16-kda prolactin down-regulates inducible nitric oxide...
TRANSCRIPT

16-kDa Prolactin Down-Regulates Inducible Nitric Oxide Synthase
Expression through Inhibition of the Signal Transducer and
Activator of Transcription 1/IFN Regulatory Factor-1 Pathway
Sok-hyong Lee,1Michiya Nishino,
2Tuhina Mazumdar,
4Gabriela E. Garcia,
5
Matthew Galfione,6Florence L. Lee,
6Cynthia L. Lee,
6Albert Liang,
7
Jeri Kim,7Lili Feng,
5N. Tony Eissa,
4Sue-Hwa Lin,
6
and Li-yuan Yu-Lee1,2,3
1Department of Immunology, 2Program in Cell and Molecular Biology, 3Immunology, Allergy, and Rheumatology Section, Department ofMedicine, 4Pulmonary and Critical Care Section, and 5Nephrology Section, Baylor College of Medicine; Departments of 6MolecularPathology and 7Genitourinary Oncology, M.D. Anderson Cancer Center, Houston, Texas
Abstract
Angiogenesis plays a key role in promoting tumorigenesis andmetastasis. Several antiangiogenic factors have been shown toinhibit tumor growth in animal models. Understanding theirmechanism of action would allow for better therapeuticapplication. 16-kDa prolactin (PRL), a NH2-terminal naturalbreakdown fragment of the intact 23-kDa PRL, exerts potentantiangiogenic and antitumor activities. The signaling mech-anism involved in 16-kDa PRL action in endothelial cellsremains unclear. One of the actions of 16-kDa PRL is toattenuate the production of nitric oxide (NO) through theinhibition of inducible NO synthase (iNOS) expression inendothelial cells. To delineate the signaling mechanism from16-kDa PRL, we examined the effect of 16-kDa PRL oninterleukin IL-1B–inducible iNOS expression, which is regu-lated by two parallel pathways, one involving IFN regulatoryfactor 1 (IRF-1) and the other nuclear factor-KB (NF-KB). Ourstudies showed that 16-kDa PRL specifically blocked IRF-1 butnot NF-KB signaling to the iNOS promoter. We found that IL-1B regulated IRF-1 gene expression through stimulation of p38mitogen-activated protein kinase (MAPK), which mediatedsignal transducer and activator of transcription 1 (Stat1)serine phosphorylation and Stat1 nuclear translocation toactivate the IRF-1 promoter. 16-kDa PRL effectively inhibitedIL-1B–inducible p38 MAPK phosphorylation, resulting inblocking Stat1 serine phosphorylation, its subsequent nucleartranslocation and activation of the Stat1 target gene IRF-1.Thus, 16-kDa PRL inhibits the p38 MAPK/Stat1/IRF-1 pathwayto attenuate iNOS/NO production in endothelial cells. (CancerRes 2005; 65(17): 7984-92)
Introduction
Prolactin (PRL) is originally identified as a lactotrophic hormonesecreted by the pituitary gland. PRL exists in several forms as aresult of post-translational modifications, such as glycosylation (1),phosphorylation (2), and proteolysis (3–5). Intact PRL (also called23-kDa PRL) is further proteolyzed into fragments of various sizes(6). One predominant proteolytic PRL fragment has an apparent
molecular weight of 16 kDa and is produced by removal of abouta quarter of the PRL molecule from the COOH terminus (7, 8).16-kDa PRL was found to be present in the hypothalamus, pituitarygland, and mammary gland and in the circulation in humans(3–5, 8–10).Rather than being an inactive breakdown product of PRL, 16-
kDa PRL has potent antiangiogenic effects. 16-kDa PRL inhibitedthe basal, basic fibroblast growth factor (bFGF)–stimulated andvascular endothelial growth factor (VEGF)–stimulated proliferationof endothelial cells (11, 12). Consistent with its antiangiogenicactivities in vitro , 16-kDa PRL inhibited the growth of newcapillaries in a chick embryo chorioallantoic membrane assay(11, 13) and blocked bFGF-induced capillary growth in a cornealangiogenesis assay (14).Angiogenesis is important not only for normal physiology but
also for pathophysiology, such as tumorigenesis and metastasis(15). Angiostatin (16) and endostatin (17), which are fragments ofplasminogen and collagen XVIII, respectively, inhibit angiogenesisand tumor growth (16–19). Similarly, 16-kDa PRL was shown tohave antitumor activities in vivo . Bentzien et al. (13) showed thatexpression of 16-kDa PRL in HCT116 human colon cancer cellsinhibited the tumorigenicity of HCT116 cells in a Rag1 mousemodel. Using a recombinant adenoviral vector containing the 16-kDa PRL gene to infect DU145 and PC-3 human prostate cancercells, Kim et al. (20) showed that overexpression of 16-kDa PRLreduced the tumorigenicity of the prostate cancer cells in axenograft nude mouse model. These studies suggest that 16-kDaPRL, through its antiangiogenic activity, inhibits tumor growthin vivo .The mechanism by which 16-kDa PRL mediates its antiangio-
genic effects is not clear. 16-kDa PRL may block tumorangiogenesis by antagonizing the effects of angiogenic factors.The cytokine interleukin (IL)-1h is one of the factors that stimulateendothelial cell proliferation in tumorigenesis (21–23). A high levelof vascularization and enhanced tumor growth was observed intumors derived from lung carcinoma cells overexpressing IL-1h(21). A fibrosarcoma cell line transfected with IL-1h exhibited rapidtumor growth, invasiveness, and metastasis to the lung, whichresulted in increased mortality in tumor-bearing animals (22). It islikely that 16-kDa PRL may antagonize the effect of IL-1h in tumorangiogenesis.IL-1h elicits its angiogenic effects in part by inducing the
production of nitric oxide (NO), which is an endothelial cellsurvival factor that inhibits apoptosis and promotes endothelialcell proliferation and migration (24, 25). NO production is regulated
Requests for reprints: Li-yuan Yu-Lee, Department of Medicine, Baylor College ofMedicine, One Baylor Plaza, Houston, TX 77030. Phone: 713-798-4770; Fax: 713-798-2050; E-mail: [email protected].
I2005 American Association for Cancer Research.doi:10.1158/0008-5472.CAN-05-0631
Cancer Res 2005; 65: (17). September 1, 2005 7984 www.aacrjournals.org
Research Article
Cancer Research. on September 13, 2018. © 2005 American Association forcancerres.aacrjournals.org Downloaded from

by NO synthases (NOS). In endothelial cells, the constitutivelyexpressed endothelial NOS (eNOS) is responsible for the low(nanomolar) concentrations of NO that regulates endothelial cellhomeostasis (26). In contrast, proinflammatory cytokines inducethe transcription of inducible NOS (iNOS), which generates high(micromolar) concentrations of NO that contributes to endothelialcell pathology (27). Proinflammatory cytokines, including IL-1h,tumor necrosis factor-a, IFN, and IL-6, regulate iNOS genetranscription through the activation of several transcriptionfactors, including signal transducer and activator of transcription1 (Stat1), IFN regulatory factor 1 (IRF-1), CAAT/enhancer bindingprotein (C/EBP), and nuclear factor-nB (NF-nB; refs. 27–29).In this study, we investigated the effects of 16-kDa PRL on IL-1h
stimulation of iNOS and NO production in endothelial cells. Ourstudies showed that 16-kDa PRL attenuated IL-1h–inducible iNOSand NO expression through inhibition of a signaling cascadeinvolving the p38 mitogen-activated protein kinase (MAPK)/Stat1/IRF-1 pathway but not the NF-nB pathway.
Materials and Methods
Reagents, antibodies, and purified recombinant 16-kDa PRL. Human
16-kDa PRL was expressed and purified from baculovirus-infected insect
cells as described previously (30). IL-1h (R&D Systems, Minneapolis, MN);anti-p65 NF-nB and anti-InBa (Santa Cruz Biotechnology, Santa Cruz, CA);
anti-human iNOS (Research and Diagnostic Antibodies); anti-phospho-Stat1
(Tyr701), anti-phospho-Stat1 (Ser727), anti-Stat1, anti-phospho-p38 MAPK,
and anti-p38 MAPK (Cell Signaling Technology, Beverly, MA); and anti-h-tubulin (Sigma-Aldrich, St. Louis, MO) antibodies were purchased from
commercial sources. Anti-rat IRF-1 antibody was generated as described
previously (31).
