20031 gmps for management: how to prepare for a systems - based inspection – understanding the...
TRANSCRIPT

2003 1
GMPs for Management: How to Prepare for a Systems -
Based Inspection – Understanding the FDA’s
Inspectional Approach and Ensuring Compliance
This product was funded by a grant awarded under the President’s High Growth Job Training Initiative as implemented by the U.S. Department of Labor’s Employment & Training Administration. The information contained in this product was created by a
grantee organization and does not necessarily reflect the official position of the U.S. Department of Labor. All references to non-governmental companies or organizations, their services, products, or resources are offered for informational purposes and should
not be construed as an endorsement by the Department of Labor. This product is copyrighted by the institution that created it and is intended for individual
organizational, non-commercial use only.

DAY ONE FDA’s Inspectional Initiatives & Tools for Regulating Drugs and Biotech Drugs
FDA Structure and Organization - Regulations and the Law - FDA Guidelines
Administrative Enforcement Tools - 483 Lists of Observations - Warning Letters
Legal Enforcement Tools FDA Inspections trends
Examine the Fundamentals of a Systems-Based Inspection Learn the concepts of modern Quality systems including the "Six-system
Inspection Model" examined in detail and each sub-system: Quality System Facilities and Equipment System Production System Materials System Lab Control Systems Packaging and labeling system
DAY ONE
Risk - base FDA Inspections Learn how FDA will select firms for inspections using the Risk base Approach Understand the FDA's intent for the systems-based inspection program Identify the differences between systems-based and product-based inspections Determine the factors that may influence which sub-systems are inspected.
Review of additional cGMPs and the concept of Modern Quality Systems (Guidance for Industry Quality System Approach to Pharmaceutical cGMP Regulations)
Quality Quality by Design Quality Risk Management The Quality Unit
Other critical SOPs of interest to the FDA Batch Control Review Deviation Handling Failure Investigations Change Control CAPA Training Validation

DAY TWO Determine what additional the requirements under will be reviewed under the quality
systems model How do you comply with the additional Four Major Sections? Examined in detail and each sub-system: Management Responsibilities Resources Manufacturing Operations Other critical SOPs of interest to the FDA
Batch Control Review Deviation Handling Failure Investigations Training Validation
Documentation Essentials Controlling Documents Document Problems GMP Records Compliance Within Established Standards

DAY TWO
Pharmaceutical Development Q8,Q9 Key Development Steps
Ensuring Good Manufacturing Practices for Drug Products History of Regulatory Enforcement FDA Policy and Program Issues Hosting An FDA Inspection
Preparing for a successful FDA inspection How to develop an Inspection strategy and an inspection Tool kits (Checklist) Learn how to save time and money by planning and focusing on the right systems
for an inspection How to conducting Mock Inspections Plan an effective strategy to prepare for an inspection Maintain compliance for facilities and equipment
Questions & Wrap-Up

Module 1
FDA’s Inspectional Initiatives & Tools for Regulating Drugs and Biotech Drugs FDA Structure and Organization
- Regulations and the Law - FDA Guidelines
Administrative Enforcement Tools - 483 Lists of Observations - Warning Letters
Legal Enforcement Tools FDA Inspections trends

Historical Perspective
1938 FD&C 1962 GMPs 1978 cGMPs 1997 FDAMA 2000 (QSIT-Devices) 2001 Pilot Program (Drugs) 2002 System Based Inspection Program

How does FDA regulate? Routine inspections Controlling product approvals for
commercialization Surveillance Programs through Adverse
event reports Undercover surveillance (drug/device
promotion, distribution) Consumer complaints/former employee
complaints Bioresearch monitoring

Forms Commonly Used During FDA Inspections
FDA 482 - Notice of of Inspection FDA 483 - List of Observations FDA 484 - Receipt for Samples FDA 463 - Affidavit
Special purpose affidavits
Various special purpose forms EIR - Establishment Inspection Report

Outcomes & Enforcement
No Violation (possible Thank-you letter) 483 Citation Warning Letter Injunction Consent Decree Legal Proceedings

Warning Letter Definition
WARNING LETTER: An informal advisory to a firm communicating the agency's position on a matter but does not commit FDA to taking enforcement action.
The agency's policy is that Warning Letters should be issued for violations which are of regulatory significance in that failure to adequately and promptly take corrections may be expected to result in enforcement action should the violation(s) continue

Reasons for the Letters GMP
Repetitive reasons Failure to follow SOPs Packaging and Labeling Controls Sampling and testing of components Inadequate Specifications Process validation Finished product sampling and testing Production and process controls

Consent Decree Definition
Consent Decree: An injunction to which the defendant has agreed and which is filed in court
Injunction: An order issued by the Court requiring a defendant to do or refrain from doing a specified act

Reasons for a Consent Decree
Continual non-conformance in the quality management systems
Repeated Form 483 observations over several audits and inspections for GMP violations
Repeated warning letters for GMP violations Repeated unsatisfactory or no response to warning letters
and 483 observations Failure of the company to make progress or correct
deficiencies

Why Does FDA Use Enforcement
To warn companies To stop continuing bad behavior To put entire industry on notice To control dangerous products To establish legal precedent To punish wrongdoers

Summary of Consent Decrees
Abbott 11/99 QSR violations 256 violations by independent consultant 100 M$ fine
Schering Plough 5/2002 GMP problems since 1998 Broad based observations 500 M$ fine

Impact of the Decree
Financial Impact Fines Forfeiture of profits Injunction on sales of marketed drugs Delays in approvals Cost of compliance

Impact of the Decree
Compliance Impact Required to hire outside consultants to
ensure compliance Compliance plans Expert certification Retraining Completion of missing or inadequate
studies

Summary of Consent Decrees
GSK 4/2005 Products withdrawn from market due to
consistency of dose problems Independent party to analyze manufacturing CAPA plan required for submission Pay up to 10M$ per year if GSK fails in it’s
commitments Post 650M$ bond to guarantee defective product
is disposed or reconditioned Pharmakon
7/2005 Firm shut by permanent injunction order by court
Module 2
Examine the Fundamentals of a Systems-Based Inspection Learn the concepts of modern Quality systems including
the "Six-system Inspection Model" examined in detail and each sub-system:
Quality System Facilities and Equipment System Production System Materials System Lab Control Systems Packaging and labeling system

What is this new “SIX-SYSTEM INSPECTIONAPPROACH”
CGMPS and the concepts of modern
Quality systems

Pharmaceutical CGMPs for the 21st Century
Reference:
Pharmaceutical CGMPs for the 21st Century – A Risk Based Approach
Final Report – Fall 2004 Latest release: September 2006

Six-system Inspection Model
This diagram shows the relationship among the six systems: the quality system and the five manufacturing systems.

THE QUALITY SYSTEMS MODEL
The quality system provides the foundation for the manufacturing systems that are linked and function within it.
Much like QSIT, ISO9000/2000, ISO13485, “The Linked-Process Approach”

The FDA’s overhaul of the pharmaceutical cGMPs encourages manufacturers to modernize their methods, equipment and facilities that will help eliminate both production inefficiencies and undue risks for consumers.
Lester M. Crawford, D.V.M., Ph.D., Acting Commissioner of Food and Drug Administration -- U.S. Pharmacopeia Convention - March 11, 2005

Quality Systems Approach to Pharmaceutical cGMPs
Release in September 2006 Integrate quality systems and risk
management approaches Harmonization of CGMPs, QSIT and
other non-US pharmaceutical regulatory systems
Go beyond quality control

The Guidance Document
What is in that guidance document?
Hint…. Systems!

Why Improve Systems?
“…we are concerned of the apparent system deficiencies that have allowed the unacceptable practices cited herein to occur. Such system deficiencies appear to involve internal quality auditing, corrective and preventive actions, management controls relating to quality planning and resource allocations (staffing of appropriately qualified individuals) and /or training of personnel….”

Why Improve Systems?
“…The specific violation noted in this letter and in form FDA 483, Inspectional Observations, issued at the conclusion of the inspection by the FDA investigators may be symptomatic of serious underlying problems in your firm’s manufacturing and quality assurance systems.”
The quality systems model
Does not treat the five manufacturing systems as discrete entities, but instead integrates them into appropriate sections of the model.
The inter-relationship should be readily apparent.
One of the important themes of the systems based inspection compliance program is to be able to assess whether each of the systems is in a state of control.
The quality system model will also serve to help firms achieve the desired state of control.

