201 familial cancer tr f
TRANSCRIPT

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 1/103
Le diagnosticmoléculaire au regard
de syndromes deprédisposition à uncancer héréditaire : ledépistage génétique etson retentissementclinique
Rapporttechnologique
numéro 41 novembre 2003

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 2/103
Citer le présent document comme suit : Ho C, Banerjee S, Mensinkai S. Le diagnostic moléculaire au
regard de syndromes de prédisposition à un cancer héréditaire : le dépistage génétique et sonretentissement clinique. Ottawa : Office canadien de coordination de l’évaluation des technologies de lasanté; 2003. Rapport technologique no 41.
La reproduction de ce document à des fins non commerciales est autorisée à condition que l’OCCETS soitdûment mentionné.
L’OCCETS est un organisme sans but lucratif financé par les gouvernements fédéral, provinciaux etterritoriaux.
Dépôt légal – 2003Bibliothèque nationale du Canada
ISBN : 1-894978-38-2 (version imprimée)ISBN : 1-894978-39-0 (version électronique)
POSTE-PUBLICATIONS CONVENTION NU. 40026386PORT DE RETOUR GARANTIE ÀOFFICE CANADIEN DE COORDINATION DE L’ÉVALUATIONDES TECHNOLOGIES DE LA SANTÉ600-865, AVENUE CARLINGOTTAWA (ONTARIO) K1S 5S8
Adresser toute demande de publications à :
OCCETS600-865, avenue Carling
Ottawa (Ontario) Canada K1S 5S8Tél. : (613) 226-2553
Téléc. : (613) 226-5392Courriel : [email protected]
ou télécharger les publications du siteWeb de l’OCCETS à :http://www.ccohta.ca

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 3/103
Office canadien de coordination de l’évaluation des technologies de la santé
Le diagnostic moléculaire au regard de syndromesde prédisposition à un cancer héréditaire :
le dépistage génétiqueet son retentissement clinique
Chuong Ho, M.D., M.Sc.1 Srabani Banerjee, Ph.D.
Shaila Mensinkai, M.A., M.B.S.I.
novembre 2003
________________________ 1 Office canadien de coordination de l’évaluation des technologies de la santé, Ottawa (Ontario) Canada

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 4/103
i
Examinateurs Les personnes mentionnées ci-dessous ont eu l’amabilité d’offrir leurs observations sur le
présent rapport.
Examinateurs externes Andrea Eisen, M.D., F.R.C.P.C.Oncologie médicaleProfesseur adjointCentre régional de cancérologie d’HamiltonHamilton (Ontario)
Sherry A.M. Taylor, Ph.D., F.C.C.M.G.Professeure agrégéeDépartement de pathologie et de
médecine moléculaireUniversité Queen’sKingston (Ontario)
Doug Horsman, M.D., F.R.C.P.C., F.C.C.M.G.Directeur, Programme sur les cancershéréditaires
British Columbia Cancer AgencyVancouver (Colombie-Britannique)
Peter Watson, M.A., M.B., B.Chir.,F.R.C.P.C.
Département de pathologie
Faculté de médecineUniversité du ManitobaWinnipeg (Manitoba)
Charmaine Kim-Sing, M.B., F.R.C.P.C.Directrice médicaleProgramme sur les cancers héréditairesBritish Columbia Cancer AgencyVancouver (Colombie-Britannique)
Examinateurs du Conseil consultatif scientifique de l’OCCETS
Kenneth Marshall, B.A., M.Sc., M.D.,F.R.C.P.C., F.C.F.P.
Professeur de médecine familiale (à la retraite)Université de Western OntarioLondon (Ontario)
Jeff Scott, M.B.Ch.B., M.H.Sc.,M.H.S.A., F.R.C.P.C.
Médecin hygiénisteProvince de Nouvelle-ÉcosseHalifax (Nouvelle-Écosse)
Le présent rapport est un examen d’articles, d’études, de documents et d’autres renseignements
publiés (regroupés sous l’appellation « documentation d’origine ») auxquels l’OCCETS a pu
avoir accès. L’OCCETS ne peut donner l’assurance, ni être tenu responsable, de l’exactitude du
contenu de la documentation d’origine sur laquelle se fonde le rapport; l’OCCETS décline
également toute responsabilité quant à la qualité, la propriété, l’inexactitude ou le bien-fondé
des énoncés, renseignements ou conclusions qui figurent dans la documentation d’origine.
L’OCCETS assume la pleine responsabilité quant à la forme et au contenu définitifs du présent
rapport. Les énoncés et conclusions qui y apparaissent reflètent l’opinion de l’OCCETS, et non
celle des membres de ses conseils ou des examinateurs.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 5/103
ii
Paternité de l’ouvrage
À titre d’auteur principal, le Dr Chuong Ho a dirigé l’élaboration du protocole du projet,supervisé l’analyse documentaire, rédigé la version préliminaire, révisé et préparé le rapport auxfins de publication. Mme Srabani Banerjee a collaboré avec le Dr Ho à la rédaction du rapport, aurassemblement des articles, à l’évaluation de leur pertinence et de leur qualité, et à l’extractiondes données. Mme Shaila Mensinkai a conçu la stratégie de recherche documentaire, effectué larecherche, rédigé la section sur la recherche documentaire et l’annexe à ce sujet, et elle a vérifiéet structuré la bibliographie.
RemerciementsLes auteurs expriment leur gratitude aux membres du personnel de l’Office canadien decoordination de l’évaluation des technologies de la santé (OCCETS) qui ont uni leurs effortsdans le cadre de ce projet. Ils remercient tout particulièrement la D re Ingeborg Blancquaert del’Agence d’évaluation des technologies et des modes d’intervention en santé (AETMIS) de ses précieuses observations sur le rapport.
Conflits d’intérêtsLes auteurs et examinateurs n’ont aucun conflit d’intérêts à déclarer.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 6/103
1
LE RAPPORT EN BREF novembre 2003
Le diagnostic moléculaire au regard de syndromes deprédisposition à un cancer héréditaire
Appellation de la technologieLes tests génétiques de dépistage des syndromes de prédisposition à un cancer héréditaire.
Maladie/troubleLa présence d’un syndrome prédisposant à un cancer héréditaire accroît le risque d’apparition du cancer.Le diagnostic de ces syndromes peut reposer sur lessignes cliniques, quoique des tests génétiques soientnécessaires dans bien des cas pour poser ou préciser le diagnostic.
Description de la technologie
En égard au diagnostic ou au dépistage, des testsgénétiques constituent le seul moyen de déceler l’altération génétique en cause. Le diagnosticmoléculaire par analyse génétique est rendu possiblegrâce aux techniques de biologie moléculairemodernes.
Le sujetIl convient de déterminer l’efficacité pratique globalede l’utilisation des tests génétiques dans la prévisionde la probabilité d’apparition du cancer, au vu deleurs limites. La capacité de détecter une altération
génétique varie selon de nombreux facteurs, dont legène en cause, la nature de la mutation, et lasensibilité et la spécificité du test. L’efficacité prédictive de l’analyse génétique est égalementinfluencée par l’interaction complexe entre la prédisposition génétique et les aspectsenvironnementaux. D’autre part, le dépistagegénétique soulève des questions sociales, éthiqueset juridiques particulières.
Objectifs de l’évaluation• Déterminer la validité, tant analytique que
clinique, la disponibilité et le coût des tests
génétiques dans le dépistage et le diagnostic des
syndromes de prédisposition à un cancer héréditaire.
• Documenter le retentissement du dépistagegénétique sur la prise en charge clinique dessyndromes de prédisposition à un cancer héréditaire.
MéthodeIl s’agit d’une étude méthodique consécutive à unerecherche documentaire structurée. Selon des critèresde sélection prédéterminés, deux examinateursindépendants ont relevé 457 documents pertinents.
Les auteurs se penchent sur la recension des testsgénétiques disponibles et leur effet sur la prise encharge clinique de 20 syndromes. Ils ont dressé laliste des techniques moléculaires utilisées pour dépister les syndromes les plus courants de prédisposition à un cancer héréditaire et examiné leur sensibilité analytique et clinique, leur coût et leur disponibilité. Ils ont également établi une liste desservices et laboratoires d’analyse génétique axée sur les cancers héréditaires au Canada.
Conclusions
• S’agissant de nombreux types de cancershéréditaires, le dépistage génétique est loind’être au point en raison des coûts relativementélevés des tests génétiques; de leur validitéanalytique et clinique variable; et de leur disponibilité limitée.
• Compte tenu de l’apparition rapide de nouvellestechniques moléculaires et de la demande à cetégard, le recours à des tests génétiques dans la prise en charge clinique est justifié danscertaines affections.
Le présent résumé est tiré d’un rapport exhaustif d’évaluation d’une technologie de la santé disponible sur le site Web del’OCCETS (www.ccohta.ca) : Ho C, Banerjee S, Mensinkai S. Le diagnostic moléculaire au regard de syndromes deprédisposition à un cancer héréditaire : le dépistage génétique et son retentissement clinique.
Office canadien de coordination de l’évaluation des technologies de la santé (OCCETS)600-865, avenue Carling, Ottawa (Ontario) Canada K1S 5S8 Tél. : (613) 226-2553 Téléc. : (613) 226-5392 www.ccohta.ca
L’OCCETS est un organisme de recherche en santé, indépendant et sans but lucratif, financé par les gouvernements fédéral, provinciaux et territoriaux.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 7/103
iv
RÉSUMÉ
Le sujet
Les cancers héréditaires posent un énorme problème de santé publique; en effet, ce sont 10 %des cancers qui auraient pour origine une mutation génétique héréditaire. Au nombre des défautsgénétiques en cause dans les syndromes de prédisposition à un cancer, mentionnons lesmutations germinales subies par des oncogènes, des gènes suppresseurs de tumeurs et des gènesdu système de réparation de l’ADN, particuliers à divers types de cancers. Le diagnosticmoléculaire de maladies génétiques offre la possibilité de recourir au dépistage génétique pour diagnostiquer les personnes atteintes de cancer et prévoir le risque de cancer chez les parentsindemnes. Le dépistage génétique soulève cependant des questions sociales, éthiques et juridiques particulières. La taille et la complexité de certains gènes reliés au cancer rendent ladétection des mutations difficile, posant des défis d’ordre technique. La capacité de prédire la probabilité d’apparition du cancer varie selon le gène et la mutation décelée. L’interprétation etl’application des résultats de l’analyse génétique peuvent donc poser problème. L’interactioncomplexe entre la prédisposition génétique et les influences environnementales restreintégalement la capacité prévisionnelle du dépistage génétique. L’utilité du dépistage et dudiagnostic moléculaires repose sur la validité du test et son retentissement sur la prise en chargeclinique. L’expansion fulgurante des techniques moléculaires de détection des mutations,l’intérêt grandissant à l’endroit des tests génétiques et l’usage répandu de l’analyse génétiqueauront des répercussions à la fois sur la santé de la population et les coûts des soins de santé.
Objectifs
Les objectifs de la présente étude consistent à effectuer une analyse documentaire méthodiquedes données probantes concernant la disponibilité, le coût et la validité analytique et clinique destests génétiques dans le dépistage et le diagnostic de syndromes de prédisposition à un cancer
héréditaire, et de documenter l’impact du dépistage génétique sur la prise en charge clinique decertains syndromes de prédisposition à un cancer héréditaire.
Les résultats
La documentation, publiée ou inédite, a été recensée par la recherche électronique dans des basesde données en fonction d’une stratégie définie, par le dépouillement manuel de la bibliographiede certains documents et en communiquant avec des laboratoires d’analyse génétique. Deuxexaminateurs ont sélectionné, de façon indépendante, 457 documents pertinents.
Les auteurs ont dressé la liste des 20 syndromes les plus courants prédisposant à un cancer héréditaire et répertorié leur prévalence, leur pénétrance et le risque oncogène connexe, ainsi queles techniques moléculaires utilisées pour déceler les mutations en cause. Ils ont ensuite examinéle diagnostic moléculaire et l’impact du dépistage génétique sur la prise en charge dessyndromes. Le présent rapport renferme par ailleurs une liste de cliniques de génétique sur lescancers héréditaires au Canada.
Les auteurs ont regroupé les syndromes de prédisposition à un cancer héréditaire dans lescatégories suivantes : les syndromes pour lesquels le dépistage génétique peut être considérécomme faisant partie de la prise en charge clinique des familles atteintes; les syndromes pour
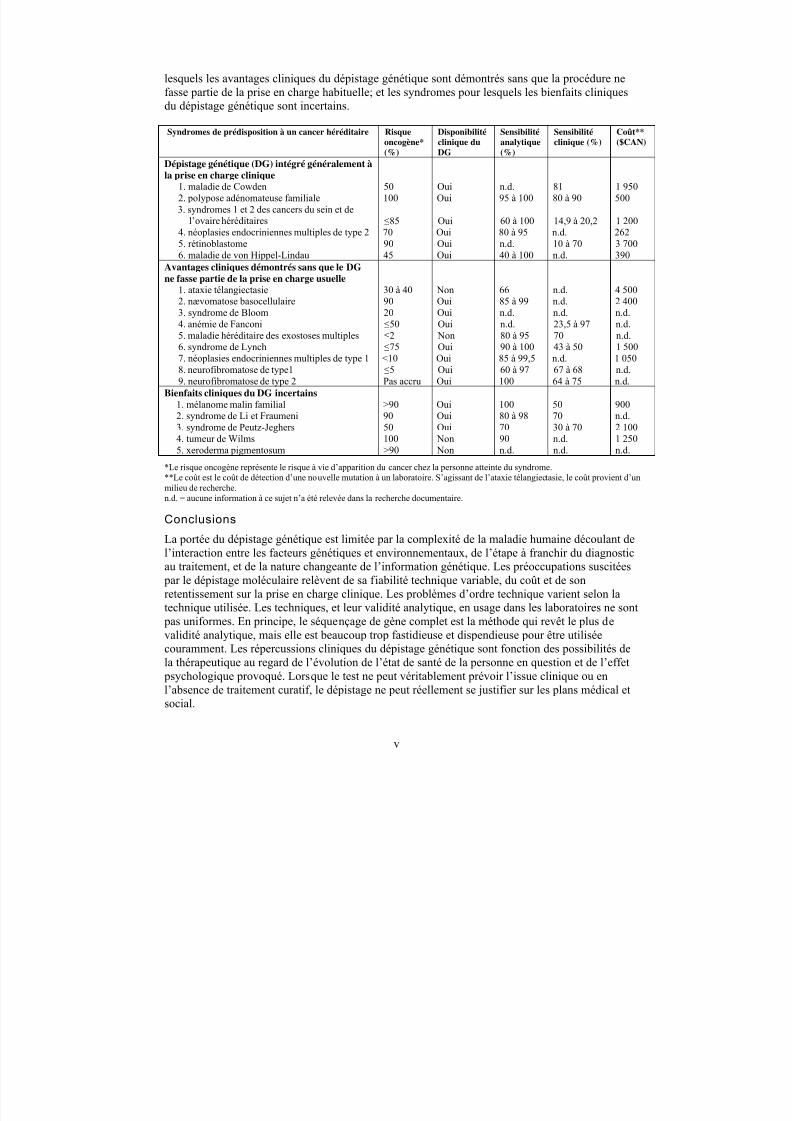
5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 8/103
v
lesquels les avantages cliniques du dépistage génétique sont démontrés sans que la procédure nefasse partie de la prise en charge habituelle; et les syndromes pour lesquels les bienfaits cliniquesdu dépistage génétique sont incertains.
Syndromes de prédisposition à un cancer héréditaire Risqueoncogène*
(%)
Disponibilitéclinique du
DG
Sensibilitéanalytique
(%)
Sensibilitéclinique (%)
Coût**($CAN)
Dépistage génétique (DG) intégré généralement àla prise en charge clinique
1. maladie de Cowden 50 Oui n.d. 81 1 9502. polypose adénomateuse familiale 100 Oui 95 à 100 80 à 90 5003. syndromes 1 et 2 des cancers du sein et de
l’ovaire héréditaires ≤85 Oui 60 à 100 14,9 à 20,2 1 2004. néoplasies endocriniennes multiples de type 2 70 Oui 80 à 95 n.d. 2625. rétinoblastome 90 Oui n.d. 10 à 70 3 7006. maladie de von Hippel-Lindau 45 Oui 40 à 100 n.d. 390
Avantages cliniques démontrés sans que le DGne fasse partie de la prise en charge usuelle
1. ataxie télangiectasie 30 à 40 Non 66 n.d. 4 500
2. nævomatose basocellulaire 90 Oui 85 à 99 n.d. 2 4003. syndrome de Bloom 20 Oui n.d. n.d. n.d.4. anémie de Fanconi ≤50 Oui n.d. 23,5 à 97 n.d.5. maladie héréditaire des exostoses multiples ≤2 Non 80 à 95 70 n.d.6. syndrome de Lynch ≤75 Oui 90 à 100 43 à 50 1 5007. néoplasies endocriniennes multiples de type 1 <10 Oui 85 à 99,5 n.d. 1 0508. neurofibromatose de type1 ≤5 Oui 60 à 97 67 à 68 n.d.9. neurofibromatose de type 2 Pas accru Oui 100 64 à 75 n.d.
Bienfaits cliniques du DG incertains1. mélanome malin familial >90 Oui 100 50 9002. syndrome de Li et Fraumeni 90 Oui 80 à 98 70 n.d.3. syndrome de Peutz-Jeghers 50 Oui 70 30 à 70 2 1004. tumeur de Wilms 100 Non 90 n.d. 1 250
5. xeroderma pigmentosum >90 Non n.d. n.d. n.d.*Le risque oncogène représente le risque à vie d’apparition du cancer chez la personne atteinte du syndrome.**Le coût est le coût de détection d’une nouvelle mutation à un laboratoire. S’agissant de l’ataxie télangiectasie, le coût provient d’unmilieu de recherche.n.d. = aucune information à ce sujet n’a été relevée dans la recherche documentaire.
Conclusions
La portée du dépistage génétique est limitée par la complexité de la maladie humaine découlant del’interaction entre les facteurs génétiques et environnementaux, de l’étape à franchir du diagnosticau traitement, et de la nature changeante de l’information génétique. Les préoccupations suscitées par le dépistage moléculaire relèvent de sa fiabilité technique variable, du coût et de sonretentissement sur la prise en charge clinique. Les problèmes d’ordre technique varient selon la
technique utilisée. Les techniques, et leur validité analytique, en usage dans les laboratoires ne sont pas uniformes. En principe, le séquençage de gène complet est la méthode qui revêt le plus devalidité analytique, mais elle est beaucoup trop fastidieuse et dispendieuse pour être utiliséecouramment. Les répercussions cliniques du dépistage génétique sont fonction des possibilités dela thérapeutique au regard de l’évolution de l’état de santé de la personne en question et de l’effet psychologique provoqué. Lorsque le test ne peut véritablement prévoir l’issue clinique ou enl’absence de traitement curatif, le dépistage ne peut réellement se justifier sur les plans médical etsocial.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 9/103
vi
Malgré l’apparition rapide de nouvelles techniques moléculaires, la mise en application dudépistage génétique dans le cadre de la prise en charge clinique de nombreuses affections estimmotivée. Avant d’être en mesure de se prononcer quant à l’intégration de tests particuliers auxservices cliniques et au financement de ces tests, il faudra obtenir d’autres renseignementsconcernant leur impact sur l’évolution de l’état de santé des patients et leur coût.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 10/103
vii
TABLE DES MATIÈRES
RÉSUMÉ ...................................................................................................................................... iv
TABLE DES MATIÈRES.......................................................................................................... vii
ABRÉVIATIONS......................................................................................................................... ix
GLOSSAIRE ................................................................................................................................ xi
1 INTRODUCTION................................................................................................................. 1
2 OBJECTIFS .......................................................................................................................... 4
3 MÉTHODE............................................................................................................................ 4
3.1 Recherche documentaire............................................................................................... 43.2 Critères de sélection et méthode ................................................................................... 43.3 Extraction des données ................................................................................................. 53.4 Limites .......................................................................................................................... 5
4 RÉSULTATS......................................................................................................................... 6 4.1 Quantité de documentation disponible.......................................................................... 6
4.2 Sommaire des syndromes de prédisposition à un cancer héréditaire et de leurs tests dedépistage génétique ................................................................................................................ 64.3 Impact du dépistage génétique sur la prise en charge clinique................................... 14
4.3.1 Ataxie télangiectasie .................................................................................... 14
4.3.2 Nævomatose basocellulaire ......................................................................... 154.3.3 Syndrome de Bloom .................................................................................... 164.3.4 Maladie de Cowden ..................................................................................... 174.3.5 Polypose adénomateuse familiale ................................................................ 184.3.6 Mélanome familial ....................................................................................... 204.3.7 Anémie de Fanconi ...................................................................................... 214.3.8 Syndromes 1 et 2 des cancers du sein et de l’ovaire héréditaires ................ 224.3.9 Maladie héréditaire des exostoses multiples................................................ 274.3.10 Syndrome de Lynch ..................................................................................... 284.3.11 Syndrome de Li et Fraumeni........................................................................ 304.3.12 Néoplasies endocriniennes multiples de type 1 ........................................... 31
4.3.13 Néoplasies endocriniennes multiples de type 2 ........................................... 324.3.14 Neurofibromatose de type 1......................................................................... 334.3.15 Neurofibromatose de type 2......................................................................... 354.3.16 Syndrome de Peutz-Jeghers ......................................................................... 364.3.17 Rétinoblastome ............................................................................................ 374.3.18 Maladie de von Hippel-Lindau .................................................................... 384.3.19 Tumeur de Wilms ........................................................................................ 394.3.20 Xeroderma pigmentosum............................................................................. 40

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 11/103
viii
5 DISCUSSION ...................................................................................................................... 42
6 CONCLUSION ................................................................................................................... 45
7 RÉFÉRENCES.................................................................................................................... 46
Annexe 1 : Stratégie de recherche documentaire.......................................................................... 75 Annexe 2 : Sélection des études.................................................................................................... 81 Annexe 3: Services et laboratoires de dépistage génétique au Canada ........................................82

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 12/103
ix
ABRÉVIATIONS
ACP amplification en chaîne par polyméraseADN acide désoxyribonucléiqueAF anémie de Fanconi
AH analyse des hétéroduplexAHI analyse d’hybridation intrachromosomiqueAIM analyse de l’instabilité de microsatelliteALF analyse de liaison factorielleASC analyse séquentielle comparativeAT ataxie télangiectasieATS analyse du transfert de SouthernBAAO bioanalyse d’allèles par oligonucléotideBAEA bioanalyse de l’expression d’allèlesBRCA1 syndrome 1 des cancers du sein et de l’ovaire héréditairesBRCA2 syndrome 2 des cancers du sein et de l’ovaire héréditaires
CCM clivage chimique de mésappariementCIH coloration immunohistochimiqueCLHP chromatographie liquide à haute performanceCLHPD chromatographie liquide à haute performance en fonction de la dénaturationDEM détection enzymatique des mutationsEE examen d’exonsEGB exploration génique bidimensionnelleEGC électrophorèse en gel en fonction de la conformationEGGD électrophorèse en gel selon le gradient de dénaturantEGGT électrophorèse en gel selon le gradient de températureHISF hybridation in situ en fluorescence
HNPCC syndrome de LynchIE immuno-essaiIM instabilité de microsatellite IM-HF IM à haute fréquenceMC maladie de CowdenMF mélanome familialMHEM maladie héréditaire des exostoses multiples NB nævomatose basocellulaire NEM1 néoplasies endocriniennes multiples de type 1 NEM2 néoplasies endocriniennes multiples de type 2 NF1 neurofibromatose de type 1
NF2 neurofibromatose de type 2PAF polypose adénomateuse familialePCR-CDNA transcription inverse suivie d’ACPPCSB polymorphisme de conformation des ADN simples brinsPCSB-F polymorphisme de conformation des ADN simples brins en fluorescencePR polymorphisme de restrictionRB rétinoblastomeSB syndrome de BloomSGC séquençage de gène complet

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 13/103
x
SLF syndrome de Li et FraumeniSPJ syndrome de Peutz-JeghersTPT test de protéine tronquéeTW tumeur de WilmsVHL maladie de von Hippel-Lindau
VPN valeur prédictive négativeVPP valeur prédictive positiveXP xeroderma pigmentosum

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 14/103
xi
GLOSSAIRE
Amplification en chaîne par polymérase (ACP) : technique d’amplification faisant en sorte quel’épreuve, selon un choix judicieux d’amorces, est dirigée sur la mutation d’intérêt ou« l’emplacement suspecté » de plusieurs mutations potentielles sur le gène, en utilisant une
quantité minime de matière première. Méthode particulièrement utile pour déceler des mutations ponctuelles ou des microdélétions, lesquelles passent souvent inaperçues dans l’analyse dutransfert de Southern.
Analyse de liaison factorielle (ALF) : méthode utilisée lorsque le gène en cause est inconnu oudans le cas d’affections dues à un grand nombre de mutations. Elle repose sur l’analysecomparative des membres de la fratrie et des parents, atteints ou non; les familles ne se prêtent pas toutes à cette technique.
Analyse du transfert de Southern (ATS) : technique de transfert d’un gel à une membrane defragments d’ADN obtenus par électrophorèse pour mettre en évidence par la suite le fragment
d’intérêt par hybridation de l’acide nucléique à l’aide d’une sonde nucléaire complémentairemarquée. Méthode de détection des délétions importantes en repérant une perte ou la réductionde taille d’un fragment cible.
Bioanalyse d’allèles par oligonucléotide (BAAO) : méthode de détection des mutations par hybridation de courtes séquences d’acide désoxyribonucléique (ADN) synthétique représentantun mutant particulier avec un jeu ordonné de séquences cibles sur des membranes de nylon.
Bioanalyse de l’expression d’allèles (BAEA) : méthode de repérage des mutations vues commeun déséquilibre de la représentation des allèles dans l’ARN issu de la transcription.
Chromatographie liquide à haute performance (CLHP) : technique de chromatographie liquide(soit une méthode de séparation en fonction des interactions différentes des molécules en phasemobile et en phase stationnaire au fur et à mesure que le prélèvement se déplace dans le solvant)de haute résolution, d’analyse rapide et de bonne reproductibilité.
Chromatographie liquide à haute performance en fonction de la dénaturation (CLHPD) : méthode chromatographique selon laquelle le prélèvement est séparé sous pression élevée àl’aide d’une phase stationnaire et d’une phase mobile. La CLHPD est en fait la technique deCLHP dans des conditions de température partiellement dénaturantes. La CLHPD est uneméthode sensible et spécifique de grand débit de traitement.
Clivage de mésappariement par RNase : méthode de clivage par RNase de bases mésappariées(provenant du prélèvement de tissu tumoral porteur de mutations) produisant des fragmentsrepérés sous rayonnement ultraviolet.
Détection enzymatique des mutations (DEM) : méthode faisant appel à une enzyme, comme larésolvase, qui parcoure un fragment d’ADN en quête d’une distorsion structurale due à unemutation ponctuelle, une insertion ou une délétion.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 15/103
xii
Électrophorèse : technique de séparation des molécules en fonction de leur différence demobilité sous l’influence d’un champ électrique.
Électrophorèse en gel en fonction de la conformation (EGC) : méthode de séparation defragments d’ADN sur une matrice de gel. La méthode repose sur la propriété intrinsèque de
flexion de l’ADN en solution et la modification de sa conformation en présence d’une séquencenucléotidique altérée (p. ex., mésappariement de bases). Les différences de conformation entreun homoduplex et un hétéroduplex sont mises en relief grâce à un agent légèrement dénaturant.En électrophorèse, l’hétéroduplex se distingue par sa mobilité réduite. Cette méthode permet dedéceler ne serait-ce qu’une seule paire de bases mésappariées.
Électrophorèse en gel selon le gradient de dénaturant (EGGD) : méthode de séparation defragments d’ADN sur une matrice en gel renfermant un agent dénaturant en gradient deconcentration. Cette méthode est fondée sur le principe voulant que le double brin d’ADN sedénature (séparation en simples brins) dans une mesure variant selon la séquence nucléotidiqueet la concentration du dénaturant. Cela modifie la conformation de l’ADN, donc sa mobilité dans
la matrice de gel. Cette méthode permet de détecter des différences de comportement dedénaturation de petits fragments d’ADN (de 200 à 700 paires de bases) qui tiennent à aussi peuqu’une paire de bases. Hausser la température peut également entraîner la dénaturation del’ADN. Lorsqu’un gradient de température remplace un gradient de concentration de dénaturant,la méthode s’appelle électrophorèse en gel selon le gradient de température (EGGT).
Électrophorèse en gel selon le gradient de température (EGGT ): se reporter à l’EGGD.
Gène : séquence de nucléotides qui occupe un locus (emplacement) précis sur un chromosome.Il constitue l’unité fonctionnelle de l’hérédité.
Génotype : ensemble des gènes d’une personne, par opposition aux caractères apparents ou phénotype.
Hybridation in situ en fluorescence (HISF) : méthode de détermination de la position d’un gène par l’utilisation d’une sonde nucléaire indicatrice et la visualisation en microscopie par fluorescence. Technique sensible dans la détection de séquences nucléotidiques précises d’un prélèvement fixé sur lamelle de microscope. L’HISF permet de repérer les vastes délétions ouinsertions et les translocations.
Lignée cellulaire germinale : lignée cellulaire provenant directement des gamètes (ovule etspermatozoïde) qui à la fécondation forment un zygote, par opposition aux cellules somatiques
(les autres cellules de l’organisme). Les mutations de la lignée germinale sont transmises à ladescendance (c.-à-d., héréditaires), alors que les mutations des cellules somatiques ne le sont pas.
Microsatellite : courte séquence de longueur variable de deux ou trois nucléotides qui se répètentle long du gène. L’instabilité du microsatellite est définie comme une modification de longueur quelle qu’elle soit en raison d’une insertion ou d’une délétion d’unités répétitives dans unmicrosatellite de tissu tumoral, par rapport au tissu normal.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 16/103
xiii
Mosaïcisme : en génétique, présence chez une même personne d’au moins deux lignéescellulaires différentes du point de vue caryotypique ou génotypique, issues du zygote.
Mutation : erreur survenue dans la séquence des bases nucléotidiques d’un gène. Elle peutentraver la capacité du gène à produire une protéine entièrement fonctionnelle. Il peut s’agir
d’une mutation ponctuelle (substitution d’une seule paire de bases), d’une délétion ou d’uneinsertion, avec ou sans changement de phase (perte ou addition d’acides aminés dans un codon),d’une amplification ou d’une séquence répétitive de trois nucléotides (hausse du nombre deséquences répétitives) ou d’une translocation (échange de grands segments chromosomiquesentre deux chromosomes). Une mutation faux-sens est une mutation qui échange un codonspécifiant un acide aminé contre un codon qui en spécifie un autre. Une mutation non-sens estune mutation qui altère la séquence codante et empêche la synthèse de la protéine.
Pénétrance : probabilité que le gène mutant s’exprime dans le phénotype de la personne porteuse de la mutation.
Phénotype : présentation clinique ou expression d’un gène particulier ou des gènes dans unmilieu environnant précis.
Polymorphisme de conformation des ADN simples brins (PCSB) : méthode qui repose sur lesdifférences de conformation, applicable aux brins simples. Elle peut déceler des mutations ponctuelles de gènes. La substitution d’une seule base peut altérer la structure secondaire d’unfragment d’ADN dont la mobilité sur gel est ainsi modifiée.
Polymorphisme de restriction (PR) : méthode selon laquelle le gène est coupé en fragments par des enzymes de restriction et les fragments d’ADN sont séparés par électrophorèse en gel avantd’être révélés par coloration au bromure d’éthidium. Les mutations du gène abolissent ou créent
des sites de clivage des enzymes de restriction, d’où le schéma altéré des fragments d’ADN. Proposant : personne atteinte qui est évaluée seule, en l’absence de la fratrie ou des parents, dansune étude génétique.
Risque oncogène : c’est le risque d’apparition du cancer pendant la vie que court une personneatteinte d’un syndrome héréditaire de prédisposition à un cancer familial.
Séquençage de gène complet (SGC) : il s’agit de la détermination de la séquence nucléotidiqued’un gène. En théorie, c’est la technique la plus sensible quant à la détection de toutes lesmutations potentielles. Dans l’état actuel de la technologie cependant, le SGC est trop fastidieux
et dispendieux pour être appliqué de façon courante en pratique clinique, et la technique peut ne pas déceler des mutations localisées hors de la région codante du gène (p. ex., dans des zonescomportant des introns, promoteurs ou renforceurs-facilitateurs).
Test de protéine tronquée (TPT) : test qui repère précisément les mutations qui entraînent lacessation de la traduction à partir d’un ARN messager et qui donnent lieu à des protéinestronquées. Il permet de déceler les mutations non-sens, les mutations de changement de phase etles mutations du site d’épissage, tout en passant outre les mutations faux-sens. En règle générale,

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 17/103
xiv
le fragment d’ADN est amplifié selon l’ACP, puis inséré dans un système de transcription-traduction. La protéine qui en découle et le produit témoin normal, fixés sur gel, sont comparéssous l’angle de la taille.
Transmission autosomique dominante : une personne atteinte d’un trouble génétique à
transmission dominante hérite d’un parent une copie normale du gène et de l’autre une copiealtérée. La seule présence de cette copie altérée (mutée) suffit à causer la maladie. Latransmission verticale (du grand-parent atteint au parent, puis à l’enfant) traduit le caractèredominant de l’affection. Le terme autosomique s’applique aux chromosomes numérotés (de 1 à 22),et non aux chromosomes sexuels (X et Y). La plupart des syndromes de prédisposition à uncancer héréditaire sont de caractère dominant.
Transmission autosomique récessive : les deux copies du gène doivent être porteuses demutations pour qu’une personne souffre d’une affection génétique transmise de façon récessive.La personne atteinte a hérité d’une copie altérée du gène de chacun des parents. La personnedont une copie du gène est altérée et l’autre normale est dite porteuse ou hétérozygote. Dans ce
cas, les symptômes ou problèmes propres à l’affection ne se manifestent pas habituellement. Latransmission horizontale (parents indemnes et au moins deux personnes de la fratrie qui ensouffrent) traduit le caractère récessif de l’affection.
Utilité clinique : cette notion est déterminée en précisant les avantages cliniques et psychologiques du test au regard des risques posés par les résultats positif ou négatif. L’utilitéclinique d’un test génétique varie selon l’état de santé et la population soumise au test. Il y a lieude démontrer l’utilité clinique d’un test avant de le rendre disponible en pratique clinique.
Validité analytique : la sensibilité analytique d’un test génétique renvoie à sa capacité de déceler les mutations pour lesquelles il a été conçu. La spécificité analytique se rapporte à son efficacité
à identifier avec précision les personnes exemptes de la mutation. La validité analytique du test,élevée lorsque la sensibilité et la spécificité sont élevées, varie selon la méthode appliquée dansl’analyse de l’ADN.
Validité clinique : la validité clinique, c’est-à-dire la précision avec laquelle le test génétique permet dediagnostiquer une maladie ou d’en prédire le risque de survenue (ici un syndrome de prédisposition àun cancer héréditaire, et non le cancer en question) est déterminée en fonction de la sensibilité et de laspécificité cliniques, et des valeurs prédictives positive et négative du test. La sensibilité cliniqueconsiste en la probabilité d’un résultat positif dans une population atteinte de la maladie, alors que laspécificité clinique est la probabilité d’un résultat négatif dans une population indemne. La valeur prédictive positive représente la probabilité que les personnes pour qui le résultat du test est positif
présenteront la maladie, tandis que la valeur prédictive négative est la probabilité que les personnes pour qui le résultat du test est négatif seront épargnées. La validité clinique d’un test génétique varieselon l’état de santé et la population soumise au test.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 18/103
1
1 INTRODUCTION
La détermination de la séquence du génome humain et des gènes humains a enrichi nosconnaissances sur la santé et la pathogenèse. Nous savons désormais que pratiquement toutes lesmaladies comportent un aspect génétique. C’est ainsi que la génétique moléculaire pourrait s’avérer
un puissant outil de diagnostic et de dépistage. L’interaction complexe entre la prédispositiongénétique et les facteurs environnementaux limite, cependant, l’efficacité prédictive du dépistagegénétique1. Scruter à la loupe la composition constitutionnelle fondamentale d’une personne soulèveen outre des questions d’ordre psychologique, éthique et professionnel2-4.
Le phénomène de vulnérabilité génétique a été étudié5-9. Tous les cancers sont causés par desmutations de gènes qui contrôlent des aspects de la biologie cellulaire comme la multiplication et ladifférenciation cellulaires. La plupart du temps, les mutations se produisent dans des cellulessomatiques (c.-à-d., à l’origine des cancers sporadiques). Habituellement, l’altération se manifestedans plusieurs oncogènes ou gènes suppresseurs de tumeurs avant que le cancer sporadiquesurvienne. Chez certaines personnes, la mutation est transmise par un parent car elle s’est produite
dans la lignée cellulaire germinale (c.-à-d., à l’origine des cancers héréditaires). La mutation héritéeainsi prédispose ces personnes à un cancer. La mutation germinale constitue l’événement initiateur de la transformation tumorale, bien qu’elle soit insuffisante pour causer à elle seule le cancer. Lessyndromes héréditaires qui entraînent un risque accru de survenue d’un cancer sont appeléssyndromes de prédisposition à un cancer héréditaire. Le présent rapport est axé sur le dépistagegénétique de syndromes de prédisposition à un cancer héréditaire.
Comparativement aux cancers sporadiques, les cancers héréditaires sont caractérisés par lesaspects cliniques suivants10 :• survenue à un âge plus jeune que la moyenne de la présentation usuelle de ce type de
cancers;• forme multifocale du cancer dans un organe simple ou atteinte bilatérale du cancer dans
une paire d’organes;• apparition de plus d’une tumeur primitive chez la même personne;• histoire familiale de cancers du même type ou de plusieurs types reliés à un syndrome de
prédisposition à un cancer chez un ou des parents proches;• taux élevé de cancers dans une famille;• apparition d’un cancer chez une personne ou dans une famille atteinte d’anomalies
congénitales.
Les gènes concernés dans la cancérogenèse sont les gènes suppresseurs de tumeurs, lesoncogènes et les gènes des systèmes de réparation de l’ADN11. Les mutations de ces gènes
entraînent une prolifération cellulaire anarchique, amorçant la formation tumorale. Les gènessuppresseurs de tumeurs codent des protéines qui inhibent la division cellulaire; les délétions oumutations empêchent la synthèse de ces protéines ou provoquent la formation de protéinesanormales ou inactives, incapables de remplir leur fonction anti-oncogène dans la cellule. Quantaux oncogènes, les mutations se traduisent par la production de protéines appelées oncoprotéines,activées de façon permanente pour stimuler la division et la multiplication cellulaires. De leur côté, les gènes des systèmes de réparation de l’ADN jouent un rôle essentiel dans la protectionde l’intégrité des gènes. Les mutations à l’origine des cancers héréditaires sont le plus souvent

