7gjxz lvaavxz fhe=h7c7 :;b b78eh7jeh?e :; …babec.org/files/pdf_maestro/amgen-bruce...
TRANSCRIPT


Prefacio

Prefacio
La importancia de la ciencia que están por aplicar a veces se da por sentada ya que los protocolos se han estudiado y se han vuelto a estudiar una y otra vez, de modo que exista una alta probabilidad de que tengan éxito y obtengan el producto molecular deseado. El trabajo que están a punto de realizar se basa en una ciencia desarrollada por científicos galardonados con el Premio Nobel. Werner Arbor, Daniel Nathans y Hamilton Smith recibieron este premio por su trabajo con enzimas de restricción. Stanley Cohen, Paul Berg y Herb Boyer lo recibieron por obtener la primera molécula de ADN recombinante. La molécula de ADN recombinante que ustedes utilizarán va más allá de su trabajo porque emplea el gen de un organismo eucariota, en lugar de uno procariota. Hace apenas unos años, en 1993, Kary Mullis recibió el Premio Nobel por el descubrimiento de la reacción en cadena de la polimerasa, una suerte de química elegante que utilizarán en el Laboratorio 8. De modo que la ciencia que aprenderán durante las próximas semanas es muy importante y continuará teniendo un papel importante en el desarrollo de la biotecnología y la medicina.
Su profesor merece buena parte del crédito por hacer que esta experiencia de laboratorio sea posible. Si bien Amgen provee los equipos y suministros necesarios para implementar los laboratorios, para que fuera posible utilizarlos, su profesor ha aportado muchas horas de preparación, a menudo durante las noches y los fines de semana. Si han disfrutado de esta experiencia de laboratorio, recuerden agradecerle a su profesor por haberla hecho realidad.
Este programa de extensión educativa es en gran medida el resultado del esfuerzo del Dr. Bruce Wallace, científico de Amgen, que creía fervientemente que la industria de la biotecnología tenía la responsabilidad de contribuir a la educación científica de nuestra sociedad. Antes de su fallecimiento, el Dr. Wallace logró ver su programa de extensión educativa crecer y evolucionar hacia la aventura del descubrimiento que usted está a punto de iniciar.
Estamos en condiciones de traer este programa a su establecimiento gracias a varias sociedades importantes: la Fundación Amgen, la Fundación de Pierce College, el Centro de Biotecnología del Condado de Orange/Los Angeles, el Instituto Bio-Bridge (biobridge.ucsd.edu/a1), New England Biolabs, Fotodyne, Invitrogen, Rainin Pipettes, VWR y Bio-Rad.
Si tienen alguna pregunta sobre estas prácticas de laboratorio, no duden en enviarme un correo electrónico a la dirección que se indica a continuación.
Mar tin IkkandaProfesor de BiologíaLos Angeles Pierce College

Tabla de Contenidos

Tabla de
Contenidos
Introducción a los microvolúmenes y el pipeteado
Análisis de restricción de pARA y pKAN-R
Digestión de restricción de pARA-R Introducción a los plásmidos y las enzimas de restricción
Ligación de fragmentos de restricción de pARA y pKAN-R Producción de un plásmido recombinante: pARA-R
Confirmación de la restricción y la ligación utilizando electroforesis en gel de agarosa
Confirmación de la digestión de restricción de pARA-R
Transformación de la Escherichia coli con un plásmido recombinante
Transformación de la Escherichia coli con pARA-R Preparación de un cultivo “overnight” de la Escherichia coli
Purificación de mFP a partir de un cultivo “overnight”
Extracción del ADN genómico de células epiteliales bucales
1.1 – 1.6
2.1 – 2.3
2a.1–2a.4
3.1–3.3
4.1–4.4
4a.1–4a.4
5.1–5.6
5a.1–5a.4
6.1 – 6.2
7.1 – 7.6
8.1 – 8.5
Tabla de Contenidos

Laboratorio 11
1.1
Introducción a los
microvolúmenes y el pipeteado
El propósito de esta práctica de laboratorio es ofrecerles experiencia en el uso de algunas de las herramientas y las técnicas importantes más comunes en la biología molecular y presentarles algunas de las mediciones volumétricas de uso más frecuente en este campo de la ciencia. Se les brindará la oportunidad de practicar algunas de las destrezas que necesitarán para construir una molécula de ADN recombinante. Los instrumentos y suministros que utilizarán durante las próximas semanas son idénticos a los que se utilizan en los laboratorios de investigación.
Si bien los fundamentos teóricos sobre los que se han construido la biotecnología y las ciencias de ADN se remontan a principios de 1900, la mayoría de las técnicas de laboratorio empleadas son relativamente recientes. Y, aunque las técnicas que irán aprendiendo en las próximas semanas se han vuelto de rutina en los modernos laboratorios de investigación, son pocos los estudiantes secundarios y universitarios que tienen la oportunidad de experimentar la biología molecular con tal grado de sofisticación.
Cada vez que asistan a una clase de química, una de las cosas que notarán rápidamente será las diferencias en las cantidades de reactivos y productos químicos que em-plean. En un laboratorio de química típico, los volúmenes se miden en grandes cilindros graduados. Por lo general, las soluciones se miden en volúmenes de 50, 100 ó 200 mililitros (ml). Generalmente, el peso de los elementos
sólidos se expresa en gramos (g). En el laboratorio de biología molecular, por lo general los volúmenes se miden en microlitros (µl); donde 1 µl equivale a 0.001 ml. El peso a menudo se expresa en términos de microgramos (µg) o nanogramos (ng), donde 1 µg equivale a 0.000001 gramo y 1 ng equivale a 0.000000001 gramo.
Quizás se estén preguntando por qué los biólogos moleculares emplean volúmenes y cantidades de material tan pequeños. El motivo se relaciona con el costo de estos materiales y lo difícil que resulta obtenerlos. Por ejemplo, en el próximo laboratorio recibirán algunos plásmidos diseñados especialmente (ADN). Si este ADN se vendiera “por libra”, ésta costaría unos 360 millones de dólares. De modo que no se sorprendan si sólo les entregamos una cantidad diminuta de moléculas de ADN. La razón por la que estos productos químicos son tan costosos se relaciona con la dificultad para prepararlos en forma pura. Muchos de estos productos químicos se producen en organismos vivos, como las bacterias, y deben purificarse y separarse de las otras miles de sustancias presentes en la célula. Sin embargo, la biología molecular realmente requiere este nivel de pureza y precisión. A medida que realicen este trabajo de laboratorio, recuerden que están realizando biología molecular como en el mundo real.

Laboratorio 11
1.2
Los protocolos de biología molecular exigen la utilización de micropipetas ajustables. Las micropipetas se emplean para verter diferentes volúmenes de líquidos. Si bien los investigadores tienen varios tipos de micropipetas en su mesa de trabajo, estos laboratorios han sido diseñados para utilizar una P-20. Esta pipeta está fabricada para verter entre 2 y 20 µl. Se trata de un instrumento de precisión de alta calidad y es fundamental que aprendan a usarlo correctamente. Les pedimos que lean y sigan estas precauciones:
La micropipeta digital
■ No definan el ajuste por debajo de 2 µl ni por encima de 20 µl, a menos que su profesor se los indique.
■ No usen la micropipeta sin la punta desechable adecuada firmemente ajustada al cilindro. Si no utilizan una punta para pipeta contaminarán el cilindro de la pipeta.
■ No dejen la micropipeta con líquido en la punta ni la sostengan con la punta hacia arriba. Si la punta desechable no está bien ajustada al cilindro, el líquido puede caer nuevamente dentro de la pipeta.
■ Eviten que el émbolo haga un “chasquido” al retirar o eyectar líquido; con el tiempo esto destruirá el pistón.
Tubos de microfuga de 1.5 mlMicropipeta P-20 (2-20 µl) Puntas para pipeta desechablesMarcador indelebleEquipo de electroforesisSuministro eléctricoGradilla plástica para tubos de microfuga
Materiales
Solución 1Solución 2Solución 3H2O destilada (dH2O)Gel de agarosa al 0.8% (preparado con anterioridad)1 x NaB (o TBE 0.5x)
Métodos
Botón del émbolo
Eyector de la punta
Ventana del visor
Cilindro

Laboratorio 11
■ Al aspirar (succionar) una solución, presionen el émbolo hacia la primera marca y bajen la punta de la pipeta por debajo del nivel de la
solución de la que están tomando una muestra. Deben sostener el tubo que contiene la solución en la mano a la altura de los ojos. Es importante ver que la solución entre realmente en la punta de la pipeta.
■ Lentamente suelten el émbolo y dejen que el líquido ingrese a la punta de la pipeta. Asegúrense de no aspirar aire en la punta.
■ Al verter (vaciar) el líquido, coloquen la punta de la pipeta en el tubo que recibirá la solución. Ubiquen la punta de manera que toque la pared interior del tubo y se acerque al fondo. Lentamente empujen el émbolo hacia la primera marca y luego hacia la segunda.
Mantengan el pulgar en el émbolo y retiren la punta del tubo en el que están vertiendo el líquido. Esto evitará que vuelvan a aspirar el líquido en la punta de la pipeta. Asegúrense de ver que la solución salga de la punta.
■ Retiren la punta: para ello utilicen el botón eyector de la pipeta y arrójenla a un recipiente de residuos. Si vierten el mismo reactivo en tubos separados y no hay peligro de contaminación cruzada, pueden utilizar la misma punta varias veces. Para evitar la contaminación, es bueno depositar cada reactivo sobre la pared lateral cerca del fondo del tubo de microfuga sin tocar ninguno de los demás reactivos. Esta técnica les permite utilizar la misma punta para verter el reactivo en varios tubos que contengan un reactivo diferente.
■ Al verter un nuevo reactivo, utilicen siempre una punta nueva para evitar la contaminación.
1.3
Ejercicio de pipeteado Nº 1Busquen la pantalla en el mango de la micropipeta y observen su configuración. Giren la perilla estriada del mango en el sentido de las agujas del reloj para disminuir el volumen, o en sentido contrario a las agujas del reloj para aumentar el volumen. Al girar esta perilla, cambia la distancia que viaja el émbolo. Las cifras a continuación representan algunas de las configuraciones de la pipeta y los volúmenes de líquido vertido.
Coloquen una punta desechable en el extremo del cilindro de la pipeta. En un movimiento de torsión con el dedo pulgar e índice, verifiquen que la punta esté firmemente ajustada al cilindro. Eviten tocar el extremo de la punta porque pueden contaminarla. Recuerden que la punta debe estar en su lugar al utilizar la pipeta.
Coloquen el pulgar sobre el botón que activa el émbolo. Presionen el botón y observen que tiene una marca de “detención”. Si ejercen un poco más de presión con el pulgar, pueden presionar el botón del émbolo hasta llegar a una segunda marca. En esta marca se introduce un pequeño volumen de aire en la punta para eyectar la solución.
20.0 L 12.4 L 5.5 L 2.0 L
20
0
12
4
05
5
02
0

Laboratorio 11
1.4
Ejercicio de pipeteado Nº 2Utilicen un marcador indeleble para rotular los tres tubos de reacción: A, B y C.
En la tabla de la página 1.4 se resumen los contenidos de cada tubo, pero sigan las instrucciones que comienzan en el paso 3 para preparar las muestras.
Coloquen la micropipeta P-20 en 2 µl y viertan dH2O en los tubos A, B y C.
Expulsen la punta en el recipiente de residuos de plástico y reemplácenla por una nueva.
Coloquen 4 µl de la solución 1 en el tubo A.
Expulsen la punta en el recipiente de residuos de plástico y reemplácenla por una nueva.
Utilicen una punta nueva y viertan 4 µl de la solución 3 en el tubo A.
Utilicen una punta nueva y viertan 8 µl de la solución 2 en el tubo B.
Utilicen una punta nueva y viertan 8 µl de la solución 3 en el tubo C.
Guarden los tres tubos para la próxima parte de la experiencia.
Comprobación de la precisión y la consistencia del pipeteado
Los tubos A, B y C deben contener 10 µl de solución.
Preparen su micropipeta P-20 en 10 µl y coloquen una punta nueva en el cilindro.
Verifiquen con cuidado el volumen de cada tubo de microfuga. Debe haber 10 µl en cada uno de ellos.
Guarden los tubos A, B y C para la próxima parte de la experiencia.
Tubo dH20 Solución 1 Solución 2 Solución 3 Volumen totalA 2 µL 4 µL – 4 µL 10 µL
2 µL – 8 µL – 10 µL2 µL – – 8 µL 10 µL
BC

Laboratorio 11
Add comb
Agarosedissolved inelecrophoresis buffer
Pipette tip TBE buffer
Well Agarose gel
Fig. 1.3
Gel de agarosa Orificio
Punta para pipetaSolución reguladora de NaB
Utilización de la electroforesis en gel para separar moléculas
La electroforesis en gel es un método en el que se utiliza corriente eléctrica y una matriz de gel (red) para separar moléculas como el ADN y las proteínas. Las moléculas que se separan tienen carga negativa o se cargan negativamente. Con la corriente eléctrica, las moléculas cargadas son atraídas y atraviesan una red del material que separará las moléculas de acuerdo con su tamaño, aunque la forma molecular y el grado de electronegatividad influirán en el movimiento al atravesar el gel. Debido a que las moléculas tienen carga negativa, se trasladarán por el gel hacia el electrodo positivo (rojo). A mayor carga negativa, mayor velocidad en la migración de la molécula.
En este laboratorio, su profesor ha preparado un gel compuesto por agarosa, un polisacárido (azúcar compuesto). La agarosa se mezcla con una solución electrolítica llamada borato de sodio (NaB). Esta solución contiene iones que son átomos cargados eléctricamente. Estos iones ayudan a conducir la corriente eléctrica a través del gel. Como las moléculas son atraídas hacia el electrodo positivo, las más pequeñas pueden moverse en esta red de agarosa mucho más rápido que las moléculas más grandes. Por lo tanto, en toda la extensión del gel, las moléculas se separan por tamaño.
Su profesor ya ha preparado un gel de agarosa, pero deberán cubrir el gel de agarosa con la cantidad adecuada de la solución reguladora de NaB para que el gel actúe adecuadamente. Dos grupos compartirán un gel. Lleven la cubeta hacia el suministro eléctrico que utilizarán para activar el gel.
Controlen que el gel esté ubicado en la cubeta de modo que los “orificios” estén ubicados hacia el electrodo negativo (negro). Los colorantes están cargados negativamente y se moverán hacia el electrodo positivo (rojo).
Rellenen la cubeta con solución reguladora de NaB 1x (hay varios envases plásticos que contienen esta solución reguladora en el laboratorio) hasta un nivel que cubra toda la superficie del gel a una profundidad entre 1 y 2 mm. Controlen que el gel esté cubierto con la solución reguladora y que no aparezcan “hoyuelos” en los orificios; agreguen más solución reguladora si es necesario.
Preparen la micropipeta en 10 µl y carguen cada muestra en un orificio separado como lo indica su profesor. Usen una punta nueva para cada muestra. Recuerden que su grupo compartirá este gel. Un grupo cargará sus muestras en tres separaciones a la izquierda mientras que el otro utilizará las tres separaciones a la derecha. Es recomendable que tomen nota de la solución que colocan en cada separación.
Al cargar cada muestra, coloquen la punta de la pipeta en el centro y suavemente bajen el émbolo de la pipeta para expulsar lentamente la muestra. Utilicen ambas manos para sostener la pipeta para evitar que se mueva. Dado que sus densidades son mayores que la solución reguladora de NaB, los colorantes se hundirán en los orificios.
Cierren la tapa con firmeza sobre la cámara de electroforesis. Conecten los cables eléctricos al suministro de energía. Asegúrense de que ambos cables estén conectados al mismo canal con el cátodo (–) al cátodo (negro con negro) y ánodo (+) al ánodo (rojo con rojo).
Enciendan el suministro eléctrico y establezcan el voltaje en 130–135 v.
Luego de dos o tres minutos, observen los colorantes para asegurarse de que se estén moviendo hacia el electrodo positivo (rojo). Comenzarán a ver que el colorante púrpura (llamado azul de bromofenol) comienza a separarse del colorante azul (xilenocianol).
En aproximadamente 10 minutos, o cuando puedan distinguir los tres colorantes, apaguen el interruptor de energía y desconecten los electrodos del suministro eléctrico. Háganlo tomando el enchufe del suministro eléctrico, no jalando el cable con fuerza. Retiren con cuidado la tapa de la cubeta de gel para que puedan ver mejor los colorantes en el gel.
En un papel, registren las bandas o esquema de colores en cada una de las franjas que contienen sus muestras. Utilicen esta información para responder las preguntas en la sección “Conclusiones”.
Dejen los geles en la cubeta de gel.
1.5

Laboratorio 11
Los colorantes que separaron utilizando la electroforesis en gel fueron: naranja G (amarillo), azul de bromofenol (púrpura) y xilenocianol (azul). ¿Qué carga eléctrica transportaron estos colorantes?
¿Qué evidencia les permitió arribar a esta conclusión?
El tamaño molecular puede cumplir un rol importante en la separación ya que las moléculas pequeñas se mueven por la matriz del gel con mayor rapidez que las moléculas más grandes. El peso formular o molecular de estos colorantes son: naranja G (452.38), azul de bromofenol (669.98) y xilenocianol (538.62). Según sus resultados, ¿les parece que estas moléculas se separaron claramente por el tamaño? ¿Qué otros factores pueden haber jugado un rol importante en la separación de estos colorantes?
¿Qué tubo contenía un solo colorante? ¿El A, B o C?
Nombren este colorante.
Al aspirar una solución, ¿por qué es importante ver que realmente la solución ingresa a la punta de la pipeta?
Luego de cargar el gel, ¿permanece alguna solución en los tubos A, B o C?
¿Cómo se puede explicar que la solución quede en estos tubos?
Conclusiones
1.6
.......................................................................................................................................
.......................................................................................................................................
.......................................................................................................................................
.......................................................................................................................................
.......................................................................................................................................
.......................................................................................................................................
.......................................................................................................................................
.......................................................................................................................................
.......................................................................................................................................
.......................................................................................................................................
.......................................................................................................................................

