c is the critical event leading to caspase activation, and hence propagation of apoptosis. The effectors of apoptosis, in particular the caspases, are redox sensitive, and the cell needs to maintain a strict reducing environment for these to function. Therefore, by reasoning it’s recorded that apoptosis cannot occur in cells subjected to excessive oxidative stress. However, the ability of oxidants to inhibit
Biomedical Research Scientist and Educating Professor – 2009 Page | 61

John J. Haddad, PhD
caspase function need not be incompatible with oxidant-dependent caspase activation. First, low levels of oxidants appear sufficient for optimal caspase activation, suggesting specific signaling pathways rather than widespread oxidation. Second, the observed ability of cells to repair or replace oxidized caspases indicates that the full complement of apoptotic effector molecules can be returned at some time after the initial oxidative stress. Elucidation of the pathways of oxidant-induced apoptosis, therefore, may provide alternate therapies to scavenging of the initial oxidant in those systems where excessive oxidative stress and redox disequilibrium leads to inevitable cell death.
Mitochondrial ‘Sensor’
Exogenous Oxidative Stress
Endogenous Oxidative Stress
External Stimulus
Cytochrome c
Pro-caspase
Active Caspase
Excessive Oxidative Stress
AntioxidantsGSH Precursors
PhosphatidylserineAsymmetry
Nuclear ChangesDNA Damage
Membrane andMitochondrial
Changes Fig. 5. Oxidant and redox regulation of apoptosis.
Accumulating evidence has linked the pathogenesis of a variety of diseases to oxidative stress. Derived O2 species, in particular, may contribute to alveolar-capillary membrane perturbations and development of lung injury. Oxidative injury involves the modification of cellular macromolecules by toxic by-products of O2 metabolism. This condition often leads to cell death and/or necrotic lysis of sensitive cells, which result in the microvascular and alveolar injury typical of pulmonary O2 toxicity. Thus, dynamic variation in alveolar pO2 and its effect on cellular redox state may impose a direct role in modulating the pattern of gene expression and, thus, could be crucial in determining cellular fate and the inflammatory process regulated by cytokines.
Reactive oxygen species (ROS) play a crucial role in the initiation and progression
of pathophysiological conditions. The signaling mediators involved in stress-induced lung injury are regulated, at least in part, by ROS that up-regulate their secretion as part of antioxidant
Biomedical Research Scientist and Educating Professor – 2009 Page | 62

John J. Haddad, PhD
and immune defence mechanisms. For example, bronchial epithelial cells produce soluble mediators on exposure to ROS, which stimulate the release of glycoconjugates in vitro. In addition, ROS can induce the production of IL-6 and IL-8 in bronchial and epithelial cells, respectively. Furthermore, ROS can be released by many cell types in response to a variety of stimuli, such as TNF-α and lipopolysaccharide, and that they serve as intracellular signals for the activation of redox-sensitive transcription factors.
Among those mediators of oxidative stress, cytokines are of particular importance as they
serve as signaling cofactors. IL-1β, for instance, is a pleiotropic cytokine produced in response to various stimuli, and acts as a modulator of redox equilibrium. IL-6 is responsive to inflammatory stimuli and oxidative stress. TNF-α, a stress-induced cytokine and a mediator of oxidative injury, has been implicated in the pathogenesis of respiratory distress. Moreover, there is substantial evidence suggesting that IL-1β, among other regulatory cytokines, participates in transducing signals in oxidative injury via ROS, and have stimulatory effects on regulatory transcription factors mediating apoptosis. Cytokines, in addition, are shown to induce oxidative stress in several cell models.
The ‘biomarkers’ of oxidative stress, such as antioxidant inefficiency, redox
disequilibrium and derivation of oxidant radicals, for instance, may arise from conditions other than hyperoxia (oxidizing signals) per se, such as hypoxia/re-oxygenation and cytokine-dependent processes. In physiological conditions, the intracellular redox status of thiols is highly reductive. GSH, for example, is present in high concentrations in lung epithelial lining fluid and has been reported to maintain the integrity of the airspace epithelium in vitro and in vivo. In contrast, GSH depletion has been linked to the pathophysiology of idiopathic pulmonary fibrosis, adult respiratory distress syndrome, bronchopulmonary dysplasia and cystic fibrosis. Exogenous/endogenous agents which induce the formation of ROS, for example, can affect redox homeostasis by up-regulating antioxidant enzymes, particularly glutathione peroxidase and enzymes involved in glutathione recycling and biosynthesis. Furthermore, ROS signaling could be mediated by cytokines, whose participation in cellular pathways is modulated by redox status. Conversely, cytokines which themselves are mediators of oxidative stress have the potential to alter redox equilibrium, thereby affecting GSH/GSSG shuttling and recycling.
Dr. Haddad indicates that the immunopharmacological potential assigned to glutathione
stems from established observations. IL-1 induced responses, for instance, occurs through modulating redox equilibrium. In addition, ROS signaling regulating the transcription of IL-4, IL-6, IL-8 and TNF-α occurs through a thiol-dependent mechanism. Interestingly, antioxidants and glutathione precursors have been shown to down-regulate cytokine synthesis, activation and downstream processes. Among several agents that were used for repletion and depletion of GSH, N-acetyl-L-cysteine (NAC) and L-buthionine-(S,R)-sulfoximine (BSO) are, respectively, of particular importance as they exhibit antagonistic effects on a pro-inflammatory signal. NAC, an antioxidant and a GSH precursor, ameliorates cytokine production and ROS-mediated lung injury. In contrast, BSO, which depletes GSH by irreversibly inhibiting γ-GCS, the rate-limiting enzyme in the biosynthesis of glutathione, has the potential to enhance cytokine secretion by up-regulating ROS. It has been reasoned that a differential manipulation of glutathione homeostasis and shuttling may antagonistically affect a pro-inflammatory signal, thus
Biomedical Research Scientist and Educating Professor – 2009 Page | 63

John J. Haddad, PhD
bearing potential consequences for the treatment of respiratory distresses, where cytokines are recognized as major participants in their pathophysiology.
Dynamic changes in pO2 constitute a potential signaling mechanism for the regulation of
the expression/activation of redox-sensitive transcription factors, apoptosis signaling and pro-inflammatory cytokines. The transition from placental to pulmonary-based respiration causes a relative hyperoxic shift, or oxidative stress, which the perinatal developing lung experiences during birth. This project, proposed by Dr. Haddad, is primarily concerned with redox signaling and gene regulation, and the role of oxygenation in determining cell fate and the downstream inflammatory state. The hypotheses, to be tested in this thesis, are as follows:
1. Dynamic variation in ΔpO2 differentially regulates the compartmentalization
and function of transcription factors HIF-1α and NF-κB. 2. O2-evoked regulation of HIF-1α and NF-κB is closely coupled with the
intracellular redox state, such that modulating redox equilibrium affect their expression/activation.
3. The differential regulation of HIF-1α and NF-κB in vitro is paralleled by O2-
and redox-dependent pathways governing the regulation of these factors during the transition from placental to pulmonary-based respiration ex vivo.
4. The birth transition period in vitro and ex vivo regulate apoptosis signaling
pathways in a redox-dependent manner, consistent with NF-κB playing an anti-apoptotic function.
5. An association is established between an oxidative stress condition and the
augmentation of a pro-inflammatory state, regulated by the O2- and redox-sensitive pleiotropic cytokines.
The molecular response of the alveolar epithelium to oxidative stress is regulated, in part, by redox-sensitive transcription factors. The abrupt change in pO2, which accompanies the transition from placental to pulmonary-based respiration, constitutes such a mechanism that allows a specific genetic regulation. Such transcription factors that form an integral part of the pathways augmented during this transition period are HIF-1α and NF-κB, both of which are sufficiently tuned to govern a specific response in hypoxia and a relative hyperoxic shift. The activation of HIF-1α is consistent with its role in coordinating adaptive homeostatic responses under hypoxic conditions. The alveolar epithelium, in particular, is genetically responsive to changes in O2 availability, as it functionally acts as more than just a barrier by participating in signaling processes. The O2 history of the perinatal epithelium is continuously lying at ≈ 23-30 Torr (3%), the O2 transfer capacity of the umbilical vein in utero. The reasoning is that a relative hyperoxic shift during the birth transition period from placental to pulmonary-based respiration would rather constitute a potential signaling mechanism for the activation of O2-sensitive transcription factors.
Biomedical Research Scientist and Educating Professor – 2009 Page | 64

