a langendorff-perfused mouse heart model for … a langendorff-perfused mouse heart model for...
TRANSCRIPT

A Langendorff-Perfused Mouse Heart Model For Delayed Remote Limb Ischemic Preconditioning Studies
By
Sagar Rohailla
A thesis submitted in conformity with the requirements for the degree of Masters of Science
Institute of Medical Science University of Toronto
© Copyright by Sagar Rohailla (2012)

ii
A Langendorff-Perfused Mouse Heart Model for
Delayed Remote Limb Ischemic Preconditioning Studies
Sagar Rohailla
Master of Science
Institute of Medical Science University of Toronto
2012
Abstract Remote ischemic preconditioning (rIPC) through transient limb ischemia induces potent
cardioprotection against ischemia reperfusion (IR) injury. I examined the delayed phase of
protection that appears 24 hours after the initial rIPC stimulus. The primary objective of this
study was to establish a mode of sedation and control treatment for delayed rIPC experiments. I
used an ex-vivo, Langendorff isolated-mouse heart preparation of IR injury to examine the
delayed effects of an intra-peritoneal (IP) injection, sodium-pentobarbital (SP), halothane and
nitrous oxide (N2O) anesthesia on post-ischemic cardiac function. Each anesthetic method
improved left-ventricular function after IR injury. SP and halothane anesthesia also reduced LV
infarct size. Delayed cardioprotection after IP injections was associated with an increase in
phosphorylated-Akt levels. The present study shows that IP injections and inhalational anesthesia
invoke cardioprotection and, therefore, indicates that these modes of sedation should not be used
as control treatments for studies examining the delayed rIPC phenotype.

iii
Acknowledgements
First and foremost I would like to thank my supervisor, Dr. Christopher Caldarone for his
support throughout this project. I am grateful for all your encouragement and guidance, and for
never hesitating to open your door for a chat. Your commitment to ensuring success among your
students and your ability to remind us of the bigger picture has always been reassuring. These are
qualities I hope to emulate in my future.
I would like to thank Dr. Andrew Redington for being a mentor and pillar over the last
year. Thank you for believing in me and for giving me the opportunity to develop as a scientist.
Working with you has had an immeasurable positive impact on me. I am enormously grateful for
the experience.
I would like to thank my committee members, Dr. Gregory Wilson and Dr. John Coles
for your insight and advice throughout the project. I hope we can work together again in the
future. To Dr. Edward Hickey, thank you for being a part of this experience and for connecting
me with the field of endotoxin preconditioning. Your PhD thesis was a friendly vision into the
world I hope to enter.
There are no adequate words to describe the support I received from Dr. Jing Li, Dr. Can
Wei and Dr. Xiao Jing Dai. Thank you Dr. Li and Dr. Wei for introducing me to the complexity
and beauty of the mouse Langendorff. Thank you for all of the hearts you mounted and cardiac
function data you helped me to collect. This project was possible because of your hard work and
generosity. I would also like to thank Alex Di Battista for all the support and numerous chats – it
has been extremely useful and fun bouncing ideas.
Lastly, thank you to Kimberly Elias, friends and my family. You all have been the best
academic counselors, companions and sources of support. I am truly thankful to have you in my
life.

iv
Table of Contents
Acknowledgements iii Table of Contents iv List of Tables vi List of Figures vii List of Appendices ix List of Abbreviations x Chapter 1: Review of Literature 1
1.1 Introduction: Unexpected Findings 1 1.2 Ischemia Reperfusion Injury 5 1.3 Inflammation and IR Injury 6 1.4 Remote Ischemic Preconditioning 8
1.4.1 Mechanism of rIPC 10 1.5 Reperfusion Injury Salvage Kinases (RISK) 12 1.6 Delayed Preconditioning 15
1.6.1 Mechanism of Delayed Preconditioning 16 1.7 Current models of 2W Preconditioning 17 1.8 Anesthetic Preconditioning 20
1.8.1 Effects of Anesthesia on Heart Function 21 1.8.2 Mechanism of APC 22 1.8.3 Isoflurane 24 1.8.4 Halothane 26 1.8.5 Sevoflurane, Enflurane and Desflurane 27 1.8.6 Nitrous Oxide 27 1.8.7 Intra-peritoneal Anesthesia: Ketamine and Barbiturates 28
1.9 Anesthetic and Ischemic Preconditioning: Clinical Utility 30 1.10 Langendorff Isolated Heart Model of IR Injury 31
Chapter 2: Research Aims and Hypotheses 33
2.1 Summary and Rationale 33 2.2 Research Aims/Objective 34 2.3 Hypotheses 34
Chapter 3: Methods 35
3.1 Ethics 35 3.2 Experimental Groups 35 3.3 Induction of rIPC Using Inguinal Tourniquet Model 38

v
3.4 A Langendorff Isolated Heart Model of Ischemia-Reperfusion Injury 39 3.5 Infarct Size Determination 43 3.6 Protein Concentration Determination 44 3.7 SDS-Page and Western Blot Analysis 45 3.8 Data and Statistical Analysis 47
Chapter 4: Results 48
4.1 The Delayed Effects of Intra-Peritoneal and Inhalational Anesthesia on Left-Ventricular Function after Global Ischemia 48 4.1.1 Baseline Function 48 4.1.2 Left Ventricular Developed Pressure 50 4.1.3 Left Ventricular End-Diastolic Pressure 55 4.1.4 Maximum Rate of Contraction 60 4.1.5 Maximum Rate of Relaxation 65
4.2 Delayed Preconditioning with Intra-Peritoneal and Inhalational Anesthesia Reduce Infarct Size after IR Injury 69
4.3 Delayed Preconditioning with Injectable and Gas Anesthesia increase phospho-Akt and phospho-p44/42 MAPK Expression 72
Chapter 5: Discussion 76
5.1 Intra-Peritoneal Injections Induce Delayed Preconditioning Against Global IR Injury 78
5.2 Halothane Anesthesia Induces Delayed Preconditioning Against Global IR Injury 82
5.3 Nitrous-Oxide Improves Post-Ischemic Cardiac Performance But Does Not Reduce Infarction Size 83
5.4 Cross-talk between signaling cascades 86
Chapter 6: Conclusions 88 Chapter 7: Future Directions 90
7.1. An In-Vivo Model of Delayed rIPC 91 7.2. Revisiting the role of TLR4 in delayed rIPC 92 7.3. The ‘Third’ Window And Exercise Preconditioning 92 7.4. Clinical Implications of A Mouse Model of Delayed rIPC 93
Chapter 8: References 95 Chapter 9: Appendices 109

vi
List of Tables
1. Baseline functional parameters in the Langendorff-isolated heart groups for IR injury
experiments 49

vii
List of Figures
1. Initial study examining the role of TLR4 in Delayed Preconditioning 4
2. Inflammatory response during IR injury: feed-forward cycle 9
3. Schematic representation of mechanisms involved in remote ischemic preconditioning
11
4. Cell signaling mechanisms involved in early and delayed ischemic preconditioning 14
5. The transition between the early and delayed phases of cardioprotection 18
6. A schematic of the study protocol 37
7. Remote ischemic preconditioning (rIPC) via transient ischemia of mouse hindlimb 38
8a. The mouse Langendorff-isolated heart model 40
8b. The mouse Langendorff-isolated heart model used in The Redington Lab 41
9a. The effects of intra-peritoneal anesthesia on post-ischemic left-ventricular developed
pressure (LVDP) 52
9b. The effects of inhalational anesthesia on post-ischemic left-ventricular developed pressure
(LVDP) 53
9c. Left-ventricular developed pressure (LVDP) at 60 min of reperfusion 54
10a. The effects of intra-peritoneal anesthesia on post-ischemic left-ventricular end-diastolic
pressure (LVEDP) 57
10b. The effects of inhalational anesthesia on post-ischemic left-ventricular end-diastolic pressure
(LVEDP) 58
10c. Post-ischemic left ventricular end-diastolic pressure (LVEDP) at 60 minutes of reperfusion
59
11a. The effects of intra-peritoneal anesthesia on post-ischemic rate of LV contraction (dP/dtmax)
62

viii
11b. The effects of inhalational anesthesia on post-ischemic rate of LV contraction (dP/dtmax)
63
11c. Post-ischemic rate of LV contraction (dP/dtmax) at 60 minutes of reperfusion 64
12a. The effects of intra-peritoneal anesthesia on post-ischemic rate of LV relaxation (dP/dtmin)
66
12b. The effects of inhalational anesthesia on post-ischemic rate of LV relaxation (dP/dtmin)
67
12c. Post-ischemic rate of LV relaxation (dP/dtmin) at 60 minutes of reperfusion 68
13. The effects of intra-peritoneal and inhalational anesthesia on left-ventricle infarct size after
IR injury 70
14. Representative cross-sections of mouse hearts from each treatment group
after IR injury 71
15. The effects of intra-peritoneal and inhalational anesthesia on phospho-Akt (Ser473) levels in
mouse heart before IR injury 74
16. The effects of intra-peritoneal and inhalational anesthesia on phospho-p44/42 MAPK
(Tyr202, Thr204) levels in mouse hearts before IR injury 75

ix
List of Appendices
1. Power-lab acquisition software: Cardiac output chart 110
2. Protein extraction-lysis-buffer 110

x
List of Abbreviations
1W First Window 2W Second Window 5-HD 5-Hydroxydecanoate Akt Serine Threonine Kinase- Protein Kinase B AMI Acute Myocardial Infarction AP-1 Activator Protein-1 APC Anesthetic Preconditioning AR Aldose-Reductase 1 ATP Adenosine Triphosphate Ca2+ Calcium Ion CaMKII Ca2+/calmodulin-dependent kinase II CAO Coronary Artery Occlussion COX-2 Cyclooxygenase-II CPB Cardiopulmonary Bypass DAMPs Danger-Associated Molecular Patterns dp/dtmax Maximum Rate of Contraction dp/dtmin Maximum Rate of Relaxation ECL Enhanced Chemiluminescence eNOS Endothelial Nitric-Oxide Synthase GPCR G-Protein Coupled Receptor GSK3β Glycogen Synthase Kinase 3β H+ Hydrogen Ion HMGB High Mobility Group Box 1 HO-I Heme-Oxygenase I HRP Horseradish Peroxidase Hsp27 Heat Shock Protein 27 kDa Hsp70 Heat Shock Protein 70 KDa IGF-1 Insulin Like Growth Factor-1 ICAM1 Intracellular Adhesion Molecule 1 IFN Interferon Il-6 Interleukin-6 Il-8 Interleukin-8 iNOS Inducible Nitric-Oxide Synthase IPC Ischemic Preconditioning IR Ischemia-Reperfusion K+ Potassium Ion K+-ATP Potassium Ion Adenosine Triphosphate Channel LAD Left Anterior Descending LPS Lipopolysaccharide Endotoxin LVDP Left Ventricular Developed Pressure LVEDP Left Ventricular End-Diastolic Pressure LVP Left Ventricular Pressure MAC Minimum Alveolar Equivalent MAO Mesenteric Artery Occlusion MAPK Mitogen Activated Protein Kinase

xi
MnSOD Manganese Superoxide Dismutase mPTP Mitochondrial Permeability Transition Pore Na+ Sodium Ion NF-κB Nuclear Factor-Kappa B nNOS Neuronal Nitric-Oxide Synthase NO Nitric Oxide N2O Nitrous Oxide ORB Orthodontic Rubber Band PAGE Polyacrylamide Gel-Electrophoresis PAMPs Pathogen-associated molecular patterns PC Preconditioning PI3K Phosphoinositide-3 Kinase PKA Protein Kinase A PKC Protein Kinase C PKG Protein Kinase G PLN Phospholamban Protein PRR Pattern recognition receptor PTK Protein Tyrosine Kinase rIPC Remote Ischemic Preconditioning RISK Reperfusion-Injury Salvage Kinases ROS Reactive Oxygen Species SDS Sodium Dodecyl Sulphate SEM Standard error of the mean SERCA2a Sarcoplasmic Reticulum Calcium ATPase 2a SNAP S-nitroso-N-acetylpenicillamine SNS Sympathetic Nervous System SP Sodium Penotobarbital STAT1 Signal Transducers and Activators of Transcription 1 TNF-α Tumor Necrosis Alpha VCAM1 Vascular Cell Adhesion Molecule 1