Six Systems Inspection Approach
Perform in-depth internal audit focusing on system approach
Review past FDA observations
Perform trend analysis Continuous Improvement
Program Review industry trends
Hot “FDA” areas

System Covers 21 CFR 211
Quality System(Assures overall compliance with cGMPs and internal procedures and specifications)
•The quality control unit and all of its review and approval duties •All product defect evaluations and evaluation of returned and salvaged drug products.
Subparts B, E, F, G, I, J, andK.
Facilities and Equipment System(Measures and activities which provide appropriate physical environment and resources used production.)
•Buildings and facilities along with maintenance;•Equipment qualifications (installation and operation); •Calibration and preventative maintenance; •Cleaning and validation of cleaning processes •Utilities
Subparts B, C, D, and J.
Materials System This system includes measures and activities to control finished products,components, containers and closures.
•Validation of computerized inventory control processes, •Drug storage, •Distribution controls and records.
Subparts B, E, H, and J.

System Covers 21 CFR 211
Production SystemProduction System(Measures and activities to control the manufacture of drugs and drug products)
Batch compounding, Dosage form production, In-process sampling and testing, Process validation. Establishing, following, and documenting performance of approved manufacturing procedure
B, F, and J.
Packaging and Labeling System(Includes measures and activities that control thepackaging and labeling of drugs and drug products.)
Written procedures, labelExamination and usage, label storage and issuance, Packaging and labeling operations controls, andValidation of these operations.
Subparts B, G, and J
Laboratory Control System (Includes measures and activities related to laboratory)
•Laboratory procedures, •Testing, •Analytical methods development and validation or verification, •Stability program.
Subparts B, I, J, and K.
Six-system Inspection Model The inspection includes coverage of 2 or
more systems, with mandatory coverage of the Quality System.
Inspecting the minimum number of systems, or more systems as deemed necessary by the District, will provide the basis for an overall CGMP decision.

Six-system Inspection Model
The FDA's Drug Manufacturing Inspection Compliance Program is based on a systems-based approach for inspections.
Purpose of the systems based inspection compliance program is to be able to assess whether each of the company’s systems is in a state of control.

System elements (roles & responsibilities) Product reviews Complaint reviews Discrepancy and failure investigations Change control Product improvement projects Reprocess/Rework Rejects Stability failures Quarantine products Validation Training/Qualification of employees in quality control unit
function.
Quality OversightQuality Oversight

QC Unit Under GMP 211.22 Responsibilities of quality control unit.
(a) Adequate laboratory facilities for the testing and approval (or rejection) of components, drug product containers, closures, packaging materials, in-process materials, and drug products shall be available to the quality control unit.
(b) The quality control unit shall have the responsibility for approving or rejecting all procedures or specifications impacting on the identity, strength, quality, and purity of the drug product.
(c) The responsibilities and procedures applicable to the quality control unit shall be in writing; such written procedures shall be followed.

Facilities And Equipment
Cleaning and cleaning validation HVAC/environmental controls Containment Changes in critical areas Water for injection/purified water system Equipment maintenance and calibration Equipment qualification (including computer
validation and security)

Materials System
Control of incoming materials and components quarantine, storage, release, and use
Validation of computerized inventory control processes, drug storage, distribution controls and records
Release procedures Retest procedures Changes in materials

Laboratory Controls
Adequacy of equipment for intended use Enough space Calibration and maintenance Security of computer systems Change control Procedures, testing, analytical methodology,
development and verification/validation Lab documentation Adherence to out-of-specification procedures

Production System
Batch record review Component preparation – e.g. , depyrogenation,
sterilization of container/closures Hold times, in-process controls, use logs Identification and documentation of critical process
parameters Contemporaneous and complete batch production
documentation Process validation

Packaging And Labeling
Controls of master copies Issuance and restriction Changes in labeling Line clearance operations Reconciliation procedures, if applicable Vision system validation

Key Concepts Quality Quality by Design and Product Development Risk Management and Risk Assessment CAPA (Corrective and Preventive Action) Change Control The Quality Unit Six-system Inspection Model
Quality Systems Approach to Pharmaceutical Current Good Manufacturing Practice Regulation
Draft – September 2004

Module 3
Risk - base FDA Inspections Learn how FDA will select firms for inspections using the
Risk base Approach Understand the FDA's intent for the systems-based
inspection program Identify the differences between systems-based and product-
based inspections Define the difference between a full inspection, an
abbreviated inspection, and determine how previous inspection results may influence the current inspection
Determine the factors that may influence which sub-systems are inspected.

FDA’s Risk-Based Approach to GMPs Inspections
Routine Inspection: FDA tries to audit Manufacturers at least every two years.
Non compliant history New product, i.e., PMA Consumer complaints Competitor complaints

FDA Risk-Based Inspections
General Statutory Inspections Submission Related Inspections
NDA / IND / BLA / IDE / PMA Problem Related Inspections
Complaints Adverse Reports
Recalls

Pilot Risk-Ranking Model to Prioritize Manufacturing Sites for GMP Inspections

GMP Initiative Announcement:August 21, 2002
Evaluate the currency of our drug quality programs so that FDA resources are used most effectively and efficiently to address the most significant public health risks.
In order to provide the most effective public health protection, FDA must match its level of effort against the magnitude of the risk.
Resource limitations prevent uniformly intensive coverage of all pharmaceutical products and production. Although the agency has been implementing risk-based programs, a more systematic and rigorous risk based approach will be developed.

ICH Q9: Defining Risk
Preliminary working definitions (March 2004) from ICH EWG on Quality Risk Management (Q9): Risk: Combination of the probability of occurrence
of harm and the severity of that harm. ISO 14971 Harm: Damage to health, including the damage
that can occur from loss of product efficacy, safety, quality or availability
Focus on quality for our purposes

ICH Q9: Defining Risk (cont’d) Quality: Degree to which a set of inherent
characteristics of a product, system or process fulfils requirements
Requirements: Needs or expectations that are stated, generally implied or obligatory by the patients or their surrogates (e.g. health care professionals, regulators and legislators)
Combining key terms: Risk to quality is the probability/severity that drug will fail to meet the needs/expectations of the patients and their surrogates

Risk to Quality
Probability Severity
HARMFailure to meet
Needs/Expectationsof Patients/Surrogates
Quality Attributes
Loss of Quality
Probability/Severityof Adverse Impact on
Quality Attributes

Identifying Risk Factors
What hazards can adversely impact drug quality attributes/surrogates?
What processes and parameters are critical for quality attributes/surrogates?
What factors may affect the identified hazards and critical parameters/processes?
Other variables predictive of drug products with or without identified quality attributes?

SITE RISK POTENTIAL
PRODUCT PROCESS FACILITY
A RISK-BASED FRAMEWORK FOR PRIORITIZING SITES FOR cGMP INSPECTION
Site risk potential (SRP):
A function of the risk potentials for the PRODUCT, PROCESS and FACILITY component risk factors.
The SRP is a weighted combination of the PRODUCT, PROCESS and FACILITY risk potentials
Semi-quantitative risk potentials represented as “high,” “medium,” or “low,” and are numerically discrete variables in calculations.

Risk Ranking Model:Facility Factors Are some manufacturing facilities (or
manufacturers) more likely to produce a product with quality problems? Effectiveness of quality systems Inspectional record and compliance history Exposure: volume produced at facility Other characteristics?
Macher and Nickerson study

Risk Ranking Model:Process Factors Are some manufacturing processes, for
particular product classes, more likely to go wrong than others? Use expert elicitation to identify risk factors and
weightings Risk of contamination or mix-ups Maintaining state of control of the process
Potential for process capability metrics?

Selecting Systems for Coverage
System(s) for coverage will be made by the District Office based on: Specific operation, History of previous coverage, History of compliance, or Other priorities determined by the District Office.