John J. Haddad, PhD
Maintenance of epithelial cells in vitro at static pO2 (23-30 Torr) allowed maximum induction of HIF-1α protein expression, followed by subsequent nuclear localization and activation, a phenomenon sustained with a shift towards the early postnatal distal lung pO2 (70-100 Torr). Of note, the observation that HIF-1α DNA binding activity ex vivo was sustained with the first respiratory movements, though lost thereafter, thereby providing functional evidence that the sensitivity of HIF-1α to sub-ambient pO2 is responsively heightened during birth. This supports the notion that HIF-1α might participate in signaling mechanisms existing beyond a strictly hypoxic environment and that its activity resides over a wider spectrum of pO2, which incorporates both fetal and early postnatal alveolar pO2. Fig. 6 shows oxygen sensing mechanisms and the role of HIF-1α.
ig. 6. The role of hypoxia and MAPK signaling pathways in the regulation of HIF-1 and HIF-1-dependent
Although the nuclear translocation and DNA binding activity of NF-κB were not
Hypoxia
CO
OxyDe-oxy
OxyDe-oxy
Low O2
OxyDe-oxy
OxyDe-oxy
High O2
Co2+
Ni2+
OxyDe-oxy
OxyDe-oxyNAD(P)H Oxidase?
ECF
ICF
O2
O2O2
O2 . -O2 . -
NADPH
NADP
H2O2
?
FentonReaction
ROS
Ubiquitin Degradation Pathway
T1/2
HIF-1α
T1/2
HIF-1α
ARNT/HIF-1βARNT/HIF-1β
HIF-1αHIF-1α
PP
Kinase(s)
MAPKERK; MAPKp38;
MAPKJNK; SAPK; PKC
p300CBP
HIF-1
Site
CREB
Site
HIF-1
Site
CREB
Site
Modulation of Hypoxia
Responsive Genes
Expression or Suppression
IP3 /DAG /ROS (+)AA (+)
Fgene transcription. detectable in epithelia maintained under steady pO2 state (23 or 152 Torr), its activity was substantially induced by shifts from fetal to early postnatal pO2 (76 Torr) and into moderate (152 Torr) and severe (722 Torr) hyperoxia. This suggests that NF-κB is primarily responsive to acute changes in O2 tension, which, at the lower end of the range, are coincident with early postnatal pO2. As with HIF-1α, the profile of NF-κB activation ex vivo follows the expected change in
Biomedical Research Scientist and Educating Professor – 2009 Page | 65

John J. Haddad, PhD
pO2 at birth becoming maximally active during the first few hours after birth, thereafter falling. Although NF-κB activation is well established as an early response to oxidative stress in the lung, studies based in fetal distal lung epithelial cultures indicate that there is a strong association between moderate hyperoxia, ROS production and NF-κB-regulated expression of epithelial Na+ channels pointing to the importance of this pathway as a modulator of developmental events in the lung at birth. Taken together, these data show that the activity profiles of both HIF-1α and NF-κB appear sufficiently tuned to mediate changeover from hypoxic to oxidative forms of genetic regulation at pO2’s, which are within the range of those expected with the first breaths at birth. The signal transduction components which link the availability of O2 to the activation of these transcription factors are poorly defined, but are broadly believed to hinge on the free abundance of intracellular oxidants (i.e. ROS). In the case of HIF-1α, for example, post-translational stability, nuclear translocation by the aryl hydrocarbon nuclear translocator, and consensus DNA binding are coupled with O2-associated changes in both conformation and activity of a ferro-heme containing protein, believed to express peroxide generation via a NADPH oxidase-type activity. Hypoxic cessation of peroxide production mediates HIF-1α stabilization, nuclear translocation and gene expression. The activation pathway for NF-κB, on the other hand, requires the disassociation from its cytosolic inhibitory subunit, IκB, an event that requires phosphorylation and favored by oxidizing conditions. Clearly, the extent to which cells express antioxidant defenses bears consequence for the efficiency with which an O2-linked signal can be transmitted from environment to effector protein.
While spanning the activation profile of HIF-1α and NF-κB at various ascending ΔpO2 regimen, an observation of immediate interest could be particularly noted. The protein expression and DNA binding activity of either factor are prominent under mild hypoxic conditions (70-100 Torr) with approximately identical kinetics. This observation strongly suggested a possible cross talk between these two factors at this particular ΔpO2, although the molecular basis of this mechanism has yet to be ascertained. In a manner similar to its activation state in vitro, NF-κB protein expression and consensus binding maximized with oxygenation, especially during the first few days after birth. Taken together, these observations, as Dr. Haddad notes, suggested that HIF-1α and NF-κB are sufficiently tuned to mediate the changeover from hypoxic to hyperoxic induction of regulatory genetic factors at different pO2 tensions resembling those falling within the range expected during the birth transition from placental to pulmonary-based respiration.
The major determinant of the redox potential in mammalian cells is glutathione (GSH; L-
γ-gluta
peroxidase coupled reaction, thus acting as an antioxidant. For example, endogenously produced
myl-L-cysteinyl-glycine), a tripeptide thiol. This ubiquitous non-essential sulfhydryl amino acid plays a major role in maintaining intracellular redox equilibrium and in regulating cellular defenses augmented by oxidative stress. Synthesized by the action of the rate-limiting enzyme γ-glutamylcysteine synthetase (γ-GCS), GSH uniquely provides a functional cysteinyl moiety that is responsible for much of the diverse properties of glutathione. Glutathione participation, therefore, in the physiology of cellular metabolism reflects the importance of this molecule in intracellular functions. Firstly, GSH is involved in the detoxification of highly reactive peroxides (ROOH) by conjugation of electrophiles and metals through the glutathione-
Biomedical Research Scientist and Educating Professor – 2009 Page | 66

John J. Haddad, PhD
radicals such as hydrogen peroxide (H2O2) are effectively reduced by the selenium-dependent GSH peroxidase in the presence of GSH as a substrate. During this reaction, GSH is converted into oxidized disulfide glutathione (GSSG), which is recycled back to 2GSH by GSSG reductase at the expense of NADPH/H+, thus forming what is known a redox cycle. Secondly, GSH participates in maintaining intracellular protein integrity by reducing their disulfide linkages and regulating their synthesis, thereby acts as an important regulator of cellular sulfhydryl status and redox equilibrium. Thirdly, GSH governs signaling pathways in neuroimmune-endocrine interactions by acting as a neurotransmitter and an immunopharmacological reducing thiol; it also facilitates membrane trafficking of reactive chemicals and, in some cases, augments the formation of essential biological mediators. Fourthly, GSH regulates the expression and activation of redox-sensitive transcription factors, whose up-regulation is a key component of the cellular pathways activated in stress-evoked responses.
The restitution of redox equilibrium in the face of an oxidative challenge requires an
adaptiv cross talk between signaling pathways sensing variations in pO2 and genetically regulat
e ed transcription factors. Dr. Haddad has shown that glutathione (GSH)-associated
metabolism is crucial for providing an equilibrium interface between oxidative stress and adaptive responses of cytoprotection. The two forms of glutathione (GSH and GSSG) do not exist in equilibrium ratio under normal conditions, since the majority (>95%) of glutathione is in the reduced form. Shifting redox equilibrium, therefore, in favor of a reducing or oxidizing state is potentially of particular significance, as this would dictate the pattern of gene expression of HIF-1α over relevant shifts in pO2. Accordingly, the significance of the glutathione redox cycle in maintaining the integrity of the biological system should not be underestimated and as such deciphering the pathways linked to glutathione homeostasis via the redox-sensitive transcription factors is of particular relevance to the developing lung.
Antioxidant/pro-oxidant equilibrium differentially regulates HIF-1α redox sensitivity.
Selective inhibition of γ-GCS by -buthionine-(S,R)-sulfoximine (BSO) abrogated hypoxia-induced
L
nuclear localization and stabilization of HIF-1α. In parallel, BSO reduced HIF-1α DNA binding activity, in a dose-dependent manner, regardless of the direction of pO2 shift. There is substantial evidence supporting the notion that γ-GCS inhibition is accompanied by intracellular accumulation of superoxide anion (O2
−.) and H2O2, believed to play a key role in destabilizing HIF-1α. Although it cannot be inferred that the effect of BSO is exclusively ROS-dependent, it appears that maintaining GSH equilibrium and, by inference, the shuttling between reduction and oxidation states, is a prerequisite for HIF-1α activation in hypoxia and under a relative, mild shift in pO2. This assumption is reinforced with the observation that N-acetyl-L-cysteine (NAC), an antioxidant and a precursor for L-cysteine, the rate-limiting amino acid in the biosynthesis of GSH, imposed a reducing environment, thereby protracting HIF-1α stability in the cytosol, and subsequently favoring its translocation and activation. Thus shifting the equilibrium ratio of GSH/GSSG in favor of a reduction potential substantiality allowed HIF-1α stabilization and activation independent of pO2, a phenomenon observed even under hyperoxic shifts, thereby uncoupled from the classical hypoxic induction of HIF-1α.
This raised the issue of whether reversing GSH/GSSG equilibrium would interfere with
the hypoxic induction of HIF-1α. To meet this end, dithiocarbamates, including pyrrolidine dithiocarbamate (PDTC), were enrolled to span the activation of HIF-1α with ascending ΔpO2
Biomedical Research Scientist and Educating Professor – 2009 Page | 67

