waardenburg - jmg.bmj.com · 5 shah kn, dalal sj, sheth pn, joshi nc, ambani lm. white forelock,...
TRANSCRIPT

5 JMed Genet 1997;34:656-665
Syndrome of the month
Waardenburg syndrome
Andrew P Read, Valerie E Newton
AbstractAuditory-pigmentary syndromes arecaused by physical absence of melano-cytes from the skin, hair, eyes, or the striavascularis of the cochlea. Dominantlyinherited examples with patchy depig-mentation are usually labelled Waarden-burg syndrome (WS). Type I WS,characterised by dystopia canthorum, iscaused by loss offunction mutations in thePAX3 gene. Type III WS (Klein-Waardenburg syndrome, with abnormali-ties of the arms) is an extremepresentation of type I; some but not allpatients are homozygotes. Type IV WS(Shah-Waardenburg syndrome with Hir-schsprung disease) can be caused bymutations in the genes for endothelin-3 orone of its receptors, EDNRB. Type II WSis a heterogeneous group, about 15% ofwhom are heterozygous for mutations inthe MITF (microphthalmia associatedtranscription factor) gene. All these formsshow marked variability even within fami-lies, and at present it is not possible topredict the severity, even when a mutationis detected. Characterising the genes ishelping to unravel important develop-mental pathways in the neural crest andits derivatives.(YMed Genet 1997;34:656-665)
Keywords: Waardenburg syndrome; auditory-pigmentary syndromes
HistoryOn 14 December 1947 the Dutch ophthal-mologist and geneticist Petrus J Waardenburgpresented a deaf-mute man with "dystopiapunctorum lacrimarum, blepharophimosis andpartial iris atrophy" to a meeting of the DutchOphthalmological Society.' The patient hadblue eyes but was bald, and Waardenburg didnot at the time make the connection betweenhearing loss, white forelock, unusual eye col-our, and dystopia canthorum. He mentioneda report of twins with the same eyeabnormality who were "coincidentally" alsodeaf-mute. The following year, while he wasvisiting Geneva, David Klein showed him a10 year old girl with a remarkably severeauditory-pigmentary syndrome who alsohad dystopia canthorum. Realising that
coincidences were multiplying, Waarden-burg was prompted to undertake a system-atic search among 1050 inmates of fiveDutch institutions for the deaf.Waardenburg's results, published in a huge
paper2 in the American Journal of HumanGenetics in 1951, defined the syndrome nownamed type I Waardenburg syndrome (WS1).He characterised the syndrome as autosomaldominant with very high penetrance (159/161)of dystopia but reduced penetrance of all otherfeatures. He appears to have taken little interestin the patients who did not have dystopia can-thorum (he noted seven people with hetero-chromia or isochromic hypoplastic irides, butwithout dystopia, whom he did not investigatefurther). It was not until 1971 that Arias' drewattention to the existence of a separate divisionof the syndrome, which he named type IIWaardenburg syndrome (WS2). WS2 hasidentical auditory and pigmentary features toWS1 but lacks dystopia canthorum. Two ofWaardenburg's original families had this vari-ant, but both were so small that Waardenburghad overlooked the familial "non-penetrance"of dystopia.
Klein's patient was very different from thosein Waardenburg's families. As well as showingthe usual features of Waardenburg syndrometype I to an exceptional degree, she alsosuffered from a severe musculoskeletal syn-drome resembling amyoplasia. At first,Waardenburg speculated that perhaps she washomozygous for the gene mutated in hissyndrome. Later he evidently came to feel thatKlein's patient represented some differentclinical entity, a view that Klein did notappreciate.4 Over the years a small number ofother patients have been described who showsimilar features to Klein's patient, but in milderform. WS with musculoskeletal abnormalitieshas been called type III WS, Klein-Waardenburg syndrome, or WS3. Already in1983 Klein' suggested that this was a variantpresentation of WS 1.
In 1981, Shah et ar described 12 babies withHirschsprung's disease, white forelocks, andwhite eyelashes, born to five families inBombay. What, if any, relation these babies hadto Waardenburg syndrome is not clear. Dysto-pia was absent, hearing was not tested becauseall babies died in the neonatal period, and the
Department ofMedical Genetics, StMary's Hospital,Hathersage Road,Manchester M13 OJH,UKA P Read
Centre for Audiology,University ofManchester,Manchester M13 9PL,UKV E Newton
Correspondence to:Professor Read.
656
on 15 May 2019 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.34.8.656 on 1 August 1997. D
ownloaded from

Waardenburg syndrome
Table 1 The four types of Waardenburg syndrome
Type MIM Inheritance Distinguishing feature Comments
I 193500 AD Dystopia canthorum W>1.95 Nearly all have PAX3 mutationsII 193510 AD No dystopia Heterogeneous; 15% have MITF
mutationsIII (Klein-Waardenburg) 148820 AD (most cases Hypoplasia of limb muscles; Variant presentation ofWS 1; mostly
sporadic) contractures of elbows, fingers PAX3 heterozygotes; some may behomozygotes
IV (Shah-Waardenburg) 277580 Mostly AR Hirschsprung's disease Heterogeneous; includeshomozygotes for EDN3 or EDNRBmutations
pigmentary disorder of the irides was reportedas "isochromia irides, light brown irides withmosaic pattern...a common inherited condi-tion in our population". However, the pre-sumed recessive combination of pigmentarydisturbance and Hirschsprung's disease hasbeen called Shah-Waardenburg syndrome, typeIV Waardenburg syndrome, or WS4. Recentdemonstration of mutations in endothelin-3and its receptor (see below) have allowed a newand more concrete definition ofWS4.
Clinical features ofWaardenburgsyndrome types I and II
Table 1 summarises the four clinical types ofWaardenburg syndrome and formal diagnosticcriteria are shown in table 2. Whereas WS1defines a specific genetic entity, the clinicaldefinition of WS2 is arbitrary, covering anyauditory-pigmentary syndrome that does notclearly belong somewhere else. The label WS2undoubtedly covers a heterogeneous collectionof melanocyte defects. Apart from dystopiacanthorum, all features ofWS 1 and WS2 show
Table 2 Diagnostic criteria for Waardenburg syndrometypes I and II
Diagnostic criteria for WS1 have been proposed by theWaardenburg Consortium.'6 In brief, to be counted as affecteda person must have two major or one major plus two minorcriteria, from the following list:
Major criteriaCongenital sensorineural hearing loss
Pigmentary disturbances of iris (a) complete heterochromiairidium: two eyes of different colour; (b) partial or segmentalheterochromia: segments of blue or brown pigmentation inone or both eyes; or (c) hypoplastic blue eyes: characteristicbrilliant blue in both eyes
Hair hypopigmentation: white forelock
Dystopia canthorum: W> 1.95 averaged over affected familymembers (note that this was modified from the originalproposal of W>2.07 in the light of experience)
Affected first degree relative
Minor criteria
Congenital leucoderma: several areas ofhypopigmented skin
Synophyrys or medial eyebrow flare
Broad and high nasal root
Hypoplasia of alae nasi
Premature greying of hair: scalp hair predominantly whitebefore age 30
Criteria for WS2 were suggested by Lui et al.6 These authorsrecommended that two major features should be present to
make the diagnosis ofWS2. The major features are as in thelist above, except for the exclusion of dystopia canthorumand inclusion of premature greying
marked interfamilial and intrafamilial variabil-ity. Table 3 shows the penetrance of the variousfeatures in a series ofWS patients reported byLiu et al,6 from our own observations, and frompublished reports. The same pigmentationdefects appear in WS1 and WS2, but with dif-ferent frequency.6 The higher reported inci-dence of hearing loss in WS2 compared toWS1 is probably mainly a consequence ofdiagnostic requirements: without dystopia can-thorum as a guide, patients need to show moreauditory-pigmentary features to be classified asaffected. Fig 1 shows typical facial appearancesofWSl and WS2.