Pilot Risk Ranking Model: Risk-Based Method for Prioritizing cGMP Inspections-Pharma
Sites
Top-Level Components for the Site Selection Model : Factor Category and Description Example(s)
Product: Factors pertaining to the intrinsic properties of drug products such that quality deficiencies could potentially and adversely impact public health - examples: Dosage form; intrinsic chemical properties
Facility: Factors relating to characteristics of a manufacturing site believed to be predictive of potential quality risks, such as the lack of effective quality systems - examples: Poor cGMP compliance history
Process: Factors pertaining to aspects of drug manufacturing operations that may predict potential difficulties with process control and/or vulnerability to various forms of contamination - examples: Measuring; mixing; compression; filling

Pilot Risk Ranking Model: Risk-Based Method for Prioritizing cGMP Inspections-Pharma Sites

Pilot Risk Ranking Model: Risk-Based Method for Prioritizing cGMP Inspections-Pharma Sites

Pilot Risk Ranking Model: Risk-Based Method for Prioritizing cGMP Inspections-Pharma
Sites
The facility component of the risk ranking model includes 4 factors:
History of violation (e.g., CGMP deficiencies have higher weights)
History of inspection (e.g., no prior inspection, newly registered/licensed or no CGMP inspection in the past 2 years have higher weights than those with recent CGMP inspection)
Estimated volume of production output (surrogate for exposure, e.g., higher volume and production output, higher weights)
Type of establishment (e.g., manufacturer, repacker, contract lab)

Drug Pilot Inspection Program: Implemented 2/1/02
This program applies to all drug manufacturing operations.
Currently there are not enough FDA resources to audit every aspect of cGMP in every manufacturing facility during every inspection visit.
Drug Pilot Inspection Program: Implemented 2/1/02
The Full Inspection Option
The Full Inspection Option is a surveillance or compliance inspection which is meant to provide a broad and deep evaluation of the firm's CGMP.
This will be done when little or no information is known about a firm's CGMP compliance (e.g., for new firms); or for firms where there is doubt about the CGMP compliance in the firm (e.g., a firm whose history has documented short-lived compliance and recidivism); or follow up to previous regulatory actions.

Drug Pilot Inspection Program: Implemented 2/1/02
The Abbreviated Inspection Option
The Abbreviated Inspection Option is a surveillance or compliance inspection which is meant to provide an efficient update evaluation of a firm's CGMP.
The abbreviated inspection will provide documentation for continuing a firm in a satisfactory CGMP compliance status.
Generally: a firm has a record of satisfactory CGMP compliance, with no significant recall, or product defect or alert incidents, or with little shift in the manufacturing profiles of the firm within the previous two years.

Drug Pilot Inspection Program: Implemented 2/1/02
Selecting Systems for Coverage
The selection of the system(s) for coverage will be made by the District Office based on such factors as a given firm's specific operation, history of previous coverage, history of compliance, or
other priorities determined by the District Office.

Drug Pilot Inspection Program: Implemented 2/1/02
Goal is to minimize consumers’ exposure to adulterated drug products. Program applies to all drug manufacturing operations.
Recent observations/findings: inadequate documentation, no procedures, not following procedures, did not establish/follow a control system for
changes, did not review/approved procedures, and failed to qualify computers.

Drug Pilot Inspection Program: System-Wide Implementation as of 2/1/02
Importance of Operating Under a State of Control
Produces finished drug products for which there is an adequate level of assurance of quality, strength, identity, and purity.
If any one system is out of control, this means the firm is out of control!

If Out of Control – What Does this Mean?
Will result in regulatory action and follow up.
Action may include the full inspection option for the next visit, a warning letter, seizure or injunction.
The follow up will be enacted once the compliance division concurs that the company has an OAI (Official Action Indicated) situation.
The OAI situation results in an unacceptable profile of all drug profile classes for a company.

Module 4
Review of additional cGMPs and the concept of Modern Quality Systems (Guidance for Industry Quality System Approach to Pharmaceutical cGMP Regulations)
Quality Quality by Design Quality Risk Management CAPA The Quality Unit
Other critical SOPs of interest to the FDA Deviation Handling Failure Investigations Change Control Training Validation

Quality System
Quality unit’s responsibilities responsibility to review all procedures
under its control although not explicitly noted in the
program, the practice of FDA is to identify the most responsible company individuals who have the responsibility, duty and power (authority) to detect, prevent and correct any manufacturing and quality problems that may exist in their facility

Quality System(cont’d)
Quality unit’s duties annual product review discrepancy reports corrective and preventive actions change control product improvement projects reprocess/reworking/rejects stability failures status of required validations training in quality unit functions

Quality Markers
Annual Product Review Stability Trends QC Profiles (raw materials, in-process and
finished product test results, etc.) Supplier Qualification Program Number of Recalls / Complaints Manufacturing Deviations Laboratory OOS/OOT

Quality Unit
Starts at the top: Management Define organizational structure Build to meet requirements Define vision: establish policies,
objectives and plans Review and tweak Resource planning

Quality by Design
Facilities and Equipment Design / Develop Product and
Process Monitor Processes and
Operations Inputs / Outputs
Address variables

Risk Assessment / Management
Identify quality issues Determine impact Define CAPA Promote Continuous
Improvement

CGMP regulations correlation to specific elements in the quality systems model
21 CFR CGMP Regulations Related to Manufacturing Operations
Quality System Element Regulatory Citation
1. Design and Develop Product and Processes Production: § 211.100(a)
2. Examine Inputs Materials: §§ 210.3(b), 211.80 – 211.94, 211.101, 211.122, 211.125
3. Perform and Monitor Operations Production: §§ 211.100, 211.103, 211.110, 211.111, 211.113
QC criteria: §§ 211.22(a-c), 211.115(b), 211.160(a), 211.165(d)
QC checkpoints: §§ 211.22 (a), 211.84(a), 211.87, 211.110(c)
4. Address Nonconformities Discrepancy investigation: §§ 211.22(a), 211.115, 211.192, 211.198 Recalls: 21 CFR Part 7

Evaluation Activities
Analyze Data for Trends: Quality systems call for continually monitoring trends and improving systems. This
can be achieved by monitoring data and information, identifying and resolving problems, and anticipating and preventing problems.
Quality systems procedures involve collecting data from monitoring, measurement, complaint handling, or other activities, and tracking this data over time, as appropriate.
Analysis of data can provide indications that controls are losing effectiveness. The information generated will be essential to achieving problem resolution or problem prevention (see IV.D.3.).
Although the annual review required in the CGMP regulations (§ 211.180(e)) call for review of representative batches on an annual basis; quality systems calls for trending on a regular basis.
Trending enables the detection of potential problems as early as possible to plan corrective and preventive actions. Another important concept of modern quality systems is the use of trending to examine processes as a whole; this is consistent with the annual review approach. These trending analyses can help focus internal audits.
Conduct Internal Audit
A quality systems approach calls for audits to be conducted at planned intervals to evaluate effective implementation and maintenance of the quality system and to determine if processes and products meet established parameters and specifications.
Quality systems recommend that procedures takes into account the relative risks of the various quality system activities, the results of previous audits and corrective actions, and the need to audit the entire system at least annually
Also describe how auditors are trained in objective evidence gathering, their responsibilities, and auditing procedures.

CAPA
Investigate the right way! Monitor for effectiveness Evaluate across the board Understand the process /
operation before initiating CAPA

Corrective Action
Corrective action is a reactive tool for system improvement to ensure that significant problems do not recur.
Both quality systems and the CGMP regulations emphasize corrective actions.
Quality systems approaches call for procedures to be developed and documented to ensure that the need for action is evaluated relevant to the possible consequences, the root cause of the problem is investigated, possible actions are determined, a selected action is taken within a defined timeframe, and the effectiveness of the action taken is evaluated.
It is essential to maintain records of corrective actions taken (CGMP also requires this; see § 211.192).

CAPA (Corrective and Preventive Action)
CAPA focuses on investigating and correcting discrepancies and attempting to prevent recurrence.
Quality system models discuss CAPA as three concepts, all of which are used in this guidance. Remedial corrections Root cause analysis with corrective action to prevent
recurrence Preventive action to prevent initial occurrence

Why are Investigations Critical: Regulatory Need
CFR 21 Part 211 References Investigations a number of times
211.22 Responsibilities of the Quality Control Unit (a) ……, if errors have occurred, they have been fully investigated.
211.192 Production Record Review …..Any unexplained discrepancy (including a percentage of theoretical yield exceeding maximum or minimum percentages established in the master production and control records) or the failure of a batch or any of its components to meet specifications shall be thoroughly investigated, whether or not the batch has already been distributed. The investigation shall extend to other batches of the same drug product and other drug products that may have been associated with the specific failure or discrepancy. A written record of the investigation shall be made and shall include conclusions and follow-up.

What is an Investigation?
A written record of the facts surrounding (an) unexpected circumstance(s) and the actions undertaken to correct the situation both immediately and in the future.

Deviation Definition and Acronyms
Deviation – Deviation is a departure from an approved instruction or established standard. Deviations are also known as:
Non Conformances Incidents Test Incidents Discrepancies

Deviation Clarity
The intent of deviations is to document issues with the system/piece of equipment and to gain a satisfactory resolution to meet the owner’s needs.
The expectation of a completed deviation is to clearly define the deviation and the steps taken to resolve the deviation. The deviation should be clear so that it can be understood on a stand alone basis.