John J. Haddad, PhD
regimen. PDTC affects redox potential by its ability to scavenge radical species (a reduction property) and to directly oxidize GSH and related thiols (an oxidation property). Although PDTC favored a GSSG/GSH equilibrium and stabilization of HIF-1α protein in the cytosolic compartment, it failed to induce its activation, ostensibly due to the generation of thiyl radicals and thiuram disulfides due to PDTC antioxidant reaction, thus leading to oxidation of GSH. Imposing an oxidizing environment by the rapid accumulation of GSSG in the nucleus adversely affects HIF-1α DNA binding, as a reducing equilibrium is necessary for its stabilization and activation. This notion is further supported by the rather unequivocal evidence that HIF-1α consensus binding was facilitated by increasing GSH/GSSG equilibrium in isolated nuclei. In keeping with this, selective inhibition of GSSG reductase by 1,3-bis-(2-chloroethyl)-1-nitrosourea (BCNU), led to the intracellular accumulation of GSSG and subsequent abrogation of hypoxia-induced HIF-1α activation. It is very likely, therefore, that HIF-1α O2 responsiveness resides over a permissive range of antioxidant buffering capacities, with the demonstration that antioxidant/pro-oxidant equilibrium effectively uncoupled HIF-1α activity from the normal pattern observed in response to variations in pO2.
The activation of HIF-1α and NF-κB is redox and O2-sensitive. To further elaborate
the underlying molecular mechanisms involved in modulating the genetic response of the fetal lung in oxidative stress, Dr. Haddad has focused his attention on whether antioxidant/pro-oxidant equilibrium specifically affects the activation of HIF-1α and NF-κB by modulating the reduction-oxidation (redox) ratio of GSH to GSSG. He investigated the role of NAC, a thiol-containing compound and a glutathione (GSH) precursor, which non-enzymatically interacts and detoxifies reactive electrophiles and free radicals, and PDTC, an antioxidant thiocarbamate and a potent oxidizing agent (pro-oxidant), owing this property to its thiuram biochemistry, in modulating the genetic response of the fetal epithelium.
The kinematics of HIF-1α and NF-κB nuclear abundance and DNA-binding activity with
NAC or PDTC pre-treatments was investigated in vitro. NAC induced a significant increase in HIF-1α abundance in a dose-dependent manner, suggesting the presence of an environment, which
ty was enzymatically assessed in vitro with NAC pre-treatment. NAC is an acetylated variant of the amino acid L-cysteine, thus forming a major source of sulfhydryl (SH)
permits post-translational stability and/or reduction in protein turnover rate of HIF-1α
after shifting cells from extreme hypoxia to normoxia and severe hyperoxia. Of particular importance, the nuclear abundance of this transcription factor was shown to be even greater at 23→152 and 23→722 Torr ΔpO2 in cell cultures pre-treated with NAC than that observed with controls (23 and 23→76 Torr), suggesting an induced stability under non-hypoxic conditions. To determine whether NAC-induced increase in HIF-1α nuclear abundance is in parallel to an elevated degree of activation, DNA-binding activity was assessed with mobility shift assays. Indeed our results point towards an association between increasing nuclear abundance and activation under conditions that are non-hypoxic, in agreement with the observations that NAC creates a reducing environment mimicking hypoxia by suppressing and/or scavenging reactive oxygen intermediates, which are crucial inhibitors of HIF-1α post-translational stability and activation.
To further elucidate the pathways leading to activation of HIF-1α, glutathione
bioavailabili
Biomedical Research Scientist and Educating Professor – 2009 Page | 68

John J. Haddad, PhD
groups and is itself capable of stimulating the biosynthesis of GSH. Therefore, there are two pathways by which NAC enters the biochemical pathways as an antioxidant involving the buffering of free radicals: i) through the reduced sulfhydryl group and ii) through metabolism to GSH. The latter mechanism has been directly implicated in our model as we showed that pre-treatment with NAC significantly elevated GSH levels above baseline values, such that this elevation occurred at the expense of GSSG, thereby affecting the intracellular redox potential. It was previously shown by Dr. Haddad that pre-treatment of hypoxic cultures with BSO, an irreversible specific inhibitor of γ-GCS, led to a dose-dependent inactivation of HIF-1α, suggesting GSH may be a key modulator of the activity of this regulatory factor. In addition, analysis with BCNU, an inhibitor of glutathione reductase (GSSG-RD), the enzyme responsible for GSH recycling via reduction of GSSG, showed that BCNU increased the concentration of GSSG 4- to 5-fold relative to control values and that it partially blocked the activation of HIF-1α, suggesting the presence of an oxidizing environment. A relevant mechanism for any protective effect of NAC exerted against toxic species may be due to its ability to act as a precursor of GSH, thus facilitating its biosynthesis. GSH will then serve as the protective agent that detoxifies reactive species, thereby creating the optimum reducing conditions necessary for HIF-1α activation. This property of NAC acting as a reductant could be partially conferred from its ability to reduce oxidized glutathione.
Pre-treatment with NAC has shown no stimulatory effects on the abundance or activation
of NF-κB (p65, the major transactivating member of the NF-κB family), rather NAC induced NF-κB decreasing nuclear abundance and inactivation in a dose-dependent manner. As oxidants and RO
radical species, and ii) an oxidation property owing to the fact that they can oxidize GSH and related thiol c
S play a key role in the biochemical pathways leading to the activation of NF-κB, it is apparent that NAC is acting as a free radical scavenger antioxidant, a role that supersede, or is distinct from, its ability to increase intracellular glutathione stores. Recent evidence suggests that regulatory transcription factors, such as NF-κB, play critical roles in the early events controlling the molecular response to ROS. It has been shown that the activation of NF-κB by a wide variety of agents can be blocked by NAC, suggesting that the production of reactive oxygen metabolites may act as a common pathway for a diverse range of stimuli. This leads to suggest that the ability of NAC to suppress the activation of NF-κB is likely to involve the antioxidant effects of this xenobiotic substance, where the circuits involved in NF-κB activation are schematized. Apparently, the mode of action of NAC may not be the same in different experimental models and clinical situations and, consequently, extensive research is warranted to investigate the mechanisms of action of NAC that may offer a clue for elucidating its differential effects.
Dithiocarbamates, including PDTC, induce differential effects on redox equilibrium
according to: i) a reduction property based on their ability to decrease single-electron
ompounds, thereby modulating glutathione recycling potential. PDTC, like other dithiocarbamates, has the molecular capacity to exert both antioxidant and pro-oxidant effects in cells. The antioxidative mode of action of PDTC is mediated through its dithiocarboxy group, which scavenges H2O2, O2
−, and .OH. The oxidized form of dithiocarbamates is formed upon the reaction with reactive oxygen and nitrogen species, thus generating dithiocarbamate thiyl radicals that form thiuram disulphides dimers, which are responsible for the pro-oxidant effects of dithiocarbamates. The pro-oxidant effect is pronounced by the ability of thiuram conjugates to directly oxidize thiols including GSH, leading to the formation of GSSG, a potent regulator of
Biomedical Research Scientist and Educating Professor – 2009 Page | 69

John J. Haddad, PhD
many transcription factors and signal transducing systems. In this report we have particularly shown regulatory differential effects of PDTC on the activation of HIF-1α and NF-κB, revealing a striking equilibrium between the antioxidant and pro-oxidant modes of action of dithiocarbamates. PDTC, like NAC, induced HIF-1α increased nuclear abundance at all oxygen tensions investigated, as compared to controls (without PDTC pre-treatment); however, PDTC pre-treatment failed to induce HIF-1α DNA-binding activity. Dr. Haddad has observed that the GSH/GSSG ratio after PDTC treatment was significantly reduced, an indication of oxidation. This decreased ratio is attributed to increasing levels of GSSG, which is very likely creating an oxidizing environment in the nuclei thus rendering HIF-1α inactive, as for this transcription factor to bind DNA and activate hypoxia-responsive genes, a reducing environment is mandatory. It is proposed that, although PDTC might be acting as an antioxidant by scavenging ROS, its failure to activate HIF-1α be rather attributed to its pro-oxidant properties, whereby it increases oxidized glutathione that favors the balance of the redox cycle towards an oxidizing equilibrium. These results are in agreement with previously reported observations showing that PDTC increases the level of GSSG at the expense of GSH, and that GSSG affects transcriptional activation by creating oxidizing conditions. Furthermore, the above assumptions are supported by the observations that pre-treatment of nuclear extracts in vitro with decreasing GSH/GSSG ratios ameliorated the activation of HIF-1α.
By all measurable criteria investigated in this study, PDTC has been proven to be a potent
inhibitor of NF-κB in alveolar epithelia. The inhibitory effects are obvious at both the levels of nuclear abundance and activation, suggesting post-translational instability and interference with the ca
ng to the possibility that NF-κB translocation and subsequent activation is mediated by ROS, which might induce a cytosolic kinase
pacity of NF-κB to bind DNA and activate oxidant-induced stress genes. The distinguishable properties of PDTC mediating its antioxidant and/or pro-oxidant effects on NF-κB could be highlighted as follows: i) PDTC is much more a potent inhibitor of NF-κB than NAC given that its inhibitory working concentrations (10-100 μM) are much lower than that of NAC (1-50 mM). In this respect, PDTC is 100- to 500-fold more potent than NAC. ii) PDTC decreases the GSH/GSSG ratio by elevating GSSG due to oxidation of GSH. Thus, it is likely that PDTC is inhibiting NF-κB activation by acting as a pro-oxidant, although the possibility of acting as an antioxidant while mediating this inhibition cannot be excluded, as it has been reported that ROS are key mediators of NF-κB activation.
This is further supported by the observation that phosphorylation of IκB at specific serine
residues can be inhibited by dithiocarbamates, pointi
activity. The assumption of GSSG-mediated inhibition of NF-κB has been verified by the observations that the oxidation of GSH to GSSG induces the formation of NF-κB/disulphide complex, thereby inhibiting DNA binding. We speculate that a derivative and/or a metabolite of PDTC metabolism is generated under these conditions that is responsible for the outcome of this effect, as it is very unlikely that PDTC would directly interact with NF-κB to inhibit its activation, since addition of PDTC (50 μM) to nuclear extracts of cell cultures exposed to various ΔpO2 failed to inhibit NF-κB activation at 23→76, 23→152 and 23→722 Torr; iii) GSSG formed by the pro-oxidant effect of PDTC drives the formation of an oxidation equilibrium that renders NF-κB inactive. Although oxidizing conditions are necessary for the activation of NF-κB in the cytosol to allow optimum translocation and dissociation from
Biomedical Research Scientist and Educating Professor – 2009 Page | 70

