00_07_verrerie_appareils_bpl_36p
TRANSCRIPT

RD-MB 1
LA VERRERIE VOLUMETRIQUE 1-La prise d'essai est placée dans un erlenmeyer (dosages volumétriques simples) ou dans un bécher (dosages avec électrodes) préalablement rincé à l'eau déminéralisée. 2-La verrerie graduée ou jaugée : a-Généralités : -Classe A ou B : tolérance garantie par le constructeur. A est meilleure que B. exemple : pipette 10 cm3 , tolérance : 0,020 cm3 classe A, 0,070 cm3 classe B. Souvent les tolérances B ne sont pas indiquées. -Température d'utilisation : en général les instruments sont calibrés à 20°C. -Viscosité du liquide mesuré : en général le volume indiqué correspond à la viscosité d'une solution aqueuse. -Mode de jaugeage : EX : le volume indiqué est le volume obtenu par écoulement (la prise est donc légèrement supérieure). IN : le volume indiqué est le volume interne (par vidange on obtient donc un volume inférieur). Souvent les fioles jaugées sont IN , les pipettes et les burettes sont EX. b-Pipette jaugée :
Elle est destinée à délivrer un volume déterminé de liquide, par exemple 10,00 mL : 10,00 mL est le volume d’eau qui à 20°C est délivré lorsqu’elle se vide du trait repère supérieur au trait repère inférieur. matériel nécessaire à l’utilisation de la pipette : erlenmeyer, bécher, pissette à eau déminéralisée. rinçages du matériel :
eau ordinaire tout. eau déminéralisée tout. solution rincer le bécher à la solution et y introduire env. 15 à 20 mL de solution. rincer la pipette par aspiration (plonger d’environ 1 cm la pointe de la pipette dans la solution, aspirer lentement tout en maintenant la pipette verticale le long de la paroi du bécher, laisser monter le liquide jusqu’à moitié du réservoir, boucher l’orifice supérieur de la pipette avec l’index et la sortir de la solution, incliner la pipette pour amener le liquide au-dessus de la boule de sécurité, se mettre au-dessus de l’évier et laisser s’écouler le liquide, recommencer l’expérience deux fois). vider le contenu du bécher et y introduire env. 40 mL de solution.
remplissage et vidange de la pipette :
Se mettre en position (1), aspirer lentement la solution jusqu’à ce que le niveau soit compris entre le trait repère supérieur et la boule de sécurité, essuyer la pointe.
Se mettre en position (2), laisser s’écouler la solution, faire l’ajustement au trait repère supérieur (oeil en face). Remettre la pointe de la pipette dans la solution et la retirer doucement sans secousse.
Transporter la pipette au-dessus de l’erlenmeyer, se mettre en position (2), laisser s’écouler le liquide jusqu’au trait repère inférieur (oeil en face).
Vider dans l’évier ou un bécher « poubelle » le reste de solution contenue dans la pipette.
Dans le cas d’une pipette à un seul trait, laisser la pipette se vider librement sans chercher à récupérer ce qui reste dans la pipette.
fin d’utilisation :
Tout rincer à l’eau ordinaire et à l’eau déminéralisée. ATTENTION : Utiliser systématiquement une poire d’aspiration. -Tolérance : env. 0,2% pour la classe A. c-Burette :
Elle est destinée à délivrer un volume de liquide indéterminé à l’avance. Elle est constituée d’un tube calibré, muni à sa partie inférieure d’un robinet et d’une pointe capillaire épaisse, affinée à son extrémité.
Pour les burettes de 25 mL, la pointe capillaire fournit en principe deux gouttes par division, ce qui permet d’apprécier la demi-division : une lecture de volume est faite à 0,05 mL près pour les burettes au 1/10e ou 0,025 mL près pour les burettes au 1/20e.
La burette est maintenue verticale sur son support. La lubrification du robinet à l’aide de graisses spéciales doit être faite avec soin. Insuffisamment graissée, la clé peut se coincer dans le boisseau du robinet; un graissage excessif peut occasionner l’obstruction de la voie de la clé ou de la pointe capillaire.
matériel nécessaire à l’utilisation de la burette : verre à pied, bécher, vase à saturation, pissette à eau déminéralisée. rinçages du matériel :
eau ordinaire tout. eau déminéralisée bécher, vase à saturation, burette (robinet ouvert). solution rincer le bécher à la solution et y mettre env. 40 mL de cette solution. Puis la burette étant robinet ouvert, faire couler 3 mL de la solution contre la paroi de la burette, attendre que le liquide s’écoule, recommencer l’opération deux autres fois. Une fois le liquide totalement écoulé, fermer le robinet.
remplissage de la burette :

RD-MB 2
Verser la solution le long de la paroi en ayant bien soin de ne pas faire mousser le liquide, dépasser la graduation zéro. Ouvrir en grand le robinet puis le refermer afin de remplir la pointe capillaire et faire l’ajustement au zéro. Si une goutte reste accrochée, la faire partir. dosage :
Placer le vase à saturation sous la burette. Régler la hauteur de celle-ci de manière à ce que la pointe soit enfoncée d’env. 1 cm dans l’ouverture du récipient. Tenir la clé de la main gauche, la main droite tient le vase à saturation et lui imprime un mouvement de rotation afin d’assurer le mélange. Au cours d’un dosage, ne pas oublier les jets de pissette d’eau déminéralisée pour ramasser la solution projetée sur la paroi du vase à saturation. Toute goutte restant accrochée doit être éliminée en faisant entrer en contact la pointe de la burette avec le col du récipient. fin d’utilisation :
Vider complètement la burette, la rincer à l’eau ordinaire et à l’eau déminéralisée. d-Fiole jaugée :
C’est un récipient destiné à contenir un volume déterminé de liquide (indication ‘ IN ’). Elle est caractérisée par un corps en forme de poire, à fond plat, surmonté d’un col cylindrique portant un trait repère circulaire. Elle peut être munie ou non d’un bouchon rôdé.
Fiole de 100 mL : 100 mL est le volume de solution contenue dans la fiole à 20°C quand elle est remplie jusqu’au trait repère, c’est à dire lorsque le plan horizontal passant par le bord supérieur du trait est tangent au ménisque de l’eau en son point le plus bas. C’est un instrument précis. -Pour diluer un liquide : rincer à l'eau déminéralisée, verser le volume de liquide nécessaire, compléter au 2/3
avec de l'eau déminéralisée, homogénéiser sans retourner, compléter au trait de jauge (remplir d’abord rapidement la fiole avec la pissette à eau déminéralisée, s’arrêter à 0,5 cm du trait ; après avoir essuyé le col avec du papier Joseph, finir de remplir la fiole avec le compte-goutte ; ajuster au bas du ménisque) ; boucher et homogénéiser par retournement ( env. 20 fois ). -Pour préparer une solution titrée à partir d'une pesée : rincer à l'eau déminéralisée, introduire quantitativement le solide pesé à l'aide d'un entonnoir et d'eau déminéralisée sans dépasser les 2/3 , agiter jusqu'à dissolution complète du solide , compléter au trait de jauge avec de l'eau déminéralisée (remplir d’abord rapidement la fiole avec la pissette à eau déminéralisée, s’arrêter à 0,5 cm du trait ; après avoir essuyé le col avec du papier Joseph, finir de remplir la fiole avec le compte-goutte ; ajuster au bas du ménisque), boucher et homogénéiser par retournement. Ne jamais chauffer même si la dissolution est lente. Pour les dissolutions difficiles opérer d'abord dans un petit bécher puis transvaser quantitativement après refroidissement la solution obtenue dans la fiole. -Rincer à l’eau ordinaire et à l’eau déminéralisée après utilisation. -Tolérance : 0,1 à 0,2 % . e-Eprouvette graduée :
C’est un tube cylindrique muni d’un bec verseur à la partie supérieure, et à la base d’un pied pour assurer sa stabilité.
Les traits de graduation tous les ″ sont circulaires, ce qui permet d’effectuer les lectures sans erreur de parallaxe. La section des éprouvettes étant plus importante que celle des autres instruments, la précision sur le volume mesuré est moins bonne. A n'utiliser que pour des mesures de volume ne nécessitant pas de précision.
LES APPAREILS Utilisation de la balance de précision : - vérifier le niveau (horizontalité) - vérifier la propreté du plateau (pinceau) - faire les lectures portes fermées - ne pas prendre la capsule de pesée avec les doigts en
cours de pesée - après utilisation éteindre la balance, la nettoyer,
fermer les portes. Utilisation des appareils électriques : consulter la notice
- vérifier le choix des électrodes (attention aux éventuelles bulles d’air dans l’électrode de référence), leur position (ne jamais serrer sur la partie en verre, placer l’électrode de verre plus haut que l’électrode de référence, attention aux chocs avec le barreau aimanté), les connexions (il peut y avoir des problèmes de contact électrique d’où une mauvaise stabilisation des mesures)
- vérifier les réglages, le choix du calibre ; attention en conductimétrie aux positions conductivité et résistivité et en potentiométrie aux positions i = 0 et i = 1µ A
- penser à étalonner les pH-mètres - respecter les consignes (vitesse d’agitation …)
RD-MB 3
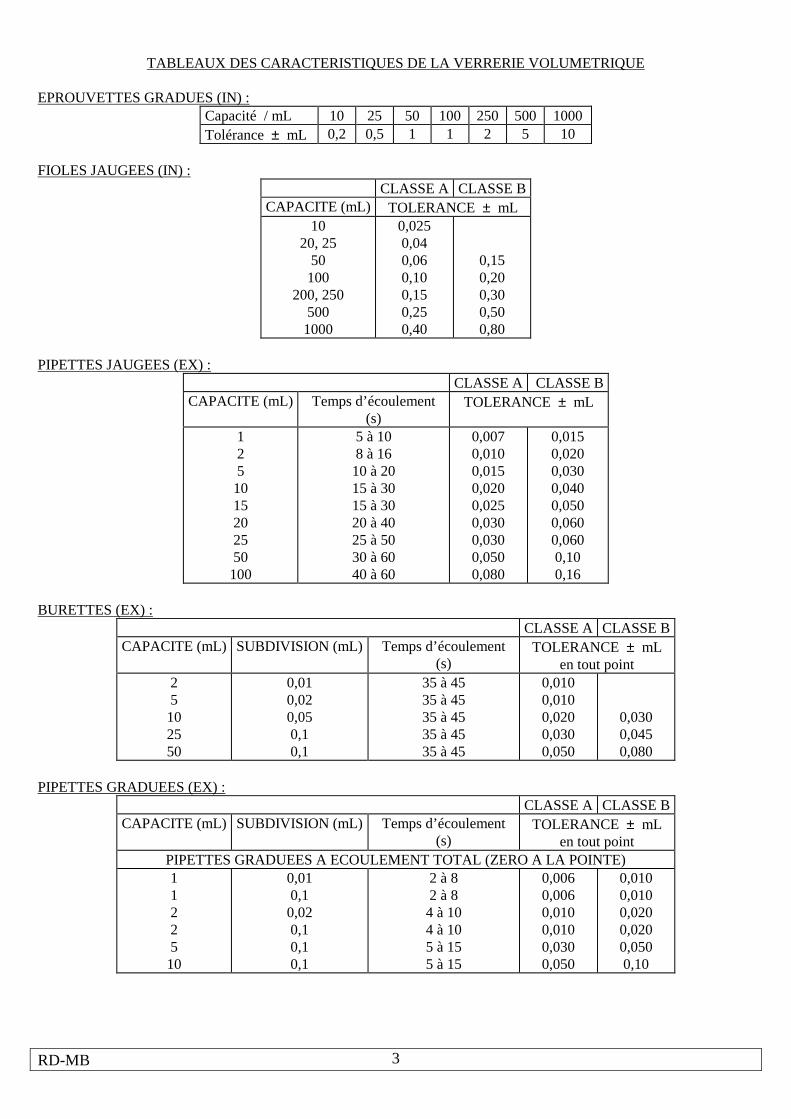
TABLEAUX DES CARACTERISTIQUES DE LA VERRERIE VOLUMETRIQUE
EPROUVETTES GRADUES (IN) :
Capacité / mL 10 25 50 100 250 500 1000 Tolérance ± mL 0,2 0,5 1 1 2 5 10
FIOLES JAUGEES (IN) :
CLASSE A CLASSE B CAPACITE (mL) TOLERANCE ± mL
10 20, 25
50 100
200, 250 500 1000
0,025 0,04 0,06 0,10 0,15 0,25 0,40
0,15 0,20 0,30 0,50 0,80
PIPETTES JAUGEES (EX) :
CLASSE A CLASSE B CAPACITE (mL) Temps d’écoulement
(s) TOLERANCE ± mL
1 2 5 10 15 20 25 50 100
5 à 10 8 à 16 10 à 20 15 à 30 15 à 30 20 à 40 25 à 50 30 à 60 40 à 60
0,007 0,010 0,015 0,020 0,025 0,030 0,030 0,050 0,080
0,015 0,020 0,030 0,040 0,050 0,060 0,060 0,10 0,16
BURETTES (EX) :
CLASSE A CLASSE B CAPACITE (mL) SUBDIVISION (mL) Temps d’écoulement
(s) TOLERANCE ± mL
en tout point 2 5 10 25 50
0,01 0,02 0,05 0,1 0,1
35 à 45 35 à 45 35 à 45 35 à 45 35 à 45
0,010 0,010 0,020 0,030 0,050
0,030 0,045 0,080
PIPETTES GRADUEES (EX) :
CLASSE A CLASSE B CAPACITE (mL) SUBDIVISION (mL) Temps d’écoulement
(s) TOLERANCE ± mL
en tout point PIPETTES GRADUEES A ECOULEMENT TOTAL (ZERO A LA POINTE) 1 1 2 2 5 10
0,01 0,1 0,02 0,1 0,1 0,1
2 à 8 2 à 8 4 à 10 4 à 10 5 à 15 5 à 15
0,006 0,006 0,010 0,010 0,030 0,050
0,010 0,010 0,020 0,020 0,050 0,10

