untersuchungen zur eignung von zellulosederivaten zum
TRANSCRIPT

Untersuchungen zur Eignung von Zellulosederivaten
zum Coaten von Partikeln mit funktionellen
Inhaltsstoffen in der Wirbelschicht
vorgelegt von
M. Sc. Adrian Kape
geb. in Rostock
von der Fakultät III - Prozesswissenschaften
der Technische Universität Berlin
zur Erlangung des akademischen Grades
Doktor der Ingenieurwissenschaften
-Dr.-Ing.-
genehmigte Dissertation
Promotionsausschuss:
Vorsitzender: Prof. Dr. rer. nat. Lothar Kroh
Gutachter: Prof. Dr. sc. agr. Stephan Drusch
Gutachter: Prof. Dr.-Ing. Eckhard Flöter
Gutachterin: PD Dr. rer. nat. habil. Regina Scherließ
Tag der wissenschaftlichen Aussprache: 28.07.2016
Berlin 2016


Danksagung
III
Danksagung
Ganz herzlich bedanken möchte ich mich bei Prof. Dr. Stephan Drusch, der mich durch die
Jahre meiner Promotion mit viel gutem Rat und konstruktiver Kritik begleitet hat.
Mein Dank gilt Frau PD Dr. Regina Scherließ und Herrn Prof. Dr. Eckhard Flöter, die sich
bereit erklärt haben, meine Dissertation zu begutachten.
Den Mitarbeitern der Bundesanstalt für Materialforschung und Prüfung in Berlin Adlershof,
Annett Zimathies und Carsten Prinz, möchte ich für die Durchführung der Messungen, die
für die Partikelcharakterisierung unerlässlich waren, danken.
Des Weiteren gilt mein Dank den Mitarbeitern der ZELMI der Technischen Universität Ber-
lin, Christoph Fahrenson und Iryna Driehorst, für die Unterstützung bei der Erstellung der
Aufnahmen mit dem Rasterelektronenmikroskop.
Meinen Kollegen des Fachgebietes für Lebensmitteltechnologie und -materialwissen-
schaften möchte ich für die Hilfestellung bei der Laborarbeit und für die vielen anregenden
Diskussionen danken. Weiterhin möchte ich meinen Studenten, Babs, Daniel, Ursula und
Alexandra, meinen Dank aussprechen.
Besonders möchte ich mich bei Janine und Freddi bedanken. Ohne euch hätte ich sowohl
mein Studium, als auch meine Promotion sicher nicht mit der gleichen Freude und dem
gleichen Erfolg gemeistert.
Meine lieben Eltern haben mich während meines Studiums und meiner Promotion stets
unterstützt und standen mir immer zur Seite. Ihnen gilt daher mein ganz herzlicher Dank!

Zusammenfassung
IV
Zusammenfassung
Coatings, die im Bereich der Mikroverkapselung zumeist in der Wirbelschicht appliziert
werden, sind häufig dazu eingesetzt, zum Schutz der verkapselten Inhaltstoffe eine Barrie-
re gegen äußere Einflüsse zu bilden. Vor dem Hintergrund des in dieser Arbeit verwende-
ten, mehrfach ungesättigte Fettsäuren enthaltenden Rapsöls und der glasübergangstem-
peratursensiblen Trägermatrix aus Maltodextrin musste ein Coatingmaterial ausgewählt
werden, welches einen guten Kompromiss bezüglich der Barriereeigenschaften gegen
Sauerstoff und Wasserdampf bildet. Die Verwendung von zellulosederivatbasierten
Coatings könnte diesen Kompromiss erfüllen, da zu diesen bereits Studien zu den Barrie-
reeigenschaften gegenüber Sauerstoff- und Wasserdampf in Form von gegossenen Fil-
men vorliegen. Daher war es das Ziel dieser Arbeit, Zellulosederivate hinsichtlich ihrer
Eignung als funktionelle Coatings auf einer maltodextrinbasierten Trägermatrix zu untersu-
chen.
Die Oberflächenspannung der Coatinglösung und die freie Oberflächenenergie der Trä-
germatrix machen einen wesentlichen Teil der Partikel-Flüssigkeits-Interaktion aus, daher
wurde diese in der vorliegenden Arbeit untersucht und ein Modell zur Charakterisierung
der Spreitungs- und Haftungseigenschaften etabliert. Dieses Modell trägt dazu bei, die
Benetzungsprozesse und somit die Coatingqualität besser vorhersagen zu können.
Durch die rheologischen Charakterisierungen ist es gelungen, für methylzellulosebasierte
Coatingmaterialien die Grenzen hinsichtlich der Prozesstemperatur im Wirbelschicht-
coatingprozess zu bestimmen. Es konnte gezeigt werden, dass unter bestimmten Pro-
zessbedingungen die Eigenschaften und damit die Funktionalität von MC-Coatings negativ
beeinflusst werden können.
Hinsichtlich der Anwendung im Wirbelschichtcoatingprozess konnten die prozessbeding-
ten Einflüsse auf die Lipidoxidation des verkapselten Rapsöls und die Funktionalität des
methylzellulosebasierten Coatings identifiziert werden. Hierbei konnte festgestellt werden,
dass die Prozesszeit und -temperatur einen wesentlichen Einfluss auf die Stabilität des
verkapselten Rapsöls, nicht jedoch auf die Partikelstruktur haben. Weiterhin zeigte sich,
dass Coatings aus Methylzellulose, entgegen der gängigen Literatur, keine Barriereeigen-
schaft gegenüber Sauerstoff aufweisen, sich jedoch sehr gut zur Anwendung als Wasser-
dampfbarriere eignen.
Zukünftige Arbeiten sollten der Fragestellung nachgehen, wie das in der Trägermatrix ver-
kapselte Öl im Wirbelschichtcoatingprozess besser gegen eine Schädigung zu schützen
ist. Dies könnte durch eine Optimierung der Prozessparameter, beispielsweise einer Kon-
ditionierung der Fluidisierungsluft, erfolgen.

Abstract
V
Abstract
In order to protect a microencapsulated ingredient against environmental impacts coatings
can be applied in a fluidised bed coating process. With regard to the raw materials used in
this study, the rapeseed oil with a high content of polyunsaturated fatty acids and the
maltodextrin based carrier matrix with a high sensitivity regarding the glass transition tem-
perature, there is the need to find a coating material that is able perform a barrier against
oxygen as well as against water vapour. As described in literature, the use of coating ma-
terials based on cellulose derivatives are a promising compromise, due to their ability to
act as a barrier against oxygen and water vapour when applied as a film. Therefore the
aim of this study was to investigate the suitability of coating materials based on cellulose
derivatives as a functional coating on a maltodextrin based carrier matrix.
The surface tension of the coating solution and the surface free energy of the core material
are accountable for an essential part of the particle-liquid-interaction. Hence, there was a
model established in order to investigate the spreading and adhesion properties of coating
materials on model surfaces. The established model contributes to the prediction of the
wetting properties and therefore the prediction of the coating quality.
By means of rheological measurements it was feasible to determine the limits of the appli-
cation of methylcellulose based coatings with regards to the process temperature in the
fluidised bed coating process. Furthermore it was shown that the use of certain process
parameters such as the process temperature can negatively influence the functionality of
methylcellulose based coatings.
With regard to the application in the fluidised bed coating process, it was possible to identi-
fy process induced changes in the lipid oxidation of the encapsulated rapeseed oil as well
as the functionality of the methylcellulose based coating. It was proved, that the duration
and the temperature of the process have a high impact on the stability of the encapsulated
rapeseed oil, but not on the structure of the coated particles. In contrast to common litera-
ture it was shown, that the methylcellulose based coating do not act as a barrier against
oxygen. Nevertheless the methylcellulose based coatings show excellent barrier properties
against water vapour.
The protection of the encapsulated oil in the core particle during the fluidised bed coating
process should be part of future research. This could be realised for example by using
conditioned fluidisation air in order to optimise the drying of the film and the temperature
regime.

Inhaltsverzeichnis
VI
Inhaltsverzeichnis
Danksagung ..................................................................................................................... III
Zusammenfassung .......................................................................................................... IV
Abstract ............................................................................................................................. V
Inhaltsverzeichnis ............................................................................................................ VI
Abbildungsverzeichnis .................................................................................................... IX
Tabellenverzeichnis ....................................................................................................... XIV
Formelverzeichnis ........................................................................................................ XXII
Abkürzungen und Symbole ........................................................................................ XXIV
1 Einleitung .................................................................................................................... 1
2 Problemstellung .......................................................................................................... 5
3 Theoretische Grundlagen ........................................................................................... 8
3.1 Technologie des Coatens .......................................................................................... 9
3.2 Parameter zur Beeinflussung des Coatingergebnisses im
Wirbelschichtcoatingprozess ................................................................................... 11
3.3 Zellulose und Zellulosederivate ................................................................................ 22
3.3.1 Chemische und technologische Eigenschaften von Zellulosederivaten ................. 23
3.3.2 Rheologische Charakterisierung und rheologische Eigenschaften von
Zellulosederivaten ................................................................................................. 27
3.4 Lipidoxidation ........................................................................................................... 29
4 Material und Methoden ............................................................................................. 32
4.1 Charakterisierung des Haftungs- und Spreitungsverhaltens von
Zellulosederivatlösungen auf maltodextrinbasierten Modelloberflächen ................... 32
4.1.1 Herstellung der Coatinglösungen .......................................................................... 35
4.1.2 Charakterisierung der Coatinglösungen ................................................................ 35

Inhaltsverzeichnis
VII
4.1.3 Herstellung und Charakterisierung maltodextrinbasierter Modelloberflächen ........ 37
4.1.4 Charakterisierung der Partikel-Flüssigkeit-Interaktion ........................................... 41
4.1.5 Statistische Auswertung ........................................................................................ 43
4.2 Charakterisierung des rheologischen Verhaltens von lebensmitteltauglichen
Zellulosederivaten ................................................................................................... 44
4.2.1 Bestimmung der Viskosität in Abhängigkeit von Temperatur, Konzentration und
Schergeschwindigkeit ........................................................................................... 45
4.2.2 Bestimmung der strukturbildenden Charakteristika von MC .................................. 45
4.2.3 Charakterisierung des Strukturwiederaufbaus von MC-Lösungen nach
Scherbeanspruchung ............................................................................................ 48
4.3 Charakterisierung der Funktionalität von MC-Coatings als Sauerstoff- und
Wasserdampfbarriere .............................................................................................. 50
4.3.1 Herstellung der Primärpartikel, Agglomerate und gecoateten Agglomerate ........... 50
4.3.2 Chemische Analysen der Primärpartikel, Agglomerate, gecoateten Agglomerate . 55
4.3.3 Physikalische Analysen der Primärpartikel, Agglomerate und gecoateten
Agglomerate ......................................................................................................... 58
5 Ergebnisse ................................................................................................................ 65
5.1 Charakterisierung des Haftungs- und Benetzungsverhaltens von
Zellulosederivatlösungen auf MDEMUL ................................................................... 65
5.1.1 Charakterisierung der Coatinglösungen ................................................................ 67
5.1.2 Charakterisierung der maltodextrinbasierten Modelloberflächen ........................... 70
5.1.3 Charakterisierung der Partikel-Flüssigkeit-Interaktion ........................................... 71
5.2 Charakterisierung des rheologischen Verhaltens von lebensmitteltauglichen
Zellulosederivaten ................................................................................................... 76
5.2.1 Bestimmung der Viskosität von MC-Lösungen in Abhängigkeit von Temperatur,
Konzentration und Schergeschwindigkeit .............................................................. 76
5.2.2 Bestimmung der strukturbildenden Charakteristika von MC .................................. 79
5.2.3 Charakterisierung des Strukturwiederaufbaus von MC-Lösungen nach
Scherbeanspruchung ............................................................................................ 84
5.3 Charakterisierung der Funktionalität von zellulosederivatbasierten Coatingmaterialien
als Sauerstoffbarriere für funktionelle Lebensmittelinhaltsstoffe ............................... 89

Inhaltsverzeichnis
VIII
5.3.1 Vorversuche zur Überprüfung der Methoden zur Charakterisierung von gecoateten
Agglomeraten und der Untersuchung der Barriereeigenschaft der MC-Coatings .. 89
5.3.2 Untersuchung des Einflusses verschiedener Prozessparameter in der
Wirbelschicht auf verkapseltes Rapsöl in Agglomeraten ....................................... 91
5.3.3 Untersuchungen der strukturellen Partikeleigenschaften vor und nach dem
Prozessieren in der Wirbelschicht und deren Einfluss auf verkapseltes Rapsöl .... 96
5.3.4 Charakterisierung der Barriereeigenschaften von MC-Coatings auf ölbeladenen
Agglomeraten ..................................................................................................... 103
5.3.5 Untersuchung der Sauerstoffbarriereeigenschaft von MC-Filmen auf
Modelloberflächen ............................................................................................... 112
6 Diskussion ............................................................................................................... 113
6.1 Haftungs- und Benetzungsverhalten von zellulosederivatbasierten
Coatingmaterialien auf maltodextrinbasierten Modelloberflächen .......................... 113
6.2 Rheologische Charakterisierung der Coatinglösungen ........................................... 118
6.3 Prozessinduzierter Einfluss auf die Partikelstruktur und die Lipidoxidation von in der
Wirbelschicht behandelten Agglomeraten .............................................................. 122
6.4 Barriereeigenschaften zellulosebasierter Coatingmaterialien während der Lagerung126
7 Schlussbetrachtungen und Ausblick .................................................................... 129
8 Literaturverzeichnis ................................................................................................ 132
A. Anhang ................................................................................................................... 143
Tabellenanhang .............................................................................................................. 143
Abbildungsanhang .......................................................................................................... 174
Eidesstattliche Erklärung ................................................................................................ 176

Abbildungsverzeichnis
IX
Abbildungsverzeichnis
Abbildung 2.1: Darstellung der drei Arbeitsteile dieser Arbeit mit den dazugehörigen
Aufgabenstellungen und Untersuchungsmethoden......................................................5
Abbildung 3.1: Schema eines Top-Spray-Wirbelschichtcoaters mit Luftzufuhr, Abluft und
der zugeführten Coatinglösung; modifiziert vom Autor, nach Duangkhamchang et al.
(2015) ........................................................................................................................ 10
Abbildung 3.2: Schematische Darstellung der verschiedenen Arten einer
Wirbelschichtcoatinganlage mit (a) Top-Spray, (b) Bottom-Spray, (c) Bottom-Spray mit
Wurstereinsatz und (d) Rotor mit seitlich angebrachter Düse nach Teunou & Poncelet
(2002) ........................................................................................................................ 11
Abbildung 3.3: Schematische Darstellung einer außenmischenden und einer
innenmischende Zweistoffdüse, vom Autor bearbeitet, nach Salman et al. (2006) .... 12
Abbildung 3.4: Schematische Darstellung des Zerfallsprozesses des Flüssigkeitsfilms in
Ligamente und Tropfen in der Mischzone einer außenmischenden Zweistoffdüse mit
Angabe der Filmdicke (dsh), der Ligamentendicke (dlg) und-länge (Llg), vom Autor
bearbeitet, nach Duangkhamchan (2012) .................................................................. 13
Abbildung 3.5: Schematische Darstellung des Druckverlustes der Fluidiserungsluft Δp im
Fließbett in Abhängigkeit der Lufteintrittsgeschwindigkeit U mit den drei
verschiedenen Zuständen des Fließbettes mit zunehmender Geschwindigkeit ......... 17
Abbildung 3.6: Klassifizierung von Partikeln für Wirbelschichtanwendungen nach Geldart
(1973), mit dem Unterschied der Dichte zwischen Partikel und umgebener Gasphase
als Funktion der Partikelgröße; dargestellt sind die Bereiche für die Partikelklassen: C
„cohesive“, A „aerateble“, B „bubbling“ und C „spouting“ ........................................... 18
Abbildung 3.7: Schematische Darstellung der Materialeigenschaften (schwarz) und der
Prozesseigenschaften (grau), die Einfluss auf die Partikel-Flüssigkeits-Interaktion
haben ........................................................................................................................ 21
Abbildung 3.8: Schematischer Aufbau von Zellulose aus Pflanzenfasern, vom Autor
bearbeitet nach Wüstenberg (2015) .......................................................................... 22
Abbildung 3.9: Schematischer Ablauf der thermisch induzierten Gelbildung von
Zellulosederivaten am Beispiel von MC, vom Autor bearbeitet, nach Haque & Morris
(1993) ........................................................................................................................ 27

Abbildungsverzeichnis
X
Abbildung 4.1: Schematische Darstellung des Tropfenkonturmessgerätes für die Messung
des Kontaktwinkels von Referenz- und Coatinglösungen auf maltodextrinbasierten
Modelloberflächen, vom Autor modifiziert, nach Tamm et al. (2012) .......................... 33
Abbildung 4.2: SE-Plot zur Bestimmung der Oberflächenenergie einer Modelloberfläche
mit der beispielhaften Kombination von Referenzlösungen WEF und der zur
Berechnung herangezogenen Ausgleichsgeraden .................................................... 40
Abbildung 4.3 Bestimmung der strukturbildenden Parameter bei der Untersuchung der
Gelbildung nach Kastner et al. (2012), hier beispielhaft vom Autor für eine 5%ige
MC15-Lösung dargestellt........................................................................................... 46
Abbildung 4.4: Schematischer Ablauf der Versuche zur Bestimmung des
Strukturwiederaufbaus der MC15- und MC383-Lösungen ......................................... 48
Abbildung 4.5: Volumetrischer Verlauf der Gasadsorption mit Va: spezifisches adsorbiertes
Volumen und p/p0: Relativdruck sowie schematische Darstellung der Messapparatur
(nach DIN ISO 9277); mit 1: Probe, 2: Dewargefäß mit Kühlbad, 3: Vakuumpumpe, 4:
Manometer, 5: kalibriertes Volumen (Gasbürette), 6: Sättigungsdruckröhrchen, 7:
Adsorptivbehälter und 8: Gasbehälter für die Totvolumenbestimmung; ..................... 61
Abbildung 4.6: Darstellung des BET-Diagramms zur Auswertung der spezifischen
Oberfläche der Probe nach DIN ISO 9277; mit 1: Mehrpunkt-BET-Gerade, 2:
Einpunkt-BET-Gerade, : experimentelle Datenpunkte, +: Datenpunkt für Einpunkt-
Berechnung, p/p0: Relativdruck des Adsorptivs, α: Ordinatenabschnitt, Δx
Abszissendifferenz und Δy Ordinatendifferenz .......................................................... 61
Abbildung 5.1: Exemplarischer Vergleich zweier Kontaktwinkelmessungen von
Diiodmethan mit einem Volumen von 2,5 µL auf Glas mit und ohne
Basisliniendetektion (BDL), dargestellt sind die gemittelten Kontaktwinkel, die über
eine Messdauer von einer Minute aufgenommen wurden .......................................... 66
Abbildung 5.2: Kontaktwinkel einer MCl Coatinglösung auf MDEMUL über die Messzeit
von 20 s in 20-fach Wiederholung mit Verteilungsbreite und Median ......................... 67
Abbildung 5.3: SE-Plot zur Bestimmung der Oberflächenenergie von MDEMUL mit den
Kombination von Referenzlösungen WEF, WDEF, WDE, WDF und DEF und den
daraus resultierenden Ausgleichgeraden mit den Datenpunkten für D, E, F und W
(von links) .................................................................................................................. 70
Abbildung 5.4: gemessener Kontaktwinkel der Coatinglösungen auf MDEMUL in den vier
verschiedenen Viskositätsstufen (sehr niedrig: vl; niedrig: l; mittel: m und hoch: h) mit

Abbildungsverzeichnis
XI
A: CMC; B: MC; C: Nutrateric® und D: HPMC; für die Tabelle: A, B, unterschiedliche
Buchstaben kennzeichnen signifikante Unterschiede zwischen verschiedenen
Viskositätsstufen ....................................................................................................... 74
Abbildung 5.5: Wetting Envelope der MDEMUL Oberfläche mit Linien gleichen
Kontaktwinkels (schmal, durchgezogen), Linien gleicher Adhäsionsarbeit (schmal,
gepunktet) sowie Linien des Adhäsionskorridors (dick, durchgezogen) und den
Punkten der einzelnen Coatinglösungen ................................................................... 75
Abbildung 5.6: Darstellung von GP und GA, IST und CST sowie SDR in Abhängigkeit von
der Konzentration der MC383 .................................................................................... 79
Abbildung 5.7: Darstellung des Speichermoduls G’ zu Beginn (t=0) der Haltezeit bei 70 °C
und nach 30 Minuten Haltezeit (t=30) von MC383 in verschiedenen Konzentrationen
.................................................................................................................................. 80
Abbildung 5.8: Darstellung von GP und GA, IST und CST sowie SDR in Abhängigkeit von
der Konzentration der MC15 ...................................................................................... 80
Abbildung 5.9: Darstellung des Speichermoduls G’ zu Beginn (t=0) der Haltezeit bei 70 °C
und nach 30 Minuten Haltezeit (t=30) von MC15 in verschiedenen Konzentrationen . 81
Abbildung 5.10: Darstellung de Strukturwiederaufbaus der MC383-Lösungen nach der
Scherbelastung bei einer Systemtemperatur von 20 °C dargestellt als Regeneration
[%] gegenüber des Ausgangswertes vor der Scherbelastung über die Zeit ............... 85
Abbildung 5.11: Darstellung des Strukturwiederaufbaus der MC15-Lösungen nach der
Scherbelastung bei einer Systemtemperatur von 20 °C dargestellt als Regeneration
[%] gegenüber des Ausgangswertes vor der Scherbelastung über die Zeit ............... 87
Abbildung 5.12: Zeitlicher Verlauf der Hydroperoxidkonzentration von sprühgetrocknetem
Pulver, Agglomeraten und gecoateten Agglomeraten bei 60 °C Prozesstemperatur
und einer Prozesszeit von 30 bzw. 60 Minuten .......................................................... 90
Abbildung 5.13: Extrahierbarer Ölgehalt und Mikroverkapselungseffizienz von
sprühgetrockneten Pulvern, unbehandelten Agglomeraten und verschiedenen in der
Wirbelschicht behandelten Agglomeraten .................................................................. 93
Abbildung 5.14: Zeitlicher Verlauf der Hydroperoxidkonzentration von unbehandelten
Agglomeraten und verschieden trocken in der Wirbelschicht behandelten
Agglomeraten mit Prozesstemperaturen von 30°C bzw. 60 °C und Prozesszeiten von
20, 40 bzw. 60 Minuten ............................................................................................. 94

Abbildungsverzeichnis
XII
Abbildung 5.15: Zeitlicher Verlauf der Hydroperoxidkonzentration von unbehandelten
Agglomeraten und verschieden in der Wirbelschicht unter Wasserzugabe behandelten
Agglomeraten mit Prozesstemperaturen von 30°C bzw. 60 °C und Prozesszeiten von
20, 40 bzw. 60 Minuten ............................................................................................. 95
Abbildung 5.16: Aufnahmen des Rasterelektronenmikroskops; A: MP-H2O-T60t60; B: MP-
T60t60; C: PP-H2O-T60t60; D: PP-T60t60 ; A, B, C: Vergrößerung 500-fach; D:
Vergrößerung 300-fach ............................................................................................. 98
Abbildung 5.17: Hydroperoxidkonzentration der unbehandelten Agglomerate und der bei
60°C über 20 bzw. 60 Minuten mit und ohne Wasser behandelten Agglomerate über
den Zeitraum von 16 Wochen mit Untersuchungen alle zwei Wochen ....................... 99
Abbildung 5.18: Porengrößenverteilung der Probe MP-Agglomerate nach der Hg-
Porosimetrie (Druckaufbau: 0,0007-402 MPa, Einwaage Probe: 0,5 g) ................... 101
Abbildung 5.19: Aufnahmen des Rasterelektronenmikroskops von MC15 gecoateten
Agglomeraten; A, B: MC15-T30t20 ; C, D: MC15-T60t60; A, C: Vergrößerung: 500-
fach; B: Vergrößerung 4000-fach; D: Vergrößerung: 3000-fach ............................... 106
Abbildung 5.20: Hydroperoxidkonzentration der unbehandelten Agglomerate und der bei
30°C und 60°C über 20 bzw. 60 Minuten mit einer 5%igen MC-Lösung gecoateten
Agglomerate über den Zeitraum von 18 Wochen mit Untersuchungen alle zwei
Wochen ................................................................................................................... 107
Abbildung 5.21: Wasserdampfsorption der ungecoateten Agglomerate und der mit
Methylzellulose in verschiedenen Zeit-Temperatur Kombinationen gecoateten
Agglomerate bei der Lagerung in verschiedener relativer Luftfeuchtigkeit bei
20 ± 0,5 °C über einen Zeitraum von 35 Tagen ....................................................... 109
Abbildung 5.22: Aufnahmen der Proben zur Wasserdampfsorptionsmessung nach
Durchlaufen von Feuchtigkeitsstufen 11, 33, 53, 75 und 85 % relativer Luftfeuchtigkeit
mit: 1: ungecoatete Agglomerate, 2: MC15-T30-20, 3: MC15-T30t60, 4: MC15-T60t20,
5: MC15-T60t60 ...................................................................................................... 111
Abbildung 5.23: Hydroperoxidkonzentration von ungecoateten und gecoateten
Modelloberflächen über einen Lagerzeitraum von zehn Wochen mit Untersuchungen
im zweiwöchigen Untersuchungsintervall ................................................................ 112
Abbildung A.1: Logarithmisches Differenzialvolumen über der Porenweite der ölbeladenen
Agglomerate und unterschiedlich in der Wirbelschicht behandelten ölbeladenen
Agglomeraten .......................................................................................................... 174

Abbildungsverzeichnis
XIII
Abbildung A.2: Kumulatives Porenvolumen über der Porenweite der ölbeladenen
Agglomerate und unterschiedlich in der Wirbelschicht behandelten ölbeladenen
Agglomeraten .......................................................................................................... 174
Abbildung A.3: Logarithmisches Differenzialvolumen über der Porenweite der
Matrixpartikel-Agglomerate und unterschiedlich in der Wirbelschicht behandelten
Matrixpartikel-Agglomeraten .................................................................................... 175
Abbildung A.4: Kumulatives Porenvolumen über der Porenweite der Matrixpartikel-
Agglomerate und unterschiedlich in der Wirbelschicht behandelten Matrixpartikel-
Agglomeraten .......................................................................................................... 175

Tabellenverzeichnis
XIV
Tabellenverzeichnis
Tabelle 3.1: Partikel- und Wirbelbettcharakteristika nach Rhodes (2008); durch Autor
ergänzt ...................................................................................................................... 20
Tabelle 3.2: Übersicht über die Wasserdampfpermeabilität von gegossenen Filmen aus
CMC, HPMC und MC aus der Literatur mit jeweiligem Molekulargewicht, falls in
Studie angegeben, teils umgerechnet nach Hagenmaier (2012) ............................... 25
Tabelle 3.3: Übersicht über die Sauerstoffpermeabilität von gegossenen Filmen von MC,
HPMC und CMC aus der Literatur mit jeweiligem Molekulargewicht, falls in Studie
angegeben, teils umgerechnet nach Hagenmaier (2012) .......................................... 26
Tabelle 4.1: Volumina der verschiedenen Referenzlösungen zur Methodenoptimierung der
Kontaktwinkelmessung auf Glas ................................................................................ 34
Tabelle 4.2: Konzentrationen [g/100g] der verschiedenen Coatingmaterialien zur
Herstellung der Coatinglösungen in verschiedenen Viskositätsstufen........................ 35
Tabelle 4.3: Einzusetzende Masse [g] für die verschiedenen Viskositätsstufen für 50 g
Nutrateric®-Lösung berechnet auf Grundlage der Auflösevorschriften der Firma
Colorcon Limited ....................................................................................................... 35
Tabelle 4.4: Viskosität η [mPa·s] der verwendeten Coatinglösungen in den jeweiligen
Viskositätsstufen ....................................................................................................... 36
Tabelle 4.5: Drehzahlen [min-1] für die Spindel 00 im UL-Adapter für die Bestimmung der
Viskosität in verschiedenen Viskositätsstufen der Coatinglösungen mittels Brookfield
LV-Rotationsviskosimeter .......................................................................................... 36
Tabelle 4.6: Oberflächenspannung mit polaren und dispersen Anteilen der
Referenzlösungen zur Bestimmung der freien Oberflächenenergie der
maltodextrinbasierten Modelloberflächen Rabel (1971) und Ström et al. (1987) ........ 41
Tabelle 4.7: Herstellerangaben zu den in dieser Arbeit verwendeten Zellulosederivaten .. 44
Tabelle 4.8: Verwendete Temperaturen, Konzentrationen und Scherraten zur
Untersuchung der Viskosität ...................................................................................... 45
Tabelle 4.9: Verwendete Methylzellulose- und Zitronensäurekonzentrationen zur
Ermittlung der strukturbildenden Charakteristika ....................................................... 47
Tabelle 4.10: Parameter zur Durchführung der Sprungversuche zur Bestimmung des
thixotropen Verhaltens von MC-Lösungen ................................................................. 49

Tabellenverzeichnis
XV
Tabelle 4.11: Einsatzmengen [g] für einen Ansatz von 100 g Emulsion bzw. Dispersion für
die Herstellung von Primär- bzw. Matrixpartikeln ....................................................... 51
Tabelle 4.12: Auftragsmengen an Methylzellulose und Prozessparameter für das Coaten
von Agglomeraten (Versuchsreihe 4) ........................................................................ 53
Tabelle 4.13: Darstellung der Probenbezeichnungen für Versuchsreihe 2 für die
Agglomerate, die in der Wirbelschicht den unterschiedlichen Prozessbedingungen
ausgesetzt wurden, mit: „mit“ (mit H2O), „ohne“ (ohne H2O), den entsprechenden
Prozesszeiten (20, 40 und 60 Minuten) und den entsprechenden
Produkttemperaturen (60 und 30 °C) ......................................................................... 53
Tabelle 4.14: Durchgeführte Versuchsreihen mit den jeweiligen Untersuchungen ............ 54
Tabelle 5.1: SFT, SFTDodecan, SFTp und SFTd der Coatinglösungen in verschiedenen
Viskositätsstufen ....................................................................................................... 69
Tabelle 5.2: SE mit SEd und SEp, ihren prozentualen Anteilen und dem Bestimmtheitsmaß
für die Kalkulation von SE der MDEMUL-Oberfläche unter Verwendung verschiedener
Kombinationen von Referenzlösungen ...................................................................... 71
Tabelle 5.3: WA der verwendeten Coatingmaterialien unter Berücksichtigung der
Verwendung von unterschiedlichen Kombinationen der Referenzlösungen zur
Bestimmung der Oberflächenenergie der MDEMUL-Oberflächen nach OWRK und der
WA berechnet nach dem Modell nach YD .................................................................. 72
Tabelle 5.4: Nach dem Modell von OWRK berechneter Kontaktwinkel von verschiedenen
Coatingmaterialien unter Verwendung unterschiedlicher Kombinationen von
Referenzlösungen zur Oberflächenenergiecharakterisierung von MDEMUL-
Oberflächen ............................................................................................................... 73
Tabelle 5.5: Mittelwerte der Viskosität der scherrheologischen Untersuchungen von
MC383 in Abhängigkeit von der Konzentration, der Scherrate und der Temperatur... 77
Tabelle 5.6: Mittelwerte der Viskosität der scherrheologischen Untersuchungen von MC15
in Abhängigkeit von der Konzentration, der Scherrate und der Temperatur ............... 78
Tabelle 5.7: IST, GP, CST, SDR, GA, Gelstärke zu Beginn der Haltezeit (t=0) sowie
Gelstärke nach 30 Minuten Haltezeit (t=30) der MC15 und MC383-Proben bei
verschiedenen Konzentrationen mit Zugabe verschiedener Konzentrationen an
Zitronensäure (ZS) .................................................................................................... 82
Tabelle 5.8: IST, GP, CST, SDR, GA, Gelstärke zu Beginn der Haltezeit (t=0) und
Gelstärke nach 20 Minuten Haltezeit (t=20) der MC15– und MC383-Proben bei

Tabellenverzeichnis
XVI
verschiedenen Konzentrationen mit Zugabe verschiedener Konzentrationen an EtOH
in der Lösemittelphase .............................................................................................. 84
Tabelle 5.9: Darstellung der prozentualen Regeneration der MC383-Proben in
Abhängigkeit von der Zeit bezogen auf den jeweiligen Wert der Viskosität am Ende
der Vorscherung ........................................................................................................ 86
Tabelle 5.10: Darstellung der prozentualen Regeneration der MC15-Proben in
Abhängigkeit von der Zeit bezogen auf den jeweiligen Wert der Viskosität am Ende
der Vorscherung ........................................................................................................ 88
Tabelle 5.11: Gesamtfettgehalt und darüber berechnete applizierte Coatingmenge vom
sprühgetrockneten Pulver, den Agglomeraten und zwei mit unterschiedlich viel (70
und 130 mL) MC15-Lösung gecoateten Agglomeraten in % bezogen auf die
Gesamtmasse der gecoateten Agglomerate .............................................................. 89
Tabelle 5.12: Charakteristische Verteilungsparameter (d10, d50 und d90) und
Verteilungsbreite (span) der Öltropfengrößenbestimmung mittels SLS von der
Grundemulsion und dem sprühgetrockneten Pulver der ersten und zweiten Woche
und den Agglomeraten .............................................................................................. 91
Tabelle 5.13: Charakteristische Verteilungsparameter (d10, d50 und d90) und
Verteilungsbreite (span) der Öltropfengrößenbestimmung mittels SLS von
verschieden in der Wirbelschicht behandelten und resolubilisierten Agglomeraten ... 92
Tabelle 5.14: Charakteristische Verteilungsparameter (d10, d50 und d90) und
Verteilungsbreite (span) der Öltropfengrößenbestimmung mittels SLS der
Primäremulsion, dem sprühgetrockneten Pulver, den Agglomeraten und
verschiedener in der Wirbelschicht behandelter und resolubilisierter Agglomerate .... 97
Tabelle 5.15: Mikroverkapselungseffizienz von sprühgetrocknetem Pulver, unbehandelten
Agglomeraten und in der Wirbelschicht verschieden behandelten Agglomeraten ...... 97
Tabelle 5.16: Oberflächengrößen der unbehandelten und behandelten Matrixpartikel-
Agglomerate sowie der unbehandelten und behandelten ölbeladenen Agglomerate
ermittelt durch Gasadsorptionsmethode mit Krypton bei 77 K bzw. durch dynamische
Dampfsorption (DVS) mit n-Octan; Auswertung erfolgte für die Gasadsorption mittels
BET bzw. BET und ESW für die DVS ...................................................................... 100
Tabelle 5.17: Gegenüberstellung der Feststoffdichten von unbehandelten Matrixpartikel-
Agglomeraten, in der Wirbelschicht behandelten Matrixpartikel-Agglomeraten,
unbehandelten ölbeladenen Agglomeraten und in der Wirbelschicht behandelten

Tabellenverzeichnis
XVII
ölbeladenen Agglomeraten, welche mittels zwei verschiedener Messverfahren
ermittelt wurden ....................................................................................................... 102
Tabelle 5.18: Charakteristische Verteilungsparameter (d10, d50 und d90) und
Verteilungsbreite (span) der Öltropfengrößenbestimmung mittels SLS der
Primäremulsion, dem sprühgetrockneten Pulver, den Agglomeraten und
verschiedener in der Wirbelschicht gecoateter und resolubilisierter Agglomerate .... 104
Tabelle 5.19: Gesamtfettgehalt und berechnete applizierte Coatingmenge von
unterschiedlich gecoateten Agglomeraten ............................................................... 105
Tabelle 5.20: Mikroverkapselungseffizienz von Agglomeraten, die mit verschieden Zeit-
Temperatur-Kombinationen gecoatet sind ............................................................... 105
Tabelle 5.21: Darstellung der Feststoffdichte der 20 bzw. 60 Minuten und bei 30 °C bzw.
60 °C gecoateten Agglomerate ................................................................................ 108
Tabelle 5.22: Restfeuchte nach Schnellfeuchtebestimmung und abgegebene
Wassermenge während der Vortrocknung bei 0% relative Luftfeuchte nach 21 Tage
von ungecoateten und in verschiedenen Zeit-Temperatur-Kombinationen gecoateten
Agglomeraten .......................................................................................................... 109
Tabelle 5.23: Werte für die Wasseraufnahme für die jeweilige relative Feuchtigkeit,
dargestellt am jeweiligen Tag der Gewichtskonstanz, Lagerung bei 20 ± 0,5 °C ..... 110
Tabelle A.1: Verwendete Geräte ..................................................................................... 143
Tabelle A.2: Verwendete Chemikalien ............................................................................ 147
Tabelle A.3: Verwendete Materialien .............................................................................. 150
Tabelle A.4: Gemessene Kontaktwinkel der Referenzlösungen auf den
maltodextrinbasierten Modelloberflächen ................................................................ 151
Tabelle A.5: Direkt gemessene Kontaktwinkel (CAm) auf den Oberflächen MDT, MDGSS
und MDOSA ............................................................................................................ 151
Tabelle A.6: Freie Oberflächenenergie mit dispersen und polaren Anteil, ihren
prozentualen Anteilen und dem Regressionskoeffizienten für die Kalkulation der
Oberflächenenergie MDT unter Verwendung verschiedener Kombinationen von
Referenzlösungen zur Oberflächenenergiecharakterisierung .................................. 152
Tabelle A.7: Freie Oberflächenenergie mit dispersen und polaren Anteil, ihren
prozentualen Anteilen und dem Regressionskoeffizienten für die Kalkulation der

Tabellenverzeichnis
XVIII
Oberflächenenergie MDGSS unter Verwendung verschiedener Kombinationen von
Referenzlösungen zur Oberflächenenergiecharakterisierung .................................. 152
Tabelle A.8: Freie Oberflächenenergie mit dispersen und polaren Anteil, ihren
prozentualen Anteilen und dem Regressionskoeffizienten für die Kalkulation der
Oberflächenenergie MDOSA unter Verwendung verschiedener Kombinationen von
Referenzlösungen zur Oberflächenenergiecharakterisierung .................................. 153
Tabelle A.9: Berechnete Adhäsionsarbeit der verwendeten Coatingmaterialien unter
Berücksichtigung der Verwendung von unterschiedlichen Kombinationen
Referenzlösungen zur Bestimmung der Oberflächenenergie von MDGSS .............. 154
Tabelle A.10: Nach dem Modell von OWRK berechneter Kontaktwinkel von verschiedenen
Coatingmaterialien unter Verwendung unterschiedlicher Kombinationen von
Referenzlösungen zur Oberflächenenergiecharakterisierung von MDGSS .............. 155
Tabelle A.11: Technische Daten des luftgelagerten Rheometers USD200 der Fima Anton
Paar Germany GmbH .............................................................................................. 155
Tabelle A.12: Technische Daten des luftgelagerten Rheometers MCR301 der Firma Anton
Paar Germany GmbH .............................................................................................. 156
Tabelle A.13: Durchschnittliche Fettsäurezusammensetzung des verwendeten
kommerziellen Rapsöls (Kaiser’s A&P); zur Verfügung gestellt am 21.05.2015 durch
Denise Erkelenz (Kaiser’s Tengelmann GmbH) ....................................................... 156
Tabelle A.14: Ergebnisse des Scherratensweep mit den Ergebnissen bei 1000 s-1, 2500 s-1
und 4000 s-1 bei 30 °C, 45 °C und 60 °C für eine 1 %ige MC-Lösung von MC383 ... 157
Tabelle A.15: Ergebnisse des Scherratensweep mit den Ergebnissen bei 1000 s-1, 2500 s-1
und 4000 s-1 bei 30 °C, 45 °C und 60 °C für eine 3 %ige MC-Lösung von MC383 ... 158
Tabelle A.16: Ergebnisse des Scherratensweep mit den Ergebnissen bei 1000 s-1, 2500 s-1
und 4000 s-1 bei 30 °C, 45 °C und 60 °C für eine 5 %ige MC-Lösung von MC383 ... 159
Tabelle A.17: Ergebnisse des Scherratensweep mit den Ergebnissen bei 1000 s-1, 2500 s-1
und 4000 s-1 bei 30 °C, 45 °C und 60 °C für eine 5 %ige MC-Lösung von MC15 ..... 160
Tabelle A.18: Ergebnisse des Scherratensweep mit den Ergebnissen bei 1000 s-1, 2500 s-1
und 4000 s-1 bei 30 °C, 45 °C und 60 °C für eine 6,5 %ige MC-Lösung von MC15 .. 161
Tabelle A.19: Ergebnisse des Scherratensweep mit den Ergebnissen bei 1000 s-1, 2500 s-1
und 4000 s-1 bei 30 °C, 45 °C und 60 °C für eine 8 %ige MC-Lösung von MC15 ..... 162

Tabellenverzeichnis
XIX
Tabelle A.20 Temperaturprofil 1 zur Bestimmung der strukturbildenden Charakteristika der
MC-Lösungen .......................................................................................................... 162
Tabelle A.21: Temperaturprofil 2 zur Bestimmung der strukturbildenden Charakteristika der
MC-Lösungen unter Einfluss eine Zugabe von Ethanol ........................................... 163
Tabelle A.22: Initiale Strukturierungstemperatur (IST), Gelpunkt (GP), kritische
Strukturierungstemperatur (CST), mittlere Strukturentwicklungsgeschwindigkeit
(SDR), Gelauflösungstemperatur (GA), Gelstärke zu Beginn der Haltezeit (t=0) und
Gelstärke nach 30 Minuten Haltezeit (t=30) der MC15– und MC383-Proben bei
verschiedenen Konzentrationen .............................................................................. 163
Tabelle A.23: Herstellungsanweisung für die Lösungen zur Bestimmung der
Hydroperoxidkonzentration ...................................................................................... 164
Tabelle A.24: Gehalte an Eisen(III)-chloridlösung für die Verdünnungsstufen der
Kalibriergeraden zur Bestimmung der Hydroperoxidkonzentration .......................... 165
Tabelle A.25: Einwaagen der Salze zur Herstellung von übersättigten Salzkonzentrationen
für die Bestimmung des Wasserdampfsorption von Pulvern .................................... 165
Tabelle A.26: Hydroperoxidkonzentration des sprühgetrockneten Pulvers, der
Agglomerate, und der mit 70 bzw. 130 mL MC-Lösung gecoateten Agglomerate über
den Zeitraum von acht Wochen mit wöchentlicher Untersuchung, mit Ausnahme von
Woche sieben .......................................................................................................... 166
Tabelle A.27: Hydroperoxidkonzentration der unbehandelten Agglomerate und der bei
30°C über 20, 40 und 60 Minuten behandelten Agglomerate über den Zeitraum von
10 Wochen mit Untersuchungen alle zwei Wochen ................................................. 166
Tabelle A.28: Hydroperoxidkonzentration der bei 60°C über 20, 40 und 60 Minuten
behandelten Agglomerate über den Zeitraum von 10 Wochen mit Untersuchungen
alle zwei Wochen .................................................................................................... 167
Tabelle A.29 Hydroperoxidkonzentration der bei 30°C über 20, 40 und 60 Minuten mit
Wasser behandelten Agglomerate über den Zeitraum von 10 Wochen mit
Untersuchungen alle zwei Wochen .......................................................................... 167
Tabelle A.30: Hydroperoxidkonzentration der bei 60°C über 20, 40 und 60 Minuten mit
Wasser behandelten Agglomerate über den Zeitraum von 10 Wochen mit
Untersuchungen alle zwei Wochen .......................................................................... 168

Tabellenverzeichnis
XX
Tabelle A.31: Hydroperoxidkonzentration der unbehandelten Agglomerate und der bei
60°C über 20 bzw. 60 Minuten mit und ohne Wasser behandelten Agglomerate über
den Zeitraum von 16 Wochen mit Untersuchungen alle zwei Wochen ..................... 168
Tabelle A.32: Hydroperoxidkonzentration der unbehandelten Agglomerate und der bei
30°C und 60°C über 20 bzw. 60 Minuten mit einer 5%igen MC-Lösung gecoateten
Agglomerate über den Zeitraum von 18 Wochen mit Untersuchungen alle zwei
Wochen ................................................................................................................... 169
Tabelle A.33: Werte für die Wasseraufnahme der ungecoateten und mit MC gecoateten
Agglomerate in fünf verschiedenen relativen Luftfeuchten und über eine Zeit von 35
Tagen ...................................................................................................................... 170
Tabelle A.34: Hydroperoxidkonzentration von ungecoateten und gecoateten
Modelloberflächen über einen Lagerzeitraum von zehn Wochen zweiwöchigem
Untersuchungsinterval ............................................................................................. 171
Tabelle A.35: Hydroperoxidkonzentrationen von ungecoateten bzw. unbehandelten
Agglomeraten der verschiedenen Versuchsreihen mit Startwerten (Woche 0) und den
Wochen 8, 10, 16 und 18 aus den jeweiligen Versuchsreihen mit: Versuch 1:
Vorversuch zur Schichtdickenvariation; Versuch 2: Versuch zur Prozessoptimierung
ohne Wasserzugabe; 4: Versuch zur Partikelanalyse ohne Wasserzugabe und
Versuch 6: Versuch zum MC- Coating ..................................................................... 171
Tabelle A.36: Hydroperoxidkonzentrationen der bei 30 °C für 20 Minuten gecoateten bzw.
behandelten Agglomeraten der verschiedenen Versuchsreihen mit Startwerten
(Woche 0) und den Wochen 8, 10, 16 und 18 aus den jeweiligen Versuchsreihen mit:
Versuch 2: Versuch zur Prozessoptimierung ohne Wasserzugabe; Versuch 3:
Versuch zur Prozessoptimierung mit Wasserzugabe und Versuch 6: Versuch zum
MC-Coating ............................................................................................................. 172
Tabelle A.37: Hydroperoxidkonzentrationen der bei 30 °C für 60 Minuten gecoateten bzw.
behandelten Agglomeraten der verschiedenen Versuchsreihen mit Startwerten
(Woche 0) und den Wochen 8, 10, 16 und 18 aus den jeweiligen Versuchsreihen mit:
Versuch 2: Versuch zur Prozessoptimierung ohne Wasserzugabe; Versuch 3:
Versuch zur Prozessoptimierung mit Wasserzugabe und Versuch 6: Versuch zum
MC- Coating ............................................................................................................ 172
Tabelle A.38: Hydroperoxidkonzentrationen der bei 60 °C für 20 Minuten gecoateten bzw.
behandelten Agglomeraten der verschiedenen Versuchsreihen mit Startwerten
(Woche 0) und den Wochen 8, 10, 16 und 18 aus den jeweiligen Versuchsreihen mit:

Tabellenverzeichnis
XXI
Versuch 2: Versuch zur Prozessoptimierung ohne Wasserzugabe; Versuch 3:
Versuch zur Prozessoptimierung mit Wasserzugabe; Versuch 5: Versuch zur
Partikelanalyse mit Wasserzugabe und Versuch 6: Versuch zum MC- Coating ....... 173
Tabelle A.39: Hydroperoxidkonzentrationen der bei 60 °C für 60 Minuten gecoateten bzw.
behandelten Agglomeraten der verschiedenen Versuchsreihen mit Startwerten
(Woche 0) und den Wochen 8, 10, 16 und 18 aus den jeweiligen Versuchsreihen mit:
Versuch 1: Vorversuch zur Schichtdickenvariation; Versuch 2: Versuch zur
Prozessoptimierung ohne Wasserzugabe; Versuch 3: Versuch zur
Prozessoptimierung mit Wasserzugabe; Versuch 4: Versuch zur Partikelanalyse ohne
Wasserzugabe; Versuch 5: Versuch zur Partikelanalyse mit Wasserzugabe und
Versuch 6: Versuch zum MC- Coating ..................................................................... 173

Formelverzeichnis
XXII
Formelverzeichnis
Formel 3.1: Empirische Abschätzung des Sauterdurchmessers d32 beim Tropfenzerfall
unter Berücksichtigung der Ohnesorg-Zahl und der Weber-Zahl [µm] ...... 14
Formel 3.2: Berechnung der Weber-Zahl .................................................................... 14
Formel 3.3: Berechnung der Ohnesorg-Zahl ............................................................... 14
Formel 3.4: Lockerungsgeschwindigkeit für Partikel mit d < 100 µm [m/s] .................. 15
Formel 3.5: Archimedes-Zahl ...................................................................................... 15
Formel 3.6: Lockerungsgeschwindigkeit für Partikel mit d > 100 µm [m/s] .................. 16
Formel 3.7: Schwebegeschwindigkeit für Partikel unter der Annahme eines komplett
kugelförmigen Partikels [m/s] ................................................................... 16
Formel 3.8: Voraussetzungen für Partikel um der Klasse A nach Geldart (1973) zu ent-
sprechen .................................................................................................. 18
Formel 3.9: Vereinfachte Voraussetzungen für Partikel um der Klasse A nach Geldart
(1973) zu entsprechen ............................................................................. 18
Formel 3.10: Voraussetzungen für Partikel um der Klasse B nach Geldart (1973) zu ent-
sprechen .................................................................................................. 18
Formel 3.11: Voraussetzungen für Partikel um der Klasse D nach Geldart (1973) zu ent-
sprechen .................................................................................................. 18
Formel 3.12: Berechnung des Schubmoduls [N/m2] ...................................................... 27
Formel 3.13: Berechnung des Speichermoduls [N/m2] .................................................. 27
Formel 3.14: Berechnung des Verlustmoduls [N/m2] ..................................................... 27
Formel 4.1: Laplace-Druck an einer gekrümmten Oberfläche [N/m2] ........................... 36
Formel 4.2: Druckdifferenz zwischen Laplace-Druck und dem Umgebungsdruck einer
gekrümmten Oberfläche [N/m2] ................................................................ 36
Formel 4.3: Geradengleichung zur Bestimmung der freien Oberflächenenergie ......... 38
Formel 4.4: Freie Oberflächenenergie mit polaren und dispersen Anteilen [mN/m] ..... 39
Formel 4.5: Adhäsionsarbeit nach dem Modell von Young-Dupré [mN/m] .................. 40
Formel 4.6: Adhäsionsarbeit nach dem Modell von Owens, Wendt, Rabel und
Kaelble [mN/m] ........................................................................................ 41

Formelverzeichnis
XXIII
Formel 4.7: Annahme der Gleichsetzung von WA-YD und WA-OWRK ............................... 41
Formel 4.8: Einsetzen von Formel 4.5 und Formel 4.6 in Formel 4.7 .......................... 41
Formel 4.9: Kosinus des berechneten Kontaktwinkels nach OWRK ............................ 41
Formel 4.10: Berechneter Kontaktwinkel nach OWRK [°] .............................................. 41
Formel 4.11: Strukturbildungsgeschwindigkeit nach Kastner et al. (2012) [Pa/s]........... 44
Formel 4.12: Fettgehalt in der Trockenmasse ungecoateter Agglomeraten [g/100 g] .... 55
Formel 4.13: Fettgehalt in der Trockenmasse gecoateter Agglomeraten [g/100 g] ........ 55
Formel 4.14: Referenzfaktor zur Bestimmung des Verhältnisses von fettfreier Trocken-
masse zu Fettgehalt in der Trockenmasse von ungecoateten Agglomeraten
................................................................................................................ 55
Formel 4.15: theoretische fettfreie Trockenmasse von gecoateten Agglomeraten ohne
das aufgetragene Coating [g/100 g] ......................................................... 55
Formel 4.16: Trockenmasse von gecoteten Agglomeraten ohne das aufgetragene
Coating [g/100 g] ...................................................................................... 55
Formel 4.17: Trockenmasse des aufgetragenen Coatings [g/100 g] ............................. 55
Formel 4.18: Hydroperoxidkonzentration des verkapselten Rapsöls in der Probe
[mmol/kg Öl] ............................................................................................. 56
Formel 4.19: Span der Öltropfenverteilung ................................................................... 58
Formel 4.20: Volumen der Probe bei der Bestimmung der Feststoffdichte [cm3] ........... 59
Formel 4.21: Feststoffdichte der Probe [g/cm3] ............................................................. 59
Formel 4.22: Allgemeine BET-Gleichung ...................................................................... 61
Formel 4.23: Spezifische Monoschichtstoffmenge [Mol] ................................................ 61
Formel 4.24: Spezifische Oberfläche [m2/g] .................................................................. 61
Formel 4.25: vereinfachte Berechnung der spezifischen Oberfläche für die Verwendung
von Krypton [m2/g] .................................................................................... 61
Formel 4.26: Washburn-Gleichung ............................................................................... 61
Formel 4.27: Oberflächenbelegungskonzentration nach Gibbs [Mol/m2] ....................... 62
Formel 4.28: Überschüssige Oberflächenarbeit [N·m]................................................... 62
Formel 6.1: D-Wert für die Bestimmung der freien Oberflächenenergie .................... 112

Abkürzungen und Symbole
XXIV
Abkürzungen und Symbole
CA Kontaktwinkel [°]
CAm direkt gemessener Kontaktwinkel [°]
CAOWRK mit Hilfe des OWRK-Modell berechneter Kontakt-
winkel
[°]
CMC Carboxymethylzellulose als Coatingmaterial
D Diodmethan als Referenzlösung
δ Verlustfaktor [°]
E Ethylenglycol als Referenzlösung
F Formamid als Referenzlösung
G* Schubmodul [Pa]
G’ Speichermodul [Pa]
G’’ Verlustmodul [Pa]
HPMC Hydroxypropyl Methylzellulose als Coatingmaterial
MC Methylzellulose als Coatingmaterial
MDT maltodextrinbasierte kompaktierte Tablette
MDGSS maltodextrinbasierte Modelloberfläche
MDOSA maltodextrinbasierte Modelloberfläche mit OSA-
Stärke
MDEMUL emulsions- und maltodextrinbasierte Modelloberflä-
che
N Nutrateric® als Coatingmaterial
OSA-Stärke n- Octenylsuccinate-acid- Stärke (mit Bernsteinsäu-
re substituierte Stärke)
OWRK Methode zur Oberflächencharakterisierung nach
Owens, Wendt, Rabel und Kaelble
SE, 𝛾𝑆 freie Oberflächenenergie eines Feststoffes an der [mN/m]

Abkürzungen und Symbole
XXV
Luft-Feststoff Phasengrenze (surface free energie)
SEd, 𝛾𝑆𝑑 disperser Anteil der freien Oberflächenenergie [mN/m]
SEp, 𝛾𝑆𝑝 polarer Anteil der freien Oberflächenenergie [mN/m]
SFT, 𝛾𝐿 Oberflächenspannung einer Flüssigkeit an der Luft-
Wasser Phasengrenze (surface tension)
[mN/m]
SFTd, 𝛾𝐿𝑑 disperser Anteil der Oberflächenspannung [mN/m]
SFTp, 𝛾𝐿𝑝 polarer Anteil der Oberflächenspannung [mN/m]
SFTdodecan Oberflächenspannung einer Flüssigkeit an der
Wasser-Dodecan-Grenzfläche
[mN/m]
W destilliertes Wasser als Referenzlösung
WA Adhäsionsarbeit (work of adhesion) [mN/m]
YD Methode zur Charakterisierung Partikel-
Flüssigkeits-Interaktion nach Young und Dupré

Einleitung
1
1 Einleitung
Coaten, das Überziehen, ist ein Prozess, der in der Lebensmittelindustrie vielseitig Anwen-
dung findet. Gecoatet wird meist mit dem Ziel, eine neue Funktionalität zu erreichen oder
eine Barriere zu bilden, um das gecoatete Gut vor äußeren Einflüssen zu schützen
(Debeaufort & Desobry, 2012; Hede, 2006; Lima et al., 2010; Madene, Jacquot, Scher, &
Desobry, 2006; Soliva-Fortuny, Rojas-Graü, & Martín-Belloso, 2012). Diese Barriere kann
zum Beispiel als Schutz gegen die Permeation von Wasserdampf oder Sauerstoff oder als
Maskierung eines unerwünschten Geschmacks fungieren (Behzadi, Toegel, & Viernstein,
2008; Bourlieu, Guillard, Vallès-Pamiès, Guilbert, & Gontard, 2009; Erdohan & Turhan,
2005; Farag & Leopold, 2011; Nazan Turhan & Şahbaz, 2004; Teunou & Poncelet, 2002;
Tong, Xiao, & Lim, 2008).
Die Anwendungen des Coatens im Lebensmittelbereich finden zumeist auf makroskopi-
scher Ebene statt. So werden beispielsweise Äpfel und andere nicht prozessierte Obstsor-
ten gecoatet, um eine Barriere gegenüber Umwelteinflüssen zu erreichen und somit eine
Haltbarkeitsverlängerung zu gewährleisten (Baloch, Bibi, & Jilani, 2013; Choi, Park, Ahn, &
Lee, 2002; Scramin et al., 2011; Velásquez, Skurtys, Enrione, & Osorio, 2011). Aber auch
für Lebensmittel tierischen Ursprungs werden Coatings angewendet, wie z.B. für die Halt-
barkeitsverlängerung von Lachs (Souza et al., 2010). Bei der Bildung von Barriereeigen-
schaften steht aber nicht nur die Verlängerung der Haltbarkeit im Fokus, sondern auch
technologisch relevante Barriereeigenschaften können durch die Verwendung von
Coatings erreicht werden. So kann durch zellulosederivatbasierte oder molkenproteinba-
sierte Coatings die Fettaufnahme während des Frittierens von Produkten reduziert werden
(Balasubramaniam, Chinnan, Mallikarjunan, & Phillips, 1997; Dragich & Krochta, 2010;
Garcia, 2004; Tavera-Quiroz, Urriza, Pinotti, & Bertola, 2012).
Um eine Barriere zu formen, gibt es die technologische Möglichkeit der Mikroverkapselung.
Die Größenordnung ist hier im Gegensatz zu den bisher genannten Beispielen jedoch
deutlich kleiner. Ursprünglich wurde die Mikroverkapselung in der pharmazeutischen In-
dustrie eingesetzt und dient dazu, Wirkstoffe zu verkapseln, um etwa eine kontrollierte
Freisetzung zu erreichen oder die Nebenwirkung eines Wirkstoffes zu minimieren (Kuang,
Oliveira, & Crean, 2010). Die Technik der Mikroverkapselung findet auch in der Lebensmit-
telindustrie Anwendung. Die Mikroverkapselung ist hier beispielsweise eine Möglichkeit,
Aromen zu verkapseln, um diese erst an einem bestimmten Punkt im Herstellungsprozess
oder während des Konsumierens durch den Verbraucher (z.B. Aromen in Kaugummis)
freizusetzen (Da Silva et al., 2014; Fuchs et al., 2006; Gibbs et al., 1999; Madene et al.,
2006; Milanovic et al., 2010). Darüber hinaus können mittels Mikroverkapselung weitere

Einleitung
2
Lebensmittelinhaltsstoffe verkapselt werden. So ist die Bedeutung von mehrfach ungesät-
tigten Fettsäuren in der Humanernährung sehr hoch (Moffatt & Stamford, 2006). In der
Vergangenheit konnten durch die Aufnahme von mehrfach ungesättigten Fettsäuren viele
positive Effekte für die Gesundheit nachgewiesen werden. Die für die Funktion der Netz-
haut und des zerebralen Cortex wichtigen mehrfach ungesättigten Fettsäuren beispielswei-
se können nur über die Nahrung aufgenommen werden (Connor & Neuringer, 1988). Des
Weiteren können Vitamine durch Mikroverkapselung vor äußeren Einflüssen wie Licht,
Temperatur oder dem pH-Wert des Magensaftes geschützt werden (Atarés, Pérez-Masiá,
& Chiralt, 2011; Janjarasskul, Min, & Krochta, 2011; Matalanis, Jones, & McClements,
2011).
Üblicherweise erfolgt die Mikroverkapselung über einen Einschluss des Wirkstoffes oder
des Lebensmittelinhaltsstoffes in eine Matrix aus Kohlenhydraten (Drusch, Serfert, &
Schwarz, 2006; Kosaraju, 2005; Serfert, Drusch, & Schwarz, 2009). Der Prozess, der dabei
genutzt wird, ist meist die Sprühtrocknung (Da Silva et al., 2014; Fuchs et al., 2006; Gibbs
et al., 1999), aber auch die Verwendung des Extrusionsprozesses ist üblich, wie Kaushik et
al. (2015) zusammengefasst haben. Allerdings ist bei diesen Prozessen, besonders bei der
Sprühtrocknung, darauf zu achten, dass die Prozessbedingungen einen Einfluss auf die
Partikelstruktur haben und diese wiederum die Barriereeigenschaften der Matrix beeinflus-
sen können (Drusch & Schwarz, 2006). Die Autoren zeigten auf, dass Parameter, wie
Öltropfengrößenverteilung, freies Oberflächenfett, Porosität und die spezifische Oberfläche
von Partikeln, durch ungünstige Prozessbedingungen nachteilig beeinflusst werden.
Auch wenn die Barriereeigenschaften gegenüber Sauerstoff in einer kohlenhydratbasierten
Matrix zu einem entscheidenden Teil von dem verwendeten Emulgator und dessen Fähig-
keiten zur Grenzflächenstabilisierung während des Prozesses abhängt (Drusch & Schwarz,
2006; Tamm et al., 2015), kann die generelle Barriereeigenschaft einer Matrix, wie in
sprühgetrockneten Partikeln, als gut angesehen werden. Der Nachteil von sprühgetrockne-
ten Partikeln ist, dass die kohlenhydratbasierte Matrix sehr hygroskopisch und somit eine
Aufnahme der Feuchtigkeit aus der Umgebungsluft sehr wahrscheinlich ist. Kohlenhydrat-
basierte Partikel zur Verkapselung von funktionellen Inhaltstoffen haben weiterhin das
Problem, dass sie, abhängig vom Wert des Dextroseäquivalent (DE-Wert) und der relativen
Feuchte in der Umgebungsluft, vom Glaszustand, den sie nach dem Sprühtrocknen haben,
durch Feuchtigkeitsaufnahme in den gummiartigen Zustand übergehen können. Der gum-
miartige Zustand hat durch eine höhere Ordnung der Moleküle eine schlechtere Barriereei-
genschaft als eine Matrix im Glaszustand. Maltodextrin mit einem DE-Wert von 36 hat eine
Glasübergangstemperatur von 6 °C bei 52 % relativer Feuchte und Maltodextrin mit einem
niedrigeren DE-Wert (20) hat bei gleicher Feuchte eine Glasübergangstemperatur von

Einleitung
3
37 °C (Roos & Karel, 1991). Hier ergibt sich das Problem, dass Kohlenhydrate mit höheren
DE-Werten zwar eine bessere Barriereeigenschaft aufweisen, jedoch durch die niedrigere
Glasübergangstemperatur die Barriereeigenschaft bei ungünstiger Luftfeuchte wieder ver-
loren gehen kann (Hogan, McNamee, O’Riordan, & O’Sullivan, 2001; Subramaniam et al.,
2013). Hinzu kommt, dass durch den Sprühtrocknungsprozess an der Oberfläche, bzw. in
immobilisierten Öltropfen in Oberflächennähe, sogenanntes freies Oberflächenfett entsteht.
Dieses Oberflächenfett kann die Haltbarkeit von verkapseltem Öl in sprühgetrockneten
Partikeln negativ beeinflussen (Drusch & Berg, 2008). Um das an der Oberfläche liegende
freie Fett besser vor äußeren Einflüssen zu schützen, kann beispielsweise ein Coating auf
die Partikel aufgetragen werden.
Das Coaten von Partikeln wird üblicherweise in der Wirbelschicht durchgeführt (Behzadi et
al., 2008; Dewettinck & Huyghebaert, 1999; Teunou & Poncelet, 2002). Dieser Prozess
besteht aus drei Phasen, der Luft (zum Fluidisieren der Partikel und Trocknen des Films),
der Flüssigkeit (Coatinglösung) und der festen Phase (zu coatender Partikel). Die drei
Phasen gehen während des Prozesses eine Vielzahl von Wechselwirkungen ein, welche
einen Einfluss auf die resultierende Coatingqualität zeigen. Diese wird häufig als Homoge-
nität des Coatings, sprich, als eine geringe Abweichung in der durchschnittlichen Schicht-
dicke des Coatings auf dem Partikel definiert (Atarés, Depypere, Pieters, & Dewettinck,
2012; Laksmana, Hartman Kok, Vromans, Frijlink, & Van der Voort Maarschalk, 2009;
Palamanit, Soponronnarit, Prachayawarakorn, & Tungtrakul, 2013; Perpar, Luštrik, Dreu,
Srčič, & Žun, 2013; Vanderroost, Ronsse, Pieters, & Dewettinck, 2010). Die Qualität ist
abhängig von den stofflichen Parametern der drei Phasen, die am Coatingprozess teilneh-
men. Die Luft, die für das Fluidisieren der Partikel und das Trocknen der Coatinglösung auf
dem Partikel verantwortlich ist, kann eine bestimmte Temperatur und Einström-
geschwindigkeit haben. Die Partikel selbst sind durch Größe und Dichte auf der einen Seite
und durch eine freie Oberflächenenergie auf der anderen Seite gekennzeichnet. Weiterhin
sind die Viskosität und die Oberflächenspannung der Coatinglösung von Bedeutung, da
diese Eigenschaften einerseits einen Einfluss auf den Prozess des Zerstäubens haben und
andererseits eine Rolle bei der Haftung und dem Spreiten der Lösung auf der Partikelober-
fläche spielen.
Die Schwierigkeit bei der Auswahl eines Coatingmaterials besteht darin, dass es zum ei-
nen gut mit dem Partikel interagieren muss und zum anderen, dass eine Vielzahl von Funk-
tionalitäten erreicht werden soll. Meist bieten Coatingmaterialien eine sehr gute Funktionali-
tät bspw. als Barriere gegen Sauerstoff, jedoch haben sie nur eine sehr geringe gegenüber
Wasserdampf. Dies bedeutet, dass grundsätzlich, sofern nicht ausschließlich eine Funktio-
nalität erreicht werden soll, ein Kompromiss zwischen den unterschiedlichen Funktionalitä-

Einleitung
4
ten erzielt werden muss. Als mögliche Coatingmaterialien kommen z.B. zellulosederivatba-
sierte Coatings in Betracht. Gegossene Filme dieser Art wurden in vielen Untersuchungen
auf ihre Sauerstoff- und Wasserdampfpermeabilität hin untersucht. Dabei zeigten sich mo-
derate Barriereeigenschaften gegenüber Sauerstoff (Ayranci & Tunc, 2003; de Moura et
al., 2011; de Moura, Avena-Bustillos, McHugh, Krochta, & Mattoso, 2008; Ghanbarzadeh,
Almasi, & Entezami, 2011; Miller & Krochta, 1997; Park, Weller, Vergano, & Testin, 1993;
Srivastava & Mishra, 2010) und gute Barriereeigenschaften gegenüber Wasserdampf (de
Moura et al., 2011, 2008; Mishra, Khatkar, Garg, & Wilson, 2010; Park et al., 1993; Tong et
al., 2008). Studien zur Untersuchung der Haftungs- und Spreitungseigenschaften von zellu-
losederivatbasierten Coatingmaterialien auf maltodextrinbasierten Trägermaterialien sind
bislang nicht bekannt. Zumeist wurden in der Vergangenheit Modellsysteme, wie etwa
Glas, genutzt (Nienaltowska et al., 2010). Auch gibt es bisher keine Studien, die sich mit
den Barriereeigenschaften gegenüber Sauerstoff und Wasserdampf von zellulosederivat-
basierten Coatings auf sprühgetrockneten Partikeln befassen.
Daher soll in der vorliegenden Arbeit untersucht werden, ob sich zellulosederivatbasierte
Coatinglösungen eignen, um auf einem maltodextrinbasierten Trägersystem als Sauerstoff-
und Wasserdampfbarriere eingesetzt zu werden.