1
Chapter 1 Review of Literature
1.1 Introduction: Unexpected Findings
Our laboratory is interested in the biology of myocardial remote ischemic preconditioning
(rIPC), an innate form of organ protection whereby brief episodes of ischemia with intermittent
reperfusion to an organ (e.g., limb) can protect the heart against subsequent lethal ischemia-
reperfusion (IR) injury. We are conducting a range of studies examining the potential triggers
released from the remote organ to the activation of intracellular signaling pathways at the heart
mediating organ survival.
The initial objective of this study was to examine the role of innate immunity in the
development of cardioprotection after rIPC. A general feature of ischemic preconditioning (IPC)
involves suppressing inflammation during IR injury. Previous work in the lab has shown that
rIPC modifies gene expression in mouse myocardium and human neutrophils towards an anti-
inflammatory portfolio[1], [2]. We have also observed that repeated rIPC inhibits human
neutrophil activity by decreasing adhesion and phagocytosis[3-6]. These findings are similar to
those produced in other labs that have observed a preconditioning-induced systemic anti-
inflammatory response [7-12].
While it is well established that inflammation is a main contributor to the damaging
effects of IR injury, emerging evidence indicates that it may also be required to generate
cardioprotection. For example, mice deficient in tumor-necrosis alpha (TNF-α) or nuclear factor
kappa-B (NF-κB), key factors in the inflammatory response, do not develop ischemic tolerance
following a preconditioning stimulus[3], [7], [8], [13]. Further evidence for the role of an
inflammatory response in preconditioning is based on findings that exogenous administration of
sub-lethal doses of a variety of pro-inflammatory cytokines (TNF-α, Interleukin-6, 12 - IL-6,12)

2
or bacterial lipopolysaccharide (LPS) endotoxin induce cardioprotection through similar
mechanisms as IPC, particularly in the late or delayed phase of protection (24 hours after the
preconditioning stimulus)[14-18]. These studies found that the cytokine-induced cardioprotection
occurred in-part through activation of toll-like receptor 4 (TLR4), an integral component of
innate immunity[19-23]. Toll-like receptors are classified as pattern recognition receptors (PRR)
and are integral for orchestrating the initial steps of the innate immune response to exogenous
pathogens by binding to pathogen-associated molecular patterns (PAMP) present on microbial
species. However, TLRs are also important for initiating host immune responses to non-
microbial challenges such as hypoxia, ischemia, heat shock and sepsis. Given the importance of
TLRs in inflammation, a process also involved in generating preconditioning, I aimed to
investigate whether TLR4-signaling plays a role in IPC.
There are some studies that have already addressed this question. In a study examining
the effects of early phase (15 min after the preconditioning stimulus) IPC, cardioprotection was
intact in a TLR4-deficient strain[12], [24-26]. This finding aligns with previous studies on
endotoxin or cytokine preconditioning that TLR4-mediated protection occurs in the delayed
phase[27-30]. Pradillo et al. investigated the role of TLR4 in delayed preconditioning against
stroke injury. They discovered that TLR4-deficiency was associated with a decrease in the
magnitude of protection when compared to wild-type controls[20]. This was also associated with
reduced NF-κB activity and lowered expression of TNF-α. Further evidence for a role of TLR4
in preconditioning comes from findings generated by our lab in which it was observed that there
is a decrease in TLR4 gene expression in the early and delayed phases of protection [1], [2].
Based on these findings, I set out to test the hypothesis that cardioprotection arising from
delayed rIPC would be diminished in TLR4-deficient mice. I proposed that rIPC conferred
protection by inducing a mild-inflammatory response involving TLR4 signaling, and that this

3
process was required to initiate the pro-survival pathways mediating protection against lethal IR
injury.
As shown in figure 1, I designed an experiment to investigate whether TLR4-deficient
mice of the C3H/HeJ strain are capable of developing delayed rIPC. However, these experiments
were interrupted after I observed that wild-type mice (C3H/HeN), receiving the standard sedation
method of sodium-pentobarbital (SP) anesthesia via an intra-peritoneal injection as a control
treatment on day 1, also seemed to develop delayed cardioprotection. Using an isolated mouse
heart model, I discovered that this control group had reduced infarction sizes and preserved
cardiac function after global IR injury twenty-four hours after sedation with SP. Furthermore,
these mice that had been treated with SP anesthesia developed a kinase response typically seen
during the early phase of IPC.
The use of SP anesthesia has been established as an appropriate control treatment for
early phase rIPC studies in mice, as it has not been shown to induce cardioprotection when
administered 60-90 minutes prior to IR injury[31], and it was assumed that this would apply to
the delayed phase as well. It is required by animal care committees that mice be sedated with
anesthesia when applying transient ischemia to the limb to induce rIPC. However, as I uncovered
in my preliminary experiments, the mode and type of sedation is critical as there is accumulating
evidence suggesting that preconditioning can occur after exposure to anesthesia (see review by
Weber et al.) [32]. Experiments in this field need to control for these ‘additional’ potential
preconditioning stimuli in order to examine the cardioprotective phenotype specifically afforded
by rIPC. Thus, the primary objective of this study changed to developing a mode of sedation that
does not induce cardioprotection in order to identify and establish a control treatment for future
delayed rIPC experiments. I began examining alternative methods of anesthesia to identify a

4
method of sedation that would not confer delayed cardioprotection. The following dissertation is
a report of those experiments.
Figure 1: Initial study examining the role of TLR4 in Delayed rIPC. C3H/HeN and C3H/HeJ
(TLR4-/-) mice were allocated to receive sodium-pentobarbital anesthesia (SP) ± rIPC (4 cycles
of 5 min ischemia and 5 min reperfusion) on day 1. Mouse hearts were excised twenty-four hours
after treatment (day 2) and mounted on Langendorff preparation for ischemia-reperfusion injury
experiments. 2W – second window/delayed phase, rIPC – remote ischemic preconditioning

5
1.2 Ischemia-Reperfusion Injury
Coronary artery disease is a leading cause of morbidity and mortality throughout the
world[33]. Acute reductions in coronary bloodflow lead to the clinical syndromes of angina and
acute myocardial infarction (AMI). A significant number of these patients also undergo coronary
balloon angioplasty with stenting, or cardiac surgery, and many will experience episodes of peri-
procedural myocardial ischemia. Such events may severely impair myocardial function, delay
effective post-operative recovery and greatly increase the risk for future complications. The
primary aim of treatment for a coronary ischemic event is to quickly restore blood flow to
preserve muscle function and limit post-infarct sequelae [34]. However, paradoxically, restoring
coronary flow to occluded regions causes pronounced tissue damage in a process known as
reperfusion injury[35-37].
The discovery of reperfusion injury in 1960 by Jennings et al., and the advent of ischemic
preconditioning (IPC) over two decades later provided a better understanding of the mechanisms
involved in ischemia-reperfusion (IR) injury [35], [38]. These important early discoveries helped
to elucidate the independent contributions of ischemia and reperfusion to resulting organ injury,
and also provided an array of targets for pharmacological-based cardioprotective strategies[35],
[38], [39].
IR injury results in a battery of biochemical, structural, and metabolic changes in the
parenchymal and endothelial cells of the heart that impair contractile capacity and ultimately
may decrease blood flow to other essential organs, such as the brain. During ischemia, obstructed
coronary blood flow causes the affected cells to switch their metabolism towards anaerobic
pathways for energy production[35], [39], [40]. This causes a dramatic shift in ion concentrations
in and out of the cell. Lactate accumulates as a metabolic byproduct from pyruvate oxidation,
increasing hydrogen (H+) ion concentration within the cytoplasm. The ensuing acidosis causes

6
sodium (Na+) and calcium (Ca2+) ion levels to increase in an attempt to reestablish normal
physiological pH[35], [41], [42]. Active transport mechanisms, normally able to correct such ion
imbalances, are non-functional due to depleting adenosine-triphosphate (ATP) levels.
Upon reperfusion, extracellular pH is rapidly normalized, forcing the extrusion of H+ ions
from within the cell via the sarcolemmal Na+/H+ exchanger which drives a further increase in
Na+ and Ca2+ ions[36], [43]. Ca2+ overload is a crucial step in the pathogenesis of IR injury[37].
Ca2+ buildup causes hypercontracture of cardiomyocyte myofibrils, activates Ca2+-dependent
proteases involved in apoptosis, and triggers the formation of the mitochondrial permeability
transition pore (mPTP) – a nonselective channel between the inner mitochondrial membrane and
the sarcoplasm[35], [44].
The mPTP is a key component in the pathology of IR injury, as its formation initiates the
eventual death of cells within the infarct zone. Inhibiting pore formation has been a major focus
of many pharmacological interventions aimed at reducing the severity of IR injury[33].
Permeabilization causes the breakdown of oxidative phosphorylation and further exhausts ATP
levels[33]. Furthermore, the mitochondria serve as a reservoir for reactive oxygen species (ROS),
which upon release can overwhelm cellular anti-oxidant defenses, promote the formation of
mPTPs in surrounding cardiomyocytes, attract circulating leukocytes and activate pro-
inflammatory pathways within the affected cells – all of which promote cell death and local
organ injury[41], [45].
1.3 Inflammation and IR Injury
A pronounced inflammatory response is a major contributor to reperfusion injury. The
endothelium plays a large role in orchestrating this process by facilitating the infiltration of
inflammatory cells to the injury site[44]. In response to accumulating ROS, endothelial cells