John J. Haddad, PhD
inhibitory IκB, NF-κB must be maintained in a reduced state in the nucleus for DNA binding to occur. This assumption is based on the ability of an increased ratio of GSSG/GSH in vitro to inhibit NF-κB activation. GSSG elevation promotes oxidation of protein cysteinyl thiols, shifting the equilibrium of thiol-disulphide exchange significantly in the direction of mixed disulphide formation and, ultimately, changing protein conformation. Consequently, the binding capacity of regulatory transcription factors is dramatically altered. The possibility that PDTC is interfering with the translocation of NF-κB to the nucleus, however, cannot be ruled out, rather what is evident is that the inhibitory effects on NF-κB supports the hypothesis that dithiocarbamates act as pro-oxidants by elevating the concentration of oxidized glutathione (GSSG), which is capable of direct (by forming a mixed NF-κB-thiol inactive complexes) or indirect (creating an oxidized environment in the nucleus) prevention of NF-κB activation (Fig. 7).
Fig. 7. NF-κB signal transduction pathway. Various incoming signals converge on the activation of the IκB kinase (IKK) complex. IKK then phosphorylates IκB at 2 N-terminal s
Incoming Signals
Cytoplasm
IκB Kinase
IκBRelAp65 p50 IκB
RelAp65 p50
P P
RelAp65 p50
Proteasome
Nucleus RelAp65 p50 DNATranscription
erines, which signals it for biquitination and proteolysis (proteasome). Freed NF-κB complex (p50/p65) enters the nucleus and activates ene expression.
ug
Biomedical Research Scientist and Educating Professor – 2009 Page | 71

John J. Haddad, PhD
The regulation of apoptosis during the transition from placental to pulmonary-based respiration is not well characterized in the perinatal epithelium. The kinetics of this process centers on the activation of redox-sensitive transcription factors, which modulate the pattern of the molecular response to oxidative stress. Oxyexcitation (ΔpO2)-dependent suppression of Bcl-2, favoring the balance of Bax, suggests the involvement of an O2-responsive pathway. The likely pathway implicated could be involving cell cycle arrest through the activation of p53, consistent with the hypothesis that hyperoxic injury is characterized by a complex pattern during which the alveolar surface is damaged, denuded and repopulated. It is postulated that chemioxyexcitation triggers a process eventually leading to suppression of Bcl-2 and up-regulation of Bax, through a p53-linked pathway.
The process of apoptosis is redox-sensitive. Selective inhibition of glutathione (GSH)
biosynthesis up-regulated Bax and p53 independent of pO2. Since the depletion of GSH, a major antioxidant, could trigger apoptosis, it’s likely that ROS are involved in the induction of Bax and p53. Of
nction as a redox buffer and/or effective ROS sc
note, GSH depletion also induced the intracellular accumulation of Bcl-2 although redox disequilibrium seems to be in favor of apoptosis. These data suggest that loss of intracellular GSH might induce oxidative stress, possibly through the formation of ROS, thereby altering redox potential in favor of oxidation equilibrium. This subsequently leads to up-regulation of the downstream agonists of apoptosis and cell cycle arrest.
Effective pharmacological intervention by NAC may indicate modulation of redox
equilibrium in association with oxyexcitation. NAC is an antioxidant thiol which, after uptake, deacylation and biochemical conversion to GSH, may fu
avenger. The inhibitory effect of NAC is evident through suppression of apoptosis in response to oxyexcitation by down-regulating Bax/p53 and inducing Bcl-2, an effect mimicked by γ-GCE, a cell-permeant GSH precursor. The antioxidant potential of NAC/γ-GCE, discovered by Dr. Haddad, therefore, may contribute to inhibition of oxidative stress-mediated apoptosis and expression of selective agonists.
The potential involvement of the redox-sensitive transcription factor, NF-κB, in apoptosis
pathways is well documented. For example, over-expression of the dominant-negative form of IκB-α, a cytosolic inhibitor of NF-κB, promoted apoptosis in vitro. Moreover, mice bearing the knockout gene RelA−/− (p65−/−) show that they are more susceptible to agents stimulating apoptosis than the wild type, suggesting a potential role for NF-κB in regulating this process. Non-selective inhibition of NF-κB by PDTC, an antioxidant/pro-oxidant molecule, which decreases the GSH/GSSG ratio, led to up-regulation of Bax independent of p53. However, selective inhibition of NF-κB by SSA induced up-regulation of Bax in a p53-depedent mechanism. Dr. Haddad suggests that NF-κB exhibits an anti-apoptotic potential, presumably through regulating responsive genes particularly involved in controlling apoptosis and cell cycle events.
The maturation of the developing lung at different stages of gestation and postnatally
occurs within widely different pO2. The successful and smooth transition from placental to pulmon ry-based respiration inca urs a relative hyperoxic shift in the lung, an event that constitutes a potential signaling mechanism for determining the potential for apoptosis in vivo and ex vivo. The anti-apoptotic potential postnatally was dramatically suppressed, especially
Biomedical Research Scientist and Educating Professor – 2009 Page | 72

John J. Haddad, PhD
during the first early hours after birth, in accord with the decline in Bcl-2 expression with ascending ΔpO2 in vitro. Of particular interest, the pro-apoptotic potential, resembled by Bax, was not prominently sufficient to exceed the anti-apoptotic potential, given that the equilibrium ratio of Bcl-2/Bax remained steadily constant at birth. This challenged the assumption that Bax is the major cofactor involved in regulating apoptosis ex vivo. Accordingly, the focus was to determine whether there is a particular involvement of p53, a cell cycle regulator, under which Bcl-2 and Bax are transcriptionally controlled. The birth transition period predominantly up-regulated the expression of p53 in a time-dependent manner, allowing us to postulate that a p53-dependent pathway is involved in apoptosis in the developing lung. Whereas the depression of apoptosis antagonists is concomitant with up-regulating the agonist expression through a p53-dependent pathway in vitro, apoptosis in the perinatal developing lung is p53-dependent and possibly Bax-insensitive.
Cytokines act as major participants in mediating molecular responses in physiology
and pathophysiology. This report, written by Dr. Haddad, shows that the conventially known ‘pro-inflammatory’ cytokines are observed as O2-sensitive mediators, providing evidence that the alveolar epithelium integrates O2-linked pathways mediated by cytokines via an ROS-dependent mechanism. A direct contact of the epithelium with fluctuations in pO2 at the alveolar space b
suggesting an integral role of endogenous ROS. As such cytokines could form a pivotal link in ROS-dependent pathwa
lood barrier during pulmonary-based ventilation is likely to up-regulate cytokines. This is strengthened by the evidence implicating the epithelium in a front-line defense strategy activated in preparation for birth into an O2-rich environment.
The generation of ROS might induce pulmonary damage, however, moderate oxidative
stress can induce a signaling mechanism mediated, at least in part, by cytokines. Chemiexcitation-derived ROS induced pro-inflammatory cytokine biosynthesis in a dose-dependent manner. This response was abrogated by selective antioxidants,
ys leading to the activation of redox-sensitive transcription factors whose up-regulation determines the specificity of the cellular response to oxidative stress. Thus dynamic variation in pO2 regulates the release of cytokines through a ROS-dependent pathway, thereby bearing consequences for the pediatric treatment of respiratory distress under conditions of clinical O2 therapy where cytokines are observed as potential participants in their pathophysiology.
The observation that intracellular ROS are major participants in cytokine signaling led
Dr. Haddad to investigate whether this responsiveness to dynamic pO2 is redox-dependent. There is growing evidence implicating an association between oxidative stress and a pro-inflammatory state, thereby placing more demand on the utilization of intracellular GSH. As such, the respiratory epithelium becomes more engaged in regulating enzymes involved in maintaining redox equilibrium. Dr. Haddad theorized that whilst the glutathione biosynthetic machinery is overwhelmed in disease, an up-regulation of cytokines may contribute to acute exacerbation of the clinical symptoms. Thiol regulation of pro-inflammatory cytokines, therefore, bears clinical relevance to the pediatric treatment of respiratory distresses, where cytokines are crucial elements in their pathophysiology.
Biomedical Research Scientist and Educating Professor – 2009 Page | 73