RD-MB 4
PIPETTES GRADUEES A ECOULEMENT PARTIEL (ENTRE
TRAITS) 1 2 5 10
0,1 0,1 0,1 0,1
10 à 20 10 à 20 15 à 30 15 à 30
0,006 0,010 0,030 0,050
0,010 0,020 0,050 0,10

RD-MB 5
VERIFICATION DES APPAREILS DE VOLUMETRIE (D’après document Blaubrand juillet 2004)
Les normes DIN EN ISO 9001, DIN EN ISO 10012 et DIN EN ISO/IEC 17025 demandent, dans le cadre de la surveillance des moyens de contrôle, la connaissance et la vérification de la précision et tolérance de mesure de tous les appareils utilisés.
Il est donc obligatoire d'effectuer un contrôle répété de tous les appareils de volumétrie à intervalles prévus (selon les cas une fois par an ou, au minimum, une fois tous les trois ans).La précision des mesures d'appareils de volumétrie peut être modifiée au cours du temps, par ex. à cause de l'utilisation de produits chimiques agressifs ou des procédés et de la fréquence du nettoyage. Le contrôle d'appareils de volumétrie est décrit dans la norme ISO 4787. Rappels :
Ajustage du ménisque en cas d'une marque circulaire. La lecture se fait au point le plus bas du
ménisque.
Ajustage du ménisque en cas d’une bande
photophore. La lecture se fait au point de contact des deux pointes.
1. Préparation pour le contrôle 1.1 Définition claire de l'appareil de mesure à contrôler Numéro de lot/de série*, marque de fabrique*, type de l'appareil, volume nominal*, marges de tolérance*, matériau. (*imprimés sur l'appareil de mesure.) Remplir le procès-verbal d'essai (voir page) 1.2 Type de l'appareil A inscrire au procès-verbal d'essai Appareils de volumétrie calibrés pour contenir "IN" : Fioles jaugées : forme trapèze , modèle standard , verre brun , bord évasé (à préciser) Eprouvettes graduées Eprouvettes bouchées Pipettes graduées, pour contenir (0,1 mL et 0,2 mL) Appareils de volumétrie calibrés pour écouler "EX" : Pipettes jaugées : 1 trait ou 2 traits Pipettes graduées . Pipettes graduées à cotonner, écoulement total (1 mL et 2 mL)
. Pipettes graduées, écoulement total
. Pipettes graduées avec bande photophore, écoulement total . Pipettes graduées, écoulement partiel . Pipettes graduées pour analyses enzymatiques, écoulement partiel Burettes . Burettes : robinet à pointeau latéral , robinet en verre latéral , robinet à pointeau droit , robinet en verre droit …. . Microburettes : robinet à pointeau latéral , robinet en verre latéral , robinet à pointeau droit , robinet en verre droit … Burettes à zéro automatique . Burettes à zéro automatique, avec robinet intermédiaire et robinet à pointeau . Burettes à zéro automatique, avec robinet intermédiaire et robinet en verre . Burettes à zéro automatique, sans robinet intermédiaire avec robinet à pointeau 1.3 Volume nominal : subdivision

RD-MB 6
Pour les appareils de volumétrie avec graduation indiquer également la subdivision, par ex. 20 : 0,1 ml. A inscrire au procès-verbal d'essai 1.4 Marges de tolérance Lire les marges de tolérance sur l'appareil correspondant. A inscrire au procès-verbal d'essai 1.5 Matériau verre de chimie (AR-Glas®) : pipettes jaugées et pipettes graduées verre borosilicaté (DURAN®) : fioles jaugées, éprouvettes graduées, burettes autre matériau : par ex. le PP, le PMP A inscrire au procès-verbal d'essai 1.6 Acitivités du client A inscrire au procès-verbal d'essai Résultat (par ex.): Numéro de série/de l’appareil : 99 080634 Marque de fabrique: BLAUBRAND® Calibrage: EX Type de l'appareil: pipette graduée à cotonner, écoulement total Volume nominal/subdivision: 2 : 0,02 ml Marge de tolérance: ±0,010 ml Matériau: verre AR-Glas® 2. Contrôle visuel 2.1 Propreté Pour obtenir la précision de volume indiquée, la surface du verre doit être propre et exempte de graisse. Si des gouttes demeurent sur la paroi du verre ou si le ménisque ne s'ajuste pas exactement, l'appareil de mesure n'est pas exempt de graisse et doit être nettoyé avec un détergent légèrement alcalin. Ensuite, rincer avec de l'eau de ville, et après avec de l'eau distillée ou déminéralisée. 2.2 Inscriptions sur les appareils de volumétrie Les signes distinctifs par ex. de l'attestation de conformité, de la classe A/AS, du volume nominal, de la marge de tolérance, de la température de référence, du calibrage IN/EX, du numéro de lot/de série etc. ainsi que les marques de volumes doivent être clairement lisibles. 2.3 Détériorations La surface du verre ne doit pas présenter de dommages significatifs comme par ex. des rayures ou ébréchures. Pour les pipettes et burettes, c'est en particulier l'orifice de la pointe qui ne doit pas être détérioré. Les robinets des burettes doivent fermer de façon étanche, sans à-coups et aisément. (Dans une période de 60 secondes aucune goutte ne doit se former à la pointe.) 3. Mise en équilibre à la température ambiante de la verrerie à tester
Durée : 1 heure 4. Equipement d'essai et accessoires - Eau déminéralisée - Récipient pour pesée : par exemple fiole Erlenmeyer 100 ml - Entonnoir - Thermomètre d'une précision de 0,2 °C - Balance de précision - Chronomètre pour l'observation exacte du temps d'attente d'une précision de ± 1 s. - Papier filtre - Poire d’aspiration - Baromètre pour le contrôle de la pression atmosphérique, d'une précision de ± 5 hPa. En utilisant des moyens de contrôle calibrés (balance et thermomètre), on satisfait à l’exigence des normes DIN EN ISO 9001, DIN EN ISO 10012 et DIN EN ISO/IEC 17025 et suivantes. Le calibrage de la balance peut être effectué par ex. par son étalonnage officiel direct ou bien à l’aide de poids officiellement étalonnés . Le calibrage du thermomètre peut également être effectué par l’étalonnage officiel ou en le comparant avec des thermomètres officiellement étalonnés (dans des conditions définies). 5. Contrôle gravimétrique 5.1 Appareils de volumétrie, calibrés pour contenir "IN" 5.1.1 Fioles jaugées, éprouvettes graduées et éprouvettes bouchées (DURAN®)
� Déterminer la température de contrôle (liquide d'essai).
� Déterminer le « poids » à vide de l'appareil . � Inscrire les valeurs au procès-verbal d'essai. � Remplir l'appareil de mesure de liquide d'essai
jusqu'à ce que celui-ci dépasse la marque circulaire de 5 mm environ.
� La paroi du verre au-dessus du ménisque ne doit pas être humectée ; si besoin est, essuyer avec un papier filtre.
� Ajuster le ménisque exactement sur la marque circulaire par soutirage de liquide à l'aide d'une pipette (la lecture étant exempte de parallaxe et pour une meilleure lecture du ménisque, placer la fiole jaugée sur du papier blanc.)
� Déterminer le « poids » de l'appareil de mesure. � Inscrire la valeur au procès-verbal d'essai.
5.1.2 Pipettes graduées, pour contenir (verre AR-Glas®)
� Déterminer la température de contrôle (liquide d'essai).
� Déterminer le poids à vide de l'appareil. � Inscrire les valeurs au procès-verbal d'essai. � Tenir la pipette graduée presque horizontalement
et avec la pointe toucher la surface de l'eau de la