Problemstellung
5
2 Problemstellung
Um die Eignung von zellulosederivatbasierten Coatinglösungen zur Verwendung auf einem
maltodextrinbasierten Trägersystem genauer zu untersuchen und festzustellen, welche
Einflussgrößen die Funktionalität eines zellulosederivatbasierten Coatings als Sauerstoff-
und Wasserdampfbarriere beeinflussen, wurde die vorliegende Arbeit in drei Teile geglie-
dert (siehe Abbildung 2.1).
Abbildung 2.1: Darstellung der drei Arbeitsteile dieser Arbeit mit den dazugehörigen
Aufgabenstellungen und Untersuchungsmethoden
Im ersten Teil wurde aus vier zellulosederivatbasierten Coatingmaterialien (MC, HPMC,
CMC und Nutrateric® als kommerzielles Beispiel für ein EC-basiertes Coating) eines identi-
fiziert, dass am besten als Coating auf einer maltodextrinbasierten Trägermatrix einzuset-
Charakterisierung des Haftungs- und Benetzungsverhaltens von Zellulose-derivatlösungen auf maltodextrinba-
sierten Modelloberflächen
Rheologische Charakterisierung der zellulosederivatbasierten Coatingma-
terialien in Lösung
Charakterisierung der Funktionalität von MC–Coatings als Sauerstoff– und Wasserdampf-barriere und Einfluss des Wirbelschichtpro-zesses auf die Partikelstruktur ölbeladener
Agglomerate
• Charakterisierung der Coatinglösungen (SFT, SFTp und SFTd)
• Charakterisierung der MDOSA (SE, SEp und SEd)
• Bestimmung der Partikel-Flüssigkeits-Interaktion (CAOWRK durch Berechnung, CAm durch direkte Messung, WA nach YD und WA nach OWRK)
• Bestimmung der Viskosität η in Abhängigkeit von Temperatur, Konzentration und Scherge-schwindigkeit
• Bestimmung der strukturbildenden Charakteristika von MC
• Charakterisierung des thixotropen Verhaltens von MC – Lösungen
• Charakterisierung von MC - gecoateten ölbe-ladenen Agglomeraten (LOX, ÖTG, MVE, Coatingmenge und REM)
• Charakterisierung verschiedener Temperatur - Zeit - Kombinationen im Wirbelschichtpro-zess auf die Partikelstruktur (LOX, ÖTG, MVE, BET, HG-Intrusionsporosemetrie, He-Pygnometrie, DVS, REM, TM)
• Ermittlung der Wasserdampfbarriere-eigenschaften

Problemstellung
6
zen ist. Um zu ermitteln, welches Coatingmaterial sich als geeignet erweist, musste zu-
nächst die Interaktion zwischen der Coatinglösung und der Partikeloberfläche bestimmt
werden. Da sich die Oberfläche eines Partikels aufgrund der geringen Partikelgröße nicht
eignet, um die Interaktion zwischen Coatinglösung und Partikeloberfläche zu untersuchen,
mussten Modelloberflächen hergestellt werden, die in der Oberflächenbeschaffenheit der
Partikeloberfläche eines sprühgetrockneten Partikels entsprachen. Zur Untersuchung der
Interaktion zwischen Modelloberfläche und Coatinglösung wurden zwei verschiedene Mo-
delle angewendet. Das erste Modell ist das Modell nach Owens, Wendt, Rabel und Kaelble
(OWRK). Bei diesem Modell muss zunächst die Oberflächenenergie (SE), inklusive ihrer
polaren und dispersen Anteile (SEp und SEd), der maltodextrinbasierten Modelloberfläche
ermittelt werden. Diese wird durch das Vermessen des Kontaktwinkels (CA) von Referenz-
lösungen mit bekannter Oberflächenspannung (SFT) bestimmt. Anschließend muss die
Oberflächenspannung, sowie deren polarer und disperser Anteile (SFTp und SFTd), der
verwendeten Coatinglösungen bestimmt werden. Aus diesen beiden Einzelcharakterisie-
rungen kann nun die Interaktion in Form der Adhäsionsarbeit (WA-OWRK) und des berechne-
ten Kontaktwinkels (CAOWRK) zwischen den Coatinglösungen und der Modelloberfläche
bestimmt werden. Das zweite Modell, welches die Bestimmung der Interaktion zwischen
Modelloberfläche und Coatinglösung ermöglicht, ist das Model nach Young-Dupré (YD).
Bei diesem Modell ist keine vorhergehende Charakterisierung der Modelloberfläche not-
wendig, sondern lediglich die Ermittlung der SFT der Coatinglösung. Zur Bestimmung der
Interaktion wird der Kontaktwinkel der Coatinglösung direkt auf der Modelloberfläche ver-
messen (CAm) und die Adhäsionsarbeit (WA-YD) über eben diesen Kontaktwinkel und die
Oberflächenspannung der Coatinglösung bestimmt. Die Abbildung 2.1 bietet einen Über-
blick über die durchgeführten Untersuchungen.
Im zweiten Teil der vorliegenden Arbeit stand das rheologische Verhalten der als Coating-
material in Frage kommenden MC-Lösung im Mittelpunkt. Hierbei wurde die Lösung hin-
sichtlich ihrer Viskosität bei verschiedenen Scherbelastungen, Temperaturen und Konzent-
rationen untersucht. Dies sollte einen Aufschluss darüber geben, wie sich die Viskosität
unter verschiedenen Prozessbedingungen verhält. Weiterhin wurden mittels Oszillations-
messung unterschiedlich konzentrierte Lösungen von MC hinsichtlich ihrer temperaturab-
hängigen Strukturausprägung untersucht. Dies war notwendig, um vorhersagen zu können,
ab welcher Temperatur-Konzentration-Kombination die Lösung beginnt eine Struktur aus-
zubilden und sich zum Gel zu verfestigen. Ebenfalls bestimmt wurden hier die Gelauflö-
sungstemperatur, die Gelstärke und eine Reihe weiterer wichtiger Strukturausbildungspa-
rameter. Die dritte Versuchsreihe innerhalb der rheologischen Charakterisierung stellte die
Untersuchung des thixotropen Strukturwiederaufbauverhaltens dar. Diese sollten klären,

Problemstellung
7
inwiefern die Struktur der MC-Lösungen durch eine Scherung geschädigt wird. Hierzu wur-
den die Lösungen nach einer Temperierungsphase kurzzeitig stark durch Scherung bean-
sprucht und der Strukturwiederaufbau anschließend mittels Oszillationsmessung bestimmt.
Der dritte und umfangreichste Teil der Arbeit beschäftigt sich mit der Funktionalität der MC-
Coatings auf einem maltodextrinbasierten Trägerpartikel und dem Einfluss der Prozesspa-
rameter des Wirbelschichtcoatingprozesses auf die Partikelstruktur. Diese Partikel wurden
aus einer Emulsion, die Rapsöl, OSA-Stärke, Wasser und Maltodextrin enthielt, hergestellt.
Dazu wurde die Emulsion sprühgetrocknet und die entstandenen Partikel anschließend zur
besseren Fluidisierbarkeit aufagglomeriert.
Zunächst wurde eine Vorversuchsreihe durchgeführt, um die Applizierbarkeit der Coating-
menge und die Methode zur Bestimmung der Hydroperoxidkonzentration (LOX) für gecoa-
tete Agglomerate zu überprüfen. Im Anschluss an den Vorversuch wurde der Einfluss von
Prozesstemperatur und -zeit auf den Verlauf der Hydroperoxidkonzentration des verkapsel-
ten Rapsöls, die Öltropfenverteilung (ÖTG), die Trockenmasse (TM) und die Mikroverkap-
selungseffizienz (MVE) der Agglomerate untersucht. Um weitere Einflüsse von Prozess-
temperatur und -zeit auf die Struktur der behandelten und unbehandelten Agglomerate zu
untersuchen, wurde eine weitere Versuchsreihe durchgeführt. Die hierfür hergestellten Ag-
glomerate wurden den gleichen Temperatur-Zeit-Kombinationen im Prozess ausgesetzt.
Zusätzlich zu den Versuchen, die auch in der vorhergehenden Versuchsreihe durchgeführt
wurden, fand für diese Proben eine Vielzahl weiterer Untersuchungen statt. Die Agglome-
rate wurden mittels BET-Gasadsorptionsmessung und dynamischer Dampfsorption auf ihre
spezifische Oberfläche hin untersucht. Mittels Hg-Porosimetrie wurden die Agglomerate auf
ihre Porosität in Abhängigkeit verschiedener Prozessbedingungen untersucht und die He-
Pyknometrie gab Aufschluss über die Feststoffdichte der Agglomerate. Zusätzlich konnte
mittels Rasterelektronenmikroskopie die Oberflächenstruktur der behandelten Agglomerate
visualisiert werden. In einer vierten Versuchsreihe wurden die hergestellten Agglomerate
mit MC in unterschiedlichen Temperatur-Zeit-Kombinationen gecoatet. Die gecoateten Ag-
glomerate wurden hinsichtlich der Sauerstoffbarriereeigenschaften des Coatings mittels
Bestimmung der Hydroperoxidkonzentration untersucht und über die Gesamtfettbestim-
mung die applizierte Coatingmenge ermittelt. Wie in den vorhergehenden Versuchsreihen
auch, wurden die Proben zusätzlich auf ihre TM, die ÖTG und die MVE hin untersucht. Um
auch die Wasserdampfbarriereeigenschaften zu ermitteln, wurden die Proben der Reihe
nach bei verschiedener relativer Luftfeuchtigkeit in einem Exsikkator gelagert und die Was-
serdampfaufnahme wurde gravimetrisch bestimmt.

Theoretische Grundlagen
8
3 Theoretische Grundlagen
Da sich die vorliegende Arbeit mit dem Coaten von Partikeln, die funktionelle Lebensmittel-
inhaltstoffe enthalten, beschäftigt, sollen zunächst theoretische Grundlagen zusammenge-
tragen werden, die für das Verständnis der Arbeit von Bedeutung sind. Zunächst wird ein
Überblick über den Prozess des Coatens in der Wirbelschicht gegeben. Weiterhin werden
die drei am Prozess beteiligten thermodynamischen Phasen und deren Parameter vorge-
stellt. Dieses Kapitel wird anschließend einen Überblick darüber geben, welche zellulose-
derivatbasierten Coatingmaterialien in dieser Arbeit zum Einsatz kommen. Darüber hinaus
soll ihre chemische Struktur und das daraus resultierende rheologische und technologische
Verhalten dargestellt werden. Zuletzt sollen die für diese Arbeit wichtigen Grundlagen zum
Prozess der Lipidoxidation dargelegt werden, da diese bei der Verkapselung von mehrfach
ungesättigten Fettsäuren eine entscheidende Rolle spielt.
Das Coaten unterscheidet sich von anderen Verkapselungstechniken in der Lebensmittel-
industrie dadurch, dass im Nachhinein auf einen bereits hergestellten Partikel oder eine
hergestellte Kapsel ein Coating aufgetragen wird. Dies bedeutet zwar einen zusätzlichen
Prozessschritt, jedoch lassen sich durch das Aufbringen eines Coatings verschiedene
Funktionalitäten erreichen, die ohne ein Coating schwer bzw. gar nicht zu erzielen sind.
Das Coaten von Partikeln in der Lebensmittelindustrie kann vielseitige Gründe haben.
Meist besteht die Funktion eines Coatings darin, den zu coatenden Partikel zusätzlich zu
schützen. Dieser Schutz kann zum Beispiel bedeuten, dass eine Barriere zwischen dem
Primärpartikel und der Umwelt geschaffen werden soll, um das Eindringen von Sauerstoff
in den Partikel zu unterbinden. Eine weitere Anwendung ist die kontrollierte Freisetzung
von aktiven Inhaltsstoffen (de S. Medeiros, Pinheiro, Carneiro-da-Cunha, & Vicente, 2012;
Kayumba et al., 2007; Nakano & Yuasa, 2001; Takeuchi, Yasuji, Yamamoto, &
Kawashima, 2000). Die kontrollierte Freisetzung von Inhaltsstoffen, beispielsweise durch
Mikroverkapselung, fand zunächst Anwendung in der Pharmazie, wo eine kontrollierte oder
modifizierte Freigabe von Wirkstoffen zu einer Verbesserung der Bioverfügbarkeit und zur
Minimierung von Nebenwirkungen dienen kann (Kuang et al., 2010). Diese Anwendungen
wurden in die Lebensmittelindustrie übernommen, wo es vor allem um die kontrollierte
Freisetzung von Aromen oder funktionellen Inhaltsstoffen, wie Probiotika oder Vitaminen
geht (Ahn et al., 2008; Kuang et al., 2010; Milanovic et al., 2010; Mokarram, Mortazavi,
Najafi, & Shahidi, 2009).

Theoretische Grundlagen
9
3.1 Technologie des Coatens
Andrade et al. (2012) haben verschiedene technologische Möglichkeiten für das Aufbrin-
gen einer Coatinglösungen vorgestellt, wovon eine das Trommelcoaten ist. Dieses ist eine
Methode, bei der das Produkt in einem rotierenden Kessel fluidisiert und mittels einer Düse
die Coatinglösung aufgesprüht wird. Durch einen Luftstrom wird die Feuchtigkeit aus dem
Kessel transportiert und der Überzug kann trocknen.
Ein weiteres Verfahren ist das Eintauchen, bei dem das zu beschichtende Produkt in einem
Tauchbad überzogen wird. Diese Methode ist gut für raue Oberflächen geeignet, die voll-
ständig bedeckt werden sollen. Nach Abfließen der überschüssigen Coatinglösung wird
das Produkt bei Raumtemperatur oder im Trockner getrocknet.
Das Sprühcoaten wird angewendet, um eine gleichmäßigere Schichtdicke zu erzeugen.
Ober- und Unterseite des Produktes können in zwei Schritten überzogen werden und es ist
möglich, verschiedene Beschichtungen aufzusprühen.
Ein viertes Verfahren um ein Coating aufzutragen, ist das Coaten in der Wirbelschicht. Der
Prozess des Wirbelschichtcoatens ist ein drei-Phasen-Prozess (Erbil, 2006; Farag, 2010;
Letellier, Mayaffre, & Turmine, 2007; Vanderroost, Ronsse, Dewettinck, & Pieters, 2011).
Hierbei treffen die Coatinglösung (flüssige Phase), die Partikel, die es zu coaten gilt (feste
Phase) und die Fluidisierungsluft aufeinander. Dieses Zusammenspiel der drei Phasen und
das Entstehen eines Coatings, vor allem dessen Homogenität und Gleichförmigkeit auf
dem Partikel, ist abhängig von einer Vielzahl von Prozessparametern, die in Kapitel 3.2
näher beschrieben werden.
Wie in Abbildung 3.1 zu erkennen ist, werden die Partikel im Wirbelschichtapparat durch
Luft, die durch eine Lochsiebscheibe von unten her eingebracht wird, fluidisiert. Dies ge-
schieht zum einen um die Partikel zu vereinzeln und somit eine große Oberfläche zum
Coaten zu haben. Zum anderen nimmt die Luft die Feuchtigkeit aus der Coatinglösung auf,
wodurch der Film auf der Oberfläche der Partikel abtrocknen kann. Die Coatinglösung wird
über eine Pumpe an eine Düse herangeführt, die die Coatinglösung zerstäubt. Die zer-
stäubten Tropfen der Coatinglösung treffen auf die Partikel, spreiten und haften auf der
Partikeloberfläche und beginnen durch das Evaporieren des Lösungsmittels einen Film zu
bilden.

Theoretische Grundlagen
10
Abbildung 3.1: Schema eines Top-Spray-Wirbelschichtcoaters mit Luftzufuhr, Abluft und der zuge-
führten Coatinglösung; modifiziert vom Autor, nach Duangkhamchang et al. (2015)
Das in Abbildung 3.1 gezeigte Top-Spray-Verfahren ist das älteste der vier Verfahren und
zeichnet sich durch eine leichte Zugänglichkeit der Düse und eine, verglichen mit den an-
deren Verfahren, hohe Ansatzmengenkapazität aus. Das Top-Spray-Verfahren bietet für
Coatingprozesse jedoch nicht die beste Coatingqualität und wird in der Regel nicht für
Coatinganwendungen im Bereich der kontrollierten Freisetzung angewendet.
Das Top-Spray-Verfahren ist nicht die einzige Möglichkeit, in der Wirbelschicht zu coaten.
In Abbildung 3.2 sind drei weitere Verfahren der Prozessführung zum Coaten in der Wir-
belschicht dargestellt. Die Vor- und Nachteile dieser insgesamt vier Verfahren wurden von
Teunou und Poncelet (2002) sehr anschaulich zusammengetragen.
Das Bottom-Spray-Verfahren (Abbildung 3.2 b) hat eine deutlich höhere Effizienz bezogen
auf die Partikel-Flüssigkeits-Kollision im Vergleich zum Top-Spray-Verfahren. Durch diese
Effizienzsteigerung kann die Materialeffizienz gegenüber dem Top-Spray-Verfahren deut-
lich gesteigert werden. Durch den Einsatz eines Rohres in ein Bottom-Spray-System konn-
te Wurster (Abbildung 3.2 c) die gezielte Zirkulation der Partikel verbessern und somit die
Agglomerationswahrscheinlichkeit gegenüber den beiden anderen Verfahren zum Wirbel-

Theoretische Grundlagen
11
schichtcoaten deutlich senken. Weiterhin konnte durch dieses Verfahren eine deutlich bes-
sere Homogenität der Coatings und damit eine sehr gute Coatingqualität erreicht werden.
Deshalb wird dieses Verfahren meist in der Herstellung von Kapseln für Anwendungen in
der kontrollierten Freisetzung verwendet.
Das Rotor-System mit seitlicher Sprühdüse (Abbildung 3.2 d) gewährleistet durch die
Kombination aus rotierender Scheibe und Luftstrom eine hohe Sphärizität und Dichte der
fertig gecoateten Partikel. Der Nachteil dieses Verfahrens ist es jedoch, dass zu poröse
und damit mechanisch anfällige Ausgangsmaterialien auf Grund der hohen physikalischen
Beanspruchung nicht für dieses Verfahren geeignet sind.
Abbildung 3.2: Schematische Darstellung der verschiedenen Arten einer Wirbelschichtcoatinganlage
mit (a) Top-Spray, (b) Bottom-Spray, (c) Bottom-Spray mit Wurstereinsatz und (d) Rotor mit seitlich
angebrachter Düse nach Teunou & Poncelet (2002)
3.2 Parameter zur Beeinflussung des Coatingergebnisses im Wir-belschichtcoatingprozess
Im Folgenden sollen die einzelnen Teilprozesse und Parameter der am Prozess teilhaben-
den Phasen (fest, flüssig, gasförmig) vorgestellt werden. Hierbei wird besonderes Augen-
merk auf die Parameter und Teilprozesse gelegt, welche einen Einfluss auf die Qualität
und Funktionalität des Coatings haben können.
Die flüssige Phase und deren Zerstäubung und Tropfengrößenverteilung
Das Zerstäuben der Coatinglösung ist ein wichtiger Teilprozess des Wirbelschichtcoatens
und hat einen bedeutenden Einfluss auf die Qualität des Coatings. Aktuelle Studien zum

Theoretische Grundlagen
12
Thema Zerstäubung zeigen, dass die Änderung der Düsengeometrie die Wirkung zwischen
Zerstäubungsluft und Flüssigkeit verbessern kann und somit eine bessere Stabilität und
homogenere Verteilung der zerstäubten Flüssigkeit erreicht wird (Portoghese, Ferrante,
Berruti, Briens, & Chan, 2010; Serfert et al., 2013; Thompson & Rothstein, 2007). Die Sta-
bilität des Sprays und die homogene Tropfengrößenverteilung der zerstäubten Flüssigkeit
sind Parameter, die einen direkten Einfluss auf die Partikel-Flüssigkeits-Interaktion haben.
Generell werden zwei Bauarten von Düsen für die Anwendung im Coatingbereich unter-
schieden: innen- und außenmischende Zweistoffdüsen (Abbildung 3.3). Die beiden Bauar-
ten unterscheiden sich in erster Linie durch die Lage ihrer Mischzonen. Die innenmischen-
de Zweistoffdüse hat ihre Mischzone innerhalb des Düsenkörpers. Der Vorteil dieser Bau-
art liegt darin, dass ein niedrigerer Zerstäubungsdruck notwendig ist, um eine homogene
Tropfengrößenverteilung im Spray zu erreichen. Ein Nachteil der innenmischenden Bauart
ist, dass die Flüssigkeit, die zerstäubt werden soll, keinerlei Feststoffanteile enthalten soll-
te, da diese die innere Mischkammer beschädigen können und damit die Lebensdauer der
Düse herabsetzen. Bei der außenmischenden Bauform findet die Zerstäubung außerhalb
der Düse statt. Das hat den Vorteil, dass der Prozess der Zerstäubung besser reguliert
werden kann, indem beide Materialströme separat voneinander gesteuert werden können
(Hede, Bach, & Jensen, 2008).
Abbildung 3.3: Schematische Darstellung einer außenmischenden und einer innenmischende Zwei-
stoffdüse, vom Autor bearbeitet, nach Salman et al. (2006)
Für die Verwendung von außenmischenden Zweistoffdüsen einfachster Geometrie haben
Hede et al. beschrieben, dass die Tropfengröße der zerstäubten Flüssigkeit bei konstan-
Pneumatische Zweistoffdüse
innenmischend außenmischend
Flüssigkeit
Zerstäubungsluft
Mischzone
Sprühkegel

Theoretische Grundlagen
13
tem Flüssigkeitsstrom über den Druck der Atomisierungsluft geregelt werden kann (Hede
et al., 2008). Der typische Zerfall des Flüssigkeitsfilms beim Verlassen der außenmischen-
den Zweistoffdüse ist in Abbildung 3.4 dargestellt. Durch die Unterschiede in Dichte und
Geschwindigkeit der beiden aufeinander treffenden Ströme kommt es zum Zerfall des
Films in Ligamente und Tropfen. Zunächst liegt die Flüssigkeit als Film mit einer definierten
Filmdicke (dsh) vor und zerfällt bei Kontakt mit der Zerstäubungsluft in Ligamente mit einer
bestimmten Ligamentdicke (dlg) und Ligamentlänge (Llg). Beim Zerfall der Ligamente
kommt es zur Entstehung der Tropfen, welche das Spray bilden. Das Aufbrechen des
Flüssigkeitsfilms ist zum einen beeinflusst durch Prozessparameter, wie dem Druck der
Atomisierungsluft oder dem Volumenstrom der zu zerstäubenden Flüssigkeit zum anderen
sind es stoffliche Parameter, die den Zerfall des Flüssigkeitsfilms beeinflussen. Seitens der
Atomisierungsluft spielt hierbei die Dichte der Luft eine bedeutende Rolle. Zur Dichte
kommt auf Seiten der zu zerstäubenden Flüssigkeit noch die Viskosität und die Oberflä-
chenspannung hinzu (Hede et al., 2008).
Abbildung 3.4: Schematische Darstellung des Zerfallsprozesses des Flüssigkeitsfilms in Ligamente
und Tropfen in der Mischzone einer außenmischenden Zweistoffdüse mit Angabe der Filmdicke
(dsh), der Ligamentendicke (dlg) und-länge (Llg), vom Autor bearbeitet, nach Duangkhamchan (2012)
Um den Tropfenzerfall bei der Zerstäubung der Coatinglösung mit einer außenmischenden
Düse vorherzusagen, kann der Sauterdurchmesser der hierbei entstehenden Tropfen
empirisch bestimmt werden werden (Mulhem, Fritsching, Schulte, & Bauckhage, 2003).
Dies geschieht unter Berücksichtigung zweier Kräfteverhältniskennzahlen, der Weber-Zahl
Weair und der Ohnesorg-Zahl Oh, dem Durchmesser der Düsenöffnung 𝑑𝑜𝑟𝑖𝑓𝑖𝑐𝑒 sowie dem
Verhältnis der beiden an der Zerstäubung beteiligten Massenströme �̇�𝑎𝑖𝑟 und �̇�𝑙𝑖𝑞𝑢𝑖𝑑
(Formel 3.1).

Theoretische Grundlagen
14
𝑑32 = 0,21 · 𝑑𝑜𝑟𝑖𝑓𝑖𝑐𝑒 · 𝑂ℎ0,0622 · (𝑊𝑒𝑎𝑖𝑟 ·�̇�𝑎𝑖𝑟
�̇�𝑙𝑖𝑞𝑢𝑖𝑑)
−0,4
(3.1)
Die Weber-Zahl der Luft, Weair, ist eine dimensionslose Kennzahl, die das Verhältnis der
Trägheitskraft einer Phase in einem mehrphasigen System gegenüber der stabilisierenden
Oberflächenkraft beschreibt (Formel 3.2). In diesem Fall ist es das Verhältnis der Trägheit
eines Tropfen der Coatinglösung, beschrieben durch die Parameter der Luftge-
schwindigkeit der Atomisierungsluft, 𝜈𝑎𝑖𝑟, der Dichte der Atomisierungsluft, 𝜌𝑎𝑖𝑟, und dem
Durchmesser der Düsenöffnung, 𝑑𝑜𝑟𝑖𝑓𝑖𝑐𝑒 , gegenüber der Oberflächenspannung der
Coatinglösung, 𝜎𝑙𝑖𝑞𝑢𝑖𝑑, die zerstäubt wird.
𝑊𝑒𝑎𝑖𝑟 =𝜈𝑎𝑖𝑟
2 ·𝜌𝑎𝑖𝑟·𝑑𝑜𝑟𝑖𝑓𝑖𝑐𝑒
𝜎𝑙𝑖𝑞𝑢𝑖𝑑 (3.2)
Die Ohnesorgzahl Oh ist ebenfalls eine dimensionslose Größe bei der Beschreibung des
Tropfenzefalls. Diese beschreibt das Verhältnis der Zähigkeit der Flüssigkeit, in diesem Fall
die Viskosität der Coatinglösung, 𝜂𝑙𝑖𝑞𝑢𝑖𝑑, gegenüber der Trägheits- und der Oberflächen-
kraft eines Tropfens. Wobei die beiden letzteren über die Wurzel aus dem Produkt von der
Oberflächenspannung der Coatinglösung, 𝜎𝑙𝑖𝑞𝑢𝑖𝑑 , der Dichte der Coatinglösung, 𝜌𝑙𝑖𝑞𝑢𝑖𝑑 ,
und dem Durchmesser der Düsenöffnung, 𝑑𝑜𝑟𝑖𝑓𝑖𝑐𝑒 , beschrieben werden können (Formel
3.3).
𝑂ℎ =𝜂𝑙𝑖𝑞𝑢𝑖𝑑
√𝜎𝑙𝑖𝑞𝑢𝑖𝑑·𝜌𝑙𝑖𝑞𝑢𝑖𝑑·𝑑𝑜𝑟𝑖𝑓𝑖𝑐𝑒 (3.3)
Wie in den Formeln 3.2 und 3.3 erkennbar ist, haben die Viskosität, die Dichte sowie die
Oberflächenspannung einen Einfluss auf die entstehende Tropfengröße. Dies sind die
Parameter, die bei der Auswahl der Coatinglösung berücksichtigt werden müssen.
Bezogen auf den Zerstäubungsprozess kann aufgrund der Simulationen von Laksmana et
al. (2009) postuliert werden, dass die Tropfengröße im Spray einen Einfluss auf die
Coatingqualität hat. Die Autoren fanden ebenfalls heraus, dass besonders kleine Tropfen
zu einem deutlich gleichmäßigeren Coating führen als größere Tropfen. Größere Tropfen
von Coatinglösung, die in den Coatingprozess eingebracht wurden, tragen auf Grund einer
Diskrepanz zwischen Partikel- und Tropfengröße zu einer höheren Agglomerationsaffinität
bei. Auch die Verteilung der Tropfengrößen ist hierbei entscheidend. Die Tropfengrößen-
verteilung sollte so schmal wie möglich sein, um ein gleichmäßiges Coating zu applizieren.

Theoretische Grundlagen
15
Die gasförmige Phase und deren Eigenschaften
Die Fluidisierungsluft erfüllt in einem Wirbelschichtcoatingprozess zwei wichtige Aufgaben.
Zum einen sollen durch sie die Partikel im Wirbelschichtbett fluidisiert werden und zum
anderen soll die Fluidisierungsluft so konditioniert sein, dass sie das Trocknen der Coating-
lösung und damit die Filmbildung durch die Coatinglösung auf der Partikeloberfläche er-
möglicht. Das Mollier-h-x-Diagramm zeigt, dass Luft mit einer höheren Temperatur und
gleicher relativer Luftfeuchte in der Lage ist, mehr Feuchtigkeit aufzunehmen. Demnach
kann mit zunehmender Temperatur mehr Lösungsmittel aus der Coatinglösung verdampft
werden. Somit ermöglicht eine höhere Temperatur die Sprührate zu erhöhen und dadurch
eine größere Menge an Coatingmaterial pro Zeiteinheit in den Prozess einzubringen, ohne
eine ungewollte Agglomeration zu verursachen (Uhlemann & Mörl, 2000). Allerdings ist bei
der Verwendung einer höheren Temperatur im System zu beachten, dass die Eigenschaf-
ten der Coatinglösung und der Partikel dadurch nicht negativ beeinflusst werden. Dies be-
deutet, bezogen auf die Coatinglösung, dass zum Beispiel zellulosederivatbasierte
Coatinglösungen bei Temperaturzunahme gelieren können. Demzufolge ist bei der Aus-
wahl der Prozesstemperatur, gesteuert durch die Temperatur der Fluidisierungsluft, auf die
Gelbildungstemperatur der Coatinglösung zu achten (Bayer & Knarr, 2012). Bezogen auf
die Partikel bedeutet eine eventuelle Schädigung, dass Inhaltsstoffe, die in der Matrix der
Partikel eingeschlossen sind, temperaturanfällig sein können und somit bei zu hohen Pro-
zesstemperaturen beschädigt werden könnten (Drusch & Berg, 2008).
Um eine Fluidisierung zu gewährleisten, muss die Luft eine gewisse Einströmge-
schwindigkeit erreichen. Hier spricht man von der minimalen Fluidisierungsgeschwindigkeit
bzw. von der Lockerungsgeschwindigkeit Umf. Bei dieser Einströmgeschwindigkeit der Luft
fängt das Festbett aus Partikeln an, sich zu einem Wirbelbett zu entwickeln. Die Ge-
schwindigkeit ist spezifisch für jede Art von Partikel (Teunou & Poncelet, 2002) und wird
über die Partikelgröße dp und die Partikeldichte 𝜌𝑝 ermittelt. Auch die Eigenschaften der
Gasphase spielen hierbei eine Rolle, wie die dynamische Gasviskosität µ𝑔 und die Gas-
dichte 𝜌𝑔.
Bei der Berechnung der Lockerungsgeschwindigkeit muss in zwei Partikelgrößenbereiche
eingeteilt werden. Zulässig für dp < 100 µm:
𝑈𝑚𝑓 =(𝜌𝑝−𝜌𝑔)
0.934𝑔0.934𝑑𝑝
1.8
111µ0.87𝜌𝑔0.066 (3.4)
Die Lockerungsgeschwindigkeit für Partikel mit einer Partikelgröße über 100 µm ist zusätz-
lich von der Archimedes-Zahl Ar abhängig.

Theoretische Grundlagen
16
𝐴𝑟 =𝜌𝑠𝑑𝑝
3(𝜌𝑝−𝜌𝑔)𝑔
µ2 (3.5)
Daraus ergibt sich für Partikel mit dp > 100 µm:
𝑈𝑚𝑓 = µ𝐺
𝜌𝐺 𝑑𝑣{(1135.7 + 0.0408 𝐴𝑟)0.5 − 33.7} (3.6)
Neben der Lockerungsgeschwindigkeit existiert als wichtige Charakterisierungsgröße die
Schwebegeschwindigkeit Us. Diese Geschwindigkeit beschreibt die Eintrittsgeschwindigkeit
der Fluidisierungsluft, ab der die Partikel nicht mehr im Wirbelbett gehalten werden, son-
dern die Partikel des Betts pneumatisch gefördert werden (Teunou & Poncelet, 2002). Die-
se Geschwindigkeit ist jedoch nicht ohne weiteres zu beschreiben und soll hier nur für den
Idealfall eines kugelförmigen Partikels gezeigt werden. Die Komplexität der Schwebege-
schwindigkeitsberechnung beruht auf ihrem physikalischen Ansatz. Zur Berechnung wird
angenommen, dass Gewichtskraft und Auftriebskraft sich gegenseitig aufheben, wodurch
die Schwebegeschwindigkeit wie folgt berechnet werden kann:
𝑈𝑠 = √4∙(𝜌𝑝−𝜌𝑔)∙𝑑𝑝∙𝑔
3∙𝜌𝑔∙𝑐𝑊 (3.7)
Hierbei steht die Größe cW für den Strömungswiderstand der Kugel. Zur Veranschauli-
chung der beiden Größen Umf und Us werden die drei Arten, die ein Wirbelbett einnehmen
kann, in Abbildung 3.5 dargestellt.

Theoretische Grundlagen
17
Abbildung 3.5: Schematische Darstellung des Druckverlustes der Fluidiserungsluft Δp im Fließbett in
Abhängigkeit der Lufteintrittsgeschwindigkeit U mit den drei verschiedenen Zuständen des Fließbet-
tes mit zunehmender Geschwindigkeit
Die feste Phase und ihre Eigenschaften
Neben den Eigenschaften der zerstäubten Flüssigkeit spielen natürlich auch die Eigen-
schaften der Partikel, die im Wirbelschichtcoatingprozess fluidisiert und gecoatet werden
sollen, eine wichtige Rolle. Die Haupteigenschaften, wenn es um das Fluidisieren der Par-
tikel geht, sind die Dichte und die Größe der Partikel. Die erste Klassifizierung von Parti-
keln bezüglich ihrer Dichte und Größe im Hinblick auf eine Anwendung in der Wirbelschicht
wurde von Geldart (1973) vorgenommen. Er teilte die Partikel bezüglich ihrer Größe und
Dichte in vier Kategorien ein, C, A, B und D. Die Kategorien sind nach dem Verhalten, dass
die Partikel der einzelnen Kategorien in einem Wirbelschichtprozess zeigen, benannt (sie-
he Abbildung 3.6).
Δp
U Umf Us
Festbett Wirbelbett pneumatischer Transport

Theoretische Grundlagen
18
Abbildung 3.6: Klassifizierung von Partikeln für Wirbelschichtanwendungen nach Geldart (1973), mit
dem Unterschied der Dichte zwischen Partikel und umgebener Gasphase als Funktion der Partikel-
größe; dargestellt sind die Bereiche für die Partikelklassen: C „cohesive“, A „aerateble“, B „bubbling“
und C „spouting“
Partikel der Klasse C werden als kohäsiv bezeichnet. Sie haben in der Regel eine Partikel-
größe, die unter 100 µm liegt. Partikel dieser Klasse können nicht, bzw. nur unter Zuhilfen-
ahme mechanischer Einbauten in das Fließbett, fluidisiert werden. Die Adhäsion zwischen
den Partikeln ist zu groß, wodurch sich nur Steigkanäle im Bett der Partikel ausbilden. Ein
typisches Beispiel für Partikel der Klasse C ist Mehl.
Partikel der Klasse A können als „belüftbar“ (engl. aerateble) bezeichnet werden. Sie besit-
zen eine Dichte von unter 1,4 g/cm3 und haben eine Partikelgröße von unter 1 mm. Diese
Partikel lassen sich mit Erreichen der minimalen Fluidisierungsgeschwindigkeit der ein-
strömenden Luft homogen fluidisieren. Jedoch brechen aus dem fluidisierten Bett schnell
Blasen aus, was die Stabilität des Bettes beeinträchtigt. Für Partikel der Klasse A muss die
Gleichung 3.8 erfüllt sein.
8∙10−4𝑔𝑑𝑠𝑣(𝜌𝑠−𝜌𝑓)
𝐾𝑀𝐵𝜇≤ 1 (3.8)
Hierbei kann KMB als Wert aus der Darstellung von Partikelgröße über der minimalen Flui-
disierungsgeschwindigkeit ermittelt werden. Der Wert für µ (Gasviskosität) kann für nicht
konditionierte Luft zur Vereinfachung mit µ=1,8·10-4 g/cm·s angenommen werden. Der
Wert dsv (sv für solid-vapour, also fest-gasförmig) kann durch d’ (die Partikelgröße) ersetzt
werden, was eine Vereinfachung der Gleichung 3.8 auf Gleichung 3.9 ermöglicht.
C
A B
D
Partikelgröße [µm]
Dic
hted
eiffe
renz
[kg·
m-3
]
500
500
1000
1000
100 50
5000

Theoretische Grundlagen
19
(𝜌𝑠 − 𝜌𝑓)𝑑′ ≤ 225 (3.9)
Als typisches Beispiel für Partikel der Klasse A gilt Milchpulver.
Der Unterschied zwischen Partikeln der Klasse B und D ist nicht so gravierend wie er zwi-
schen A und C ist (Geldart, 1973). In Klasse B werden Partikel eingeordnet, die unter Be-
rücksichtigung der Gasphasendichte und der Partikeldichte die Gleichung 3.10 erfüllen.
(𝜌𝑠 − 𝜌𝑓)1.17
≥ 9.06 ∙ 105 (3.10)
Die Partikelgröße von Partikeln der Klasse B bewegt sich in der Regel zwischen 40 und
500 µm und die Dichte dieser Partikel rangiert zwischen 1,4 und 4 g/cm3. Typischer Vertre-
ter dieser Klasse sind Partikel in der Größe von Kies.
Partikel der Klasse D sind sehr viel dichter als Partikel der vorherigen Klassen und ihre
Partikelgröße ist auch deutlich höher. Generell muss für eine Einordnung in die Klasse D
die Gleichung 3.11 erfüllt sein.
(𝜌𝑠 − 𝜌𝑓)(𝑑′)2 ≥ 106 (3.11)
Typische Vertreter für Partikel der Klasse D sind Kaffeebohnen.
Weitere Eigenschaften der Partikel in den Klassen A-D bzw. Eigenschaften eines aus die-
sen Partikeln bestehenden Wirbelbetts sind in Tabelle 3.1 aufgeführt.

Theoretische Grundlagen
20
Tabelle 3.1: Partikel- und Wirbelbettcharakteristika nach Rhodes (2008); durch Autor ergänzt
Klasse C Klasse A Klasse B Klasse D
wichtigste
Charakteristika
sehr kohäsiv,
schwer zu
fluidisieren
ideale Wirbel-
schicht-
anwendungen
bildet kaum Bla-
sen während des
Fluidisierens
Blasenbildung
startet bei Umf
sehr grobe
Partikel
Wirbelbett-
ausdehnung
niedrig, wegen
Steigkanälen
hoch moderat niedrig
Entlüftungsrate zunächst
schnell, dann
exponentiell
langsam,
linear
schnell schnell
Blasen-
eigenschaften
keine Blasen,
nur Steig-
kanäle
Blasen platzen
und koaleszieren,
max. Blasengröße
keine max.
Blasengröße
keine max.
Blasengröße
Partikelvermischung sehr niedrig hoch moderat niedrig
Gasvermischung sehr niedrig hoch moderat niedrig
„spouting“ -
spritzen
nein nein nur in flachen
Partikelbetten
ja, auch in
tiefen Parti-
kelbetten
Für die Partikel, die in der Wirbelschicht gecoatet werden sollen, müssen noch weitere Ei-
genschaften berücksichtigt werden. Dazu zählt die Oberflächenbeschaffenheit, die durch
die Porosität, ermittelt durch Porengrößen, Porenverteilung und Feststoffdichte, charakteri-
siert werden kann und einen wichtigen Einfluss auf die Coatingqualität hat (Börjesson,
Innings, Trägårdh, Bergenståhl, & Paulsson, 2014; Hay, Dragila, & Liburdy, 2008; Wiącek,
2011). Ein weiterer wichtiger Aspekt ist die Oberflächenenergie der Partikeloberfläche.
Diese entscheidet mit darüber, wie sich die Coatinglösung auf der Partikeloberfläche ver-
hält.

Theoretische Grundlagen
21
Partikel-Flüssigkeits-Interaktion
Wie bislang gezeigt wurde, sind die Eigenschaften der drei Phasen entscheidend für die
Qualität des applizierten Coatings.
Abbildung 3.7: Schematische Darstellung der Materialeigenschaften (schwarz) und der Prozessei-
genschaften (grau), die Einfluss auf die Partikel-Flüssigkeits-Interaktion haben
Um die Abhängigkeit des Coatingergebnisses der drei interagierenden Phasen zu be-
schreiben, wurden diese in Abbildung 3.7 zusammengefasst.
ZERSTÄUBUNG
TROCKNUNG PARTIKEL – FLÜSSIGKEITS -
INTERAKTION
FLUIDISIERUNG
Luft
Coatinglösung Viskosität, Oberflächenspannung, Zusammensetzung
Partikel Partikeldichte, Partikelgröße, Oberflächenenergie
rel. Luftfeuchte, Temperatur
Tropfen
Düsendesign, Zerstäubungsdruck
Tropfengröße, Tropfengeschwindigkeit
Lockerungs- geschwindigkeit, Gasdichte, Gasviskosität
I II
III IV
Adhäsionsarbeit Kontaktwinkel
Bislang wurde gezeigt, dass die Eigenschaften der flüssigen Phase, also der Coatinglö-
sung, von Bedeutung für den Zerstäubungsprozess sind. So können die Viskosität und die
Oberflächenspannung den Tropfenzerfall beeinflussen. Die entstehende Tropfengröße
wiederum spielt für die Coatingqualität eine Rolle. Die Partikeleigenschaften, wie etwa die
Partikeldichte und Partikelgröße, zeigen einen Einfluss auf die Fluidisierung und stehen
somit in direkter Wechselwirkung mit der Fluidisierungsluft, welche je nach Partikelbeschaf-
fenheit eine definierte Einströmgeschwindigkeit braucht, um die Partikel zu fluidisieren. Die
Partikel haben aber auf Grund ihrer Zusammensetzung eine weitere wichtige Eigenschaft,
nämlich die Oberflächenenergie. Diese trägt in Kombination mit der Oberflächenspannung
zu einer Wechselwirkung bei, die in dieser Arbeit als Partikel-Flüssigkeits-Interaktion be-
zeichnet wird. In dieser Interaktion spielen Adhäsions- und Spreitungsphänomene eine
Rolle. Diese können über unterschiedliche Modelle, wie dem Modell nach Owens-Wendt-
Rabel und Kaelble oder dem Modell nach Young-Dupré, beschrieben werden um somit
einen Beitrag zur Vorhersage der Coatingqualität zu liefern.

Theoretische Grundlagen
22
3.3 Zellulose und Zellulosederivate
Zellulose ist ein nachwachsender Rohstoff, der zum Beispiel aus der Zellwand von Pflan-
zenfasern gewonnen werden kann. Wie in Abbildung 3.8 zu erkennen ist, ist Zellulose aus
sequenziellen Cellobioseeinheiten aufgebaut. Jeder dieser Bausteine besteht aus zwei
Anhydroglucoseeinheiten (AGUs, β-D-Glukose-Monomer) und jede AGU besitzt drei Hyd-
roxylgruppen, an denen eine Modifikation durch Addition ablaufen kann (Cash & Caputo,
2010). Weiterhin ist zu erkennen, dass die Elementarfibrillen kristalline Bereiche, in denen
mehrere Fibrillen zusammenlaufen, aufweisen. Jede einzelne Elementarfibrille besteht aus
mehreren Cellulosefäden, die jeweils aus den oben genannten Cellbioseeinheiten besteht.
Abbildung 3.8: Schematischer Aufbau von Zellulose aus Pflanzenfasern, vom Autor bearbeitet nach
Wüstenberg (2015)
Eine Modifikation ist für den Einsatz in wässrigen Systemen notwendig, da native Zellulose
nicht wasserlöslich ist. Die Derivate der Zellulose werden im Allgemeinen in die Gruppe der
Hydrokolloide eingegliedert. Das bedeutet, dass sie eine Affinität zu Wasser haben. Man
unterscheidet hierbei in zwei Arten von Hydrokolloiden. Einige können Wasser binden, an-
dere können in Wasser quellen und es so immobilisieren (Wüstenberg, 2015).
Im Folgenden soll ein Überblick über die chemischen und technologischen Eigenschaften
gegeben werden.

Theoretische Grundlagen
23
3.3.1 Chemische und technologische Eigenschaften von Zellulosederiva-ten
Hydroxypropyl-Methylzellulose (HPMC) ist ein lineares, wasserlösliches, nichtionisches
Polysaccharid, das in die Gruppe der Zelluloseether gehört (Cash & Caputo, 2010;
Embuscado, Huber, & Nieto, 2009; Soliva-Fortuny et al., 2012). HPMC wird aus aufgerei-
nigter Zellulose, die mit Methylchlorid bei basischem pH-Wert Methylzellulose bildet, her-
gestellt. Dabei werden die kristallinen Strukturen der Zellulose aufgebrochen und diese
aktivierte Matrix (Alkalizellulose) reagiert über eine stufenweise Addition mit Propylenoxid
zu HPMC (Embuscado et al., 2009).
HPMC ist gut in kaltem Wasser löslich (Cash & Caputo, 2010; Embuscado et al., 2009).
Die Löslichkeitseigenschaften sind stark vom Substitutionsgrad (DS) und dem Polymerisa-
tionsgrad (DP) abhängig. Ein hoher DS und ein niedriger DP führen zu einer besseren Lös-
lichkeit des HPMCs in Wasser (Cash & Caputo, 2010).
Bei der Verwendung als Coatingmaterial bildet es transparente, ölresistente, wasserlösli-
che Filme (Debeaufort & Desobry, 2012; Miller & Krochta, 1997; Soliva-Fortuny et al.,
2012). Des Weiteren stellen sie eine moderate Barriere gegen Sauerstoff (Tabelle 3.3) und
Kohlenstoffdioxid sowie gegen Lipide dar und eine effiziente Barriere gegen Wasserdampf
(Debeaufort & Desobry, 2012), siehe auch Tabelle 3.2. Die Inkorporation von Nano- oder
auch Mikropartikeln in zellulosebasierte Filme zeichnet sich als vielversprechender Ansatz
ab, wenn eine Verbesserung der Barriereeigenschaften erzielt werden soll. Bilbao-Sainz et
al. (2010) haben diesen Ansatz weiterverfolgt und in ihrer Studie den Versuch unternom-
men, mikrokristalline Zellulosepartikel in HPMC einzubetten. Die Autoren haben als Ziel
zwar nicht die Untersuchung der Sauerstoffpermeabilität an die erste Stelle gestellt, jedoch
die Barriereeigenschaften in Bezug auf Wasserdampf untersucht. Dabei stellten sie fest,
dass mit abnehmender Größe der Nanopartikel, aber auch mit zunehmender Konzentration
die Permeabilität deutlich erniedrigt werden kann. Dies ist laut den Autoren darauf zurück-
zuführen, dass die Nanopartikel eine polare Charakteristik aufweisen und sie so mit den
hydrophilen Gruppen des HPMC-Hauptgerüstes in Wechselwirkung treten können. Damit
wird eine engere Zusammenlagerung der HPMC-Moleküle erreicht (siehe auch Tabelle 3.2
und Tabelle 3.3). Eine weitere Möglichkeit der Verbesserung der Barriereeigenschaften
wurde von de Moura et al. (2011) untersucht. Die Sauerstoffbarriereeigenschaften von
HPMC-basierten Filmen lassen sich erheblich verbessern, wenn in das relativ weitläufige
Geflecht von HPMC Molekülen zerkleinerte Zellulosefasern eingebracht werden. Hierbei
wurden die Zellulosefasern als Dispersion in einer HPMC-basierten Lösung in einem Micro-
fluidizer zerkleinert und die Partikelgröße vor und nach einer variablen Anzahl von Durch-

Theoretische Grundlagen
24
gängen bestimmt. De Moura et al. stellten fest, dass die Partikelgröße nach zwanzig
Durchgängen von anfangs 37 µm auf 2,5 µm sank. Diese Zerkleinerung und Einlagerung in
die Zwischenräume des HPMC-Geflechts von Zellulosefasern scheint ausreichend für eine
Verbesserung der Barriereeigenschaften von HPMC-Filmen gegenüber Sauerstoff zu sein.
Ebenfalls hat sich die Studie von de Moura et al. (2011) der Einbringung von Füllpartikeln
in zellulosebasierte Filme angenommen.
Von Atarés et al. (2011) wurde die Verbesserung der Barriereeigenschaften gegenüber
Sauerstoff von HPMC-basierten Filmen durch Zugabe von Ascorbin- und Zitronensäure
untersucht. Hierbei wurde Mandelöl in Probengefäße gegeben und mit einem Film aus
HPMC abgedeckt. Das Mandelöl wurde regelmäßig während der 85 Tagen Lagerzeit auf
den Hydroperoxidgehalt untersucht. Die von Ayranci et al. (2003) und Atarés et al. (2011)
postulierte Quervernetzung zwischen Säure- und Zellulosemolekülen wurde von Ghan-
brarzadeh et al. (2011) genauer untersucht. Die Autoren zeigten, dass es zwischen der
Carboxylgruppe der Säure, in diesem Fall Zitronensäure, und der Hydroxylgruppe des Zel-
lulosederivates zu einer Ausprägung von Wasserstoffbrücken kommt und somit zu einer
dichteren Anlagerung der Zellulosemoleküle untereinander.
Neben HPMC gehört Methylzellulose (MC) zur Gruppe der nichtionischen, wasserlöslichen
Zelluloseether und verfügt über ähnliche Coating-Eigenschaften. MC ist verglichen mit
HPMC schwerer wasserlöslich und MC-Coatings weisen eine niedrigere Wasserdampf-
permeabilität im Vergleich zu anderen hydrophilen Coatings auf (Erdohan & Turhan, 2005).
Filme aus HPMC und MC zeigen moderate Barriereeigenschaften gegenüber Sauerstoff
(Miller & Krochta, 1997), siehe auch Tabelle 3.3. Bereits Park et al. (1993) stellten fest,
dass das Molekulargewicht von MC einen großen Einfluss auf die Barriereeigenschaften
der MC-basierten Filme zeigt. Hierbei stellten sie heraus, dass MC-basierte Filme aus MC
mit niedrigem Molekulargewicht (13 kDa) die geringste Sauerstoffpermeabilität im unter-
suchten Molekulargewichtsspektrum aufwiesen.
Die Barriereeigenschaften von zellulosederivatbasierten Coatings lassen sich weiter ver-
bessern, um längerfristige Lagerzeiten von oxidationsanfälligen Produkten zu gewährleis-
ten. Eine Möglichkeit der Modifizierung von Barriereeigenschaften zeigten Ayranci et al.
(2003). Hier wurde der MC-Lösung Ascorbinsäure oder Zitronensäure beigemengt. Die
Barriereeigenschaften wurden in dieser Studie indirekt gemessen, indem die Bräunung von
Pilzen und Blumenkohl, welche auf Anwesenheit von Sauerstoff zurückzuführen ist, vor
und nach einer Lagerzeit mittels L-a-b-Messung ermittelt wurde.

Theoretische Grundlagen
25
Tabelle 3.2: Übersicht über die Wasserdampfpermeabilität von gegossenen Filmen aus CMC, HPMC
und MC aus der Literatur mit jeweiligem Molekulargewicht, falls in Studie angegeben, teils umge-
rechnet nach Hagenmaier (2012)
Temperatur für
0-100% rel. Feuchte
Permeabilität
[g·mm/m2·h·kPa]
Quelle
CMC 20 °C 1,3 (Tong et al., 2008)
HPMC 25 °C 0,79 (de Moura et al., 2008)
HPMC 25 °C 0,89 (de Moura et al., 2011)
MC (13 kDa) 30 °C 0,31 (Park et al., 1993)
MC (86 kDa) 30 °C 0,448 (Park et al., 1993)
Carboxy-Methylzellulose (CMC) ist ein wasserlösliches, lineares Polysaccharid, das zu den
anionischen Zelluloseethern zählt (Bifani et al., 2007; Cash & Caputo, 2010; Embuscado et
al., 2009; Soliva-Fortuny et al., 2012). Hergestellt wird das Hydrokolloid aus der Reaktion
von Alkalizellulose mit Natriumchloracetat. Dabei bestimmt der Substitutionsgrad die Was-
serlöslichkeit sowie die Filmbildungseigenschaften (Cash & Caputo, 2010; Soliva-Fortuny
et al., 2012). Unter einem pH-Wert von 3,0 wird CMC in Wasser unlöslich und verliert die
Eigenschaft der Wasserbindung. Dabei erniedrigt ein hoher Substitutionsgrad den hydroly-
tischen Abbau (Cash & Caputo, 2010; Embuscado et al., 2009). Carboxy-Methylzellulose
kann beispielsweise als Coating für Früchte eingesetzt werden um deren Haltbarkeit zu
verlängern (Hussain, Meena, Dar, & Wani, 2010).
Riveros et al. (2013) haben herausgefunden, dass MC- und CMC-basierte Filme den oxi-
dativen Verderb von gerösteten Erdnüssen zu minimieren. Sie fanden heraus, dass die
Erdnüsse, die mit CMC-basierten Filmen überzogen waren, den niedrigsten Peroxidgehalt
nach 55 Tagen Lagerdauer aufwiesen. CMC-basierte Filme wurden von Mishra et al.
(2010) untersucht und die Ergebnisse deuteten darauf hin, dass sich CMC-basierte Filme
gut für die Verwendung als Sauerstoffbarriere eignen. Die Sauerstoffpermeabilität von
CMC-basierten Filmen lag laut Mishra et al. (2010) in der gleichen Größenordnung, wie
Filme aus Bienenwachs, welche ebenfalls in dieser Studie charakterisiert wurden. Bienen-
wachs hat laut den Autoren jedoch den Nachteil der Off-Flavour-Bildung, den CMC-
basierte Filme nicht aufweisen. Weiter geben die Autoren zu bedenken, dass die Verwen-
dung von Plastifizierern zur Erhöhung der Sauerstoffpermeabilität führt.