Cell culture. Rat aortic endothelial cells (RAEC; refs. 32, 33)8 weremaintained in DMEM with 10% fetal bovine serum (Invitrogen, Carlsbad,
CA) and 5% gentamicin (Sigma-Aldrich). Cells were grown to confluence,
placed in serum-free medium for 24 hours, and treated with 20 nmol/L16-kDa PRL for 1 hour before the addition of 10 ng/mL IL-1h in the
continued presence of 16-kDa PRL. Lipopolysaccharide (LPS; Sigma-Aldrich;
5 Ag/mL)–treated or IFN-g (R&D Systems; 100 ng/mL)–treated RAEC cells
were used as positive controls for the activation of NF-nB and IRF-1,respectively.
Nitric oxide and citrulline conversion assay. NOS activity in RAEC
was determined by measuring the accumulation of nitrite in the culture
medium. The culture medium (100 AL) was mixed with 100 AL Griessreagents (Sigma-Aldrich) for 10 minutes at room temperature, and the
absorbance at 543 nm was measured. Serial dilutions of sodium nitrite were
used as standards (34). NOS activity in cell lysates was analyzed by the
L-arginine to L-citrulline conversion assay (35) using 1 ACi 3H-L-arginine(53.4 Ci/mmol, Sigma-Aldrich) in the NOS detection kit (Stratagene, La Jolla,
CA). The protein extract (30 Ag) was used for each assay and the conversion
rate was expressed as counts per minute per microgram protein.Western blot analysis. RAECs were lysed in buffer containing 20 mmol/L
Tris (pH 7.4), 100 mmol/L NaCl, 5 mmol/L EDTA, 0.5% Triton X-100,
1 mmol/L phenylmethylsulfonyl fluoride (Sigma), and a protease-inhibitor
cocktail (Sigma-Aldrich). Total protein (10-20 Ag) was resolved by NuPAGE4% to 12% Bis-Tris gradient gels (Invitrogen) and transferred to
nitrocellulose membranes (Bio-Rad, Hercules, CA), and the filters were
blocked with 5% nonfat milk in TBST [10 mmol/L Tris (pH 7.5), 150 mmol/L
NaCl, 0.1% Tween 20]. The filters were blocked with 5% bovine serumalbumin (BSA) in TBST when blotting with anti-phospho-antibodies. The
blots were incubated with antibodies against iNOS (1:70), InBa (1:500),
phospho-Stat1 (Ser727; 1:1,000), phospho-Stat1 (Tyr701; 1:1,000), Stat1(1:1,000), phospho-p38 MAPK (1:1,000), p38 MAPK (1:1,000), or h-tubulin
(1:1,000) followed by goat anti-mouse or rabbit horseradish peroxidasesecondary antibodies (1:2,000; Santa Cruz Biotechnology) and developed by
enhanced chemiluminescence (Pierce, Rockford, IL).
Reverse transcription-PCR. Total RNA (2 Ag; RNA-Bee, Tel-Test, Inc.,Friendswood, TX) was used for cDNA synthesis. The PCR primers (Sigma-Genosys, The Woodlands, TX) used were as follows: Rat iNOS sense
5V-GGAGCGAGTTGTGGATTGTT-3V and antisense 5V-CTTCGGGCTTCAGG-TTATTG-3V (405-bp PCR product); rat IRF-1 sense 5V-CATTCACACAGGCC-GATACA-3V and antisense 5V-AGAGAGACTGCTGCTGACGAC-3V (406-bpPCR product); and histone H3.3 sense 5V-GACTGCCCGCAAATCCAC-3V andantisense 5V-GCACCAGACGCTGAAAGG-3V (200-bp PCR product). For iNOS,
denaturation was at 95jC for 60 seconds, annealing at 55jC for 60 seconds,
and extension at 72jC for 60 seconds followed by a final extension for5 minutes at 72jC; for IRF-1 and H3.3, the same conditions were used, except
that annealing was at 56jC for 60 seconds. The PCR reaction was carried out
for 35 cycles. Fold induction was normalized against H3.3 PCR controls.Electrophoretic mobility shift assay. Rat iNOS promoter sequences
(27) were used to generate the NF-nB and IRF-1 gel shift oligonucleo-
tides. iNOS NF-nB oligonucleotides: forward 5V-ATAATGGAAAATCC-CATGCC-3V and reverse 5V-ACATGGCATGGGATTTTCCATTAT-3V; iNOS
IRF-1 oligonucleotides: forward 5V-CAATATTTCACTTTCATAAT-3V and
reverse 5V-TTCCATTATGAAAGTGAAATATTG-3V, which contain a compos-
ite IRF-1 (underlined) and C/EBP (italicized) binding site. The gel shift
oligonucleotide pairs (Sigma-Genosys) were labeled with 40 ACi[32P]dGTP and [32P]dATP (3,000 Ci/mmol, ICN, Costa Mesa, CA) by a
3V fill-in reaction using Klenow polymerase (Promega Corporation,
Madison, WI). For the NF-nB gel shift, nuclear extracts (10 Ag; NuclearExtract kit, Active Motif, Carlsbad, CA) were incubated with anti-p65 NF-
nB antibody for 30 minutes at room temperature before the addition of
labeled probes. For the IRF-1 gel shift, whole-cell extracts (10 Ag) were
incubated with labeled probes first before the addition of anti-IRF-1
antibody (36, 37). Reactions were resolved on a 5% nondenaturing
polyacrylamide gel in 0.5� Tris/boric acid/EDTA. Gels were dried,
analyzed by autoradiography, and quantified using a Storm960 Phos-
phorImager (Molecular Dynamics, Inc., Sunnyvale, CA).
Transient transfection and promoter analysis. The 1.7-kb rat IRF-1promoter, 1.7-kb mutant IRF-1 promoter containing site-directed mutations
in the IFN-g-activated sequence (GAS) element (mut-IRF-1), three-copy
GAS-thymidine kinase (TK) promoter, and three-copy mutant GAS-TK
promoter (mut-GAS) were described previously (36) and subcloned frompBL-CAT into pGL3-Basic luciferase reporter (Promega). The three-copy
Sp1-TK promoter luciferase reporter was generated by multimerizing the
IRF-1 Sp1 element as described previously (37) and subcloning into pGL3-
Basic. The pGL3 NF-nB-TK promoter luciferase reporter containing twocopies of the NF-nB element was provided by Dr. Natarajan Sivasubrama-
nian (Baylor College of Medicine, Houston, TX). RAEC cells (1 � 106) at 80%
to 90% cell confluence were cultured in antibiotic-free DMEM in six-wellplates (Becton Dickinson, Franklin Lakes, NJ) and transfected with 4 Agreporter construct in 10 AL LipofectAMINE 2000 (Invitrogen). After
24 hours, 16-kDa PRL (20 nmol/L) was added for 1 hour before the
addition of 10 ng/mL IL-1h in the continued presence of 16-kDa PRL. After4 hours, luciferase activity in 20 Ag lysate per sample was assayed in
triplicates using the Luciferase Assay System (Promega) in a TD20/20
luminometer (Turner Design, Sunnyvale, CA). Background luminescence
from transfection with pGL3-Basic vector alone was used to normalizerelative luciferase units (RLU) in each experiment.