Laboratorio 12
Análisis de restricción de
pARA y pKAN-R
Los plásmidos son piezas circulares de ADN que se encuentran naturalmente en las células bacteriales. Los plásmidos utilizados en la biología molecular han sido modificados a través de la ingeniería genética para facilitar la clonación de genes y la producción de proteínas (expresión del gen) en bacterias. Los genes resistentes a los antibióticos han sido diseñados en estos plásmidos y funcionan como marcadores seleccionables; es decir, estos genes nos permiten seleccionar las bacterias que albergan los plásmidos de aquéllas que no lo hacen. Si una bacteria contiene un plásmido con un gen resistente a los antibióticos, la bacteria podrá crecer y reproducirse en presencia de ese antibiótico; aquellas bacterias sin el plásmido no podrán crecer. Por lo tanto, se pueden utilizar antibióticos para seleccionar las bacterias que son resistentes y que probablemente tienen un plásmido con el gen resistente de aquellas bacterias que no portan el plásmido. En este laboratorio se utilizarán dos plásmidos: el pARA contiene un gen de resistencia a la ampicilina, ampr, y el pKAN-R contiene un gen con resistencia a la kanamicina, kanr.
El objetivo de este laboratorio tiene tres aspectos: 1) presentar un método comúnmente utilizado para analizar los elementos genéticos del plásmido de ADN; 2) analizar el papel y la naturaleza de las enzimas de restricción; y 3) realizar los primeros pasos para producir una molécula de ADN recombinante.
El plásmido pARA tiene 4058 pares de base (pb) de tamaño. Un “par de base” sería la adenina:timina o guanina:citosina y es el método más común que se utiliza para expresar el tamaño de las moléculas de ADN. El plásmido tiene el gen ampr, que codifica la proteína betalactamasa, una enzima que destruye el antibiótico de la ampicilina. Así, la betalactamasa le permite a las bacterias reproducirse en presencia de la ampicilina. Además, el pARA porta un gen para la proteína AraC, una proteína que ayuda a la bacteria a hacer proteínas codificadas por genes insertados en este plásmido. Un gen, incluso uno extraño, se puede expresar (producir) si está dentro de una
ubicación específica en este plásmido. La región del pARA rotulada como pBAD, en el mapa del plásmido, indica el lugar donde la ARN polimerasa necesita unirse para iniciar la transcripción. Los sitios rotulados como “BamH I” y “Hind III” representan los lugares de restricción de estas dos enzimas de restricción. Analicen el mapa del plásmido a continuación y ubiquen estos componentes del plásmido.
El plásmido pKAN-R porta el gen resistente a la kanamicina, kanr, que codifica una fosfotransferasa, enzima que transfiere un grupo de fosfato a la molécula de kanamicina destruyendo sus efectos antibióticos. La kanamicina es un antibiótico que mata a las bacterias evitando que se hagan proteínas. Si una célula no puede sintetizar proteínas, morirá. El gen kanr le otorga resistencia a la kanamicina a las bacterias que absorbió este gen. Además de kanr, el plásmido tiene el gen para la proteína fluorescente mutada, mFP, llamada proteína fluorescente roja “rfp”. El plásmido pKAN-R tiene aproximadamente 5,408 pb de tamaño.
El gen de la proteína fluorescente originalmente fue aislado del Discosoma sp, una anémona de mar encontrada en el océano Indo-Pacífico. El gen en estado natural ha sido mutado mediante un proceso llamado evolución dirigida, de modo que produzca colores que son mucho más brillantes que las proteínas en estado natural. El término “en estado natural” se refiere al gen original, el que encontraría en la naturaleza. El gen rfp ha sido transformado en un plásmido pKAN-R. Observen que el gen rfp de la mFP tiene ambos sitios de restricción: BamH I y Hind III en cada lado. Un “sitio de restricción” marca la ubicación específica donde la enzima cortará al plásmido de ADN. Si pKAN-R se digiere con BamH I y Hind III, el gen rfp se cortará físicamente del plásmido. Durante este laboratorio, sacarán el gen rfp del pKAN-R y sacarán el pequeño fragmento de 40 pb del plásmido pARA utilizando las mismas enzimas. Durante el próximo laboratorio, insertarán el gen rfp en el pARA generando una molécula de ADN recombinante.
pARA4058 bpam
p r
PBAD
BamH IHind III
araC
pKAN-R5408 bpKan r
rfp702 bp
BamH I
Hind III
rfp
2.1

Laboratorio 12
2.2
A + = pARA + enzimas + solución reguladora
Micropipeta P-20 y puntasTubos de microfuga de 1.5 mlMinicentrifugador Baño María a 37 °C Marcador indeleble
Materiales
En este protocolo de laboratorio se utilizan las enzimas de restricción BamH I y Hind III para digerir los plásmidos pARA y pKAN-R. Este es el primer paso para fabricar una molécula recombinante de ADN.
Preparación de la digestión de restricción de pARA-R
pARA (80 ng/µl) pKAN-R (80 ng/ µL) Enzimas de restricción (BamH I + Hind III)
Solución reguladora de restricción 2.5x Agua destilada, dH2O
Métodos
Tubo Solución reguladora 2.5x dH20 pARA
A+
A-
K+
K-
4µL
4µL
4µL
4µL
2µL
–
–
2µL
4µL
4µL
–
–
–
–
4µL
4µL
–
2µL
2µL
–
10µL
10µL
10µL
10µL
pKAN-R Enzimas Volumen Total
Consigan los siguientes cuatro tubos de microfuga: pARA, pKAN-R, BamH I y Hind III (mezcla de enzimas) y solución reguladora 2.5x.
Tomen cuatro tubos de microfuga de 1.5 ml limpios y utilicenun marcador para rotularlos de la siguiente manera:
A + = pARA + BamH I y Hind IIIA - = pARA no digerido (pARA sin enzima) K+ = pKAN-R+ BamH I y Hind IIIK- = pKAN-R no digerido (pKAN-R sin enzima)
La matriz de reacción resume los reactivos utilizados en la digestión de restricción. Para preparar la solución de digestión, sigan las instrucciones específicas a partir del paso 4.
Utilicen una punta nueva y agreguen 4µl de solución reguladora de restricción 2.5x en todos los tubos.
Agreguen 2µl de dH2O a los tubos rotulados A- y K -. ¿Cuál es el objetivo de este paso?
Utilicen una punta nueva y agreguen 4µl de pARA a los tubos rotulados con A+ y A-.
Utilicen una punta nueva y agreguen 4µl de pKAN-R a los tubos rotulados con K + y K -.
Agreguen 2µl de la mezcla de enzimas que contiene BamH I y Hind III, a los tubos A+ y K +. Agreguen las enzimas directamente a la solución en el fondo del tubo de microfuga. Asegúrense de utilizar una punta nueva para cada tubo para evitar la contaminación. Luego de agregar las enzimas, bombeen suavemente la solución hacia adentro y hacia afuera con la pipeta para que se mezclen los reactivos y tapen los tubos.
Si hay un minicentrifugador disponible, coloquen los tubos en el rotor, asegurándose de que los tubos estén en una configuración equilibrada y centrifuguen los tubos durante cuatro segundos. Este breve giro combinará todos los reactivos en el fondo de cada tubo.
Coloquen los cuatro tubos a baño María a 37 °C e incuben durante por lo menos 60 minutos.
Luego de los 60 minutos de incubación, la solución de digestión se puede mantener congelada, a –20 °C, hasta que sea el momento de la electroforesis.

Laboratorio 12
5,.......................G G A T C C.......................3
,
3,.......................C C T A G G.......................5
,
5,.......................A 3
,
3,.......................T T C G A 5
,
BamH I
5, A G C T T......................3
,
3, A .................... 5
,Extremo cohesivo
5,.......................A A G C T T.......................3
,
3,......................T T C G A A.......................5
,Hind III
Extremo cohesivo
Analicen los mapas de restricciones de los plásmidos pARA y pKAN-R. Dado que BamH I y Hind III son endonucleasas de restricción específica, cortarán de manera consistente el ADN cuando éste se encuentre con las secuencias de reconocimiento de base seis como se indica a continuación. La ubicación exacta donde se corta se llama sitio de restricción. La molécula de ADN consta de dos cade-nas de componentes básicos de nucleótidos. Estos componentes básicos están orientados en dirección opuesta en cada cadena. Por lo tanto, se dice que las dos cadenas que constituyen la molécula de ADN son “antiparalelas”. Para comodidad, podemos decir que una cadena está orientada en dirección de 5’ (“5 primo”) a 3’ (“tres primo”), mientras que la otra cadena está orientada de 3’ a 5’. Un cuidadoso análisis de las secuencias de restricción revelará que la secuencia de los nucleótidos es un palíndromo, es decir, dice lo mismo en ambas cadenas cuando se lee en dirección 5’ 3’.
Por lo tanto, cuando el Hind III se encuentra con esta secuencia de base seis, cortará la hélice de ADN entre las bases de adenina adyacentes. Esto deja cuatro bases no apareadas que forman un “extremo cohesivo”.
¿Cuál es la secuencia de reconocimiento del BamH I? En una dirección de 5’ 3’, ¿qué secuencia de bases representa los “extremos cohesivos de cada uno”? Analicen los mapas de plásmidos pARA y pKAN-R y completen lo siguiente:
la digestión pARA producirá fragmentos y tendrá pares de base de largo. la digestión pKAN-R producirá fragmentos y tendrá pb y pb de largo. Supongan que reciben un cultivo de bacterias que tiene uno o ambos plásmidos. Diseñen un experimento simple que puedan usar para determinar cuál de estos plásmidos, pARA o pKAN-R, portaba las bacterias del cultivo.
Conclusiones
2.3

Laboratorio 12

Laboratorio 12a
2a.1
Digestión de restricción de pARA-R Introducción a los plásmidos y las enzimas de restricción
Dos herramientas poderosas pero fundamentales que se utilizan en la biotecnología son las enzimas de restricción y los plásmidos bacteriales. Las enzimas de restricción les permiten a los biólogos moleculares cortar las moléculas de ADN de diferentes organismos y recombinar las piezas moleculares para producir moléculas de ADN recombinantes. Los plásmidos son piezas circulares de ADN que se encuentran naturalmente en las bacterias. Mediante la tecnología del ADN recombinante y las enzimas de restricción, los plásmidos de ADN recombinante se pueden diseñar para que clonen genes o expresen proteínas codificadas por genes.
Las enzimas de restricción fueron observadas por primera vez por Werner Arbor en 1962. Arbor descubrió que, al parecer, algunas bacterias utilizaban un sistema inmunológico primitivo que no dejaba que el ADN viral se duplicara dentro de la célula huésped infectada. Algunos años más tarde, se descubrió que este mecanismo inmunológico involucraba una clase de proteínas ahora conocidas como enzimas de restricción. El nombre deriva de la capacidad de las enzimas de restringir el crecimiento de los virus en las células bacteriales. Las enzimas de restricción logran esto al romper un enlace en la cadena fosfato-azúcar del ADN viral; las enzimas cortan el ADN viral en pequeños fragmentos.
Las enzimas de restricción que se identificaron en primer lugar, al parecer, realizaron la digestión de la molécula de ADN al azar. Posteriormente, se encontraron y purificaron las enzimas de restricción que cortaban la cadena de fosfato-azúcar en un lugar específico o dentro de una secuencia de nucleótidos específica, por lo general, de cuatro a seis nucleótidos de largo. En la Tabla 1 se identifican algunas de estas enzimas de restricción específicas, su origen y las secuencias de nucleótidos que cada una reconoce. En 1978, Daniel Nathans (Universidad Johns Hopkins), Hamilton Smith (Universidad Johns Hopkins) y Werner Arbor recibieron el Premio Nobel de Medicina por su trabajo con las enzimas de restricción.
Tabla 1. Enzimas de restricción utilizadas en este laboratorio. indica los sitios donde se corta o se separa la cadena fosfato-azúcar.
Cuando las enzimas de restricción cortan o digieren el ADN, los fragmentos que se obtienen, llamados fragmentos de restricción, poseen varias bases no apareadas que se extienden desde los extremos de corte. Estos son llamados “extremos cohesivos”. Si las moléculas de ADN de origen diferente se digieren utilizando la misma enzima de restricción, las bases no apareadas de cada pieza deben poder unirse (o recocerse), dado que las bases no apareadas en los extremos cohesivos serán complementarias, A:T y G:C. Este es el único atributo de las enzimas de restricción que les permite a los ingenieros genéticos combinar fragmentos de ADN de diferentes organismos para producir moléculas de ADN recombinante.
(a)
(b)
Figura 1. (a) molécula de ADN con sitios de restricción BamH I y Hind III (en negritas). Las flechas indican los sitios donde las enzimas cortarán la cadena fosfato-azúcar de la molécula de ADN. (b) La molécula de ADN inferior indica la ubicación de los “extremos cohesivos” (negritas).
Los plásmidos bacteriales son piezas circulares relativamente pequeñas de ADN que las bacterias pueden portar además de su ADN genómico (cromosoma único). En la naturaleza, el plásmido de ADN por lo general lleva de uno a varios genes que ayudan a que la bacteria sobreviva, tal vez brindándole resistencia a un antibiótico. Las bacterias pueden pasar a lo largo de los plásmidos durante la conjugación (apareamiento). Las bacterias que utilizamos en el laboratorio han sido mutadas, de manera que no pueden intercambiar plásmidos durante la reproducción sexual.
Los plásmidos producidos en forma natural han sido modificados de modo que realicen funciones específicas: por lo general, la clonación y la expresión de genes. En este laboratorio se examina el pARA-R, un plásmido de ADN recombinante que ha sido modificado para que exprese el gen rfp a fin producir una proteína fluorescente roja mutante (mFP). El plásmido contiene varios elementos de control que le permiten a una bacteria portar este
Origen Enzima de restricción
BamH I 5, G G A T C C 3,3, C C T A G G 5,Bacillus
amyloliquefaciens
Secuencia de reconocimiento
EcoR I 5, G A A T T C 3,3, C T T A A G 5,Escherichia coli
Hind III 5, A A G C T T 3,3, T T C G A A 5,Haemophilus
influenzae
Origen Enzima de restricción
BamH I 5, G G A T C C 3,3, C C T A G G 5,Bacillus
amyloliquefaciens
Secuencia de reconocimiento
EcoR I 5, G A A T T C 3,3, C T T A A G 5,Escherichia coli
Hind III 5, A A G C T T 3,3, T T C G A A 5,Haemophilus
influenzae

Laboratorio 12a
pARA (70 ng/µl)Enzimas de restricción (Bamh I + Hind III)Solución reguladora de restricción 2.5x Agua destilada (dH2O)
plásmido para expresar un gen extraño. El gen se obtuvo originalmente del genoma del Discosoma sp., una anémona de mar encontrada en el océano Indo-Pacífico. En el mapa de plásmido a continuación se indican algunas de las regiones de control más importantes, araC and PBAD, y la ubicación del gen rfp. Además, se indica la ubicación de los dos sitios de restricción: uno para Hind III y otro para BamH I. ¿Cómo podrían proceder en el corte del gen rfp?
También tengan en cuenta que el plásmido posee un gen de resistencia a los antibióticos: ampr. Este gen le permitirá a la bacteria que porta el plásmido vivir en un ambiente que contenga el antibiótico ampicilina.
Micropipeta P-20 y puntasTubos de microfuga de 1.5 mlMinicentrifugadorBaño María a 37 °C
Materiales
pARA-R4720 bpam
p r
rfp702 bp
BamH I
Hind III
rfp
araC
PBAD
2a.2

Laboratorio 12a
Métodos
Soliciten a su profesor los siguientes tres tubos de microfuga de 1.5 ml: pARA-R, mezcla de enzimas y solución reguladora de restricción 2.5x.
Coloquen dos tubos de microfuga de 1.5 ml limpios y utilicen un marcador para rotular los tubos de la siguiente manera: “A+” y “A-”. Incluyan su número de grupo y horario de clase en cada tubo de manera que puedan ubicarlos en la próxima clase de laboratorio.
La matriz de reacción resume los reactivos utilizados en la digestión de restricción. Para preparar la solución de digestión, sigan las instrucciones específicas a partir del paso 4.
Utilicen una punta nueva y agreguen 4µl de solución reguladora de restricción 2.5x para ambos tubos.
Agreguen 2µl de dH2O al tubo rotulado A-. ¿Cuál es el objetivo de este paso?
Utilicen una punta nueva y agreguen 4µl de pARA-R a los tubos rotulados A+ y A-.
Lleven el tubo A+ a su profesor, quien verterá la mezcla de enzimas en el tubo o, si se les proporcionó esta mezcla de enzimas, agreguen con cuidado 2 µl de la mezcla de enzimas directamente en la solución del tubo A+ que contiene el plásmido y la solución reguladora. Luego de agregar las enzimas, cubran el tubo y den golpecitos suaves en la parte inferior de cada tubo para mezclar los contenidos.
Si hay un minicentrifugador disponible, coloquen los tubos en el rotor, asegurándose de que los tubos estén en una configuración equilibrada y centrifuguen los tubos durante cuatro segundos. Este breve giro combinará todos los reactivos en el fondo de cada tubo.
Coloquen ambos tubos a baño María a 37 °C e incuben durante por lo menos 60 minutos.
Luego de la incubación de 60 minutos, su profesor puede colocar los tubos en el congelador hasta que estén listos para la electroforesis (Laboratorio 4a).
Los plásmidos digeridos se pueden conservar a -20 °C de manera indefinida.
El objetivo de este laboratorio tiene dos aspectos: 1) analizar el rol de las enzimas de restricción y su importancia en la ingeniería genética; y 2) analizar un plásmido bacterial y cómo se utiliza en la biotecnología.
En este protocolo de laboratorio se utilizan las enzimas de restricción BamHI y Hind III para digerir el plásmido recombinante pARA-R. La digestión de
restricción aislará del pARA el gen rfp de un fragmento más grande del plásmido que contiene ampr, araC y PBAD. En el protocolo se utiliza un pARA-R de control no digerido, junto con un marcador de tamaño o escala de ADN que lo ayudará a identificar y confirmar el tamaño de los fragmentos de restricción.
Digestión de restricción de pARA-R
Preparación de la digestión de restricción de pARA-R
Tubo
A+
A-
4µL
4µL
–
2µL
4µL
4µL
2µL
–
10µL
10µL
Solución reguladora 2.5x
Mezcla de enzimas Volumen totalH2O pARA-R
2a.3

Laboratorio 12a
5,.......................A A G C T T.......................3
,
3,.......................T T C G A A.......................5
,
5,.......................A 3
,
3,.......................T T C G A 5
, 5, A G C T T......................3
,
3, A .................... 5
,Extremo cohesivo
Extremo cohesivo
¿Cuáles son las secuencias de reconocimiento de Hind III y BamH I?
En una dirección de 5’ 3’, ¿qué secuencia de bases representa los “extremos cohesivos”?
Analicen el mapa del plásmido pARA-R y completen lo siguiente: ¿Cuántos fragmentos de restricción resultarán de la digestión de pARA con BamH I y Hind III?
¿Cuáles serán las longitudes aproximadas, en pares de base, de estos fragmentos de restricción?
¿Qué fragmento de restricción llevará el gen ampr?
¿Qué fragmento de restricción llevará el gen rfp?
Supongan que su profesor les entrega un cultivo de bacterias. El cultivo puede contener bacterias que portan el plásmido pARA-R o un cultivo que contenga bacterias sin el plásmido. Diseñen un experimento simple que puedan usar para determinar cuál de los dos cultivos les entregó.
Analicen el mapa de restricción del plásmido pARA-R. BamH I y Hind III son enzimas de restricción específicas y cortarán consistentemente el ADN bicatenario cuando se encuentren con su respectiva secuencia de reconocimiento de base seis que se muestra en la tabla de la página 2a.1. Estas ubicaciones de corte se llaman sitios de restricción. La molécula de ADN consta de dos cadenas de componentes básicos de nucleótidos. Estos componentes básicos están orientados en dirección opuesta en cada cadena. Por lo tanto, se dice que las dos cadenas que constituyen la molécula de ADN son “antiparalelas”. Por convención, podemos decir que una cadena está orientada en dirección de 5’ (“5 primo”) a 3’ (“tres primo”), mientras que la otra cadena está orientada de 3’ a 5’. Un cuidadoso análisis de las secuencias de restricción BamH I y Hind III revelará que las secuencias de nucleótidos son palíndromos; es decir, dicen lo mismo en ambas cadenas cuando se leen en sentido 5’ 3’
Por lo tanto, cuando el Hind III se encuentra con esta secuencia de base seis, cortará la hélice de ADN entre las bases de adenina adyacentes. Esto deja cuatro bases no apareadas que forman un “extremo cohesivo”.
Conclusiones
2a.4
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................