John J. Haddad, PhD
Glutathione biosynthesis is selectively blocked through a specific and irreversible inhibition of γ-GCS (Please refer to Fig.1, proposed by Dr. Haddad). This inhibition led to ROS accumulation and, as expected, their inappropriate disposition and intracellular localization augmented a pro-inflammatory signal. Replenishing intracellular GSH stores favored the reduction of ROS, subsequently suppressing the downstream cytokine-dependent pathway. In summary, selective inhibition of GSH biosynthesis up-regulated cytokines in a ROS-dependent manner; blockage of glutathione recycling uncoupled the ROS/cytokine pathway, and GSH precursors suppress intracellular ROS formation and the down-stream cytokine-dependent pathway. Thus, shifting redox potential in favor of reduction equilibrium negatively interferes with the capacity to up-regulate a pro-inflammatory signal, thereby bearing consequences for determining the survivorship of epithelial cells under conditions mimicking clinical O2 therapy.
In conclusion, the perinatal epithelium responds to dynamic variations in pO2 by
regulating the expression and activation of the redox-sensitive transcription factors, HIF-1α and NF-κB. This responsiveness is coupled to up-regulating glutathione biosynthesis, as a major intracellular thiol bearing an antioxidant potential. Modulating the antioxidant/pro-oxidant equilibrium selectively interferes with the regulatory pathways controlling HIF-1α and NF-κB. A novel equilibrium among signaling agonists and antagonists of apoptosis is exhibited in response to chemioxyexcitation (ΔpO2/ROS), an event associated with an inflammatory signal through up-regulating pleiotropic cytokines, which are recognized as O2-resposnive and redox-sensitive signaling molecules that might participate in the physiology and pathophysiology of the developing lung.
Dr. Haddad possesses all the necessary expertise for the proper scientific conduct of this work. Indeed, he has substantial outstanding practical (and published) experience with several of the assays mentioned in the proposal that he gained whilst attaining his Masters and doctoral degrees. He has also played an active role in conceiving the experiments designed to test each of the stated aims. This creates confidence that his input on this and other acclaimed projects will go a long way towards ensuring their success and projecting them to the benefit of the community.
The following is a partial list of aforementioned “Research Proposals and Grants”
authored and proposed by Dr. Haddad as the principle investigator:
1. “The Role of MAPK Signaling Pathways in Determining Cellular Death or
. “The Kinetics of Cytokine-mediated Activation of HIF-1α and NF-κB over
to oxidative stress”.
Survival in Hypoxia: Emerging Pharmacological Targets for Therapeutic Intervention”.
2. “Kinetics of Cytokine-Mediated Activation of HIF-1α and NF-κB in Alveolar Epithelia: Signaling Pathways of Apoptosis in Oxidative Stress”.
3fetal-to-neonatal oxygen tensions: The role of thymulin and receptor antagonists in modulating the apoptotic response of alveolar cells exposed
Biomedical Research Scientist and Educating Professor – 2009 Page | 74

John J. Haddad, PhD
4. “The involvement of thymulin in ICE and IL-1β mediated apoptosis in alveolar cells exposed to oxidative stress: Implications for respiratory distress syndrome in pre-term infants”.
erging Targets for Therapeutic Intervention in Respiratory Distress Syndrome”.
6.
erapeutic Intervention in Bronchopulmonary Dysplasia”.
7.
ctors HIF-1α and NF-κB”.
5. “Co-ordinate Regulation of Mitogen-Activated Protein Kinases and Inhibitory-κB in NF-κB-Driven Gene Transcription of Pro-inflammatory Cytokines in the Alveolar Epithelium: Em
“Selective Phosphodiesterase Inhibition Ameliorates Endotoxin-Induced Transcription and Biosynthesis of Pro-inflammatory Cytokines in the Alveolar Epithelium: A Clinical Model for Th
“Thiol Regulation of Pro-inflammatory Cytokines in the Alveolar Epithelium: Potential Immunopharmacological Role of Glutathione and Redox-Sensitive Transcription Fa
The aforementioned and below mentioned indicate that the ability of Dr. Haddad to accrue extramural funds reflects the high caliber of his biomedical research. Of note, Dr. Haddad’s p high profile rior achievements and past and current record of specific andbiomedica achievements justify projections of future benefits to the interests of the l mentioned fields.
♣
Biomedical Research Scientist and Educating Professor – 2009 Page | 75

John J. Haddad, PhD
Given the caliber of Dr. Haddad’s research in the fields of stroke, coronary heat attack, respiratory distress and brain injury, he was a recipient of several recognized awards (cting few examples).
• National Institutes of Health (NIH) postdoctoral research scholar (Molecular Neurosciences, Immunology and Anesthesiology Medicine); Severenghaus-Radiometer Research Laboratories, Molecular Neuroscience Research Division, Department of Anesthesia and Perioperative Care, School of Medicine, University of California at San Francisco (UCSF), San Francisco, California, USA.
• Postdoctoral research fellow (Molecular Biology and Pulmonary
Physiology); The Tayside Institute of Child Health, Faculty of Medicine, Ninewells Hospital and Medical School, University of Dundee (UoD), Dundee, Scotland, UK.
• Doctor of Science; Ph.D. (Molecular Pharmacology, Cell Physiology
and Biochemistry); The Tayside Institute of Child Health, Faculty of Medicine, Ninewells Hospital and Medical School, University of Dundee (UoD), Dundee, Scotland, UK.
• Masters of Science, with honors; M.Sc., Biology (Neuroimmunology),
Department of Biology, Faculty of Arts and Sciences, in affiliation with the Departments of Human Morphology and Physiology, Faculty of Medicine, American University of Beirut (AUB), Beirut, Lebanon (New York, USA).
• Bachelor of Science, with distinction; B.Sc., Pre-medical Biology,
Department of Biology, American University of Beirut (AUB), Beirut, Lebanon (New York, USA).
• Advisory member on the board of Current Drugs Ltd.: Investigational
Drugs database company affiliated with PharmaPress specialized in pharmaceutical industry and headquartered in London, England, UK.
• The Georges John J. Livanos (London) prize at the Tayside Institute of
Child Health, Faculty of Medicine, Ninewells Hospital and Medical School, University of Dundee, Dundee, Scotland, UK, received in recognition of scientific achievements made during the masters years.
• The University of Dundee, Ninewells Hospital and Medical School,
annual travel award to outstanding young scientists. Awarded to attend Experimental Biology-2000 in San Diego, California, USA.
Biomedical Research Scientist and Educating Professor – 2009 Page | 76

John J. Haddad, PhD
• The American Physiological Society, Bethesda, MD (USA), travel award for attending the joint annual meeting with the Scandinavian Physiological Society in Amsterdam, Sweden.
• The Wellcome Trust, London (UK), travel award for attending the joint
annual meeting of the Federal Association for the Societies of Experimental Biology (FASEB) in Orlando, Florida, USA.
• The Physiological Society, London (UK), travel award, under the
Affiliate Travel Grant Scheme, for attending the joint annual meeting of the Federal Association for the Societies of Experimental Biology (FASEB) in Orlando, Florida, USA.
• The National Institute for Biological Standards and Controls (NIBSC)
(NIBSC, England, UK) grant award ($10,000), in collaboration with the American University of Beirut, Beirut, Lebanon (New York, USA).
• Tenovus-Scotland for Supporting Medical Research (Scotland, UK)
grant award ($15,000).
Dr. Haddad may have been an active member of the following accredited and recognized societies. His nomination was based on the merit of his achievements and his substantial contributions to the field of Biological Sciences.
• The American Chemical Society (ACS), Washington, D.C., USA. • The Physiological Society (PS), London, UK. • Federation of the American Societies for Experimental Biology
(FASEB), Bethesda, Maryland, USA.
• The American Physiological Society (APS), Bethesda, Maryland, USA.
• The Society for Experimental Biology (SEB), Piccadilly, London, UK. • The National Geographic Society (NGS), Washington, D.C., USA.
• The International Cytokine Society (ICS), Augusta, GA, USA.
• The Society for Neuroscience (SFN), Washington, D.C., USA.
• The New York Academy of Science (NYAS), New York City, New
York, USA.
Biomedical Research Scientist and Educating Professor – 2009 Page | 77