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 19/103
2
localisées dans les gènes suppresseurs de tumeurs, quoiqu’elles puissent également s’attaquer parfois à quelques oncogènes et gènes des systèmes de réparation de l’ADN.
L’essor des techniques de la génétique moléculaire nous a permis en grande partie de mieux connaîtrele processus de prédisposition au cancer. Grâce à ces techniques, les gènes prédisposant à un cancer ont
pu être cernés, isolés, caractérisés sur le plan fonctionnel et cartographiés afin de relever leur emplacement dans le génome humain. Le diagnostic moléculaire de maladies génétiques n’est pas pour autant dénué de complexité.
La génétique moléculaire à des fins diagnostiques suppose l’utilisation de toute la gamme destechniques modernes de la biologie moléculaire, notamment le polymorphisme de conformation desADN simples brins (PCSB), la chromatographie liquide à haute performance en fonction de ladénaturation (CLHPD), l’électrophorèse en gel selon le gradient de dénaturant (EGGD), le test de protéine tronquée (TPT), l’analyse du transfert de Southern (ATS), l’amplification en chaîne par polymérase (ACP), la bioanalyse d’allèles par oligonucléotide (BAAO) faisant intervenir au préalablel’ACP, l’hybridation in situ en fluorescence (HISF) et le séquençage de l’ADN.
Le choix de la technique varie selon que le gène est connu et son degré d’hétérogénéité12. Les troublesgénétiques peuvent être répartis en deux catégories : les troubles pour lesquels le gène en cause a étécerné et les troubles pour lesquels le gène en cause est inconnu. La méthode de dépistage des troublesde la première catégorie consiste en l’analyse directe de la mutation génique, tandis que la méthodeutilisée dans la seconde catégorie est l’analyse de liaison factorielle à l’aide de sondes polymorphesd’ADN, agissant à titre de marqueurs, situées à proximité sur le même chromosome, pourvu que lacartographie des gènes ait révélé que l’affection trouve son origine dans ce chromosome. Le degréd’hétérogénéité renvoie au nombre de gènes ou à la diversité des mutations du même gène qui participent à l’étiologie d’une seule et même maladie. Plus l’hétérogénéité est élevée, plus le test estexigeant en main-d’œuvre et coûteux. En présence d’une très grande hétérogénéité, l’analyse directe de
la mutation devient impensable et il faut s’en remettre à l’analyse de liaison, même si le gène en cause aété cloné.
Si bon nombre des mutations d’un gène reliées à un cancer sont connues et que ces mutations sont peunombreuses, l’analyse directe de ces mutations est alors simple et abordable. Si le gène est de grandetaille et que les mutations causant le cancer sont nombreuses et dispersées ou se produisent dans deszones peu accessibles du gène, l’analyse des mutations reposera alors sur des techniques permettantd’examiner de grands fragments d’ADN. Ces tests, tels l’EGGD, le PCSB, le TPT, la CLHPD et leséquençage de gène complet, sont longs et dispendieux. Une fois la mutation cernée chez une personneatteinte, le dépistage subséquent parmi les membres de sa famille est plus simple, car le processus estaxé sur l’anomalie en question. La mutation d’intérêt peut alors être étudiée en recourant à l’ACP. Dans
le cas de troubles pour lesquels le gène en cause est inconnu ou les mutations trop nombreuses, lediagnostic prédictif par l’analyse de liaison demeure possible dans certaines familles. Les stratégies dedétection des mutations sont illustrées à la figure 1.
Les tests génétiques peuvent être utilisés à des fins diagnostiques ou de dépistage. Les tests génétiquesdiagnostiques permettent de poser ou de préciser le diagnostic d’un trouble ou d’un syndrome chez une personne symptomatique ou chez le fœtus. Les tests génétiques de dépistage permettent de relever la présence du trouble chez une personne saine, sans égard aux antécédents familiaux de la maladie.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 20/103
3
Confirmation• Analyse
séquentielle
Dépistagepréliminaire• EGGD• PCSB• TPT• CLHPD• Autres
Confirmation directe• SGC
Mutation ponctuelle• ACP et électrophorèse
en gel• ACP et BAAO• ACP quantitative
Vastes délétions• ATS• HISF• ACP quantitative
Examiner le gène pour détecter les mutations
Étudier des mutations précises
ANALYSE MUTATIONNELLE DIRECTE
Le gène en cause est connu
Figure 1 : Stratégies de détection des mutations
3

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 21/103
4
2 OBJECTIFS
1. Effectuer une analyse documentaire méthodique des données probantes concernant ladisponibilité, le coût et la validité analytique et clinique des tests génétiques dans ledépistage et le diagnostic de syndromes de prédisposition à un cancer héréditaire.
2. Documenter l’impact du dépistage génétique sur la prise en charge clinique de certainssyndromes de prédisposition à un cancer héréditaire.
3 MÉTHODE
3.1 Recherche documentaire
La documentation, publiée ou inédite, a été recensée en effectuant une recherche dans plusieurs bases de données (pour connaître la stratégie de recherche documentaire détaillée, voir l’annexe1). Par le système DIALOG®, MEDLINE®, EMBASE®, Cancerlit, BIOSIS Previews® etPASCAL® ont été consultées en août 2002 en fonction des vedettes-matières MeSH, dedescripteurs, du nom des gènes et de mots-de-textes. La stratégie de recherche documentaire aété établie d’après les objectifs du rapport et un filtre a restreint la recherche aux études pertinentes. Seuls les documents portant sur les humains ont été pris en considération. Des alerteset mises à jour à intervalles réguliers ont été mises sur pied en utilisant les mêmes vedettes-matières et mots-clés que ceux de la recherche initiale pour répertorier de nouvelles mentions jusqu’en avril 2003. Les autres bases de données consultées sont The Cochrane Library sur cédérom, la base de données OMIM (Online Mendelian Inheritance in Man) du National Center for Biotechnology Information, PubMed et GENETests. De plus, nous avons consulté desregistres d’essais cliniques, le site Web d’organisations d’évaluation de technologies de la santéet d’organisations connexes. Pour obtenir les renseignements concernant les laboratoiresexécutant des tests particuliers, nous avons eu recours au moteur de recherche Google MC. Enfin,nous avons dépouillé manuellement certaines bibliographies et communiqué avec deslaboratoires de génétique pour obtenir des renseignements supplémentaires.
3.2 Critères de sélection et méthode
Dans le cadre de la présente étude, nous avons établi deux critères de sélection.
• L’étude devait être pertinente du point de vue des objectifs du projet et être axée sur l’utilisation de tests génétiques dans le dépistage et le diagnostic des formes familiales decancers et sur la technologie, à savoir les tests utilisés pour déceler les mutationsd’oncogènes, de gènes suppresseurs de tumeurs et de gènes des systèmes de réparation del’ADN.
• Aucune restriction de langue ne limite la sélection des résumés et l’admissibilité desarticles.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 22/103
5
Les documents publiés sous forme de lettre, d’éditorial ou de bref précis ainsi qu’une seconde publication d’une même étude présentant les mêmes résultats ont été rejetés.
3.3 Extraction des données
Deux examinateurs (CH et SB) ont sélectionné les articles conformément aux critères desélection. Les données extraites portent sur :
• la validité analytique et clinique des tests génétiques dans la détection de cancershéréditaires;
• les aspects techniques et le coût du dépistage génétique;• le retentissement du dépistage génétique sur la prise en charge clinique de syndromes de
prédisposition à un cancer héréditaire.
3.4 LimitesLes principales études sur le rendement des tests génétiques ne sont pas à répartition aléatoire, desorte qu’il a été impossible d’effectuer une analyse critique de la qualité de l’information.D’autre part, la diversité des affections examinées ainsi que l’hétérogénéité des groupes participant aux études ont rendu impossible l’analyse quantitative des données.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 23/103
6
4 RÉSULTATS
4.1 Quantité de documentation disponible
Par la recherche documentaire initiale, nous avons relevé 1 786 résumés. Nous avons obtenu laversion intégrale de 637 articles présumément pertinents afin de les évaluer de façonapprofondie. Nous avons également récupéré la version intégrale de 50 articles à la suite dudépouillement des alertes et des bibliographies. Dans l’ensemble, 230 articles ont été écartés pour motif de non-conformité aux critères de sélection. Le présent rapport tient donc compte de457 articles pertinents (le schéma de sélection des documents figure à l’annexe 2).
4.2 Sommaire des syndromes de prédisposition à un cancerhéréditaire et de leurs tests de dépistage génétique
Le tableau 1 présente les techniques de biologie moléculaire utilisées pour détecter lessyndromes les plus courants de prédisposition à un cancer héréditaire ainsi que leur sensibilitéanalytique et clinique, leur coût et leur disponibilité. La prévalence des affections, de même quele risque oncogène et la pénétrance, sont également mentionnés. La section 4.3 couvre de façon plus exhaustive le dépistage moléculaire de chacun des syndromes et son incidence sur la priseen charge clinique. Les services de dépistage génétique et les laboratoires d’analyse des cancershéréditaires au Canada sont énumérés à l’annexe 3.
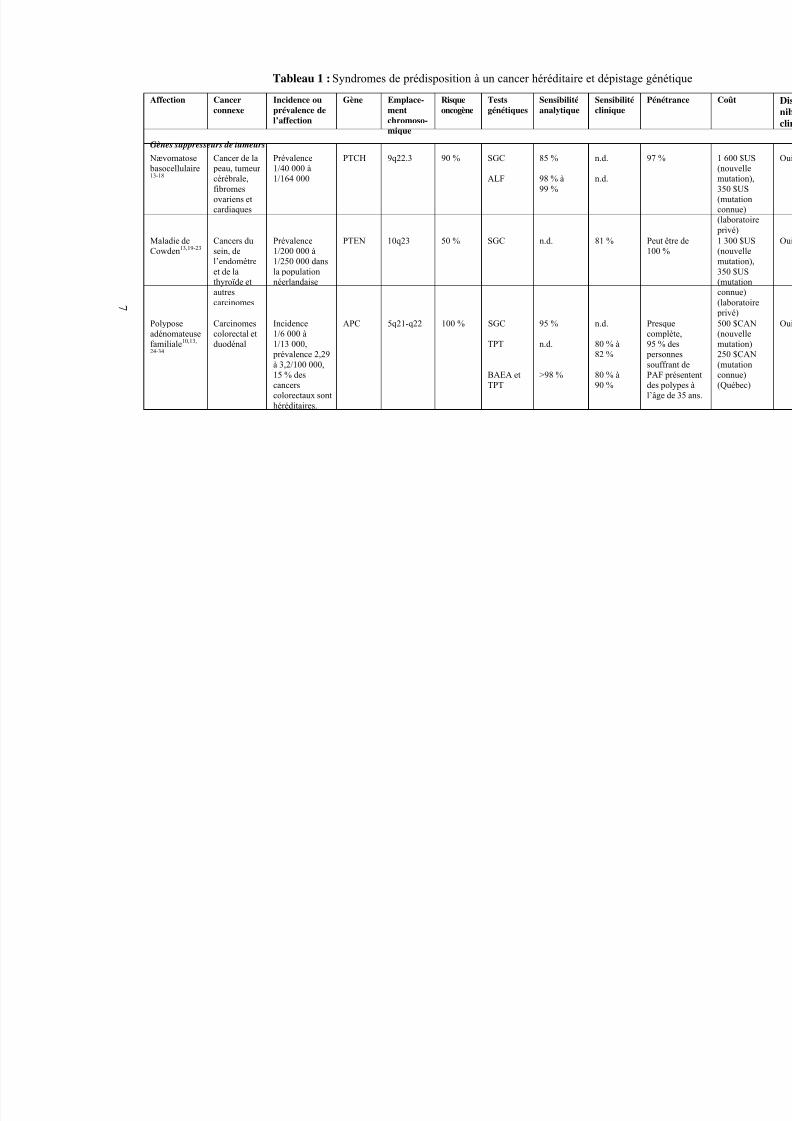
5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 24/103
7
Tableau 1 : Syndromes de prédisposition à un cancer héréditaire et dépistage g
Affection Cancerconnexe
Incidence ouprévalence del’affection
Gène Emplace-mentchromoso-mique
Risqueoncogène
Testsgénétiques
Sensibilitéanalytique
Sensibilitéclinique
P
Gènes suppresseurs de tumeurs
Nævomatose basocellulaire13-18
Cancer de la peau, tumeur cérébrale,fibromesovariens etcardiaques
Prévalence1/40 000 à1/164 000
PTCH 9q22.3 90 % SGC
ALF
85 %
98 % à99 %
n.d.
n.d.
9
Maladie deCowden13,19-23
Cancers dusein, del’endomètreet de lathyroïde etautres
carcinomes
Prévalence1/200 000 à1/250 000 dansla populationnéerlandaise
PTEN 10q23 50 % SGC n.d. 81 % P
Polyposeadénomateusefamiliale10,13,
24-34
Carcinomescolorectal etduodénal
Incidence1/6 000 à1/13 000,
prévalence 2,29à 3,2/100 000,15 % descancerscolorectaux sonthéréditaires.
APC 5q21-q22 100 % SGC
TPT
BAEA etTPT
ALF
CLHPD
PCSB
95 %
n.d.
>98 %
100 %
n.d.
n.d.
n.d.
80 % à82 %
80 % à90 %
n.d.
n.d.
n.d.
Pc9
psPdl
7
5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 25/103
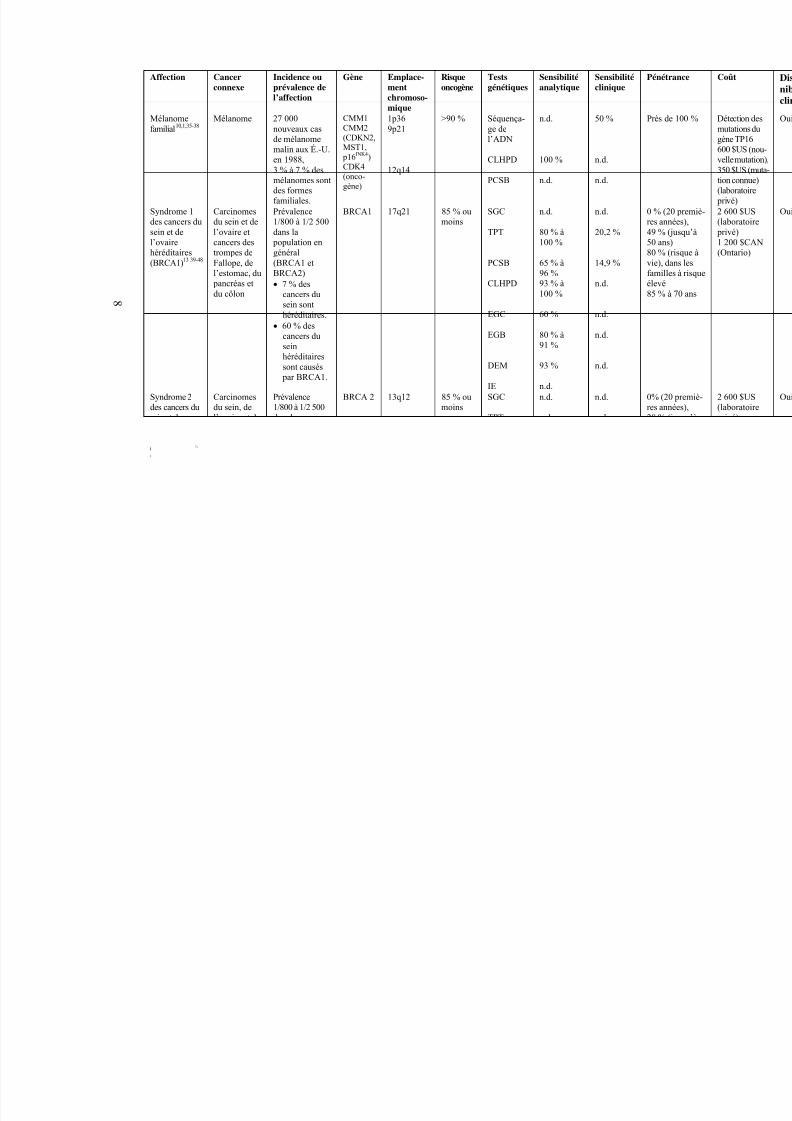
8
Affection Cancerconnexe
Incidence ouprévalence del’affection
Gène Emplace-mentchromoso-mique
Risqueoncogène
Testsgénétiques
Sensibilitéanalytique
Sensibilitéclinique
P
Mélanomefamilial10,1,35-38
Mélanome 27 000nouveaux casde mélanome
malin aux É.-U.en 1988,3 % à 7 % desmélanomes sontdes formesfamiliales.
CMM1CMM2(CDKN2,
MST1, p16INK4)CDK4(onco-gène)
1p369p21
12q14
>90 % Séquença-ge del’ADN
CLHPD
PCSB
n.d.
100 %
n.d.
50 %
n.d.
n.d.
P
Syndrome 1des cancers dusein et del’ovairehéréditaires(BRCA1)13 39-48
Carcinomesdu sein et del’ovaire etcancers destrompes deFallope, del’estomac, du
pancréas etdu côlon
Prévalence1/800 à 1/2 500dans la
population engénéral(BRCA1 etBRCA2)• 7 % des
cancers du
sein sonthéréditaires.
• 60 % descancers duseinhéréditairessont causés
par BRCA1.
BRCA1 17q21 85 % oumoins
SGC
TPT
PCSB
CLHPD
EGC
EGB
DEM
IE
n.d.
80 % à100 %
65 % à96 %93 % à100 %
60 %
80 % à91 %
93 %
n.d.
n.d.
20,2 %
14,9 %
n.d.
n.d.
n.d.
n.d.
0r458vfé8
Syndrome 2des cancers dusein et del’ovairehéréditaires(BRCA2)13,40,
42-44,47
Carcinomesdu sein, del’ovaire et du
pancréas
Prévalence1/800 à 1/2 500dans la popu-lation en général(BRC1 et BRC2)•
7% des cancersdu sein sonthéréditaires.
• 20 % descancers du seinhéréditairessont causés par BRCA2.
BRCA 2 13q12 85 % oumoins
SGC
TPT
IE
n.d.
n.d.
n.d.
n.d.
n.d.
n.d.
0r25dà8
8
5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 26/103
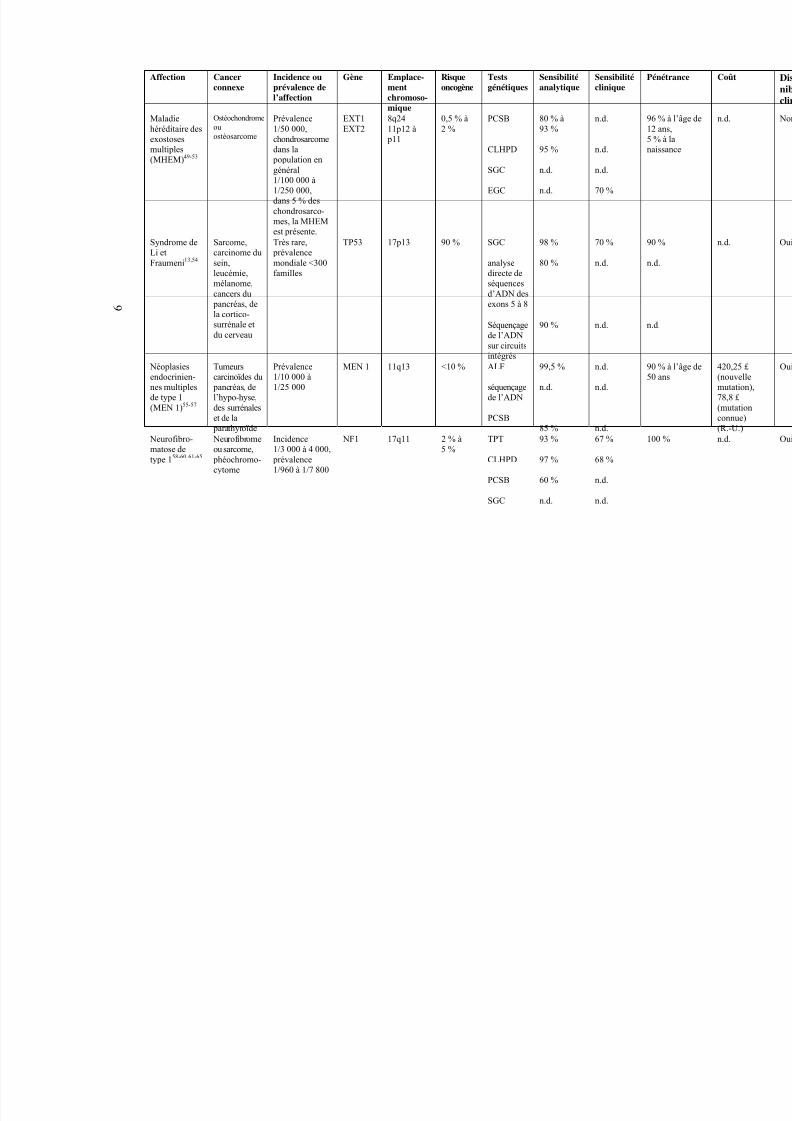
9
Affection Cancerconnexe
Incidence ouprévalence del’affection
Gène Emplace-mentchromoso-mique
Risqueoncogène
Testsgénétiques
Sensibilitéanalytique
Sensibilitéclinique
P
Maladiehéréditaire desexostoses
multiples(MHEM)49-53
Ostéochondromeouostéosarcome
Prévalence1/50 000,chondrosarcome
dans la population engénéral1/100 000 à1/250 000,dans 5 % deschondrosarco-mes, la MHEMest présente.
EXT1EXT2
8q2411p12 à
p11
0,5 % à2 %
PCSB
CLHPD
SGC
EGC
80 % à93 %
95 %
n.d.
n.d.
n.d.
n.d.
n.d.
70 %
9
5
n
Très rare, prévalencemondiale <300familles
90 % SGC
analysedirecte deséquencesd’ADN des
exons 5 à 8
98 %
80 %
70 %
n.d.
9
n
Syndrome deLi etFraumeni13,54
Sarcome,carcinome dusein,leucémie,mélanome,cancers du
pancréas, dela cortico-surrénale etdu cerveau
TP53 17p13
Séquençagede l’ADNsur circuitsintégrés
90 % n.d. n
Néoplasiesendocrinien-nes multiplesde type 1(MEN 1)55-57
Tumeurscarcinoïdes du
pancréas, del’hypo-hyse,des surrénaleset de la
parathyroïde
Prévalence1/10 000 à1/25 000
MEN 1 11q13 <10 % ALF
séquençagede l’ADN
PCSB
99,5 %
n.d.
85 %
n.d.
n.d.
n.d.
95
Neurofibro-
matose detype 158-60 61-65
Neurofibrome
ou sarcome, phéochromo-cytome
Incidence
1/3 000 à 4 000, prévalence1/960 à 1/7 800
NF1 17q11 2 % à
5 %
TPT
CLHPD
PCSB
SGC
93 %
97 %
60 %
n.d.
67 %
68 %
n.d.
n.d.
9
5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 27/103
10
Affection Cancerconnexe
Incidence ouprévalence del’affection
Gène Emplace-mentchromoso-mique
Risqueoncogène
Testsgénétiques
Sensibilitéanalytique
Sensibilitéclinique
P
Neurofibro-matose de
type 213,61,63,
66-69
Neurofibrome,méningiome,
schwannome
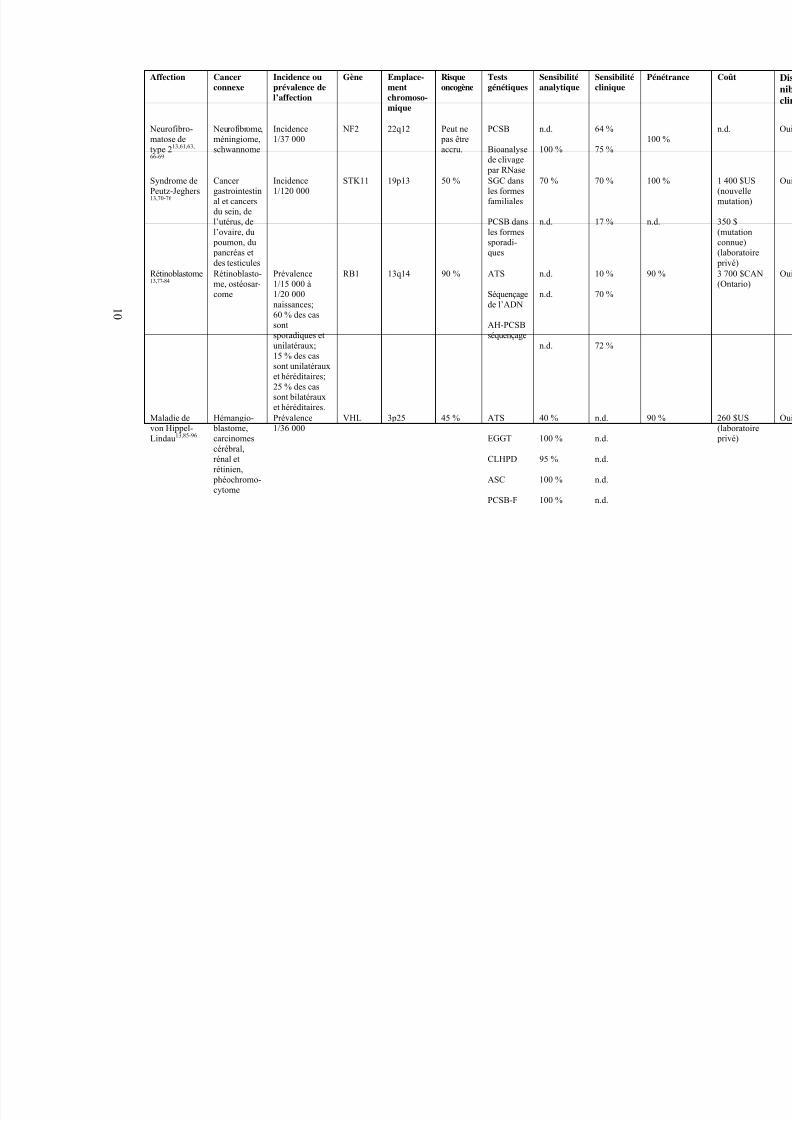
Incidence1/37 000
NF2 22q12 Peut ne pas être
accru.
PCSB
Bioanalysede clivage par RNase
n.d.
100 %
64 %
75 %
Syndrome dePeutz-Jeghers13,70-76
Cancer gastrointestinal et cancersdu sein, del’utérus, del’ovaire, du
poumon, du pancréas etdes testicules
Incidence1/120 000
STK11 19p13 50 % SGC dansles formesfamiliales
PCSB dansles formessporadi-ques
70 %
n.d.
70 %
17 % n
Rétinoblastome13,77-84
Rétinoblasto-me, ostéosar-come
Prévalence1/15 000 à1/20 000
naissances;60 % des cassontsporadiques etunilatéraux;15 % des cassont unilatérauxet héréditaires;25 % des cassont bilatérauxet héréditaires.
RB1 13q14 90 % ATS
Séquençage
de l’ADN
AH-PCSBséquençage
n.d.
n.d.
n.d.
10 %
70 %
72 %
9
Maladie devon Hippel-Lindau13,85-96
Hémangio- blastome,carcinomes
cérébral,rénal etrétinien,
phéochromo-cytome
Prévalence1/36 000
VHL 3p25
45 % ATS
EGGT
CLHPD
ASC
PCSB-F
40 %
100 %
95 %
100 %
100 %
n.d.
n.d.
n.d.
n.d.
n.d.
9
1 0
5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 28/103
11
Affection Cancerconnexe
Incidence ouprévalence del’affection
Gène Emplace-mentchromoso-mique
Risqueoncogène
Testsgénétiques
Sensibilitéanalytique
Sensibilitéclinique
P
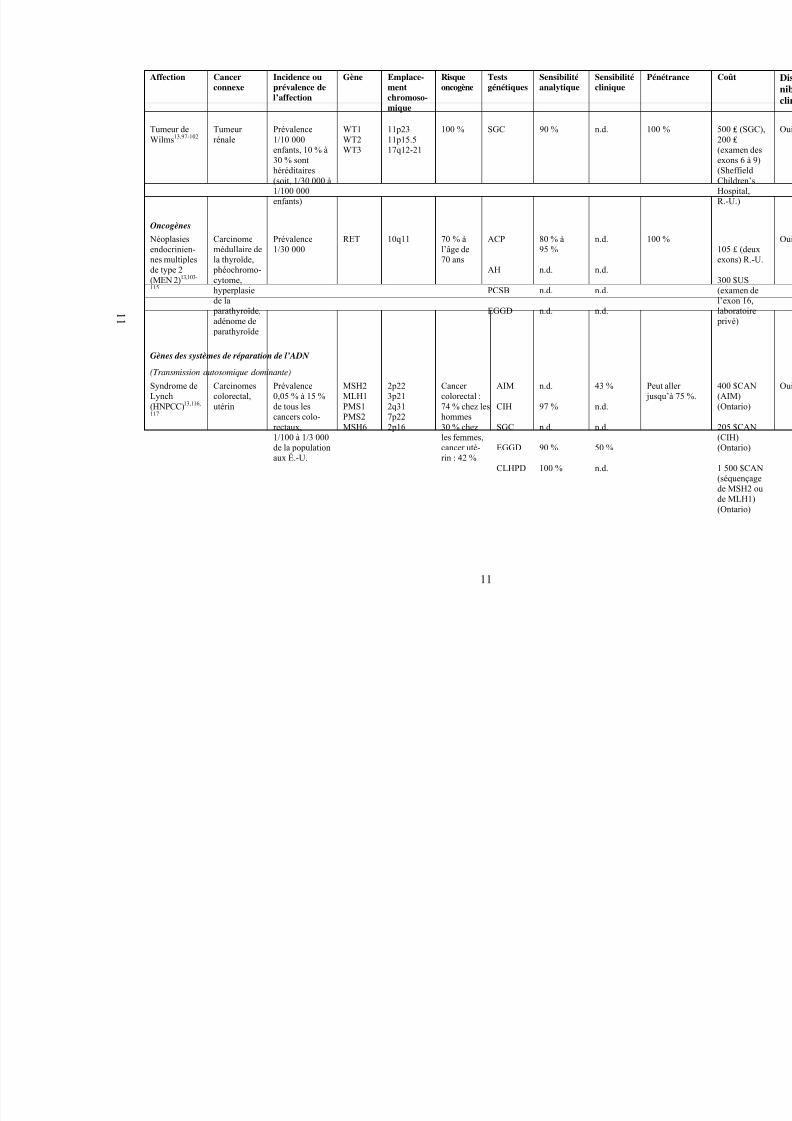
Tumeur deWilms13,97-102
Tumeur rénale
Prévalence1/10 000
enfants, 10 % à30 % sonthéréditaires(soit, 1/30 000 à1/100 000enfants)
WT1WT2
WT3
11p2311p15.5
17q12-21
100 % SGC 90 % n.d.
Oncogènes
Néoplasiesendocrinien-nes multiplesde type 2(MEN 2)13,103-
115
Carcinomemédullaire dela thyroïde,
phéochromo-cytome,hyperplasie
de la parathyroïde,adénome de
parathyroïde
Prévalence1/30 000
RET 10q11 70 % àl’âge de70 ans
ACP
AH
PCSB
EGGD
80 % à95 %
n.d.
n.d.
n.d.
n.d.
n.d.
n.d.
n.d.
Gènes des systèmes de réparation de l’ADN
(Transmission autosomique dominante)
Syndrome deLynch(HNPCC)13,116,
117
Carcinomescolorectal,utérin
Prévalence0,05 % à 15 %de tous lescancers colo-rectaux,1/100 à 1/3 000de la populationaux É.-U.
MSH2MLH1PMS1PMS2MSH6
2p223p212q317p222p16
Cancer colorectal :74 % chez leshommes30 % chezles femmes,cancer uté-rin : 42 %
AIM
CIH
SGC
EGGD
CLHPD
n.d.
97 %
n.d.
90 %
100 %
43 %
n.d.
n.d.
50 %
n.d.
P j
1 1
5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 29/103
12
Affection Cancerconnexe
Incidence ouprévalence del’affection
Gène Emplace-mentchromoso-mique
Risqueoncogène
Testsgénétiques
Sensibilitéanalytique
Sensibilitéclinique
P
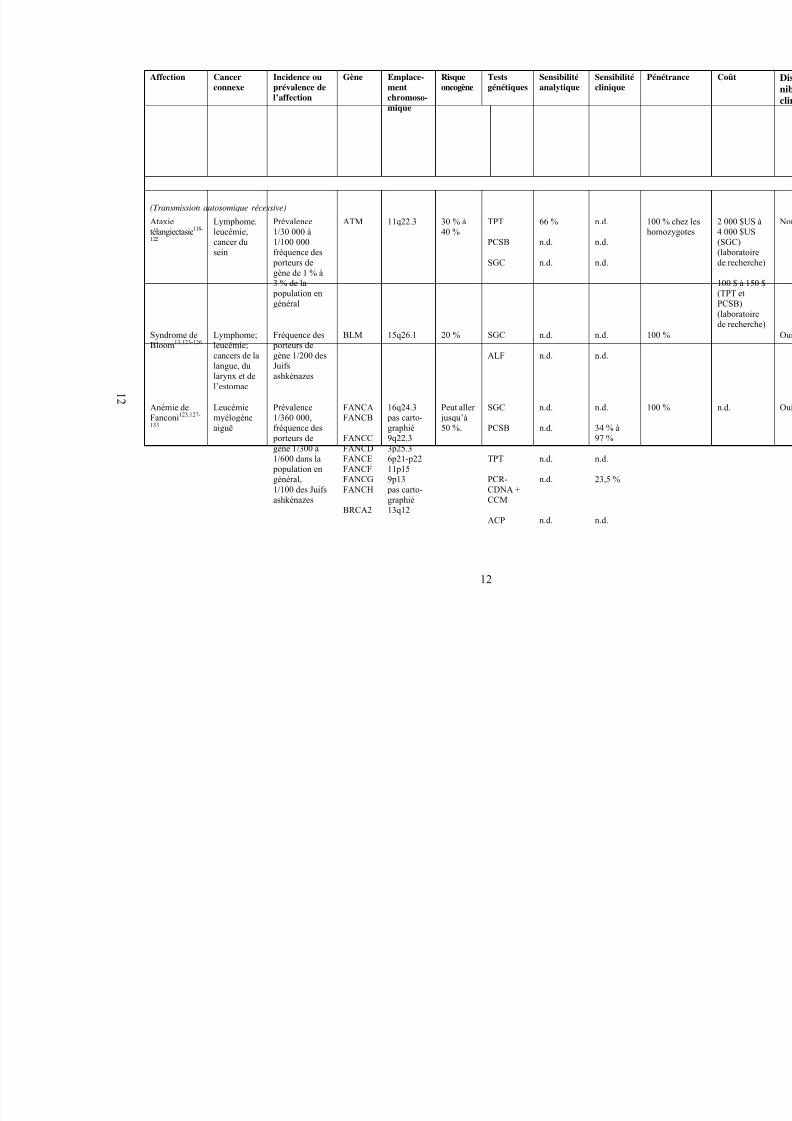
(Transmission autosomique récessive)
Ataxietélangiectasie118-
122
Lymphome,leucémie,cancer dusein
Prévalence1/30 000 à1/100 000fréquence des
porteurs degène de 1 % à3 % de la
population en
général
ATM 11q22.3 30 % à40 %
TPT
PCSB
SGC
66 %
n.d.
n.d.
n.d.
n.d.
n.d.
h
Syndrome deBloom13,123-126
Lymphome;leucémie;cancers de lalangue, dularynx et del’estomac
Fréquence des porteurs degène 1/200 desJuifsashkénazes
BLM 15q26.1 20 % SGC
ALF
n.d.
n.d.
n.d.
n.d.
Anémie deFanconi123,127-
133
Leucémiemyélogèneaiguë
Prévalence1/360 000,fréquence des
porteurs degène 1/300 à1/600 dans la
population engénéral,1/100 des Juifsashkénazes
FANCAFANCB
FANCCFANCDFANCEFANCFFANCGFANCH
BRCA2
16q24.3 pas carto-graphié9q22.33p25.36p21-p2211p159p13
pas carto-graphié13q12
Peut aller jusqu’à50 %.
SGC
PCSB
TPT
PCR-CDNA +CCM
ACP
n.d.
n.d.
n.d.
n.d.
n.d.
n.d.
34 % à97 %
n.d.
23,5 %
n.d.
1 2

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 30/103
13
Affection Cancerconnexe
Incidence ouprévalence del’affection
Gène Emplace-mentchromoso-mique
Risqueoncogène
Testsgénétiques
Sensibilitéanalytique
Sensibilitéclinique
P
multiplexen fluores-cence
Le risque oncogène est le risque d’apparition du cancer connexe pendant la vie de la personne atteinte du syndrome.La disponibilité clinique renvoie à la disponibilité des tests génétiques de dépistage de l’affection à des fins cliniques par opposition à l’utiliLa pénétrance représente la probabilité que la personne porteuse d’un gène mutant en particulier exprimera le phénotype altéré correspondan.d. = information non disponible
ACP = amplification en chaîne par polyméraseAH = analyse des hétéroduplexAHI = analyse d’hybridation intrachromosomiqueAIM = analyse de l’instabilité du microsatelliteALF = analyse de liaison factorielleASC = analyse séquentielle comparativeATS = analyse du transfert de Southern
BAAO = bioanalyse d’allèles par oligonucléotideBAEA = bioanalyse de l’expression d’allèlesCCM = clivage chimique de mésappariementCIH = coloration immunohistochimique
CLHPD = chromatographie liquide à haute performance en fonction de la dénaturationDEM = détection enzymatique des mutationsEE = examen d’exonsEGB = exploration génique bidimensionnelleEGC = électrophorèse en gel en fonction de laconformation
EGGD = électrophorèse en gel selon le gradient dedénaturantEGGT = électrophorèse en gel selon le gradient detempérature
IE = imPCR-Caprès trPCSB =simplesPCSB-Fsimples
PR = poSGC = TPT = t
1 3

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 31/103
14
4.3 Impact du dépistage génétique sur la prise en chargeclinique
4.3.1 Ataxie télangiectasie
L’ataxie télangiectasie (AT) est une affection neurologique rare et progressive de transmissionautosomique récessive, ATM (11q22.3) étant le gène en cause141-145. La protéine ATMcontribuerait à l’expression du phénotype de l’AT146,147, comme elle jouerait un rôle dans laréparation et la vérification148, la réaction devant la détérioration oxydative149,150 et la résistanceau rayonnement151,152. L’AT se caractérise par l’ataxie cérébelleuse (soit une diminution de lacoordination musculaire causée par une lésion du cervelet), la perturbation des mouvementsoculaires, un trouble de l’élocution, un dysfonctionnement endocrinien, l’immunodéficience, lavulnérabilité accrue au rayonnement ionisant, le vieillissement prématuré et la télangiectasie(apparition de traînées rouges dues à la dilatation de certains petits vaisseaux superficiels de la peau exposée au soleil)120,153; l’affection prédispose aux lymphomes, leucémies et autrescancers121,154,155. La plupart des personnes atteintes en sont réduites au fauteuil roulant dès
l’adolescence et vivent rarement plus longtemps que 45 ans. Parce que le diagnostic moléculairede l’AT est complexe, le diagnostic repose sur les signes cliniques et des analyses de laboratoireconcernant la concentration sérique d’alphaprotéines et la synthèse d’ADN radiorésistante156.
a) Dépistage moléculaire
Le dépistage moléculaire du gène ATM est limité à la recherche. Sous l’angle technique, ledépistage moléculaire de l’AT est complexe. La grande taille du gène ATM ainsi que la diversitédes mutations (la plupart étant uniques et réparties uniformément le long du gène) réduisent les possibilités de la détection directe des mutations en tant qu’outil diagnostique157. Les mutationsdu gène ATM ont été cernées par la méthode exhaustive d’ACP suivant la transcription inverse(PCR-CDNA), la cartographie peptidique de restriction par endonucléase158 ou le PCSB159. Une
bioanalyse d’un jeu ordonné d’oligonucléotides à haute densité a été utilisée pour déceler lesvariations de séquence dans le gène ATM160. La détection de mutations particulières seraitfaisable dans des sous-groupes de la population, porteurs d’une mutation précise. Compte tenude la fréquence élevée (70 %) des mutations par troncature dans le gène ATM, le dépistage pourrait être fondé sur le TPT, capable de déceler 66 % des mutations122. Le TPT etl’électrophorèse en gel en fonction de la conformation (EGC) ont permis de déceler des protéinestronquées et d’autres mutations d’ATM dans certains groupes ethniques122. En ce qui a trait à ladétection des mutations d’ATM chez les porteurs, des experts prétendent qu’il conviendrait derelever les mutations les plus courantes dans certains groupes ethniques, puis de mettre au pointdes bioanalyses rapides ne nécessitant qu’une petite quantité d’ADN génomique et des méthodesmoins coûteuses122. Le coût du dépistage moléculaire de l’AT dans un laboratoire privé aux
États-Unis varie de 2 000 $US à 4 000 $US pour le SGC et de 100 $ à 150 $ pour le TPT ou lePCSB122.
b) Impact du dépistage moléculaire sur la prise en charge clinique
Les avantages cliniques du dépistage génétique de l’AT ont été mis en évidence, mais la procédure ne fait pas partie intégrante de la prise en charge usuelle. On conseille aux personnesatteintes d’AT de subir de façon périodique une numération globulaire, un examen cutanéapprofondi et un examen d’imagerie de l’appareil gastrointestinal supérieur si des symptômes se