Laboratorio 3
3.1
Ligación de fragmentos de restricción pARA/pKAN-R que
producen un plásmido recombinante pARA-R
En este laboratorio los fragmentos de restricción producidos durante el Laboratorio 2 se ligarán o se unirán, utilizando ADN ligasa, formando nuevos plásmidos recombinantes. Estos plásmidos recientemente formados representarán moléculas de ADN recombinante porque los cuatro fragmentos de restricción han sido recombinados de diferentes maneras para producir nuevas construcciones. Por ejemplo, suponga que los cuatro fragmentos de plásmidos se representaron con las letras A, A’, K y R, en los que A y A’ representan los fragmentos pARA, y K y R representan los dos fragmentos que resultan de la digestión de pKAN-R. Los plásmidos se pueden representar con cualquier combinación de dos letras, como AK o A’R, y cualquier combinación incluso de fragmentos numerados, como por ejemplo AKA’R o ARAAKK y así sucesivamente. Como pueden ver, hay muchos tipos de moléculas recombinantes que pueden resultar de mezclar estos fragmentos de restricción.
Como recordarán, las enzimas de restricción que estamos utilizando son BamH I y Hind III. Al cortar los plásmidos en los sitios de restricción BamH I y Hind III se dejan “extremos cohesivos”. Los extremos cohesivos del ADN cortado pueden ligarse a cualquier otro fragmento de ADN que tenga un extremo cohesivo adicional. Analicen el mapa del plásmido pARA a continuación para ver las ubicaciones de los sitios de restricción de BamH I y Hind III y los extremos cohesivos que se forman en los extremos de 5’ de su fragmento de restricción.
Dado que pARA tiene un sitio de restricción BamH I y otro Hind III, la digestión dejará dos fragmentos. A continuación se representan los fragmentos de restricción. Es importante
recordar que el fragmento de restricción más grande porta el gen ampr, el gen que suministra la resistencia a la ampicilina.
El fragmento más pequeño no porta ningún gen.
El plásmido pKAN-R tiene un sitio de restricción BamH I y otro Hind III que flanquea el gen rfp. La digestión de pKAN-R dejará dos fragmentos, uno será de 4706 pb y el otro, de 702 pb.
Con la ligación se unirá cualquiera de los dos extremos cohesivos BamH I con cualquiera de los dos extremos cohesivos Hind III. Deberían poder observar que son posibles combinaciones diferentes de fragmentos. La combinación que nos interesa es la del fragmento recombinado de 4018 pb pARA (que contiene el gen ampr) con el fragmento de 702 pb pKAN-R (gen rfp). La combinación de estos dos fragmentos proporcionará un plásmido recombinante que llamaremos pARA-R.
La ligación del fragmento de 702 pb pKAN-R colocará al gen rfp del plásmido en un lugar que le permitirá a la bacteria sintetizar (expresar) la proteína fluorescente mutante, mFP.
Inicialmente, los fragmentos de restricción se mantienen juntos por el enlace de hidrógeno entre las bases de los
5, G A T C C A 3
,
3,
G T T C G A 5, 4018 pb
5’ A G C T T G 3,
3’ A C C T A G 5’ 40 pb
pARA4058 pbam
p r
PBAD
BamH IHind III
araC
5, G A T C C A 3
,
3,
G T T C G A 5, 4706 pb
5’ A G C T T G 3,
3’ A C C T A G 5’ 702 pb
pKAN-R5408 pbKan r
rfp702 pb
BamH I
Hind III
rfp
pARA4720 pbam
p r
rfp 702 pb
BamH I
Hind III
rfp
araC
PBAD

Laboratorio 3
3.2
Ligación de fragmentos de restricción pARA/pKAN-R que
producen un plásmido recombinante pARA-R
nucleótidos que forman los extremos cohesivos. Cabe recordar que la adenina y la timina comparten dos enlaces de hidrógeno mientras que la citosina y la guanina comparten tres. Esto asegura que solamente coincidirán los extremos cohesivos complementarios.
Los enlaces de hidrógeno son enlaces químicos débiles y no son adecuados para mantener los extremos cohesivos juntos de manera permanente. La enzima ADN ligasa, con la energía suministrada por ATP, formará enlaces covalentes entre el azúcar y los grupos de fosfato del esqueleto del ADN. En el diagrama a continuación pueden ver las ubicaciones de estos enlaces en cada lado de la molécula de ADN. Cuando se forman los enlaces covalentes, estos completan la unión fosfodiéster entre los dos azúcares y el grupo de fosfato de cada cadena. Los enlaces químicos que resultan constituyen un enlace relativamente fuerte.
Tomen los tubos A+ y K+ de la gradilla que se encuentra en el frente de la clase. Coloquen los dos tubos a baño María a 70 °C durante 30 minutos. Esta exposición al calor desnaturalizará (inactivará) cualquier BamH I y Hind III que pudiera estar activo. ¿Por qué es importante esto?
Mientras sus tubos se encuentren a baño María, solicítenle a su profesor la solución reguladora 5x y un tubo de ligasa. El tubo de ligasa contiene 2 µl de ADN ligasa. Rotulen este tubo con sus iniciales.
Luego de 30 minutos, duración del paso de incubación a 70 °C, agreguen 4 µl de A+ directamente en la ligasa de ADN en el fondo del tubo de Ligasa.
Utilizando una punta nueva, agreguen 4 µl de K+ a la solución en el tubo de Ligasa.
Utilizando una punta nueva, agreguen 3 µl de solución reguladora de ligación 5x directamente en la solución en el fondo del tubo Ligasa. Desechen el tubo de la solución reguladora.
Agreguen 2 µl de dH2O al tubo de Ligasa, utilizando una punta limpia. Suave y lentamente muevan el émbolo hacia adentro y hacia afuera para mezclar los reactivos. Realicen este paso sin salpicar la solución en los laterales del tubo de microfuga. En la tabla a continuación se resumen los contenidos del tubo de Ligasa.
Si hay gotitas de líquido en los laterales del tubo, centrifuguen brevemente el tubo para combinar los reactivos.
Coloquen sus tubos de ligasa, A+ y K+ en las gradillas en el frente de la clase. Su tubo de ligasa permanecerá durante la noche a temperatura ambiente.
Métodos
Materiales
pARA digerido (A+ del laboratorio 2) pKAN-R digerido (K+ del laboratorio 2) Solución reguladora de ligación 5x con ATP ADN ligasa T4 en el tubo rotulado “Lig” Agua destilada
A+
4µL4µL 3µL 2µL 2µL 15µL
K+ dH2O LigasaSolución reguladora de ligación 5x Volumen total
53
,
3,
5
G C
A T
A T
T A
C G
T AADN ligasa+ ATP
ADN ligasa+ ATP
53
,
3,
5
G C
A T
A T
T A
C G
T A
Micropipeta P-20 y puntasBaño María a 70 °C Gradilla plástica para tubos de microfuga Marcador indeleble

Laboratorio 3
¿Por qué es importante colocar los tubos A+ y K+ a baño María a 70 °C antes de preparar la reacción de ligación?
¿Qué creen que podría pasar si se omitiera este paso?
Realicen un diagrama para mostrar cómo se unieron los siguientes extremos cohesivos. (: = enlace de hidrógeno) Consulten la página 3.2 para ver el ejemplo de apareamiento de bases.
Si bien son muchos los plásmidos recombinantes, dibujen tres plásmidos recombinantes posibles. Incluyan dentro de esos tres, la combinación en la que estamos más interesados, la que combina pARA con el fragmento pKAN-R que porta el gen rfp.
¿Pueden dos fragmentos rfp unirse y circular en el tubo de ligasa?
En la molécula de ADN, hay dos tipos de enlaces químicos: enlaces químicos covalentes y enlaces de hidrógeno. Describan brevemente cómo difieren estos enlaces en cuanto a qué tan fuertes son y en qué parte de la molécula de ADN los encontrará.
Durante la ligación, ¿cuáles de estos enlaces (hidrógeno o covalente) se forma primero? ¿Dónde se forman? ¿Qué enlaces se forman después y dónde?
¿Para formar qué enlaces se necesita ADN ligasa?
Conclusiones
A
T T C G A
A G C T T
A . .. . . .. .
3.3
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................
...........................................................................................................................................

Laboratorio 3

Laboratorio 14
4.1
Confirmación de la restricción y la ligaciónUso de electroforesis en gel de agarosa
Es importante en esta etapa de nuestro procedimiento experimental que confirmemos que el BamH I y Hind III han digerido los plásmidos pKAN-R y pARA originales, y que los fragmentos de restricción han sido ligados por el ADN ligasa. En este laboratorio se demostrará que tenemos moléculas de ADN recombinantes.
La electroforesis en gel es un procedimiento comúnmente utilizado para separar fragmentos de ADN según su tamaño molecular. Como los colorantes que separaron en el Laboratorio 1, los fragmentos de ADN migrarán por el laberinto de la agarosa. El ADN, debido a los grupos de fosfato, tiene carga negativa y se moverá hacia el electrodo positivo (rojo). Dado que es más fácil que las moléculas pequeñas se muevan por la matriz de agarosa, éstas migrarán más rápido que los fragmentos más grandes. Imagínense un grupo de corredores de campo traviesa cruzando una espesa selva tropical. Con todos los demás factores iguales, los corredores de menos altura podrán circular por la espesura de enredaderas que cuelgan y el denso follaje más rápido que los corredores más altos. De manera que los fragmentos más pequeños de ADN se moverán más rápido por la espesura de las moléculas de agarosa que los fragmentos más grandes.
Tomaremos todas las muestras de plásmidos: digeridos, no digeridos y ligados, y utilizaremos la electroforesis para separar estas piezas. Tal vez predijeron que sus plásmidos sin cortar producirían solamente una banda de ADN; no hay motivo para pensar lo contrario. Sin embargo, es posible que aparezcan dos o tres líneas en las franjas de plásmidos no digeridos. El motivo de esto es que los plásmidos aislados de las células existen de diversas formas. Una de las formas del plásmido se llama “superenrollada”. Es posible visualizar esta forma pensando en una pieza circular de tubería plástica retorcida. La torcedura o enrollamiento tiene como resultado una molécula muy compacta, que se moverá por el gel rápidamente debido a su tamaño.
Una segunda forma de plásmido es llamada “círculo mellado” o “círculo abierto”. Con frecuencia, un plásmido experimentará el rompimiento de uno de los enlaces covalentes ubicados en la cadena de fosfato-azúcar a lo largo de una de las dos cadenas de nucleótidos. El congelamiento y descongelamiento repetido del plásmido u otro tratamiento severo puede causar el rompimiento. Cuando esto ocurre, la tensión acumulada en el plásmido superenrollado se libera a medida que el plásmido enrollado se desenrolla. Esta forma de plásmido circular no se moverá tan fácilmente por el gel de agarosa como la forma superenrollada. Si bien tiene el mismo tamaño, en términos de pares de base, estará ubicado más cerca del orificio que la forma superenrollada.
La última forma del plásmido que podemos ver se llama “multímero”. Cuando las bacterias duplican los plásmidos, éstos por lo general se duplican tan rápido que terminan unidos como los eslabones de una cadena. Si los dos plásmidos se unen, el multímero será dos veces más grande que un solo plásmido y migrará muy lentamente por el gel. De hecho, se moverá más lentamente que el círculo mellado. Sus muestras de pKAN-R – y pARA –, luego, pueden tener tres bandas que aparecen en el gel. Comenzando muy cerca del orificio, pueden observar que el multímero se desplaza primero, seguido de una banda con forma circular mellada y finalmente una banda superenrollada.
Utilizaremos una técnica de tinción especial que nos permite ver los fragmentos incrustados en el gel; luego realizaremos un registro fotográfico del gel para documentar este importante paso.

Laboratorio 14
4.2
Muestras de plásmidos: K-, K+ A-, A+ Plásmido ligado (tubo “LIG”) Gel de agarosa al 0.8% Colorante de carga 5x NaB 1x (o TBE 0.5x) Marcador de tamaño del ADN (25 ng/µl)
Confirmación de la restricción y la ligaciónUso de electroforesis en gel de agarosa
Micropipeta P-20 y puntasTubos de microfuga de 1.5 ml Aparato de electroforesisSuministro eléctricoLapicera para marcarGradilla plástica para tubos de microfuga
Materiales
Reúnan las cinco muestras de plásmidos y el marcador de ADN que su profesor les brindará y colóquenlos en la gradilla plástica para tubos. Deben tener seis tubos.
Consigan cinco tubos de microfuga de 1.5 ml limpios y rotúlenlos de la siguiente manera: A-, A+, K-, K+, y L. El tubo de microfuga con el marcador ya debe estar rotulado.
En la siguiente tabla se resume la preparación de la muestra de plásmidos para la electroforesis. Vean los “Consejos” antes de preparar estos tubos.
Consejos:■ Por ejemplo, al tubo rotulado con “A-”, agréguenle 4 µl de pARA-, 4 µl de dH2O
y 2 µl de colorante de carga. El colorante de carga debe ubicarse en la gradilla plástica para tubos de microfuga al lado del tubo de dH2O.
■ Si estudian esta tabla, verán que pueden agregar agua a los cinco tubos, luego agregar el colorante de carga a todos los tubos sin cambiar la punta. Luego, viertan la muestra de plásmido en cada tubo, cambiando la punta cada vez para evitar la contaminación.
■ Guarden el tubo “LIG” que contiene su plásmido ligado; debe haber alrededor de 10µl restante en este tubo. Importante: regresen el tubo “LIG” a la gradilla con los tubos, en el frente del salón, porque lo necesitarán para el próximo laboratorio.
■ Centrifuguen todas las muestras para combinar los reactivos en el fondo de cada tubo. Asegúrense de que los tubos estén ubicados en una configuración equilibrada.
Preparen el gel y la cubeta de electroforesis para recibir las muestras de plásmidos.
■ Asegúrense de que los orificios de gel estén orientados hacia el electrodo negativo (negro).
■ Viertan la solución reguladora de NaB 1x (o TBE 0.5x) sobre el gel hasta que no haya “hoyuelos” que rompan la superficie de la solución reguladora sobre los orificios. Es importante que el gel esté completamente por debajo de la solución reguladora de NaB. No obstante, es recomendable que no utilicen demasiada solución en la cubeta para permitir que la corriente eléctrica vaya por la solución reguladora y no por el gel.
Coloquen sus muestras de plásmidos y el marcador al gel, con la pipeta y las puntas.
Métodos
Continúa en la página siguiente...
Tubo
A+
A–
K–
K+
L
4 L
4 L
4 L
3 L
4 L 4 L
2 L
2 L
2 L
2 L
2 L
– –
–
–
–
–
–
–
–
–
–
– –
4 L
4 L–
–
–
–
–
–
4 L–
10 L
10 L
10 L
10 L5 L
10 L
dH20 K- Volumentotal
Colorantede carga K+ A+ LIG A-

Laboratorio 14
Add comb
Agarosedissolved inelecrophoresis buffer
Pipette tip TBE buffer
Well Agarose gel
Fig. 1.3
Gel de agarosa Orificio
Punta para pipetaSolución reguladora de NaB
Compartirán este gel con otro grupo.A menos que su profesor les haya hecho cargar sus muestras con un esquema diferente, carguen sus muestras como se indica a continuación. Sigan las instrucciones de carga que comienzan en el paso siete. Si cargan su muestra en diferente orden, asegúrense de registrarlo en su cuaderno para tomarlo como referencia más tarde.
Utilizando un punta limpia, preparen su micropipeta P-20 a 10 µl. Aspiren 10 µl de su “marcador de tamaño de ADN” y lentamente viértanlo en el orificio.
■ A medida que hacen esto, bajen suavemente la punta de la pipeta por debajo de la superficie de la solución reguladora directamente sobre, pero no dentro, del orificio. Si colocan la punta dentro del mismo pueden dañar la pared del orificio o perforar el fondo del mismo.Esto no es aconsejable.
■ Utilicen ambas manos para estabilizar la pipeta. Lentamente viertan la muestra empujando hasta la primera marca de detención de la pipeta. Debido al colorante de carga, la muestra tendrá una densidad superior a la solución reguladora de electroforesis. Esto le permitirá a la muestra hundirse en el orificio.
■ Importante: Mientras sostienen el botón en la primera marca, retiren lenta-mente la punta de la pipeta de la cubeta de gel. Si han cargado su muestra cor-rectamente, el orificio se llenará de una solución de color azul.
Realicen este mismo procedimiento con las muestras de plásmidos, siguiendo el orden in-dicado en la página 4.3. Para cada muestra cambien la punta. Si eligen cargar sus muestras en un orden diferente, asegúrense de registrar el orden de las muestras en su cuaderno.
Cierren la tapa de la cubeta de gel con firmeza sobre la cámara de electroforesis. Conecten los cables eléctricos al suministro de energía. Asegúrense de que ambos cables estén conectados al mismo canal (mismo lado), el negativo (negro) con el negativo (negro) y el positivo (rojo) con el positivo (rojo).
En el suministro eléctrico, coloquen el voltaje en 130-135 v.
Luego de dos o tres minutos, observen el gel y asegúrense de que el colorante púrpura (azul de bromofenol) se mueva hacia el electrodo positivo. Si se mueve en otra dirección, hacia el electrodo negativo (negro), verifiquen los cables eléctricos para ver que estén conectados al suministro eléctrico correctamente.
Asegúrense de regresar su tubo “LIG” al frente de la clase. Este tubo debe contener los plásmidos recombinantes que utilizarán en el próximo laboratorio.
Su profesor explicará qué hacer con los geles: escuchen con atención. Si el tiempo de laboratorio es breve, no les alcanzará para completar la electroforesis. El colorante amarillo deberá llegar justo hasta el final del gel, en alrededor de 40 ó 50 minutos.
Continuación desde la página anterior...
marcador K+ K- A+ A- L
4.3

Laboratorio 14
¿Cómo comparan sus resultados reales con sus predicciones sobre el gel?
¿Aparecen algunas bandas en la fotografía del gel que no esperaban?
¿Cómo pueden explicar el origen de estas bandas inesperadas?
¿Observan evidencia de las tres formas de plásmidos en las franjas sin cortes?
¿Hay pruebas de que hay más de una forma de multímero?
¿Por qué los plásmidos ligados están tan cerca del orificio?
Dos de los fragmentos pKAN-R de 702 pb, los fragmentos del gen rfp, pueden formar un fragmento circular porque cada extremo de los fragmentos termina en un extremo cohesivo de BamH I y Hind III. ¿Hay pruebas de que haya un fragmento circular de 1404 pb en la franja ligada?
ConclusionesRespondan a estas preguntas después de que tengan la oportunidad de analizar la fotografía del gel.
4.4
.............................................................................................................
.............................................................................................................
.............................................................................................................
.............................................................................................................
.............................................................................................................
.............................................................................................................
.............................................................................................................
.............................................................................................................
.............................................................................................................
.............................................................................................................
.............................................................................................................
.............................................................................................................
.............................................................................................................

