wide thermal range, exclusive occurrence of technically
TRANSCRIPT

Bull. Mater. Sci. (2020) 43:188 © Indian Academy of Scienceshttps://doi.org/10.1007/s12034-020-02120-8
Wide thermal range, exclusive occurrence of technically significantchiral nematic phase: synthesis and mesomorphism ofcholesterol-based non-symmetric dimers
RASHMI ASHWATHAMA NAYAK1,2, SACHIN A BHAT1, D S SHANKAR RAO1
and C V YELAMAGGAD1,∗1Centre for Nano and Soft Matter Sciences, Bengaluru 560 013, India2Present address: CSIR-National Chemical Laboratory, Pune 411 008, India∗Author for correspondence ([email protected]; [email protected])
MS received 6 September 2019; accepted 7 February 2020
Abstract. Fifteen new non-symmetric chiral dimers belonging to three different series have been synthesized and evaluatedfor their mesomorphic properties. They are formed by interlinking cholesterol with salicylaldimine (SAN) cores (with reverseimine groups) via an ω-oxyalkanoyloxy spacer. Within a series, the length of the terminal n-alkoxy tails has been varied fora fixed even-parity spacer. Three even-parity spacers such as 4-oxybutanoyloxy, 6-oxyhexanoyloxy and 8-oxyoctanoyloxyhave been used to join two cores, whereas the terminal tails such as n-butyloxy, n-hexyloxy, n-octyloxy, n-decyloxy andn-dodecyloxy chains have been attached to the SAN core. Microscopic and calorimetric experimental results show that allthe dimers behave identically exhibiting the chiral nematic (N*) phase solely, which was authenticated by powder X-raydiffraction studies carried out on some selected samples. In the vast majority of the cases, this phase is thermodynamicallystable, and while cooling, it exists over a wide thermal range covering room temperature (RT) due to supercooling. Thisfinding is notable given the fact that the N* phase possesses technologically significant optical properties. At RT, the N*phase displayed one of the iridescent colours characteristically caused by interference and diffraction of the reflected andscattered light. A comparative study reveals that the lengths of both the terminal chain and central spacer influence theclearing temperature of the dimers, and also the temperature range of the N* phase. The selective reflection measurementsrevealed that the pitch of the N* phase is either temperature sensitive or temperature insensitive. Temperature-dependentcircular dichroism (CD) spectra were recorded for the planar texture of the N* phase formed by a dimer, as a representativecase. The presence of an intense negative CD band suggests the left-handed screw sense of the N* phase helix.
Keywords. Liquid crystals; dimers; cholesteric phase; selective reflection; CD activity.
1. Introduction
In recent years, salicylaldimines [N -(2-hydroxy-4-alkoxy-benzylidene)anilines] (SANs), which are nothing but o-hydroxy Schiff bases, have attracted the attention of not onlyfundamental researchers but also technologists [1–21] due totheir unique chemical as well as physical properties. A facileone-step synthesis involving the condensation of salicylalde-hydes with primary amines (Chart 1) in the presence of acatalytic amount of a weak acid generates SANs in quan-titative yields. Owing to the presence of an inherent quasisix-membered ring, resulting from the intramolecular hydro-gen (H)-bonding between the H-atom of the aromatic hydroxygroup and the N-atom of the imine linking group [1,2], SANsare quite stable to heat and moisture, which is not the case withtheir corresponding Schiff base compounds. Notably, they areelectron-rich molecules and have the natural tendency/abilityof exhibiting a reversible proton-transfer phenomenon both insolid-state and solutions yielding two tautomers, enol–imine(OH) (Chart 1: Ia) and keto–enamine (NH) (Chart 1: Ib)
forms; the latter form, which is supposed to be in equilib-rium with the (NH) form, is typically found, and hence, ithas been investigated in detail [1–7]. The abovementionedreversible proton-transfer process enables these materials toexhibit technologically significant phenomena such as pho-tochromism and thermochromism [1–8].
Because the SAN core is highly robust and intrinsicallyfunctional, it is often incorporated in a variety of ther-motropic liquid crystals (LCs) [8–32]. In particular, it has beenused in calamitics [8,19,20], bent-core LCs [9–15], metallo-mesogens [8–10] and achiral dimeric bidentate ligands[21,25–27], and some of these LCs have shown remarkableLC properties that can be attributed to the presence of theSAN core. Indeed, this core has also used in the synthesisof dimers [22–24,28–31]. In particular, it is used to realizethermally and hydrolytically stable non-symmetric, opticallyactive dimers derived from cholesterol [22–24,28–31]; weshall return to this subject in the section to follow. Notably,the SAN core has been incorporated in ferroelectric meso-gens [15–17] to realize technologically important materials
0123456789().: V,-vol

188 Page 2 of 18 Bull. Mater. Sci. (2020) 43:188
Chart 1. Salicylaldimine (SAN), formed by treating salicylaldehyde with 1◦-amine, generally exists inenol–imine (OH) form (Ia) which can tautomerize to yield keto–enamine (NH) form (Ib). Molecular struc-tures of the previously studied cholesterol-based dimers II and III comprising SAN segment [22–24].
that are insensitive to heat and moisture. The SAN core, asa pendant unit in an achiral side-chain polymer, has playeda vital role in the first discovery of Soto-Bustamante/Blinovanticlinic bilayer antiferroelectrics [18]. Given these inter-esting characteristics of SANs, and in continuation to ourwork on this core [19–29,31], we intended to incorporate SANmesogen in cholesterol-based non-symmetric dimers.
Over the last two decades or so, it has been shown thatcholesterol-based non-symmetric (optically active) dimers,constituted by covalently linking cholesterol with a conven-tional rod-like (calamitic)/disc-like (discotic) mesogenic unitvia a flexible central spacer of varying parity and length, areimmensely significant from the viewpoints of both funda-mental research and applied science [22–24,28–31,33–70].They exhibit vibrant and exciting phase sequences, as well asdifferent frustrated and reentrant LC phases [31]. Moreover,they enable modulation of the characteristics of technologi-cally significant LC phases through the choice of mesogenic(mostly calamitic) cores (joined to cholesterol) and/or theparity (and length) of the spacer [31]. The fundamentalresearch interest in such dimers originated from a notablefinding in 1994 by Jin and others [34]. They observed thatan LC dimer, formed by covalently linking cholesterol witha Schiff base core via a central flexible spacer, shows anincommensurate smectic A (SmAic) phase. Subsequent inves-tigations demonstrated the significance of the Schiff baseunit, besides cholesterol, in determining the phase transi-tional behaviour mentioned above, especially the occurrence
of SmAic phase [31,33,35–38]. Because Schiff bases are heat-and moisture-sensitive compounds, the SAN core, whichstructurally resembles the Schiff base segment to a largeextent, was incorporated by our group in dimers derivedfrom [22–24]; the molecular structures of these novel dimers,II [22,23] and III [24], are shown in Chart 1. This study wasspecially undertaken not only to realize stable cholesterol-based o-hydroxy Schiff bases, but also to gain insights intothe structure–property correlation. The dimers II and IIImen-tioned above not only exhibited stability towards heat andmoisture, but also was observed to present a wide varietyof LC phases between the solid and isotropic liquid states.The mesophases displayed by these dimers comprise chiralnematic (N*), twist grain boundary (TGB), smectic A (SmA)and chiral smectic C (SmC*) phases. The occurrence of tech-nologically relevant SmC* and N* phases in these dimers isnoteworthy. Most importantly, some of these dimers showedferroelectric switching characteristics in the SmC* phase [23].
Given these exciting findings, we synthesized similardimers where the imine (–CH=N–) linking group of the SANcore is reversed (–N=CH–). Precisely, we intended to syn-thesize and characterize new chiral, non-symmetric dimerswhere SAN (with a modified imine linkage) and choles-terol entities are joined together via an ω-oxyalkanoyloxyspacer. We chose to incorporate an even-parity spacer pur-posely to unveil the effect of reversing the imine linkageof the SAN core on the thermal behaviour of cholesterol-based dimers such as II explicitly. Accordingly, three series
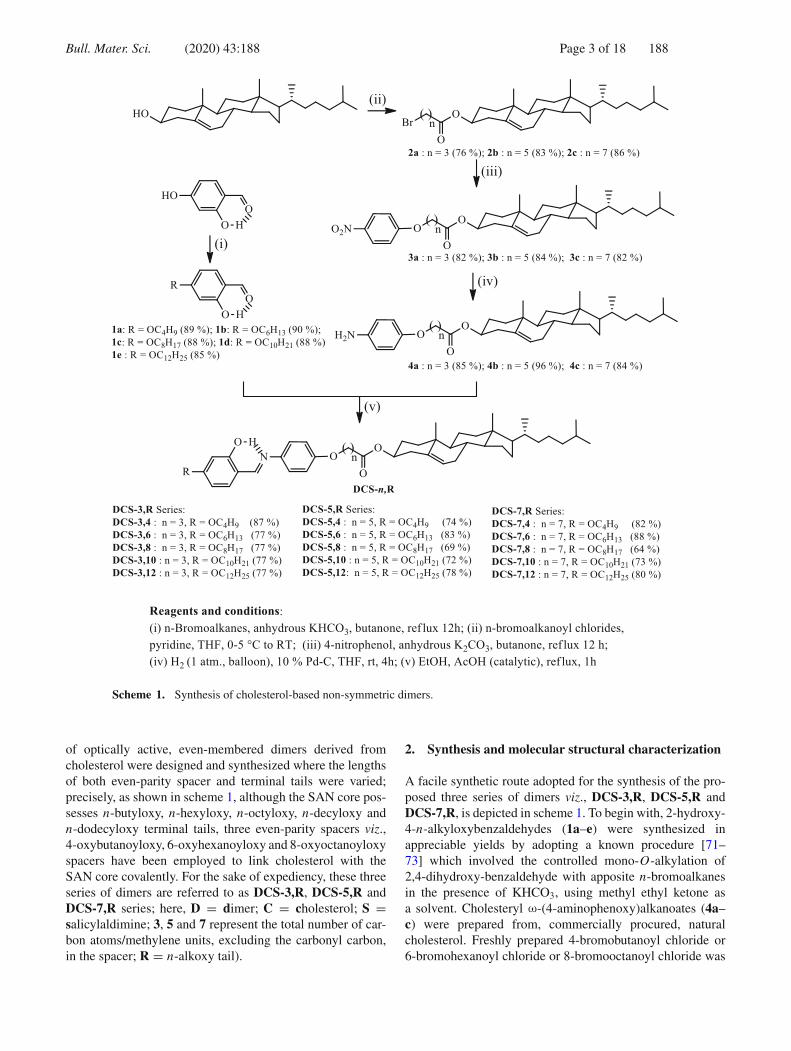
Bull. Mater. Sci. (2020) 43:188 Page 3 of 18 188
Scheme 1. Synthesis of cholesterol-based non-symmetric dimers.
of optically active, even-membered dimers derived fromcholesterol were designed and synthesized where the lengthsof both even-parity spacer and terminal tails were varied;precisely, as shown in scheme 1, although the SAN core pos-sesses n-butyloxy, n-hexyloxy, n-octyloxy, n-decyloxy andn-dodecyloxy terminal tails, three even-parity spacers viz.,4-oxybutanoyloxy, 6-oxyhexanoyloxy and 8-oxyoctanoyloxyspacers have been employed to link cholesterol with theSAN core covalently. For the sake of expediency, these threeseries of dimers are referred to as DCS-3,R, DCS-5,R andDCS-7,R series; here, D = dimer; C = cholesterol; S =salicylaldimine; 3, 5 and 7 represent the total number of car-bon atoms/methylene units, excluding the carbonyl carbon,in the spacer; R = n-alkoxy tail).
2. Synthesis and molecular structural characterization
A facile synthetic route adopted for the synthesis of the pro-posed three series of dimers viz., DCS-3,R, DCS-5,R andDCS-7,R, is depicted in scheme 1. To begin with, 2-hydroxy-4-n-alkyloxybenzaldehydes (1a–e) were synthesized inappreciable yields by adopting a known procedure [71–73] which involved the controlled mono-O-alkylation of2,4-dihydroxy-benzaldehyde with apposite n-bromoalkanesin the presence of KHCO3, using methyl ethyl ketone asa solvent. Cholesteryl ω-(4-aminophenoxy)alkanoates (4a–c) were prepared from, commercially procured, naturalcholesterol. Freshly prepared 4-bromobutanoyl chloride or6-bromohexanoyl chloride or 8-bromooctanoyl chloride was

188 Page 4 of 18 Bull. Mater. Sci. (2020) 43:188
treated with cholesterol in the presence of pyridine usingtetrahydrofuran (THF) as a solvent to obtain the crucialintermediates namely, cholesteryl ω-bromoalkanoates (2a–c) [39,68,72]; n-alkanoylchlorides were in turn obtained inquantitative yields by treating the correspondingω-bromoalkanoic acids with oxalyl chloride at room tem-perature (RT). The Williamson etherification reaction of 4-nitrophenol with bromoalkanoates 2a–d afforded cholesterylω-(4-nitrophenoxy)alkanoates (3a–c) which upon subjectingto the catalytic hydrogenation using 1 atm of H2 (balloon)and 10% palladium (by wt%) adsorbed on carbon (Pd–C) afforded the corresponding 1◦-amines 4a–c in excellentyields. The desired target LC dimers belonging to DCS-3,R, DCS-5,R and DCS-7,R series were obtained by themild acid-catalysed condensation reaction of amines 4a–cwith salicylaldehydes 1a–e in refluxing ethanol medium. Thestructural characterization data collected for the intermediatesmentioned above were in good agreement with the reportedones in [71–73] and [68,74], respectively. The protocol ofthe syntheses and the structural characterization data, derivedfrom spectroscopic methods and elemental analyses of thefinal compounds have been presented in section 3.