7
increase the expression of cell-adhesion molecules (intracellular adhesion molecule-1, ICAM-1,
vascular cell adhesion molecule-1, VCAM-1) and release chemoattractants to recruit cells of the
innate immune system [45], [46]. Inflammation during IR injury is mediated initially by tissue
residing macrophages and later by infiltrating neutrophils[42], [44], [47]. Neutrophils migrate to
the region within the first 6 hours from the onset of reperfusion, and invade the myocardium over
the next 24 hours [47], [48]. Mobilization of inflammatory cells is accelerated by the marked
levels of cell necrosis that occurs during IR injury due to shifts in cell osmolality that induce
swelling and rupture [34], [49].
Necrosis liberates a plethora of intra-cellular molecules, often referred to as danger-
associated molecular patterns (DAMPs), that can serve as potential ligands for Toll-like receptors
(TLRs) present on innate immune cells, endothelium and cardiomyocytes[36], [42], [50-52].
DAMPs can include high-mobility group box-1 (HMGB1), heat shock proteins (Hsps), uric acid,
S100 proteins, hyaluronic acid, and oxidized lipoproteins [36], [51]. Cells that are undergoing
necrosis release their internal components into the extracellular space where they bind to and
activate TLRs present on macrophages and neutrophils[36], [53]. TLR signaling initiates a
cellular stress response involving nuclear localization of NF-κB and the transcription of genes
coding for pro-inflammatory cytokines (TNF-α, IL-6, IL-8) [46], [50], [53-55]. Activation of
TLRs on circulating neutrophils, causes the release of degradative enzymes, ROS, and other
cytokines that further exacerbate injury and promote continued DAMP release [36], [46], [53-
55]. This ultimately results in a feed-forward cycle of tissue injury (figure 2)

8
1.4 Remote Ischemic Preconditioning
Despite a greater understanding of the mechanisms underlying IR injury, interventions
aimed at improving clinical outcomes after acute myocardial infarction (AMI) or perioperative
IR injury have been suboptimal. This may largely be due to the likelihood that most interventions
target a specific component of IR injury, whereas the pathology of this disease involves a large
portfolio of destructive pathways. Furthermore, the heterogeneity of IR injury in a clinical setting
and the existence of co-morbidities in patients provide additional challenges in developing a
panacea treatment[56], [57].
However, over the last two decades it has become apparent from numerous studies that
exposing organs to brief periods of non-lethal ischemia, with intermittent reperfusion, termed
ischemic preconditioning (IPC), can protect against future lethal IR injury and limit irreversible
tissue damage. This seminal observation first made by Murry et al in 1986, in which canine
hearts that underwent local coronary IPC had nearly a 75% reduction in myocardial infarct size,
was later expanded to include endogenous protection derived from transient ischemia to distant
organs, known as remote IPC (rIPC) [37], [38], [58]. Initial experiments described rIPC of
myocardium through renal[59], [60], cerebral[61] or mesenteric artery occlusion[62-64] –
procedures that are limited in a clinical setting because of the need for invasive surgery. It was
later discovered in animal models that protection against a sustained coronary artery occlusion
(CAO) could be generated following transient ischemia of rat or rabbit skeletal muscle [65], [66].
In 2002, the lab group described a simpler, non-invasive method of inducing rIPC by applying 3-
4 cycles of transient ischemia and reperfusion to a limb using a standard blood pressure cuff in
patients or tourniquet in animals[67].
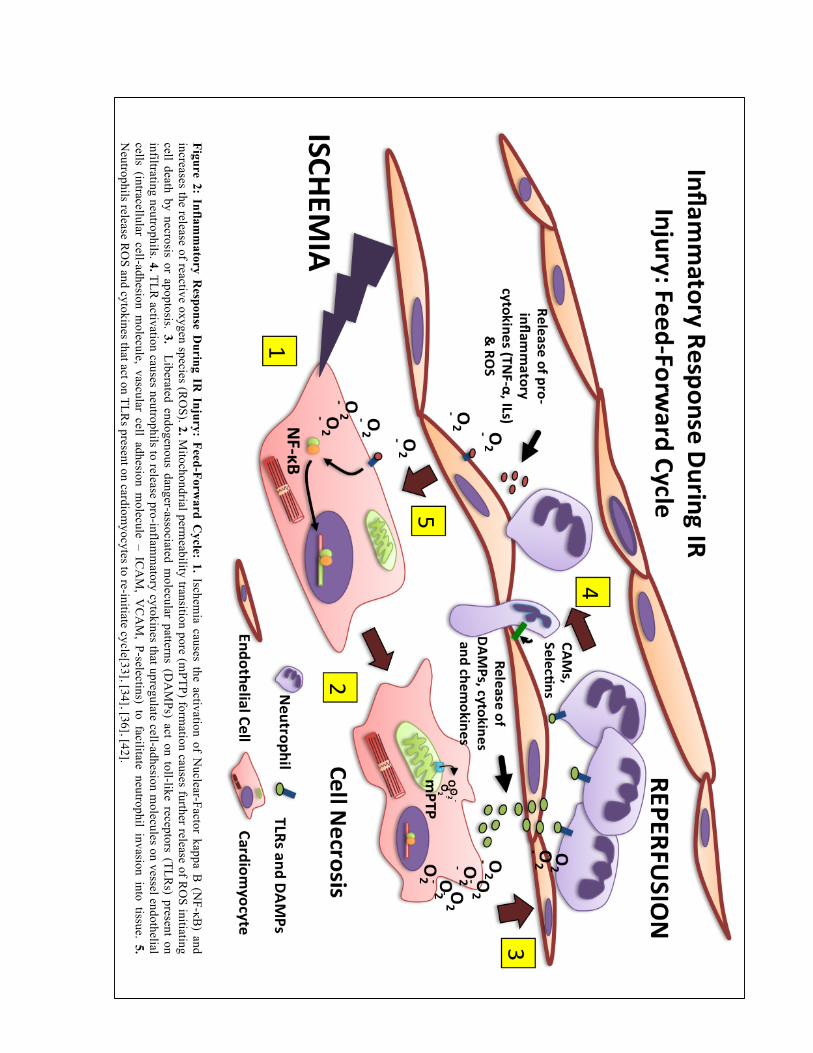
Figure 2: Inflam
matory R
esponse During IR
Injury: Feed-Forward C
ycle: 1. Ischemia causes the activation of N
uclear-Factor kappa B (N
F-κB) and
increases the release of reactive oxygen species (RO
S). 2. Mitochondrial perm
eability transition pore (mPTP) form
ation causes further release of RO
S initiating cell death by necrosis or apoptosis. 3. Liberated endogenous danger-associated m
olecular patterns (DA
MPs) act on toll-like receptors (TLR
s) present on infiltrating neutrophils. 4. TLR
activation causes neutrophils to release pro-inflamm
atory cytokines that upregulate cell-adhesion molecules on vessel endothelial
cells (intracellular cell-adhesion molecule, vascular cell adhesion m
olecule – ICA
M, V
CA
M, P-selectins) to facilitate neutrophil invasion into tissue. 5.
Neutrophils release R
OS and cytokines that act on TLR
s present on cardiomyocytes to re-initiate cycle[33], [34], [36], [42].

10
rIPC through limb ischemia has been shown to invoke a similar magnitude of protection
compared to local coronary IPC, and because of its ease of application, can realistically be
translated into clinical practice[68], [69].
1.4.1 Mechanism of rIPC
Since its discovery, there have been major advancements in our understanding of the
mechanisms underlying inter-organ protection. It is well established that rIPC activates a similar
intracellular signaling response as local IPC[49], [68], [70]. Transient ischemia of an organ
causes the release of endogenous substances from cells, such as adenosine, opioids, bradykinin,
and NO, which can enter the circulation and travel to the distant organ or act on nearby afferent
nerves[71] (figure 3). In either case, these preconditioning triggers can bind to their respective
receptors that are coupled to G proteins or directly activate intracellular mediators of
preconditioning [41], [49], [72], [73]. Activation of G-protein coupled receptors (GPCRs)
induces an intracellular signaling response involving a cascade of pro-survival kinases that
results in cardioprotection (figure 4). While the preconditioning phenotype encompasses
numerous proteins, a key step of the signal involves the activation of phosphatidylinositol-3-
kinase (PI3K) and its associated serine-threonine kinase (Akt) and the mitogen activated protein
kinase (MAPK) family, specifically the extracellular regulated kinases 1/2 (Erk 1/2) [74], [75].
While there is still some debate regarding the putative end effectors of preconditioning, it
is generally accepted that protection involves (1) limiting Ca2+ overload, (2) maintaining an open
state of sarcolemmal and mitochondrial K+-ATP sensitive channels, and (3) inhibiting the
formation of the mPTP[71]. As previously mentioned, Ca2+ overload initiates the process that
ultimately disables cell function and leads to death by necrosis or apoptosis.
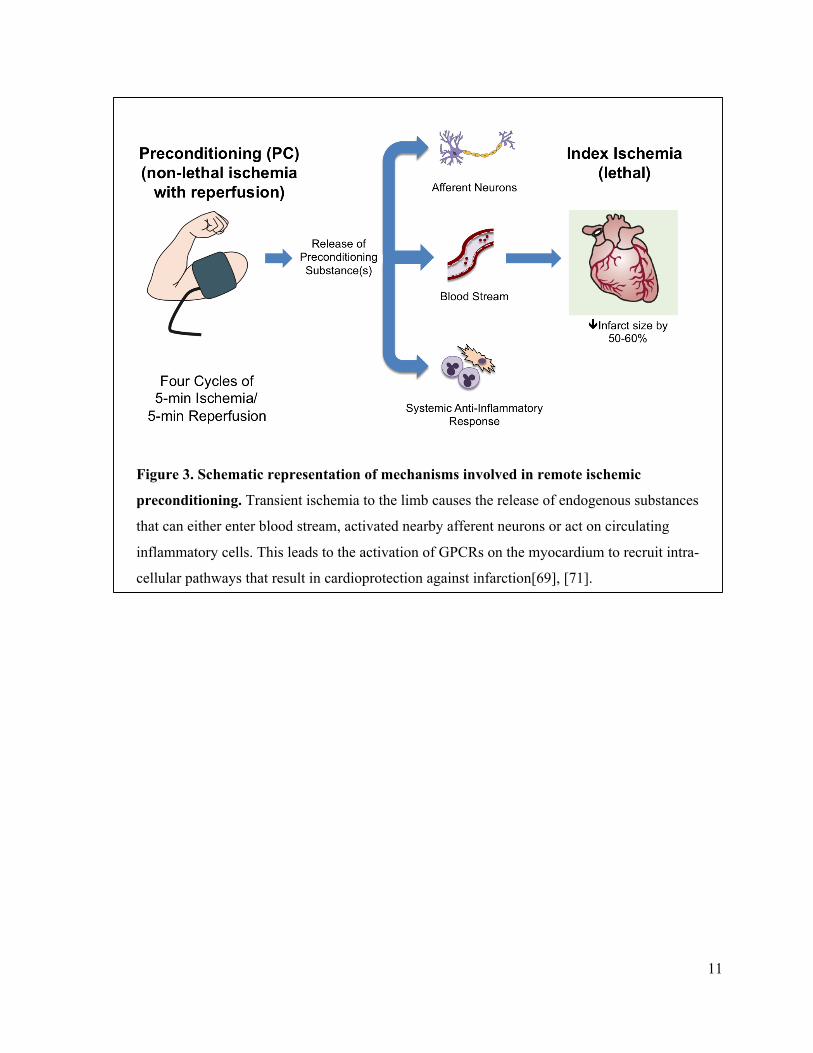
11
Figure 3. Schematic representation of mechanisms involved in remote ischemic
preconditioning. Transient ischemia to the limb causes the release of endogenous substances
that can either enter blood stream, activated nearby afferent neurons or act on circulating
inflammatory cells. This leads to the activation of GPCRs on the myocardium to recruit intra-
cellular pathways that result in cardioprotection against infarction[69], [71].