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 32/103
15
manifestent. Vu que des études démontrent une incidence accrue de cancer du sein chez les porteuses de mutations d’ATM, les femmes hétérozygotes et les femmes homozygotes sont priées de se soumettre à un programme de surveillance161. Comme les porteurs du gène ATMmuté sont plus vulnérables au rayonnement, certains se demandent si les rayons X, de faibleintensité de rayonnement, sont nocifs pour ces personnes152,162. Les anomalies d’ATM,
cependant, ne représentent pas la principale cause des complications de la radiothérapie chez lesfemmes souffrant de cancer du sein.163. Les rayons X ne devraient être utilisés qu’à des finsdiagnostiques, et non à des fins de dépistage13. On préconise une prévention énergique desinfections chez les femmes homozygotes.
4.3.2 Nævomatose basocellulaire
Rare, la nævomatose basocellulaire (NB) ou syndrome de Gorlin est une affection autosomiquedominante causée par la mutation du gène PTCH (9q22.3)13. Le gène PTCH code pour une protéine transmembranaire, dont la disparition (par suite de la mutation) peut modifier le cyclenormal de la division cellulaire et stimuler la multiplication cellulaire. Les mutations germinales
englobent des insertions, des délétions et des mutations ponctuelles entraînant la terminaison dela chaîne prématurée ou le déphasage. Le risque de survenue de l’affection chez la descendanced’une personne atteinte est de 50 %18. Les principales caractéristiques de la NB sont la formationde multiples kystes kératosiques maxillaires, habituellement à la vingtaine, l’apparition de godetscutanés palmaires et plantaires ou la survenue d’un carcinome basocellulaire, habituellement àcompter de la trentaine. Une calcification ectopique se manifeste chez plus de 90 % des maladesavant l’âge de 20 ans. Outre le risque élevé de cancer de la peau, les personnes atteintes sont àrisque accru d’une tumeur cérébrale pendant l’enfance, de fibromes ovariens et de fibromescardiaques18. Environ 5 % des enfants atteints de NB souffriront de médulloblastome, l’incidencela plus élevée étant observée à l’âge de deux ans18. Les fibromes cardiaques et ovarienssurviennent dans respectivement 2 % et 20 % des cas de NB18. Le diagnostic moléculaire, soit ladétection de la mutation pathogène, vient confirmer le diagnostic clinique de la NB.
a) Dépistage moléculaire
En pratique clinique, le dépistage moléculaire du gène PTCH est disponible. L’analyseséquentielle de la zone codante de PTCH, qui permet de détecter près de 85 % des mutations, estla méthode utilisée pour déceler les mutations chez les personnes présentant les signes cliniquestypes de la NB18. Si les résultats de cette analyse sont négatifs, elle peut être suivie de l’ATS afinde déceler de vastes délétions. L’analyse de liaison factorielle (ALF), de très grande précisiondans les familles comptant plus d’une personne atteinte, permet de détecter près de 99 % desmutations18. Le dépistage prénatal chez le fœtus pour qui le risque de NB est de 50 % estdisponible en pratique clinique si la mutation a été repérée chez un membre atteint de la famille
ou si l’analyse de liaison dans la famille a été révélatrice.
Le coût de la détection des mutations par l’examen et le séquençage d’exons chez un nouveau patient dans un laboratoire privé est de 1 600 $US. Le séquençage de l’ADN des personnesapparentées lorsque la mutation est connue coûte 350 $US. Le coût du diagnostic prénatal fondésur deux prélèvements (prélèvement de villosités choriales et cellules amniotiques en milieu deculture) s’élève à 700 $US. Le délai d’exécution de l’analyse de nouveaux patients est d’environhuit semaines, tandis qu’il est de deux semaines lorsque la mutation dans la famille est connue14.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 33/103
16
On ne peut écarter la possibilité de la NB sur la seule foi du dépistage génétique moléculaire, puisque les mutations de PTCH ne sont pas décelables chez toutes les personnes manifestant lessignes cliniques de la maladie. Les mutations faux-sens sont fréquentes et il peut être difficiled’interpréter leur présence lorsque l’histoire familiale de la personne ne révèle aucun cas de NBet que les signes cliniques ne correspondent pas aux critères diagnostiques18.
b) Impact du dépistage moléculaire sur la prise en charge clinique
Les avantages cliniques du dépistage génétique de la NB ont été mis en évidence, mais la procédure ne fait pas partie intégrante de la prise en charge usuelle. Des rapports préliminaires ne peuvent préciser de corrélations entre le génotype et le phénotype des personnes atteintes de NB164. D’autre part, certains experts estiment qu’il s’avère approprié de déterminer le statutgénétique des enfants à risque au vu de la nécessité de surveiller la survenue éventuelle pendantl’enfance des complications de la maladie, plus particulièrement le médulloblastome18. Soit leséquençage du gène, soit l’analyse de liaison sera envisagé selon que la mutation pathogène dansla famille touchée est connue ou en présence de marqueurs reliés dans la famille.
4.3.3 Syndrome de BloomTrouble rare transmis selon le mode autosomique récessif, le syndrome de Bloom (SB) estcaractérisé par un grave retard de croissance, un érythème facial exacerbé au soleil ou de latélangiectasie, des anomalies immunitaires graves, de l’infertilité et un risque élevé de tumeursmalignes, et une brève survie menant rarement à l’âge adulte165-167. Environ 20 % des personnesatteintes du SB souffriront d’un cancer, qui surviendra avant l’âge de 20 ans dans la moitié descas123,168. Les types de cancers vont du lymphome non hodgkinien et de la leucémie aiguë auxcarcinomes de la langue, du larynx ou de l’estomac. Le gène mutant en cause est BLM(15q26.1), qui code pour une hélicase de l’ADN, laquelle participe à la surveillance desanomalies de bases dans l’ADN169. Le gène mutant BLM est plus fréquent dans la population
juive ashkénaze
126
. Habituellement, le diagnostic de l’affection repose sur la présentationclinique couplée aux analyses de laboratoire de confirmation démontrant le taux accru d’échangede chromatides sœurs, qui entraîne des cassures indésirables et la perte possible del’hétérozygosité.
a) Dépistage moléculaire
Le dépistage moléculaire du gène BLM est disponible en pratique clinique. Environ un Juif ashkénaze sur 231 est porteur de la même délétion 6-bp insertion 7-bp à la position 2281 deBLM (blm Ash)124. Cette mutation, à l’origine de la plupart des cas, voire de tous, du SB danscette population, peut être cernée par une méthode fondée sur la digestion par une enzyme derestriction d’un segment contenant la mutation ayant fait l’objet d’une ACP125. L’analyse
génétique des porteurs est offerte aux couples des vieilles familles juives (ashkénazes) del’Europe de l’Est dans le cadre d’une procédure de dépistage prénatal13.
b) Impact du dépistage moléculaire sur la prise en charge clinique
Les avantages cliniques du dépistage génétique du SB ont été mis en évidence, mais la procédurene fait pas partie intégrante de la prise en charge usuelle. Les personnes hétérozygotes sousl’angle du gène mutant BLM sont priées de se soumettre à un examen physique et à unenumération globulaire périodiques dès l’enfance13. Les personnes atteintes du SB (homozygotes

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 34/103
17
quant au gène mutant) devraient se prêter à une procédure de surveillance axée sur les cancers dela peau et de l’appareil gastrointestinal. Les femmes qui en souffrent devraient être suivies enraison du risque accru de cancers du col de l’utérus et du sein. Enfin, les personnes atteintesdevraient éviter l’exposition solaire directe13.
4.3.4 Maladie de CowdenRare, la maladie de Cowden (MC), ou syndrome des hamartomes multiples, est un troubledécoulant de mutations germinales du gène suppresseur de tumeurs PTEN (10q23)170,171. Cegène code pour la phosphatase PTEN, qui intervient à titre de médiatrice de l’arrêt du cyclecellulaire et de l’apoptose (mort cellulaire programmée). Les mutations comprennent desmutations non-sens et des mutations faux-sens. Des études génétiques ont confirmé latransmission autosomique dominante de la MC, dont la pénétrance est élevée chez les deux sexeset l’expression des symptômes diffère modérément d’une famille à une autre ou au sein de lamême famille172. Même si la maladie frappe parfois les hommes, elle survient principalementchez les femmes (6:1)172,173. Bien que la MC soit caractérisée surtout par des lésionsmucocutanées bénignes et des hamartomes gastrointestinaux, elle comporte également un risqueà vie allant de 25 % à 50 % de cancer du sein, de 10 % de cancer de la thyroïde et un risqued’autres cancers, notamment le cancer de l’endomètre, l’hypernéphrome, le mélanome et leglioblastome13,21,174. Les lésions cutanées, dont les trichilemmomes faciaux multiples, précèdentla transformation maligne; elles permettraient d’identifier les femmes à risque élevé de cancer dusein ou de la thyroïde175.
a) Dépistage moléculaire
Au nombre des méthodes de détection des mutations du gène PTEN figurent le séquençagedirect, disponible en règle générale, et d’autres méthodes (ATS, ACP-PCSB, ACP-EGGD)disponibles dans les milieux de recherche21,176-179. La méthode de l’ACP-PCSB par coloration àl’argent non isotopique est plus sensible que le séquençage direct automatisé dans la détectiondes mutations ponctuelles de PTEN176. L’analyse séquentielle de chacun des neuf exons dePTEN est dotée d’une sensibilité clinique de 81 %19,20. Il est nécessaire que le prélèvementcontienne de l’ADN tumoral en proportion de 10 % pour procéder à l’ACP-PCSB, mais en proportion variant de 30 % à 70 % s’agissant du séquençage direct.
Le coût de la détection des mutations chez un nouveau patient par l’examen et le séquençage desexons dans un laboratoire privé est de 1 300 $US. Le coût de l’analyse génétique des membresde la famille lorsque la mutation est connue s’élève à 350 $US 20. Le délai d’exécution est de sixà huit semaines concernant un nouveau patient et de deux semaines pour les personnesapparentées lorsque la mutation est connue.
À ce jour, 82 mutations germinales du gène PTEN en cause dans le phénotype clinique de la MC(et plus de 300 mutations somatiques du gène PTEN à l’origine des cancers sporadiques) ont étérépertoriées23,178. Selon les estimations, 80 % des familles touchées par la MC portent unemutation décelable du gène PTEN21,177,180. Des mutations germinales de PTEN ont également étérepérées chez 50 % à 60 % des familles présentant un syndrome de Bannayan-Zonana ou unsyndrome de Protée19,21,181.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 35/103
18
La MC est d’expression variable, réduite souvent à des signes cutanés discrets, de sorte qu’elleest difficile à diagnostiquer. La cartographie des mutations sur le gène PTNE est essentielle audiagnostic de la maladie182. La corrélation entre les mutations de PTEN et la MC, du génotype au phénotype, est toutefois incertaine. L’association constatée entre la présence d’une mutation dePTEN et le cancer du sein n’est pas toujours confirmée. La présentation clinique de certains cas
de MC sans mutation de PTEN ne diffère pas de celle de la maladie découlant d’une mutation dePTEN22. Par contre, les données démontrent que la présence ou l’absence d’une mutation dePTEN permet de déterminer le type de tumeurs du sein (c.-à-d., malignes ou bénignes)19,175.
b) Impact du dépistage moléculaire sur la prise en charge clinique
Le dépistage génétique est intégré à la prise en charge clinique courante de la MC. Le diagnostic préliminaire est fondé sur les signes cliniques. Par définition, le diagnostic de la MC n’estcorroboré que lorsqu’une mutation de PTEN est repérée. Le dépistage moléculaire des mutationsdu gène PTEN est donc essentiel au diagnostic et à la prise en charge de la MC183. Le dépistage précoce des manifestations cutanées caractéristiques de la MC, particulièrement le critère pathognomonique des trichilemmomes multiples de la face, revêt de l’importance car il permet
d’identifier les femmes qui bénéficieraient du dépistage moléculaire des mutations de PTEN. Il yaurait, semble-t-il, un lien entre les mutations en amont et le motif fondamental de la phosphatase (qui renferme la plupart des mutations faux-sens) et l’apparition de la maladiemultiorganique19. Les femmes dont le diagnostic de MC est confirmé devraient se soumettre à unexamen des seins fréquent, à une mammographie périodique dès l’âge de 30 ou 35 ans, à la biopsie des lésions suspectes et au dépistage du cancer de la thyroïde13.
4.3.5 Polypose adénomateuse familiale
La polypose adénomateuse familiale (PAF) est un syndrome de prédisposition au cancer colorectal héréditaire, de transmission autosomique dominante. Le gène en cause, APC(adenomatous polyposis coli) (5q21-q22), code pour la protéine APC qui contrôle l’apoptose184.Une fois muté, le gène APC entraîne une instabilité chromosomique et la formation d’une protéine APC tronquée qui provoque une prolifération cellulaire anarchique et la formationtumorale185. La PAF se caractérise par la survenue hâtive de plus de 100 polypes adénomateuxcolorectaux et des manifestations extracoliques. Le cancer est inévitable, car au moins un des polypes colorectaux se transformera en tumeur maligne, en l’absence de colectomie prophylactique186. Le cancer peut survenir à tout âge, de la fin de l’enfance à l’âge de 70 ans,l’âge médian au diagnostic clinique étant de 40 ans. La PAF est une affection de pénétranceélevée (95 % des personnes atteintes présentent des polypes à l’âge de 35 ans) 28. Le diagnosticrepose sur la présentation clinique. Le dépistage génétique moléculaire intervient le plus souventdans le diagnostic précoce de la maladie chez les membres de la famille à risque et dans laconfirmation de la PAF lorsque les signes cliniques sont équivoques 187,188.
a) Dépistage moléculaire
Le dépistage moléculaire de la PAF aux fins diagnostiques est disponible en pratique clinique.Le Groupe de travail canadien sur les soins de santé préventifs a recommandé en 2001 d’intégrer le dépistage génétique ou la sigmoïdoscopie à l’examen médical périodique des membres desfamilles aux prises avec la PAF189. L’analyse du gène APC est difficile en raison de sa grandetaille (15 exons) et du nombre de mutations connues dispersées le long du gène. Le séquençage

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 36/103
19
de gène complet (SGC), la bioanalyse de l’expression d’allèles (BAEA) couplée au TPT, le TPTseul et l’ALF comptent au nombre des techniques utilisées pour préciser la mutation du gèneAPC190. La méthode du TPT améliorée accroît la sensibilité clinique du test191. La CLHPD a faitses preuves en décelant 100 % des mutations (soit, une sensibilité analytique de 100 %) 24. Laméthode du PCSB modifiée, le PCSB à froid, s’avèrerait une technique fiable et moins coûteuse
de détection des mutations connues du gène APC25
. La bioanalyse TPT appliquée au gène APCdans le dépistage de la PAF produit environ 20 % de résultats faux négatifs, c’est-à-dire que le patient est porteur d’une mutation mais le test est négatif 192. Compte tenu que le cancer colorectal survient chez toutes les personnes, pour ainsi dire, souffrant de PAF, un résultat fauxnégatif peut avoir des conséquences dévastatrices. Le dépistage prénatal chez le fœtus à risque de50 % de PAF est disponible en clinique si la mutation a été cernée chez un membre de la familleou si l’analyse de liaison est révélatrice dans la famille.
Au Québec (Canada), le coût de l’analyse du gène APC selon la méthode du TPT suivi duséquençage de l’ADN (concernant un nouveau patient) et du TPT (s’agissant des personnesapparentées à risque dans une famille où la mutation est connue) est évalué à respectivement
500 $CAN et 250 $CAN l’analyse26
.
Étant donné que le dépistage génétique des mutations du gène APC est long et dispendieux, lerecours au dépistage génétique prédictif plutôt qu’au dépistage clinique classique de la PAF (àsavoir, l’examen périodique de la présence de sang dans les selles et les sigmoïdoscopies oucolonoscopies à répétition) ne fait pas l’unanimité193-195. Vu que la probabilité de ne pas déceler la PAF diminue avec l’âge, l’ampleur des économies varie selon l’âge auquel le dépistagecommence. Une étude méthodique récente26 démontre que les économies découlant du dépistageclinique cessent lorsque celui-ci commence après l’âge de 36 ans. L’économie moyenne par personne lorsque le dépistage commence à l’âge de 12 ans est de plus de 900 $CAN, mais ellechute à 200 $CAN à l’âge de 30 ans. Une étude comparant le coût du dépistage génétique
prédictif et celui du dépistage clinique classique de la PAF indique que le dépistage génétique prédictif par l’analyse des hétéroduplex (AH) et le TPT le cas échéant (4 975 $CAN par famille)est moins dispendieux que le dépistage clinique classique par la sigmoïdoscopie flexible(8 031 $CAN), pour autant que la fréquence de la surveillance clinique soit la même dans lesdeux stratégies196.
b) Impact du dépistage moléculaire sur la prise en charge clinique
Le dépistage génétique est intégré à la prise en charge clinique courante de la PAF. La détectionmoléculaire d’une mutation du gène APC chez un membre d’une famille comptant des cas dePAF permet de distinguer les porteurs des non-porteurs de l’allèle mutant d’APC parmi lesapparentés à risque. Étant donné que les mutations d’APC surviennent tôt dans la cancérogenèse
colorectale197
, le dépistage moléculaire des mutations du gène APC aux fins diagnostiques, s’ils’avère positif, favoriserait la détection et le traitement précoces de la PAF. Comme les stratégiesde prise en charge classiques des membres d’une famille à risque prévoient la sigmoïdoscopie àintervalles réguliers accompagnée de la colectomie préventive lorsque les polypes apparaissent,le diagnostic repose encore sur l’endoscopie dans la plupart des cas198. Le diagnostic moléculairede la PAF est particulièrement utile quant le résultat est véritablement négatif (c.-à-d., quand il permet d’identifier les personnes libres de mutations d’APC) en permettant aux non-porteurs dese soustraire à la surveillance clinique et sigmoïdoscopique199.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 37/103
20
Bien des efforts ont été déployés afin de cerner les corrélations entre le génotype et le phénotype pour orienter la prise en charge27,200. La polypose profuse (5 000 polypes en moyenne) découlerait demutations dans les codons 1250 à 1464201. Les mutations aux codons 1309 et 1328 seraient à l’origined’un phénotype de polypose toujours grave202. Les mutations entre les codons 1444 et 1581 sontassociées à une incidence accrue de tumeurs desmoïdes203,204. Différentes mutations du gène APC se
traduisent par différentes expressions de la PAF. Les mutations de 5’ (5’ du codon 158) et 3’ (3’ ducodon 1596) produiraient un phénotype moins grave de PAF (PAF atténuée)205,206. L’emplacement dela mutation sur le gène APC influence l’évolution de la PAF, de sorte que le dépistage génétiquemoléculaire permettrait d’orienter la prise en charge chirurgicale207.
4.3.6 Mélanome familial
Le mélanome familial (MF) ou syndrome du nævus dysplasique est un mélanome malin detransmission autosomique dominante13,208-212. Le diagnostic de MF est posé en présence de 10 à100 nævi dysplasiques (lésions cutanées visibles) ou si la personne est apparentée au premier degré à deux personnes souffrant de mélanome213. Selon les estimations, de 3 % à 7 % des cas demélanome proviennent de familles à risque génétique élevé10,36. Trois gènes de prédisposition aumélanome ont été cernés : CMM1 (1p36); CMM2, appelé également TP16 ou MTS1 ouCDKN2A (9p21); CDK4 (12q14)10,209. Le gène TP16 code pour la protéine TP16, qui agit entant que médiatrice de la régulation du cycle cellulaire. Diverses mutations du gène TP16 ont étérelevées, dont des mutations non-sens, l’épissage du site donneur, des mutations faux-sens, par insertion ou délétion10,38. L’âge moyen au moment du diagnostic de MF est de 34 ans, soit 20 ans plus tôt que le moment type d’apparition de la maladie dans la population en général. En cas deMF, les nævi suspects dont la taille ou la couleur change, se transformeront presque à coup sûr (risque de près de 100 %) en tumeurs malignes, à moins d’être enlevés sans délai. L’astrocytomeet le cancer du pancréas ont été rapportés chez des personnes souffrant de MF.
a) Dépistage moléculaire
Le dépistage moléculaire du MF est disponible en pratique clinique. Le séquençage de l’ADN, laCLHPD et le PCSB comptent au nombre des méthodes de détection des mutations des gènes de prédisposition au mélanome, la CLHPD se démarquant du lot par sa rapidité, sa sensibilité et sarentabilité35,214. Tandis qu’il est facile d’identifier les membres d’une famille à risque, il s’estavéré difficile d’élucider le fondement moléculaire du MF. CDK4 est un oncogène, alors queCMM1 et TP16 sont des gènes suppresseurs de tumeurs. Une mutation germinale de TP16 est àl’origine d’environ 50 % des cas de MF. Il peut s’agir d’une mutation non-sens, d’une mutationfaux-sens, de l’épissage du site donneur ou d’insertions entraînant principalement la troncaturede la protéine p1613. Le gène CDK4 mute rarement, seules quelques familles sont porteuses demutations germinales de ce gène. Contrairement à d’autres syndromes de prédisposition à uncancer héréditaire, la prévalence des mutations germinales de CDKN2A et de CDK4 est basse(2 %) en cas de mélanome d’apparition précoce35,215.
Le coût de la détection des mutations par l’analyse séquentielle à un laboratoire privé est de600 $US dans le cas du gène CDKN2A et de 150 $US pour ce qui est du gène CDK4 chez la personne atteinte. L’analyse de l’ADN des personnes apparentées coûte 350 $US s’agissantd’une mutation connue. Le délai d’exécution de l’analyse génétique est de huit semainesconcernant un nouveau patient et de deux semaines pour ce qui est des personnes apparentéeslorsque la mutation est connue38.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 38/103
21
b) Impact du dépistage moléculaire sur la prise en charge clinique
Les avantages cliniques du dépistage génétique du MF sont incertains. Il importe de dépister et decontrôler le MF en raison de sa prévalence élevée, du fait qu’il découle presque invariablement d’unelésion cutanée visible et du pronostic sombre si la maladie est décelée à un stade avancé. Les risqueset les bienfaits du dépistage du cancer en rapport avec ce syndrome ne sont pas établis. Les porteurs
de mutations du gène TP16 voient le risque de mélanome s’accroître d’un facteur de 75 et le risquede cancer du pancréas d’un facteur de 13. Les personnes libres de mutations du gène TP16, issues defamilles comptant des cas de MF, voient le risque de mélanome augmenter d’un facteur de 38209. Lastratégie de surveillance préconisée en présence de MF prévoit l’auto-examen mensuel de la peau,une évaluation dermatologique accompagnée de la photographie de la peau deux fois l’an et l’exérèseimmédiate des lésions suspectes10. L’usage quotidien d’un écran solaire et l’évitement des coups desoleil sont également recommandés.
4.3.7 Anémie de Fanconi
Rare, l’anémie de Fanconi (AF) est une affection de transmission autosomique récessive caractérisée par
divers symptômes cliniques, notamment l’aplasie médullaire progressive et un risque oncogène accru, principalement de leucémie myéloïde aiguë216. En règle générale, l’âge au diagnostic est de six ans, les principaux signes étant l’anémie, le saignement et les contusions, et l’âge moyen au décès de 16ans10,13,217,218. L’AF englobe au moins huit sous-types, de l’AF(A) à l’AF(H), correspondant aux huitgènes en cause connus, soit les gènes FANCA à FANCH129,130,219-223, FANCA, FANCC et FANCE étantà l’origine de, respectivement, 66 %, 13 % et 13 % des cas d’AF224-226. Des études indiquent que les protéines AF de groupes différents participent à un mécanisme cellulaire commun128,227,228. Une étuderécente révèle que des personnes atteintes d’AF(B) ou d’AF(D) sont porteuses de mutations du gèneBRCA2127, ce qui donne à penser que le gène BRCA2 et les protéines AF collaborent dans le cadre d’unmécanisme commun de réaction à l’altération de l’ADN. Les cellules de l’AF sont très vulnérablesdevant l’action d’agents de réticulation de l’ADN dont le diépoxybutane et la mitomycine C. Par conséquent, le diagnostic de l’AF est fondé sur l’évaluation du degré de brisure chromosomique(invisible sur un caryotype habituel) dans le cadre d’un test de résistance chromosomique au stress provoqué par la mitomycine C ou le diépoxybutane229,230.
a) Dépistage moléculaire
Le dépistage moléculaire de l’AF est disponible en pratique clinique. La bioanalyse par buvardage,associée à la correction phénotypique selon la technique rétrovirale de cellules d’AF, constitue uneméthode de sous-typage rapide130. L’analyse ACP-PCSB a été utilisée pour déterminer les variationsde séquence dans les gènes de l’AF de divers sous-types; La sensibilité clinique de l’épreuve varieselon les groupes de sous-types, de 34 % à 97 %131,231,232. L’ACP suivant la transcription inverse(PCR-CDNA) et le clivage chimique de mésappariement (CCM) ont également été utilisés pour
cerner l’emplacement de mutations par épissage distinctes du gène FANCC, qui pourraient être àl’origine de la plupart des cas d’AF dans la population juive ashkénaze132. Dans cette population,l’ACP-CCM fait preuve d’une sensibilité clinique de 23,5 %. La technique PCR-CDNA et le CCMen fluorescence permettent de détecter de petites mutations du gène FANCA. Ces épreuves sontcombinées à l’ACP multiplex quantitative en fluorescence dans le cas de grossières délétions133. Desmutations par épissage, des mutations non-sens et des mutations faux-sens du gène FANCG ont étédécelées par l’association de la bioanalyse de fusion des cellules somatiques, de la complémentationrétrovirale et de la méthode de buvardage233.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 39/103
22
b) Impact du dépistage moléculaire sur la prise en charge clinique
Les avantages cliniques du dépistage génétique de l’AF sont établis, mais la procédure ne fait pas partie intégrante de la prise en charge usuelle. Des corrélations entre le génotype et le phénotypeont été précisées. Ainsi, l’expression d’une protéine liée à FAC, FRP-50, correspond à l’anémie
de sous-type AF(C) d’intensité légère. La concentration endogène de PRP-50 dans les cellules porteuses de la mutation delG322 représente une indication mesurable de la protection contre lescaractéristiques phénotypiques de l’AF grave234. La perte d’un chromosome 7 (monosomie 7)favoriserait l’évolution de l’AF vers la leucémie, d’où le pronostic sombre235. La détection de lamonosomie 7 par hybridation in situ en fluorescence (HISF) peut être adoptée en pratiquecourante afin de prédire la survenue de la leucémie myéloïde aiguë en présence d’AF236. On saitégalement qu’un rétrovirus actif sur le plan fonctionnel agissant à titre de vecteur peut transférer un gène FANCC fonctionnel dans les lignées cellulaires lymphoïdes établies d’un patient atteintd’AF(C)237. Cela laisse entrevoir l’utilité potentielle de la thérapie génique dans le traitement del’AF.
4.3.8 Syndromes 1 et 2 des cancers du sein et de l’ovaire héréditairesLe cancer du sein représente la deuxième cause de mortalité par suite de cancer chez la femmeau Canada. Sept pour cent des cancers du sein sont héréditaires, et 60 % de ces cas sontimputables à des mutations prédisposant au cancer du gène BRCA1 (17q21), alors que 20 % sontattribuables à des mutations prédisposant au cancer du gène BRCA2 (13q12), tous les deux detransmission autosomique dominante43,238-245. Ces chiffres révèlent que la fréquence globale des porteuses de mutations de l’un ou l’autre de ces gènes serait de l’ordre de 1/800 à 1/2 500 danstous les groupes13,246-249. Les mutations du gène BRCA1 sont à l’origine de 30 % à 45 % des casde cancer du sein dans les familles caractérisées par l’incidence élevée de cancer du seind’apparition précoce et de près de 90 % des cas dans les familles où l’incidence des cancers dusein et de l’ovaire est élevée41,242. Les mutations du gène BRCA2 causeraient environ 35 % descas dans les familles où le cancer du sein précoce est de fréquence élevée243-245. Les mutations deBRCA1 prédisposent également au cancer ovarien, au cancer de la prostate, au cancer destrompes de Fallope et aux cancers de l’estomac, du pancréas et du côlon250,251. En Ontario dans la période allant de 1990 à 1998, 11 % des femmes chez qui l’on a diagnostiqué un cancer destrompes de Fallope étaient porteuses de mutations de BRCA1, tandis que 5 % étaient porteusesde mutations de BRCA2252. Les mutations de BRCA1 et de BRCA2 les plus courantes sontdécelées à une fréquence respective de 1,09 % et de 1,52 % dans la population juive ashkénazeen général253-255.
a) Dépistage moléculaire
Il est possible de détecter les mutations des gènes BRCA1 et BRCA2 à l’aide des techniquesdisponibles en pratique clinique. Le SGC, doté de la plus haute sensibilité, est considéré commeétant la méthode par excellence, mais elle est également la plus dispendieuse et la plus longue.Le TPT et le PCSB constituent les deux techniques de dépistage les plus couramment utilisées.Au nombre des autres méthodes, mentionnons l’EGC, l’examen d’exons (EE), l’EGGD, ladétection enzymatique des mutations (DEM), la CLHPD, l’EGB, l’immuno-essai (IE)39,47,256-259,la BAAO et l’ACP d’allèles précis et la bioanalyse de distinction des allèles suivant l’ACP,d’automatisation récente260-262, lorsque la mutation est connue.
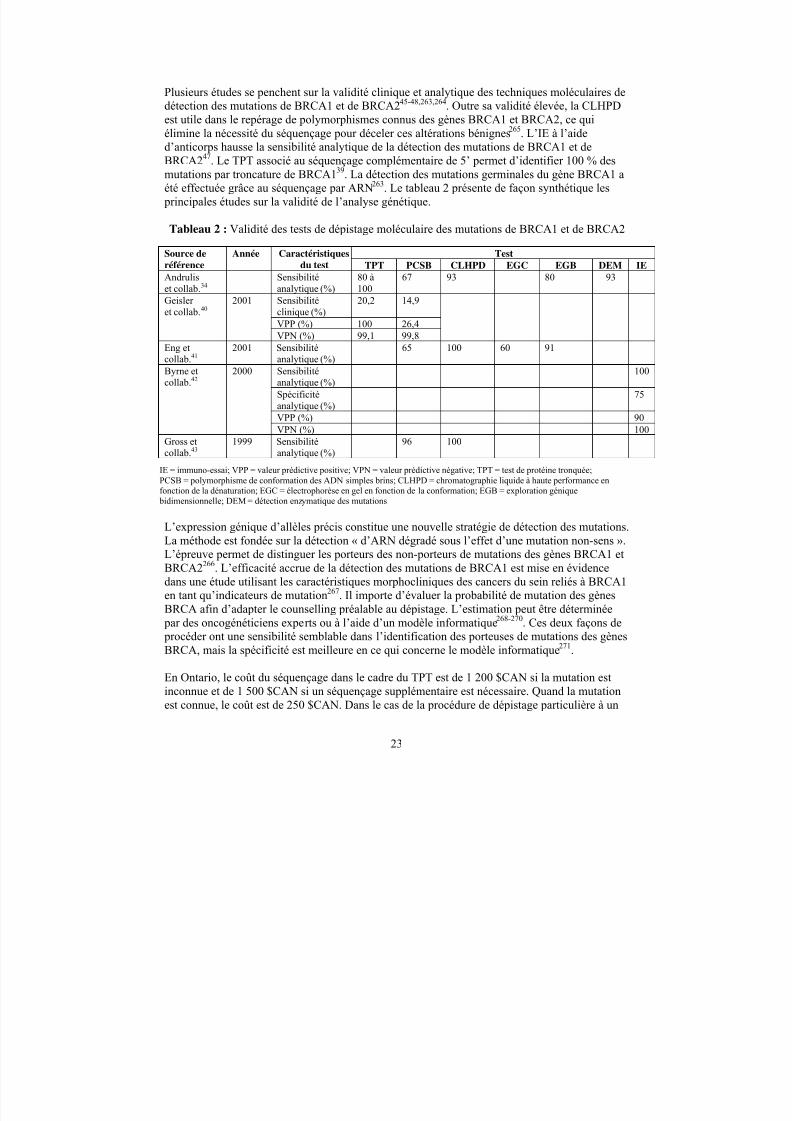
5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 40/103
23
Plusieurs études se penchent sur la validité clinique et analytique des techniques moléculaires dedétection des mutations de BRCA1 et de BRCA245-48,263,264. Outre sa validité élevée, la CLHPDest utile dans le repérage de polymorphismes connus des gènes BRCA1 et BRCA2, ce quiélimine la nécessité du séquençage pour déceler ces altérations bénignes265. L’IE à l’aide
d’anticorps hausse la sensibilité analytique de la détection des mutations de BRCA1 et deBRCA247. Le TPT associé au séquençage complémentaire de 5’ permet d’identifier 100 % desmutations par troncature de BRCA139. La détection des mutations germinales du gène BRCA1 aété effectuée grâce au séquençage par ARN263. Le tableau 2 présente de façon synthétique les principales études sur la validité de l’analyse génétique.
Tableau 2 : Validité des tests de dépistage moléculaire des mutations de BRCA1 et de BRCA2
TestSource deréférence
Année Caractéristiquesdu test TPT PCSB CLHPD EGC EGB DEM IE
Andruliset collab.34
Sensibilitéanalytique (%)
80 à100
67 93 80 93
Sensibilitéclinique (%) 20,2 14,9
VPP (%) 100 26,4
Geisler et collab.40 2001
VPN (%) 99,1 99,8Eng etcollab.41
2001 Sensibilitéanalytique (%)
65 100 60 91
Sensibilitéanalytique (%)
100
Spécificitéanalytique (%)
75
VPP (%) 90
Byrne etcollab.42
2000
VPN (%) 100Gross et
collab.43
1999 Sensibilité
analytique (%)
96 100
IE = immuno-essai; VPP = valeur prédictive positive; VPN = valeur prédictive négative; TPT = test de protéine tronquée;PCSB = polymorphisme de conformation des ADN simples brins; CLHPD = chromatographie liquide à haute performance enfonction de la dénaturation; EGC = électrophorèse en gel en fonction de la conformation; EGB = exploration génique
bidimensionnelle; DEM = détection enzymatique des mutations
L’expression génique d’allèles précis constitue une nouvelle stratégie de détection des mutations.La méthode est fondée sur la détection « d’ARN dégradé sous l’effet d’une mutation non-sens ».L’épreuve permet de distinguer les porteurs des non-porteurs de mutations des gènes BRCA1 etBRCA2266. L’efficacité accrue de la détection des mutations de BRCA1 est mise en évidencedans une étude utilisant les caractéristiques morphocliniques des cancers du sein reliés à BRCA1en tant qu’indicateurs de mutation267. Il importe d’évaluer la probabilité de mutation des gènesBRCA afin d’adapter le counselling préalable au dépistage. L’estimation peut être déterminée par des oncogénéticiens experts ou à l’aide d’un modèle informatique268-270. Ces deux façons de procéder ont une sensibilité semblable dans l’identification des porteuses de mutations des gènesBRCA, mais la spécificité est meilleure en ce qui concerne le modèle informatique271.
En Ontario, le coût du séquençage dans le cadre du TPT est de 1 200 $CAN si la mutation estinconnue et de 1 500 $CAN si un séquençage supplémentaire est nécessaire. Quand la mutationest connue, le coût est de 250 $CAN. Dans le cas de la procédure de dépistage particulière à un

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 41/103
24
groupe ethnique, le coût s’élève à 325 $CAN (entretien personnel du 5 septembre 2003 avec Nancy Carson de l’Hôpital des enfants de l’est de l’Ontario à Ottawa). Le coût du SGC à unlaboratoire privé est d’environ 2 600 $US. Les résultats sont connus en deux à trois semaines.L’application d’un modèle décisionnel révèle que le dépistage génétique au regard de BRCA estrentable, semble-t-il, chez les femmes à risque élevé, le coût incrémental par année de vie
pondérée par la qualité variant de 3 500 $US à 4 900 $US272
. Au vu du coût de l’analysegénétique de BRCA et des aspects éthiques et juridiques connexes, la sélection judicieuse des patientes est primordiale.
La sensibilité et la spécificité des méthodes de détection des mutations de BRCA1 et de BRCA2influencent l’exactitude du diagnostic moléculaire. Quoique les oncogénéticiens du monde entier abordent la prise en charge d’après la probabilité des mutations génétiques, l’exactitude de cetteinformation est mal définie. Le SGC représente la technique de dépistage la plus sensible, maisla procédure est fastidieuse sur le plan technique, coûteuse et longue, particulièrement lorsqu’ils’agit de gros gènes comme BRCA1 et BRCA2 qui comptent de nombreux exons. En outre, leSGC ne permet pas de repérer les vastes délétions, les petites mutations affectant les sites de
reconnaissance de l’amorce et les sites d’épissage intron-exon ou les mutations de point de bifurcation pouvant entraîner des sauts d’exon273. L’application à grande échelle du SGC est fort peu pratique. Le TPT et le PCSB constituent les deux techniques de dépistage les plus courantes,mais leur validité analytique laisse à désirer. Le TPT ne décèle que les altérations débouchant sur des produits protéiniques tronqués de façon prématurée. Le PCSB, l’AH, l’EGGD, l’EGC ne peuvent distinguer les mutations causales des polymorphismes48,274-277. Une évaluationcomparative à l’aveugle des méthodes utilisées par les laboratoires pour estimer la prévalencedes mutations de BRCA1 constate des résultats faux négatifs en nombre remarquable ainsi quedes degrés variables de sensibilité et de spécificité, même dans les laboratoires experts(sensibilité allant de 60 % à 100 %)46.
Le dépistage de mutations dans les régions codantes de BRCA1 et de BRCA2 dans les famillesoù l’incidence de cancer du sein est élevée, combinée à l’analyse de liaison factorielle, ne permet pas de repérer le nombre escompté de mutations dans ces deux gènes, ce qui donne à penser qu’ilexiste au moins un autre gène de prédisposition au cancer du sein278,279. Le résultat négatif del’analyse génétique axée sur BRCA1 et BRCA2 peut traduire l’absence de prédispositionhéréditaire au cancer du sein (le test est véritablement négatif); l’incapacité à déceler la mutation parce qu’il s’agirait de grandes délétions ou d’altérations génomiques complexes; ou l’existenced’un autre gène de prédisposition inconnu280. Nous savons qu’au moins deux autres gènes, TP53dans le syndrome de Li et Fraumeni et PTEN dans la maladie de Cowden, sont mutés dans lesfamilles aux prises avec un cancer du sein transmissible par hérédité42. À moins qu’une mutation précise n’ait été cernée chez un autre membre de la famille, un résultat négatif à l’analyse
génétique de BRCA1 et de BRCA2 peut ne pas être assurément négatif et s’avérer inutile dans la plupart des cas281-283. D’autre part, un résultat positif débouche sur une évaluation du risque pluséclairée, sans pour autant constituer une prédiction individuelle quant à la survenue de lamaladie, parce que la pénétrance de BRCA1 et de BRCA2 est encore méconnue240,284,285.
b) Impact du dépistage moléculaire sur la prise en charge clinique
Le dépistage génétique fait partie intégrante de la prise en charge clinique des cancers du seinfamiliaux 1 et 2. Le conseil génétique dans le cadre d’une discussion éclairée sur les aspects