Laboratorio 14a
4a.1
Confirmacion de
la digestión de restricción de pARA-R
El objetivo de este protocolo es revisar los fragmentos de restricción que resultan de la doble digestión de pARA-R por BamH I y Hind III (Laboratorio 2a). La electroforesis en gel es un procedimiento comúnmente utilizado para separar fragmentos de ADN según el tamaño molecular de los fragmentos de restricción o el número de pares de base. Como los colorantes que separaron en el Laboratorio 1, los fragmentos de ADN migrarán por el laberinto de la agarosa. El ADN, debido a los grupos de fosfato, tiene carga negativa y migrará hacia el electrodo positivo (rojo). Dado que es más fácil que las moléculas pequeñas se muevan por la matriz de agarosa, éstas migrarán más rápido que los fragmentos más grandes. Imagínense un grupo de corredores de campo traviesa cruzando una espesa selva tropical. Con todos los demás factores iguales, los corredores de menos altura podrían circular por la espesura de enredaderas que cuelgan y el denso follaje más rápido que los corredores más altos. De manera que los fragmentos más pequeños de ADN se moverán más rápido por la espesura de las moléculas de agarosa que los fragmentos más grandes.
Tomaremos ambas muestras de plásmido, digerido y no digerido, y utilizaremos la electroforesis para separar estos fragmentos de restricción. Tal vez predijeron que sus plásmidos sin cortar producirían solamente una sola banda de ADN: no hay motivo para pensar lo contrario. Sin embargo, es posible que aparezcan dos o tres líneas en la franja de plásmidos no digeridos (control). Este es el motivo: los plásmidos aislados de las células tienen varias formas. Una de las formas del plásmido se llama “superenrollada”. Es posible visualizar esta forma pensando en una pieza circular de tubería plástica torcida. La torcedura o enrollamiento del plásmido tiene como resultado una molécula muy compacta, que se moverá por el gel rápidamente debido a su tamaño.
Una segunda forma de plásmido es llamada “círculo mellado” o “círculo relajado”. Con frecuencia, un plásmido experimentará el rompimiento de uno de los enlaces covalentes ubicados en la cadena de fosfato-azúcar a lo largo de una de las dos cadenas de nucleótidos. El congelamiento y descongelamiento repetido del plásmido u otro tratamiento severo puede causar el rompimiento. Cuando esto ocurre, la tensión acumulada en el plásmido superenrollado se libera a medida que el plásmido enrollado se desenrolla. Esta forma de plásmido circular no se moverá tan fácilmente por el gel de agarosa como la forma superenrollada. Si bien tiene el mismo tamaño, en términos de pares de base, estará ubicado más cerca del orificio que la forma superenrollada.
La última forma de plásmido que pueden observar se llama “multímero”. Cuando las bacterias duplican plásmidos, estos por lo general se duplican tan rápido que terminan unidos como los eslabones de una cadena. Si los dos plásmidos se unen, el multímero será dos veces más grande que un solo plásmido y migrará muy lentamente por el gel. De hecho, se moverá más lentamente que el círculo mellado. La muestra de plásmido no digerido, pARA-R, puede tener tres bandas que aparecen en el gel. Comenzando muy cerca del orificio, pueden observar que el multímero se desplaza primero, seguido de una banda con forma circular mellada y finalmente una banda superenrollada.
Utilizaremos una técnica de tinción especial que nos permite visualizar los fragmentos incrustados en el gel; luego realizaremos un registro fotográfico del gel para documentar este importante paso.

Laboratorio 14a
4a.2
Confirmacion de
la digestión de restricción de pARA-R
Muestras de plásmidos:A– y A+ (del laboratorio 2a)
Gel de agarosa al 0.8%Colorante de carga 5x NaB 1x (o TBE 0.5x) Marcador de tamaño del ADN (25 ng/µl)
Micropipeta P-20 y puntasTubos de microfuga de 1.5 ml Aparato de electroforesisSuministro eléctricoGradilla plástica para tubos de microfugaLapicera para marcar
Materiales
Reúnan ambas muestras de plásmidos y el marcador de ADN que su profesor les brindará y colóquenlos en la gradilla plástica para tubos. Deben tener tres tubos.
Agreguen 2 µl de colorante de carga a los tubos A+ y A-. Tengan cuidado de no contaminar las muestras de plásmido. El colorante de carga aumentará la densidad de cada muestra para que el ADN se hunda en el orificio del gel. El colorante de carga también contiene colorantes visibles para que podamos seguir el progreso de las muestras durante la electroforesis. El marcador de tamaño del ADN ya contiene colorante de carga. Sin hacer burbujas, bombeen suavemente la pipeta varias veces para mezclar el colorante de carga con las muestras de plásmido. Asegúrense de utilizar una punta nueva por cada muestra de plásmido para evitar la contaminación.
Preparen el gel y la cámara de electroforesis para recibir las muestras de plásmidos.
■ Asegúrense de que los orificios de gel estén orientados hacia el electrodo negativo (negro).
■ Viertan la solución reguladora de NaB 1x (o TBE 0.5x) dentro de la cubeta de electroforesis hasta que no haya “hoyuelos” que rompan la superficie de la solución reguladora sobre los orificios en el gel. Es importante que el gel esté completamente sumergido debajo de la solución reguladora, pero es recomendable que no haya demasiada solución reguladora en la cubeta, dado que la electricidad corre solamente a través de la solución reguladora y no a través del gel.
Coloquen sus muestras de plásmidos y el marcador al gel, con la pipeta y las puntas. Pueden compartir este gel con otro grupo.
A menos que su profesor les haya hecho cargar sus muestras con un esquema diferente, carguen sus muestras como se indica a continuación. Sigan las instrucciones de carga que comienzan en el paso seis. Si cargan su muestra en diferente orden, asegúrense de registrarlo en su cuaderno para tomarlo como referencia más tarde.
Métodos
marcador A – A +
Continúa en la página siguiente...

Laboratorio 14a
Utilizando una punta limpia, preparen su micropipeta P-20 a 10 µl. Aspiren 10 µl de su “marcador de tamaño de ADN” y lentamente viértanlo en el orificio.
■ A medida que hacen esto, bajen lentamente la punta de la pipeta por debajo de la solución reguladora directamente sobre, no dentro, del orificio. Si colocan la punta dentro del orificio pueden dañar la pared o perforar el fondo del mismo.
■ Utilicen ambas manos para estabilizar la pipeta. Lentamente viertan la muestra empujando hasta la primera marca de detención de la pipeta. Debido al colorante de carga, la muestra tendrá una densidad superior a la solución reguladora de NaB o TBE. Esto le permitirá a la muestra hundirse en el orificio.
■ Importante: mientras sostienen el botón en la primera marca, retiren lentamente la punta de la pipeta de la cubeta de gel. Si han cargado su muestra correctamente, el orificio se llenará de una solución de color azul que contiene la muestra.
Cambien la punta de la pipeta y carguen 12 µl de la muestra A- enel próximo orificio.
Cambien la punta de la pipeta y carguen 12 µl de la muestra A+ en el próximo orificio.
Cierren la tapa de la cubeta de gel con firmeza sobre la cámara de electroforesis. Conecten los cables eléctricos al suministro de energía. Asegúrense de que ambos cables estén conectados al mismo canal (mismo lado), el negativo (negro) con el negativo (negro) y el positivo (rojo) con el positivo (rojo).
En el suministro eléctrico, coloquen el voltaje en 130-135 v.
Luego de dos o tres minutos, observen el gel y asegúrense de que el colorante púrpura (azul de bromofenol) se mueva hacia el electrodo positivo. Si se mueve en otra dirección, hacia el electrodo negativo (negro), verifiquen los cables eléctricos para ver que estén conectados al suministro eléctrico correctamente.
Su profesor explicará qué hacer con los geles, escuchen con atención. Si el tiempo de laboratorio es corto, no les alcanzará para completar la electroforesis. El colorante amarillo deberá llegar justo hasta el final del gel, en alrededor de 30 ó 40 minutos.
Add comb
Agarosedissolved inelecrophoresis buffer
Pipette tip TBE buffer
Well Agarose gel
Fig. 1.3
Gel de agarosa Orificio
Punta para pipetaSolución reguladora de NaB
Continuación desde la página anterior...
4a.3

Laboratorio 14a
Además de utilizarla para separar fragmentos de ADN, la electroforesis puede utilizarse para calcular su tamaño real. Por ejemplo, podríamos estar buscando un gen y sospechamos que tiene determinado tamaño. La electroforesis se puede utilizar para ubicar fragmentos dentro de ese rango de tamaño. Para hacerlo, necesitaríamos utilizar un gel con una mezcla de fragmentos de ADN de tamaños conocidos. La mezcla, llamada “marcador” o “escala”, funciona como control o estándar con el que podemos comparar las ubicaciones de otras bandas de ADN en el mismo gel.
En el diagrama a continuación, la franja “marcador” contiene 10 bandas de ADN de tamaños conocidos. El tamaño de los fragmentos se brinda a continuación. Utilizando esta información y el mapa del plásmido de pARA-R, pronostiquen las ubicaciones de las bandas de ADN producidas en A- y A+. Es aconsejable revisar las diferentes formas de plásmidos descritas en la página 4a.1 y el mapa del plásmido pARA-R descrito en la página 2a.3.
Comparen la fotografía de gel con su predicción. ¿Ven alguna banda de ADN inesperada?
En relación con la escala de ADN, ¿entre qué dos bandas está ubicado el gen rfp? ¿Aquí es dónde predijeron que estaría ubicado?
En la franja A-, ¿ven algún indicio de que haya formas de plásmidos diferentes? ¿Qué forma de conformación de plásmido migra más rápido? ¿Cuál es la más lenta?
La franja A+, ¿indica una digestión completa? Expliquen su respuesta.
¿Qué fragmento de ADN contiene el gen ampr? ¿Cuál es el tamaño de este fragmento de restricción de ADN?
Conclusiones
marcador A- A+
1
2
3
4
5
6
7
8
9
10
Fragmentos marcadores10.0 par de kilobase8.0 6.0 5.0 4.0 3.0 (banda ancha)2.01.51.00.5
1234
5
7
8
9
10
6
Las preguntas 1 y 2 deben responderse antes o durante la electroforesis.
Preguntas respondidas luego de la documentación de la electroforesis.
4a.4
....................................................................................................................................................
....................................................................................................................................................
....................................................................................................................................................
....................................................................................................................................................
....................................................................................................................................................
....................................................................................................................................................
....................................................................................................................................................
....................................................................................................................................................

Laboratorio 15
5.1
Transformación de Escherichia colicon un plásmido recombinante
Hasta aquí, han producido plásmidos recombinantes ligados. Con suerte, algunos de estos ADN recombinantes tendrán fragmentos de pKAN-R de 702 pb, el gen rfp, ligados a un fragmento de restricción pARA más grande. A este plásmido se le conoce como pARA-R. Ahora, queremos que estos plásmidos recombinantes pasen a las células bacteriales para que podamos obtener las células que expresen el gen rfp y generar la proteína fluorescente mutante.
El proceso de incluir partes extrañas de ADN, como un plásmido, en una célula bacterial se denomina transformación. La transformación es un proceso que ocurre en la naturaleza, aunque probablemente es algo poco frecuente. El médico británico Frederick Griffith fue el primero en estudiar el proceso en 1928. Por lo general, las bacterias transfieren un exceso de material genético cromosomático, como los plásmidos, durante la conjugación (sexo bacterial) en lugar de dejarse libradas a su suerte. Pero incluir plásmidos puede suministrar a las bacterias determinados genes que confieren una ventaja selectiva, por ejemplo, la resistencia a los antibióticos. Sin embargo, bajo condiciones experimentales, es posible preparar células de modo que alrededor de una célula en mil tome un plásmido del medio circundante.
Existen diversos factores que determinan la eficiencia de la transformación. Dos factores están relacionados directamente con el plásmido utilizado para la transformación. Cuanto más grande sea éste, menos probabilidades habrá de que ingrese a la bacteria. Recuerden que para que la bacteria tome ADN extraño, el plásmido debe pasar por la membrana plasmática y la pared celular de las bacterias.
Por lo tanto, los plásmidos pequeños tienen más posibilidades de pasar por las membranas plasmáticas de la bacteria (E. coli tiene dos) y la pared celular que los plásmidos grandes.
Los plásmidos pueden tener diferentes formas. La forma superenrollada entra con mayor facilidad a la célula, en tanto las formas de círculo mellado o multímero, es decir, dos o más plásmidos unidos, tienen mayor dificultad para hacerlo. El tubo de ligación que contiene los plásmidos recombinantes que prepararon, no contiene ningún plásmido superenrollado. El superenrollado de un plásmido requiere una enzima que se encuentra en la célula bacterial; ésta no estaba incluida dentro de su tubo de ligación. Los plásmidos recombinantes que prepararon son principalmente de círculo mellado, pero hay una gran variedad en el tamaño.
En la naturaleza, la transformación es relativamente poco frecuente. Para aumentar las posibilidades de que plásmidos recombinantes ingresen a las células bacteriales, utilizaremos células “competentes”. Esto significa que las células están preparadas para recibir plásmidos. En su mayor parte, no se encuentran células competentes en la naturaleza; éstas deben hacerse competentes en el laboratorio. Una forma común de hacerlo es embebiendo las células en cloruro de calcio.
Recuerden que el ADN tiene carga negativa. ¿Recuerdan por qué? Las membranas plasmáticas que rodean la célula bacterial también contienen grupos de fosfato y poseen carga negativa. El problema que se presenta al intentar pasar ADN con carga negativa por una membrana con carga negativa es que las cargas eléctricas iguales tienden a rechazarse. Cuando las células se vuelven competentes, se suspenden en una solución de cloruro de calcio porque los iones del calcio (átomos de calcio con carga positiva, Ca 2+) ayudan a neutralizar las cargas eléctricas negativas de la membrana plasmática y el plásmido. Neutralizando las cargas repulsivas mediante iones de calcio, el ADN plasmídico pasa con mayor facilidad por la membrana plasmática de la célula bacterial. Las células ya fueron hechas competentes y su profesor les entregará una alícuota. No obstante, deberán realizar el siguiente paso.
Ahora que hemos neutralizado las cargas negativas del ADN y de las membranas plasmáticas, necesitamos crear una diferencia de presión entre la parte interna y externa de la célula bacterial. Esto se logra, en primer lugar, enfriando las bacterias y luego sumergiéndolas rápidamente en agua tibia. Esto se denomina “choque térmico” y crea una situación en la que la presión externa de la célula es apenas mayor que la presión interna de la misma. Este gradiente de presión ayudará a trasladar el ADN plasmídico desde el exterior hasta el interior de la célula bacterial. Luego de este duro tratamiento, tendremos que alimentar a nuestras bacterias y dejarlas recuperarse durante algunos minutos antes de diseminarlas en placas de agar.
Una vez que las células se han recuperado, se tomarán muestras de estas células y se dispondrán en una serie
Paredcelular
ADN genómico
Membranasplasmáticas
Plásmido
Plásmidos
Pared celular
ADN genómico
Membranasplasmáticas

Laboratorio 15
5.2
Transformación de Escherichia colicon un plásmido recombinante
Micropipeta P-20 y puntasMicropipeta P-200 y puntasBaño María a 42 °C1 paquete de esparcidores de células (compartido)Gradilla plástica para tubos de microfugaTubos de microfuga de 1.5 mlLapicera para marcar
Materiales
Tubo LIG (plásmidos recombinantes)100 µl de células competentes (LMG)350 µl de caldo de LB (esterilizado) Hielo molido (en una taza de poliestireno)Placas de agar, esterilizadas1 LB, 1 LB/Amp, 1 Lb/amp/ara
de placas de agar esterilizadas. Una de estas placas contendrá solamente alimentos bacteriales; la placa no contiene antibióticos. Rotulen esta placa como “LB”. La segunda, debe contener LB y ampicilina y deben rotularla como “amp”. La tercera placa debe contener LB, ampicilina y un monosacárido llamado arabinosa, y deben rotularla como “ara”.
La ampicilina es un antibiótico que evita que las bacterias formen completamente su pared celular. Las células que no resisten la ampicilina no pueden crecer en su presencia, las células nuevas simplemente se rompen o se lisan. Si una célula recibe un gen resistente a la ampicilina, ampr, producirá una proteína que destruirá químicamente la ampicilina y, de esa forma, podrá crecer con ampicilina en su medio.
La bacteria necesita arabinosa, un monosacárido, para expresar el gen rfp. Si una bacteria incluye pARA-R, la arabinosa ayuda a la enzima ARN polimerasa, necesaria para transcribir el gen rfp, para que se alinee correctamente en el plásmido. Esta relación se tratará en el próximo laboratorio.
Aunque la cepa de E. coli que se utiliza en estos laboratorios es relativamente benigna, es importante que utilicen técnicas adecuadas al momento de manipularlas.