12
Maintaining intracellular ion concentrations can ameliorate this destructive process. The
role of K+-ATP channels in limiting cell damage during IR injury is not completely understood,
yet they may be involved in reducing Ca2+ overload and stabilizing mitochondrial structure and
function, and thus maintaining ATP generation[43], [71], [76].
Additionally, IPC is known to inhibit circulating neutrophil function and causes a shift
towards the production of anti-inflammatory and anti-apoptotic mediators within the
myocardium[2], [3], [21], [22], [77]. A similar response also occurs with sub-lethal doses of pro-
inflammatory cytokines or endotoxin. Some studies suggest that cytokine preconditioning
involves suppression of the inflammatory response via negative feedback inhibitors of TLR
signaling[2], [3], [21], [22], [77]. This is thought to involve the PI3K-Akt signaling pathway
[17], [78], [79]. Following an increase in NF-κB activity, the p85 subunit of PI3K is recruited to
the cytoplasmic domain of TLR4 to modulate its activity (figure 4). This inhibits the TLR4 pro-
inflammatory pathway and initiates the pro-survival signaling pathways mediated through
Akt[17], [78]. Negative feedback on TLR4 may also be involved in rIPC-induced immune
suppression, but this remains to be studied.
1.5 Reperfusion-Injury Salvage Kinases (RISK)
In 2005, the Yellon group provided strong evidence that IPC involves the activation of a
set of kinases that collectively mediate cardioprotection against IR injury. These enzymes have
been termed the reperfusion-injury salvage kinases (RISK) and their activation has been
consistently associated with reduced cell death after IR injury[75], [80]. The RISK pathway is
also a convergence point for many forms of pharmacological preconditioning, as has been shown
in a number of preclinical studies evaluating the cardioprotective effects of insulin, glucagon-like
peptide 1, erythropoietin and adenosine (reviewed in Hausenloy & Yellon 2007)[81]. The Akt

13
and Erk1/2 kinases are important elements of the RISK pathway and can be considered markers
for the preconditioning phenotype, as they play a critical role in the cellular basis of IPC that
ultimately enhances cell survival.
Akt – Protein Kinase B
PI3K activation of Akt leads to the phosphorylation of several downstream targets
responsible for preconditioning the myocardium, some of which include endothelial nitric oxide
synthase (eNOS)[80], [82], [83], inactivation of glycogen-synthase kinase-3β (GSK-3β)[84] and
pro-apoptotic signaling proteins (caspases, Bad, Bax)[70], and the different isoforms of protein
kinase C (PKC)[85], [86] (figure 4). Akt is activated by the inositol lipid byproduct of PI3K, a
pathway that is well characterized in insulin-like growth factor-1 (IGF-1) signaling, promoting
cell survival and protein synthesis, while limiting apoptosis.
A number of studies indicate that Akt is activated after the preconditioning phase and is later
phosphorylated during reperfusion with antecedent IPC[75], [86], [87]. The Redington Lab
provided strong evidence for Akt activation after rIPC in a study examining the effects of four
cycles of unilateral hindlimb ischemia on cardiac function and infarction size in isolated mouse
hearts. It was found that hind limb ischemia led to nearly a two-fold increase in phosphorylated
–Akt levels in mouse hearts, which was also associated with improved cardiac recovery and
reduced infarction after IR injury. These cell signaling and cardiprotective effects of rIPC were
abrogated when co-adminstered with the PI3K inhibitor, wortmannin, indicating a critical role
for PI3K-Akt signaling [31].

14
Figure 4: Cell signaling mechanisms involved in early and delayed ischemic
preconditioning. 1. Early preconditioning. Initial triggers released into circulation from
remote tissue may include, GPCR agonists or endogenous DAMPs stimulating TLR4.
Intracellular mediators involve the PI3K-Akt pathway acting on downstream kinases, most
notably PKC. End-effectors include mitochondrial and sarcolemmal K+ATP
channels. 2. Delayed
preconditioning. Nuclear translocation of NF-κB and subsequent transcription of a battery of
genes leads to the development of delayed preconditioning[22], [32], [43], [59], [68], [88].

15
Extracellular Regulated Kinase (Erk1/2) – MAPK p44/42
The Erk 1/2 or p44/42 proteins are members of the MAPK protein family responsible for
regulating a number of proliferative pathways that are important for cell survival[81]. Similar to
Akt, p44/42 activation via GPCR signaling, or through PKC-espilon, occurs after
preconditioning and results in the inhibition of pro-apoptotic enzymes that normally orchestrate
cell death processes that occur during reperfusion[89], [90]. A number of studies have implicated
phosphorylated-p44/42 in IPC as a key element of the RISK pathway[75], [81], [89]. Further
studies would also confirm a role for MAPK in rIPC. Heidbreder et al. performed a study
comparing the role of p44/42 signaling in cardioprotection after both IPC and rIPC. They
observed an increase in phospho-p44/42 in rat hearts after preconditioning via transient coronary
artery occlusions (CAO) or mesenteric artery occlusion (MAO)[89]. Shimizu et al. observed a
similar increase in phospho-p44/42 levels after rIPC via transient limb ischemia in rabbits[85].
They found that rabbits subjected to four-cycles of hind limb ischemia with intermittent
reperfusion showed an increase in myocardial levels of phospho-p44/42, which was associated
with reduced infarction sizes in in-vivo and ex-vivo isolated hearts.
1.6 Delayed Preconditioning
A unique property of the conditioning response is that the target organ possesses the
ability to regain the preconditioned state long after the initial stimulus. In both local and remote
IPC, there is an initial phase of protection arising within minutes that can last for up to 2-4 hours,
termed a first window (1W) or early preconditioning response[77]. The early phase relies on the
activation of pro-survival signaling pathways mostly through post-translational modification of
existing proteins within cells[91]. Shortly after the discovery of rIPC, it was revealed that the
protective phenotype reappears 12-24 hours later, known as the second window (2W) or delayed

16
preconditioning response[92],[93]. The delayed phase is less robust but may have greater clinical
utility given its broader range and longer duration of protection (up to 3-4 days)[94]. Delayed
preconditioning has also been observed to limit post-ischemic myocardial stunning, a period in
which the heart’s contractile capacity is below pre-ischemic values[34]. Interestingly, recent
studies suggest that a third or chronic preconditioning effect may also exist with repeated
exposure to transient ischemia, operating through an entirely different signaling response to that
of both early and delayed preconditioning[95].
1.6.1 Mechanism of Delayed Preconditioning
Whether through non-pharmacological (ischemia, heat stress, exercise) or
pharmacological (adenosine/opioid receptor agonists, NO, ROS, LPS, TNF-α) stimuli, delayed
preconditioning develops through similar signal transduction pathways responsible for early
local and remote IPC[49]. The differences in the protective phenotype lie in the end-effectors
(figure 5). While early preconditioning involves protein modification, the late phase is largely
mediated by gene induction and de novo synthesis of cardioprotective proteins[91], [96]. The
prevailing hypothesis for delayed preconditioning is that the intracellular mediators of the early
response, most notably PKC-ε[97], protein tyrosine kinase (PTK)[98] and p44/42 MAPK, lead to
increased transcriptional activity of NF-κB, activator protein-1 (AP-1) and signal transducers and
activators of transcription 1 (STAT1) [9], [88], [99]. There is also some evidence to suggest that
Akt activation can also lead to an increase in NF-κB transcriptional activity[100]. These
activated transcription factors migrate to the nucleus and increase the expression of genes
responsible for delayed preconditioning.
In addition to its well-known detrimental role in inflammation, NF-κB modulates a host
of genes involved in the cell stress response to a variety of pathological stimuli[54]. It is