INCIDENCE AND PREVALENCEWaardenburg' estimated the prevalence of hissyndrome to be 1/42 000 of the population and1.43% of the congenitally deaf. Fraser7 found aprevalence of 2.12-3.01/100 000 in his schoolstudy of 2355 deaf children, and estimated aprevalence of 1.44-2.05/100 000 in the generalpopulation. WS1 and WS2, as defined by thediagnostic criteria in table 2, are about equallycommon. In addition there are many familiesascertained through a proband with hearingloss, in which one or more relatives have singlefeatures of WS2, without anybody satisfyingthe full diagnostic criteria. Without moleculardata it is impossible to say whether or not thesefamilies should be added to the figures forWS2. The mutation rate has been estimated as0.4/100 000 gametes' or 0.39/100 000gametes.7
DYSTOPIA CANTHORUMDystopia canthorum is the most penetrant fea-ture of WS1, being present in 99% of thoseaffected.8 Dystopia presents with the appear-ance of blepharophimosis and with fusion ofthe inner eyelids medially leading to a reduc-tion in the medial sclerae (fig 1). The inferiorlachrymal ducts are displaced laterally, with thepunctae opposite the cornea. Not only theinner canthal, but also the interpupillary andouter canthal distances are greater than nor-mal, indicating a degree of hypertelorism.
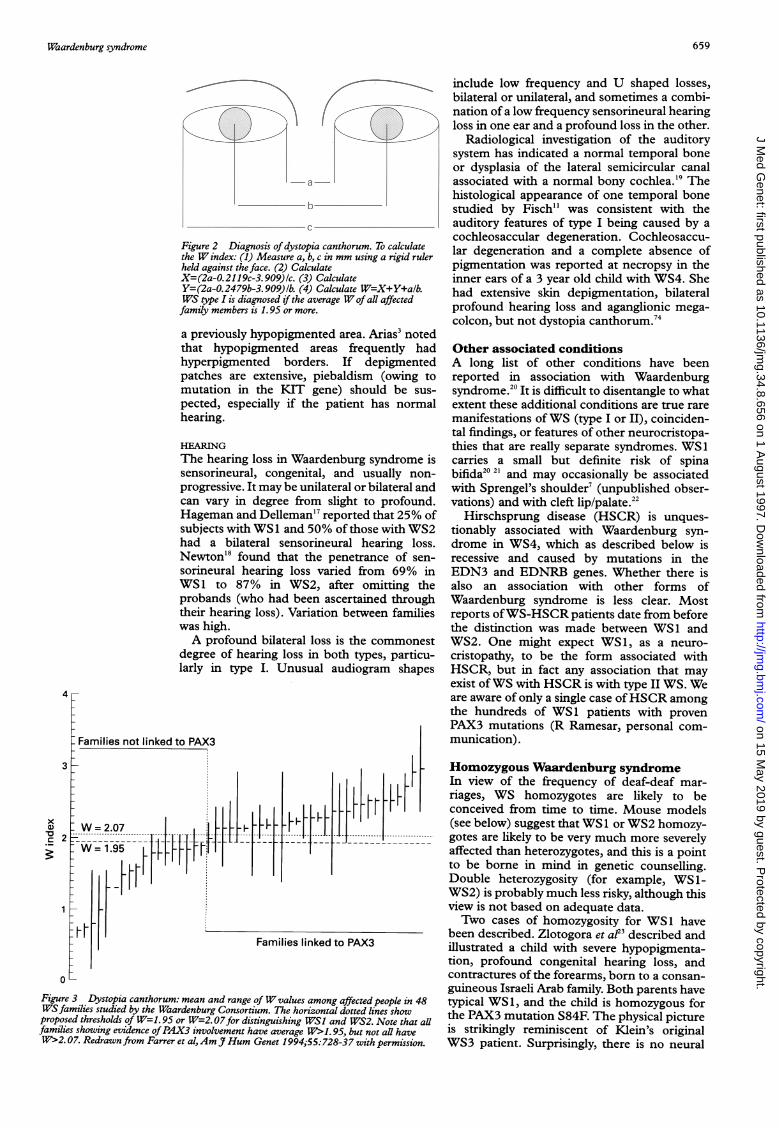
In the majority of affected subjects dystopiais readily recognised, but our experience showsthat clinical impression is not altogetherreliable. Subjects with WS2 risk being misdiag-nosed as WS 1 if there is mild hypertelorism. Itis much better to rely on a biometric index.The W index (fig 2) is the best of severalbroadly equivalent indices9 (the bizarre num-bers in the formula come from a discriminantanalysis). Such indices are based upon theinner canthal, interpupillary, and outer canthal
657
on 15 May 2019 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.34.8.656 on 1 August 1997. D
ownloaded from

658 Read, Newton
Table 3 Penetrance (%) of clinical features of type I and type II Waardenburg syndrome.Data from Liu et alr (where references to published cases are given)
Type Source No SNHL HetI HypE WF EG Skin HNR Eyb
WSI Liu et al 60 58 15 15 48 38 36 100 63Other reports 210 57 31 18 43 23 30 52 70
WS2 Liu et al 81 78 42 3 23 30 5 0 7Other reports 43 77 54 23 16 14 12 14 7
SNHL=sensorineural hearing loss, HetI= heterochromia irides, HypE=hypoplastic blue eyes,WF=white forelock, EG=early greying, Skin=white skin patches, HNR=high nasal root,Eyb=medial eyebrow flare.
Figure 1 Facial appearance of Waardenburg syndrome. (A) Type I WS. Note dystopiacanthorum. (B) Type I WS. Typical features include profound hearing loss, dystopiacanthorum, pale blue eyes, white forelock, and white eyelashes on the left. This boy has a
deletion of 7-8 Mb ofDNA including the entire PAX3 gene. Other clinicalfeatures (mentalretardation, growth retardation) are attributed to deletion of other genes. (C) Type II WScaused by a splice site mutation in the MITF gene. Note eye colour, with blue left eye andbrown right eye with a sharply demarcated radial blue segment; note normal build offace withno dystopia canthorum. She has mild unilateral hearing loss. Other affected relatives withthe same mutation show white forelock, early greying, and varying degrees of hearing loss.
distances. Indices based on these three meas-urements are more reliable than those basedupon two measures alone, although if there isstrabismus the interpupillary distance cannotbe used. The indices depend upon therelationship between the measurements ratherthan absolute measures, and so should beunaffected by age, race, or sex.