John J. Haddad, PhD
Dr. Haddad has been invited to be an independent reviewer for the following journals. He is acting on independent panels and boards to judge the work of other scientists and make recommendations that fit his experienced and prowess profile.
• Cytokine (CYTO), 103 Baughman’s Lane, Frederick, MD 21702, USA. • Proceedings of the National Academy of Sciences of the United States
of America (PNAS), 2101 Constitution Avenue, NW, NAS 340, Washington, D.C. 20418, USA.
• Journal of Neuroimmunology (JNI), Albert Einstein College of
Medicine, Department of Pathology, F140, 1300 Morris Park Avenue, New York, NY 10461, USA.
• Free Radical Biology and Medicine (FRBM), Elsevier Science
Incorporation, 275 Washington Street, Newton, MA 02458, USA.
• Antioxidants and Redox Signaling (ARS), ARS Central Editorial Office, Cardiovascular Research Division, Department of Surgery / MC 1110, University of Connecticut, School of Medicine, Farmington, CT 06030, USA.
• Current Opinion in Investigational Drugs (COID), PharmaPress Ltd,
London, England, UK.
• Critical Care (CC), the official journal of the Critical Care Forum, affiliated to the International Symposium on Intensive Care and Emergency Medicine (ISICEM), Brussels, Belgium.
Dr. Haddad has also acted on independent boards that review and judge the work of
scientists and clinicians who have submitted their current research proposals (Biological Sciences) to prominent granting agencies.
• The Wellcome Trust (WT), London, England, UK: “Targeting NF-κB
in the lung to prevent viral-induced asthma.” Dr. R. Garofalo, Department of Pediatrics, University of Texas Medical Branch, Galveston, Texas, USA.
• The Arthritis Research Campaign (ARC), Chesterfield, Derbyshire,
England, UK: “Hypoxia-driven changes in synovial cell function: Clinical implications in arthritis.” Professor S. W. Edwards, School of Biological Sciences, University of Liverpool, Liverpool, England, UK.
• The American Heart Association (AHA), Washington, D.C., USA: “The
role of MAPK signaling pathways in determining death or survival
Biomedical Research Scientist and Educating Professor – 2009 Page | 78

John J. Haddad, PhD
in hypoxic neurons.” Professor C. S. Fahlman, School of Medicine, University of California, San Francisco, CA, USA.
Dr. Haddad has also received several invitations to present his outstanding biomedical research in conferences, symposia and nationally recognized annual meetings.
• The Physiological Society semi-annual meeting in Manchester, England,
UK. • The 9th Postgraduate Research Symposium in Dundee, Scotland, UK.
• The Experimental Biology (FASEB) annual meeting in San Diego,
California, USA.
• The 10th Postgraduate Research Symposium in Dundee, Scotland, UK.
• The American Physiological Society and the Scandinavian Physiological Society joint annual meeting in Stockholm, Sweden.
• The Physiological Society annual meeting in London, England, UK.
• The Experimental Biology (FASEB) annual meeting in Orlando,
Florida, USA. • The Biochemical Society annual meeting in Bristol, England, UK.
• The International Society of Neuroimmunology 6th International
Congress in Edinburgh, Scotland, UK. • The 17th Transatlantic Airway Conference in Lucerne, Switzerland.
• The University of California multi-campus workshop on ‘Oxygen
Sensing and Adaptation Mechanisms: High Altitude Biology’ in Riverside, Ontario, California, USA.
• The Society for Neuroscience annual meeting in Orlando, Florida, USA.
In recognition of his pioneering research, Dr. Haddad received many invitations from prominent academic institutions to debate and discuss with peers his vision of current understanding of the molecular basis of lung and brain injury in oxidative stress and their clinical implications.
• Departments of Human Morphology and Physiology, Faculty of Medicine, and Department of Biology, Faculty of Arts and Sciences, American University of Beirut, Beirut, Lebanon (New York, USA).
Biomedical Research Scientist and Educating Professor – 2009 Page | 79

John J. Haddad, PhD
• Tayside Institute of Child Health, Faculty of Medicine, University of Dundee, Dundee, Scotland, UK.
• Departments of Human Morphology and Physiology, Faculty of
Medicine, and Department of Biology, Faculty of Arts and Sciences, American University of Beirut, Beirut, Lebanon (New York, USA).
• Department of Medicine, National Jewish Medical and Research Center,
global leader in lung, allergic and immune diseases, Denver, Colorado, USA.
• Developmental Biology Program, Children’s Hospital Los Angeles
Research Institute, Smith Research Tower, University of Southern California School of Medicine (UCLA), Los Angeles, California, USA.
• Division of Molecular Cardiovascular Biology, The Children’s Hospital
Research Foundation, Children’s Hospital Medical Center, Cincinnati, Ohio, USA.
• Boehringer Ingelheim International GmbH, Transatlantic Airway
Conference (17th TAC annual meeting), Lucerne, Switzerland.
• Division of Pulmonary, Critical Care and Occupational Medicine, Department of Internal Medicine, Health Sciences Center, School of Medicine, Saint Louis University, Saint Louis, Missouri, USA.
• Programs in Pharmacology and Toxicology, Department of
Pharmaceutical Sciences, College of Pharmacy, Health Sciences Center, University of Oklahoma, Oklahoma City, Oklahoma, USA.
• Center for Environmental Health Sciences, Department of
Pharmaceutical Sciences, University of Montana, Missoula, Montana, USA.
• Spinal Cord and Brain Injury Research Center, Sanders Brown Center in
Aging, University of Kentucky, Lexington, Kentucky, USA.
Dr. Haddad has meticulously developed indispensable techniques and strategies in molecular biology (Biochemistry and Pharmacology), which helped disseminate scientific knowledge of outstanding merit to other researchers and biologists in the United States. The below mentioned is a detailed list of the techniques conceived and created by Dr. Haddad.
• Cell Tissue Culture Perchloric Acid (PCA) Extraction for Cellular
metabolites. • Cell Tissue Culture Total Protein Extraction Assay.
Biomedical Research Scientist and Educating Professor – 2009 Page | 80

John J. Haddad, PhD
• Cell Tissue Culture Nuclear Protein Extraction Assay.
• Whole Organ Total Protein Extraction Assay.
• Colorimetric Bradford Protein Assay.
• DNA Radioactive Labeling Solutions and Protocols.
• DNA TdT-mediated dUTP Nick End Labeling (TUNEL) Assay.
• Electrophoretic Mobility Shift Assay (EMSA).
• Protein Extraction to Autoradiography and Phosphorimaging.
• Detailed Protocols for DNA Binding Reaction.
• Radioactive RNase Protection Assay.
• Western Blotting for Protein Analysis and Separation by Gel
Electrophoresis.
• Adenosine-5′-Triphosphate (ATP) Determination with Phosphoglycerate Kinase Assay. (This assay is very crucial for determining cellular bioenergetics).
• Principles of the Alkaline Phosphatase (ALP) Assay: Determination
with p-Nitrophenylphosphate (pNPP).
• Isolation of Mitochondria for In Vitro Analysis of Cytochrome c Release.
• Extraction of Intact DNA Assay. • Principles of the Lactate Dehydrogenase (LDH) Release Assay. • Tetrazolium MTT Colorimetric Cytotoxic and Proliferation Assay.
• γ-Glutamylcysteine Synthetase Enzyme Activity Assay.
• Glutathione Synthetase Enzyme Activity Assay.
• Glutathione Peroxidase Enzyme Activity Assay.
• Glutathione Reductase Enzyme Activity Assay.
Biomedical Research Scientist and Educating Professor – 2009 Page | 81

John J. Haddad, PhD
• Principles of the Immuno-cytochemistry Assay.
• Mitogen-activated Protein Kinase (MAPKp38) Indirect Kinase Activity by Hsp27 Phosphorylation.
• Exposure to LPS and IFN-γ, X/XO and H2O2 in the Presence of
Pentoxifylline (PTX) or Lysofylline (LSF) Assay.
• Glutathione (GSH:GSSG) Determination with Methylglyoxal.
• Nicotinamide Adenine Dinucleotide (NAD) Determination Assay.
• NADP Determination Assay.
• Analysis Of Reactive Oxygen Species Generation By The Fenton Reaction: Principles Of The Fenton Reaction.
• Colorimetric Method for the Measurement of Hydrogen Peroxide.
• Measurement of Superoxide (O2
−.) Generation.
• Enzyme-linked Immunosorbent Assay (ELISA) for the Measurement of Cytokines in Blood Sera, Tissues and Cultures.
At the molecular and diagnostic levels, Dr. Haddad has cloned and/or created several gene plasmids for the investigation of cellular effects of mutated genes in the lung and brain. These genetic vectors are indispensable to other researchers and biologists in the United States, especially while investigating cells death during injury and stroke. The Table below mentioned represents a detailed list of the genes conceived and created by Dr. Haddad.
Genetic Nomenclature of the Cloned Vector
The Gene’s Purpose and Role
pLNCX38
Cloned MAPKp38 gene, an enzyme that plays a major role in cell death and injury.
MKK6
Cloned upstream kinase that phosphorylates MAPKp38 enzyme.
pCFS-neo
Cloned kinase that is responsible for the phosphorylation of a class of G-coupled membrane receptors (Tyrosine Phosphatase) in a variety of tissues.
pCFS-PTPeM-Flag
Cloned kinase that is responsible for the phosphorylation
Biomedical Research Scientist and Educating Professor – 2009 Page | 82