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 42/103
25
éthiques, juridiques et psychosociaux s’avère essentiel avant l’analyse génétique. Une étude par entrevue révèle que les femmes sont plus enclines à consentir à l’analyse génétique lorsqu’un testdont la valeur prédictive est élevée peut être suivi de véritables mesures préventives noneffractives, ce qui n’est pas le cas pour les cancers du sein et de l’ovaire héréditaires286. Lesfemmes dont le risque de cancer du sein est élevé en raison d’une prédisposition familiale ont le
choix entre la surveillance à intervalles réguliers et des traitements préventifs287
. Une étude prospective sur des porteuses de mutations de BRCA indique que le conseil et l’analysegénétiques accroissent la surveillance globale du cancer par des examens physiques et desexamens d’imagerie288. Les programmes de surveillance proposés aux femmes à risque élevé enraison de leurs antécédents familiaux (les femmes porteuses de mutations de BRCA1 ou deBRCA2 ne sont pas considérées de façon distincte dans l’analyse) permet de déceler 75 % destumeurs289,290. Les études axées seulement sur les porteuses de mutations de BRCA1 ou demutations de BRCA2 ne sont toutefois pas concluantes quant à l’efficacité de la surveillance ducancer ou d’autres mesures visant à réduire le risque de cancer du sein291. Des études démontrentque l’IRM du sein pourrait être supérieure à la mammographie et à l’échographie dans ledépistage chez les femmes à risque élevé de cancer du sein héréditaire292. La surveillance permet
de détecter plus de cas de cancer chez les porteuses de mutations de BRCA1 ou de BRCA2 quechez des femmes non porteuses du même groupe d’âge293. Le dépistage des cancers du sein et del’ovaire précoce est préconisé chez les porteuses de mutations de BRCA1. Le dépistage ducancer du sein précoce est recommandé chez les porteuses de mutations de BRCA2291. Par contre, une étude récente met en relief que la mammographie occasionne beaucoup plus (p=0,01)de résultats faux négatifs chez les porteuses que chez les personnes témoins (62 % contre 29 %) bien que la taille de la tumeur et la densité du tissu mammaire soient comparables294. Le clichédes porteuses révèle beaucoup moins souvent (p=0,01) de spiculation (réaction type de latransformation maligne de la tumeur mammaire), parce que ces signes histologiques particulierssont localisés à la marge de la tumeur, d’où l’image mieux définie. Cette constatation indiquequ’il faut y regarder de près avant de se prononcer devant l’interprétation de la mammographie
des porteuses de mutations.Les mesures préventives englobent la chirurgie ou la chimiothérapie prophylactique295. Aucunerecommandation ne plaide en faveur ou contre la chirurgie prophylactique (p. ex., lamastectomie, l’ovariectomie, l’hystérectomie); ces interventions constituent des choix pour les porteuses de mutations296,297. Une étude mentionne que 51 % du groupe de femmes saines porteuses d’une mutation déterminée optent pour la mastectomie bilatérale et 64 % pour l’ovariectomie298. En Ontario, 16 femmes porteuses de mutations de BRCA1 ou de BRCA2 ontsubi une ovariectomie préventive dans la période de 1992 à 1998299. Selon des données récentes,la mastectomie et l’ovariectomie bilatérales prophylactiques réduisent le risque de cancer du seinet de cancer de l’ovaire chez les porteuses de mutations de BRCA1 ou de BRCA2300-304. Les
études préconisent de tenir compte de l’âge au diagnostic du cancer du sein primaire relié àBRCA1 au moment d’envisager la mastectomie prophylactique305,306. Le nombre de cas decancer des trompes de Fallope observés dans les études sur des porteuses de mutations deBRCA1 ou de BRCA2 pourrait inciter à recommander l’hystérectomie en plus des autreschirurgies préventives252,307. De l’avis d’un groupe comparant, à l’aide d’un modèleinformatique, des stratégies chirurgicales prophylactiques chez des porteuses de mutations deBRCA1, la mastectomie et l’ovariectomie prophylactiques sont surtout efficaces sur le plan del’espérance de vie308. Selon ce modèle, la mastectomie accompagnée de l’ovariectomie bilatérale

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 43/103
26
en prophylaxie à l’âge de 30 ans peut prolonger la survie de 4,9 ans par rapport à la surveillanceseule en présence de mutations de BRCA1 ou de BRCA2309,310. Cependant, des études sur d’autres groupes appuient le traitement conservateur du sein et indiquent que rien ne vientdémontrer que la radiothérapie a des effets indésirables chez les porteuses de mutations deBRCA1 ou de BRCA2311,312. Le modèle d’analyse décisionnelle d’une étude examinant le
retentissement du dépistage génétique de trois mutations sur la survie et le rapport coût-efficacitédans la population juive ashkénaze313 révèle que le dépistage génétique peut augmenter remarquablement la survie moyenne (intervalle de probabilité de 95 %). L’étude indiqueégalement que le dépistage génétique suivi de l’une ou l’autre des trois interventionschirurgicales prophylactiques (ovariectomie, mastectomie ou ovariectomie et mastectomie) chezles femmes dont le résultat est positif améliorent le rapport coût-efficacité comparativement à lasurveillance (test génétique, examen physique, examen gynécologique, échographie, mesure deCA 125, mammographie et test de Papanicolaou). Le dépistage génétique est potentiellementrentable comparé aux divers programmes de surveillance seuls si toutes les femmes dont lerésultat est positif subissent une chirurgie préventive313.
La chimioprévention, notamment par le tamoxifène, a été appliquée à des porteuses de mutationsde BRCA1 ou de BRCA2314-317. Un essai clinique de petite envergure, randomisé et à doubleinsu, met en évidence que, tel qu’il est escompté en raison de son caractère anti-œstrogénique, letamoxifène réduit de façon notable l’incidence de cancer du sein chez les porteuses saines demutations de BRCA2 positives aux récepteurs des oestrogènes, mais pas chez les porteuses demutations de BRCA1 négatives aux récepteurs des oestrogènes318. Une étude cas-témoinsconstate de même que le tamoxifène aurait un effet protecteur quant au risque de cancer du seincontralatéral319. Une étude méthodique se penchant sur des essais cliniques où le tamoxifène estadministré pendant au moins trois ans estime son effet prophylactique probable, à savoir uneréduction de 13 % du risque de cancer du sein chez les porteuses de mutations de BRCA1 et de27 % chez les porteuses de mutations de BRCA2320. Toutefois, ces études ne répondent pas à la
question de savoir quelle est la durée du traitement préventif par le tamoxifène chez les porteusesde mutations de BRCA1 ou de BRCA2. D’autre part, voilà soulevée la question éthique desavoir s’il faut exposer des femmes en santé aux effets indésirables potentiellement nocifs de lachimiothérapie. Dans une étude, l’analyse selon un modèle décisionnel démontre que letamoxifène prescrit à des femmes de 30 ans atteintes du cancer du sein lié à BRCA1 ou àBRCA2 accroît l’espérance de vie dans une mesure allant de 0,5 an (mutations de faible pénétrance) à 1,3 an (mutations de pénétrance élevée)321. L’effet des contraceptifs oraux dans laréduction du risque de cancer ovarien chez les porteuses de mutations de BRCA1 ou de BRCA2est incertain322,323. Les porteuses de mutations de BRCA1 ou de BRCA2 ayant mené desgrossesses à terme sont plus à risque de souffrir du cancer du sein à l’âge de 40 ans que les porteuses nullipares44.
Des femmes souffrant de cancer du sein de même stade peuvent réagir différemment autraitement, comme leur état de santé peut évoluer différemment. Les indicateurs prévisionnelsconnus de l’apparition de métastases, telle l’adénopathie et le degré de différenciationhistopathologique, ne peuvent bien souvent pas prévoir l’issue avec justesse. Une basseconcentration de BRCA2-ARNm serait un signe de réponse favorable au traitement du cancer dusein par le docétaxel324. La recherche se penche sur la corrélation entre le génotype et le phénotype ou sur un schéma d’expression génétique des femmes souffrant de cancer du sein

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 44/103
27
familial qui pourrait être utilisé comme indicateur prévisionnel de l’issue clinique. Des donnéesrecueillies pendant un suivi de dix ans illustrent une tendance vers le pronostic sombre lorsqueles tumeurs des porteuses de mutations de BRCA1 surexpriment le gène suppresseur de tumeurs p53325. Des études ont cerné un schéma d’expression génétique qui laisse fortement présager l’apparition de métastases éloignées à brève échéance (la signature d’un « pronostic sombre »).
La signature des tumeurs des porteuses de mutations de BRCA1 a également été décryptée326
.Ces tumeurs seraient envahissantes, de degré de différenciation élevé, négatives aux récepteursoestrogéniques (indicateurs de pronostic sombre) et augmenteraient le risque de cancer du seincontralatéral, mais les données sur leur agressivité clinique portent à la controverse327-330. Lesmutations germinales de BRCA1 et de BRCA2 constituent en elles-mêmes des prédicteurs del’évolution de l’état de santé après la survenue du cancer du sein331-334. Chez les jeunes femmes porteuses de mutations de BRCA1 ou de BRCA2 qui présentent un cancer du sein et quisubissent une chirurgie conservatrice du sein (tumorectomie du sein) suivie de radiothérapie, letaux de cancer du sein homolatéral ou contralatéral dans les 12 ans est plus élevé que chez lesfemmes atteintes d’un cancer du sein sporadique. La prise en charge clinique des porteuses ensanté de mutations de BRCA1 ou de BRCA2 devrait prendre en considération le fait que le
schéma d’expression génétique et les corrélations entre le génotype et le phénotype peuvent permettrent d’orienter la sélection des patientes qui bénéficieraient des traitements préventifs. Unsoutien psychologique personnalisé peut s’avérer nécessaire.
4.3.9 Maladie héréditaire des exostoses multiples
La maladie héréditaire des exostoses multiples (MHEM) est une affection rare dont la principalecaractéristique clinique est la croissance de protubérances osseuses (exostoses) sur les os longs produisant une dysplasie épiphysaire. Les exostoses peuvent ralentir la croissance squelettique,déformer les os, restreindre la mobilité articulaire, limiter la taille et causer de l’arthrose prématurée335. La complication la plus grave consiste en la transformation maligne desexostoses. L’affection se transmet de façon autosomique dominante. La pénétrance est d’environ95 %. La proportion des personnes souffrant de la MHEM qui présentent des signes cliniques passe d’environ 5 % à la naissance à 96 % à l’âge de 12 ans52. L’âge médian au diagnostic est detrois ans; à toutes fins utiles, le diagnostic est toujours posé à l’âge de 12 ans. Le risque dedégénérescence maligne vers l’ostéochondrosarcome augmente avec l’âge, quoique le risque àvie de transformation maligne soit d’environ 1 %336,337.
a) Dépistage moléculaire
Le dépistage génétique moléculaire de la MHEM n’est disponible qu’à des fins de recherche.L’analyse de liaison a permis de repérer trois loci, à savoir 8q24.11-q24.13 (EXT1), 11p12-p11(EXT2) et 19q (EXT3), en cause dans l’apparition de la maladie. EXT3 n’a pas encore été cloné,
tandis que EXT1 et EXT2 ont été clonés et séquencés. Au moins 49 mutations ont été cernées sur EXT1 et 25 sur EXT249,338. La plupart des mutations provoquent la formation d’une protéineEXT1 tronquée ou non fonctionnelle. La détection de mutations des gènes EXT1 ou EXT2 par l’EGC dans des familles comptant des cas de MHEM est fructueuse dans 70 % des familles53.Des taux de détection semblables ont été obtenus par le PCSB-F et la CLHPD (respectivementde 80 % à 93 % et de 95 %)50. L’analyse de liaison révèle que 60 % des familles aux prises avecla MHEM présentent un lien avec la région 8q24339.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 45/103
28
L’efficacité de la détection des mutations varie selon les études. Ainsi, une étude ayant recoursaux techniques de PCSB-F et de CLHPD indique que 76 % des familles sont porteuses demutations du gène EXT1, alors que 24 % d’entre elles portent des mutations du gène EXT250.Par opposition, deux autres études de détection de mutations rapportent que la fréquencerespective des mutations de EXT1 et de EXT2 est de 58 % et 42 %, et de 50 % et 50 % selon les
méthodes de l’EGC53
et du PCSB51
.b) Impact du dépistage moléculaire sur la prise en charge clinique
Les avantages cliniques du dépistage génétique de la MHEM ont été mis en évidence, mais la procédure ne fait pas partie intégrante de la prise en charge usuelle. Les diverses manifestations dela MHEM pourraient découler de l’effet particulier des différents gènes mutants. Un récentexamen indique que les liaisons chromosomiques de la MHEM joueraient le rôle d’indicateurs prévisionnels de l’emplacement, de l’ampleur et du type d’ostéochondromes340. Les personnessouffrant de MHEM dont la mutation en cause est localisée au chromosome 8 présentent dessignes radiographiques et cliniques différents de ceux des malades exempts de mutations à cechromosome. La proportion des lésions osseuses sessiles (plus dévastatrices que les lésions
pédonculées) et la gravité de la déformation angulaire diffèrent sous l’angle statistique entre lesdeux groupes (p<0,00001) en faveur du groupe dont le chromosome 8 est indemne. Cettecorrélation entre le génotype et le phénotype rendrait le dépistage génétique moléculaire encore plus utile dans l’optimisation de la prise en charge clinique et l’amélioration du pronostic de laMHEM.
4.3.10 Syndrome de Lynch
Le syndrome de Lynch (HNPCC) est une affection courante caractérisée par une prédisposition,transmise de façon autosomique dominante, au cancer colorectal d’apparition précoce (âgemoyen de 44 ans), un risque accru de cancer de l’endomètre utérin et une incidence accrued’autres cancers. Il s’agit de la forme la plus courante de cancer colorectal héréditaire341,342. Lediagnostic de HNPCC repose sur les antécédents familiaux en matière de cancer conformémentaux critères d’Amsterdam ou de Bethesda13,343, qui, s’ils sont tous remplis, laissent entrevoir unefréquence élevée de mutations des gènes MSH2 ou MLH1344-346.
Le syndrome de Lynch est causé par des mutations germinales de cinq gènes des systèmes deréparation de l’ADN : MSH2, MLH1, PMS1, PMS2 et MSH6. Les mutations vont de latroncature à la perte du site d’épissage, de mutations faux-sens aux petites délétions116,347. Lesmutations de MSH2 et de MLH1 sont à l’origine de la majorité des cas de HNPCC348 chez 28 %des personnes aux prises avec un cancer colorectal avant l’âge de 30 ans349. Les mutations deMSH2 et de MLH1 sont plus fréquentes dans les cas de cancer colorectal précoce survenant dansdes familles à risque élevé que dans les familles exemptes de risque350. L’analyse de l’expressiondes protéines MSH2 et MLH1 par des méthodes de coloration immunohistochimique (CIH)démontre que la disparition des protéines MSH2 et MLH1 représente un des premiers incidentsde la cancérogenèse endométriale351.
a) Dépistage moléculaire
Le dépistage moléculaire des gènes MLH1 et MSH2 au regard du syndrome de Lynch estdisponible en pratique clinique. La détection des mutations de MSH2 et de MLH1 chez des

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 46/103
29
personnes atteintes du syndrome et chez les personnes qui leur sont apparentées est possible par diverses méthodes, notamment l’EGGD, la PCR-CDNA, le TPT, le PCSB, la CLHPD etl’analyse de l’instabilité des microsatellites (AIM)352-358. L’EGGD peut détecter de petitesaltérations de la séquence des bases avec une sensibilité supérieure à 90 %; la méthode estappropriée pour déceler des mutations du site d’épissage, des mutations faux-sens ou non-sens et
des mutations de changement de phase. Les techniques fondées sur l’ARN, comme la PCR-CDNA et le TPT, permettent de détecter de grandes délétions, rares dans le syndrome deLynch359. L’analyse selon la CLHPD des mutations de MSH2 et de MLH1, décelées initialement par l’analyse séquentielle, constitue une méthode hautement sensible (sensibilité analytique de près de 100 %) et rentable360. La plupart des programmes de dépistage en Ontario se sont dotésd’un appareil de CLHPD, ce qui pourrait accélérer la procédure (soit, un mois pour la plupart desmutations).
La coloration immunohistochimique (CIH) et l’analyse de l’instabilité des microsatellites (AIM)sont deux techniques de dépistage préliminaire appliquées au tissu tumoral. La CIH pour déterminer l’expression de MLH1 et de MSH2 représente une méthode rapide de dépistage
préliminaire des tumeurs dans la recherche de mutations causales du syndrome de Lynch
361,362
.Dans plus de 90 % des cancers colorectaux survenant chez des personnes porteuses de mutationsde MSH2 ou de MLH1 existent des mutations de changement de longueur de plusieursséquences de nucléotides répétitives. Ce phénomène est désigné par l’expression instabilité demicrosatellite à haute fréquence (IM-HF)363,364. L’IM-HF se produit dans 43 % des famillesinscrites dans le registre des familles à risque élevé de cancer colorectal. Le dépistage desmutations germinales de MSH2 et de MLH1 par l’EGGD confirme de nouveau les mutationschez 50 % des porteurs d’IM-HF365. L’une des stratégies consiste à exécuter d’abord une AIMchez les membres de la famille atteinte présentant une tumeur colorectale. Si l’IM est présentedans la tumeur, on peut alors envisager le dépistage des mutations germinales de MSH2 ou deMLH1, pour lesquels le dépistage moléculaire commercial est disponible366.
En Ontario, le coût du séquençage du gène MSH2 ou du gène MLH1 est de 1 500 $CAN chacunlorsque la mutation est inconnue et de 250 $CAN si elle est connue (entretien personnel du 5septembre 2003 avec Nancy Carson).
b) Impact du dépistage moléculaire sur la prise en charge clinique
Les avantages cliniques du dépistage génétique du syndrome de Lynch ont été mis en évidence,mais la procédure ne fait pas partie intégrante de la prise en charge usuelle. Pour la déterminationdes mutations dans les familles respectant les critères diagnostiques cliniques, le dépistagegénétique est une option importante dans la prise en charge clinique du syndrome 347. Il est possible de réduire la morbidité et la mortalité chez les personnes à risque de HNPCC par le
dépistage précoce et approfondi367
. S’agissant des porteurs de mutations reliées au HNPCC, lacolectomie prophylactique demeure la méthode la plus efficace pour réduire le risque de cancer colorectal. Un modèle d’analyse décisionnelle démontre que les avantages de l’intervention vontde 13,5 années de vie gagnées grâce à la surveillance endoscopique à 15,6 années gagnées par lacolectomie prophylactique, comparativement à l’absence d’intervention368.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 47/103
30
4.3.11 Syndrome de Li et Fraumeni
Le syndrome de Li et Fraumeni (SLF) est un syndrome de prédisposition rare (moins de300 familles dans le monde) au sarcome des tissus mous, au cancer du sein, à la leucémie, àl’ostéosarcome, au mélanome et aux cancers du côlon, du pancréas, de la corticosurrénale et du
cerveau
54
. Le syndrome prédispose à un cancer de survenue précoce. Les tumeurs malignes peuvent apparaître tant chez l’enfant que l’adulte369,370. Le risque de cancer à vie va d’environ85 % à 90 %; Il est plus élevé chez la femme que chez l’homme en raison du risque élevé decancer du sein13,54.
Le SLF est de transmission autosomique dominante. Dans environ 70 % des diagnosticscliniques, on constate une mutation du gène TP53 (appelé également p53) (17p13). Le gèneTP53 code pour la protéine TP53, qui exerce un rôle crucial dans la réparation cellulaire,l’apoptose et la stabilité génomique.
a) Dépistage moléculaire
Les mutations germinales de p53 peuvent être détectées par l’analyse directe de séquencesd’ADN, limitée souvent aux exons cinq à huit, disponible en pratique clinique à certainslaboratoires. Le séquençage de l’ADN sur circuits intégrés est disponible également, mais à desfins de recherche seulement. Les mutations sont surtout des mutations ponctuelles touchant lesexons cinq à huit et des mutations non-sens et du site d’épissage. La sensibilité analytique duSGC de p53 est de 98 %, de l’examen des exons cinq à huit de 80 % et du séquençage sur circuits intégrés de 90 %54. La sensibilité clinique des techniques courantes est de 70 %. On neconnaît pas le coût du dépistage moléculaire du SLF.
La plupart des mutations de p53 sont localisées dans les exons cinq à huit, quoique des mutationsgerminales puissent être repérées dans tout le gène, y compris dans les exons codants et non
codants, dans les familles atteintes du SLF. La présence des mutations est vérifiée par leséquençage du brin complémentaire et l’analyse de restriction371. C’est pourquoi l’analyse dugène entier est essentielle dans le dépistage des familles atteintes du SLF.
Le séquençage direct de tous les exons du gène p53 ne décèle pas de mutations germinales chezenviron 30 % de 21 familles atteintes du SLF372. D’où la question de savoir s’il existe desdifférences phénotypiques entre les familles porteuses de mutations de p53 décelables et lesfamilles sans mutations décelables.
b) Impact du dépistage moléculaire sur la prise en charge clinique
Les avantages cliniques du dépistage génétique du SLF sont incertains. Une étude sur les
porteurs de mutations du gène p53 dans des familles atteintes du SLF révèle que plus de lamoitié des cancers et que près du tiers des cancers du sein ont été diagnostiqués avant l’âge de30 ans373. Le jeune âge à l’apparition du cancer du sein chez les porteuses de mutationsgerminales de p53 justifie la nécessité d’appliquer des procédures de dépistage de ce cancer (mammographie annuelle et examen clinique des seins aux six mois)54 aux porteusesasymptomatiques de mutations germinales de p53 dès l’âge de 25 ou 30 ans. Le recours à lamammographie porte, cependant, à la controverse au vu de la vulnérabilité accrue des porteusesde mutations germinales de p53 aux rayons X374. En outre, des experts proposent la tomographie

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 48/103
31
assistée par ordinateur de la tête et de l’abdomen, la numération globulaire annuelle et l’examenmanuel d’un frottis de sang périphérique pour détecter les signes de leucémie chez les personnesatteintes du SLF10.
4.3.12 Néoplasies endocriniennes multiples de type 1
Les néoplasies endocriniennes multiples de type 1 (NEM1) forment un syndrome familial detransmission autosomique dominante375. Le défaut génétique prédisposant des NEM1 a été cerné par l’analyse de liaison factorielle57. Le gène MEN1 (11q13) a été cloné, séquencé et reconnucomme étant le gène en cause dans l’apparition du syndrome de cancer familial des NEM1376-378.Le gène MEN1 code pour la ménine, une protéine qui se lie à JUND, un facteur de transcription.Les membres des familles atteintes sont prédisposés à diverses combinaisons de lésionsnéoplastiques des glandes endocrines : la parathyroïde (95 % des personnes atteintes), le pancréas (73 %), l’hypophyse (44 %) et les glandes surrénales (16 %)55,56,379. La pénétrance des NEM1 est élevée, environ 99 % des porteurs du gène mutant présentant le syndrome à lacinquantaine56. Les symptômes varient selon la substance sécrétée par le tissu néoplastique;néanmoins, 90 % des malades présentent de l’hypercalcémie, un ulcère gastroduodénal, del’hypoglycémie et des manifestations indicatrices d’une masse hypophysaire.
a) Dépistage moléculaire
Le dépistage génétique des NEM1 est disponible en pratique clinique. Au nombre des méthodesde détection des mutations du gène MEN1 figurent le séquençage de l’ADN, l’ALF et le PCSB,cette dernière technique pouvant déceler 85 % des mutations et l’ALF 99,5 % (sensibilitéanalytique)56,57. L’association du PCSB et de l’AH aurait une sensibilité analytique de 100 %380.Les mutations répertoriées sont des mutations de changement de phase, des mutations non-sensou faux-sens et des mutations de décalage par délétion. Des études de cartographie génétique quiexaminent les tumeurs en rapport avec MEN1 révèlent la perte de l’hétérozygosité dans 70 % des prélèvements de tissu tumoral étudiés.
Diverses mutations germinales dans la région codante du gène MEN1 ont été repérées dans les NEM1 familiales et les NEM1 sporadiques377,381. Il n’y aurait pas, semble-t-il, de corrélationentre les mutations de MEN1 et les manifestations cliniques de l’affection382. De 5 % à 20 % des personnes souffrant de NEM1 seraient exemptes de mutations dans la région codante du gèneMEN156,377,382,383. L’analyse des mutations à des fins diagnostiques est longue et dispendieuse,en raison du fait que les mutations germinales n’engendrent pas de corrélations manifestes entrele génotype et le phénotype.
Au Royaume-Uni, le coût du dépistage moléculaire des NEM1 est de 420 £ (1 050 $CAN)lorsque la mutation est inconnue et de 78,8 £ (197 $CAN) si la mutation est connue, le délaid’exécution variant de une à huit semaines384.
b) Impact du dépistage moléculaire sur la prise en charge clinique
Les avantages cliniques du dépistage génétique des NEM1 ont été mis en évidence, mais la procédure ne fait pas partie intégrante de la prise en charge usuelle. Le dépistage génétique, quienglobe le dépistage phénotypique (histoire médicale, examen clinique, radiographie de la selleturcique [hypophyse] et analyse sanguine biochimique) et l’analyse de l’ADN, a confirmé la

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 49/103
32
liaison avec le gène MEN1 chez les personnes atteintes, tout en écartant d’autres385,386. Ledépistage génétique est, par conséquent, utile pour identifier les personnes prédisposées.
L’histoire familiale (apparition du phénotype particulier, pénétrance selon l’âge) révèle que la plupart des personnes atteintes présenteront au moins une tumeur endocrinienne avant l’âge de
30 ans. Le recours au dépistage génétique allié au dépistage dans des organes précis a ramenél’âge auquel les signes organiques cliniques des NEM1 sont détectés de la quarantaine à la fin del’adolescence387.
4.3.13 Néoplasies endocriniennes multiples de type 2
Les néoplasies endocriniennes multiples de type 2 (NEM2) constituent un syndrome génétiquerare transmis selon le mode autosomique dominant. Le syndrome comprend deux sous-types : NEM2A et NEM2B105-107. Les NEM2A sont caractérisées par la combinaison d’un carcinomemédullaire de la thyroïde, de phéochromocytome et de tumeurs bénignes de la parathyroïde. Lamanifestation des symptômes cliniques des NEM2A va de l’enfance à la fin de l’âge adulte. Les NEM2B sont caractérisées par la présence d’un carcinome médullaire de la thyroïde, d’un phéochromocytome, d’une neuromatose muqueuse de la langue, des lèvres et des paupières, deganglioneuromes à l’appareil gastrointestinal et d’anomalies musculosquelettiques et oculaires.Les NEM2B surviennent à un plus jeune âge que les NEM2A et évoluent en général plusrapidement en occasionnant plus de complications. Le gène de prédisposition des NEM2 estl’oncogène RET (10q11.2), un récepteur de la tyrosine-kinase, dont la mutation entraînel’activation constitutionnelle de la tyrosine-kinase (NEM2A) ou l’altération de la spécificité àl’égard du substrat (NEM2B)107,109. Outre les NEM2A et NEM2B, les mutations de l’oncogèneRET sont à l’origine du cancer médullaire de la thyroïde familial (CMTF)388 et de la maladie deHirschsprung ou mégacôlon congénital389,390.
a) Dépistage moléculaire
Le dépistage moléculaire des NEM2 est disponible en pratique clinique. L’analyse prédictive dela transmission des allèles mutants chez les personnes à risque des NEM2 a d’abord été exécutée par l’application de marqueurs liés à l’ADN391, pour être améliorée ensuite par l’utilisation deséquences de dinucléotides répétitives flanquantes et du polymorphisme de restriction (PR)392.Les méthodes de détection des mutations de RET comprennent le séquençage de l’ADN, l’ACPaccompagnée de la digestion des produits de l’amplification par une enzyme de restriction110,393,l’analyse des hétéroduplex111, le PCSB113,394 et l’EGGD consécutive à l’ACP115,395. Dans ladétection des mutations de RET, ces épreuves ont une sensibilité supérieure à 80 %.
Au Royaume-Uni, le coût du séquençage des exons 10 et 11 (dans le cas des NEM2A) et desexons 15 et 16 (en ce qui concerne les NEM2B) est de 105 £ (262 $CAN) s’agissant d’unenouvelle mutation et de 78,8 £ (197 $CAN) si la mutation est connue. Le délai d’exécution del’analyse va de une à huit semaines384. Le coût de l’examen de l’exon 16 du gène RET en vue dedéceler les mutations à l’origine des NEM2B à un laboratoire privé selon la technique de l’ACPsuivie du séquençage de l’ADN est de 300 $US. Le diagnostic prénatal par l’examen de deux prélèvements (prélèvement des villosités choriales, cellules amniotiques fraîches ou en milieu deculture) coûte 700 $US. Le délai d’exécution est d’environ trois ou quatre semaines en ce qui atrait à un nouveau patient et de deux semaines lorsque la mutation est connue104.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 50/103
33
b) Impact du dépistage moléculaire sur la prise en charge clinique
Le dépistage génétique fait partie intégrante de la prise en charge clinique des NEM2. L’analysedes mutations est utile dans l’identification des personnes à risque au sein d’une famille porteusede mutations, dans le repérage de mutations chez les membres d’une famille manifestant dessignes cliniques des NEM2, pour déterminer si un cas apparemment sporadique de carcinome
médullaire de la thyroïde ou de phéochromocytome constitue l’une des manifestations des NEM2 et dans la détection d’une corrélation entre le génotype et le phénotype qui faciliterait la prise en charge de l’affection.
Le dépistage génétique chez les personnes à risque est une procédure clinique établie396. Entenant compte des limites du dépistage biochimique397, le dépistage moléculaire des mutations deRET dès que possible représente la démarche de dépistage de premier choix chez toutes les personnes à risque ou chez qui l’on soupçonne les NEM2114. En présence d’un résultat positif, ilfaudrait envisager la thyroïdectomie préventive et le dépistage du phéochromocytome et del’hyperparathyroïdie. La thyroïdectomie prophylactique est recommandée dans les plus brefsdélais étant donné que le cancer médullaire de la thyroïde chez les personnes atteintes des
NEM2B est fulgurant et rapidement envahissant112,398
.
Le dépistage moléculaire occupe une place importante dans la détermination de la proportion descas supposément sporadiques qui sont héréditaires. S’agissant des personnes souffrant d’uncarcinome médullaire de la thyroïde apparemment sporadique, les données rétrospectives sur lescas de NEM2A démontrent que le phéochromocytome survient de deux à 11 ans suivantl’apparition du carcinome médullaire de la thyroïde dans plus de 40 % des cas399. C’est pourquoil’analyse des mutations de RET doit être effectuée, que le carcinome médullaire de la thyroïdesoit familial ou sporadique108. L’étroitesse de la gamme de mutations fait en sorte que la procédure est techniquement faisable dans la plupart des cas. En l’absence d’antécédentsfamiliaux et de signes d’hyperplasie dans la thyroïde prélevée, l’impossibilité à détecter une
mutation dans les exons 10, 11, 13, 14 et 16 du gène RET écarte l’éventualité des NEM2A selonune probabilité supérieure à 99 %, et celle des NEM2B en l’absence de phénotype anormal400.
Les mutations du gène RET donnent lieu à différentes présentations cliniques des NEM2, ce quilaisse supposer qu’il existe une relation entre le génotype et le phénotype401. Il existe ainsi uneassociation statistiquement significative (p<0,001) entre les mutations au codon 634 et le phénotype caractérisé par les phéochromocytomes ou l’hyperparathyroïdie400. Les mutations auxcodons 768 et 804 ne sont observées que dans les cas de carcinome médullaire de la thyroïdefamilial, ce qui laisse présager que ces personnes ne souffriront pas de phéochromocytomes oud’affection de la parathyroïde. Les mutations au codon 918 sont particulières aux NEM2B400.L’association est trop faible pour offrir la chirurgie prophylactique aux personnes à risque élevé
de mutations ou pour omettre le dépistage chez les personnes exemptes de mutations.4.3.14 Neurofibromatose de type 1
La neurofibromatose de type 1 (NF1) (appelée également maladie de von Recklinghausen) estl’un des troubles autosomiques les plus courants (prévalence de 1/960 à 1/7 800). Le gène NF1(17q11.2) est en cause dans la NF1. La neurofibromine, protéine codée par NF1, freine l’activitédu proto-oncogène p21-RAS. La mutation du gène NF1 entraîne l’extinction de la protéineneurofibromine, ce qui a pour effet d’activer l’oncogène p21-RAS et de provoquer une

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 51/103
34
prolifération cellulaire anarchique62. Dans une proportion de 70 % à 80 %, les mutationsoccasionnent la troncature de la protéine. La NF1 est caractérisée par les neurofibromesmultiples, lesquels peuvent se transformer en tumeurs malignes (neurofibrosarcomes), les tachescafé au lait, l’apparition d’éphélides axillaires et inguinales, les gliomes du nerf optique et lesnodules Lisch de l’iris. Les manifestations cliniques sont diverses402,403, dont la taille réduite, la
macrocéphalie, les troubles d’apprentissage et les crises épileptiques. La NF1 peut se manifester à n’importe quel âge dans n’importe quel système organique. La pénétrance du trouble est de près de 100 % à l’âge de cinq ans et des nouvelles mutations sont en cause dans la moitié descas. Le diagnostic de la NF1 repose sur l’évaluation clinique64,404.
a) Dépistage moléculaire
Le dépistage moléculaire des mutations de NF1 par le TPT est disponible en pratique clinique.Plusieurs méthodes permettent de déceler les mutations du gène NF1, notamment le PCSB, laCLHPD, l’EGGD et le séquençage direct, la CLHPD faisant preuve d’une sensibilité analytiquede 97 % et d’une sensibilité clinique de 68 %58,59. Selon la technique TPT, l’ARN provenant deleucocytes subit une transcription inverse pour être converti en fragments d’ADN
complémentaire se superposant au gène NF1, devenant les modèles de la synthèse in vitro defragments de neurofibromine. Chacun des fragments de neurofibromine produits est séparé enfonction de sa taille sur un gel de polyacrylamide dénaturant pour détecter la présence d’un peptide tronqué. Le TPT a une sensibilité clinique de 67 % dans la détection des mutations pathogènes du gène NF1 et une sensibilité analytique de 93 %65.
Les méthodes de détection des mutations causales chez les personnes souffrant de NF1 sontlimitées en raison de la grande taille du gène NF1 (59 exons) et de la diversité des mutations. Ladétection des mutations chez les personnes atteintes de NF1 fait intervenir des techniqueslongues et coûteuses. Malgré la prévalence de la NF1 et les connaissances acquises en biologiemoléculaire, la pathogenèse de l’affection n’est pas complètement élucidée. Ce qui laisse
perplexe au sujet de la NF1, c’est son hétérogénéité clinique, qui ne peut être expliquée par unemutation du gène NF1. Il est improbable que le diagnostic de NF1 puisse être écarté par laméthode moléculaire405.
b) Impact du dépistage moléculaire sur la prise en charge clinique
Les avantages cliniques du dépistage génétique de la NF1 ont été mis en évidence, mais la procédure ne fait pas partie intégrante de la prise en charge usuelle. Bien que de 70 % à 80 % desmutations du gène NF1 provoquent la formation d’une protéine tronquée, environ 10 % desmutations prennent la forme de délétions sur tout le gène, associées à une déficienceintellectuelle ou à des troubles d’apprentissage, à des anomalies faciales et à l’élargissement ducou406. La sensibilité, la spécificité et la VPP du TPT disponible sur le marché appliqué à un
vaste groupe ne sont pas connus60
. Le diagnostic prénatal est possible dans la plupart des cas oùl’un des parents est atteint de NF1. Ce diagnostic peut être effectué par analyse directe si unemutation précise a été cernée dans la famille ou par analyse de liaison s’il est possible del’appliquer au nombre suffisant de membres de la famille et que l’information génétique de lafamille est suffisante pour étudier la coségrégation de la maladie et des marqueurs génétiques407.L’impossibilité de prévoir la gravité de la NF1 limite l’utilité pratique du dépistage prénatal.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 52/103
35
4.3.15 Neurofibromatose de type 2
La neurofibromatose de type 2 (NF2), ou neurofibromatose acoustique bilatérale, est uneaffection héréditaire caractérisée par l’apparition de multiples tumeurs, bénignes ou malignes, dusystème nerveux – méningiomes, schwannomes, neurofibromes – au début de l’âge adulte,
transmise sur le mode autosomique dominant et dont la pénétrance est complète à l’âge de60 ans61,408,13,63,409. Le gène NF2 (22q12) code pour la protéine merline, l’une des protéines ducytosquelette qui modèle l’architecture des cellules et participe à la stabilité membranaire410.Avant l’âge de 30 ans, les personnes atteintes de NF2 souffriront toutes, pour ainsi dire, d’unedéficience auditive due à la présence de schwannomes des nerfs vestibulaires bilatéraux. Desgliomes malins ont été détectés chez des personnes souffrant de NF2, sans que le risque decancer s’en trouve augmenté. Des études révèlent que l’incidence dans la population est d’une personne sur 37 000 et que 50 % des cas sont causés par de nouvelles mutations67.
a) Dépistage moléculaire
Le dépistage moléculaire de la NF2 est disponible en pratique clinique. Il prend la forme de deux
analyses de l’ADN : l’analyse des mutations du gène NF2 et l’ALF. Au nombre des méthodes dedétection moléculaire des mutations du gène NF2 figurent le PCSB, l’EGGD, le séquençagedirect et le clivage de mésappariement d’ARN66,69. L’analyse des mutations permet de cerner lesmutations germinales du gène NF2 chez 64 % des patients par l’examen d’exons selon le polymorphisme de conformation des ADN simples brins (PCSB) suivi du séquençage direct68.Une nouvelle bioanalyse d’après la méthode de clivage de mésappariement d’ARN a fait preuved’une sensibilité analytique de 100 % et d’une sensibilité clinique de 75 % dans la détection desmutations du gène NF269. Les études de liaison ne sont fiables que dans la mesure où lediagnostic clinique de NF2 est exact concernant les membres de la famille atteinte et que lesrapports génétiques dans la famille sont connus avec précision. La coségrégation trompeuse de lamaladie et des marqueurs génétiques de familles atteintes de la NF2 est toujours possible en
raison de la fréquence de mosaïcisme élevée chez les fondateurs (c.-à-d., la première personnedans une famille à souffrir de la NF2)411.
Au même titre que celui de la NF1, le diagnostic de la NF2 repose sur l’évaluation clinique. Ladétection des mutations est longue et dispendieuse, et peut être impuissante à déceler la mutationcausale. Les techniques courantes, comme le PCSB, peuvent déceler les deux tiers des mutationsdu gène NF2. Pour confirmer les mutations détectées par l’épreuve de PCSB, il faut procéder auséquençage direct de l’ADN. Le dépistage moléculaire chez les enfants à risque pose problèmecar on ne sait pas si des mesures de protection adoptées durant l’enfance modifieront l’issuefinale.
b) Impact du dépistage moléculaire sur la prise en charge clinique
Les avantages cliniques du dépistage génétique de la NF2 ont été mis en évidence, mais la procédure ne fait pas partie intégrante de la prise en charge usuelle. Les stratégies de prise encharge de la NF2 sont multidisciplinaires412-414. Comme on ne sait pas si la NF2 accroît le risqueoncogène, aucun programme de surveillance étroite n’est préconisé. La détermination d’unecorrélation entre le génotype et le phénotype permettrait de préciser le pronostic et d’orienter la prise en charge de la NF2. Bien que la maladie suive essentiellement le même cours chezl’homme et la femme, il y a une différence sur le plan de l’histoire naturelle selon que le gène