Fig 3 shows the justification for placing suchemphasis on dystopia. In this data set, everyfamily with mean W>1.95 (averaged over allaffected family members), but no family withmean W< 1.95, shows evidence of PAX3involvement from linkage or mutation analysis.One family had a W value between 1.95 and
2.07, the threshold first adopted by theWaardenburg Consortium, and was publishedas a type II family with a PAX3 mutation.' Thedata in fig 3 suggest that a threshold of 1.95,averaged across all affected family members,gives the best distinction, and reclassifies thisfamily as WS1. As the figure shows, the eyemeasurements in persons with WS1 and WS2can overlap, so the threshold cannot be taken asabsolute, but it has proven a very useful guide.Its practical value is as a guide to whether ornot it is worth asking the laboratory to look fora PAX3 mutation (see below).
OTHER FACIAL FEATURESA broad, high nasal root, medial hypertrichosisand synophyrys, and hypoplasia of the alae nasiare features associated with dystopia cantho-rum in WS1 (table 3). Other facial featuresdescribed include a patent metopic suture andsquare jaw."l Strabismus may be more commonwith WS 1 than normally. 12
EYE COLOURIris heterochromia may be complete or partial.In complete heterochromia each iris is a differ-ent colour, while in partial heterochromia thedifferently coloured area of the iris is sharplydemarcated from the remainder and is usually,but not invariably, a radial segment (fig 1C).Partial heterochromia may be unilateral orbilateral and, if bilateral, may be symmetrical orasymmetrical. Partial heterochromia was foundin 4.2% of subjects with WS1 and 27.5% ofthose with WS2 in our study.6
Hypoplastic blue irises are found wherethere is deficient iris stroma, and mainly inassociation with a severe or profound hearingloss.6 We found that hypoplastic blue iriseswere significantly more common in childrenwith WS 1 than WS2. The fundus is reported toshow pigmentary changes that correspond tothose found on the retina.'2 13
HAIR COLOURA distinctive white forelock is usually de-scribed, but the forelock may be red orblack.3 1' The site of the forelock is usually inthe midline but it may be elsewhere on thehead. It may vary in size from a few hairs to aclump of hair and, if present at birth, may per-sist or disappear only to reappear later, usuallyin the teens, when it is considered to representearly greying.'5 Complete depigmentation ofthe hair may occur in the teens and the hairmay be sparse and of poor quality.7 Thepremature greying signifying WS is defined bythe Waardenburg Consortium as predomi-nance of white hairs appearing before the ageof 30 years with the white hairs appearing inthe midline.'6 Pigmentation defects can affectthe eyebrows and eyelashes as well as scalphair.7
SKIN SIGNSHypopigmentation of the skin is congenital andmay be found on the face, trunk, or limbs. Itmay be associated with an adjacent white fore-lock. Hyperpigmentation has also beendescribed" and this may develop after birth in
_A*
.-' I,J..-1P-Alow,
AW'
on 15 May 2019 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.34.8.656 on 1 August 1997. D
ownloaded from
Waardenburg syndrome
Al 'A
a--a
b._
Figure 2 Diagnosis of dystopia canthorum. To calculatethe W index: (1) Measure a, b, c in mm using a rigid rulerheld against the face. (2) CalculateX=(2a-0.2119c-3.909)1c. (3) CalculateY=(2a-0.2479b-3.909)1b. (4) Calculate W=X+Y+alb.WS type I is diagnosed if the average Wof all affectedfamily members is 1.95 or more.
a previously hypopigmented area. Arias3 notedthat hypopigmented areas frequently hadhyperpigmented borders. If depigmentedpatches are extensive, piebaldism (owing tomutation in the KIT gene) should be sus-pected, especially if the patient has normalhearing.
HEARINGThe hearing loss in Waardenburg syndrome issensorineural, congenital, and usually non-progressive. It may be unilateral or bilateral andcan vary in degree from slight to profound.Hageman and Delleman'7 reported that 25% ofsubjects with WS1 and 50% of those with WS2had a bilateral sensorineural hearing loss.Newton'8 found that the penetrance of sen-sorineural hearing loss varied from 69% inWS1 to 87% in WS2, after omitting theprobands (who had been ascertained throughtheir hearing loss). Variation between familieswas high.A profound bilateral loss is the commonest
degree of hearing loss in both types, particu-larly in type I. Unusual audiogram shapes
Figure 3 Dystopia canthorum: mean and range ofW values among affected people in 48WSfamilies studied by the Waardenburg Consortium. The horizontal dotted lines showproposed thresholds of W=1. 95 or W=2.07for distinguishing WSI and WS2. Note that allfamilies showing evidence ofPAX3 involvement have average W>1. 95, but not all haveW>2. 07. Redrawn from Farrer et al,Am Hum Genet 1994;55:728-37 with permission.
include low frequency and U shaped losses,bilateral or unilateral, and sometimes a combi-nation of a low frequency sensorineural hearingloss in one ear and a profound loss in the other.
Radiological investigation of the auditorysystem has indicated a normal temporal boneor dysplasia of the lateral semicircular canalassociated with a normal bony cochlea.'9 Thehistological appearance of one temporal bonestudied by Fisch" was consistent with theauditory features of type I being caused by acochleosaccular degeneration. Cochleosaccu-lar degeneration and a complete absence ofpigmentation was reported at necropsy in theinner ears of a 3 year old child with WS4. Shehad extensive skin depigmentation, bilateralprofound hearing loss and aganglionic mega-colcon, but not dystopia canthorum.74
Other associated conditionsA long list of other conditions have beenreported in association with Waardenburgsyndrome.20 It is difficult to disentangle to whatextent these additional conditions are true raremanifestations ofWS (type I or II), coinciden-tal findings, or features of other neurocristopa-thies that are really separate syndromes. WS1carries a small but definite risk of spinabifida20 21 and may occasionally be associatedwith Sprengel's shoulder7 (unpublished obser-vations) and with cleft lip/palate.22
Hirschsprung disease (HSCR) is unques-tionably associated with Waardenburg syn-drome in WS4, which as described below isrecessive and caused by mutations in theEDN3 and EDNRB genes. Whether there isalso an association with other forms ofWaardenburg syndrome is less clear. Mostreports ofWS-HSCR patients date from beforethe distinction was made between WS 1 andWS2. One might expect WS1, as a neuro-cristopathy, to be the form associated withHSCR, but in fact any association that mayexist ofWS with HSCR is with type II WS. Weare aware of only a single case ofHSCR amongthe hundreds of WS1 patients with provenPAX3 mutations (R Ramesar, personal com-munication).
Homozygous Waardenburg syndromeIn view of the frequency of deaf-deaf mar-riages, WS homozygotes are likely to beconceived from time to time. Mouse models(see below) suggest that WS 1 or WS2 homozy-gotes are likely to be very much more severelyaffected than heterozygotes, and this is a pointto be borne in mind in genetic counselling.Double heterozygosity (for example, WS1-WS2) is probably much less risky, although thisview is not based on adequate data.Two cases of homozygosity for WS1 have
been described. Zlotogora et a!23 described andillustrated a child with severe hypopigmenta-tion, profound congenital hearing loss, andcontractures of the forearms, born to a consan-guineous Israeli Arab family. Both parents havetypical WS1, and the child is homozygous forthe PAX3 mutation S84F. The physical pictureis strikingly reminiscent of Klein's originalWS3 patient. Surprisingly, there is no neural
4
3
xa)
23._
1
659
on 15 May 2019 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.34.8.656 on 1 August 1997. D
ownloaded from

Read, Newton
tube defect, although in the Splotch mousemodel homozygotes have lethal neural tubedefects. Ayme and Philip24 described a fetusthat was the product of brother-sister incest ina French Gypsy family with typical WS 1. Thepregnancy was terminated because of anen-cephaly diagnosed on scan; the fetus had majorabnormalities very reminiscent of homozygousSp mouse embryos, including severe contrac-tures and webbing of the limbs. No materialwas available from the fetus, but we have founda PAX3 mutation N269L in a heterozygousmember of the family (M Tassabehji, A PRead, unpublished data). This mutation istypical of those found in WS 1.