Immunofluorescence imaging. RAEC cells were cultured on glass
coverslips coated with poly-D-lysine (Sigma). Cells were maintained inserum-free DMEM for 24 hours before treatment with 50 nmol/L 16-kDa
PRL for 1 hour followed by the addition of either 200 ng/mL IL-1h or 500
ng/mL IFN-g for 30 minutes. Cells were fixed with 4% paraformaldehyde
(Polysciences, Inc., Warrington, PA) in PEM buffer [80 mmol/L PIPES (pH7.0), 1 mmol/L EGTA, 1 mmol/L MgCl2] for 30 minutes and permeabilized
with 0.5% Triton X-100 in PEM buffer for 20 minutes. The coverslips were
blocked in 2% BSA and 2% goat serum in TBST containing 0.2% sodium
azide for 1 hour and incubated with anti-Stat1 antibody (1:500) overnight at4jC followed by goat anti-mouse IgG conjugated Alexa Fluor 488 (1:1,000;8 http://cellapplications.com/RAOEC.htm
16-kDa Prolactin Inhibits p38 MAPK/Stat1/IRF-1 Signaling to iNOS/NO
www.aacrjournals.org 7985 Cancer Res 2005; 65: (17). September 1, 2005
Cancer Research. on September 13, 2018. © 2005 American Association forcancerres.aacrjournals.org Downloaded from

Molecular Probes, Inc., Eugene, OR) for 1 hour and counterstained with 4V,6-diamidino-2-phenylindole (DAPI) using SlowFade Light Antifade kit with
DAPI (Molecular Probes). Images were obtained using an Olympus IX71
Microscope (Olympus America, Inc., Melville, NY) and figures were
compiled using Adobe Photoshop 7.0 (Adobe Systems, Inc., San Jose, CA).Statistical analysis. All results were confirmed in multiple independent
experiments, with each time point or condition assayed in duplicates or
triplicates within each experiment. Densitometry data were analyzed by
using the Student’s t test and expressed as mean F SE. P < 0.05 wasconsidered statistically significant.
Results
Interleukin-1B–inducible inducible nitric oxide synthaseexpression and nitric oxide production in aortic endothelialcells. To examine whether IL-1h stimulates NO production, RAECswere stimulated with 10 ng/mL IL-1h and the time course of NOaccumulation in the culturemediumwas determined (Fig. 1A). IL-1hstimulated a robust increase in NO production. Preincubation ofRAEC with 20 nmol/L recombinant human 16-kDa PRL for 1 hourresulted in a reproducible and significant reduction in basal as wellas IL-1h–induced increase in NO production. The reduction in NOproduction was accompanied by a reduction in IL-1h–inducibleiNOS protein expression in response to 16-kDa PRL (Fig. 1B) and IL-1h–inducible iNOS enzyme activity (Fig. 1C). Further analysisshowed that 16-kDa PRL attenuated iNOS gene expression asdetermined by reverse transcription-PCR (RT-PCR; Fig. 1D). IL-1hstimulated an increase in iNOS RNA expression between 1 and4 hours, reaching a maximum of 20-fold induction at 24 hours (Fig.1D, lanes 1-5). Preincubation of RAEC for 1 hour with 16-kDa PRLresulted in a pronounced inhibition of basal as well as the IL-1h–inducible increase in iNOS gene expression (Fig. 1D, lanes 6-10).Control histone H3.3 expression remained unchanged. 16-kDa PRLinhibition of IL-1h–inducible iNOS mRNA levels was further
confirmed by RNase protection analysis (data not shown). Thus, inRAEC, 16-kDa PRL attenuated IL-1h–inducible NO production byreducing iNOS gene expression.Nuclear factor-KB signaling to the inducible nitric oxide
synthase gene. Multiple cytokines and signaling pathwaysregulate the transcription of the iNOS gene (29). IL-1h is knownto activate both the NF-nB and the IRF-1 signaling pathways toregulate iNOS gene transcription (27–29). To determine which ofthe IL-1h–inducible signaling pathways might be a target of 16-kDa PRL inhibition, we analyzed NF-nB or IRF-1 binding to therat iNOS promoter (27) in electrophoretic mobility shift assays(EMSA). Using the NF-nB oligonucleotide, a complex of threebands was transiently induced at 30 minutes of IL-1h stim-ulation in RAEC (Fig. 2A, lane 2). This complex was specific as itwas competed by increasing molar excess of unlabeled probe(Fig. 2A, lanes 11 and 12). This complex contained p65 NF-nB asit was supershifted by anti-p65 NF-nB antibody (Fig. 2A, lane 13)and by anti-p50 NF-nB antibody (data not shown). The twofaster migrating bands represent p50/p65 NF-nB heterodimersand p50/p50 NF-nB homodimers, respectively (data not shown).Interestingly, this NF-nB-containing complex was not altered by16-kDa PRL treatment (Fig. 2A, lanes 6-10). These results showthat 16-kDa PRL did not affect IL-1h–inducible NF-nB binding tothe iNOS promoter in RAEC. Consistent with a lack of 16-kDaPRL effect on NF-nB DNA binding at the iNOS promoter, 16-kDaPRL treatment also did not affect IL-1h–inducible transientdown-regulation of the NF-nB inhibitor InBa (Fig. 2B). Theseresults show that 16-kDa PRL did not affect IL-1h–inducibleInBa degradation or NF-nB signaling to the iNOS promoterin RAEC.16-kDa Prolactin inhibits IFN regulatory factor 1 signaling
to the inducible nitric oxide synthase gene. We next analyzedIRF-1 binding to the composite IRF-1/C/EBP binding site in the rat
Figure 1. 16-kDa PRL attenuatesIL-1h–inducible NO production and iNOSexpression in aortic endothelial cells.RAECs were pretreated for 1 hour with20 nmol/L 16-kDa PRL followed byincubation with 10 ng/mL IL-1h in thecontinued presence of 16-kDa PRL.A, NO (nitrite) levels that haveaccumulated in the culture medium weredetermined by the Griess reaction (34).Columns, mean of two independentexperiments; bars, F SE. *, P < 0.05; **,P < 0.01. B, Western blot analysis of iNOS(131-kDa) protein expression. h-tubulin(55-kDa) was used as a protein loadingcontrol. Lane C, positive control fromLPS-treated RAW247.6 cells. Columns,mean of four independent experiments;bars, F SE. *, P < 0.05; **, P < 0.005.C, iNOS enzyme levels were determinedby the [3H]L-arginine to [3H]L-citrullineconversion assay (35). D, RT-PCRanalysis of iNOS RNA expression. HistoneH3.3 was used as a RT-PCR control.Points, mean of four independentexperiments; bars, F SE. *, P < 0.005; **,P < 0.001.
Cancer Research
Cancer Res 2005; 65: (17). September 1, 2005 7986 www.aacrjournals.org
Cancer Research. on September 13, 2018. © 2005 American Association forcancerres.aacrjournals.org Downloaded from

iNOS promoter (27). Incubation of RAEC with IL-1h for 1 hourinduced the formation of a complex (Fig. 3A, lane 2), which wasspecific as it was competed by increasing molar excess of unlabeledprobe (Fig. 3A, lanes 3 and 4). This IL-1a-inducible complexcontains IRF-1 as it was supershifted by anti-IRF-1 antibody(Fig. 3A, lane 5) and exhibited similar mobility with the IFN-g-inducible IRF-1-positive control (Fig. 3A, lane 6). The darker, slowermigrating complex represented C/EBP binding to the IRF-1/C/EBPcomposite site (data not shown; ref. 27). IL-1h stimulated atransient increase in IRF-1 binding at 1 hour to the rat iNOSpromoter (Fig. 3B, lanes 1-4), with binding returning to basal levelsby 8 hours. Interestingly, IL-1h–inducible IRF-1 binding wasinhibited by 16-kDa PRL treatment (Fig. 3B, lanes 5-8). Thus, 16-kDa PRL selectively blocked only the IL-1h–inducible IRF-1 but notNF-nB binding to the iNOS promoter in RAEC.16-kDa Prolactin inhibits signal transducer and activator of
transcription 1 signaling to the IFN regulatory factor 1 gene.We further examined whether 16-kDa PRL inhibited IL-1h–inducible IRF-1 gene expression. RAECs were pretreated with or
without 20 nmol/L 16-kDa PRL for 1 hour before stimulation with10 ng/mL IL-1h, and IRF-1 gene expression was analyzed by RT-PCR (Fig. 3C). IL-1h stimulated a rapid but transient increase inIRF-1 gene expression, which was maximal at 1 hour and declined
Figure 2. 16-kDa PRL does not inhibit IL-1h–inducible NF-nB bindingto the iNOS promoter. A, NF-nB binding to a rat iNOS specific NF-nBoligonucleotide. RAECs were either treated with 10 ng/mL IL-1h alone(lanes 1-5 ) or pretreated for 1 hour with 20 nmol/L 16-kDa PRL followed byincubation with IL-1h in the continued presence of 16-kDa PRL (lanes 6-10 ).Nuclear extracts (10 Ag) were used in EMSA to examine IL-1h–inducible NF-nBbinding to the rat iNOS promoter. Using the 30-minute IL-1h–stimulated nuclearextracts, the specificity of the NF-nB complex (bracket ) was determined bycompetition with 10- or 40-fold molar excess of cold oligonucleotides (lanes 11and 12) and by supershift (S.S. ) analysis with anti-p65 NF-nB antibody(lane 13). Lane 14, positive control for NF-nB binding using LPS-treated RAECextracts. Columns, mean of three independent experiments; bars, F SE.B, 16-kDa PRL does not inhibit the IL-1h–inducible transient down-regulation ofInBa (37-kDa). Whole-cell extracts (20 Ag) from RAEC treated as in A wereimmunoblotted with anti-InBa antibody. h-tubulin was used as a protein loadingcontrol. Representative blot of two independent experiments.