17
generally accepted that NF-κB signaling is obligatory for the development of delayed
preconditioning[13],[77]. In its quiescent state, NF-κB remains in the cytoplasm bound to its
inhibitor protein, I-Kappa B (IκB) that masks it nuclear localization sequence. When the cell is
under stress, such as following a preconditioning stimulus, IκB is degraded allowing NF-κB to
translocate to the nucleus, most often as a heterodimer consisting of the p50 and p65 subunits.
During IR injury, nuclear binding peaks at approximately 15 min and 3 hours after the onset of
reperfusion, likely from exposure to ROS or pro-inflammatory cytokines[46], [54]. However,
there is also an increase in NF-κB promoter binding activity at 30min following preconditioning,
which seems to occur through an increase in nitric-oxide (NO)-induced-PKC-ε activation[13].
NF-κB enters the nucleus and promotes the transcription of a variety of genes shown to be
important for delayed PC, which include heat shock proteins (Hsp70, Hsp27), manganese
superoxide dismutase (MnSOD), aldose reductase (AR), hemo-oxygenase-1 (HO-1)
cyclooxygenase-2 (COX-2) and inducible nitric-oxide synthase (iNOS). Similar to early
preconditioning, the protective phenotype converges on restricting pore formation and
maintaining an open state of mitochondrial and sarcolemmal K+-ATP channels (figure 5) [101],
[101], [102].
1.7 Current Models of Delayed Limb rIPC
Much of our initial understanding of the physiology of myocardial delayed
preconditioning is derived from local IPC studies. The Bolli group has conducted a number of
elegant experiments using a working model of delayed local IPC to characterize the mechanisms
behind the late phase of protection[93],[103]. Their work has provided evidence for the currently
established mediators of delayed preconditioning. In particular, they have demonstrated
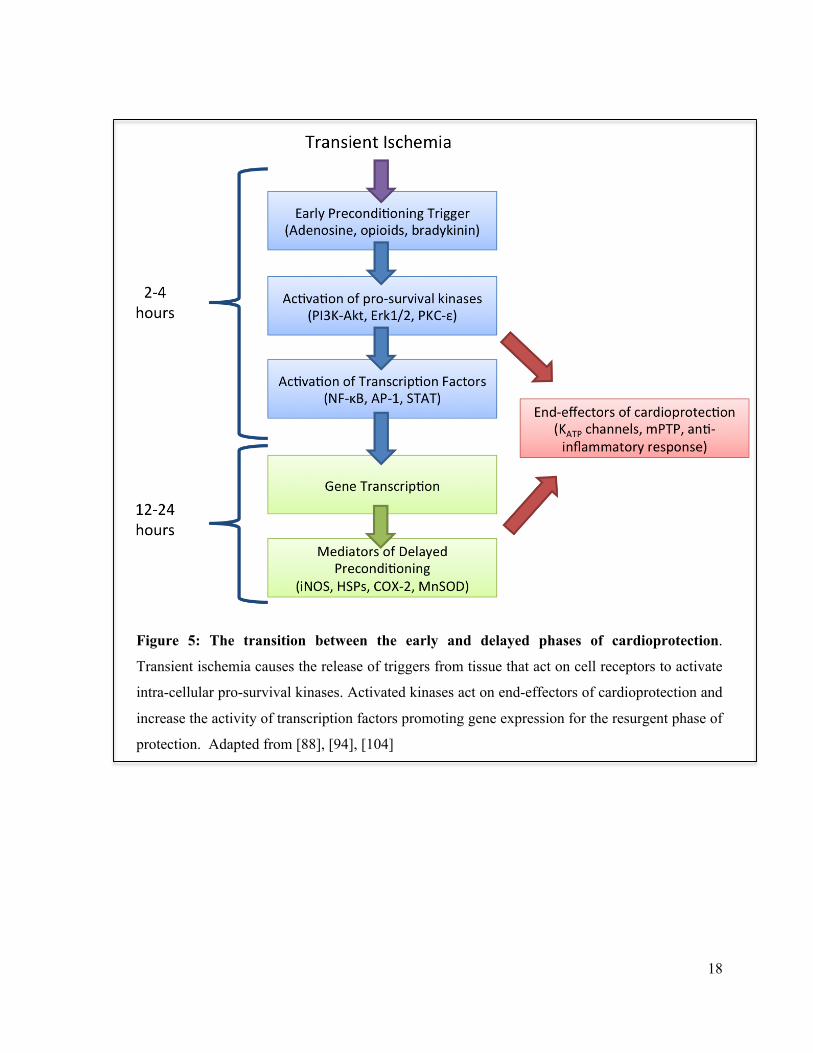
18
Figure 5: The transition between the early and delayed phases of cardioprotection.
Transient ischemia causes the release of triggers from tissue that act on cell receptors to activate
intra-cellular pro-survival kinases. Activated kinases act on end-effectors of cardioprotection and
increase the activity of transcription factors promoting gene expression for the resurgent phase of
protection. Adapted from [88], [94], [104]

19
an essential role for cytokine signaling, validating the growing hypothesis that a mild
inflammatory response precedes ischemic tolerance.
The Redington group and many others have demonstrated in numerous studies that transient
limb ischemia provides potent early cardioprotection in animal and human models of IR injury
[4-6], [8-12], [105], [106]. While there have been major strides in elucidating the mechanism of
early rIPC via limb ischemia, evidence of its delayed and chronic preconditioning effects is
sparse. Li et al. provided some initial evidence of delayed rIPC in a mouse isolated heart model
of global ischemia. They found that six 5-minute cycles of hindlimb ischemia intercalated with
periods of reperfusion administered twenty-four hours prior to injury reduced infarct size by
34%. They also discovered that the delayed phenotype was absent in NF-κB or iNOS deficient
mice[7], [8], [13]. However, it is unclear what their control sedation treatment was for limb
ischemia – an important consideration as will be discussed in later sections.
In human studies, 10 days of repeated limb rIPC has been shown to reduce neutrophil
adhesion and phagocytosis activity from day one onwards[3], [15-18]. Furthermore,
Loukogeorgakis et al. found that limb rIPC generated a late phase of protection against
endothelial IR injury, which was blocked with administration of trimetaphan (an autonomic
ganglion blocker) at the time of limb ischemia and reperfusion, indicating the requirement for
intact neural signaling[14], [20-23]. However, some clinical trials have shown no observable
benefit from delayed limb rIPC. In a recent study by Pavione et al., it was shown that four cycles
of 5 min limb ischemia applied a day before children were placed on cardiopulmonary bypass
(CPB) for congenital heart disease surgery had no effect on reducing cardiac troponin I plasma
concentrations, a commonly used surrogate marker of myocardial damage[12], [19].
Part of the difficulty in examining the delayed phase of preconditioning comes from
emerging evidence of a protective effect arising from exposure to anesthesia used during

20
experiments [24-26], [28], [30]. Mice are sedated with intra-peritoneal anesthetics, such as
pentobarbital, in order to apply transient ischemia to the hind limb. This is also done to minimize
the stress of the stimulus. As such, control treatments for rIPC involve sedating mice with
anesthesia without applying unilateral hind limb ischemia. In the Redington lab, this protocol
has been effective for examining the first window or early phase of preconditioning after rIPC, as
pentobarbital does not give rise to early preconditioning in mice[31]. However, the preliminary
data of this study suggest that exposure to an intra-peritoneal injection of pentobarbital does give
rise to delayed cardioprotection. This and additional evidence describing a delayed
preconditioning effect from other forms of anesthesia has limited the effectiveness of studies
specifically examining the biology of delayed limb preconditioning in mice. This may be
because the threshold for eliciting benefits from additional preconditioning stimuli may be
greater after exposure to anesthesia. Furthermore, the recent clinical studies showing no benefits
from rIPC highlight the possibility that certain substances used during surgery may inhibit the
effects of antecedent rIPC[20], [27], [29].
1.8 Anesthetic Preconditioning
Cardioprotection from exposure to anesthesia, known as anesthetic preconditioning
(APC), may be of profound importance in clinical settings as an adjunct that can safely elicit
protection during high-risk cardiac surgery[107], [108]. The beneficial role of anesthesia in
minimizing myocardial injury during IR has been known for decades, described by several
groups in the late 1960s [109], [110]. It was later discovered that exposure to anesthesia could
mimic preconditioning by attenuating post-ischemic damage and improving cardiac functional
recovery [111].

21
The following sections will be provide an overview of the effects of anesthesia on heart
function, the signaling pathways involved in APC and a summary of the accumulated evidence
on inhalational and intra-peritoneal APC. This will provide some background information on
potential anesthetics that may used to develop a model of delayed rIPC.
1.8.1 Effects of Anesthesia on Heart Function
In today’s clinical settings, the currently used inhalational anesthetics include: nitrous
oxide (N2O), halothane, enflurane, isoflurane, sevoflurane and desflurane – all of which (with the
exception of N2O) share the common feature of possessing a halogenated atom on a carbon
backbone[112]. It is well known that inhalational anesthesia causes a dose-dependent depressive
effect on myocardial function[108], manifested through reduced myocardial oxygen demand,
metabolic activity and vasodilation of coronary arteries – all of which may protect the heart
during IR injury[113]. All of the modern volatile anesthetics reduce cardiac output and mean
arterial pressure (mostly via a decrease in systemic vascular resistance) to some extent. This
mainly occurs through changes in Ca2+-handling within the cardiomyocytes at both the
sarcolemmal and sarcoplasmic reticulum (SR) level [114]. Volatile anesthesia decreases calcium
entry through L-type Ca2+ channels, which reduces Ca2+-dependent-Ca2+ release from ryanodine
receptors located on the SR. As a result, intracellular Ca2+ levels fall which reduces cardiac
inotropy and output[108], [110].
Halothane is thought to have the greatest negative effect on cardiac output, but causes
minimal changes in vascular resistance[115]. Conversely, isoflurane has a minimal effect on
cardiac inotropy, but invokes a large decrease in systemic vascular resistance[113], [116].
Interestingly, it has been suggested that isoflurane can cause mild ischemic episodes within the
coronary vasculature by producing varied levels of vasodilation. This process has been termed,

22
“coronary steal, ” which describes how under isoflurane anesthesia, non-diseased vessels may
dilate and ‘steal’ blood flow from vessels incapable of maximal dilation[113]. This may explain
some of the preconditioning-like effects that develop after isoflurane exposure.
Ketamine and sodium-pentobarbital (SP) are commonly used intra-peritoneal anesthetics
in animal models of myocardial IR injury. Similar to volatile anesthesia, ketamine and SP are
also known to depress cardiac function through changes in SR Ca2+ release and uptake and by
reducing extracellular Ca2+ entry [117], [118]. Jiang et al. showed that both ketamine and SP
reduced heart rate, left-ventricle systolic pressure and the rate of contractility in isolated rat
hearts[118]. In the same study, it was observed that SP showed a pronounced respiratory
depressive effect compared to ketamine and chloral hydrate anesthesia. In-vivo, ketamine has
been shown to markedly decrease heart rate and coronary flow and may induce pronounced
hypotension[103].
1.8.2 Mechanism of APC
The signal transduction pathways involved in APC are strikingly similar to those
activated during IPC. APC acts through opioid and adenosine GPCRs, which amplify the initial
signal and lead to the activation of the protective intracellular signalling response[32], [119],
[120]. As it occurs in IPC, the protective phenotype involves activating PKC, maintaining
intracellular Ca2+ ion concentration and stabilizing mitochondrial function via K+-ATP
channels[59], [121]. ROS production from the mitochondria is also integral for APC [110]. The
transition between early and delayed APC is also bridged via NF-κB activation[122], [123],
which upregulates the production of many of the same mediators responsible for delayed IPC,
such as iNOS and NO, MnSOD and Hsps[32]. There even exists some resemblance between
both forms of preconditioning at the genomic level. Sergeev et al. found that both IPC and APC