Change Control
Have a management system for handling change
Understand the impact of the change
Evaluate the change
Quality and Change Control
Quality reviews proposed changes for adequacy of rationale for the decision to make the change, compliance to the change control procedure, assurance that adequate documentation of the change is present, impact on internal and external customers or cGMP, required changes to other procedures, specifications, batch records, etc.
Although Quality manages change control, other departments coordinate change control
Quality must have a system for tracking and monitoring changes
Quality is the last department to review and approve change control - responsible for ensuring everything has been completed as required/agreed to.

Change Control Change control and periodic reviews are critical to
ensuring deviations are minimized and that systems and processes are maintained in a “state of control”.
Purpose of change control is to ensure that all changes (planned and emergency) are documented, evaluated, approved, and implemented in a timely manner.
There should be a time period in which changes are considered “void” if not implemented within a specified time period.
Changes can apply to equipment, utilities, processes, and documents.

CGMP regulations correlation to specific elements in the quality systems model
21 CFR CGMP Regulations Related to Management Responsibilities
Quality System Element Regulatory Citations
1. Leadership
—
2. Structure
Establish quality function: § 211.22 (a) (see definition § 210.3(b)(15))
Notification: § 211.180(f)
3. Build QS
QU procedures: § 211.22(d)
QU procedures, specifications: § 211.22(c), with reinforcement in: §§ 211.100(a), 211.160(a)
QU control steps: § 211.22(a), with reinforcement in §§: 211.42(c), 211.84(a), 211.87, 211.101(c)(1), 211.110(c), 211.115(b), 211.142, 211.165(d), 211.192
QU quality assurance; review/investigate: § 211.22(a), 211.100(a-b) 211.180(f), 211.192, 211.198(a)
Record control: § 211.180(a-d), 211.180(c), 211.180(d), 211.180(e), 211.186, 211.192, 211.194, 211.198(b)
4. Establish Policies, Objectives and Plans
Procedures: § 211.22(c-d), 211.100(a)
5. System Review
Record review: § 211.180(e), 211.192, 211.198(b)(2)

A documented quality system
For example, according to the model, when documenting a quality system, the following should be included: The scope of the quality system, including any
outsourcing The manufacturer’s policies to implement the quality
systems criteria, and the supporting objectives The procedures needed to establish and maintain
the quality system
CGMP regulations correlation to specific elements in the quality systems model
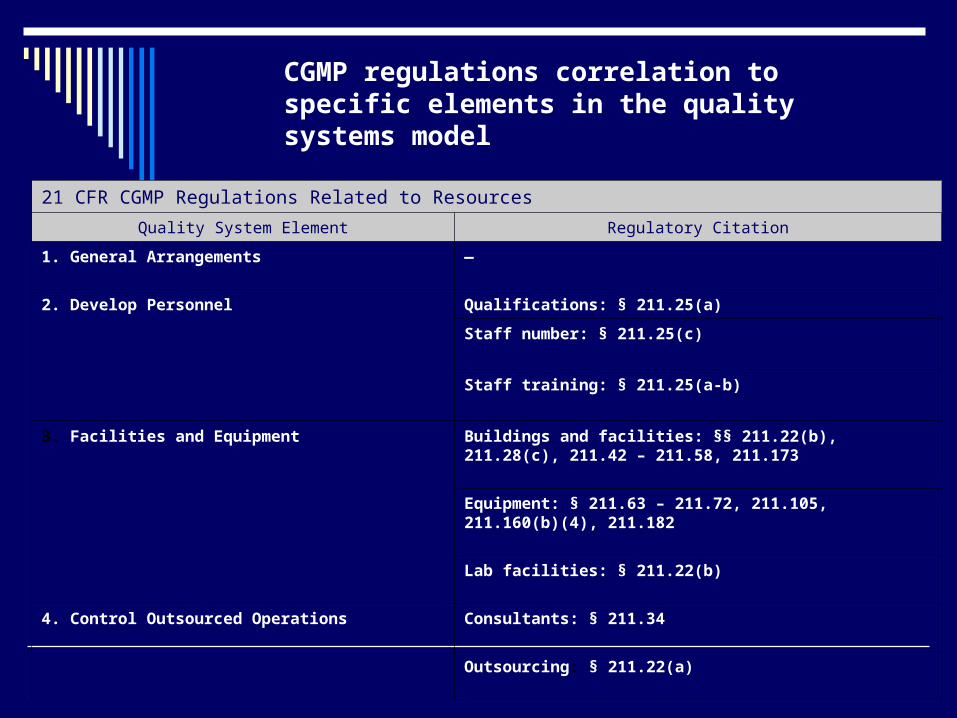
21 CFR CGMP Regulations Related to Resources
Quality System Element Regulatory Citation
1. General Arrangements
—
2. Develop Personnel Qualifications: § 211.25(a)
Staff number: § 211.25(c)
Staff training: § 211.25(a-b)
3. Facilities and Equipment Buildings and facilities: §§ 211.22(b), 211.28(c), 211.42 – 211.58, 211.173
Equipment: § 211.63 – 211.72, 211.105, 211.160(b)(4), 211.182
Lab facilities: § 211.22(b)
4. Control Outsourced Operations Consultants: § 211.34
Outsourcing: § 211.22(a)

Resources
Under a robust quality system, sufficient allocation of resources for quality system and operational activities is critical.
Senior management, is responsible for providing adequate resources for the following:
To supply and maintain the appropriate facilities and equipment to consistently manufacture a quality product
To acquire and receive materials that are suitable for their intended purpose
For processing the materials to produce the finished drug product
For laboratory analysis of the finished drug product, including collection, storage, and examination of in-process,
stability, and reserve samples

Training
Continued training is critical to ensure that the employees remain proficient in their operational functions and in their understanding of CGMP regulations.
Under a quality system (and the CGMP regulations), training is expected to focus on both the employees’ specific job functions and the related CGMP regulatory requirements.
Managers are expected to establish training programs that include the following: Evaluation of training needs Provision of training to satisfy these needs Evaluation of effectiveness of training Documentation of training and/or re-training

211.25 Personnel Qualifications.
(a) Each person engaged in the manufacture, processing, packing, or holding of a drug product shall have education, training, and experience, or any combination thereof, to enable that person to perform the assigned functions.
(b) Training shall be in the particular operations that the employee performs and in current good manufacturing practice (including the current good manufacturing practice regulations in this chapter and written procedures required by these regulations) as they relate to the employee's functions.
(c) Training in current good manufacturing practice shall be conducted by qualified individuals on a continuing basis and with sufficient frequency to assure that employees remain familiar with CGMP requirements applicable to them.

Things to consider….
What are the education, training, skills and experience required by this job/task?
How does this person meet those qualifications?
Do you Train the entire organization? Starting from the top?
Determine needs…..
How do you determine the necessary education, training, skills and experience for people performing work affecting product quality?
What training or other actions do you provide to satisfy the needs of personnel?

Facilities and Equipment
Under a quality system, the technical experts (e.g., engineers, development scientists), who have an understanding of pharmaceutical science, risk factors, and manufacturing processes related to the product, are responsible for specific facility and equipment requirements.
According to the CGMP regulations, equipment must be qualified, calibrated, cleaned, and maintained to prevent contamination and mix-ups (§§ 211.63, 211.67, 211.68).
The CGMP regulations place as much emphasis on process equipment as on testing equipment (§ 211.42(b)), while most quality systems focus only on testing equipment.

Manufacturing Operations
There is significant overlap between the elements of a quality system and the CGMP regulation requirements for manufacturing operations.
It is important to emphasize again that FDA’s enforcement programs and inspectional coverage remain based on the CGMP regulations.
Design and Develop Product and Processes: In a modern quality systems manufacturing environment, the significant characteristics
of the product being manufactured should be defined, from design to delivery, and control should be exercised over all changes.
Quality and manufacturing processes and procedures — and changes to them — should be defined, approved, and controlled (CGMP also requires this; see § 211.100).

Process Validation
The design concept established during product development typically matures into a commercial design after process experimentation and progressive modification.
Areas of process weakness should be identified, and factors that are influential on critical quality attributes should receive increased scrutiny.
Process validation provides initial proof that the design of the process produces the intended product quality.

Definition of Process Validation
‘87 Process Validation Guidance: establishing documented evidence which
provides a high degree of assurance that a specific process will consistently produce a product meeting its predetermined specifications and quality attributes.

Process Validation
Although initial commercial batches can provide evidence to support the validity and consistency of the process, the entire life-cycleentire life-cycle should be addressed by the establishment of continuous improvement mechanisms in the quality system.
Process validation is not a one time event, but an on-going activity.