Hulten et ar3 described a profoundly deafand severely depigmented child born to firstcousin parents who both had white forelocksand white skin patches but normal hearing.The child might have been a WS2 homozygote,although homozygosity for piebaldism (KITmutation) is perhaps more likely.
The neural crest in auditory-pigmentarysyndromesThe association of hearing loss and pigmentaryabnormalities has long been known in a varietyof mammals.26 In his Origin of Species, CharlesDarwin asked "What can be more singular thanthe relation between blue eyes and deafness incats?" In all these auditory-pigmentary syn-dromes the underlying cause of the hearing lossis a still unexplained requirement for melano-cytes in the stria vascularis of the cochlea.27There is no requirement for melanin (albinoshave normal hearing), but in the absence ofmelanocytes the stria is abnormally thin, noendocochlear potential is generated, and laterin development Reissner's membrane collapsesleading to destruction of the organ of Corti.26 27Thus auditory-pigmentary syndromes arecaused by a physical absence of melanocyteswhich may affect skin, hair, eyes, or the striavascularis. Usually the melanocyte deficiency ispatchy, but alternatively a general dilution ofpigmentation may be seen. In man, hearing losswith uniform dilution of pigmentation isusually described as Tietz-Smith syndrome(MIM 103500) rather than Waardenburg syn-drome, but in both man and mouse, differentalleles of the mi/MITF gene can cause eitherspotty or uniformly diluted depigmentation(see below).
All melanocytes except those in the retinaoriginate in the embryonic neural crest.Absence of melanocytes could be because of afailure of differentiation in the neural crest, afailure of melanoblasts to migrate, or a failureto terminally differentiate and survive in theirfinal location. Countless genes must be in-volved in these processes, and so the genetics ofauditory-pigmentary syndromes is likely to becomplex.28 29 A distinction might be madebetween those syndromes where only melano-cytes are involved and those where there is abroader malfunction of the embryonic neuralcrest. A condition affecting both skin and reti-nal melanocytes is likely to be melanocyte spe-cific, since retinal melanocytes are not derivedfrom the neural crest. In Waardenburg syn-
drome, some WS2 may be melanocyte specific,whereas WS 1 and the rare variants WS3 andWS4 are neurocristopathies, involving thefrontal bone, limb muscles, and enteric ganglia,respectively. All these extra tissues are neuralcrest derivatives.
PAX3 and Waardenburg syndrome type IFoy et at0 mapped WS 1 to the distal long armof chromosome 2 in 1990, using a clueprovided by a Japanese patient with de novoWS 1 and a chromosomal inversioninv(2)(q35q37.3).31 The marker showing link-age was ALPP, the placental alkaline phos-phatase. ALPP was said32 to map to 2q37, butthis was based on radiolabelled in situ hybridi-sation results which were not totally unambigu-ous. ALPP is a difficult marker to use for in situhybridisation because distal 2q contains at leastthree highly homologous alkaline phosphataseloci, ALPP, ALPI, and ALPPL2, which allcross react. Physically, the WS 1 gene must belocated at one of the inversion breakpoints inthe Japanese patient, that is, 2q35 or 2q37.3.Fluorescent in situ hybridisation has nowlocated the WS1 gene at 2q35.33On the basis of this map position, Foy et aP"
suggested that WS1 might be homologous tothe Splotch mouse mutant. This speculationproved correct when three groups identifiedPAX3 (originally called HuP2) and its mousehomologue Pax-3 as the gene mutated in WS 1and Sp.34 36Our knowledge of PAX3 expression mostly
comes from studies of the mouse Pax-3gene,37 39 although preliminary studies inhuman embryos suggest a similar pattern.4"Summarising from the extensive studies ofGruss's group,3' no Pax-3 transcripts weredetected in any tissues of adult mouse, buttranscripts were present in embryos from day 8to day 17, peaking at days 9 to 12 during neu-rulation. Transcripts were concentrated inneuroepithelium, in the dorsal part of the neu-ral groove and in the recently closed neuraltube. Pax-3 is expressed longitudinally downthe length of the neural tube from thehindbrain, but only in mitotically active cells ofthe alar and roof plates, dorsal to the sulcuslimitans. These cells are the source of the neu-ral crest. Among neural crest derivatives, Pax-3expression was seen in the spinal ganglia andsome craniofacial cells (nasal process and somefirst and second branchial arch derivatives), butnot in melanocytes, chromaffin granule cells,the developing heart, or sympathetic ganglia. Inaddition to neural tissue, segmented mesodermalso contained Pax-3 transcripts between 8.5and 11 days of gestation. Onset of Pax-3expression coincided closely with the divisionof presegmented mesoderm into discretesomites, and preceded formation of the dermo-myotome and sclerotome; as the somites disso-ciate, Pax-3 expression is switched off. Note,however, that postnatal appearance or disap-pearance of a white forelock and greying in theteens or twenties have repeatedly been docu-mented in WS1 families with defined PAX3mutations, so not all effects of PAX3 areconfined to neurulating embryos. Another site
660
on 15 May 2019 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.34.8.656 on 1 August 1997. D
ownloaded from

Waardenburg syndrome
A
434del(1 6)
364del(5)
358del(1)
288del(1)
191 del(17)
m-- rm;mm2o-I Hiw
Q254X
E251X
R223XA196T 87
R195X \ W2
556del(2) W2
SS
G99D
G81A
V78M
II185del(18)F45L
91 6del(1)
Y305X
74ins(1) x2!74X
!66X
r /lii_-
"W266C
W269L
rR270C"IR270L
" R271C x2
R271H
Truncatingmutations
Non-truncatingmutations
Paired box 1 Homeobox
B
Spliceuonor x2 Spi ice
acceptor
R214X
R259X
nt944dell
1 2 3 4 5 6 7 8 9~~~~~~~.