Laboratorio 15
Tomen dos tubos de microfuga limpios. Rotulen uno “P+” y el otro “P-”.
Tomen una taza de poliestireno con hielo molido y coloquen un tubo que contenga 100 µl de células competentes dentro del hielo. Es impor tante que las células permanezcan a 0 °C. Además, coloquen los tubos P+ y P- en el hielo.
Tomen los plásmidos ligados de la gradilla para tubos de microfuga rotulada “tubos LIG”. Su tubo “LIG” deberá estar rotulado con su número de grupo y horario de clase.
Preparen la pipeta P-200 en 50 µl (configurada en “0-5-0”) y coloquen una punta limpia en su cilindro. Con mucho cuidado, vuelvan a suspender las células moviéndolas suavemente hacia adentro y hacia fuera dos veces. Sostengan el tubo por el borde superior para evitar calentar las células con los dedos.
Repar tan una alícuota de 50 µl de las células suspendidas en los tubos de microfuga P+ y P- previamente enfriados. Inmediatamente regresen las células al refrigerante. Sostengan los tubos por el borde superior para evitar calentar las células con los dedos.
Utilizando la pipeta P-20, agreguen 10 µl del plásmido ligado al tubo rotulado “P+”. Mezclen suavemente el plásmido con la suspensión de células bombeando la suspensión de células dos veces. Regresen inmediatamente el tubo P+ al hielo. No agreguen plásmido al tubo P–. Las células de este tubo ser virán para el “control de plásmidos”.
Mantengan las células en el hielo durante 15 minutos.
Mientras las células incuban en el hielo, consigan lo siguiente:Cada una de estas placas de agar:LB, LB/amp (LB + ampicilina) yLB/amp/ara (LB + amp + arabinosa)
Métodos
Rotulen las bases de las tres placas que contienen agar con su número de grupo y horario de la clase. Escriban con letra pequeña y en el borde de la placa. Luego tracen dos líneas en las placas LB y amp por la mitad. Rotulen una de las mitades de cada placa “P+” y la otra mitad “P-”. Vean el diagrama a continuación. No dividan la placa ara.
Luego de la incubación de 15 minutos en hielo, lleven la taza de hielo que contiene las células a baño María a 42 °C. Retiren los tubos del hielo y sométanlos a baño María durante 45 segundos. Luego de un choque de calor de 45 segundos, colóquenlos otra vez en el refrigerante de inmediato durante por lo menos un minuto.
Luego de un minuto, utilicen la pipeta P-200 para agregar 150 µl (configurada en “1-5-0”) de caldo de LB al tubo P-. Tapen el tubo y den golpecitos suaves en la parte inferior del tubo dos o tres veces para mezclar.
Utilicen una nueva punta y transfieran 150 µl de caldo de LB al tubo P+. Cierren la tapa y den golpecitos suaves en el tubo para mezclar.
Soliciten a su profesor un paquete de esparcidores de células esterilizados. Trabajarán con el paquete cada dos grupos.
Ahora están listos para esparcir sus células bacteriales en las placas de agar esterilizadas.
Utilizando una pipeta P–200 (configurada en “0-5-0”), muevan suavemente la pipeta dos o tres veces para volver a suspender las células; luego aspiren 50 µl de células del tubo P-. Abran la tapa de la placa LB como una “almeja”. Viertan estas células en la mitad de la placa marcada “P-”. Cierren la tapa.
Para que este laboratorio funcione sin dificultades, es importante que sepan qué tareas se les asignaron a cada miembro del grupo antes de comenzar. Un miembro debe preparar el hielo y conseguir las células competentes, otro puede recuperar los plásmidos ligados y otro puede conseguir las placas de agar, el caldo de LB y los tubos de microfuga limpios.
P+ P-
P-
Placa LB
P+ P-
Placa LB/amp
P+ P+
LB/amp/ara
5.3

Laboratorio 15
Mango Superficie donde se esparcen
Vuelvan a suspender las células bombeando suavemente con la pipeta, luego aspiren una segunda alícuota de 50 µl para la placa LB/amp. Recuerden que deben depositar las células P– en la mitad de la placa rotulada “P-”. Tapen la placa.
Abran el paquete de esparcidores de células esterilizados en la terminación más cercana a los mangos del esparcidor. Compartirán este paquete con otro grupo. Retiren sólo un esparcidor y mantengan los demás esterilizados. Sostengan el esparcidor por el mango y no permitan que la terminación doblada toque ninguna superficie, ya que podría contaminar el esparcidor. Cierren el paquete para evitar que los demás esparcidores se contaminen.
Abran la tapa de la placa LB como una almeja y, suavemente, con un leve desplazamiento,diseminen las células por la superficie del agar, manteniendo las células en el lado P– de la placa. Intenten esparcirlas de forma pareja y a los lados de la placa.
Con cuidado, diseminen las células P- en la placa LB/amp utilizando el mismo esparcidor y la misma técnica.
Coloquen el esparcidor usado en la bolsa para riesgos biológicos.
Repitan los pasos 14-a hasta 14-e para sembrar las placas LB y LB/amp con el cultivo P+. Asegúrense de utilizar la pipeta “+” y un nuevo esparcidor para evitar la contaminación.
Ahora están listos para sembrar la placa LB/amp/ara.
Utilizando la pipeta P-200 (configurada en “1-0-0”), transfieran 100 µl del cultivo P+ a la superficie de la placa LB/amp/ara. Coloquen 100 µl de células en varias áreas de la superficie del agar en vez de depositarlas en un solo lugar.
Levanten la tapa como una almeja y diseminen las células uniformemente sobre la superficie de la placa.
Giren suavemente la placa por debajo del esparcidor P+ de manera que las células se puedan diseminar por toda la superficie de esta placa. Intenten diseminar las células uniformemente también a lo largo de la pared de la placa.
Cuando terminen, tapen la placa.
Coloquen las tres placas del lado derecho hacia arriba durante cinco minutos.
Utilizando una cinta adhesiva de color, aseguren las placas y colóquenlas en la incubadora, con el gel boca arriba. Asegúrense de haber rotulado claramente las placas con su número de grupo y el horario de clase. Pueden marcar la cinta adhesiva para encontrarlas rápidamente el próximo laboratorio.
Desechen el desecho celular contaminado: esparcidores, tubos de células y puntas para pipetas, colocándolas en la bolsa para desechos celulares contaminados que su profesor les entregará.
P-
Placa LB
50 µL 50 µL
P+ P-
Placa LB/amp
P+
P-
Placa LB/amp/ara
100 µL de células
P+
P+
5.4

Laboratorio 15
Anticipen el crecimiento, si ocurre, en las siguientes placas. Recuerden que las células del cultivo P+ recibieron los plásmidos recombinantes, mientras que las del cultivo P– no. Utilicen un “+” si anticipan el crecimiento y un “–” si anticipan que no crecerán.
¿Qué tienen en común todas las células que crecen en las placas LB/amp y LB/amp/ara?
¿Qué fragmento de restricción deben tener todas para crecer en las placas con ampicilina?
¿Esperan que todas las células que crezcan en la placa LB/amp/ara se transformen con el mismo plásmido? Expliquen por qué.
¿Cómo podrían determinar qué células de la placa LB/amp/ara contienen pARA-R, el plásmido recombinante que han creado ligando el gen rfp con el fragmento de restricción pARA?
Conclusiones
P-
Placa LB
P+ P-
Placa LB/amp
P+ P+
LB/amp/ara
Respondan a las preguntas 1 a 3 antes de ver los resultados de la transformación.
5.5
.................................................................................................................................................
.................................................................................................................................................
.................................................................................................................................................
.................................................................................................................................................
.................................................................................................................................................
.................................................................................................................................................
.................................................................................................................................................

Laboratorio 15
Utilicen la siguiente tabla para comparar el resultado de la transformación real con los resultados previstos. Vean los resultados “pronosticados” en la página 5.5.
Si sus resultados reales resultaron diferentes de lo esperado, propongan razones que expliquen estas diferencias.
¿Cuántas colonias rojas estuvieron presentes en la placa LB/amp/ara?
¿Por qué las colonias rojas sólo aparecen en esta placa y no en la placa LB/amp?
¿Esperaban que algunas de las bacterias de la placa LB/amp se transformaran con pARA-R? Describan brevemente cómo podrían probar su respuesta.
Placa Resultados pronosticados Resultados reales
LB
LB/Amp
LB/Amp/Ara
Respondan a estas preguntas después de ver los resultados de la transformación.
5.6
....................................................................................................................................................
....................................................................................................................................................
....................................................................................................................................................
....................................................................................................................................................
....................................................................................................................................................
....................................................................................................................................................
....................................................................................................................................................

Laboratorio 15a
5a.1
Transformación de Escherichia colicon pARA-REl proceso de incluir partes extrañas de ADN, como un plásmido, en una célula bacterial se denomina transformación. La transformación es un proceso que ocurre en la naturaleza, aunque probablemente es algo poco frecuente. El médico británico Frederick Griffith fue el primero en estudiar el proceso en 1928. Por lo general, las bacterias transfieren un exceso de material genético cromosomático, como plásmidos, durante la conjugación (sexo bacterial) en lugar de dejarse libradas a su suerte. Pero incluir plásmidos puede suministrar a las bacterias determinados genes que confieren una ventaja selectiva, por ejemplo, la resistencia a los antibióticos. Sin embargo, bajo condiciones experimentales, es posible preparar células de modo que alrededor de una célula en mil tome un plásmido del medio circundante.
Existen diversos factores que determinan la eficiencia de la transformación. Dos factores están relacionados directamente con el plásmido utilizado para la transformación. Cuanto más grande sea éste, menos probabilidades habrá de que ingrese a la bacteria. Recuerden que para que la bacteria tome ADN extraño, el plásmido debe pasar por la membrana plasmática y la pared celular de las bacterias. Por lo tanto, los plásmidos pequeños tienen más posibilidades de pasar por las membranas plasmáticas de la bacteria (E. coli tiene dos) y la pared celular que los plásmidos grandes.
Los plásmidos pueden tener diferentes formas. La forma superenrollada entra con mayor facilidad a la célula, en tanto las formas de círculo mellado o multímero, es decir, dos o más plásmidos unidos, tienen mayor dificultad para hacerlo.
En la naturaleza, la transformación es relativamente poco frecuente. Para aumentar las posibilidades de que plásmidos recombinantes ingresen a las células bacteriales, utilizaremos células “competentes”. Esto significa que las células están preparadas para recibir plásmidos. En su mayor parte, no se encuentran células competentes en la naturaleza; éstas deben hacerse competentes en el laboratorio. Una forma común de hacerlo es embebiendo las células en cloruro de calcio.
Recuerden que el ADN tiene carga negativa. ¿Recuerdan por qué? Las membranas plasmáticas que rodean la célula bacterial también contienen grupos de fosfato y poseen carga negativa. El problema que se presenta al intentar pasar ADN con carga negativa por una membrana con carga negativa es que las cargas eléctricas iguales tienden a rechazarse. Cuando las células se vuelven competentes, se suspenden en una solución de cloruro de calcio porque los iones del calcio (átomos de calcio con carga positiva, Ca 2+) ayudan a neutralizar las cargas eléctricas negativas de la membrana plasmática y el plásmido. Neutralizando las cargas repulsivas mediante iones de calcio, el ADN plasmídico pasa con mayor facilidad por la membrana plasmática de la célula bacterial.
Ahora que hemos neutralizado las cargas negativas del ADN y de las membranas plasmáticas, necesitamos crear una diferencia de presión entre la parte interna y externa de la célula bacterial. Esto se logra, en primer lugar, enfriando las bacterias y luego sumergiéndolas rápidamente en agua tibia. Esto se denomina “choque térmico” y crea una situación en la que la presión externa de la célula es apenas mayor que la presión interna de la misma. Este gradiente de presión ayudará a trasladar el ADN plasmídico desde el exterior hasta el interior de la célula bacterial.
Una vez que las células se han recuperado, se tomarán muestras de estas células y se dispondrán en una serie de placas de agar esterilizadas. Una de las placas contendrá sólo el alimento bacterial. Esta placa no debe contener antibióticos. Rotulen esta placa como “LB”. La segunda, debe contener LB y ampicilina, y deben rotularla como “amp”. La tercera placa debe contener LB, ampicilina y un monosacárido llamado arabinosa, y deben rotularla como “ara”.
La ampicilina es un antibiótico que evita que las bacterias formen completamente su pared celular. Las células que no resisten la ampicilina no pueden crecer en su presencia, las células nuevas simplemente se rompen o se lisan. Si una célula recibe un gen resistente a la ampicilina, ampr, producirá una proteína que destruirá químicamente la ampicilina y, de esa forma, podrá crecer con ampicilina en su medio.
La bacteria necesita arabinosa, un monosacárido, para expresar el gen rfp. Si una bacteria incluye pARA-R, la arabinosa ayuda a la enzima ARN polimerasa, necesaria para transcribir el gen rfp, para que se alinee correctamente en el plásmido. Esta relación se tratará en el próximo laboratorio.
Aunque la cepa de E. coli que se utiliza en estos laboratorios es relativamente benigna, es importante que utilicen técnicas adecuadas al momento de manipularlas.
Paredcelular
ADN genómico
Membranasplasmáticas
Plásmido
Plásmidos
Pared celular
ADN genómico
Membranasplasmáticas

Laboratorio 1
5a.2
5a
P-
Placa LB
P+ P-
Placa LB/amp
P+ P+
LB/amp/ara
Transformación de Escherichia colicon pARA-R
La transformación bacterial exige técnicas estériles. Es fundamental que sigan las instrucciones con precisión.
Utilicen el marcador para rotular como P+ uno de los tubos de microfuga esterilizados de 1.5 ml y el otro tubo como P-. El ADN plasmídico se agregará sólo al tubo P+. El tubo P- representará un control plasmídico negativo.
Tomen el tubo que contiene CaCl2. Utilicen la pipeta P-200 y una punta limpia para transferir 250 µl de CaCl2 a los tubos P+ y P-. Indicación: preparen la pipeta P-200 en 125 µl y transfieran dos alícuotas a cada tubo rotulado. La pipeta debe indicar “1-2-5” en la pantalla.
El instructor entregará a la clase un plato Petri con colonias de células de E. coli. Utilicen un asa de siembra esterilizada para raspar suavemente dos o tres colonias grandes de bacterias del plato Petri y transferirlas al tubo P+. Den un golpecito con el asa contra la pared del tubo para quitar las colonias del asa. Tapen el tubo y muévanlo enérgicamente por la superficie de la gradilla para tubos de microfuga para suspender las células en CaCl2. Continúen haciendo esto hasta que no se vean aglomeraciones visibles de bacterias. Coloquen este tubo en el hielo triturado.
Tubos de microfuga de 1.5 ml Asa de siembra esterilizada Esparcidores desechables de células (2)Gradilla para tubos de microfugaMarcador indeleble Micropipeta y puntas P-20Micropipeta y puntas P-200Vaso de precipitados con desinfectanteBaño María a 42 °C
Materiales
Placa con E. coli (LMG)Hielo triturado10 µl de pARA-R (10 ng/µl)1 ml 50 mm de CaCl21 placa con LB1 placa con LB y amp1 placa con LB y amp y ara
Métodos
Preparación de células competentes para la transformación.
Repitan este procedimiento usando la misma asa de siembra, pero transfieran la colonia y suspendan las células en el tubo P-. Coloquen este tubo en el hielo triturado. Coloquen el asa de siembra en desinfectante o en una bolsa de desechos celulares para su correcta eliminación.
Transfieran 10 µl de plásmido (pARA-R) directamente en la suspensión celular del tubo P+. Agiten brevemente la mezcla dando golpecitos suaves en la base del tubo de microfuga con el dedo índice. Eviten salpicar la mezcla en la superficie lateral del tubo de transformación. Regresen el tubo P+ al hielo triturado.
Incuben ambos tubos en hielo triturado durante 15 minutos. Asegúrense de que los tubos estén en contacto con el hielo. Es importante que las células se enfríen bien.
Tomen una de cada una de las siguientes placas: LB, LB/amp y LB/amp/ara. Usando el marcador y una regla, dibujen una línea en el medio de las placas LB y LB/amp, pero no en la placa LB/amp/ara. Realicen esta división en la base de gel de ambas placas. Rotulen con una “P-” y una “P+” las mitades de las bases de las placas LB y LB/amp y con un “+” la base de la placa LB/amp/ara.