John J. Haddad, PhD
of a class of G-coupled membrane receptors (Tyrosine Phosphatase) in a variety of tissues.
pCFS-PTPeM-DA-Flag
Cloned kinase that is responsible for the phosphorylation of a class of G-coupled membrane receptors (Tyrosine Phosphatase) in a variety of tissues.
pCFS-PTPeC-Flag
Cloned kinase that is responsible for the phosphorylation of a class of G-coupled membrane receptors (Tyrosine Phosphatase) in a variety of tissues.
pCFS-PTPeC-DA-Flag
Cloned kinase that is responsible for the phosphorylation of a class of G-coupled membrane receptors (Tyrosine Phosphatase) in a variety of tissues.
pBF-PTPeM-Flag
Cloned kinase that is responsible for the phosphorylation of a class of G-coupled membrane receptors (Tyrosine Phosphatase) in a variety of tissues.
pBF-PTPeM-DA-Flag
Cloned kinase that is responsible for the phosphorylation of a class of G-coupled membrane receptors (Tyrosine Phosphatase) in a variety of tissues.
pBF-PTPeC-Flag
Cloned kinase that is responsible for the phosphorylation of a class of G-coupled membrane receptors (Tyrosine Phosphatase) in a variety of tissues.
pBF-PTPeC-DA-Flag
Cloned kinase that is responsible for the phosphorylation of a class of G-coupled membrane receptors (Tyrosine Phosphatase) in a variety of tissues.
pcDNA3.1 (+/-) MAD-3
Cloned mutated gene that encodes IκB-α, a cytosolic inhibitor of the transcription factor, NF-κB.
Dr. Haddad has published more than 20 conference abstracts, skillfully presented at symposia and meetings in the USA and abroad (Please see the detailed list in the c.v.).
Biomedical Research Scientist and Educating Professor – 2009 Page | 83

John J. Haddad, PhD
As noted, Dr. Haddad has also published more than 70 peer-reviewed articles in prestigious journals and further afield, in addition to textbooks and major references (please see detailed list in the c.v.). Since his return to Lebanon back in 2004, Dr. Haddad has been meticulously involved with teaching instruction in Biology and Biomedical Sciences at various venues, including University of Balamand.
♣
Biomedical Research Scientist and Educating Professor – 2009 Page | 84
John J. Haddad, PhD
The Following Table Enlists The Expertise Of Dr. Haddad.
Exceptional Ability, Expertise and Professionalism
Projection of Benefits in the National
Scientific Interest
1. Pain and hyperalgesia molecular mechanisms.
1. Perception of pain and its conception by
higher centers in brain-spinal cord zone of the nervous system.
Benefit: Alleviation of pain in patients with debilitating ailments, such as advanced cancer and coronary stroke.
2. Thermal and mechanical kinetics of hyperalgesia.
2. Toleration of pain at various levels of higher
nervous system reception areas. Benefit: Physiotherapy and psychological training for terminal illness patients with incurable kind of diseases.
3. Surgical procedures, including thymectomy,
sympathectomy, cordotomy, vagotomy and brain electrolytic lesions.
3. Brain surgery in the treatment of pain and
hyperalgesia. Benefit: Neuro-surgery in patients for the location of pain areas in the brain for the injection of pharmacological drugs at specific target areas.
4. Inflammatory pain and nociception neurogenic
inflammation molecular mechanisms.
4. Peptide cytokine role in exacerbating pain
transmitted from peripheral tissues to the central nervous system.
Benefit: Pharmacological treatment for blocking the activity of deleterious cytokines using analgesic drugs, such as steroidal and non-steroidal compounds in patients with inflammatory and infectious diseases.
5. Lung inflammation associated with oxidative
stress.
5. Analysis of the degree of inflammation in the
lung in association with oxygen therapy. Benefit: Treatment for respiratory distress syndrome (RDS) in pre-term infants, young children and adults with lung inflammation and inflammatory edema. The deleterious effects of oxygen therapy are reversible and preventable.
Biomedical Research Scientist and Educating Professor – 2009 Page | 85
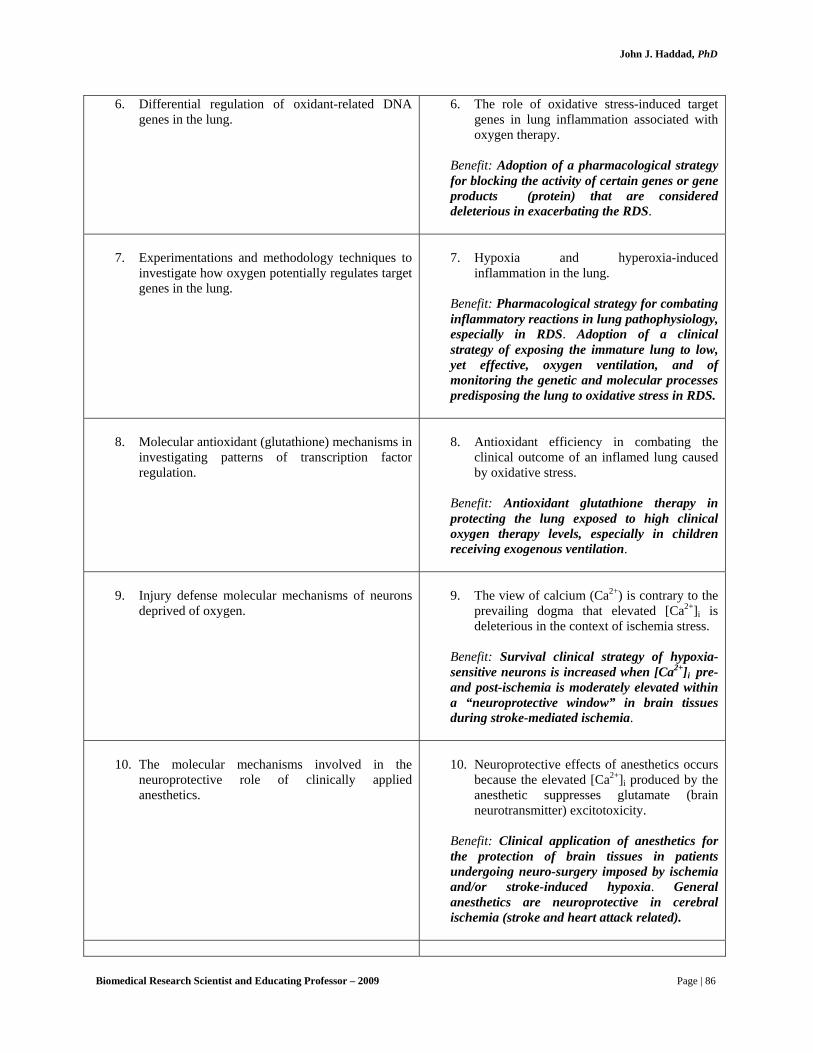
John J. Haddad, PhD
6. Differential regulation of oxidant-related DNA genes in the lung.
6. The role of oxidative stress-induced target genes in lung inflammation associated with oxygen therapy.
Benefit: Adoption of a pharmacological strategy for blocking the activity of certain genes or gene products (protein) that are considered deleterious in exacerbating the RDS.
7. Experimentations and methodology techniques to
investigate how oxygen potentially regulates target genes in the lung.
7. Hypoxia and hyperoxia-induced
inflammation in the lung. Benefit: Pharmacological strategy for combating inflammatory reactions in lung pathophysiology, especially in RDS. Adoption of a clinical strategy of exposing the immature lung to low, yet effective, oxygen ventilation, and of monitoring the genetic and molecular processes predisposing the lung to oxidative stress in RDS.
8. Molecular antioxidant (glutathione) mechanisms in
investigating patterns of transcription factor regulation.
8. Antioxidant efficiency in combating the
clinical outcome of an inflamed lung caused by oxidative stress.
Benefit: Antioxidant glutathione therapy in protecting the lung exposed to high clinical oxygen therapy levels, especially in children receiving exogenous ventilation.
9. Injury defense molecular mechanisms of neurons
deprived of oxygen.
9. The view of calcium (Ca2+) is contrary to the
prevailing dogma that elevated [Ca2+]i is deleterious in the context of ischemia stress.
Benefit: Survival clinical strategy of hypoxia-sensitive neurons is increased when [Ca2+]i pre- and post-ischemia is moderately elevated within a “neuroprotective window” in brain tissues during stroke-mediated ischemia.
10. The molecular mechanisms involved in the neuroprotective role of clinically applied anesthetics.
10. Neuroprotective effects of anesthetics occurs
because the elevated [Ca2+]i produced by the anesthetic suppresses glutamate (brain neurotransmitter) excitotoxicity.
Benefit: Clinical application of anesthetics for the protection of brain tissues in patients undergoing neuro-surgery imposed by ischemia and/or stroke-induced hypoxia. General anesthetics are neuroprotective in cerebral ischemia (stroke and heart attack related).
Biomedical Research Scientist and Educating Professor – 2009 Page | 86