3. Experimental
3.1 General information
The bulk organic solvents obtained from local chemical sup-pliers were purified by distillation over appropriate dryingreagents. Necessary chemicals such as 4-bromobutyric acid,6-bromocaproic acid, 8-bromocaprylic acid, 4-hydroxyben-zaldehyde, 4-nitrophenol and cholesterol were purchasedfrom an overseas chemical supplier and were used as received.Most of the reactions were performed under a nitrogen atmo-sphere using glassware dried in an oven for 12 h at 90◦C.The progress and completion of reactions were monitoredby employing analytical thin-layer chromatography plates,which comprise a thin layer of silica gel (Merck, Kieselgel60,F254) on an aluminium foil backing; they were also used todetermine the purity of the compounds. The organic spotswere evidenced using UV light at 254 nm and/or KMnO4
stains. Silica gel (100–200 mesh) and neutral alumina wereused as stationary phases while purifying crude materialsusing column chromatography technique. The organic solu-tions collected during the column chromatography processwere concentrated under reduced pressure using a Büchirotary evaporator, having a vertical condenser circulated withcold water. Optical absorption spectra were recorded at RTwith the aid of a Perkin Elmer Lambda 20 UV–Vis spec-trophotometer. Fourier-transform infrared (FTIR) spectra ofthe samples, taken in a KBr matrix and pressed to form apellet, were recorded using a Perkin Elmer Spectrum 1000FT-IR spectrometer; the X -axes of IR spectra are plottedin wavenumber (cm−1) unit. Elemental analyses were per-formed on a Perkin Elmer Elemental Analyzer Series II 2400
elemental analyzer. Solution nuclear magnetic resonance(NMR) experiments were carried out on a Bruker AMX-400(400 MHz) spectrometer operating at 400 MHz for 1H and at100 MHz for 13C. While reporting the data, chemical shifts(δ) are presented in parts per million (ppm) relative to tetram-ethylsilane (TMS) using the residual CHCl3 peak in CDCl3solution as an internal standard (δ H 7.26 and δ C 77.0 rela-tive to TMS); coupling constants (J ) are given in hertz (Hz);multiplicities of the NMR peaks are presented as s (singlet),d (doublet), t (triplet) and m (multiplet). Fast atom bombard-ment (FAB) mass spectra, using 3-nitrobenzyl alcohol as aliquid matrix, were recorded using a JEOL JMS-600H massspectrometer.
The mesomorphic behaviour of the target dimers andsome of the key intermediates were ascertained using anOlympus BX50 (model BX50F4) optical polarizing micro-scope, equipped with a calibrated Mettler FP82HT hot stageprogrammed by an FP90 Central Processor. The micropho-tographs of the LC textures were captured with the aid ofa digital camera attached to the microscope. Enthalpies ofphase transition (expressed in kJ mol−1) were derived fromthermograms recorded on a Perkin-Elmer Diamond differ-ential scanning calorimeter operated at a scanning rate of5◦C min−1. Powder XRD patterns, with CuKα wavelength(λ) = 0.15418 nm radiation, were recorded with the helpof a PANalytical X’Pert PRO MP X-ray diffractometer con-sisting of a focusing elliptical mirror and a fast-resolutiondetector (PIXCEL). Randomly selected dimers for the mea-surements contained in Lindemann glass capillary tubes(0.5 mm diameter, procured from Capillary Tube SuppliesLtd.) were carefully flame sealed at both the ends such thatthe samples are intact.
3.2 General procedure for the synthesis of4-n-alkoxy-2-hydroxybenzaldehyde (1a–e)
A mixture of 2,4-dihydroxybenzaldehyde (7.24 mmol,1 equiv.), n-alkylbromide (7.96 mmol, 1.1 equiv.) and anhy-drous KHCO3 (7.96 mmol, 1.1 equiv.) in butanone was heatedat reflux with constant stirring for 12 h under a nitrogen atmo-sphere. The reaction mixture was poured into ice cold waterand extracted with ethyl acetate (20 ml × 3). The collectedorganic layer was washed with a cold 5% (aq.) NaOH solu-tion (30 ml × 3), water (25 ml × 3) and brine, and driedover anhydrous Na2SO4. The solution was concentrated undervacuum and the pure product was obtained after column chro-matography (100–200 mesh silica gel; hexanes–ethyl acetatefrom 95:5 and then 92:8 (v/v)). In view of the fact that thesealdehydes 1a–e are well-known [71–73], they have been char-acterized only by FTIR, 1H NMR and FAB mass techniques.
1a– 4-n-Butoxy-2-hydroxybenzaldehyde: Rf = 0.53 in30% CH2Cl2–hexanes; pale yellow liquid; yield: 1.3 g, 89%;IR (neat): νmax in cm−1 3346, 2928, 2856, 1740, 1294, 1113;1H NMR (400 MHz, CDCl3): δ 10.33 (s, 1H, 1×CHO), 7.80(d, J = 8.8 Hz, 1H, Ar), 6.53 (dd, J1 = 8.4 Hz, J2 = 2.4 Hz,1H, Ar), 6.43 (d, J = 2.4 Hz, 1H, Ar), 4.06–4.01 (m, 2H,

Bull. Mater. Sci. (2020) 43:188 Page 5 of 18 188
1×OCH2), 1.86–1.47 (m, 4H, 2×CH2), 1.01 (t, J = 7.4 Hz,3H, 1 × CH3); MS (FAB+) : m/z Calcd. for C11H14O3
(M + 1): 195.19; Found: 195.2.1b – 4-n-Hexyloxy-2-hydroxybenzaldehyde: Rf = 0.53 in
30% CH2Cl2–hexanes; pale yellow liquid; yield: 1.5 g, 90%;IR (neat): νmax in cm−1 3339, 2924, 2857, 1678, 1292, 1113;1H NMR (400 MHz, CDCl3): δ 10.33 (s, 1H, 1×CHO), 7.80(d, J = 8.4 Hz, 1H, Ar), 6.53 (dd, J1 = 8.4 Hz, J2 = 2.4 Hz,1H, Ar), 6.42 (d, J = 2.0 Hz, 1H, Ar), 4.05–4.00 (m, 2H,1×OCH2), 1.87–1.32 (m, 8H, 4×CH2), 0.93 (t, J = 7.0 Hz,3H, 1×CH3); MS (FAB+) : m/z Calcd. for C13H18O3(M+1):223.13; Found: 223.3.1c – 4-n-Octyloxy-2-hydroxybenzaldehyde: Rf = 0.53 in
30% CH2Cl2–hexanes; pale yellow liquid; yield: 1.6 g, 88%;IR (neat): νmax in cm−1 3350, 2928, 2857, 1679, 1295, 1112;1H NMR (400 MHz, CDCl3): δ 11.47 (s, 1H, 1 × OH), 9.70(s, 1H, 1 × CHO), 7.42 (d, J = 8.8 Hz, 1H, Ar), 6.54 (dd,J1 = 8.8 Hz, J2 = 2.4 Hz, 1H, Ar), 6.41 (d, J = 2.4 Hz,1H, Ar), 4.02 (t, J = 6.8 Hz, 2H, 1 × OCH2), 1.83–1.28(m, 12H, 6 × CH2), 0.91 (t, J = 7.1 Hz, 3H, 1 × CH3); MS(FAB+) : m/z Calcd. for C15H22O3: 250.2; Found: 250.9.
1d – 4-n-Decyloxy-2-hydroxybenzaldehyde: Rf = 0.54 in30% CH2Cl2–hexanes; pale yellow liquid; yield: 1.7 g, 88%;IR (neat): νmax in cm−1 3346, 2928, 2856, 1740, 1294, 1113;1H NMR (400 MHz, CDCl3): δ 10.33 (s, 1H, 1×CHO), 7.80(d, J = 8.7 Hz, 1H, Ar), 6.52 (dd, J1 = 8.7 Hz, J2 = 1.5 Hz,1H, Ar), 6.42 (d, J = 2.2 Hz, 1H, Ar), 4.06–3.99 (m, 2H,1×OCH2), 1.87–1.28 (m, 16H, 8×CH2), 0.90 (t, J = 6.7 Hz,3H, 1×CH3); MS (FAB+) : m/z Calcd. for C17H26O3: 278.2;Found: 278.9.1e – 4-n-Dodecyloxy-2-hydroxybenzaldehyde: Rf = 0.54
in 30% CH2Cl2–hexanes; pale yellow liquid; yield: 2.0 g,85%; IR (neat): νmax in cm−1 3344, 2928, 2856, 1740, 1294and 1113; 1H NMR (400 MHz, CDCl3): δ 10.26 (s, 1H, 1 ×CHO), 7.73 (d, J = 8.4 Hz, 1H, Ar), 6.45 (dd, J1 = 8.8 Hz,J2 = 2.0 Hz, 1H, Ar), 6.35 (d, J = 2.4 Hz, 1H, Ar), 3.96–3.92 (m, 2H, 1×OCH2), 1.80–1.23 (m, 20H, 10×CH2), 0.83(t, J = 6.8 Hz, 3H, 1 × CH3); MS (FAB+) : m/z Calcd. forC19H30O3: 306.2; Found: 306.5.
3.3 General procedure for the synthesis of cholesterylω-bromoalkanoates (2a–c)
A mixture of ω-bromoalkanoic acid (6.5 mmol, 1 equiv.) in6 ml of oxalyl chloride was stirred at RT under a nitrogenatmosphere for 4 h. Unreacted oxalyl chloride was removedcompletely from the reaction mixture by distillation undermild vacuum and the pale yellow residue of ω-bromoalkanoylchloride obtained in almost pure form was added drop-wiseto a solution of cholesterol (2.5 g, 12.9 mmol, 1 equiv.) and inpyridine (4 ml) in dry THF (30 ml) at 0–5◦C. The reaction mix-ture was allowed to warm-up to RT and stirred overnight. Theclear reaction mixture (yellow solution), obtained after filter-ing the reaction mixture through a Celite bed, was evaporatedto dryness. The resulting pale yellow mass was dissolved in amixture of CH2Cl2 and Et2O (20:80) (80 ml) and the organic
layer was thoroughly washed with water and brine; the organiclayer thus obtained was dried over anhydrous Na2SO4. Thecrude compound obtained after the evaporation of the mixtureof solvents was purified by column chromatography: neutralalumina; hexanes–CH2Cl2 90:10 and then 80:20 (v/v). Giventhe fact that these compounds are known [39,68,72], theyhave been characterized with only a couple of spectroscopictechnique as detailed below.2a–Cholesteryl 4-bromobutanoate: Rf = 0.33 in hexanes–
CH2Cl2, 70:30 (v/v); a white solid; yield: 2.8 g, 76%; m.p.:87–88◦C; IR (KBr pellet): νmax in cm−1 2948, 2863, 1734,1465, 1172, 1027; 1H NMR (400 MHz, CDCl3): δ 5.39 (brd,J = 4.0 Hz, 1H, 1×olefinic), 4.67 (m, 1H, 1×CHOCO), 3.48(t, J = 6.4 Hz, 2H, 1×CH2Br), 2.49 (m, 4H, 2×allylic CH2),2.03–1.05 (m, 28H, 11×CH2, 6×CH), 1.02 (s, 3H, 1×CH3),0.93 (d, J = 6.8 Hz, 3H, 1 × CH3), 0.88 (d, J = 1.6 Hz, 3H,1×CH3), 0.86 (d, J = 2.0 Hz, 3H, 1×CH3) and 0.68 (s, 3H,1×CH3); MS (FAB+) : m/z Calcd. for C31H53BrO2(M+2):536.3; Found: 536.21.2b–Cholesteryl 6-bromohexanoate: Rf = 0.33 in hexanes–
CH2Cl2, 70:30 (v/v); a white solid; yield: 3 g, 83%; m.p.:100–101◦C; IR (KBr pellet): νmax in cm−1 2949, 2863, 1733,1465, 1175, 1020; 1H NMR (400 MHz, CDCl3): δ 5.38 (brd,J = 4.0 Hz, 1H, 1×olefinic), 4.65 (m, 1H, 1×CHOCO), 3.42(t, J = 8.0 Hz, 2H, 1×CH2Br), 2.32 (m, 4H, 2×allylic CH2),2.03–1.04 (m, 32H, 13×CH2, 6×CH), 1.02 (s, 3H, 1×CH3),0.92 (d, J = 4.0 Hz, 3H, 1 × CH3), 0.87 (d, J = 1.6 Hz, 3H,1 × CH3), 0.86 (d, J = 1.6 Hz, 3H, 1 × CH3), 0.68 (s, 3H,1 × CH3); MS (FAB+) : m/z Calcd. for C33H56BrO2: 563.3;Found: 563.4.2c – Cholesteryl 8-bromooctanoate: Rf = 0.33 in hexanes–
CH2Cl2, 70:30 (v/v); a white solid; yield: 3.3 g, 86%; m.p.:86–87◦C; IR (KBr pellet): νmax in cm−1 2947, 2866, 1733,1465, 1171, 1026; 1H NMR (400 MHz, CDCl3): δ 5.38 (brd,J = 3.4 Hz, 1H, 1×olefinic), 4.63 (m, 1H, 1×CHOCO), 3.42(t, J = 8.0 Hz, 2H, 1×CH2Br), 2.32 (m, 4H, 2×allylic CH2),2.02–1.04 (m, 36H, 15×CH2, 6×CH), 1.02 (s, 3H, 1×CH3),0.92 (d, J = 4.0 Hz, 3H, 1 × CH3), 0.87 (d, J = 1.6 Hz, 3H,1 × CH3), 0.86 (d, J = 1.6 Hz, 3H, 1 × CH3), 0.68 (s, 3H,1 × CH3); MS (FAB+): m/z Calcd. for C35H59BrO2: 590.37;Found: 590.11.
3.4 General procedure for the synthesis of cholesterylω-(4-nitrophenoxy)alkanoates (3a–c)
A mixture of cholesteryl ω-bromoalkanoate (2a–c) (0.5 g,0.95 mmol, 1 equiv.), 4-nitrophenol (2 mmol, 2.1 equiv.),anhydrous K2CO3 (1.05 mmol, 1.1 equiv.) and butanone(50 ml) was refluxed for 12 h under nitrogen. After cool-ing the reaction mixture to RT, it was poured into ice coldwater and extracted with ethyl acetate. The organic layer waswashed with cold 5% (aq.) NaOH solution (20 ml×3), water(30 ml × 3) and brine, and dried over anhydrous Na2SO4.The organic layer was concentrated and the crude productobtained was purified by column chromatography: 100–200mesh silica gel; hexanes–ethyl acetate from 90:10 (v/v).