Figure 3. 16-kDa PRL inhibits IL-1h–inducible IRF-1 expression. A, IRF-1binding to the iNOS promoter using a rat iNOS-specific oligonucleotide thatcontains a compound IRF-1/C/EBP binding site as probe (27). RAECs werestimulated with 10 ng/mL IL-1h for 30 minutes (lane 2 ). The specificity of theIL-1h–inducible IRF-1 complex was determined by competition with 10- or 40-foldmolar excess of cold oligonucleotides (lanes 3 and 4) and by supershift analysiswith anti-IRF-1 antibody (lane 5). Lane 6, positive control for IRF-1 bindingusing IFN-g–stimulated RAEC extracts. B, Time course of IL-1h–inducible IRF-1binding to the iNOS promoter (lanes 1-4) and its inhibition by 16-kDa PRLtreatment (lanes 5-8). Columns, mean of three independent experiments; bars,F SE. *, P < 0.01. C, RT-PCR analysis of IRF-1 RNA expression in response toIL-1h stimulation in the absence (lanes 1-5) or presence (lanes 6-10 ) of 1-hourpretreatment with 16-kDa PRL. Histone H3.3 was employed as a RT-PCR control.Points, mean of four independent experiments; bars, F SE. *, P < 0.002.
16-kDa Prolactin Inhibits p38 MAPK/Stat1/IRF-1 Signaling to iNOS/NO
www.aacrjournals.org 7987 Cancer Res 2005; 65: (17). September 1, 2005
Cancer Research. on September 13, 2018. © 2005 American Association forcancerres.aacrjournals.org Downloaded from
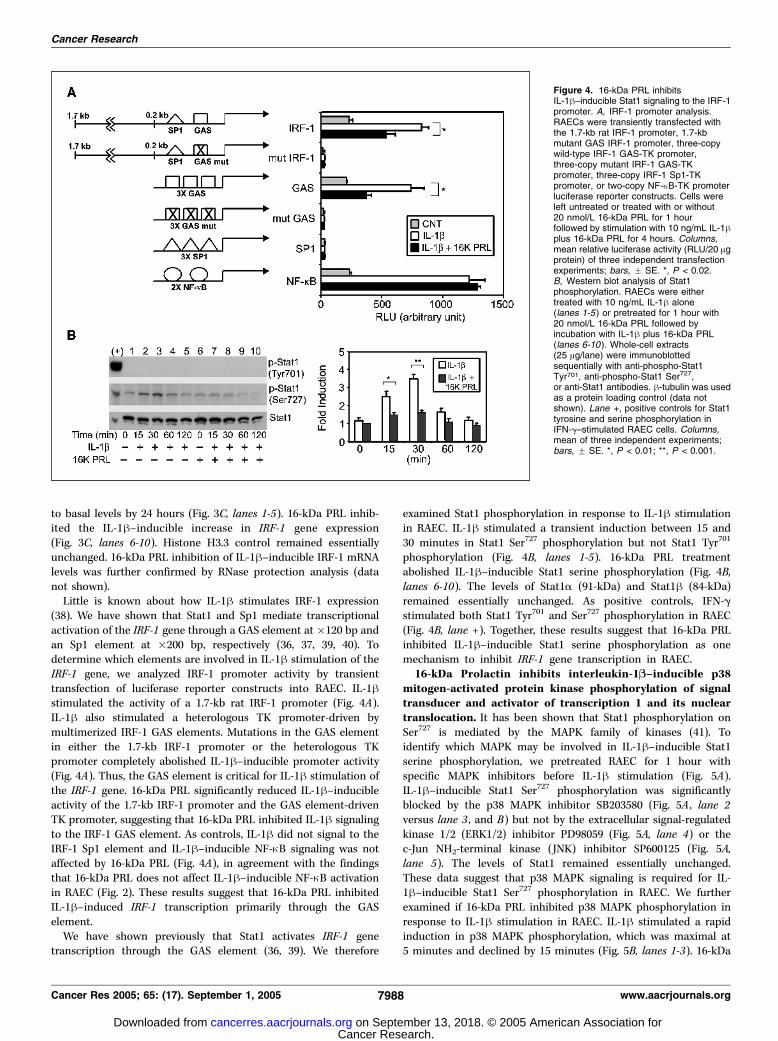
to basal levels by 24 hours (Fig. 3C, lanes 1-5). 16-kDa PRL inhib-ited the IL-1h–inducible increase in IRF-1 gene expression(Fig. 3C, lanes 6-10). Histone H3.3 control remained essentiallyunchanged. 16-kDa PRL inhibition of IL-1h–inducible IRF-1 mRNAlevels was further confirmed by RNase protection analysis (datanot shown).Little is known about how IL-1h stimulates IRF-1 expression
(38). We have shown that Stat1 and Sp1 mediate transcriptionalactivation of the IRF-1 gene through a GAS element at �120 bp andan Sp1 element at �200 bp, respectively (36, 37, 39, 40). Todetermine which elements are involved in IL-1h stimulation of theIRF-1 gene, we analyzed IRF-1 promoter activity by transienttransfection of luciferase reporter constructs into RAEC. IL-1hstimulated the activity of a 1.7-kb rat IRF-1 promoter (Fig. 4A).IL-1h also stimulated a heterologous TK promoter-driven bymultimerized IRF-1 GAS elements. Mutations in the GAS elementin either the 1.7-kb IRF-1 promoter or the heterologous TKpromoter completely abolished IL-1h–inducible promoter activity(Fig. 4A). Thus, the GAS element is critical for IL-1h stimulation ofthe IRF-1 gene. 16-kDa PRL significantly reduced IL-1h–inducibleactivity of the 1.7-kb IRF-1 promoter and the GAS element-drivenTK promoter, suggesting that 16-kDa PRL inhibited IL-1h signalingto the IRF-1 GAS element. As controls, IL-1h did not signal to theIRF-1 Sp1 element and IL-1h–inducible NF-nB signaling was notaffected by 16-kDa PRL (Fig. 4A), in agreement with the findingsthat 16-kDa PRL does not affect IL-1h–inducible NF-nB activationin RAEC (Fig. 2). These results suggest that 16-kDa PRL inhibitedIL-1h–induced IRF-1 transcription primarily through the GASelement.We have shown previously that Stat1 activates IRF-1 gene
transcription through the GAS element (36, 39). We therefore
examined Stat1 phosphorylation in response to IL-1h stimulationin RAEC. IL-1h stimulated a transient induction between 15 and30 minutes in Stat1 Ser727 phosphorylation but not Stat1 Tyr701
phosphorylation (Fig. 4B, lanes 1-5). 16-kDa PRL treatmentabolished IL-1h–inducible Stat1 serine phosphorylation (Fig. 4B,lanes 6-10). The levels of Stat1a (91-kDa) and Stat1h (84-kDa)remained essentially unchanged. As positive controls, IFN-gstimulated both Stat1 Tyr701 and Ser727 phosphorylation in RAEC(Fig. 4B, lane +). Together, these results suggest that 16-kDa PRLinhibited IL-1h–inducible Stat1 serine phosphorylation as onemechanism to inhibit IRF-1 gene transcription in RAEC.16-kDa Prolactin inhibits interleukin-1B–inducible p38
mitogen-activated protein kinase phosphorylation of signaltransducer and activator of transcription 1 and its nucleartranslocation. It has been shown that Stat1 phosphorylation onSer727 is mediated by the MAPK family of kinases (41). Toidentify which MAPK may be involved in IL-1h–inducible Stat1serine phosphorylation, we pretreated RAEC for 1 hour withspecific MAPK inhibitors before IL-1h stimulation (Fig. 5A).IL-1h–inducible Stat1 Ser727 phosphorylation was significantlyblocked by the p38 MAPK inhibitor SB203580 (Fig. 5A , lane 2versus lane 3 , and B) but not by the extracellular signal-regulatedkinase 1/2 (ERK1/2) inhibitor PD98059 (Fig. 5A, lane 4) or thec-Jun NH2-terminal kinase (JNK) inhibitor SP600125 (Fig. 5A,lane 5). The levels of Stat1 remained essentially unchanged.These data suggest that p38 MAPK signaling is required for IL-1h–inducible Stat1 Ser727 phosphorylation in RAEC. We furtherexamined if 16-kDa PRL inhibited p38 MAPK phosphorylation inresponse to IL-1h stimulation in RAEC. IL-1h stimulated a rapidinduction in p38 MAPK phosphorylation, which was maximal at5 minutes and declined by 15 minutes (Fig. 5B, lanes 1-3). 16-kDa