--- S29;R203K
N278DNorn-tru ncati ngm utati o ns
dei R217]Y253C
S250P
::: .Basic donmain HLH domair
Figure 4 (A) PAX3 mutations in Waardenburg syndrome type I and III.434del(16) and 916del(1) mutations werefound in patients with type III I
were in type I. Note that non-truncating mutations are concentrated in the.paired box and in the third helix of the homeobox, the two regions criticalfoiDNA recognition. The A196T mutation is likely to affect splicing because itconserved G at the 3' end ofexon 447 (unpublished data from our laborator3groups have reported similar data.'6"13 (B) MITF mutations. Thefamily sdel(R217) mutation (which is identical to the original microphthalmia moshad Tietz-Smith rather than Waardenburg syndrome47; all otherfamilies haII WS. The R203K mutation may be a neutral change: it was seen in the pifour generation family with typical WS2, but did not track with WS throug)pedigree.47 Data from our laboratory47 (unpublished data), Nobokuni et alR259X),4 and Morell et al (944de1l1)).65
of expression was the undifferentichyme of the limb buds,4' exj
phenotype ofWS3.PAX3 encodes a DNA binding
factor, one of a family of nine humteins defined by the presence of Eacid paired domain. The prototypsophila paired (prd) gene. PAX3,PAX7 proteins additionally contadomain. An important research go
100% Effective level of PAX3 protein
Dystopia,,WS1'
50% Melanocytes
Limb buds "WS3"
0% I Neural tube defects
rhgure S A hypothesis to explain PAX3 dosage effects.The effective level ofPAX3 protein depends both on theamount offfunctional PAX3 protein and on variations inthe cellular systems that respond to PAX3 signalling.Dystopia canthorum is always seen when PAX3 dosage isreduced, melanocyte defects are common in people with 50%dosage, limb defects (WS3) are seen only in heterozygoteswho have relatively inefficient PAX3 response systems, or inpeople homozygous for loss offfunction PAX3 mutations.
tify the DNA targets to which the PAX3protein binds. Optimal binding sites for PAX3protein in vitro have been determined byrepeated cycles of immunoprecipitation andPCR ofpanels ofrandom oligonucleotides. Forthe paired domain optimal binding wasachieved with the oligonucleotideCGTCACG(G/G)TT,4 and for the homeodo-main (of the Drosophila paired protein)CCTGAGTCTAATTGATTACTGTACAG.43
Truncating Within these longer sequences, consensus coremutations binding sequences of GTTCC or GTT-AC
(paired domain) and ATTA (homeodomain)have been recognised."
Exons It is worth reflecting, however, that masterswitches in development probably depend onlow affinity interactions. Downstream of themaster genes, the effector mechanisms need
38P high affinity binding to make them specific andreliable. However, the master switch, the deci-sion point that sends only some of a populationof cells down a certain developmental pathway,needs a finely balanced affinity to work. As withhaemoglobin and oxygen, binding that is toostrong is just as bad as binding that is too weak.
zip Thus, the natural targets may be deliberatelynon-optimal. Most in vitro studies of PAX3
WST all others DNA binding have used the e5 sequence from5'part of the the promoter of the Drosophila even-skipped?r protein gene; however, it should be noted thaty)r other Drosophila mutants deficient in prd, or ectopi-with the cally expressing prd, show no changes inuse mutation) even-skipped regulation.45 To date, only onezd typical type natural target of PAX3 is known, the METroband of a 42rh the oncogene.(R214X, Over 50 different PAX3 mutations have been
described in patients withWS 1 or WS3. Fig 4Asummarises mutations detected by our
:lated mesen- group'0 35 46 47 (unpublished data). Others haveplaining the reported similar findings.36 4853 The mutations
are almost always different in different affectedtranscription families; the only mutations seen in more thanan PAX pro- one family are 874ins(G) which inserts aa 128 amino seventh G into a run of six Gs, and substitu-e is the Dro- tions ofR270 or R271 attributable to deamina-,PAX6, and tion of CpG. Mutations fall into three classes.in a homeo- (1) Deletions, fErameshifts, splice site muta-al is to iden- tions, or nonsense mutations that are expected
39r77777 .-N
661
... Lx2u
on 15 May 2019 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.34.8.656 on 1 August 1997. D
ownloaded from

Read, Newton
to act as null alleles. These include completedeletion of the PAX3 gene. Mutations in thisclass are scattered across exons 2-6 of thePAX3 gene, though in contrast to PAX6 (andfor unknown reasons), they are rare in the 3'part of the gene.
(2) Amino acid substitutions in the 5' part ofthe paired box. These all affect amino acidsknown to make important DNA contacts in theprd-DNA complex.54
(3) Amino acid substitutions in the thirdalpha helix of the homeodomain. This helix(the recognition helix) is known to be criticalfor recognition by homeodomain proteins oftheir DNA target.55These findings suggest that the mutational
mechanism is loss of function and that thepathogenesis of WS1 depends on haploinsuffi-ciency. We favour the model in fig 5. Develop-ment of the frontal bone must be uniquely sen-sitive to PAX3 dosage, so that it is virtuallyalways disturbed by loss of function mutationsin PAX3. Differentiation and survival ofmelanocytes is less sensitive, so that pigmen-tary changes and hearing loss are much morevariable features of the syndrome. Develop-ment of limb buds is relatively insensitive toPAX3 dosage and is normally disturbed only inhomozygotes. Occasional heterozygotes, how-ever, do show signs of limb involvement,usually minor, and these are the mild WS3patients discussed below. The effect of reducedPAX3 protein level could be milder or moresevere, depending on variations in the un-known protein or DNA targets of PAX3 actionin development. These are the modifier genesfor PAX3 effects.
In general there is no clear correlationbetween genotype and phenotype in WS 1. Thesymptoms are very variable even within fami-lies, which is perhaps only to be expected if themechanism is haploinsufficiency; genetic back-ground will have important modifying effects.However, mutation of asparagine 47 in thepaired domain of PAX3 might have a specialeffect. Two families have been described thathave atypically severe phenotypes segregatingwith mutations at this site. In the only knownfamily with more than one case of WS3,56 fouraffected members have the mutation N47H,46while a small family having a phenotypedescribed as craniofacial-deafness-hand syn-drome (MIM 122880) have the mutationN47K.57 The three affected people in this fam-ily all have sensorineural hearing loss, dystopiacanthorum, flexion contractures of the fingers,and an almost complete absence of nasalbones, but no pigmentary disturbances.
MITF and Waardenburg syndrome typeIIHughes et at8 mapped the mutation in onelarge WS2 family to 3p12-pl4. At the sametime, Tachibana et ar5 mapped MITF, thehuman homologue of the mouse microphthal-mia gene to the same location. Microphthalmiahad long been seen as a good candidate homo-logue for WS60 and mutations in MITF weresoon found in several WS2 families.6'
MITF and its mouse homologue mi encodeproteins belonging to the well known family ofb-HLH-Zip (basic helix-loop-helix leucine zip-per) transcription factors. These proteinsdimerise as homo- or heterodimers throughtheir HLH-Zip regions and bind DNA throughtheir basic regions. Mi/MITF is one of the fewloci at which more alleles and a richer molecu-lar pathology have been found in mice than inman. Mutations in the basic region generallyproduce molecules that can cause dominantnegative effects by sequestering wild type mol-ecules in dimers that cannot bind DNAcorrectly. Mice heterozygous for these muta-tions have white spotting or, in some cases,dilution of the coat colour. Homozygotes forthese, or for the recessive alleles with dimerisa-tion defects, are mainly or entirely white, andsome alleles produce microphthalmia, mast celldefects, osteopetrosis, or dental defects. Com-pound heterozygotes sometimes have pheno-types unexpectedly more severe or less severethan expected on this model. The mi proteindimer has been shown to bind to a DNAsequence AGTCATGTGCT (the M box)found upstream of several melanocyte specificgenes. 2 The CATGTG core of this sequence isa target for binding by several b-HLH-ZIPproteins. Expression of mi in mouse 3T3fibroblasts can cause them to undergomelanocyte-like differentiation.63 It seems pos-sible that mi/MITF is a master gene switchingon melanocyte development, although it is alsoexpressed in heart, and the phenotypes of somemi mutants suggest additional functions.