Theoretische Grundlagen
26
Tabelle 3.3: Übersicht über die Sauerstoffpermeabilität von gegossenen Filmen von MC, HPMC und
CMC aus der Literatur mit jeweiligem Molekulargewicht, falls in Studie angegeben, teils umgerechnet
nach Hagenmaier (2012)
Testtemperatur,
(relative Feuchte)
Permeabilität
[cm3·µm/m2·d·kPa]
Quelle
MC 24 °C, (50 %) 97,0 (Miller & Krochta, 1997)*
MC 25 °C, (52 %) 90,0 (Miller & Krochta, 1997)*
MC (13 kDa) 25 °C, (50%) 276,9 (Park et al., 1993)
MC (86 kDa) 25 °C, (50%) 440,8 (Park et al., 1993)
HPMC 24 °C, (50 %) 272 (Miller & Krochta, 1997)*
HPMC 23 °C, (55 %) 182,4 (de Moura et al., 2011)
HPMC 23 °C, (55 %) 182,3 (de Moura et al., 2008)
CMC 14 °C, (55 %) 230,7 (Mishra et al., 2010)
*: Daten wurden nicht gemessen, sondern als Herstellerangaben von „The Dow Chemical Co., Mid-
land, MI, USA“ wiedergegeben.
Ethylzellulose (EC) ist ein nichtionischer Zelluloseether. Im Gegensatz zu HPMC, MC und
CMC ist EC zumeist nicht in Wasser, sondern nur in organischen Lösungsmitteln löslich.
Eine Ausnahme bilden hier Produkte mit einem DS ≤ 1,3. Die freien Hydroxylgruppen in
der Kette sind je nach Grad mit Ethylgruppen substituiert. Maximal ist ein Wert von 2,8
Ethylgruppen pro AGU möglich, wobei im Lebensmittelbereich ein DS von 2,25 - 2,6 üblich
ist (Wüstenberg, 2015).
EC ist beständig gegen Änderungen des pH-Wertes, besonders im alkalischen Bereich.
Das Molekulargewicht beeinflusst die mechanischen Eigenschaften der daraus gebildeten
Filme, wie etwa Zug, Dehnung und Flexibilität (Cash & Caputo, 2010). (Cash & Caputo,
2010; Debeaufort & Desobry, 2012; Wüstenberg, 2015). EC wird für die Stabilisierung von
Aromen und als Bindemittel und Coating für Vitaminpräparate (Cash & Caputo, 2010), aber
auch als Maskierungsmittel für sensorisch unerwünschte Aromen eingesetzt wird.

Theoretische Grundlagen
27
3.3.2 Rheologische Charakterisierung und rheologische Eigenschaften von Zellulosederivaten
Aus der Literatur ist bekannt, dass Zellulosederivate eine Vielzahl von technologisch rele-
vanten rheologischen Eigenschaften besitzen. Die meisten Zellulosederivate, so auch MC,
CMC und HPMC, zeigen ein scherverdünnendes Fließverhalten (Wüstenberg, 2015). Dies
bedeutet, dass die Viskosität 𝜂 mit zunehmender Scherbelastung abnimmt. Diese Eigen-
schaft erlaubt es, bei einer höheren Scherbelastung, wie sie z.B. in einer Düse zur Zer-
stäubung auftritt, eine höhere Konzentration der Zellulosederivatlösung einzusetzen und so
in einem Coatingprozess mehr Material auf die zu coatenden Partikel zu bringen.
Eine weitere Eigenschaft von Zellulosederivaten ist die Gelbildung. MC, HPMC und CMC
beispielsweise können durch Anhebung der Temperatur thermoreversible Gele bilden.
Hierbei spricht man von thermisch induzierter Gelbildung (Haque & Morris, 1993).
Abbildung 3.9: Schematischer Ablauf der thermisch induzierten Gelbildung von Zellulosederivaten
am Beispiel von MC, vom Autor bearbeitet, nach Haque & Morris (1993)
Über den exakten Mechanismus der Gelbildung bei Zellulosederivaten gibt es unterschied-
liche Ansätze (Knarr, 2003). In Abbildung 3.9 ist ein schlüssiger Mechanismus dargestellt.
Wenn sich die MC in Lösung bei niedriger Temperaturen befindet, legen sich Wassermole-
Fibrillenbündel in Lösung bei niedrigen Temperaturen
Erhitzen auf ca. 30°C
Fibrillenbündel lösen sich aus starrer Struktur
Trennung der Fibrillen an ihren Enden
Hydrophobe Assoziation der Fibrillenenden in Wechselwirkung mit anderen Kettenbündeln
Erhitzen auf ca. 55–60°C
Erhitzen auf ca. 80°C

Theoretische Grundlagen
28
küle um die Methoxy-Substituenten und bilden eine Käfigstruktur um diese aus (Sarkar,
1995). Mit zunehmender Temperatur wird diese Struktur und damit die Ordnung der Ele-
mentarfibrillenbündel der MC aufgebrochen. Die Fibrillen bewegen sich aus ihrer engen
linearen Lage hin zu einer lockereren Anordnung. Wird dem System mehr Energie durch
Temperaturerhöhung auf ca. 55-60 °C zugeführt, fangen die Ketten an, sich durch die Auf-
lösung der Gitterstrukturen an den Enden voneinander zu lösen. Eine weitere Tempera-
turerhöhung auf bis zu 80 °C führt dazu, dass sich die Ketten der MC über hydrophobe
Wechselwirkungen an Ketten von anderen MC-Strukturen legen, wodurch unter Bindung
von Wasser ein Gel ausgeprägt wird (Haque & Morris, 1993).
Die Bildung eines Gels lässt sich mittels periodischer Oszillation mit einem Doppelspalt-
Messsystem in einem Rheometer erfassen (Mezger, 2010). Hierbei wird mit einem Pelle-
tier-Heizelement die Temperatur der Zellulosederivatlösung langsam erhöht, wobei niedri-
ge Heizraten von 1K/min bis 2K/min üblich sind (Balaghi, Edelby, & Senge, 2014; Cellesi,
Tirelli, & Hubbell, 2002; Chenite, Buschmann, Wang, Chaput, & Kandani, 2001; Kastner et
al., 2012; Lin Li, 2002; Verbeken, Neirinck, Van Der Meeren, & Dewettinck, 2005; Weng,
Zhang, Ruan, Shi, & Xu, 2004). Während des Versuches wird eine Temperaturrampe ge-
fahren und eine festgelegte Haltezeit bei einer geeigneten Temperatur eingehalten. Bei
Bedarf kann die Temperaturrampe im Anschluss wieder zurückgefahren werden. Über die
Zeit der Messung wird eine spezifische Auslenkung des Doppelspaltmesskörpers
(φ(t) [mrad]) bzw. eine Deformation (γ(t) [%]) festgelegt. Über das aufgenommene Dreh-
moment (M(t) [mNm]) und den detektierten Verlustfaktor (δ [°]) wird die Schubspannung
(τ(t) [Pa]) bestimmt. Daraus wird der Schubmodul (G*), der Speichermodul (G’) und der
Verlustmodul (G’’) berechnet. Der Schubmodul wird über:
𝐺∗ =𝜏(𝑡)
𝛾(𝑡) (3.12)
bestimmt, wobei hier für jeden Messpunkt der zugehörige Wert der Schubspannung und
der Deformation herangezogen wird. Der Speichermodul wird wie folgt errechnet:
𝐺′ =𝜏(𝑡)
𝛾(𝑡)∙ cos 𝛿. (3.13)
Der Verlustmodul berechnet sich über:
𝐺′′ =𝜏(𝑡)
𝛾(𝑡)∙ sin 𝛿. (3.14)
Diese Werte ermöglichen es nun, den Gelpunkt (die Temperatur, an dem die Gelbildung
einsetzt) und weitere Charakteristika der Zellulosederivatlösung zu bestimmen (Mezger,
2010). Der Gelpunkt wird als die Temperatur definiert, bei dem Speicher- und Verlustmodul
gleich groß sind. Eine andere Methode zur Bestimmung des Gelpunktes wurde von Kast-

Theoretische Grundlagen
29
ner et al. (2012) beschrieben und für Zellulosederivate von Balaghi et al. (2014) durchge-
führt. Diese Methode und weitere Größen zur Charakterisierung der strukturbildenden Pa-
rameter, wie etwa die Strukturentwicklungsgeschwindigkeit, die initiale Strukturierungstem-
peratur und die kritische Strukturierungstemperatur, werden in Kapitel 4.2.2 dargestellt.
3.4 Lipidoxidation
Ungesättigte Fettsäuren, wie Ölsäure (18:1), Linolsäure (18:2) oder Linolensäure (18:3),
die bspw. in Rapsöl enthalten sind, haben aufgrund ihrer mehrfach ungesättigten Doppel-
bindungen für die Ernährungsphysiologie des Menschen eine große Bedeutung (Minihane
& Lovegrove, 2007). Mit zunehmender Anzahl der Doppelbindungen nimmt die Reaktivität
und damit die Oxidationsgeschwindigkeit (Lipidoxidation) der Fettsäuren zu (Frankel,
2005).
Die Lipidoxidation von Fetten, die ungesättigte Fettsäuren enthalten, wird auch Autoxidati-
on genannt. Diese findet in Form einer radikalischen Kettenreaktion mit Sauerstoff statt
(Belitz, Grosch, & Schieberle, 2008; Choe & Min, 2006; Jacobsen & Let, 2007). Die Reakti-
on von Sauerstoff und ungesättigter Fettsäure findet jedoch nicht direkt statt, sondern über
den Umweg nach der Bildung von Lipidalkylradikalen. Die Bildung dieser wird durch die
Anwesenheit von Initiatoren in Gang gesetzt. Diese Initiatoren sind entweder Schwer-
metallionen, Peroxidradikale oder Hydroperoxide, welche ubiquitär vorkommen (Frankel,
2005) oder durch Fotooxigenierung entstehen (Belitz et al., 2008). Zunächst wird bei einer
ungesättigten Fettsäure das Wasserstoffatom in Allylstellung abgespalten (Wasserstoffabs-
traktion). Dies ist möglich, da die Bindungsenergie des Wasserstoffatoms in der Allylstel-
lung am geringsten ist. Durch die Abspaltung des Wasserstoffs kommt es zur Bildung von
Lipidalkylradikalen (R·, siehe Startreaktion, 1). Diese Startreaktion verläuft relativ langsam
im Gegensatz zu den nachfolgenden Kettenreaktionen, weshalb man in dieser Phase auch
von der Induktionsphase (lag-phase) spricht.

Theoretische Grundlagen
30
Induktionsphase:
(1) Initiator + RH InitiatorH +R·
(2) R· + O2 ROO·
Kettenwachstum:
(2) ROO· + RH ROOH + R·
(3) RO· + RH ROH + R·
Kettenverzweigung:
(4) ROOH RO· + ·OH
(5) 2 ROOH ROO· + RO· + H2O
Kettenabbruch:
(6) 2 R· stabile Produkte
(7) R· + ROO· stabile Produkte
(8) 2 ROO· stabile Produkte
Aus Alkylperoxidradikalen (ROO·) oder Alkoxyradikalen (RO·) entstehen durch Wasser-
stoff-Abstraktion an den ungesättigten Fettsäuren Fettsäurealkohole und Hydroperoxide
(ROOH) und weitere Lipidalkylradikale (R·). Die Anwesenheit von Sauerstoff hält das ex-
ponentielle Kettenwachstum aufrecht (2) (Frankel, 2005). Die entstandenen Hydroperoxide
sind die primären Produkte der Autoxidation (Jacobsen & Let, 2007). Diese können in se-
kundäre Edukte zerfallen, wie z.B. Carbonylverbindungen oder Semialdehyde, welche zu
Geruchs- und Geschmacksverderb im Öl führen können (Frankel, 2005). Auch kann es bei
den gebildeten Hydroperoxiden zu einer Kettenverzweigung kommen, wodurch weitere
Radikale gebildet werden (4 und 5).
Das Kettenwachstum kann durch die Reaktion zweier Radikale abgebrochen werden, da
stabile (nicht-radikalische) Produkte entstehen (Kettenabbruch). Steht in dem System kein
Sauerstoff zur Verfügung, können bereits nach der Induktionsphase aus den gebildeten
oder bereits im System vorhandenen Radikalen stabile Produkte entstehen, welche nicht
weiter zu einem Fettverderb führen (Belitz et al., 2008).
Die analytische Bestimmung des Fettverderbs kann über unterschiedliche Methoden erfol-
gen. Zum einen kann die Peroxidzahl bestimmt werden, indem die Miliäquivalente aktiven
Sauerstoffs quantitativ ermittelt wird. Weiterhin möglich ist die Bestimmung des Anisidin-

Theoretische Grundlagen
31
wertes. Hierbei werden die Carbonylgruppen, die während der Autoxidation entstehen,
quantitativ bestimmt (BVL L 13.00-15:2001-07; DIN EN ISO 6885:2000-10). Die sekundä-
ren Zerfallsprodukte (wie Propanal) können mittels einer Headspace-GC detektiert und so
quantifiziert werden (Drusch, Serfert, Scampicchio, Schmidt-Hansberg, & Schwarz, 2007).
Oder aber die Hydroperoxide werden mittels der Thiocyanatmethode (IDF Standard Me-
thode 74A:1991) bestimmt. Hierbei werden die während der Autoxidation entstandenen
Hydroperoxide quantitativ erfasst. Drusch und Berg (2008) stellten fest, dass die Werte bei
der Lagerung von sprühgetrockneten Partikeln, die verkapseltes Fischöl enthielten, für die
Anisidinzahl, den Propanalgehalt und die Hydroperoxidkonzentration sehr gut (𝑟 ≥ 0,906)
korrelierten. Daher kann die Methode zur Bestimmung des oxidativen Verderbs in Form
von der Bestimmung der Hydroperoxidkonzentration mittels Thiocyanatmethode als ver-
lässlich angesehen werden.

Material und Methoden
32
4 Material und Methoden
Die in dieser Arbeit verwendeten Geräte sind in Tabelle A.1 im Anhang aufgelistet und
werden im Folgenden nur mit ihren Gerätebezeichnungen verwendet. Materialien und
Chemikalien, die in dieser Arbeit zum Einsatz kommen, sind in Tabelle A.2 im Anhang mit
Herstellern, Bestell- und Chargennummern aufgelistet. Die in der vorliegenden Arbeit ver-
wendeten Coatingmaterialien waren Muster, die dankenswerter Weise von den Herstellern
bzw. Händlern der Rohstoffe kostenfrei zur Verfügung gestellt wurden. Die für diese Arbeit
verwendeten Materialien sind in Tabelle A.3 im Anhang aufgeführt.
Im Folgenden werden die verwendeten Materialien und Chemikalien nur mit ihren Abkür-
zungen bzw. Trivialnamen bezeichnet.
4.1 Charakterisierung des Haftungs- und Spreitungsverhaltens von Zellulosederivatlösungen auf maltodextrinbasierten Modell-oberflächen
Die Haftungs- und Spreitungseigenschaften von Coatinglösungen auf Modelloberflächen
lassen sich z.B. über die Messung des Kontaktwinkels der Coatinglösung auf der Modell-
oberfläche ermitteln. Alle Messungen zur Ermittlung des Kontaktwinkels wurden mit Hilfe
eines Kontaktwinkel- und Tropfenkonturmessgerätes (OCA-20) der Firma Data Physics
Instruments GmbH, Filderstadt, Deutschland (Abbildung 4.1) durchgeführt. Das Grundprin-
zip dieser Messung beruht auf einer optischen Auswertung der Kontur eines Tropfens. Für
die Bestimmung der Oberflächenenergie der Modelloberflächen wurde hierbei die Methode
der Kontaktwinkelmessung verwendet (sessile drop). Für die Bestimmung der Oberflä-
chenspannung der Coatinglösungen wurde die Methode des hängenden Tropfens verwen-
det (pendant drop). Bei beiden Methoden fand die Tropfenerzeugung mittels einer compu-
tergesteuerten Dosiereinheit statt. Im Fall der Kontaktwinkelmessung wurde der Tropfen
auf den jeweiligen Modellberflächen abgesetzt. Für die Messung der Oberflächenspannung
wurde die Kontur des hängenden Tropfens erfasst und daraus über die Young-Laplace-
Gleichung die Oberflächenspannung ermittelt. Bei der Erzeugung der Tropfen wurden
Hohlnadeln mit definierten Außendurchmessern verwendet. Für die Messungen zur Kon-
taktwinkelermittlung betrug der Außendurchmesser der Hohlnadeln 0,91 mm und für die
Ermittlung der Oberflächenspannung der Coatinglösungen hatten die Nadeln einen Au-
ßendurchmesser von 1,65 mm. Aufgrund der Fertigungsunterschiede bei der Herstellung
der Hohlnadeln musste jede Nadel vor der Messung auf ihre Breite hin überprüft werden
und der Außendurchmesser als Referenzbreite für die optische Auswertung in die
OCA-20 Software eingetragen werden.

Material und Methoden
33
Die Kamera zur Erfassung der Kontur des hängenden und liegenden Tropfens erfasst hier-
bei das Bild in Graustufen. Durch eine hinter dem Tropfen liegende Lichtquelle wird der
Kontrast zwischen der wässrigen Phase des Tropfens und der Luftphase der Umgebung
verstärkt. Die Software erkennt, wo ein Unterschied zwischen den Phasen besteht und
kann so die Kontur des Tropfens erfassen.
Abbildung 4.1: Schematische Darstellung des Tropfenkonturmessgerätes für die Messung des Kon-
taktwinkels von Referenz- und Coatinglösungen auf maltodextrinbasierten Modelloberflächen, vom
Autor modifiziert, nach Tamm et al. (2012)
Den Messungen der Kontaktwinkel zur Charakterisierung der maltodextrinbasierten Mo-
delloberflächen ging eine Methodenvalidierung und -optimierung voraus. Als Oberfläche für
die Methodenvalidierung wurde die einfache Oberfläche Glas (Objektträger) verwendet.
Für die Kontaktwinkel der in dieser Arbeit verwendeten Referenzlösungen auf Glas existie-
ren eine Vielzahl von Literaturwerten. Somit können die Ergebnisse der Kontaktwinkelmes-
sung anhand dieser Literaturwerte überprüft werden. Als Referenzlösungen wurden destil-
liertes Wasser (W), Diiodmethan (D), Ethylenglykol (E) und Formamid (F) verwendet.
Weiterhin wurden verschiedene Parameter der Messung optimiert um eine gut reprodu-
zierbare Messung zu gewährleisten. Im Folgenden werden diese Parameter genauer vor-
gestellt.
Die OCA-20-Software bietet die Möglichkeit einer automatischen Basisliniendetektion. Die
Basislinie ist die Linie oberhalb welcher der Kontaktwinkel durch die Software ermittelt wird.

Material und Methoden
34
Somit ist darauf zu achten, dass die Basislinie möglichst genau der Oberfläche der zu cha-
rakterisierenden Modelloberfläche entspricht. Bei der automatischen Basisliniendetektion
wird eine Basislinie automatisch vor jeder Messung des Kontaktwinkels (hier alle 4 s) neu
auf die Grundlinie der Oberfläche, auf welcher der Kontaktwinkel zu bestimmen ist, gelegt.
Das Volumen des gemessenen Tropfens, der auf der Oberfläche abgesetzt wird, hat einen
Einfluss auf die Messung des Kontaktwinkels. Zum einen besteht der Einfluss darin, dass
es bei zu geringen Volumina unmöglich ist, den Tropfen auf der Oberfläche abzusetzen, da
dieser an der Nadel verbleibt. Zum anderen besteht der Einfluss auch in der unterschiedli-
chen Größe der Oberfläche des Tropfens und des dadurch veränderten Verdunstungspro-
zesses. In den Messungen zur Methodenvalidierung wurden die Kontaktwinkel von Tropfen
mit verschiedenen Volumina für die unterschiedlichen Referenzlösungen verwendet (siehe
Tabelle 4.1).
Tabelle 4.1: Volumina der verschiedenen Referenzlösungen zur Methodenoptimierung der Kontakt-
winkelmessung auf Glas
Volumen in µL
Wasser 2,5 5,0 7,5
Ethylenglykol 2,5 5,0 7,5
Diiodmethan 1,5 2,5
Formamid 5,0
Auch der Messzeitpunkt der Kontaktwinkelmessung hat einen Einfluss auf die Ergebnisse
des Kontaktwinkels zwischen Referenzlösung und Glasoberfläche. Erfolgt die Messung zu
rasch nach dem Absetzen des Tropfens, so ist die Abweichung zwischen den gemessenen
Kontaktwinkeln zu hoch, um eine Wiederholbarkeit der Messung sicherzustellen. Des wei-
teren zittert der Tropfen auf der Oberfläche direkt nach dem Absetzen, so dass eine Mes-
sung des Kontaktwinkels nicht möglich ist. Verbleibt der Tropfen andererseits zu lange auf
der Oberfläche des Deckglases bevor die Messung gestartet wird, so besteht die Gefahr
des Verdunstens. Durch das Verdunsten des Tropfens zeigen sich starke Variationen in
den gemessenen Kontaktwinkeln.
Um den bestmöglichen Zeitpunkt für die Messung des Kontaktwinkels zu ermitteln, wurde
jeweils ein Tropfen der Referenzlösungen in 20-facher Wiederholung auf Glasoberflächen
abgesetzt und der Kontaktwinkel über den Zeitraum von 60 s mit einem Messintervall von
2 s gemessen und aufgezeichnet.

Material und Methoden
35
4.1.1 Herstellung der Coatinglösungen
Alle in dieser Arbeit verwendeten Coatinglösungen wurden mit destilliertem Wasser ange-
setzt. Um unterschiedliche Viskositätsbereiche zu erreichen (Tabelle 4.2), wurden ver-
schiedene Konzentrationen der Zellulosederivate auf Basis ihrer mitgelieferten Spezifikati-
onen ermittelt, die zur jeweils gewünschten Viskosität (Tabelle 4.4) führt.
Tabelle 4.2: Konzentrationen [g/100g] der verschiedenen Coatingmaterialien zur Herstellung der
Coatinglösungen in verschiedenen Viskositätsstufen
sehr niedrig (vl) niedrig (l) mittel (m) hoch (h)
MC 0,50 1,20 2,20 3,20
CMC 0,10 0,50 1,20 2,00
HPMC 1,20 3,00 4,75 7,25
Nutrateric® wurde gemäß der Auflösevorschrift der Firma Colorcon Limited, Dartford Kent,
Vereinigtes Königreich, aus zwei Komponenten hergestellt, Surelease® und NS Enteric®.
In Tabelle 4.3 ist der Mischungsansatz der jeweiligen Viskositätsstufe für 50 g Coatinglö-
sung dargestellt. Dabei besteht Komponente 1 aus einer 60 %igen (w/w) Lösung von Su-
release® mit destilliertem Wasser und Komponente 2 aus einer 1,4 %igen (w/w) Lösung
von NS Enteric®. NS Enteric® besteht hauptsächlich aus Na-Alginat.
Tabelle 4.3: Einzusetzende Masse [g] für die verschiedenen Viskositätsstufen für 50 g Nutrateric®-
Lösung berechnet auf Grundlage der Auflösevorschriften der Firma Colorcon Limited
sehr niedrig (vl) niedrig (l) mittel (m) hoch (h)
Komponente 1 1,42 5,67 11,33 17,00
Komponente 2 2,75 11,00 22,00 33,00
Dest. Wasser 45,83 33,33 16,67 -
4.1.2 Charakterisierung der Coatinglösungen
Die Viskosität der Coatinglösung (Tabelle 4.4) wurde mit einem LV-Rotationsviskosimeter
Brookfield Engineering Laboratories Vertriebs GmbH, Lorch, Deutschland bestimmt. Als
Rotationskörper kam die Spindel 00 und als Probenkörper der gerätespezifische UL-
Adapter zum Einsatz. Für die Messungen wurden jeweils 16 g Probe in den Messbehälter

Material und Methoden
36
gefüllt. Die Messung der Viskosität fand in Doppelbestimmung statt. Die Messzelle wurde
auf 24 °C temperiert und für eine Messung jeweils 20 s Messzeit angesetzt.
Tabelle 4.4: Viskosität η [mPa·s] der verwendeten Coatinglösungen in den jeweiligen Viskositätsstu-
fen
sehr niedrig (vl) niedrig (l) mittel (m) hoch (h)
Nutrateric 2,7 8,0 28,1 85,3
MC 2,8 7,5 25,3 86,2
CMC 2,8 8,2 25,3 85,2
HPMC 2,8 9,2 25,2 84,5
Die Drehzahlen und damit die Scherraten wurden innerhalb einer Viskositätsstufe jeweils
konstant gehalten. Jedoch mussten die Drehzahlen, um den Drehmomentsbereich des
Viskosimeters bei 20 % Auslastung zu halten, für die unterschiedlichen Viskositätsbereiche
entsprechend Tabelle 4.5 angepasst werden.
Tabelle 4.5: Drehzahlen [min-1] für die Spindel 00 im UL-Adapter für die Bestimmung der Viskosität
in verschiedenen Viskositätsstufen der Coatinglösungen mittels Brookfield LV-Rotationsviskosimeter
sehr niedrig (vl) niedrig (l) mittel (m) hoch (h)
Drehzahl 50 50 20 4
Für die Bestimmung der Oberflächenspannung (SFT) und ihrer polaren und dispersen An-
teile wurden zunächst die Coatinglösungen in verschiedenen Viskositätsstufen hergestellt
und anschließend mittels pendant-drop-Methode vermessen. Für die Bestimmung der pola-
ren und dispersen Anteile der SFT musste zusätzlich zur Messung der SFT an der Luft-
Wasser-Phasengrenze eine Messung der SFT an der Grenzfläche von Coatinglösung zu
unpolaren Lösungsmittel durchgeführt werden.
Für die Beschreibung gekrümmter Grenzflächen werden die Young-Gleichung sowie die
Laplace-Gleichung verwendet. Die Young-Gleichung beschreibt das Verhalten von Kräften
an gekrümmten Oberflächen mit der Hauptaussage, dass diese sich proportional zum Mit-
telwert der Krümmung ändern. Demnach gilt für eine sphärisch gekrümmte Oberfläche:
p=p0+2γL
r (4.1)

Material und Methoden
37
Wird die Grenzfläche eines Tropfens betrachtet, so besteht ein Druckunterschied zwischen
der konkaven und der konvexen Seite des Tropfens. Auf der konkaven Seite herrscht der
Druck p0+Δp und auf der konvexen Seite p0. Die Druckdifferenz Δp=p-p0 entspricht dem
Krümmungsdruck (Runke, Song, & Springer, 1994). Dieser wird durch die Laplace-
Gleichung beschrieben:
∆𝑝 = 𝛾𝐿 ∙ (1
𝑅1+
1
𝑅2) (4.2)
Sind die beiden Krümmungsradien und die Druckdifferenz zwischen der umgebenden Pha-
se und der Tropfenphase bekannt, kann die Oberflächenspannung bestimmt werden. Der
Druck der Tropfenphase wird durch die Kontur des Tropfens ermittelt. Zur Bestimmung der
polaren und dispersen Anteile der SFT der Coatinglösungen musste ein Tropfen der
Coatinglösung (V=5 µL) in einer Küvette mit n-Dodecan vermessen werden. Über die SFT
an der Luft-Wasser-Phasengrenzfläche und die SFT an der n-Dodecan-Grenzfläche konn-
ten mittels OCA-20-Software die polaren und dispersen Anteile der SFT bestimmt werden.
Die Summe der polaren und dispersen Anteile der SFT ergeben die Gesamt-SFT der
Coatinglösung.
4.1.3 Herstellung und Charakterisierung maltodextrinbasierter Modell-oberflächen
Für die Bestimmung der Partikel-Flüssigkeits-Interaktion konnten keine Glasoberflächen
verwendet werden, da sie den Oberflächen während des Coatingprozesses von malto-
dextrinbasierten Primärpartikeln nicht genügend entsprechen. Demzufolge wurden unter-
schiedliche Modelloberflächen hergestellt, deren Oberfläche eher der von maltodextrinba-
sierten Primärpartikeln ähnelt.
Die maltodextrinbasierte Tablette (MDT) wurde am Fachgebiet Pharmazeutische Techno-
logie der Freien Universität Berlin hergestellt. Die Herstellung der Tablette erfolgte mittels
einer Exzenterpresse (EK0, Korsch, Berlin, Deutschland). Die Tabletten wurden in drei
Chargen hergestellt mit einem jeweiligen Ansatz von 300 g Maltodextrin und 1% w/w Mag-
nesiumstearat. Die Zugabe von Magnesiumstearat ist notwendig, da dieses als „Schmier-
mittel“ während des Kompaktierungsprozesses fungiert und somit eine Kompaktierung von
Maltodextrin erst ermöglicht (Wurster, Likitlersuang, & Chen, 2005). Magnesiumstearat ist
als Zusatzstoff E470b für den Einsatz in Lebensmitteln zugelassen (Art. 1 §5 ZZulV, 2012).
Die hergestellten Tabletten hatten die Form „F-Type“ (Standardform flach mit abgesetztem
Rand), einen Durchmesser von 9 mm ± 0,2 mm und eine Härte von ca. 45 N (vor Ort mit-
tels eines Härtetesters der Firma Erweka GmbH, Heusenstamm, Deutschland untersucht).

Material und Methoden
38
Auf einer Tablette konnte jeweils ein Tropfen der Referenz- bzw. der Coatinglösung abge-
setzt werden.
Für die Modelloberfläche aus Maltodextrin im glasartigen Zustand (MDGSS) wurde zu-
nächst eine 50% w/w Lösung aus Maltodextrin und destilliertem Wasser hergestellt. Diese
wurde 16 Stunden bei Raumtemperatur mit ca. 300 rpm auf einem Magnetrührer gerührt,
damit sich das Maltodextrin vollständig lösen kann. Anschließend wurde die Lösung mittels
Einweg-Pasteurpipette auf einen Objektträger aufgetragen, bis der gesamte Objektträger
mit einer gleichmäßig hohen Schicht an Maltodextrinlösung bedeckt war. Hierbei war un-
bedingt dafür Sorge zu tragen, dass die Lösung nicht an der Seite vom Objektträger herun-
terläuft, da sich ansonsten keine plane Oberfläche ausbilden konnte. Die Objektträger wur-
den nach dem Auftragen der Lösung in einem Trockenschrank über 16 Stunden bei 70 °C
getrocknet. Oberflächen, die nach dem Trocknen Risse oder Furchen auf der Oberfläche
zeigten, wurden verworfen. Für die Messung der Kontaktwinkel der Referenz- und Coating-
lösungen wurden pro Oberfläche jeweils vier Tropfen abgesetzt.
Die Modelloberflächen aus Maltodextrin im glasartigen Zustand mit OSA-Stärke (MDOSA)
wurden analog zu den MDGSS-Oberflächen hergestellt. Lediglich die Rezeptur der auf den
Objektträger aufgebrachten Lösung wurde verändert. Die Rezeptur für die MDOSA-
Oberflächen bestand aus einer 35 % w/w Lösung aus Maltodextrin und destilliertem Was-
ser. Dieser wurde nach 16 Stunden Rührzeit, analog zur MDGSS-Oberfläche, 5 % w/w
OSA Stärke beigemengt und weitere 2 Stunden bei gleichen Bedingungen gelöst.
Die MDEMUL Oberflächen wurden analog zu den MDGSS-Oberflächen hergestellt. Die
Rezeptur wurde hierbei jedoch um MCT-Öl erweitert. Auch hier wurde eine 35 % w/w Lö-
sung aus Maltodextrin und destilliertem Wasser hergestellt und unter den gleichen Bedin-
gungen gerührt, wie die Maltodextrinlösung für die MDGSS-Oberflächen. Anschließend
wurde die OSA-Stärke (5% w/w) hinzugegeben und die Lösung zwei weitere Stunden ge-
rührt. Im Anschluss daran wurde der Maltodextrin/OSA-Stärke-Lösung 10 % w/w MCT-Öl
beigemengt. Die Emulsion wurde mittels Ultra Turrax bei 13500 rpm 1 Minute voremulgiert
und im Anschluss daran mit 300 bar/ 50 bar in zwei Durchläufen im Hochdruckhomogeni-
sator (GEA NIRO Soavi, Lübeck, Deutschland) emulgiert. Das Auftragen der Emulsion er-
folgte analog zur MDGSS-Oberfläche. Die Trocknungstemperaturen wurden für die Emul-
sion auf 35 °C herabgesetzt und die Trocknungszeit auf 18 Stunden erhöht. Im Anschluss
an die Trocknungszeit wurde noch eine Schicht der Emulsion auf die Modelloberflächen
aufgetragen und 30 Minuten im Trockenschrank getrocknet. Dies war nötig, um Trock-
nungsrisse zu vermeiden und eine gleichmäßige Oberfläche zu erzeugen.

Material und Methoden
39
Das für die Herstellung der Emulsion verwendete MCT-Öl wurde im Vorhinein von oberflä-
chenaktiven Substanzen, wie z.B. freien Fettsäuren, befreit (Wulff-Pérez, Torcello-Gómez,
Martín-Rodríguez, Gálvez-Ruiz, & de Vicente, 2011), um eine Beeinflussung der Kontakt-
winkelmessung zu vermeiden. Die Aufreinigung erfolgte mittels Magnesiumsilikat analog zu
der in der Literatur von Wüstneck et al. (1999) beschriebenen Methode. Die Methode wur-
de durch das Herabsetzen des Verhältnisses von MCT-Öl zu Florisil auf 1:4 leicht abge-
wandelt. In Vorversuchen zur Etablierung dieser Methode im Fachgebiet Lebensmitteltech-
nologie und -materialwissenschaften zeigte sich, dass dieses Verhältnis ausreichend ist,
um keine oberflächenaktiven Substanzen mehr im Öl nachweisen zu können. Auch die
Zentrifugationszeit und -intensität wurde den Möglichkeiten am Fachgebiet angepasst. Zum
Einsatz kam die Zentrifuge (Sigma Laborzentrifugen GmbH, Osterode am Harz, Deutsch-
land) mit dem Rotor 12155 und einer eingestellten Schwere von 10062 g. Die Zeit des
Zentrifugierens wurde auf 10 Minuten festgelegt.
Die Berechnung der freien Oberflächenenergie wurde auf Basis des Models nach Owens,
Wendt, Rabel und Kaelble durchgeführt (Owens & Wendt, 1969). Hierbei werden die Kon-
taktwinkel von mindestens zwei Referenzlösungen auf der zu charakterisierenden Oberflä-
che abgesetzt. Die Oberflächenspannung der Referenzlösungen sowie deren disperse und
polare Anteile müssen hierbei bekannt sein.
Über den gemessenen Kontaktwinkel und die bekannte SFT der Referenzlösungen kann
aus einem Koordinatensystem die Oberflächenenergie der Modelloberfläche mit polaren
und dispersen Anteilen ermittelt werden. Die Formel 4.3 ist in diesem Fall die Grundlage für
die Achsenbeschriftung.
1
2(1 + 𝑐𝑜𝑠𝜃) ∙
𝛾𝐿𝑉
√𝛾𝐿𝑑
= √𝛾𝑆𝑝
∙ √𝛾𝐿
𝑝
𝛾𝐿𝑑 + √𝛾𝑆
𝑑 (4.3)
Abbildung 4.2 zeigt exemplarisch den SE-Plot für die Kombination der Referenzlösungen
WEF. Über die dargestellte Ausgleichsgeradengleichung lassen sich nun die Werte für den
Achsenabschnitt und den Anstieg ermitteln. Der Achsenabschnitt entspricht dem Wert √𝛾𝑆𝑑
durch den der disperse Anteil der freien Oberflächenenergie ermittelt werden kann. Der
Wert für den Anstieg der Ausgleichsgeraden entspricht dem Wert √𝛾𝑆𝑝
durch den der polare
Anteil der freien Oberflächenenergie errechnet werden kann.
𝛾𝑆 = 𝛾𝑆𝑝
+ 𝛾𝑆𝑑 (4.4)

Material und Methoden
40
Die Gesamtoberflächenenergie der Modelloberflächen wird durch die Summe der polaren
und der dispersen Komponente der Oberflächenenergie berechnet (siehe Formel 4.4).
Abbildung 4.2: SE-Plot zur Bestimmung der Oberflächenenergie einer Modelloberfläche mit der bei-
spielhaften Kombination von Referenzlösungen WEF und der zur Berechnung herangezogenen
Ausgleichsgeraden
Für die Berechnung der freien Oberflächenenergie der maltodextrinbasierten Modelober-
flächen wurden vier verschiedene Referenzlösungen in unterschiedlichen Kombinationen
aus mindestens drei Lösungen verwendet.
Die Werte für die Oberflächenspannung sowie ihrer polaren und dispersen Anteile für
Wasser, Diiodmethan und Ethylenglykol wurden aus der OCA 20 Software entnommen
(siehe Tabelle 4.6) und basieren auf einer Veröffentlichung von Ström et al. (Ström,
Fredriksson, & Stenius, 1987). Die Werte für die Oberflächenspannung und ihre dispersen
und polaren Anteile für Formamid sind ebenfalls der OCA-20-Software entnommen worden
und beruhen auf einer Veröffentlichung von Rabel (1971).
y = 8,9024x + 1,3013
0
5
10
15
20
25
0 0,5 1 1,5 2 2,5
1/2(
1+co
s(θ)
)·(SF
T/sq
rt(S
FTd)
)
sqrt(SFTp/SFTd)
W
E
F

Material und Methoden
41
Tabelle 4.6: Oberflächenspannung mit polaren und dispersen Anteilen der Referenzlösungen zur
Bestimmung der freien Oberflächenenergie der maltodextrinbasierten Modelloberflächen Rabel
(1971) und Ström et al. (1987)
SFT
[mN/m]
SFTp
[mN/m]
SFTd
[mN/m]
Destilliertes Wasser 72,8 51,0 21,8
Diiodmethan 50,8 0,0 50,8
Ethylenglykol 47,7 16,8 30,9
Formamid 58,2 29,5 28,7
4.1.4 Charakterisierung der Partikel-Flüssigkeit-Interaktion
Die Partikel-Flüssigkeit-Interaktion wird in dieser Arbeit durch zwei Größen beschrieben,
zum einen über das Spreiten (Kontaktwinkel) einer Coatinglösung auf der Modelloberflä-
che, zum anderen über das Haften (Adhäsionsarbeit) der Coatinglösung auf einer Modell-
oberfläche. Im Folgenden werden die in dieser Arbeit verwendeten Modelle zur Ermittlung
beider Größen vorgestellt.
Zur Bestimmung des direkt gemessenen Kontaktwinkels CAm der Coatinglösung auf der
Modeloberfläche wurde die jeweilige Modelloberfläche auf dem justier- und temperierbaren
Tisch mit einer Messkammer darauf positioniert. Mittels einer softwaregesteuerten Dosie-
reinheit wurde ein Tropfen der Coatinglösung mit einem Volumen von 5 µL erzeugt. Dieser
wurde auf der Modelloberfläche abgesetzt und die Kontur des Tropfens wurde über 20s
hinweg erfasst. Pro Modelloberfläche erfolgte das Absetzen von vier Tropfen und es wur-
den pro Coatinglösung fünf Oberflächen verwendet. Daraus ergibt sich eine 20-fache Wie-
derholung der Kontaktwinkelmessung pro Coatinglösung. Für die Bestimmung des CAm der
Coatinglösung auf der MDT-Oberfläche wurde auf jeder Tablette nur ein Tropfen abgesetzt
und pro Coatinglösung 20 Tabletten verwendet, was auch hier zu einer 20-fachen Wieder-
holung führt. Angegeben wird der CAm in [°].
Die Berechnung der Adhäsionsarbeit nach dem Modell von Young und Dupré (YD) beruht
auf einer Messung des direkten Kontaktwinkels einer Coatinglösung auf der Modelloberflä-
che.
𝑊𝐴−𝑌𝐷 = 𝛾𝐿𝑉(1 + 𝑐𝑜𝑠𝜃) (4.5)

Material und Methoden
42
Die Adhäsionsarbeit wird mittels der Formel 4.5 berechnet. Hierbei ist ersichtlich, dass die
Oberflächenspannung der Coatinglösung 𝛾𝐿𝑉 in die Berechnung eingeht. Als Interaktions-
größe zwischen der Modelloberfläche und der Coatinglösung geht hier der direkt gemes-
sene Kontaktwinkel in die Berechnung ein. Als Einheit wird die Adhäsionsarbeit in [mN/m]
angegeben.
Die Adhäsionsarbeit nach dem Modell von Owens, Wendt, Rabel und Kaelble (OWRK)
wird nicht über den CAm bestimmt. Für die Berechnung der Adhäsionsarbeit nach OWRK
wird der polare und disperse Anteil der Oberflächenspannung der Coatinglösungen genau-
so wie der polare und der disperse Anteil der freien Oberflächenenergie der Modelloberflä-
che verwendet. Über diese beiden Charakteristika lässt sich dann die Adhäsionsarbeit zwi-
schen Coatinglösung und Modelloberfläche berechnen (siehe Formel 4.6).
𝑊𝐴−𝑂𝑊𝑅𝐾 = 𝑊𝐴𝑝
+ 𝑊𝐴𝑑 = 2 (√𝛾𝑆
𝑝∙ 𝛾𝐿
𝑝+ √𝛾𝑆
𝑑 ∙ 𝛾𝐿𝑑) (4.6)
In Formel 4.6 stehen 𝛾𝑆𝑝 und 𝛾𝑆
𝑑 für den polaren bzw. dispersen Anteil der freien Oberflä-
chenenergie der Modelloberfläche und 𝛾𝐿𝑝 und 𝛾𝐿
𝑑 für den polaren bzw. dispersen Anteil der
Oberflächenspannung der Coatinglösung. Angegeben wird die Adhäsionsarbeit in [mN/m].
Die Software des OCA 20 erlaubt eine Berechnung des Kontaktwinkels zwischen Modell-
oberfläche und Coatinglösung, ohne diesen direkt zu messen. Die Berechnung erfolgt auf
der Annahme, dass die Adhäsionsarbeit unabhängig vom verwendeten Modell gleich ist
und somit auch mathematisch gleich gesetzt werden kann (siehe Formel 4.7).
𝑊𝐴−𝑌𝐷 = 𝑊𝐴−𝑂𝑊𝑅𝐾 (4.7)
Durch Einsetzen der Formel 4.5 und 4.6 in 4.7 ergibt sich Formel 4.8.
𝛾𝐿𝑉(1 + cos(𝜃)) = 2 · (√𝛾𝑆𝑝
· 𝛾𝐿𝑝
+ √𝛾𝑆𝑑 · 𝛾𝐿
𝑑) (4.8)
Durch Umformen ergibt sich der Zwischenschritt für die Berechnung des Kontaktwinkels
(Formel 4.9).
cos (θ )=2(√γS
p ∙γLp+√γS
d ∙γLd)
γLV-1 (4.9)
Durch Anwendung der Invertierungsfunktion des Cosinus erhält man die Berechnungsfor-
mel (Formel 4.10) für den berechneten Kontaktwinkel (CAOWRK).
θ= arccos (2(√γS
p ∙γLp+√γS
d ∙γLd)
γLV-1) (4.10)

Material und Methoden
43
Die Berechnung des Kontaktwinkels CAOWRK beruht somit ausschließlich auf separat ermit-
telten Charakteristika für die Modelloberfläche und die Coatinglösung. CAOWRK wird in [°]
angegeben.
4.1.5 Statistische Auswertung
Alle Messergebnisse, die im Rahmen dieses ersten Versuchsblockes erzeugt wurden, sind
im Vorfeld mit dem Ausreißertest nach Grubbs untersucht worden.
Im Anschluss an den Ausreißertest wurden die Messwerte mittels Shapiro-Wilk-Test auf
Normalverteilung untersucht. Dieser Test bietet im Vergleich zu anderen Normalvertei-
lungstests eine sehr gute Teststärke bei einer Anzahl von Wiederholungen von n<50
(Razali & Wah, 2010). Als Signifikanzniveau wurde stets 0,01 gewählt.
Für den Fall, dass die Werte normalverteilt waren, wurde eine ANOVA mit einem Signifi-
kanzniveau von 0,01 durchgeführt. Ergab sich mittels der ANOVA ein signifikanter Unter-
schied zwischen den Mittelwerten, wurde der Post-hoc-Test nach Scheffé durchgeführt, um
die signifikanten Unterschiede in den Mittelwerten zu identifizieren. Auch hier betrug das
Signifikanzniveau 0,01.
Für nicht normalverteilte Messwertreihen kann keine herkömmliche ANOVA durchgeführt
werden. Um dennoch zu überprüfen, ob es in den Mittelwerten der Messwertreihen signifi-
kante Unterschiede gibt, kann eine Kruskal-Wallis-ANOVA durchgeführt werden
(Kankaanranta, Mwegerano, & Hämeenoja, 2012). Auch hierbei wurde das Signifikanzni-
veau auf 0,01 festgelegt.
Sämtliche statistische Untersuchungen wurden mit der Software „OriginPro“ Version 8G
SR4 v8.0951 Built 951 der Firma OriginLab Corporation, Northampton, Vereinigte Staaten
durchgeführt.

Material und Methoden
44
4.2 Charakterisierung des rheologischen Verhaltens von lebensmit-teltauglichen Zellulosederivaten
Zur rheologischen Charakterisierung von MC wurden zwei verschiedene Zellulosederivate
der Produktreihe MethocelTM verwendet, die die The DOW Chemical Company AG,
Bomlitz, Deutschland als Muster zur Verfügung stellte. Die Charakterisierung umfasst die
Messung der Viskosität in Abhängigkeit von Temperatur, Konzentration und Scherbelas-
tung, die Bestimmung der strukturbildenden Charakteristika von MC und die Ermittlung der
Strukturwiederaufbaufähigkeit nach einer Scherbelastung.
Die Versuche zur rheologischen Charakterisierung wurden in einem luftgelagerten Rheo-
meter (Anton Paar Germany GmbH, Ostfildern-Scharnhausen, Deutschland) durchgeführt.
Dazu wurde das Temperiersystem C-PTD 200 und das Doppelspalt-Messsystem DG26.7
verwendet. Zur Durchführung der Scher- und Oszillationsversuche sowie zur Berechnung
der Messwerte wurde das Programm RHEOPLUS/32 Multi6 V3.62 verwendet.
Tabelle 4.7: Herstellerangaben zu den in dieser Arbeit verwendeten Zellulosederivaten
Hersteller-
bezeichnung Typ Abkürzung
η bei 20 °C, 2 %
[mPa·s]
Methoxyl-
gruppen [%]
Methocel A4C MC MC383 383 29,6
Methocel A15
Premium LV MC MC15 15 28,5
Die MC-Lösungen (Herstellerangaben aus Analysezertifikaten in Tabelle 4.7) wurden in
den entsprechenden Konzentrationen hergestellt und mindestens 18 Stunden gerührt. An-
schließend erfolgte die Entgasung der Lösungen in einem Ultraschallbad für 5 Minuten. In
einem Vorversuch wurde ermittelt, welche Konzentration die höchstmögliche ist, die bla-
senfrei in den Doppelspaltzylinder eingefüllt und vermessen werden konnte.
Um die Verdunstung des Lösungsmittels aus der Probe während der Messungen zu ver-
ringern, wurde eine dünne Schicht Silikonöl auf die Probenoberfläche im Rheometer aufge-
tragen.

Material und Methoden
45
4.2.1 Bestimmung der Viskosität in Abhängigkeit von Temperatur, Kon-zentration und Schergeschwindigkeit
Die Viskosität der MC-Proben (zwei verschiedene MC-Proben in je drei unterschiedlichen
Konzentrationen) wurde in Abhängigkeit von den drei Faktoren Temperatur, Konzentration
und Schergeschwindigkeit ermittelt. Dazu wurden Untersuchungen mit einem linearen
Scherratenanstieg von 100 - 4000 s-1 durchgeführt. Die Werte für die Scherrate wurden so
gewählt, dass sie erstens einen möglichst weiten Bereich der auftretenden Scherbelastung
beim Zerstäuben wiederspiegeln (1000 - 10000 s-1) und zweitens in dem Bereich liegen, in
dem das verwendete Rheometer Drehmomente korrekt erfassen kann.
Pro Messung wurden 40 Messpunkte mit einer Messpunktdauer von 3 s aufgezeichnet. Die
weiteren Parameter für die Viskositätsbestimmungen sind der Tabelle 4.8 zu entnehmen.
Die technischen Daten des für die Viskositätsmessungen verwendeten Rheometers kön-
nen der Tabelle A.11 im Anhang entnommen werden.
Tabelle 4.8: Verwendete Temperaturen, Konzentrationen und Scherraten zur Untersuchung der Vis-
kosität
Zellulosederivat Temperatur [°C] Konzentration [%] Scherrate [s.1]
MC15 60 45 30 8 6,5 5 3000 2000 1000
MC383 60 45 30 5 3 1 4000 2500 1000
4.2.2 Bestimmung der strukturbildenden Charakteristika von MC
Um das Verhalten unter verschiedenen Temperaturbedingungen und in unterschiedlichen
Konzentrationen im Prozess besser beurteilen zu können, wurden verschiedene Gelstruk-
tureigenschaften der beiden MC-Materialien untersucht. Die Berechnung dieser Eigen-
schaften erfolgte auf Grundlage der Studie von Kastner et al. (2012). Hierbei wurde die
Ableitung des Speichermoduls nach der Zeit über der Temperatur aufgetragen und somit
konnten die Parameter der initialen Strukturbildungstemperatur (IST), der kritischen Struk-
turbildungstemperatur (CST), der Gelpunktstemperatur (GP) und der Strukturbildungsrate
(SDR) siehe Formel 4.11, ermittelt werden (siehe Abbildung 4.3).
𝑆𝐷𝑅 =𝐺𝑒𝑛𝑑
′ −𝐺𝐼𝑆𝑇′
𝑡𝑒𝑛𝑑−𝑡𝐼𝑆𝑇 (4.11)

Material und Methoden
46
Abbildung 4.3 Bestimmung der strukturbildenden Parameter bei der Untersuchung der Gelbildung
nach Kastner et al. (2012), hier beispielhaft vom Autor für eine 5%ige MC15-Lösung dargestellt
Die strukturbildenden Charakteristika der Proben wurden in den gleichen Konzentrationen
bestimmt wie bei den Untersuchungen zur Viskosität (siehe Kapitel 4.2.1). Bei den Unter-
suchungen zu den strukturbildenden Charakteristika wurden zwei verschiedene Tempera-
turprofile verwendet. Das Temperaturprofil 1 wurde für die Untersuchung der strukturbil-
denden Charakteristika der MC-Proben ohne und mit Zitronensäurezusatz benutzt und
Temperaturprofil 2 wurde für die Vermessung der Proben mit Ethanolzusatz angewendet.
Das Temperaturprofil 1 beinhaltet eine Aufwärmphase mit einer Heizrate von 1K/min von
20 °C auf 70 °C. Anschließend wird eine 30-minütige Haltezeit bei 70 °C durchgeführt, die
gefolgt ist von einer kontrollierten Abkühlphase von 70 °C auf 20 °C mit einer Abkühlrate,
analog zur Aufheizrate, von 1K/min. Das Temperaturprofil 2 beinhaltet eine kontrollierte
Aufwärmphase von 20 °C auf 60 °C mit einer Heizrate von 1K/min gefolgt von einer 20
minütigen Haltezeit bei 60 °C. Anschließenden wurde eine kontrollierten Abkühlung auf
20 °C mit 1K/min, wie in Temperaturprofil 1, gefahren. Die Temperaturprofile 1 und 2 sind
in den Tabelle A.20 und Tabelle A.21 im Tabellenanhang genauer beschrieben.
Als Methode zur Aufnahme des Drehmoments und Bestimmung der strukturbildenden Cha-
rakteristika wurde dabei die Oszillationsrheologie mit einer Frequenz von f = 1 Hz und einer
0 5 10 15 20 25 30 35 40 45 50
0
1
2
3
4
dG'/d
t [P
a/m
in]
Zeit [min]
CSTIST
end
GP
0
200
400
600
800
1000
1200
1400
Spe
iche
rmod
ul [P
a]
20 25 30 35 40 45 50 55 60 65 70
X
Temperatur [°C]
Material und Methoden
47
Scherbelastung von γ = 0,001 gewählt. Alle Versuche wurden in Doppelbestimmung
durchgeführt.
Wie in der Literatur beschrieben, kann die Zugabe von Zitronensäure zu MC zu einer Ver-
besserung der Barriereeigenschaften führen (Ayranci & Tunc, 2003). Um den Einfluss einer
Zitronensäurezugabe auch auf die strukturbildenden Charakteristika zu untersuchen wur-
den Oszillationsversuche mit unterschiedlichen Temperaturprofilen durchgeführt. Die Fre-
quenz wurde hierbei ebenso bei f = 1 Hz und die Scherbelastung bei γ = 0,001 eingestellt.
Die MC-Lösungen und ihre Zusammensetzung, die in diesem Versuch verwendet wurden,
sind in Tabelle 4.9 dargestellt. Alle Versuche wurden ebenfalls in Doppelbestimmung und
unter Verwendung des Temperaturprofils 1 durchgeführt.
Tabelle 4.9: Verwendete Methylzellulose- und Zitronensäurekonzentrationen zur Ermittlung der
strukturbildenden Charakteristika
Methylzellulose Konz. [%] ZS [%] Anteil ZS bzgl. MC-Einwaage [%]
MC383 3 0,15 5
3 0,3 10
MC15
5 0,25 5
5 0,5 10
5 2 40
6,5 0,65 10
Die Zugabe von Ethanol in die wässrige Phase einer Coatinglösung hat einen Einfluss auf
das Verdunstungsverhalten und damit auf das Filmbildungsverhalten der Coatinglösung.
Um einen Einfluss dieses Lösungsmittels auf die strukturbildenden Charakteristika der MC-
Lösungen zu untersuchen, wurden zwei Lösungen mit einem Ethanol-Wasser-Gemisch
hergestellt. Der Anteil an Ethanol betrug dabei 5 % (v/v) in der wässrigen Phase. Unter-
sucht wurden MC 383 mit 5 % (w/w) und MC 15 mit 10 % (w/w) in Doppelbestimmung. Die
Proben durchliefen anschließend das Temperaturprofil 2. Die Haltezeit wurde auf
20 Minuten und die Temperatur auf 60 °C begrenzt, um einer übermäßigen Verdunstung
des Ethanols entgegenzuwirken. Das Temperaturprofil ist in Tabelle A.21 im Anhang
ausführlich dargestellt. Die Oszillationsparameter wurden auch mit einer Frequenz von
f = 1 Hz und einer Scherbelastung von γ = 0,001 gewählt.

Material und Methoden
48
4.2.3 Charakterisierung des Strukturwiederaufbaus von MC-Lösungen nach Scherbeanspruchung
Um den Strukturwiederaufbau der Coatinglösungen nach einer Scherbelastung zu untersu-
chen, wurden Sprungversuche im Rotationsmodus durchgeführt. Dabei simuliert eine Vor-
scherung bei geringer Scherbelastung das Ruheverhalten der Lösung und ist notwendig,
um eine konstante Ausgangsviskosität zu bestimmen. Die Moleküle in den strukturviskosen
Lösungen richten sich zunächst nach der Fließrichtung aus, wobei die Viskosität abnimmt
und im Idealfall schließlich auf einem konstanten Level verbleibt. In der Belastungsphase
erfolgt ein Strukturabbau bei kurzzeitiger, hoher Scherbelastung. In der dritten Phase kann
ein Strukturwiederaufbau bei identischer Scherbelastung wie in der 1. Phase beobachtet
werden. Der schematische Ablauf dieser Versuche ist in Abbildung 4.4 dargestellt.
Abbildung 4.4: Schematischer Ablauf der Versuche zur Bestimmung des Strukturwiederaufbaus der
MC15- und MC383-Lösungen
Die Untersuchungen hierfür wurden bei 20, 30 und 40 °C in Dreifachbestimmung durchge-
führt. Für diese Untersuchungen wurden die gleichen Konzentrationen für die MC-
Lösungen verwendet, wie sie bereits in Kapitel 4.2.1 eingeführt wurden.
Die Parameter, nach denen die Sprungversuche durchgeführt wurden, sind in Tabelle 4.10
dargestellt.
Sch
erge
schw
indi
gkei
t [s-
1 ]
5
1000
Visk
ositä
t [m
Pa·
s] unbelastet
regeneriert
belastet
1.Phase 2. Phase 3. Phase
Strukturwiederaufbau

Material und Methoden
49
Tabelle 4.10: Parameter zur Durchführung der Sprungversuche zur Bestimmung des thixotropen
Verhaltens von MC-Lösungen
1. Phase 2. Phase 3. Phase
Bezeichnung Vorscherung Belastung Strukturwiederaufbau
Schergeschwindigkeit
�̇� [s-1]
5 1000 5
Dauer [s] 300 3 180
Messpunkte 60 3 900
Messpunktdauer [s] 5 1 0,2

Material und Methoden
50
4.3 Charakterisierung der Funktionalität von MC-Coatings als Sau-erstoff- und Wasserdampfbarriere
Für die folgenden Untersuchungen wurde eine Emulsion aus Maltodextrin, OSA-Stärke,
Wasser und Rapsöl hergestellt, die anschließend sprühgetrocknet und aufagglomeriert
wurde. Als Coating wurde hierbei das im Kapitel 4.2 charakterisierte MC 15 verwendet.
Im Folgenden werden die Herstellung von Primärpartikeln, Agglomeraten zum Coaten in
der Wirbelschicht und die Herstellung gecoateter Partikel für die Charakterisierung der
Sauerstoff- und Wasserdampfbarriereeigenschaften von MC-basierten Filmen beschrieben.
Weiterhin werden die chemischen und physikalischen Methoden zur Analyse der Primär-
partikel, der unbehandelten bzw. ungecoateten Agglomerate und der behandelten bzw.
gecoateten Agglomerate beschrieben.
4.3.1 Herstellung der Primärpartikel, Agglomerate und gecoateten Ag-glomerate
Die Herstellung der gecoateten Partikel kann in folgende Arbeitsschritte unterteilt werden:
Emulsionsherstellung mittels Hochdruckhomogenisator
Herstellung von Primärpartikeln mittels Sprühtrocknung
Aufagglomerieren der Primärpartikel zu fließfähigen Agglomeraten
Coaten der Agglomerate mittels Wirbelschichtcoater
Für einige der Untersuchungen, die im folgenden Kapitel beschrieben werden, wurden zu-
sätzlich zu den Primärpartikeln auch Matrixpartikel hergestellt. Diese wurden in der Rezep-
tur im Gegensatz zu den Primärpartikeln ohne Ölbeladung hergestellt. Bei diesen wurde
der Wasseranteil um genau den Prozentsatz erhöht, den bei den ölbeladenen Primärparti-
keln das Öl einnahm. Die Rezeptur für 100 g Dispersionsansatz zur Herstellung der Mat-
rixpartikel ist der Tabelle 4.11 zu entnehmen. Die Herstellung der Matrixpartikel verlief ana-
log zur Herstellung der Primärpartikel, mit der Ausnahme, dass der flüssige Ansatz nicht
homogenisiert wurde. Die Trocknung der Dispersion erfolgte analog zur Trocknung der
Primärpartikel-Emulsion und auch der Schritt der Herstellung von Agglomeraten wurde
analog zur Herstellung von Agglomeraten der Primärpartikel durchgeführt.
Die Ansätze für die Versuche waren meist deutlich höher, können jedoch problemlos hoch-
gerechnet werden.