188 Page 6 of 18 Bull. Mater. Sci. (2020) 43:188
3a – Cholesteryl 4-(4-nitrophenoxy)butanoate: Rf = 0.43in hexanes–CH2Cl2, 70:30 (v/v); white solid; yield: 0.43 g,82%; heating: Cr 134 I; cooling: I 130.6 N* 117.7 TGB-SmA91 Cr; IR (neat): νmax in cm−1 2944, 2866, 1733, 1593, 1510,1378, 1339, 1178, 1112; 1H NMR (400 MHz, CDCl3): δ 8.21(d, 2H, J = 6.8 Hz, Ar), 6.96 (d, J = 7.2 Hz, 2H, Ar),5.38 (brd, J = 3.6 Hz, 1H, 1 × olefinic), 4.68–4.63 (m, 1H,1 × CHOCO), 4.14 (t, 2H, J = 6.4 Hz, 1 × OCH2), 2.52–2.30 (m, 4H, 2 × allylic CH2); 2.16–0.68 (m, 43H, 6 × CH,11 × CH2, 5 × CH3); Anal. calcd. for C37H55NO5: C, 74.83;H, 9.34; N, 2.36; Found: C, 74.68; H, 9.17; N, 2.38.3b – Cholesteryl 6-(4-nitrophenoxy)hexanoate: Rf = 0.44
in hexanes–CH2Cl2, 70:30 (v/v); white solid; yield: 0.47 g,84%; heating: Cr 137 I; cooling: I 124 N* 102 Cr; IR (neat):νmax in cm−1 2946, 2870, 1734, 1592, 1512, 1388, 1338,1178, 1112; 1H NMR (400 MHz, CDCl3): δ 8.21 (d, 2H,J = 7.2 Hz, Ar), 6.96 (d, J = 6.8 Hz, 2H, Ar), 5.37(brd, J = 4.0 Hz, 1H, 1 × olefinic), 4.65–4.61 (m, 1H,1 × CHOCO), 4.07 (t, 2H, J = 6.4 Hz, 1 × OCH2), 2.34–2.30 (m, 4H, 2 × allylic CH2), 2.00–0.68 (m, 47H, 6 × CH,13 × CH2, 5 × CH3); Anal. calcd. for C39H59NO5: C, 75.32;H, 9.56; N, 2.25; Found: C, 76.26; H, 9.41; N, 2.19.3c – Cholesteryl 8-(4-nitrophenoxy)octanoate: Rf = 0.45
in hexanes–CH2Cl2, 70:30 (v/v); white solid; yield: 0.43 g,82%; Cr 96 N* 107 (2) I; cooling I 106.5 (1.2) N 54.0 Cr;IR (neat): νmax in cm−1 2944, 2868, 1732, 1593, 1510, 1337,1180, 1110; 1H NMR (400 MHz, CDCl3): δ 8.20 (d, 2H, J =7.2 Hz, Ar), 6.95 (d, J = 7.2 Hz, 2H, Ar), 5.37 (brd, J =4.0 Hz, 1H, 1 × olefinic), 4.65–4.57 (m, 1H, 1 × CHOCO),4.06 (t, 2H, J = 6.4 Hz, 1 × OCH2), 2.32–2.26 (m, 4H,2 × allylic CH2), 1.99–0.68 (m, 51H, 6 × CH, 15 × CH2,5 × CH3); Anal. calcd. for C41H63NO5: C, 75.77; H, 9.77; N,2.16; Found: C, 75.35; H, 9.8; N, 2.28.
3.5 General procedure for the synthesis of cholesterylω-(4-aminophenoxy)alkanoates (4a–c)
A mixture of nitro compound 3a/3b/3c (0.5 g), 10% Pd–C(10 wt%) and THF (25 ml) taken in a round flask was degassedthoroughly and then hydrogen gas was introduced using itsballoon (1 atm). The reaction mixture was stirred for 4 hat RT. The reaction mixture was filtered carefully (caution:flammable) using a Celite bed and the filtrate obtained wasconcentrated. The resulting crude solid product was recrys-tallized from hexanes to obtain an off-white solid compound.The characterization of three compounds 4a–c is given below.
4a – Cholesteryl 4-(4-aminophenoxy)butanoate: Rf =0.48 in hexanes–CH2Cl2, 50:50 (v/v); white solid; yield:0.41 g, 85%; heating: Cr 122 SmA-TGB-N* 127 I; cooling I126 N*-TGB 126 SmA 102 Cr; IR (neat): νmax in cm−1 3376,2937, 2868, 1726, 1179, 1103; 1H NMR (400 MHz, CDCl3):δ 6.74 (d, 2H, J = 6.8 Hz, Ar), 6.65 (d, J = 6.8 Hz, 2H,Ar), 5.37 (brd, J = 3.6 Hz, 1H, 1 × olefinic), 4.63–4.60 (m,1H, 1 × CHOCO), 3.91 (t, 2H, J = 6.0 Hz, 1 × OCH2), 3.42(brs, 2H, 1 × NH2), 2.35–2.30 (m, 4H, 2 × allylic CH2), 2.0–0.68 (m, 43H, 6 × CH, 11 × CH2, 5 × CH3); Anal. calcd. for
C37H57NO3: C, 78.81; H, 10.19; N, 2.48; Found: C, 78.67; H,10.12; N, 2.52.4b–Cholesteryl 6-(4-aminophenoxy)hexanoate: Rf = 0.50
in hexanes–CH2Cl2, 50:50 (v/v); white solid; yield: 0.46 g,96.2%; heating: Cr 96 SmA-TGB-N* 107 I; cooling I 106 N*-TGB 101 SmA 46 Cr; IR (neat): νmax in cm−1 3374, 2940,2869, 1726, 1179, 1102; 1H NMR (400 MHz, CDCl3): δ 6.65(d, 2H, J = 6.4 Hz, Ar), 6.56 (d, J = 6.4 Hz, 2H, Ar),5.31 (brd, J = 4.0 Hz, 1H, 1 × olefinic), 4.6–4.5 (m, 1H,1 × CHOCO), 3.87 (t, 2H, J = 6.4 Hz, 1 × OCH2), 2.28–2.23 (m, 4H, 2 × allylic CH2), 1.92–0.61 (m, 47H, 6 × CH,13 × CH2, 5 × CH3); Anal. calcd. for C39H61NO3: C, 79.14;H, 10.39; N, 2.37; Found: C, 79.3; H, 10.41; N, 2.44.4c – Cholesteryl 8-(4-aminophenoxy)octanoate: Rf = 0.51
in hexanes–CH2Cl2, 50:50 (v/v); white solid; yield: 0.4 g,84%; heating: Cr 109 SmA-TGB-N* 113 I; cooling I 112 N*-TGB 101 SmA 62 Cr; IR (neat): νmax in cm−1 3379, 2941,2869, 1726, 1179, 1106; 1H NMR (400 MHz, CDCl3): δ 6.75(d, 2H, J = 6.8 Hz, Ar), 6.67 (d, J = 6.4 Hz, 2H, Ar),5.38 (brd, J = 3.6 Hz, 1H, 1 × olefinic), 4.7–4.6 (m, 1H,1 × CHOCO), 3.90 (t, 2H, J = 6.4 Hz, 1 × OCH2), 2.35–2.3(m, 4H, 2×allylic CH2), 2.0–0.68 (m, 51H, 6×CH, 15×CH2,5 × CH3); Anal. calcd. for C41H65NO3: C, 79.43; H, 10.57;N, 2.26; Found: C, 79.51; H, 10.63; N, 2.19.
3.6 General procedure for the synthesis ofcholesterol-based dimers DCS-n,R
A mixture of 4-n-alkoxy-2-hydroxybenzaldehyde (1a–e)(0.63 mmol, 1.5 equiv.), cholesteryl ω-(4-aminophenoxy)alkanoate (4a–c) (0.126 mmol, 1 equiv.) and a catalyticamount of glacial acetic acid (three drops) in absolute ethanolwas refluxed for 1 h. The yellow bright solid obtained oncooling was filtered, washed with hot ethanol and furtherpurified by repeated recrystallizations from a mixture ofCH2Cl2:EtOH (10:90 v/v) to obtain the pure compound.
DCS-3,4 – Cholesteryl 4-(4-((4-(butoxy)-2-hydroxyben-zylidene)amino)phenoxy)butanoate: pale yellow solid; yield:0.068 g, 87%; IR (KBr pellet): νmax in cm−1 3429, 2927,2851, 1724, 1622, 1192, 1111; UV–Vis: λmax = 346 nm,ε = 8.1 × 103 l mol−1 cm−1; 1H NMR (400 MHz, CDCl3): δ
8.81 (s, 1H, 1 × CH=N), 8.07 (d, J = 8.4 Hz, 1H, Ar), 7.18(d, J = 9.2 Hz, 2H, Ar), 6.91 (d, J = 8.8 Hz, 2H, Ar), 6.56(dd, J1 = 8.8 Hz, J2 = 2.0 Hz, 1H, Ar), 6.45 (d, J = 2.4 Hz,1H, Ar), 5.38 (brd, J = 4.8 Hz, 1H, olefinic), 4.68 (m, 1H,1 × CHOCO), 4.03 (m, 4H, 2 × OCH2), 2.52–2.31 (m, 4H,2 × allylic CH2), 2.14–0.68 (m, 50H, 6 × CH, 13 × CH2,6 × CH3); 13C NMR (100 MHz): 172.6, 163.0, 160.3 157.0,154.4, 146.3, 139.7, 128.6, 122.7, 122.1, 115.0, 106.1, 99.3,74.1, 68.1, 67.9, 67.1, 56.7, 56.2, 50.1, 42.3, 39.8, 39.5, 38.2,37.0, 36.6, 36.2, 35.8, 31.9, 31.3, 31.2, 28.2, 28.0, 27.8, 24.8,24.3, 23.8, 22.8, 22.6, 21.0, 19.3, 19.2, 18.7, 13.8, 11.9; Anal.calcd. for C48H69NO5: C, 77.90; H, 9.40; N, 1.89; Found: C,79.32; H, 9.74; N, 2.21.DCS-3,6 – Cholesteryl 4-(4-((4-(hexyloxy)-2-hydroxy-
benzylidene)amino)phenoxy)butanoate: pale yellow solid;

Bull. Mater. Sci. (2020) 43:188 Page 7 of 18 188
yield: 0.063 g, 77%; IR (neat): νmax in cm−1 3429, 2928,2851, 1724, 1622, 1174, 1131; UV–Vis: λmax = 345 nm,ε = 2.51 × 103 l mol−1 cm−1; 1H NMR (400 MHz, CDCl3):δ 8.82 (s, 1H, 1×CH=N), 8.07 (d, J = 8.8 Hz, 1H, Ar), 7.18(d, J = 9.2 Hz, 2H, Ar), 6.91 (d, J = 8.8 Hz, 2H, Ar), 6.56(dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H, Ar), 6.45 (d, J = 2.4 Hz,1H, Ar), 5.38 (brd, J = 4.8 Hz, 1H, olefinic), 4.65 (m, 1H,1 × CHOCO), 4.03 (m, 4H, 2 × OCH2), 2.52–2.31 (m, 4H,2 × allylic CH2), 2.14–0.68 (m, 54H, 6 × CH, 15 × CH2,6 × CH3); 13C NMR (100 MHz): 172.6, 163.0, 160.3 157.0,154.4, 146.3, 139.7, 128.6, 122.7, 122.1, 118.4, 114.9, 106.2,99.3, 74.1, 68.5, 68.2, 67.1, 56.7, 56.2, 50.1, 42.3, 39.8, 39.5,38.2, 37.0, 36.6, 36.2, 35.8, 31.9, 31.6, 31.5, 31.2, 29.2, 29.1,28.2, 28.0, 27.8, 25.8, 25.7, 24.8, 24.3, 23.8, 22.8, 22.6, 21.0,19.3, 19.2, 18.7, 14.0, 11.9; Anal. calcd. for C50H73NO5: C,78.38; H, 9.58; N, 1.82; Found: C, 78.93; H, 9.91; N, 1.9.DCS-3,8 – Cholesteryl 4-(4-((4-(octyloxy)-2-hydroxyben-
zylidene)amino)phenoxy)butanoate: pale yellow solid; yield:0.072 g, 85%; IR (neat): νmax in cm−1 3429, 2927, 2867, 1724,1622, 1192, 1111; UV–Vis: λmax = 352 nm, ε = 2.81 ×103 l mol−1 cm−1; 1H NMR (400 MHz, CDCl3): δ 8.81 (s,1H, 1 × CH=N), 8.07 (d, J = 8.4 Hz, 1H, Ar), 7.18 (d,J = 6.8 Hz, 2H, Ar), 6.91 (d, J = 6.8 Hz, 2H, Ar), 6.56 (dd,J1 = 8.4 Hz, J2 = 2.0 Hz, 1H, Ar), 6.45 (d, J = 2.4 Hz,1H, Ar), 5.38 (brd, J = 4.8 Hz, 1H, olefinic), 4.68 (m, 1H,1 × CHOCO), 4.03 (m, 4H, 2 × OCH2), 2.52–2.31 (m, 4H,2 × allylic CH2), 2.13–0.68 (m, 58H, 6 × CH, 17 × CH2,6 × CH3); 13C NMR (100 MHz): 172.6, 163.7, 163.3, 159.7,157.7, 141.6, 139.7, 133.1, 122.7, 122.0, 115.2, 113.1, 107.4,101.6, 74.1, 68.2, 67.2, 56.7, 56.2, 50.1, 42.3, 39.8, 39.5, 38.2,37.0, 36.6, 36.2, 35.8, 31.9, 31.8, 31.1, 29.3, 29.2, 29.1, 28.2,28.0, 27.8, 26.0, 24.7, 24.3, 23.9, 22.8, 22.7, 22.6, 21.1, 19.3,18.7, 14.1, 11.9; Anal. calcd. for C52H77NO5: C, 78.44; H,9.75; N, 1.76; Found: C, 77.91; H, 9.80; N, 1.96.DCS-3,10 – Cholesteryl 4-(4-((4-(decyloxy)-2-hydroxy-