In humans (fig 4B), MITF mutations havebeen found in a modest number of familieswith WS2,47 61 64 65 and in one family with thephenotype of Tietz-Smith syndrome.47 Tietz-Smith syndrome (MIM 103500) shows hear-ing loss combined with uniform non-patchydilution of pigmentation; as mentioned above,some mi mouse mutants also show dilutionrather than spotting. In the dominant familiesthat attract the label of WS2, haploinsuffi-ciency rather than a dominant negative effectseems the most likely pathogenicmechanism.47 64 The wide range of recessivephenotypes seen in mice prompted a search forMITF mutations in patients with severe reces-sive pigmentary syndromes, but so far none hasbeen found. All the human families aredominant, and with the exception of the singleTietz-Smith family, all have typical WS2.Perhaps the incidence of hearing loss is ratherhigher than in non-MITF WS2 families, but itis not strikingly different, and we cannotpredict which WS2 families will carry MITFmutations. MITF mutations have been foundin only about 15% of families fitting thediagnostic criteria for WS2, and the majorWS2 locus or loci remains to be found.
PAX3 and Waardenburg syndrome typeIIIWS3 remains something of an anomaly. Threerather separate combinations of auditory-pigmentary symptoms with hypoplasia or con-tractures of the upper limbs can be seen. (1)Klein's original patient and the PAX3 homozy-
662
on 15 May 2019 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.34.8.656 on 1 August 1997. D
ownloaded from

Waardenburg syndrome
gote of Zlotogora et alf have profound hearingloss, depigmentation much more severe than inWS 1, and a severe amyoplasia-like conditionaffecting the arms. (2) In the family reportedby Sheffer and Zlotogora,56 people hetero-zygous for the PAX3 mutation N47H havetypical WS 1 plus significant amyoplasia, inher-ited as a dominant condition. The amyoplasiais identical in pattern to that of Klein's patient,but less severe. (3) Finally, most cases labelledWS3 are sporadic or part ofWS 1 families, andhave WS 1 plus quite minor contractures of theelbows or fingers. Two cases we have tested areheterozygous for a 16 bp deletion and a 1 bpdeletion, respectively, in the PAX3 gene, muta-tions typical ofWS 1.
Endothelin 3, endothelin receptor B, andWaardenburg syndrome type IVRecent progress in identifying HSCR suscepti-bility genes has shed considerable light on WS4and allowed a new definition of the syndrome.Patients with mutations in the endothelin 3gene, EDN3, or the gene for its receptor,EDNRB, occasionally show a WS-HSCR phe-notype, especially if homozygous.66 67 75 76 Het-erozygotes are usually unaffected or haveisolated HSCR.61-70 A family reported byHofstra et al" is interesting: there is a pattern oflow penetrance isolated deafness or pigmentarydisturbances, resembling many families wehave seen that do not quite meet criteria forWS2. In the family of Hofstra et al,7' whencousins married they produced children withtypical WS4 who were homozygous for anEDN3 mutation. However, we have soughtEDN3 or EDNRB mutations in our families invain (M Tassabehji, A P Read, unpublisheddata). They are certainly not a common causeofWS in the absence ofHSCR. Some patientswith chromosomal deletions or translocationsaffecting the sites of EDNRB at 13q22 orEDN3 at 20q13 have pigmentary disturbanceswithout dystopia and with72 or without73HSCR. Mutations in other unidentified neuralcrest genes may also produce HSCR with pig-mentary disturbances and maybe hearing loss,with or without dystopia. However, HSCRpatients with RET mutations do not havemelanocyte defects (M Seri, personal commu-nication), nor probably do those with muta-tions in the RET ligand, GDNF.
Conclusions and summaryResearch into Waardenburg syndrome pro-vides some ofthe best examples ofthe interplaybetween mouse and human genetic research.PAX3 was investigated independently as a can-didate gene for Splotch and Waardenburgsyndrome on the basis of its map location ineach species and expression pattern in themouse. The mouse mi, Ednrb, and Edn3 geneswere each cloned when mice with randomtransgene insertions unexpectedly showed thephenotypes of microphthalmia, piebald lethal,and lethal spotting respectively. MITF andEDNRB then became positional candidates forthe 3pl4 linked WS2 and 13q linked WS4respectively, while EDN3 was investigated as apure (non-positional) candidate gene for WS4.
PAX3 and MITF illustrate how mutationanalysis can help elucidate mechanisms ofdominance. For PAX3 the evidence for hap-loinsufficiency is convincing, with the caveatthat mutation of asparagine 47 may have someadditional effect. For MITF the mechanismsare less certain, but it appears that here toohaploinsufficiency is a major factor in produc-ing the relatively mild abnormalities of WS2among heterozygous mutation carriers. Domi-nant negative effects and pure recessive effectsmay also be found.
Clinically, the payoff from identifying thegenes and characterising the mutations has sofar been relatively modest. Diagnostic labelshave been refined. WS 1 and WS4 are now welldefined genetic entities, while the label WS3 islargely redundant. WS2 remains a heterogene-ous mix. Only a small proportion of type IIfamilies are accounted for by mutations in anyof the genes defined so far. Moreover, the clini-cal definition ofWS2 as a dominantly inheritedpatchy phenotype seems likely to exclude somepatients with MITF mutations, for example,patients with dominant partial albinism of theTietz-Smith type or with major recessivesyndromes that include severe depigmentation.We can offer families an explanation of whythey have Waardenburg syndrome, but we arestill unable to predict what features ofWS anyparticular PAX3, MITF, EDNRB, or EDN3mutation will produce in a given person.Fortunately there is little interest in prenataldiagnosis among WS families. Nor is theremuch prospect for gene therapy, given that theabnormalities ofWS arise in the early embryo.
Research into WS has allowed definition oftranscription factors important in humanembryonic development, not to mention find-ing the first homeobox gene to be implicated ina human inherited disease. PAX3 and MITFbetween them exemplify three major families oftranscription factors, the paired domain,homeodomain, and b-HLH-Zip proteins.Many questions remain, particularly about theprecise developmental pathways in which thesegenes act, and the identity of modifier genesresponsible for the highly variable expression infamilies. It was unexpected that defects in theEDN3-EDNRB system would produce devel-opmental abnormalities, and the role ofendothelins in development has yet to be eluci-dated. Auditory-pigmentary syndromes, asdefects in cell differentiation, always promisedto be biologically interesting, and research sofar has amply borne out this promise.
We wish to thank May Tassabehji for her superb laboratorywork, and members of the Waardenburg Consortium forsharing data and ideas. Our work was supported by the HearingResearch Trust and Wellcome Trust (grant 035301).
1 Waardenburg PJ. Dystopia punctorum lachrimarum,blepharophimosis en partiele irisatrophie bij een doofs-tomme. Ned Tschr Geneeskd 1948;92:3463-5.
2 Waardenburg PJ. A new syndrome combining developmen-tal anomalies of the eyelids, eyebrows and nose root withpigmentary defects of the iris and head hair and with con-genital deafiess. Am J Hum Genet 195 1;3:195-253.
3 Arias S. Genetic heterogeneity in the Waardenburgsyndrome. Birth Defects 1971;7(4):87-101.
4 Klein D. Historical background and evidence for dominantinheritance of the Klein-Waardenburg syndrome (type II[).AmJrMed Genet 1983;14:231-9.