Process Development and Optimization (or Process R&D)
Build Quality into a product & Build Robustness into the process
Robustness- ability to produce the same result under the reasonably expected and inherent variation in materials, environment, personnel, equipment, etc.
To make a process robust, you must know what variation may be encountered and how that variation affects the process.

Production System

Production & Process Controls
Production System This system includes measures and activities to
control the manufacture of drugs and drug products including batch compounding, dosage form production, in-process sampling and testing, and process validation.
It also includes establishing, following, and documenting performance of approved manufacturing procedures. See the CGMP regulation, 21 CFR 211 Subparts B, F, and J.

Production and Process Controls
Production and process changes
Environmental control
Personnel
Contamination control
Buildings
Equipment (PM, cleaning)
Manufacturing material
Automated processes
Inspection, measuring, and test equipment; calibration
Process validation

Production & Process Controls
equipment cleaning and use logs master production and control records batch production and control records process validation, including validation and security
of computerized or automated processes change control; the need for revalidation evaluated documented investigation into any unexpected
discrepancy

Process Monitoring FDA expects the manufacturer to develop procedures for
monitoring, measuring, and analyzing operations When implementing data collection procedures, consider the
following: Are collection methods documented? When in the product life-cycle will the data be collected? How and to whom will measurement and monitoring
activities be assigned? When should analysis and evaluation (e.g. trending) of
laboratory data be performed (see V.E.1.)? What records are needed?
Must apply appropriate analytical methods Must apply appropriate analytical methods and/or statistical techniquesand/or statistical techniques

Laboratory Control System:
Resources, measures and activities related to laboratory procedures, testing, analytical methods development and validation or verification, and the stability program.
….Provide “Test Data”
Lab Control SystemLab Control System

QualitySystem
Lab Control System
Stability Program
CAPA &Investigations
Lab Investigations
Systems CGMP Quality System Element
(Address Nonconformities)
QUALITY SYSTEMSQUALITY SYSTEMSInterrelationsInterrelations

Lab Control SystemCGMP vs. Quality System element(s)
CGMP system elements Training/qualification Staffing Facility and Equipment Instrument calibration Reference Standards System Suitability Validation/verification Change Control Investigations Record keeping Testing and Release Testing and Release Stability Testing
Modern “Quality System” Elements Develop Personnel Develop Personnel Facilities and Equipment Facilities and Equipment Perform & Monitor Operations Perform & Monitor Operations Perform & Monitor Operations Build Quality System Address Nonconformities Build Quality System Perform & Monitor Operations Examine all inputs Perform & Monitor Operations

Module 5
Documentation Essentials Controlling Documents Document Problems Batch Records & Review GMP Records Compliance Within Established
Standards

What does this mean?
There must be adequate written procedures Procedures must be approved by appropriate
persons Procedures must be followed
Concept of cGMP

General Rules of Documentation
Procedures are designed so that, if followed the drug products will comply with: Specifications Assure compliance with regulations

General Rules of Documentation
Verify that dates reflect correct manufacturing sequence
Actual conditions comply with manufacturing parameters Processing temperature Mixing time/speed Filter integrity test pressure

Regulatory Requirements for Documentation
“Each manufacturer shall establish and maintain procedures to control all documents that are required by this part. The procedures shall provide for the following:”
(a) Document approval and distribution. Review and approve prior to issuance; signature and
date Document availability for use Obsolete documents not available for use
- 21 CFR 820.40, Document Controls, Quality System Regulations

GMP Procedures
Create a documentation strategy Write clear, concise, logical procedures Associate procedures with applicable
records Evaluate sample procedures

Create a documentation strategy
Policies
StandardOperating
Procedures
StandardTest Methods
MasterBatch Records
SOP Forms FormsProduction
Batch Records

Write clear, concise, logical procedures
Put steps in the procedure in the order in which they occur. If this principle is followed, there’s no need to refer to the
next step (go to step 10.4) if it immediately follows the previous step.
Balance the following two principles: Don’t make the procedure too long to be understood by a
trainee, but Don’t make too many short procedures that have to be
followed – too many documents to train on and follow.

GMP Records
Design records to match the process Ensure records include required data Use good documentation practices Complete records neatly and legibly Evaluate sample records

Design records to match the process
Start with a flowchart of the process, then write the SOP.
Attach the flowchart to the SOPUse the flowchart as a training tool (many
people learn best visually)Flowcharts from SOPs are useful to
summarize how a procedure works in an audit or inspection
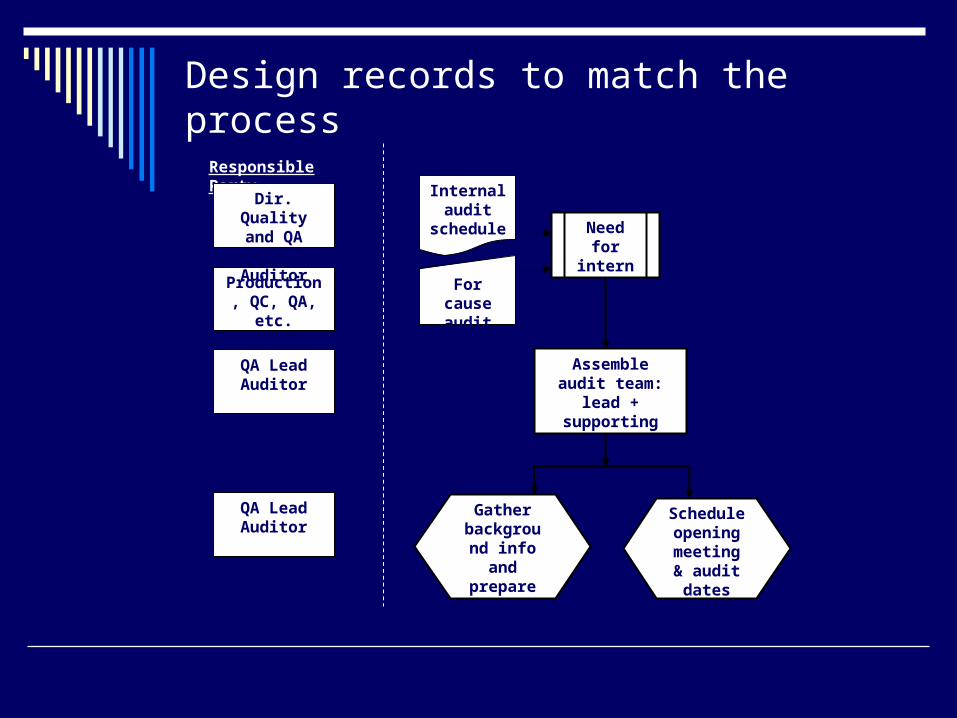
Design records to match the process
Production, QC, QA, etc.
Need for
internal audit
Internal audit
schedule
For cause audit
Assemble audit team: lead + supporting
auditors + SMEs
Gather background info and prepare
audit plan
Schedule opening
meeting & audit dates
Responsible Party
Dir. Quality and QA Lead
Auditor
QA Lead Auditor
QA Lead Auditor

Ensure records include required data
2. Check regulatory requirements for records:
Master Batch Records: Name of product and reference code List of raw materials and intermediates with
quantity to be used, including unit of measure
Production location and major production equipment to be used…
- Q7A, 6.41

Ensure records include required data
2. Check regulatory requirements for records:
Production Batch Records Actual results recorded for critical process
parameters Any sampling performed Signatures of persons performing…and checking
critical operation steps Actual yield at appropriate phases Representative label
- Q7A, 6.52

Use good documentation practices
Regulatory requirements: Entries in records are:
Legible Indelible Made in designated spaces Identify person responsible Include dates of tasks performed Made directly after performing task (real-time) Detailed enough for clear understanding Laid out in orderly fashion Easy to check Reproduction process of master should not
introduce errors

Use good documentation practices
Regulatory requirements: Corrections to entries in records are:
Dated Signed Leave original entry legible Reason for correction is indicated

Use good documentation practices When an error is made on a record, DO NOT: Obscure the data by:
scribbling over it, trying to change the written data, erasing it, using correction fluid
Copy it to a new record and discard the original data,
Backdate (fill in an earlier date on a later date with no explanation that it was added later)

Regulatory Inspection of Documents
Follow written procedures and complete records as required
Ensure quick and accurate retrieval of requested documentation
Review documentation before presentation to regulatory body

Adequacy of SOPs
SOPs are the first thing asked for during audits and provide an inspector with the following information: Are the SOPs current, accurate, and is there a procedure
for writing SOPs? Do operators follow both the steps and order of steps
as detailed? Do procedures exist not only for operation, but also for
maintenance, change control, and cleaning? Is there a routine review of procedures?