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 53/103
36
NF2 provient de la mère ou du père. Les personnes ayant hérité du gène maternel souffre de lamaladie à un plus jeune âge (à l’âge de 18 ans comparativement à 24 ans dans le cas du gène paternel; p=0,027) et la progression de la maladie est accélérée (l’âge moyen au décès étant de24 ans comparativement à 39 ans dans le cas du gène paternel)67. Des corrélations entre desmutations germinales du gène NF2 et la gravité de la NF2 et des anomalies rétiniennes ont été
précisées. Une étude met en relief le fait que les porteurs de mutations non-sens ou dechangement de phase sont plus jeunes à la survenue de la maladie et souffrent d’un plus grandnombre moyen de tumeurs (p≤0,05) que les porteurs de mutations du site d’épissage415. Celacorrobore la corrélation existant entre les mutations non-sens et de changement de phase et la NF2 grave.
4.3.16 Syndrome de Peutz-Jeghers
Le syndrome de Peutz-Jeghers (SPJ) est un trouble rare transmis selon le mode autosomiquedominant. L’affection est caractérisée par la combinaison d’une polypose gastrointestinale etd’une lentiginose de la peau et des muqueuses. L’hybridation génomique comparative etl’analyse de liaison ciblée ont permis d’établir le lien entre le SPJ et des mutations du gèneSTK11 (19p13.3), l’un des gènes de la sérine thréonine kinase71,72,416. Au nombre des mutationsfigurent les remaniements intragéniques et les délétions, des mutations non-sens et des mutationsdu site d’épissage. Environ 50 % des cas sont héréditaires (un parent atteint) et environ 50 %seraient sporadiques. Les polypes peuvent se loger partout dans l’appareil gastrointestinal (le plus souvent à l’intestin grêle) et se transformer en tumeurs malignes. Les taches brunes, quiapparaissent pendant l’enfance, sont concentrées sur les lèvres et la muqueuse orale. Elles sont présentes dans plus de 95 % des cas. Les personnes atteintes du SPJ ont un risque élevé decancers gastrointestinaux et de cancer d’autres organes, dont le sein, l’utérus, les ovaires, les poumons et les testicules13,70,73,417-419.
a) Dépistage moléculaire
Le dépistage moléculaire du SPJ est disponible en pratique clinique. L’examen des exons etl’analyse séquentielle de l’ADN génomique obtenu du prélèvement oral permettent de détecter les mutations du gène STK11. Le coût de la détection des mutations s’agissant d’un nouveau patient à un laboratoire privé est de 1 400 $US et celui concernant les personnes apparentéeslorsque la mutation est connue, de 350 $US74. Le diagnostic prénatal par l’examen de deux prélèvements (prélèvement des villosités choriales et cellules amniotiques fraîches ou en milieude culture) coûte 700 $US. Dans le cas d’un nouveau patient, le délai d’exécution de l’analyseest d’environ six à huit semaines. Quant au diagnostic prénatal, lorsque la mutation dans lafamille est connue, le délai d’exécution est d’environ deux semaines74.
Le séquençage de l’ADN est d’une sensibilité analytique d’environ 70 % dans les familles où laliaison avec le gène STK11 est établie. Le séquençage de bout en bout du gène STK11 est d’unesensibilité clinique telle qu’il permet d’identifier 70 % des proposants ayant des antécédentsfamiliaux de SPJ75. En l’absence d’antécédents familiaux de SPJ, l’EGC suivie du séquençagedirect permet de repérer 17 % des patients76. Ces données laissent entrevoir l’hétérogénéitégénique du SPJ et la participation d’autres loci.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 54/103
37
b) Impact du dépistage moléculaire sur la prise en charge clinique
Les avantages cliniques du dépistage génétique du SPJ sont incertains. Si l’on veut identifier lesnon-porteurs au sein de grandes familles dont la liaison au gène STK11 est établie, on peutrecourir à l’analyse de liaison factorielle. Un expert propose que les porteurs du gène STK11mutant (et les personnes présentant les taches brunes caractéristiques du SPJ) soient soumis au
dépistage des polypes gastrointestinaux et des cancers du sein, de l’ovaire et de l’utérus420
.
4.3.17 Rétinoblastome
Le rétinoblastome (RB) est un cancer de la rétine survenant pendant l’enfance. Il frappe environune naissance vivante sur 20 000 dans les pays industrialisés; dans 60 % des cas, il est unilatéralet sporadique; dans 15 % des cas, unilatéral et héréditaire; et dans 25 % des cas, bilatéral ethéréditaire. La prédisposition héréditaire au RB, qui relève de mutations germinales du gènesuppresseur de tumeurs RB1 (13q 14.1), est transmise selon le mode autosomique dominant et de pénétrance élevée79-82. Le RB se manifeste le plus souvent par une leucocorie (pupille blanche),suivie de strabisme. Lorsqu’il y a délétion du chromosome 13q et que la bande 13q14 est en
cause, les patients présentent un phénotype particulier sur le plan morphologique et un troubleneurologique421. Dans la plupart des cas, le diagnostic est posé avant l’âge de cinq ans. Lesmutations germinales du gène RB1 accroissent le risque de tumeurs extra-oculaires, tell’ostéosarcome, le sarcome d’Ewing, la leucémie, le lymphome, le mélanome, le cancer du poumon et le cancer de la vessie.
a) Dépistage moléculaire
Le dépistage moléculaire du RB est disponible en pratique clinique. La détection des mutationsgerminales du long (27 exons) gène RB1 s’est avérée ardue. Les laboratoires qui effectuent ladétection des mutations par diverses techniques ne peuvent donner l’assurance d’une sensibilitéde 100 %. Le diagnostic de RB héréditaire est encore posé en fonction de critères cliniques13.
La méthode classique de détection d’une délétion partielle du gène RB1 est l’ATS, capable dedéceler la mutation chez 10 % des patients. Quant à la détection de mutations ponctuelles, lestechniques de dépistage fondées sur l’analyse multiplex de fragments d’ADN en fluorescence, suiviedu séquençage direct, ont une sensibilité clinique de 70 %78. L’AH et le PCSB non isotopique permettent de détecter 72 % des mutations84. L’exploration génique bidimensionnelle, faisantintervenir l’ACP et l’EGGD, constitue une solution de rechange abordable au séquençage 77. Lesdélétions localisées à des loci polymorphes peuvent être décelées par l’analyse de liaison factoriellesi la constellation d’allèles des parents est révélatrice. Dernièrement, la stratégie combinant l’ACPmultiplex quantitative, le séquençage d’exons doubles et l’ACP sensible à la méthylation et dirigéesur l’agent promoteur s’est révélée sensible et efficace dans la détection des mutations de RB1422.
En Ontario, le coût du séquençage du gène RB1 est de 3 700 $CAN. Une étude comparative descoûts du dépistage moléculaire (extraction de l’ADN, analyse des fragments, séquençage, conseilgénétique) et du dépistage traditionnel (examen de la rétine complet à intervalles réguliers)auprès de personnes apparentées à risque de RB en 1994 révèle que le dépistage moléculaireamène des économies de coûts importantes423. Selon un rapport de 2003, le dépistagemoléculaire du gène RB1 se traduit par des économies moyennes de 6 591 $CAN par famille endépenses de santé dans un échantillon représentatif de 20 familles ontariennes422.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 55/103
38
b) Impact du dépistage moléculaire sur la prise en charge clinique
Le dépistage génétique fait partie intégrante de la prise en charge clinique du RB. Il s’agit là d’unexcellent exemple illustrant les avantages économiques du diagnostic moléculaire. Les enfantsexempts de mutations ne sont pas soumis inutilement à des procédures effractives, alors que les porteurs de mutations bénéficient d’évaluations ophtalmologiques fréquentes et d’un traitement en
temps opportun. Compte tenu qu’un enfant atteint d’un RB unilatéral peut être le premier signed’une mutation de RB1 de faible pénétrance dans la famille424,425, le dépistage moléculaire seraitutile dans ce cas. Une étude récente sur des familles ayant subi le dépistage génétique du RB1indique que l’information et le conseil génétiques sont importants et que les conséquences préjudiciables à long terme du dépistage génétique sont rares83. La thérapie génique du RB à titreexpérimental fait ressortir que le transfert du gène de la thymidine kinase de l’herpès simplex virusdans les cellules du rétinoblastome accroît la sensibilité de ces cellules à l’égard du gancyclovir etde l’acyclovir in vitro
426, plutôt encourageant comme début pour la thérapie génique du RB.
4.3.18 Maladie de von Hippel-Lindau
La maladie de von Hippel-Lindau (VHL) est un trouble de transmission autosomique dominante, prédisposant aux carcinomes cérébral et rétinien, et à l’hypernéphrome, aux angioblastomes de lamoelle épinière et aux kystes pancréatiques. Les angioblastomes cérébelleux provoquent descéphalées et vomissements, perturbent la démarche ou causent de l’ataxie. Les angioblastomesrétiniens peuvent être les premières manifestations de la VHL et entraîner la cécité.L’hypernéphrome survient chez 40 % des patients et constitue la principale cause de mortalité86.La maladie englobe deux sous-types : la VHL de type 1 (absence de phéochromocytomes) et laVHL de type 2 (apparition éventuelle de phéochromocytomes)13. Le diagnostic est souvent établidans la vingtaine, quoique des symptômes, habituellement les angioblastomes rétiniens, puissentse manifester pendant l’enfance. Une étude démontre que la maladie frappe 40 % des personnes présentant des angioblastomes427. Le gène suppresseur de tumeurs VHL (3p25), en cause dans lesyndrome de prédisposition VHL, code pour une protéine qui inhiberait, semble-t-il, l’élongation pendant la traduction. L’extinction de la protéine favoriserait la multiplication cellulaireanarchique. Les mutations sont des délétions, des insertions, des mutations faux-sens ou non-sens et des mutations du site d’épissage88,90.
a) Dépistage moléculaire
Le dépistage génétique moléculaire est offert en pratique clinique aux cas conformes aux critèresdiagnostiques cliniques de la VHL87. L’ATS et l’ATS quantitative sont utilisées pour détecter,respectivement, les délétions partielles et les délétions complètes du gène, tandis que l’analyseséquentielle permet de déceler les mutations ponctuelles. Les délétions cernées par l’ATSquantitative sont confirmées par l’HISF96. L’EGGT en parallèle constitue une méthode de
détection rapide de sensibilité analytique élevée lorsqu’il s’agit d’analyser un grand nombre de prélèvements95. La CLHPD a été présentée comme étant une méthode hautement productive dedétection des mutations. Une étude illustre que la CLHPD et le PCSB en fluorescence font preuve d’une sensibilité analytique élevée (de 95 % à 100 %) et d’une spécificité (100 %)comparables dans la détection des mutations de VHL92. La CLHPD par le système Wave Nucleic
Acid Fragment Analysis a pu identifier 93 % des porteurs de VHL mutant dans des famillesatteintes où les mutations précises avaient été relevées94. Le séquençage direct permet de déceler 100 % des mutations, mais il est dispendieux, particulièrement dans les laboratoires de grande

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 56/103
39
production. L’analyse séquentielle comparative a fait ses preuves comme méthode rapideet précise (sensibilité et spécificité analytiques de 100 %) dans la détection des mutationsde VHL78.
Le coût de l’analyse moléculaire de la VHL par le séquençage direct était de 260 $US en 1998. Le
coût du dépistage annuel (examen ophtalmologique, dosage des catécholamines urinaires) est de650 $US (sans tenir compte de l’absentéisme au travail). Parce que les symptômes peuvent semanifester dès l’âge de cinq ans ou ne survenir qu’à 25 ans, le calcul prend en considération unsuivi régulier pendant 20 ans. Le coût du dépistage pendant 20 ans s’élève à 13 000 $. Si ledépistage moléculaire est véritablement négatif, les économies de coûts seront de l’ordre de12 000 $US par personne sur 20 ans89. Une autre étude examinant l’impact du dépistage génétiquemoléculaire du gène VHL chez des personnes présentant des angioblastomes cérébelleux428 établitque le coût du diagnostic moléculaire par l’ATS et le PCSB est de 960 euros (1 500 $CAN) ou de1 070 euros (1 700 $CAN) si le séquençage est nécessaire. Le coût du programme de dépistageclinique en Allemagne, y compris l’imagerie par résonance magnétique (IRM) du cerveau, ducanal vertébral et de l’abdomen, l’examen ophtalmologique, l’angiographie en fluorescence de la
rétine et le dosage de l’excrétion urinaire des catécholamines pendant 24 heures, est de 2 570 euros(4 100 $CAN) par an428.
b) Impact du dépistage moléculaire sur la prise en charge clinique
Le dépistage génétique fait partie intégrante de la prise en charge clinique usuelle de la VHL.Outre le fait d’offrir une valeur prédictive exacte, la caractérisation des mutations germinales deVHL permet également de préciser le phénotype probable. Ces renseignements facilitent la priseen charge des personnes atteintes de la VHL. Des corrélations entre le génotype et le phénotypedans la VHL ont été déterminées429-432. Les mutations germinales dont l’effet escompté consiste àinactiver la protéine codée par VHL sont reliées à l’hypernéphrome et aux angioblastomes dusystème nerveux central sans phéochromocytomes (VHL de type 1), alors que les mutations
germinales dont l’effet prévu est de produire des protéines VHL de pleine longueur provoqueraientdes phéochromocytomes en plus des autres manifestations de la VHL (VHL de type 2). Lesgrandes délétions et les mutations occasionnant selon toute probabilité la formation de protéinestronquées sont associées à un risque plus bas de phéochromocytomes que les mutations faux-sens.Une mutation faux-sens au codon 167 s’accompagne d’un risque élevé de phéochromocytomes(respectivement de 53 % et de 82 % à 30 ans et à 50 ans). Comme les mutations au codon 167 sontfréquentes, la détection de cette mutation dans les familles présentant des phéochromocytomes pourrait être utile. Le dépistage moléculaire de la VHL offre non seulement des renseignements de première importance, mais permet également de dépister les tumeurs extra-neurologiques chez le patient et d’étudier sa famille. Lors d’une réunion consensuelle en 1998 aux Pays-Bas, les participants ont affirmé dans une proportion de 56 %, dans le cadre d’un scrutin interactif, préférer
que le dépistage moléculaire soit effectué avant l’âge de cinq ans; dans la période allant de l’âge decinq ans à l’âge de 10 ans, dans une proportion de 15 %; et à l’âge où l’enfant est apte à prendredes décisions le concernant, dans une proportion de 18 %433.
4.3.19 Tumeur de Wilms
La tumeur de Wilms (TW) ou néphroblastome est une tumeur maligne du rein survenant pendantl’enfance chez un enfant sur 10 000. Tumeur solide la plus fréquente dans l’enfance, elle esthabituellement diagnostiquée à l’âge de cinq ans100. Ce diagnostic est rare à l’âge adulte101. Selon

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 57/103
40
les estimations, 1 % des TW découlent d’un gène mutant transmis par l’un des parents. La TWhéréditaire est due à des mutations germinales des gènes suppresseurs de tumeurs WT1 (11p13),WT2 (11p15.5) et WT3 (17q12-21), dont la transmission est autosomique dominante98,99. La perted’hétérozygosité du chromosome 16q est une modification structurale observée dans environ20 % à 30 % des cas de TW434. Le gène WT1 code pour une protéine qui est un facteur de
transcription97
. Certaines mutations ponctuelles du gène WT1 entraînent de graves anomaliesgénito-urinaires, dont le pseudohermaphrodisme et la glomérulosclérose pouvant entraîner l’insuffisance rénale tôt dans l’enfance (syndrome de Drash)102. Contrairement à d’autres gènessuppresseurs de tumeurs qui ne perdent rien sur le plan fonctionnel en hétérozygosité,l’hémizygosité pour le WT1 provoque des anomalies du développement des voies génito-urinaires,dont la cryptorchidie et l’hypospadias.
a) Dépistage moléculaire
Le dépistage génétique de la TW est disponible en pratique clinique. Les mutations de WT1 sontdétectées par le séquençage de l’ADN, dont les jonctions intron-exon (entretien personnel du11 juillet 2002 avec Ann Dalton, Sheffield Children’s Hospital, Sheffield [R.-U.]). La sensibilité
analytique de la technique est supposément d’au moins 90 %. Lorsqu’une modification est relevée,on doit savoir si elle relève du polymorphisme courant dans la population locale. Quand lamodification est localisée à proximité d’une jonction intron-exon, il faut également déterminer s’il ya eu perte du site d’épissage. Le coût du séquençage direct de l’ADN est de 500 ₤ (1 250 $CAN) pour la pleine détection des mutations de tous les exons (de un à 10). Le coût de l’examen des exonssix à neuf (où les mutations se produisent le plus souvent) est de 200 ₤ (500 $CAN). Le délaid’exécution varie et peut être de quatre ou cinq mois s’il s’agit de la détection complète. L’ATS etl’AIM dans l’analyse de l’ADN des cas de TW sporadiques révèlent la participation de deux régions1p à l’étiologie des tumeurs de Wilms435.
b) Impact du dépistage moléculaire sur la prise en charge clinique
Les avantages cliniques du dépistage génétique de la TW sont incertains. Il existe une corrélationentre la perte de l’hétérozygosité de 16q et le taux de survie. Une analyse statistique fait ressortir unécart notable d’échec subséquent au traitement entre les enfants ayant conservé l’hétérozygosité 16qet ceux l’ayant perdu. L’absence d’hétérozygosité 16q est constatée trois fois plus souvent dans letissu tumoral des enfants décédés que dans le tissu tumoral des survivants (p=0,0024)434.
4.3.20 Xeroderma pigmentosum
Le xeroderma pigmentosum (XP), affection rare qui se transmet selon le mode autosomique récessif,est dû à une insuffisance de la capacité de réparation de l’ADN endommagé par le rayonnementultraviolet. Le XP est caractérisé par une extrême sensibilité au rayonnement solaire qui engendre
des anomalies pigmentaires et le cancer (incidence élevée) de la peau exposée au soleil
135,436,437
. Lerisque de carcinomes basocellulaires et d’épithéliomas spinocellulaires serait plus élevé d’un facteur 2 000 que celui de la population en général. L’âge moyen à l’apparition du cancer cutané est dehuit ans (comparativement à 60 ans dans la population en général). Près de 20 % des enfants atteintssouffrent également de troubles neurologiques, dont la déficience mentale, la microcéphalie et laspasticité. La survie est brève en raison de la transformation tumorale, seulement 70 % des personnes atteintes de XP vivant jusqu’à l’âge de 40 ans 135. En règle générale, le diagnostic est posé

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 58/103
41
en fonction des manifestations cliniques observées dans l’enfance. Quelques laboratoires sont enmesure de confirmer la défaillance du mécanisme de réparation de l’excision de l’ADN obtenu par excision.
a) Dépistage moléculaire
Le dépistage moléculaire du XP n’est disponible qu’à des fins de recherche. Sept gènes mutants, de XPAà XPG, sont en cause dans les sept groupes de complémentation du XP134. La méthode de détection desmutations du site de restriction, fondée sur l’ADN, d’efficacité démontrée dans la détection desmutations de fréquence élevée à différents loci, est utilisée dans le dépistage génétique des cellulesincapables de réparer l’ADN du XP438. La PCR-CDNA, la PCR-PR, l’ATS et le PCSB sont lestechniques utilisées pour déceler les mutations de XP, notamment les mutations faux-sens, les délétionsengendrant des protéines tronquées, des mutations d’épissage et des mutations non-sens439-441. Lediagnostic prénatal du XP s’effectue par l’analyse de la capacité de réparation de l’excision desfibroblastes du fœtus ou des cellules amniotiques442,443.
b) Impact du dépistage moléculaire sur la prise en charge clinique
Les avantages cliniques du dépistage génétique du XP sont incertains. Les porteurs de mutations génétiquesdu groupe G (XPG) sont ceux qui, selon toute probabilité, présenteront les symptômes les plus divers sur le plan clinique, accompagnés de troubles neurologiques. Les protéines XPG tronquées sont incapables deréparer les nucléotides excisés et sont à l’origine de l’extrême vulnérabilité de la peau au rayonnementultraviolet qui caractérise le XP du groupe G439. Les mutations du gène XPD sont reliées à une gamme designes cliniques de gravité diverse ayant trait à des aspects du dommage à l’ADN et à la sensibilitécellulaire à cet égard444. Des mutations d’épissage du gène XP dans le groupe C occasionneraient del’autisme et de l’hypoglycémie445. Cette corrélation entre le génotype et le phénotype serait utile dans lecadre du diagnostic moléculaire pour optimiser la prise en charge et améliorer le pronostic du XP. Letransfert par rétrovirus de gènes des systèmes de réparation de l’ADN dans les cellules du XP permettrait àcelles-ci de reconquérir une survie normale au regard du rayonnement ultraviolet, ce qui donne à penser
que la thérapie génique serait possible dans ce cas446
.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 59/103
42
5 DISCUSSION
L’expansion fulgurante des connaissances sur la génétique en quelques années, couplée à l’essor technologique, nous a permis d’en savoir davantage au sujet des cancers héréditaires. Ladécouverte et le repérage des gènes en cause dans l’apparition de cancers héréditaires dans les
familles à risque élevé laissent entrevoir la possibilité d’accélérer les processus du diagnostic etde la prise en charge.
Le dépistage génétique, dans toutes ses facettes complexes, suscite un intérêt grandissant de la part du public. Les médias447,448 rapportent que des experts déplorent la situation actuelle auCanada quant au dépistage génétique. Ils proposent que les tests de dépistage des cancershéréditaires, particulièrement le cancer du sein et le cancer colorectal, soient facilement mis à ladisposition de tous les Canadiens dans le cadre de programmes régionaux et provinciaux. Uneenquête effectuée en 2000 auprès de Canadiens révèle que la plupart d’entre eux sont en faveur du dépistage génétique à des fins médicales précises, notamment la détermination du risque detransmission de la maladie aux enfants (91 % des répondants) et du risque futur de souffrir d’uneaffection médicale (91 % des répondants)449. Selon un rapport de Santé Saskatchewan publié en2001, la demande anticipée au regard du dépistage génétique dans le système de santé de la province varie, celle relative au cancer du sein et au cancer colorectal étant considérée de niveaumoyen (le dépistage génétique du cancer du sein représente la demande médicale la pluscourante concernant le dépistage génétique en Saskatchewan)450. Un rapport présentant etévaluant des trousses d’éducation sur le dépistage génétique d’affections d’apparition tardiveconçues à l’intention des omnipraticiens et des patients a été préparé pour le compte de SantéCanada451.
Plusieurs organisations ont précisé des paramètres d’évaluation des tests génétiques et des critères demise en application de ces tests. L’American Society of Clinical Oncology452 est d’avis que le dépistaged’une prédisposition génétique à un cancer ne devrait être offert que lorsque :
• la personne a de lourds antécédents familiaux de cancer ou qu’elle est jeune à l’apparition de lamaladie;
• le test peut être interprété avec justesse;• les résultats influencent la prise en charge médicale du patient ou du membre de la famille.
Pour qu’un test génétique corresponde aux critères du Blue Cross and Blue Shield AssociationTechnology Evaluation Center aux États-Unis453 :
•
il doit avoir été approuvé en bout de ligne par les organismes gouvernementaux appropriés;• les faits scientifiques sont concluants quant à l’effet de la technologie sur l’évolution de l’étatde santé (soit, la validité analytique et clinique);
• il doit améliorer l’issue clinique nette (soit, l’utilité clinique);• il est aussi bénéfique que les solutions de rechange établies;• l’amélioration des résultats cliniques doit être possible hors des milieux de la recherche.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 60/103
43
Les principales préoccupations que suscite le dépistage génétique concernent la validité des testset les problèmes éthiques qu’ils soulèvent pour les professionnels de la santé. Les lignesdirectrices sur le dépistage génétique définies par la Société canadienne de pédiatrie en 2003 sontconformes aux principes éthiques applicables aux services génétiques proposés par l’Organisation mondiale de la Santé454,455. La validité analytique, la validité clinique, l’utilité
clinique et les conséquences sociales constituent les paramètres en vertu desquels sont évaluésles avantages et les risques des tests génétiques456. Comme c’est le cas de n’importe quelleépreuve diagnostique, l’utilité clinique du dépistage génétique varie selon sa capacité à produiredes résultats assurés, qu’ils soient positifs ou négatifs. L’utilité clinique d’un test génétique n’est pas déterminée exclusivement en fonction de son effet sur le patient, mais également en fonctionde son effet sur les membres de la famille. Il se peut que la famille, après avoir pris connaissancedu résultat d’un test, éprouve un apaisement émotif l’incitant à tort à passer outre la surveillancemédicale, ou de l’anxiété et adopte des mesures préventives inutiles. Dans les familles oùl’analyse séquentielle débouche sur des résultats véritablement positifs (détection de la mutation pathogène), la surveillance intensive et la possibilité d’une chirurgie prophylactique peuvent êtreofferts seulement aux personnes porteuses de la mutation. Les membres de la famille exempts de
la mutation pourraient se soumettre à la même surveillance que celle préconisée dans la population en général.
Lorsqu’une mutation germinale est repérée chez une personne atteinte, le dépistage pourrait alorsêtre offert aux membres de la famille à risque. Advenant qu’aucune mutation ne soit déceléechez le membre de la famille atteint, cela n’écarte pas nécessairement la possibilité d’une prédisposition héréditaire au cancer, mais peut vouloir dire que la technologie n’est passuffisamment sensible pour détecter la mutation. Il se pourrait, d’autre part, que la famille soit porteuse d’une mutation d’un gène pour lequel le dépistage génétique n’est pas disponible ouqu’elle soit porteuse d’une mutation d’un gène de prédisposition au cancer encore inconnu. Lerésultat négatif d’un test de détection de mutations peut également tenir au fait que, par hasard, le
cancer dont souffre la personne examinée est sporadique, alors même que d’autres membres decette famille sont porteurs de mutations germinales. Si aucune mutation germinale n’est déceléechez un membre atteint d’une famille, le dépistage ne devrait pas être offert aux membres àrisque. Ceux-ci auraient tout de même un risque accru de cancer en raison de leur histoirefamiliale et devraient se plier à la procédure de dépistage du cancer recommandée dans ce cas.
La pertinence et l’opportunité de la mise en œuvre d’innovations en matière de dépistagegénétique dans le cadre du système de santé soulèvent des questions d’ordre technique etclinique, et des questions ayant trait à la prestation des services.
Les questions d’ordre technique portent sur le rendement du test. Les techniques de laboratoire et
leur validité analytique sont diverses. Le type d’analyses varie selon le gène en question et selonque la mutation dans la famille est connue ou inconnue. Le dépistage génétique chez lesmembres d’une famille où les mutations sont connues est de sensibilité et de spécificité plusélevée que lorsque les mutations sont inconnues. Le séquençage de gène complet est la méthodede validité analytique la plus élevée en principe, mais elle est trop fastidieuse et dispendieuse pour être appliquée couramment en pratique clinique. Il importe d’accélérer le processus de

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 61/103
44
dépistage vu que de vastes études sont nécessaires pour évaluer la susceptibilité de mutations particulières à l’éclosion et la pénétrance des cancers familiaux. Il faut donc disposer detechniques efficientes capables d’analyser un grand nombre de prélèvements en toute diligence.
Les questions d’ordre clinique ou le retentissement clinique du dépistage génétique varie selon
l’avancée thérapeutique possible sous l’angle de l’évolution de l’état de santé sous l’impulsiondu dépistage génétique et les répercussions psychologiques du dépistage. Les corrélations entrele génotype et le phénotype déterminées par le dépistage génétique des cancers familiaux ouvrentla porte à l’amélioration de la prise en charge et du pronostic. Lorsque le test ne peutvéritablement prévoir l’issue clinique ou en l’absence de traitement curatif, le dépistage ne peutréellement se justifier sur les plans médical et social. Le conseil génétique et un suivi appropriéss’avèrent également essentiels.
Les questions ayant trait à la prestation des services portent sur l’assurance de la qualité enlaboratoire, la qualité des programmes de dépistage, le coût des tests, l’organisation et lacoordination des services, la planification de la main-d’œuvre et la formation. Le coût du
dépistage moléculaire peut varier de centaines de dollars à des milliers de dollars. Les régimesd’assurance-maladie peuvent ne pas couvrir les frais du dépistage génétique, considéré commen’étant pas nécessaire.
Si le dépistage moléculaire suppose la détection de nouvelles mutations, son coût s’en trouveaugmenté. Quand la mutation est connue, le dépistage subséquent de cette mutation chez lesmembres de la famille coûte moins cher. Les décideurs devraient tenir compte de cet élément.
Les répercussions économiques d’un test génétique prédictif fluctuent selon la technique, sonapplication et l’effet des résultats sur la prise en charge clinique. Un rapport publié en 2002 par le ministère de la Santé et des Soins de longue durée de l’Ontario 457 précise que l’impact du
dépistage génétique sur les coûts des soins de santé varie en fonction de la répartition proportionnelle des personnes suivant le résultat du test, positif ou négatif, et de la façon dont ces personnes modifient leur schéma de consommation de soins de santé sous l’influence du résultat.Les décisions quant à la couverture des tests génétiques prédictifs sont fondées sur la volonté demettre ces tests à la disposition des personnes qui en ont besoin tout en évitant un usageinapproprié susceptible d’enfler les coûts de façon injustifiée.
Le présent rapport a une portée limitée parce qu’il ne se fonde pas avant tout sur une analysecritique de la qualité de l’information disponible, impossible en raison de la natureobservationnelle des principales études sur le rendement du dépistage génétique. En outre, ladiversité des affections dont il est question ici, ainsi que l’hétérogénéité des groupes de la
population touchés, ont empêché l’analyse quantitative des données.Avant de se prononcer quant à l’intégration de tests particuliers aux services cliniques et aufinancement de ces tests, il faudra obtenir d’autres données sur l’impact du dépistage génétiquesur l’évolution de l’état de santé des patients au regard de chacune des affections et desrenseignements supplémentaires sur le coût du dépistage génétique.
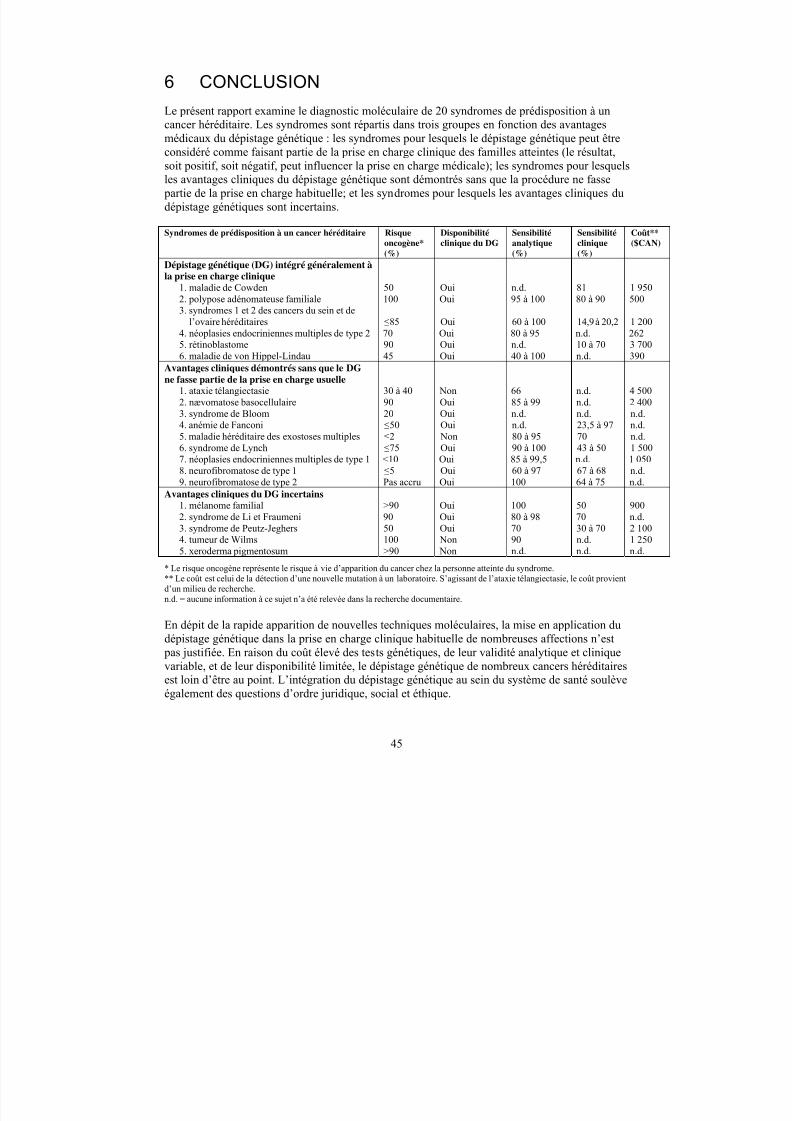
5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 62/103
45
6 CONCLUSION
Le présent rapport examine le diagnostic moléculaire de 20 syndromes de prédisposition à uncancer héréditaire. Les syndromes sont répartis dans trois groupes en fonction des avantagesmédicaux du dépistage génétique : les syndromes pour lesquels le dépistage génétique peut être
considéré comme faisant partie de la prise en charge clinique des familles atteintes (le résultat,soit positif, soit négatif, peut influencer la prise en charge médicale); les syndromes pour lesquelsles avantages cliniques du dépistage génétique sont démontrés sans que la procédure ne fasse partie de la prise en charge habituelle; et les syndromes pour lesquels les avantages cliniques dudépistage génétiques sont incertains.
Syndromes de prédisposition à un cancer héréditaire Risqueoncogène*(%)
Disponibilitéclinique du DG
Sensibilitéanalytique(%)
Sensibilitéclinique(%)
Coût($CA
Dépistage génétique (DG) intégré généralement àla prise en charge clinique
1. maladie de Cowden 50 Oui n.d. 81 1 95
2. polypose adénomateuse familiale 100 Oui 95 à 100 80 à 90 5003. syndromes 1 et 2 des cancers du sein et del’ovaire héréditaires ≤85 Oui 60 à 100 14,9 à 20,2 1 20
4. néoplasies endocriniennes multiples de type 2 70 Oui 80 à 95 n.d. 2625. rétinoblastome 90 Oui n.d. 10 à 70 3 706. maladie de von Hippel-Lindau 45 Oui 40 à 100 n.d. 390
Avantages cliniques démontrés sans que le DGne fasse partie de la prise en charge usuelle
1. ataxie télangiectasie 30 à 40 Non 66 n.d. 4 502. nævomatose basocellulaire 90 Oui 85 à 99 n.d. 2 403. syndrome de Bloom 20 Oui n.d. n.d. n.d.4. anémie de Fanconi ≤50 Oui n.d. 23,5 à 97 n.d.5. maladie héréditaire des exostoses multiples ≤2 Non 80 à 95 70 n.d.6. syndrome de Lynch ≤75 Oui 90 à 100 43 à 50 1 507. néoplasies endocriniennes multiples de type 1 <10 Oui 85 à 99,5 n.d. 1 058. neurofibromatose de type 1 ≤5 Oui 60 à 97 67 à 68 n.d.9. neurofibromatose de type 2 Pas accru Oui 100 64 à 75 n.d.
Avantages cliniques du DG incertains1. mélanome familial >90 Oui 100 50 9002. syndrome de Li et Fraumeni 90 Oui 80 à 98 70 n.d.3. syndrome de Peutz-Jeghers 50 Oui 70 30 à 70 2 104. tumeur de Wilms 100 Non 90 n.d. 1 255. xeroderma pigmentosum >90 Non n.d. n.d. n.d.
* Le risque oncogène représente le risque à vie d’apparition du cancer chez la personne atteinte du syndrome.** Le coût est celui de la détection d’une nouvelle mutation à un laboratoire. S’agissant de l’ataxie télangiectasie, le coût provientd’un milieu de recherche.n.d. = aucune information à ce sujet n’a été relevée dans la recherche documentaire.
En dépit de la rapide apparition de nouvelles techniques moléculaires, la mise en application dudépistage génétique dans la prise en charge clinique habituelle de nombreuses affections n’est pas justifiée. En raison du coût élevé des tests génétiques, de leur validité analytique et cliniquevariable, et de leur disponibilité limitée, le dépistage génétique de nombreux cancers héréditairesest loin d’être au point. L’intégration du dépistage génétique au sein du système de santé soulèveégalement des questions d’ordre juridique, social et éthique.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 63/103
46
7 RÉFÉRENCES1. Cancer epidemiology. In: Schneider K, editor. Counseling about cancer: strategies for genetic
counseling. 2nd ed. New York: Wiley-Liss; 2002. p.1-17.
2. Psychological aspects of cancer counseling. In: Schneider K, editor. Counseling about cancer: strategiesfor genetic counseling. 2nd ed. New York: Wiley-Liss; 2002. p.207-47.
3. Predisposition testing and counseling. In: Schneider K, editor. Counseling about cancer: strategies forgenetic counseling. 2nd ed. New York: Wiley-Liss; 2002. p.249-90.
4. The ethical issues. In: Schneider K, editor. Counseling about cancer: strategies for genetic counseling.2nd ed. New York: Wiley-Liss; 2002. p.291-312.
5. Valentin J. Genetic susceptibility to cancer: ICRP publication 79. Ann ICRP 1998;28(1-2):1-157.
6. Seemayer TA, Cavenee WK. Biology of disease. Molecular mechanisms of oncogenesis. Lab Invest 1989;60(5):585-99.
7. Hsu TC. Genetic predisposition to cancer with special reference to mutagen sensitivity. In Vitro Cell DevBiol 1987;23(9):591-603.
8. Knudson AG, Jr. Genetics of human cancer. Annu Rev Genet 1986;20:231-51.
9. Hussain SP, Harris CC. Molecular epidemiology of human cancer. Recent Results Cancer Res 1998;154:22-36.
10. Lindor NM, Greene MH, Mayo Familial Cancer Program. The concise handbook of family cancer syndromes. J Natl Cancer Inst 1998;90(14):1039-71.
11. Cancer biology. In: Schneider K, editor. Counseling about cancer: strategies for genetic counseling. 2nded. New York: Wiley-Liss; 2002. p.51-71.
12. Grody WW, Noll WW. Molecular diagnosis of genetic diseases. In: Henry JB, editor. Clinical diagnosisand management by laboratory methods. 20th ed. Philadelphia: W.B. Saunders; 2001. p.1372-89.
13. Schneider,K, editor. Counseling about cancer: strategies for genetic counseling. 2nd ed. New York:Wiley-Liss; 2002.
14. PTCH testing for Gorlin syndrome [information sheet]. Rockville (MD): GeneDx; 2002.
15. Gorlin RJ. Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore) 1987;66(2):98-113.
16. Shanley S, Ratcliffe J, Hockey A, Haan E, Oley C, Ravine D, et al. Nevoid basal cell carcinoma syndrome:review of 118 affected individuals. Am J Med Genet 1994;50(3):282-90.
17. Evans DG, Farndon PA, Burnell LD, Gattamaneni HR, Birch JM. The incidence of Gorlin syndrome in 173consecutive cases of medulloblastoma. Br J Cancer 1991;64(5):959-61.
18. Evans GD, Farndon PA. Nevoid basal cell carcinoma syndrome. In: GENETests [database online]. Seattle:University of Washington; 2002. Available:http://www.geneclinics.org/serv1et/access?id=24673&key=Gt3XtrpCoDhr0&gry=INSER.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 64/103
47
19. Marsh DJ, Coulon V, Lunetta KL, Rocca-Serra P, Dahia PL, Zheng Z, et al. Mutation spectrum andgenotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartomasyndromes with germline PTEN mutation. Hum Mol Genet 1998;7(3):507-15.
20. PTEN testing for Cowden Syndrome [information sheet]. Rockville (MD): GeneDx; 2002.
21. Pilarski RT, Hampel H, Charis E. PTEN hamartoma tumor syndrome (PHTS). In: GENETests [databaseonline]. Seattle: University of Washington; 2001. Available:http://www.geneclinics.org/servlet/access?id=24673&key=ZNQx2Z0Y2CWKV&gry=INSERTGRY&fcn=y&fw=L41L&filename=/profiles/phts/details.html (accessed 2003 July).
22. Nelen MR, Kremer H, Konings IB, Schoute F, van Essen AJ, Koch R, et al. Novel PTEN mutations in patients with Cowden disease: absence of clear genotype-phenotype correlations. Eur J Hum Genet 1999;7(3):267-73.
23. Bonneau D, Longy M. Mutations of the human PTEN gene. Hum Mutat 2000;16(2):109-22.
24. Wu G, Wu W, Hegde M, Fawkner M, Chong B, Love D, et al. Detection of sequence variations in theadenomatous polyposis coli ( APC ) gene using denaturing high-performance liquid chromatography.Genet Test 2001;5(4):281-90.
25. Strippoli P, Sarchielli S, Santucci R, Bagnara GP, Brandi G, Biasco G. Cold single-strand conformation polymorphism analysis: optimization for detection of APC gene mutations in patients with familialadenomatous polyposis. Int J Mol Med 2001;8(5):567-72.
26. Chikhaoui Y, Gélinas H, Joseph L, Lance JM. Cost-minimization analysis of genetic testing versus clinicalscreening of at-risk relatives for familial adenomatous polyposis. Int J Technol Assess Health Care 2002;18(1):67-80.
27. Friedl W, Caspari R, Sengteller M, Uhlhaas S, Lamberti C, Jungck M, et al. Can APC mutation analysiscontribute to therapeutic decisions in familial adenomatous polyposis? Experience from 680 FAP families.Gut 2001;48(4):515-21.
28. Bisgaard ML, Fenger K, Bülow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat 1994;3(2):121-5.
29. Järvinen HJ. Epidemiology of familial adenomatous polyposis in Finland: impact of family screening onthe colorectal cancer rate and survival. Gut 1992;33(3):357-60.
30. Burn J, Chapman P, Delhanty J, Wood C, Lalloo F, Cachon-Gonzalez MB, et al. The UK Northern regiongenetic register for familial adenomatous polyposis coli: use of age of onset, congenital hypertrophy of theretinal pigment epithelium, and DNA markers in risk calculations. J Med Genet 1991;28(5):289-96.
31. Solomon CH, Burt RW. Familial adenomatous polyposis. In:GENETests [database online]. Seattle: University of Washington; 2002. Available:http://www.geneclinics.org/servlet/access?id=24673&key=ZNQx2Z0Y2CWKV&gry=INSERTGRY&fcn=y&fw=nAU3&filename=/profiles/fap/details.html (accessed 2003 July).
32. Burt RW, Ward K, Spirio L, Groden J, Joslyn G, Lapport M, et al. Acurate identification of familialadenomatous polyposis coli using newly developed genetic markers. Gastroenterology 1992;102:A347.
33. Powell SM, Petersen GM, Krush AJ, Booker S, Jen J, Giardiello FM, et al. Molecular diagnosis of familialadenomatous polyposis. N Engl J Med 1993;329(27):1982-7.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 65/103
48
34. Powell SM. Direct analysis for familial adenomatous polyposis mutations. Mol Biotechnol 2002;20(2):197-207.
35. Tsao H, Zhang X, Kwitkiwski K, Finkelstein DM, Sober AJ, Haluska FG. Low prevalence of germlineCDKN2A and CDK4 mutations in patients with early-onset melanoma. Arch Dermatol 2000;136(9):1118-22.
36. Lynch HT, Krush AJ. Heredity and malignant melanoma: implications for early cancer detection. CMAJ 1968;99(1):17-21.
37. Fountain JW, Bale SJ, Housman DE, Dracopoli NC. Genetics of melanoma. Cancer Surv 1990;9(4):645-71.
38. p16 Gene testing in familial melanoma [information sheet]. Rockville (MD): GeneDx; 2002.
39. Andrulis IL, Anton-Culver H, Beck J, Bove B, Boyd J, Buys S, et al. Comparison of DNA- and RNA-basedmethods for detection of truncating BRCA1 mutations. Hum Mutat 2002;20(1):65-73.
40. Culver JB, Hull J, Levy-Lahad E, Daly M, Burke W. Breast cancer genetics: an overview. In:
GENEReviews [database online]. Seattle: University of Washington; 2000. Available:http://www.geneclinics.org/servlet/access?db=geneclinics&site=gc&id=24673&key=7GIQ5dFnInN83&gr y=&fcn=y&fw=Memc&filename=/profiles/brca/index.html (accessed 2001 Dec 12).
41. Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE, Breast Cancer Linkage Consortium. Risks of cancer in BRCA1-mutation carriers. Lancet 1994;343(8899):692-5.
42. National Cancer Institute. Genetics of breast and ovarian cancer (PDQ®). Natl Cancer Inst Website 2002.Available: http://www.cancer.gov/cancerinfo/pdq/genetics/breast-and-ovarian (accessed 2002 Aug 16).
43. Breast cancer. In: National Health and Medical Research Council, Australian Cancer Network, editors.Familial aspects of cancer: a guide to clinical practice. Canberra: Commonwealth of Australia; 1999.
p.43-62. Available: http://www.health.gov.au/nhmrc/publications/pdf/cp67.pdf.
44. Jernström H, Lerman C, Ghadirian P, Lynch HT, Weber B, Garber J, et al. Pregnancy and risk of early breast cancer in carriers of BRCA1 and BRCA2. Lancet 1999;354(9193):1846-50.
45. Geisler JP, Hatterman-Zogg MA, Rathe JA, Lallas TA, Kirby P, Buller RE. Ovarian cancer BRCA1mutation detection: protein truncation test (PTT) outperforms single strand conformation polymorphismanalysis (SSCP). Hum Mutat 2001;18(4):337-44.
46. Eng C, Brody LC, Wagner TM, Devilee P, Vijg J, Szabo C, et al. Interpreting epidemiological research: blinded comparison of methods used to estimate the prevalence of inherited mutations in BRCA1. J MedGenet 2001;38(12):824-33.
47. Byrne TJ, Reece MT, Adams LA, Hoffman DE, Lane MA, Cohn GM. A rapid immunoassay predictsBRCA1 and BRCA2 mutations in buccal cells. Oncol Rep 2000;7(6):1203-7.
48. Gross E, Arnold N, Goette J, Schwarz-Boeger U, Kiechle M. A comparison of BRCA1 mutation analysis by direct sequencing, SSCP and DHPLC. Hum Genet 1999;105(1-2):72-8.
49. Chansky HA, Raskind WH. Hereditary multiple exostoses. In:GENETests [database online]. Seattle: University of Washington; 2000. Available:http://www.geneclinics.org/servlet/access?id=24673&key=7GIQ5dFnInN83&gry=INSERTGRY&fcn=y&f w=CjF-&filename=/profiles/ext/details.html (accessed 2003 July).