Figure 4. 16-kDa PRL inhibitsIL-1h–inducible Stat1 signaling to the IRF-1promoter. A, IRF-1 promoter analysis.RAECs were transiently transfected withthe 1.7-kb rat IRF-1 promoter, 1.7-kbmutant GAS IRF-1 promoter, three-copywild-type IRF-1 GAS-TK promoter,three-copy mutant IRF-1 GAS-TKpromoter, three-copy IRF-1 Sp1-TKpromoter, or two-copy NF-nB-TK promoterluciferase reporter constructs. Cells wereleft untreated or treated with or without20 nmol/L 16-kDa PRL for 1 hourfollowed by stimulation with 10 ng/mL IL-1hplus 16-kDa PRL for 4 hours. Columns,mean relative luciferase activity (RLU/20 Agprotein) of three independent transfectionexperiments; bars, F SE. *, P < 0.02.B, Western blot analysis of Stat1phosphorylation. RAECs were eithertreated with 10 ng/mL IL-1h alone(lanes 1-5 ) or pretreated for 1 hour with20 nmol/L 16-kDa PRL followed byincubation with IL-1h plus 16-kDa PRL(lanes 6-10 ). Whole-cell extracts(25 Ag/lane) were immunoblottedsequentially with anti-phospho-Stat1Tyr701, anti-phospho-Stat1 Ser727,or anti-Stat1 antibodies. h-tubulin was usedas a protein loading control (data notshown). Lane +, positive controls for Stat1tyrosine and serine phosphorylation inIFN-g–stimulated RAEC cells. Columns,mean of three independent experiments;bars, F SE. *, P < 0.01; **, P < 0.001.
Cancer Research
Cancer Res 2005; 65: (17). September 1, 2005 7988 www.aacrjournals.org
Cancer Research. on September 13, 2018. © 2005 American Association forcancerres.aacrjournals.org Downloaded from

PRL inhibited IL-1h–inducible p38 MAPK phosphorylation inRAEC (Fig. 5B, lanes 4-6). The levels of p38 MAPK remainedessentially unchanged. These data show that 16-kDa PRLinhibited IL-1h–inducible p38 MAPK phosphorylation in RAEC.To examine the functional consequence of p38 MAPK
inhibition on IL-1h–inducible Stat1 signaling, we analyzed IRF-1promoter activity by transient transfection assays (Fig. 5C).Addition of 16-kDa PRL, the p38 MAPK inhibitor SB203580, orboth all significantly reduced IL-1h signaling to the IRF-1promoter. The extent of inhibition was similar for 16-kDa PRLand/or p38 MAPK inhibitor, suggesting that 16-kDa PRL and thep38 MAPK inhibitor acted in the same pathway to inhibit IL-1h–
inducible IRF-1 promoter activity. On the other hand, neither16-kDa PRL nor the p38 MAPK inhibitor reduced IL-1h–inducibleNF-nB-dependent TK promoter activity, in agreement with ourobservation that 16-kDa PRL does not affect the NF-nB signalingpathway in RAEC (Figs. 2 and 4A). Together, the data suggest that16-kDa PRL inhibited IL-1h–inducible p38 MAPK phosphorylationof Stat1 and blocked Stat1-mediated activation of the IRF-1 genein RAEC.As a further step in understanding how 16-kDa PRL inhibits
Stat1 signaling, we examined Stat1 nuclear translocation byimmunofluorescence microscopy (Fig. 5D). For these experiments,higher levels of cytokines were used to maximize Stat1 nuclear
Figure 5. 16-kDa PRL inhibits IL-1 h–inducible p38 MAPK activation of Stat1 and Stat1 nuclear translocation in RAEC. A, Western blot analysis of Stat1 serinephosphorylation in the presence of MAPK inhibitors. RAECs were incubated with medium alone (lane 1), 20 ng/mL IL-1h (lane 2), or 20 Amol/L of each of threeMAPK inhibitors for 1 hour followed by 30 minutes of IL-1h. These include the p38 MAPK inhibitor SB203580 (SB ; lane 3), ERK1/2 inhibitor PD98059 (PD ; lane 4 ), andJNK inhibitor SP600125 (SP ; lane 5 ). Whole-cell extracts (25 Ag/lane) were immunoblotted with anti-phospho-Stat1 (Ser727; pStat1) and reblotted with anti-Stat1antibodies. h-tubulin was used as a protein loading control (data not shown). Columns, mean of three independent experiments; bars, F SE. *, P < 0.001. B, Western blotanalysis of p38 MAPK phosphorylation. RAECs were either treated without (lane 1 ) or with 10 ng/mL IL-1h for 5 to 15 minutes (lanes 2 and 3) or pretreated 1 hour with 20nmol/L 16-kDa PRL followed by incubation without (lane 4) or with IL-1h in the continued presence of 16-kDa PRL (lanes 5 and 6). Whole-cell extracts (25 Ag/lane) wereimmunoblotted with anti-phospho-p38 MAPK first and then reblotted with anti-p38 MAPK antibodies. h-tubulin was used as protein loading control (data not shown).Columns, mean of three independent experiments; bars, F SE. *, P < 0.05; **, P < 0.01. C, IRF-1 promoter analysis. RAECs were transiently transfected with the 1.7-kbrat IRF-1 promoter or the two-copy NF-nB-TK promoter luciferase reporter constructs. Cells were left untreated or treated with 10 ng/mL IL-1h for 4 hours, 20 nmol/L16-kDa PRL for 1 hour followed by IL-1h, 20 Amol/L SB203590 for 1 hour followed by IL-1h, or 16-kDa PRL plus SB203590 for 1 hour followed by IL-1h. Columns,mean relative luciferase activity (RLU/20 Ag protein) of three independent transfection experiments; bars, F SE. *, P < 0.001. D, RAECs (7. 5 � 104 cells) were culturedin 12-well plates and incubated in serum-free medium for 24 hours. Cells were left untreated, treated with 200 ng/mL IL-1h for 30 minutes, pretreated with 50 nmol/L16-kDa PRL for 1 hour followed by IL-1h plus 16-kDa PRL for 30 minutes, 50 nmol/L 16-kDa PRL for 1.5 hours, or 500 ng/mL IFN-g for 30 minutes as a positive control.Cells were fixed, permeabilized, incubated with anti-Stat1 antibody (green ), and counterstained with DAPI for DNA (blue ), and the images were merged. Representativeof four independent experiments. The cytokine concentrations used were empirically determined for optimal Stat1 nuclear translocation.