23
modulate genes involved in cell defense, growth, metabolism and inflammation. This is also
consistent with the changes in gene expression after rIPC[1], [2]. Similarly to IPC, APC
upregulates the expression of Hsps, inhibits proteins from the Bcl-2 family involved in initiating
apoptosis, and also activates NF-κB and its associated downstream products[124].
There are several levels at which APC intervenes to protect the myocardium. As in other
forms of preconditioning, maintaining Ca2+ homeostasis and minimizing overload is of critical
importance to preserving mitochondrial and cell function[59]. The mitochondrial K+ATP channel
has consistently shown to be an important component of the preconditioning phenotype. There is
still some debate regarding the importance of the sarcolemmal potassium channel, although some
studies confirm its involvement in APC[125], [126]. Both in-vivo and in-vitro studies have
shown that administration of 5-hydroxydecanoate (5-HD - a mitochondrial K+ATP channel
antagonist) or HMR-1098 (sarcolemmal K+-ATP channel antagonist) abolishes the benefits from
APC[126]. The mechanism of how K+-ATP channel opening protects the myocardium against IR
injury is not completely understood. It appears that opening of mitochondrial K+ATP channels
depolarizes the mitochondrial membrane, the immediate effect of which limits Ca2+ buildup in
the organelle and in the greater sarcoplasm. Depolarization is believed to initially cause swelling
of the mitochondria and a transient decrease in ATP production, which in turn activates complex
compensatory mechanisms that ultimately optimize oxidative phosphorylation and ATP
generation [59].
A key component of the APC phenotype involves an increase in ROS activity. It is not
entirely clear when the ROS burst occurs during the development of APC, however it is known
that a surge is obligatory as evidenced by loss of cardioprotection with the use of free-radical
scavengers[127]. Transient depolarization of the mitochondrial membrane leads to ROS release
from the mitochondria, which amplify the signaling cascade even further by spreading

24
throughout myocardium through intercellular gap junctions. ROS activate PKC in surrounding
cardiomycytes and ultimately lead to the opening of more mitochondrial K+ATP channels and
further ROS bursts[110].
APC is also known to suppress the inflammatory response during IR injury[128].
Sevoflurane and isoflurane have been shown to precondition isolated rat hearts by inhibiting
neutrophil adherence to cells[129], [130]. This is partly explained by an anesthetic-induced
decrease in neutrophil expression of CD11b adhesion molecules[130]. Several other studies have
shown that pretreatment with isoflurane anesthesia reduces neutrophil superoxide production in
addition to decreasing their adherence to the coronary vasculature [131], [132].
1.8.3 Isoflurane
Exposure to isoflurane anesthesia prior to myocardial IR injury is perhaps the most well
established method of APC. As a more soluble halogenated anesthetic than halothane and
enflurane, isoflurane is commonly used in experimental animal studies, owing to its stability and
quick clearance[116]. In 1988, Warltier et al. produced one of the earliest observations of a
protective effect of anesthesia on the myocardium during IR injury. They discovered that
isoflurane and halothane, when given during ischemia, lead to rapid improvements in post-
ischemic myocardial contractile function in dogs[111]. This set the stage for investigations into
whether isoflurane could mimic IPC. In 1996, Kersten et al. found that 1 minimum alveolar
concentration (MAC – defined as the minimum concentration of anesthesia at 1 atm that prevents
movement in response to stimulation in 50% of subjects[116]) administered for 30 min, thirty
minutes prior to CAO in a canine model, led to an 80% recovery of cardiomyocyte cell
shortening compared to 16% in non-isoflurance treated dogs [133]. This effect was abolished
when isoflurane was co-administered with glibenclamide, a non-specific K+ATP channel

25
antagonist. Further studies have confirmed the preconditioning effects of isoflurane in both in-
vivo [133] and in-vitro isolated heart models of IR injury [107], [134], [135].
Isoflurane preconditioning occurs through similar pathways as local and remote IPC.
Early isoflurane preconditioning involves the activation of PKC-ε, with subsequent maintenance
of mitochondrial K+ATP channels [107], [121], [134]. Other studies have shown that isoflurane
may also activate the PI3K-Akt signaling pathway. Raphael et al. found that 30 min of isoflurane
administration prior to CAO, reduced infarct size in rabbits, and this effect abolished with the use
of the PI3K inhibitors, wortmanin or LY294002 [136]. Inhibition of PI3K was also associated
with a decrease in phospho-Akt levels, and an increase in the pro-apoptotic proteins, Bad, Bax
and caspase-3.
Isoflurane preconditioning is also known to cause a delayed protective effect[122], [137-
140]. Chen et al. found that delayed isoflurane preconditioning in rats occurs through local NO
release, which triggers the PKC-induced increase in NF-κB activity. They discovered that NF-κB
amplifies the initial NO signal through upregulation of iNOS and further NO synthesis, which
then spreads the signal throughout the myocardium [122]. Ultimately, protection manifests by
maintaining an open state of K+ATP channels. Tonkovic-Capin et al. provided evidence for an
essential role for both the sarcolemmal and mitochondrial K+-ATP channels in delayed isoflurane
preconditioning of isolated rabbit hearts. They observed that 1 MAC of isoflurane administered
24 hour prior to global ischemia reduced infarct size and improved left-ventricular develop
pressure recovery, an effect that disappears with the use of either 5-HD or HMR-1098 K+ATP
channel antagonists [138].

26
1.8.4 Halothane
Since its introduction in 1956, halothane has been viewed as an anesthetic with the
greatest cardiodepressant effects, producing a decrease in blood pressure, cardiac output and
heart rate[113], [115], [116], [141]. Halothane is effective at reducing myocardial injury when
applied at the onset of reperfusion [24], [141], [142]. Similar to other anesthetics, halothane
attenuates post-ischemic cardiac dysfunction by reducing Ca2+ cycling and overload[24], [26]. A
halothane-induced anti-inflammatory effect during reperfusion injury has also been described.
Kowalski et al. showed that halothane and isoflurane administration decreased post-ischemic
neutrophil adhesion in isolated guinea pig hearts that received 6x105 neutrophils in the coronary
perfusate prior to ischemia[27].
Despite considerable evidence describing the benefits of halothane during IR injury, its
role as a preconditioning stimulus is still uncertain. Preliminary findings indicate that the
protective effects of halothane anesthesia may be confined to mitigating reperfusion injury.
Roscoe et al. showed that halothane exerted no cardioprotective benefits on human atrial muscle
when given 30 min before anoxia, and may have even inhibited the effects of prior transient
anoxia preconditioning[107]. However, there is some evidence that halothane may exert
cardioprotective benefits[109], [111]. In a study by Piriou et al examining the effects of
halothane preconditioning on rabbit myocardium, it was shown that 30 minutes of halothane
exposure 15 minutes before IR injury reduced LV infarct size compared to untreated
controls[109]. In other systems of preconditioning, such as in the brain, halothane has been the
choice of anesthesia in mice undergoing transient middle cerebral artery occlusion, producing no
demonstrable protection in non-preconditioned controls[143], [144]. To our knowledge, there are
no previous studies describing a role for halothane anesthesia in the development of delayed
tolerance to IR injury.

27
1.8.5 Sevoflurane, Enflurane and Desflurane
Other halogenated anesthetics have been found to exert cardioprotective effects through
similar mechanisms as isoflurane, including the activation of PKC, mitochondrial K+ATP channels
and increased NO bioavailability [141], [145], [146]. Lu et al. showed that a critical step of
sevoflurane-APC involves ROS-dependent activation of NF-κB. In isolated rat hearts, they found
that 2.5% sevoflurane reduced infarct size, preserved cardiac function and reduced pro-apoptotic
protein activation [147]. The importance of ROS-induced Nf-κB activation has been confirmed
in several studies of sevoflurane preconditioning [148].
Additionally, there are several reports of a delayed preconditioning effect with
administration of sevoflurane, enflurane and desflurane. Chiari et al. found that 24 hours after
intravenous administration of emulsified enflurane and sevoflurane, infarct size after CAO was
reduced in APC rabbits by approximately 50% compared to lipid vehicle and saline treated
controls[149]. Furthermore, Lotz et al. found that 1MAC of desflurane administered 24 hours
before CAO, reduced infarction sizes in rabbits. They identified an essential role for peroxisome-
proliferator-activated receptor γ activation and increased levels of NO in desflurane
preconditioned hearts[150].
1.8.6 Nitrous Oxide
Nitrous oxide (N2O) is one of the oldest methods of anesthesia and is still in clinical use
today as a potent analgesic[28], [30], [113]. Nitrous oxide is often used with other halogenated
anesthetics and is known to potentiate their myocardial depressant effects[151]. The nitrous
oxide anti-nociceptive effects are known to be mediated by activation of κ-opioid receptors in the
periaqueductal gray region of the brain[152], [153]. Given the well known role of opioid

28
signaling in preconditioning, it is possible that nitrous oxide may induce cardioprotection by
binding to opioid receptors located on the myocardium[6], [33], [72].
Weber et al. conducted a study to examine whether nitrous oxide could precondition rat
hearts in a coronary artery occlusion (CAO) model of IR injury[154]. Rats received three 5 min
cycles of 60% N2O (with 20% O2 and 20% N2) interspersed with 5-min washout periods prior to
sustained CAO. This was compared this with the effects of APC via N2O+isoflurane delivered in
the same manner. They found that exposure to nitrous oxide provided no protection against IR
injury, as N2O-treated mice displayed similar infarct sizes as untreated controls. Not surprisingly,
isoflurane+N2O reduced infarction sizes by approximately 40%. As such, they concluded that
nitrous oxide might be the first inhalational anesthetic without preconditioning effects[154]. In
another study examining nitrous oxide preconditioning against hypoxia-reoxygenation injury, it
was found that N2O improved LVDP and reduced L-type Ca2+ channel currents in isolated rat
hearts when administered during hypoxia and had no effect when provided as a preconditioning
stimulus [155].
Therefore, it appears that like other inhalational anesthetics, nitrous oxide improves
myocardial function when administered during low oxygen challenges, but may not induce a
cardioprotective phenotype when given prior to lethal IR injury.
1.8.7 Intra-peritoneal Anesthesia: Ketamine and Barbiturates
Ketamine
There is some indication that intra-peritoneal anesthetics may induce or even inhibit the
effects of IPC. Ko et al. discovered that ketamine hydrochloride anesthesia decreases K+ATP
channel activity in a dose-dependent manner in rat ventricular myocytes [156]. Another study
extended this finding by showing that both the sarcolemmal and mitochondrial K+ATP channels

29
are inhibited with exposure to ketamine[157]. These findings suggested that ketamine might also
block the benefits of IPC. Later studies confirm this hypothesis, but showed that the ketamine-
blockade of IPC appears to be isomer specific. A number of studies discovered that treatment
with the optical isomer R(–) ketamine prior to local IPC blocked its infarct sparing effects in an
isolated heart or in-vivo animal model[158], [159]. With regards to delayed IPC, Müllenheim et
al. showed that a racemic mixture of the R and S enantiomers inhibited the cardioprotective
effects of late IPC via transient CAO. In each report, it was shown that S(+)Ketamine had no
effects on the preconditioning stimulus, further establishing that the inhibition is specific to the R
isomer.
Hanouz et al. sought to investigate whether the racemic mixture was in fact blocking
cardioprotection or inducing its own preconditioning-like phenotype[160]. They examined the
effects of racemic and S(+) ketamine on human atrial myocardium subjected to hypoxic injury.
Interestingly, they discovered that exposure to both the racemic and S isomer of ketamine
enhanced contractile recovery after hypoxia when compared to untreated controls[160]. It was
also discovered that these protective effects were abolished with co-administration of 5-HD,
HMR 1098 or α/β adrenergic receptor antagonists. These findings indicate that what was initially
believed to be a ketamine-induced blockade of IPC, may have actually been a preconditioning-
like effect overriding benefits from additional cardioprotective stimuli.
Pentobarbital
Pentobarbital is often the standard method of sedation used for isolated heart or in-vivo
APC experiments. SP has been shown to cause a large decrease in cardiac output, left-ventricle
systolic pressure and heart rate in rats in comparison to ketamine and chloral hydrate anesthesia
[35], [44], [118]. Barbiturates are also known to act as free radical scavengers and thus may limit