Laboratorio 15a
Transformación de E. coli con pARA-RDespués del enfriamiento en hielo de 15 minutos, realicen un choque térmico en las células de ambos tubos mediante los siguientes procedimientos:
■ Trasladen el contenedor de hielo con los tubos de transformación P+ y P- del paso 7 a baño María a 42 °C. Es importante que las bacterias experimenten un cambio bien abrupto de temperatura.
■ Mantengan ambos tubos a baño María durante 45 segundos exactamente. ■ Devuelvan ambos tubos al hielo triturado de forma inmediata durante al menos un minuto.
Después del enfriamiento de un minuto, vuelvan a colocar los dos tubos en la gradilla de tubos de ensayo y manténganlos a temperatura ambiente.
Esparcimiento de las células transformadas en placas de agarColoquen las tres placas en el siguiente orden: LB, LB/amp, LB/amp/ara.
Preparen la pipeta P-200 en 50µl. La pipeta indicará “0-5-0”. Sostengan las células P- (control) entre el dedo pulgar e índice. Den golpecitos suaves en la base del tubo para volver a suspender las células. Depositen 50 µl de estas células en la mitad “-” de las placas LB y LB/amp. No depositen estas células en la placa LB/amp/ara.
Abran el paquete de esparcidores esterilizados de células en la terminación más cercana a los mangos del esparcidor. Compartirán este paquete con otro grupo. Retiren sólo un esparcidor y mantengan los demás esterilizados. Sostengan el esparcidor por el mango y no permitan que la terminación doblada toque ninguna superficie, ya que podrían contaminar el esparcidor. Cierren el paquete para evitar que los demás esparcidores se contaminen.
Abran la tapa de la placa LB como una almeja y, con movimientos suaves y deslizantes, esparzan las células por la superficie del agar, manteniendo las células sólo en el lado “–” de la placa. Intenten esparcirlas de forma pareja y a los lados de la placa.
Mango Superficie donde se esparcen
Utilizando el mismo esparcidor, repitan el procedimiento de esparcimiento en la placa LB/amp.
Después de utilizarlo en la placa LB/amp, desechen el esparcidor de células en desinfectante o en una bolsa de desechos celulares.
Una vez que hayan vuelto a suspender las células en el tubo “P+”, utilicen una punta nueva y depositen 50 µl de células en el lado “+” de las placas LB y LB/amp; luego depositen 150 µl de las células en la placa LB/amp/ara.
Utilizando un esparcidor de células nuevo, repitan los pasos 4 y 5 esparciendo las células en el lado “+” de las placas LB y LB/amp y sobre toda la superficie de la placa LB/amp/ara.
Desechen este esparcidor de células en desinfectante o en una bolsa de desechos celulares.
Utilicen cinta adhesiva para unir las placas. Escriban su nombre en la cinta para poder reconocer las placas más adelante. Coloquen las placas boca abajo en una incubadora a 37 °C.
Incuben las placas entre 24 y 36 horas a 37 °C.
5a.3

Laboratorio 15a
Anticipen el crecimiento, si ocurre, en las siguientes placas. Recuerden que las células del cultivo P+ recibieron el plásmido, mientras que las del cultivo P– no. Utilicen un “+” si anticipan el crecimiento y un “–” si anticipan que no crecerán.
¿Qué tienen en común todas las células que crecen en las placas LB/amp y LB/amp/ara?
¿Qué fragmento del plásmido pARA-R permite que estas células crezcan en estas placas?
¿Qué tamaño tiene este fragmento en bases apareadas?
¿En qué placa/s anticiparían que las células expresarán el gen rfp?
Utilicen la siguiente tabla para comparar el resultado de la transformación real con los resultados previstos.
Si sus resultados reales resultaron diferentes de lo esperado, propongan razones que expliquen estas diferencias.
¿Cuántas colonias “rojas” estuvieron presentes en la placa LB/amp/ara?
¿Por qué las colonias rojas sólo aparecen en esta placa y no en la placa LB/amp?
Conclusiones
P-
Placa LB
P+ P-
Placa LB/amp
P+ P+
LB/amp/ara
Placa Resultados pronosticados Resultados reales
LB
LB/Amp
LB/Amp/Ara
Respondan las preguntas 1 a 3 antes de ver los resultados de la transformación.
Respondan estas preguntas después de ver los resultados de la transformación.
5a.4
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................

Laboratorio 16
6.1
Preparación de
Preparación de un cultivo “overnight” de Escherichia coli
El propósito de este laboratorio es iniciar un cultivo de bacterias que produzca una cantidad suficiente de proteína fluorescente mutante que les permita aislar y purificar la proteína. El profesor seleccionará una de las colonias rojas de la placa LB/amp/ara transformadas con el plásmido pARA-R y la usará para sembrar un cultivo “overnight”.
El gen para mFP fue aislado originalmente de una anémona de mar. La mFP se usa con mucha frecuencia en investigaciones porque la proteína se puede fusionar con otras proteínas y luego se puede hacer un seguimiento usando un microscopio fluorescente. El gen original de la proteína fluorescente se mutó para producir una molécula que tiene un brillo fluorescente muchas veces mayor. El plásmido pARA-R fue diseñado luego para la expresión del gen.
En el siguiente diagrama se ilustra la región de pARA-R que contiene los principales elementos de control requeridos para expresar el gen rfp. Es fundamental que tengan en cuenta que sólo una pequeña parte del plásmido pARA-R está representada en este diagrama y que el ADN está ilustrado como una línea recta en lugar de un círculo. El diagrama identifica tres regiones importantes: 1) el gen araC; 2) la región promotora (PBAD); y 3) la ubicación del gen rfp en relación con los demás elementos de control en el plásmido.
El gen araC representa una proteína reguladora conocida como “proteína AraC”. La proteína AraC participa en la activación y desactivación del gen rfp. En el diagrama anterior se resume la relación entre el gen AraC, la transcripción, el ARN mensajero, el desplazamiento y la proteína araC.
El sitio promotor es la parte de ADN donde ocurre la regulación de la expresión del rfp. Cuando no hay arabinosa en el medio donde se encuentra la bacteria, la proteína AraC se une físicamente con dos regiones del plásmido, el sitio promotor y una región cercana al gen araC. Esto hace que la molécula de ADN se doble, formando un bucle. Cuando
el ADN se encuentra en esta configuración, la transcripción del ARNm no puede ocurrir porque evita que la ARN polimerasa se una al sitio promotor. Sin el ARNm, la bacteria no puede producir mFP.
Cuando la arabinosa está presente en el medio de la bacteria, la arabinosa se une a la proteína AraC, formando un complejo. Esto evita que se forme el bucle de ADN. La unión de la arabinosa también origina un cambio en la conformación de la proteína (forma), que resulta en la formación de un pequeño bolsillo que ayudará a una tercera molécula, la ARN polimerasa, a unirse al complejo.
Este complejo de tres moléculas se une al sitio promotor, y la ARN polimerasa se alinea en la molécula de ADN de manera que pueda transcribir el gen rfp. Esta transcripción produce el ARNm, que se traduce como mFP. Por lo tanto, la proteína AraC tiene una función doble: puede inhibir la síntesis de mFP al permitir que el ADN forme un bucle y al evitar que la ARN polimerasa se una a la región promotora, y puede activar la transcripción del gen rfp y, por lo tanto, la producción de mFP, si se une a la arabinosa.
genaraC rfpPromotor
Proteína AraC
mRNA
Transcripción
Traducción
gen araC
genrfp
Promotor
La proteína AraC evita la transcripción del rfp causando la formación de un bucle en la región del gen rfp
mRNA
mFP
genaraC rfp
Promotor
arabinosa – Complejo de proteína araC
proteína araC
arabinosa
ARN polimerasa
Transcripción
El complejo de proteína AraC-arabinosa evita la formación de bucles en el ADN y ayuda a alinear la ARN polimerasa en el sitio promotor

Laboratorio 16
6.2
Agitador palillo de dientes limpio
Materials
Placa LB/amp/ara (del laboratorio 5 ó 5a) Frasco esterilizado con caldo LB/amp/ara
Dado que trabajará con bacterias, es impor tante que lo haga rápido para evitar la contaminación.
El instructor utilizará un palillo de dientes limpio para trans-ferir algunas células desde una colonia que exprese el mFP (rojo) a un frasco que contenga caldo LB/amp/ara.
El frasco se ubicará en un agitador y se agitará durante toda la noche para provocar la división de células y la expre-sión del gen rfp.
Métodos
Conclusiones
Aunque el instructor haya trabajado rápidamente para transferir una muestra de bacterias que expresan el gen rfp, hay probabilidad de que también se hayan transferido algunas bacterias que no expresan este gen.¿Qué evitaría el crecimiento de estas bacterias en el caldo LB/amp/ara?
El objetivo de este cultivo overnight es clonar las bacterias que expresan el gen rfp y lograr que éstas produzcan suficiente mFP para purificar la proteína de las demás proteínas de la célula. Como las células son cultivadas, ¿esperaría encontrar el mFP dentro de las células bacteriales o en el caldo de nutrientes que rodea a las células?
Cuando la bacteria expresa el gen rfp y produce la proteína fluorescente mutante, la célula toma estas moléculas de mFP y las concentra en cuerpos de inclusión. Los cuerpos de inclusión son gránulos concentrados de moléculas de mFP y no están unidos mediante una membrana.
Pared celularOtras proteínasCitoplasma
Moléculas de mFP de los cuerpos de inclusiónADN genómico
Membranas plasmáticas
rpARA
Pared celularOtras proteínas
ADN genómico
Membranas plasmáticas
pARA-R
pARA-R
Membranas plasmáticas
Moléculas de mFP de los cuerpos de inclusión
Pared celular
Moléculas de mFP de los cuerpos de inclusión
ADN genómico Otras proteínas
Citoplasma
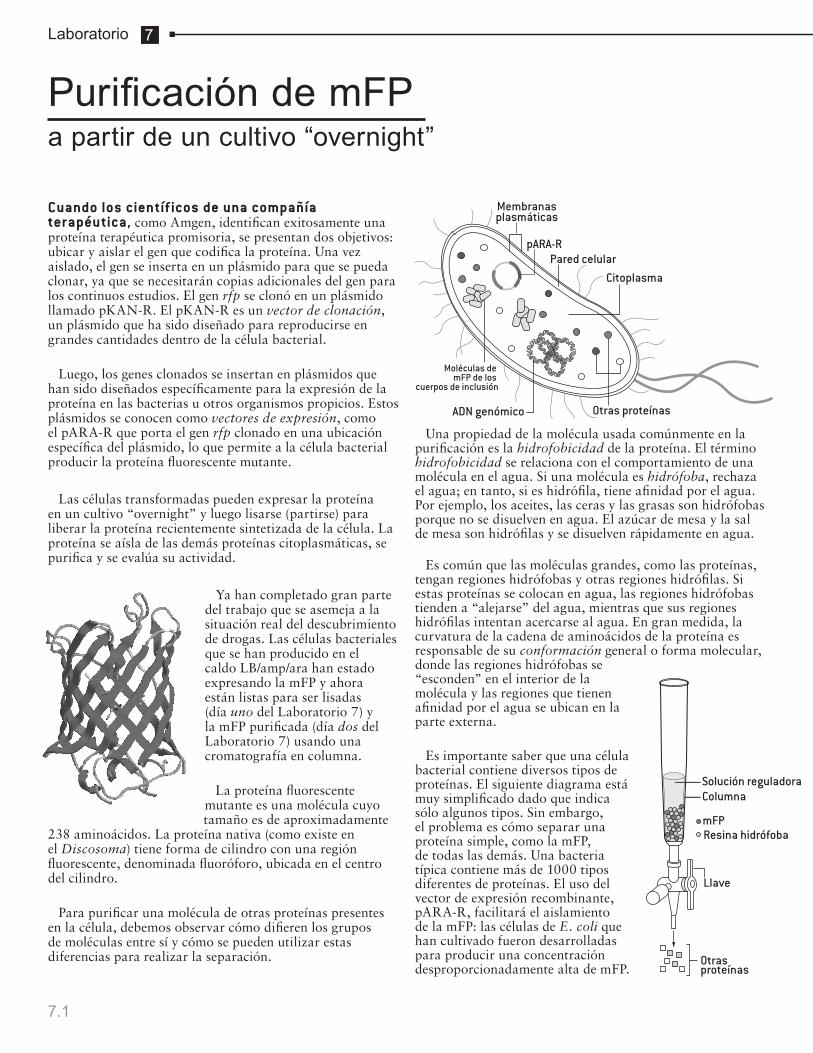
Laboratorio 17
7.1
Purificación de mFP a partir de un cultivo “overnight”
Cuando los científ icos de una compañía terapéutica, como Amgen, identifican exitosamente una proteína terapéutica promisoria, se presentan dos objetivos: ubicar y aislar el gen que codifica la proteína. Una vez aislado, el gen se inserta en un plásmido para que se pueda clonar, ya que se necesitarán copias adicionales del gen para los continuos estudios. El gen rfp se clonó en un plásmido llamado pKAN-R. El pKAN-R es un vector de clonación, un plásmido que ha sido diseñado para reproducirse en grandes cantidades dentro de la célula bacterial.
Luego, los genes clonados se insertan en plásmidos que han sido diseñados específicamente para la expresión de la proteína en las bacterias u otros organismos propicios. Estos plásmidos se conocen como vectores de expresión, como el pARA-R que porta el gen rfp clonado en una ubicación específica del plásmido, lo que permite a la célula bacterial producir la proteína fluorescente mutante.
Las células transformadas pueden expresar la proteína en un cultivo “overnight” y luego lisarse (partirse) para liberar la proteína recientemente sintetizada de la célula. La proteína se aísla de las demás proteínas citoplasmáticas, se purifica y se evalúa su actividad.
Ya han completado gran parte del trabajo que se asemeja a la situación real del descubrimiento de drogas. Las células bacteriales que se han producido en el caldo LB/amp/ara han estado expresando la mFP y ahora están listas para ser lisadas (día uno del Laboratorio 7) y la mFP purificada (día dos del Laboratorio 7) usando una cromatografía en columna.
La proteína fluorescente mutante es una molécula cuyo tamaño es de aproximadamente
238 aminoácidos. La proteína nativa (como existe en el Discosoma) tiene forma de cilindro con una región fluorescente, denominada fluoróforo, ubicada en el centro del cilindro.
Para purificar una molécula de otras proteínas presentes en la célula, debemos observar cómo difieren los grupos de moléculas entre sí y cómo se pueden utilizar estas diferencias para realizar la separación.
Una propiedad de la molécula usada comúnmente en la purificación es la hidrofobicidad de la proteína. El término hidrofobicidad se relaciona con el comportamiento de una molécula en el agua. Si una molécula es hidrófoba, rechaza el agua; en tanto, si es hidrófila, tiene afinidad por el agua. Por ejemplo, los aceites, las ceras y las grasas son hidrófobas porque no se disuelven en agua. El azúcar de mesa y la sal de mesa son hidrófilas y se disuelven rápidamente en agua.
Es común que las moléculas grandes, como las proteínas, tengan regiones hidrófobas y otras regiones hidrófilas. Si estas proteínas se colocan en agua, las regiones hidrófobas tienden a “alejarse” del agua, mientras que sus regiones hidrófilas intentan acercarse al agua. En gran medida, la curvatura de la cadena de aminoácidos de la proteína es responsable de su conformación general o forma molecular, donde las regiones hidrófobas se “esconden” en el interior de la molécula y las regiones que tienen afinidad por el agua se ubican en la parte externa.
Es importante saber que una célula bacterial contiene diversos tipos de proteínas. El siguiente diagrama está muy simplificado dado que indica sólo algunos tipos. Sin embargo, el problema es cómo separar una proteína simple, como la mFP, de todas las demás. Una bacteria típica contiene más de 1000 tipos diferentes de proteínas. El uso del vector de expresión recombinante, pARA-R, facilitará el aislamiento de la mFP: las células de E. coli que han cultivado fueron desarrolladas para producir una concentración desproporcionadamente alta de mFP.
Resina hidrófobamFP
Llave
Columna
Otras proteínas
Solución reguladora
Cell wallOther proteinsCytoplasm
Inclusion bodymFP moleculesGenomic DNA
Plasmamembranes
pARA-R
Otras proteínas
Citoplasma
Moléculas de mFP de los
cuerpos de inclusión
Pared celularpARA-R
ADN genómico
Membranasplasmáticas

Laboratorio 17
7.2
2 ml de Cultivo LB/amp/ara de E. coli (laboratorio 6)Lisozima (10 mg/ml)Solución reguladora de unión, 4 M (NH4)2SO4Solución reguladora de equilibrio de columna, 2 M (NH4)2SO4Solución reguladora de lavado de columna, 1.3 M (NH4)2SO4Solución reguladora de extracción, 10 mM TEEtanol al 20% 10% de blanqueador u otro desinfectante TE (igual al amortiguador de extracción)
Pipeta y puntas P-200Pipeta y puntas P-1000Columna de cromatografíaGradilla para tubos de microfugaTubos de microfuga de 1.5 mlMarcador indelebleTubo de recolección de residuos de 6 mlBolsa para desechos celulares contaminados
Materiales
La purificación de proteínas puede utilizar la hidrofobicidad para separar y purificar las moléculas proteicas. Un procedimiento de purificación común que utiliza las diferencias de hidrofobicidad para separar las proteínas se denomina cromatografía en columna. En la cromatografía en columna se utiliza un cilindro de plástico o vidrio donde se coloca un medio separador conocido como “resina”. El tipo específico de resina utilizado puede variar dependiendo del tipo de proteína que se desea purificar. En este laboratorio, utilizaremos una cama de resina que consta de pequeñas cuentas hidrófobas. La proteína fluorescente mutante es altamente hidrófoba y cuando la mFP se coloca en una solución de alta concentración salina, la molécula de mFP se distorsiona haciendo que las regiones hidrófobas de la molécula se adhieran a la resina hidrófoba de la columna de cromatografía. Las proteínas hidrófilas originadas por la célula continúan bajando por la columna a través de la resina sin adherirse a la cama de resina y luego se eliminan.
Una vez que la mFP queda atrapada en la cama de resina, la columna se puede lavar con una solución con menor concentración salina para extraer (lavar) las moléculas moderadamente hidrófobas de la columna. Esta
solución reguladora de lavado para la columna tendrá una concentración relativamente menor de sal que la solución utilizada para unir la mFP con la resina, pero la salinidad será lo suficientemente alta para lavar la mFP de la resina. Finalmente, podemos usar una solución con una concentración muy baja de sal para extraer o liberar la mFP de las cuentas de resina. Bajo una concentración salina baja, las regiones hidrófobas de la molécula de mFP apuntan al interior de la molécula, liberando de esa forma la mFP de la resina hidrófoba de la columna.
La purificación industrial de proteínas es mucho más compleja que este protocolo de purificación de la mFP, pero los principios aplicados por la industria son similares. La muestra de mFP que obtuvieron de esta purificación contiene otras proteínas. No obstante, el procedimiento ha retirado muchas de las demás proteínas presentes en el citoplasma de la bacteria.