benzylidene)amino)phenoxy)butanoate: pale yellow solid;yield: 0.068 g, 78%; IR (neat): νmax in cm−1 3429, 2927,2851, 1724, 1622, 1192, 1111; UV–Vis: λmax = 348 nm,ε = 1.44 × 103 l mol−1 cm−1; 1H NMR (400 MHz, CDCl3):δ 8.81 (s, 1H, 1×CH=N), 8.07 (d, J = 8.8 Hz, 1H, Ar), 7.18(d, J = 6.8 Hz, 2H, Ar), 6.91 (d, J = 6.8 Hz, 2H, Ar), 6.56(dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H, Ar), 6.45 (d, J = 2.0 Hz,1H, Ar), 5.38 (brd, J = 4.0 Hz, 1H, olefinic), 4.65 (m, 1H,1 × CHOCO), 4.03 (m, 4H, 2 × OCH2), 2.52–2.30 (m, 4H,2 × allylic CH2), 2.14–0.68 (m, 62H, 6 × CH, 19 × CH2,6 × CH3); 13C NMR (100 MHz): 172.9, 163.0, 160.4, 157.1,154.4, 146.2, 128.6, 122.6, 122.2, 114.9, 106.2, 99.4, 73.9,68.5, 68.3, 67.7, 56.7, 56.2, 50.1, 42.3, 39.8, 39.6, 38.2, 37.0,36.2, 35.8, 34.4, 31.9, 31.6, 31.5, 29.2, 29.1, 28.8, 28.3, 28.0,27.9, 25.8, 25.7, 24.3, 23.9, 22.8, 22.6, 21.8, 21.1, 19.3, 18.7,14.0, 11.9; Anal. calcd. for C54H81NO5: C, 78.69; H, 9.91; N,1.70; Found: C, 79.02; H, 10.40; N, 1.75.DCS-3,12 – Cholesteryl 4-(4-((4-(dodecyloxy)-2-hydr-
oxybenzylidene)amino)phenoxy)butanoate: pale yellowsolid; yield: 0.078 g, 86%; IR (neat): νmax in cm−1 3429, 2927,2867, 1724, 1622, 1192, 1111; UV–Vis: λmax = 346 nm,
ε = 1.55 × 103 l mol−1 cm−1; 1H NMR (400 MHz, CDCl3):δ 8.81 (s, 1H, 1×CH=N), 8.07 (d, J = 8.4 Hz, 1H, Ar), 7.18(d, J = 9.2 Hz, 2H, Ar), 6.91 (d, J = 8.8 Hz, 2H, Ar), 6.56(dd, J1 = 8.8 Hz, J2 = 2.4 Hz, 1H, Ar), 6.45 (d, J = 2.4 Hz,1H, Ar), 5.38 (brd, J = 4.4 Hz, 1H, olefinic), 4.67 (m, 1H,1 × CHOCO), 4.03 (m, 4H, 2 × OCH2), 2.52–2.31 (m, 4H,2 × allylic CH2), 2.13–0.68 (m, 54H, 6 × CH, 21 × CH2,6 × CH3); 13C NMR (100 MHz): 172.8, 163.7, 163.3, 159.6,157.8, 141.4, 139.7, 133.1, 122.7, 122.0, 115.2, 107.4, 101.6,73.9, 68.2, 67.7, 56.7, 56.2, 50.0, 42.3, 39.7, 39.5, 38.2, 37.0,36.6, 36.2, 35.8, 34.3, 31.9, 31.8, 29.3, 29.2, 29.1, 28.6, 28.2,28.0, 27.8, 26, 24.3, 23.8, 22.8, 22.6, 22.5, 21.7, 21.0, 19.3,14.1, 11.9; Anal. calcd. for C56H85NO5: C, 78.92; H, 10.05;N, 1.64; Found: C, 79.37; H, 9.87; N, 2.02.DCS-5,4 – Cholesteryl 6-(4-((4-(butoxy)-2-hydroxyben-
zylidene)amino)phenoxy)hexanoate: pale yellow solid; yield:0.048 g, 74%; IR (neat): νmax in cm−1 3440, 2927, 2867,1724, 1622, 1192, 1111; UV–Vis: λmax = 348 nm, ε =1.77 × 103 l mol −1 cm−1; 1H NMR (400 MHz, CDCl3): δ
8.81 (s, 1H, 1 × CH=N), 8.07 (d, J = 8.4 Hz, 1H, Ar), 7.18(d, J = 8.8 Hz, 2H, Ar), 6.91 (d, J = 8.8 Hz, 2H, Ar), 6.56(dd, J1 = 8.8 Hz, J2 = 2.0 Hz, 1H, Ar), 6.45 (d, J = 2.0 Hz,1H, Ar), 5.38 (brd, J = 4.8 Hz, 1H, olefinic), 4.63 (m, 1H,1 × CHOCO), 4.04 (m, 4H, 2 × OCH2), 2.34–2.30 (m, 4H,2 × allylic CH2), 2.02–0.68 (m, 54H, 6 × CH, 15 × CH2,6 × CH3); 13C NMR (100 MHz): 173.1, 163.0, 154.3, 146.2,139.7, 128.6, 122.6, 112.2, 114.9, 106.2, 99.3, 73.8, 68.2,67.9, 56.7, 56.2, 50.1, 42.3, 39.8, 39.6, 38.2, 37.0, 36.6, 36.2,35.8, 34.6, 31.9, 31.3, 31.2, 29.0, 28.3, 28.0, 27.9, 25.7, 24.8,24.3, 23.9, 22.8, 22.6, 22.1, 19.3, 18.7, 13.8, 11.9; Anal. calcd.for C50H73NO5: C, 78.18; H, 9.58; N, 1.82; Found: C, 78.39;H, 10.13; N, 2.23.DCS-5,6 – Cholesteryl 6-(4-((4-(hexyloxy)-2-hydroxy-
benzylidene)amino)phenoxy)hexanoate: pale yellow solid;yield: 0.055 g, 83%; IR (neat): νmax in cm−1 3429, 2927,2851, 1724, 1622, 1192, 1111; UV–Vis: λmax = 348 nm,ε = 6.10 × 103 l mol−1 cm−1; 1H NMR (400 MHz, CDCl3):δ 8.82 (s, 1H, 1×CH=N), 8.07 (d, J = 8.4 Hz, 1H, Ar), 7.18(d, J = 8.8 Hz, 2H, Ar), 6.90 (d, J = 8.8 Hz, 2H, Ar), 6.56(dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H, Ar), 6.45 (d, J = 2.4 Hz,1H, Ar), 5.38 (brd, J = 4.8 Hz, 1H, olefinic), 4.66 (m, 1H,1 × CHOCO), 4.03 (m, 4H, 2 × OCH2), 2.34–2.30 (m, 4H,2 × allylic CH2), 2.02–0.68 (m, 58H, 6 × CH, 17 × CH2,6 × CH3); 13C NMR (100 MHz): 173.1, 163.0, 160.4, 157.2,154.4, 146.1, 139.7, 128.8, 122.6, 122.2, 118.4, 114.9, 106.2,99.4, 73.8, 68.5, 68.3, 68.0, 56.7, 56.2, 50.1, 42.4, 39.8, 39.6,38.2, 37.0, 36.6, 36.2, 35.8, 34.6, 31.9, 31.6, 31.5, 29.2, 29.1,29.0, 28.3, 28.0, 27.9, 25.8, 25.7, 24.9, 24.3, 23.9, 22.8, 22.6,21.1, 19.3, 18.7, 14.0, 11.9; Anal. calcd. for C52H77NO5: C,78.44; H, 9.75; N, 1.76; Found: C, 77.83; H, 10.13; N, 2.17.DCS-5,8 – Cholesteryl 6-(4-((4-(octyloxy)-2-hydroxy-
benzylidene)amino)phenoxy)hexanoate: pale yellow solid;yield: 0.048 g, 69%; IR (neat): νmax in cm−1 3418, 2935,2868, 1724, 1622, 1173, 1128; UV–Vis: λmax = 348 nm,ε = 3.49×103 l mol−1 cm−1; 1H NMR (400 MHz, CDCl3):δ 8.82 (s, 1H, 1 × CH=N), 8.07 (d, J = 8.4 Hz, 1H,

188 Page 8 of 18 Bull. Mater. Sci. (2020) 43:188
Ar), 7.18 (d, J = 8.8 Hz, 2H, Ar), 6.90 (d, J = 8.8 Hz,2H, Ar), 6.56 (dd, J1 = 8.8 Hz, J2 = 2.0 Hz, 1H, Ar),6.45 (d, J = 2.4 Hz, 1H, Ar), 5.38 (brd, J = 3.6 Hz, 1H,olefinic), 4.78 (m, 1H, 1×CHOCO), 4.03 (m, 4H, 2×OCH2),2.34–2.29 (m, 4H, 2 × allylic CH2), 2.02–0.67 (m, 62H,6 × CH, 19 × CH2, 6 × CH3); 13C NMR (100 MHz): 172.7,163.0, 160.4, 157.0, 154.5, 146.3, 139.7, 128.6, 122.7, 122.2,118.4, 115.0, 106.2, 99.4, 74.1, 68.5, 68.3, 67.1, 56.7, 56.2,50.1, 42.4, 39.8, 39.6, 38.2, 37.0, 36.6, 36.2, 35.8, 31.9, 31.2,29.7, 29.6, 29.4, 29.3, 29.2, 28.3, 28.0, 27.8, 26.1, 26.0, 24.8,24.3, 23.8, 23.9, 22.8, 22.7, 22.6, 21.1, 19.3, 18.7, 14.1, 11.9;Anal. calcd. for C54H81NO5: C, 78.69; H, 9.91; N, 1.70;Found: C, 79.19; H, 10.35; N, 1.86.DCS-5,10 – Cholesteryl 6-(4-((4-(decyloxy)-2-hydroxy-
benzylidene)amino)phenoxy)hexanoate: pale yellow solid;yield: 0.052 g, 72%; IR (neat): νmax in cm−1 3440, 2926,2853, 1732, 1608, 1179, 1112; UV–Vis: λmax = 348 nm,ε = 2.35×103 l mol−1 cm−1; 1H NMR (400 MHz, CDCl3):δ 8.82 (s, 1H, 1×CH=N), 8.07 (d, J = 8.8 Hz, 1H, Ar), 7.18(d, J = 8.8 Hz, 2H, Ar), 6.90 (d, J = 8.8 Hz, 2H, Ar), 6.56(dd, J1 = 8.4 Hz, J2 = 2.4 Hz, 1H, Ar), 6.44 (d, J = 2.4 Hz,1H, Ar), 5.38 (brd, J = 4.8 Hz, 1H, olefinic), 4.70 (m, 1H,1 × CHOCO), 4.02 (m, 4H, 2 × OCH2), 2.34–2.30 (m, 4H,2 × allylic CH2), 1.97–0.67 (m, 66H, 6 × CH, 21 × CH2,6 × CH3); 13C NMR (100 MHz): 173.1, 163.0, 160.3, 157.2,154.3, 146.1, 139.7, 128.6, 122.6, 122.2, 118.4, 114.9, 106.2,99.3, 73.8, 68.5, 68.2, 67.9, 56.7, 56.2, 54.3, 50.1, 44.7, 42.3,40.0, 39.8, 39.5, 38.2, 37.0, 36.8, 36.6, 36.2, 35.8, 34.6, 34.1,32.0, 31.9, 29.6, 29.4, 29.3, 29.2, 29.1, 29.0, 28.2, 28.0, 27.8,26.1, 26.0, 25.7, 24.8, 24.3, 23.9, 22.8, 22.7, 22.6, 21.1, 19.3,18.7, 14.1, 11.9; Anal. calcd. for C56H85NO5: C, 78.92; H,10.05; N, 1.64; Found: C, 79.26; H, 10.51; N, 2.03.DCS-5,12 – Cholesteryl 6-(4-((4-(dodecyloxy)-2-hydr-
oxybenzylidene)amino)phenoxy)hexanoate: pale yellowsolid; yield: 0.058 g, 78%; IR (neat): νmax in cm−1 3429, 2928,2851, 1724, 1622, 1174, 1132; UV–Vis: λmax = 348 nm,ε = 3.73×103 l mol−1 cm−1; 1H NMR (400 MHz, CDCl3):δ 8.82 (s, 1H, 1 × CH=N), 8.07 (d, J = 8.4 Hz, 1H, Ar),7.18 (d, J = 8.8 Hz, 2H, Ar), 6.90 (d, J = 8.8 Hz, 2H,Ar), 6.56 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H, Ar), 6.44 (d,J = 2.4 Hz, 1H, Ar), 5.38 (brd, J = 3.6 Hz, 1H, olefinic),4.70 (m, 1H, 1 × CHOCO), 4.02 (m, 4H, 2 × OCH2), 2.30–2.28 (m, 4H, 2 × allylic CH2), 2.02–0.67 (m, 70H, 6 × CH,23 × CH2, 6 × CH3); 13C NMR (100 MHz): 173.0, 163.8,163.3, 159.5, 157.9, 141.4, 139.7, 133.1, 122.6, 122.0, 115.2,113.1, 107.4, 101.6, 73.8, 68.2, 68.0, 56.7, 56.2, 50.1, 42.3,39.8, 39.5, 38.2, 37.0, 36.6, 36.2, 35.8, 34.6, 31.9, 29.7, 29.6,29.4, 29.1, 29.0, 28.2, 28.0, 27.8, 26.0, 25.6, 24.8, 24.3, 23.9,22.8, 22.7, 22.6, 21.1, 19.3, 18.7, 14.1, 11.9; Anal. calcd. forC58H89NO5: C, 79.13; H, 10.19; N, 1.59; Found: C, 79.70; H,10.46; N, 2.01.DCS-7,4 – Cholesteryl 8-(4-((4-(butoxy)-2-hydroxyben-
zylidene)amino)phenoxy)octanoate: pale yellow solid; yield:0.068 g, 82%; IR (neat): νmax in cm−1 3456, 2950, 2866,1735, 1608, 1181, 1098; UV–Vis: λmax = 348 nm, ε =1.64 × 103 l mol−1 cm−1; 1H NMR (400 MHz, CDCl3): δ
8.82 (s, 1H, 1 × CH=N), 8.07 (d, J = 8.8 Hz, 1H, Ar),7.18 (d, J = 8.8 Hz, 2H, Ar), 6.92 (d, J = 8.8 Hz, 2H,Ar), 6.56 (dd, J1 = 8.8 Hz, J2 = 2.0 Hz, 1H, Ar), 6.45 (d,J = 2.4 Hz, 1H, Ar), 5.38 (brd, J = 4.8 Hz, 1H, olefinic),4.66 (m, 1H, 1 × CHOCO), 4.03 (m, 4H, 2 × OCH2), 2.30–2.28 (m, 4H, 2 × allylic CH2), 1.99–0.68 (m, 58H, 6 × CH,17 × CH2, 6 × CH3); 13C NMR (100 MHz): 173.2, 163.0,160.3, 157.2, 154.3, 146.1, 139.7, 128.5, 122.6, 122.1, 118.4,116.4, 114.9, 106.1, 99.3, 73.7, 68.5, 68.2, 56.7, 56.2, 50.0,42.3, 39.7, 39.5, 38.2, 37.0, 36.6, 36.2, 35.8, 34.7, 31.9, 31.6,31.5, 29.3, 29.2, 29.1, 29.0, 28.2, 28.0, 27.8, 25.9, 25.8, 25.7,25.0, 24.3, 23.8, 22.8, 22.6, 21.0, 19.3, 18.7, 14.1, 11.9; Anal.calcd. for C52H77NO5: C, 78.44; H, 9.75; N, 1.76; Found: C,78.84; H, 10.21; N, 2.12.DCS-7,6 – Cholesteryl 8-(4-((4-(hexyloxy)-2-hydroxy-