663
on 15 May 2019 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.34.8.656 on 1 August 1997. D
ownloaded from

Read, Newton
5 Shah KN, Dalal SJ, Sheth PN, Joshi NC, Ambani LM.White forelock, pigmentary disorder of the irides and longsegment Hirschsprung disease: possible variant ofWaardenburg syndrome. _7 Pediatr 198 1;99:432-5.
6 Liu XZ, Newton VE, Read AP. Waardenburg syndrome type2: phenotypic findings and diagnostic criteria. Am _7 MedGenet 1995;55:95-100.
7 Fraser GR. The causes of profound deafness in childhood.Baltimore: Johns Hopkins University Press, 1976.
8 Arias S, Mota M. Apparent non-penetrance for dystopia inWaardenburg syndrome type 1 with some hints on thediagnosis of dystopia canthorum. 7 Genet Hum 1978;26:101-31.
9 Newton VE. Waardenburg's syndrome: a comparison ofbiometric indices used to diagnose lateral displacement ofthe inner canthi. ScandAudiol 1989;18:221-3.
10 Tassabehji M, Read AP, Newton VE, et al. Mutations in thePAX3 gene causing Waardenburg syndrome type 1 andtype 2. Nat Genet 1993;3:26-30.
11 Fisch L. Deafness as part of an hereditary syndrome. .7Laryngol Otol 1959;73:355-82.
12 Delleman JW, Hageman MJ. Ophthalmological findings in34 patients with Waardenburg syndrome. _7 PaediatrOphthalmol Strab 1978;15:341-5.
13 Goldberg MF. Waardenburg's syndrome with fundus andother abnormalities. Arch Ophthalmol 1966;76:797-809.
14 Reed WB, Stone VM, Boder E, Ziprkowski L. Pigmentarydisorders in association with congenital deafness. Arch Der-matol 1967;95:176-86.
15 Di George AM, Olmsted RW, Harley RD. Waardenburg'ssyndrome. I Pediatr 1960;57:649-69.
16 Farrer LA, Grundfast KM, Amos J, et al. Waardenburg syn-drome (WS) type 1 is caused by defects at multiple loci,one of which is near ALPP on chromosome 2: first reportof the WS Consortium. AmJ7Hum Genet 1992;50:902-13.
17 Hageman M, Delleman J. Heterogeneity in Waardenburgsyndrome. Am _7 Hum Genet 1977;29:468-85.
18 Newton VE. Hearing loss and Waardenburg syndrome:implications for genetic counselling. . Laryngol Otol 1990;104:97-103.
19 Nemansky J, Hageman MJ. Tomographic findings of theinner ears of 24 patients with Waardenburg's syndrome.A.7R 1975;124:250-5.
20 da-Silva EO. Waardenburg I syndrome: a clinical andgenetic study of two large Brazilian kindreds, and literaturereview. Am J Med Genet 199 1;40:65-74.
21 Pantke OA, Cohen MM. The Waardenburg syndrome. BirthDefects 1971;7(7):147-52.
22 Giacola JP, Klein SW Waardenburg's syndrome withbilateral cleft lip. AmI Dis Child 1969;117:344-8.
23 Zlotogora J, Lerer I, Bar-David S, Ergaz Z, Abielovich D.Homozygosity for Waardenburg syndrome. Am 7 HumGenet 1995;56:1173-8.
24 Ayme S, Philip N. Possible homozygous Waardenburg syn-drome in a fetus with exencephaly. Am .7 Med Genet 1995;59:263-5.
25 Hulten M, Honeyman MM, Mayne AJ, Tarlow MJ.Homozygosity in piebald trait. .7 Med Genet 1987;24:568-71.
26 Steel KP, Bock GR. Hereditary inner-ear abnormalities inanimals. Arch Otolaryngol 1983;109:22-9.
27 Steel KP, Barkway C. Another role for melanocytes: theirimportance for normal stria vascularis development in themammalian inner ear. Development 1989;107:453-63.
28 Hearing VI. Unraveling the melanocyte. Am _7 Hum Genet1993;52: 1-7.
29 Barsh GS. Pigmentation, pleiotropy, and genetic pathwaysin humans and mice. Am J Hum Genet 1995;57:743-7.
30 Foy C, Newton VE, Wellesley D, Harris R, Read AP.Assignment of WS1 locus to human 2q37 and possiblehomology between Waardenburg syndrome and theSplotch mouse. Am _
Hum Genet 1990;46:1017-23.31 Ishikiriyama S, Tonoki H, Shibuya Y, et al. Waardenburg
syndrome type I in a child with de novo inversion (2)(q35q37.3). Am _Med Genet 1989;33:505-7.
32 Martin D, Tucker DF, Gorman P, Sheer D, Spurr NK,Trowsdale J. The human alkaline phosphatase gene andrelated sequences map to chromosome 2 band 2q37. AnnHum Genet 1987;51:145-52.
33 Tsukamoto K, Tohma T, Ohta T, et al. Cloning and charac-terization of the inversion breakpoint at chromosome 2q35in a patient with Waardenburg syndrome type 1. Hum MolGenet 1992;1:315-17.
34 Epstein DJ, Vekemans M, Gros P. Splotch (Sp2H), a muta-tion affecting development of the mouse neural tube, showsa deletion within the paired homeodomain of Pax-3. Cell1991;67:767-74.
35 Tassabehji M, Read AP, Newton VE, et al. Waardenburgsyndrome patients have mutations in the human homo-logue of the Pax-3 paired box gene. Nature 1992;355:635-6.
36 Baldwin CT, Hoth CF, Amos JA, da-Silva EO, Milunsky A.An exonic mutation in the HuP2 paired domain genecauses Waardenburg's syndrome. Nature 1992;355:637-8.
37 Goulding MD, Chalepakis G, Deutsch U, Erselius JR,Gruss P. Pax-3, a novel murine DNA-binding proteinexpressed during early neurogenesis. EMBO 7 1991;10:1135-47.
38 Gruss P, Walther C. Pax in development. Cell 1992;69:719-22.
39 Stuart ET, Kioussi C, Gruss P. Mammalian PAX genes.Annu Rev Genet 1994;28:219-36.
40 Gerard M, Abitbol M, Delezoide AL, Dufier JL, Mallet J,Vekemans M. PAX-genes expression during human
embryonic development, a preliminary report. C R AcadSci Paris 1995;318:57-66.
41 Bober E, Franz T, Arnold HH, Gruss P, Tremblay P. Pax-3is required for the development of limb muscles: a possiblerole for migration of dermomyotomal muscle progenitorcells. Development 1994;120:603-12.
42 Epstein JA, Shapiro DN, Cheng J, Lam PYP, Maas RL.Pax3 modulates expression of the c-Met receptor duringlimb muscle development. Proc Natl Acad Sci USA1996;93:4213-18.
43 Underhill DA, Vogan KJ, Gros P. Analysis of the mouseSplotch-delayed mutation indicates that the Pax-3 paireddomain can influence homeodomain DNA-binding activ-ity. Proc NatlAcad Sci USA 1995;92:3692-6.
44 Chalepakis G, Jones FS, Edelman GM, Gruss P. Pax-3 con-tains domains for transcription activation and transcriptioninhibition. Proc NatlAcad Sci USA 1994;91:12745-9.