Material und Methoden
51
Tabelle 4.11: Einsatzmengen [g] für einen Ansatz von 100 g Emulsion bzw. Dispersion für die Her-
stellung von Primär- bzw. Matrixpartikeln
Primärpartikel Matrixpartikel
dest. H2O 52,5 62,5
Maltodextrin 35,0 35,0
Rapsöl 10,0 0
OSA-Stärke 2,5 2,5
Für die Herstellung von 200 g gecoateten bzw. behandelten Agglomeraten musst ein
Emulsions- bzw. Dispersionsansatz mit einer Trockenmasse von 628,1 g hergestellt wer-
den. Dies bedeutet bezugnehmend auf Tabelle 4.11, dass für jede hergestellte Probe ein
Ansatz 973,6 g Lösung entsprechend der Rezeptur angesetzt werden musste.
Für die Herstellung der Emulsion wurde zunächst Maltodextrin für 16h in dem gesamten in
der Rezeptur vorgeschriebenen destillierten Wasser gelöst. Dies war nötig, um eine voll-
ständige Lösung des Maltodextrins zu gewährleisten. Im Anschluss wurde die entspre-
chende Menge OSA-Stärke zur Maltodextrinlösung hinzugegeben und für 2 Stunden in die
Maltodextrinlösung eingerührt. Im Anschluss daran wurde unter Rühren der Maltodext-
rin/OSA-Stärke-Lösung das Rapsöl (Zusammensetzung siehe Tabelle A.13) hinzugege-
ben. Der fertige Ansatz wurde mittels Ultra Turrax für eine Minute bei 13500 U/min vor-
emulgiert. Für die Herstellung der Emulsion wurde ein Hochdruckhomogenisator (Pan-
daPlus 1000, GEA Niro Soavi AG, Lübeck, Deutschland) mit zwei Druckstufen verwendet.
Die Druckstufen wurden auf 300 bzw. 50 bar eingestellt und zwei Durchläufe vorgenom-
men.
Zur Herstellung der Primärpartikel für die Weiterverarbeitung zu Agglomeraten wurde die
Emulsion, um diese später in der Wirbelschicht zu coaten, sprühgetrocknet. Hierzu wurde
ein Mobile Minor der Firma GEA Niro AG, Soeberg, Dänemark verwendet. Die Eingangs-
temperatur wurde auf 180 °C und die Gebläseleistung auf 80 % festgelegt. Über den Feed
von ca. 2,4 L/h wurde die Ausgangstemperatur des Turms auf ca. 70 °C eingestellt. Zur
Zerstäubung wurde eine Zweistoffdüse verwendet, an die ein Zerstäubungsdruck von
1,9 bar angelegt wurde.
Für eine bessere Fluidisierbarkeit der Partikel wurden die Primärpartikel mit Hilfe eines
Trommelcoaters (AR401, Erweka GmbH, Heusenstamm, Deutschland) aufagglomeriert.
Hierzu wurden jeweils 100 g Primärpartikel in den Trommelcoater gefüllt, unter mittelgroßer

Material und Methoden
52
Drehzahl fluidisiert und mit einer Sprühflasche intervallweise destilliertes Wasser zugege-
ben. Mittels eines Heißluftgebläses wurde die Feuchtigkeit aus dem Prozess abgeführt. Die
Primärpartikel wurden auf eine Größe von 0,5 - 1 mm aufagglomeriert und lagen somit von
der Partikelgröße her im Bereich der Klasse A bzw. B (Geldart, 1973). Die Größenklassifi-
zierung wurde mittels Siebkette erreicht. Primärpartikel, die kleiner als 0,5 mm waren, wur-
den dem Agglomerationsprozess erneut zugeführt, um insgesamt eine bessere Ausbeute
zu erreichen. Dennoch traten beim Agglomerieren Verluste von ca. 80 % bezogen auf die
sprühgetrockneten Partikel in der gewünschten Größenklasse auf. Die entstandenen Ag-
glomerate werden im folgenden Verlauf der Arbeit als unbehandelte bzw. ungecoatete Ag-
glomerate bezeichnet.
Das Coaten der Agglomerate fand in einem modifizierten Wirbelschichttrockner (Aeroma-
tic-Fielder new Strea-1, GEA Process Engineering Inc., Columbia, Vereinigte Staaten)
statt. In den Trockner wurde über die verschließbaren Seitenöffnungen eine Stativstange
eingeführt, an der eine Düseneinheit mit einer außenmischenden Zweistoffdüse (Multi-
spray, Modell 920, Düsen-Schlick GmbH, Untersiemau/Coburg, Deutschland) angebracht
wurde. Die Düse wurde mit einer Düsensteuereinheit (ebenfalls Düsen-Schlick GmbH, Un-
tersiemau/Coburg, Deutschland) betrieben. Hierbei wurde der Feed auf 18 U/min einge-
stellt und es wurde intervallartig Coatinglösung in das System eingebracht, um ein zu star-
kes Agglomerieren der Partikel zu verhindern. Es wurden in einem 2-minütigen Intervall
jeweils 20 g Coatinglösung in das System gesprüht. Der Zerstäubungsdruck wurde auf
2 bar festgelegt und der Düsenspalt der außenmischenden Zweistoffdüse wurde auf die
Stufe 2 eingestellt. Die Eingangstemperatur wurde je nach Versuch variiert. Ziel war es,
jeweils eine Temperatur von 30 °C bzw. 60 °C im Fließbett zu erreichen. Die Verweilzeit
wurde zwischen 20 Minuten und 60 Minuten variiert. Dadurch konnten verschiedene Auf-
tragsmengen von Coatingmaterial auf die Agglomerate realisiert werden.
Als Coatingmaterial wurde MC 15 in Form einer 5 % (w/w) Lösung verwendet. Die ver-
schiedenen Auftragsmengen und Prozessparameter sind der Tabelle 4.12 zu entnehmen.

Material und Methoden
53
Tabelle 4.12: Auftragsmengen an Methylzellulose und Prozessparameter für das Coaten von Ag-
glomeraten (Versuchsreihe 4)
Probenmasse
[g]
Coatinglösung
[g]
Temperatur
[°C]
Verweilzeit
[min]
MC15-T30-t20 200 100 30 20
MC15-T30-t60 200 200 30 60
MC15-T60-t20 200 100 60 20
MC15-T60-t60 200 200 60 60
Die Agglomerate wurden entweder in der Wirbelschicht mit MC gecoatet oder in der Wir-
belschicht bestimmten Prozessbedingungen ausgesetzt. Die Prozessbedingungen sollten
das Coaten der Agglomerate simulieren. Demzufolge wurden einige Proben mit destillier-
tem Wasser, statt Coatinglösung besprüht und andere Proben wurden nur bei entspre-
chender Prozesszeit und -temperatur in der Wirbelschicht behandelt. Bei den Proben, die
mit destilliertem Wasser besprüht wurden, wurden Auftragsvolumen und Auftragsintervall
des destillierten Wassers genauso wie bei den gecoateten Agglomeraten gewählt.
Tabelle 4.13: Darstellung der Probenbezeichnungen für Versuchsreihe 2 für die Agglomerate, die in
der Wirbelschicht den unterschiedlichen Prozessbedingungen ausgesetzt wurden, mit: „mit“ (mit
H2O), „ohne“ (ohne H2O), den entsprechenden Prozesszeiten (20, 40 und 60 Minuten) und den ent-
sprechenden Produkttemperaturen (60 und 30 °C)
60 °C 30 °C
20 min 40 min 60 min 20 min 40 min 60 min
mit H2O-
T60-t20
H2O-
T60-t40
H2O-
T60-t60
H2O-
T30-t20
H2O-
T30-t40
H2O-
T30-t60
ohne T60-t20 T60-t40 T60-t60 T30-t20 T30-t40 T30-t60
Die in Tabelle 4.13 eingeführte Nomenklatur für die Proben gilt sowohl für die aus Primär-
partikeln als auch für die Proben aus Matrixpartikeln hergestellten Agglomeraten. Für die
Agglomerate aus Matrixpartikeln wird das Kürzel MP der Probenbezeichnung vorangestellt,
für die Proben aus Primärpartikeln das Kürzel PP. Sollte vor einer Probenbezeichnung kei-
nes der Kürzel stehen, so ist die Probe stets aus Primärpartikeln hergestellt.

Material und Methoden
54
Die Herstellung der Primärpartikel, Agglomerate und gecoateten Agglomerate zog sich
meist über drei Tage hin. Die Proben wurden zwischen den einzelnen Herstellungsprozes-
sen in verschlossenen Gefäßen bei -10 °C gelagert.
Um die Einordnung der durchgeführten Untersuchungen zu erleichtern, werden in Tabelle
4.14 die Untersuchungen, die zu den jeweiligen Versuchen durchgeführt wurden, darge-
stellt.
Tabelle 4.14: Durchgeführte Versuchsreihen mit den jeweiligen Untersuchungen
Versuchsreihen 1 2 3 4 5
Trockenmassebestimmung X X X
Messen der Öltropfengröße X X X
Untersuchung der Lipidoxidation (Wochen) X (8) X (10) X (14) X (14) X (10)
Ermitteln der Mikroverkapselungseffizienz X X X
Bestimmung des Fettgehaltes nach Soxhlet X X
Untersuchung der Wasserdampfsorption X
Erstellung von REM-Aufnahmen X X
Charakterisierung der Gasadsorption X
Partikelporositätsmessung mit Hg-Porosimetrie X
Feststoffdichtenmessung mit He-Pyknometrie X X
Oberflächencharakterisierung mittels
dynamischer Dampfsorpionsmessung X
1. Vorversuch zur Applizierbarkeit der Coatingmenge; 2. Analyse des Temperatur- und Zeiteinflus-
ses im Prozess des Wirbelschichtcoatens auf den Verlauf der Lipidoxidation; 3. Analyse der Parti-
keleigenschaften von PP- und MP-Agglomeraten nach Durchführung des Prozesses mit verschiede-
nen Zeit-Temperatur-Kombinationen; 4. Charakterisierung der Eigenschaften von MC-gecoateten
Agglomeraten aus ölbeladenen Primärpartikeln und 5. Charakterisierung der Sauerstoffbarriere von
MC-Filmen auf Modelloberflächen
Nach der Probenherstellung wurden alle Untersuchungen durchgeführt. Zusätzlich wurden
Proben für Lagerversuche vorbereitet, um den Verlauf der Lipidoxidation des in den Ag-
glomeraten verkapselten Rapsöls zu untersuchen. Hierzu wurden Proben der unbehandel-
ten bzw. ungecoateten Agglomerate und der behandelten bzw. gecoateten Agglomerate in
einem verdunkelten Exsikkator eingelagert, in dem die Luftfeuchtigkeit mittels übersättigter

Material und Methoden
55
Magnesiumchlorid-Lösung auf 33% relative Feuchte eingestellt wurde, während die Tem-
peratur auf 20 °C ± 3 °C gehalten wurde. Die Proben wurden in Rollrandgläser gefüllt und
offen im Exsikkator gelagert, wobei jeweils 1,5 g pro Probe je Rollrandglas eingewogen
wurde.
4.3.2 Chemische Analysen der Primärpartikel, Agglomerate, gecoateten Agglomerate
Mittels Extraktion nach Soxhlet kann die Gesamtfettmenge bestimmt werden und somit
unter Berücksichtigung der Trockenmassemessung die fettfreie Trockenmasse bestimmt
werden. Der Unterschied in der fettfreien Trockenmasse zwischen einer ungecoateten und
einer gecoateten Probe kann als Coatingmenge angenommen werden. Der Extraktion
nach Soxhlet zur Gesamtfettbestimmung ging ein Säureaufschluss nach Weibull-Stoldt
voran. Hierzu wurden 5 g Probe in ein 600 mL Becherglas eingewogen. Das Becherglas
wurde mit Siedesteinchen, einem Glasstab zum Rühren und einem Uhrglas als Deckel
versehen. Die Probe wurde mit 100 mL dest. H2O und 50 mL 37%iger HCl versetzt und
anschließend auf einer Heizplatte zum Sieden gebracht. Nach 45-minütigem Sieden wer-
den zur Probe 100 mL destilliertes Wasser hinzugegeben und die Probenflüssigkeit abfil-
triert (Filtrak 488, Durchmesser 20 cm). Die Filter wurden mit destilliertem Wasser bis zur
Chloridfreiheit gespült. Die Chloridfreiheit wurde während des Spülens regelmäßig mittels
Silbernitrattest überprüft. Anschließend wurden die Filterpapiere im Trockenschrank bei
105 °C zwei Stunden getrocknet und in eine Soxhlethülse gegeben. Bis zur Extraktion nach
Sohxlet wurden die Hülsen mit den Filterpapieren in einem Exsikkator aufbewahrt.
Für die Soxhlet-Extraktion wurde zunächst das Leergewicht von Stehkolben (V = 250 mL)
inklusive Siedesteinchen bestimmt. Die Stehrundkolben wurden mit ca. 200 mL Petrolether
gefüllt und anschließend mit dem Sohxlet-Kühler verbunden, in den die Hülse mit dem Fil-
terpapier aus dem Weibull-Stoldt-Aufschluss platziert wurde. Die Temperatur für das Was-
serbad, in dem sich die Stehrundkolben befanden, wurde auf 80 °C und das Wasserbad,
an das die Rückflusskühler angeschlossen waren, auf 20 °C eingestellt. Die Dauer der Ex-
traktion war 3 Stunden, was ungefähr 20-30 Durchläufe des Extraktionsmittels durch die
Probe bedeutet. Nach dem Ende des Extraktionsvorgangs wurde das verbleibende Lö-
sungsmittel aus den Stehrundkolben bei 65 °C, 700 - 900 mbar abgedampft und die
Stehrundkolben bis zur Gewichtskonstanz bei 105 °C im Trockenschrank getrocknet. Die
Extraktion nach Soxhlet wurde ebenfalls in Dreifachbestimmung durchgeführt.

Material und Methoden
56
Mit Hilfe des bestimmten Fettgehaltes und der ermittelten Trockenmasse (siehe Kapitel
4.3.3) konnte der Fettgehalt in der Trockenmasse (Fett i.Tr.Agglomerate) der Agglomerate und
der gecoateten Agglomerate bestimmt werden.
𝐹𝑒𝑡𝑡 𝑖. 𝑇𝑟.𝐴𝑔𝑔𝑙𝑜𝑚𝑒𝑟𝑎𝑡𝑒 =𝐹𝑒𝑡𝑡𝑔𝑒ℎ𝑎𝑙𝑡 [%]𝐴𝑔𝑔𝑙𝑜𝑚𝑒𝑟𝑎𝑡𝑒
𝑇𝑟𝑜𝑐𝑘𝑒𝑛𝑚𝑎𝑠𝑠𝑒 [%]𝐴𝑔𝑔𝑙𝑜𝑚𝑒𝑟𝑎𝑡𝑒 (4.12)
𝐹𝑒𝑡𝑡 𝑖. 𝑇𝑟.𝑔𝑒𝑐𝑜𝑎𝑡𝑒𝑡𝑒 𝐴𝑔𝑔𝑙𝑜𝑚𝑒𝑟𝑎𝑡𝑒 =𝐹𝑒𝑡𝑡𝑔𝑒ℎ𝑎𝑙𝑡 [%]𝑔𝑒𝑐𝑜𝑎𝑡𝑒𝑡𝑒 𝐴𝑔𝑔𝑙𝑜𝑚𝑒𝑟𝑎𝑡𝑒
𝑇𝑟𝑜𝑐𝑘𝑒𝑛𝑚𝑎𝑠𝑠𝑒 [%]𝑔𝑒𝑐𝑜𝑎𝑡𝑒𝑡𝑒 𝐴𝑔𝑔𝑙𝑜𝑚𝑒𝑟𝑎𝑡𝑒 (4.13)
Im Anschluss daran musste für die Agglomerate ein Referenzfaktor (RF) für das Verhältnis
von fettfreier Trockenmasse (FFTM) zu Fettgehalt in der Trockenmasse (Fett i.Tr.) errech-
net werden, wobei sich die fettfreie Trockenmasse aus dem Anteil von Maltodextrin und
OSA-Stärke zusammensetzt.
𝑅𝐹 =𝐹𝐹𝑇𝑀𝐴𝑔𝑔𝑙𝑜𝑚𝑒𝑟𝑎𝑡𝑒
𝐹𝑒𝑡𝑡 𝑖.𝑇𝑟.𝐴𝑔𝑔𝑙𝑜𝑚𝑒𝑟𝑎𝑡𝑒 (4.14)
Mittels des Faktor RF kann nun der Gehalt an fettfreier Trockenmasse für die gecoateten
Agglomerate (FFTMgecoatete Agglomerate) bestimmt werden.
𝐹𝐹𝑇𝑀𝑔𝑒𝑐𝑜𝑎𝑡𝑒𝑡𝑒 𝐴𝑔𝑔𝑙𝑜𝑚𝑒𝑟𝑎𝑡𝑒 = 𝐹𝑒𝑡𝑡 𝑖. 𝑇𝑟.𝑔𝑒𝑐𝑜𝑎𝑡𝑒𝑡𝑒 𝐴𝑔𝑔𝑙𝑜𝑚𝑒𝑟𝑎𝑡𝑒· 𝑅𝐹 (4.15)
Anschließend wird die theoretische Trockenmasse des gecoateten Agglomerats ohne
Coating (TMAgglomerat ohne Coating) berechnet. Hierzu wird die fettfreie Trockenmasse des gecoa-
teten Agglomerates (FFTMgecoatetes Agglomerat) mit dem Fettgehalt in der Trockenmasse des
gecoateten Agglomerates (Fett i.Tr.gecoatete Agglomerate) aufsummiert.
𝑇𝑀𝐴𝑔𝑔𝑙𝑜𝑚𝑒𝑟𝑎𝑡 𝑜ℎ𝑛𝑒 𝐶𝑜𝑎𝑡𝑖𝑛𝑔 = 𝐹𝐹𝑇𝑀𝑔𝑒𝑐𝑜𝑎𝑡𝑒𝑡𝑒𝑠 𝐴𝑔𝑔𝑙𝑜𝑚𝑒𝑟𝑎𝑡 + 𝐹𝑒𝑡𝑡 𝑖. 𝑇𝑟.𝑔𝑒𝑐𝑜𝑎𝑡𝑒𝑡𝑒𝑠 𝐴𝑔𝑔𝑙𝑜𝑚𝑒𝑟𝑎𝑡 (4.16)
Anschließend kann die Trockenmasse des aufgetragenen Coatings (TMCoating) bestimmt
werden:
𝑇𝑀𝐶𝑜𝑎𝑡𝑖𝑛𝑔 = 100 − 𝑇𝑀𝐴𝑔𝑔𝑙𝑜𝑚𝑒𝑟𝑎𝑡 𝑜ℎ𝑛𝑒 𝐶𝑜𝑎𝑡𝑖𝑛𝑔. (4.17)
Die Lipidoxidation (LOX) des Rapsöls in den unbehandelten bzw. ungecoateten Agglome-
raten und den behandelten bzw. gecoateten Agglomeraten wurde mittels Bestimmung der
Hydroperoxidkonzentration ermittelt. Das Untersuchungsintervall auf die Hydroperoxidkon-
zentration wurde für die ersten vier Wochen auf sieben Tage festgelegt. Danach fanden die
Untersuchungen alle zwei Wochen.
Die Hydroperoxidkonzentration wurde mittels der Thiocyanat-Methode nach ISO
3976:2006 (IDF 74:2006) in einer modifizierten Form über die Konzentration von Eisen(III)
in der Probe photometrisch bestimmt. Die für diese Methode benötigten Lösungen und
deren Herstellungsanleitung sind in Tabelle A.23 im Anhang aufgelistet. Der Bestimmung

Material und Methoden
57
geht eine Fettextraktion voraus. Dazu wurden die Proben in Doppelbestimmung in Kul-
turröhrchen in 2,5 mL Leitungswasser redispergiert. Es folgte die Zugabe von je 7 mL
Isooctan/2-Propanol-Gemisch (1:1, v/v). Nachdem die Proben mit einem Reagenzglas-
schüttler durchmischt wurden, wurden diese bei 2000 U/min (666 g) für 2 Minuten mit einer
Sigma 3K12 (Sigma Laborzentrifugen GmbH, Osterode am Harz, Deutschland) zentrifu-
giert. Dem Überstand, der das Öl enthält, wurden 2 x ca. 500-700 µL entnommen und in
zuvor ausgewogene Reagenzgläser mit Schliff überführt. Das Volumen des abpipettierten
Überstandes wurde so variiert, dass er mindestens 10 mg Öl enthielt. Das Lösungsmittel
wurde unter Stickstoff bei 40 °C über 30 Minuten abgedampft. Durch Rückwiegen der Rea-
genzgläser mit dem aus dem Überstand verbliebenem Öl wurde der Ölgehalt für die an-
schließende Untersuchung bestimmt.
Um den Hydroperoxidgehalt des Öls zu bestimmen, wurde das aus dem Überstand ver-
bliebene Öl mit 5 mL 2-Propanol, 50 µL Ammoniumthiocyanatlösung und 50 µL Eisen(II)-
chloridlösung versetzt. Nach dem Whirlen der Proben wurden diese bei 60 °C für
30 Minuten im Wasserbad temperiert und anschließend für 10 Minuten in einem Wasser-
bad mit Eis auf Raumtemperatur abgekühlt. Die Lösungen wurden in Einweg-Küvetten
überführt und die Extinktion bei einer Wellenlänge von 485 nm am Photometer (Helios
Omega UV- VIS, Thermo Fisher Scientific Germany BV & Co KG, Braunschweig Deutsch-
land) gemessen. Die Hydroperoxidkonzentration wird anschließend über die Geradenglei-
chung einer Kalibriergerade bestimmt.
Zum Erstellen der Kalibriergeraden wurden in einer Vierfachbestimmung 10 - 550 µL Kalib-
rierlösung (Eisen(III)-chlorid-Hexahydrat-Lösung) in Reagenzgläser mit Schliff pipettiert und
diese mit 2-Propanol auf 5 mL aufgefüllt (die Zusammensetzung der Kalibrierlösungen ist
in Tabelle A.24 im Anhang aufgeführt). Für den Kalibrierblindwert wurden in einer Zwei-
fachbestimmung 5 mL 2-Propanol in ein Reagenzglas mit Schliff pipettiert. In die so vorbe-
reiteten Proben wurden jeweils 50 µL Ammoniumthiocyanatlösung und 50 µL Eisen(II)-
chloridlösung pipettiert.
Die Hydroperoxidkonzentration des Öls in der Probe wurde wie folgt berechnet:
Hydroperoxidkonzentration in [mmol/kg Öl] = Fe(III)-gehalt (µg)Einwaage (g) ∙55,84
(4.18)

Material und Methoden
58
4.3.3 Physikalische Analysen der Primärpartikel, Agglomerate und gecoa-teten Agglomerate
Mittels Bestimmung des extrahierbaren Ölgehaltes aus Primärpartikeln, unbehandelten
bzw. ungecoateten Agglomeraten und behandelten bzw. gecoateten Agglomeraten kann
die Mikroverkapselungseffizienz bestimmt werden. Hierzu wird die Menge an extrahierba-
rem Öl pro g Pulver als Maß für die Mikroverkapselungseffizienz (MVE) bestimmt. Die Me-
thode wurde von Westergaard (Westergaard, 2004) beschrieben und nach (Serfert et al.,
2009) angepasst.
Als Vorbereitung wurden 100 mL Rundkolben mind. 90 Minuten im Trockenschrank bei
104 °C getrocknet und anschließend zum Abkühlen in einen Exsikkator überführt. Nach-
dem die Rundkolben abgekühlt wurden, wurde das Leergewicht der Rundkolben bestimmt.
Im Anschluss daran wurden 10 g Pulverprobe in eine 100 mL Schottflasche eingewogen.
Der Probe wurden 50 mL Petrolether zugesetzt und die Probe mit dem Petrolether wurde
für 15 Minuten bei 90 U/min geschüttelt. Anschließend wurde das Gemisch filtriert (Filtrak
488, Durchmesser 15 cm, Spezialpapier FILTRAK GmbH, Niederschlag Deutschland) und
es wurden sofort 25 mL des Filtrats in einen zuvor ausgewogenen Rundkolben pipettiert.
Der Petrolether wurde bei 65 °C, 700 mbar mittels eines Rotationsverdampfers (RV 10,
IKA-Werke GmbH & CO. KG, Staufen Deutschland) abgedampft und die Rundkolben wur-
den für 90 Minuten erneut in den Trockenschrank gelegt, um das Lösungsmittel vollständig
zu verdampfen. Im Anschluss daran konnte die Ölmenge über Differenzwägung bestimmt
werden. Die Angabe der Menge extrahierbaren Öls wird in g Öl/g Pulver angegeben. Die
Bestimmung des extrahierbaren Ölgehaltes wurde in Dreifachbestimmung pro Probe
durchgeführt.
Die Öltropfengröße wurde von der Emulsion, den Primärpartikeln, den unbehandelten bzw.
ungecoateten Agglomeraten und den behandelten bzw. gecoateten Agglomeraten be-
stimmt. Von den Primärpartikeln und den Agglomeraten wurden jeweils 3 g resolubilisiert.
Hierbei wurde die Wassermenge der Probe hinzugegeben, die durch die Trocknung im
Sprühturm zuvor entfernt worden war. Zur Bestimmung der Öltropfengröße und deren Ver-
teilung wurde ein Partikelmessgerät mit statischer Laserbeugung (LA 950 Horiba, Retsch
GmbH, Haan, Deutschland) verwendet. Die Wellenlänge der Lichtquelle lag bei
LD = 655 nm und LED = 705 nm. Der Brechungsindex der Probe von 1,53 - 0,00i wurde
zuvor durch ein softwaregestütztes SOP manuell bestimmt. Der Transmissionswertbereich,
der mit dem Einbringen der Probe in das Messsystem erreicht werden muss, wurde auf
98 % - 90 % festgelegt. Der Füllstand der Messzelle wurde auf „mittel“ gestellt und die
Rühr- und Zirkulationsgeschwindigkeit auf die Stufe 7 festgelegt. Mit einer Kolbenhubpipet-

Material und Methoden
59
te (1 - 10 µL) wurde die Probe in die Messzelle eingebracht. Nach dem Anschalten des
Rührwerks und der Umwälzpumpe wurde eine Minute Verteilungszeit eingehalten, bevor
die Messung gestartet wurde. Als Kennwerte für die Partikelgrößenverteilung wurden der
Median D50, der D10/D90 als Maß für die Verteilungsparameter und der Span als Maß für die
Verteilungsbreite ermittelt.
Der Span berechnet sich wie folgt:
Span = D90-D10D50
(4.19)
Die Trockenmasse wurde über die Restfeuchte der Primärpartikel, der unbehandelten bzw.
ungecoateten Agglomerate und der behandelten bzw. gecoateten Agglomerate mittels des
Infrarot-Feuchtebestimmers (Mettler-Toledo GmbH, Giessen, Deutschland) ermittelt. Für
die Dreifachbestimmung wurde jeweils 1 g der Probe gleichmäßig in einer tarierten Alumi-
niumschale verteilt und bis zur Gewichtskonstanz getrocknet. Die Messung wurde beendet,
wenn die Gewichtsdifferenz über eine Zeit von 120 s kleiner als 1 mg war. Die prozentuale
Trockenmasse konnte direkt am Gerät abgelesen werden.
Die Feststoffdichte wurde in einem Heliumpyknometer (AccyPyc 1330, Micromeritics
GmbH, Aachen, Deutschland) gemessen. Dabei wird das Volumen der Prüfsubstanz auf-
grund der Druckänderung des Inertgases Helium in einer Zelle mit veränderbarem Volu-
men gemessen. Die Volumenänderung kann durch Öffnen eines Ventils zu einer zweiten
Kammer erfolgen. Alle Zellenvolumina wurden durch Kalibrierung mit einem Kalibrierstan-
dard (zwei Stahlkugeln, 6,3722 cm3) ermittelt. Zur Bestimmung der Feststoffdichte wurde
der Messzylinder zu 2/3 mit der Probe befüllt und die Masse der Proben bestimmt (Drei-
fachbestimmung). Der Messzylinder wurde anschließend in das Gerät eingesetzt, die Pro-
benmasse zur automatischen Kalkulation der Dichte eingegeben und die Analyse gestartet.
Vor der eigentlichen Messung wurde die Messkammer samt Probe durch mehrfaches Spü-
len mit dem Messgas von Verunreinigungen (z.B. eingeschlossene Luſt) befreit. Anschlie-
ßend erfolgte die vollautomatische Messung des Probenvolumens, pro Einwaage wurden
fünf Messwerte bestimmt. Dafür wurde im ersten Schritt durch das Öffnen eines Ventils
Messgas in die Messkammer (VZell) mit der Probe (VProbe) geleitet. Das Ventil wurde nach
Erreichen des Messdrucks geschlossen. Der Gleichgewichtsdruck p1 wurde bestimmt. Für
die Einstellung der Druckkonstanz wurde festgelegt, dass die Druckänderung innerhalb
einer Minuten nicht größer als 0,005 psi betragen darf. Im zweiten Schritt erfolgte durch
das Öffnen eines zweiten Ventils die Expansion des Gases in die Expansionszelle (VExp)
und somit ein Druckabfall. Der Gleichgewichtsdruck p2 wurde nach dem oben festgelegten
Kriterium der Druckkonstanz aufgenommen.

Material und Methoden
60
Das Volumen der Probe wird wie folgt berechnet:
VProbe (i) = VZellVExp
(p1 (i)p2 (i)
--1)
(4.20)
mit i = 1 bis 5 (entsprechend der Messungen je Einwaage)
Daraus folgt für die Feststoffdichte:
ρ = EV̅Probe
(4.21)
mit E = Einwaage der Probe in g und V̅Probe = Mittelwert aus den Messungen einer Einwaa-
ge in cm³. Die Feststoffdichte einer Probe ergibt sich aus dem Mittelwert der vorgenomme-
nen Dreifachbestimmung.
Zur Visualisierung der Oberfläche wurden die unbehandelten bzw. ungecoateten Agglome-
rate, die behandelten bzw. gecoateten Agglomerate sowie Agglomerate auf Basis der Mat-
rixpartikel mit einem Rasterelektronenmikroskop (S4000, Hitachi Ltd., Tokio, Japan) an der
ZELMI der Technischen Universität Berlin untersucht. Mit einem Sputter (SCD 030,
Oerlikon Balzers Coating AG, Balzers, Liechtenstein) wurden die Proben vor der Untersu-
chung im Rasterelektronenmikroskop mit einer Goldschicht bedampft. Die Bilderfassung
fand online mit einer Kamera (A3 Data S Pentax, Ricoh Imaging Co., Ltd., Tokio, Japan)
statt. Die Beschleunigungsspannung wurde auf 20 kV und der Arbeitsabstand auf ca.
20 mm festgelegt. Die Auswertung erfolgte mit der Software DISS5 und DIPS (point
electronic GmbH, Halle (Saale), Deutschland).
Die Gasadsorptionsanalyse wurde in Kooperation mit der Bundesanstalt für Materialfor-
schung in Berlin (BAM) durchgeführt. Diese Methode ermöglicht die Berechnung der spezi-
fischen Oberfläche von porösen Feststoffen. Die Vortrocknung der Proben fand unter Va-
kuum für 24 Stunden bei 40 °C statt. Die Messung mit dem Edelgas Krypton wurde an ei-
nem Gassorptionsmessgerät (ASAP 2020, Micromeritics GmbH, Aachen, Deutschland)
durchgeführt. Während der Messung wurde die Probenkammer mit Flüssigstickstoff auf
77 K gekühlt. Zur Bestimmung der spezifischen Oberfläche wurde das volumetrisch-
statische Multipoint-Verfahren (BET), benannt nach Brunauer, Emmett und Teller (1938).
Dabei wird die Messkammer mit der darin gelagerten Probe evakuiert und das Messgas
(Adsorbat) in die Messkammer geleitet und adsorbiert an der Oberfläche der Probe. Der in
der Messkammer entstehende Druck wird gegen den Druck vor der Evakuierung ins Ver-
hältnis gesetzt (p/p0). Über diese Druckdifferenz kann die Menge des adsorbierten Mess-
gases bestimmt und die spezifische Oberfläche ermittelt werden (siehe Abbildung 4.5).

Material und Methoden
61
Abbildung 4.5: Volumetrischer Verlauf der Gasadsorption mit Va: spezifisches adsorbiertes Volumen
und p/p0: Relativdruck sowie schematische Darstellung der Messapparatur (nach DIN ISO 9277); mit
1: Probe, 2: Dewargefäß mit Kühlbad, 3: Vakuumpumpe, 4: Manometer, 5: kalibriertes Volumen
(Gasbürette), 6: Sättigungsdruckröhrchen, 7: Adsorptivbehälter und 8: Gasbehälter für die Totvolu-
menbestimmung;
Die Auswertung erfolgt unter Zuhilfenahme eines BET-Diagramms (siehe Abbildung 4.6)
Abbildung 4.6: Darstellung des BET-Diagramms zur Auswertung der spezifischen Oberfläche der
Probe nach DIN ISO 9277; mit 1: Mehrpunkt-BET-Gerade, 2: Einpunkt-BET-Gerade, : experimen-
telle Datenpunkte, +: Datenpunkt für Einpunkt-Berechnung, p/p0: Relativdruck des Adsorptivs, α:
Ordinatenabschnitt, Δx Abszissendifferenz und Δy Ordinatendifferenz

Material und Methoden
62
Die allgemeine BET-Gleichung lautet wie folgt:
𝑝 𝑝0⁄
𝑛𝑎(1−𝑝 𝑝0⁄ )=
1
𝑛𝑚∙𝐶+
𝐶−1
𝑛𝑚∙𝐶∙ 𝑝 𝑝0⁄ (4.22)
Wobei na die adsorbierte Gasmenge, nm die spezifische Monoschichtstoffmenge ist und C
als spezifischer BET-Parameter sowie p/p0 als Ausdruck für den Relativdruck gilt. Für die
Mehrpunktbestimmung können einige Vereinfachungen vorgenommen werden, die aus
dem BET-Diagramm (Abbildung 4.6) entnommen werden können. Der Anstieg (b) und der
Ordinatenabschnitt (a) der Mehrpunktgerade wird entsprechend der allgemeinen Geraden-
gleichung (y = a + b · x) ermittelt. Somit gilt 𝑏 = Δ𝑦 Δ𝑥⁄ = (𝐶 − 1) (𝑛𝑚 · 𝐶)⁄ bzw.
𝑎 = 1 (𝑛𝑚 · 𝐶)⁄ . Mit den nun zur Verfügung stehenden Werten kann die spezifische Mono-
schichtstoffmenge nm ermittelt werden:
𝑛𝑚 =1
𝑎+𝑏 (4.23)
Um aus der spezifischen Monoschichtmenge die auf die Probenmasse bezogene spezifi-
sche Oberfläche zu bestimmen, sind noch die Avogadro-Konstante 𝐿 (mit
𝐿 = 6,022 · 1023 𝑚𝑜𝑙−1) und der molekulare Flächenbedarf von Krypton bei 77K 𝑎𝑚 (mit
𝑎𝑚 = 0,202 𝑛𝑚2) nötig. Die spezifische Oberfläche wird anschließend wie folgt berechnet:
𝑎𝑠 = 𝑛𝑚 · 𝑎𝑚 · 𝐿 (4.24)
oder vereinfacht ausgedrückt:
𝑎𝑠 = 1,22 · 105 · 𝑛𝑚 (4.25)
Der Prüfbereich dieser Methode liegt zwischen 0,1 m2/g und 1400 m2/g.
Die Quecksilber-Intrusionsporosimetrie (ebenfalls in Kooperation mit der BAM) wurde an
einem Gerät der Firma Micromeritics GmbH, Aachen, Deutschland durchgeführt. Die Pro-
ben wurden auch für diese Untersuchung bei 40 °C unter Vakuum für 24 Stunden vorge-
trocknet. Die Probe wurde zu Beginn der Messung evakuiert und das Quecksilber wurde
unter langsamer Druckerhöhung in die Poren gepresst. Der Porenradius (r) wird über die
Washburn-Gleichung (4.18) bestimmt.
𝑝 =−2∙𝛾∙cos 𝜃
𝑟 (4.26)
Hier ist zu erkennen, dass der in der Messzelle aufgebaute Druck (p) umgekehrt proportio-
nal zur Porengröße (r) ist. Weiterhin gehen in die Gleichung die Oberflächenspannung (γ)
des Quecksilbers und dessen Benetzungswinkel (θ) ein (Washburn, 1921).
Mittels dynamischer Dampfsorptionsmethode (DVS) kann die Aufnahme von Gasen, wie
Wasserdampf oder Dampf von Lösemitteln untersucht werden. Diese Methode wurde

Material und Methoden
63
ebenfalls mit Unterstützung der BAM durchgeführt. Hierbei wird die Probe zunächst bis zur
Massenkonstanz getrocknet. Anschließend werden die unterschiedlichen Gehalte an Sorp-
tionsmittel (Wasserdampf oder n-Oktan in diesem Fall) über einen Volumenmischer einge-
stellt. Hierbei wird der Volumenstrom des gesättigten Sorptionsmittels mit trockenem Stick-
stoff bis zum gewünschten Gehalt an Sorptionsmittel gemischt und sowohl in eine Kammer
ohne Probe (Referenzmesskammer) als auch in die Kammer mit der Probe geleitet. Über
die integrierte Mikrowaage können Differenzen von 0,1 µg detektiert werden. Die ge-
wünschten Gehalte an Sorptionsmittel werden solange in der Messzelle aufrecht gehalten,
bis ein Abbruchkriterium greift. Dieses Abbruchkriterium ist in dieser Arbeit bei einem Wert
von dm/dt = 0,002 g/10 min festgelegt worden. Das bedeutet, dass sobald sich die Masse
der Probe über einen Zeitraum von 10 Minuten nicht um mehr als 0,002 g verändert, wird
die nächsthöhere Sorptionsmittelstufe (Adsorptionsuntersuchung) bzw. die nächst niedrige-
re Sorptionsmittelstufe (Desorptionsuntersuchung) eingestellt. Die Masse der Probe wird
hierbei im Intervall von 2 Minuten aufgezeichnet.
Die über die ansteigende und abnehmende Sorptionsmittelkonzentration aufgenommenen
Sorptionsisotherme können auch Aufschluss über die Oberfläche der Probe geben. Hierbei
wird nach der ESW-Methode der Anstieg zwischen zwei nicht linearen Sorptionsmittelkon-
zentrationen ermittelt und das Minimum des chemischen Potentials in diesem Bereich er-
mittelt. Dem liegt die Theorie von Gibbs zugrunde, die besagt, dass sich die Sorption bzw.
Oberflächenbelegungskonzentration Γ aus einem Verhältnis der Oberflächenenergie 𝛾𝑠𝑣
und einer Änderung des chemischen Potentials ∆µ ergibt (J. Adolphs, 2007):
Γ = − (𝑑𝛾𝑠𝑣
𝑑∆µ) (4.27)
Adolphs und Setzer (1996) beschrieben schon zuvor die Größe 𝜙 als excess surface work
(ESW, überschüssige Oberflächenarbeit). Dies lässt sich aus der Gleichung 4.27 ableiten
und kann wie folgt beschrieben werden:
Überschüssige Oberflächenarbeit = Oberflächenenergie + Sorptionsarbeit
oder über:
𝑑𝜙 = Δµ𝑑Γ𝑎𝑑𝑠 + Γ𝑎𝑑𝑠𝑑(Δ𝜇) (4.28)
Physikalisch bedeutet dies, dass jedes Molekül, das auf der Oberfläche adsorbiert, zu-
gleich die Oberflächenspannung verringert und die Sorptionsarbeit erhöht (J. Adolphs,
2007). Liegen diese beiden Prozesse im Gleichgewicht zu einander, zeigt das chemische
Potential ein Minimum. Vor dem Gleichgewicht überwiegt die Oberflächenenergie der un-
belegten Probenoberfläche. Mit Erreichen eines Monolayers an Adsorbat auf der Oberflä-
che überwiegen die Wechselwirkungen des Adsorbats und das chemische Potential steigt

Material und Methoden
64
wieder an. Somit kann die Menge an Adsorbat, die notwendig ist, um einen Monolayer
auszubilden, bestimmt werden. Über die Rückrechnung des molekularen Flächenbedarfes
von n-Oktan (0,646 nm2) und der Avogadro-Konstante 𝐿 lässt sich analog zur BET-
Methode die spezifische Oberfläche bestimmen.
Die statische Wasserdampfsorption wurde von MC15-gecoateten und von ungecoateten
Primärpartikeln in einer Zweifachbestimmung durch das Aufstellen einer Adsorptionsiso-
therme untersucht. Das Adsorptionsverhalten wurde bei Raumtemperatur mit dem Durch-
laufen von fünf Feuchtigkeitsstufen (11 %, 33 %, 53 %, 75 %, 85 %) getestet. Die Vor-
trocknung der Proben erfolgte in einem Exsikkator über stark hygroskopischem Phosphor-
pentoxid (P2O5). Pro Probe wurden ca. 1 g Partikel in Petrischalen eingewogen und diese
in dem Exsikkator platziert. Dazu wurden die Petrischalen aus dem Exsikkator entnommen
und die Proben rückgewogen.
Für die Untersuchung der Wasserdampfsorption wurde von jeder vorgetrockneten Probe
jeweils 0,3 g in zwei vorgetrocknete Petrischalen eingewogen. Die Proben wurden in zwei
Exsikkatoren gleicher Geometrie platziert, die mit einer übersättigten Lithiumchloridlösung
auf einen relativen Feuchtegehalt von 11% eingestellt waren. Die relative Luftfeuchte in
den Exsikkatoren wurde vor dem Befüllen mit den Proben mittels eines Feuchtemessgeräts
überprüft. Die Proben wurden jeden Tag bis zur Gewichtskonstanz rückgewogen. Es folgte
das Überführen der Proben in zwei Exsikkatoren, die mit Magnesiumchlorid auf eine relati-
ve Luftfeuchte von 33 % eingestellt waren. Die Proben wurden ebenfalls jeden Tag bis zur
Gewichtskonstanz rückgewogen. Für die übrigen Feuchtigkeitsstufen wurde genauso ver-
fahren. Die Konzentrationen der Salze zur Herstellung der übersättigten Salzlösungen fin-
den sich in Tabelle A.25 im Anhang.

Ergebnisse
65
5 Ergebnisse
Die Ergebnisse zur Charakterisierung der Modelloberflächen und der Partikel-Flüssigkeits-
Interaktion beschränken sich in diesem Kapitel auf die emulsionsbasierte Modelloberfläche
(MDEMUL). Die Ergebnisse zur Charakterisierung der Modelloberflächen und der Partikel-
Flüssigkeit-Interaktion für die maltodextrinbasierte kompaktierte Tablette (MDT) und die
maltodextrinbasierte Modelloberfläche mit OSA-Stärke (MDOSA) befinden sich im Tabel-
lenanhang (Tabelle A.1-Tabelle A.10). Die Ergebnisse zur Partikel-Flüssigkeits-Interaktion
für MDOSA waren sehr ähnlich zu denen der MDEMUL-Oberfläche und werden in diesem
Teil der Arbeit nicht vorgestellt. Die MDT-Oberfläche entspricht der Oberfläche eines
sprühgetrockneten Partikels nicht so sehr wie die Oberfläche von MDEMUL und wurde in
erster Linie vermessen, um die Tauglichkeit der Kontaktwinkelmessung auf einem hetero-
genen System zu überprüfen. Da sich diese Arbeit nicht mit dem Coaten von Tabletten
beschäftigen soll, sondern Einblicke in das Coaten von sprühgetrockneten und aufagglo-
merierten Partikeln liefern soll, werden die Ergebnisse der Partikel-Flüssigkeits-Interaktion
für die MDT-Oberfläche nicht vorgestellt. Die Ergebnisse und deren Interpretation der Par-
tikel-Flüssigkeits-Interaktion für die maltodextrinbasierte Modelloberfläche (MDGSS) sind
der vorab veröffentlichen Studie von Kape et al. (2016) zu entnehmen.
Im zweiten Teil dieses Kapitels werden die Ergebnisse zur rheologischen Charakterisie-
rung von zwei MC-Coatingmaterialien vorgestellt. Beide Materialien unterscheiden sich im
Molekulargewicht, nicht jedoch im Methylierungsgrad.
Abschließend werden die Ergebnisse zum Einfluss der Prozessparameter, wie Prozess-
temperatur und –zeit auf die Partikelstruktur gezeigt. Darüber hinaus werden die Ergebnis-
se hinsichtlich der Barriereeigenschaften eines MC-Coatings gegenüber Sauerstoff und
Wasserdampf dargestellt.
5.1 Charakterisierung des Haftungs- und Benetzungsverhaltens von Zellulosederivatlösungen auf MDEMUL
Die Methodenoptimierung für die Kontaktwinkelmessungen wurde zuvor auf Objektträgern
durchgeführt. Die Ergebnisse dieser Methodenoptimierung sind im Folgenden zusammen-
gefasst.
Bei der Bestimmung des Kontaktwinkels der Referenzlösungen auf Deckglas konnte fest-
gestellt werden, dass es erhebliche Unterschiede zwischen den Ergebnissen für den Kon-
taktwinkel gab. Diese Unterschiede konnten unter anderem auf die Verwendung der auto-
matischen Basisliniendetektion zurückgeführt werden.

Ergebnisse
66
Wie in Abbildung 5.1 exemplarisch gezeigt, war die Spannbreite der Werte der Kontaktwin-
kels teilweise sehr groß. Diese Fehlmessungen konnten im optischen Messfenster der
OCA-20-Software nachvollzogen werden. Über die automatische Basisliniendetektion wur-
de die Oberfläche des Deckglases nicht immer einwandfrei erkannt und daher häufig auch
nicht auf die Kontur der Deckglasoberfläche gelegt. Dies führte dazu, dass im weiteren
Verlauf der Arbeit die Basislinie manuell zu Beginn der Messung gelegt wurde und diese
während einer Messung nicht mehr verändert wurde (vgl. Abbildung 5.1).
Abbildung 5.1: Exemplarischer Vergleich zweier Kontaktwinkelmessungen von Diiodmethan mit ei-
nem Volumen von 2,5 µL auf Glas mit und ohne Basisliniendetektion (BDL), dargestellt sind die ge-
mittelten Kontaktwinkel, die über eine Messdauer von einer Minute aufgenommen wurden
Bei der Erprobung unterschiedlicher Volumina des gemessenen Tropfens der einzelnen
Referenzlösungen stellte sich heraus, dass für die unterschiedlichen Referenzlösungen die
Dosierung unterschiedlicher Tropfenvolumina notwendig war. So war es nicht möglich ei-
nen Tropfen Diiodmethan mit einem Tropfenvolumen über 2,5 µL zu erzeugen, da dieser
von der Nadel abriss. Auf der anderen Seite konnten zu kleine Tropfen von der Kamera
nicht erfasst werden. Ein sehr starkes Heranzoomen wurde nötig, womit die Kontur des
Tropfens jedoch nicht mehr scharf dargestellt werden konnte. Bei den anderen drei Refe-
renzlösungen (destilliertes Wasser, Ethylenglykol und Formamid) war es nicht möglich,
Tropfen von der Größe von 2,5 µL auf der Messoberfläche abzusetzen, da sie zu klein wa-
ren und an der Nadel verblieben. Dies führte dazu, dass für diese drei Referenzlösungen
ein Tropfenvolumen von 5 µL ausgewählt wurde.
45
50
55
60
65
70
mit BLD ohne BLD
Kon
takt
win
kel [
°]

Ergebnisse
67
Auch der Messzeitpunkt der Kontaktwinkelmessung zeigte einen Einfluss auf die Werte der
Kontaktwinkel. Mittels ANOVA konnte festgestellt werden, dass es über 60 s Messdauer zu
keiner signifikanten Volumenänderung des abgesetzten Tropfens kam. Weiterhin war wäh-
rend der Messungen zu beobachten, dass der Tropfen unmittelbar nach dem Absetzen
durch das experimentelle Prozedere (das Herausfahren der Nadel aus dem Tropfen, mit
der dieser abgesetzt wurde) zitterte und es einige Sekunden dauerte, bis sich der Tropfen
in Ruhe befand. Aufgrund des experimentellen Prozedere und der hohen Abweichung der
Kontaktwinkelwerte am Anfang einer Messung (siehe Abbildung 5.2) wurde der Zeitpunkt,
bei dem die Messung des Kontaktwinkels durchgeführt wird, auf 16 s festgelegt. Auch
wenn der Wert nach 20 s in Abbildung 5.2 eine niedrigere Streuung zeigt, so ist das Volu-
men des abgesetzten Tropfens nach 20 s bereits deutlich reduziert, was auf eine Verduns-
tung zurückzuführen ist.
Abbildung 5.2: Kontaktwinkel einer MCl Coatinglösung auf MDEMUL über die Messzeit von 20 s in
20-fach Wiederholung mit Verteilungsbreite und Median
5.1.1 Charakterisierung der Coatinglösungen
Die Charakterisierung der Coatinglösung umfasst die Messung der Oberflächenspannung
der Coatinglösungen in verschiedener Viskosität an der Luft-Wasser-Phasengrenzfläche
zur Bestimmung der Gesamtoberflächenspannung (SFT). Weiterhin wurde die Oberflä-
35
45
55
65
75
85
95
0 4 8 12 16 20
Kon
takt
win
kel [
°]
Zeit [s]

Ergebnisse
68
chenspannung an der Dodecan-Wasser-Grenzfläche (SFTDodecan) bestimmt, um die disper-
sen (SFTd) und polaren (SFTp) Anteile der Oberflächenspannung zu ermitteln.
In Bezug auf die Oberflächenspannung sind die Werte für die unterschiedlichen Coating-
materialien normal verteilt, so dass eine ANOVA zulässig ist und die Unterschiede in den
Mittelwerten auch mit dem Scheffé-Post-Hoc-Test eingehender untersucht werden können.
Dabei lassen sich bei der SFT der Coatinglösungen signifikante Unterschiede zwischen
allen Coatingmaterialien feststellen (siehe Tabelle 5.1).
Bezüglich der Viskosität zeigt sich kein signifikanter Einfluss der unterschiedlichen Viskosi-
tätsstufen und damit der Konzentrationen auf SFTDodecan oder SFTd bzw. SFTp (siehe Ta-
belle 5.1). Für SFTDodecan zeigten die Werte zwischen den Coatingmaterialien signifikante
Unterschiede, mit Ausnahme der Kombination MC-HPMC. Eine Normalverteilung lag auch
hier laut Shapiro-Wilk-Test vor. Bezüglich des polaren Anteils der Oberflächenspannung
der Coatinglösungen unterscheiden sich lediglich die Werte von Nutrateric ® und HPMC
nicht signifikant voneinander. Die Werte des dispersen Anteils der Oberflächenspannung
zwischen CMC und Nutrateric® zeigten keine signifikanten Unterschiede, wohingegen an-
dere Kombinationen aus Coatingmaterialien signifikante Unterschiede zeigen.

Ergebnisse
69
Tabelle 5.1: SFT, SFTDodecan, SFTp und SFTd der Coatinglösungen in verschiedenen Viskositätsstu-
fen
SFT
[mN/m]
SFTDodecan
[mN/m]
SFTp
[mN/m]
SFTd
[mN/m]
CMCvl 74,2 ± 0,1a 46,7 ± 0,3a 46,6a 27,6a
CMCl 73,9 ± 0,5a 46,8 ± 0,2a 46,8a 27,1a
CMCm 72,8 ± 0,1a 43,6 ± 0,2a 43,4a 29,4a
CMCh 74,0 ± 0,4a 47,3 ± 1,1a 47,3a 26,7a
MCvl 50,3 ± 0,4b 17,4 ± 0,3b 16,9b 33,5b
MCl 50,3 ± 0,9b 17,1 ± 0,2b 16,5b 33,8b
MCm 49,8 ± 0,5b 16,7 ± 0,2b 16,1b 33,7b
MCh 50,0 ± 0,2b 16,5 ± 0,1b 15,9b 34,1b
Nvl 34,6 ± 0,1c 8,3 ± 0,1c 8,3c 26,3a
Nl 33,0 ± 0,3c 5,4 ± 0,1c 5,3c 27,7a
Nm 31,7 ± 0,2c 4,0 ± 0,0c 3,9c 27,8a
Nh 31,0 ± 0,5c 3,4 ± 0,4c 3,4c 27,6a
HPMCvl 59,4 ± 0,7d 15,2 ± 0,1b 10,0c 49,3c
HPMCl 58,4 ± 1,5d 14,2 ± 0,0b 8,1c 50,2c
HPMCm 57,0 ± 1,5d 13,8 ± 0,1b 9,7c 47,4c
HPMCh 57,5 ± 1,4d 13,1 ± 0,0b 7,9c 49,6c
a, b, c, d unterschiedliche Buchstaben kennzeichnen signifikante Unterschiede zwischen unter-
schiedlichen Coatingmaterialien

Ergebnisse
70
5.1.2 Charakterisierung der maltodextrinbasierten Modelloberflächen
Die Charakterisierung der MDEMUL beinhaltet die Vermessung des Kontaktwinkels der
vier verschiedenen Referenzlösungen auf der MDEMUL. Aus diesen Kontaktwinkeln kön-
nen die polaren (SEp) und dispersen (SEd) Anteile der freien Oberflächenenergie (SE) und
durch aufsummieren der beiden Anteile die SE selbst bestimmt werden.
Die Ergebnisse zur Berechnung der SE der MDEMUL-Oberfläche sind in Tabelle 5.2 dar-
gestellt. Unter Verwendung von allen vier Referenzlösungen wurde eine SE von
58,1 mN/m mit SEp = 28,1 mN/m und SEd = 30,0 mN/m berechnet. Eine Kombination aus
W, E und F für die Referenzlösungen führte zum höchsten Wert für die SE, wobei SEd für
diese Kombination aus Referenzlösungen zu einem deutlich niedrigerem Wert berechnet
wurde, als bei allen anderen Kombinationen von Referenzlösungen.
Abbildung 5.3: SE-Plot zur Bestimmung der Oberflächenenergie von MDEMUL mit den Kombination
von Referenzlösungen WEF, WDEF, WDE, WDF und DEF und den daraus resultierenden Aus-
gleichgeraden mit den Datenpunkten für D, E, F und W (von links)
Die DEF-Kombination aus Referenzlösungen führte zum niedrigsten Ergebnis für die SE.
Zwischen den Kombinationen für Referenzlösungen WDEF, WDE, und WDF zeigte sich
kein deutlicher Unterschied zwischen den berechneten Werten für SE und SEp. Für die
Berechnung von SEd jedoch weicht das Ergebnis für die Kombination WDF im Gegensatz
zu WDEF und WDE deutlich ab. Um dies zu verdeutlichen, sind in Abbildung 5.3 die Aus-
gleichsgeraden zur Bestimmung der SE unter Verwendung verschiedener Referenzlösun-
gen dargestellt. Hier ist zu erkennen, dass die Anstiege und die Achsenabschnitte teils
erheblich variieren, was zu den Unterschieden in der berechneten SE führt.
0
5
10
15
20
25
0 0,5 1 1,5 2 2,5 3
1/2(
1+co
s(θ)
) ·(S
FT/s
qrt(S
FTd)
)
sqrt(SFTp/SFTd)
WEF
WDEF
WDE
WDF
DEF

Ergebnisse
71
Tabelle 5.2: SE mit SEd und SEp, ihren prozentualen Anteilen und dem Bestimmtheitsmaß für die
Kalkulation von SE der MDEMUL-Oberfläche unter Verwendung verschiedener Kombinationen von
Referenzlösungen
WEF WDEF WDE WDF DEF
SE [mN/m] 65,0 58,1 59,5 62,1 51,9
SEd [mN/m] 7,7 30,0 30,6 35,1 35,7
SEd [% SE] 11,8 51,7 51,5 56,5 68,8
SEp [mN/m] 57,4 28,1 28,9 27,0 16,2
SEp [% SE] 88,2 48,3 48,5 43,5 31,1
R2 1,0 0,94 0,94 0,98 0,93
5.1.3 Charakterisierung der Partikel-Flüssigkeit-Interaktion
Für die Charakterisierung der Partikel-Flüssigkeits-Interaktion werden im Folgenden die
Ergebnisse für die Berechnung der Adhäsionsarbeit (WA) unter Berücksichtigung von ver-
schiedenen Kombinationen aus Referenzlösungen dargestellt. Weiterhin sind die Ergeb-
nisse für den CAOWRK für verschiedene Kombinationen aus Referenzlösungen für zwei
Coatinglösungen in vier Viskositätsstufen nachzuvollziehen. Ebenso wurde der direkt ge-
messene Kontaktwinkel (CAm) der Coatinglösungen in verschiedener Viskosität auf der
MDEMUL-Oberfläche bestimmt.
Bezüglich der Auswahl von Referenzlösungen bzw. der Auswahl des verwendeten Modells
zur Bestimmung von WA zeigte sich, dass es keinen signifikanten Unterschied zwischen
der WEF-Kombination (OWRK) an Referenzlösungen und der Verwendung des YD-
Modells gab (siehe Tabelle 5.3). Für alle anderen Kombinationen aus Referenzlösungen
und der Verwendung des YD-Modells ergab sich ein signifikanter Unterschied zwischen
den Werten für die WA zwischen der Coatinglösung und MDEMUL. Innerhalb der Kombina-
tionen aus Referenzlösungen zur Bestimmung der Adhäsionsarbeit zeigten sich mehrere
signifikante Unterschiede. So unterschied sich WA-WDEF signifikant von WA-DEF. Ebenfalls
signifikant verschieden waren die Kombinationen WDF und DEF für die Werte von WA. Die
Werte für WA für die Kombinationen WDEF, WDE und WDF waren nicht signifikant unter-
schiedlich.
Die Werte für WA, unabhängig von der Kombination der Referenzlösungen oder des ver-
wendeten Modells, zeigten keine signifikanten Unterschiede zwischen HPMC und MC. Für

Ergebnisse
72
alle anderen Coatinglösungen unterschieden sich die Werte für WA signifikant voneinander
(siehe Tabelle 5.3).
Tabelle 5.3: WA der verwendeten Coatingmaterialien unter Berücksichtigung der Verwendung von
unterschiedlichen Kombinationen der Referenzlösungen zur Bestimmung der Oberflächenenergie
der MDEMUL-Oberflächen nach OWRK und der WA berechnet nach dem Modell nach YD
WEF WDEF WDE WDF DEF YD
CMCvl 132,5c,A 129,9c,B,C 131,5c,B,C 133,2c,B 117,7c,C 136,1c,A
CMCl 132,4c,A 129,6c,B,C 131,1c,B,C 132,8c,B 117,3c,C 132,5c,A
CMCm 129,8c,A 129,3c,B,C 130,8c,B,C 132,8c,B 117,8c,C 128,0c,A
CMCh 132,8c,A 129,5c,B,C 131,1c,B,C 132,7c,B 117,1c,C 128,9c,A
MCvl 94,2a,A 106,9a,B,C 108,1a,B,C 111,2a,B 102,2a,C 92,7a,A
MCl 93,7a,A 106,8a,B,C 108,0a,B,C 111,1a,B 102,2a,C 91,1a,A
MCm 93,0a,A 106,2a,B,C 107,4a,B,C 110,5a,B 101,7a,C 88,4a,A
MCh 92,7a,A 106,2a,B,C 107,5a,B,C 110,6a,B 101,9a,C 86,4a,A
Nvl 72,1d,A 86,8d,B,C 87,7d,B,C 90,7d,B 84,5d,C 65,4d,A
Nl 64,1d,A 82,1d,B,C 83,0d,B,C 86,3d,B 81,5d,C 61,6d,A
Nm 59,1d,A 78,7d,B,C 79,5d,B,C 83,0d,B 78,9d,C 57,6d,A
Nh 56,9d,A 77,0d,B,C 77,9d,B,C 81,3d,B 77,6d,C 55,0d,A
HPMCvl 86,9a,A 110,6a,B,C 111,8a,B,C 116,2a,B 109,5a,C 109,3a,A
HPMCl 82,5a,A 108,0a,B,C 109,2a,B,C 113,7a,B 107,7a,C 104,8a,A
HPMCm 85,2a,A 108,4a,B,C 109,6a,B,C 113,9a,B 107,3a,C 99,7a,A
HPMCh 81,6a,A 107,0a,B,C 108,2a,B,C 112,7a,B 106,8a,C 98,3a,A
a, b, c, d unterschiedliche Buchstaben kennzeichnen signifikante Unterschiede zwischen unter-
schiedlichen Coatingmaterialien
A, B, C unterschiedliche Buchstaben kennzeichnen signifikante Unterschiede zwischen unterschied-
lichen Kombinationen aus Referenzlösungen bzw. unterschiedlichen Berechnungsmodellen für die
Adhäsionsarbeit
Für die berechneten Kontaktwinkel zwischen Coatinglösungen und MDEMUL nach OWRK
(CAOWRK) in Tabelle 5.4 lag keine Normalverteilung in Bezug auf die Auswahl der Coating-
materialien und der Kombinationen verschiedener Referenzlösungen zu Oberflächenener-

Ergebnisse
73
giecharakterisierung der MDEMUL-Oberflächen vor. Die Werte für CAOWRK in Bezug auf die
Viskosität waren normalverteilt. Die Viskosität und die Auswahl der Referenzlösungen für
die Oberflächenenergiecharakterisierung zeigten keinen signifikanten Einfluss auf CAOWRK
bei einem Signifikanzlevel von 0,01 in der einfaktoriellen ANOVA bzw. der Kruskal-Wallis-
ANOVA. Die Auswahl der Coatingmaterialien zeigte einen signifikanten Einfluss auf den
CAOWRK in der Kruskal-Wallis-ANOVA bei einem Signifikanzlevel von 0,01.
Tabelle 5.4: Nach dem Modell von OWRK berechneter Kontaktwinkel von verschiedenen Coating-
materialien unter Verwendung unterschiedlicher Kombinationen von Referenzlösungen zur Oberflä-
chenenergiecharakterisierung von MDEMUL-Oberflächen
WEF WDEF WDE WDF DEF
CMCvl 38,1 41,3 39,4 37,2 54,0
CMCl 37,6 41,1 39,2 37,1 54,0
CMCm 38,4 39,1 37,2 34,5 51,8
CMCh 37,3 41,3 39,5 37,4 54,3
HPMCvl 62,3 30,4 27,9 16,8 32,4
HPMCl 65,5 31,8 29,5 18,6 32,3
HPMCm 60,4 25,7 22,8 4,1 28,1
HPMCh 65,3 30,7 28,3 16,4 31,0
Um die Ergebnisse der Kontaktwinkelmessung der zellulosederivatbasierten Coatinglösun-
gen auf MDEMUL hinsichtlich des signifikanten Einflusses von Viskosität oder Art des
Coatingmaterials auf den CAm zu untersuchen, musste zunächst ein Shapiro-Wilk-Test auf
Normalverteilung durchgeführt werden. Hiermit konnte festgestellt werden, dass die Werte
für den direkt gemessenen Kontaktwinkel hinsichtlich der Viskosität und der Auswahl des
Coatingmaterials normalverteilt waren. Es konnte nach anschließend durchgeführter
ANOVA kein signifikanter Einfluss der Auswahl der Coatingmaterialien auf den sich ausbil-
denden Kontaktwinkel zwischen Modelloberfläche und Coatinglösung festgestellt werden.
Bezüglich des Einflusses der Viskosität ist zu erkennen, dass die Werte für CAm mit zu-
nehmender Viskosität ansteigen (siehe Abbildung 5.4). Ein signifikanter Anstieg ist jedoch
nur zwischen der niedrigsten Viskositätsstufe und der mittleren bzw. hohen Viskositätsstufe
zu verzeichnen.