SOP Content SOPs should be concise, easy to manage and updated
as necessary. SOPs should provide sufficient detail to allow an
operator the ability to run the unit if doing it the first time with training.
SOPs should reference operating limits determined during validation studies, applicable diagrams for the placement of equipment components or sampling locations used for environmental sampling.
SOPs should be kept to a minimum number of pages since the greater the number of pages, the less an operator reads it.

Records and Reports
Batch / Production Records Relates to In-Process / Finished Product Production History for Each Lot Critical to Decision Analysis – Accept / Reject Availability for Review, Analysis, Copying Retained in Hard Copy, Electronic, Microfilm,
Microfiche Retention – 1 Year post expiration date
OTC 3 Years after last distribution of lot

Master Production and Control Records
Written Procedures Master Production Record Contents
Product Name / Dosage Form / Strength
Detailed Formula Theoretical Weight / Yield Packaging / Labeling Manufacturing / Processing Instructions

Batch Production and Control Records
Reproduction of Master Production Record Process Documentation
Formulation – Identity, Weights, Measures Equipment Packaging / Labeling Operation (Labeling Control) Sampling Responsible Personnel / Supervision Examination / Test Results
Production / Batch Record Review - QC

Complaint Investigations
Recalls Returned Goods Salvage Operations Corrective Actions Management Reporting Reports to FDA
Records and Reports

Module 7
Pharmaceutical Development Q8,Q9 Key Development Steps
Ensuring Good Manufacturing Practices for Drug Products
History of Regulatory Enforcement FDA Policy and Program Issues

CGMPS AND THE CONCEPTS OF MODERN QUALITY SYSTEMS
Key concepts which are critical for any discussion of modern quality systems.
The following concepts are used throughout this guidance as they relate to the manufacture of pharmaceutical products.

Quality Every pharmaceutical product has
established identity, strength, purity, and other quality characteristics designed to ensure the required levels of safety and effectiveness.
For the purposes of the draft guidance document, the phrase achieving quality means achieving these characteristics for the product.

Quality by Design and Product Development
Quality by design means designing and developing manufacturing processes during the product development stage to consistently ensure predefined quality at the end of the manufacturing process.
A quality system provides a sound framework for the transfer of process knowledge from development to the commercial manufacturing processes and for post-development changes and optimization

What is “Quality by Design”?
Quality “Good pharmaceutical quality represents an acceptably low
risk of failing to achieve the desired clinical attributes.” Quality by Design (QbD)
“Means that product and process performance characteristics are scientifically designed to meet specific objectives, not merely empirically derived from performance of test batches.”

Quality by Design
Design features and decisions that deliver a high degree of confidence that manufactured lots will have the intended product quality and performance
Design to specifications (not setting specifications after the fact)
Redundant (i.e., an additional layer of security –risk based) end product testing (not testing to document quality)

ICH – Q8
An ability to describe and justify why proposed design features deliver with confidence the intended quality (not awaiting test results to be submitted in a post approval supplement)
More fully defined, developed pharmaceutical development
Directed at product rationale instead of simple data reporting
Better understanding of critical aspect effects on quality attributes

Adoption of Q8 delivers a new state:
Product quality and performance achieved and assured by design of effective and efficient manufacturing processes
Product specifications based on mechanistic understanding of how formulation and process factors impact product performance
An ability to effect Continuous Improvement and Continuous "real time" assurance of quality

Q8 applicable to all products -but at the applicant’s discretion
Not all information “mandatory” Guideline constructed to avoid potential
misunderstanding that may evolve from this
Guideline describes one system with different levels of design focus we use the “process understanding – predictive
ability” principles as a means to create a continuous framework and avoid “two different systems”
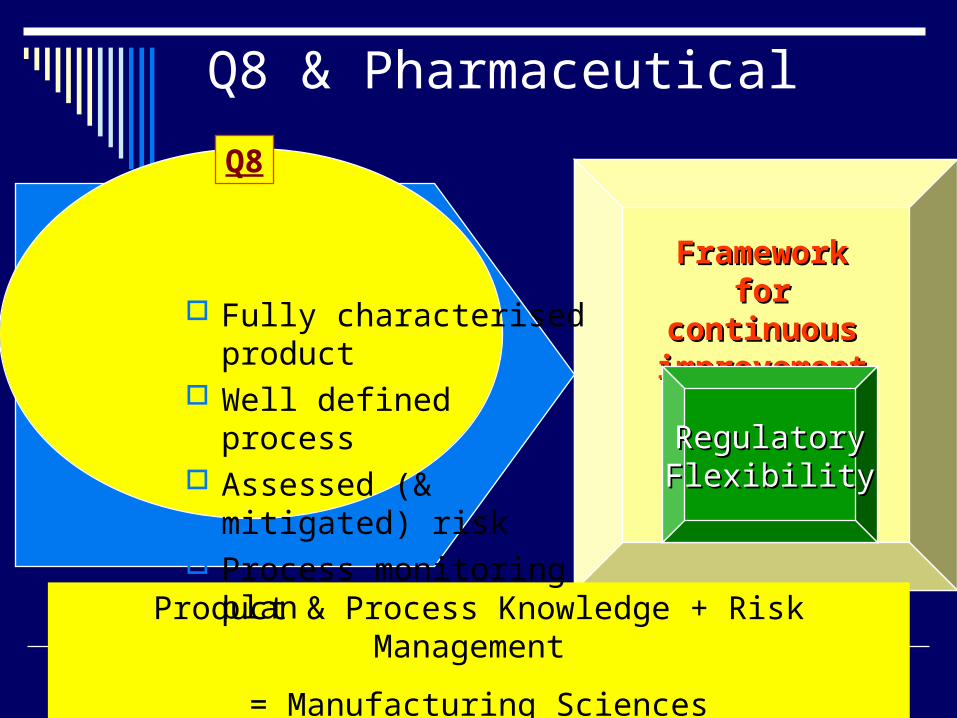
Q8 & Pharmaceutical
Framework Framework for for
continuous continuous improvementimprovement
Product & Process Knowledge + Risk Management
= Manufacturing Sciences
RegulatoryRegulatoryFlexibilityFlexibility
Q8
Fully characterised product
Well defined process Assessed (& mitigated)
risk Process monitoring plan

Risk Management and Risk Assessment
The concept risk management is a major focus of the Pharmaceutical CGMPs for the 21st Century Initiative.
Risk management can guide the setting of specifications and process parameters.
Risk assessment is also used in determining the need for discrepancy investigations and corrective action and how to prioritize them.
As risk assessment is used more formally by manufacturers, it can be implemented within the quality system framework.

ICH Q9: Defining Risk Preliminary working definitions
(March 2004) from ICH EWG on Quality Risk Management (Q9): Risk: Combination of the probability of
occurrence of harm and the severity of that harm. ISO 14971
Harm: Damage to health, including the damage that can occur from loss of product efficacy, safety, quality or availability
Focus on quality for our purposes

ICH Q9: Defining Risk (cont’d)
Quality: Degree to which a set of inherent characteristics of a product, system or process fulfils requirements
Requirements: Needs or expectations that are stated, generally implied or obligatory by the patients or their surrogates (e.g. health care professionals, regulators and legislators)
Combining key terms: Risk to quality is the probability/severity that drug will fail to meet the needs/expectations of the patients and their surrogates

Identify Predicted/Known Hazards to Quality Attributes: Risk Factors
Risks to pharmaceutical quality can be identified based on the probability and severity of adverse impact on these quality attributes Explicitly include factors that mitigate
probability/severity of adverse effects or factors that have a positive impact

Risk ManagementRisk Management Includes:
Risk Assessment Risk Control
Option analysis Implementation Residual risk Analysis Over-all risk acceptance
Option: Additional Risk Analysis Design Mitigations Verification and Validation Testing

Risk Management Risk Management PlanningIdentify risk management processAssign responsibilitiesDefine Risk acceptability criteria
Risk AnalysisDefine intended use Describe deviceIdentify hazards Estimate risks
Risk EvaluationDetermine/decide risk acceptability
Risk ControlAnalyze options Implement measuresEvaluate residual risk Decide overall risk acceptability
Re-assess residual risks,
design changes or new hazards
Post Production ControlEvaluate production results Review risk management resultsControl design changes
Risk Management
Risk Assessment
Hazard AnalysisRisk EstimationRisk Evaluation Risk Control