benzylidene)amino)phenoxy)octanoate: pale yellow solid;yield: 0.076 g, 88%; IR (neat): νmax in cm−1 3456, 2931,2854, 1732, 1608, 1177, 1112; UV–Vis: λmax = 347 nm,ε = 2.06×103 l mol−1 cm−1; 1H NMR (400 MHz, CDCl3):δ 8.82 (s, 1H, 1 × CH=N), 8.07 (d, J = 8.8 Hz, 1H, Ar),7.18 (d, J = 8.8 Hz, 2H, Ar), 6.91 (d, J = 8.8 Hz, 2H,Ar), 6.56 (dd, J1 = 8.8 Hz, J2 = 2.4 Hz, 1H, Ar), 6.45 (d,J = 2.0 Hz, 1H, Ar), 5.38 (brd, J = 3.6 Hz, 1H, olefinic),4.68 (m, 1H, 1 × CHOCO), 4.03 (m, 4H, 2 × OCH2), 2.32–2.26 (m, 4H, 2 × allylic CH2), 1.98–0.68 (m, 62H, 6 × CH,19 × CH2, 6 × CH3); 13C NMR (100 MHz): 173.2, 163.0,160.3, 157.2, 154.3, 146.1, 139.7, 128.6, 122.6, 122.2, 118.4,114.9, 106.1, 99.3, 73.7, 68.2, 68.1, 67.9, 56.7, 56.2, 50.0,42.3, 39.7, 39.5, 38.2, 37.0, 36.8, 36.6, 36.2, 35.8, 34.7, 32.0,31.9, 31.3, 31.2, 29.3, 29.0, 28.2, 28.0, 27.8, 25.9, 25.0, 24.3,23.8, 22.8, 22.6, 21.2, 21.0, 19.3, 19.2, 18.7, 13.8, 11.9; Anal.calcd. for C54H81NO5: C, 78.69; H, 9.91; N, 1.70; Found: C,79.14; H, 10.32; N, 2.02.DCS-7,8 – Cholesteryl 8-(4-((4-(octyloxy)-2-hydroxyben-
zylidene)amino)phenoxy)octanoate: pale yellow solid; yield:0.057 g, 64%; IR (neat): νmax in cm−1 3456, 2931, 2855, 1732,1608, 1177, 1112; UV–Vis: λmax = 347 nm, ε = 2.67 ×103 l mol−1 cm−1; 1H NMR (400 MHz, CDCl3): δ 8.82 (s,1H, 1 × CH=N), 8.07 (d, J = 8.4 Hz, 1H, Ar), 7.18 (d,J = 8.8 Hz, 2H, Ar), 6.91 (d, J = 8.8 Hz, 2H, Ar), 6.56(dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H, Ar), 6.45 (d, J =2.4 Hz, 1H, Ar), 5.38 (brd, J = 4.0 Hz, 1H, olefinic), 4.66(m, 1H, 1 × CHOCO), 4.02 (m, 4H, 2 × OCH2), 2.32–2.26(m, 4H, 2 × allylic CH2), 2.03–0.67 (m, 66H, 6 × CH, 21 ×CH2, 6 × CH3); 13C NMR (100 MHz): 173.2, 163.0, 160.3,157.2, 154.4, 154.2, 146.1, 139.7, 128.5, 122.7, 122.0, 118.4,115.0, 106.1, 99.4, 73.7, 68.5, 68.2, 50.0, 42.3, 39.5, 38.2,36.6, 35.8, 34.6, 31.9, 29.2, 29.0, 28.2, 27.8, 26.0, 25.0, 23.8,22.9, 22.6, 22.5, 21.0, 14.2, 11.8; Anal. calcd. for C56H85NO5:C, 78.92; H, 10.05; N, 1.64; Found: C, 79.31; H, 10.48; N,2.03.DCS-7,10 – Cholesteryl 8-(4-((4-(hexyloxy)-2-hydroxy-
benzylidene)amino)phenoxy)octanoate: pale yellow solid;yield: 0.067 g, 73%; IR (neat): νmax in cm−1 3456, 2950,2866, 1735, 1608, 1181, 1098; UV–Vis: λmax = 348 nm,ε = 2.13×103 l mol−1 cm−1; 1H NMR (400 MHz, CDCl3):

Bull. Mater. Sci. (2020) 43:188 Page 9 of 18 188
δ 8.82 (s, 1H, 1×CH=N), 8.07 (d, J = 8.8 Hz, 1H, Ar), 7.18(d, J = 8.8 Hz, 2H, Ar), 6.91 (d, J = 9.2 Hz, 2H, Ar), 6.56(dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H, Ar), 6.45 (d, J = 2.0 Hz,1H, Ar), 5.38 (brd, J = 3.6 Hz, 1H, olefinic), 4.68 (m, 1H,1 × CHOCO), 4.03 (m, 4H, 2 × OCH2), 2.32–2.26 (m, 4H,2 × allylic CH2), 1.86–0.68 (m, 70H, 6 × CH, 23 × CH2,6 × CH3); 13C NMR (100 MHz): 173.2, 163.0, 160.3, 157.2,154.3, 146.1, 139.7, 128.5, 122.6, 122.1, 118.4, 114.9, 106.1,99.3, 73.7, 68.5, 68.2, 56.7, 56.3, 56.2, 50.0, 39.7, 39.5, 38.2,37.0, 36.8, 36.6, 36.2, 35.8, 34.7, 31.9, 29.6, 29.5, 29.4, 29.3,29.2, 29.1, 29, 28.6, 28.2, 28.0, 27.8, 26.1, 26.0, 25.9, 25.0,24.3, 23.8, 22.8, 22.7, 22.6, 21.2, 21.0, 19.3, 18.7, 14.1, 11.8;Anal. calcd. for C58H89NO5: C, 79.13; H, 10.19; N, 1.59;Found: C, 78.80; H, 10.46; N, 2.09.DCS-7,12 – Cholesteryl8-(4-((4-(dodecyloxy)-2-hydroxy-
benzylidene)amino)phenoxy)octanoate: pale yellow solid;yield: 0.076 g, 80%; IR (neat): νmax in cm−1 3456, 2931,2855, 1732, 1608, 1177, 1112; UV–Vis: λmax = 352 nm,ε = 4.64×103 l mol−1 cm−1; 1H NMR (400 MHz, CDCl3):δ 8.82 (s, 1H, 1×CH=N), 8.07 (d, J = 8.8 Hz, 1H, Ar), 7.18(d, J = 8.8 Hz, 2H, Ar), 6.91 (d, J = 8.8 Hz, 2H, Ar), 6.55(dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 1H, Ar), 6.44 (d, J = 2.4 Hz,1H, Ar), 5.38 (brd, J = 4.8 Hz, 1H, olefinic), 4.71 (m, 1H,1 × CHOCO), 4.02 (m, 4H, 2 × OCH2), 2.32–2.26 (m, 4H,2 × allylic CH2), 2.02–0.64 (m, 74H, 6 × CH, 25 × CH2,6 × CH3); 13C NMR (100 MHz): 172.9, 163.0, 160.3, 157.1,154.4, 146.2, 139.7, 128.6, 122.6, 122.2, 118.4, 114.9, 106.2,99.3, 73.9, 68.5, 68.2, 67.7, 56.7, 56.2, 50.1, 42.3, 39.8, 39.5,38.2, 37.0, 36.6, 36.2, 35.8, 34.3, 31.9, 29.7, 29.6, 29.4, 29.3,29.2, 29.1, 28.7, 28.2, 28.0, 27.8, 26.1, 26.0, 24.3, 23.8, 22.8,22.7, 22.6, 21.8, 21.0, 19.3, 18.7, 14.1, 11.9; Anal. calcd. for
C60H93NO4: C, 79.33; H, 10.32; N, 1.54; Found: C, 79.71; H,10.48; N, 1.98.
4. Results and discussion
4.1 Microscopic and calorimetric studies
Table 1 depicts the transition temperatures along with transi-tion enthalpies and phase sequences of the three (DCS-3,R,DCS-5,R and DCS-7,R) series of dimers synthesized in thisstudy. The vertical bar graphs presented in figure 1 portraynot only the phase sequences recorded during the heating–cooling cycle (figure 1a), but also depict the dependence ofthermal properties of the dimers on the number of methyleneunits (carbon atoms) both in alkoxy terminal chains and cen-tral flexible spacer (figure 1b). The data presented in table 1and figure 1 are derived from both calorimetric and micro-scopic studies. The phase transition temperatures noted fromthe peak values of the differential scanning calorimetry (DSC)thermograms of the first heating–cooling cycles at a rate of5◦C min−1 were found to be in good agreement with thosepointed out during the polarized optical microscopy (POM)study. The samples contained in clean glass cells, each pre-pared by placing a clean coverslip over a glass slide, wereused for microscopic studies; here, basically, the optical tex-tures, that are characteristic of each of the different LC phasesserving as a diagnostic technique for assigning mesophasetype, were examined. The textures obtained for the mesophaseexhibited by these mesogens, under different conditions, areshown in figure 2. The DSC experiments confirmed that all
Table 1. Transition temperatures (◦C)a and enthalpies of transitions (kJ mol−1) of DCS-3,R, DCS-5,R and DCS-7,R series of dimers.
Phase sequence
Compound Heating Cooling
DCS-3,4 Cr 113.4 (32.1) N*114.5 (0.43) I I 113.5 (0.8) N∗b
DCS-3,6 Cr 115.9 (36.6) I I 104.7 (0.8) N∗b
DCS-3,8 Cr 57.3 (21) N* 98.2 (0.9) I I 97.8 (0.8) N∗b
DCS-3,10 Cr 73.4 (31.5) N* 95.8 (0.9) I I 95.2 (0.8) N∗b
DCS-3,12 Cr 62.2 (29.8) N* 93.4 (1.5) I I 92.7 (1.3) N∗b
DCS-5,4 Cr 121.6 (53.2) I I 99.1 (1.3) N* 71 (27.4) CrDCS-5,6 Cr 85.3 (35.3) N* 93 (1.3) I I 92.2 (0.9) N∗b
DCS-5,8 Cr 84.1 (41.3) N* 88.3 (1.3) I I 87.3 (0.9) N∗b
DCS-5,10 Cr 89.7 (57.1) I I 86.2 (1.5) N∗b
DCS-5,12 Cr 64.4 (45.4) N* 82.6 (1.7) I I 82.0 (1.7) N∗b
DCS-7,4 Cr 102.6 (50.7) I I 92.0 (2.5) N* 61.3 (28.3) CrDCS-7,6 Cr 88.2 (40.7) I I 85 (2.4) N∗b
DCS-7,8 Cr 77.6 (42.6) N* 82.2 (1.6) I I 81.5 (1.3) N∗b
DCS-7,10 Cr 70.6 (44.9) N* 80 (1.8) I I 79.3 (1.5) N∗b
DCS-7,12 Cr 64.2 (47) N* 77.2 (2.1) I I 77.1 (1.9) N∗b
C, crystal; N*, chiral nematic phase; I, isotropic phase.aTransition temperatures determined by both POM and peak values of the DSC traces during the first heating/cooling cycles at a rate of 5◦C min−1.bCrystallization was not observed until −40◦C.
188 Page 10 of 18 Bull. Mater. Sci. (2020) 43:188
0
20
40
60
80
100
120
140
DCS-
3,4
DCS-
3,4
DCS-
3,6
DCS-
3,6
DCS-
3,8
DCS-
3,8
DCS-
3,10
DCS-
3,10
DCS-
3,12
DCS-
3,12
DCS-
5,4
DCS-
5,4
DCS-
5,6
DCS-
5,6
DCS-
5,8
DCS-
5,8
DCS-
5,10
DCS-
5,10
DCS-
5,12
DCS-
5,12
DCS-
7,4
DCS-
7,4
DCS-
7,6
DCS-
7,6
DCS-
7,8
DCS-
7,8
DCS-
7,10
DCS-
7,10
DCS-
7,12
DCS-
7,12
(erutarep
meTºC
)
N* Cr
4 6 8 10 1275
80
85
90
95
100
105
110
115
erutarepmeT
(°°C
)
Number of carbon atoms (n)
DCS-3,n DCS-5,n DCS-7,n
(a)
(b)
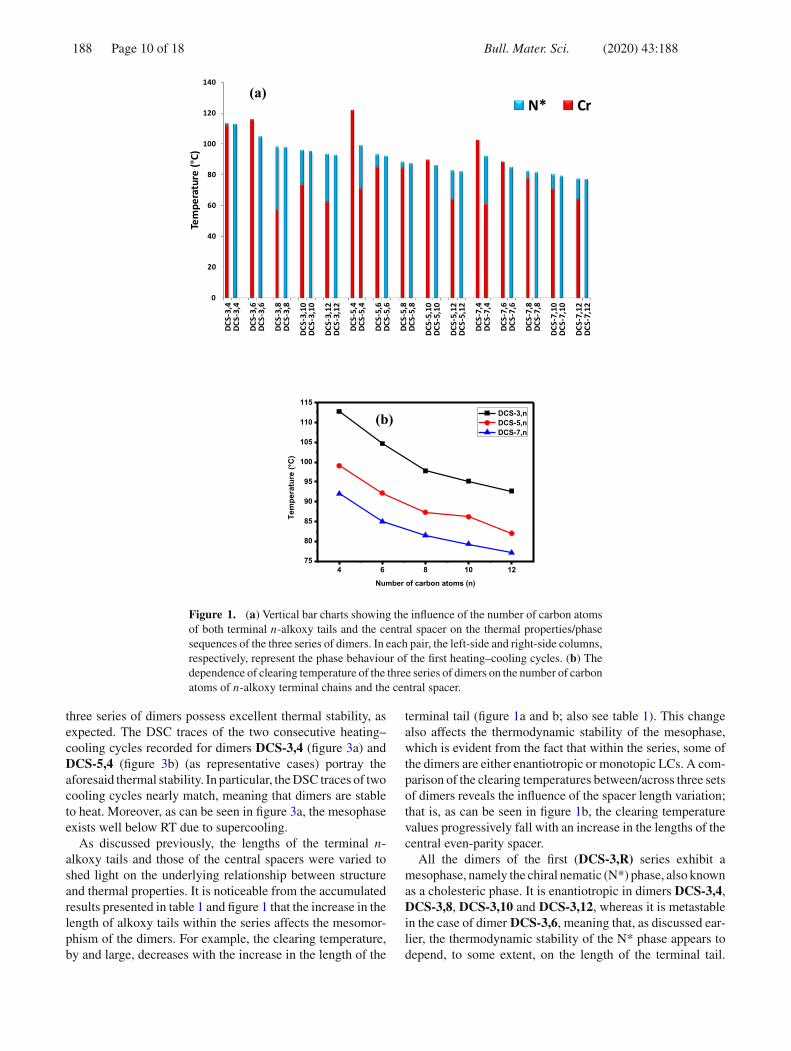
Figure 1. (a) Vertical bar charts showing the influence of the number of carbon atomsof both terminal n-alkoxy tails and the central spacer on the thermal properties/phasesequences of the three series of dimers. In each pair, the left-side and right-side columns,respectively, represent the phase behaviour of the first heating–cooling cycles. (b) Thedependence of clearing temperature of the three series of dimers on the number of carbonatoms of n-alkoxy terminal chains and the central spacer.
three series of dimers possess excellent thermal stability, asexpected. The DSC traces of the two consecutive heating–cooling cycles recorded for dimers DCS-3,4 (figure 3a) andDCS-5,4 (figure 3b) (as representative cases) portray theaforesaid thermal stability. In particular, the DSC traces of twocooling cycles nearly match, meaning that dimers are stableto heat. Moreover, as can be seen in figure 3a, the mesophaseexists well below RT due to supercooling.