RD-MB 7
fiole à pesée rempli de liquide d'essai ; la pipette se remplira tout seule à cause de la capillarité.
� Remplir l'appareil de mesure de liquide d'essai exactement jusqu'à la marque circulaire du volume nominal.
� Essuyer l'extérieure de la pointe de la pipette avec du papier filtre.
� Déterminer le « poids » de l'appareil de mesure. � Inscrire la valeur au procès-verbal d'essai.
5.2 Appareils de volumétrie, calibrés pour écouler "EX" 5.2.1 Pipettes jaugées et pipettes graduées (verre AR-Glas®)
� Déterminer la température de contrôle (liquide d'essai).
� Déterminer le « poids» du récipient pour pesée. � Inscrire les valeurs au procès-verbal d'essai. � Remplir la pipette à l'aide d'un auxiliaire de
pipetage jusqu'à ce que le liquide dépasse la marque circulaire du volume nominal de 5 mm environ.
� Essuyer l'extérieure de la pointe de la pipette avec du papier filtre.
� Ajuster exactement l'appareil de mesure par évacuation de liquide, la lecture étant exempte de parallaxe ; si une goutte demeure à la pointe, l'essuyer contre la paroi intérieure du récipient pour pesée.
� Ensuite, laisser le liquide s'écouler dans le récipient à pesée, la pointe de la pipette étant appuyée contre la paroi inclinée du récipient. Le temps d'attente commence dès que le ménisque s'immobilise dans la pointe de la pipette pour les pipettes à 1 trait *.
� Après le temps d'attente de 15 secondes (lire le temps sur le chronomètre), essuyer la pointe contre la paroi intérieure du récipient.
� Si une goutte demeure à la pointe, l'essuyer contre la paroi intérieure du récipient à pesée.
� Déterminer à nouveau le « poids » du récipient. � Inscrire les valeurs au procès-verbal d'essai.
* Pour les pipettes à 2 traits : laisser l'eau s'écouler jusqu'à 10 mm environ au-dessus du trait de division le plus bas, la pointe de la pipette étant appuyée contre la paroi inclinée du récipient à pesée. Après le temps d'attente de 15 secondes, ajuster exactement sur le trait de division. 5.2.2 Burettes et burettes à zéro automatique (DURAN®)
� Déterminer la température de contrôle (liquide d'essai).
� Déterminer le « poids » du récipient pour pesée. � Inscrire les valeurs au procès-verbal d'essai. � Fixer la burette verticalement dans le statif. � Remplir la burette à environ 5 mm au-dessus de
la marque zéro, ensuite la laisser s'écouler jusqu'au volume nominal pour désaérer le robinet
de burette. Après le premier remplissage, une petite bulle d'air peut se trouver dans le robinet de burette. Pour éliminer la bulle, pencher la burette et tapoter légèrement avec le doigt sur l'endroit où se trouve celle-ci. Remplir la burette à environ 5 mm au-dessus de la marque zéro ; la paroi du verre au-dessus de la marque zéro ne doit pas être humectée (si besoin est, l'essuyer avec un papier filtre).
� Ajuster exactement sur le point zéro par évacuation de liquide la lecture étant exempte de parallaxe.
� Ensuite, laisser l'eau s'écouler librement dans le récipient à pesée jusqu'à 5 mm environ au-dessus du trait de division le plus bas, la pointe de la burette ne devant pas toucher la paroi du récipient.
� Après le temps d'attente de 30 secondes (lire le temps sur le chronomètre), ajuster le ménisque exactement sur le trait de division du volume nominal et essuyer la pointe contre la paroi intérieure du récipient. Si une goutte demeure à la pointe, l'essuyer contre la paroi intérieure du récipient à pesée.
� Déterminer à nouveau le poids du récipient � Inscrire les valeurs au procès-verbal d'essai.
6. Evaluation Le nombre de mesures à effectuer dépend en premier lieu de l'aptitude du vérificateur. Normalement, un seul contrôle suffit, au moins pour tous les appareils de mesure calibrés pour contenir "IN". Pour les appareils de mesure calibrés pour écouler "EX", il est recommandé pour plus de sûreté d'utiliser la valeur moyenne résultant de 3 valeurs mesurées. La dispersion des valeurs individuelles mesurées ne doit pas dépasser un quart de la marge de tolérance admise pour l'appareil de mesure correspondante. (Exemple: la marge de tolérance admise pour une pipette jaugée de 10 mL est de ± 0,020 mL. La dispersion des valeurs individuelles doit, en ce cas, être inférieure à ± 0,005 mL. Si cette valeur est dépassée, il est nécessaire de vérifier la méthode d'essai et d'effectuer à nouveau le contrôle.) Le contrôle d'appareils de volumétrie est décrit dans la norme ISO 4787 où est indiquée également la règle à calcul générale suivante:
(((( )))) (((( ))))(((( ))))L20 2 1
L G
1V W W 1 1 t 20 C
ω
ργ
ρ ρ ρ
= − − − − °= − − − − °= − − − − °= − − − − ° −−−−
Vu l'application très compliquée de cette règle et le grand nombre de tables nécessaires, le calcul a été simplifié. On écrit :
(((( ))))20 2 1V W W z= −= −= −= −
V20 [mL]: volume de l'appareil de mesure à 20 °C .

RD-MB 8
W1 [g]: valeur obtenue de la pesée de l'appareil de mesure vide/ou bien avant l’écoulement du contenu. W2 [g]: valeur obtenue de la pesée de l'appareil de mesure rempli/ou bien après l’écoulement du contenu. z [mL/g]: facteur des paramètres de contrôle. Le facteur "z" comprend des paramètres suivants:
� Masse volumique du poids de calibrage de la balance (ρG) : par exemple 8,000 g/mL (voir mode d'emploi du fabricant de la balance).
� Masse volumique de l’air qui dépend de la pression atmosphérique, de la température et de l’humidité relative de l'air de 40 - 90 % (ρL) : Pour tous les appareils de volumétrie, à l'exception de fioles jaugées > 250 mL, l'influence de la pression atmosphérique est relativement réduite par rapport aux marges de tolérance données ; il faut alors utiliser le coefficient de correction "z" de la table "gamme de pression atmosphérique moyenne". Pour les appareils de volumétrie > 250 ml, il faut choisir la table correspondante se référant à la gamme de pression atmosphérique supérieure, moyenne ou inférieure.
� Masse volumique de l'eau en fonction de la
température (ρω).
� Coefficient de dilatation de l'appareil de volumétrie en fonction du matériau: DURAN®: γ = 9,9 10-6 °C-1 AR-Glas®: γ = 27 10-6 °C-1 PP: γ = 450 10-6 °C-1 (données des fabricants, valeur moyenne résultant de γ = 300 · 10-6 °C-1 jusqu'à γ = 600 · 10-6 °C-1) PMP: γ = 351 · 10-6 °C-1 (données du fabricant: Mitsui)
Un exemple de calcul de volume V20 : Numéro de série/ de l’appareil: 99 080634 ; Marque de fabrique: BLAUBRAND® ; Type de l'appareil: fiole jaugée, modèle standard ; Calibrage: IN ; Volume nominal/subdivision: 100 mL ; Marges de tolérance : ± 0,1 mL ; Température de contrôle: 23 °C ; Matériau: DURAN® ; Activité du client: laboratoire de tests FT
« Poids » à vide de la fiole jaugée: W1 = 25,456 g ; « Poids » de la fiole jaugée remplie: W2 = 125,124 g Facteur "z" de la table 1, pression atmosphérique moyenne, vu le volume de la fiole jaugée < 250 mL: z(23 °C) , DURAN® = 1,00348 mL/g V20 = (W2 - W1) z = (125,124 g - 25,456 g)* 1,00348 = 100,01 mL Tables pour le facteur "z" Table 1 : le facteur "z" peut être lu en fonction de la température (de 15 ºC à 30 ºC) et de la pression atmosphérique (de 980 hPa à 1040 hPa), concernant les verres AR-Glas® et DURAN®. Table 2 : si on veut contrôler des appareils de volumétrie en matière plastique, c'est la table 2 qui porte sur le facteur "z" pour le PP et PMP. Remarque et études complémentaires envisageables (recherche sur internet) : Des méthodes spectrophotométriques alternatives à la gravimétrie existent. Elles sont très performantes pour les essais sur les petits volumes de l'ordre du µL (par exemple pour vérifier des pipettes à piston pour des volumes du µL) là où les méthodes gravimétriques sont d'application très délicates (nécessité de balances au µg et difficultés pour gérer l'évaporation). Dans ces méthodes, il s'agit en fait de mesurer des facteurs de dilution en utilisant des substances absorbantes et de les relier au volume inconnu soumis à l'essai. Pour être convenables, les résultats obtenus doivent pouvoir être associés à une détermination rigoureuse d’incertitude : un calcul analytique d'incertitude-type composée combinant les caractéristiques métrologiques du spectrophotomètre utilisé, les déviations à la loi de Beer-lambert, les effets des variations de température, les caractéristiques de la verrerie de classe A utilisée…. Il existe deux méthodes spectrophotométriques décrites dans la norme ISO 8655-7 :2005(F) et le rapport technique ISO/TR 16153 :2004(F) : • la méthode dite « en cellule d'écoulement » (cuve à circulation) ; • la méthode dite « à deux colorants et changements de cuves ».