30
the damage from the ROS bursts occurring during IR injury. [110]. However, it is evident from a
number of studies that SP does not block the effect of early local and remote IPC in a number of
species [5], [8], [31], [103], [161-165]. The Bolli group has shown that SP anesthesia does not
induce cardioprotection in an early and late mouse model of local IPC[9], [103], [166-168]. Their
initial study in which they developed their model involved pilot work with various forms of
anesthesia and they found that barbiturate anesthetized mice exhibited heart rates closest to
normal physiological levels (avg. 650 beats/min)[103]. They reported that additional
experimental adjustments needed to be made to compensate for the cardiopulmonary depressive
effects of SP in mice, such as the constant monitoring of body temperature and proper tidal
ventilation [103]. Although the Bolli group has shown pentobarbital does not induce
preconditioning during local IPC, an evaluation into the effects of SP during late rIPC has not
been undertaken.
1.9 Anesthetic and Ischemic Preconditioning: Clinical Utility
APC represents a clinically useful and safer method of inducing cardioprotection when
compared to local coronary IPC, eliminating the requirement for invasive surgery that can
damage the coronary vasculature and increase the risk for future complications. The advent of
rIPC by transient limb ischemia overcomes this obstacle. Findings generated by our group and
others demonstrate the powerful clinical benefits of rIPC [69], [105], [106], [169]. In the last
decade, accumulating evidence has shown that making rIPC a part of the clinical management of
AMI can result in reduced patient mortality and morbidity [47], [48], [69], [106].
However, these important findings have met a fair share of conflicting results. Several
recent studies suggest that rIPC may provide no additional benefit against IR injury during
surgery. Rahman et al. conducted a large trial examining the effects of three-5-min cycles of

31
upper arm ischemia in patients undergoing coronary bypass surgery. They found that rIPC
provided no benefits in reducing troponin T levels or in improving cardiac function compared to
placebo sham-treated controls[170]. In a recent clinical trial, Kottentburg et al. found that the
effects of rIPC were negligible when applied with propofol anesthesia[29]. It should be noted
that neutral findings generated in these clinical studies may reflect differences in the settings in
which rIPC is applied. Our lab group is interested in examining the possibility that certain agents
used in patient care may block rIPC – a necessary investigation to further optimize the
preconditioning response.
Nonetheless, the usefulness of APC and the recent neutral trials does not eliminate the
need to better describe rIPC through limb ischemia, as it can be used outside of the clinical arena
where access to anesthesia is not possible. This is especially important when considering rIPC
may benefit cardiovascular function in a number of ways, other than minimizing IR injury. For
instance, our group has shown that rIPC can increase coronary blood flow and lower coronary
vascular resistance[171]. These and other findings highlight that intermittent ischemia may have
potential blood-pressure lowering effects, which could be of profound importance for exercise
therapy in patients with cardiovascular disease[172]. Our lab group has also shown that repeated
transient limb ischemia for 28 days can reduce post-infarct adverse remodeling[173], can induce
a sustained anti-inflammatory phenotype when repeated for several days[1-3] and can even
improve exercise performance in highly-trained athletes[174].
1.10 Langendorff Isolated-Heart Model of IR Injury
Anesthetic preconditioning, both in clinical and experimental settings, adds a challenge to
examining the biology of rIPC beyond the early phase of protection. Given that animals need to
be sedated with anesthesia in order to apply unilateral transient limb ischemia, it is imperative to

32
employ a mode of sedation without cardioprotective effects. This will also serve as part of a
control treatment for delayed rIPC. Based on preliminary findings that intra-peritoneal
pentobarbital, when administered twenty-four hours prior to IR injury, is associated with
cardioprotection in mice, I began to investigate the effects of various inhalational and injectable
anesthetics in order to develop a mouse model of delayed rIPC.
The isolated Langendorff mouse heart model has been an invaluable tool for examining
cardiac physiology and has played an important role in identifying the signaling pathways
involved in preconditioning. It is a highly reproducible preparation that can be operated at a
relatively low cost and provides a wealth of data describing the myocardial response to various
stimuli [175]. Advancing transgenic mouse species are making murine Langendorff models a
frequently used tool for studying basic cardiac physiology. The isolated heart preparation allows
measurement of a broad spectrum of physiological and biochemical parameters during a variety
of challenges, such as IR injury, hypoxia, drug-dose responses, hypo-/hyperthermia and
electrophysiological alterations[176], [177], the main advantage being that the acquired
responses are cardiac specific, devoid of any influence from systemic circulation, neuro-
hormonal or immunological influences.
The small size of the mouse heart makes the isolated-heart model technically challenging
in this species. As a result, initial studies employing this method showed a high degree of
baseline variability in their results. The Headrick group has conducted a number of valuable
studies aimed at characterizing the murine Langendorff model[175], [178]. They have provided
baseline assessments of various cardiac parameters that can be used as normal criteria for the
mouse isolated-heart preparation. Our lab has made use of their experimental approaches and
setups in developing our system.

33
Chapter 2 Research Aims and Hypotheses
2.1 Summary and Rationale
It is clear that further studies are required to improve our understanding of rIPC,
especially relating to its delayed and potential chronic effects. Investigating these additional
phases will provide a more general knowledge base of cardioprotection and help to identify
potential clinical settings in which the intervention can benefit individuals.
However, as with any potential clinical intervention, a sound physiological model must
first be developed to properly understand the mechanisms behind delayed preconditioning. This
is particularly relevant to studies of delayed preconditioning, as animal care committees currently
mandate anesthesia for the initial preconditioning stimulus. Thus an important component of this
model requires that it be able to separately examine the effects of anesthetic and ischemic PC. As
discussed earlier, there is a wealth of evidence showing that most modernly used volatile
anesthetics invoke potent early and delayed cardioprotection. However, it is still unknown
whether halothane and nitrous oxide cause delayed cardioprotection. Furthermore, while there is
a substantial body of literature in animal and human studies describing an early preconditioning
effect arising after transient limb ischemia, evidence of a late phase of protection is scarce.
Delayed preconditioning may have a strong impact in settings beyond the operating room, as it
can potentially be used to improve various parameters of cardiovascular function. It may also be
important in populations where maintaining a baseline preconditioned state can ameliorate the
negative side effects of certain cardiovascular diseases, such as diabetes, hypertension and
pathological cardiac hypertrophy.

34
Using the isolated heart model is an important and necessary first step in examining the
delayed preconditioning phenotype. Taking advantage of the cardiac specific responses will
expand our understanding of the late phase protection and may provide insight into future areas
of investigation required to optimize this powerful method of organ protection.
2.2 Research Aims/Objectives
Primary Objective
To identify and establish a method of sedation in mice for delayed rIPC experiments
using the Langendorff-model of IR injury
Specific Aims
1. To examine the delayed/second window (2W) effects of intra-peritoneal (IP) injection of
sodium-pentobarbital (SP) anesthesia or saline, on post-ischemic heart function, and to
compare the results with the early (first window-1W) control and rIPC groups
2. To investigate the delayed/2W effects of inhalational halothane and nitrous-oxide
anesthesia on post-ischemic heart function in order to assess their suitability as alternative
methods of sedation for delayed rIPC studies.
2.3 Hypotheses
i. It was hypothesized that intra-peritoneal injections of saline will induce delayed
cardioprotection against IR injury in isolated-hearts.
ii. It was hypothesized that halothane will induce delayed preconditioning against IR injury
whereas nitrous oxide will not confer cardioprotection.

35
Chapter 3 Methods
3.1 Ethics
All animal protocols were approved by the Animal Care and Use Committee of the
Hospital for Sick Children in Toronto and conformed to the Guide for the Care and Use of
Laboratory Animals published by the National Institutes of Health (NIH publication No. 85–23,
revised 1996).
3.2 Experimental Groups
Male C57BL/6 mice (9-11 weeks of age) were used for all treatment groups. Animals
were maintained on a 12h dark/light cycle and housed in single cages at room temperature and
were provided with food and water ad libitum. Mice were divided into intra-peritoneal and
inhalational anesthesia treatment categories to assess the delayed/second window (2W) effects of
these drugs with an established model of early/first window (1W) preconditioning studies used in
our lab. 6-8 mice were used in each group (figure 6).
First window model of rIPC
The established mode of preconditioning using the first window model of rIPC was
compared with the various delayed stimuli examined in this study to evaluate their potential
cardioprotective effects. In our model of early rIPC, mice were divided into first window control
(1WSP) and rIPC (1WSP+rIPC) groups. Mice in the 1WSP group received SP (60mg/kg of body
weight) via an intra-peritoneal (IP) injection and were kept under anesthesia for 40 minutes (to
match rIPC treatment duration). The 1WSP+rIPC group received SP and rIPC (four cycles of 5

36
min of hind-limb ischemia and 5 min of reperfusion). To examine the early phase of
preconditioning, hearts from the 1WSP and 1WSP+rIPC were isolated 15 minutes after each
treatment for mouse Langendorff IR injury or Western blot protein experiments (figure 3).
Intra-peritoneal anesthesia – the delayed effects of sodium-pentobarbital
To assess the delayed effects of SP alone and with additional rIPC, mice were divided
into 2WSP and 2WSP+rIPC groups. Mice in the 2WSP and 2WSP+rIPC groups received SP
anesthesia (60mg/kg of body weight; IP) on day1 ± rIPC (as per method in 1WSP+rIPC). We
also developed a delayed saline treated group (2W Saline) to test for the potential
preconditioning effects of an apparently ‘benign’ IP injection. Mice in this group received
0.12mL saline via IP injection as previously described[179]. To examine the delayed phase of
preconditioning, mice from these groups re-anesthetized with SP twenty-four hours later
(60mg/kg of body weight; IP) and hearts were isolated for global ischemia or for Western blot
protein experiments.
Inhalational Anesthesia – the delayed effects of halothane and N2O
To examine the delayed effects halothane and nitrous oxide inhalational anesthesia, mice
were divided into 2W Halothane and 2W N2O groups. The 2W Halothane group underwent
sedation with halothane anesthesia for 40 minutes at a 2% halothane/98% oxygen induction
concentration and was maintained at 1% halothane/99% oxygen for duration of the experiment.
These concentrations were determined from pilot experiments examining the amount of
halothane that was required to eliminate any response from hindlimb ischemia via a tourniquet.
Mice in the 2W N2O group were exposed to 40 minutes of N2O delivered at a 2:1 nitrous oxide
to oxygen ratio while placed in a 12 cm x 24 cm container. Twenty-four hours after sedation,