As discussed previously, the lengths of the terminal n-alkoxy tails and those of the central spacers were varied toshed light on the underlying relationship between structureand thermal properties. It is noticeable from the accumulatedresults presented in table 1 and figure 1 that the increase in thelength of alkoxy tails within the series affects the mesomor-phism of the dimers. For example, the clearing temperature,by and large, decreases with the increase in the length of the
terminal tail (figure 1a and b; also see table 1). This changealso affects the thermodynamic stability of the mesophase,which is evident from the fact that within the series, some ofthe dimers are either enantiotropic or monotopic LCs. A com-parison of the clearing temperatures between/across three setsof dimers reveals the influence of the spacer length variation;that is, as can be seen in figure 1b, the clearing temperaturevalues progressively fall with an increase in the lengths of thecentral even-parity spacer.
All the dimers of the first (DCS-3,R) series exhibit amesophase, namely the chiral nematic (N*) phase, also knownas a cholesteric phase. It is enantiotropic in dimers DCS-3,4,DCS-3,8, DCS-3,10 and DCS-3,12, whereas it is metastablein the case of dimer DCS-3,6, meaning that, as discussed ear-lier, the thermodynamic stability of the N* phase appears todepend, to some extent, on the length of the terminal tail.

Bull. Mater. Sci. (2020) 43:188 Page 11 of 18 188
α
P/2
PDark line
Dark line
Dark line
Dark line
S S
2P/2 3P/2 4P/2 5P/2 6P/2
Sca�ered LightIncident Light
Sca�ered LightNo alignment- Sca�ering State(Gives focal-conic texture)
(d)
)
(f)
(a)
(b)
(c)
(e
Figure 2. Left panels: Microphotographs of the optical textures observed under different conditions for the N* phase:(a) focal conic fans growing from the isotropic melt (113.2◦C) and (b) planar texture formed when the latter, fully grownfan-like pattern of dimer DCS-3,4 is pressed gently (112◦C). (c) The GC dislocation lines observed for the N* phase ofDCS-3,12 (92◦C). Right panels: Schematic representation of ambient light incident on the focal conic texture (for thesample held between untreated glass plates) showing some light scattering (d) and the oily streak texture (for the sampleplaced between glass plates treated for planar orientation) showing selectively reflected light as well as transmission (e)Schematic diagram of the N* phase confined in a wedge cell (f).
188 Page 12 of 18 Bull. Mater. Sci. (2020) 43:188
-30 0 30 60 90 120-2
0
2
4
6
8
108 109 110 111 112 113 114 115 116-1
0
1
2
3
4
5
6
7
puodn
E)
Wm(
wol ftaeH
Temperature (°C)
Cr = CrystalN* = Chiral nematic phaseIso = Isotropic liquid state
Iso
Iso
Cr
Iso
Cr
Cr
N*
Heating Cooling Heating Coolingpu
odnE)
Wm(
wolftaeH
Temperature (°C)
Heating Cooling
N*
(a)
50 100-2
0
2
4
6
8
10
12
CrIso
Iso
N*
puodnE
)W
m(wolftae
H
Temperature (°C)
Heating Cooling
Cr
(b)
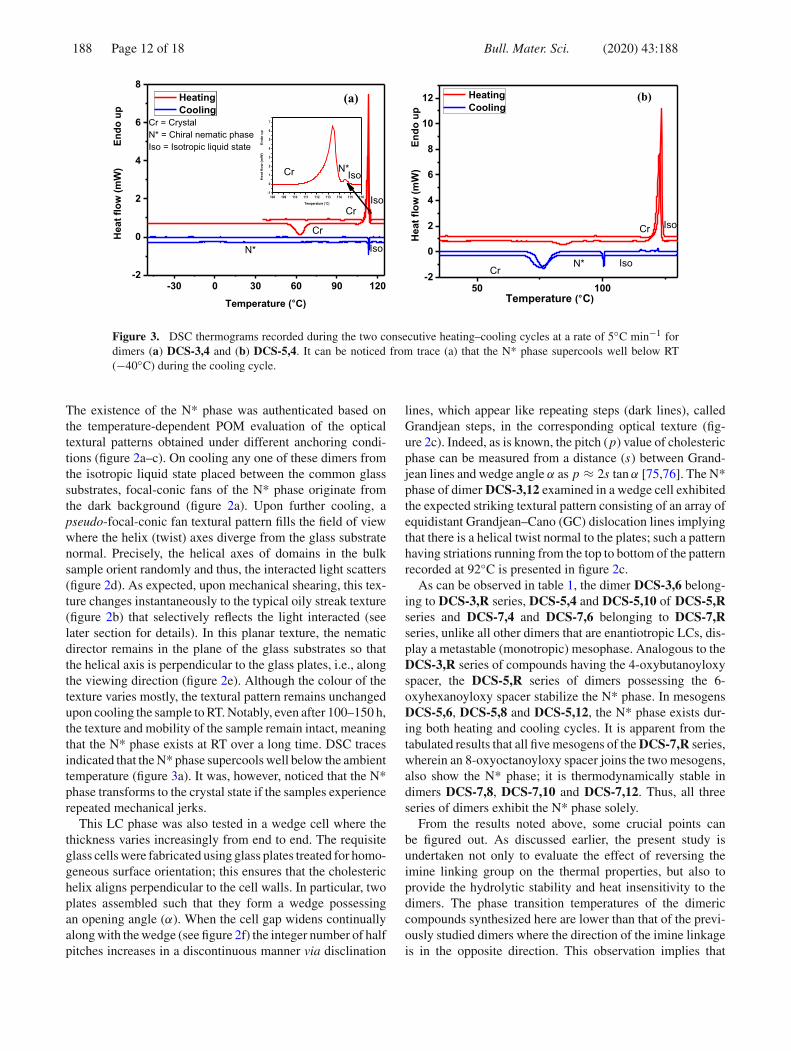
Figure 3. DSC thermograms recorded during the two consecutive heating–cooling cycles at a rate of 5◦C min−1 fordimers (a) DCS-3,4 and (b) DCS-5,4. It can be noticed from trace (a) that the N* phase supercools well below RT(−40◦C) during the cooling cycle.
The existence of the N* phase was authenticated based onthe temperature-dependent POM evaluation of the opticaltextural patterns obtained under different anchoring condi-tions (figure 2a–c). On cooling any one of these dimers fromthe isotropic liquid state placed between the common glasssubstrates, focal-conic fans of the N* phase originate fromthe dark background (figure 2a). Upon further cooling, apseudo-focal-conic fan textural pattern fills the field of viewwhere the helix (twist) axes diverge from the glass substratenormal. Precisely, the helical axes of domains in the bulksample orient randomly and thus, the interacted light scatters(figure 2d). As expected, upon mechanical shearing, this tex-ture changes instantaneously to the typical oily streak texture(figure 2b) that selectively reflects the light interacted (seelater section for details). In this planar texture, the nematicdirector remains in the plane of the glass substrates so thatthe helical axis is perpendicular to the glass plates, i.e., alongthe viewing direction (figure 2e). Although the colour of thetexture varies mostly, the textural pattern remains unchangedupon cooling the sample to RT. Notably, even after 100–150 h,the texture and mobility of the sample remain intact, meaningthat the N* phase exists at RT over a long time. DSC tracesindicated that the N* phase supercools well below the ambienttemperature (figure 3a). It was, however, noticed that the N*phase transforms to the crystal state if the samples experiencerepeated mechanical jerks.
This LC phase was also tested in a wedge cell where thethickness varies increasingly from end to end. The requisiteglass cells were fabricated using glass plates treated for homo-geneous surface orientation; this ensures that the cholesterichelix aligns perpendicular to the cell walls. In particular, twoplates assembled such that they form a wedge possessingan opening angle (α). When the cell gap widens continuallyalong with the wedge (see figure 2f) the integer number of halfpitches increases in a discontinuous manner via disclination
lines, which appear like repeating steps (dark lines), calledGrandjean steps, in the corresponding optical texture (fig-ure 2c). Indeed, as is known, the pitch (p) value of cholestericphase can be measured from a distance (s) between Grand-jean lines and wedge angle α as p ≈ 2s tan α [75,76]. The N*phase of dimer DCS-3,12 examined in a wedge cell exhibitedthe expected striking textural pattern consisting of an array ofequidistant Grandjean–Cano (GC) dislocation lines implyingthat there is a helical twist normal to the plates; such a patternhaving striations running from the top to bottom of the patternrecorded at 92◦C is presented in figure 2c.
As can be observed in table 1, the dimer DCS-3,6 belong-ing to DCS-3,R series, DCS-5,4 and DCS-5,10 of DCS-5,Rseries and DCS-7,4 and DCS-7,6 belonging to DCS-7,Rseries, unlike all other dimers that are enantiotropic LCs, dis-play a metastable (monotropic) mesophase. Analogous to theDCS-3,R series of compounds having the 4-oxybutanoyloxyspacer, the DCS-5,R series of dimers possessing the 6-oxyhexanoyloxy spacer stabilize the N* phase. In mesogensDCS-5,6, DCS-5,8 and DCS-5,12, the N* phase exists dur-ing both heating and cooling cycles. It is apparent from thetabulated results that all five mesogens of theDCS-7,R series,wherein an 8-oxyoctanoyloxy spacer joins the two mesogens,also show the N* phase; it is thermodynamically stable indimers DCS-7,8, DCS-7,10 and DCS-7,12. Thus, all threeseries of dimers exhibit the N* phase solely.
From the results noted above, some crucial points canbe figured out. As discussed earlier, the present study isundertaken not only to evaluate the effect of reversing theimine linking group on the thermal properties, but also toprovide the hydrolytic stability and heat insensitivity to thedimers. The phase transition temperatures of the dimericcompounds synthesized here are lower than that of the previ-ously studied dimers where the direction of the imine linkageis in the opposite direction. This observation implies that

Bull. Mater. Sci. (2020) 43:188 Page 13 of 18 188
the orientation/direction of the imine linkage influences thedipole moment/polarizability and perhaps the geometricalconformation of the dimeric compounds.
4.2 Selective reflection experiments
The N* phase possesses a helical macroscopic structure wherethe preferred direction of the long molecular axis (calledthe director) remains normal to the helix axis. The helicaltwist can be either right-handed or left-handed that primarilydepends on the stereochemical structure/absolute configura-tion of the constituent chiral mesogens of the phase. The pitch(p) of the helix, which is the distance over which the direc-tor rotates by 360◦, is temperature dependent. The N* phasescatters or reflects the incident white light through Braggreflection as the p of the helix is comparable with the wave-length of light falling within the visible range. The light withthe wavelength of λ = np (where n is the mean refractiveindex of the LC phase) is reflected selectively, and light ofother wavelengths is transmitted (figure 2e). The pitch andhence the wavelength of the reflected light (λmin) is temper-ature sensitive. In other words, the temperature dependenceof colour is an outcome of the temperature variation of thepitch of the helix that is defined by the twist elastic constants.This exceptional property of the N* phase has been used inreflective display devices and thermal sensors [77].
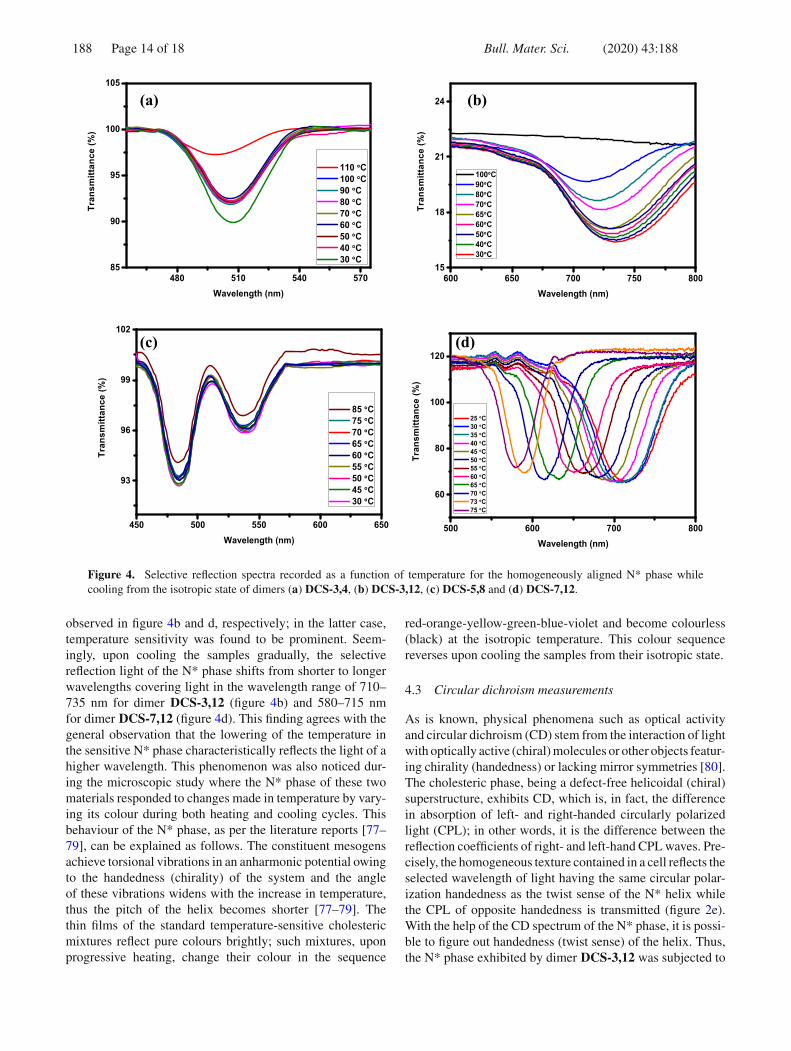
Thus, the N* phase of the randomly chosen dimeric mate-rials, viz., DCS-3,4, DCS-3,12, DCS-5,8 and DCS-7,12, wassubjected to the temperature-dependent selective reflectionstudy using a UV–Vis spectrophotometer coupled with atemperature controllable hot stage (table 2). The solid sam-ple under examination, held between two quartz substrates(2 × 2 cm2 plates), was heated to ~5◦C above the isotropiza-tion temperature, and at this juncture, the top substrate waspressed firmly not only to ensure the uniform distributionof the sample, but also to remove most of the air pockets.None of the UV–Vis spectra recorded in this liquid state fordimers DCS-3,4, DCS-3,12 and DCS-7,12 showed absorp-tion peaks, as expected. However, interestingly, the isotropicliquid state of dimer DCS-5,8 exhibited a band at around485 nm that can be ascribed to an electronic absorption band.Providing reasons for the absence of such a peak in UV–Vis profiles recorded for the isotropic state of other dimericmaterials DCS-3,4, DCS-3,12 and DCS-7,12 are difficult astheir basic molecular structures are analogous. The samplewas slowly cooled to obtain the N* phase just below theisotropic phase, and at this point, it was subjected to mechan-ical shearing to attain the Grandjean (planar) texture wherethe helix lies perpendicular to the quartz substrate and thus,the phase reflects the incident light. Figure 4a, b, c and d,respectively, depicts the spectra recorded as a function of tem-perature for compounds DCS-3,4, DCS-3,12, DCS-5,8 andDCS-7,12; these spectra characteristically comprise a Braggreflection band centred at λmin, which is related to the pitchp. These profiles show a general characteristic that beingthe peak intensity of the selective reflection phenomenon,
Table 2. Photophysical data of the cholesteric phase of dimersDCS-3,4, DCS-3,12, DCS-5,8 and DCS-7,12 derived from theirrespective UV–Vis spectra recorded as a function of temperature.