RD-MB 9

RD-MB 10

RD-MB 11

RD-MB 12
LE VOCABULAIRE LIE A LA QUALITE EN CHIMIE ANALYTIQU E (ordre non alphabétique)
QUALITE : Mesure selon laquelle un ensemble de caractéristiques inhérentes satisfait aux exigences (ISO 9000 année 2000) (les exigences d’un client) ACCREDITATION : Procédure selon laquelle un organisme qui fait autorité délivre une reconnaissance formelle stipulant qu’un organisme ou une personne est compétent pour effectuer des tâches spécifiques. CERTIFICATION : Procédure selon laquelle un tiers délivre une garantie écrite stipulant qu’un produit, procédé ou un service est conforme aux exigences spécifiées. La certification diffère essentiellement de l’accréditation par le fait que la compétence technique n’est pas spécifiquement abordée. ASSURANCE QUALITE (AQ) : L’assurance qualité décrit l’ensemble des mesures qu’un laboratoire met en place pour garantir la qualité de ses activités. Classiquement, celle-ci peut comprendre :
� Un système qualité ; � Un environnement approprié pour le laboratoire ; � Un personnel diplômé, formé et qualifié ; � Des procédures et des dossiers de formation ; � Un matériel convenablement entretenu et
étalonné ; � Des procédures de contrôle de la qualité ; � Des méthodes documentées et validées ; � Traçabilité et incertitudes des mesures ; � Des procédures de compte-rendu ; � Des mesures préventives et correctives ; � Des tests de compétences ; � Un audit interne et des procédures de revue ; � Des procédures de réclamation ; � Exigences concernant les réactifs, les solutions
d’étalonnage, les étalons de mesures et les matériaux de référence.
CONTROLE QUALITE (CQ) : Les techniques opératoires et les activités qui sont utilisées pour répondre aux exigences en matière de qualité. Les procédures du contrôle qualité ont trait à ce qui garantit la qualité des échantillons spécifiques ou des lots d’échantillons et comprennent :
� Analyses de matériaux de référence / des étalons de mesures ;
� Analyses des échantillons en aveugle ; � Utilisation d’échantillons pour le contrôle
qualité ; � Analyses des blancs ; � Analyses des échantillons dopés ; � Analyses en double.
AUDIT ET REVUE : En pratique, les audits qualité sont de deux formes. Lorsqu’un audit est effectué par un organisme externe indépendant (pour un processus d’accréditation), on parle d’évaluation. Des audits « qualité » effectués en interne sont parfois subdivisés en audit interne (lequel permet de vérifier que les procédures qualité sont en place et bien appliquées) et en revue (laquelle veille à garantir que le système qualité soit effectif et réponde aux objectifs attendus). STANDARD : En anglais ce mot a plusieurs significations. Dans le passé, il était couramment utilisé pour désigner des documents normatifs, puis il a par la suite désigné des étalons chimiques ou physiques. On emploie aujourd’hui le mot standard pour se référer à des normes écrites. L’expression étalon de mesure est employée pour des étalons chimiques ou physiques utilisée à des fins d’étalonnage ou de validation, tels que : des produits chimiques d’une pureté déterminée et leurs solutions correspondantes ayant des concentrations connues ; filtres U.V. ; masses etc. Les matériaux de référence constituent une catégorie d’étalons de mesures. MATERIAU DE REFERENCE (MR) : Matériau ou substance dont une ou plusieurs valeurs d’une propriété donnée sont suffisamment homogènes et bien établies pour permettre de l’utiliser pour l’étalonnage d’un appareil, l’évaluation d’une méthode de mesurage ou l’attribution de valeurs à des matériaux. MATERIAU DE REFERENCE CERTIFIE (MRC) : Matériau de référence, accompagné d’un certificat, dont une ou plusieurs valeurs d’une propriété donnée sont certifiées par une procédure, qui établit son raccordement à une réalisation exacte de l’unité dans laquelle on exprime les valeurs de la propriété et pour laquelle chaque valeur certifiée s’accompagne d’une incertitude à un niveau de confiance indiqué. TRACABILITE : Propriété du résultat d’un mesurage ou d’un étalon tel qu’il puisse être relié à des références déterminées, généralement des étalons nationaux ou internationaux, par l’intermédiaire d’une chaîne ininterrompue de comparaisons, ayant toutes des incertitudes déterminées. INCERTITUDE DE LA MESURE : un paramètre associé au résultat d’un mesurage qui caractérise la dispersion des valeurs attribuées au mesurande.

RD-MB 13
LE COFRAC Le COFRAC est le COmité FRançais d'ACcréditation.
Les pouvoirs publics français ont choisi de mettre en
place un système unique d'accréditation pour toute la
France. Le COFRAC a été constitué sous la forme d'une
association à but non lucratif en 1994.
Les missions du COFRAC sont :
� procéder à l'accréditation conformément aux
normes françaises (NF), européennes (EN) ou
internationales (ISO) de tous les organismes
intervenant pour l'évaluation de la conformité à
un référentiel et dans tous les domaines où une
accréditation est utile.
� développer la confiance du marché envers les
organismes accrédités.
� assurer une reconnaissance européenne et
internationale du système d'accréditation
français.
� négocier des accords avec d'autres organismes
accréditeurs ; assurer la représentation des
intérêts français dans les instances et
organismes européens et internationaux
traitant de l'accréditation.
Le COFRAC et l'accréditation des laboratoires
d'étalonnages ou d'essais
Un laboratoire n'est pas accrédité dans sa "globalité de
laboratoire" mais sur des méthodes précises validées.
Les méthodes de références (par exemple les
méthodes des normes NF, EN, ISO) sont évidemment
validées. Mais un laboratoire peut très bien développer
des méthodes originales internes ou des variantes des
méthodes de référence. Si le laboratoire les valide, il
pourra prétendre à l'accréditation pour de telles
méthodes.
Les exigences générales d'accréditation du COFRAC
pour les laboratoires d'étalonnages ou d'essais sont
décrites dans l'incontournable norme ISO/CEI 17025
"Prescriptions générales concernant la compétence des
laboratoires d'étalonnages et d'essais" (année 2005).
Le COFRAC a élaboré des programmes d'accréditation.
Il évalue les demandes d'accréditation au travers
d'audits de l'organisation qualité et d'audits de
compétence technique.
L'accréditation COFRAC bénéficie d'un accord de
reconnaissance mutuelle signé avec les autres pays
européens dans le cadre de l'EAL (European
Cooperation for Accreditation of Laboratories)
L'intérêt de l'accréditation COFRAC des
laboratoires d'étalonnages ou d'essais
L'accréditation COFRAC fait reconnaître, dans le
cadre d'exigences définies au niveau international (ISO
17025), la compétence, l'impartialité et la mise en
oeuvre d'un système qualité pour les méthodes
accréditées d'un laboratoire. Et l'accréditation
COFRAC est une accréditation reconnue au plan
international.
Les pouvoirs publics français et européens s'appuient
de plus en plus sur l'accréditation (une démarche
volontaire pour le laboratoire qui désire se faire
accréditer) comme préalable à de nombreux agréments.
COFRAC et BPL OCDE
En France c'est le COFRAC qui est chargé des
inspections et des vérifications entrant dans le cadre
des "BPL OCDE" (bonnes pratiques de laboratoire,
référentiel OCDE). Les résultats sont utilisés par le
GIPC (groupement interministériel des produits
chimiques) pour prononcer la reconnaissance BPL d'un
laboratoire dans une activité donnée du domaine des
BPL OCDE MESURES 766 - JUIN 2004 Les dix ans du Cofrac
Etre connu et reconnu
1994. Création le 22 juin du Comité Français
d’Accréditation
1995. Après audit, le Cofrac entre dans l’accord EAL
(European cooperation for Accreditation of
Laboratories). Auparavant, le RNE et BNM avaient été
audités, respectivement pour les activités essais et
étalonnages
1996. Après audit, le Cofrac entre dans l’accord EAC
(European Accreditation of Certification)
2003. Signature de l’accord multilatéral (MLA) pour
l’inspection et signature d’une convention avec les
pouvoirs publics qui prévoit que l’Etat reconnaît le
Cofrac comme organisme national d’accréditation. Une
reconnaissance officielle.
Etre réactif et anticiper les besoins
1997. Premier laboratoire d’analyses en biologie
médicale (LABM) accrédité (aujourd’hui, 70 LABM sur
5 000 sont accrédités dans une démarche purement
volontaire, un marché potentiel important mais qui a du
mal à décoller…)
1998. Les Etats-Unis imposent subitement
l’accréditation à tous les laboratoires d’essais pour les
éléments de fixations destinés à l’aéronautique. Le
Cofrac relève le défi de la FQA (Fastener Quality Act)