Laboratorio 17
Métodos
Soliciten al profesor 1 ml del cultivo LB/amp/ara.
Analicen este cultivo. ¿De qué color es?
Coloquen este tubo en el centrifugador. Importante: ustedes o el profesor deberán asegurarse de que los tubos hayan sido colocados en el rotor en una configuración equilibrada antes de encender el centrifugador. Centrifuguen los tubos de microfuga durante 5 minutos.
Una vez que el rotor se haya detenido, retiren con cuidado el tubo para no dañar el sedimento celular.
Determinen la ubicación de la mFP. ¿Está en el sedimento de la célula bacterial o en el sobrenadante (el líquido sobre el sedimento celular)?
Una vez que hayan determinado la ubicación de la mFP, decanten cuidadosamente (extraigan) el sobrenadante en el vaso de precipitados con desinfectante. Realicen esto sin dañar el sedimento celular.
Obtengan una segunda medida de 1 ml del cultivo “overnight” y repitan los pasos del 3 al 6.
Pidan al profesor un tubo con “solución reguladora de extracción” y “lisozima”.
Inviertan el tubo de microfuga que contiene el sedimento celular y, usando papel secante, intenten eliminar la mayor cantidad de líquido
Preparación del lisado celular a partir del cultivo “overnight” líquido
que sea posible del tubo de microfuga sin tocar el sedimento celular. Desechen el papel utilizado en la bolsa para desechos celulares contaminados.
Usando la pipeta P-1000 (preparada en “0-2-5”) y una punta limpia, transfieran 250 µl de solución reguladora de extracción al sedimento celular. Cierren y ajusten bien la tapa.
Vuelvan a suspender las células moviendo enérgicamente el tubo de microfuga bien cerrado por la superficie de la gradilla para tubos de microfuga. Es posible que deban realizar esto varias veces para volver a suspender las células. Examinen el tubo cuidadosamente para asegurarse de que no haya aglomeraciones visibles de células.
Usando la pipeta P-200 (preparada en “0-4-0”), transfieran 40 µl de lisozima a las células en suspensión. La lisozima es una enzima que digiere la pared celular bacterial. Esta digestión enzimática de la pared celular debilita en gran medida a la célula, y las células comienzan a lisarse (se parten). Agiten o muevan el tubo de microfuga por la superficie de la gradilla para tubos para mezclar bien. Si el tiempo lo permite, incuben muestras a 50 °C durante 30 a 60 minutos.
Asegúrense de haber rotulado este tubo con su número de grupo y el horario de clase. Entreguen el tubo al profesor. Estas células se
colocarán en el congelador después de la incubación.
Las células se pueden conservar congeladas a –20 °C, hasta el siguiente laboratorio.
7.3

Laboratorio 17
Ahora la columna de cromatografía está lista para la muestra de mFP. Mientras esperan la muestra de mFP, asegúrense de que el fluido no esté drenando desde la columna. Si el tubo de recolección de residuos está lleno, es momento de vaciar el líquido en el fregadero.
Preparación de la muestra de mFP
Centrifuguen el lisado celular durante cinco minutos para sedimentar los restos celulares. Ustedes o el profesor deberán asegurarse de que el rotor esté equilibrado antes de cerrar la tapa y centrifugar. Es muy importante equilibrar estos tubos antes del centrifugado.
Después del centrifugado, tomen el tubo de microfuga. Examínenlo. ¿Dónde está la mFP? ¿en el sobrenadante o en el sedimento celular?
Sin dañar el sedimento de restos celulares, retiren cuidadosamente 200 µl de sobrenadante usando la pipeta P-200 (preparada en “2-0-0”) y una punta limpia. Realicen esto sin arrastrar ningún resto celular. Si desplazan el sedimento, deberán centrifugar el tubo nuevamente. Viertan los 200 µl de sobrenadante limpio y sin restos en un tubo de microfuga de 1.5 ml con el rótulo “sobrenadante” (uno de los miembros del grupo rotuló este tubo).
Reemplacen la punta de la pipeta P-200 y agreguen 200 µl de solución reguladora de unión al sobrenadante que vertieron en el tubo correspondiente. Mezclen la solución reguladora de unión con el sobrenadante bombeando suavemente las soluciones hacia afuera y adentro usando la pipeta.
Usando la pipeta P-1000 (preparada en “0-4-0”) y una punta limpia, agreguen 400 µl de esta solución, sobrenadante de mFP y solución reguladora de unión, a la columna preparada usando la misma pipeta que utilizaron para mezclar las soluciones. Realicen esto sin dañar la superficie de la cama de resina, vertiendo la solución por el costado de la columna.
Sin dejar secar la columna, abran la llave de paso y dejen que la solución de la columna drene hacia el tubo de recolección de residuos. Dejen aproximadamente 1 ó 2 mm de solución reguladora sobre la cama de resina.
Obtención de materiales
Dividan el grupo para realizar diversas tareas.
■ Persona A deberá verificar que los siguientes reactivos estén en el lugar de trabajo. Estos reactivos se compartirán con otro grupo.
Solución reguladora de unión Solución reguladora de equilibrio Solución reguladora de lavado Etanol al 20% (si su grupo es el último en usar la columna) ■ Persona B deberá pedir al profesor las células lisadas;
estas células estuvieron congeladas durante la noche. Esta persona deberá llevar las células al centrifugador para sedimentar los restos celulares.
■ Persona C deberá reunir los siguientes suministros: 2• tubos de microfuga de 1.5 ml. Rotulen un tubo como
“mFP” y otro como “sobrenadante”.
1• tubo de recolección de residuos de 6 ml (es probable que ya esté en la gradilla plástica para tubos).
Preparación de la columna
Ubiquen la columna de cromatografía según las indicaciones del profesor, tengan cuidado de no desplazar la llave de paso de la parte inferior del tubo.
Coloquen el tubo de recolección de residuos en la gradilla plástica para tubos de microfuga. Abran cuidadosamente la columna girando la válvula de la llave de paso y permitan que el fluido comience a drenar desde la columna. Dejen aproximadamente 1 mm de este líquido sobre la cama de resina para evitar que la columna se seque.
Usando la pipeta P-1000 (preparada en “1-0-0”) y una punta limpia, agreguen 3000 µl (=3 ml) de solución reguladora de equilibrio a la columna de cromatografía. Agreguen la solución reguladora lentamente por el costado de la columna para que no desestabilice la superficie de la cama de resina. Dejen que la solución reguladora fluya lentamente por el costado de la columna.
Permitan que la solución reguladora de equilibrio drene desde la columna hacia el tubo de recolección de residuos, pero dejen aproximadamente 1 ó 2 milímetros de solución reguladora sobre la cama de resina para evitar que se seque.
Purificación de la proteína fluorescente mutante a partir del lisado celular
Continúa en la página siguiente...
7.4

Laboratorio 17
Examinen la columna y coloquen la mFP. ¿La mFP está esparcida por toda la cama de resina o parece haber quedado limitada a una sola banda?
Usando la pipeta P-1000 (preparada en “1-0-0”), agreguen con cuidado 1000 µl (= 1ml) de solución reguladora de lavado por el costado de la columna. Sin dejar secar la columna, permitan que la solución reguladora drene desde la columna, dejando 1 ó 2 mm de solución reguladora sobre la cama de resina.
Examinen la columna y coloquen la mFP. ¿Ha cambiado la ubicación de la mFP en la cama de resina? La solución reguladora de lavado extraerá algunas de las proteínas menos hidrófobas de la columna. La concentración salina de la solución reguladora de lavado es menor que la de la solución reguladora de unión, pero lo suficientemente alta para hacer que la mFP se libere de la resina.
Usando la pipeta P-1000 (preparada en “1-0-0”) y una punta limpia, agreguen con cuidado 1000 µl dos veces (= 2ml en total) de solución reguladora de extracción por el costado de la columna. A medida que la mFP comience a gotear desde la punta de la llave de paso, recojan la proteína en el tubo con el rótulo “mFP”. En este tubo, recojan sólo el eluato rojo. Cuando hayan recogido toda la mFP, tapen el tubo.
Después de haber recogido toda la mFP, agreguen 2000 µl (= 2 ml) de solución reguladora de equilibrio a la columna usando la pipeta P-1000 y una punta limpia. Esto ayudará a preparar la columna para la próxima clase. Si es el último grupo en usar las columnas, agreguen 4000 µl (= 4 ml) de etanol al 20% a la columna. Drenen aproximadamente 2 ml de etanol desde la columna hacia el tubo de recolección de residuos.
Tapen y ajusten bien la columna.
La solución del tubo de recolección de residuos se puede desechar en el fregadero.
Todos los tubos de microfuga, excepto el que contiene la mFP, se deben desechar en la bolsa para desechos celulares contaminados.
Comparen su tubo con los tubos con mFP de los demás grupos. ¿Existe alguna diferencia de intensidad de color entre las muestras?
Continúa desde la página anterior...
7.5

Laboratorio 17
¿Qué característica de la mFP se utiliza como base para la separación mediante la cromatografía en columna?
Después del centrifugado del lisado celular, ¿la mFP se ubicó en el sobrenadante o en el sedimento de restos celulares?
¿Cuándo se habrían extraído las proteínas hidrófilas de la columna?
¿El eluato que contiene la mFP parece más o menos brillante que en el lisado celular después del centrifugado?
Si existe una diferencia notable en la intensidad del color rojo, ¿cuál sería la razón de la diferencia?
¿Cómo se podría ajustar o modificar la columna para aumentar la pureza de la muestra de mFP?
Aunque este laboratorio trató sobre la expresión de un gen de la anémona de mar, citen algunos ejemplos de proteínas humanas que se podrían expresar y purificar potencialmente usando métodos similares.
Conclusiones
7.6
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................

Laboratorio 18
8.1
Extracción de ADN genómico
de células epiteliales bucales
El propósito de este laboratorio es recoger una muestra de ADN de las células que recubren la parte interna de la boca y utilizar esta muestra para explorar una de las técnicas más poderosas de la biología molecular: la reacción en cadena de la polimerasa (RCP). Aunque la RCP tiene muchas aplicaciones, se utiliza comúnmente para producir numerosas copias de un segmento seleccionado del gen o locus de ADN. Por ejemplo, en la ciencia forense criminal, la RCP se utiliza para amplificar la evidencia de ADN de muestras pequeñas que podrían haber quedado en la escena del crimen. Un técnico experto hasta puede obtener una muestra de ADN de la saliva que quedó en la parte posterior de una estampilla postal que se utilizó para enviar una carta. Las muestras de ADN obtenidas de esta forma se han utilizado para la RCP en diversos casos criminales de alto perfil.
Para obtener la muestra de ADN, deberán usar un palillo de dientes y recoger células epiteliales bucales. Las células se transferirán a una solución con cuentas de Chelex. Las cuentas de Chelex ligarán los iones de magnesio bivalentes (Mg2+). Estos iones sirven a menudo como cofactores para las nucleasas que degradarán la muestra de ADN y podrían interferir con la enzima (polimerasa Taq) utilizada en la reacción. Retirando los iones de magnesio, se reduce la degradación del ADN genómico que realizan las nucleasas. Esta mezcla se colocará en agua hirviendo para lisar las células y liberar el ADN.
Luego se centrifugan la mezcla de ADN genómico, los restos celulares y las cuentas de Chelex para que sedimenten los restos celulares y el Chelex, mientras se mantiene el ADN genómico en el sobrenadante. Esta es una forma rápida y sencilla de separar el ADN genómico de los restos celulares. Sin embargo, la muestra de ADN no es pura, ya que contiene proteínas y ácidos nucleicos de los organismos que estaban en la boca al momento de tomar la muestra (mayormente bacterias y alimentos). Por lo general, estos contaminantes no inhiben la RCP porque el proceso utiliza cebadores específicos, es decir, segmentos cortos de ADN de aproximadamente 25 nucleótidos de largo que pueden seleccionar sólo el ADN genómico humano. Por lo tanto, si el sobrenadante porta ADN extraño, no debería interferir en la selección de los cebadores específicamente humanos. Más adelante, en este laboratorio se brindará una descripción más detallada de la RCP y de la función de los cebadores.
Aunque estamos utilizando epitelio bucal como fuente de ADN, también se pueden usar otros tejidos. Aquí presentamos la producción de ADN de otros tejidos humanos: la sangre produce 40 µg/ml; la raíz del cabello aproximadamente 250 ng/ml; el músculo aproximadamente 3 µg/ml; y el esperma 3.3 pg/célula.
La segunda parte de este laboratorio trata sobre la RCP real. Utilizarán la muestra de ADN genómico que han recogido como objetivo para la reacción RCP.

Laboratorio 18
8.2
Tomen un tubo con Chelex. Tengan en cuenta que este tubo está identificado con un número y una letra. Registren este número y esta letra en sus cuadernos. Sólo ustedes conocerán este código anónimo.
Para recoger células epiteliales bucales, utilicen palillos de dientes esterilizados o una punta de pipeta amarilla para raspar suavemente la parte interna de ambas mejillas. Este procedimiento no debe ser invasivo, de manera que no se debe extraer sangre.
Transfieran las células que han recogido del palillo de dientes al tubo con Chelex. Hagan girar enérgicamente el palillo de dientes en la resina Chelex para quitar las células. Es importante que retiren la mayor cantidad de células posible del palillo de dientes y las coloquen en el tubo con Chelex.
Cierren y ajusten bien el tubo con Chelex. Llévenlo a baño María hirviendo o a un bloque caliente a 100 °C. Hiervan o calienten las células durante 10 minutos. Este calentamiento lisará las células y ayudará a destruir algunas de las nucleasas que degradan el ADN.
Utilicen el centrifugador a alta velocidad para centrifugar el Chelex y los restos celulares.
Usando la pipeta P-20 y una punta de pipeta limpia, retiren cuidadosamente 20 µl de sobrenadante y colóquenlo en un tubo de microfuga limpio de 1.5 ml. Eviten aspirar las cuentas de Chelex, ya que aspirarlas inhibiría el proceso de RCP de salida. Rotulen este tubo con su código personal y anónimo (número).
Si no van a utilizar la muestra inmediatamente, déjenla en el frente del salón sobre la gradilla con el rótulo “Muestras de ADN genómico”. Estas muestras se colocarán en el refrigerador durante la noche y las tendrán en el siguiente laboratorio.
Las muestras de ADN genómico se pueden conservar en el refrigerador a 4 °C o en el congelador a –20 °C hasta que estén listas para
ejecutar la reacción RCP.
0.5 ml de solución de Chelex al 10%Mezclas maestras I y II
Extracción de ADN genómico
de células epiteliales bucales
Baño María hirviendoMicrocentrifugador Ciclador térmicoTubo de microfugaMarcador indeleblePalillos de dientes esterilizados
Materiales
Métodos
Preparación de la muestra...

Laboratorio 18
8.3
La reacción en cadena de la polimerasa, RCP, es una técnica de la biología molecular que fue descubierta por Kary Mullis a comienzos de la década de 1980. La técnica utiliza una suerte de química elegante y un ciclado termal exacto de reactivos para dirigir y amplificar una ubicación específica (locus) a lo largo de la molécula de ADN. Desde un ADN objetivo único, la RCP puede producir más de mil millones de copias del objetivo en aproximadamente una hora y media. Se comprobó que esta poderosa química es tan importante que Mullis recibió el Premio Nobel de Química en 1993. Hoy en día, la RCP es considerada un protocolo estándar en la biología molecular y todos los años se publican cientos de documentos científicos que utilizan esta técnica.
El locus que amplificaremos está ubicado en el gen Activador Plasminógeno del tejido (tPA). Este gen se encuentra en el cromosoma 8; el gen representa una proteína que participa de la disolución de coágulos de sangre. El tPA es una proteína administrada a las víctimas de ataques cardíacos para reducir la incidencia de los ataques. No obstante, la región que amplificaremos está ubicada en un intrón (región no traducida) del gen tPA.
El intrón que seleccionaremos para la amplificación es dimorfo, lo que significa que el locus tiene dos formas. Una de las formas porta un fragmento de ADN de 300 pb conocido como el elemento Alu y la segunda forma del locus no porta este fragmento. Por lo tanto, cuando examinamos
el locus, encontramos que puede o no portar un elemento Alu. En la figura a continuación se indica el intrón que seleccionaremos para la RCP.
Los elementos Alu son fragmentos cortos, de alrededor de 300 pb de ADN que se distribuyen a lo largo de nuestro genoma. Se estima que podemos portar más de 1,000,000 de copias de este fragmento. Al parecer, el elemento Alu forma parte de la codificación de ADN de una molécula ARN que ayuda a la secreción de polipéptidos recientemente formados de la célula. A menos que se inserte en un exón o región de codificación, tiene poco o ningún efecto sobre la función de la proteína.
La amplificación de ADN por la RPC depende de los cebadores que se dirigen a algunos loci específicos. Los dos cebadores que utilizaremos poseen secuencias de nucleótidos únicas que se complementan con sólo un locus en el genoma humano. Las secuencias de los cebadores son:
Amplificación del locus tPA utilizando
la reacción en cadena de la polimerasa
Cromosoma 8
5,
3,
3,
5,
gen tPA
Alu+ Alelo
Alu- Alelo
300bp
elemento Alu
400bp
100bp
Cebador
ExónIntrón
Cebador
e i e i e i e
Cebador Cebador
Cebador sentido: 5’ G T A A G A G T T C C G T A A C G G A C A G C T 3’ Cebador antisentido: 5’ C C C C A C C C T A G G A G A A C T T C T C T T 3’

Laboratorio 18
8.4
Código numérico
Amplificación del locus tPA utilizando
la reacción en cadena de la polimerasaMétodos
Tomen la muestra de ADN genómico con su número y el tubo de RCP rotulado con su código numérico anónimo.
El tubo de RCP ya contiene la mezcla maestra I. Ésta contiene los dos cebadores que se dirigirán al locus tPA, los dNTP (trifosfatos desoxinucleótidos: ATP, TTP, CTP y GTP), la solución reguladora de la RCP, el agua de grado molecular (muy pura) y la polimerasa Taq.
Utilizando una punta de pipeta limpia, agreguen 5 µl del ADN genómico a este tubo de RCP. Añadan con cuidado la muestra de ADN directamente a la mezcla maestra I de 10µl. Realicen esto sin crear burbujas.
Tapen cuidadosamente el tubo de RCP. Éste es un tubo con paredes muy delgadas, así que eviten romperlo, pero asegúrense de que la tapa esté bien colocada sobre la apertura del tubo.
Coloquen el tubo de RCP en la cubeta con hielo mediante el ciclador térmico.
Antes de colocar las muestras en el ciclador térmico, el instructor agregará 10µl de la mezcla maestra II que contiene MgCl2. La polimerasa Taq, una enzima de la bacteria Thermus aquaticus, requiere iones de Mg2+ como cofactores para activarla.
Desechen la muestra de ADN genómico.
El instructor realizará la reacción RCP en otro momento.
Después de realizar la RCP, se cargarán 15 µl del producto de la RCP en un gel de agarosa al 2%. El gel se teñirá y se documentará mediante fotografías. Estos pasos los realizará el instructor.

Laboratorio 18
Conclusiones
¿Cuál es su genotipo con respecto al gen tPA?
¿Cuántos genotipos son posibles con respecto al gen tPA?
Completen la siguiente tabla usando la información de la clase. Comiencen determinando la cantidad de cada genotipo presente en la clase.
Calculen la frecuencia (porcentaje) de cada genotipo presente.
Calculen la frecuencia de cada alelo presente en la clase.
Comparen sus resultados con otras clases. ¿Existe alguna diferencia en la frecuencia relativa de los genotipos y los alelos?
Genotipos
* El término “alelos” se refiere a las formas de un gen o una secuencia de ADN
Alu+ Alu+
Alu+ Alu–
Alu– Alu–
Alu+ alelos* Alu– alelos
Total Alu+ alelos Total Alu– alelos
Respondan estas preguntas después de ver los resultados de la RCP.
8.5
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................
.....................................................................................................................................