Ergebnisse
74
Coatinglösung CAm [°] Coatinglösung CAm [°]
CMCvl 33,3 ± 0,9A MCvl 32,6 ± 1,1A
CMCl 37,4 ± 1,0A,B MCl 35,7 ± 0,9A,B
CMCm 40,7 ± 1,0B MCm 39,2 ± 1,3B
CMCh 42,0 ± 1,1B MCh 43,2 ± 1,3B
Nvl 26,9 ± 0,6A HPMCvl 32,8 ± 0,8A
Nl 29,9 ± 0,7A,B HPMCl 37,2 ± 0,7A,B
Nm 34,9 ± 0,8B HPMCm 41,6 ± 1,0B
Nh 38,9 ± 0,5B HPMCh 44,9 ± 0,8B
Abbildung 5.4: gemessener Kontaktwinkel der Coatinglösungen auf MDEMUL in den vier verschie-
denen Viskositätsstufen (sehr niedrig: vl; niedrig: l; mittel: m und hoch: h) mit A: CMC; B: MC; C:
Nutrateric® und D: HPMC; für die Tabelle: A, B, unterschiedliche Buchstaben kennzeichnen signifi-
kante Unterschiede zwischen verschiedenen Viskositätsstufen
Abbildung 5.5 zeigt den Wetting Envelope, der die Interaktion zwischen den Coatinglösun-
gen und der MDEMUL-Modelloberfläche darstellt. Aufgetragen sind hier die Oberflächen-
spannungen der Coatinglösungen über dem polaren Anteil der Oberflächenspannung. So-
0
10
20
30
40
50
vl l m h
Kon
takt
win
kel [
°]
A
0
10
20
30
40
50
vl l m h
Kon
takt
win
kel [
°]
B
0
10
20
30
40
50
vl l m h
Kon
takt
win
kel [
°]
C
0
10
20
30
40
50
vl l m h
Kon
takt
win
kel [
°]
D
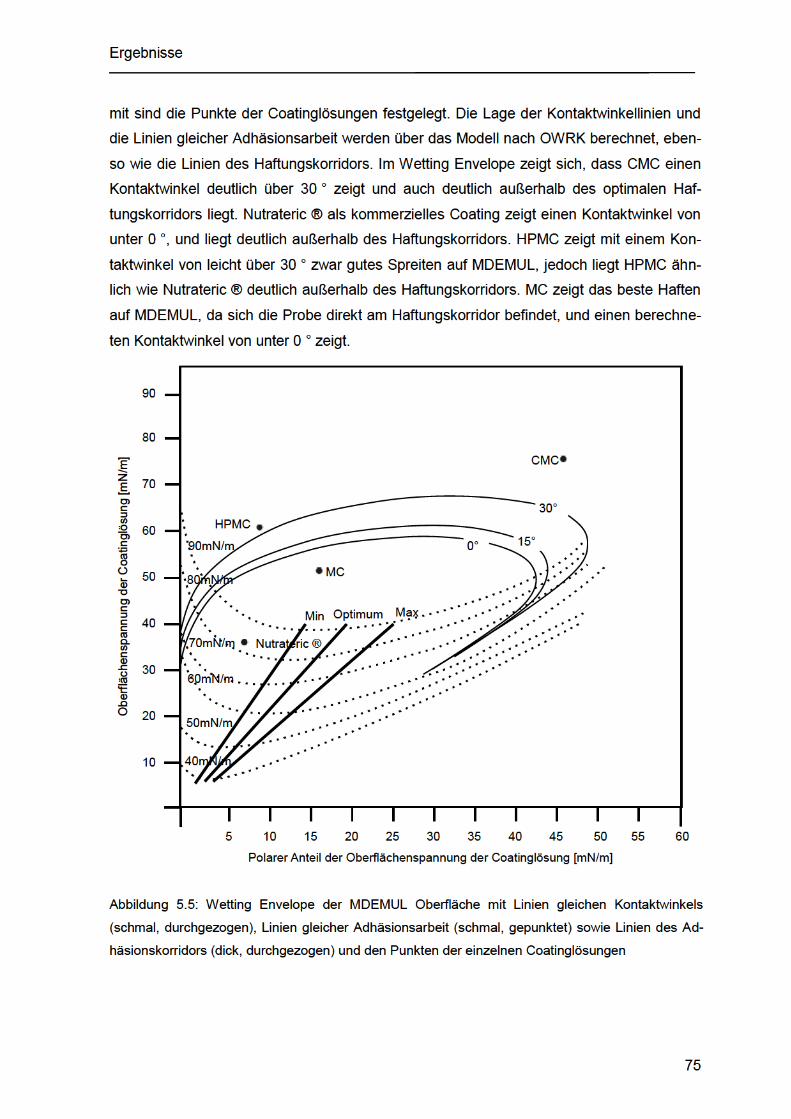
Ergebnisse
75
mit sind die Punkte der Coatinglösungen festgelegt. Die Lage der Kontaktwinkellinien und
die Linien gleicher Adhäsionsarbeit werden über das Modell nach OWRK berechnet, eben-
so wie die Linien des Haftungskorridors. Im Wetting Envelope zeigt sich, dass CMC einen
Kontaktwinkel deutlich über 30 ° zeigt und auch deutlich außerhalb des optimalen Haf-
tungskorridors liegt. Nutrateric ® als kommerzielles Coating zeigt einen Kontaktwinkel von
unter 0 °, und liegt deutlich außerhalb des Haftungskorridors. HPMC zeigt mit einem Kon-
taktwinkel von leicht über 30 ° zwar gutes Spreiten auf MDEMUL, jedoch liegt HPMC ähn-
lich wie Nutrateric ® deutlich außerhalb des Haftungskorridors. MC zeigt das beste Haften
auf MDEMUL, da sich die Probe direkt am Haftungskorridor befindet, und einen berechne-
ten Kontaktwinkel von unter 0 ° zeigt.
Abbildung 5.5: Wetting Envelope der MDEMUL Oberfläche mit Linien gleichen Kontaktwinkels
(schmal, durchgezogen), Linien gleicher Adhäsionsarbeit (schmal, gepunktet) sowie Linien des Ad-
häsionskorridors (dick, durchgezogen) und den Punkten der einzelnen Coatinglösungen
80
70
60
50
40
30
20
10
90
5 10 15 20 25 30 35 40 45 50 55 60
HPMC
MC
CMC
Nutrateric ®
30°
15° 0°
Oberflächenspannung der Coatinglösung [mN/m]
Polarer Anteil der Oberflächenspannung der Coatinglösung [mN/m]
90mN/m
80mN/m
70mN/m
60mN/m
50mN/m
40mN/m
Optimum Max Min

Ergebnisse
76
5.2 Charakterisierung des rheologischen Verhaltens von lebensmit-teltauglichen Zellulosederivaten
Für die rheologischen Untersuchungen wurden ausschließlich MC-Lösungen in den Kon-
zentrationen 1 %, 3 % und 5 % (m/m) für MC383 und 5 %, 6,5 % und 8 % (m/m) für MC15
verwendet. Die Ergebnisse der rheologischen Charakterisierung dieser beiden Materialien
sollen im Folgenden in Abhängigkeit von Temperatur, Scherbelastung, Zugabe von Zitro-
nensäure bzw. Ethanol und der Konzentration dargestellt werden.
5.2.1 Bestimmung der Viskosität von MC-Lösungen in Abhängigkeit von Temperatur, Konzentration und Schergeschwindigkeit
Die Messergebnisse für die Bestimmung der Viskosität in Abhängigkeit der Temperatur,
Konzentration und Scherbeanspruchung sind im Anhang zu finden. Für die Probe MC383
sind die Werte in Tabelle A.14 (1 %ige Lösung), Tabelle A.15 (3 %ige Lösung) und Tabelle
A.16 (5 %ige Lösung) aufgelistet. Für die Probe MC15 befindet sich die Auflistung nach
Konzentrationen in Tabelle A.17 (5 %ige Lösung), in Tabelle A.18 (6,5 %ige Lösung) und in
Tabelle A.19 (8 %ige Lösung). Die beiden MC-Materialien (MC383 und MC15) zeigten ein
sehr ähnliches Verhalten bezüglich der Einflussfaktoren Temperatur, Konzentration und
Schergeschwindigkeit. Beide Proben zeigten mit zunehmender Konzentration eine
Zunahme in der Viskosität. So lagen die Viskositäten für die niedrigste Konzentration, 1 %
(m/m), der MC383-Probe zwischen 4,2 ± 0,9 mPa·s (60 °C und 4000 s-1) und
18,3 ± 1,5 mPa·s (30 °C und 1000 s-1). Für die höchste Konzentration, 5 % (m/m), lagen
die Werte für die Viskosität, abhängig von Temperatur und Scherrate, zwischen
51,6 ± 9,2 mPa·s (60 °C und 4000 s-1) und 922, 4 ± 69,0 mPa·s (30 °C und 1000 s-1). Wie
in Tabelle 5.5 zu erkennen ist, sinkt die Viskosität der MC383-Lösungen mit zunehmender
Temperatur. Ebenso ist ein Viskositätsabfall mit zunehmender Scherbelastung zu erken-
nen, so dass die Probe MC383 mit dem Begriff scherverdünnend beschrieben werden
kann.
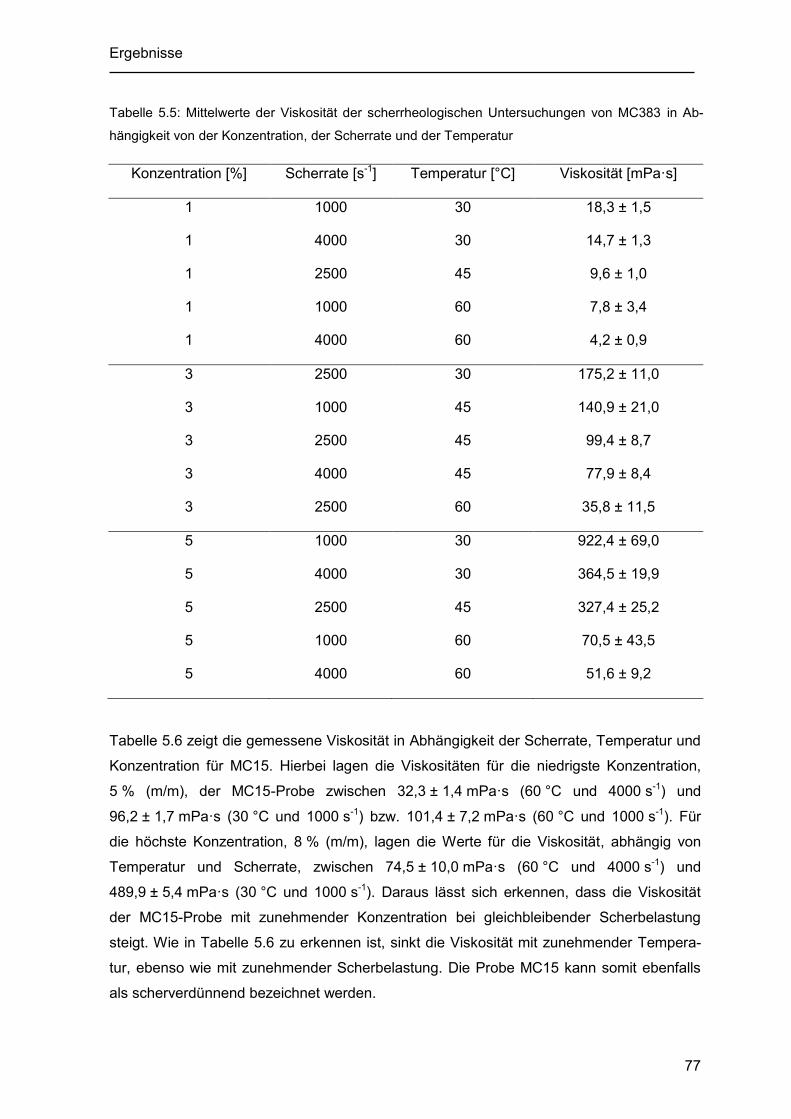
Ergebnisse
77
Tabelle 5.5: Mittelwerte der Viskosität der scherrheologischen Untersuchungen von MC383 in Ab-
hängigkeit von der Konzentration, der Scherrate und der Temperatur
Konzentration [%] Scherrate [s-1] Temperatur [°C] Viskosität [mPa·s]
1 1000 30 18,3 ± 1,5
1 4000 30 14,7 ± 1,3
1 2500 45 9,6 ± 1,0
1 1000 60 7,8 ± 3,4
1 4000 60 4,2 ± 0,9
3 2500 30 175,2 ± 11,0
3 1000 45 140,9 ± 21,0
3 2500 45 99,4 ± 8,7
3 4000 45 77,9 ± 8,4
3 2500 60 35,8 ± 11,5
5 1000 30 922,4 ± 69,0
5 4000 30 364,5 ± 19,9
5 2500 45 327,4 ± 25,2
5 1000 60 70,5 ± 43,5
5 4000 60 51,6 ± 9,2
Tabelle 5.6 zeigt die gemessene Viskosität in Abhängigkeit der Scherrate, Temperatur und
Konzentration für MC15. Hierbei lagen die Viskositäten für die niedrigste Konzentration,
5 % (m/m), der MC15-Probe zwischen 32,3 ± 1,4 mPa·s (60 °C und 4000 s-1) und
96,2 ± 1,7 mPa·s (30 °C und 1000 s-1) bzw. 101,4 ± 7,2 mPa·s (60 °C und 1000 s-1). Für
die höchste Konzentration, 8 % (m/m), lagen die Werte für die Viskosität, abhängig von
Temperatur und Scherrate, zwischen 74,5 ± 10,0 mPa·s (60 °C und 4000 s-1) und
489,9 ± 5,4 mPa·s (30 °C und 1000 s-1). Daraus lässt sich erkennen, dass die Viskosität
der MC15-Probe mit zunehmender Konzentration bei gleichbleibender Scherbelastung
steigt. Wie in Tabelle 5.6 zu erkennen ist, sinkt die Viskosität mit zunehmender Tempera-
tur, ebenso wie mit zunehmender Scherbelastung. Die Probe MC15 kann somit ebenfalls
als scherverdünnend bezeichnet werden.

Ergebnisse
78
Tabelle 5.6: Mittelwerte der Viskosität der scherrheologischen Untersuchungen von MC15 in Abhän-
gigkeit von der Konzentration, der Scherrate und der Temperatur
Konzentration [%] Scherrate [s-1] Temperatur [°C] Viskosität [mPa·s]
5 1000 30 96,2 ± 1,7
5 4000 30 70,5 ± 0,4
5 2500 45 48,0 ± 0,8
5 1000 60 101,4 ± 7,2
5 4000 60 32,3 ± 1,4
6,5 2500 30 173,4 ± 4,0
6,5 1000 45 155,9 ± 4,6
6,5 2500 45 107,8 ± 1,9
6,5 4000 45 89,2 ± 1,3
6,5 2500 60 73,0 ± 9,2
8 1000 30 489,9 ± 5,4
8 4000 30 255,7 ± 0,5
8 2500 45 210,0 ± 2,2
8 1000 60 213,8 ± 22,4
8 4000 60 74,5 ± 10,0

Ergebnisse
79
5.2.2 Bestimmung der strukturbildenden Charakteristika von MC
In Tabelle A.22 im Anhang sind die initiale Strukturierungstemperatur (IST), die Gelpunkt-
temperatur (GP), die kritische Strukturierungstemperatur (CST), die mittlere Strukturent-
wicklungsgeschwindigkeit (SDR), die Gelauflösungstemperatur (GA), die Gelstärke zu Be-
ginn der Haltezeit (t = 0) und die Gelstärke nach 30 Minuten Haltezeit (t = 30) angegeben.
Für die hochviskose MC383 Probe ist zu erkennen, dass die Gelpunkttemperatur mit zu-
nehmender Konzentration in der gleichen Größenordnung bleibt. Auch der Gelauflösungs-
punkt verringert sich nur geringfügig bei einer Konzentration von 5 % gegenüber den bei-
den niedrigeren Konzentrationen der MC383. Die IST hingegen sinkt von 44,9 °C (1 %ige
Lösung) auf 39,8 °C (5 %ige Lösung). Ebenfalls deutlich niedriger zeigt sich die CST der
MC383 - Probe mit zunehmender Konzentration, nämlich 61,6 °C für die 1 %ige Lösung
bzw. 51,1 °C für die 5 %ige Lösung. Die SDR steigt mit zunehmender Konzentration je
Konzentrationsstufe um ca. eine Zehnerpotenz an und liegt für die 1 %ige Lösung bei
0,07 Pa/s bzw. bei 2,29 Pa/s für die 5 %ige Lösung (siehe Abbildung 5.6.
Abbildung 5.6: Darstellung von GP und GA, IST und CST sowie SDR in Abhängigkeit von der Kon-
zentration der MC383
Die Gelstärke, also der Speichermodul, nimmt mit zunehmender Konzentration sowohl am
Anfang als auch am Ende der Haltezeit zu (siehe Tabelle A.22 im Anhang). Für jede Kon-
zentration lagen die Werte für die Gelstärke zu Beginn und am Ende der Haltezeit in ähnli-
chen Größenordnungen (vgl. Abbildung 5.7).
0
0,5
1
1,5
2
2,5
0
10
20
30
40
50
60
70
1,0 3,0 5,0
SDR [Pa/s]
Temperatur [°C]
Konzentration MC383 [%]
IST [°C] GP [°C] CST [°C] GA [°C] SDR [Pa/s]

Ergebnisse
80
Abbildung 5.7: Darstellung des Speichermoduls G’ zu Beginn (t=0) der Haltezeit bei 70 °C und nach
30 Minuten Haltezeit (t=30) von MC383 in verschiedenen Konzentrationen
Im Vergleich zur MC383-Probe sind die Unterschiede bei der MC15-Probe zwischen den
einzelnen Konzentrationsstufen eindeutiger. Die Strukturparameter für die MC15-Probe
sind im Einzelnen in Tabelle A.22 im Anhang zu finden. Die Gelpunkttemperatur der MC15-
Probe nimmt mit zunehmender Konzentration deutlich ab, ähnlich wie die Gelauflösungs-
temperatur (vgl. Abbildung 5.8) und 42,9 °C (8 %ige Lösung) für die Gelpunkttemperatur.
Für die Gelauflösungstemperatur liegen die Werte zwischen 31,4 °C (5 %ige Lösung) und
26,5 °C (8 %ige Lösung).
Abbildung 5.8: Darstellung von GP und GA, IST und CST sowie SDR in Abhängigkeit von der Kon-
zentration der MC15
Die IST sinkt mit zunehmender Konzentration im gleichen Größenumfang wie bei der
MC383-Probe von 38,2 °C für die 5 %ige Lösung auf 34,2 °C für die 8 %ige Lösung. Die
0
1000
2000
3000
4000
5000
6000
1 3 5
Speichermodul [Pa]
Konzentration MC383 [%]
G' (t=0) MC383
G' (t=30) MC383
0
0,5
1
1,5
2
2,5
3
3,5
4
0
10
20
30
40
50
60
5,0 6,5 8,0
SDR [Pa/s]
Temperatur [°C]
Konzentration MC15 [%]
IST [°C] GP [°C] CST [°C] GA [°C] SDR [Pa/s]

Ergebnisse
81
CST verringert sich im Vergleich zur MC383-Probe nur geringfügig und zeigt für die 5 %ige
Lösung einen Wert von 53,5 °C bzw. 50,1 °C für die 8 %ige Lösung. Die SDR steigt von
der 5 %igen Lösung mit 0,72 Pa/s im Vergleich zur 6,5 %igen Lösung (2,17 Pa/s) ver-
gleichsweise stark an, wohingegen der Unterschied zur 8 %igen Lösung (3,40 Pa/s) nicht
mehr so gravierend ausfällt.
Die Gelstärke, also der Speichermodul, steigt mit zunehmender Konzentration in der
MC15-Lösung stark an (siehe auch Tabelle A.22). Auch bei dieser Probe liegt die Gelstär-
ke zu Beginn und am Ende der Haltezeit in vergleichbaren Größenordnungen. Im Gegen-
satz zur MC383 liegt die Gelstärke der Probe mit der niedrigsten Konzentration hier jedoch
schon bei Werten von 1356 Pa (t=0) bzw. 1905 Pa (t=30), welche denen der mittleren Kon-
zentration an MC383 in etwa entsprechen. Die höchsten Werte für die Gelstärke werden
bei der MC15-Probe bei einer Konzentration von 8 % nach der Haltezeit (9574 Pa) gemes-
sen.
Abbildung 5.9: Darstellung des Speichermoduls G’ zu Beginn (t=0) der Haltezeit bei 70 °C und nach
30 Minuten Haltezeit (t=30) von MC15 in verschiedenen Konzentrationen
Einfluss von Zitronensäure auf strukturbildende Charakteristika von MC
In Tabelle 5.7 im Anhang sind die initiale Strukturierungstemperatur (IST), die Gelpunkt-
temperatur (GP), die kritische Strukturierungstemperatur (CST), die mittlere Strukturent-
wicklungsgeschwindigkeit (SDR), die Gelauflösungstemperatur (GA), die Gelstärke zu Be-
ginn der Haltezeit (t=0) und die Gelstärke nach 20 Minuten Haltezeit (t=30) der MC383-
Proben bei verschiedenen Konzentrationen mit Zugabe verschiedener Konzentrationen an
Zitronensäure (ZS) angegeben.
0
2000
4000
6000
8000
10000
12000
5 6,5 8
Spei
cher
mod
ul [P
a]
Konzentration MC15 [%]
G' (t=0) MC15G' (t=30) MC15

Ergebnisse
82
Die Gelbildungstemperatur der MC383-Probe (siehe Tabelle 5.7) wird nur in sehr geringem
Maße durch die Zugabe von 0,15 % bzw. 0,3 % Zitronensäure bezogen auf die MC-
Einwaage beeinflusst. Die Werte liegen hier bei 50,2 °C bzw. 50,4 °C für 0,15 % bzw.
0,3 % Zitronensäurezugabe und zeigen somit keinen Unterschied. Die Gelauflösungstem-
peratur wird durch die Zugaben von Zitronensäure nur leicht angehoben und liegt bei
32,3 °C bzw. 32,7 °C für 0,15 % bzw. 0,3 % Zitronensäurezugabe. Auch die kritische Struk-
turierungstemperatur bleibt von der Zitronensäurezugabe unbeeinflusst. Im Vergleich zur
3 %igen Lösung von MC383 erniedrigt die Zugabe von Zitronensäure die IST nur leicht.
Der Wert liegt hierbei für eine Zugabe von 0,3 % Zitronensäure bezogen auf die MC-
Einwaage ebenfalls bei 39,8 °C. Die Zugabe von 0,15 % Zitronensäure bezogen auf die
MC-Einwaage führt zu einer Erniedrigung von 1 °C in der IST gegenüber der 3 %igen
MC383-Lösung und liegt bei einem Wert von 42,4 °C. Die Strukturausbildungsgeschwin-
digkeit hingegen nimmt durch die Zugabe von 0,15 % Zitronensäure bezogen auf die MC-
Einwaage zu und liegt bei 0,81 Pa/s, während die Zugabe von 0,3 % Zitronensäure den
Wert auf 0,69 Pa/s sinken lässt, womit dieser annähernd gleich der SDR der 3 %igen
MC383 Lösung ohne Zitronensäure ist.
Tabelle 5.7: IST, GP, CST, SDR, GA, Gelstärke zu Beginn der Haltezeit (t=0) sowie Gelstärke nach
30 Minuten Haltezeit (t=30) der MC15 und MC383-Proben bei verschiedenen Konzentrationen mit
Zugabe verschiedener Konzentrationen an Zitronensäure (ZS)
IST
[°C]
GP
[°C]
CST
[°C]
SDR
[Pa/s]
GA
[°C]
Gelstärke
(t=0) [Pa]
Gelstärke
(t=30) [Pa]
3 % MC383
+ 0,15 % ZS 42,4 50,2 53,4 0,81 32,3 1325 1789
3 % MC383
+ 0,3 % ZS 39,8 50,4 53,9 0,69 32,7 1239 1696
5 % MC15
+ 0,25 38,3 49,3 53,0 0,99 30,5 1866 2562
5 % MC15
+ 0,5 % ZS 41,4 49,8 53,1 1,01 30,8 1736 2376
5 % MC15
+ 2% ZS 40,3 50,2 54,1 0,67 32,1 1181 865

Ergebnisse
83
Bei der Betrachtung der Gelstärke (Tabelle 5.7) vor und nach der Haltezeit von 30 Minuten
zeigt sich, dass die Zugabe von 0,15 % Zitronensäure die Gelstärke ansteigen lässt, eine
Zugabe von 0,3 % Zitronensäure jedoch wieder zu einer Erniedrigung der Gelstärke führt.
Diese Tendenz ist sowohl vor der Haltezeit als auch danach zu beobachten. Auffällig ist
auch, dass die Gelstärke nach der Haltezeit von 30 Minuten ebenfalls stärker ausgeprägt
ist als vor dieser. Darin stimmen die Werte der Gelstärke tendenziell für die MC383-Proben
mit und ohne Zitronensäure überein.
Die Strukturierungsparameter der MC15-Probe sind in der Tabelle 5.7 aufgelistet. Die Zu-
gabe von Zitronensäure hat auf die Gelpunkttemperatur bzw. auf die Gelauflösungstempe-
ratur nur einen sehr geringen Einfluss. Auch die IST der MC15-Proben mit Zitronensäure
steigt gegenüber der MC15-Proben ohne Zitronensäure nur leicht an. Gleiches gilt für die
Werte der CST.
Die SDR ändert sich durch die Zugabe von Zitronensäure bei den MC15-Proben nur ge-
ringfügig. Ein deutlicher Einfluss der Zugabe von Zitronensäure ist jedoch bei der Gelstärke
vor und nach der Haltezeit von 30 Minuten zu beobachten. Hier zeigt sich, dass sich mit
der Zugabe von 0,25 % bzw. 0,5 % Zitronensäure bezogen auf die Einwaage von MC15
eine Erhöhung der Gelstärke sowohl vor als auch nach der Haltezeit erzielen lässt. Die
Zugabe von 2 % Zitronensäure bezogen auf die MC15-Einwaage lässt die Gelstärke je-
doch vor der Haltezeit leicht und die Gelstärke nach der Haltezeit sehr deutlich abfallen.
Einfluss von Ethanol auf strukturbildende Charakteristika von MC
In Tabelle 5.8 sind die initiale Strukturierungstemperatur (IST), die Gelpunkttemperatur
(GP), die kritische Strukturierungstemperatur (CST), die mittlere Strukturentwicklungsge-
schwindigkeit (SDR), die Gelauflösungstemperatur (GA), die Gelstärke zu Beginn der Hal-
tezeit (t=0) und die Gelstärke nach 20 Minuten Haltezeit der MC15– und MC383-Proben
bei verschiedenen Konzentrationen mit Zugabe verschiedener Konzentrationen an Ethanol
(EtOH) in der Lösemittelphase aufgeführt. Hierbei wurde die Haltezeit auf 20 Minuten her-
abgesetzt, um der Verdunstung des EtOHs entgegenzuwirken. Für die MC383-Proben
konnte durch die Zugabe von 5 % EtOH (v/v) im Lösungsmittelanteil eine Erhöhung der
Gelpunkttemperatur von 3,0 °C und eine Erhöhung der Gelauflösungstemperatur von
3,5 °C erreicht werden. Auch die IST und die CST erhöhten sich mit 3,1 °C bzw. 0,2 °C
leicht. Durch die Zugabe von EtOH wurde die SDR um 0,7 Pa/s erniedrigt. Der größte Ein-
fluss der EtOH-Zugabe ist bei der Gelstärke zu verzeichnen. Hier reduzierte sie die Gel-
stärke vor und nach der Haltezeit um 841 Pa bzw. 1732 Pa.
Die Effekte durch die Zugabe von Ethanol ähneln sich bei der MC15-Probe sehr. Lediglich
die CST wurde durch die Ethanolzugabe um 1,1 °C erniedrigt, im Gegensatz zur MC383-

Ergebnisse
84
Probe, bei der die Zugabe zu einer Erhöhung von 0,2 °C führte. Auch die Erniedrigung der
Gelstärke folgt bei der Zugabe von 5 % EtOH (v/v) im Lösungsmittelanteil dem gleichen
Trend wie bei der MC383-Probe.
Tabelle 5.8: IST, GP, CST, SDR, GA, Gelstärke zu Beginn der Haltezeit (t=0) und Gelstärke nach
20 Minuten Haltezeit (t=20) der MC15– und MC383-Proben bei verschiedenen Konzentrationen mit
Zugabe verschiedener Konzentrationen an EtOH in der Lösemittelphase
IST
[°C]
GP
[°C]
CST
[°C]
SDR
[Pa/s]
GA
[°C]
Gelstärke
(t=0) [Pa]
Gelstärke
(t=20) [Pa]
5 % MC383 41,0 48,3 53,4 0,9 28,9 1085 2385
5 % MC383
+ 5 % EtOH 44,1 51,3 53,7 0,2 32,4 244 653
Δ mit/ohne
EtOH 3,1 3,0 0,2 -0,7 3,5 -841 -1732
10 % MC15 29,7 39,2 49,4 2,5 24,3 4440 6745
10 % MC15
+ 5 % EtOH 33,8 41,6 48,3 0,8 25,8 1139 1848
Δ mit/ohne
EtOH 4,1 2,4 -1,1 -1,8 1,4 -3301 -4897
5.2.3 Charakterisierung des Strukturwiederaufbaus von MC-Lösungen nach Scherbeanspruchung
In diesen Versuchen wurde der Strukturwiederaufbau der MC-Lösungen nach einer Scher-
beanspruchung im Rheometer untersucht. In Abbildung 5.10 sind die Ergebnisse dieser
Untersuchung exemplarisch für die MC383-Lösung bei einer Temperatur von 20 °C darge-
stellt. Wie zu erkennen ist, kann keine der Proben den Strukturwiederaufbau vollkommen
vollziehen. Jedoch liegen die Werte für alle drei Proben nach 180 s sehr dicht beieinander
(siehe Tabelle 5.9).

Ergebnisse
85
Abbildung 5.10: Darstellung de Strukturwiederaufbaus der MC383-Lösungen nach der Scherbelas-
tung bei einer Systemtemperatur von 20 °C dargestellt als Regeneration [%] gegenüber des Aus-
gangswertes vor der Scherbelastung über die Zeit
Des Weiteren sind in Tabelle 5.9 die Ergebnisse für den prozentualen Strukturwiederauf-
bau der Untersuchungen von MC383 bei 30 °C und 40 °C dargestellt. Jedoch liegen die
Werte für die Proben, die bei 20 °C und 30°C untersucht wurden, sehr nahe an einer
100 %igen Strukturregeneration. Lediglich die Proben, die bei 40 °C untersucht wurden (2
bzw. 5 %), zeigen mit Werten von 97,5 ± 0,9 % bzw. 95,2 ± 1,6 % niedrigere Werte, als die
Proben, die bei 20 °C bzw. 30 °C untersucht wurden. Die Probe mit 3 % ließ sich nicht bei
40 °C vermessen, da hier ein zu starker Anstieg der Viskosität während der Vorscherung
stattfand und somit kein konstanter Bezugswert vor der Scherbelastung festgelegt werden
konnte. Auffällig ist weiterhin, dass die Proben, die bei 40 °C untersucht wurden, nach 0,8 s
deutlich niedrigere Werte beim prozentualen Strukturwiederaufbau zeigten als die übrigen
Proben mit gleicher Konzentration.
70
75
80
85
90
95
100
0,1 1 10 100 1000
Reg
ener
atio
n [%
]
Zeit [s]
MC383 5% 20 °CMC383 3% 20°CMC383 2% 20°C

Ergebnisse
86
Tabelle 5.9: Darstellung der prozentualen Regeneration der MC383-Proben in Abhängigkeit von der
Zeit bezogen auf den jeweiligen Wert der Viskosität am Ende der Vorscherung
Regeneration [%] in Abhängigkeit von der Zeit [s]
Konzentra-
tion [%]
Tempera-
tur [°C] 0,8 1 5 60 180
2 20 95,7 ± 3,0 95,8 ± 3,2 96,9 ± 2,8 98,2 ± 2,1 98,1 ± 1,9
2 30 96,8 ± 1,3 97,1 ± 1,7 98,3 ± 1,1 99,3 ± 0,7 99,6 ± 0,6
2 40 91,9 ± 2,6 92,4 ± 2,4 94,1 ± 2,2 96,7 ± 1,4 97,5 ± 0,9
3 20 90,8 ± 1,5 91,6 ± 1,6 95,9 ± 1,8 98,1 ± 1,3 98,1 ± 1,2
3 30 89,1 ± 1,2 90,1 ± 1,3 95,0 ± 1,3 97,9 ± 0,7 98,4 ± 0,6
5 20 93,7 ± 0,7 94,7 ± 0,8 97,6 ± 0,3 98,8 ± 0,1 99,0 ± 0,1
5 30 93,3 ± 0,3 94,0 ± 0,5 96,9 ± 0,8 98,7 ± 0,9 99,0 ± 0,7
5 40 77,8 ± 2,2 78,7 ± 2,0 85,5 ± 1,5 91,6 ± 1,6 95,2 ± 1,6
In Abbildung 5.11 ist exemplarisch der Strukturaufbauverlauf für die MC15-Proben, die bei
20 °C untersucht wurden, dargestellt. Dabei ist zu erkennen, dass der Verlauf des Struk-
turwiederaufbaus stark mit zunehmender Konzentration verlangsamt wird. Des Weiteren
wird deutlich, dass die Probe mit der höchsten Konzentration an MC15 den niedrigsten
prozentualen Strukturwiederaufbauwert erreicht. In der Darstellung ist zu erkennen, dass
die Graphen zu unterschiedlichen Messzeitpunkten beginnen. Dies ist der Tatsache ge-
schuldet, dass für die Proben mit 5 und 6,5 % zu Beginn der Messung, trotz mehrfachen
Wiederholungen, starke Schwankungen in der Viskosität auftauchten, die eine Auswertung
nicht möglich machten. Demzufolge sind die Graphen für diese beiden Proben erst ab dem
Zeitpunkt dargestellt, ab dem die Messung störungsfrei verlief. Ab 0,8 s zeigten alle Proben
der Abbildung 5.11 störungsfreie Signale.

Ergebnisse
87
Abbildung 5.11: Darstellung des Strukturwiederaufbaus der MC15-Lösungen nach der Scherbelas-
tung bei einer Systemtemperatur von 20 °C dargestellt als Regeneration [%] gegenüber des Aus-
gangswertes vor der Scherbelastung über die Zeit
Weiterhin sind in Tabelle 5.10 die Werte der Viskosität während des Strukturwiederaufbaus
der MC15-Probe bei weiteren Temperaturbedingungen aufgelistet. Hierbei fällt besonders
die Probe mit einer Konzentration von 8,0 % und einer Temperatur von 40 °C auf. Diese
Probe zeigt während des Strukturwiederaufbaus deutlich niedrigere Werte in der Viskosität
bezogen auf die Anfangsviskosität als alle anderen Proben. Die Vermessung der beiden
anderen Konzentrationen war bei 40 °C nicht möglich, da diese bereits einen zu starken
Anstieg in der Viskosität zeigten, so dass eine Ermittlung des Startwertes und damit die
Bestimmung des strukturellen Wiederaufbaus nicht möglich war. Weiterhin ist zu erkennen,
dass die Werte für den prozentualen Strukturwiederaufbau zu Beginn der Messzeit, also
bei 0,8 s, für die Probe mit 8 % MC-Konzentration, die bei 40 °C untersucht wurde, deutlich
niedriger sind als für die Proben mit gleicher Konzentration.
70
75
80
85
90
95
100
0,1 1 10 100 1000
Reg
ener
atio
n [%
]
Zeit [s]
MC15 8% 20°CMC15 6,5% 20°CMC15 5% 20 °C

Ergebnisse
88
Tabelle 5.10: Darstellung der prozentualen Regeneration der MC15-Proben in Abhängigkeit von der
Zeit bezogen auf den jeweiligen Wert der Viskosität am Ende der Vorscherung
Regeneration[%] in Abhängigkeit von der Zeit [s]
Konzent-
ration [%]
Tempera-
tur [°C] 0,8 1 5 60 180
5,0 20 100,4 ± 1,6 99,4 ± 1,2 99,2 ± 0,6 99,6 ± 0,5 99,4 ± 0,5
5,0 30 98,7 ± 0,1 98,2 ± 0,5 99,1 ± 0,1 99,6 ± 0,2 99,7 ± 0,1
6,5 20 93,7 ± 0,9 93,7 ± 0,9 95,7 ± 1,0 97,4 ± 0,7 97,6 ± 0,8
6,5 30 92,7 ± 3,9 92,7 ± 3,9 94,4 ± 2,6 97,7 ± 1,4 98,3 ± 0,8
8,0 20 83,8 ± 5,3 83,8 ± 3,8 88,5 ± 6,5 92,4 ± 5,9 92,9 ± 5,4
8,0 30 83,3 ± 3,8 83,3 ± 3,8 87,4 ± 4,2 92,1 ± 4,1 93,9 ± 3,7
8,0 40 36,4 ± 3,9 36,4 ± 3,9 41,2 ± 2,8 54,5 ± 1,7 67,9 ± 3,2

Ergebnisse
89
5.3 Charakterisierung der Funktionalität von zellulosederivatbasier-ten Coatingmaterialien als Sauerstoffbarriere für funktionelle Lebensmittelinhaltsstoffe
Die Untersuchungen zur Funktionalität der zellulosederivatbasierten Coatingmaterialien
wurden in mehrere Versuchsblöcke unterteilt. Zur besseren Verständlichkeit sind die fol-
genden Unterkapitel zur Darstellung der Ergebnisse nach diesen Versuchsreihen unterteilt.
Die Untersuchungen, die in jeder Versuchsreihe durchgeführt wurden, sind in Tabelle 4.14
aufgeführt. Die Fehlerindikatoren in den Abbildungen zum zeitlichen Verlauf der Hydroper-
oxidkonzentrationen zeigen die Standardabweichung der chemischen Analytik.
5.3.1 Vorversuche zur Überprüfung der Methoden zur Charakterisierung von gecoateten Agglomeraten und der Untersuchung der Barriere-eigenschaft der MC-Coatings
Mittels Gesamtfettbestimmung nach Soxhlet mit vorhergehendem Weibull-Aufschluss
konnte über Rückrechnung die applizierte Coatingmenge nachgewiesen werden (siehe
Tabelle 5.11). Dies erfolgte durch Berechnen der fettfreien Trockenmasse in den ungecoa-
teten Partikeln und die Änderung dieser nach dem Auftragen eines Coatings. Hierbei zeigte
sich, dass mit verschiedenen Mengen an Coatinglösung verschiedene Coatingmengen
appliziert werden können. Diese entsprechen jedoch nicht den theoretisch berechneten
Coatingmengen, welche für 70 mL Coatinglösung bei 1,6 % und für 130 mL bei 3 % liegen.
In Tabelle 5.11 zeigt sich allerdings, dass die tatsächlich aufgetragene Coatingmenge auf
den Agglomeraten, die mit 130 mL Coatinglösung besprüht wurden, deutlich über der theo-
retischen Coatingmenge liegt.
Tabelle 5.11: Gesamtfettgehalt und darüber berechnete applizierte Coatingmenge vom sprühge-
trockneten Pulver, den Agglomeraten und zwei mit unterschiedlich viel (70 und 130 mL) MC15-
Lösung gecoateten Agglomeraten in % bezogen auf die Gesamtmasse der gecoateten Agglomerate
Gesamtfettgehalt [%] berechnete Coatingmenge [%]
Pulver 21,7 ± 0,1 -
Agglomerate 21,2 ± 0,1 -
MC15-70-T60-t30 21,0 ± 0,3 1,8 ± 0,9
MC15-130-T60-t60 17,0 ± 1,0 20,1 ± 4,7

Ergebnisse
90
Aus Abbildung 5.12 kann der Verlauf der Lipidoxidation anhand der über die Zeit abgetra-
genen Hydroperoxidkonzentration abgelesen werden. Dieser Versuch wurde durchgeführt,
um zu überprüfen, ob die Methode der Bestimmung der Hydroperoxidkonzentration für
Agglomerate, die mit Zellulosederivatlösungen gecoatet sind, durchführbar ist. Dies konnte
bestätigt werden, da aus den ungecoateten Agglomeraten ebenso das Öl im Zuge der Un-
tersuchung extrahiert werden konnte wie aus den gecoateten Agglomeraten.
Es zeigt sich hierbei, dass die Startwerte der Hydroperoxidkonzentration (siehe Tabelle
A.26) zwar in der gleichen Größenordnung liegen, sich jedoch leicht voneinander unter-
scheiden. Der Startwert für die Proben des sprühgetrockneten Pulvers lag mit
5,2 ± 0,7 mmol/kg Öl deutlich unter der Hydroperoxidkonzentration der in der Wirbelschicht
mit 70 bzw. 130 mL MC-Coatinglösung gecoateten Agglomerate (mit ca. 8,8 ± 1,0 bzw. 8,9
± 0,5 mmol/kg). Für das Rapsöl konnte keine Konzentration an Hydroperoxiden nachge-
wiesen werden. Die Hydroperoxidkonzentration der Proben nach 8 Wochen Lagerzeit zeig-
te keinen Unterschied zwischen den Werten des sprühgetrockneten Pulvers und den un-
gecoateten Agglomeraten (17,3 ± 0,3 und 17,2 ± 2,1 mmol/kg Öl). Die Werte für die Hydro-
peroxidkonzentration der mit 70 bzw. 130 mL MC-Coatinglösung gecoateten Agglomerate
lagen bei 40,5 ± 0,3 bzw. 51,2 ± 5,8 mmol/kg Öl und damit deutlich höher als die Hydro-
peroxidkonzentrationen der beiden anderen Proben. Sie unterschieden sich auch unterei-
nander deutlich.
Abbildung 5.12: Zeitlicher Verlauf der Hydroperoxidkonzentration von sprühgetrocknetem Pulver,
Agglomeraten und gecoateten Agglomeraten bei 60 °C Prozesstemperatur und einer Prozesszeit
von 30 bzw. 60 Minuten
0
10
20
30
40
50
60
70
0 1 2 4 5 6 8
Hyd
rope
roxi
dkon
zent
ratio
n [m
mol
/kg
Öl]
Zeit [Wochen]
PulverAgglomerateMC15-70-T60-t30MC15-130-T60-t60

Ergebnisse
91
5.3.2 Untersuchung des Einflusses verschiedener Prozessparameter in der Wirbelschicht auf verkapseltes Rapsöl in Agglomeraten
Die Werte der Hydroperoxidkonzentration der gecoateten Agglomerate aus dem Vorver-
such stiegen schneller an und zeigten höhere Werte am Ende der Lagerzeit als die unge-
coateten Agglomerate. Deswegen wurden zunächst Untersuchungen durchgeführt, die den
Einfluss der Prozessparameter auf den Verlauf der Hydroperoxidbildung und der Partikel-
struktur klären sollen.
Die Untersuchungen wurden auf zwei Wochen verteilt durchgeführt, wobei in jeder Woche
ein neuer Ansatz der Proben erfolgte. In der ersten Woche wurden die Agglomerate ohne
Wasserzugabe und in der zweiten Versuchswoche mit Wasser in der Wirbelschicht pro-
zessiert. Für beide Versuchswochen wurden jeweils die Emulsionen, das sprühgetrocknete
Pulver und die Agglomerate separat hergestellt.
Bei der Auswertung der Öltropfengröße zeigt sich, dass der d10 und d50 der Emulsion und
des sprühgetrockneten Pulvers in beiden Herstellungswochen sehr ähnlich sind (siehe Ta-
belle 5.12). Nur im d90 und damit auch im span unterscheiden sich die Werte zwischen den
Herstellungswochen. Die Werte für die Verteilungsparameter und Verteilungsbreite der
unbehandelten Agglomerate liegen in der gleichen Größenordnung wie die Werte für die
Öltropfengröße des sprühgetrockneten Pulvers.
Tabelle 5.12: Charakteristische Verteilungsparameter (d10, d50 und d90) und Verteilungsbreite (span)
der Öltropfengrößenbestimmung mittels SLS von der Grundemulsion und dem sprühgetrockneten
Pulver der ersten und zweiten Woche und den Agglomeraten
d10 [µm] d50 [µm] d90 [µm] span
Emulsion (1. Woche) 1,52 ± 0,07 3,08 ± 0,27 7,65 ± 2,29 1,97 ± 0,65
Emulsion (2. Woche) 1,55 ± 0,01 2,98 ± 0,04 5,91 ± 0,11 1,47 ± 0,01
Pulver (1. Woche) 1,20 ± 0,02 2,15 ± 0,01 4,03 ± 0,12 1,31 ± 0,06
Pulver (2. Woche) 1,21 ± 0,01 2,54 ± 0,02 6,07 ± 0,11 1,91 ± 0,04
Agglomerate 1,18 ± 0,01 2,27 ± 0,06 4,47 ± 0,20 1,44 ± 0,05
Die Auswertung der Verteilungsparameter und Verteilungsbreite der Öltropfengröße der in
der Wirbelschicht behandelten Agglomerate (siehe Tabelle 5.13) zeigt, dass sich die Werte
zwischen den Proben, die trocken behandelt wurden, nicht maßgeblich unterscheiden.

Ergebnisse
92
Auch innerhalb der Gruppe der mit Wasser behandelten Agglomerate zeigten sich kaum
Unterschiede durch die unterschiedlichen Behandlungstemperaturen und -zeiten.
Ein Unterschied ist jedoch erkennbar zwischen den Verteilungsparametern und der Vertei-
lungsbreite, wenn die Werte der Gruppe der ohne Wasser in der Wirbelschicht behandel-
ten, den Werten der Gruppe, die mit Wasser in der Wirbelschicht behandelt wurden, ge-
genübergestellt werden. Die Verteilungsparameter und die Verteilungsbreite sind grund-
sätzlich höher in der Gruppe der mit Wasser behandelten Agglomerate gegenüber der tro-
cken behandelten Gruppe.
Tabelle 5.13: Charakteristische Verteilungsparameter (d10, d50 und d90) und Verteilungsbreite (span)
der Öltropfengrößenbestimmung mittels SLS von verschieden in der Wirbelschicht behandelten und
resolubilisierten Agglomeraten
d10 [µm] d50 [µm] d90 [µm] span
T30-t20 1,20 ± 0,00 2,33 ± 0,03 4,63 ± 0,13 1,47 ± 0,04
T30-t40 1,21 ± 0,04 2,41 ± 0,17 5,17 ± 0,90 1,63 ± 0,25
T30-t60 1,19 ± 0,01 2,20 ± 0,05 4,26 ± 0,20 1,40 ± 0,06
T60-t20 1,23 ± 0,07 2,40 ± 0,22 4,92 ± 0,92 1,53 ± 0,20
T60-t40 1,23 ± 0,05 2,18 ± 0,10 4,00 ± 0,39 1,27 ± 0,15
T60-t60 1,19 ± 0,01 2,27 ± 0,11 4,38 ± 0,38 1,40 ± 0,10
H2O-T30-t20 1,36 ± 0,01 2,80 ± 0,14 6,67 ± 1,02 1,89 ± 0,27
H2O-T30-t40 1,38 ± 0,01 2,71 ± 0,03 5,52 ± 0,16 1,52 ±0,04
H2O-T30-t60 1,37 ± 0,01 2,72 ± 0,15 5,57 ± 0,21 1,54 ± 0,05
H2O-T60-t20 1,34 ± 0,02 2,65 ± 0,08 5,43 ± 0,30 1,54 ± 0,06
H2O-T60-t40 1,39 ± 0,05 2,74 ±0,13 5,55 ± 0,32 1,52 ± 0,03
H2O-T60-t60 1,48 ± 0,04 2,97 ± 0,22 6,56 ± 1,37 1,69 ± 0,32
Bei der Auswertung der Werte der Mikroverkapselungseffizienz von verschieden behandel-
ten Agglomeraten zeigten sich deutliche Unterschiede zwischen den Werten der Proben,
die mit bzw. ohne Wasser behandelt wurden.
Zur Verdeutlichung der Unterschiede zwischen den einzelnen Prozessparametern wurde
der Gehalt an extrahierbarem Öl in Abbildung 5.13 dargestellt. Die Werte der Mikroverkap-
selungseffizienz für das sprühgetrocknete Pulver lagen bei 97,5 ± 0,1 % und 95,8 ± 0,8 %

Ergebnisse
93
für die beiden separat hergestellten Ansätze. Die Mikroverkapselungseffizienz für die Ag-
glomerate betrug 97,8 ± 0,1 %.
ohne Wasser mit Wasser
Prozessparameter Mikroverkapselungseffizienz [%]
T30-t20 96,6 ± 0,5 98,2 ± 0,1
T30-t40 96,6 ± 0,4 98,5 ± 0,0
T30-t60 97,2 ± 0,5 98,7 ± 0,0
T60-t20 97,0 ± 0,3 98,0 ± 0,1
T60-t40 95,5 ± 0,3 98,4 ± 0,1
T60-t60 96,0 ± 0,2 98,2 ± 0,3
Abbildung 5.13: Extrahierbarer Ölgehalt und Mikroverkapselungseffizienz von sprühgetrockneten
Pulvern, unbehandelten Agglomeraten und verschiedenen in der Wirbelschicht behandelten Agglo-
meraten
In Abbildung 5.14 ist der zeitliche Verlauf der Lipidoxidation für Agglomerate, die unbehan-
delt sind und die bei 30 °C bzw. 60 °C und 20, 40 bzw. 60 Minuten in der Wirbelschicht
behandelt wurden, nachzuvollziehen. Die Startwerte der Hydroperoxidkonzentration liegen
zwischen 2,8 ± 0,1 mmol/kg Öl für die unbehandelten Agglomerate und 7,1 ± 3 mmol/kg Öl
für die Agglomerate T30t40.
Die Werte für die Hydroperoxidkonzentration nach 10 Wochen Lagerzeit zeigen einen Un-
terschied zwischen den Werten der unbehandelten Agglomerate (47,5 ± 4,2 mmol/kg Öl)
0,00
1,00
2,00
3,00
4,00
5,00
6,00
T30-t20 T30-t40 T30-t60 T60-t20 T60-t40 T60-t60
extr
ahie
rbar
er Ö
lgeh
alt [
%]
Behandlungsparameter der Agglomerate in der Wirbelschicht
ohne Wasser mit Wasser

Ergebnisse
94
und den Werten der behandelten Agglomerate, die unabhängig von der Temperatur für 40
bzw. 60 Minuten in der Wirbelschicht behandelt wurden und deren Werte zwischen
210,6 ± 22,2 und 266,1 ± 14,7 mmol/kg Öl variieren. Die Werte für die Hydroperoxidkon-
zentration der behandelten Agglomerate mit einer Prozesstemperatur von 30 °C unter-
schieden sich zwischen den Prozesszeiten 20 bzw. 40 Minuten und 20 bzw. 60 Minuten.
Die gleichen Unterschiede zwischen den Hydroperoxidkonzentrationen der behandelten
Agglomerate in den Prozesszeiten zeigen sich bei einer Prozesstemperatur von 60 °C.
Temperaturübergreifend unterscheiden sich die Werte für die Hydroperoxidkonzentrationen
nur innerhalb der Prozesszeiten von 20 bzw. 40 Minuten und 20 bzw. 60 Minuten.
Abbildung 5.14: Zeitlicher Verlauf der Hydroperoxidkonzentration von unbehandelten Agglomeraten
und verschieden trocken in der Wirbelschicht behandelten Agglomeraten mit Prozesstemperaturen
von 30°C bzw. 60 °C und Prozesszeiten von 20, 40 bzw. 60 Minuten
Der in Abbildung 5.15 dargestellte zeitliche Verlauf der Lipidoxidation zeigt die Hydroper-
oxidkonzentration von unbehandelten Agglomeraten und den mit Wasser in der Wirbel-
schicht behandelten Agglomeraten, die für 20, 40 bzw. 60 Minuten und bei 30 °C bzw.
60 °C behandelt wurden.
Unterschiedlich sind demnach die Hydroperoxidkonzentrationen der unbehandelten Ag-
glomerate und der 40 bzw. 60 Minuten bei 30 °C mit Wasser behandelten Agglomerate.
Die Werte für die Hydroperoxidkonzentration für die Prozesszeiten von 40 bzw. 60 Minuten
zwischen den beiden Prozesstemperaturen unterscheiden sich ebenfalls. Auch der Ver-
gleich von T60t60 gegen T30t60 bzw. T60t40 gegen T30t60 zeigt einen Unterschied in der
Hydroperoxidkonzentration. Kein deutlicher Unterschied in der Hydroperoxidkonzentration
0
20
40
60
80
100
120
140
0 2 4 6 8 10
Hyd
rope
roxi
dkon
zent
ratio
n [m
mol
/kg
Öl]
Zeit [Wochen]
AgglomerateT30-t20T30-t40T30-t60T60-t20T60-t40T60-t60

Ergebnisse
95
kann hingegen in der Variation der Prozesszeit bei 60°C Prozesstemperatur gefunden
werden. Ebenfalls ohne erkennbaren Unterschied in der Hydroperoxidkonzentration sind
die Agglomerate, die 20 Minuten prozessiert wurden, unabhängig von Prozesstemperatur.
Die Werte des zeitlichen Verlaufes der Lipidoxidation anhand der Hydroperoxidkonzentrati-
onen sind der Tabelle A.27, Tabelle A.28, Tabelle A.29 und Tabelle A.30 zu entnehmen.
Abbildung 5.15: Zeitlicher Verlauf der Hydroperoxidkonzentration von unbehandelten Agglomeraten
und verschieden in der Wirbelschicht unter Wasserzugabe behandelten Agglomeraten mit Prozess-
temperaturen von 30°C bzw. 60 °C und Prozesszeiten von 20, 40 bzw. 60 Minuten
Wie in Abbildung 5.14 und Abbildung 5.15 zu erkennen ist, hat die Behandlung mit und
ohne Wasser einen sehr deutlichen Einfluss auf den Verlauf der Hydroperoxidbildung. Die
ohne Wasser behandelten Agglomerate, die unabhängig von der Temperatur 40 Minuten
bzw. 60 Minuten behandelt wurden zeigen nach 8 Wochen Lagerzeit bereits Werte von
deutlich über 140 mmol/kg Öl. Die Werte für die Hydroperoxidkonzentration für die Agglo-
merate, die mit Wasser behandelt wurden, zeigen nach 8 Wochen Lagerzeit jedoch Werte
zwischen 30 mmol/kg Öl und 43 mmol/kg Öl. Erst nach 10 Wochen Lagerzeit steigen die
Werte für die Hydroperoxidkonzentration der für 20 Minuten bzw. 40 Minuten mit Wasser
behandelten Agglomerate auf ca. 96 mmol/kg Öl. Zu beachten ist hierbei, dass die höchs-
ten Werte für die Hydroperoxidkonzentration bei den Proben, die mit Wasser behandelt
wurden, diejenigen waren, die bei niedriger Temperatur für kurze Zeit behandelt wurden.
Bei den trocken behandelten Agglomeraten zeigten jedoch die Proben mit der längsten
Behandlungszeit und der höchsten Temperatur auch die höchsten Werte für die Hydroper-
oxidkonzentration.
0
20
40
60
80
100
120
140
0 2 4 6 8 10
Hyd
rope
roxi
dkon
zent
ratio
n [m
mol
/kg
Öl]
Zeit [Wochen]
Agglomerate
H2O-T30-t20
H2O-T30-t40
H2O-T30-t60
H2O-T60-t20
H2O-T60-t40
H2O-T60-t60

Ergebnisse
96
5.3.3 Untersuchungen der strukturellen Partikeleigenschaften vor und nach dem Prozessieren in der Wirbelschicht und deren Einfluss auf verkapseltes Rapsöl
Die Ergebnisse der Untersuchungen zum Einfluss der Prozesszeit und -temperatur zeigten,
dass die Werte der Hydroperoxidkonzentration der mit Wasser behandelten Agglomerate
einen flacheren Verlauf aufwiesen und niedriger waren als die der Agglomerate, die ohne
Wasser behandelt wurden. Daraus ergab sich der nächste Versuchsblock, in dem unter-
sucht werden sollte, ob die Unterschiede in der Hydroperoxidkonzentration in den ver-
schiedenen Behandlungsparametern ihre Ursachen in der Struktur der Agglomerate haben.
Hierzu wurde neben den bereits bekannten Methoden der Untersuchung der Öltropfengrö-
ße und -verteilung, der Mikroverkapselungseffizienz und der Bestimmung der Hydroper-
oxidkonzentration über eine Lagerzeit von 16 Wochen auch die Struktur der Agglomerate
mittels verschiedener Gasadsorptionsverfahren und REM-Untersuchungen charakterisiert.
Um zu erkennen, ob die strukturellen Unterschiede in den Agglomeraten von der Ölbela-
dung herrühren, wurden neben den bislang untersuchten Agglomeraten mit verkapseltem
Rapsöl auch Agglomerate ohne Ölbeladung hergestellt. Diese wurden jedoch ausschließ-
lich den Untersuchungen zur Strukturaufklärung unterzogen. Zur besseren Unterscheidung
werden bei den Untersuchungen, die an Agglomeraten mit und ohne Ölbeladung durchge-
führt wurden, die ölbeladenen Proben in diesem Unterkapitel mit dem Zusatz PP (für Pri-
märpartikel) und die ohne Ölbeladung hergestellten Proben mit MP (für Matrixpartikel) ver-
sehen.
Die Auswertung der Ergebnisse der Verteilungsparameter und Verteilungsbreite der Öltrop-
fengröße der zur Strukturanalyse verwendeten Proben (siehe Tabelle 5.14) zeigt hinsicht-
lich des Prozesses (Sprühtrocknung, Agglomeration und Behandlung der Agglomerate in
der Wirbelschicht) eine Abhängigkeit des d10 vom Prozess an sich. Während der d10 für die
Emulsion 1,38 ± 0,13 µm ist, zeigen die resolubilisierten behandelten Agglomerate nur d10-
Werte von 1,22 ± 0,03 µm bzw. 1,20 ± 0,03 µm. Der d50-Wert für die Emulsion ist mit
2,41 ± 0,31 µm am höchsten. Das weitere Prozessieren durch Sprühtrocknen, Agglomerie-
ren und Behandeln in der Wirbelschicht zeigt eine Verringerung des d50-Wertes. Die beiden
übrigen Parameter (d90 und span) zeigten keinen ersichtlichen Einfluss durch das Sprüh-
trocknen, Agglomerieren und Behandeln in der Wirbelschicht. Auch die Parameter Pro-
zesszeit und Behandlung mit und ohne Wasser lassen in dieser Versuchsreihe keinen Ein-
fluss auf die Verteilungsbreite bzw. die Verteilungsparameter erkennen. Die Werte für die
Öltropfengröße ähneln denen der Versuchsreihe zuvor.