RD-MB 14
et met en place en quelques mois un programme
d’accréditation(depuis, les Etats-Unis sont revenus sur
leur décision).
Harmoniser les accréditations
1999. Fusion des sections Etalonnage et Essai.
2000. Fusion des sections Certification d’entreprises
et de personnels avec Environnement (règlement
Ecoaudit)
2001. Fusion des sections Certification des produits
industriels et services avec Certification des produits
agricoles et alimentaires.
2002. Pour les laboratoires d’étalonnage et d’essai,
passage à la norme internationale ISO 17025 (la date
butoir tombait à la fin de l’année) pour l’accréditation
des organismes d’inspection.
Le Cofrac en chiffres
66 personnes dans la structure permanente ; 150
auditeurs qualiticiens ; 800 experts techniques ; 300
bénévoles
Budget 2003 : 8 millions d’euros
1600 accréditations délivrées dont :
1100 laboratoires d’essais ; 300 laboratoires
d’étalonnage ; 110 organismes d’inspection ; 29
certificateurs de systèmes qualité, système de
management environnemental et de personnes ; 43
certificateurs de produits et services
1400 audits réalisés par an (une entreprise accréditée
est auditée par le Cofrac en moyenne tous les 15 mois)
Répartition des activités du Cofrac
Laboratoires (essais + étalonnage) 75%
Inspections 8 %
Certifications (entreprises, personnes,produits) 14 %
Divers 3 %
A l’occasion des dix ans du Cofrac, son directeur Daniel Pierre rappelle les grands principes de l’accréditation. L’organisme accréditeur se doit d’être indépendant, impartial, compétent, sans but lucratif. Appliquant des procédures, l’accréditeur doit aussi chercher à répondre aux besoins et à la demande. Au niveau international, Daniel Pierre défend une accréditation non soumise aux règles de la concurrence. (Propos recueillis par Marie-Pierre Vivarat-Perrin) Mesures. Vous êtes directeur du Cofrac depuis 1994, année de la mise en place d’un système national l’accréditation unifié. Vous avez donc suivi toute son évolution. Dix ans après, quand vous vous retournez, quelle impression avez-vous?
Daniel Pierre. L’impression d’un immense chemin parcouru. Non seulement en termes quantitatifs - aujourd’hui 1600 laboratoires ou organismes de certification ou d’inspections sont accrédités par le Cofrac -mais également en termes de connaissance et de reconnaissance. L’accréditation Cofrac est reconnue comme un gage de confiance. Aujourd’hui, un industriel sait ce que représente l’accréditation Cofrac du prestataire auquel il s’adresse pour réaliser un essai sur un produit, pour étalonner un instrument ou encore pour certifier son système de management de la qualité. Les consommateurs finaux sont eux aussi de plus en plus attachés aux certifications produits même s’ils ne savent pas que les étiquettes “NF” ou “Label Rouge” cachent une accréditation du certificateur par le Cofrac. Enfin, les pouvoirs publics qui délèguent de plus en plus les contrôles font appel à nous pour accréditer des organismes dans lesquels ils tiennent à avoir confiance. Cette confiance, nous l’avons gagnée dès l’origine du Cofrac avec la volonté de rassembler toujours les différentes parties intéressées. Ainsi, les accrédités eux-mêmes (laboratoires, organismes d’inspection, certificateurs), leurs clients, les pouvoirs publics et les donneurs d’ordre ou les consommateurs finaux sont toujours représentés au sein du conseil d’administration, des comités de section ou encore des commissions de travail. Mesures. Une confiance qui coûte cher…Les industriels se plaignent souvent du prix de l’accréditation répercutée sur la facture des prestations… Daniel Pierre. Oui, je sais, c’est toujours trop cher. C’est le prix de la confiance. Il faut que les industriels sachent ce qu’ils veulent : le mieux ou le moins disant. Par ailleurs, les accrédités reconnaissent eux-mêmes que ce qui leur coûte le plus cher, ce n’est pas tant la prestation du Cofrac en elle-même, mais le temps passé pour formaliser leurs procédures, finaliser leur système qualité, répondre aux exigences de l’accréditation… Je tiens à souligner que l’activité du Cofrac est à but non lucratif, nos tarifications sont revues chaque année dans le but d’équilibrer notre budget. Nous le faisons dans la transparence. Nous nous autofinançons à 94 %, l’aide de l’Etat ne représentant qu’environ 6 % de notre budget. Mesures. Quels ont été les grands chantiers du Cofrac au cours de ces dix ans? Daniel Pierre. En 1994, le Cofrac a intégré deux activités existantes d’accréditation, celles du RNE (Réseau National d’Essais) pour l’accréditation des laboratoires d’essais et celles du BNM (Bureau National de Métrologie) pour l’accréditation des laboratoires d’étalonnage. En revanche, il n’existait rien pour l’accréditation des organismes de certification

RD-MB 15
d’entreprises, de personnes, de produits, ni pour l’inspection. A l’époque, le certificateur qui auditait une entreprise pour son système d’assurance ISO 9000 n’était lui-même jamais contrôlé. Nous nous sommes donc attachés à mettre en place ces nouvelles activités. 8 comités de sections ont été alors créés. Dans un second temps, et dans une logique d’harmonisation et d’efficacité nous avons cherché autant que possible à fusionner ces activités parce que finalement les exigences posées sont les mêmes, qu’il s’agisse de l’accréditation d’un laboratoire d’étalonnage ou celle d’un laboratoire d’essai. D’ailleurs, la norme ISO 17011 qui devrait être adoptée prochainement est un texte unique pour toutes les activités d’accréditation. Notre organisation s’articule aujourd’hui autour de 4 comités de sections : la section Laboratoires (essais + étalonnage), la section Inspection, la section Certification d’entreprises et de personnels et environnement, la section Certification de produits. La première fusion, en 1999, celle entre les anciennes sections essais et étalonnage a sans doute été la plus longue à mettre en place. La dernière, celle entre les anciennes sections certification de produits industriels et de services et certification de produits agricoles et alimentaires en 2001, n’a pas été la plus facile. Il n’a pas été sans peine de réunir autour d’une même table le monde industriel (marques NF de l’Afnor, certification LNE…) et le monde agricole (Label rouge, agriculture biologique…). Pourtant, les deux utilisent le même référentiel international… Mesures. Il a fallu aussi faire connaître le Cofrac au niveau international. Vous êtes d’ailleurs également président de l’EA (European cooperation for Accreditation) qui rassemble les accréditeurs nationaux pour l’Europe… Daniel Pierre. Oui, dès le début, nous nous sommes investis pour une reconnaissance européenne du Cofrac .Je termine actuellement mon troisième et dernier mandat (de deux ans) à la présidence de l’EA. Mesures. Qu’en est-il de l’accréditation dans les autres pays? Daniel Pierre. La volonté européenne est la mise en place d’un seul organisme d’accréditation par pays. La plupart des pays européens respectent cette volonté, à l’exception de l’Italie qui en a conservé trois (un pour les essais, un autre pour les étalonnages, un troisième pour les certifications) et l’Allemagne qui est un cas très compliqué avec plusieurs accréditeurs dans le domaine volontaire et plusieurs accréditeurs dans le domaine réglementaire, tous n’étant pas membres d’EA. Mesures. On entend dire parfois que tous les pays ne sont pas au même niveau, que certains accréditeurs sont moins exigeants que d’autres. Le Cofrac aurait
une réputation d’être assez sévère. Le niveau de compétence entre les pays est-il toujours le même? Daniel Pierre. Les accréditeurs appliquent les mêmes règles basées sur les normes internationales et leurs guides d’application. Tout est toujours perfectible. Mais il est faux de dire que certains accréditeurs sont laxistes et d’autres sont plus durs. C’est le rôle de l’EA d’assurer une bonne homogénéité et de vérifier que les règles sont bien les mêmes. Nous nous attachons à gommer les différences et nous sommes en progression constante. Mesures. Les dix pays qui viennent de rentrer dans la communauté européenne ont ils eux aussi leur organisme d’accréditations? Daniel Pierre. Oui, bien sûr, et certains pays, comme les pays baltes, la République Tchèque ou la Slovaquie sont même en avance par rapport à d’autres pays depuis longtemps dans la Communauté, comme le Portugal. Mesures. Va-t-on vers l’ouverture d’un marché de l’accréditation avec une concurrence commerciale entre accréditeurs? Daniel Pierre. On peut comprendre qu’une entreprise souhaite par exemple faire accréditer toutes ses filiales par le même organisme. Le marché de l’accréditation est donc ouvert mais sous certaines conditions. Ainsi, un accréditeur peut travailler à l’étranger à condition d’en informer l’accréditeur local. Nous avons l’exemple d’un laboratoire espagnol qui s’est adressé au Cofrac parce que sa demande d’accréditation avait été refusée dans son pays. Dans ce cas-là, nous avons évidement rejeté sa demande. La position européenne est claire. Il n’est pas question de mettre en concurrence les accréditeurs nationaux. Un organisme d’accréditation doit être indépendant, impartial et sans but lucratif. Ces principes sont déjà stipulés mais l’EA travaille avec la commission européenne pour entériner l’idée qu’un accréditeur doit être un service économique d’intérêt général non soumis aux règles de la concurrence. Il ne doit pas y avoir de compétition entre les organismes. De ce point de vue, la vision européenne se heurte à la vision américaine. Les Etats-Unis soutiennent une approche mercantile et concurrentielle du marché de l’accréditation. Si on suivait cette politique, il faudrait, pour conserver une indépendance et une qualité des prestations, mettre en place une surveillance des accréditeurs. Accréditer les accréditeurs…, on n’en finirait plus. Mesures. Quel chemin reste-t-il encore à parcourir? Daniel Pierre. Nous sommes en haut de la pyramide de l’établissement de la confiance et nous devons maintenir notre niveau de compétence, toujours dans un esprit d’indépendance. Dans un souci constant d’amélioration,