John J. Haddad, PhD
11. Biochemistry and electrophysiology of N-methyl-D-aspartate (NMDA) receptor functioning in the brain.
11. NMDA-mediated calcium regulation in ageing (senile) neurons in the brain.
Benefit: Aging alters [Ca2+]i homeostasis in hippocampal neurons during and following ischemia. NMDA suppression in anoxia is also critical for the survival of neonatal mammalian neurons during hypoxia. Pharmacological regulation of NMDA receptors in the elderly is of immense significance in delaying the natural onset of the ageing process.
12. The molecular interaction between operating room
anesthetics and the elevation of [Ca2+]i.
12. Anesthetics, because they elevate [Ca2+]i and
alter [Ca2+]i homeostasis during or post ischemia, negatively influence the survival of senile neurons.
Benefit: Anesthetics have a direct role in regulating the survival (life span) of brain neurons in elderly patients. Increased frequency of postoperative cognitive deficits in the elderly is related to anesthetics increasing [Ca2+]I in senile neurons to toxic concentrations. Nitrous oxide, the most commonly used vapor anesthetic in the USA, for instance, significantly changes [Ca2+]i homeostasis, with even sub-anesthetic levels having effects larger than other vapor anesthetics.
13. The biochemistry and pharmacology of [Ca2+]i disequilibrium and its molecular significance in regulating neuronal cell death.
13. The functional decline and death of senile
neurons is that with aging, neurons lose the ability to tightly regulate [Ca2+]i and that the Ca2+ dishomeostasis suppresses normal excitability and accelerates cell loss.
Benefit: Pharmacological intervention for the manipulation of calcium homeostasis in the brain of elderly patients.
14. The molecular pharmacology of mitogen-activated
protein kinases (MAPKs), enzymes that regulate cell death and survival.
14. The balance between brain survival and death
as regulated by the MAPK pathways. Benefit: Pharmacological inhibition and/or activation of selective MAPKs is crucial for neuroprotection in patients suffering heart attack and imposed hypoxia or ischemia. Selective inhibition of MAPKs reduces brain injury and neurological deficits in cerebral focal ischemia. The patterns of MAPKs in the brain are crucial in neuroprotection during ischemia and in stroke-mediated oxygen deprivation.
Biomedical Research Scientist and Educating Professor – 2009 Page | 87

John J. Haddad, PhD
15. Molecular mechanisms involving the role of oxygen in lung development and gene regulation.
15. In pre-term infants, sustained therapy with high-inspired oxygen tensions (e.g. for respiratory distress syndrome) causes epithelial damage, septal fibrosis and a scattered occlusion of distal airways characteristic of broncho-pulmonary dysphasia.
Benefit: It can be clinically demonstrated that oxygen, its poisonous metabolites and cytokines co-operate in sending vital messages, which regulate lung cell survival, and then a foundation is laid for developing therapeutic strategies for the treatment of infant respiratory distress conditions.
16. The biochemistry of the role of oxygen in
developing an inflammatory signal that is associated with oxidative stress, a condition that is suppressed with the supplementation of antioxidants.
16. The activation of genetic factors mediated by
reactive oxygen species (ROS) is a potential determinant of programmed cell death. Moreover, ROS contribution to cell death has been attenuated by the antioxidant capacity of reduced glutathione.
Benefit: As epithelial apoptosis and necrosis found in bronchopulmonary dysplasia and respiratory distress syndrome (RDS) are intimately linked to oxidative ROS production, and as glutathione ROS-buffering capacity is substantially depressed in the perinatal period, it would appear that the oxygen shift in the lung at birth, or indeed elevations in lung lumen Po2 by oxygen therapy, may evoke programmed cell death in the perinatal lung. Pharmacological protection by glutathione thus provides an ideal approach to treating RDS.
17. The molecular mechanisms involved in cytokine-
mediated oxidative stress.
17. Crucial role for specific genes in mediating
inflammation imposed by oxygen-induced up-regulation of cytokines.
Benefit: Since lung vital sensitivity is controlled at the gene level, it’s anticipated that cytokine-mediated signaling via regulatory genetic factors may constitute a major target for therapeutic intervention in the treatment of respiratory diseases.
Biomedical Research Scientist and Educating Professor – 2009 Page | 88

John J. Haddad, PhD
The Following Summarizes Dr. Haddad’s Profile:
• Designed molecular and biochemical techniques of transfection and cloning to investigate how oxygen potentially regulates target genes involved in lung physiology and pathophysiology, with particular emphasis on oxidative stress.
• Proposed diagnostic clinical strategy of exposing the immature lung to low, yet
effective, oxygen ventilation, and of monitoring the genetic and molecular processes predisposing the lung to respiratory distress syndrome.
• Theorized that brain damage from asphyxia or ischemia is an important cause of
human mortality and neurological impairment, conditions for which no adequate therapy exists in the USA.
• Suggested that therapies directed strictly at preventing [Ca2+]I increases
throughout the peri-ischemic period during heart attack (stroke) might backfire. Indeed, toxicity from brain receptor antagonists has been a frustrating problem in pre-clinical and clinical trials, a problem perhaps related to too little Ca2+ at critical times.
• Proposed studies for examining the role of brain receptor phosphorylation in
anoxia-tolerant neurons to help clarify the significance of these processes in injury and disease and help guide new approaches to neuroprotection.
• Proposed that general anesthetics are neuroprotective in cerebral ischemia.
Although he suggested that an increase in [Ca2+]i is a key factor in producing suppression of neuronal synaptic transmission (i.e. the anesthetic state), the anesthetic might also have the effect of reducing glutamate release and glutamate toxicity in the context of hypoxia or ischemia.
• Since understanding and preventing the functional decline of the aging brain has
huge significance for the American society, especially for patients with Alzheimer’s, he theorized that while ascertaining the functional decline and death of senile neurons is associated with aging, neurons may lose the ability to tightly regulate [Ca2+]i and that the Ca2+ dishomeostasis suppresses normal excitability and accelerates cell loss (degeneration).
• Suggested that increased frequency of postoperative cognitive deficits in the
elderly are related to anesthetics increasing [Ca2+]i in senile neurons to toxic concentrations. Anesthetics that cause the largest increases in [Ca2+]i may produce the greatest problems with Ca2+ homeostasis in aged/senile neurons. Therefore, exploring the cellular effects of anesthetics on aged neurons is critical to understanding the basis of increased cognitive deficits following anesthesia in the elderly in the United States.
Biomedical Research Scientist and Educating Professor – 2009 Page | 89

John J. Haddad, PhD
Biomedical Research Scientist and Educating Professor – 2009 Page | 90
• Proposed that disregulation of [Ca2+]i clearly plays a key role in the pathogenesis of brain hypoxia/ischemia, but its role may be considerably more complex than merely transducing injury in a linear fashion. The changing role of [Ca2+]i during development and during senility may suggest new ways to protect the aging brain from additional cell loss. Of note, insights provided would identify mechanisms by which anesthetics protect neurons and ways in which the neuroprotective efficacy of anesthetic management could be maximized.
• Theorized that neuroprotection with isoflurane or nitrous oxide anesthetics will
wane with age, and possibly be related to interference with [Ca2+]i homeostasis in aged neurons. The epidemiological value of this observation is that it might be a clue as to why so many elderly individuals suffer neurological (primarily cognitive) deficits following anesthesia.
• Indicated that degeneration and death of neurons of the central nervous system are
responsible, at least in part, for the clinical manifestations of stroke- and ischemia-mediated injury. In patients suffering from stroke, the level of oxygen reaching for the brain decreases; those particular cortical areas suffer from a severe hypoxic condition to an extent cells can no longer support effective brain functioning, due in part to differential expression of specific enzymatic MAPKs.
• Proposed that sustained therapy with high-inspired oxygen tensions (e.g. for
respiratory distress syndrome) causes epithelial damage, septal fibrosis and a scattered occlusion of distal airways characteristic of broncho-pulmonary dysphasia.
• Indicated that accumulating evidence has linked the pathogenesis of a variety of
diseases to oxidative stress. Theorized that unraveling the pathways of oxidant-induced cell death may provide alternate effective therapies to scavenging the initial oxidant in those systems where excessive oxidative stress and redox disequilibrium leads to inevitable brain and lung injury and death.
• Proposed that dynamic variations in oxygen levels regulate the release of
inflammatory/infectious mediators, thereby bearing consequences for the pediatric treatment of respiratory distress under conditions of clinical oxygen therapy, whereby those mediators are observed as potential participants in brain and lung pathophysiology.