Module 8
Hosting An FDA Inspection Preparing for a successful FDA inspection How to develop an Inspection strategy and an inspection
Tool kits (Checklist) Learn how to save time and money by planning and
focusing on the right systems for an inspection How to conducting Mock Inspections Plan an effective strategy to prepare for an inspection Maintain compliance for facilities and equipment
Questions & Wrap-Up

They’re Here
After verification of credentials and issuance of FD482, the FDA investigator is immediately escorted to a conference room that will be set aside for exclusive use during the inspection Post a notice on the door that an
inspection is in progress

QA Management
Verify Credential
Accept Notice of Inspection
(FDA Form 482)Escort to Conference Room
Review Company Policies

Mock Systems based inspection agenda!QUALITY SYSTEM
Evaluate Quality Control Unit. (This also includes the associated recordkeeping systems)
Preparedness for “Product reviews” at least annually; should include information from areas listed below as appropriate; batches reviewed, for each product, are representative of all batches manufactured; trends are identified; refer to 21 CFR 211.180(e).
- Complaint reviews (quality and medical): documented; evaluated; investigated in a timely manner; includes corrective action where appropriate.
- Discrepancy and failure investigations related to manufacturing and testing: documented; evaluated; investigated in a timely manner; includes corrective action where appropriate. S0P.
- Change Control: documented; evaluated; approved; need for revalidation assessed.
- Product Improvement Projects: for marketed products
- Reprocess/Rework: evaluation, review and approval; impact on validation and stability.
- Returns/Salvages: assessment; investigation expanded where warranted; disposition.
- Rejects: investigation expanded where warranted; corrective action where appropriate.
- Stability Failures: investigation expanded where warranted; need for field alerts evaluated; disposition.
- Quarantine products.
- Validation: status of required validation/revalidation (e.g., computer, manufacturing process, laboratory methods).
- Training/qualification of employees in quality control unit functions.

Mock Systems based inspection agenda!
FACILITIES AND EQUIPMENT SYSTEM
1. Facilities
- cleaning and maintenance
- facility layout and air handling systems for prevention of cross-contamination (e.g. penicillin, beta-lactams, steroids, hormones, cytotoxics, etc.)
- specifically designed areas for the manufacturing operations performed by the firm to prevent contamination or mix-ups
- general air handling systems
- control system for implementing changes in the building
- lighting, potable water, washing and toilet facilities, sewage and refuse disposal
- sanitation of the building, use of rodenticides, fungicides, insecticides, cleaning and sanitizing agents
2. Equipment
- equipment installation and operational qualification where appropriate
- adequacy of equipment design, size, and location
- equipment surfaces should not be reactive, additive, or absorptive
- appropriate use of equipment operations substances, (lubricants, coolants, refrigerants, etc.) contacting products/containers/etc.
- cleaning procedures and cleaning validation
- controls to prevent contamination, particularly with any pesticides or any other toxic materials, or other drug or non-drug chemicals
- qualification, calibration and maintenance of storage equipment, such as refrigerators and freezers for ensuring that standards, raw materials, reagents, etc. are stored at the proper temperatures
- equipment qualification, calibration and maintenance, including computer qualification/validation and security
- control system for implementing changes in the equipment
- equipment identification practices (where appropriate)
- documented investigation into any unexpected discrepancy

Mock Systems based inspection agenda!MATERIALS SYSTEM
- training/qualification of personnel
- identification of components, containers, closures
- inventory of components, containers, closures
- storage conditions
- storage under quarantine until tested or examined and released
- representative samples collected, tested or examined using appropriate means
- at least one specific identity test is conducted on each lot of each component
- a visual identification is conducted on each lot of containers and closures
- testing or validation of supplier's test results for components, containers and closures
- rejection of any component, container, closure not meeting acceptance requirements. Investigate fully the firm's procedures for verification of the source of components.
- appropriate retesting/reexamination of components, containers, closures
- first in-first out use of components, containers, closures
- quarantine of rejected materials
- water and process gas supply, design, maintenance, validation and operation
- containers and closures should not be additive, reactive, or absorptive to the drug product
- control system for implementing changes in the materials handling operations
- qualification/validation and security of computerized or automated processes
- finished product distribution records by lot
- documented investigation into any unexpected discrepancy

Mock Systems based inspection agenda!PRODUCTION SYSTEM
- training/qualification of personnel
- control system for implementing changes in processes
- adequate procedure and practice for charge-in of components
- formulation/manufacturing at not less than 100%
- identification of equipment with contents, and where appropriate phase of manufacturing and/or status
- validation and verification of cleaning/sterilization/ depyrogenation of containers and closures
- calculation and documentation of actual yields and percentage of theoretical yields
- contemporaneous and complete batch production documentation
- established time limits for completion of phases of production
- implementation and documentation of in-process controls, tests, and examinations (e.g., pH, adequacy of mix, weight variation, clarity)
- justification and consistency of in-process specifications and drug product final specifications
- prevention of objectionable microorganisms in non-sterile drug products
- adherence to preprocessing procedures (e.g., set-up, line clearance, etc.)
- equipment cleaning and use logs
- master production and control records
- batch production and control records
- process validation, including validation and security of computerized or automated processes
- change control; the need for revalidation evaluated
- documented investigation into any unexpected discrepancy

Mock Systems based inspection agenda! PACKAGING AND LABE LING SYSTEM
- training/qualification of personnel
- acceptance operations for packaging and labeling materials
- control system for implementing changes in packaging and labeling operations
- adequate storage for labels and labeling, both approved and returned after issued
- control of labels which are similar in size, shape, and color for different products
- finished product cut labels for immediate containers which are similar in appearance without some type of 100 percent electronic or visual verification system or the use of dedicated lines
- gang printing of labels is not done, unless they are differentiated by size, shape, or color
- control of filled unlabeled containers that are later labeled under multiple private labels
- adequate packaging records that will include specimens of all labels used
- control of issuance of labeling, examination of issued labels and reconciliation of used labels
- examination of the labeled finished product
- adequate inspection (proofing) of incoming labeling
- use of lot numbers, destruction of excess labeling bearing lot/control numbers
- physical/spatial separation between different labeling and packaging lines
- monitoring of printing devices associated with manufacturing lines
- line clearance, inspection and documentation
- adequate expiration dates on the label
- conformance to tamper-evident (TEP) packaging requirements (see 21CFR 211.132 and Compliance Policy Guide, 7132a.17)
- validation of packaging and labeling operations including validation and security of computerized processes
- documented investigation into any unexpected discrepancy

LABORATORY CONTROL SYSTEM
- training/qualification of personnel
- adequacy of staffing for laboratory operations
- adequacy of equipment and facility for intended use
- calibration and maintenance programs for analytical instruments and equipment
- validation and security of computerized or automated processes
- reference standards; source, purity and assay, and tests to establish equivalency to current official reference standards as appropriate
- system suitability checks on chromatographic systems (e.g., GC or HPLC)
- specifications, standards, and representative sampling plans
- adherence to the written methods of analysis
- validation/verification of analytical methods
- control system for implementing changes in laboratory operations
- required testing is performed on the correct samples
- documented investigation into any unexpected discrepancy
- complete analytical records from all tests and summaries of results
- quality and retention of raw data (e.g., chromatograms and spectra)
- correlation of result summaries to raw data; presence of unused data
- adherence to an adequate Out of Specification (OOS) procedure which includes timely completion of the investigation
- adequate reserve samples; documentation of reserve sample examination
- stability testing program, including demonstration of stability indicating capability of the test methods
D. Sampling
Samples of defective product constitute persuasive evidence that significant CGMP problems exist.
F. Reporting
The Summary of Findings will identify and explain in the body of the report the rationale for inspecting the profile classes covered. Report and discuss in full any adverse findings by systems under separate captions.

Developing an Effective SOP for FDA Inspections
Inspection preparedness is essential for a successful outcome
A well-written Standard Operating Procedure Defines the roles Describes the process

Who is Involved?
Establish the roles and responsibilities Senior management Audit host Department representatives Legal department

Documents
Anticipate what will be needed Regulatory documents Organizational charts Floor plans Company documents Standard Operating Procedures

Logistics
Prepare in advance of an audit Locate a suitable room Personal comfort matters Stage documents Anticipate audit topics

Training the Staff
Who has authority? Who provides answers? How to get information? Who approves information?

Training the Staff
Providing documents Give what is asked for, and no more Be sure that it is OK to provide it Have someone review it first Provide documents promptly

Providing Documents
Have someone review these first Right documents? Complete? Post-it notes removed? Organized? What was asked for?