RD-MB 16
nous avons aussi entrepris une importante réforme de l’accréditation des laboratoires. Mesures. En quoi consiste cette réforme? Daniel Pierre. Le processus d’accréditation reste toujours le même : l’examen d’une demande, un audit et une décision sur base du rapport d’audit. Pour cela, nous appliquons des procédures. Par nature, nous sommes donc procéduriers. Mais nous devons nous attacher à le paraître le moins possible. Auparavant, nous travaillions exclusivement sur la base de «programmes» d’accréditation. Si l’entreprise, le laboratoire qui faisait la demande d’une accréditation correspondait à un de ces programmes, tant mieux pour lui. Sinon, il était difficile de répondre à sa demande. Nous souhaitons offrir aujourd’hui davantage de souplesse en examinant tous les types de demandes, au cas par cas si nécessaire. Cette approche moins rigide demande une meilleure évaluation de la demande et par conséquent une plus grande compétence des ingénieurs qui examinent les demandes et des auditeurs plus spécialisés. Notre réforme va dans ce sens-là. Mesures. En termes quantitatifs, le champ de compétences de l’accréditation peut-il encore s’élargir? Daniel Pierre. On s’en tient toujours à des procédures. Ce qui veut dire qu’on ne peut pas tout accréditer. Par exemple, on ne sait accréditer des laboratoires que pour des méthodes validées. Ainsi, on ne sait pas accréditer un laboratoire de recherche. D’autre part, l’accréditation reste par principe une démarche volontaire même si dans certains cas elle peut être imposée par les pouvoirs publics ou des donneurs d’ordres. Dans le domaine de la certification de systèmes d’entreprise, on peut accréditer le certificateur pour les systèmes de management de la qualité comme l’ISO 9000, les systèmes de management de l’environnement comme l’ISO 14000 mais pas les systèmes de management de la sécurité car le Conseil d’administration du Cofrac a considéré inopportun d’accréditer une telle certification qui d’ailleurs n’est pas basée sur une norme ISO. Néanmoins, dans toutes les sections, de nouveaux domaines d’accréditation se développent : je pense par exemple à la certification des produits issus de l’agriculture raisonnée. En étalonnage, les perspectives de croissance sont limitées car elles sont basées sur les grandeurs physiques fondamentales. Notre plus important potentiel se situe dans les essais et analyses et l’inspection. Tous les jours, de nouveaux produits, de nouvelles machines, de nouveaux matériaux apparaissent sur le marché. Mais là encore, nous avons besoin de référentiels, si possibles internationaux. Nous n’avons pas assez de normes internationales d’essais et d’analyses. Et l’évolution de l’accréditation se fait avec celle de la normalisation. Nous attendons aussi le développement de l’accréditation des laboratoires
d’analyses de biologie médicale (LABM).Aujourd’hui, quelque 70 LABM seulement sont accrédités sur plus de 5 000 existants. Evidemment, le jour où ils feront la démarche, notre activité connaîtra une croissance très importante ! D’après un article paru en janvier 2004 dans Compétences (édité par le Cofrac) et écrit par Philippe Delmas, membre et président sortant de la commission d‘audit interne du Cofrac. Il existe aujourd’hui trois textes, valables pour le monde entier, qui régissent les organismes d’accréditation :
� guide ISO/CEI 58 (NF EN 45003) pour l’accréditation des laboratoires
� guide ISO/CEI 61 (NF EN 45010) pour l’accréditation des certificateurs
� document ISO TR 17010 (FD TR 17010) Les raisons de cette diversité sont historiques. Les laboratoires furent les premiers à justifier la mise en place de procédures d’accréditation alors que les organismes d’inspection ne s’y sont intéressés que récemment. Par ailleurs, les organismes d’accréditation étaient spécialisés. Cependant, ils sont progressivement devenus multivalents. Ainsi, en Europe, il n’existe plus qu’un seul accréditeur dans presque tous les États membres. Ces organismes multivalents doivent respecter les trois textes de base, lesquels, si on les analyse correctement, traitent finalement des mêmes sujets et posent les mêmes exigences, mais selon des termes légèrement différents. Ce qui ne manque pas de soulever quelques problèmes pratiques...y compris au Cofrac. Le besoin d’une norme unique est devenu évident. C’est ainsi que l’ISO, depuis 1999, travaille sur un projet de norme sous la référence17011. Le projet Final Draft International Standard, dernier stade avant l’adoption, est actuellement à l’enquête. Des organismes qui se créent à l’étranger s’y réfèrent déjà. La norme ISO 17011 innove-t-elle ? En ce qui concerne les concepts, la réponse est non. La norme reprend les concepts connus d’indépendance, d’impartialité, de compétence et de confidentialité, les règles de participation des parties intéressées, etc. En ce qui concerne les exigences elles-mêmes, leurs principes demeurent inchangés mais le texte est plus précis, plus explicite, plus détaillé. A titre d’illustration, on remarque que la norme 45003 comporte 6 pages alors que l’ISO 17011 en compte 29. C’est ainsi qu’est affirmée très clairement l’interdiction, pour un organisme d’accréditation, de réaliser des activités effectuées par des organismes d’évaluation de la conformité (ses clients) ou de proposer des missions de conseil. A noter aussi l’obligation faite aux accréditeurs de rechercher de façon systématique la satisfaction des besoins des organismes accrédités, la nécessité d’agir à titre préventif et de veiller à l’amélioration continuelle, le devoir d’analyser les

RD-MB 17
risques de conflits d’intérêts avec des organismes apparentés (s’il en existe). Remarque : ISO 17011 a été remplacée par ISO 17025 en 2005. Les instances internationales d’accréditation Au niveau européen EA (European cooperation for Accreditation), entité juridique enregistrée aux Pays-Bas : elle rassemble les organismes accréditeurs nationaux européens pour les laboratoires (essais, étalonnages), les organismes d’inspection.
Au niveau international ILAC (International Laboratory Accreditation Cooperation), entité juridique enregistrée aux Pays-Bas : elle rassemble les organismes accréditeurs nationaux pour les laboratoires (essais, étalonnages) et les organismes d’inspection. IAF (International Accreditation Forum) , entité juridique enregistrée aux Etats-Unis (état du Delaware) : elle rassemble les accréditeurs de certificateurs (entreprises, personnels, produits) et les organismes d’inspection.

RD-MB 18
COMPLEMENT DE VOCABULAIRE et OUTILS STATISTIQUES (ordre non alphabétique)
Ces définitions sont issues du :
� Vocabulaire International des Termes Fondamentaux et Généraux de Métrologie (VIM)
� Guide pour l’Expression de l’Incertitude de Mesure (GUM)
� Ou autre MESURANDE : Grandeur particulière soumise à mesurage La définition du mesurande peut nécessiter des indications de température, pression …. MESURAGE : Ensemble d’opérations ayant pour but de déterminer une valeur d’une grandeur. Le déroulement des opérations peut être automatique. MODE OPERATOIRE : Ensemble des opérations, décrites d’une manière spécifique, mises en œuvre lors de l’exécution de mesurages particuliers selon une méthode donnée. GRANDEUR D’INFLUENCE : Grandeur qui n’est pas le mesurande mais qui a un effet sur le résultat du mesurage. FIDELITE : Etroitesse de l’accord entre différents résultats de mesures. JUSTESSE : Aptitude d’un instrument de mesure à donner des indications exemptes d’erreur systématique (VIM) ; Etroitesse de l’accord entre la valeur moyenne obtenue à partir d’une large série de résultats d’essai et une valeur de référence acceptée (NF ISO 5725).
Justesse et fidélité
ETALONNAGE : Ensemble des opérations établissant, dans des conditions spécifiées, la relation entre les valeurs de la grandeur indiquées par un appareil de mesure ou un système de mesure, ou les valeurs représentées par une mesure matérialisée ou par un matériau de référence, et les valeurs correspondantes de la grandeur réalisées par des étalons.

RD-MB 19
CALIBRAGE : Positionnement matériel de chaque repère d’un instrument de mesure en fonction de la valeur correspondante du mesurande
Calibrage d’un appareil
Remarque : il y a souvent confusion entre calibrage et étalonnage même dans les polycopiés. AJUSTAGE : Opération interne à l’appareil destinée à amener un instrument de mesure à un état de fonctionnement convenant à son utilisation. REGLAGE : Opération faite par l’utilisateur pour amener un instrument de mesure à un état de fonctionnement convenant à son utilisation. REPETABILITE : Etroitesse de l’accord entre les résultats des mesurages successifs du même mesurande, mesurages effectués dans les mêmes conditions de mesure. Ces conditions sont appelées conditions de répétabilité ; elles comprennent :
� Même mode opératoire � Même opérateur � Même instrument de mesure utilisé dans les
mêmes conditions � Même lieu � Répétition durant une courte période de temps
La répétabilité peut s’exprimer quantitativement à l’aide des caractéristiques de dispersion des résultats (écart-type de répétabilité). REPRODUCTIBILITE : Etroitesse de l’accord entre les résultats des mesurages du même mesurande, mesurages effectués en faisant varier les conditions de mesure. Il faut préciser les conditions que l’on fait varier ; elles comprennent :
� Le principe de mesure � La méthode de mesure � L’opérateur � L’instrument de mesure � L’étalon de référence
� Le lieu � Le temps ….
La reproductibilité peut s’exprimer quantitativement à l’aide des caractéristiques de dispersion des résultats. INCERTITUDE DE MESURE : Paramètre associé au résultat d’un mesurage, qui caractérise la dispersion des valeurs attribuées au mesurande. INCERTITUDE-TYPE : Incertitude du résultat d’un mesurage exprimée sous la forme d’un écart-type. INCERTITUDE ELARGIE : Grandeur définissant un intervalle, autour du résultat d’un mesurage, dont on puisse s’attendre à ce qu’il comprenne une fraction élevée de la distribution des valeurs attribuées au mesurande. FACTEUR D’ELARGISSEMENT : Facteur numérique utilisé comme multiplicateur de l’incertitude-type composée pour obtenir l’incertitude élargie. Un facteur d’élargissement k a le plus souvent une valeur comprise entre 2 et 3. SENSIBILITE : Rapport de la variation de la réponse instrumentale à la variation de la concentration pour différentes solutions étalons ou pour des ajouts de l’analyte dans une matrice. LINEARITE : Capacité à fournir des réponses proportionnelles à la concentration en analyte dosé. LIMITES DE LINEARITE : Limites expérimentales entre lesquelles un modèle linéaire peut être appliqué avec un niveau de confiance connu (régression linéaire cf cours de mathématiques). LIMITES DE DETECTION : Plus petite concentration de l’analyte pouvant être détectée mais non quantifiée, avec un risque d’erreur connu. LIMITE DE QUANTIFICATION : Plus petite concentration de l’analyte pouvant être quantifiée, avec un risque d’erreur connu. Remarque : des outils statistiques permettent de déterminer les limites de linéarité, de détection et de quantification d’une méthode (il faut suivre les indications d’une norme et utiliser Excel). FONCTIONS DE DISTRIBUTION (cf cours de mathématiques):
concentration
Signal mesuré