Ergebnisse
97
Tabelle 5.14: Charakteristische Verteilungsparameter (d10, d50 und d90) und Verteilungsbreite (span)
der Öltropfengrößenbestimmung mittels SLS der Primäremulsion, dem sprühgetrockneten Pulver,
den Agglomeraten und verschiedener in der Wirbelschicht behandelter und resolubilisierter Agglo-
merate
d10 [µm] d50 [µm] d90 [µm] span
Emulsion 1,38 ± 0,13 2,41 ± 0,31 4,39 ± 0,75 1,24 ± 0,11
Pulver 1,23 ± 0,01 2,04 ± 0,00 3,63 ± 0,05 1,17 ± 0,03
Agglomerate 1,27 ± 0,04 2,03 ± 0,05 3,40 ± 0,32 1,05 ± 0,15
H2O-T60-t20 1,22 ± 0,03 2,12 ± 0,03 3,88 ± 0,23 1,25 ± 0,10
H2O-T60-t60 1,20 ± 0,03 2,15 ± 0,06 4,07 ± 0,31 1,33 ± 0,12
T60-t60 1,20 ± 0,02 2,08 ± 0,02 3,85 ± 0,17 1,27 ± 0,08
Die Ergebnisse für die Mikroverkapselungseffizienz der in der Wirbelschicht behandelten
Agglomerate belegen, dass die mit Wasser behandelten Agglomerate (98,4 ± 0,0 bzw.
98,7 ± 0,1 %) eine leicht erhöhte Mikroverkapselungseffizienz gegenüber den trocken be-
handelten Agglomeraten (97,5 ± 0,5 %) zeigen. Die Werte für Mikroverkapselungseffizienz
der behandelten Proben liegen jedoch in ähnlicher Größenordnung wie die der unbehan-
delten und des sprühgetrockneten Pulvers (siehe Tabelle 5.15).
Tabelle 5.15: Mikroverkapselungseffizienz von sprühgetrocknetem Pulver, unbehandelten Agglome-
raten und in der Wirbelschicht verschieden behandelten Agglomeraten
Verkapselungseffizienz [%]
Pulver 97,3 ± 0,1
Agglomerate 98,7 ± 0,3
H2O-T60-t20 98,4 ± 0,0
H2O-T60-t60 98,7 ± 0,1
T60-t60 97,5 ± 0,5
Bei der Auswertung der REM-Aufnahmen der mit und ohne Wasser behandelten, ölbela-
denen Agglomerate (PP) und der ohne Ölbeladung hergestellten Agglomerate (MP) zeigen
sich zwischen den Proben sowohl strukturelle Unterschiede als auch Gemeinsamkeiten.

Ergebnisse
98
Bei den MP-Agglomeraten, die in der Wirbelschicht mit Wasser behandelt wurden, zeigt
sich eine deutlich glattere Oberfläche als bei den ohne Wasser in der Wirbelschicht behan-
delten MP-Agglomeraten (A & B). Gleiches gilt für die ölbeladenen Agglomerate (C & D).
Zwischen den in der Wirbelschicht mit Wasser behandelten Agglomeraten (A & C) zeigt
sich in der Oberflächenbeschaffenheit kein deutlich erkennbarer Unterschied hinsichtlich
der Porosität. Auf beiden Oberflächen sind runde Strukturen erkennbar. Diese stammen
von Wassertropfen, die auf die Oberfläche treffen und das Maltodextrin in diesem Bereich
lösen. Nach dem Trocknen bleiben eben diese runden Strukturen zurück. Ein Unterschied
bezüglich der Oberflächenbeschaffenheit lässt sich jedoch zwischen den beiden ohne
Wasser prozessierten Proben erkennen. Hier ist erkennbar, dass die agglomerierten Mat-
rixpartikel (B) eine deutlich rauere Oberfläche aufweisen, als die trocken behandelten ölbe-
ladenen Agglomerate (D).
Abbildung 5.16: Aufnahmen des Rasterelektronenmikroskops; A: MP-H2O-T60t60; B: MP-T60t60; C:
PP-H2O-T60t60; D: PP-T60t60 ; A, B, C: Vergrößerung 500-fach; D: Vergrößerung 300-fach
A
D
B
C

Ergebnisse
99
In Abbildung 5.17 ist der zeitliche Verlauf der Hydroperoxidkonzentration der bei
verschiedenen Prozesszeit-Temperatur-Kombinationen behandelten Agglomerate und der
unbehandelten Agglomerate dargestellt. Die Werte der Hydroperoxidkonzentration zu
Beginn der Lagerung weisen eine Streuung von 3,4 ± 0,5 - 3,8 ± 0,5 mmol/kg Öl auf, sie
liegen somit alle in der gleichen Größenordnung.
Die Hydroperoxidkonzentrationen nach 16 Wochen Lagerzeit zeigen deutliche Unterschie-
de. Einzig die Proben mit 20 Minuten Prozesszeit und einer Behandlung mit Wasser
(26,4 ± 1,3 mmol/kg Öl) und die der unbehandelten Agglomerate (24,9 ± 3,2 mmol/kg Öl)
liegen in der gleichen Größenordnung. Ein sehr deutlicher Unterschied zeigt sich zwischen
den Proben, die mit Wasser behandelt wurden, jedoch unterschiedlichen Prozesszeiten
ausgesetzt waren (20 bzw. 60 Minuten). Hierbei lässt sich ein Einfluss der Prozesszeit auf
die Hydroperoxidkonzentration erkennen. Die Hydroperoxidkonzentrationen dieser Proben
variierten zwischen 26,4 ± 1,3 mmol/kg Öl (20 Minuten) und 67,9 ± 2,7 mmol/kg Öl
(60 Minuten). Auch die Proben, die bei 60 Minuten Prozesszeit mit bzw. ohne Wasser be-
handelt wurden, zeigen einen deutlichen Unterschied in der Hydroperoxidkonzentration.
Die Werte für die Hydroperoxidkonzentration lagen hier bei 67,9 ± 2,7 mmol/kg Öl für die
mit Wasser behandelten Agglomerate bzw. 235,5 ± 2,7 mmol/kg Öl für die trocken behan-
delten Agglomerate. Die gesamten Werte zur Abbildung 5.17 finden sich im Anhang in der
Tabelle A.31.
Abbildung 5.17: Hydroperoxidkonzentration der unbehandelten Agglomerate und der bei 60°C über
20 bzw. 60 Minuten mit und ohne Wasser behandelten Agglomerate über den Zeitraum von 16 Wo-
chen mit Untersuchungen alle zwei Wochen
Um die spezifische Oberfläche der unbehandelten und behandelten MP-Agglomerate und
der unbehandelten und behandelten PP-Agglomerate zu bestimmen, wurden vier ver-
0
5
10
15
20
25
30
35
40
45
50
0 2 4 6 8 10 12 14 16
Hyd
rope
roxi
dgeh
alt [
mm
ol/k
g Ö
l]
Zeit [Wochen]
AgglomerateH2O-T60-t20H2O-T60-t60T60-t60

Ergebnisse
100
schiedene Messverfahren durchgeführt. Die Gasadsorptionsmessung mit Stickstoff führte
genauso wie die dynamische Dampfsorptionsmessung zu keinem Ergebnis. Die Gasad-
sorptionsmessung mit Krypton und anschließender BET-Auswertung zeigte, dass die Wer-
te für die spezifische Oberfläche der Proben durchweg sehr niedrig waren, die Ergebnisse
lagen zwischen 0,028 und 0,08 m2/g. Die gravimetrischen Ergebnisse der dynamischen
Dampfsorptionsmessung unter Verwendung von n-Octan ließen die Berechnung der spezi-
fischen Oberfläche auf Grundlage zweier verschiedener Modelle zu. Zum einen die BET-
Auswertung und zum anderen die ESW-Auswertung. Für beide Methoden der Auswertung
ergaben sich Werte für die spezifische Oberfläche der Proben, die im ähnlichen Größen-
verhältnis zueinander standen. Gegenüber den Werten, die mittels Gasadsorptionsmes-
sung mit Krypton ermittelt wurden, lagen die Ergebnisse für die spezifische Oberfläche, die
über dynamische Dampfsorption ermittelt wurden, eine Zehnerpotenz höher.
Tabelle 5.16: Oberflächengrößen der unbehandelten und behandelten Matrixpartikel-Agglomerate
sowie der unbehandelten und behandelten ölbeladenen Agglomerate ermittelt durch Gasadsorpti-
onsmethode mit Krypton bei 77 K bzw. durch dynamische Dampfsorption (DVS) mit n-Octan; Aus-
wertung erfolgte für die Gasadsorption mittels BET bzw. BET und ESW für die DVS
Gassorption dynamische Dampfsorption
BET [m2/g] BET [m2/g] ESW [m2/g]
MP-Agglomerate 0,0521 1,0755 0,9401
MP-H2O-T60-t60 0,0484 1,5786 1,37335
MP-T60-t60 0,0477 1,3744 1,19585
PP-Agglomerate 0,0429 3,2209 2,74545
PP-H2O-T60-t20 0,0425 1,4473 1,34065
PP-H2O-T60-t60 0,0276 2,42325 3,7397
PP-T60-t60 0,0791 3,7778 3,0331
In Abbildung 5.18 ist die Porengrößenverteilung der Hg-Porosimetrie der unbehandelten
Matrixpartikel-Agglomerate exemplarisch dargestellt. Die drei in der Kurve des kumulativen
Porenvolumens zu erkennenden Peaks stellen jeweils in der Probe auftretende Porengrö-
ßen dar. Die Peaks liegen bei ca. 205 µm, bei ca. 15 µm und bei ca. 5 nm. Die Darstellun-
gen der behandelten Matrixpartikel-Agglomerate und der behandelten und unbehandelten
ölbeladenen Agglomerate ähneln dieser Darstellung sehr stark und sind deswegen im An-
hang in Abbildung A.1 und Abbildung A.2 für die unbehandelten und behandelten Matrix-
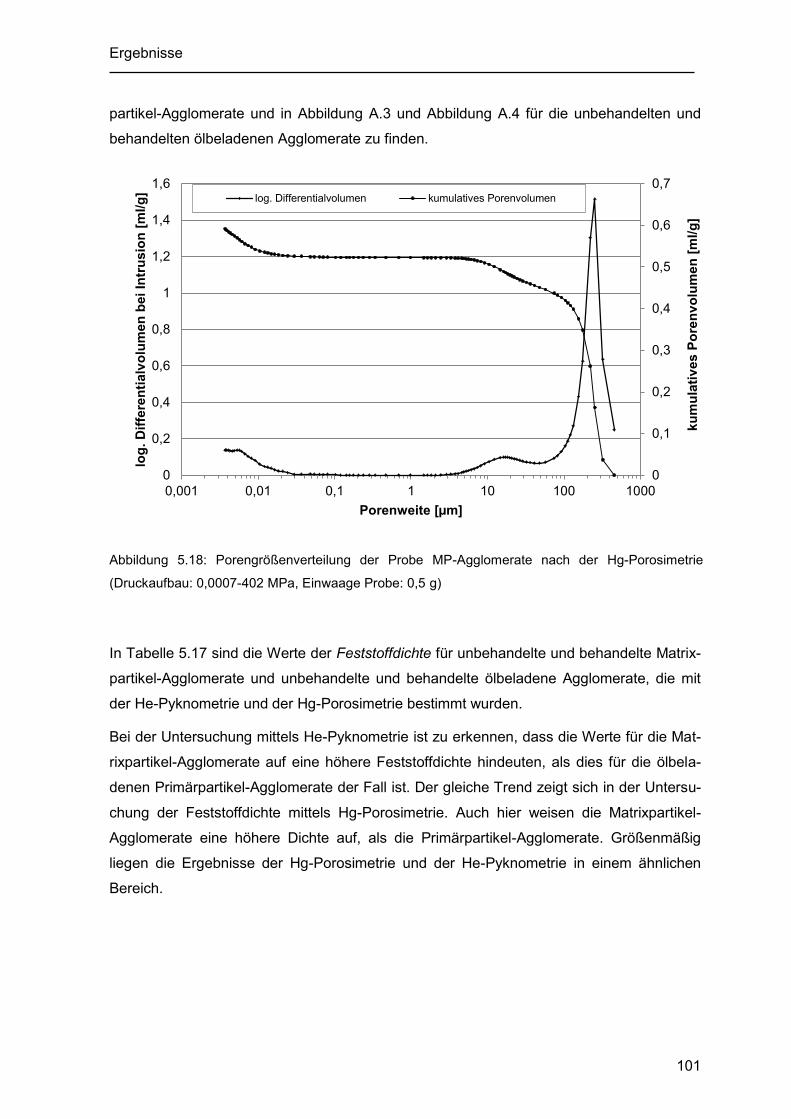
Ergebnisse
101
partikel-Agglomerate und in Abbildung A.3 und Abbildung A.4 für die unbehandelten und
behandelten ölbeladenen Agglomerate zu finden.
Abbildung 5.18: Porengrößenverteilung der Probe MP-Agglomerate nach der Hg-Porosimetrie
(Druckaufbau: 0,0007-402 MPa, Einwaage Probe: 0,5 g)
In Tabelle 5.17 sind die Werte der Feststoffdichte für unbehandelte und behandelte Matrix-
partikel-Agglomerate und unbehandelte und behandelte ölbeladene Agglomerate, die mit
der He-Pyknometrie und der Hg-Porosimetrie bestimmt wurden.
Bei der Untersuchung mittels He-Pyknometrie ist zu erkennen, dass die Werte für die Mat-
rixpartikel-Agglomerate auf eine höhere Feststoffdichte hindeuten, als dies für die ölbela-
denen Primärpartikel-Agglomerate der Fall ist. Der gleiche Trend zeigt sich in der Untersu-
chung der Feststoffdichte mittels Hg-Porosimetrie. Auch hier weisen die Matrixpartikel-
Agglomerate eine höhere Dichte auf, als die Primärpartikel-Agglomerate. Größenmäßig
liegen die Ergebnisse der Hg-Porosimetrie und der He-Pyknometrie in einem ähnlichen
Bereich.
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0
0,2
0,4
0,6
0,8
1
1,2
1,4
1,6
0,001 0,01 0,1 1 10 100 1000
kum
ulat
ives
Por
envo
lum
en [m
l/g]
log.
Diff
eren
tialv
olum
en b
ei In
trus
ion
[ml/g
]
Porenweite [µm]
log. Differentialvolumen kumulatives Porenvolumen

Ergebnisse
102
Tabelle 5.17: Gegenüberstellung der Feststoffdichten von unbehandelten Matrixpartikel-
Agglomeraten, in der Wirbelschicht behandelten Matrixpartikel-Agglomeraten, unbehandelten ölbe-
ladenen Agglomeraten und in der Wirbelschicht behandelten ölbeladenen Agglomeraten, welche
mittels zwei verschiedener Messverfahren ermittelt wurden
Feststoffdichte [g/cm³]
Probe He-Pyknometrie Hg-Porosimetrie
MP-Agglomerate 1,39 ± 0,01 1,41
MP-T60-t60 1,39 ± 0,00 1,41
MP-H2O-T60-t60 1,39 ± 0,00 1,40
PP-Agglomerate 1,31 ± 0,00 1,33
PP-T60-t60 1,30 ± 0,00 1,30
PP-H2O-T60-t20 1,29 ± 0,00 1,33
PP-H2O-T60-t60 1,27 ± 0,00 1,26

Ergebnisse
103
5.3.4 Charakterisierung der Barriereeigenschaften von MC-Coatings auf ölbeladenen Agglomeraten
Bislang wurden die Einflüsse der unterschiedlichen Prozessparameter (Zeit-Temperatur-
Kombinationen und Behandlung mit und ohne Wasser) auf den Verlauf der Hydroperoxid-
konzentration, auf die Verteilung des verkapselten Rapsöls und die Partikelstruktur hin un-
tersucht. Nun sollen die Ergebnisse der Untersuchungen dargestellt werden, mit denen die
Barriereeigenschaften von MC gegenüber Sauerstoff und Wasserdampf charakterisiert
wurden. Auch hierbei werden die Ergebnisse der Untersuchungen zum Verlauf der Hydro-
peroxidkonzentration, der Öltropfengröße und –verteilung und der Mikroverkapselungseffi-
zienz, Feststoffdichte und der REM-Untersuchungen gezeigt. Weiterhin werden zur Cha-
rakterisierung der Barriereeigenschaften gegenüber Wasserdampf die Ergebnisse der sta-
tischen Wasserdampfsorption dargestellt. Der Ablauf des Coatingprozesses konnte vor der
Durchführung dieser Versuchsreihe gegenüber dem Vorversuch durch Optimierung der
Düseneinstellung deutlich verbessert werden.
Die Auswertung der Verteilungsparameter und Verteilungsbreite der Öltropfengrößenbe-
stimmung der Primäremulsionen, sprühgetrockneten Partikel, Agglomerate und der mit
verschiedenen Prozessbedingungen gecoateten Agglomerate zeigen diverse Unterschiede
(siehe Tabelle 5.18). Für die gecoateten Agglomerate zeigt sich ein Einfluss der Coating-
dauer auf d50, d90 und den span. Je länger die Proben (t60 gegenüber t20) in der Wirbel-
schicht gecoatet werden, desto kleiner sind die Werte für d50, d90 und span.
Auch ist ein Einfluss der Probenherstellung auf die Verteilungsparameter und die Vertei-
lungsbreite feststellbar. Gegenüber der Emulsion weist das sprühgetrocknete Pulver deut-
lich niedrigere Werte für die Verteilungsparameter bzw. die Verteilungsbreite auf. Durch
das anschließende Agglomerieren sind leicht erhöhte Werte für die Verteilungsparameter
und Verteilungsbreite zu erkennen.

Ergebnisse
104
Tabelle 5.18: Charakteristische Verteilungsparameter (d10, d50 und d90) und Verteilungsbreite (span)
der Öltropfengrößenbestimmung mittels SLS der Primäremulsion, dem sprühgetrockneten Pulver,
den Agglomeraten und verschiedener in der Wirbelschicht gecoateter und resolubilisierter Agglome-
rate
d10 [µm] d50 [µm] d90 [µm] span
Emulsion 1,15 ± 0,0 2,09 ± 0,1 4,00 ± 0,3 1,36 ± 0,1
Pulver 0,90 ± 0,0 1,49 ± 0,0 2,57 ± 0,0 1,12 ± 0,0
Agglomerate 1,08 ± 0,0 1,68 ± 0,0 2,71 ± 0,0 0,97 ± 0,0
MC15-T30-t20 1,20 ± 0,0 2,35 ± 0,1 5,03 ± 0,4 1,62 ± 0,1
MC15-T30-t60 1,07 ± 0,2 1,86 ± 0,2 3,35 ± 0,4 1,22 ± 0,1
MC15-T60-t20 1,22 ± 0,0 2,21 ± 0,1 4,27 ± 0,6 1,38 ± 0,2
MC15-T60-t60 1,18 ± 0,1 1,98 ± 0,1 3,53 ± 0,3 1,18 ± 0,1
Die Ergebnisse der Gesamtfettbestimmung der mit einer 5%igen MC-15-Coatinglösung
gecoateten Agglomerate sind der Tabelle 5.19 zu entnehmen. Hierbei ist zu erkennen,
dass es keinen Einfluss der Prozesstemperatur auf die berechnete Coatingmenge gibt. Die
Prozessdauer und damit die Zeit, in der die Coatinglösung auf die Agglomerate aufgetra-
gen wurde, weist einen Einfluss auf die berechnete Coatingmenge auf. Hierbei zeigt sich,
dass Agglomerate, die länger gecoatet wurden, eine deutlich höhere aufgetragene
Coatingmenge enthielten. Die theoretische Coatingmenge, die für die 20 Minuten lang ge-
coateten Agglomerate berechnet wurde, liegt bei 2,5 % und zeigt somit nur leichte Abwei-
chungen von der tatsächlich aufgebrachten Coatingmenge. Für die 60 Minuten lang gecoa-
teten Agglomerate wurde die theoretische Coatingmenge mit 5 % berechnet, somit liegt die
tatsächlich aufgetragene Coatingmenge mit 4,9 ± 2,6 % sehr nah am theoretisch berechne-
ten Wert. Die Agglomerate, die in diesem Versuch verwendet wurden, wiesen einen Ge-
samtfettgehalt von 21,3 ± 1,0 % auf.

Ergebnisse
105
Tabelle 5.19: Gesamtfettgehalt und berechnete applizierte Coatingmenge von unterschiedlich ge-
coateten Agglomeraten
Gesamtfettgehalt [%] berechnete Coatingmenge [%]
MC15-T30-t20 21,0 ± 0,0 1,6 ± 0,1
MC15-T30-t60 20,3 ± 0,3 4,9 ± 1,2
MC15-T60-t20 21,0 ± 0,2 1,8 ± 1,1
MC15-T60-t60 20,3 ± 0,6 4,9 ± 2,6
Die Werte für die Mikroverkapselungseffizienz des sprühgetrockneten Pulvers lagen bei
97,5 ± 0,7 % und die der Agglomerate bei 98,7 ± 0,1 % Die Betrachtung der Werte der
Mikroverkapselungseffizienz für die gecoateten Agglomerate lässt keinen Einfluss von Pro-
zesszeit oder -temperatur erkennen. Alle Werte liegen im Bereich von 99,2 ± 0,1 % bis
99,8 ± 0,0 % (Tabelle 5.20). Damit zeigen die Werte der gecoateten Agglomerate eine
deutlich höhere Mikroverkapselungseffizienz als die ungecoateten Agglomerate.
Tabelle 5.20: Mikroverkapselungseffizienz von Agglomeraten, die mit verschieden Zeit-Temperatur-
Kombinationen gecoatet sind
Verkapselungseffizienz [%]
MC15-T30-t20 99,7 ± 0,0
MC15-T30-t60 99,8 ± 0,0
MC15-T60-t20 99,2 ± 0,1
MC15-T60-t60 99,6 ± 0,0
Die Aufnahmen mittels Rasterelektronenmikroskop in Abbildung 5.19 zeigen, dass der ap-
plizierte Film an MC auf den Partikeln bei einer Auftragungszeit (Prozesszeit) von 20 Minu-
ten (Bild A und B) etwas dünner ist, als bei Agglomeraten, die für 60 Minuten in der Wirbel-
schicht mit MC gecoatet wurden (C und D).

Ergebnisse
106
Abbildung 5.19: Aufnahmen des Rasterelektronenmikroskops von MC15 gecoateten Agglomeraten;
A, B: MC15-T30t20 ; C, D: MC15-T60t60; A, C: Vergrößerung: 500-fach; B: Vergrößerung 4000-
fach; D: Vergrößerung: 3000-fach
Die Abbildung 5.20 zeigt den zeitlichen Verlauf der Hydroperoxikonzentrationen von
ungecoateten und mit einer 5%igen MC-Lösung gecoateten Agglomeraten über einen
zeitlichen Verlauf von 18 Wochen.
Der Startwert der Hydroperoxidkonzentration des sprühgetrockneten Pulvers zu Beginn der
Untersuchung (0,66 ± 0,2 mmol/kg Öl) lag in einer ähnlichen Größenordnung wie der der
ungecoateten und gecoateten Agglomerate, die zwischen 1,1 ± 0,5 mmol/kg Öl (unge-
coatet) und 3,3 ± 1,0 mmol/kg Öl (T60-t60) lagen.
Die Hydroperoxidkonzentrationen zeigen nach 18 Wochen Lagerzeit Werte, die eine Grup-
pierung der Proben erlaubt. So zeigen die unbehandelten Agglomerate und die 20 Minuten
lang bei 30 °C gecoateten Agglomeraten mit 20,1 ± 0,8 bzw. 21,6 ± 0,2 mmol/kg Öl sehr
ähnliche Werte der Hydroperoxidkonzentration. Die bei 60°C für 20 Minuten und die bei
A B
D C

Ergebnisse
107
30 °C und 60 Minuten gecoateten Agglomerate zeigen Werte von 31,5 ± 0,7 bzw.
33,2 ± 3,7 mmol/kg Öl und liegen somit auch in einem ähnlichen Bereich. Die 60 Minuten
lang bei 60 °C gecoateten Agglomerate weisen einen Wert für die Hydroperoxidkonzentra-
tion von 45,5 ± 3,0 mmol/kg Öl auf und zeigen damit den höchsten Wert der Hydroperoxid-
konzentration. Die gesamten Werte zur Abbildung 5.20 finden sich im Anhang in der Tabel-
le A.32.
Abbildung 5.20: Hydroperoxidkonzentration der unbehandelten Agglomerate und der bei 30°C und
60°C über 20 bzw. 60 Minuten mit einer 5%igen MC-Lösung gecoateten Agglomerate über den Zeit-
raum von 18 Wochen mit Untersuchungen alle zwei Wochen
In Tabelle 5.21 sind die Ergebnisse der Feststoffdichtenbestimmung der bei 30 °C bzw.
60 °C und 20 bzw. 60 Minuten gecoateten Agglomerate dargestellt.
Hier zeigt sich, dass die Agglomerate, die bei 30 °C für 20 Minuten gecoatet wurden mit
1,30 ± 0,0 g/cm3 den höchsten Wert der Feststoffdichte aufweisen. Die niedrigste Fest-
stoffdichte mit 1,26 ± 0,0 g/cm3 zeigen die Agglomerate, die bei 60 °C für 60 Minuten ge-
coatet wurden. Eine Unterscheidung der bei 30 °C 60 Minuten lang bzw. der bei 60 °C für
20 Minuten gecoateten Agglomerate ist nicht möglich.
0
10
20
30
40
50
60
0 2 4 6 8 10 14 18
Hyd
rope
roxi
dgeh
alt [
mm
ol/k
g Ö
l]
Zeit [Wochen]
Agglomerate
MC15-T30-t20
MC15-T30-t60
MC15-T60-t20
MC15-T60-t60

Ergebnisse
108
Tabelle 5.21: Darstellung der Feststoffdichte der 20 bzw. 60 Minuten und bei 30 °C bzw. 60 °C ge-
coateten Agglomerate
Feststoffdichte [g/cm3]
MC15-T30-t20 1,30 ± 0,0
MC15-T30-t60 1,28 ± 0,0
MC15-T60-t20 1,28 ± 0,0
MC15-T60-t60 1,26 ± 0,0
Der Untersuchung des Wasseraufnahmeverhaltens der ungecoateten und gecoateten Ag-
glomerate ging eine Vortrocknung voraus, um die Proben möglichst gut für den anschlie-
ßenden Versuch zur Wasseraufnahme vorzubereiten. Es ist in Tabelle 5.22 zu erkennen,
dass die Agglomerate den höchsten Wert der Wasserabgabe zeigen. Die gecoateten Ag-
glomerate zeigen eine Staffelung der Werte. So liegen die bei 30 °C Prozesstemperatur
gecoateten Agglomerate in derselben Größenordnung für die Wasserabgabe. Gleiches ist
für die Agglomerate, die bei 60 °C gecoatet wurden, zu erkennen. Darüber hinaus kann
zusammengefasst werden, dass je höher die Prozesstemperatur während des Coatingpro-
zesses ist, desto weniger Feuchtigkeit gaben die Proben während des Vortrocknens ab.
Die Beobachtungen des Vortrocknungsvorganges lassen sich auch auf die Werte der mit-
tels Schnellfeuchtebestimmer bestimmte Trockenmassen übertragen. Um dies zu veran-
schaulichen, sind die Werte für die Restfeuchte und die Werte für die abgegebe Wasser-
menge während der 21 Tage Vortrocknungsdauer in Tabelle 5.22 gegenübergestellt. Hier
zeigt sich, dass die abgegebene Wassermenge während der Vortrocknung in etwa mit der
durch den Schnellfeuchtebestimmer aus den Proben entfernten Wassermenge überein-
stimmt.

Ergebnisse
109
Tabelle 5.22: Restfeuchte nach Schnellfeuchtebestimmung und abgegebene Wassermenge wäh-
rend der Vortrocknung bei 0% relative Luftfeuchte nach 21 Tage von ungecoateten und in verschie-
denen Zeit-Temperatur-Kombinationen gecoateten Agglomeraten
Restfeuchte [%] Wasserabgabe in Vortrocknung [%]
Agglomerate 8,33 10,34
MC15-T30-t20 6,41 6,50
MC15-T30-t60 5,11 7,38
MC15-T60-t20 3,78 4,15
MC15-T60-t60 3,12 3,73
In Abbildung 5.21 ist der zeitliche Verlauf der Wasserdampfsorption der ungecoateten und
in verschiedenen Zeit-Temperatur-Kombinationen mit MC gecoateten Agglomerate darge-
stellt. Dabei ist zu erkennen, dass die Agglomerate bis Tag 22 (75 % rel. Feuchte) konstant
den höchsten Wert an Wasserdampfsorption zeigen.
Abbildung 5.21: Wasserdampfsorption der ungecoateten Agglomerate und der mit Methylzellulose in
verschiedenen Zeit-Temperatur Kombinationen gecoateten Agglomerate bei der Lagerung in ver-
schiedener relativer Luftfeuchtigkeit bei 20 ± 0,5 °C über einen Zeitraum von 35 Tagen
Ab dem Zeitpunkt zeigen die MC15-T30t20 gecoateten Agglomerate eine leicht höhere
Wasserdampfsorption als die Agglomerate. Nach 35 Tagen liegen eben diese beiden Pro-
ben ungefähr in der gleichen Größenordnung mit Werten von 18,0 bzw. 18,2 % Wasser-
0
0,02
0,04
0,06
0,08
0,1
0,12
0,14
0,16
0,18
0,2
0 1 2 3 4 7 8 9 10 11 12 13 14 15 16 20 21 22 23 24 28 29 30 31 32 35
11 33 53 75 85
Was
sera
ufna
hme
[g/g
]
Lagerzeit [Tage] mit jeweiliger relativer Luftfeuchtigkeit
Agglomerate
MC15-T30-t20
MC15-T30-t60
MC15-T60-t20
MC15-T60-t60

Ergebnisse
110
dampfsorption. Die geringste Menge an Wasserdampf nach 35 Tagen Lagerzeit hat die
Probe aufgenommen (16,7 %), die bei 60 °C 60 Minuten lang im Wirbelschichtprozess ge-
coatet wurde. Die Proben, die bei 30 °C 60 Minuten und bei 60 °C 20 Minuten lang ge-
coatet wurden, liegen mit Werten von 17,4 bzw. 17,8 % Wasserdampfsorption im Mittelfeld
der Proben. Die Werte für die Wasserdampfsorption der einzelnen Untersuchungstage
während der Lagerzeit sind in Tabelle A.33 im Tabellenanhang zu finden. Die Werte der
Tage, an denen die Proben die Gewichtskonstanz in der jeweiligen relativen Feuchtigkeit
erreicht haben, sind in Tabelle 5.23 dargestellt. Hier ist noch einmal deutlich erkennbar,
dass die Wasseraufnahme der bei 60 °C für 60 Minuten gecoateten Agglomerate von An-
fang an am niedrigsten war.
Tabelle 5.23: Werte für die Wasseraufnahme für die jeweilige relative Feuchtigkeit, dargestellt am
jeweiligen Tag der Gewichtskonstanz, Lagerung bei 20 ± 0,5 °C
Wasseraufnahme (g/g Probe)
11 %
(Tag 8)
33 %
(Tag 12)
53 %
(Tag 20)
75 %
(Tag 28)
85 %
(Tag 35)
Agglomerate 0,024 0,050 0,079 0,106 0,180
MC15-T30-t20 0,020 0,047 0,075 0,108 0,182
MC15-T30-t60 0,015 0,041 0,068 0,101 0,174
MC15-T60-t20 0,022 0,044 0,069 0,103 0,178
MC15-T60-t60 0,018 0,044 0,061 0,097 0,167
In Abbildung 5.22 sind Bilder der Proben aus dem Lagerversuch zur Bestimmung der Was-
serdampfsorption von ungecoateten und mit MC in verschiedenen Zeit-Temperatur-
Kombinationen gecoateten Agglomeraten nach 30 Tagen Lagerzeit zusammengetragen.
Die Proben haben zu diesem Zeitpunkt die Vortrocknung und die Stufen der relativen Luft-
feuchtigkeit von 11%, 33%, 53%, 75% und zwei Tage bei 85% relativer Luftfeuchte durch-
laufen. Die gecoateten Agglomerate (Bild 2 und 3; bei 30 °C; Bild 3 und 4; bei 60 °C) sind
in dieser Phase noch als vereinzelte Partikel zu erkennen und waren rieselfähig. Die unge-
coateten Agglomerate zeigten bei 85 % relativer Luftfeuchte bereits Auflösungserschei-
nungen und waren nicht mehr als einzelne Partikel zu erkennen bzw. rieselfähig. Der La-
gerversuch musste nach dem Erreichen des Ausgleichszustandes bei 85 % relativer Luft-
feuchte abgebrochen werden, da sich auf den ungecoateten Agglomeraten bei dieser Luft-
feuchte anfing Schimmel auf der Probe zu bilden.

Ergebnisse
111
Abbildung 5.22: Aufnahmen der Proben zur Wasserdampfsorptionsmessung nach Durchlaufen von
Feuchtigkeitsstufen 11, 33, 53, 75 und 85 % relativer Luftfeuchtigkeit mit: 1: ungecoatete Agglomera-
te, 2: MC15-T30-20, 3: MC15-T30t60, 4: MC15-T60t20, 5: MC15-T60t60
1
2 3
4 5

Ergebnisse
112
5.3.5 Untersuchung der Sauerstoffbarriereeigenschaft von MC-Filmen auf Modelloberflächen
Abbildung 5.23 zeigt die Hydroperoxidkonzentration von gecoateten und ungecoateten
Modelloberflächen über einen Zeitraum von 10 Wochen.
Die Werte für die Hydroperoxidkonzentration der Startwoche der 10-wöchigen Lagerung
liegen mit Werten von 1,4 ± 0,5 mmol/kg Öl für die ungecoateten Modelloberflächen und
2,3 ± 0,9 mmol/kg Öl sehr dicht beieinander. Nach 10 Wochen Lagerzeit zeigen die gecoa-
teten Modelloberflächen mit 16,9 ± 0,9 mmol/kg Öl einen leicht höheren Wert als die unge-
coateten Modelloberflächen mit 14,0 ± 0,7 mmol/kg Öl.
Die Werte für die Hydroperoxidkonzentration der gecoateten und ungecoateten Objektträ-
ger sind in Tabelle A.34 im Tabellenanhang aufgeführt.
Abbildung 5.23: Hydroperoxidkonzentration von ungecoateten und gecoateten Modelloberflächen
über einen Lagerzeitraum von zehn Wochen mit Untersuchungen im zweiwöchigen Untersuchungs-
intervall
0
5
10
15
20
0 2 4 6 8 10
Hyd
rope
roxi
dgeh
alt [
mm
ol/k
g Ö
l]
Zeit [Wochen]
ungecoatete Modelloberfläche
gecoatete Modeloberfläche

Diskussion
113
6 Diskussion
6.1 Haftungs- und Benetzungsverhalten von zellulosederivatbasier-ten Coatingmaterialien auf maltodextrinbasierten Modellober-flächen
Bezüglich der Interaktion von zellulosederivatbasierten Coatinglösungen und maltodextrin-
basierten Oberflächen (z.B. MDEMUL) ist wenig in der einschlägigen wissenschaftlichen
Literatur beschrieben. Auch über Untersuchungen zur Oberflächenspannung (SFT) von
zellulosederivatbasierten Coatinglösungen ist wenig bekannt. Lediglich für MC und CMC
liegen Daten aus der Fachliteratur vor. So nennen Schmidt et al. (2012) für eine 1 %ige
(m/m) CMC-Lösung, was in etwa der CMCm-Lösung entspricht, 32,8 mN/m und 28,9 mN/m
für den polaren bzw. dispersen Anteil der SFT. Für eine 2 %ige w/w MC-Lösung, was in
etwa der MCm-Lösung in dieser Arbeit entspricht, berichten Schmidt et al. (2012) von
16,6 mN/m für den dispersen und von 33,0 mN/m für den polaren Anteil der SFT. Diese
Werte liegen in etwa im Rahmen der in dieser Arbeit ermittelten Werte.
Die Ergebnisse für die Bestimmung der freien Oberflächenenergie (SE) von MDEMUL vari-
ieren je nach Kombination der Referenzlösungen, die für die Bestimmung verwendet wur-
den. Die Werte liegen im Bereich von 65 mN/m (SEd=7,7 MN/m bzw. SEp=57,4 mN/m) für
die WEF-Kombination und 51,9 mN/m (SEd=35,7 mN/m bzw. SEp=16,2 mN/m) für die DEF-
Kombination. Die Berechnung der SE, wie sie von Owens und Wendt (1969) beschrieben
wurde, wird in der aktuellen Literatur auch die Zwei-Flüssigkeits-Methode (two-liquid-
method) genannt (Karbowiak, Debeaufort, & Voilley, 2006). Dies rührt daher, dass für die
Bestimmung mindestens zwei Referenzlösungen verwendet werden müssen, die einen
größtmöglichen Unterschied in den polaren und dispersen Eigenschaften aufweisen. Um
eine Eignung von Referenzlösungen für die Berechnung der SE zu bestimmen, hat bereits
Kaelble (1970) einen Wert eingeführt, der sich aus den polaren und dispersen Eigenschaf-
ten der Referenzlösungen ergibt, den D-Wert.
𝐷 =||(√𝛾𝐿
𝑑)𝑖
(√𝛾𝐿𝑝
)𝑖
(√𝛾𝐿𝑑)
𝑗
(√𝛾𝐿𝑝
)𝑗
|| (5.1)
In seiner Studie beschreibt Kaelble, dass die Verwendung von Referenzlösungen nur dann
zulässig ist, wenn |D| oberhalb von 10 mN/m liegt. Im Rahmen der vorliegenden Arbeit
konnte jedoch gezeigt werden, dass die Verwendung von nur zwei Referenzlösungen nicht
zu verlässlichen Ergebnissen bezüglich der SE-Berechnung führte, da hierbei nur die In-

Diskussion
114
teraktion von zwei Referenzlösungen mit unterschiedlichen polaren und dispersen Anteilen
und der Modelloberfläche berücksichtigt wird. Dies ist aber in der Regel nicht ausreichend,
um die Interaktion zwischen Referenzlösungen mit einer breiten Variation von polaren und
dispersen Eigenschaften mit der Modelloberfläche zu charakterisieren und somit einen
verlässlichen Wert für die SE zu berechnen. Die Daten, bei denen zwei Referenzlösungen
zur SE-Bestimmung verwendet wurden, sind in der Arbeit aufgrund ihrer unzureichenden
Präzision nicht gezeigt. Die Werte der SE streuen noch weiter über den Bereich der mit
drei bzw. vier Referenzlösungen bestimmten SE und zeigen Werte von 70,1 mN/m
(SEd=5,7 MN/m bzw. SEp=64,4 mN/m) für die EF-Kombination und 46,4,9 mN/m
(SEd=37,9 mN/m bzw. SEp=8,5 mN/m) für die DE-Kombination. So erreicht z.B. die Kombi-
nation von W und E zwar einen D-Wert von 38,7 mN/m und wäre somit für die SE-
Charakterisierung geeignet. Jedoch zeigt die mit dieser Kombination errechnete SE einen
Wert von 65,1 mN/m (SEd=7,3 mN/m bzw. SEp=57,8 mN/m) und liegt somit am obersten
Ende der Ergebnisse, die unter Verwendung von drei bzw. vier Referenzlösungen für die
SE errechnet wurden. Dies zeigt eindeutig, dass beide Voraussetzungen in Betracht gezo-
gen werden müssen: Ein D-Wert von über 10mN/m, aber auch ein Unterschied in polaren
und dispersen Anteilen der SFT der Referenzlösungen. Diese Annahme wird gestützt
durch die Werte für die SE bezüglich der Kombinationen WEF und DEF, welche die nied-
rigsten und höchsten Werte für die SE zeigen. Offensichtlich führt das Fehlen einer Refe-
renzlösung mit niedrigem polaren und hohen dispersen Anteil der SFT zu einem über-
schätzt hohen Wert für die SE und einem sehr niedrigen dispersen Anteil der SE (siehe
WEF). Dem entgegen führt das Fehlen einer Referenzlösung mit einem hohen polaren und
einem niedrigen dispersen Anteil der SFT zu einer Unterschätzung der SE und ihrem pola-
ren Anteil. Alle Kombinationen, die W und D enthalten, führen zu vergleichbaren Ergebnis-
sen für die SE und können dabei als verlässlich angesehen werden. Mit einer SE von ca.
60 mN/m und einer relativ gleichmäßigen Verteilung an polaren und dispersen Anteilen
(27,0 - 28,9 mN/m und 30,0 - 35,1 mN/m) für MDEMUL sind die Werte der von kompaktier-
ter Laktose relativ ähnlich. Die Werte für kompaktierte Laktose, die ein Disaccharid ist, wie
ein gewisser Anteil im hier verwendeten Maltodextrin auch, wurden mittels Wilhelmy-Platte
von Pinto et al. (1995) erfasst und liegen in deren Studie bei 17,3 mN/m bzw. 44,9 mN/m
für die polaren und dispersen Anteile der SE. Für Laktose konnte hierbei gezeigt werden,
dass die SE sehr stark variiert, je nach verwendeter Modelloberfläche und es zu sogenann-
ten „hot-spots“ (Bereiche mit hochgradig abweichender SE) kommen kann. Zusätzlich kann
die elektrostatische Wechselwirkung zwischen den einzelnen Partikeln, wie sie bei kom-
paktierten Partikeln häufig vorkommen, die SE beeinflussen (Kou, Chan, Steckel, & Heng,
2012). Schlussendlich führen Unterschiede in der Anzahl der reduzierenden Gruppen und

Diskussion
115
die unterschiedliche Struktur zu dem Fakt, dass ein Vergleich mit den Literaturwerten eine
Aussage über die Größenordnung der SE geben, aber nicht im Detail verglichen werden
kann.
Die anderen Modelloberflächen die im Rahmen dieser Arbeit untersucht wurden, wiesen
sehr ähnliche Werte in der SE im Vergleich zu MDEMUL auf (siehe Tabelle A.6 - Tabelle
A.8). Der Unterschied zwischen der MDGSS-Oberfläche und der MDOSA-Oberfläche kann
durch die geringen Mengenverhältnisse der OSA-Stärke in der Oberfläche erklärt werden.
Der Unterscheid zwischen den SE-Werten der MDEMUL- und der MDGSS-Oberfläche
kann dadurch erklärt werden, dass zwar eine Ölbeladung in der Oberfläche vorhanden war,
jedoch die Verkapselung durch die OSA-Stärke so effizient war, dass durch die geringe
Menge an Oberflächenfett keine Veränderung der SE festgestellt werden konnte. Mit dem
gleichen Ansatz kann auch der fehlende Unterschied zwischen MDT und MDEMUL erklärt
werden. Da in die Berechnung der WA nach OWRK nur die SE als Parameter der Modell-
oberflächen eingeht, kann somit auf eine weitere detaillierte Betrachtung der Partikel-
Flüssigkeits-Interaktion zwischen den in dieser Arbeit verwendeten Coatingmaterialien und
Modelloberflächen mit Ausnahme MDEMUL verzichtet werden. Gleiches gilt für die Be-
rechnung des Kontaktwinkels nach OWRK (CAOWRK) für die Parameter der verschiedenen
Modelloberflächen.
Für die Charakterisierung der Partikel-Flüssigkeits-Interaktion geht auf Seiten der Modell-
oberfläche die SE ein, während auf Seiten der Coatinglösung die SFT eingeht. Genau aus
dieser Kombination ergibt sich hinsichtlich der Berechnung des Benetzungs- und Haf-
tungsverhaltens der Coatinglösung auf der Modelloberfläche mittels OWRK-Modell eine
Problematik. Bezüglich der Benetzung der Modelloberfläche berechnete die OCA-Software
über das OWRK-Modell CAOWRK-Werte von 0° für MC und N. Wie aus Formel 4.10 ersicht-
lich ist, beruht die Berechnung des CAOWRK auf der Winkelfunktion arcos. Hier wird der ar-
cos aus einem Term, der Werte für die polaren und dispersen Anteile der SFT der Coating-
lösungen und die polaren und dispersen Anteile der SE von MDEMUL enthält, berechnet.
Daraus ergibt sich ein Nachteil des OWRK-Modells. Der arcos ist nur in den Grenzen von
[-1;1] definiert. Somit wird die Berechnung des CAOWRK im Falle einer unpassenden Kombi-
nation aus polaren und dispersen Anteilen von SFT und SE keine Werte liefern. Der Wert
0 ° ist in diesem Fall mathematisch unkorrekt, jedoch gibt die OCA-20-Software in diesem
Fall keinen n.d. (nicht definiert) Ausdruck an, weshalb hier der Wert 0 ° angezeigt wird.
Als Konsequenz daraus, dass die Auswahl der Referenzlösungen den Wert der SE beein-
flusst, wird auch der Wert von CAOWRK durch diese verändert. Auch für den CAOWRK liefert
die Berechnung nach OWRK für Kombinationen, die W und D enthalten, vergleichbare

Diskussion
116
Ergebnisse mit Ausnahme von HPMC für die WDF-Kombination. Diese Ausnahme kann
mit einer ungünstigen Kombination aus polaren und dispersen Anteilen von SFT und SE
erklärt werden, da die SFT von HPMC verglichen mit der von CMC relativ niedrig ist, je-
doch der disperse Anteil der SFT bei HPMC mit ca. 50 mN/m deutlich höher ist als der von
CMC mit ca. 28 mN/m. Die Unterschiede zwischen CAm und CAOWRK fallen für CMC gene-
rell sehr gering aus (33,3 ° - 42 °für CAm; 34,5 ° - 41,3 ° für CAOWRK). Die Unterschiede zwi-
schen CAm und CAOWRK (unter Ausklammerung der WDF-Kombination) für HPMC sind
deutlich höher (32,8 ° - 44,9 ° für CAm; 22,8 ° - 31,8 ° für CAOWRK). Diese Unterschiede las-
sen sich zum einen auf die Kombination von SE und SFT mit polaren und dispersen Antei-
len zurückführen und zum anderen auch auf Phänomene der physikalischen Struktur der
Modelloberfläche, welche im Modell nach OWRK nicht berücksichtigt werden können. Um
die Berechnung der SE so robust wie möglich zu machen, empfiehlt es sich für die Be-
rechnung des CAOWRK in diesem Fall die Kombination WDEF zu nutzen. Wie von Young
(1805) bereits beschrieben, bedeutet ein CA zwischen 0 ° und 90 ° eine partielle Benet-
zung. Somit zeigen alle in dieser Arbeit verwendeten Coatinglösungen ein Verhalten von
partiellem Benetzen auf MDEMUL.
Die Berechnung der WA-OWRK basiert auf der Bestimmung der SE inklusive ihrer dispersen
und polaren Anteile. Daher ist die Berechnung der WA-OWRK auch abhängig von der Aus-
wahl der Referenzlösungen zur Bestimmung der SE. Die bereits oben diskutierten Kombi-
nationen WDE und WDF führen genauso wie die Kombination WDEF, führen auch bei der
WA-OWRK zu vergleichbaren Ergebnissen. Hier kann die Studie von Planinsek et al. (2000)
herangezogen werden. Dabei wurde die WA für HPMC-Lösungen und kompaktierter Lakto-
se mittels der Modelle nach Wu, Good-van Oss und Della Volpe bestimmt und die Werte
lagen mit 104,5 mN/m, 89,1 mN/m und 94,6 mN/m zumindest in vergleichbarer Größen-
ordnung zu den Werten der WA, die in dieser Arbeit ermittelt wurden. Des Weiteren stellten
Karbowiak et al. (2006) fest, dass das Modell nach OWRK vergleichbare Werte in der Ad-
häsionsarbeit wie die Modelle nach Fowkes und Zizman liefern.
Vergleiche zwischen WA-OWRK und WA-YD legen nahe, dass Unterschiede zwischen beiden
Werten je nach verwendeter Coatinglösung auftreten. Eine allgemeine Erklärung hierfür ist,
dass die Berechnung von WA-YD auf einer direkten Messung des CA der Coatinglösung auf
MDEMUL basiert. Die Berechnung der WA-OWRK hingegen basiert auf der SFT der Coating-
lösung und deren polaren und dispersen Anteile und der Bestimmung der SE. Die Berech-
nung der SE hängt, wie gezeigt wurde, stark von der Auswahl der Referenzlösungen ab
und berücksichtigt lediglich die Interaktion von MDEMUL und Referenzlösung, nicht jedoch
die Interaktion von MDEMUL und Coatinglösung. Wie zuerst von Young (1805) beschrie-
ben, sind CA-Messungen nur für den Fall verlässlich, wenn gewisse Parameter erfüllt sind.

Diskussion
117
Ergebnisse von CA-Messungen zeigen nur dann den „echten“ CA, wenn die Oberfläche,
auf der gemessen wird, glatt, flach, homogen, inert, nicht reaktiv gegenüber der Flüssigkeit
und nicht verformbar ist. Jedoch haben Chau et al. (2009) zusammengetragen, dass die
meisten der applikationsrelevanten Oberflächen diesen Ansprüchen natürlich nicht gerecht
werden. Demzufolge sollte der gemessene CA als „scheinbarer“ CA angegeben werden,
um überhaupt eine Aussage über die Partikel-Flüssigkeits-Interaktion treffen zu können.
Die in dieser Arbeit verwendeten Modelloberflächen erfüllen natürlich nicht alle Anforde-
rung von Young. Allerdings kann durch die hohe Anzahl an Wiederholungen von jeweils
vier Tropfen auf fünf unterschiedlichen MDEMUL Oberflächen und einer niedrigen Stan-
dardabweichung festgestellt werden, dass der „scheinbare“ CA, der in dieser Arbeit be-
stimmt wurde ein hohes Maß an Reproduzierbarkeit aufweist.
Ein detaillierterer Blick auf die Werte von WA-OWRK und WA-YD zeigt, dass es keinen allge-
meinen Trend für die Werte der WA für die unterschiedlichen Coatinglösungen gibt. Die
Werte für WA-YD und WA-OWRK (für WDEF, WDE und WDF) sind für CMC und HPMC ähnlich,
während für N und MC die Werte zwischen WA-YD und WA-OWRK (für WDEF, WDE und WDF)
stark variieren. Der Grund hierfür mag in den Differenzen der polaren und dispersen Antei-
le der SFT der von N und MC bzw. HPMC und CMC liegen. Auch in der Literatur gibt es
keine Klarheit darüber, woher diese Unterschiede genau rühren. Michalski et al. (1998)
untersuchten verschiedene Öle auf ihre WA auf Oberflächen, wie Glas, PET, LDPE und
Stahl und verwendeten dabei sechs unterschiedliche Modelle, unter anderem auch YD und
Owens-Wendt. Die Autoren dieser Studie konnten jedoch auch keinen Trend in Hinblick auf
Unterschiede der WA verschiedener Öle auf o.g. Oberflächen feststellen, vermuten aber
einen Einfluss der rheologischen Eigenschaften der Öle.
Der Wetting Envelope zeigt, dass Nutrateric® und MC unterhalb der 0 °-Linie liegen. Dies
zeigt, dass aufgrund von unpassender Kombination von polaren und dispersen Anteilen
von SE und SFT mittels OWRK-Modell kein Kontaktwinkel für beide Lösungen berechnet
werden konnte. Der direkt gemessene Kontaktwinkel zeigt für alle vier zellulosederivatba-
sierten Oberflächen einen Wert, der auf ein gutes Spreiten hinweist. Nutzt man den direkt
gemessenen Kontaktwinkel also als ergänzende Information zum mittels OWRK erstellten
Wetting Envelope, kann eine ganzheitliche Betrachtung zum Haftungs- und Spreitungsver-
halten der Coatinglösungen vorgenommen werden. So zeigt sich, dass alle vier Materialien
einen niedrigen Kontaktwinkel von ungefähr 30 ° aufwiesen. Zusammen mit der Betrach-
tung des Haftungskorridors im Wetting Envelope zeigt sich, dass MC neben Nutrateric®
diesem am nächsten ist und sich somit gegenüber der HPMC und der CMC absetzt.

Diskussion
118
Zusammenfassend kann gesagt werden, dass aufgrund der Partikel-Flüssigkeits-
Interaktion, MC auf einem maltodextrinbasierten Trägersystem am besten von den unter-
suchten Coatingmaterialien geeignet ist, um auf diesem gut zu spreiten und zu haften. Ge-
paart mit den vielversprechenden Eigenschaften bezüglich der Barriere gegen Sauerstoff
und Wasserdampf aus der Literatur soll für die nächsten Versuchsreihen die Tauglichkeit
von MC weiter charakterisiert werden.
6.2 Rheologische Charakterisierung der Coatinglösungen
Beide in dieser Arbeit rheologisch charakterisierten MC-Proben zeigen scherverdünnendes
Verhalten. Bei einer Scherbelastung, wie sie beim Zerstäuben auftritt, kann eine höhere
Trockenmasse und damit eine größere Menge an Coatingmaterial in der Lösung eingesetzt
werden. Dies führt bei gleicher Auftragsmenge auf die Agglomerate zu einer Verkürzung
des Coatingprozesses. Serfert et al. (2013) fanden heraus, dass mit zunehmender Viskosi-
tät der zu zerstäubenden Lösung die Tropfengröße des Sprays zunimmt. Bei einer höheren
Tropfengröße kann es zur Ausbildung von Flüssigkeitsbrücken zwischen den Agglomera-
ten kommen, zur ungewollten Agglomeration und somit die Qualität des Coatings abneh-
men (Dewettinck & Huyghebaert, 1999; Laksmana et al., 2009). Demzufolge ist bei der
Zerstäubung von Coatinglösungen im Prozess des Wirbelschichtcoatings eine niedrige
Viskosität bei gleichzeitig hoher Trockenmasse erwünscht.
Um bei gleicher Scherbeanspruchung und gleicher Temperatur eine vergleichbare Viskosi-
tät einzustellen, konnten bei der MC15-Probe mit 8 % und bei der MC383-Probe mit 5 %
TM eingesetzt werden. Dies rührt von einem niedrigeren Molekulargewicht der MC15 her,
sodass diese Probe bei gleicher Trockenmasse in einer Lösung die Viskosität im Vergleich
zur MC383-Probe nicht in gleichem Maße erhöhen kann (Bayer & Knarr, 2012). Für die
Werte der scherbelastungsabhängigen Viskosität zeigt sich bei niedrigeren Temperaturen
(30 °C bzw. 45 °C) eine deutlich höhere Viskosität als bei einer Temperatur von 60 °C.
Dies zeigt, dass auch die Verwendung einer höheren Temperatur im Prozess die maximal
mögliche Beladung an Coatingmaterial in der Coatinglösung erhöht.
Um die Prozessstabilität der MC-Lösungen, also den Strukturwiederaufbau nach einer
Scherbelastung wie dem Zerstäubungsprozess, zu überprüfen, wurden Sprungversuche
mit unterschiedlichen Scherbeanspruchungen durchgeführt. Die Probe wurde dabei nach
einer Vorscherungsphase mit niedriger Scherbelastung einer kurzen starken Scherbean-
spruchung ausgesetzt, um im Anschluss daran bei einer niedrigen Scherbelastung den
Strukturwiederaufbau zu ermitteln. Die Scherbelastung wurde der in einer Düse herrschen-

Diskussion
119
den nachempfunden. Der Strukturwiederaufbau nach einer Scherbelastung durch Zerstäu-
bung wurde bislang für MC-Lösungen noch nicht untersucht. Demnach ist eine Einordnung
der in dieser Arbeit ermittelten Werte nicht möglich. Floury et al. (2002) stellten jedoch fest,
dass nach einer Behandlung in einem Hochdruckhomogenisator die Ausgangsviskosität
der MC-Lösungen nicht wieder erreicht wurde. Die Scherbeanspruchung konnten die Auto-
ren unter Zuhilfenahme des semiempirischen Modells von Lander et al. (2000) bei einem
Homogenisierungsdruck von 350 bar zu 109 s-1 bestimmen. Die Größenordnung der Bean-
spruchung ist nicht vergleichbar mit den 103 s-1, die in dieser Arbeit für die Scherbeanspru-
chung in einer Düse beim Zerstäuben angenommen wurden (Mezger, 2010). Allerdings
konnten Floury et al. (2002) auch schon bei der Behandlung mit niedrigerem Homogenisie-
rungsdruck eine anschließende Abnahme der Viskosität der MC-Lösungen feststellen. Die
Autoren konnten weiterhin nachweisen, dass durch die Scherbeanspruchung im Homo-
genisator das Molekulargewicht der MC-Proben abnahm. Dadurch kann die niedrigere Vis-
kosität gegenüber der der unbehandelten MC-Proben erklärt werden. Die Erkenntnisse von
Floury et al. (2002) können auf die in dieser Arbeit durchgeführten Messungen übertragen
werden. Die Werte der Viskosität der MC-Proben erreichten in Abhängigkeit von Tempera-
tur und Konzentration die Ausgangsviskosität erst nach mehr als 5 s bzw. bei höheren
Konzentrationen und Temperaturen nie.
Die Untersuchungen von Floury et al. (2002) konnten durch DSC-Messungen weiterhin
belegen, dass die Scherbeanspruchung keinen Einfluss auf das Gelbildungsverhalten
zeigt. Dies ist kohärent zu den Untersuchungen, die belegen, dass die Gelbildung durch
die Anzahl der Methylgruppen und nicht durch die Molekülgröße beeinflusst wird
(Desbrières, Hirrien, & Ross-Murphy, 2000; Haque & Morris, 1993; Hirrien, Chevillard,
Desbrières, Axelos, & Rinaudo, 1998; Sarkar, 1995)
Neben der Temperatur der Lösung beim Zerstäuben hat auch die Temperatur der Luft im
Wirbelschichtprozess einen Einfluss auf die MC-Lösungen, da ab einer bestimmten Tem-
peratur Strukturierungsmechanismen einsetzen und die MC-Lösung in den Gelzustand
übergeht. Hinsichtlich der Gelpunkttemperatur (GP) zeigten sich zwischen den verschiede-
nen Materialien kaum Unterschiede. Bei beiden Materialien nimmt die GP mit zunehmen-
der Konzentration ab, wie es auch von Tomšič et al. (2008) beschrieben wurde. Diese
Konzentrationsabhängigkeit begründen Li et al. (2002) damit, dass mit zunehmender Kon-
zentration die Energie, die für eine hydrophobe Assoziation zwischen den Fibrillen notwen-
dig ist, abnimmt. Dies drückt sich auch in den Werten für die initiale Strukturierungstempe-
ratur aus. Diese nimmt ebenfalls mit zunehmender Konzentration ab.

Diskussion
120
Hinsichtlich der Strukturentwicklungsgeschwindigkeit der beiden Proben zeigt sich, dass
die Struktur in den niedrigen Konzentrationen bei MC15 um eine Zehnerpotenz schneller
ausgebildet wird als bei MC383. Dieser Unterschied relativiert sich mit zunehmender Kon-
zentration. Letzteres Phänomen ist, analog zu der Konzentrationsabhängigkeit der Gel-
punkttemperatur, darauf zurückzuführen, dass mit zunehmender Konzentration die Ener-
gie, die dem Gesamtsystem zugeführt werden muss, um in den Gelzustand überzugehen,
niedriger ist (L. Li et al., 2002). Damit ist bei vergleichbarer Menge an zugeführter Energie
in Form von Wärme bei höheren Konzentrationen eine höhere Strukturausbildungsge-
schwindigkeit zu beobachten. Die höhere Strukturausbildungsgeschwindigkeit von MC15
gegenüber MC383 ist durch das niedrigere Molekulargewicht zu erklären.
Die Gelfestigkeit zu Beginn der Haltezeit und am Ende der Haltezeit korreliert mit dem Mo-
lekulargewicht. Mit niedrigerem Molekulargewicht nimmt die Gelfestigkeit zu. Beide Materi-
alien haben eine sehr ähnlichen Methylierungsgrad (MC383 29,6% bzw. MC15 28,5%),
wodurch ein Einfluss des Methylierungsgrades auf die Gelfestigkeit nicht untersucht wer-
den konnte.
Die Gelwiederauflösungstemperaturen liegen für beide Materialien unabhängig von der
Konzentration unterhalb der Gelpunkttemperatur. Dieses Phänomen konnte von Tomšič et
al. (2008) ebenfalls mittels Oszillationsversuchen in einem Rheometer, mittels SAXS und
mittels DSC bestätigt werden. Li et al. (2002) beschreiben, dass beim Übergang von einem
Gel in den Lösungszustand zwei Energielevel überwunden werden müssen, was durch
DSC-Messungen bestätigt wurde. Dies bedeutet, dass die Temperatur im Prozess ent-
sprechend gewählt werden muss, damit es zu keiner Gelbildung der Coatinglösung kommt.
Es muss jedoch berücksichtig werden, dass selbst wenn es zu Schwankungen in der Pro-
zesstemperatur kommt, die Gelwiederauflösungstemperatur beachtet werden muss.
In der Literatur wird beschrieben, dass eine Verknüpfung zwischen der Carboxylgruppe der
Zitronensäure und den Hydroxylgruppen des Zellulosederivates stattfindet (Ghanbarzadeh
et al., 2011). Darüber hinaus konnten die Autoren durch die Zugabe von Zitronensäure die
Gaspermeabilität von Maisstärkefilmen minimieren. Der Einfluss der Zitronensäure auf die
Strukturierungseigenschaften zeigt sich bei beiden Proben nicht. Dies steht im Wider-
spruch zu Shimokawa et al. (2009) und Iso & Yamamoto (1970), die beschreiben, dass die
Zitronensäure die Gelbildungstemperatur erniedrigt. Da der Einfluss auf die Strukturie-
rungsparameter der MC-Proben nicht nachgewiesen werden konnte, wird in den Versu-
chen zur Funktionalisierung des Coatings als Barriere gegen Sauerstoff auf Untersuchun-
gen des Einflusses von Zitronensäure verzichtet.