16-kDa Prolactin Inhibits p38 MAPK/Stat1/IRF-1 Signaling to iNOS/NO
www.aacrjournals.org 7989 Cancer Res 2005; 65: (17). September 1, 2005
Cancer Research. on September 13, 2018. © 2005 American Association forcancerres.aacrjournals.org Downloaded from

entry (42). In control RAEC, Stat1 (green) was localized primarilyin the cytoplasm as shown by the lack of Stat1 staining in thenucleus (DAPI in blue) in the merged image. Stimulation of RAECwith IL-1h for 30 minutes induced the nuclear translocation ofStat1. Interestingly, 16-kDa PRL effectively blocked IL-1h–inducible Stat1 nuclear translocation. Neither 16-kDa PRL alonenor p38 MAPK inhibitor SB203580 (data not shown) had anyeffect on Stat1 cytoplasmic localization. IFN-g, which inducedStat1 phosphorylation on both Tyr701 and Ser727 (Fig. 4B),stimulated Stat1 nuclear translocation (Fig. 5D). These resultsreveal for the first time that 16-kDa PRL, via a mechanism thatinvolved p38 MAPK inhibition, blocked Stat1 serine phosphory-lation and nuclear translocation. The inhibition of Stat1activation blocked IL-1h–inducible IRF-1 gene expression andattenuated iNOS/NO expression in RAEC.Taken together, our data indicate that 16-kDa PRL modulates
endothelial cell activity through the inhibition of a specificintracellular pathway (Fig. 6). Although IL-1h regulates iNOSgene expression and NO production in aortic endothelial cells byactivating both IRF-1 and NF-nB signaling pathways, 16-kDa PRLattenuates IL-1h effects by antagonizing one of these signalingpathways. 16-kDa PRL effectively inhibits IL-1h–inducible p38MAPK phosphorylation, resulting in blocking Stat1 Ser727
phosphorylation, its subsequent nuclear translocation andactivation of Stat1 target genes, such as IRF-1 . In contrast, 16-kDa PRL does not affect IL-1h–inducible NF-nB signaling to theiNOS gene. This pathway results in 16-kDa PRL attenuation ofIL-1h–induced iNOS expression and NO production in aorticendothelial cells.
Discussion
We have shown that 16-kDa PRL attenuates IL-1h–inducibleiNOS gene expression in RAECs, and this effect is mediated inpart through the inhibition of the expression of IRF-1, a factornecessary for iNOS expression. IRF-1 is a transcription factorthat regulates the expression of genes involved in immune andinflammatory responses (43, 44). However, the role andregulation of IRF-1 in endothelial cells are not known. Resultsfrom this study suggest that IRF-1 may play a role in theregulation of angiogenesis through modulating iNOS expressionin endothelial cells. Although 16-kDa PRL has been shown toinhibit angiogenesis and tumorigenesis in both in vitro andin vivo studies (11–14, 20), the mechanism leading to theantiangiogenic activity of 16-kDa PRL remains largely unknown.Our study elucidates one of the molecular mechanisms thatmay be involved in the antiangiogenic and antitumor effect of16-kDa PRL.IL-1h–inducible Stat1 Ser727 phosphorylation plays an impor-
tant role in activating IRF-1 expression (Figs. 3 and 4). Althoughboth Tyr701 and Ser727 phosphorylation of Stat1 occurs in re-sponse to cytokine and growth factor stimulation (41, 45), IL-1hdid not induce Stat1 Tyr701 phosphorylation but stimulatedStat1 Ser727 phosphorylation through activation of p38 MAPK inRAEC (Figs. 4 and 5; refs. 41, 46). The phosphorylated Ser727
residue on Stat1 has been shown to mediate the association ofStat1 with coactivators to regulate chromatin remodeling attarget genes (47). We have shown previously that extensivehistone acetylation and chromatin remodeling at the IRF-1promoter is required for IRF-1 gene transcription (48). Becausemutations in the GAS element abolished IRF-1 promoter activity(Fig. 4A), it is likely that serine-phosphorylated Stat1 regulatesIL-1h–inducible IRF-1 expression through the GAS element inthe IRF-1 promoter. We thus suggest that 16-kDa PRL inhibitsIRF-1 gene expression through down-regulation of the Stat1signaling pathway via inhibition of Stat1 Ser727 phosphorylationand Stat1 nuclear translocation. Consistent with our observationthat Stat1 is involved in antiangiogenic signaling mechanisms,recent studies also show that down-regulation of Stat1 levelscontributes to the antiangiogenic effects of endostatin (49), butit is not known whether Tyr701 and/or Ser727 phosphorylation ofStat1 is involved.Our studies indicate that the inhibition of iNOS expression by
16-kDa PRL in aortic endothelial cells is not mediated throughthe NF-nB pathway (Fig. 2). This is in contrast to the studies byMacotela et al. (50) and Tabruyn et al. (51), which showed that16-kDa PRL activates NF-nB activity in primary mouse embryoniclung fibroblasts and bovine brain capillary endothelial cells,respectively. Because the IL-1h–inducible NF-nB pathway isessentially unperturbed by 16-kDa PRL in RAEC, these resultsmay explain why 16-kDa PRL only inhibited f40% iNOS genetranscription, which is likely contributed by the IL-1h–inducibleIRF-1 pathway. Interestingly, 16-kDa PRL also inhibited basalIRF-1 (Fig. 3C) and basal iNOS (Fig. 1D) gene expression in theabsence of IL-1h stimulation, suggesting that other pathwaysmight be involved. Thus, 16-kDa PRL regulates multiple signalingpathways, including Stat1 (Figs. 4 and 5), IRF-1 (Fig. 3), Ras, Raf,ERK1/2 MAPK (12, 52), p38 MAPK (Fig. 5), and NF-nB (50, 51), ina cell type– and tissue-specific manner.Several antiangiogenic proteins (i.e., angiostatin and endo-
statin) are fragments of larger protein counterparts (16, 17). Both
Figure 6. Model of 16-kDa PRL inhibition of IL-1h signaling to iNOS/NOexpression in RAECs. IL-1h binding to the IL-1h receptor (IL-1R ) activatesp38 MAPK that leads to Stat1 Ser727 phosphorylation, Stat1 nucleartranslocation, and activation of Stat1 target genes, such as IRF-1 . IL-1h alsostimulates NF-nB activation and nuclear translocation. Both IRF-1 and NF-nBbind to specific sites on the iNOS promoter and together stimulate iNOS genetranscription and NO production in aortic endothelial cells. 16-kDa PRLbinding to a yet unidentified 16-kDa PRL receptor (16-kDa PRL-R) inhibitsIL-1h–inducible p38 MAPK activation, resulting in inhibition of Stat1 Ser727
phosphorylation, Stat1 nuclear translocation, and IRF-1 gene expression. InRAEC, 16-kDa PRL does not affect IL-1h–inducible NF-nB signaling to theiNOS promoter. Thus, 16-kDa PRL down-regulates IL-1h–inducible iNOS/NOexpression in aortic endothelial cells through the inhibition of the p38 MAPK/Stat1/IRF-1 pathway.