RD-MB 20
Distribution rectangulaire Forme A utiliser quand : Incertitude-type
Un certificat donne des limites sans spécifier le niveau de confiance (par exemple 25 mL ± 0,05 mL. Une estimation est faite sous la forme d’un intervalle maximum (± a) sans connaître la forme de la distribution
au(v)
3====
Distribution triangulaire
Forme A utiliser quand : Incertitude-type
Les informations sur x sont moins limitées que pour une distribution rectangulaire. Des valeurs proches de v sont plus probables que celles situées près des bornes. Une estimation est faite sous la forme d’un intervalle maximum (± a) décrit à l’aide d’une distribution symétrique.
au(v)
6====
0,5a
x
densité de probabilité
v-a v+a v
0,5a
v-a v v+a x
densité de probabilité

RD-MB 21
Distribution normale Forme A utiliser quand : Incertitude-type
Une estimation est faite à partir des observations répétées d’un processus variant de manière aléatoire. On a obtenu n résultats de mesures notés xi.
Une estimation de µ (la moyenne) est
n
ii 1
xx
n========∑∑∑∑
Une estimation de l’écart type σ est :
(((( ))))n
2
ii 1
x xs
n 1====
−−−−====
−−−−
∑∑∑∑
L’incertitude-type est donnée par u(x) = s
s2 est une estimation de la variance et s
CVx
==== est le
coefficient de variation.
Intervalle de confiance pour la moyenne : loi de Student
1 / 2, 1 / 2,
s sx t x t
n nα ν α νµ− −− −− −− −− × ≤ ≤ + ×− × ≤ ≤ + ×− × ≤ ≤ + ×− × ≤ ≤ + ×
t1-α/2 est la valeur de t obtenue dans une table de Student avec ν = n-1 degrés de liberté au risque 1-α/2. On prend un risque bilatéral de valeur α de se tromper. On prend un risque α/2 côté borne inférieure et α/2 côté borne supérieure de se tromper.
Exemple : Soit n = 10, x 0,1050==== et s = 0,0020
Au risque α = 0,05 (5%), pour n-1 = 9 une table de t (loi de Student) donnera t 1-α/2 = t 0,975 = 2,262
On a : 1 / 2,
s 0,002t 2,262 0,00143
n 10α ν−−−− × = × =× = × =× = × =× = × = ; les 2 limites sont 0,1036 et 0,1064 .Il y a 5 chances sur 100 de se tromper en affirmant que la
moyenne parente µ est comprise entre 0,1036 et 0,1064. On dit aussi : l'intervalle de confiance à p = 0,95 de la moyenne parente est [0,1036 ; 0,1064]
x
Densité de probabilité
µ µ-σ µ+σ

RD-MB
22
22
ESTIMATION DES INCERTITUDES EN CHIMIE ANALYTIQUE I- Etape 1 : spécifier le mesurande un pH, une masse, un volume mais le plus souvent une concentration qui est le résultat d’une expérience suivie de l’application d’une formule. II- Etape 2 : identifier les sources d’incertitude Echantillonnage : l’échantillon prélevé est-il représentatif ? Les analyses menées sur plusieurs prélèvements effectués dans les mêmes conditions ne vont pas conduire à des résultats identiques. Conditions de conservation : les conditions et durée de stockage des échantillons sont des sources d’incertitudes. Effets des appareils utilisés : balance, spectrophotomètre, verrerie volumétrique ….mais aussi un dispositif de régulation de température qui peut imposer une température différente de la température de consigne…. sont sources d’incertitudes. La pureté des réactifs utilisés Conditions de mesures : par exemple la verrerie volumétrique peut être utilisée à une autre température que sa température de calibration. Effets dus à la composition de l’échantillon :
échantillon = substance à analyser + matrices (le reste)
Dosage de Pb2+ dans l’eau de mer ; la matrice est « tout » sauf Pb2+ ; la matrice contient H2O , Na+ , Cl-, des traces d’hydrocarbures, d’autres cations métalliques…. Les autres cations métalliques peuvent fausser le dosage, c’est ce que l’on nomme effet de matrice. Effets dus aux calculs : l’utilisation du modèle de la régression linéaire par exemple, les arrondis de calculs… sont des sources d’incertitude. Correction du blanc : par exemple un mauvais choix de solution pour faire le zéro optique en S.A.M. est source d’incertitudes. Effets dus à l’opérateur : « nobody is perfect ». Effets aléatoires : tout ce que l’on n’a pas réussi à identifier. Remarques : les sources ne sont pas forcément indépendantes. III- Etape 3 : « quantifier » l’incertitude Les différentes approches :
L’approche intra-laboratoire consiste à utiliser les résultats de mesures obtenus uniquement au sein du laboratoire. Cette approche comprend :
� La définition du mesurande, l’analyse du processus de mesure et l’écriture de la formule qui conduit au calcul du mesurande (approche analytique*)
� L’estimation des incertitudes-types � L’estimation de l’incertitude composée � L’incertitude élargie et l’expression du résultat
final *Il existe aussi l’approche MSP (Maîtrise Statistique des Procédés) qui consiste à utiliser des cartes de contrôles réalisées au laboratoire) L’approche inter-laboratoires consiste à utiliser les résultats obtenus par plusieurs laboratoires (ISO 5725) IV- Approche analytique intra-laboratoire : étude d’un exemple (il est exceptionnellement permis de ne pas tout comprendre) Etalonnage d’une solution de soude par pesées d’hydrogénophtalate de potassium (noté KHP) 1- Principe On utilise KHP « pur » et anhydre comme étalon. Une pesée exacte (m) de KHP aux alentours de 0,39 g avec une balance au 1/10 mg est mise en solution dans environ 50 mL d'eau déminéralisée. On titre par la solution de soude à étalonner afin de déterminer le volume (Vb) nécessaire pour l'équivalence acidobasique. A l'équivalence on a la relation nKHP = nOH. Pour déterminer Vb, le laboratoire utilise une burette automatique informatisée couplée à la mesure du pH soit un titrateur automatique qui donne la courbe pH = f(volume NaOH versé). Le volume Vb est alors déterminé par la méthode de la dérivée première. Soit m la masse exacte de KHP. Soit MKHP la masse molaire de KHP. Soit c la concentration de la solution
soude ; on a : KHP
KHP b
1000 m c
M V==== avec c en mol.L-1 ; m en
g ; MKHP en g.mol-1 ; Vb en mL
2- c est le mesurande.

RD-MB
23
23
3- Identification et analyse des sources d'incertitudes. C'est la relation d'étalonnage qui va nous donner le départ pour le recensement des sources d’incertitudes.
Première étape dans la construction d’un diagramme des causes et des effets La combinaison terminale ne sera pas complexe dans le cadre de l'exemple proposé puisque m, M et Vb sont des variables indépendantes.
4- Détermination des composantes de l’incertitude
a) Incertitude-type sur la masse molaire MKHP Il suffit d'utiliser la table des masses molaires avec les incertitudes absolues associées. Cette table est disponible auprès de l'IUPAC (International Union of Physical and Applied Chemistry). Ainsi on trouve :
Elément Masse Molaire /g.mol-1
Incertitude indiquée
Incertitude-type
C 12,0107 ± 0,0008 0,00046 H 1,00794 ± 0,00007 0,000040 O 15,9994 ± 0,0003 0,00017 K 39,0983 ± 0,0001 0,000058
Pour chaque élément, on considère que l’incertitude indiquée par l’IUPAC constitue les limites d’une distribution rectangulaire. On obtient l’incertitude type en
divisant ces valeurs par 3 .
La formule KHP est C8H5O4K , on peut ainsi calculer la masse molaire de KHP et appliquer les propriétés de propagation des écart-types. On obtient :
MKHP = 8 * 12,0107 + 5 * 1,00794 + 4 * 15,9994 + 39,0983 = 204,2212 g.mol-1
Et en désignant par u(MKHP) l'incertitude-type sur MKHP.
u2(MKHP) = 82 * 0,000462 + 52 * 0,0000402 + 42 * 0,000172 + 0,000058 2 = 0,00001405
qui donne u(M KHP ) = 0,0038 g.mol-1 (expression d'une incertitude-type sur MKHP sous forme conventionnelle d'un écart-type)
b) Simplification du problème et répétabilité
La pesée de KHP: la répétabilité de la balance, l'incertitude sur l'étalonnage de la balance, la question des pesées dans l'air qui ne fournissent que des masses conventionelles.
� Le volume (Vb) nous renvoie aussi à plusieurs sous problèmes :
� L'incertitude d'étalonnage sur la burette ; La répétabilité de la burette ; La température et les variations de température dans le laboratoire ; � L’existence d’un biais systématique sur la détermination du point d'équivalence ainsi que la répétabilité sur la détermination de Vb (liée au
matériel et à la technique de mise en évidence de l'équivalence acido-basique).
Reportons toutes ces nouvelles problématiques
dans le "diagramme initial des causes et des effets" qui devient :
On s'intéresse alors à la question de la répétabilité. On peut conduire une étude expérimentale statistique de répétabilité globale du dosage qui englobera en fait tous les termes de répétabilité qu'ils soient liés à la pesée, à la mesure de volume Vb où à la détermination du point d'équivalence.
On est ainsi conduit à un nouveau diagramme des "causes et des effets" associé à "une nouvelle formule" de calcul de détermination de la solution NaOH à étalonner :