Providing Documents
Make a list of requested items Check them off when presented Log them back when returned Complete review and return Avoid leaving a lot of documents
Copying Documents
Limit what is copied if possible Make duplicate copies Keep one set for your files Verify that copies match original
Complete Legible Nothing cut off

If You Cannot Find a Document
Be open and honest Limit the scope of what’s missing Don’t be evasive It is not the end of the world Do not stall or delay the audit

Daily Summaries
Ask how things are going Identify what was reviewed Record the observations Plan for the following day Request and prepare documents

Close Out Meeting
Who should attend? Management Department heads Quality assurance Regulatory affairs Legal

Close Out Meeting
What to say/ what not to say Listen carefully, it is OK to clarify You are listening, not agreeing Do not become defensive Be respectful during the meeting Plan to respond in writing

Unwritten Observations
Suggestions may be offered These are good advice Record these items for review Implement suggestions if appropriate Share this information with others

Preparing a Response
Respond in writing Address each item List corrective actions taken Describe changes to systems Commit to times and dates Be positive in your response

FDA Readiness Kit

Anticipate Having Available
Floor Plan Organizational Chart (Current) Company History Master Schedule SOP Table of Contents CV’s Training Records

FDA Inspection Follow up Activities
Retrieve copy of final issued FD481 or Establishment Inspection Report (EIR), which is the audit “narrative” the FDA investigator writes and states what records and reports were reviewed, questions asked, responses received, tours of the facility.
The EIR is a very useful document to review to understand the inspectional approach scheme for future inspection planning.

FDA Inspection Follow up Activities
The Establishment Inspection Report (EIR), a diary of the inspection, is subject to the Freedom of Information Act (FOIA) disclosure, except during an ongoing investigation
Try to get the EIR If FDA doesn’t give it to you, this could be a bad
sign Might also serve as a useful road map of other
FDA concerns not cited in a 483

Staff Readiness for Inspections
FDA Inspection procedure in place.
- Plan on a recording secretary. Meeting place. Work out rough schedule with investigator. Disclosable/nondisclosable information. Photographs/recording devices and
affidavits.

Readiness for Inspections
Documentation review and discussion area. Set up a room where documentation can
be reviewed prior to presenting to the FDA. Need to be sure it is the right document,
pages are not missing, post it notes are not attached etc.
Room is a place where discussions can be held for response to complex questions.

Facility Appearance
Facility should always appear neat and orderly. Areas that appear out of control usually are!
Materials labeled and in the proper locations Large quarantine areas are a target for inspection. Housekeeping creates first impression of anyone
touring facility.

GMP Training
Not enough to have policies and procedures, staff must understand and follow.
Train on the FDA inspection procedure. Train on the GMP’s. Train on Defect Awareness.

Preparation
1. Tour of the process flow2. Preparatory audits of
impacted areas3. Retrieval and staging of any
pre-requested documentation4. Familiarization of audit
responsibilities given to the selected personnel for the various audit roles

Training on General Interview Guidelines:
1. Don’t Panic!1. Don’t Panic!
2. Be Honest and Direct2. Be Honest and Direct
3. Be Polite 3. Be Polite
4. Control Emotions4. Control Emotions
5. Do NOT volunteer information; answer questionsto the best of your ability
5. Do NOT volunteer information; answer questionsto the best of your ability
Remember first impressions arelasting impressions
Remember first impressions arelasting impressions
• You represent the company
• Ultimate goal – leave the inspector with a
positive mental picture of yourself
• Confidence and attitude are key
• There are NO second chances in making
first impression
• You represent the company
• Ultimate goal – leave the inspector with a
positive mental picture of yourself
• Confidence and attitude are key
• There are NO second chances in making
first impression

Preparation – Preparation – Training the ParticipantsTraining the Participants
Preparation – Preparation – Training the ParticipantsTraining the Participants
Know company policies and proceduresKnow company policies and procedures
Are SOPs in your area:
1. Current?2. In Compliance?3. Followed?4. Defined clearly?
Are SOPs in your area:
1. Current?2. In Compliance?3. Followed?4. Defined clearly?

Preparation – Training the Participants
Preparation – Training the Participants
Know how to answer the questionKnow how to answer the question
1. Listen Carefully1. Listen Carefully
2. Ask for clarification if needed2. Ask for clarification if needed
3. Provide only what is asked for3. Provide only what is asked for
4. Be honest and direct. If you don’t know – say so!4. Be honest and direct. If you don’t know – say so!
5. Be confident in your answers5. Be confident in your answers
6. Back up verbal info. with documentation6. Back up verbal info. with documentation
7. STOP when the question is fully answered7. STOP when the question is fully answered

Preparation – Preparation – Training the ParticipantsTraining the Participants
Preparation – Preparation – Training the ParticipantsTraining the Participants
Understand the various interview techniques
Understand the various interview techniques
• Direct Questions – “Do you have”• Direct Questions – “Do you have”
• Open ended questions – “Tell me about”• Open ended questions – “Tell me about”
• The “Silence” Technique• The “Silence” Technique
• The “Buddy” Technique• The “Buddy” Technique

Answering Questions: The Basic Rules
Tell the TRUTH Know your rights (and FDA’s) Listen to the question Answer the question that is asked Refer to others what you do not know STOP when the question is fully answered Always tell the truth

Do’s During Inspections Listen carefully and repeat the question or ask it to be
repeated, if necessary Answer completely, directly and honestly Speak slowly and clearly Speak only for your area of expertise Know your procedures Be able to verify everything you say Expect what you say to be documented in FDA field
notebook

Dont’s During Inspections
Do not make casual conversation Do not assume the investigator is your “buddy” Do not guess or make up an answer Do not lie Do not volunteer more information than
necessary to completely answer the question
Never say “That’s not how we do things around here” or “We’ve always done it that way”.

Dont’s During Inspections
Do not speculate. Don’t be afraid to find information and give it to the investigator later
If not certain how to answer, ask to confer with the escort privately
Do not assume to know what the investigator means Ask for clarification, examples or specifies
Do not conduct the inspection for the investigator Never question the investigator’s authority Never argue or raise your voice

Preparation – Preparation – Training the ParticipantsTraining the Participants
Preparation – Preparation – Training the ParticipantsTraining the Participants
Do NOT :Do NOT :
Volunteer information Volunteer information
Provide inaccurate information Provide inaccurate information
Provide “hearsay” answers Provide “hearsay” answers
React negatively to a question/comment React negatively to a question/comment

Preparation – Training the Participants
Preparation – Training the Participants
Do NOT :Do NOT :
Make the auditor’s wait for information Make the auditor’s wait for information
Override a Liaison’s decision in front of the auditors
Override a Liaison’s decision in front of the auditors
Have a BAD attitude Have a BAD attitude

FDA Investigators ...
Are aware that interviewees may be nervous
Understand that firms prepare their employees for inspections
Know that firms do not want to divulge adverse information
Are trained to detect deception Know that true deception is rare

FDA Investigators ... Want the inspection to go smoothly Do not seek to “trap” anyone Expect most people to be fully
honest and forthcoming Want to “get it right the first time”

Conduct during the audit
Hold debriefing sessions at the end of each day.
Discuss any observations or concerns expressed by the investigator.
Obtain clarification of outstanding issues. Discuss conversations that occurred.

The closing meeting
Closing meeting, there should be no surprises. Assemble those that were key during the inspection and key
members of top management. A FDA form 483 may have been prepared – A Notice of
Objectionable Conditions. If the observation is unclear, ask for clarification. Discuss the findings, tactfully debate misinterpretations. Usually not a good time to make any major commitments.

Conclusion
Implementation of a comprehensive quality systems model for pharmaceutical products, including biological products, will facilitate compliance with 21 CFR parts 210 and 211.
The central goal of a quality system is to ensure consistent production of safe and
effective products and that these activities are sustainable.
A robust quality system will promote process consistency by integrating effective knowledge-building mechanisms into daily operational decisions.
Successful quality systems share the following:
Science-based approaches Decisions are based on an understanding of the intended use
of a product Proper identification and control of areas of potential process
weakness Responsive deviation and investigation systems that lead to
timely remediation Sound methods for assessing risk Well-defined processes and products, starting from
development and extending throughout the product life cycle Systems for careful analyses of product quality Supportive management (philosophically and financially)

Good Business Sense!
Both good manufacturing practice and good business practice require a robust quality system.
When fully developed and effectively managed, a quality system will lead to consistent, predictable processes that ensure that pharmaceuticals are safe, effective.