Diskussion
121
Die Zugabe von Ethanol zu einer MC-Coatinglösung hat den Effekt, dass der Film auf der
Oberfläche des Partikels schneller gebildet werden kann, da Ethanol mit einer Siedetempe-
ratur von 78 °C flüchtiger ist als Wasser (Nazan Turhan & Şahbaz, 2004). Die Erhöhung
der Gelpunkttemperatur kann auf eine Störung in der Ausprägung der hydrophoben Wech-
selwirkungen zwischen den Filamenten bei einer Zugabe von Ethanol zurückgeführt wer-
den (Turhan, Şahbaz, & Güner, 2001). Vor dem Hintergrund dieser Arbeit ist es wichtig, für
eine Coatinganwendung auch den Einfluss einer Zugabe von Ethanol auf die Gel-
punkttemperatur und die weiteren strukturbildenden Charakteristika zu untersuchen. Hier-
bei zeigte sich, dass die initiale Strukturierungstemperatur, die Gelpunkttemperatur, sowie
die Gelauflösungstemperatur bei der Zugabe von 5 % bezogen auf die wässrige Phase
ansteigen und die Strukturierungsgeschwindigkeit und die Gelfestigkeit abnahmen. Dies
ermöglicht ein nach oben hin größeres Fenster bei der Temperaturführung in einem Wir-
belschichtcoatingprozess, da die Temperatur im Prozess höher gewählt werden kann, oh-
ne dass die Coatinglösung im Prozess geliert.
Zusammenfassend muss festgestellt werden, dass die rheologischen Eigenschaften der
MC-Lösungen von den äußeren Prozessbedingungen und der Konzentration der Proben
abhängen. Das Auftragen des Coatingmaterials als Gel ist zum einen hinderlich für das
Spreiten des Coatingmaterials auf der Partikeloberfläche und zum anderen nachteilig für
die Barriereeigenschaften, da die Struktur eines Gelnetzwerkes von MC nicht so dicht ge-
legt ist, wie die eng aneinander liegenden Fibrillen in einer Lösung vorliegen (Haque &
Morris, 1993). Daher konnte durch die Bestimmung der Gelpunkttemperatur eine maximal
zulässige Prozesstemperatur ermittelt werden, die keine nachteiligen Auswirkungen auf die
Struktur und damit die Barriereeigenschaften der MC-Materialien haben sollte. Darüber
hinaus wurde herausgefunden, dass durch eine mechanische Beanspruchung der MC-
Lösung strukturelle Veränderungen in der Lösung erzeugt werden können und dass die
Fähigkeit des Strukturwiederaufbaus der MC-Lösungen von der Temperatur und der Kon-
zentration abhängig ist. Daher sollte im Folgenden untersucht werden, inwiefern sich die
vermuteten Einflüsse der Prozessbedingungen auf die Barriereeigenschaften von MC aus-
wirken. Die Proben der MC15 zeigen eine höhere Konzentration bei gleichbleibender Vis-
kosität und es kann somit mehr Material in den Coatingprozess eingebracht werden. Wei-
terhin konnten Park et al. (1993) feststellen, dass ein niedrigeres Molekulargewicht von MC
die Barriereeigenschaften positiv beeinflussen kann. Aus diesen Gründen wird in den Ver-
suchen zur Funktionalisierung des Coatings nur MC15 als Coatingmaterial eingesetzt.

Diskussion
122
6.3 Prozessinduzierter Einfluss auf die Partikelstruktur und die Lip-idoxidation von in der Wirbelschicht behandelten Agglomeraten
Im Vorversuch zeigte das Pulver nach dem Sprühtrocknen einen deutlichen Anstieg der
Hydroperoxidkonzentration gegenüber dem Rapsöl. Dies kann zum einen an dem Luftein-
trag während des Voremulgierens mittels Ultra Turrax liegen, zum anderen auch an der
mechanischen Belastung und der damit einhergehenden Oberflächenvergrößerung wäh-
rend des Homogenisierens (Serfert et al., 2009). Hinzu kommt die Vorschädigung durch
die Prozesstemperaturen, den hohen Luft- und damit Sauerstoffeintrag im Sprühturm und
der eventuellen Freilegung von Oberflächenfett (Baik et al., 2004). Weiterhin zeigte sich,
dass die Agglomerate, die mit MC gecoatet wurden, nach zehn Wochen Lagerung eine
deutlich höhere Hydroperoxidkonzentration aufwiesen als die Agglomerate, die nicht in der
Wirbelschicht gecoatet wurden.
Daraus ergab sich die Hypothese, dass der Prozess des Wirbelschichtcoatens selbst einen
Einfluss auf den Verlauf der Hydroperoxidkonzentration hat. Dieser Einfluss sollte im Fol-
genden untersucht werden.
Zum einen kann der Einfluss auf die Hydroperoxidkonzentration von einer Änderung der
Mikroverkapselungseffizienz und der Öltropfengrößenverteilung herrühren. Diese beiden
Parameter können sich verändern, indem die Oberfläche der Agglomerate durch die
Coatinglösung angelöst und im Anschluss durch die Trocknung im Prozess eine Schicht
bildet, in der eventuell eine Koaleszenz der Öltropfen stattgefunden haben könnte. Die
Mikroverkapselungseffizienz kann für den Verlauf der Lipidoxidation entscheidend sein, da
nicht verkapseltes Öl an der Oberfläche der Atmosphäre und somit dem Sauerstoff ausge-
liefert sein kann. Dieser Einfluss von Mikroverkapselungseffizienz wurde auch in der Litera-
tur mehrfach untersucht und beschrieben (Ahn et al., 2008; Drusch & Berg, 2008; Vega &
Roos, 2006).
Zum anderen kann der Verlauf der Hydroperoxidkonzentration auch durch eine Verwen-
dung unterschiedlicher Prozessparameter zustande kommen. Um diese beiden möglichen
Ursachen der veränderten Hydroperoxidkonzentration zu klären, wurden die Agglomerate
bei verschiedenen Zeit-Temperatur-Kombinationen in der Wirbelschicht behandelt, ohne
ein Coating aufzutragen. Um den Einfluss der Coatinglösung und das damit verbundene
Anlösen der Oberfläche zu simulieren, wurden die Agglomerate zusätzlich entweder mit
Wasser während des Prozesses besprüht oder trocken den Prozessbedingungen ausge-
setzt. Während des Wirbelschichtprozesses verdunstet das Wasser an der Oberfläche der
Agglomerate. Für diese Verdunstung wird der Luft der Umgebung Energie entzogen. Durch
diesen Energiefluss wird die Oberfläche des Agglomerates gekühlt (Uhlemann & Mörl,

Diskussion
123
2000). Durch die permanente Verdunstung der Flüssigkeit kann somit die Temperatur des
Agglomerats niedriger als die Temperatur der Prozessluft gehalten werden (Grassmann,
Widmer, & Sinn, 1997). Somit wird auch das in den Agglomeraten verkapselte Öl nicht den
gleichen Temperaturen ausgesetzt, wie es bei den ohne Wasser behandelten Agglomera-
ten der Fall ist.
Die Agglomerate, die mit Wasser behandelt wurden, weisen eine höhere Mikroverkapse-
lungseffizienz als die ohne Wasser behandelten Agglomerate auf. Jedoch ist die Erhöhung
der Mikroverkapselungseffizienz mit ca. 1 % so gering, dass dieser Parameter nicht als
alleinige Ursache für den niedrigeren Verlauf der Hydroperoxidkonzentration angesehen
werden kann. Die Verteilungsparameter und Verteilungsbreite der Öltropfengröße der mit
Wasser behandelten Agglomerate erhöhen sich gegenüber der trocken behandelten Ag-
glomerate nicht. Demzufolge können die deutlichen Unterschiede zwischen den Proben mit
und ohne Wasserbehandlung auch nicht über die Veränderung der Verteilungsparameter
der Öltropfengröße erklärt werden.
Bei der Betrachtung der Hydroperoxidkonzentrationen der mit und ohne Wasser behandel-
ten Agglomerate zeigte sich, dass eine kürzere Behandlung der Agglomerate mit Wasser
bei 60 °C deutlich niedrigere Hydroperoxidkonzentrationen als die längere Behandlung
liefert, unabhängig davon, ob die Agglomerate mit oder ohne Wasser behandelt wurden.
Generell muss festgestellt werden, dass die Hydroperoxidkonzentrationen der mit Wasser
behandelten Proben einen niedrigeren Verlauf zeigten.
Da die Öltropfengrößenverteilung und Mikroverkapselungseffizienz zwischen den mit und
ohne Wasser behandelten Proben und auch unabhängig von Temperatur und Behand-
lungszeit nicht variierte, konnten diese Parameter als Gründe für einen unterschiedlichen
Verlauf der Hydroperoxidkonzentration ausgeschlossen werden. Andererseits sind die Pro-
zesstemperatur und -zeit sowie die Behandlung mit und ohne Wasser offensichtliche Ein-
flussgrößen auf den Verlauf der Lipidoxidation. Um zu überprüfen, ob die Prozessparame-
ter einen Einfluss auf die Partikelstruktur haben, wurden erneut ausgewählte Proben aus
dem vorherigen Set hergestellt und auf den Verlauf der Hydroperoxidkonzentration hin un-
tersucht. Darüber hinaus wurden Untersuchungen der Oberflächenbeschaffenheit in Form
von Porengröße und -verteilung und die visuelle Beurteilung mittels Rasterelektronenmik-
roskopie durchgeführt. Um die Oberflächenbeschaffenheit weiter zu quantifizieren, wurde
die spezifische Oberfläche und Feststoffdichte untersucht. Hierbei wurde vermutet, dass
das Anlösen der Oberfläche zu einem Verschließen der Poren auf der Oberfläche der Ag-
glomerate führt und somit die Porengröße und -verteilung beeinflusst. Sollte sich dies be-
stätigen, wäre davon auszugehen, dass die trocken behandelten Agglomerate eine höhere

Diskussion
124
Porosität besitzen und somit eine größere Oberfläche. In den vorhandenen Poren wäre
damit auch mehr Oberflächenfett zu finden. Drusch & Schwarz (2006) bestätigen, dass
eine erhöhte Porosität der Partikel zu einer schlechteren Oxidationsstabilität führen kann.
Die Untersuchungen der Feststoffdichte sollten zeigen, ob es durch die Behandlung in der
Wirbelschicht zu einer Bildung von Hohlräumen innerhalb der Partikel gekommen ist, die
für Quecksilber (Porositätsmessung) und Petrolether (Mikroverkapselungseffizienzbestim-
mung) nicht zugänglich sind und somit bei der Messung der Porosität und des freien Ober-
flächenfettes nicht erfasst worden sind.
Die Aufnahmen der Rasterelektronenmikroskopie zeigen, dass die Oberfläche der mit
Wasser behandelten Agglomerate deutlich glatter erscheint als die Oberfläche der trocken
behandelten Proben. Die Rauigkeit der trocken behandelten Agglomerate lässt sich in ers-
ter Linie über statische Aufladung erklären. Die kleinen Anhaftungen auf der Oberfläche
der Agglomerate sind feinste Pulverpartikel, die im Prozess der Wirbelschichtbehandlung
von den Agglomeraten abgerieben wurden und sich anschließend auf der Oberfläche ab-
setzen.
Die Gasadsoptionsmessung mit Stickstoff brachte keine Ergebnisse, so dass die Gasad-
sorption mit Krypton wiederholt werden musste, um mittels BET-Auswertung eine spezifi-
sche Oberfläche zu bestimmen. Die Auswertung ergab jedoch keinen Unterschied in der
Oberfläche und lag zudem mit ca. 0,1 m2/g unterhalb des für das Gerät angegeben Mess-
bereiches. Eine anschließend durchgeführte Sorptionsmessung mittels dynamischer
Dampfsorption mit n-Oktan und Auswertung mittels ESW-Modell (J. Adolphs, 2007), die
einen Messbereich von unter 0,1 m2/g zuließ, bestätigte die sehr kleine spezifische Ober-
fläche der Agglomerate. Ein Unterschied zwischen den Behandlungsarten, -temperaturen
oder -zeiten war nicht erkennbar. Serfert et al. (2009) konnten für sprühgetrocknete Partikel
mit ähnlicher Matrix bestehend aus Glukosesirup, Fischöl und OSA-Stärke spezifische
Oberflächen von 0,5 m2/g feststellen. Die im Vergleich zu Serfert et al. (2009) niedrigen
Werte der spezifischen Oberfläche lassen sich durch den Agglomerationsprozess, bei dem
sehr kompakte Agglomerate entstanden sind, erklären. Zusätzlich wurde die Dichte der
behandelten und unbehandelten Agglomerate bestimmt. Diese zeigen Werte zwischen
1,3 g/cm3 und 1,4 g/cm3 und unterscheiden sich daher nicht voneinander, lassen also auch
keine Schlussfolgerungen auf eventuell eingeschlossene Hohlräume zu.
Mittels Hg-Porosimetrie wurden die verschieden behandelten und unbehandelten Agglome-
rate auf ihre Porosität hin untersucht und die Größe der auftretenden Poren konnte ermit-
telt werden. Die größten hierbei auftretenden Poren sind zwischen 15 µm und 205 µm groß
und stellen das sogenannte Zwischenkornvolumen dar. Das ist das Volumen, das bei nied-

Diskussion
125
rigem Überdruck durch das Quecksilber zwischen den Partikeln gefüllt wird. Erst mit An-
steigen des Druckes füllen sich kleinere Poren. In der Studie von Drusch et al. (2007)
konnten in den Partikeln, die mit OSA-Stärke verkapseltes Fischöl und Glukosesirup ent-
hielten, Poren mit eine Porenweite von ca. 130 nm detektiert werden. Diese Poren sind in
den Proben der behandelten und unbehandelten Agglomerate nicht nachweisbar. Hier
zeigt sich, dass die Agglomerate vermutlich durch den vorherigen Agglomerationsprozess
ihre Porosität verloren haben und eine glatte, geschlossene Oberfläche besitzen. Die Po-
renweiten, die in den Agglomeraten im Bereich von 0,012 nm und 0,0013 nm detektiert
wurden, lassen sich durch das Zusammenpressen der Agglomerate bei ca. 400 MPa erklä-
ren und sind demzufolge Artefakte.
Die Untersuchungen mittels unterschiedlicher Gasadsorptionsverfahren zur Aufklärung der
strukturellen Veränderungen der Agglomerate zeigten keinen Unterschied in der Oberflä-
chenbeschaffenheit und in den Parametern, wie der Porosität und Porengröße. Demzufol-
ge können die unterschiedlichen Verläufe der Hydroperoxidkonzentration nicht auf struktu-
relle Unterschiede in den behandelten Agglomeraten zurückgeführt werden.
Anwar & Kunz (2011) untersuchten den Einfluss unterschiedlicher Verkapselungsverfah-
ren, unter anderem Sprühtrocknung und Sprühgranulation, auf die oxidative Stabilität von
Fischöl. Sie stellten fest, dass über einen Lagerzeitraum von acht Wochen die Hydroper-
oxidkonzentration im verkapselten Fischöl in den Mikrokapseln, die durch Sprühtrocknung
hergestellt wurden, deutlich höher war, als in den Mikrokapseln, die mittels Sprühgranulati-
on hergestellt wurden. Die Startwerte der Hydroperoxidkonzentration zu Beginn der Lage-
rung hingegen zeigten keine deutlichen Unterschiede. Dies kann ein Beleg dafür sein, dass
eine Vorschädigung im Prozess stattgefunden hat, da die Prozesstemperaturen und -zeiten
beider Prozesse sich maßgeblich unterscheiden.
Zusammenfassend muss festgestellt werden, dass die unterschiedlichen Verläufe der Hyd-
roperoxidkonzentration nicht durch strukturell bedingte Änderungen der Agglomerate her-
vorgerufen werden und die Behandlung mit Wasser den eindeutigsten Einfluss auf die Lip-
idoxidation des verkapselten Rapsöls hat. Demzufolge lässt sich die Verdunstungswärme,
die durch das Evaporieren des Wassers von der Agglomeratoberfläche abgeführt wird, als
wahrscheinlichster Faktor für einen niedrigeren Verlauf der Hydroperoxidkonzentration be-
nennen.

Diskussion
126
6.4 Barriereeigenschaften zellulosebasierter Coatingmaterialien während der Lagerung
Im Versuch zur Untersuchung der Barriereeigenschaften gegenüber Sauerstoff und Was-
serdampf sollte geklärt werden, inwiefern die Coatings in Abhängigkeit von der Auftrags-
menge, welche durch die Prozesszeit variiert wurde, und der Prozesstemperatur diese Bar-
riere ausbilden können. Die Proben wurden zusätzlich zur Charakterisierung des Lipidoxi-
dationsverlaufes und der Wasserdampfsorption auf Mikroverkapselungseffizienz und
Öltropfengrößenverteilung untersucht, um zu überprüfen, ob diese einen Einfluss auf die
Barriereeigenschaften zeigen. Darüber hinaus wurden von den Proben Rasterelektronen-
mikroskopaufnahmen angefertigt, um visuell zu überprüfen, ob das applizierte Coating
gleichmäßig ist.
Die Verteilungsparameter und die Verteilungsbreite der Öltropfengröße der mit MC gecoa-
teten Agglomerate liegen in der gleichen Größenordnung wie die der bei vergleichbaren
Bedingungen ohne Coatinglösung behandelten Agglomerate. Somit kann ein Einfluss der
Öltropfengrößenverteilung auf die Lipidoxidation und Wasserdampfsorption bei den unter-
schiedlich gecoateten Agglomeraten ausgeschlossen werden.
Die Werte für die Mikroverkapselungseffizienz der mit MC gecoateten Agglomerate lagen
mit ca. 99,6 % nur leicht über denen der ungecoateten Agglomerate mit ca. 97,5 %. Dies
ist darauf zurückzuführen, dass das Oberflächenfett durch das MC-Coating maskiert wurde
und somit über die Methode des Abwaschens des Oberflächenfettes, wie es hier nach
Westergaard (2004) durchgeführt wurde, nicht mehr nachweisbar ist. Ein Einfluss der
Mikroverkapselungseffizienz auf die Barriereeigenschaften kann somit ausgeschlossen
werden.
Die Aufnahmen des Rasterelektronenmikroskops der gecoateten Partikel zeigen, dass
Filmdicken, die exemplarisch bemaßt sind, sich voneinander unterscheiden und somit die
Berechnung der aufgetragenen Coatingmenge stützen. Es sind neben der Filmdicke aller-
dings auch Blasen zu erkennen, die entstehen, wenn das Lösungsmittel an einer Stelle
noch nicht vollständig evaporiert ist, sich jedoch darüber eine weitere Schicht des
Coatingmaterials bildet. Allerdings ist auf den Aufnahmen des Rasterelektronenmikroskops
auch zu erkennen, dass es Bruchkanten gibt, die entstehen, wenn die mechanische Bean-
spruchung der Agglomerate in der Wirbelschicht zu stark wird. Diese Bruchkanten, an de-
nen das Öl nicht mehr durch eine Coatinghülle vor der Atmosphäre geschützt ist, könnten
ein Durchlass für Atmosphärenluft oder Wasserdampf sein und somit die Barriereeigen-
schaft der MC-Filme nachteilig beeinflussen.

Diskussion
127
Barriereeigenschaften gegenüber Sauerstoff
Die Auswertung der Lipidoxidation zeigte, dass die Proben, die mit einer Prozesstempera-
tur von 60 °C gecoatet wurden, nach 18 Wochen den höchsten Wert in der Hydroperoxid-
konzentration aufwiesen. Somit ist der Einfluss einer thermischen Vorschädigung auf das
verkapselte Öl unabhängig von der aufgetragenen Coatingmenge nachgewiesen. Auch
Choe & Min (2006) haben in ihrem Review für den Fettverderb die Temperatur, wenn auch
während der Lagerung, als wichtigen Faktor für eine beschleunigte Lipidoxidation identifi-
ziert. Anwar et al. (2010) konnten für verschiedene verkapselte Öle nachweisen, dass die
Hydroperoxidkonzentration in gecoateten Partikeln stärker bei einer Lagerungszeit von
sechs Wochen zunahm, als in ungecoateten Partikeln. Bei dieser Studie wurde eine Pro-
zesstemperatur (Produkttemperatur) von 30 - 40 °C eingestellt, was den Bedingungen der
niedrigen Prozesstemperatur in dieser Arbeit entspricht. Zu bemerken bleibt jedoch, dass
Anwar et al. (2010) die Partikel vor dem Coatingprozess unter hohen Temperaturen (70°C)
granuliert haben, was eine zusätzliche Vorschädigung des verkapselten Öls mit sich brin-
gen kann. Da in der vorliegenden Arbeit dieser Schritt nicht durchgeführt wurde, konnte
nachgewiesen werden, dass die Vorschädigung des Öls auch durch niedrigere Temperatu-
ren erfolgen kann. Let et al. (2003) untersuchten Emulsionen mit Ölen, die sich in ihrer
Fettsäurezusammensetzung unterschieden. Die Emulsionen wurden über eine Lagerzeit
von zwei Wochen regelmäßig auf ihren Hydroperoxidgehalt untersucht. Hierbei konnte
nachgewiesen werden, dass ein leicht höherer Startgehalt an Hydroperoxiden, unabhängig
von der Fettsäurezusammensetzung des Öls, einen schnelleren oxidativen Verderb nach
sich zieht. Auch die Startwerte der gecoateten Agglomerate lagen leicht erhöht über denen
der ungecoateten Agglomerate, somit konnte der Einfluss einer Vorschädigung nachge-
wiesen werden.
Generell liegen die Werte der Hydroperoxidkonzentration für die gecoateten Agglomerate
unter den Werten der trocken behandelten Agglomerate, was nicht unbedingt auf eine Bar-
rierefunktion des MC-Coatings zurückzuführen ist, sondern eventuell auf den Effekt der
Verdunstungskälte (siehe 6.3).
Da die Aufnahmen des Rasterelektronenmikroskops Bruchkanten zeigten und somit bei
der Diskussion der Barriereeigenschaften nicht davon ausgegangen werden kann, dass
das Coating auf den Agglomeraten gleichmäßig ist, waren weitere Untersuchungen not-
wendig. Hierbei wurden MDEMUL-Modelloberflächen mit einem MC-Coating überzogen,
um eine konstante Schichtdicke zu formen und ein vollständiges Coating zu gewährleisten.
Hierbei konnte nach 10 Wochen Lagerzeit kein eindeutiger Unterschied in der Hydroper-
oxidkonzentration der Proben nachgewiesen werden.

Diskussion
128
Es wurde somit, entgegen der Literatur (Ayranci & Tunc, 2003; Bonilla, Atarés, Vargas, &
Chiralt, 2012; Erdohan & Turhan, 2005; Miller & Krochta, 1997; Park et al., 1993) nachge-
wiesen, dass die MC-Coatings keine Barriere gegen Sauerstoff ausbilden.
Barriereeigenschaften gegenüber Wasserdampf
Die Untersuchung der Wasserdampfsorption der ungecoateten und gecoateten Agglomera-
te zeigte, dass die Agglomerate, die 60 Minuten bei 60 °C gecoatet wurden, am wenigsten
Feuchtigkeit in den unterschiedlichen Stufen von relativer Feuchtigkeit aufgenommen ha-
ben. Die ungecoateten Agglomerate haben sich bereits bei einer Luftfeuchte von 85 % auf-
gelöst. Eine weitere Betrachtung ihrer Wasserdampfsorption ist damit nicht mehr möglich
und auch hinfällig. Die Wasserdampfbarriereeigenschaften von MC wurden bereits an ge-
gossenen Filmen untersucht und bestätigt (Erdohan & Turhan, 2005). Da eine Vergleich-
barkeit von gegossenen Filmen und einem auf einer Trägermatrix applizierten Coating je-
doch nicht uneingeschränkt gegeben war (siehe die Untersuchungen zur Barriere gegen
Sauerstoff), war es notwendig, die Barriereeigenschaft gegenüber Wasserdampf in diesem
Versuch zu prüfen. Der Mechanismus der Wasserdampfbarriereeigenschaft ähnelt vermut-
lich der von HPMC. Dieses quillt beim Befeuchten auf und verzögert so den weiteren Aus-
tauschprozess zwischen der Atmosphäre und der Trägermatrix (Siepmann & Peppas,
2012).
Zusammenfassend kann postuliert werden, dass hinsichtlich der Barriere gegenüber Was-
serdampf die Nutzung einer höheren Prozesstemperatur von Vorteil ist, da sie ein schnelle-
res Trocknen des Films auf den Agglomeraten ermöglicht. In den Aufnahmen des Raster-
elektronenmikroskops konnte festgestellt werden, dass die Hohlräume im Coating kleiner
waren, wenn eine höhere Prozesstemperatur verwendet wurde. Aussagen zur Struktur des
Films und damit ein Einfluss der Prozessparameter auf den Film selbst konnten nicht
nachgewiesen werden.

Schlussbetrachtungen und Ausblick
129
7 Schlussbetrachtungen und Ausblick
Ziel der vorliegenden Arbeit war es, die Eignung von Zellulosederivaten als funktionelle
Coatings auf einer maltodextrinbasierten Trägermatrix im Wirbelschichtcoatingprozess zu
untersuchen. Dabei waren drei verschiedene Teilaspekte zu berücksichtigen. Zum ersten
musste die Interaktion der zellulosederivatbasierten Coatinglösungen mit einer malto-
dextrinbasierten Trägermatrix charakterisiert werden. Zum zweiten wurden Lösungen aus
Methylzellulose auf ihre rheologischen Eigenschaften bezüglich der im Wirbelschicht-
coatingprozess wichtigen Prozess- und Materialparameter hin untersucht. Abschließend
wurde die Funktionalität eines methylzellulosebasierten Coatings als Sauerstoff- und Was-
serdampfbarriere in Abhängigkeit verschiedener Prozessparameter untersucht.
Im ersten Teil der Arbeit gelang es, eine Methodik zu etablieren, die eine Interaktion von
Coatinglösungen und Modelloberflächen messbar macht. Hinzu kommt, dass die Zusam-
mensetzung der Modelloberflächen viel besser der von maltodextrinbasierten Trägersys-
temen entspricht, als es die bislang in der Literatur verwendeten Glasoberflächen tun.
Mittels der angewandten Methodik konnten Wetting Envelopes erstellt werden, die für alle
untersuchten zellulosederivatbasierten Coatingmaterialien ein unterschiedliches Verhalten
auf der Modelloberfläche zeigten. Unter Zuhilfenahme dieser Wetting Envelopes kann zu-
künftig die Eignung von Coatings im Vorfeld auf der Trägermatrix überprüft werden. Fest-
zustellen bleibt jedoch, dass das direkte Messen des Kontaktwinkels verlässlichere Ergeb-
nisse als die Berechnung des entstehenden Kontaktwinkels hervorbringt. Deswegen sollten
in zukünftigen Untersuchungen beide in dieser Arbeit vorgestellten Verfahren verwendet
werden.
Bezüglich der rheologischen Untersuchungen konnte über die Viskosität ermittelt werden,
welche maximal einsetzbare Trockenmasse in einer Sprühlösung möglich ist, um somit in
kurzer Zeit eine höhere Trockensubstanz in den Prozess einzubringen. Darüber hinaus
wurde das Verhalten der zellulosederivatbasierten Coatinglösung nach starker Scherbean-
spruchung untersucht. Durch diese Untersuchung konnte festgestellt werden, dass der
Grad des Strukturwiederaufbaus von der Konzentration der Coatinglösung abhängt, aber
auch von der Temperatur. Dies führt dazu, dass das Verhalten der Coatinglösungen nach
der Zerstäubung im Prozess abhängig von der Prozesstemperatur vorhergesagt werden
kann. Durch die in dieser Arbeit durchgeführten Messungen konnte das Verhalten der
Coatinglösungen ab einem Zeitpunkt nach 0,8 s charakterisiert werden. Diese Zeit scheint
in Anbetracht der Verweilzeit des Tropfens im Prozess, bevor er auf ein Partikel trifft, je-
doch recht groß. Um diesen Zeitraum zu verkleinern, sollte eventuell ein anderes Messsys-

Schlussbetrachtungen und Ausblick
130
tem verwendet werden, das die durch die Trägheit der Probenmasse hervorgerufenen Stö-
rungen unterbinden kann.
Die Konzentrations- und Temperaturabhängigkeit der strukturbildenden Charakteristika der
Coatinglösungen konnte mit Hilfe von Oszillationsversuchen überprüft werden. Die Ergeb-
nisse dieser Untersuchungen lassen es zu, die maximal zulässige Temperatur der Luft im
Wirbelschichtprozess zu definieren. Diese Information trägt dazu bei, dass der Prozess des
Wirbelschichtcoatens mit zellulosederivatbasierten Coatings besser designt werden kann
und die mit einer Gelierung der Coatinglösung einhergehenden nachteiligen Phänomene
verhindert werden können.
Es konnte festgestellt werden, dass die Prozesse des Sprühtrocknens, Agglomerierens
und Wirbelschichtcoatens bzw. -behandelns keinen Einfluss auf die Startwerte der Hydro-
peroxidkonzentration haben. Andererseits zeigte sich, dass während der Lagerung der
Proben die Parameter der Prozesstemperatur und -zeit einen sehr deutlichen Einfluss aus-
übten. Daraus können negative Einflüsse des Prozesses auf die Lagerstabilität von ver-
kapseltem Öl mit mehrfachungesättigten Fettsäuren abgeleitet werden.
Die in dieser Arbeit durchgeführten Untersuchungen zur Funktionalisierung eines zellulo-
sederivatbasierten Coatings konnten die in der Literatur beschriebenen Barriereeigenschaf-
ten gegenüber Sauerstoff nicht nachweisen. Beim Vergleich der Versuchsreihen zwischen
dem Behandeln mit Wasser und dem Auftragen eines zellulosederivatbasierten Coatings
deutet sich eine wirksame Barriere an. Allerdings wurden, um die Inhomogenität des
Coatings als Ursache für die fehlenden Barriereeigenschaften auszuschließen, Versuche
mit komplett überzogenen Modelloberflächen durchgeführt. Diese Versuche belegen, dass
Methylzellulosecoatings keine Barriere gegen Sauerstoff bilden.
Nachgewiesen werden konnte hingegen eine Wasserdampfbarriere des zellulosederivat-
basierten Coatings. Dies bedeutet, dass die in dieser Arbeit charakterisierten Methylzellu-
losecoatings einen Schutz gegen glasübergangstemperaturinduzierten Strukturänderungen
innerhalb der Trägermatrix bilden. Der Einsatz von zellulosederivatbasierten Coatings be-
schränkt sich demnach auf Kapseln, die temperaturstabile Inhaltstoffe enthalten und bei
denen Probleme bezüglich der Glasübergangstemperatur vorgebeugt und in Abhängigkeit
des Zellulosederivates kontrollierte Freisetzungsfunktionalitäten erreicht werden sollen.
Zukünftige Untersuchungen können die in dieser Arbeit gewonnenen Erkenntnisse nutzen,
um den Schutz von verkapselten Ölen, die mehrfach ungesättigte Fettsäuren enthalten,
durch zellulosederivatbasierte Coatings weiter zu verbessern. Eine Möglichkeit ist dabei,
die in dieser Arbeit verwendete OSA-Stärke gegen einen Emulgator mit antioxidativen Ei-
genschaften auszutauschen. Dies hat den Vorteil, dass die im Prozess des Wirbelschicht-

Schlussbetrachtungen und Ausblick
131
coatens entstandene Vorschädigung unterbunden werden könnte und das verkapselte
Rapsöl so während der Lagerung weniger stark oxidieren kann.
Ein anderer Ansatz wäre die Luft die im Prozess eingesetzt wird, zu trocknen. Durch eine
besonders trockene Luft könnte die Temperaturabsenkung bei der Verdunstung des Was-
sers aus der Coatinglösung zusätzlich verstärkt werden, wodurch eine geringere Vorschä-
digung im Prozess auftreten würde. Die Temperatur der Prozessluft selbst sollte dabei
nicht weiter herabgesetzt werden, da die Trocknung ansonsten zu lange dauern würde und
eine Anwendung im industriellen Maßstab nicht mehr sinnvoll wäre.
Eine weitere Möglichkeit wäre, den Eintrag von Sauerstoff in das System zu minimieren,
indem die Fluidisierungsluft gegen ein Gasgemisch ausgetauscht wird, welches einen ge-
ringeren Anteil an Sauerstoff hat, als die Atmosphärenluft.

Literaturverzeichnis
132
8 Literaturverzeichnis
Adolphs, J. (2007). Excess surface work-A modelless way of getting surface energies and specific surface areas directly from sorption isotherms. Applied Surface Science, 253(13 SPEC. ISS.), 5645–5649. http://doi.org/10.1016/j.apsusc.2006.12.089
Adolphs, J. J., & Setzer, M. J. (1996). A Model to Describe Adsorption Isotherms. Journal of Colloid and Interface Science, 180(1), 70–76. http://doi.org/10.1006/jcis.1996.0274
Ahn, J.-H., Kim, Y.-P., Lee, Y.-M., Seo, E.-M., Lee, K.-W., & Kim, H.-S. (2008). Optimization of microencapsulation of seed oil by response surface methodology. Food Chemistry, 107(1), 98–105. http://doi.org/10.1016/j.foodchem.2007.07.067
Andrade, R., Skurtys, O., & Osorio, F. (2012). Experimental study of drop impacts and spreading on epicarps: Effect of fluid properties. Journal of Food Engineering, 109(3), 430–437. http://doi.org/10.1016/j.jfoodeng.2011.10.038
Anwar, S. H., & Kunz, B. (2011). The influence of drying methods on the stabilization of fish oil microcapsules: Comparison of spray granulation, spray drying, and freeze drying. Journal of Food Engineering, 105(2), 367–378. http://doi.org/10.1016/j.jfoodeng.2011.02.047
Anwar, S. H., Weissbrodt, J., & Kunz, B. (2010). Microencapsulation of fish oil by spray granulation and fluid bed film coating. Journal of Food Science, 75(6). http://doi.org/10.1111/j.1750-3841.2010.01665.x
Atarés, L., Depypere, F., Pieters, J. G., & Dewettinck, K. (2012). Coating quality as affected by core particle segregation in fluidized bed processing. Journal of Food Engineering, 113(3), 415–421. http://doi.org/10.1016/j.jfoodeng.2012.06.012
Atarés, L., Pérez-Masiá, R., & Chiralt, A. (2011). The role of some antioxidants in the HPMC film properties and lipid protection in coated toasted almonds. Journal of Food Engineering, 104(4), 649–656. http://doi.org/10.1016/j.jfoodeng.2011.02.005
Ayranci, E., & Tunc, S. (2003). A method for the measurement of the oxygen permeability and the development of edible films to reduce the rate of oxidative reactions in fresh foods. Food Chemistry, 80(3), 423–431. http://doi.org/10.1016/S0308-8146(02)00485-5
Baik, M.-Y., Suhendro, E. L., Nawar, W. W., McClements, D. J., Decker, E. a., & Chinachoti, P. (2004). Effects of antioxidants and humidity on the oxidative stability of microencapsulated fish oil. Journal of the American Oil Chemists’ Society, 81(4), 355–360. http://doi.org/10.1007/s11746-004-0906-7
Balaghi, S., Edelby, Y., & Senge, B. (2014). Evaluation of thermal gelation behavior of different cellulose ether polymers by rheology. In AIP Conference Proceedings (Vol. 1593, pp. 755–761). http://doi.org/10.1063/1.4873886
Balasubramaniam, V. M., Chinnan, M. S., Mallikarjunan, P., & Phillips, R. D. (1997). The Effect of Edible Film on Oil Uptake and Moisture Retention of a Deep-Fat Fried Poultry Product. Journal of Food Process Engineering, 20(1), 17–29. http://doi.org/10.1111/j.1745-4530.1997.tb00408.x
Baloch, M. K., Bibi, F., & Jilani, M. S. (2013). Effect of Coatings Over the Quality and Shelf Life of Mango ( Mangifera Indica L.) Fruit. Journal of Food Processing and Preservation, 37(1), 66–73. http://doi.org/10.1111/j.1745-4549.2011.00614.x
Bayer, R., & Knarr, M. (2012). Thermal precipitation or gelling behaviour of dissolved methylcellulose (MC) derivatives-Behaviour in water and influence on the extrusion of

Literaturverzeichnis
133
ceramic pastes. Part 1: Fundamentals of MC-derivatives. Journal of the European Ceramic Society, 32(5), 1007–1018. http://doi.org/10.1016/j.jeurceramsoc.2011.11.025
Behzadi, S. S., Toegel, S., & Viernstein, H. (2008). Innovations in coating technology. Recent Patents on Drug Delivery & Formulation, 2(3), 209–30.
Belitz, H. D., Grosch, W., & Schieberle, P. (2008). Lehrbuch der Lebensmittelchemie (6. Auflage). Berlin, Heidelberg: Springer-Verlag Berlin Heidelberg. http://doi.org/10.1007/978-3-540-73202-0
Bifani, V., Ramírez, C., Ihl, M., Rubilar, M., García, A., & Zaritzky, N. (2007). Effects of murta (Ugni molinae Turcz) extract on gas and water vapor permeability of carboxymethylcellulose-based edible films. LWT - Food Science and Technology, 40(8), 1473–1481. http://doi.org/10.1016/j.lwt.2006.03.011
Bilbao-Sáinz, C., Avena-Bustillos, R. J., Wood, D. F., Williams, T. G., & Mchugh, T. H. (2010). Composite edible films based on hydroxypropyl methylcellulose reinforced with microcrystalline cellulose nanoparticles. Journal of Agricultural and Food Chemistry, 58, 3753–3760. http://doi.org/10.1021/jf9033128
Bonilla, J., Atarés, L., Vargas, M., & Chiralt, a. (2012). Edible films and coatings to prevent the detrimental effect of oxygen on food quality: Possibilities and limitations. Journal of Food Engineering, 110(2), 208–213. http://doi.org/10.1016/j.jfoodeng.2011.05.034
Börjesson, E., Innings, F., Trägårdh, C., Bergenståhl, B., & Paulsson, M. (2014). Evaluation of particle measures relevant for powder bed porosity-A study of spray dried dairy powders. Powder Technology, 253, 453–463. http://doi.org/10.1016/j.powtec.2013.11.050
Bourlieu, C., Guillard, V., Vallès-Pamiès, B., Guilbert, S., & Gontard, N. (2009). Edible moisture barriers: how to assess of their potential and limits in food products shelf-life extension? Critical Reviews in Food Science and Nutrition, 49(5), 474–99. http://doi.org/10.1080/10408390802145724
Brunauer, S., Emmett, P. H., & Teller, E. (1938). Adsorption of Gases in Multimolecular Layers. Journal of the American Chemical Society, 60(2), 309–319. http://doi.org/10.1021/ja01269a023
Cash, M. J., & Caputo, S. J. (2010). Cellulose Derivatives. In Food Stabilizers, Thickeners and Gelling Agents (pp. 95–115). Retrieved from http://blog.ub.ac.id/fawzy/files/2013/07/Food-Stabilizer.pdf#page=66
Cellesi, F., Tirelli, N., & Hubbell, J. A. (2002). Materials for cell encapsulation via a new tandem approach combining reverse thermal gelation and covalent crosslinking. Macromolecular Chemistry and Physics, 203(10-11), 1466–1472. http://doi.org/10.1002/1521-3935(200207)203:10/11<1466::AID-MACP1466>3.0.CO;2-P
Chau, T. T., Bruckard, W. J., Koh, P. T. L., & Nguyen, A. V. (2009). A review of factors that affect contact angle and implications for flotation practice. Advances in Colloid and Interface Science, 150(2), 106–15. http://doi.org/10.1016/j.cis.2009.07.003
Chenite, A., Buschmann, M., Wang, D., Chaput, C., & Kandani, N. (2001). Rheological characterisation of thermogelling chitosan / glycerol-phosphate solutions. Carbohydrate Polymers, 46, 39–47.
Choe, E., & Min, D. B. (2006). Comprehensive Reviews in Food Science and Food Safety Mechanisms and Factors for Edible Oil Oxidation. Comprehensive Reviews in Food Science and Food Safety, 5, 169–186. http://doi.org/10.1111/j.1541-

Literaturverzeichnis
134
4337.2006.00009.x
Choi, W., Park, H., Ahn, D., & Lee, J. (2002). Wettability of chitosan coating solution on “Fuji”apple skin. Journal of Food, 67(7), 3–7.
Connor, W. E., & Neuringer, M. (1988). Importance of Dietary Omega-3 Fatty Acids in Retinal Function and Brain Chemistry. In Nutritional Modulation of Neural Function (pp. 191–201). Elsevier. http://doi.org/10.1016/B978-0-12-506455-2.50021-6
Da Silva, P. T., Fries, L. L. M., Menezes, C. R. De, Holkem, A. T., Schwan, C. L., Wigmann, É. F., … Silva, C. D. B. Da. (2014). Microencapsulation: concepts, mechanisms, methods and some applications in food technology. Ciência Rural, 44(7), 1304–1311. http://doi.org/10.1590/0103-8478cr20130971
de Moura, M. R., Avena-Bustillos, R. J., McHugh, T. H., Krochta, J. M., & Mattoso, L. H. C. (2008). Properties of novel hydroxypropyl methylcellulose films containing chitosan nanoparticles. Journal of Food Science, 73(7). http://doi.org/10.1111/j.1750-3841.2008.00872.x
de Moura, M. R., Avena-Bustillos, R. J., McHugh, T. H., Wood, D. F., Otoni, C. G., & Mattoso, L. H. C. (2011). Miniaturization of cellulose fibers and effect of addition on the mechanical and barrier properties of hydroxypropyl methylcellulose films. Journal of Food Engineering, 104(1), 154–160. http://doi.org/10.1016/j.jfoodeng.2010.12.008
de S. Medeiros, B. G., Pinheiro, A. C., Carneiro-da-Cunha, M. G., & Vicente, A. a. (2012). Development and characterization of a nanomultilayer coating of pectin and chitosan – Evaluation of its gas barrier properties and application on “Tommy Atkins” mangoes. Journal of Food Engineering, 110(3), 457–464. http://doi.org/10.1016/j.jfoodeng.2011.12.021
Debeaufort, S., & Desobry, F. (2012). Encapsulation of flavors, nutraceuticals, and antibacterials. In E. A. Baldwin, R. Hagemaier, & J. Bai (Eds.), Edible Coatings and Films to Improve Food Quality (Vol. 6, pp. 334–366). Boca Raton, London, New York: CRC Press.
Desbrières, J., Hirrien, M., & Ross-Murphy, S. B. (2000). Thermogelation of methylcellulose: Rheological considerations. Polymer, 41(7), 2451–2461. http://doi.org/10.1016/S0032-3861(99)00413-9
Deutsches Institut für Normung e.V., D. (2012). DIN ISO 9277. Berlin.
Dewettinck, K., & Huyghebaert, A. (1999). Fluidized bed coating in food technology. Trends in Food Science & Technology, 10(4-5), 163–168. http://doi.org/10.1016/S0924-2244(99)00041-2
Dragich, A. M., & Krochta, J. M. (2010). Whey protein solution coating for fat-uptake reduction in deep-fried chicken breast strips. Journal of Food Science, 75(1), 43–47. http://doi.org/10.1111/j.1750-3841.2009.01408.x
Drusch, S., & Berg, S. (2008). Extractable oil in microcapsules prepared by spray-drying: Localisation, determination and impact on oxidative stability. Food Chemistry, 109(1), 17–24. http://doi.org/10.1016/j.foodchem.2007.12.016
Drusch, S., & Schwarz, K. (2006). Microencapsulation properties of two different types of n-octenylsuccinate-derivatised starch. European Food Research and Technology, 222(1-2), 155–164. http://doi.org/10.1007/s00217-005-0020-3
Drusch, S., Serfert, Y., Scampicchio, M., Schmidt-Hansberg, B., & Schwarz, K. (2007). Impact of physicochemical characteristics on the oxidative stability of fish oil microencapsulated by spray-drying. Journal of Agricultural and Food Chemistry,

Literaturverzeichnis
135
55(26), 11044–11051. http://doi.org/10.1021/jf072536a
Drusch, S., Serfert, Y., & Schwarz, K. (2006). Microencapsulation of fish oil with n-octenylsuccinate-derivatised starch: Flow properties and oxidative stability. European Journal of Lipid Science and Technology, 108(6), 501–512. http://doi.org/10.1002/ejlt.200500312
Duangkhamchan, W., Ronsse, F., Depypere, F., Dewettinck, K., & Pieters, J. G. (2012). CFD study of droplet atomisation using a binary nozzle in fluidised bed coating. Chemical Engineering Science, 68(1), 555–566. http://doi.org/10.1016/j.ces.2011.10.022
Duangkhamchan, W., Ronsse, F., Siriamornpun, S., & Pieters, J. G. (2015). Numerical study of air humidity and temperature distribution in a top-spray fluidised bed coating process. Journal of Food Engineering, 146, 81–91. http://doi.org/10.1016/j.jfoodeng.2014.09.005
Embuscado, M., Huber, K., & Nieto, M. B. (2009). Edible Films and Coatings for Food Applications. In K. C. Huber & M. E. Embuscado (Eds.), Edible Films and Coatings for Food Applications (p. 410). New York, NY: Springer New York. Retrieved from http://link.springer.com/10.1007/978-0-387-92824-1
Erbil, Y. (2006). Surface chemstry of solid and liquid interfaces. Oxford: Blackwell Publishing.
Erdohan, Ö. Z., & Turhan, N. K. (2005). Barrier and mechanical properties of methylcellulose-whey protein films. Packaging Technology and Science, 18(6), 295–302. http://doi.org/10.1002/pts.700
Farag, Y. (2010). Characterization of Different Shellac Types and Development of Shellac-Coated Dosage Forms.
Farag, Y., & Leopold, C. S. (2011). Development of shellac-coated sustained release pellet formulations. European Journal of Pharmaceutical Sciences : Official Journal of the European Federation for Pharmaceutical Sciences, 42(4), 400–5. http://doi.org/10.1016/j.ejps.2011.01.006
Floury, J., Desrumaux, A., Axelos, M. A. V, & Legrand, J. (2002). Degradation of methylcellulose during ultra-high pressure homogenisation. Food Hydrocolloids, 16(1), 47–53. http://doi.org/10.1016/S0268-005X(01)00039-X
Frankel, E. N. (2005). Lipid Oxidation (Second Edi). Bridgwater: The Oily Press.
Fuchs, M., Turchiuli, C., Bohin, M., Cuvelier, M. E., Ordonnaud, C., Peyrat-Maillard, M. N., & Dumoulin, E. (2006). Encapsulation of oil in powder using spray drying and fluidised bed agglomeration. Journal of Food Engineering, 75(1), 27–35. http://doi.org/10.1016/j.jfoodeng.2005.03.047
Garcia, M. A. (2004). Methylcellulose Coatings Applied to Reduce Oil Uptake in Fried Products. Food Science and Technology International, 10(5), 339–346. http://doi.org/10.1177/1082013204047564
Geldart, D. (1973). Types of gas fluidization. Powder Technology, 7(5), 285–292. http://doi.org/10.1016/0032-5910(73)80037-3
Ghanbarzadeh, B., Almasi, H., & Entezami, A. a. (2011). Improving the barrier and mechanical properties of corn starch-based edible films: Effect of citric acid and carboxymethyl cellulose. Industrial Crops and Products, 33(1), 229–235. http://doi.org/10.1016/j.indcrop.2010.10.016
Gibbs, B. F., Kermasha, S., Alli, I., Mulligan, C. N., Gibbs, F., Kermasha, S., & Inteaz Al, B.

Literaturverzeichnis
136
(1999). Encapsulation in the food industry: a review. International Journal of Food Sciences and Nutrition, 50(3), 213–24. http://doi.org/10.1080/096374899101256
Grassmann, P., Widmer, F., & Sinn, H. (1997). Einführung in die thermische Verfahrenstechnik (3. vollstä). Berlin, Boston: De Gruyter.
Hagenmaier, R. (2012). Gas-exchange properties of edible films and coatings. In E. A. Baldwin, R. Hagenmaier, & J. Bai (Eds.), Edible Coatings and Films to Improve Food Quality (Second Edi, p. 450). Boca Raton, London, New York: CRC Taylor & Francis.
Haque, A., & Morris, E. R. (1993). Thermogelation of methylcellulose. Part I: molecular structures and processes. Carbohydrate Polymers, 22(3), 161–173. http://doi.org/10.1016/0144-8617(93)90137-S
Hay, K. M., Dragila, M. I., & Liburdy, J. (2008). Theoretical model for the wetting of a rough surface. Journal of Colloid and Interface Science, 325(2), 472–7. http://doi.org/10.1016/j.jcis.2008.06.004
Hede, P. D. (2006). Fluid Bed Particle Processing. Bookboon, Copenhagen. Retrieved from http://80.191.221.11/files/research/site/ebooks/chemistry-chemical-engineering/fluid-bed-particle-processing.pdf
Hede, P. D., Bach, P., & Jensen, A. D. (2008). Two-fluid spray atomisation and pneumatic nozzles for fluid bed coating/agglomeration purposes: A review. Chemical Engineering Science, 63(14), 3821–3842. http://doi.org/10.1016/j.ces.2008.04.014
Hirrien, M., Chevillard, C., Desbrières, J., Axelos, M. A. V, & Rinaudo, M. (1998). Thermogelation of methylcelluloses: new evidence for understanding the gelation mechanism. Polymer, 39(25), 6251–6259. http://doi.org/10.1016/S0032-3861(98)00142-6
Hogan, S. a., McNamee, B. F., O’Riordan, E. D., & O’Sullivan, M. (2001). Emulsification and microencapsulation properties of sodium caseinate/carbohydrate blends. International Dairy Journal, 11(3), 137–144. http://doi.org/10.1016/S0958-6946(01)00091-7
Hussain, P. R., Meena, R. S., Dar, M. A., & Wani, A. M. (2010). Carboxymethyl cellulose coating and low-dose gamma irradiation improves storage quality and shelf life of pear (Pyrus communis L., Cv. Bartlett/William). Journal of Food Science, 75(9), M586–96. http://doi.org/10.1111/j.1750-3841.2010.01868.x
Iso, N., & Yamamoto, D. (1970). Effects of Sucrose and Citric Acid on the Sol-Gel Transformation of Methylcellulose in Water. Agricultural and Biological Chemistry, 34(12), 1867–1869. http://doi.org/10.1080/00021369.1970.10859863
Jacobsen, C., & Let, M. B. (2007). Using Polyunsaturated Fatty Acids (PUFAs) as Functional Ingredients. In C. Williams & J. Buttriss (Eds.), Improving the fat content of foods (pp. 428–453). Cambridge, England: Woodhead Publishing Limited.
Janjarasskul, T., Min, S. C., & Krochta, J. M. (2011). Storage stability of ascorbic acid incorporated in edible whey protein films. Journal of Agricultural and Food Chemistry, 59(23), 12428–12432. http://doi.org/10.1021/jf201629r
Kaelble, D. H. (1970). Dispersion-Polar Surface Tension Properties of Organic Solids. The Journal of Adhesion, 2(2), 66–81. http://doi.org/10.1080/0021846708544582
Kankaanranta, J., Mwegerano, A., & Hämeenoja, O. (2012). The importance of having mobile terminal samples for analyzing and verifying customer issues. African Journal of Business Management, 6(44), 11088–11094. http://doi.org/10.5897/AJBM12.1001
Kape, A., Ruick, B., & Drusch, S. (2016). Characterisation of the work of adhesion of food

Literaturverzeichnis
137
grade coating materials on a maltodextrin model surface. Chemical Engineering Research and Design, 7, 6–13. http://doi.org/10.1016/j.cherd.2016.01.023
Karbowiak, T., Debeaufort, F., Champion, D., & Voilley, A. (2006). Wetting properties at the surface of iota-carrageenan-based edible films. Journal of Colloid and Interface Science, 294(2), 400–10. http://doi.org/10.1016/j.jcis.2005.07.030
Karbowiak, T., Debeaufort, F., & Voilley, A. (2006). Importance of surface tension characterization for food, pharmaceutical and packaging products: a review. Critical Reviews in Food Science and Nutrition, 46(5), 391–407. http://doi.org/10.1080/10408390591000884
Kastner, H., Einhorn-Stoll, U., & Senge, B. (2012). Structure formation in sugar containing pectin gels - Influence of Ca2+ on the gelation of low-methoxylated pectin at acidic pH. Food Hydrocolloids, 27(1), 42–49. http://doi.org/10.1016/j.foodhyd.2011.09.001
Kaushik, P., Dowling, K., Barrow, C. J., & Adhikari, B. (2015). Microencapsulation of omega-3 fatty acids: A review of microencapsulation and characterization methods. Journal of Functional Foods, 19, 868–881. http://doi.org/10.1016/j.jff.2014.06.029
Kayumba, P. C., Huyghebaert, N., Cordella, C., Ntawukuliryayo, J. D., Vervaet, C., & Remon, J. P. (2007). Quinine sulphate pellets for flexible pediatric drug dosing: formulation development and evaluation of taste-masking efficiency using the electronic tongue. European Journal of Pharmaceutics and Biopharmaceutics : Official Journal of Arbeitsgemeinschaft Für Pharmazeutische Verfahrenstechnik e.V, 66(3), 460–5. http://doi.org/10.1016/j.ejpb.2006.11.018
Knarr, M. (2003). Rheologische Untersuchung und Modellierung der Sol-Gel-Charakteristika von Methylcellulose und Carrageenan.
Kosaraju, S. L. (2005). Colon targeted delivery systems: review of polysaccharides for encapsulation and delivery. Critical Reviews in Food Science and Nutrition, 45(4), 251–8. http://doi.org/10.1080/10408690490478091
Kou, X., Chan, L. W., Steckel, H., & Heng, P. W. S. (2012). Physico-chemical aspects of lactose for inhalation. Advanced Drug Delivery Reviews, 64(3), 220–232. http://doi.org/10.1016/j.addr.2011.11.004
Kuang, S. S., Oliveira, J. C., & Crean, a M. (2010). Microencapsulation as a tool for incorporating bioactive ingredients into food. Critical Reviews in Food Science and Nutrition, 50(10), 951–68. http://doi.org/10.1080/10408390903044222
Laksmana, F. L., Hartman Kok, P. J. a, Vromans, H., Frijlink, H. W., & Van der Voort Maarschalk, K. (2009). Development and application of a process window for achieving high-quality coating in a fluidized bed coating process. AAPS PharmSciTech, 10(3), 732–42. http://doi.org/10.1208/s12249-009-9250-1
Lander, R., Manger, W., Scouloudis, M., Ku, A., Davis, C., & Lee, A. (2000). Gaulin homogenization: A mechanistic study. Biotechnology Progress, 16(1), 80–85. http://doi.org/10.1021/bp990135c
Let, M. B., Jacobsen, C., Frankel, E. N., & Meyer, A. S. (2003). Oxidative flavour deterioration of fish oil enriched milk. European Journal of Lipid Science and Technology, 105(9), 518–528. http://doi.org/10.1002/ejlt.200300821
Letellier, P., Mayaffre, A., & Turmine, M. (2007). Drop size effect on contact angle explained by nonextensive thermodynamics. Young’s equation revisited. Journal of Colloid and Interface Science, 314(2), 604–14. http://doi.org/10.1016/j.jcis.2007.05.085

Literaturverzeichnis
138
Li, L. (2002). Thermal gelation of methylcellulose in water: Scaling and thermoreversibility. Macromolecules, 35(15), 5990–5998. http://doi.org/10.1021/ma0201781
Li, L., Shan, H., Yue, C. Y., Lam, Y. C., Tam, K. C., & Hu, X. (2002). Thermally induced association and dissociation of methylcellulose in aqueous solutions. Langmuir, 18(20), 7291–7298. http://doi.org/10.1021/la020029b
Lima, Á. M., Cerqueira, M. a., Souza, B. W. S., Santos, E. C. M., Teixeira, J. a., Moreira, R. a., & Vicente, A. a. (2010). New edible coatings composed of galactomannans and collagen blends to improve the postharvest quality of fruits – Influence on fruits gas transfer rate. Journal of Food Engineering, 97(1), 101–109. http://doi.org/10.1016/j.jfoodeng.2009.09.021
Madene, A., Jacquot, M., Scher, J., & Desobry, S. (2006). Flavour encapsulation and controlled release - a review. International Journal of Food Science and Technology, 41(1), 1–21. http://doi.org/10.1111/j.1365-2621.2005.00980.x
Matalanis, A., Jones, O. G., & McClements, D. J. (2011). Structured biopolymer-based delivery systems for encapsulation, protection, and release of lipophilic compounds. Food Hydrocolloids, 25(8), 1865–1880. http://doi.org/10.1016/j.foodhyd.2011.04.014
Mezger, T. G. (2010). Das Rheologie Handbuch (3. überarb). Hannover, Deutschland: Vincentz Network Hannover.
Michalski, M.-C., Desobry, S., Pons, M.-N., & Hardy, J. (1998). Adhesion of edible oils to food contact surfaces. Journal of the American Oil Chemists’ Society, 75(4), 447–454. http://doi.org/10.1007/s11746-998-0247-9
Milanovic, J., Manojlovic, V., Levic, S., Rajic, N., Nedovic, V., & Bugarski, B. (2010). Microencapsulation of Flavors in Carnauba Wax. Sensors, 10(1), 901–912. http://doi.org/10.3390/s100100901
Miller, K. S., & Krochta, J. M. (1997). Oxygen and aroma barrier properties of edible films: A review. Trends in Food Science & Technology, 81(July), 228–237. Retrieved from http://www.sciencedirect.com/science/article/pii/S0924224497010510
Minihane, A. M., & Lovegrove, J. A. (2007). Health benefits of polyunsaturated fatty acids (PUFAs). In C. Williams & J. Buttriss (Eds.), Improving the fat content of foods (pp. 107–140). Cambridge, England: Woodhead Publishing Limited.
Mishra, B., Khatkar, B. S., Garg, M. K., & Wilson, L. A. (2010). Permeability of edible coatings. Journal of Food Science and Technology, 47(1), 109–13. http://doi.org/10.1007/s13197-010-0003-7
Moffatt, R. J., & Stamford, B. (2006). Lipid Metabolism and Health. (R. J. Moffatt & B. Stamford, Eds.). Boca Raton, London, New York: CRC Taylor & Francis.
Mokarram, R. R., Mortazavi, S. A., Najafi, M. B. H., & Shahidi, F. (2009). The influence of multi stage alginate coating on survivability of potential probiotic bacteria in simulated gastric and intestinal juice. Food Research International, 42(8), 1040–1045. http://doi.org/10.1016/j.foodres.2009.04.023
Mulhem, B., Fritsching, U., Schulte, G., & Bauckhage, K. (2003). Effect of solid particle characteristics on suspension atomization. Atomization and Sprays: Journal of the International Institutions for Liquid Atomization and Spray Systems.
Mura, S., Corrias, F., Stara, G., Piccinini, M., Secchi, N., Marongiu, D., … Greppi, G. F. (2011). Innovative Composite Films of Chitosan, Methylcellulose, and Nanoparticles. Journal of Food Science, 76(7), 54–60. http://doi.org/10.1111/j.1750-3841.2011.02295.x