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 66/103
49
50. Dobson-Stone C, Cox RD, Lonie L, Southam L, Fraser M, Wise C, et al. Comparison of fluorescent single-strand conformation polymorphism analysis and denaturing high-performance liquid chromatography for detection of EXT1 and EXT2 mutations in hereditary multiple exostoses. Eur J Hum Genet 2000;8(1):24-32.
51. Wuyts W, Van Hul W, De Boulle K, Hendrickx J, Bakker E, Vanhoenacker F, et al. Mutations in the EXT1
and EXT2 genes in hereditary multiple exostoses. Am J Hum Genet 1998;62(2):346-54.52. Legeai-Mallet L, Munnich A, Maroteaux P, Le Merrer M. Incomplete penetrance and expressivity skewing
in hereditary multiple exostoses. Clin Genet 1997;52(1):12-6.
53. Philippe C, Porter DE, Emerton ME, Wells DE, Simpson AH, Monaco AP. Mutation screening of theEXT1 and EXT2 genes in patients with hereditary multiple exostoses. Am J Hum Genet 1997;61(3):520-8.
54. Schneider KA, Li F. Li-Fraumeni syndrome. In:GENETests [database online]. Seattle: University of Washington; 1999. Available:http://www.geneclinics.org/servlet/access?id=24673&key=ZNQx2Z0Y2CWKV&gry=INSERTGRY&fcn=y&fw=PUAi&filename=/profiles/li-fraumeni/details.html (accessed 2002 May 29).
55. Schimke RN. Genetic aspects of multiple endocrine neoplasia. Annu Rev Med 1984;35:25-31.
56. Bassett JH, Forbes SA, Pannett AA, Lloyd SE, Christie PT, Wooding C, et al. Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet 1998;62(2):232-44.
57. Larsson C, Shepherd J, Nakamura Y, Blomberg C, Weber G, Werelius B, et al. Predictive testing for multiple endocrine neoplasia type 1 using DNA polymorphisms. J Clin Invest 1992;89(4):1344-9.
58. Friedman JM. Neurofibromatosis 1. In:GENETests [database online]. Seattle: University of Washington; 1998. Available:http://www.geneclinics.org/servlet/access?id=24673&key=7GIQ5dFnInN83&gry=INSERTGRY&fcn=y&f w=Uy7h&filename=/profiles/nf1/details.html (accessed 2002 May 29).
59. Han S, Cooper DN, Upadhyaya M. Evaluation of denaturing high performance liquid chromatography(DHPLC) for the mutational analysis of the neurofibromatosis type 1 ( NF1 ) gene. Hum Genet 2001;109(5):487-97.
60. Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol 2000;151(1):33-40.
61. Lefkowitz IB, Obringer AC, Meadows AT. Neurofibromatosis and cancer: incidence and management. In:Rubenstein AE, Korf BR, editors. Neurofibromatosis: a handbook for patients, families, and health-care professionals. New York: Thieme Medical Publishers; 1990. p.99-110.
62. Shen MH, Harper PS, Upadhyaya M. Molecular genetics of neurofibromatosis type 1 (NF1). J Med Genet 1996;33(1):2-17.
63. Korf BR, Carey JC. Molecular genetics of neurofibromatosis. In: Rubenstein AE, Korf BR, editors.Neurofibromatosis: a handbood for paitents, families, and health-care professionals. New York:Thieme Medical Publishers; 1990. p.178-200.
64. Obringer AC, Meadows AT, Zackai EH. The diagnosis of neurofibromatosis-1 in the child under the age of 6 years. Am J Dis Child 1989;143(6):717-9.
65. Heim RA, Kam-Morgan LN, Binnie CG, Corns DD, Cayouette MC, Farber RA, et al. Distribution of 13truncating mutations in the neurofibromatosis 1 gene. Hum Mol Genet 1995;4(6):975-81.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 67/103
50
66. MacCollin M. Neurofibromatosis 2. In:GENETests [database online]. Seattle: University of Washington; 2001. Available:http://www.geneclinics.org/servlet/access?id=24673&key=7GIQ5dFnInN83&gry=INSERTGRY&fcn=y&f w=Y9eY&filename=/profiles/nf2/details.html (accessed 2002 May 29).
67. Evans DG, Huson SM, Donnai D, Neary W, Blair V, Teare D, et al. A genetic study of type 2
neurofibromatosis in the United Kingdom. I. Prevalence, mutation rate, fitness, and confirmation of maternal transmission effect on severity. J Med Genet 1992;29(12):841-6.
68. MacCollin M, Ramesh V, Jacoby LB, Louis DN, Rubio MP, Pulaski K, et al. Mutational analysis of patients with neurofibromatosis 2. Am J Hum Genet 1994;55(2):314-20.
69. Faudoa R, Xue Z, Lee F, Baser ME, Hung G. Detection of novel NF2 mutations by an RNA mismatchcleavage method. Hum Mutat 2000;15(5):474-8.
70. Amos CI, Frazier ML, McGarrity TJ. Peutz-Jeghers syndrome. In:GENETests [database online]. Seattle: University of Washington; 2002. Available:http://www.geneclinics.org/servlet/access?id=24673&key=7GIQ5dFnInN83&gry=INSERTGRY&fcn=y&f w=gEOk&filename=/profiles/pjs/details.html (accessed 2002 Jan 24).
71. Amos CI, Bali D, Thiel TJ, Anderson JP, Gourley I, Frazier ML, et al. Fine mapping of a genetic locus for Peutz-Jeghers syndrome on chromosome 19p. Cancer Res 1997;57(17):3653-6.
72. Mehenni H, Blouin JL, Radhakrishna U, Bhardwaj SS, Bhardwaj K, Dixit VB, et al. Peutz-Jegherssyndrome: confirmation of linkage to chromosome 19p13.3 and identification of a potential second locus,on 19q13.4. Am J Hum Genet 1997;61(6):1327-34.
73. Spigelman AD, Murday V, Phillips RK. Cancer and the Peutz-Jeghers syndrome. Gut 1989;30(11):1588-90.
74. STK11 testing in Peutz-Jeghers syndrome [information sheet]. Rockville (MD): GeneDx; 2002.
75. Amos CI, Frazier ML, Lynch PM, McGarrity TJ. Genetic studies of Peutz-Jeghers syndrome [abstract].Am J Hum Genet 2000;67:S83.
76. Boardman LA, Couch FJ, Burgart LJ, Schwartz D, Berry R, McDonnell SK, et al. Genetic heterogeneity inPeutz-Jeghers syndrome. Hum Mutat 2000;16(1):23-30.
77. Dhanda RK, van Orsouw NJ, Sigalas I, Eng C, Vijg J. Critical factors in the performance and cost of two-dimensional gene scanning: RB1 as a model. Biotechniques 1998;25(4):664-6, 668, 670, 672-5.
78. Lohmann DR, Bornfeld N, Horsthemke B, Passarge E. Retinoblastoma. In:GENETests [database online]. Seattle: University of Washington; 2000. Available:http://www.geneclinics.org/servlet/access?id=24673&key=7GIQ5dFnInN83&gry=INSERTGRY&fcn=y&f w=gTAc&filename=/profiles/retinoblastoma/details.html (accessed 2002 Jan 24).
79. Moll AC, Kuik DJ, Bouter LM, Den Otter W, Bezemer PD, Koten JW, et al. Incidence and survival of retinoblastoma in the Netherlands: a register based study 1862-1995. Br J Ophthalmol 1997;81(7):559-62.
80. Suckling RD, Fitzgerald PH, Stewart J, Wells E. The incidence and epidemiology of retinoblastoma in NewZealand: a 30-year survey. Br J Cancer 1982;46(5):729-36.
81. Devesa SS. The incidence of retinoblastoma. Am J Ophthalmol 1975;80(2):263-5.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 68/103
51
82. Bunin GR, Emanuel BS, Meadows AT, Buckley JD, Woods WG, Hammond GD. Frequency of 13qabnormalities among 203 patients with retinoblastoma. J Natl Cancer Inst 1989;81(5):370-4.
83. Cohen JG, Dryja TP, Davis KB, Diller LR, Li FP. RB1 genetic testing as a clinical service: a follow-upstudy. Med Pediatr Oncol 2001;37(4):372-8.
84. Lohmann DR, Brandt B, Höpping W, Passarge E, Horsthemke B. The spectrum of RB1 germ-linemutations in hereditary retinoblastoma. Am J Hum Genet 1996;58(5):940-9.
85. Schimke NR, Collins DR, Stolle CA. Von Hippel-Lindau Syndrome. In:GENETests [database online]. Seattle: University of Washington; 2000. Available:http://www.geneclinics.org/servlet/access?id=24673&key=7GIQ5dFnInN83&gry=INSERTGRY&fcn=y&f w=gVmh&filename=/profiles/vhl/details.html (accessed 2003 July).
86. Maher ER, Yates JR, Harries R, Benjamin C, Harris R, Moore AT, et al. Clinical features and naturalhistory of von Hippel-Lindau disease. Q J Med 1990;77(283):1151-63.
87. Glenn GM, Choyke PL, Zbar B, Linehan M. Von Hippel-Lindau disease: clinical review and molecular genetics. Probl Urol 1990;4:312-30.
88. Shuin T, Kondo K, Ashida S, Okuda H, Yoshida M, Kanno H, et al. Germline and somatic mutations invon Hippel-Lindau disease gene and its significance in the development of kidney cancer. ContribNephrol 1999;128:1-10.
89. Atuk NO, Stolle C, Owen JA, Carpenter JT, Vance ML. Pheochromocytoma in von Hippel-Lindau disease:clinical presentation and mutation analysis in a large, multigenerational kindred. J Clin Endocrinol Metab 1998;83(1):117-20.
90. Richards FM, Webster AR, McMahon R, Woodward ER, Rose S, Maher ER. Molecular genetic analysis of von Hippel-Lindau disease. J Intern Med 1998;243(6):527-33.
91. Maher ER, Iselius L, Yates JR, Littler M, Benjamin C, Harris R, et al. Von Hippel-Lindau disease: agenetic study. J Med Genet 1991;28(7):443-7.
92. Ellis LA, Taylor CF, Taylor GR. A comparison of fluorescent SSCP and denaturing HPLC for highthroughput mutation scanning. Hum Mutat 2000;15(6):556-64.
93. Mattocks C, Tarpey P, Bobrow M, Whittaker J. Comparative sequence analysis (CSA): a new sequence- based method for the identification and characterization of mutations in DNA. Hum Mutat 2000;16(5):437-43.
94. Klein B, Weirich G, Brauch H. DHPLC-based germline mutation screening in the analysis of the VHLtumor suppressor gene: usefulness and limitations. Hum Genet 2001;108(5):376-84.
95. Hernández A, Meyer A, Enczmann J, Ackermann R, Wernet P. Establishment of experimental conditionsfor the rapid detection of mutations in the von Hippel-Lindau gene by parallel temperature gradient gelelectrophoresis. Electrophoresis 1999;20(10):1958-61.
96. Stolle C, Glenn G, Zbar B, Humphrey JS, Choyke P, Walther M, et al. Improved detection of germlinemutations in the von Hippel-Lindau disease tumor suppressor gene. Hum Mutat 1998;12(6):417-23.
97. Little MH, Prosser J, Condie A, Smith PJ, Van Heyningen V, Hastie ND. Zinc finger point mutationswithin the WT1 gene in Wilms tumor patients. Proc Natl Acad Sci U S A 1992;89(11):4791-5.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 69/103
52
98. Feinberg AP. A developmental context for multiple genetic alterations in Wilms' tumor. J Cell Sci 1994;107 Suppl 18:7-12.
99. Schwartz CE, Haber DA, Stanton VP, Strong LC, Skolnick MH, Housman DE. Familial predisposition toWilms tumor does not segregate with the WT1 gene. Genomics 1991;10(4):927-30.
100. Breslow NE, Beckwith JB. Epidemiological features of Wilms' tumor: results of the National Wilms'Tumor Study. J Natl Cancer Inst 1982;68(3):429-36.
101. Babaian RJ, Skinner DG, Waisman J. Wilms' tumor in the adult patient: diagnosis, management, andreview of the world medical literature. Cancer 1980;45(7):1713-9.
102. Hata J. Wilms' tumor and the WT1 gene. Contrib Nephrol 1999;128:62-74.
103. Wiesner GL, Snow K. Multiple endocrine neoplasia type 2. In: GENETests [database online]. Seattle:University of Washington; 1999. Available:http://www.geneclinics.org/servlet/access?db=geneclinics&site=gc&id=24673&key=7GIQ5dFnInN83&gr y=&fcn=y&fw=NHyw&filename=/profiles/men2/index.html (accessed 2002 Jan 25).
104. RET gene testing in multiple endocrine neoplasia 2B [information sheet]. Rockville (MD): GeneDx;2002.
105. Jackson CE, Norum RA. Genetic mechanisms of neoplasia in MEN 2. Henry Ford Hosp Med J 1989;37(3-4):116-9.
106. Raue F, Frank-Raue K, Grauer A. Multiple endocrine neoplasia type 2. Clinical features and screening.Endocrinol Metab Clin North Am 1994;23(1):137-56.
107. Ponder BA. Multiple endocrine neoplasia type 2. In: Vogelstein B, Kinzler KW, editors. The genetic basisof human cancer. New York: McGraw-Hill; 1998. p.475-87.
108. Calender A. Genetic testing in multiple endocrine neoplasia and related syndromes. Forum (Genoa, Italy) 1998;8(2):146-59.
109. Hansford JR, Mulligan LM. Multiple endocrine neoplasia type 2 and RET: from neoplasia to neurogenesis.J Med Genet 2000;37(11):817-27.
110. Xue F, Yu H, Maurer LH, Memoli VA, Nutile-McMenemy N, Schuster MK, et al. Germline RETmutations in MEN 2A and FMTC and their detection by simple DNA diagnostic tests. Hum Mol Genet 1994;3(4):635-8.
111. Kambouris M, Jackson CE, Feldman GL. Diagnosis of multiple endocrine neoplasia [MEN] 2A, 2B andfamilial medullary thyroid cancer [FMTC] by multiplex PCR and heteroduplex analyses of RET proto-oncogene mutations. Hum Mutat 1996;8(1):64-70.
112. Morrison PJ, Nevin NC. Multiple endocrine neoplasia type 2B (mucosal neuroma syndrome, Wagenmann-Froboese syndrome). J Med Genet 1996;33(9):779-82.
113. Ceccherini I, Hofstra RM, Luo Y, Stulp RP, Barone V, Stelwagen T, et al. DNA polymorphisms andconditions for SSCP analysis of the 20 exons of the ret proto-oncogene. Oncogene 1994;9(10):3025-9.
114. Vieira AEF, Mello MP, Elias LLK, Lau IF, Maciel LMZ, Moreira AC, et al. Molecular and biochemicalscreening for the diagnosis and management of medullary thyroid carcinoma in multiple endocrineneoplasia type 2A. Horm Metab Res 2002;34(4):202-6.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 70/103
53
115. Decker RA, Peacock ML. Update on the profile of multiple endocrine neoplasia type 2a RET mutations: practical issues and implications for genetic testing. Cancer 1997;80(3 Suppl):557-68.
116. Xia L, Shen W, Ritacca F, Mitri A, Madlensky L, Berk T, et al. A truncated hMSH2 transcript occurs as acommon variant in the population: implications for genetic diagnosis. Cancer Res 1996;56(10):2289-92.
117. Wikman FP, Katballe N, Christensen M, Laurberg S, Ørntoft TF. Efficient mutation detection in mismatchrepair genes using a combination of single-strand conformational polymorphism and heteroduplex analysisat a controlled temperature. Genet Test 2000;4(1):15-21.
118. Swift M. Genetics and epidemiology of ataxia-telangiectasia. In: Gatti RA, Swift M, editors. Ataxia-telangiectasia: genetics, neuropathology, and immunology of a degenerative disease of childhood.
New York: Alan R. Liss; 1985. p.133-46.
119. Swift M. Genetic aspects of ataxia-telangiectasia. Immunodefic Rev 1990;2(1):67-81.
120. Taylor AMR, McConville CM, Woods GW, Byrd PJ, Hernandez D. Clinical and cellular heterogeneity inataxia-telangiectasia. In: Gatti RA, Painter RB, editors. Ataxia-telangiectasia. Vol H 77 [NATO ASISeries]. Heidelberg: Springer-Verlag; 1993. p.209-31.
121. Swift M, Chase CL, Morrell D. Cancer predisposition of ataxia-telangiectasia heterozygotes. CancerGenet Cytogenet 1990;46(1):21-7.
122. Telatar M, Teraoka S, Wang Z, Chun HH, Liang T, Castellvi-Bel S, et al. Ataxia-telangiectasia:identification and detection of founder-effect mutations in the ATM gene in ethnic populations. Am J HumGenet 1998;62(1):86-97.
123. German J. Bloom's syndrome: incidence, age of onset, and types of leukemia in the Bloom's syndromeregistry. In: Bartsocas CS, Loukopoulos D, editors. Genetics of hematological disorders. Washington;
New York: Hemisphere Publishers; 1992. p.241-58.
124. Oddoux C, Clayton CM, Nelson HR, Ostrer H. Prevalence of Bloom syndrome heterozygotes amongAshkenazi Jews [letter]. Am J Hum Genet 1999;64(4):1241-3.
125. Straughen JE, Johnson J, McLaren D, Proytcheva M, Ellis N, German J, et al. A rapid method for detectingthe predominant Ashkenazi Jewish mutation in the Bloom's syndrome gene. Hum Mutat 1998;11(2):175-8.
126. Ellis NA, German J. Molecular genetics of Bloom's syndrome. Hum Mol Genet 1996;5 Spec No:1457-63.
127. Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, Die-Smulders C, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002;297(5581):606-9.
128. Joenje H, Patel KJ. The emerging genetic and molecular basis of Fanconi anaemia. Nat Rev Genet 2001;2(6):446-57.
129. Carreau M, Buchwald M. Fanconi's anemia: What have we learned from the genes so far? Mol Med Today 1998;4(5):201-6.
130. Pulsipher M, Kupfer GM, Naf D, Suliman A, Lee JS, Jakobs P, et al. Subtyping analysis of Fanconi anemia by immunoblotting and retroviral gene transfer. Mol Med 1998;4(7):468-79.
131. Levran O, Erlich T, Magdalena N, Gregory JJ, Batish SD, Verlander PC, et al. Sequence variation in theFanconi anemia gene FAA. Proc Natl Acad Sci U S A 1997;94(24):13051-6.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 71/103
54
132. Whitney MA, Saito H, Jakobs PM, Gibson RA, Moses RE, Grompe M. A common mutation in the FACCgene causes Fanconi anaemia in Ashkenazi Jews. Nat Genet 1993;4(2):202-5.
133. Morgan NV, Tipping AJ, Joenje H, Mathew CG. High frequency of large intragenic deletions in theFanconi anemia group A gene. Am J Hum Genet 1999;65(5):1330-41.
134. Cleaver JE, Thompson LH, Richardson AS, States JC. A summary of mutations in the UV-sensitivedisorders: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. Hum Mutat 1999;14(1):9-22.
135. Nance MA, Berry SA. Cockayne syndrome: review of 140 cases. Am J Med Genet 1992;42(1):68-84.
136. Kobayashi T, Kuraoka I, Saijo M, Nakatsu Y, Tanaka A, Someda Y, et al. Mutations in the XPD geneleading to xeroderma pigmentosum symptoms. Hum Mutat 1997;9(4):322-31.
137. McDowell ML, Nguyen T, Cleaver JE. A single-site mutation in the XPAC gene alters photoproductrecognition. Mutagenesis 1993;8(2):155-61.
138. Cleaver JE, McDowell M, Jones C, Wood R, Karentz D. Mutation and expression of the XPA gene in
revertants and hybrids of a xeroderma pigmentosum cell line. Som Cell Mol Genet 1994;20(4):327-37.139. Moriwaki S, Nishigori C, Imamura S, Yagi T, Takahashi C, Fujimoto N, et al. A case of xeroderma
pigmentosum complementation group F with neurological abnormalities. Br J Dermatol 1993;128(1):91-4.
140. Flejter WL, McDaniel LD, Askari M, Friedberg EC, Schultz RA. Characterization of a complexchromosomal rearrangement maps the locus for in vitro complementation of xeroderma pigmentosumgroup D to human chromosome band 19q13. Genes Chromosomes Cancer 1992;5(4):335-42.
141. Swift M, Morrell D, Cromartie E, Chamberlin AR, Skolnick MH, Bishop DT. The incidence and genefrequency of ataxia-telangiectasia in the United States. Am J Hum Genet 1986;39(5):573-83.
142. Shiloh Y. Ataxia-telangiectasia: closer to unraveling the mystery. Eur J Hum Genet 1995;3(2):116-38.
143. Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia telangiectasia genewith a product similar to PI-3 kinase. Science 1995;268(5218):1749-53.
144. Gatti RA, Berkel I, Boder E, Braedt G, Charmley P, Concannon P, et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22-23. Nature 1988;336(6199):577-80.
145. Meyn MS. Ataxia-telangiectasia, cancer and the pathobiology of the ATM gene. Clin Genet 1999;55(5):289-304.
146. Soares HD, Morgan J, I, McKinnon PJ. Atm expression patterns suggest a contribution from the peripheralnervous system to the phenotype of ataxia-telangiectasia. Neuroscience 1998;86(4):1045-54.
147. Brown KD, Tagle DA. Molecular perspectives on cancer, the cell cycle and the inherited disorder ataxia-telangiectasia. Prog Clin Biol Res 1997;396:101-13.
148. Jeggo PA, Carr AM, Lehmann AR. Splitting the ATM: distinct repair and checkpoint defects in ataxia-telangiectasia. Trends Genet 1998;14(8):312-6.
149. Shackelford RE, Innes CL, Sieber SO, Heinloth AN, Leadon SA, Paules RS. The Ataxia telangiectasiagene product is required for oxidative stress-induced G1 and G2 checkpoint function in human fibroblasts.J Biol Chem 2001;276(24):21951-9.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 72/103
55
150. Ward AJ, Olive PL, Burr AH, Rosin MP. Response of fibroblast cultures from ataxia-telangiectasia patientsto reactive oxygen species generated during inflammatory reactions. Environ Mol Mutagen 1994;24(2):103-11.
151. Tribius S, Pidel A, Casper D. ATM protein expression correlates with radioresistance in primaryglioblastoma cells in culture. Int J Radiat Oncol Biol Phys 2001;50(2):511-23.
152. Ramsay J, Birrell G, Baumann K, Bodero A, Parsons P, Lavin M. Radiosensitive melanoma cell line withmutation of the gene for ataxia telangiectasia. Br J Cancer 1998;77(1):11-4.
153. Bundey S. Clinical and genetic features of ataxia-telangiectasia. Int J Radiat Biol 1994;66(6):S23-S29.
154. Haidar MA, Kantarjian H, Manshouri T, Chang CY, O'Brien S, Freireich E, et al. ATM gene deletion in patients with adult acute lymphoblastic leukemia. Cancer 2000;88(5):1057-62.
155. Rodriguez C, Theillet C. Ataxie-télangiectasie et cancer : une question ouverte. Bull Cancer 1997;84(7):763-6.
156. Uhrhammer N, Bay JO, Bignon YJ. Seventh international workshop on ataxia-telangiectasia. Cancer Res
1998;58(15):3480-5.157. Concannon P, Gatti RA. Diversity of ATM gene mutations detected in patients with ataxia-telangiectasia.
Hum Mutat 1997;10(2):100-7.
158. Gilad S, Khosravi R, Harnik R, Ziv Y, Shkedy D, Galanty Y, et al. Identification of ATM mutations usingextended RT-PCR and restriction endonuclease fingerprinting, and elucidation of the repertoire of A-Tmutations in Israel. Hum Mutat 1998;11(1):69-75.
159. Vorechovsky I, Luo L, Lindblom A, Negrini M, Webster AD, Croce CM, et al. ATM mutations in cancer families. Cancer Res 1996;56(18):4130-3.
160. Hacia JG, Sun B, Hunt N, Edgemon K, Mosbrook D, Robbins C, et al. Strategies for mutational analysis of the large multiexon ATM gene using high-density oligonucleotide arrays. Genome Res 1998;8(12):1245-58.
161. Swift M, Morrell D, Massey RB, Chase CL. Incidence of cancer in 161 families affected by ataxia-telangiectasia. N Engl J Med 1991;325(26):1831-6.
162. Busch D. Genetic susceptibility to radiation and chemotherapy injury: diagnosis and management. Int JRadiat Oncol Biol Phys 1994;30(4):997-1002.
163. Shayeghi M, Seal S, Regan J, Collins N, Barfoot R, Rahman N, et al. Heterozygosity for mutations in theataxia telangiectasia gene is not a major cause of radiotherapy complications in breast cancer patients. Br JCancer 1998;78(7):922-7.
164. Wicking C, Shanley S, Smyth I, Gillies S, Negus K, Graham S, et al. Most germ-line mutations in thenevoid basal cell carcinoma syndrome lead to a premature termination of the PATCHED protein, and nogenotype-phenotype correlations are evident. Am J Hum Genet 1997;60(1):21-6.
165. German J. Bloom's syndrome. I. Genetical and clinical observations in the first twenty-seven patients. AmJ Hum Genet 1969;21(2):196-227.
166. Passarge E. Bloom's syndrome: the German experience. Ann Genet 1991;34(3-4):179-97.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 73/103
56
167. German J, Bloom D, Passarge E. Bloom's syndrome XI. Progress report for 1983. Clin Genet 1984;25(2):166-74.
168. Kaneko H, Inoue R, Fukao T, Kasahara K, Tashita H, Teramoto T, et al. Two Japanese siblings with Bloomsyndrome gene mutation and B-cell lymphoma. Leuk Lymphoma 1997;27(5-6):539-42.
169. Imamura O, Fujita K, Shimamoto A, Tanabe H, Takeda S, Furuichi Y, et al. Bloom helicase is involved inDNA surveillance in early S phase in vertebrate cells. Oncogene 2001;20(10):1143-51.
170. Marsh DJ, Dahia PL, Coulon V, Zheng Z, Dorion-Bonnet F, Call KM, et al. Allelic imbalance, includingdeletion of PTEN/MMACI, at the Cowden disease locus on 10q22-23, in hamartomas from patients withCowden syndrome and germline PTEN mutation. Genes Chromosomes Cancer 1998;21(1):61-9.
171. Perren A, Weng LP, Boag AH, Ziebold U, Thakore K, Dahia PLM, et al. Immunohistochemical evidenceof loss of PTEN expression in primary ductal adenocarcinomas of the breast. Am J Pathol 1999;155(4):1253-60.
172. Starink TM, van der Veen JP, Arwert F, de Waal LP, de Lange GG, Gille JJ, et al. The Cowden syndrome:a clinical and genetic study in 21 patients. Clin Genet 1986;29(3):222-33.
173. Fackenthal JD, Marsh DJ, Richardson AL, Cummings SA, Eng C, Robinson BG, et al. Male breast cancer in Cowden syndrome patients with germline PTEN mutations. J Med Genet 2001;38(3):159-64.
174. Haibach H, Burns TW, Carlson HE, Burman KD, Deftos LJ. Multiple hamartoma syndrome (Cowden'sdisease) associated with renal cell carcinoma and primary neuroendocrine carcinoma of the skin (Merkelcell carcinoma). Am J Clin Pathol 1992;97(5):705-12.
175. Brownstein MH, Wolf M, Bikowski JB. Cowden's disease: a cutaneous marker of breast cancer. Cancer 1978;41(6):2393-8.
176. Fan X, Furnari FB, Cavenee WK, Castresana JS. Non-isotopic silver-stained SSCP is more sensitive thanautomated direct sequencing for the detection of PTEN mutations in a mixture of DNA extracted fromnormal and tumor cells. Int J Oncol 2001;18(5):1023-6.
177. Marsh DJ, Kum JB, Lunetta KL, Bennett MJ, Gorlin RJ, Ahmed SF, et al. PTEN mutation spectrum andgenotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity withCowden syndrome. Hum Mol Genet 1999;8(8):1461-72.
178. Scala S, Bruni P, Lo Muzio L, Mignogna M, Viglietto G, Fusco A. Novel mutation of the PTEN gene in anItalian Cowden's disease kindred. Int J Oncol 1998;13(4):665-8.
179. Zhou XP, Gimm O, Hampel H, Niemann T, Walker MJ, Eng C. Epigenetic PTEN silencing in malignantmelanomas without PTEN mutation. Am J Pathol 2000;157(4):1123-8.
180. Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, et al. Germline mutations of the PTEN gene inCowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 1997;16(1):64-7.
181. Zhou XP, Marsh DJ, Hampel H, Mulliken JB, Gimm O, Eng C. Germline and germline mosaic PTENmutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry,arteriovenous malformations and lipomatosis. Hum Mol Genet 2000;9(5):765-8.
182. Eng C. Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet 2000;37(11):828-30.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 74/103
57
183. Eng C. Genetics of Cowden syndrome: through the looking glass of oncology (Review). Int J Oncol 1998;12(3):701-10.
184. Chung DC. The genetic basis of colorectal cancer: insights into critical pathways of tumorigenesis.Gastroenterology 2000;119(3):854-65.
185. Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol 2001;3(4):433-8.
186. Tomlinson I, Ilyas M, Novelli M. Molecular genetics of colon cancer. Cancer Metastasis Rev 1997;16(1-2):67-79.
187. Ahnen DJ, Lynch KL. Colorectal cancer screening in average- and high-risk groups. Adv Intern Med 2001;46:77-106.
188. Grandjouan S. Données pratiques en oncogénétique digestive. Ann Med Interne (Paris) 2001;152(4):262-6.
189. Canadian Task Force on Preventive Health Care. Colorectal cancer screening. Recommendation statementfrom the Canadian Task Force on Preventive Health Care. CMAJ 2001;165(2):206-8.
190. Villa E. Molecular screening for colon cancer detection. Dig Liver Dis 2000;32(2):173-7.
191. Kahmann S, Herter P, Kuhnen C, Müller K-M, Muhr G, Martin D, et al. A non-radioactive protein truncationtest for the sensitive detection of all stop and frameshift mutations. Hum Mutat 2002;19(2):165-72.
192. Giardiello FM, Brensinger JD, Petersen GM, Luce MC, Hylind LM, Bacon JA, et al. The use andinterpretation of commercial APC gene testing for familial adenomatous polyposis. N Engl J Med 1997;336(12):823-7.
193. Burt RW. Colon cancer screening. Gastroenterology 2000;119(3):837-53.
194. Demols A, Van Laethem JL, Gay F, Franchimont D, Adler M, Van Gossum A. Dépistage individuel ducancer colorectal : quelle stratégie pour quel patient ? Rev Med Brux 2001;22(4):A203-A209.
195. Ahlquist DA, Shuber AP. Stool screening for colorectal cancer: evolution from occult blood to molecular markers. Clin Chim Acta 2002;315(1-2):157-68.
196. Bapat B, Noorani H, Cohen Z, Berk T, Mitri A, Gallie B, et al. Cost comparison of predictive genetictesting versus conventional clinical screening for familial adenomatous polyposis. Gut 1999;44(5):698-703.
197. Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, et al. APC mutationsoccur early during colorectal tumorigenesis. Nature 1992;359(6392):235-7.
198. Colorectal cancer. In: National Health and Medical Research Council, Australian Cancer Network, editors.Familial aspects of cancer: a guide to clinical practice. Canberra: Commonwealth of Australia; 1999.
p.63-76. Available: http://www.health.gov.au/nhmrc/publications/pdf/cp67.pdf.
199. American Gastroenterological Association medical position statement: hereditary colorectal cancer andgenetic testing. Gastroenterology 2001;121(1):195-7.
200. Wallis YL, Morton DG, McKeown CM, Macdonald F. Molecular analysis of the APC gene in 205families: extended genotype-phenotype correlations in FAP and evidence for the role of APC amino acidchanges in colorectal cancer predisposition. J Med Genet 1999;36(1):14-20.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 75/103
58
201. Nagase H, Miyoshi Y, Horii A, Aoki T, Ogawa M, Utsunomiya J, et al. Correlation between the location of germ-line mutations in the APC gene and the number of colorectal polyps in familial adenomatous
polyposis patients. Cancer Res 1992;52(14):4055-7.
202. Wu JS, Paul P, McGannon EA, Church JM. APC genotype, polyp number, and surgical options in familialadenomatous polyposis. Ann Surg 1998;227(1):57-62.
203. Caspari R, Olschwang S, Friedl W, Mandl M, Boisson C, Böker T, et al. Familial adenomatous polyposis:desmoid tumours and lack of ophthalmic lesions (CHRPE) associated with APC mutations beyond codon1444. Hum Mol Genet 1995;4(3):337-40.
204. Gebert JF, Dupon C, Kadmon M, Hahn M, Herfarth C, von Knebel Doeberitz M, et al. Combinedmolecular and clinical approaches for the identification of families with familial adenomatous polyposiscoli. Ann Surg 1999;229(3):350-61.
205. Spirio L, Olschwang S, Groden J, Robertson M, Samowitz W, Joslyn G, et al. Alleles of the APC gene: anattenuated form of familial polyposis. Cell 1993;75(5):951-7.
206. Giardiello FM, Brensinger JD, Petersen GM. AGA technical review on hereditary colorectal cancer andgenetic testing. Gastroenterology 2001;121(1):198-213.
207. Vasen HF, van der Luijt RB, Slors JF, Buskens E, de Ruiter P, Baeten CG, et al. Molecular genetic tests asa guide to surgical management of familial adenomatous polyposis. Lancet 1996;348(9025):433-5.
208. Wallace DC, Exton LA, McLeod GR. Genetic factor in malignant melanoma. Cancer 1971;27(5):1262-6.
209. Goldstein AM, Fraser MC, Struewing JP, Hussussian CJ, Ranade K, Zametkin DP, et al. Increased risk of pancreatic cancer in melanoma-prone kindreds with p16INK4 mutations. N Engl J Med 1995;333(15):970-4.
210. Smith FE, Henly WS, Knox JM, Lane M. Familial melanoma. Arch Intern Med 1966;117(6):820-3.
211. Wallace DC, Beardmore GL, Exton LA. Familial malignant melanoma. Ann Surg 1973;177(1):15-20.
212. Kopf AW, Hellman LJ, Rogers GS, Gross DF, Rigel DS, Friedman RJ, et al. Familial malignant melanoma.JAMA 1986;256(14):1915-9.
213. Anderson DE, Smith JL, Jr., McBride CM. Hereditary aspects of malignant melanoma. JAMA 1967;200(9):741-6.
214. Orlow I, Roy P, Barz A, Canchola R, Song Y, Berwick M. Validation of denaturing high performanceliquid chromatography as a rapid detection method for the identification of human INK4A gene mutations.J Mol Diagn 2001;3(4):158-63.
215. Nagore E, Climent J, Planelles MD, Ledesma E, Rubio-Moscardo F, Fortea JM, et al. Analysis of theCDKN2A and CDK4 genes and HLA-DR and HLA-DQ alleles in two Spanish familial melanomakindreds. Acta Derm Venereol 2000;80(6):440-2.
216. Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia. Blood 2003;101(3):822-6.
217. Evans DG, Rees HC, Spreadborough A, Campbell DJ, Gau GS, Pickering E, et al. Radial ray defects, renalectopia, duodenal atresia and hydrocephalus: the extended spectrum for Fanconi anaemia. ClinDysmorphol 1994;3(3):200-6.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 76/103
59
218. Frikha M, Mseddi S, Elloumi M, Bouaziz M, Khanfir A, Mnif J, et al. La maladie de Fanconi : étude de 43cas dans le sud tunisien. Arch Pediatr 1998;5(11):1200-5.
219. Timmers C, Taniguchi T, Hejna J, Reifsteck C, Lucas L, Bruun D, et al. Positional cloning of a novelFanconi anemia gene, FANCD2. Mol Cell 2001;7(2):241-8.
220. Fanconi Anaemia/Breast Cancer Consortium. Positional cloning of the Fanconi anaemia group A gene. NatGenet 1996;14(3):324-8.
221. Whitney M, Thayer M, Reifsteck C, Olson S, Smith L, Jakobs PM, et al. Microcell mediated chromosometransfer maps the Fanconi anaemia group D gene to chromosome 3p. Nat Genet 1995;11(3):341-3.
222. Moustacchi E. Biologie cellulaire et moléculaire de l'anémie de Fanconi. MS Med Sci 1994;10(10):979-85.
223. Dos Santos CC, Gavish H, Buchwald M. Fanconi anemia revisited: old ideas and new advances. StemCells 1994;12(2):142-53.
224. Buchwald M. Complementation groups: One or more per gene? Nat Genet 1995;11(3):228-30.
225. Joenje H, Oostra AB, Wijker M, di Summa FM, van Berkel CG, Rooimans MA, et al. Evidence for at leasteight Fanconi anemia genes. Am J Hum Genet 1997;61(4):940-4.
226. Joenje H, Lo ten Foe JR, Oostra AB, van Berkel CG, Rooimans MA, Schroeder-Kurth T, et al.Classification of Fanconi anemia patients by complementation analysis: evidence for a fifth geneticsubtype. Blood 1995;86(6):2156-60.
227. Siddique MA, Nakanishi K, Taniguchi T, Grompe M, D'Andrea AD. Function of the Fanconi anemia pathway in Fanconi anemia complementation group F and D1 cells. Exp Hematol 2001;29(12):1448-55.
228. Tipping AJ, Mathew CG. Advances in the genetics and biology of Fanconi anaemia. Hematology 2000;5(1):1-13.
229. Pearson T, Jansen S, Havenga C, Stones DK, Joubert G. Fanconi anemia: a statistical evaluation of cytogenetic results obtained from South African families. Cancer Genet Cytogenet 2001;126(1):52-5.
230. Carreau M, Alon N, Bosnoyan-Collins L, Joenje H, Buchwald M. Drug sensitivity spectra in Fanconianemia lymphoblastoid cell lines of defined complementation groups. Mutat Res 1999;435(1):103-9.
231. Demuth I, Wlodarski M, Tipping AJ, Morgan NV, de Winter JP, Thiel M, et al. Spectrum of mutations inthe Fanconi anaemia group G gene, FANCG/XRCC9. Eur J Hum Genet 2000;8(11):861-8.
232. Wijker M, Morgan NV, Herterich S, van Berkel CG, Tipping AJ, Gross HJ, et al. Heterogeneous spectrumof mutations in the Fanconi anaemia group A gene. Eur J Hum Genet 1999;7(1):52-9.
233. Nakanishi K, Moran A, Hays T, Kuang Y, Fox E, Garneau D, et al. Functional analysis of patient-derivedmutations in the Fanconi anemia gene, FANCG/XRCC9. Exp Hematol 2001;29(7):842-9.
234. Yamashita T, Wu N, Kupfer G, Corless C, Joenje H, Grompe M, et al. Clinical variability of Fanconianemia (type C) results from expression of an amino terminal truncated Fanconi anemia complementationgroup C polypeptide with partial activity. Blood 1996;87(10):4424-32.
235. Yunis JJ, Brunning RD. Prognostic significance of chromosomal abnormalities in acute leukaemias andmyelodysplastic syndromes. Clin Haematol 1986;15(3):597-620.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 77/103
60
236. Thurston VC, Ceperich TM, Vance GH, Heerema NA. Detection of monosomy 7 in bone marrow byfluorescence in situ hybridization. A study of Fanconi anemia patients and review of the literature. CancerGenet Cytogenet 1999;109(2):154-60.
237. Walsh CE, Grompe M, Vanin E, Buchwald M, Young NS, Nienhuis AW, et al. A functionally activeretrovirus vector for gene therapy in Fanconi anemia group C. Blood 1994;84(2):453-9.
238. Claus EB, Risch N, Thompson WD. Autosomal dominant inheritance of early-onset breast cancer.Implications for risk prediction. Cancer 1994;73(3):643-51.
239. Biesecker BB, Boehnke M, Calzone K, Markel DS, Garber JE, Collins FS, et al. Genetic counseling for families with inherited susceptibility to breast and ovarian cancer. JAMA 1993;269(15):1970-4.
240. Newman B, Mu H, Butler LM, Millikan RC, Moorman PG, King MC. Frequency of breast cancer attributable to BRCA1 in a population-based series of American women. JAMA 1998;279(12):915-21.
241. Szabo CI, King MC. Inherited breast and ovarian cancer. Hum Mol Genet 1995;4 Spec No:1811-7.
242. Easton DF, Bishop DT, Ford D, Crockford GP, Breast Cancer Linkage Consortium. Genetic linkage
analysis in familial breast and ovarian cancer: results from 214 families. Am J Hum Genet 1993;52(4):678-701.
243. Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995;378(6559):789-92.
244. Tavtigian SV, Simard J, Rommens J, Couch F, Shattuck-Eidens D, Neuhausen S, et al. The completeBRCA2 gene and mutations in chromosome 13q-linked kindreds. Nat Genet 1996;12(3):333-7.
245. Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science 1994;265:2088-90.
246. Arver B, Claro A, Langerød A, Børresen-Dale AL, Lindblom A. BRCA1 screening in patients with afamily history of breast or ovarian cancer. Genet Test 1999;3(2):223-6.
247. Ganguly A, Leahy K, Marshall AM, Dhulipala R, Godmilow L, Ganguly T. Genetic testing for breastcancer susceptibility: frequency of BRCA1 and BRCA2 mutations. Genet Test 1997;1(2):85-90.
248. Peto J, Easton DF, Matthews FE, Ford D, Swerdlow AJ. Cancer mortality in relatives of women with breastcancer: the OPCS Study (Office of Population Censuses and Surveys). Int J Cancer 1996;65(3):275-83.
249. Szabo CI, King MC. Population genetics of BRCA1 and BRCA2. Am J Hum Genet 1997;60(5):1013-20.
250. Brose MS, Rebbeck TR, Calzone KA, Stopfer JE, Nathanson KL, Weber BL. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J Natl Cancer Inst 2002;94(18):1365-72.
251. Thompson D, Easton DF, Breast Cancer Linkage Consortium. Cancer incidence in BRCA1 mutationcarriers. J Natl Cancer Inst 2002;94(18):1358-65.
252. Aziz S, Kuperstein G, Rosen B, Cole D, Nedelcu R, McLaughlin J, et al. A genetic epidemiological studyof carcinoma of the fallopian tube. Gynecol Oncol 2001;80(3):341-5.
253. Struewing JP, Abeliovich D, Peretz T, Avishai N, Kaback MM, Collins FS, et al. The carrier frequency of the BRCA1 185delAG mutation is about 1 percent in Ashkenazi Jewish individuals. Nat Genet 1995;11(2):198-200.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 78/103
61
254. Roa BB, Boyd AA, Volcik K, Richards CS. Ashkenazi Jewish population frequencies for commonmutations in BRCA1 and BRCA2. Nat Genet 1996;14(2):185-7.
255. Lalloo F, Cochrane S, Bulman B, Varley J, Elles R, Howell A, et al. An evaluation of common breastcancer gene mutations in a population of Ashkenazi Jews. J Med Genet 1998;35(1):10-2.
256. Blesa JR, Hernández-Yago J. Adaptation of conformation-sensitive gel electrophoresis to an ALFexpressDNA sequencer to screen BRCA1 mutations. Biotechniques 2000;28(5):1019-25.
257. Wagner T, Stoppa-Lyonnet D, Fleischmann E, Muhr D, Pagès S, Sandberg T, et al. Denaturing high- performance liquid chromatography detects reliably BRCA1 and BRCA2 mutations. Genomics 1999;62(3):369-76.
258. Gayther SA, Ponder BA. Mutations of the BRCA1 and BRCA2 genes and the possibilities for predictivetesting. Mol Med Today 1997;3(4):168-74.
259. Hacia JG, Brody LC, Chee MS, Fodor SP, Collins FS. Detection of heterozygous mutations in BRCA1using high density oligonucleotide arrays and two-colour fluorescence analysis. Nat Genet 1996;14(4):441-7.
260. Abbaszadegan MR, Struewing JP, Brown KM, Snider JV, Goodsaid F, Gore-Langton R, et al. Automateddetection of prevalent mutations in BRCA1 and BRCA2 genes using a fluorogenic PCR allelicdiscrimination assay. Genet Test 1997;1(3):171-80.
261. Robinson MD, Chu CE, Turner G, Bishop DT, Taylor GR. Exon deletions and duplications in BRCA1detected by semiquantitative PCR. Genet Test 2000;4(1):49-54.
262. Pals G, Pindolia K, Worsham MJ. A rapid and sensitive approach to mutation detection using real-time polymerase chain reaction and melting curve analyses, using BRCA1 as an example. Mol Diagn 1999;4(3):241-6.
263. Jakubowska A, Górski B, Byrski T, Huzarski T, Gronwald J, Menkiszak J, et al. Detection of germlinemutations in the BRCA1 gene by RNA-based sequencing. Hum Mutat 2001;18(2):149-56.
264. Markoff A, Sormbroen H, Bogdanova N, Preisler-Adams S, Ganev V, Dworniczak B, et al. Comparison of conformation-sensitive gel electrophoresis and single-strand conformation polymorphism analysis for detection of mutations in the BRCA1 gene using optimized conformation analysis protocols. Eur J HumGenet 1998;6(2):145-50.
265. Gross E, Arnold N, Pfeifer K, Bandick K, Kiechle M. Identification of specific BRCA1 and BRCA2variants by DHPLC. Hum Mutat 2000;16(4):345-53.
266. Montagna M, Agata S, De Nicolo A, Menin C, Sordi G, Chieco-Bianchi L, et al. Identification of BRCA1and BRCA2 carriers by allele-specific gene expression (AGE) analysis. Int J Cancer 2002;98(5):732-6.
267. Lidereau R, Eisinger F, Champème MH, Noguès C, Bièche I, Birnbaum D, et al. Major improvement in theefficacy of BRCA1 mutation screening using morphoclinical features of breast cancer. Cancer Res 2000;60(5):1206-10.
268. Euhus DM. Understanding mathematical models for breast cancer risk assessment and counseling. Breast J 2001;7(4):224-32.
269. Bonadona V, Sinilnikova OM, Lenoir GM, Lasset C, Euhus DM, Esserman L, et al. Re: Pretest predictionof BRCA1 or BRCA2 mutation by risk counselors and the computer model BRCAPRO [multiple letters]. JNatl Cancer Inst 2002;94(20):1582-4.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 79/103
62
270. Berry DA, Iversen ES, Gudbjartsson DF, Hiller EH, Garber JE, Peshkin BN, et al. BRCAPRO validation,sensitivity of genetic testing of BRCA1/BRCA2, and prevalence of other breast cancer susceptibility genes.J Clin Oncol 2002;20(11):2701-12.
271. Euhus DM, Smith KC, Robinson L, Stucky A, Olopade OI, Cummings S, et al. Pretest prediction of BRCA1 or BRCA2 mutation by risk counselors and the computer model BRCAPRO. J Natl Cancer Inst
2002;94(11):844-51.272. Tengs TO, Berry DA. The cost effectiveness of testing for the BRCA1 and BRCA2 breast-ovarian cancer
susceptibility genes. Dis Manage Clin Outcomes 2000;2(1-2):15-24.
273. Ozcelik H, Nedelcu R, Chan VW, Shi XH, Murphy J, Rosen B, et al. Mutation in the coding region of theBRCA1 gene leads to aberrant splicing of the transcript [letter]. Hum Mutat 1999;14(6):540-1.
274. Hogervorst FB, Cornelis RS, Bout M, van Vliet M, Oosterwijk JC, Olmer R, et al. Rapid detection of BRCA1 mutations by the protein truncation test. Nat Genet 1995;10(2):208-12.
275. Arnold N, Gross E, Schwarz-Boeger U, Pfisterer J, Jonat W, Kiechle M. A highly sensitive, fast, andeconomical technique for mutation analysis in hereditary breast and ovarian cancers. Hum Mutat 1999;14(4):333-9.
276. Ozcelik H, Antebi YJ, Cole DE, Andrulis IL. Heteroduplex and protein truncation analysis of the BRCA1185delAG mutation. Hum Genet 1996;98(3):310-2.
277. Serova O, Montagna M, Torchard D, Narod SA, Tonin P, Sylla B, et al. A high incidence of BRCA1mutations in 20 breast-ovarian cancer families. Am J Hum Genet 1996;58(1):42-51.
278. Serova OM, Mazoyer S, Puget N, Dubois V, Tonin P, Shugart YY, et al. Mutations in BRCA1 and BRCA2in breast cancer families: are there more breast cancer-susceptibility genes? Am J Hum Genet 1997;60(3):486-95.
279. Malone KE, Daling JR, Thompson JD, O'Brien CA, Francisco LV, Ostrander EA. BRCA1 mutations and breast cancer in the general population: analyses in women before age 35 years and in women before age45 years with first-degree family history. JAMA 1998;279(12):922-9.
280. Antoniou AC, Pharoah PD, McMullan G, Day NE, Ponder BA, Easton D. Evidence for further breastcancer susceptibility genes in addition to BRCA1 and BRCA2 in a population-based study. GenetEpidemiol 2001;21(1):1-18.
281. Couch FJ, DeShano ML, Blackwood MA, Calzone K, Stopfer J, Campeau L, et al. BRCA1 mutations inwomen attending clinics that evaluate the risk of breast cancer. N Engl J Med 1997;336(20):1409-15.
282. Taylor MR. Genetic testing for inherited breast and ovarian cancer syndromes: important concepts for the primary care physician. Postgrad Med J 2001;77(903):11-5.
283. Greene MH. Genetics of breast cancer. Mayo Clin Proc 1997;72(1):54-65.
284. Dørum A, Heimdal K, Møller P. Clinical implications of BRCA1 genetic testing. Acta Obstet GynecolScand 1998;77(4):458-61.
285. Shattuck-Eidens D, Oliphant A, McClure M, McBride C, Gupte J, Rubano T, et al. BRCA1 sequenceanalysis in women at high risk for susceptibility mutations. Risk factor analysis and implications for genetic testing. JAMA 1997;278(15):1242-50.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 80/103
63
286. Press NA, Yasui Y, Reynolds S, Durfy SJ, Burke W. Women's interest in genetic testing for breast cancer susceptibility may be based on unrealistic expectations. Am J Med Genet 2001;99(2):99-110.
287. Elwood JM. Public health aspects of breast cancer gene testing in Canada. Part 1: risks and interventions.Chronic Dis Can 1999;20(1):3-13. Available: http://www.hc-sc.gc.ca/hpb/lcdc/publicat/cdic/cdic201/cd201b_e.html.
288. Scheuer L, Kauff N, Robson M, Kelly B, Barakat R, Satagopan J, et al. Outcome of preventive surgery andscreening for breast and ovarian cancer in BRCA mutation carriers. J Clin Oncol 2002;20(5):1260-8.
289. Møller P, Reis MM, Evans G, Vasen H, Haites N, Anderson E, et al. Efficacy of early diagnosis andtreatment in women with a family history of breast cancer. Dis Markers 1999;15(1-3):179-86.
290. Tilanus-Linthorst MMA, Obdeijn IMM, Bartels KCM, de Koning HJ, Oudkerk M. First experiencesscreening women at high risk for breast cancer with MR imaging. Breast Cancer Res Treat 2000;63(1):53-60.
291. Burke W, Daly M, Garber J, Botkin J, Kahn MJ, Lynch P, et al. Recommendations for follow-up care of individuals with an inherited predisposition to cancer. II. BRCA1 and BRCA2. Cancer Genetics StudiesConsortium. JAMA 1997;277(12):997-1003.
292. Warner E, Plewes DB, Shumak RS, Catzavelos GC, Di Prospero LS, Yaffe MJ, et al. Comparison of breastmagnetic resonance imaging, mammography, and ultrasound for surveillance of women at high risk for hereditary breast cancer. J Clin Oncol 2001;19(15):3524-31.
293. Brekelmans CT, Seynaeve C, Bartels CC, Tilanus-Linthorst MM, Meijers-Heijboer EJ, Crepin CM, et al.Effectiveness of breast cancer surveillance in BRCA1/2 gene mutation carriers and women with highfamilial risk. J Clin Oncol 2001;19(4):924-30.
294. Tilanus-Linthorst M, Verhoog L, Obdeijn IM, Bartels K, Menke-Pluymers M, Eggermont A, et al. ABRCA1/2 mutation, high breast density and prominent pushing margins of a tumor independentlycontribute to a frequent false-negative mammography. Int J Cancer 2002;102(1):91-5.
295. Hughes KS, Roche CA, Whitney T, McLellan R. The management of women at high risk of experiencinghereditary breast and ovarian cancer: the Lahey guidelines. Dis Manage Health Outcomes 2000;7(4):201-15.
296. Vasen HF, Haites NE, Evans DG, Steel CM, Moller P, Hodgson S, et al. Current policies for surveillanceand management in women at risk of breast and ovarian cancer: a survey among 16 European family cancer clinics. Eur J Cancer 1998;34(12):1922-6.
297. Eisinger F, Alby N, Bremond A, Dauplat J, Espie M, Janiaud P, et al. Expertise collective INSERM-FNCLCC. Recommandations portant sur la prise en charge des femmes ayant un risque d'origine génétiquede développer un cancer du sein et/ou de l'ovaire [INSERM-FNCLCC Expert Conference.Recommendations based on the management of women with genetic risk of developing breast and/or ovarian cancer]. Reprod Hum Horm 1999;12(9-10):827-33.
298. Meijers-Heijboer EJ, Verhoog LC, Brekelmans CTM, Seynaeve C, Tilanus-Linthorst MMA, Wagner A, etal. Presymptomatic DNA testing and prophylactic surgery in families with a BRCA1 or BRCA2 mutation.Lancet 2000;355(9220):2015-20. Available:http://pdf.thelancet.com/pdfdownload?uid=llan.355.9220.original_research.1295.1&x=x.pdf (accessed2003 Apr 9).
299. Elit L, Rosen B, Goel V, McLaughlin J, Fung MKF, Shime J, et al. Prophylactic oophorectomy in Ontario.Fam Cancer 2001;1(3-4):143-8.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 81/103
64
300. Hartmann LC, Schaid DJ, Woods JE, Crotty TP, Myers JL, Arnold PG, et al. Efficacy of bilateral prophylactic mastectomy in women with a family history of breast cancer. N Engl J Med 1999;340(2):77-84.
301. Meijers-Heijboer H, Van Geel B, Van Putten WLJ, Henzen-Logmans SC, Seynaeve C, Menke-PluymersMBE, et al. Breast cancer after prophylactic bilateral mastectomy in women with a BRCA1 or BRCA2mutation. N Engl J Med 2001;345(3):159-64.
302. Hartmann LC, Sellers TA, Schaid DJ, Frank TS, Soderberg CL, Sitta DL, et al. Efficacy of bilateral prophylactic mastectomy in BRCA1 and BRCA2 gene mutation carriers. J Natl Cancer Inst 2001;93(21):1633-7.
303. Rebbeck TR, Lynch HT, Neuhausen SL, Narod SA, Van't Veer L, Garber JE, et al. Prophylacticoophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med 2002;346(21):1616-22.
304. Kauff ND, Satagopan JM, Robson ME, Scheuer L, Hensley M, Hudis CA, et al. Risk-reducing salpingo-oophorectomy in women with a BRCA1 or BRCA2 mutation. N Engl J Med 2002;346(21):1609-15.
305. Verhoog LC, Brekelmans CTM, Seynaeve C, Meijers-Heijboer EJ, Klijn JGM. Contralateral breast cancer risk is influenced by the age at onset in BRCA1-associated breast cancer. Br J Cancer 2000;83(3):384-6.
306. Turner BC, Harrold E, Matloff E, Smith T, Gumbs AA, Beinfield M, et al. BRCA1/BRCA2 germlinemutations in locally recurrent breast cancer patients after lumpectomy and radiation therapy: implicationsfor breast-conserving management in patients with BRCA1/BRCA2 mutations. J Clin Oncol 1999;17(10):3017-24.
307. Paley PJ, Swisher EM, Garcia RL, Agoff SN, Greer BE, Peters KL, et al. Occult cancer of the fallopiantube in BRCA-1 germline mutation carriers at prophylactic oophorectomy: a case for recommendinghysterectomy at surgical prophylaxis. Gynecol Oncol 2001;80(2):176-80.
308. van Roosmalen MS, Verhoef LCG, Stalmeier PFM, Hoogerbrugge N, van Daal WAJ. Decision analysis of prophylactic surgery or screening for BRCA1 mutation carriers: a more prominent role for oophorectomy.J Clin Oncol 2002;20(8):2092-100.
309. Grann VR, Jacobson JS, Thomason D, Hershman D, Heitjan DF, Neugut AI. Effect of prevention strategieson survival and quality-adjusted survival of women with BRCA1/2 mutations: an updated decisionanalysis. J Clin Oncol 2002;20(10):2520-9.
310. Grann VR, Panageas KS, Whang W, Antman KH, Neugut AI. Decision analysis of prophylacticmastectomy and oophorectomy in BRCA1- positive or BRCA2-positive patients. J Clin Oncol 1998;16(3):979-85.
311. Leonard CE, Sedlacek S, Shapiro H, Hey D, Liang X, Howell K, et al. Lumpectomy and breastradiotherapy in breast cancer patients with a family history of breast cancer, ovarian cancer, or both. BreastJ 2002;8(3):154-61.
312. Pierce LJ, Strawderman M, Narod SA, Oliviotto I, Eisen A, Dawson L, et al. Effect of radiotherapy after breast-conserving treatment in women with breast cancer and germline BRCA1/2 mutations. J Clin Oncol 2000;18(19):3360-9.
313. Grann VR, Whang W, Jacobson JS, Heitjan DF, Antman KH, Neugut A, I. Benefits and costs of screeningAshkenazi Jewish women for BRCA1 and BRCA2. J Clin Oncol 1999;17(2):494-500.
314. Powles T, Eeles R, Ashley S, Easton D, Chang J, Dowsett M, et al. Interim analysis of the incidence of breast cancer in the Royal Marsden Hospital tamoxifen randomised chemoprevention trial. Lancet 1998;352(9122):98-101.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 82/103
65
315. Eeles RA, Powles TJ. Chemoprevention options for BRCA1 and BRCA2 mutation carriers. J Clin Oncol 2000;18(21 Suppl):93s-9s.
316. Cain S. Breast cancer genetics and the role of tamoxifen in prevention. J Am Acad Nurse Pract 2000;12(1):21-8.
317. Eisinger F, Charafe-Jauffret E, Jacquemier J, Birnbaum D, Julian-Reynier C, Sobol H. Tamoxifen and breast cancer risk in women harboring a BRCA1 germline mutation: computed efficacy, effectiveness andimpact. Int J Oncol 2001;18(1):5-10.
318. King MC, Wieand S, Hale K, Lee M, Walsh T, Owens K, et al. Tamoxifen and breast cancer incidenceamong women with inherited mutations in BRCA1 and BRCA2: National Surgical Adjuvant Breast andBowel Project (NSABP-P1) Breast Cancer Prevention Trial. JAMA 2001;286(18):2251-6. Available:http://jama.ama-assn.org/cgi/reprint/286/18/2251.pdf (accessed 2003 Apr 4).
319. Narod SA, Brunet JS, Ghadirian P, Robson M, Heimdal K, Neuhausen SL, et al. Tamoxifen and risk of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers: a case-control study. Lancet 2000;356(9245):1876-81. Available:http://pdf.thelancet.com/pdfdownload?uid=llan.356.9245.original_research.14368.1&x=x.pdf (accessed2003 Apr 8).
320. Duffy SW, Nixon RM. Estimates of the likely prophylactic effect of tamoxifen in women with high risk BRCA1 and BRCA2 mutations. Br J Cancer 2002;86(2):218-21.
321. Schrag D, Kuntz KM, Garber JE, Weeks JC. Life expectancy gains from cancer prevention strategies for women with breast cancer and BRCA1 or BRCA2 mutations. JAMA 2000;283(5):617-24. Available:http://jama.ama-assn.org/cgi/reprint/283/5/617.pdf (accessed 2003 Apr 9).
322. Narod SA, Risch H, Moslehi R, Dorum A, Neuhausen S, Olsson H, et al. Oral contraceptives and the risk of hereditary ovarian cancer. N Engl J Med 1998;339(7):424-8.
323. Modan B, Hartge P, Hirsh-Yechezkel G, Chetrit A, Lubin F, Beller U, et al. Parity, oral contraceptives, andthe risk of ovarian cancer among carriers and noncarriers of a BRCA1 or BRCA2 mutation. N Engl J Med 2001;345(4):235-40.
324. Egawa C, Miyoshi Y, Takamura Y, Taguchi T, Tamaki Y, Noguchi S. Decreased expression of BRCA2mRNA predicts favorable response to docetaxel in breast cancer. Int J Cancer 2001;95(4):255-9.
325. Goffin JR, Chappuis PO, Bégin LR, Wong N, Brunet JS, Hamel N, et al. Impact of germline BRCA1mutations and overexpression of p53 on prognosis and response to treatment following breast carcinoma:10-year follow up data. Cancer 2003;97(3):527-36.
326. van 't Veer LJ, Dai H, van de Vijver M, He YD, Hart AA, Mao M, et al. Gene expression profiling predictsclinical outcome of breast cancer. Nature 2002;415(6871):530-6.
327. Lakhani SR, Jacquemier J, Sloane JP, Gusterson BA, Anderson TJ, Van de Vijver MJ, et al. Multifactorialanalysis of differences between sporadic breast cancers and cancers involving BRCA1 and BRCA2mutations. J Natl Cancer Inst 1998;90(15):1138-45.
328. Verhoog LC, Brekelmans CTM, Seynaeve C, Van den Bosch LMC, Dahmen G, Van Geel AN, et al.Survival and tumour characteristics of breast-cancer patients with germline mutations of BRCA1. Lancet 1998;351(9099):316-21. Available:http://pdf.thelancet.com/pdfdownload?uid=llan.351.9099.original_research.7724.1&x=x.pdf (accessed2002 Apr 9).