RD-MB
24
24
KHP
KHP b
1000 m rc
M V××××====
et r le facteur global de répétabilité.
r = 1 (sans unité) mais avec une incertitude-type u(r). m, M, Vb et r sont des variables indépendantes.
Pour évaluer u(r), on effectue une analyse statistique de résultats expérimentaux en conditions de répétabilité ... Dans le cas étudié (titrateur automatique), la répétabilité est de 0,1% soit un écart-type, u(r) = 0,001.
Remarque : pour le protocole avec indicateur coloré de pH et burette de 25 mL, la répétabilité est de 0,25% soit u(r) = 0,0025.
c) Incertitude-type sur le volume Vb La répétabilité a été prise en compte dans r. Etalonnage de la burette : on utilise une burette automatique. On suppose qu'on a employé une burette de 20 mL annoncée à ± 0,03 mL par le fabricant. La loi de densité de probabilité recommandée est la loi triangulaire isocèle, on estime donc l'incertitude-type liée à la calibration de la burette à
0,03
6 = 0,012 mL soit u(cal.bur.) = 0,012 mL.
Température : La température du laboratoire n'est pas constante fixée à 20°C. - La verrerie est étalonnée à 20°C, et le verre, comme tous les matériaux se dilate ou se contracte selon la température (le coefficient de dilation volumique des verres se situe entre 1 et 30 10-6 °C-1). - On va supposer que la solution à étalonner est destinée à
une utilisation dans le laboratoire, équilibrée à la température du laboratoire : elle peut donc se dilater où se contracter selon les variations de température dans le laboratoire, ce qui modifie sa concentration volumique et donc le volume pour obtenir l'équivalence.
Le paramètre température n’a pas été pris en compte par l'étude de répétabilité. L'étude de répétabilité à été conduite dans un laps de temps pour lequel on a vérifié que la température du laboratoire était demeurée peu variable (par exemple moins de 0,3 °C) devant les variations possibles répertoriées (par exemple 18°C à 22°C avec climatisation).
Nous supposons un laboratoire climatisé à 20°C ± 2°C. Les régulations en température de locaux donnent des variations sensiblement sinusoïdales entre les 2 extremums. La fonction de densité de probabilité correspond alors à la une allure de type dérivée d'arc
sinus, et on aura un écart-type de 2
2 = 1,414 °C =
u(δT).
La verrerie de laboratoire est étalonnée à 20°C (la température moyenne du laboratoire avec un incertitude-type de 1,414 °C).
Pour le verre borosilicaté, on considère que le volume de la verrerie va varier avec la température d'environ 1.10-5 * V * δT avec V le volume "cible" et δT l'écart de la température à la référence 20°C. On peut donc écrire que pour un volume V voisin de 18 mL délivré par la burette : V # V20 + 1.10-5 * 18 * δT avec V20 le volume délivré pour une température de 20°C . Soit V # V20 + 18.10-5 δT avec V20 et δT deux variables indépendantes. D'où en appliquant les lois simples de propagation des variances : u2(V) = u2(V20) + (18.10-5)2 * u2(δT) Or u2(V20) n'est autre que l'incertitude-type liée à l'étalonnage de la burette et nous connaissons sa valeur (voir ci-dessus), u(cal.bur.) = 0,012 mL. Et nous connaissons u(δT) = 1,414 °C.
Donc on a : u2(V) = 0,0122 + ((18.10-5)2 * 1,4142) (le terme d'effet de température est négligeable en fait devant le terme d'étalonnage) qui donne u2(V)= 0,000144 mL qui donne u(V) = 0,012 mL.

RD-MB
25
25
Conclusion, l'incertitude-type sur le volume délivré par la burette à l'équivalence acido-basique se résume à la seule incertitude-type sur l'étalonnage de la burette avec un effet du facteur température tout à fait négligeable. u(V) = 0,012 mL.
Si maintenant, on considère l'effet température sur la dilatation ou la contraction du volume de la solution de NaOH à étalonner. Le coefficient volumique de dilatation de l'eau est de 2,1 10-4 °C-1. Le volume de chute de burette Vb attendu est de 18 mL. On obtient ainsi une incertitude-type sur le volume Vb liée au non contrôle de la température de : u(temp_liquid) # 18 * 2,1 10-4 * 1,414 = 0,0053 mL. u(Vb effet temp.) = 0,006 mL.
Réalisons le bilan sur le volume (étalonnage de la burette et effet température) :
2 2b bu(V ) u(cal bur) u(V effet temp)= += += += +
2 2bu(V ) 0,012 0,006 0,013 mL= + == + == + == + =
On décide de s'intéresser maintenant au biais éventuel de la méthode sur la détermination du volume Vb. Il n'y a pas de biais pour le protocole informatisé avec utilisation de la dérivée première. On suppose donc un biais et une incertitude-type de biais négligeable pour le protocole automatisé.
Remarque : pour un protocole manuel avec indicateur coloré de pH permettant de visualiser l'équivalence, il serait légitime de se demander si il n'y a pas un biais sur le volume équivalent l'indicateur n’a pas un virage exactement centré sur l'équivalence).
Ce calcul conduit au nouveau "diagramme des causes et des effets" qui devient :
KHP
KHP b
1000 m rc
M V××××====
avec, c en mol/L ; m en g ; M en g/mol ; Vb en mL et r le facteur global de répétabilité. r = 1 (sans unité) mais avec une incertitude-type u(r). m, M, Vb et r sont des variables indépendantes.
d) Incertitude-type sur m
Deux paramètres d'étalonnage de la balance sont à considérer, la sensibilité et la linéarité, dans le cas d’une pesée en double pesée (tarage et pesée finale). On utilise une balance analytique pour la pesée en double pesée d'une masse très faible (vers 0,38 g) devant la masse maximale possible, l'incertitude-type liée au facteur sensibilité est négligeable devant l'incertitude-type liée au facteur de linéarité qui est donc le seul à considérer. Soit la valeur de linéarité ± 0,15 mg fournie par le certificat d'étalonnage de la balance avec la recommandation d'utilisation d'une loi de densité de probabilité rectangulaire. Exprimée en incertitude-type, on a :
u(balance_linéarité) = 0,15
3 = 0,087 mg.
Pour une pesée en 2 étapes : la pesée du récipient de pesée (tarage, mi) puis la pesée du récipient avec le contenu KHP (valeur mf) et en considérant que ces 2 étapes sont indépendantes et entachées de l'incertitude-type de linéarité et que la masse pesée est la différence entre ces 2 masses i et f. On peut écrire :
2 2u(m étalonnage)= u(balance linéarité) u(balance linéarité)++++
2 2u(m étalonnage) 0,087 0,087 0,13 mg= + == + == + == + =
La pesée dans l'air ne nous fournit qu'une masse conventionnelle. La formule de correction de ce biais systématique (qui

RD-MB
26
26
donne donc la vraie masse) est très simple mais elle implique de connaître la masse volumique de KHP. Si sa masse volumique était faible devant la masse volumique standardisée des étalons à 8000 kg/m3, la correction ne serait pas si négligeable que ça.
Considérons les caractéristiques du KHP utilisé.
La pureté (en % m/m) a été déterminée sur une pesée conventionelle (dans l'air). Une conclusion s'impose : la pureté ainsi déterminée concerne des pesées de masses conventionelles .On vient donc ainsi de regrouper la question de la pesée conventionelle et de la pureté.
Et on peut réécrire le diagramme des "causes et effets" ainsi :
KHP KHP
KHP b
1000 m P rc
M V× ×× ×× ×× ×
====
avec, c en mol/L ; m en g ; M en g/mol ; Vb en mL Et r le facteur global de répétabilité. r = 1 (sans unité) mais avec une incertitude-type u(r). PKHP ; pureté exprimée avec un nombre < 1 m, P, M, Vb et r sont des variables indépendantes.
P KHP = 0,9997 avec un écart-type de 0,0004 et u(PKHP )=0,0004.
L'étape d'analyse et d'identification des sources d'incertitude est terminée.
5- Construction d'un tableau de calcul de résultat d'étalonnage avec l'incertitude-type
On suppose une manipulation qui a mis en oeuvre une pesée de 0,3888 g et qui a conduit à une valeur Vb = 18,64 mL.

RD-MB
27
27
Symbole de la variable
Signification de la variable
Valeur (x)
Incertitude-type u(x)
Incertitude-type relative u(x)/x
r répétabilité 1 0,0010 0,0010
m (g) masse pesée
(conventionnelle) 0,3888 0,00013
0,00034
PKHP titre 0,9997 0,00040 0,00040
M KHP (g.mol-1)
masse molaire de KHP
204,2212 0,0038 0,000019
Vb (mL) volume pour
l'équivalence acido-basique
18,64 0,013 0,00070
c(mol.L-1) Concentration en NaOH
(le mesurande) 0,10211 0,00014
0,0013
6- Calcul de l’incertitude composée
Il s'agit maintenant de déterminer u(c). Comme la formule pratique de l'étalonnage ne fait intervenir que des produits ou des quotients et que les variables sont indépendantes, on va utiliser la formule suivante :
222 2 2bKHP
2 2 2 2 2KHP b
u(V )u(P )u(c) u(r) u(m) u(M)c r P m V M
= + + + += + + + += + + + += + + + +
On peut calculer u(c) puisqu'on connaît u(c)/c et c. Le résultat permet de déterminer le nombre de chiffres significatifs pour c.
Le résultat peut finalement s'exprimer par : c = 0,10211 mol.L-1 avec une incertitude-type estimée à 0,00014 mol.L-1 ou
c = 0,1021 mol.L-1 avec une incertitude-type estimée à 0,0001 mol.L-1
V- Approche inter-laboratoires (cf ISO 5725)
Cette approche est basée sur un traitement statistique de résultats obtenus par plusieurs laboratoires.