LCs Temperature (◦C) Absorption [λabs (nm)]
DCS-3,4 110 499100 50690 50580 50470 50260 50650 50540 50430 507
DCS-3,12 90 71080 71970 72365 72560 72855 73050 73340 73430 738
DCS-5,8 75 57773 59370 61665 63460 65055 66250 68145 68840 70135 70730 71025 715
DCS-7,12 85 484, 53775 486, 53770 485, 53365 484, 53555 481, 53450 484, 53845 483, 53730 481, 534
more or less, increases with a decrease in temperature. Itis perceptible from figure 4a that the change (the gradualdecrease) in temperature does not affect the selective reflec-tion light (λmin) of the N* phase stabilized by dimer DCS-3,4implying that the pitch of the phase does not diverge as afunction of temperature. Thus, in this dimer, the selectivereflection of light over the entire thermal range of the N*phase occurs at a constant wavelength. Similar behaviourwas observed for the N* phase of dimer DCS-5,8 (seefigure 4c). So far as is known, such temperature-independent(temperature-insensitive) chiral nematic materials can be usedin shear-sensitive applications [77–79].
In contrast, the N* phase of dimers DCS-3,12 and DCS-7,12 showed temperature dependence, which can be evidently
188 Page 14 of 18 Bull. Mater. Sci. (2020) 43:188
Figure 4. Selective reflection spectra recorded as a function of temperature for the homogeneously aligned N* phase whilecooling from the isotropic state of dimers (a) DCS-3,4, (b) DCS-3,12, (c) DCS-5,8 and (d) DCS-7,12.
observed in figure 4b and d, respectively; in the latter case,temperature sensitivity was found to be prominent. Seem-ingly, upon cooling the samples gradually, the selectivereflection light of the N* phase shifts from shorter to longerwavelengths covering light in the wavelength range of 710–735 nm for dimer DCS-3,12 (figure 4b) and 580–715 nmfor dimer DCS-7,12 (figure 4d). This finding agrees with thegeneral observation that the lowering of the temperature inthe sensitive N* phase characteristically reflects the light of ahigher wavelength. This phenomenon was also noticed dur-ing the microscopic study where the N* phase of these twomaterials responded to changes made in temperature by vary-ing its colour during both heating and cooling cycles. Thisbehaviour of the N* phase, as per the literature reports [77–79], can be explained as follows. The constituent mesogensachieve torsional vibrations in an anharmonic potential owingto the handedness (chirality) of the system and the angleof these vibrations widens with the increase in temperature,thus the pitch of the helix becomes shorter [77–79]. Thethin films of the standard temperature-sensitive cholestericmixtures reflect pure colours brightly; such mixtures, uponprogressive heating, change their colour in the sequence
red-orange-yellow-green-blue-violet and become colourless(black) at the isotropic temperature. This colour sequencereverses upon cooling the samples from their isotropic state.
4.3 Circular dichroism measurements
As is known, physical phenomena such as optical activityand circular dichroism (CD) stem from the interaction of lightwith optically active (chiral) molecules or other objects featur-ing chirality (handedness) or lacking mirror symmetries [80].The cholesteric phase, being a defect-free helicoidal (chiral)superstructure, exhibits CD, which is, in fact, the differencein absorption of left- and right-handed circularly polarizedlight (CPL); in other words, it is the difference between thereflection coefficients of right- and left-hand CPL waves. Pre-cisely, the homogeneous texture contained in a cell reflects theselected wavelength of light having the same circular polar-ization handedness as the twist sense of the N* helix whilethe CPL of opposite handedness is transmitted (figure 2e).With the help of the CD spectrum of the N* phase, it is possi-ble to figure out handedness (twist sense) of the helix. Thus,the N* phase exhibited by dimer DCS-3,12 was subjected to
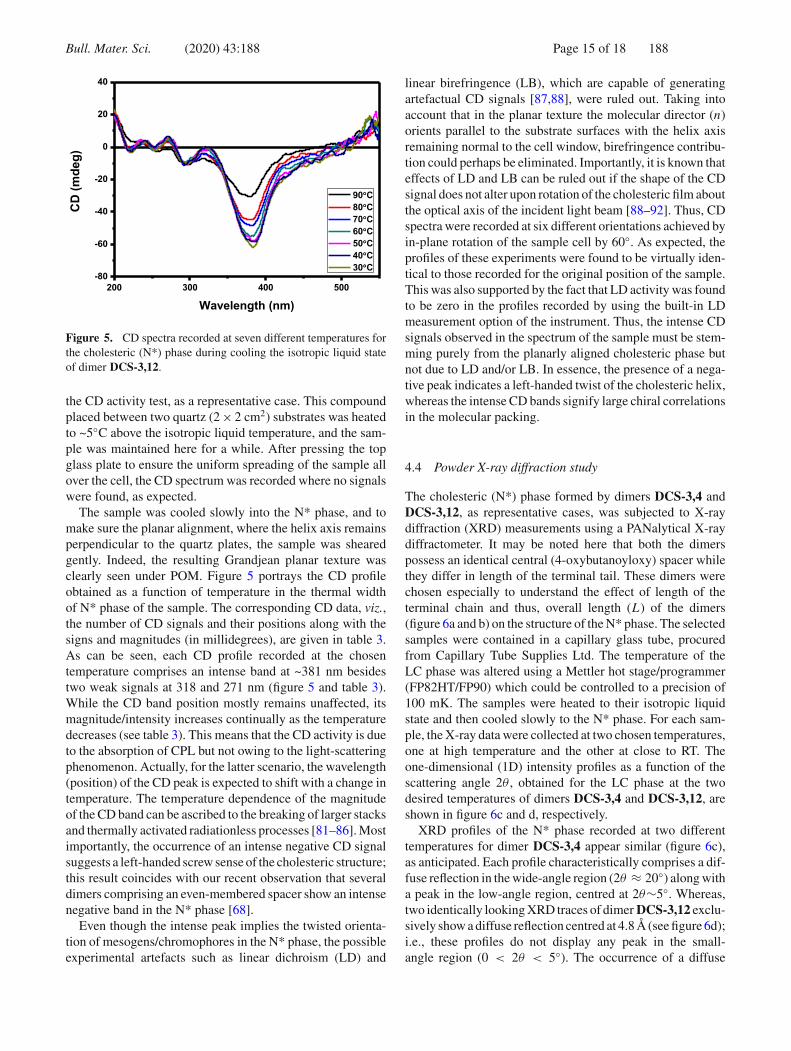
Bull. Mater. Sci. (2020) 43:188 Page 15 of 18 188
Figure 5. CD spectra recorded at seven different temperatures forthe cholesteric (N*) phase during cooling the isotropic liquid stateof dimer DCS-3,12.
the CD activity test, as a representative case. This compoundplaced between two quartz (2 × 2 cm2) substrates was heatedto ~5◦C above the isotropic liquid temperature, and the sam-ple was maintained here for a while. After pressing the topglass plate to ensure the uniform spreading of the sample allover the cell, the CD spectrum was recorded where no signalswere found, as expected.
The sample was cooled slowly into the N* phase, and tomake sure the planar alignment, where the helix axis remainsperpendicular to the quartz plates, the sample was shearedgently. Indeed, the resulting Grandjean planar texture wasclearly seen under POM. Figure 5 portrays the CD profileobtained as a function of temperature in the thermal widthof N* phase of the sample. The corresponding CD data, viz.,the number of CD signals and their positions along with thesigns and magnitudes (in millidegrees), are given in table 3.As can be seen, each CD profile recorded at the chosentemperature comprises an intense band at ~381 nm besidestwo weak signals at 318 and 271 nm (figure 5 and table 3).While the CD band position mostly remains unaffected, itsmagnitude/intensity increases continually as the temperaturedecreases (see table 3). This means that the CD activity is dueto the absorption of CPL but not owing to the light-scatteringphenomenon. Actually, for the latter scenario, the wavelength(position) of the CD peak is expected to shift with a change intemperature. The temperature dependence of the magnitudeof the CD band can be ascribed to the breaking of larger stacksand thermally activated radiationless processes [81–86]. Mostimportantly, the occurrence of an intense negative CD signalsuggests a left-handed screw sense of the cholesteric structure;this result coincides with our recent observation that severaldimers comprising an even-membered spacer show an intensenegative band in the N* phase [68].
Even though the intense peak implies the twisted orienta-tion of mesogens/chromophores in the N* phase, the possibleexperimental artefacts such as linear dichroism (LD) and
linear birefringence (LB), which are capable of generatingartefactual CD signals [87,88], were ruled out. Taking intoaccount that in the planar texture the molecular director (n)orients parallel to the substrate surfaces with the helix axisremaining normal to the cell window, birefringence contribu-tion could perhaps be eliminated. Importantly, it is known thateffects of LD and LB can be ruled out if the shape of the CDsignal does not alter upon rotation of the cholesteric film aboutthe optical axis of the incident light beam [88–92]. Thus, CDspectra were recorded at six different orientations achieved byin-plane rotation of the sample cell by 60◦. As expected, theprofiles of these experiments were found to be virtually iden-tical to those recorded for the original position of the sample.This was also supported by the fact that LD activity was foundto be zero in the profiles recorded by using the built-in LDmeasurement option of the instrument. Thus, the intense CDsignals observed in the spectrum of the sample must be stem-ming purely from the planarly aligned cholesteric phase butnot due to LD and/or LB. In essence, the presence of a nega-tive peak indicates a left-handed twist of the cholesteric helix,whereas the intense CD bands signify large chiral correlationsin the molecular packing.
4.4 Powder X-ray diffraction study
The cholesteric (N*) phase formed by dimers DCS-3,4 andDCS-3,12, as representative cases, was subjected to X-raydiffraction (XRD) measurements using a PANalytical X-raydiffractometer. It may be noted here that both the dimerspossess an identical central (4-oxybutanoyloxy) spacer whilethey differ in length of the terminal tail. These dimers werechosen especially to understand the effect of length of theterminal chain and thus, overall length (L) of the dimers(figure 6a and b) on the structure of the N* phase. The selectedsamples were contained in a capillary glass tube, procuredfrom Capillary Tube Supplies Ltd. The temperature of theLC phase was altered using a Mettler hot stage/programmer(FP82HT/FP90) which could be controlled to a precision of100 mK. The samples were heated to their isotropic liquidstate and then cooled slowly to the N* phase. For each sam-ple, the X-ray data were collected at two chosen temperatures,one at high temperature and the other at close to RT. Theone-dimensional (1D) intensity profiles as a function of thescattering angle 2θ , obtained for the LC phase at the twodesired temperatures of dimers DCS-3,4 and DCS-3,12, areshown in figure 6c and d, respectively.
XRD profiles of the N* phase recorded at two differenttemperatures for dimer DCS-3,4 appear similar (figure 6c),as anticipated. Each profile characteristically comprises a dif-fuse reflection in the wide-angle region (2θ ≈ 20◦) along witha peak in the low-angle region, centred at 2θ∼5◦. Whereas,two identically looking XRD traces of dimer DCS-3,12 exclu-sively show a diffuse reflection centred at 4.8 Å (see figure 6d);i.e., these profiles do not display any peak in the small-angle region (0 < 2θ < 5◦). The occurrence of a diffuse

188 Page 16 of 18 Bull. Mater. Sci. (2020) 43:188
Table 3. CD spectroscopic data derived from the CD spectra of dimer DCS-3,12.
CD
Mesogens LC phase Temperature (◦C) λmax (nm) CD (mdeg)
DSC-3,12 N* 90 379, 317, 270 −30.5, −6.9, −1.380 379, 318, 271 −44.6, −4.3, 3.170 382, 319, 272 −45.3, −4.1, 3.460 380, 320, 272 −55.6, −1.8, 4.250 383, 321, 270 −57.6, −0.09, 5.140 384, 318, 270 −61.7, −0.05, 6.630 383, 317, 271
Figure 6. Energy optimized space-filled molecular models of dimers (a) DCS-3,4 and (b) DCS-3,12. 1D intensity vs.2θ profiles of the N* phase exhibited by dimers (c) DCS-3,4 and (d) DCS-3,12.
reflection in the wide-angle region of all four scans impliesthe liquid-like ordering of the constituent mesogens in thedirection perpendicular to the chiral nematic director. Thepresence of a diffuse peak in the wide-angle region is indeedexpected of the N* mesophase. However, the presence of a rel-atively sharp but weak (low intensity) peak in the small-anglearea of the aforementioned XRD profiles of dimer DCS-3,4has been argued to be characteristic of the weak short-rangeorder of the smectic kind in the nematic/cholesteric phase[93,94]. Considering the fact that the spacing correspond-ing to the low-angle peak is ~16 Å, and the length of dimerDCS-3,4 in its all-trans conformation is 38.5 Å (figure 6a),the observations suggest that the short-range ordering couldbe arising from intercalated smectic regions having a short-range correlation. Foregoing observations clearly indicate thatthe length of the terminal chains dictates the structure ofthe N* phase where the cybotactic order is either present orabsent.
Apparently, in this study, we have made an attempt tounderstand the effect of the lengths of even-membered spac-ers and those of the terminal chains on the thermal propertiesof three series of new non-symmetric dimers comprisingcholesterol and SAN core. In each series, the length of aneven-spacer has been held constant while that of the termi-nal n-alkoxy tails has been varied. In particular, although theterminal chains such as n-butyloxy, n-hexyloxy, n-octyloxy,n-decyloxy and n-dodecyloxy tails have been attached tothe SAN mesogen, three even-parity spacers such as 8-oxyoctanoyloxy, 6-oxyhexanoyloxy and 4-oxybutanoyloxyspacers have been used to join the two mesogenic segmentscovalently. The results of our systematic studies, as presentedin detail in previous sections, clearly reveal the followingimportant facts:
• The lengths of both central spacer and terminal tail influ-ence the clearing temperature and hence, the thermal range

Bull. Mater. Sci. (2020) 43:188 Page 17 of 18 188
of the cholesteric phase formed by the newly synthesizeddimers.