Cancer Research
Cancer Res 2005; 65: (17). September 1, 2005 7990 www.aacrjournals.org
Cancer Research. on September 13, 2018. © 2005 American Association forcancerres.aacrjournals.org Downloaded from

angiostatin and endostatin exert their antiangiogenic effects inpart by affecting endothelial cell NO production (53, 54).Recently, Gonzalez et al. (55) showed that 16-kDa PRL atten-uated VEGF-induced constitutive eNOS activation and NO pro-duction in endothelial cells. This action led to an inhibition ofVEGF-induced proliferation of endothelial cells and endothelium-dependent vasorelaxation. Thus, blockage of NO production isone of the mechanisms of 16-kDa PRL action in endothelialcells. Our data extend these observations to show that 16-kDaPRL attenuated IL-1h–inducible iNOS expression and NOproduction in endothelial cells. Interestingly, in other cell types,such as embryonic lung fibroblasts and alveolar type II cells, 16-kDa PRL stimulated cytokine-inducible iNOS and NO production,suggesting that 16-kDa PRL may play additional roles in inflam-matory and immune responses in a cell type– and tissue-specificmanner (56).16-kDa PRL likely functions through a cell surface receptor that is
distinct from the 23-kDa PRL receptor. Clapp and Weiner (57)showed the presence of specific, high-affinity (Kd = 9.9 nmol/L), andsaturable (Bmax = 4.8 pmol/mg protein) binding sites for 16-kDa PRLon bovine brain capillary endothelial cells. However, the receptor(s)for 16-kDa PRL has not been identified. Our observation that 16-kDaPRL regulates signaling in RAEC suggests that the RAEC express the
16-kDa PRL receptors and will provide an excellent source forfurther receptor studies.That 16-kDa PRL has antiangiogenic and antitumor properties
has been observed for many years (8, 11–14, 20, 52, 58, 59).However, the lack of understanding of 16-kDa PRL signalingmechanisms has hampered the possibility of applying 16-kDa PRLin therapy. Understanding the signaling mechanism of 16-kDa PRLin endothelial cells will allow the design of better therapeuticstrategies targeting the angiogenic process. Our studies raise thepossibility that 16-kDa PRL can be used for the treatment ofdiseases characterized by abnormal angiogenesis, such as tumor-igenesis, metastasis, and proliferative retinopathies, and/or bychronic angiogenesis, such as rheumatoid arthritis and inflamma-tory bowel disease (15, 60, 61).
Acknowledgments
Received 2/22/2005; revised 6/18/2005; accepted 6/22/2005.Grant support: NIH grants P50 DK064233 (G.E. Garcia and L. Feng), RO1 DK54674
(L. Feng), RO1 HL75421 (N.T. Eissa), and RO1 CA86342 (S-H. Lin) and Elsa U. PardeeFoundation for Cancer Research (L-Y. Yu-Lee).
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
We thank Dr. Pawel Kolodziejski for critical comments and Alice Su for technicalsupport.
16-kDa Prolactin Inhibits p38 MAPK/Stat1/IRF-1 Signaling to iNOS/NO
www.aacrjournals.org 7991 Cancer Res 2005; 65: (17). September 1, 2005
References1. Lewis UJ, Singh RN, Sinha YN, et al. Glycosylatedhuman PRL. Endocrinology 1985;116:353–63.
2. Oetting WS, Tuazon PT, Traugh JA, et al. Phosphor-ylation of PRL. J Biol Chem 1986;261:1649–52.
3. Mittra I. A novel ‘‘cleaved PRL’’ in the rat pituitary. I.Biosynthesis, characterization and regulatory control.Biochem Biophys Res Commun 1980;95:1750–9.
4. Sinha YN, Gilligan TA. A cleaved form of PRL in themouse pituitary gland: identification and comparison ofin vitro synthesis and release in strains with high andlow incidences of mammary tumors. Endocrinology1984;114:2046–53.
5. Sinha YN, Gilligan TA, Lee DW, et al. Cleaved PRL:evidence for its occurrence in human pituitary glandand plasma. J Clin Endocrinol Metab 1985;60:239–43.
6. Sinha YN. Structural variants of PRL: occurrence andphysiological significance. Endocr Rev 1995;16:354–69.
7. Baldocchi RA, Tan L, King DS, et al. Mass spectro-metric analysis of the fragments produced by cleavageand reduction of rat PRL: evidence that the cleavingenzyme is cathepsin D. Endocrinology 1993;133:935–8.
8. Piwnica D, Touraine P, Struman I, et al. Cathepsin Dprocesses human PRL into multiple 16k-like N-terminalfragments: study of their antiangiogenic properties andphysiological relevance. Mol Endocrinol 2004;18:2522–42.
9. Clapp C. Analysis of the proteolytic cleavage of PRL bythe mammary gland and liver of the rat. Characteriza-tion of the cleaved and 16k forms. Endocrinology 1987;121:2055–64.
10. Clapp C, Torner L, Gutierrez-Ospina G, et al. The PRLgene is expressed in the hypothalamic-neurohypophy-seal system and the protein is processed into a 14-kDafragment with activity like 16-kDa PRL. Proc Natl AcadSci 1994;91:10384–8.
11. Clapp C, Martial JA, Guzman RC, et al. The 16-kilodalton N-terminal fragment of human PRL is apotent inhibitor of angiogenesis. Endocrinology 1993;133:1292–9.
12. Struman I, Bentzien F, Lee H, et al. Opposing actionsof intact and N-terminal fragments of the human PRL/growth hormone family members on angiogenesis: anefficient mechanism of the regulation of angiogenesis.Proc Natl Acad Sci 1999;96:1246–51.
13. Bentzien F, Struman I, Martini J, et al. Expression ofthe antiangiogenic factor 16K hPRL in human HCT116
colon cancer cells inhibits tumor growth in Rag1�/�mice. Cancer Res 2001;61:7356–62.
14. Duenas Z, Torner L, Corbacho AM, et al. Inhibition ofrat corneal angiogenesis by 16-kDa PRL and byendogenous PRL-like molecules. Invest Ophthalmol VisSci 1999;40:2498–505.
15. Carmeliet P. Angiogenesis in health and disease. NatMed 2003;9:653–60.
16. O’Reilly MS, Holmgren L, Shing Y, et al. Angiostatin: anovel angiogenesis inhibitor that mediates the suppres-sion of metastases by a Lewis lung carcinoma. Cell 1994;79:315–28.
17. O’Reilly MS, Boehm T, Shing Y, et al. Endostatin: anendogenous inhibitor of angiogenesis and tumorgrowth. Cell 1997;79:315–28.
18. O’Reilly MS, Holmgren L, Chen C, et al. Angiostatininduces and sustains dormancy of human primarytumors in mice. Nat Med 1996;2:689–92.
19. Dhanabal M, Ramchandran R, Waterman MJ, et al.Endostatin induces endothelial cell apoptosis. J BiolChem 1999;274:11721–6.
20. Kim J, Luo W, Chen D-T, et al. Antitumor activity ofthe 16-kDa PRL fragment in prostate cancer. Cancer Res2003;63:386–93.
21. Saijo Y, Tanaka M, Miki M, et al. Proinflammatorycytokine IL-1b promotes tumor growth of Lewis lungcarcinoma by induction of angiogenic factors: in vivoanalysis of tumor-stromal interaction. J Immunol 2002;169:469–75.
22. Song X, Voronov E, Dvorkin T, et al. Differentialeffects of IL-1a and IL-1b on tumorigenicity patternsand invasiveness. J Immunol 2003;171:6448–56.
23. Bar D, Apte RN, Voronov E, et al. A continuousdelivery system of IL-1 receptor antagonist reducesangiogenesis and inhibits tumor development. FASEB J2004;18:161–3.
24. Ziche M, Parenti A, Ledda F, et al. Nitric oxidepromotes proliferation and plasminogen activatorproduction by coronary venular endothelium throughendogenous bFGF. Circ Res 1997;80:845–52.
25. Murohara T, Witzenbichler B, Spyridopoulos I, et al.Role of endothelial nitric oxide synthase in endothelial cellmigration. Arterioscler ThrombVasc Biol 1999;19:1156–61.
26. Walford G, Loscalzo J. Nitric oxide in vascularbiology. J Thromb Haemost 2003;1:2112–8.
27. Teng X, Zhang H, Snead C, et al. Molecularmechanisms of iNOS induction by IL-1b and IFN-g in
rat aortic smooth muscle cells. Am J Physiol Cell Physiol2002;282:C144–52.