45 Epstein JA, Cai J, Glaser T, Jepeal L, Maas R. Identificationof a Pax paired domain recognition sequence and evidencefor DNA-dependent conformational changes. _7 Biol Chen1994;269:8355-61.
46 Tassabehji M, Newton VE, Leverton K, et al. PAX3 genestructure and mutations: close analogies between Waarden-burg syndrome type 1 and the Splotch mouse. Hum MolGenet 1994;3:1069-74.
47 Tassabehji M, Newton VE, Liu XZ, et al. The mutationalspectrum in Waardenburg syndrome. Hum Mol Genet1995;4:2131-7.
48 Hoth CF, Milunsky A, Lipsky N, Sheffer R, Clarren SK,Baldwin CT. Mutations in the paired domain of the humanPAX3 gene cause Klein-Waardenburg syndrome (WS-III)as well as Waardenburg syndrome type 1 (WS-1). Am _
Hum Genet 1993;52:455-62.49 Baldwin CT, Hoth CF, Macina RA, Milunsky A. Mutations
in PAX3 that cau,se Waardenburg syndrome type 1: ten newmutations and review of the literature. Am _7 Med Genet1995;58:115-22.
50 Morell R, Friedman TB, Moeljopawiro S, Hartono,Soewito, Asher JH. A frameshift mutation in the HuP2paired domain of the probable human homolog of murinePax-3 is responsible for Waardenburg syndrome type 1 inan Indonesian family. Hum Mol Genet 1992;1:243-7.
51 Morell R, Carey ML, Lalwani AK, Friedman TB, Asher JH.Three mutations in the paired homeodomain of PAX3 thatcause Waardenburg syndrome type 1. Hum Hered 1997;47:38-41.
52 Lalwani AK, Brister JR, Fex J, et al. Further elucidation ofthe structure of PAX3, and identification of two differentpoint mutations within the PAX3 homeobox that causeWaardenburg syndrome type 1 in two families. Am .7 HumGenet 1995;56:75-83.
53 Butt J, Greenberg J, Winship I, Sellars S, Beighton P, Ram-esar R. A splice junction mutation in PAX3 causes
Waardenburg syndrome in a South African family. HumMol Genet 1994;3:197-8.
54 Xu W, Rould MA, Jun S, Desplan C, Pabo CO. Crystalstructure of a paired domain-DNA complex at 2.5Aresolution reveals structural basis for Pax developmentalmutations. Cell 1995;80:639-50.
55 Kissinger CR, Liu B, Martin-Blanco B, Kornberg TB, PaboCO. Crystal structure of an engrailed homeodomain-DNAcomplex at 2.8A resolution: a framework for understandinghomeodomain-DNA interactions. Cell 1990;63:579-90.
56 Sheffer R, Zlotogora J. Autosomal dominant inheritance ofKlein-Waardenburg syndrome. Am 7 Med Genet 1992;42:320-2.
57 Asher JH, Sommer A, Morell R, Friedman TB. Missensemutation in the paired domain of PAX3 causes
craniofacial-deafness-hand syndrome. Humn Mutat 1996;7:30-5.
58 Hughes A, Newton VE, Lu XZ, Read AP. A gene forWaardenburg syndrome type 2 maps to human chromo-some 3p12-p14.1. Nat Genet 1994;7:509-12.
59 Tachibana M, Perez-Jurado LA, Nakayama A, et al. CloningofMITF, the human homolog of the mouse mzicrophthalmiagene and assignment to chromosome 3pi4. 1-pl2.3. HumMol Genet 1994;3:553-7.
60 Asher JH, Friedman TB. Mouse and hamster mutants as
models for Waardenburg syndrome in humans. .7 MedGenet 1990;27:618-26.
61 Tassabehji M, Newton VE, Read AP. Waardenburgsyndrome type 2 caused by mutations in the human micro-phthalmia (MITF) gene. Nat Genet 1994;8:251-5.
62 Hemesath TJ, Steingrimsson E, McGill G, et al. Microph-thalmia, a critical factor in melanocyte development,defines a discrete transcription factor family. Genes Dev1994;8:2770-80.
63 Tachibana M, Takeda K, Nobokuni Y, et al. Ectopic expres-sion of MITF, a gene for Waardenburg syndrome type 2,converts fibroblasts to cells with melanocyte characteris-tics. Nat Genet 1996;14:50-4.
64 Nobokuni Y, Watanebe A, Takeda K, Skarka H, TachibanaM. Analyses of loss-of-function mutations of the MITFgene suggest that haploinsufficiency is a cause of Waarden-burg syndrome type 2a. AmI Hum Genet 1996;59:76-83.
65 Morell R, Spritz RA, Ho L, et al. Apparent digenic inherit-ance of Waardenburg syndrome type 2 (WS2) andautosomal recessive ocular albinism (AROA). Hum MolGenet 1997;6:659-64.
66 Puffenberger EG, Hosoda K, Washington SS, et al. Amissense mutation of the endothelin-B receptor gene inmultigenic Hirschsprung's disease. Cell 1994;79:1257-66.
67 Chakravarti A. Endothelin receptor-mediated signaling inHirschsprung disease. Hum Mol Genet 1996;5:303-8.
664
on 15 May 2019 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.34.8.656 on 1 August 1997. D
ownloaded from

Waardenburg syndrome
68 Kusafuka T, Wang Y, Puri P. Novel mutations ofthe endothelin B receptor gene in isolated patientswith Hirschsprung's disease. Hum Mol Genet 1996;5:347-9.
69 Auricchio A, Casari G, Staiano A, Ballabio A. Endothelin-Breceptor mutations in patients with isolated Hirschsprungdisease from a non-inbred population. Hum Mol Genet1996;5:351-4.
70 Amiel J, Attie T, Jan D, et al. Heterozygous endothelinreceptor B (EDNRB) mutations in isolated Hirschsprungdisease. Hum Mol Genet 1996;5:355-7.
71 Hofstra RM, Osinga J, Tan-Sindhunata G, et al. Ahomozygous mutation in the endothelin-3 gene associatedwith a combined Waardenburg type 2 and Hirschsprungphenotype (Shah-Waardenburg syndrome). Nat Genet1996;12:445-7.
72 Hood OJ, Doyle M, Hebert AA, Oelberg DG. Association ofWaardenburg syndrome type II and a de novo balanced 7;20translocation. Dysmorphol Clin Genet 1989;3:122-3.
73 Van Camp G, Van Thienen MN, Handig I, et al.Chromosome 13q deletion with Waardenburg syndrome:further evidence for a gene involved in neural crest functionon 13q. YMed Genet 1995;32:531-6.
74 Nakashima S, Sando I, Takahashi H, Hashida Y Temporalbone histopathologic findings ofWaardenburg's syndrome:a case report. Laryngoscope 1992;102;563-7.
75 Attie T, Till M, Pelet A, et al. Mutation of theendothelin-receptor B gene in Waardenburg-Hirschsprungdisease. Hum Mol Genet 1995;4;2407-9.
76 Edery P, Attie T, Amiel J, et al. Mutation ofthe endothelin-3gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome). Nat Genet 1996;12:442-4.
665
on 15 May 2019 by guest. P
rotected by copyright.http://jm
g.bmj.com
/J M
ed Genet: first published as 10.1136/jm
g.34.8.656 on 1 August 1997. D
ownloaded from