Literaturverzeichnis
139
Nakano, T., & Yuasa, H. (2001). Suppression of agglomeration in fluidized bed coating. IV. Effects of sodium citrate concentration on the suppression of particle agglomeration and the physical properties of HPMC film. International Journal of Pharmaceutics, 215(1-2), 3–12.
Nazan Turhan, K., & Şahbaz, F. (2004). Water vapor permeability, tensile properties and solubility of methylcellulose-based edible films. Journal of Food Engineering, 61(3), 459–466. http://doi.org/10.1016/S0260-8774(03)00155-9
Nienaltowska, K., Perfetti, G., Meesters, G. M. H., Ronsse, F., Pieters, J. G., Dewettinck, K., & Depypere, F. (2010). Attrition strength of water-soluble cellulose derivatives coatings. Powder Technology, 198(2), 298–309. http://doi.org/10.1016/j.powtec.2009.11.026
Owens, D., & Wendt, R. (1969). Estimation of the Surface Free Energy of Polymers. Journal of Applied Polymer Science, 13, 1741–1747.
Palamanit, A., Soponronnarit, S., Prachayawarakorn, S., & Tungtrakul, P. (2013). Effects of inlet air temperature and spray rate of coating solution on quality attributes of turmeric extract coated rice using top-spray fluidized bed coating technique. Journal of Food Engineering, 114(1), 132–138. http://doi.org/10.1016/j.jfoodeng.2012.07.014
Park, H. J., Weller, C. L., Vergano, P. J., & Testin, R. F. (1993). Permeability and Mechanical Properties of Cellulose-Based Edible Films. Journal of Food …, 58(6), 1361–1364. Retrieved from http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2621.1993.tb06183.x/abstract
Perpar, M., Luštrik, M., Dreu, R., Srčič, S., & Žun, I. (2013). Estimating coating quality parameters on the basis of pressure drop measurements in a Wurster draft tube. Powder Technology, 246, 41–50. http://doi.org/10.1016/j.powtec.2013.05.005
Pinto, J. F., Buckton, G., & Newton, J. M. (1995). A relationship between surface free energy and polarity data and some physical properties of spheroids. International Journal of Pharmaceutics, 118(1), 95–101. http://doi.org/10.1016/0378-5173(94)00357-B
Planinsek, O., Pisek, R., Trojak, A., & Srcic, S. (2000). The utilization of surface free-energy parameters for the selection of a suitable binder in fluidized bed granulation. International Journal of Pharmaceutics, 207, 77–88. http://doi.org/10.1016/S0378-5173(00)00535-4
Portoghese, F., Ferrante, L., Berruti, F., Briens, C., & Chan, E. (2010). Effect of the injection-nozzle geometry on the interaction between a gas–liquid jet and a gas–solid fluidized bed. Chemical Engineering and Processing: Process Intensification, 49(6), 605–615. http://doi.org/10.1016/j.cep.2010.04.011
Rabel, W. (1971). Einige Aspekte der Benetzungstheorie und ihre Anwendung auf die Untersuchung und Veränderung der Oberflächeneigenschaften von Polymeren. Farbe Und Lack, 77(10), 997–1006.
Razali, N. M., & Wah, Y. B. (2010). Power Comparisons of some Selected Normality Tests. Regional Conference on Statistical Sciences, 2010(June), 126–138. Retrieved from http://instatmy.org.my/downloads/RCSS’10/Proceedings/13P.pdf
Rhodes, M. (2008). Geldart Sheme. In Particle Technology (p. 475).
Riveros, C. G., Mestrallet, M. G., Quiroga, P. R., Nepote, V., & Grosso, N. R. (2013). Preserving sensory attributes of roasted peanuts using edible coatings. International Journal of Food Science & Technology, 48(4), 850–859. http://doi.org/10.1111/ijfs.12036

Literaturverzeichnis
140
Roos, Y., & Karel, M. (1991). Phase-Transitions of Mixtures of Amorphous Polysaccharides and Sugars. Biotechnol. Progr., 7(1), 49–53. http://doi.org/10.1021/bp00007a008
Runke, T., Song, B., & Springer, J. (1994). Surface- and Interfacial Tensions of Liquid Crystaline Polymers. Berliner Bunsengesellschaft, 3(98), 508–511.
Salman, A. D., Hounslow, M. J., & Seville, J. P. K. (2006). Handbook of Powder Technology. (A. D. Salman, M. J. Hounslow, & J. P. K. Seville, Eds.) (Vol. Volume 11). Elsevier Science.
Sarkar, N. (1995). Kinetics of thermal gelation of methylcellulose and hydroxypropylmethylcellulose in aqueous solutions. Carbohydrate Polymers, 26(3), 195–203. http://doi.org/10.1016/0144-8617(94)00107-5
Schmidt, M. C., Müller, M., Oehr, C., & Hirth, T. (2012). Influence of semi-solid fluid’s surface tension and rheological properties on the residues at packaging materials. Journal of Food Engineering, 108(1), 211–215. http://doi.org/10.1016/j.jfoodeng.2011.02.022
Scramin, J. a., de Britto, D., Forato, L. a., Bernardes-Filho, R., Colnago, L. a., & Assis, O. B. G. (2011). Characterisation of zein-oleic acid films and applications in fruit coating. International Journal of Food Science & Technology, 46(10), 2145–2152. http://doi.org/10.1111/j.1365-2621.2011.02729.x
Serfert, Y., Drusch, S., & Schwarz, K. (2009). Chemical stabilisation of oils rich in long-chain polyunsaturated fatty acids during homogenisation, microencapsulation and storage. Food Chemistry, 113(4), 1106–1112. http://doi.org/10.1016/j.foodchem.2008.08.079
Serfert, Y., Schröder, J., Mescher, A., Laackmann, J., Rätzke, K., Shaikh, M. Q., … Schwarz, K. (2013). Spray drying behaviour and functionality of emulsions with beta-lactoglobulin/pectin interfacial complexes. Food Hydrocolloids, 31(2), 438–445. http://doi.org/10.1016/j.foodhyd.2012.11.037
Shimokawa, K. ichi, Saegusa, K., & Ishii, F. (2009). Rheological properties of reversible thermo-setting in situ gelling solutions with the methylcellulose-polyethylene glycol-citric acid ternary system (2): Effects of various water-soluble polymers and salts on the gelling temperature. Colloids and Surfaces B: Biointerfaces, 74(1), 56–58. http://doi.org/10.1016/j.colsurfb.2009.06.022
Siepmann, J., & Peppas, N. a. (2012). Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Advanced Drug Delivery Reviews, 64(SUPPL.), 163–174. http://doi.org/10.1016/j.addr.2012.09.028
Soliva-Fortuny, R., Rojas-Graü, M. A., & Martín-Belloso, O. (2012). Polysaccharide coatings. In Edible Coatings and Films to Improve Food Quality (Vol. 6, pp. 103–136). http://doi.org/10.1016/S0924-2244(00)88935-9
Souza, B. W. S., Cerqueira, M. a., Ruiz, H. a., Martins, J. T., Casariego, A., Teixeira, J. a., & Vicente, A. a. (2010). Effect of Chitosan-Based Coatings on the Shelf Life of Salmon (Salmo salar). Journal of Agricultural and Food Chemistry, 58(21), 11456–11462. http://doi.org/10.1021/jf102366k
Srivastava, S., & Mishra, G. (2010). Fluid Bed Technology : Overview and Parameters for Process Selection. International Journal of Pharmaceutical Sciences and Drug Research, 2(4), 236–246.
Ström, G., Fredriksson, M., & Stenius, P. (1987). Contact angles, work of adhesion, and interfacial tensions at a dissolving Hydrocarbon surface. Journal of Colloid and Interface Science, 119(2), 352–361. http://doi.org/10.1016/0021-9797(87)90280-3

Literaturverzeichnis
141
Subramaniam, A., Veazey, R. L., Schober, A., Rada, A., Rong, Y., Van Sleeuwen, R. M. T., … Normand, V. (2013). Orange oil stability in spray dry delivery systems. Carbohydrate Polymers, 97(2), 352–357. http://doi.org/10.1016/j.carbpol.2013.05.005
Takeuchi, H., Yasuji, T., Yamamoto, H., & Kawashima, Y. (2000). Spray-dried lactose composite particles containing an ion complex of alginate-chitosan for designing a dry-coated tablet having a time-controlled releasing function. Pharmaceutical Research, 17(1), 94–9.
Tamm, F., Gies, K., Diekmann, S., Serfert, Y., Strunskus, T., Brodkorb, A., & Drusch, S. (2015). Whey protein hydrolysates reduce autoxidation in microencapsulated long chain polyunsaturated fatty acids. European Journal of Lipid Science and Technology, 1960–1970. http://doi.org/10.1002/ejlt.201400574
Tamm, F., Sauer, G., Scampicchio, M., & Drusch, S. (2012). Pendant drop tensiometry for the evaluation of the foaming properties of milk-derived proteins. Food Hydrocolloids, 27(2), 371–377. http://doi.org/10.1016/j.foodhyd.2011.10.013
Tavera-Quiroz, M. J., Urriza, M., Pinotti, A., & Bertola, N. (2012). Plasticized methylcellulose coating for reducing oil uptake in potato chips. Journal of the Science of Food and Agriculture, 92(7), 1346–53. http://doi.org/10.1002/jsfa.4704
Teunou, E., & Poncelet, D. (2002). Batch and continuous fluid bed coating – review and state of the art. Journal of Food Engineering, 53(4), 325–340. http://doi.org/10.1016/S0260-8774(01)00173-X
Thompson, J. C., & Rothstein, J. P. (2007). The atomization of viscoelastic fluids in flat-fan and hollow-cone spray nozzles. Journal of Non-Newtonian Fluid Mechanics, 147(1-2), 11–22. http://doi.org/10.1016/j.jnnfm.2007.06.004
Tomšič, M., Prossnigg, F., & Glatter, O. (2008). A thermoreversible double gel: Characterization of a methylcellulose and κ-carrageenan mixed system in water by SAXS, DSC and rheology. Journal of Colloid and Interface Science, 322(1), 41–50. http://doi.org/10.1016/j.jcis.2008.03.013
Tong, Q., Xiao, Q., & Lim, L.-T. (2008). Preparation and properties of pullulan–alginate–carboxymethylcellulose blend films. Food Research International, 41(10), 1007–1014. http://doi.org/10.1016/j.foodres.2008.08.005
Turhan, K. N., Şahbaz, F., & Güner, A. (2001). A Spectrophotometric Study of Hydrogen Bonding in Methylcellulose-based Edible Films Plasticized by Polyethylene Glycol. Journal of Food Science, 66(1), 59–62. http://doi.org/10.1111/j.1365-2621.2001.tb15581.x
Uhlemann, H., & Mörl, L. (2000). Wirbelschicht-Sprühgranulation (Erste Ausg). Berlin, Heidelberg: Springer-Verlag Berlin Heidelberg.
Vanderroost, M., Ronsse, F., Dewettinck, K., & Pieters, J. (2011). Modelling coating quality in fluidised bed coating: Spray sub-model. Journal of Food Engineering, 106(3), 220–227. http://doi.org/10.1016/j.jfoodeng.2011.05.013
Vanderroost, M., Ronsse, F., Pieters, J., & Dewettinck, K. (2010). A global coating quality model for top-spray fluidized beds: spray sub model, (figure 2).
Vega, C., & Roos, Y. H. (2006). Invited Review: Spray-Dried Dairy and Dairy-Like Emulsions—Compositional Considerations. Journal of Dairy Science, 89(2), 383–401. http://doi.org/10.3168/jds.S0022-0302(06)72103-8
Velásquez, P., Skurtys, O., Enrione, J., & Osorio, F. (2011). Evaluation of Surface Free Energy of Various Fruit Epicarps Using Acid–Base and Zisman Approaches. Food

Literaturverzeichnis
142
Biophysics, 6(3), 349–358. http://doi.org/10.1007/s11483-011-9209-0
Verbeken, D., Neirinck, N., Van Der Meeren, P., & Dewettinck, K. (2005). Influence of k-carrageenan on the thermal gelation of salt-soluble meat proteins. Meat Science, 70(1), 161–166. http://doi.org/10.1016/j.meatsci.2004.12.007
Washburn, E. W. (1921). The dynamics of capillary flow. Physical Review, 17(3), 273–283. http://doi.org/10.1103/PhysRev.17.273
Weng, L., Zhang, L., Ruan, D., Shi, L., & Xu, J. (2004). Thermal Gelation of Cellulose in a NaOH/Thiourea Aqueous Solution. Langmuir, 20(6), 2086–2093. http://doi.org/10.1021/la035995o
Westergaard, V. (2004). Milk powder technology: evaporation and spray drying. Niro A/S, Copenhagen, Denmark. Copenhagen.
Wiącek, A. E. (2011). The wetting and interfacial properties of alumina surface treated with dipalmitoylphosphatidylcholine and lipase enzyme. Powder Technology, 212(2), 332–339. http://doi.org/10.1016/j.powtec.2011.06.006
Wulff-Pérez, M., Torcello-Gómez, A., Martín-Rodríguez, A., Gálvez-Ruiz, M. J., & de Vicente, J. (2011). Bulk and interfacial viscoelasticity in concentrated emulsions: The role of the surfactant. Food Hydrocolloids, 25(4), 677–686. http://doi.org/10.1016/j.foodhyd.2010.08.012
Wurster, D. E., Likitlersuang, S., & Chen, Y. (2005). The influence of magnesium stearate on the Hiestand Tableting Indices and other related mechanical properties of maltodextrins. Pharmaceutical Development and Technology, 10(4), 461–6. http://doi.org/10.1080/10837450500299693
Wüstenberg, T. (2015). Cellulose and Cellulose Derivatives in the Food Industry: Fundamentals and Applications. http://doi.org/10.1002/9783527682935
Wüstneck, R., Moser, B., & Muschiolik, G. (1999). Interfacial dilational behaviour of adsorbed β-lactoglobulin layers at the different fluid interfaces. Colloids and Surfaces B: Biointerfaces, 15(3-4), 263–273. http://doi.org/10.1016/S0927-7765(99)00093-4
Young, T. (1805). An essay on the cohesion of fluids. Philosophical Transactions of the Royal Society of, 95, 65–87.

Anhang
143
A. Anhang
Tabellenanhang
Tabelle A.1: Verwendete Geräte
Geräte Typ Hersteller
Analysewaage MC BA 100 Sartorius AG, Göttingen
Deutschland
Digitale Bügelmessschraube 820918 Toolcraft
Düsenstand LP-471.2 Düsen-Schlick GmbH, Unter-
siemau/Coburg Deutschland
Düsenstand - Zerstäu-
bungsdüse Multispray Mod. 930
Düsen-Schlick GmbH, Unter-
siemau/Coburg Deutschland
Evaporator V.649.061.820 VLM GmbH
Fön HT 4 AKA electrics, DDR
Gasadsorptionsmessgerät ASAP 2020 Micromeritics GmbH, Aachen
Deutschland
Heizplatte (Weibull-Stoldt)
Heliumpyknometer AccyPyc 1330 Micromeritics GmbH, Aachen
Deutschland
Hochdruckhomogenisator Panda Plus100 GEA Group Aktiengesellschaft,
Düsseldorf Deutschland
Kontaktwinkelmessgerät OCA 20 DataPhysics Instruments
GmbH, Filderstadt, Deutschland
Laboratory Fluid Bed Coater Strea 1 GEA Process Engineering Inc.,
Columbia, USA
Magnetrührer Variomag HP6 2mag, Daytona Beach, USA

Anhang
144
Geräte Typ Hersteller
Magnetrührer VMS-C7 VWR International GmbH,
Darmstadt, Deutschland
Multipette (1-50 µL) Eppendorf AG, Hamburg
Deutschland
Particle Size Analyzer LA-950 Horiba Retsch GmbH & Co. KG, Haan
Deutschland
Photometer Helios Omega UV-VIS
Thermo Fisher Scientific Ger-
many BV & Co KG, Braun-
schweig Deutschland
Pipetten 10-100 µL/100-1000
µL/1-5 mL
Eppendorf AG, Hamburg
Deutschland
Quecksilberporosimeter Auto Pore III Micromeritics GmbH, Aachen
Deutschland
Rasterelektronenmikroskop S4000 Hitachi Ltd. Corporation, Tokio,
Japan
Rasterelektronenmikroskop
Kamera A3 Data S Pentax
Ricoh Imaging Co., Ltd., Tokio,
Japan
Rasterelektronenmikroskop
Sputter SCD 030
Oerlikon Balzers Coating AG,
Balzers, Liechtenstein
Reagenzglasschüttler MS2 Minishaker IKA-Werke GmbH & CO. KG,
Staufen Deutschland
Reihenwasserbad (Soxhlet-
apparatur) 1041
GFL Gesellschaft für Labortech-
nik GmbH, Burgwedel Deutsch-
land
Rotationsverdampfer (Be-
stimmung der Hydroperoxid-
bestimmung)
BÜCHI Labortechnik AG, Flawil
Schweiz

Anhang
145
Geräte Typ Hersteller
Rührwerk (Blattrührer) RZR 2050 electronic
Heidolph Instruments GmbH &
Co.KG, Schwabach Deutsch-
land
Schlauchpumpe TL medorex e. K., Nörten-
Hardenberg Deutschland
Schnellfeuchtebestimmer LP16 Mettler-Toledo GmbH, Gießen
Deutschland
Schüttelbank Celloshaker Variospeed Chemetron Products
Siebturm 304.030.010 Retsch GmbH & Co. KG, Haan
Deutschland
Sprühturm Mobil Minor GEA Group Aktiengesellschaft,
Düsseldorf Deutschland
Trockenschrank UFP800DW Memmert GmbH + Co. KG,
Schwabach Deutschland
Trockenschrank T6120
Thermo Fisher Scientific Ger-
many BV & Co KG, Braun-
schweig Deutschland
Trommelcoater AR 401 ERWEKA GmbH, Heusen-
stamm Deutschland
Ultra-Turrax T25 basic IKA-Werke GmbH & CO. KG,
Staufen Deutschland
Umlaufkühler (Soxhletappa-
ratur) Multi Temp II Pharmacia LKB

Anhang
146
Geräte Typ Hersteller
Varipette (1-10 mL) Eppendorf AG, Hamburg
Deutschland
Viscosimeter DV-II + Pro Extra Brookfield E. L. V. GmbH,
Lorch, Deutschland
Waage L 610D Sartorius AG, Göttingen,
Deutschland
Waage MC 1 Analytic AC 210
P
Sartorius AG, Göttingen,
Deutschland
Wasserbad (Hydroperoxid-
bestimmung) 1003
GFL Gesellschaft für Labortech-
nik GmbH, Burgwedel Deutsch-
land
Dynamisches Dampfsorpti-
onsmessgerät DVS-1
Surface Measurement Systems
Ltd., London, England
Zentrifuge Sigma 3K12 Sigma Laborzentrifugen GmbH,
Osterode am Harz

Anhang
147
Tabelle A.2: Verwendete Chemikalien
Chemikalie Hersteller Chargen-Nr. Bestellnr.
2-Propanol Carl Roth GmbH & Co. KG,
Karlsruhe Deutschland 6752,5
Ammoniumthiocyanat Carl Roth GmbH & Co. KG,
Karlsruhe Deutschland 4477,2
Bariumchlorid-Dihydrat Carl Roth GmbH & Co. KG,
Karlsruhe Deutschland 4453,1
Carboxymethyl-Zellulose
CMC
Harke FoodTec GmbH,
Mühlheim, Deutschland 1012086 40-2
Diiodmethan Merck KGaA, Darmstadt,
Deutschland S6803853350
8.181.530.0
25
Eisen-(II)-sulfat-
Heptahydrat
Carl Roth GmbH & Co. KG,
Karlsruhe Deutschland P015.1
Eisen(III)-chlorid-
Hexahydrat
Carl Roth GmbH & Co. KG,
Karlsruhe Deutschland P742.1
Ethylcellulose Dispersi-
on Type D/NF
Colorcon Limited, Dartford
Kent, United Kingdom IN 526123
Ethylenglykol Carl Roth GmbH & Co. KG,
Karlsruhe Deutschland 134212798 2441,1
Florisil, 60-100 mesh Carl Roth GmbH & Co. KG,
Karlsruhe Deutschland 323 20 3800 101,2
Formamid Merck KGaA, Darmstadt,
Deutschland K43521884
1.096.841.0
00
Hydroxypropyl-
Methyzellulose HPMC
Harke Pharma GmbH,
Mühlheim, Deutschland 1109-215 HSE-6

Anhang
148
Chemikalie Hersteller Chargen-Nr. Bestellnr.
Isooctan Carl Roth GmbH & Co. KG,
Karlsruhe Deutschland 6889,2
Kaliumdihydrogen-
phosphat
Carl Roth GmbH & Co. KG,
Karlsruhe Deutschland 40255070 3904,1
Lithiumchlorid Carl Roth GmbH & Co. KG,
Karlsruhe Deutschland P007.2
Magnesiumchlorid Carl Roth GmbH & Co. KG,
Karlsruhe Deutschland 2189,3
Maltodextrin National
M2
Ingredion Germany GmbH,
Hamburg Deutschland KDB8617/CDB8602 10235709
Maltodextrin M3 Ingredion Inc. Illinois,USA LH 8631
Methylzellulose MC Harke FoodTec GmbH,
Mühlheim, Deutschland AFF 015-320008
Any Addy
AN15
MCT-Öl Cremer Oleo GmbH & Co.
KG, Hamburg, Deutschland
MUV-25.06.13-
M105-06-13
Cremercoor
60/40
Methocel A15 Premium
LV Methylcellulose
Dow Wolff Cellulosics
GmbH, Bomlitz Deutsch-
land
2G02012N22
Natriumnitrat Carl Roth GmbH & Co. KG,
Karlsruhe Deutschland 8601,2
NS Enteric Colorcon Limited, Dartford
Kent, United Kingdom 29Z19241 DT 618989
OSA-Stärke C*EmCap
12633
Cargill Deutschland GmbH,
Krefeld Deutschland R1A014A 23.826.327

Anhang
149
Chemikalie Hersteller Chargen-Nr. Bestellnr.
Petrolether 40-60 °C VWR International GmbH,
Darmstadt Deutschland 9076,2
Phosphorpentoxid Carl Roth GmbH & Co. KG,
Karlsruhe Deutschland
Rapsöl Kaiser’s Tengelmann
GmbH (A&P)
Salzsäure, 25 % Carl Roth GmbH & Co. KG,
Karlsruhe Deutschland 202 52.290
Salzsäure, 37 % VWR International GmbH,
Darmstadt Deutschland
Silbernitratlösung,
0,1 mol/L
Carl Roth GmbH & Co. KG,
Karlsruhe Deutschland K713.2

Anhang
150
Tabelle A.3: Verwendete Materialien
Materialien Bezeichnung Hersteller
Aluminiumschale
Extraktionshülse Carl Roth GmbH & Co. KG, Karlsruhe
Deutschland
Filterpapier (MVE) 388, 12,5 cm Spezialpapier FILTRAK GmbH, Nieder-
schlag Deutschland
Filterpapier
(Weibull- Stoldt) 488/N, 20 cm
Spezialpapier FILTRAK GmbH, Nieder-
schlag Deutschland
Küvetten 2,5 mL makro Brand GmbH & Co. KG
Nadeln
1 mm, Pink,
PN 7018169,
LOT/SN
4000121662
GLT Dosiertechnik, Pforzheim, Deutschland
Objektträger,
geschliffen 90°
76 x 26 mm,
Art. Nr. H869,
LOT 1532572
Carl Roth GmbH & Co. KG, Karlsruhe
Deutschland
Reinigungstücher Kim Wipes Kimberly Clark AG, Dallas, USA
Spritze Injekt-F, 1mL B. Braun Melsungen AG, Melsungen,
Deutschland
Wägepinsel 5481,1 Carl Roth GmbH & Co. KG, Karlsruhe
Deutschland

Anhang
151
Tabelle A.4: Gemessene Kontaktwinkel der Referenzlösungen auf den maltodextrinbasierten Mo-
delloberflächen
MDT MDGSS MDOSA
Dest. Wasser 34,11 ± 1,73 25,71 ± 0,84 31,35 ± 1,68
Diiodmethan 33,47 ± 4,31 42,87 ± 1,61 39,61 ± 0,67
Ethylenglykol 34,79 ± 1,98 13,00 ± 1,16 17,87 ± 0,48
Formamid 40,58 ± 2,86 14,97 ± 1,33 24,60 ± 0,61
Tabelle A.5: Direkt gemessene Kontaktwinkel (CAm) auf den Oberflächen MDT, MDGSS und MDO-
SA
MDT MDGSS MDOSA
CMCvl 32,34 ± 1,31 18.1 ± 2.1 33,09 ± 0,92
CMCl 34,95 ± 1,59 20.1 ± 2.7 36,75 ± 0,57
CMCm 38,30 ± 1,37 22.7 ± 2.8 40,07 ±1,35
CMCh 40,59 ± 1,09 26.6 ± 3.3 42,32 ± 1,51
MCvl 34,28 ± 1,76 16.8 ± 1.7 30,61 ± 0,96
MCl 36,33 ± 1,51 18.8 ± 2.5 35,40 ± 1,04
MCm 38,18 ± 0,87 20.0 ± 3.2 38,41 ± 1,33
MCh 39,70 ± 1,36 22.2 ± 1.9 42,10 ± 0,55
Nvl 40,48 ± 1,75 15.9 ± 2.6 26,65 ± 0,95
Nl 41,25 ± 2,80 19.6 ± 3.1 30,08 ± 0,83
Nm 43,35 ± 2,49 20.2 ± 4.3 35,39 ± 0,82
Nh 47,82 ± 1,64 24.8 ± 3.0 38,91 ± 0,96
HPMCvl 37,60 ± 1,22 20.5 ± 1.5 33,91 ± 1,11
HPMCl 39,60 ± 1,07 21.7 ± 0.9 37,96 ± 1,06
HPMCm 41,40 ± 1,53 23.5 ± 1.7 42,27 ± 0,98
HPMCh 44,30 ± 1,40 26.6 ± 1.3 45,28 ± 0,96

Anhang
152
Tabelle A.6: Freie Oberflächenenergie mit dispersen und polaren Anteil, ihren prozentualen Anteilen
und dem Regressionskoeffizienten für die Kalkulation der Oberflächenenergie MDT unter Verwen-
dung verschiedener Kombinationen von Referenzlösungen zur Oberflächenenergiecharakterisierung
WEF WDEF WDE WDF DEF
SE [mN/m] 70,36 54,47 58,24 59,62 48,12
SEd [mN/m] 2,31 30,69 32,42 36,99 40,71
SEd [% SE] 3,28 56,33 55,66 62,05 84,60
SEp [mN/m] 68,05 23,79 25,82 22,63 7,41
SEp [% SE] 96,72 43,67 44,34 37,95 15,40
r^2 0,99 0,85 0,88 0,91 0,89
Tabelle A.7: Freie Oberflächenenergie mit dispersen und polaren Anteil, ihren prozentualen Anteilen
und dem Regressionskoeffizienten für die Kalkulation der Oberflächenenergie MDGSS unter Ver-
wendung verschiedener Kombinationen von Referenzlösungen zur Oberflächenenergiecharakterisie-
rung
WEF WDEF WDE WDF DEF
SE [mN/m] 70.8 61.0 62.9 65.0 53.5
SEd [mN/m] 6.5 29.8 30.6 34.9 36.1
SEd [% SE] 9.2 48.8 48.6 53.7 67.5
SEp [mN/m] 64.3 31.2 32.3 30.1 17.4
SEp [% SE] 90.8 51.2 51.4 46.3 32.5
R2 1.000 0.936 0.942 0.974 0.941

Anhang
153
Tabelle A.8: Freie Oberflächenenergie mit dispersen und polaren Anteil, ihren prozentualen Anteilen
und dem Regressionskoeffizienten für die Kalkulation der Oberflächenenergie MDOSA unter Ver-
wendung verschiedener Kombinationen von Referenzlösungen zur Oberflächenenergiecharakterisie-
rung
WEF WDEF WDE WDF DEF
SE [mN/m] 66,35 58,58 60,78 62,66 52,04
SEd [mN/m] 6,97 31,14 32,11 36,23 37,92
SEd [% SE] 10,50 53,15 52,83 57,81 72,87
SEp [mN/m] 59,38 27,45 28,67 26,44 14,12
SEp [% SE] 89,50 46,85 47,17 42,19 27,13
R2 0,999 0,926 0,936 0,965 0,940

Anhang
154
Tabelle A.9: Berechnete Adhäsionsarbeit der verwendeten Coatingmaterialien unter Berücksichti-
gung der Verwendung von unterschiedlichen Kombinationen Referenzlösungen zur Bestimmung der
Oberflächenenergie von MDGSS
WA-WEF
[mN/m]
WA-WDEF
[mN/m]
WA-WDE
[mN/m]
WA-WDF
[mN/m]
WA-DEF
[mN/m]
WA-YD
[mN/m]
CMCvl 136.3 a,A 133.5 a,BC 135.6 a,BC 136.9 a,B 120.0 a,C 144.7
CMCl 136.3 a,A 133.2 a,BC 135.3 a,BC 136.5 a,B 119.6 a,C 143.4
CMCm 133.3 a,A 132.8 a,BC 134.8 a,BC 136.3 a,B 120.1 a,C 140.2
CMCh 136.7 a,A 133.2 a,BC 135.3 a,BC 136.5 a,B 119.4 a,C 140.0
MCvl 95.4 b,A 109.0 b,BC 110.6 b,BC 113.4 b,B 103.7 b,C 98.5
MCl 94.8 b,A 108.8 b,BC 110.4 b,BC 113.3 b,B 103.7 b,C 97.9
MCm 94.0 b,A 108.2 b,BC 109.8 b,BC 112.6 b,B 103.2 b,C 96.6
MCh 93.7 b,A 108.2 b,BC 109.9 b,BC 112.7 b,B 103.4 b,C 96.2
Nvl 72.5 c,A 88.2 c,BC 89.5 c,BC 92.2 c,B 85.6 c,C 67.8
Nl 63.9 c,A 83.2 c,BC 84.4 c,BC 87.5 c,B 82.5 c,C 64.1
Nm 58.6 c,A 79.6 c,BC 80.7 c,BC 83.9 c,B 79.8 c,C 61.4
Nh 56.3 c,A 77.8 c,BC 78.9 c,BC 82.2 c,B 78.4 c,C 59.0
HPMCvl 86.7 b,A 112.0 b,BC 113.7 b,BC 117.8 b,B 110.8 b,C 115.0
HPMCl 82.1 b,A 109.2 b,BC 110.8 b,BC 115.1 b,B 109.0 b,C 112.6
HPMCm 85.0 b,A 109.8 b,BC 111.4 b,BC 115.4 b,B 108.6 b,C 109.3
HPMCh 81.1 b,A 108.3 b,BC 109.8 b,BC 114.1 b,B 108.1 b,C 109.0
a, b, c, d unterschiedliche Buchstaben kennzeichnen signifikante Unterschiede zwischen unter-
schiedlichen Coatingmaterialien
A, B, C unterschiedliche Buchstaben kennzeichnen signifikante Unterschiede zwischen unterschied-
lichen Kombinationen aus Referenzlösungen bzw. unterschiedlichen Berechnungsmodellen für die
Adhäsionsarbeit

Anhang
155
Tabelle A.10: Nach dem Modell von OWRK berechneter Kontaktwinkel von verschiedenen Coating-
materialien unter Verwendung unterschiedlicher Kombinationen von Referenzlösungen zur Oberflä-
chenenergiecharakterisierung von MDGSS
WEF WDEF WDE WDF DEF
CMCvl 33,1 36,8 34,0 32,2 51,8
CMCl 32,4 36,6 33,8 32,0 51,8
CMCm 33,7 34,6 31,6 29,2 49,5
CMCh 32,0 36,8 34,0 32,3 52,1
HPMCvl 62,5 27,4 23,8 10,3 29,9
HPMCl 66,0 29,3 26,0 13,6 29,9
HPMCm 60,6 22,2 17,4 0 25,2
HPMCh 65,8 28,1 24,6 10,6 28,5
Tabelle A.11: Technische Daten des luftgelagerten Rheometers USD200 der Fima Anton Paar Ger-
many GmbH
Minimaler Wert Maximaler Wert
Drehmoment (DSO) 0.5 µNm 150 mNm
Drehmomentauflösung 0.01 µNm
Drehzahl (CSR) 10-4 min-1 1000 min-1
Frequenz 10-4 Hz 100 Hz
Winkelauflösung < 1 µrad
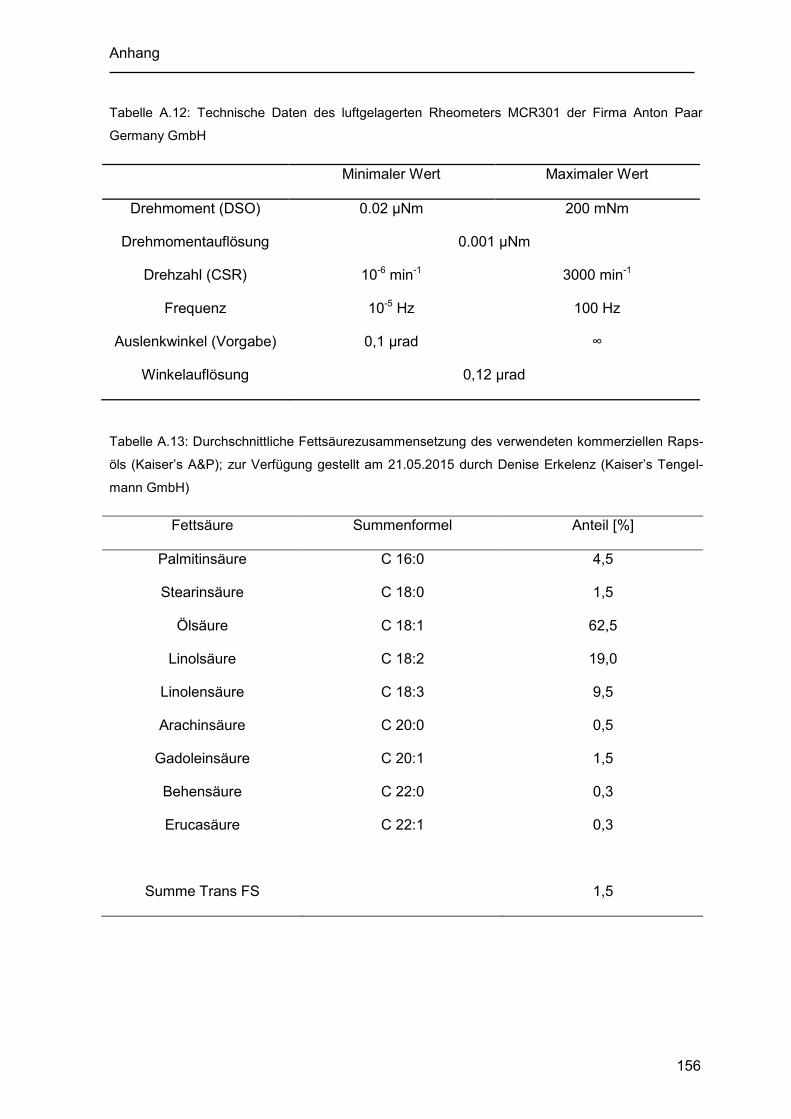
Anhang
156
Tabelle A.12: Technische Daten des luftgelagerten Rheometers MCR301 der Firma Anton Paar
Germany GmbH
Minimaler Wert Maximaler Wert
Drehmoment (DSO) 0.02 µNm 200 mNm
Drehmomentauflösung 0.001 µNm
Drehzahl (CSR) 10-6 min-1 3000 min-1
Frequenz 10-5 Hz 100 Hz
Auslenkwinkel (Vorgabe) 0,1 µrad ∞
Winkelauflösung 0,12 µrad
Tabelle A.13: Durchschnittliche Fettsäurezusammensetzung des verwendeten kommerziellen Raps-
öls (Kaiser’s A&P); zur Verfügung gestellt am 21.05.2015 durch Denise Erkelenz (Kaiser’s Tengel-
mann GmbH)
Fettsäure Summenformel Anteil [%]
Palmitinsäure C 16:0 4,5
Stearinsäure C 18:0 1,5
Ölsäure C 18:1 62,5
Linolsäure C 18:2 19,0
Linolensäure C 18:3 9,5
Arachinsäure C 20:0 0,5
Gadoleinsäure C 20:1 1,5
Behensäure C 22:0 0,3
Erucasäure C 22:1 0,3
Summe Trans FS 1,5

Anhang
157
Tabelle A.14: Ergebnisse des Scherratensweep mit den Ergebnissen bei 1000 s-1, 2500 s-1 und 4000
s-1 bei 30 °C, 45 °C und 60 °C für eine 1 %ige MC-Lösung von MC383
Scherrate [s-1] Temperatur [°C] Viskosität [mPa·s]
1000 30 19,86
1000 30 16,81
1000 30 18,22
4000 30 16,05
4000 30 13,5
4000 30 14,45
1000 60 11,22
1000 60 4,517
1000 60 7,576
4000 60 5,143
4000 60 3,366
4000 60 4,033
2500 45 10,56
2500 45 8,571
2500 45 9,781

Anhang
158
Tabelle A.15: Ergebnisse des Scherratensweep mit den Ergebnissen bei 1000 s-1, 2500 s-1 und 4000
s-1 bei 30 °C, 45 °C und 60 °C für eine 3 %ige MC-Lösung von MC383
Scherrate [s-1] Temperatur [°C] Viskosität [mPa·s]
1000 45 117,8
1000 45 146,2
1000 45 158,7
4000 45 68,56
4000 45 80,3
4000 45 84,85
2500 30 187,6
2500 30 166,9
2500 30 171
2500 60 29,78
2500 60 28,53
2500 60 49,12
2500 45 84,53
2500 45 100,3
2500 45 108,6
2500 45 107,1
2500 45 96,14
2500 45 99,67

Anhang
159
Tabelle A.16: Ergebnisse des Scherratensweep mit den Ergebnissen bei 1000 s-1, 2500 s-1 und 4000
s-1 bei 30 °C, 45 °C und 60 °C für eine 5 %ige MC-Lösung von MC383
Scherrate [s-1] Temperatur [°C] Viskosität [mPa·s]
1000 30 867,8
1000 30 899,4
1000 30 1000
4000 30 346,7
4000 30 360,9
4000 30 386
1000 60 37,05
1000 60 54,72
1000 60 119,7
4000 60 47,03
4000 60 62,19
4000 60 45,51
2500 45 307,5
2500 45 319
2500 45 355,7

Anhang
160
Tabelle A.17: Ergebnisse des Scherratensweep mit den Ergebnissen bei 1000 s-1, 2500 s-1 und 4000
s-1 bei 30 °C, 45 °C und 60 °C für eine 5 %ige MC-Lösung von MC15
Scherrate [s-1] Temperatur [°C] Viskosität [mPa·s]
1000 30 94,34
1000 30 96,71
1000 30 97,56
4000 30 70,75
4000 30 70,68
4000 30 70,04
1000 60 104,1
1000 60 93,32
1000 60 106,9
4000 60 30,92
4000 60 32,27
4000 60 33,62
2500 45 47,15
2500 45 48,55
2500 45 48,33

Anhang
161
Tabelle A.18: Ergebnisse des Scherratensweep mit den Ergebnissen bei 1000 s-1, 2500 s-1 und 4000
s-1 bei 30 °C, 45 °C und 60 °C für eine 6,5 %ige MC-Lösung von MC15
Scherrate [s-1] Temperatur [°C] Viskosität [mPa·s]
1000 45 161,1
1000 45 152,6
1000 45 154
4000 45 87,64
4000 45 90,09
4000 45 89,72
2500 30 177,8
2500 30 170
2500 30 172,5
2500 60 82,09
2500 60 63,79
2500 60 73,14
2500 45 108,6
2500 45 105,3
2500 45 110
2500 45 108
2500 45 105,6
2500 45 109

Anhang
162
Tabelle A.19: Ergebnisse des Scherratensweep mit den Ergebnissen bei 1000 s-1, 2500 s-1 und 4000
s-1 bei 30 °C, 45 °C und 60 °C für eine 8 %ige MC-Lösung von MC15
Scherrate [s-1] Temperatur [°C] Viskosität [mPa·s]
1000 30 496,1
1000 30 487,3
1000 30 486,4
4000 30 256,3
4000 30 255,3
4000 30 255,5
1000 60 302,7
1000 60 229,6
1000 60 197,9
4000 60 85,87
4000 60 67,01
4000 60 70,55
2500 45 211,6
2500 45 207,5
2500 45 211
Tabelle A.20 Temperaturprofil 1 zur Bestimmung der strukturbildenden Charakteristika der MC-
Lösungen
1. Phase 2. Phase 3. Phase
Aufheizung Haltezeit Abkühlung
Temperatur [°C] 20-70 70 70-20
Dauer [min] 50 30 50
Messpunkte 50 30 50
Messpunktdauer [min] 1 1 1

Anhang
163
Tabelle A.21: Temperaturprofil 2 zur Bestimmung der strukturbildenden Charakteristika der MC-
Lösungen unter Einfluss eine Zugabe von Ethanol
1. Phase 2. Phase 3. Phase
Aufheizung Haltezeit Abkühlung
Temperatur [°C] 20-60 60 60-20
Dauer [min] 40 20 40
Messpunkte 40 20 40
Messpunktdauer [min] 1 1 1
Tabelle A.22: Initiale Strukturierungstemperatur (IST), Gelpunkt (GP), kritische Strukturierungstem-
peratur (CST), mittlere Strukturentwicklungsgeschwindigkeit (SDR), Gelauflösungstemperatur (GA),
Gelstärke zu Beginn der Haltezeit (t=0) und Gelstärke nach 30 Minuten Haltezeit (t=30) der MC15–
und MC383-Proben bei verschiedenen Konzentrationen
IST [°C] GP [°C] CST [°C] SDR [Pa/s] GA [°C]
Gelstärke
(t=0) [Pa]
Gelstärke
(t=30) [Pa]
1,0 %
MC383 44,9 48,4 61,6 0,07 31,9 116 173
3,0 %
MC383 43,9 51,9 54,8 0,66 31,9 1110 1613
5,0 %
MC383 39,8 47,6 51,1 2,29 28,9 4125 5423
5,0 %
MC15 38,3 49,7 53,5 0,72 31,4 1356 1905
6,5 %
MC15 36,3 46,0 51,8 2,17 27,9 4371 5851
8,0 %
MC15 34,2 42,9 50,1 3,40 26,5 7262 9574

Anhang
164
Tabelle A.23: Herstellungsanweisung für die Lösungen zur Bestimmung der Hydroperoxidkonzentra-
tion
Lösungen Herstellung
Ammoniumthiocyanatlösung 30,0 g in 100 mL dest. Wasser lösen
Eisen(II)-sulfat-Heptahydratlösung 0,5 g in 50 mL dest. Wasser lösen
Bariumchloridlösung 0,4 g in 50 mL dest. Wasser lösen
Eisen(II)-chloridlösung Eisensulfat- und Bariumchloridlösung
mischen und 2 mL 25 %ige HCl zugeben,
über Nacht im Kühlschrank aufbewahren
und entstandenen Bariumsulfatnieder-
schlag abfiltrieren
Eisen(III)-chlorid-Hexahydratlösung 0,0150 g in 50 mL dest. Wasser lösen
Isooctan/2-Propanol-Gemisch Beide Lösungsmittel zu gleichen Teilen
mischen
2-Propanolrückstände Bei 370 mbar und 80 °C abdestillieren

Anhang
165
Tabelle A.24: Gehalte an Eisen(III)-chloridlösung für die Verdünnungsstufen der Kalibriergeraden zur
Bestimmung der Hydroperoxidkonzentration
Verdünnungsstufe Eisen(III)chloridlösung [µL] 2-Propanol[µL]
1 10 4990
2 50 4950
3 100 4900
4 150 4850
5 200 4800
6 250 4750
7 300 4700
8 350 4650
9 450 4550
10 550 4450
Tabelle A.25: Einwaagen der Salze zur Herstellung von übersättigten Salzkonzentrationen für die
Bestimmung des Wasserdampfsorption von Pulvern
Salz relative Luftfeuchte [%] Einwaage in dest. Wasser [g/l]
Lithiumchlorid 11 915,2
Magnesiumchlorid 33 1837
Magnesiumnitrat 53 462
Natriumnitrat 75 961,4
Kaliumchlorid 85 363

Anhang
166
Tabelle A.26: Hydroperoxidkonzentration des sprühgetrockneten Pulvers, der Agglomerate, und der
mit 70 bzw. 130 mL MC-Lösung gecoateten Agglomerate über den Zeitraum von acht Wochen mit
wöchentlicher Untersuchung, mit Ausnahme von Woche sieben
Zeit (Wochen) Pulver Agglomerate MC15-70-T60-t30 MC15-130-T60-t60
0 5,2 ± 0,7 7,5 ± 1,4 8,8 ± 1,0 8,9 ± 0,5
1 8,7 ± 2,4 8,7 ± 2,4 16,2 ± 1,1 15,1 ± 0,7
2 7,9 ± 0,5 9,5 ± 0,5 22,1 ± 2,1 22,0 ± 0,5
4 9,9 ± 0,9 9,0 ± 2,7 23,9 ± 0,3 30,7 ± 0,7
5 16,1 ± 0,3 17,0 ± 0,6 32,7 ± 0,8 47,4 ± 1,1
6 12,3 ± 0,3 12,7 ± 0,3 33,2 ± 1,5 59,3 ± 2,6
8 17,3 ± 0,3 17,2 ± 2,1 40,5 ± 0,3 51,2 ± 5,8
Tabelle A.27: Hydroperoxidkonzentration der unbehandelten Agglomerate und der bei 30°C über 20,
40 und 60 Minuten behandelten Agglomerate über den Zeitraum von 10 Wochen mit Untersuchun-
gen alle zwei Wochen
Zeit (Wochen) Agglomerate T30-t20 T30-t40 T30-t60
0 3,1 ± 0,1 4,2 ± 1,7 7,1 ± 3,0 6,5 ± 1,1
2 6,2 ± 1,1 12,7 ± 1,2 15,2 ± 3,9 14,9 ± 0,7
4 14,1 ± 1,3 20,1 ± 1,7 19,6 ±1,1 23,3 ± 0,0
6 22,6 ± 0,6 28,1 ± 0,5 28,1 ± 0,8 115,9 ± 4,2
8 31,6 ± 2,0 37,9 ± 1,7 38,1 ± 4,3 176,3 ± 11,2
10 47,5 ± 4,2 81,3 ± 10,1 227,9 ± 13,7 210,6 ± 22,2

Anhang
167
Tabelle A.28: Hydroperoxidkonzentration der bei 60°C über 20, 40 und 60 Minuten behandelten
Agglomerate über den Zeitraum von 10 Wochen mit Untersuchungen alle zwei Wochen
Zeit (Wochen) T60-t20 T60-t40 T60-t60
0 4,1 ± 0,1 6,0 ± 0,4 4,5 ± 0,3
2 12,7 ± 0,9 15,4 ± 0,1 12,8 ± 0,6
4 17,1 ± 0,4 20,4 ± 2,1 17,6 ± 0,8
6 23,3 ± 0,6 31,4 ± 1,6 42,9 ± 0,7
8 28,6 ± 1,0 94,4 ± 4,3 190,1 ± 17,0
10 47,1 ± 8,9 266,1 ± 14,7 251,8 ± 21,9
Tabelle A.29 Hydroperoxidkonzentration der bei 30°C über 20, 40 und 60 Minuten mit Wasser be-
handelten Agglomerate über den Zeitraum von 10 Wochen mit Untersuchungen alle zwei Wochen
H2O-T30-t20 H2O-T30-t40 H2O-T30-t60
0 4,1 ± 0,5 6,4 ± - 4,0 ± 0,1
2 11,8 ± 0,3 13,0 ± 0,6 12,3 ± 0,2
4 16,4 ± 2,3 16,6 ± 0,5 17,1 ± 1,1
6 27,6 ± 0,3 29,5 ± 0,6 29,6 ± 2,3
8 37,5 ± 1,3 41,9 ± 0,6 44,8 ± 2,1
10 50,2 ± 6,9 96,4 ± 10,4 97,7 ± 8,0

Anhang
168
Tabelle A.30: Hydroperoxidkonzentration der bei 60°C über 20, 40 und 60 Minuten mit Wasser be-
handelten Agglomerate über den Zeitraum von 10 Wochen mit Untersuchungen alle zwei Wochen
H2O-T60-t20 H2O-T60-t40 H2O-T60-t60
0 6,4 ± 2,1 6,6 ± 0,8 8,1 ± 1,1
2 12,9 ± 0,3 13,8 ± 0,3 18,7 ± 2,7
4 16,5 ± 2,7 20,6 ± 3,8 24,8 ± 2,5
6 30,5 ± 2,8 27,0 ± 0,5 31,1 ± 0,7
8 31,3 ± 1,2 34,1 ± 2,2 37,9 ± 1,8
10 52,5 ± 9,9 60,1 ± 2,7 61,2 ± 8,1
Tabelle A.31: Hydroperoxidkonzentration der unbehandelten Agglomerate und der bei 60°C über 20
bzw. 60 Minuten mit und ohne Wasser behandelten Agglomerate über den Zeitraum von 16 Wochen
mit Untersuchungen alle zwei Wochen
Zeit (Wochen) Agglomerate H2O-T60-t20 H2O-T60-t60 T60-t60
0 3,5 ± 1,2 3,4 ± 0,5 3,8 ± 0,5 3,7 ± 0,4
2 3,6 ± 0,4 6,6 ± 0,3 7,6 ± 0,4 8,8 ± 0,2
4 5,3 ± 0,4 8,4 ± 0,2 10,1 ± 0,4 11,7 ± 0,7
6 8,1 ± 0,2 11,5 ± 0,4 13,7 ± 0,7 15,6 ± 0,1
8 9,9 ± 0,9 13,6 ± 0,5 19,4 ± 1,2 20,6 ± 0,9
10 11,2 ± 0,3 12,9 ± 3,2 18,1 ± 0,3 19,6 ± 0,8
12 14,5 ± 0,4 16,5 ± 1,2 23,3 ± 1,2 31,1 ± 0,5
14 17,8 ± 0,2 19,2 ± 0,9 32,1 ± 4,8 89,4 ± 12,7
16 24,9 ± 3,2 26,4 ± 1,3 67,9 ± 2,7 235,5 ± 2,7

Anhang
169
Tabelle A.32: Hydroperoxidkonzentration der unbehandelten Agglomerate und der bei 30°C und
60°C über 20 bzw. 60 Minuten mit einer 5%igen MC-Lösung gecoateten Agglomerate über den Zeit-
raum von 18 Wochen mit Untersuchungen alle zwei Wochen
Zeit
(Wochen) Agglomerate
MC15-
T30-t20
MC15-
T30-t60
MC15-
T60-t20
MC15-
T60-t60
0 1,1 ± 0,5 2,9 ± 0,0 3,0 ± 0,3 2,6 ± 0,4 3,3 ± 1,0
2 2,1 ± 0,4 5,4 ± 0,0 7,0 ± 0,7 6,8 ± 0,2 8,7 ± 0,3
4 5,0 ± 0,5 9,9 ± 0,6 13,9 ± 0,5 10,5 ± 0,9 18,5 ± 0,9
6 6,3 ± 0,9 11,9 ± 0,5 18,2 ± 0,2 13,4 ± 0,3 22,5 ± 0,3
8 10,7 ± 0,7 15,8 ± 0,7 24,8 ± 0,4 18,6 ± 0,4 29,5 ± 0,5
10 13,1 ± 0,9 19,9 ± 1,1 28,8 ± 0,8 21,5 ± 0,5 35,4 ± 0,8
14 16,2 ± 0,4 20,3 ± 1,2 30,1 ± 3,5 25,1 ± 2,5 42,8 ± 3,8
18 20,1 ± 0,8 21,6 ± 0,2 33,2 ± 3,7 31,5 ± 0,7 45,5 ± 3,0

Anhang
170
Tabelle A.33: Werte für die Wasseraufnahme der ungecoateten und mit MC gecoateten Agglomerate
in fünf verschiedenen relativen Luftfeuchten und über eine Zeit von 35 Tagen
rel.
Feuchte Tage Agglomerate
MC15-
T30t20
MC15-
T30t60
MC15-
T60t20
MC15-
T60t60
11 0 0 0 0 0 0
1 0,020 0,017 0,015 0,015 0,010
2 0,023 0,021 0,018 0,019 0,014
3 0,024 0,023 0,020 0,020 0,016
4 0,028 0,021 0,015 0,022 0,017
7 0,028 0,020 0,014 0,022 0,018
8 0,024 0,020 0,015 0,022 0,018
33 9 0,048 0,043 0,038 0,041 0,039
10 0,050 0,046 0,039 0,043 0,040
11 0,050 0,046 0,041 0,044 0,041
12 0,050 0,047 0,041 0,045 0,041
13 0,050 0,047 0,041 0,044 0,044
53 14 0,080 0,075 0,069 0,069 0,063
15 0,079 0,076 0,069 0,069 0,061
16 0,079 0,075 0,069 0,069 0,061
20 0,079 0,075 0,068 0,069 0,061
75 21 0,111 0,107 0,101 0,105 0,097
22 0,111 0,109 0,101 0,106 0,099
23 0,102 0,107 0,098 0,104 0,096
24 0,105 0,108 0,100 0,103 0,096
28 0,106 0,108 0,101 0,103 0,097
85 29 0,162 0,164 0,156 0,159 0,153
30 0,171 0,175 0,165 0,166 0,159
31 0,172 0,175 0,166 0,170 0,162
32 0,173 0,171 0,166 0,172 0,165
35 0,180 0,182 0,174 0,178 0,167

Anhang
171
Tabelle A.34: Hydroperoxidkonzentration von ungecoateten und gecoateten Modelloberflächen über
einen Lagerzeitraum von zehn Wochen zweiwöchigem Untersuchungsinterval
Hydroperoxidkonzentration [mmol/kg Öl]
Wochen ungecoatete Modelloberfläche gecoatete Modelloberfläche
0 1,4 ± 0,5 2,3 ± 0,9
2 5,8 ± 0,7 7,2 ± 0,5
4 7,1 ± 1,0 8,8 ± 0,1
6 12,5 ± 0,1 13,6 ± 0,7
8 10,3 ± 2,6 12,1 ± 0,3
10 14,0 ±0,7 16,9 ± 0,9
Tabelle A.35: Hydroperoxidkonzentrationen von ungecoateten bzw. unbehandelten Agglomeraten
der verschiedenen Versuchsreihen mit Startwerten (Woche 0) und den Wochen 8, 10, 16 und 18 aus
den jeweiligen Versuchsreihen mit: Versuch 1: Vorversuch zur Schichtdickenvariation; Versuch 2:
Versuch zur Prozessoptimierung ohne Wasserzugabe; 4: Versuch zur Partikelanalyse ohne Wasser-
zugabe und Versuch 6: Versuch zum MC- Coating
Woche Versuch 1 Versuch 2 Versuch 4 Versuch 6
0 7,5 ± 1,4 3,1 ± 0,1 3,5 ± 1,2 1,1 ± 0,5
8 17,2 ± 2,1 31,6 ± 2,0 9,9 ± 0,9 10,7 ± 0,7
10
47,5 ± 4,2 11,2 ± 0,3 13,1 ± 0,9
16
24,9 ± 3,2 18,2 ± 0,6
18
20,2 ± 0,8

Anhang
172
Tabelle A.36: Hydroperoxidkonzentrationen der bei 30 °C für 20 Minuten gecoateten bzw. behandel-
ten Agglomeraten der verschiedenen Versuchsreihen mit Startwerten (Woche 0) und den Wochen 8,
10, 16 und 18 aus den jeweiligen Versuchsreihen mit: Versuch 2: Versuch zur Prozessoptimierung
ohne Wasserzugabe; Versuch 3: Versuch zur Prozessoptimierung mit Wasserzugabe und Versuch
6: Versuch zum MC-Coating
Woche Versuch 2 Versuch 3 Versuch 6
0 4,2 ± 1,7 4,1 ± 0,5 2,9 ± 0,0
8 37,9 ± 1,7 37,5 ±1,3 15,8 ± 0,7
10 81,3 ± 10,1 50,2 ± 6,9 19,9 ± 1,1
16
18,2 ± 0,8
18
21,6 ± 0,2
Tabelle A.37: Hydroperoxidkonzentrationen der bei 30 °C für 60 Minuten gecoateten bzw. behandel-
ten Agglomeraten der verschiedenen Versuchsreihen mit Startwerten (Woche 0) und den Wochen 8,
10, 16 und 18 aus den jeweiligen Versuchsreihen mit: Versuch 2: Versuch zur Prozessoptimierung
ohne Wasserzugabe; Versuch 3: Versuch zur Prozessoptimierung mit Wasserzugabe und Versuch
6: Versuch zum MC- Coating
Woche Versuch 2 Versuch 3 Versuch 6
0 6,6 ± 1,1 4,0 ± 0,1 3,0 ± 0,3
8 176,3 ± 11,2 44,8 ± 2,1 24,8 ± 0,4
10 210,6 ± 22,6 97,7 ± 8,0 28,8 ± 0,8
16
28,5 ± 6,9
18
33,2 ± 3,7

Anhang
173
Tabelle A.38: Hydroperoxidkonzentrationen der bei 60 °C für 20 Minuten gecoateten bzw. behandel-
ten Agglomeraten der verschiedenen Versuchsreihen mit Startwerten (Woche 0) und den Wochen 8,
10, 16 und 18 aus den jeweiligen Versuchsreihen mit: Versuch 2: Versuch zur Prozessoptimierung
ohne Wasserzugabe; Versuch 3: Versuch zur Prozessoptimierung mit Wasserzugabe; Versuch 5:
Versuch zur Partikelanalyse mit Wasserzugabe und Versuch 6: Versuch zum MC- Coating
Woche Versuch 2 Versuch 3 Versuch 5 Versuch 6
0 4,1 ± 0,1 6,4 ± 2,1 3,4 ± 0,5 2,6 ± 0,4
8 28,6 ± 1,0 31,3 ± 1,2 13,6 ± 0,5 18,6 ± 0,4
10 47,1 ± 8,9 52,5 ± 9,9 12,9 ± 3,2 21,5 ± 0,5
16
26,4 ± 1,3 27,5 ± 2,6
18
31,5 ± 0,7
Tabelle A.39: Hydroperoxidkonzentrationen der bei 60 °C für 60 Minuten gecoateten bzw. behandel-
ten Agglomeraten der verschiedenen Versuchsreihen mit Startwerten (Woche 0) und den Wochen 8,
10, 16 und 18 aus den jeweiligen Versuchsreihen mit: Versuch 1: Vorversuch zur Schichtdickenvari-
ation; Versuch 2: Versuch zur Prozessoptimierung ohne Wasserzugabe; Versuch 3: Versuch zur
Prozessoptimierung mit Wasserzugabe; Versuch 4: Versuch zur Partikelanalyse ohne Wasserzuga-
be; Versuch 5: Versuch zur Partikelanalyse mit Wasserzugabe und Versuch 6: Versuch zum MC-
Coating
Woche Versuch 1 Versuch 2 Versuch 3 Versuch 4 Versuch 5 Versuch 6
0 8,9 ± 0,5 4,5 ± 0,3 8,1 ± 1,1 3,7 ± 0,4 3,8 ± 0,5 3,3 ± 1,1
8 51,2 ± 5,8 190,1 ± 17,0 37,9 ± 1,8 20,6 ± 0,9 19,4 ± 1,2 29,6 ± 0,5
10
251,8 ± 21,9 61,2 ± 8,1 19,6 ± 0,8 18,1 ± 0,3 35,4 ± 0,8
16
235,5 ± 2,7 67,9 ± 2,7 45,9 ± 4,1
18
45,5 ± 3,0

Anhang
174
Abbildungsanhang
Abbildung A.1: Logarithmisches Differenzialvolumen über der Porenweite der ölbeladenen Agglome-
rate und unterschiedlich in der Wirbelschicht behandelten ölbeladenen Agglomeraten
Abbildung A.2: Kumulatives Porenvolumen über der Porenweite der ölbeladenen Agglomerate und
unterschiedlich in der Wirbelschicht behandelten ölbeladenen Agglomeraten
0
0,5
1
1,5
2
2,5
0,001 0,01 0,1 1 10 100 1000
log. Differentialvolumen bei Intrusion [ml/g]
Porenweite [µm]
Agglomerate
H2O-T60t20
H2O-T60t60
T60t60
0
0,2
0,4
0,6
0,8
1
0,001 0,01 0,1 1 10 100 1000
kumulatives Porenvolumen [mL/g]
Porenweite [µm]
Agglomerate
H2O-T60t20
H2O-T60t60
T60t60

Anhang
175
Abbildung A.3: Logarithmisches Differenzialvolumen über der Porenweite der Matrixpartikel-
Agglomerate und unterschiedlich in der Wirbelschicht behandelten Matrixpartikel-Agglomeraten
Abbildung A.4: Kumulatives Porenvolumen über der Porenweite der Matrixpartikel-Agglomerate und
unterschiedlich in der Wirbelschicht behandelten Matrixpartikel-Agglomeraten
0
0,5
1
1,5
2
2,5
0,001 0,01 0,1 1 10 100 1000
log.
Diff
eren
tialv
olum
en b
ei In
trus
ion
[ml/g
]
Porenweite [µm]
Agglomerate
H2O-T60t60
T60t60
0
0,2
0,4
0,6
0,8
1
0,001 0,01 0,1 1 10 100 1000
kum
ulat
ives
Por
envo
lum
en [m
L/g]
Porenweite [µm]
Agglomerate
H2O-T60t60
T60t60

Anhang
176
Eidesstattliche Erklärung
Hiermit versichere ich, Adrian Kape, an Eides statt, dass ich die vorliegende Arbeit selbst-
ständig und eigenhändig sowie ausschließlich unter Verwendung der aufgeführten Quellen
und Hilfsmittel angefertigt habe.
Berlin, den 19. Mai 2016
Adrian Kape