28. Saura M, Zaragoza C, Bao C, et al. Interaction ofinterferon regulatory factor-1 and nuclear factor nBduring activation of inducible nitric oxide synthasetranscription. J Mol Biol 1999;289:459–71.
29. Taylor BS, Geller DA. Molecular regulation of thehuman inducible nitric oxide synthase (iNOS) gene.Shock 2000;13:413–24.
30. Galfione M, Luo W, Kim J, et al. Expression andpurification of the antiangiogenesis inhibitor 16-kDaPRL fragment from insect cells. Protein Expr Purif 2003;28:252–9.
31. Stevens AM, Yu-Lee Ly. The transcription factorinterferon regulatory factor-1 is expressed during bothearly G1 and the G1-S transition in the PRL-inducedlymphocyte cell cycle. Mol Endocrinol 1992;6:2236–43.
32. Diglio CA, Grammas P, Giacomelli F, et al. Angiogen-esis in rat aorta ring explant cultures. Lab Invest 1988;60:523–31.
33. Garcia GE, Xia Y, Chen S, et al. NF-nB-dependentfractalkine induction in rat aortic endothelial cellsstimulated by IL-1h, TNF-a, and LPS. J Leukoc Biol2000;67:577–84.
34. Green LC, Wagner DA, Glogowski J, et al. Analysis ofnitrate, nitrite, and [15N] nitrate in biological fluids. AnalBiochem 1982;126:131–8.
35. Guo FH, Comhair SA, Zheng S, et al. Molecularmechanisms of increased nitric oxide (NO) in asthma:evidence for transcriptional and post-translationalregulation of NO synthesis. J Immunol 2000;164:5970–80.
36. Wang Y, O’Neal KD, Yu-Lee Ly. Multiple PRL receptorcytoplasmic residues and Stat1 mediate PRL signaling tothe IRF-1 promoter. Mol Endocrinol 1997;11:1353–64.
37. McAlexander MB, Yu-Lee Ly. Sp1 is required for PRLactivation of the interferon regulatory factor 1 gene. MolCell Endocrinol 2001;184:135–41.
38. Fujita T, Reis LF, Watanabe N, et al. Induction ofthe transcription factor IRF-1 and interferon-hmRNAs by cytokines and activators of second-messenger pathways. Proc Natl Acad Sci 1989;86:9936–40.
39. Stevens AM, Wang Y, Sieger KA, et al. Biphasictranscriptional regulation of the interferon regulatoryfactor-1 gene by PRL: involvement of g-interferonactivated sequence and Stat-related proteins. MolEndocrinol 1995;9:513–25.
Cancer Research. on September 13, 2018. © 2005 American Association forcancerres.aacrjournals.org Downloaded from

Cancer Research
Cancer Res 2005; 65: (17). September 1, 2005 7992 www.aacrjournals.org
40. Wang Y, Yu-Lee Ly. Multiple Stat complexes interactat the IRF-1 GAS in PRL-stimulated Nb2 T cells. Mol CellEndocrinol 1996;121:19–28.
41. Decker T, Kovarik P. Serine phosphorylation of Stats.Oncogene 2000;19:2628–37.
42. Strehlow I, Schindler C. Amino-terminal signaltransducer and activator of transcription (STAT)domains regulate nuclear translocation and STATdeactivation. J Biol Chem 1998;273:28049–56.
43. Taniguchi T, Ogasawara K, Takaoka A, et al. IRFfamily of transcription factors as regulators of hostdefense. Annu Rev Immunol 2001;19:623–55.
44. Yu-Lee Ly. PRL modulation of immune andinflammatory responses. Recent Prog Horm Res 2002;57:435–55.
45. Levy DE, Darnell JE Jr. Stats: transcriptional controland biological impact. Nat Rev Mol Cell Biol 2002;3:651–62.
46. Nguyen H, Chatterjee-Kishore M, Jiang Z, et al.IRAK-dependent phosphorylation of Stat1 on serine727 in response to interleukin-1 and effects ongene expression. J Interferon Cytokine Res 2003;23:183–92.
47. Varinou L, Ramsauer K, Karaghiosoff M, et al.Phosphorylation of the Stat1 transactivation domain isrequired for full-fledged IFNg-dependent innate immu-nity. Immunity 2003;19:793–802.
48. McAlexander MB, Yu-Lee Ly. PRL activation of IRF-1transcription involves changes in histone acetylation.FEBS Lett 2001;488:91–4.
49. Abdollahi A, Hahnfeldt P, Grone H-J, et al. Endo-statin’s antiangiogenic signaling network. Mol Cell 2004;13:649–63.
50. Macotela Y, Mendoza C, Corbacho AM, et al. 16k PRLinduces NFnB activation in pulmonary fibroblasts. JEndocrinol 2002;175:R13–8.
51. Tabruyn SP, Sorlet CM, Rentier-Delrue F, et al. Theantiangiogenic factor 16K human PRL induces caspase-dependent apoptosis by a mechanism that requiresactivation of nuclear factor-nB. Mol Endocrinol 2003;17:1815–23.
52. D’Angelo G, Martini J, Iiri T, et al. 16K human PRLinhibits vascular endothelial growth factor-inducedactivation of Ras in capillary endothelial cells. MolEndocrinol 1999;13:692–704.
53. Koshida R, Ou J, Matsunaga T, et al. Angiostatin: anegative regulator of endothelial-dependent vasodila-tion. Circulation 2002;107:803–6.
54. Urbich C, Reissner A, Chavakis E, et al. Dephosphor-ylation of endothelial nitric oxide synthase contributesto the antiangiogenic effects of endostatin. FASEB J2002;16:706–8.
55. Gonzalez C, Corbacho AM, Eiserich JP, et al. 16K PRLinhibits activation of endothelial nitric oxide synthase,
intracellular calcium mobilization and endothelium-dependent vasorelaxation. Endocrinology 2004;145:5714–22.
56. Corbacho AM, Nava G, Eiserich J, et al. Proteolyticcleavage confers nitric oxide synthase inducing activityupon PRL. J Biol Chem 2000;275:13183–6.
57. Clapp C, Weiner RI. A specific, high affinity, saturablebinding site for the 16-kilodalton fragment of PRL oncapillary endothelial cells. Endocrinology 1992;130:1380–6.
58. Martini J, Piot C, Humeau LM, et al. The antiangio-genic factor 16K PRL induces programmed cell death inendothelial cells by caspase activation. Mol Endocrinol2000;14:1536–49.
59. Tabruyn SP, Nguyen N-Q, Cornet AM, et al. Theantiangiogenic factor 16K hPRL induces endothe-lial cell cycle arrest by acting at both the G0-G1
and the G2-M phases. Mol Endocrinol 2005;19:1932–42.
60. Duenas Z, Rivera JC, Quiroz-Mercado H, et al. PRL ineyes of patients with retinopathy of prematurity:implications with vascular regression. Invest Ophthal-mol Vis Sci 2004;45:2049–55.
61. Pan H, Nguyen N-QN, Yoshida H, et al. Moleculartargeting of antiangiogenic factor 16k hPRL inhibitsoxygen-induced retinopathy in mice. Invest OphthalmolVis Sci 2004;45:2413–9.
Cancer Research. on September 13, 2018. © 2005 American Association forcancerres.aacrjournals.org Downloaded from

2005;65:7984-7992. Cancer Res Sok-hyong Lee, Michiya Nishino, Tuhina Mazumdar, et al. Factor-1 PathwayTransducer and Activator of Transcription 1/IFN RegulatorySynthase Expression through Inhibition of the Signal 16-kDa Prolactin Down-Regulates Inducible Nitric Oxide
Updated version
http://cancerres.aacrjournals.org/content/65/17/7984
Access the most recent version of this article at:
Cited articles
http://cancerres.aacrjournals.org/content/65/17/7984.full#ref-list-1
This article cites 58 articles, 17 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/65/17/7984.full#related-urls
This article has been cited by 2 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://cancerres.aacrjournals.org/content/65/17/7984To request permission to re-use all or part of this article, use this link
Cancer Research. on September 13, 2018. © 2005 American Association forcancerres.aacrjournals.org Downloaded from