Laboratorio 18

Glosario
Glosario
Ácido nucleico: molécula grande que consta de subunidades ligadas al núcleo; polímero de nucleótidos.Ver también: ADN, nucleótido, polímero, ARN.
Adenina: base que contiene nitrógeno, miembrodel par de bases AT (adenina-timina). La adenina se encuentra tanto en el ADN como en el ARN.
ADN (ácido desoxirribonucleico): molécula quecodifica la información genética. El ADN es una molécula bicatenaria unida mediante enlaces de hidrógeno entre los pares de bases de los nucleótidos. Es el material hereditario.Ver también: enlaces de hidrógeno, pares de base, nucleótido.
ADN ligasa: enzima que une las cadenas de ADNformando enlaces químicos covalentes en la cadena de azúcar-fosfato.Ver también: enlace químico covalente.
ADN polimerasa: enzima utilizada para reproducir moléculas de ADN. La RCP utiliza la ADN polimerasa de la bacteria Thermus aquaticus que se denomina polimerasa Taq.Ver también: reproducción de ADN, reacción en cadena de la polimerasa.
Agarosa: polisacárido altamente purificado (carbohidrato complejo) que se usa comúnmente en la electroforesis en gel.Ver también: electroforesis.
Alelo: forma alternativa de un locus genético; se hereda un alelo simple por cada locus de cada padre (por ej: en un locus para el color de una flor, el resultado podría ser pétalos púrpura o blancos).Ver también: locus.
Alícuota: muestra de una parte de un todo (Por ej.: una muestra de un plásmido o una muestra de células).
Aminoácido: cualquiera de más de 20 moléculas que se combinan para formar proteínas. La secuencia de aminoácidos de una proteína y por lo tanto, la función de la misma son determinadas por el código genético.
a Ampicilina: antibiótico usado comúnmente que pertenece a la familia de la penicilina. La ampicilina impide que el material nuevo de la pared celular se una adecuadamente en la bacteria. Esta pared celular debilitada impedirá el crecimiento de nuevas bacterias.
Amplificación: aumento en la cantidad de copias de un fragmento específico de ADN.Ver también: clonación, reacción en cadena de la polimerasa.
Ampr: símbolo utilizado para denominar al genresistente a la ampicilina. Este símbolo se ubica en el plásmido pARA.Ver también: pARA, plásmido.
Antibiótico: sustancia que mata o impide la producción de células.Ver también: ampicilina, kanamicina.
ARN mensajero (ARNm): molécula de ácido nucleico que transporta información genética desde los genes hacia el resto de la célula para la síntesis de proteínas.
Aspirar: extraer o tomar.
Autosoma: cromosoma que aparece en pares homólogos tanto en machos como hembras y no porta los genes que determinan el sexo.
ß-lactamasa: enzima codificada por el gen amprque destruye la ampicilina.
Base: una de las moléculas que contienen nitrógeno que distingue un nucleótido de otro. En el ADN, las bases son la adenina, la guanina, la citosina y la timina.Ver también: nucleótido, par de bases, secuencia base.
Base que contiene nitrógeno: componente molecular de los nucleótidos del ADN y el ARN. En el ADN existen cuatro bases que contienen nitrógeno: A (adenina), G (guanina), T (timina), C (citosina).
Biotecnología: conjunto de técnicas biológicasdesarrolladas a través de investigaciones básicas y aplicadas actualmente en el desarrollo de investigaciones y productos. Particularmente,
b
Glosario A - B

Glosario
la biotecnología se refiere al uso industrial del ADN recombinante, la fusión celular y las nuevas técnicas de bioprocesamiento.
Cáncer: enfermedad en la que células anormales se dividen y crecen sin control.
Cancerígeno: causante del cáncer que origina cambios en el ADN de una célula.
Cebador: cadena corta y preexistente de polinucleótidos a la cual se pueden agregar nuevos nucleótidos mediante la ADN polimerasa. Se utilizan dos cebadores diferentes para dirigir el locus tPA amplificado mediante la RCP.
Célula: unidad básica de cualquier organismo viviente que porta los procesos bioquímicos de la vida.
Células somáticas: células que conforman el cuerpo y no son células sexuales o gametos. Estas células generalmente son diploides y tienen dos conjuntos de cromosomas.Ver también: gametos
Ciencia forense: ciencia especializada en la aplicación de conocimientos científicos en asuntos legales. El ADN se utiliza con frecuencia como evidencia en cuestiones legales.
Citosina: una de las bases que contienen nitrógenoy se encuentra en el ADN y el ARN. Se complementa con la guanina.Ver también: par de bases, nucleótido.
Clonación: utilización de tecnología especializada de
ADN para producir varias copias exactas de un solo gen u otro segmento de ADN. Un segundo tipo de clonación utiliza el proceso natural de división celular para realizar muchas copias de una célula entera. La composición genética de estas células clonadas, denominada línea celular, es idéntica a la célula original. Un tercer tipo de clonación produce animales y plantas completa y genéticamente idénticos.Ver también: vector de clonación.
Código genético: secuencia de nucleótidos codificados en tríos (codones) junto con el ARNm, que determina la secuencia de aminoácidos en la síntesis
de proteínas.Ver también: codones, ARNm.
Codón: grupo de tres bases de ARNm que codifica un aminoácido simple.Ver también: aminoácido, ARNm.
Columna de cromatografía: instrumento que seutiliza para separar una mezcla de moléculas.
Competentes: células capaces de incluir ADNde plásmido.Ver también: transformación.
Cromosoma: en las células eucariotas, una cadena lineal compuesta de ADN y proteína, ubicada en el núcleo de la célula que contiene los genes; en las células procariotas, una cadena circular compuesta únicamente de ADN. En los humanos, existen 23 pares de cromosomas en las células del cuerpo.
Cromosomas homólogos: cromosoma que contiene la misma secuencia genética lineal que otra, cada una derivada de un padre. Aunque la secuencia genética es (generalmente) la misma, los alelos precisos pueden diferir entre cromosomas.Ver también: alelo, cromosoma, gen.
Degradar: disminuir la calidad; convertir en un componente más simple; descomponer.
Desnaturalización: fundición del ADN a altas temperaturas en cadenas de nucleótidos simples; cambiar la forma tridimensional de una molécula de proteína.
Desoxirribosa: tipo de azúcar de cinco carbonos que se encuentra en ADN.Ver también: ADN.
Doble hélice: estructura en la cual dos cadenas deADN están torcidas en espiral alrededor una de la otra.
e
Electroforesis: movimiento de las moléculas cargadas hacia un electrodo de carga opuesta; se utiliza para separar ácidos nucleicos y proteínas. Cuando se utiliza para separar fragmentos de ADN, la electroforesis
d
Glosario C - E
c

Glosario
separa los fragmentos por tamaño, ya que los fragmentos más pequeños se mueven más rápido que los más grandes.
Elemento Alu: pieza de 300 pares de bases de ADN que ha sido insertada de forma aleatoria en todo el genoma. Ya no parece tener ninguna función en el genoma humano. Este fragmento específico de ADN se denomina elemento “Alu” porque porta una secuencia de reconocimiento de la enzima de restricción Alu.Ver también: genoma, secuencia de reconocimiento, enzima de restricción.
Eluato: solución que lava (por ej: solucionesque gotean desde la columna de cromatografía)Ver también: columna de cromatografía.
Enlace de hidrógeno: fuerza débil que resulta de la atracción de un átomo de hidrógeno positivo hacia las regiones con carga negativa de otros átomos. Ésta es la fuerza que mantiene unidas las dos cadenas de nucleótidos en la molécula de ADN. Los pares de base GC forman tres enlaces de H, en tanto los pares AT forman dos.
Enlace químico covalente: una de las fuerzas que mantiene unidos a los átomos de una molécula. Se
considera un enlace fuerte y se forma al compartir los electrones entre dos átomos.
Enzima: proteína que actúa para acelerar las reacciones químicas.
Enzima de restricción: enzima que une y corta el ADN en una secuencia base específica(por ej:
BamH I y Hind III). Ver también: secuencia de reconocimiento
Escala de kilobase: Conjunto de fragmentos de ADN estándar con longitudes que difieren en un kilobase; se utiliza como tamaño estándar en la electroforesis.Ver también: electroforesis.
Esherichia coli: bacteria común utilizada ennumerosos protocolos de biología molecular. La cepa de E. coli utilizada en los protocolos de Amgen es relativamente inofensiva y no se reproduce con facilidad fuera del medio del laboratorio.
Estado natural: la producción original o natural de un gen o una proteína.
Eucariota: organismo que alberga sus genes dentrode un núcleo y cuenta con numerosos cromosomas lineales.
Evolución dirigida: procedimiento experimentalque utiliza la RCP que causa la producción de
numerosas mutaciones aleatorias. Esas mutaciones luego se examinan o analizan en cuanto a sus propiedades.
Expresar: crear una proteína.Ver también: vector de expresión
Expresión génica: proceso mediante el cual la información codificada del gen se convierte en las estructuras presentes y operativas de la célula. Los genes expresados incluyen aquellos que se transcriben en el ARNm y luego se traducen como proteínas.Ver también: ARNm.
Ex tremos cohesivos: extremos de una molécula de ADN cortada con determinadas enzimas de restricción. Estos extremos tienen bases no apareadas.
Exón: segmento de un gen que codifica las regiones de una proteína.Ver también: intrón.
Fago: virus cuyo huésped natural es una célula bacterial.
Fenotipo: característica que se debe a la expresión de nuestros genes; generalmente se refiere a las propiedades visibles, pero se puede referir a las características reveladas mediante exámenes de laboratorio.
Fragmento de restricción: pieza de ADN que se origina cuando la molécula de ADN se corta con una enzima de restricción. Los fragmentos a menudo se separan en un gel mediante la electroforesis.
Fluorescencia: producción de luz de una molécula (por ej: la proteína fluorescente roja libera luz roja cuando se expone a la luz ultravioleta). La proteína utilizada en los Laboratorios Amgen se puede ver con luz ambiente.
Gameto: célula reproductora masculina o femenina madura.
Gen: unidad física y funcional fundamental de la herencia; secuencia ordenada de nucleótidos ubicada en un locus que codifica un producto funcional específico.Ver también: expresión génica.
Genética: estudio de la herencia.
Genoma: todo el material genético de los cromosomas
f
g
Glosario E - G

Glosario
de un organismo en particular; su tamaño se expresa generalmente mediante su cantidad total de pares de base.
Genotipo: combinación de alelos que porta un individuo para un rasgo específico.Ver también: alelo, fenotipo.
Guanina: una de las bases que contiene nitrógeno que se encuentra en el ADN y el ARN. Se complementa con la citosina.Ver también: par de bases, nucleótido.
Haploide: conjunto simple de cromosomas presente en los gametos de las plantas y los animales. El número haploide en los humanos es de 23.Ver también: diploide.
Heterócigo: las dos copias diferentes, o alelos, del mismo gen.Ver también: alelo, gen, homócigo.
Hidrófilo: que tiene afinidad por el agua; se disuelve en agua; polar. Algunos ejemplos incluyen el azúcar y la sal.
Hidrófobo: que rechaza el agua; no se disuelve en agua; no polar. Algunos ejemplos incluyen aceite, cera y proteína fluorescente mutante.
Homócigo: que tiene dos copias idénticas, o un alelo del mismo gen.Ver también: alelo, gen, heterócigo.
Ingeniería genética: alteración del material genético de las células u organismos para permitirles crear sustancias nuevas o desempeñar nuevas funciones.
Intrón: segmento de un gen que no codifica una proteína. Los intrones se transcriben en el ARNm pero se eliminan antes de ser traducidos como proteínas.Ver también: exones, ARNm, transcripción, traducción.
Kanamicina: antibiótico que mata las células no resistentes inhibiendo la síntesis de proteínas.
kanr: símbolo del gen resistente a la kanamicina que se encuentra en el pKAN plasmídico; codifica una enzima llamada fosfotransferasa que desactiva la kanamicina.
Kilobase (kb): unidad de longitud de los fragmentos de ADN igual a 1000 pares de nucleótidos.
Ligasa: enzima requerida para unir covalentemente dos fragmentos de ADN.
Ligación: reacción que une químicamente dos fragmentos de ADN dando como resultado una molécula de ADN recombinante.
Lisis: abrirse.
Locus: lugar o ubicación en un cromosoma, puede ser un gen o simplemente cualquier sitio con variaciones mensurables. (por ej: Alu+, Alu- en el intrón tPA).
Mapa de restricción: diagrama de ADN, como un plásmido, que indica los sitios de restricción para las enzimas de restricción.
Marcador: Ver: escala de kilobase.
Molécula de ADN recombinante: combinación de moléculas de ADN de diferente origen que se unen mediante técnicas de recombinación de ADN.Ver también: ligación.
Mutágeno: agente que puede causar mutaciones.Ver también: cancerígeno
Nucleasa: familia de enzimas que degradan los ácidos nucleicos.Ver también: degradar, enzima
Nucleótido: subunidad o monómero del ADN y el ARN. Cada nucleótido consta de una base con nitrógeno, un azúcar de cinco carbonos y un grupo de fosfato.
Operón: aglomeración de genes transcriptos para dar una sola molécula de ARNm. El operón arabinosa se utiliza en el vector de expresión pARA para transcribir el gen rfp.
Pared celular: estructura que brinda a las célulasapoyo físico; rodea las células bacteriales.
h
k
l
m
l
Glosario H - P
n
o
p

Glosario
Par de bases: dos bases de nitrógeno (adenina ytimina o guanina y citosina) unidas por enlaces de hidrógeno. Dos cadenas de ADN están unidas en forma de doble hélice por los enlaces entre los pares de base. La suma de los pares de base en una molécula de ADN se utiliza frecuentemente para expresar el tamaño de la molécula.
Par de bases complementarias: bases que contienen nitrógeno y que se encuentran enfrentadas entre sí en una molécula de ADN bicatenaria. La complementariedad es el resultado del tamaño (una base grande debe estar enfrentada a una base pequeña) y la cantidad de enlaces de hidrógeno entre las bases adyacentes del par (A y T forman dos enlaces de hidrógeno, G y C forman tres). La adenina se complementa con la timina y la guanina se complementa con la citosina.
Plásmido: molécula circular de ADN que se reproduce independientemente del ADN genómico del huésped (por ej: pKAN, pARA).
Polimerasa Taq: enzima con estabilidad térmica comúnmente utilizada en la RCP; la polimerasa que se encuentra en la bacteria Thermus aquaticus.Ver también: reacción en cadena de la polimerasa.
Polímero: molécula grande formada por subunidades similares o idénticas unidas entre sí (por ej: ADN, ARN, proteínas).
Polimorfismo: diferencia en la secuencia de ADN en un locus genético particular. Las diferencias pueden resultar o no en un fenotipo distinto.Ver también: locus, fenotipo.
Polimorfismo genético: diferencia en la secuencia de ADN entre individuos, grupos o poblaciones (por ej: alelos para el locus tPA amplificados por la RCP, o la altura en las plantas de guisantes, altas o bajas).
Por tador: individuo que posee un alelo no expresado y recesivo.
Procariota: célula u organismo con un solo cromosoma y sin membrana nuclear; bacterias.
Promotor: región del ADN al frente de un gen queune la ARN polimerasa y promueve de esta forma la expresión génica; en el operón ara, la región de pARA que une el complejo de proteína AraC-arabinosa y la ARN polimerasa antes de la expresión del gen rfp.
Proteina: polímero grande de aminoácidos. Algunos ejemplos incluyen: enzimas, proteína fluorescente mutante y algunas hormonas.
Proteína AraC: proteína necesaria para la expresión de la proteína fluorescente mutante en las células
transformadas con pARA-R.
Proteína fluorescente verde: proteína producida por la medusa marina, Aequoria victoria; proteína codificada por el gen gfp.
Quimera: animal mítico formado por numerososanimales diferentes. En la biología molecular, se usa frecuentemente para describir una molécula de ADN recombinante.
Reacción en cadena de la polimerasa (RCP): procedimiento químico utilizado para amplificar una secuencia de ADN mediante ciclos repetidos de reproducción y desnaturalización.
Reproducción de ADN: uso del ADN existente como plantilla para la síntesis de cadenas nuevas de ADN.
Ribosa: azúcar de cinco carbonos que se encuentra en los nucleótidos del ARN.
Ribosoma: sitio dentro de la célula que reúne los aminoácidos en moléculas de proteína.
Secuencia base: orden de las bases de nucleótidos en una molécula de ADN; determina la estructura de las proteínas codificadas por dicho ADN.
Secuencia de reconocimiento (sitio de reconocimiento): secuencia base específica de nucleótidos reconocida por una enzima de restricción. La enzima corta el ADN dentro de esta secuencia de nucleótidos.
Solución reguladora: solución utilizada para mantener un óptimo medio fisicoquímico para que se produzca una reacción química. Las soluciones reguladoras se utilizan en las digestiones de restricción, las ligaciones y para la RCP.
Solución reguladora de NaB: solución de borato de sodio utilizada como reguladora en la electroforesis. Es una solución que conduce una corriente eléctrica y mantiene un pH relativamente constante.Ver también: TBE
Superenrollada: mayor nivel de contorsión del ADN que se encuentra a menudo en los plásmidos.
s
Glosario P - T
q
r

Glosario
TBE (ácido tris-borato-EDTA): solución utilizada para la electroforesis en gel y en la preparación de geles de agarosa. La solución ayuda a conducir la corriente eléctrica mientras que mantiene un pH constante.
Timina: una de las bases que se encuentran en el ADN; la base que se complementa con la adenina.
Traducción: proceso químico que convierte una secuencia de nucleótidos de ARNm en una secuencia de aminoácidos; el sitio de esta reacción es el ribosoma.
Transcripción: proceso químico que convierte una secuencia de nucleótidos de ADN en una secuencia de nucleótidos de ARNm; proceso que utiliza ARN polimerasa para convertir una plantilla de ADN en una cadena de ARNm.
Transformación: proceso que coloca ADN extraño, como un plásmido, dentro de una célula.
Vector: Ver: vector de clonación, vector de expresión.
Vector de clonación: molécula de ADN que se origina a partir de un virus, un plásmido o la célula de un organismo superior, en el que otro fragmento de ADN de tamaño adecuado se puede integrar sin perder la capacidad del vector de autorreproducirse; los vectores son diseñados para introducir ADN extraño a las células huésped, donde el ADN se puede reproducir en grandes cantidades. En los laboratorios Amgen, se utiliza el pKAN para clonar varias copias del gen rfp.Ver también: vector de expresión, plásmido.
Vector de expresión: plásmido diseñado genéticamente para expresar de forma específica los genes (por ej: pARA).Ver también: vector de clonación, plásmido.
t
v
Glosario T - V