• The reversal of imine linkage of SAN segment not onlylowers the transition temperatures, but also eliminates therich mesomorphism of dimers. This means that reversingthe imine linkage decreases anisotropy of electronic polar-izability, which leads to a decrease in molecular (van derWaals) interactions; indeed, such weak molecular interac-tions disfavour liquid crystal behaviour of mesogens.
5. Conclusion
In this paper, we report on the synthesis, molecular structuralcharacterization and thermal behaviour of three different setsof non-symmetric dimers derived from cholesterol. They areprepared by covalently joining cholesterol and SAN (witha reverse imine linkage) core through an ω-oxyalkanoyloxyspacer. The lengths of the central flexible spacer and that ofthe terminal tail attached to the SAN core have been variedto uncover the structure–property correlations. Interestingly,although the analogously known mesogens exhibit multipleLC phases, these newly synthesized dimers exclusively showa mesophase that has been identified as a chiral nematic (N*)phase. These observations demonstrate the effect of reversingthe imine linkage on the LC behaviour of SAN-based dimericmesogens derived from cholesterol. The N* phase, which iswell recognized for its technological potential and implica-tions, not only exists over a wide thermal range in almost allthe materials realized, but also supercooled well below RT.The clearing temperatures, and thus the thermal range of theLC phase of these dimers show dependence on the lengths ofboth spacer and terminal chain. Notably, the planarly orientedcholesteric thin films of some dimers show the temperature-dependent selective reflection of light according to the Bragglaw, as expected; however, in some cases, the reflection of lightoccurs at a constant wavelength over the entire thermal rangeof the phase. This observation implies that the lengths of thespacer and/or terminal chain govern the selective reflectionproperty of the N* phase. The chiroptical investigation usingCD spectroscopy not only confirmed the helicoidal structureformally, but also indicated the left-handed screw sense ofthe cholesteric structure formed by a sample. Overall, thisstudy demonstrates the effect of reversing the imine linkageas well as variations in the lengths of both terminal tail andspacer on the thermal behaviour of cholesterol-based dimerscomprising the SAN-mesogenic core.
Acknowledgements
CVY gratefully acknowledges the Science and EngineeringResearch Board (SERB), Department of Science and Tech-nology, Government of India, for providing funds to this studythrough a research Project No. EMR/2017/000153.
References
[1] Dominiak P M, Grech E, Barr G, Tear S, Mallinson P andWozniak K 2003 Chem. Eur. J. 9 963
[2] Krygowski T M, Zachara-Horeglad J E, Palusiak M, Pelloni Sand Lazzeretti P 2008 J. Org. Chem. 73 2138
[3] Hadjoudis E and Mavridis I M 2004 Chem. Soc. Rev. 33 579[4] Harada J, Uekusa H and Ohashi Y 1999 J. Am. Chem. Soc. 121
5809[5] Samat A and Lokshin V 1992 in Guglielmetti R and Crano J C
J (eds) Organic photochromic and thermochromic compounds(New York: Kluwer Academic) vol 2 p 415
[6] Nassau K 2001 The physics and chemistry of color 2nd ed.(New York: Wiley)
[7] Durr H and Bouas-Laurent H (eds) 2003 Photochromism.Molecules and Systems rev. ed. (Amsterdam: Elsevier)
[8] Serrano J L (ed) 1998 Metallomesogens (synthesis, proper-ties and applications) (Weinheim: Wiley-VCH) and referencescited therein
[9] Yelamaggad C V, Nagamani S A, Nair G G, Rao D S S, PrasadS K and Jakli A 2002 Liq. Cryst. 29 1181
[10] Amaranatha Reddy R and Tschierske C 2006 J. Mater.Chem. 16 907
[11] Yelamaggad C V, Hiremath U S, Nagamani S A, Rao D S Sand Prasad S K 2001 J. Mater. Chem. 11 1818
[12] Walba D M, Korblova E, Shao R and Clark N A 2001 J. Mater.Chem. 11 2743
[13] Yelamaggad C V, Mathews M, Nagamani S A, Rao D S S,Prasad S K, Findeisen S et al 2007 J. Mater. Chem. 17 284
[14] Tandel S F, Weissflog W, Baumeister U, Pelzl G, Shreeni-vasa Murthy H N and Yelamaggad C V 2012 Beilstein J. Org.Chem. 8 129
[15] Ostrovskii B I, Rabinovich A Z, Sonin A S, Sorkin E L, StrukovB A and Taraskin S A 1980 Ferroelectrics 24 309
[16] Hallsby A, Nilsson M and Otterholm B 1982 Mol. Cryst. Liq.Cryst. 82 61
[17] Otterholm B, Nilsson M, Lagerwall S T and Skarp K 1987 Liq.Cryst. 2 757
[18] Soto Bustamante E A, Yablonskii S V, Ostrovskii B I, BeresnevL A, Blinov L M and Haase W 1996 Liq. Cryst. 21 829
[19] Veerabhadraswamy B N, Shankar Rao D S and YelamaggadC V 2018 Chem. Asian J. 13 1012
[20] Veerabhadraswamy B N, Shankar Rao D S and YelamaggadC V 2015 J. Phys. Chem. B 119 4539
[21] Padmini V, Nani Babu P, Nair G G, Shankar Rao D S andYelamaggad C V 2016 Chem. Asian J. 11 2897
[22] Yelamaggad C V, Hiremath U S and Shankar Rao D S 2001Liq. Cryst. 28 351
[23] Yelamaggad C V, Hiremath U S, Anitha Nagamani S, ShankarRao D S and Krishna Pasad S 2003 Liq. Cryst. 30 681
[24] Yelamaggad C V, Mathews M, Hiremath U S, Rao D S S andPrasad S K 2005 Tetrahedron Lett. 46 2623
[25] Yelamaggad C V, Shashikala I, Rao D S S, Nair G G and PrasadS K 2006 J. Mater. Chem. 16 4099
[26] Yelamaggad C V, Shashikala I, Shankar Rao D S, Nair G Gand Prasad S K 2008 J. Mater. Chem. 18 2096
[27] Yelamaggad C V, Prasad S K, Nair G G, Shashikala I, Rao DS S, Lobo C V et al 2004 Angew. Chem. Int. Ed. 43 3429
[28] Yelamaggad C V, Shashikala I S, Liao G, Rao D S S, Prasad SK, Li Q et al 2006 Chem. Mater. 18 6100

188 Page 18 of 18 Bull. Mater. Sci. (2020) 43:188
[29] Liao G, Shashikala I, Yelamaggad C V, Shankar Rao D S,Krishna Prasad S and Jakli A 2006 Phys. Rev. E 73 051701
[30] Sarkar D D, Deb R, Chakraborty N, Mohiuddin G, Kanti NathR and Rao N V S 2013 Liq. Cryst. 40 468
[31] Yelamaggad C V, Shanker G, Hiremath Uma S and KrishnaPrasad S 2008 J. Mater. Chem. 18 2927
[32] Imrie C T and Henderson P A 2007 Chem. Soc. Rev. 36 2096[33] Pal S K and Kumar S 2017 Liquid crystal dimers (Cambridge:
Cambridge University Press)[34] Hardouin F, Achard M F, Jin J-I, Shin J W and Yun Y K 1994
J. Phys. II Fr. 4 627[35] Cha S-W, Jin J-I, Laguerre M, Achard M F and Hardouin F
1999 Liq. Cryst. 26 1325[36] Cha S-W, Jin J-I, Achard M F and Hardouin F 2002 Liq.
Cryst. 29 755[37] Lee J-W, Park Y, Jin J-I, Achard M F and Hardouin F 2003 J.
Mater. Chem. 13 1367[38] Kim K-N, Do E-D, Kwon Y-W and Jin J-I 2005 Liq. Cryst. 32
229[39] Surendranath V 1999 Mol. Cryst. Liq. Cryst. 332 135[40] Yelamaggad C V 1999 Mol. Cryst. Liq. Cryst. 326 149[41] Rao D S S, Prasad S K, Raja V N, Yelamaggad C V and Naga-
mani S A 2001 Phys. Rev. Lett. 87 085540[42] Yelamaggad C V, Shashikala I, Hiremath U S, Shankar Rao D
S and Prasad S K 2007 Liq. Cryst. 34 799[43] Pandeya V S, Dhar R A, Singha K R, Achalkumar A S and
Yelamaggad C V 2010 Phase Transit. 83 1049[44] Hiremath Uma S, Sonar Girija M, Shankar Rao D S and Yela-
maggad C V 2011 J. Mater. Chem. 21 4064[45] Pandey A S, Dhar R, Achalkumar A S and Yelamaggad C V
2011 Liq. Cryst. 38 775[46] Marcelis A T M, Koudijs A, Karczmarzyk Z and Sudholter E
J R 2003 Liq. Cryst. 30 1357[47] Shanker G and Yelamaggad C V 2012 New J. Chem. 36 918[48] Marcelis A T M, Koudijs A and Sudholter E J R 2000 Liq.
Cryst. 27 1515[49] Marcelis A T M, Koudijs A and Sudholter E J R 2004 Mol.
Cryst. Liq. Cryst. 411 193[50] Donaldson T, Staesche H, Lu Z B, Henderson P A, Achard M
F and Imrie C T 2010 Liq. Cryst. 37 1097[51] Ewa G, Vaupotic N, Zep A, Pociecha D, Yoshioka J, Yamamoto
J et al 2015 Angew. Chem. Int. Ed. 54 10155[52] Ajay Mallia V and Tamaoki N 2004 Chem. Commun. 24 2538[53] Ajay Mallia V and Tamaoki N 2003 J. Mater. Chem. 13 219[54] Tamaoki N, Aoki Y, Moriyama M and Kidowaki M 2003Chem.
Mater. 15 719[55] Tamaoki N, Purfenov A V, Masaki A and Matsuda H 1997 Adv.
Mater 9 1102[56] Tian Y, Xu X, Zhao Y, Tang X and Li T 1997 Liq. Cryst. 22 87[57] Zep A, Aya S, Aihara K, Ema K, Pociecha D, Madrak K et al
2013 J. Mater. Chem. C 1 46[58] Aya S, Zep A, Aihara K, Ema K, Pociecha D, Gorecka E et al
2014 Opt. Mater. Express 4 662[59] Wu C 2007 Mater. Lett. 61 1380[60] Zhang C, Jin L, Yin B, Jamil M and Jeon Y-J 2008Liq. Cryst. 35
39[61] Abraham S, Ajay Mallia V, Ratheesh K V, Tamaoki N and Das
S 2006 J. Am. Chem. Soc. 128 7692
[62] Chan T-N, Lu Z, Yam W-S, Yeap G-Y and Imrie C T 2012 Liq.Cryst. 39 393
[63] Majumdar K C, Mondal S, Pal N and Sinha R K 2009 Tetra-hedron Lett. 50 1992
[64] Shanker G, Bindushree A, Chaithra K, Pratap P, Gupta R K,Achalkumar A S et al 2019 J. Mol. Liq. 275 849
[65] Majumdar K C, Chakravorty S, Pal S N and Sinha R K 2009Tetrahedron 65 7998
[66] Lee H-C, Lu Z, Henderson P A, Achard M F, Mahmood K,Yeap W A et al 2012 Liq. Cryst. 39 259
[67] Majumdar K C, Ghosh T, Rao D S S and Prasad S K 2011 Liq.Cryst. 38 1269
[68] Nayak R, Bhat S A, Shanker G and Yelamaggad C V 2019NewJ. Chem. 43 2148
[69] Majumdar K C, Shyam P K, Shankar Rao D S and KrishnaPrasad S 2011 J. Mater. Chem. 21 556
[70] Pradhan B, Chakraborty N, Gupta R K, Shanker G andAchalkumar A S 2017 New J. Chem. 41 879
[71] Van Deun R, Parac-Vogt T N, Van Hecke K, Van Meervelt L,Binnemans K, Guillon D et al 2003 J. Mater. Chem. 13 1639
[72] Consiglio G, Failla S, Finocchiaro P, Oliverib I P and Di BellaS 2012 Dalton Trans. 41 387
[73] Chen L N, Kuo C C, Chiu L C and Chen W-C 2014 RSC Adv. 445345
[74] Hamley I W, Castelletto V, Parras P, Lu Z B, Imrie C T andItoh T 2005 Soft Matter 1 355
[75] Dierking I 2003 Textures of liquid crystals (Weinheim: Wiley-VCH) p 78
[76] Dierking I 2014 Symmetry 6 444[77] Bahadur B (ed) 1991 Liquid crystals – applications and uses,
Vol III (Singapore: World Scientific Publ. Co.) p 76[78] Keating P N 1969 Mol. Cryst. 8 315[79] Parmar D S and Singh J J 1992 Appl. Phys. Lett. 61 2039[80] Khanikaev A B, Arju N, Fan Z, Purtseladze D, Lu F, Lee J et al
2016 Nat. Commun. 7 12045[81] Yelamaggad C V, Achalkumar A S, Rao D S S and Prasad S K
2004 J. Am. Chem. Soc. 126 6506[82] Yelamaggad C V, Achalkumar A S, Rao D S S and Prasad S K
2007 J. Mater. Chem. 17 4521[83] Yelamaggad C V, Achalkumar A S, Rao D S S and Prasad S K
2007 J. Org. Chem. 72 8308[84] Yelamaggad C V, Achalkumar A S, Rao D S S and Prasad S K
2009 J. Org. Chem. 74 3168[85] Achalkumar A S, Hiremath U S, Rao D S S, Prasad S K and
Yelamaggad C V 2013 J. Org. Chem. 78 527[86] Achalkumar A S, Veerabhadraswamy B N, Hiremath U S, Rao
D S S, Prasad S K and Yelamaggad C V 2016 Dyes Pigm. 132291
[87] Schellman J and Jensen H P 1987 Chem. Rev. 87 1359[88] Disch R L and Sverdlik D I 1969 Anal. Chem. 41 82[89] Bjiirling S C, Coldbeck R A, Milder S J, Randall C E, Lewis
J W and Kliger D S 1991 J. Phys. Chem. 95 4685[90] Jane M B, Schneider T and Maestre I F 1970 J. Mol. Biol. 52
521[91] Narushima T and Okamoto H 2016 Sci. Rep. 6 35731[92] Freudenthal J H, Hollis E and Kahr B 2009 Chirality 21 E20[93] de Vries A 1970 Mol. Cryst. Liq. Cryst. 10 219[94] de Vries A 1975 J. Phys. Colloq. 36 C1-1