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 83/103
66
329. Møller P, Borg A, Heimdal K, Apold J, Vallon-Christersson J, Hovig E, et al. The BRCA1 syndrome andother inherited breast or breast-ovarian cancers in a Norwegian prospective series. Eur J Cancer 2001;37(8):1027-32.
330. Pericay C, Brunet J, Díez O, Sanz J, Cortès J, Baiget M, et al. Clinical and pathological findings of BRCA1/2 associated breast cancer. Breast 2001;10(1):46-8.
331. Chappuis PO, Kapusta L, Bégin LR, Wong N, Brunet JS, Narod SA, et al. Germline BRCA1/2 mutationsand p27(Kip1) protein levels independently predict outcome after breast cancer. J Clin Oncol 2000;18(24):4045-52.
332. Robson M, Gilewski T, Haas B, Levin D, Borgen P, Rajan P, et al. BRCA-associated breast cancer inyoung women. J Clin Oncol 1998;16(5):1642-9.
333. Jóhannsson ÓT, Ranstam J, Borg Å, Olsson H. Survival of BRCA1 breast and ovarian cancer patients: a population-based study from southern Sweden. J Clin Oncol 1998;16(2):397-404.
334. Stoppa-Lyonnet D, Ansquer Y, Dreyfus H, Gautier C, Gauthier-Villars M, Bourstyn E, et al. Familialinvasive breast cancers: worse outcome related to BRCA1 mutations. J Clin Oncol 2000;18(24):4053-9.
335. Hennekam RC. Hereditary multiple exostoses. J Med Genet 1991;28(4):262-6.
336. Wicklund CL, Pauli RM, Johnston D, Hecht JT. Natural history study of hereditary multiple exostoses. AmJ Med Genet 1995;55(1):43-6.
337. Schmale GA, Conrad EU, Raskind WH. The natural history of hereditary multiple exostoses. J Bone JointSurg Am 1994;76(7):986-92.
338. Wuyts W, Van Hul W. Molecular basis of multiple exostoses: mutations in the EXT1 and EXT2 genes.Hum Mutat 2000;15(3):220-7.
339. Raskind WH, Conrad EU, Matsushita M, Wijsman EM, Wells DE, Chapman N, et al. Evaluation of locusheterogeneity and EXT1 mutations in 34 families with hereditary multiple exostoses. Hum Mutat 1998;11(3):231-9.
340. Carroll KL, Yandow SM, Ward K, Carey JC. Clinical correlation to genetic variations of hereditarymultiple exostosis. J Pediatr Orthop 1999;19(6):785-91.
341. Aaltonen LA, Salovaara R, Kristo P, Canzian F, Hemminki A, Peltomäki P, et al. Incidence of hereditarynonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med 1998;338(21):1481-7.
342. Ponz de Leon M. Prevalence of hereditary nonpolyposis colorectal carcinoma (HNPCC). Ann Med 1994;26(3):209-14.
343. Syngal S, Fox EA, Eng C, Kolodner RD, Garber JE. Sensitivity and specificity of clinical criteria for hereditary non-polyposis colorectal cancer associated mutations in MSH2 and MLH1. J Med Genet 2000;37(9):641-5.
344. Wijnen J, Khan PM, Vasen H, van der Klift H, Mulder A, van Leeuwen-Cornelisse I, et al. Hereditarynonpolyposis colorectal cancer families not complying with the Amsterdam criteria show extremely lowfrequency of mismatch-repair-gene mutations. Am J Hum Genet 1997;61(2):329-35.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 84/103
67
345. Wijnen JT, Vasen HF, Khan PM, Zwinderman AH, van der Klift H, Mulder A, et al. Clinical findings withimplications for genetic testing in families with clustering of colorectal cancer. N Engl J Med 1998;339(8):511-8.
346. Park JG, Vasen HF, Park YJ, Park KJ, Peltomaki P, de Leon MP, et al. Suspected HNPCC and Amsterdamcriteria II: evaluation of mutation detection rate, an international collaborative study. Int J Colorectal Dis
2002;17(2):109-14.347. Syngal S, Fox EA, Li C, Dovidio M, Eng C, Kolodner RD, et al. Interpretation of genetic test results for
hereditary nonpolyposis colorectal cancer: implications for clinical predisposition testing. JAMA 1999;282(3):247-53.
348. Froggatt NJ, Koch J, Davies R, Evans DG, Clamp A, Quarrell OW, et al. Genetic linkage analysis inhereditary non-polyposis colon cancer syndrome. J Med Genet 1995;32(5):352-7.
349. Farrington SM, Lin-Goerke J, Ling J, Wang Y, Burczak JD, Robbins DJ, et al. Systematic analysis of hMSH2 and hMLH1 in young colon cancer patients and controls. Am J Hum Genet 1998;63(3):749-59.
350. Terdiman JP, Levin TR, Allen BA, Gum JR, Fishbach A, Conrad PG, et al. Hereditary nonpolyposiscolorectal cancer in young colorectal cancer patients: high-risk clinic versus population-based registry.Gastroenterology 2002;122(4):940-7.
351. Berends MJ, Hollema H, Wu Y, van Der Sluis T, Mensink RG, ten Hoor KA, et al. MLH1 and MSH2 protein expression as a pre-screening marker in hereditary and non-hereditary endometrial hyperplasia andcancer. Int J Cancer 2001;92(3):398-403.
352. Holinski-Feder E, Müller-Koch Y, Friedl W, Moeslein G, Keller G, Plaschke J, et al. DHPLC mutationanalysis of the hereditary nonpolyposis colon cancer (HNPCC) genes hMLH1 and hMSH2. J BiochemBiophys Methods 2001;47(1-2):21-32.
353. Wijnen J, Khan PM, Vasen H, Menko F, van der Klift H, van den Broek M, et al. Majority of hMLH1mutations responsible for hereditary nonpolyposis colorectal cancer cluster at the exonic region 15-16. AmJ Hum Genet 1996;58(2):300-7.
354. Liu B, Farrington SM, Petersen GM, Hamilton SR, Parsons R, Papadopoulos N, et al. Genetic instabilityoccurs in the majority of young patients with colorectal cancer. Nat Med 1995;1(4):348-52.
355. Nyström-Lahti M, Wu Y, Moisio AL, Hofstra RM, Osinga J, Mecklin JP, et al. DNA mismatch repair genemutations in 55 kindreds with verified or putative hereditary non-polyposis colorectal cancer. Hum MolGenet 1996;5(6):763-9.
356. Kohonen-Corish M, Ross VL, Doe WF, Kool DA, Edkins E, Faragher I, et al. RNA-based mutationscreening in hereditary nonpolyposis colorectal cancer. Am J Hum Genet 1996;59(4):818-24.
357. Beck NE, Tomlinson IP, Homfray T, Frayling I, Hodgson SV, Harocopos C, et al. Use of SSCP analysis toidentify germline mutations in HNPCC families fulfilling the Amsterdam criteria. Hum Genet 1997;99(2):219-24.
358. Froggatt NJ, Brassett C, Koch DJ, Evans DG, Hodgson SV, Ponder BA, et al. Mutation screening of MSH2and MLH1 mRNA in hereditary non-polyposis colon cancer syndrome. J Med Genet 1996;33(9):726-30.
359. Wahlberg S, Liu T, Lindblom P, Lindblom A. Various mutation screening techniques in the DNAmismatch repair genes hMSH2 and hMLH1. Genet Test 1999;3(3):259-64.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 85/103
68
360. Kurzawski G, Safranow K, Suchy J, Chlubek D, Scott RJ, Lubiñski J. Mutation analysis of MLH1 andMSH2 genes performed by denaturing high-performance liquid chromatography. J Biochem BiophysMethods 2002;51(1):89-100.
361. Thibodeau SN, French AJ, Roche PC, Cunningham JM, Tester DJ, Lindor NM, et al. Altered expression of hMSH2 and hMLH1 in tumors with microsatellite instability and genetic alterations in mismatch repair
genes. Cancer Res 1996;56(21):4836-40.362. Marcus VA, Madlensky L, Gryfe R, Kim H, So K, Millar A, et al. Immunohistochemistry for hMLH1 and
hMSH2: a practical test for DNA mismatch repair-deficient tumors. Am J Surg Pathol 1999;23(10):1248-55.
363. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute Workshop on Microsatellite Instability for Cancer Detection and Familial Predisposition:development of international criteria for the determination of microsatellite instability in colorectal cancer.Cancer Res 1998;58(22):5248-57.
364. Dietmaier W, Wallinger S, Bocker T, Kullmann F, Fishel R, Rüschoff J. Diagnostic microsatelliteinstability: definition and correlation with mismatch repair protein expression. Cancer Res 1997;57(21):4749-56.
365. Terdiman JP, Gum JR, Conrad PG, Miller GA, Weinberg V, Crawley SC, et al. Efficient detection of hereditary nonpolyposis colorectal cancer gene carriers by screening for tumor microsatellite instability
before germline genetic testing. Gastroenterology 2001;120(1):21-30.
366. Genetic testing for colon cancer: joint statement of the American College of Medical Genetics andAmerican Society of Human Genetics. Joint Test and Technology Transfer Committee Working Group.Genet Med 2000;2(6):362-6.
367. Järvinen HJ, Mecklin JP, Sistonen P. Screening reduces colorectal cancer rate in families with hereditarynonpolyposis colorectal cancer. Gastroenterology 1995;108(5):1405-11.
368. Syngal S, Weeks JC, Schrag D, Garber JE, Kuntz KM. Benefits of colonoscopic surveillance and prophylactic colectomy in patients with hereditary nonpolyposis colorectal cancer mutations. Ann InternMed 1998;129(10):787-96.
369. Chompret A, Brugières L, Ronsin M, Gardes M, Dessarps-Freichey F, Abel A, et al. P53 germlinemutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer 2000;82(12):1932-7.
370. Lustbader ED, Williams WR, Bondy ML, Strom S, Strong LC. Segregation analysis of cancer in familiesof childhood soft-tissue-sarcoma patients. Am J Hum Genet 1992;51(2):344-56.
371. Varley JM, McGown G, Thorncroft M, Santibanez-Koref MF, Kelsey AM, Tricker KJ, et al. Germ-linemutations of TP53 in Li-Fraumeni families: an extended study of 39 families. Cancer Res 1997;57(15):3245-52.
372. Varley JM, Evans DG, Birch JM. Li-Fraumeni syndrome--a molecular and clinical review. Br J Cancer 1997;76(1):1-14.
373. Birch JM, Hartley AL, Tricker KJ, Prosser J, Condie A, Kelsey AM, et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res 1994;54(5):1298-304.
374. Limacher JM, Frebourg T, Natarajan-Ame S, Bergerat JP. Two metachronous tumors in the radiotherapyfields of a patient with Li-Fraumeni syndrome. Int J Cancer 2001;96(4):238-42.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 86/103
69
375. Larsson C, Weber G, Teh BT, Lagercrantz J. Genetics of multiple endocrine neoplasia type 1. Ann N YAcad Sci 1994;733:453-63.
376. Lemmens I, Van de Ven WJ, Kas K, Zhang CX, Giraud S, Wautot V, et al. Identification of the multipleendocrine neoplasia type 1 (MEN1) gene. Hum Mol Genet 1997;6(7):1177-83.
377. Agarwal SK, Kester MB, Debelenko LV, Heppner C, Emmert-Buck MR, Skarulis MC, et al. Germlinemutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum MolGenet 1997;6(7):1169-75.
378. Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, et al.Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997;276(5311):404-7.
379. Skogseid B. Multiple endocrine neoplasia type I. Clinical genetics and diagnosis. Cancer Treat Res 1997;89:383-406.
380. Crepin M, Escande F, Pigny P, Buisine MP, Calender A, Porchet N, et al. Efficient mutation detection inMEN1 gene using a combination of single-strand conformation polymorphism (MDGA™) andheteroduplex analysis. Electrophoresis 2003;24(1-2):26-33.
381. Cebrián A, Herrera-Pombo JL, Díez JJ, Sánchez-Vilar O, Lara JI, Vázquez C, et al. Genetic and clinicalanalysis in 10 Spanish patients with multiple endocrine neoplasia type 1. Eur J Hum Genet 1999;7(5):585-9.
382. Thakker RV. Editorial: multiple endocrine neoplasia--syndromes of the twentieth century. J ClinEndocrinol Metab 1998;83(8):2617-20.
383. Teh BT, Kytölä S, Farnebo F, Bergman L, Wong FK, Weber G, et al. Mutation analysis of the MEN1 genein multiple endocrine neoplasia type 1, familial acromegaly and familial isolated hyperparathyroidism. JClin Endocrinol Metab 1998;83(8):2621-6.
384. Complete test list. In: Peninsula molecular genetics laboratory [database online]. Exeter (UK): RoyalDevon and Exeter NHS Healthcare Trust; 2003. Available:http://www.ex.ac.uk/diabetesgenes/geneticslab/fulltestlist.htm (accessed 2003 Sep 9).
385. Waterlot C, Porchet N, Bauters C, Decoulx M, Wémeau JL, Proye C, et al. Type 1 multiple endocrineneoplasia (MEN1): contribution of genetic analysis to the screening and follow-up of a large Frenchkindred. Clin Endocrinol 1999;51(1):101-7.
386. Pape UF, Höcker M, Seuss U, Wiedenmann B. New molecular aspects in the diagnosis and therapy of neuroendocrine tumors of the gastroenteropancreatic system. Recent Results Cancer Res 2000;153:45-60.
387. Learoyd DL, Twigg SM, Marsh DJ, Robinson BG. The practical management of multiple endocrineneoplasia. Trends Endocrinol Metab 1995;6(8):273-8.
388. Mulligan LM, Eng C, Healey CS, Clayton D, Kwok JB, Gardner E, et al. Specific mutations of the RET proto-oncogene are related to disease phenotype in MEN 2A and FMTC. Nat Genet 1994;6(1):70-4.
389. Edery P, Lyonnet S, Mulligan LM, Pelet A, Dow E, Abel L, et al. Mutations of the RET proto-oncogene inHirschsprung's disease. Nature 1994;367(6461):378-80.
390. Eng C. Seminars in medicine of the Beth Israel Hospital, Boston. The RET proto-oncogene in multipleendocrine neoplasia type 2 and Hirschsprung's disease. N Engl J Med 1996;335(13):943-51.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 87/103
70
391. Telenius H, Mathew CG, Nakamura Y, Easton DF, Clark J, Neumann HP, et al. Application of linked DNAmarkers to screening families with multiple endocrine neoplasia type 2A. Eur J Surg Oncol 1990;16(2):134-40.
392. Benhattar J, Cerottini JP, Saraga E, Metthez G, Givel JC. p53 mutations as a possible predictor of responseto chemotherapy in metastatic colorectal carcinomas. Int J Cancer 1996;69(3):190-2.
393. McMahon R, Mulligan LM, Healey CS, Payne SJ, Ponder M, Ferguson-Smith MA, et al. Direct, non-radioactive detection of mutations in multiple endocrine neoplasia type 2A families. Hum Mol Genet 1994;3(4):643-6.
394. Neumann HP, Eng C, Mulligan LM, Glavac D, Zäuner I, Ponder BA, et al. Consequences of direct genetictesting for germline mutations in the clinical management of families with multiple endocrine neoplasia,type II. JAMA 1995;274(14):1149-51.
395. Decker RA, Peacock ML, Borst MJ, Sweet JD, Thompson NW. Progress in genetic screening of multipleendocrine neoplasia type 2A: Is calcitonin testing obsolete? Surgery 1995;118(2):257-64.
396. Wells SA, Donis-Keller H. Current perspectives on the diagnosis and management of patients with multipleendocrine neoplasia type 2 syndromes. Endocrinol Metab Clin North Am 1994;23(1):215-28.
397. Lips CJ, Landsvater RM, Hoppener JW, Geerdink RA, Blijham G, van Veen JM, et al. Clinical screeningas compared with DNA analysis in families with multiple endocrine neoplasia type 2A. N Engl J Med 1994;331(13):828-35.
398. Niccoli-Sire P, Murat A, Baudin E, Henry JF, Proye C, Bigorgne JC, et al. Early or prophylacticthyroidectomy in MEN 2/FMTC gene carriers: results in 71 thyroidectomized patients. Eur J Endocrinol 1999;141(5):468-74.
399. Conte-Devolx B, Schuffenecker I, Niccoli P, Maes B, Boneu A, Barbot N, et al. Multiple endocrineneoplasia type 2: management of patients and subjects at risk. Horm Res 1997;47(4-6):221-6.
400. Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, Gagel RF, et al. The relationship between specificRET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. InternationalRET mutation consortium analysis. JAMA 1996;276(19):1575-9.
401. Ponder BA. The phenotypes associated with ret mutations in the multiple endocrine neoplasia type 2syndrome. Cancer Res 1999;59(7 Suppl):1736s-41s.
402. Friedman JM, Birch PH. Type 1 neurofibromatosis: a descriptive analysis of the disorder in 1,728 patients.Am J Med Genet 1997;70(2):138-43.
403. Zöller M, Rembeck B, Åkesson HO, Angervall L. Life expectancy, mortality and prognostic factors inneurofibromatosis type 1. A twelve-year follow-up of an epidemiological study in Göteborg, Sweden. ActaDerm Venereol 1995;75(2):136-40.
404. Wolkenstein P, Frèche B, Zeller J, Revuz J. Usefulness of screening investigations in neurofibromatosistype 1. A study of 152 patients. Arch Dermatol 1996;132(11):1333-6.
405. Riccardi VM. Pathogenesis: molecular biology. In: Neurofibromatosis: phenotype, natural history, andpathogenesis. 2nd ed. Baltimore: Johns Hopkins University Press; 1992. p.251-7.
406. Tonsgard JH, Yelavarthi KK, Cushner S, Short MP, Lindgren V. Do NF1 gene deletions result in acharacteristic phenotype? Am J Med Genet 1997;73(1):80-6.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 88/103
71
407. Lazaro C, Gaona A, Ravella A, Volpini V, Estivill X. Prenatal diagnosis of neurofibromatosis type 1: fromflanking RFLPs to intragenic microsatellite markers. Prenat Diagn 1995;15(2):129-34.
408. Parry DM, Eldridge R, Kaiser-Kupfer MI, Bouzas EA, Pikus A, Patronas N. Neurofibromatosis 2 (NF2):clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet 1994;52(4):450-61.
409. Martuza RL, Eldridge R. Neurofibromatosis 2 (bilateral acoustic neurofibromatosis). N Engl J Med 1988;318(11):684-8.
410. Welling DB, Akhmametyeva EM, Daniels RL, Lasak JM, Zhu L, Miles-Markley BA, et al. Analysis of thehuman neurofibromatosis type 2 gene promoter and its expression. Otolaryngol Head Neck Surg 2000;123(4):413-8.
411. Bijlsma EK, Wallace AJ, Evans DG. Misleading linkage results in an NF2 presymptomatic test owing tomosaicism. J Med Genet 1997;34(11):934-6.
412. Gutmann DH, Aylsworth A, Carey JC, Korf B, Marks J, Pyeritz RE, et al. The diagnostic evaluation andmultidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997;278(1):51-7.
413. Briggs RJ, Brackmann DE, Baser ME, Hitselberger WE. Comprehensive management of bilateral acousticneuromas. Current perspectives. Arch Otolaryngol Head Neck Surg 1994;120(12):1307-14.
414. Short PM, Martuza RL, Huson SM. Neurofibromatosis 2: clinical features, genetic counselling andmanagement issues. In: Huson SM, Huges RAC, editors. The Neurofibromatoses: a pathogenetic andclinical overview. 1st. London: Chapman & Hall Medical; 1994. p.414-44.
415. Parry DM, MacCollin MM, Kaiser-Kupfer MI, Pulaski K, Nicholson HS, Bolesta M, et al. Germ-linemutations in the neurofibromatosis 2 gene: correlations with disease severity and retinal abnormalities. AmJ Hum Genet 1996;59(3):529-39.
416. Hemminki A, Tomlinson I, Markie D, Järvinen H, Sistonen P, Björkqvist AM, et al. Localization of asusceptibility locus for Peutz-Jeghers syndrome to 19p using comparative genomic hybridization andtargeted linkage analysis. Nat Genet 1997;15(1):87-90.
417. Boardman LA, Thibodeau SN, Schaid DJ, Lindor NM, McDonnell SK, Burgart LJ, et al. Increased risk for cancer in patients with the Peutz-Jeghers syndrome. Ann Intern Med 1998;128(11):896-9.
418. Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV, et al. Very highrisk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000;119(6):1447-53.
419. Giardiello FM, Welsh SB, Hamilton SR, Offerhaus GJ, Gittelsohn AM, Booker SV, et al. Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med 1987;316(24):1511-4.
420. Tomlinson IP, Houlston RS. Peutz-Jeghers syndrome. J Med Genet 1997;34(12):1007-11.
421. Baud O, Cormier-Daire V, Lyonnet S, Desjardins L, Turleau C, Doz F. Dysmorphic phenotype andneurological impairment in 22 retinoblastoma patients with constitutional cytogenetic 13q deletion. ClinGenet 1999;55(6):478-82.
422. Richter S, Vandezande K, Chen N, Zhang K, Sutherland J, Anderson J, et al. Sensitive and efficientdetection of RB1 gene mutations enhances care for families with retinoblastoma. Am J Hum Genet 2003;72(2):253-69.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 89/103
72
423. Noorani HZ, Khan HN, Gallie BL, Detsky AS. Cost comparison of molecular versus conventionalscreening of relatives at risk for retinoblastoma. Am J Hum Genet 1996;59(2):301-7.
424. Bremner R, Du DC, Connolly-Wilson MJ, Bridge P, Ahmad KF, Mostachfi H, et al. Deletion of RB exons24 and 25 causes low-penetrance retinoblastoma. Am J Hum Genet 1997;61(3):556-70.
425. Lohmann DR, Gerick M, Brandt B, Oelschläger U, Lorenz B, Passarge E, et al. Constitutional RB1-genemutations in patients with isolated unilateral retinoblastoma. Am J Hum Genet 1997;61(2):282-94.
426. Hayashi N, Ido E, Ohtsuki Y, Ueno H. An experimental application of gene therapy for humanretinoblastoma. Invest Ophthalmol Vis Sci 1999;40(2):265-72.
427. Sora S, Ueki K, Saito N, Kawahara N, Shitara N, Kirino T, et al. Incidence of von Hippel-Lindau disease inhemangioblastoma patients: the University of Tokyo Hospital experience from 1954-1998. ActaNeurochir (Wien ) 2001;143(9):893-6.
428. Gläsker S, Bender BU, Apel TW, Natt E, van Velthoven V, Scheremet R, et al. The impact of molecular genetic analysis of the VHL gene in patients with haemangioblastomas of the central nervous system. JNeurol Neurosurg Psychiatry 1999;67(6):758-62.
429. Chen F, Kishida T, Yao M, Hustad T, Glavac D, Dean M, et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat 1995;5(1):66-75.
430. Crossey PA, Richards FM, Foster K, Green JS, Prowse A, Latif F, et al. Identification of intragenicmutations in the von Hippel-Lindau disease tumour suppressor gene and correlation with disease
phenotype. Hum Mol Genet 1994;3(8):1303-8.
431. Maher ER, Webster AR, Richards FM, Green JS, Crossey PA, Payne SJ, et al. Phenotypic expression invon Hippel-Lindau disease: correlations with germline VHL gene mutations. J Med Genet 1996;33(4):328-32.
432. Zbar B. Von Hippel-Lindau disease and sporadic renal cell carcinoma. Cancer Surv 1995;25:219-32.
433. Lips CJ. Clinical management of the multiple endocrine neoplasia syndromes: results of a computerizedopinion poll at the Sixth International Workshop on Multiple Endocrine Neoplasia and von Hippel-Lindaudisease. J Intern Med 1998;243(6):589-94.
434. Skotnicka-Klonowicz G, Rieske P, Bartkowiak J, Szymik-Kantorowicz S, Daszkiewicz P, Debiec-Rychter M. 16q heterozygosity loss in Wilms' tumour in children and its clinical importance. Eur J Surg Oncol 2000;26(1):61-6.
435. Steenman M, Redeker B, de Meulemeester M, Wiesmeijer K, Voûte PA, Westerveld A, et al. Comparativegenomic hybridization analysis of Wilms tumors. Cytogenet Cell Genet 1997;77(3-4):296-303.
436. Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH, Robbins JH. Cockayne syndrome and xeroderma pigmentosum. DNA repair disorders with overlaps and paradoxes. Neurology 2000;55(10):1442-9.
437. Nagai A, Mimaki T, Tanaka K, Mino M. Clinical symptoms and molecular basis of group a xeroderma pigmentosum. Pathophysiology 1994;1(4):241-6.
438. Steingrimsdottir H, Beare D, Cole J, Leal JFM, Kostic T, Lopez-Barea J, et al. Development of newmolecular procedures for the detection of genetic alterations in man. Mutat Res Fundam Mol MechMutagenesis 1996;353(1-2):109-21.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 90/103
73
439. Lalle P, Nouspikel T, Constantinou A, Thorel F, Clarkson SG. The founding members of xeroderma pigmentosum group G produce XPG protein with severely impaired endonuclease activity. J InvestDermatol 2002;118(2):344-51.
440. Sumitani S, Ishikawa Y, Ishikawa Y, Minami R. Molecular studies of Japanese patients with group Axeroderma pigmentosum using polymerase chain reaction and restriction fragment length polymorphism
and nonradioactive single strand conformation polymorphism analyses. J Child Neurol 1999;14(3):168-72.
441. Mondello C, Nardo T, Giliani S, Arrand JE, Weber CA, Lehmann AR, et al. Molecular analysis of the XP-D gene in Italian families with patients affected by trichothiodystrophy and xeroderma pigmentosum groupD. Mutat Res 1994;314(2):159-65.
442. Cleaver JE, Volpe JP, Charles WC, Thomas GH. Prenatal diagnosis of xeroderma pigmentosum andCockayne syndrome. Prenat Diagn 1994;14(10):921-8.
443. Arase S, Bohnert E, Fischer E, Jung EG. Prenatal exclusion of Xeroderma pigmentosum (XP-D) byamniotic cell analysis. Photodermatol 1985;2(3):181-3.
444. Broughton BC, Berneburg M, Fawcett H, Taylor EM, Arlett CF, Nardo T, et al. Two individuals withfeatures of both xeroderma pigmentosum and trichothiodystrophy highlight the complexity of the clinicaloutcomes of mutations in the XPD gene. Hum Mol Genet 2001;10(22):2539-47.
445. Khan SG, Levy HL, Legerski R, Quackenbush E, Reardon JT, Emmert S, et al. Xeroderma pigmentosumgroup C splice mutation associated with autism and hypoglycinemia. J Invest Dermatol 1998;111(5):791-6.
446. Zeng L, Sarasin A, Mezzina M. Retrovirus-mediated DNA repair gene transfer into xeroderma pigmentosum cells: perspectives for a gene therapy. Cell Biol Toxicol 1998;14(2):105-10.
447. Munro M. Are you sure you really want to know? Ignorance is not bliss, but is it better or worse to learnthat you are genetically doomed? Two sufferers tell their story. Natl Post [serial online] 2000:A16.
448. Munro M. Private lab offers quicker screening of mutant gene linked to breast cancer: $3,850test alarms MDs. Natl Post [serial online] 2000:A1.
449. Martin S. Most Canadians welcome genetic testing. CMAJ 2000;163(2):200. Available:http://www.cmaj.ca.
450. Health Services Utilization and Research Commission. Preparing for future possibilities in genetictesting [Background paper]. Saskatoon (SK): The Commission; 2001.
451. PICEPS Consultants. Educational tools which relate to adult-onset disorders for which genetic testingis available: final report. Ottawa: Health Canada; 2001. Available: http://www.hc-sc.gc.ca/pphb-dgspsp/publicat/genetics/ (accessed 2003 Jun 12).
452. American Society of Clinical Oncology. Statement of the American Society of Clinical Oncology: genetictesting for cancer susceptibility. J Clin Oncol 1996;14(5):1730-40.
453. Genetic testing for inherited susceptibility of colorectal cancer: part II. Hereditary nonpolyposis colorectalcancer (HNPCC). Tecnologica MAP Suppl 1998;10-5.
454. Bioethics Committee, Canadian Paediatric Society. Guidelines for genetic testing of healthy children.Paediatr Child Health 2003;8(1):42-5.

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 91/103
74
455. Proposed international guidelines on ethical issues in medical genetics and genetic services: report of a WHO meeting on ethical issues in medical genetics. Geneva, Switzerland: World Health Organization;2001. Available: http://www.who.int/ncd/hgn/hgnethic.htm (accessed 2003 Jun 12).
456. Secretary's Advisory Committee on Genetic Testing. Enhancing the oversight of genetic tests:recommendations of the SACGT. In: National Institutes of Health [database online]. Bethesda (MD): The
Committee; 2000. Available: http://www4.od.nih.gov/oba/sacgt/ (accessed 2002 Oct 23).457. Miller F, Hurley J, Morgan S, Goeree R, Collins P, Blackhouse G, et al. Predictive genetic tests and
health care costs: final report prepared for the Ontario Ministry of Health and Long Term Care.Toronto: Ontario Ministry of Health and Long Term Care; 2002. Available:http://www.gov.on.ca/health/english/pub/ministry/geneticsrep02/chepa_rep.pdf (accessed 2002 Oct 25).

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 92/103
75
Annexe 1 : Stratégie de recherche documentaire
Guide to Search Syntax (DIALOG®, Cochrane Library)
! Explode the search term. Retrieves the search concept plus all narrower terms.? Truncation symbol, single character. Retrieves plural and variant endings of search terms.* Truncation symbol, any number of characters.“ “ Used to search phrases.(w) Proximity operator. Words must be adjacent.() Proximity operator. Words must be adjacent.(n) Proximity operator. Words must be near each other in any order.ab Search in article abstract.de Descriptor i.e., subject heading ( a controlled, thesaurus term)ME Medical subject headingti Search in titles
tw Text word
BASES DEDONNÉES
DATES/ LIMITES
VEDETTES-MATIÈRES/MOTS-CLÉS
DIALOG®OneSearch
Cancerlit
EMBASE®
BIOSIS®
MEDLINE®
PASCAL®
Human
1975-2002/Jun
1974-2002/AugW2
1969-2002/AugW2
1966-2002/Aug
W2
1973-2002/AugW2
Neoplastic Syndromes, Hereditary!/de OR ((Hereditary or Familial) () (Cancer? OR Neoplasm?))/ti,ab OR AdenomatousPolyposis Coli/de OR Familial Adenomatous Polyposis/de OR Genes, APC/de OR FAP/ti,ab OR APC()Gene/ti, ab OR Sq21/ti,ab
OR Familial()Polyposis(3N)Colon/ti,ab OR Polyposis()Adenomatous()Intestinal/ti,ab OR Gardner()Syndrome/ti,ab OR Adenomatous()Polyposis()Coli/ti,abOR (Adenomatous Polyp/de AND Familial/ti,ab) OR Gardner Syndrome/de
OR
Basal Cell Nevus Syndrome/de OR Basal Cell Carcinoma/de OR Basal()Cell()Nevus()Syndrome/ti,ab OR Nevoid()Basal()Cell()Carcinoma/ti,ab OR Gorlin()Syndrome OR
PTCH()Gene OR 9q22/ti,ab OR Gorlin()Gotz()Syndrome/ti,ab OR Basal()Cell()Carcinoma/ti,ab OR Multiple()Basal()Cell()Nevi/ti,abOR Gorlin(1N)Gotz()syndrome/ti,abOR

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 93/103
76
BASES DEDONNÉES
DATES/ LIMITES
VEDETTES-MATIÈRES/MOTS-CLÉS
((Familial OR Hereditary) AND (Breast Neoplasms/de OR BreastCancer/de OR Breast(2N)Cancer?/ti,ab OR
Breast(2N)Neoplasm?/ti,ab)) OR Genes, BRCA1/de OR Genes,BRCA2/de OR BRCA1()Gene/ti,ab OR BRCA2()Gene/ti,ab OR Breast()Ovarian()Cancer?/ti,ab OR Breast?(2N)Neoplasm?/ti,ab))OR Hereditary Breast Cancer/de
OR
Cowden()Syndrome/ti,ab OR Hamartoma Syndrome, Multiple/deOR Cowdens()Syndrome/ti,ab OR PTEN()gene OR 10q23/ti,ab OR Cowden Syndrome/de OR Cowden Disease/de OR Cowden()Disease/ti,ab
OR
Li-Fraumeni Syndrome/de OR Genes, p53/de OR Li()Fraumeni()Syndrome/ti,ab OR p53()Gene/ti,ab OR 17p13/ti,abOR Li(1N)Fraumeni/ti,ab OR CDH1()Gene OR Cadherin()1/ti,abOR 16q22/ti,ab
OR
((Familial OR Hereditary) AND (Stomach Neoplasms/de OR
Gastric Cancer/de OR Gastric Carcinoma/de OR Melanoma OR Gastic()Cancer/ti,ab OR Gastric()Carcinoma/ti,ab OR Stomach()Cancer?/ti,ab OR Stomach() Neoplasm?/ti,ab))
OR
CDKN2()Gene/ti,ab OR CDK4()Gene/ti,ab OR p16/ti,ab OR 9p21/ti,ab OR 12q13/ti,ab OR Gorlin(1N)Gotz()Syndrome/ti,ab OR (Dysplastic()Nevus()Syndrome AND Hereditary)
OR Multiple Endocrine Neoplasia Type 1/de OR Multiple Endocrine Neoplasia!/de OR Multiple Endocrine Neoplasia/deMEN1()GENE/ti,ab OR 11q13/ti,ab OR Endocrine()Adenomatosis()Multiple/ti,ab OR

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 94/103
77
BASES DEDONNÉES
DATES/ LIMITES
VEDETTES-MATIÈRES/MOTS-CLÉS
OR
Zollinger(1N)Ellison()syndrome/ti,ab OR Multiple Endocrine Neoplasia Type 2a/de OR Multiple()Endocrine()Neoplasia()Type()2/ti,ab OR Men()2()Gene/ti,ab OR Multiple()Endocrine()Neoplasia/ti,ab OR Medullary()Thyroid()Carcinoma/ti,ab OR PTC()syndrome/ti,ab OR Sipple()Syndrome/ti,ab OR Sipple Syndrome/de
OR
Exostoses, Multiple Hereditary/de OR Osteochondroma/ti,ab OR Osteosarcoma/ti,ab OR EXT1()Gene/ti,ab OR EXT2()Gene/ti,ab OR
8q24/ti,ab OR11p12-p11/ti,ab OR Multiple(1N)Exostoses/ti,ab OR Hereditary Multiple Exostosis/de OR Hereditary MultipleExostoses/de OR Hereditary()Multiple()Exostos?s/ti,ab
OR
Neurofibromatoses/de OR Neurofibromatosis/ti,ab OR Neurofibroma/ti,ab OR Neurosarcoma/ti,ab OR NF1()gene/ti,ab OR Von()Recklinghausen()Syndrome/ti,ab OR Neurofibromatosis/deOR Neurofibromatosis()Type()2/ti,ab OR Genes, NeurofibromatosisType 2/de
OR
Peutz-Jeghers Syndrome/de OR Peutz(1N)Jeghers()Syndrome/ti,abOR Gastrointestinal()Tumo?r?/ti,ab OR STK11()Gene/ti,ab OR 19p13/ti,ab OR Polyposis()Hamartomatous()Intestinal/ti,ab OR Polyps(3N)Spots()Syndrome?/ti,ab
OR
Retinoblastoma/de OR Retinoblastoma/ti,ab OR RB1()gene/ti,ab OR
13q14/ti,abOR

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 95/103
78
BASES DEDONNÉES
DATES/ LIMITES
VEDETTES-MATIÈRES/MOTS-CLÉS
Hippel-Lindau Disease/de OR Von() Hippel(1N)Lindau()Syndrome/ti,ab OR Hemangioblastoma/ti,ab OR VHL()Gene/ti,ab
OR 3p25/ti,ab OR Von Hippel-Lindau Disease/de OR Von Hippel-Lindau Gene/de
OR
Wilms()Tumo?r?/ti,ab OR Nephroblastoma/de OR WT1()Gene/ti,abOR 11p13/ti,ab OR WT3()Gene/ti,ab OR WT4()Gene/ti,ab
OR
((Hereditary OR Papillary) AND (Carcinoma, Renal Cell/de OR
Renal()Cell()Carcinoma)) OR Hereditary()Renal()Carcinoma/ti,ab OR Papillary()Renal()Carcinoma/ti,ab OR MET()Gene/ti,ab OR 7q31/ti,ab OR Hereditary()Papillary()Renal()Carcinoma/ti,ab OR Renal()Cell()Carcinoma()Papillary/ti,ab OR Hereditary Non-Polyposis Colorectal Cancer/de OR Hereditary NonpolyposisColorectal Cancer/de OR
((Colonic Neoplasms/de OR Colon()Cancer/ti,ab) AND(Hereditary()Non(1N)Polyposis)) OR Colorectal()Cancer/ti,ab OR Lynch()Cancer()Family()Syndrome/ti,ab OR
Colorectal()Endometrial()Carcinoma/ti,ab OR MSH2()Gene/ti,abOR MLH1()Gene/ti,ab OR PMS1()Gene/ti,ab OR PMS2()Gene/ti,abOR MSH6()Gene/ti,ab
OR
Ataxia Telangiectasia/de OR Ataxia(1N)Telangiectasia/ti,ab OR ATM()Gene/ti,ab OR Louis(1N)Bar()Syndrome/ti,ab
OR
Bloom Syndrome/de OR Bloom()Syndrome/ti,ab OR BLM()Gene/ti,ab OR 15p26/ti,ab OR Bloom’s Syndrome/de OR Bloom?s()Syndrome/ti,ab
OR

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 96/103
79
BASES DEDONNÉES
DATES/ LIMITES
VEDETTES-MATIÈRES/MOTS-CLÉS
Fanconi Anemia/de OR Fanconi’s Anemia/de OR FanconiSyndrome/de OR Fanconi?()An?emia/ti,ab OR FACA()Gene/ti,ab
OR FA()gene/ti,ab OR FAA()gene/ti,ab OR Fanconi()Pancytopenia()type()1/ti,ab FANCD()GENE/ti,ab OR FANCE()Gene/ti,ab OR FANCF()Gene/ti,ab OR FANCA()Gene/ti,ab
OR
Xeroderma Pigmentosum/de OR Xeroderma()Pigmentosa/ti,ab OR Basa()Squamous()Skin()Carcinoma/ti,ab OR XPA(1N)XPE()Gene/ti,ab
AND
(combining results from disease search with genetic tests’ terms) Genetic Techniques!/de OR Genetic Predisposition to Disease!/deOR Genetic Techniques/de OR Genetic Predisposition to Disease/deOR Genetic Screening/de OR Penetrance/de OR Mutation/de OR Gene Frequency/de OR Gene Privacy/de or Genetic Counselling/deOR Genetic Predisposition/de OR Mutational Analysis/de OR Genetic Testing/de OR Mutation Detection/de OR Genetic()Technique?/ti,ab OR Genetic()Predisposition?/ti,ab OR Genetic()Screening/ti,ab OR Mutation(3N)Detection?/ti,ab OR
Genetic()Test?/ti,ab OR Molecular()Diagnos?s/ti,ab OR Gene()Privacy/ti,ab OR Genetic()Counselling/ti,ab OR Genetic()Analys?s?/ti,ab or Genetic Analysis/de OR Genetic()Susceptibilit?/ti,ab
AND
(combining disease terms + genetic tests + terms to pick diagnostic
studies relevant to the research questions)Predictive Value of Tests/de OR Sensitivity and Specificity/de OR Analytical()Validit?/ti,ab OR Analytical()Sensitivit?/ti,ab OR
Diagnostic Errors!/de OR Sensitivit?/ti,ab OR Specificit?/ti,ab OR Predictive()Value?/ti,ab

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 97/103
80
BASES DE DONNÉES DATES/
LIMITESVEDETTES-MATIÈRES/MOTS-CLÉS
Total = 1786 unique hits
Medline = 1501 hits
Cancerlit = 2 hitsPascal = 90 hits
Biosis = 53 hits
Performed 08/15/2002
DIALOG® Alerts
MEDLINE®
EMBASE®
BIOSIS Previews®
Weekly/bi-weekly alerts set up to capture new studies
Same search strategy as the full search
Cochrane Collaboration &Update Software Ltd.The Cochrane Library,Issue 2 & 3, 2002; 1, 2003
MeSH headings and keywords as the originalDIALOG® (excluding those that contain numbers asthey are ignored by the software). Appropriate searchsyntax for Cochrane Library used.
National Center for Biotechnology Information
OMIM Database
(Online MendelianInheritance in Man)
MeSH headings and keywords
Children’s Health System& University of Washington
GENETests
Searched by keywords or gene
National Library of Medicine
PubMed
MeSH headings and keywords to capture additionalstudies (pre-medline and other studies not covered byDIALOG®). Appropriate search syntax for PubMed
was used.Websites of HTA andrelated agencies; clinicaltrial registries; other databases
NICE;ECRI; National Research Register; Universityof York NHS Centre for Reviews and Dissemination – CRD databases

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 98/103
81
Annexe 2 : Sélection des études
1 786 résumés recensés
Version intégrale de 637documents supposément pertinents
Rejet de 230 documents
• études portant sur des aspects techniques non pertinents(160)
• synthèses ne comportant pas d’informationsupplémentaire ou pertinente (70)
457 documents pertinentssélectionnés
50 articlesrepérés dansalertes et
bibliographies
5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 99/103
82
Annexe 3 : Services et laboratoires de dépistage génétique auCanada
Province Ville CliniqueAlberta Calgary Alberta Cancer Genetics Program
Division of EpidemiologyRm. AE173B, Tom Baker Cancer Centre1331 - 29 St. NWCalgary AB T2N 4N2Site Web : http://www.acgp.ca/Genetic_Counselling
Calgary Cancer Genetics Research ClinicTom Baker Cancer CentreRm. CC110, 1331 - 29 St NWCalgary AB T2N 4N2Tél. : (403) 670-2438Téléc. : (403) 283-1651
Edmonton Cancer Genetics ClinicRm. 8-53 Medical Sciences BldgUniversity of AlbertaEdmonton AB T6G 2B7Tél. : (780) 407-7333Téléc. : (780) 407-6845
Colombie-Britannique
Vancouver BC Cancer Agency600 West 10th AveVancouver BC V5Z 4E6Site Web : http://www.bccancer.bc.ca
Vancouver Hereditary Cancer ProgramB.C. Cancer Agency PFC600 West 10th AveVancouver BC V5Z 4E6Tél. : (604) 877-6000, poste 2118Téléc. : (604) 872-4596
Victoria Medical GeneticsVictoria General Hospital1 Hospital WayVictoria BC V8Z 6R5Tél. : (250) 727-4212Téléc. : (250) 370-8750
Manitoba Winnipeg Hereditary Breast Cancer ClinicWHRA Breast Health Centre100 - 400 Taché AveWinnipeg MB R2H 3C3Tél. : (204) 235-3674Téléc. : (204) 231-3842
5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 100/103
83
Province Ville CliniqueTerre-Neuve-et-Labrador
St. John’s Medical Genetics ProgramHealth Care Corporation of St. John’sHealth Science Centre300 Prince Philip Dr
St. John's NL A1B 3V6Tél. : (709) 777-6223 ou 777-4363Téléc. : (709) 777 4190 ou 777- 7317
Nouvelle-Écosse / Nouveau-Brunswick /Île-du-Prince-Édouard
Halifax Maritime Medical Genetics ServiceIWK Health Centre5850-5980 University AvePO Box 3070Halifax NS B3J 3G9Tél. : (902) 428-8754Téléc. : (902) 428-8709
Ontario Hamilton Cancer Risk Assessment Clinic
Hamilton Regional Cancer Centre699 Concession StHamilton ON L8V 5C2Tél. : (905) 387-9495 ou 9711, poste 65920Téléc. : (905) 575-6326
Hamilton McMaster University Medical CentreHamilton Health SciencesRm 3N20 Genetic Services1200 Main St WHamilton ON L8S 4J9Tél. : (905) 521-5085
Téléc. : (905) 521-2651Kingston Familial Oncology ProgramKingston Regional Cancer Centre25 King St WKingston ON K7P 2N7Tél. : (613) 544-2631, poste 4124Téléc. : (613) 544-9708
London Cancer GeneticsLondon Regional Cancer Centre790 Commissioners RdLondon ON N6A 4L6
Tél. : (519) 685-8727Téléc. : (519) 685-8534Ottawa Département de génétique
Hôpital des enfants de l’est de l’Ontario401, chemin SmythOttawa ON K1H 8L1Tél. : (613) 738-3979Téléc. : (613) 738-4822

5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 101/103
84
Province Ville CliniqueOttawa Programme sur le cancer du côlon héréditaire
Institut Moses et Rose Loeb pour la recherche médicaleHôpital Civic d’Ottawa725, avenue Parkdale
Ottawa ON K1Y 4E9Tél. : (613) 798-5555, poste 7805Téléc. : (613) 761-5365
Sudbury Familial Cancer Familial Cancer Risk Northeastern Ontario Regional Cancer Centre116 - 41 Ramsey Lake RdSudbury ON P3E 5J1Tél. : (705) 522-6237, poste 2060Téléc. : (705) 523-7328
Thunder Bay Thunder Bay District Health Unit Northwestern Ontario Regional Cancer Centre
290 Munro StThunder Bay ON P7A 7T1Tél. : (807) 343-1610Téléc. : (807) 345-2630
Toronto Ontario Cancer Genetics Network Division of Preventive OncologyCancer Care Ontario620 University AveToronto ON M5G 2L7Site Web :http://www.cancercare.on.ca/prevention/ocgn.html
Toronto Genetics DepartmentCredit Valley Hospital1860 - 2200 Eglinton Ave WMississauga ON L5M 2N1Tél. : (905) 813-4104Téléc. : (905) 813-4347
Toronto Familial Breast Cancer ClinicMount Sinai Hospital1286 - 600 University AveToronto ON M5G 1X5Tél. : (416) 586-3244
Téléc. : (416) 586-8659Toronto Familial GI Cancer RegistryMount Sinai Hospital1157 - 600 University AveToronto ON M5G 1X5Tél. : (416) 586-8334Téléc. : (416) 586-8644Site Web : www.mtsinai.on.ca/familialgican
5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 102/103
85
Province Ville CliniqueToronto Familial Ovarian Cancer Clinic
Princess Margaret Hospital610 University Ave Rm M-700Toronto ON M5G 2M9
Tél. : (416) 946-2270Téléc. : (416) 946-2288Toronto Breast Cancer Clinic
University Health Network Princess Margaret Hospital8-502A - 610 University AveToronto ON M5G 2M9Tél. : (416) 946-4409Téléc. : (416) 946-4410
Toronto Genetics Programme North York General Hospital
391 - 4001 Leslie St North York ON M2K 1E1Tél. : (416) 756-6345Téléc. : (416) 756-6727
Toronto Familial Breast Cancer Research UnitThe Centre For Research in Women’s HealthWomen’s College Hospital750A - 790 Bay St., 7th Floor Toronto ON M5G 1N8Tél. : (416) 351-3765Téléc. : (416) 351-3767
Site Web : http://www.utoronto.ca/crwhToronto Department of Preventive OncologyToronto-Sunnybrook Regional Cancer Centre2075 Bayview AveToronto ON M4N 3M5Tél. : (416) 480-6835Téléc. : (416) 480-6002Site Web : http://www.swchsc.on.ca/
Windsor Windsor Regional Cancer Centre2220 Kildare RdWindsor ON N8W 2X3
Tél. : (519) 253-5253Téléc. : (519) 253-4204
5/9/2018 201 Familial Cancer Tr f - slidepdf.com
http://slidepdf.com/reader/full/201-familial-cancer-tr-f 103/103
86
Province Ville CliniqueQuébec Montréal Division de génétique médicale
Hôpital général de Montréal1650, avenue Cedar, pièce L10-120Montréal QC H3G 1A4
Tél. : (514) 937-6011, poste 4067Téléc. : (514) 934-8273Montréal Clinique des cancers familiaux de
Montréal / CHUMPavillon Masson de l’Hôtel-Dieu 8-0313850, rue Saint-UrbainMontréal QC H2W 1T8Tél. : (514) 890-8104Téléc. : (514) 412-7131
Québec Département de génétique humaineCentre hospitalier de l’Université Laval
2705, boul. Laurier Québec QC G1V 4G2Tél. : (418) 654-2103Téléc. : (418) 654-2748
Saskatchewan Saskatoon Saskatoon Cancer Centre20 Campus Dr Saskatoon SK S7N 4H4Tél. : (306) 655-6717Téléc. : (306) 655-2639
Saskatoon Division of Medical GeneticsUniversity of Saskatchewan
Royal University HospitalSaskatoon SK S7N 0X0Tél. : (306) 966-1692Téléc. : (306) 966-1736