37
Figure 6: A schematic of the study protocol. Mice were divided into seven groups based on method of anesthesia±rIPC. Groups were also divided into intra-peritoneal and inhalational anesthesia for comparison with established 1W model of rIPC. For 1W groups, IR injury experiments occurred on the same day as preconditioning treatment. For 2W groups, IR injury experiments occurred on day 2, twenty-four hours after preconditioning treatment. Hearts were collected for western blot experiments at indicated points (WB). At the end of Langendorff IR injury, hearts were stained with TTC for LV infarct size analysis. 1W-first window/early, 2W-second window/delayed, rIPC-remote ischemic preconditioning, SP-sodium pentobarbital, N2O-nitrous oxide, TTC-triphenyltetrazolium chloride, WB-western blot protein analysis

38
mice were re-anesthetized with SP (60mg/kg of body weight via an IP injection) and hearts were
isolated for global ischemia or for Western blot protein experiments.
3.3 Induction of rIPC Using Inguinal Tourniquet Model
Mice were sedated with a method of anesthesia depending on the treatment group. rIPC
was induced by four cycles of 5 minutes of femoral artery occlusion intercalated with 5 minutes
of reperfusion. This was done using a tourniquet tied around the left hindlimb at the inguinal
level to apply unilateral ischemia to the femoral vasculature (figure 7). Ischemia was marked by
limb paleness and reperfusion by rapid hyperemia as previously described[31]. Hearts were then
isolated for Langendorff preparation or Western blot analysis 15 min (1W) or 24 hours (2W)
after rIPC.
Figure 7: Remote ischemic preconditioning (rIPC) via transient ischemia of mouse
hindlimb. A tourniquet is tied at the inguinal level to apply unilateral ischemia. Ischemia was
marked by distal limb paleness. Reperfusion was marked by rapid hyperemia.
5 min Ischemia
5 min Reperfusion
Tourniquet
Tourniquet
Tourniquet Tourniquet

39
3.4 A Langendorff Isolated Heart Model of Ischemia-Reperfusion Injury
IR injury was induced by 30 min of global ischemia followed by 60 min reperfusion in
isolated mouse hearts using a constant hydrostatic pressure, non-circulating, isovolumic
Langendorff preparation (Radnotti Technologies) as previously described in our lab [31], [178]
(figure 8a,b).
Preparation of Krebs-Heinseleit buffer
Hearts were perfused with modified Krebs-Heinseleit Buffer (KHB) at 80mmHg at 37°C
containing (mM): NaCl 120mM, NaHCO3 25mM, KCl 4.7mM, MgSO4 1.2 mM, KH2PO4 1.2
mM, CaCl2 2.5mM, EDTA 0.5mM and glucose 15mM. Buffer was prepared in a 2L Erlenmeyer
flask and subsequently filtered through a 0.2 µm Nalgene bottle top filter (Nalgene Cat#595-
4520, Rochester, NY). Buffer was adjusted to a pH of 7.4 by 6N HCl and oxygenated (95% O2,
5% CO2) in a water-jacketed reservoir via a sintered glass gas distributor (Radnotti
Technologies) for 60 minutes prior to experiment. Perfusion fluid was delivered to hearts
through water-jacketed tubes at 37°C.
Excision of Mouse Hearts and Aortic Cannulation
Mice were anesthetized with SP (60mg/kg of body weight) and received heparin to
prevent coagulation on apparatus (200 units – Sigma Inc.) via IP injections 5 min prior to heart
removal. Due to the respiratory depressant effects of SP[176], mice were then intubated and
ventilated at a respiratory rate of 100 breaths/min with a tidal volume of 1.5 mL per stroke. A
trans-abdominal incision was then made through the peritoneum to expose the diaphragm, which
is then cut to access the thoracic cavity and heart via a bilateral thoracotomy. Hearts were rapidly
excised and placed in a 100 mm dish containing oxygenated KHB at 37°C where it was trimmed

40
of excess connective, lung and thymus tissue. Hearts were then placed in a 4°C cannulate buffer
(NaCl 140mM, KCL 4.2mM, KH2PO4 1.2 mM, MgCl2 0.5mM, Hepes 10mM – pH 7.4) and the
aorta was cannulated with a 20-gauge metal cannula under magnification to the point just before
the aortic valves to allow for perfusate to enter through the coronary ostia. Sutures were tied
around aorta-cannula junction to secure aorta and to prevent leakage of KHB. Once the aorta was
cannulated, the three-way stopcock on the mounting position was turned to perfuse the coronary
circulation at 80mmHg with KHB.
Figure 8a. The mouse Langendorff-isolated heart model. Mouse coronary arteries are
perfused with modified Krebs-Heinseleit Buffer at a constant pressure of 80mmHg at 37°C, in
retrograde fashion through a cannula inserted into the aorta. An intra-ventricular balloon is
inserted into the left-ventricle through the mitral valve and connected to a pressure transducer
hooked up to bio-lab software to measure peak left-ventricle pressure (LVP), left-ventricular
end-diastolic pressure (LVEDP), left-ventricle developed pressure (LVDP), rate of contraction,
rate of relaxation and heart rate

41
Figure 8b. The mouse Langendorff-isolated heart model used in the Redington laboratory.
Langendorff setup can operate two mouse hearts using 2L of Krebs-Heinseleit buffer. Heart is
mounted on cannula inserted through aorta via a one way in-flow valve, which can readily be
closed to induce global ischemia.

42
Hearts were then mounted by attaching metal cannula to three-way stopcock on the Langendorff
apparatus. The enitre process from heart excision to mounting takes approximately 3-5 minutes.
Additional sutures were knotted around aorta to secure it to the cannula. To facilitate drainage of
perfusate from coronary circulation, a small incision was made at the base of pulmonary artery
using micro-scissors.
Insertion of An Intra-Ventricular Balloon
Once perfusion is initiated, hearts were submerged in a water-jacketed container at 37°C
throughout the duration of experiment. The auricular appendage of the left atrium was removed
to expose the atrium. A saran wrap balloon, with PE60 polyethylene tubing connected to a
microsyringe and pressure transducer (AD instruments (ADI)-ML844, Colorado Springs, CO),
was placed in the left ventricle through the mitral valve. The balloon volume was initially
deflated upon insertion and then adjusted once placed within the ventricle to give a preload left-
ventricular end-diastolic pressure (LVEDP) of 7-10mmHg (contains <20µL of distilled water).
Balloons were fastened to cannula using tape to maintain a straight entry into the ventricle. The
balloon volume was kept constant throughout the duration of the experiment.
Measurement of Cardiac Function and Induction of Global Ischemia
Hearts were stabilized on the apparatus for 20 min prior to global ischemia. Using Power-
lab acquisition software (ADI instruments, Colorado Springs, CO – see appendix 1) we measured
hemodynamic parameters of cardiac function throughout the experiment, which included peak
left ventricular pressure (LVP), maximum rate of ventricular contraction (+dP/dtmax) and
maximum rate of ventricular relaxation (-dP/dtmin), LVEDP and heart rate. Left-ventricular
developed pressure (LVDP) was determined as the difference between peak systolic and diastolic

43
pressures. Coronary flow rate was measured by collecting effluent produced in 1 min via the
coronary sinus at two time points: before global ischemia and at the end of 60 minutes of
reperfusion. Thirty minutes of global ischemia was induced by turning the three-way stopcock to
the closed position to cease perfusion with KHB and was set to the open position during
reperfusion (60min). At the end of the experiment, hearts were collected and immersed in 10%
KCl to induce diastole. Hearts were then weighed and frozen in liquid nitrogen and stored at
-80°C and later measured for infarct size.
Special Considerations
Given the technical challenges involved in excising and perfusing mouse hearts and the
various important details involved with the apparatus setup, it was important to use established
criteria for determining the suitability of hearts for the experiment[178]. At the end of the 20 min
stabilization, hearts were excluded if they acquire: (1) bradycardic heart rate < 300 bpm, (2) flow
rate > 5 mL/min, (3) LVP <80 mmHg (4) high level of arrhythmia.
3.5 Infarct Size Determination
After reperfusion, the frozen heart was transversely cut into six 1-mm thick slices using a
Mouse Heart Slicer Matrix (Zivic Instruments) which were stained with 1.25% 2,3,5-
triphenyltetrazolium chloride (TTC) in 200 mM Tris/HCL solution (pH 7.4) for 15 min in a 37°C
water bath. After staining, heart slices were fixed for 2-4 hours in 10% neutral buffered
formaldehyde. Both sides of each slice was then photographed at 1200 DPI resolution using a
computer scanner (CanoScan 4400F). Images were processed with Adobe Photoshop® CS2
software to measure infarct size and left-ventricle area using automated planimetry. Viable
myocardium stains red due to the reaction of tetrazolium salts with NADH and dehydrogenase

44
enzymes while infarcted tissue, that does not possess enzymes, appears pale[180]. Infarct sizes of
each slice were expressed as the percentage of the total left ventricle area. The reported infarct
size is the mean of infarct size measurements from both sides of all the individual slices.
3.6 Protein Extraction
For protein analysis experiments, hearts were harvested either 15 min (1W groups) or 24
hours (2W groups) after exposure to anesthesia ± rIPC without IR injury. Each heart was frozen
in liquid nitrogen and stored at –80°C. For determining protein concentration, hearts were cut
into small pieces (totaling 50mg) and shaken in 1 mL micro-tubes (Dia-Med Lab Supplies Inc.
Mississauga, ON) using two metals beads in order to homogenize tissue in lysis buffer (see
appendix). Homogenate was incubated on ice for 30 min to allow for tissue lysing. Homegenate
was then centrifuged at 10,000 rpm at 4°C for 30 min to obtain whole tissue supernatant and the
discarded pellet. Collected supernatant was then centrifuged at 10,000 rpm at 4°C for 10 min to
further separate supernatant and discarded pellet. Protein concentration was determined using a
bovine serum albumin (BSA) standard (25 µg/mL) (Sigma-Aldrich Co., St. Louis, MO). Protein
concentration was determined by preparing a solution consisting of 1-2 µL of resulting sample
supernatant, 200 µL of dye reagent protein assay concentrate (Bio-Rad Laboratories Catalog,
Hercules, CA) and 800 µL of dd-H2O. The solution was vortexed and allowed to react for a
minimum of 15min. Using a UV visible spectrophotometer (Ultrospec 3000, Fisher Scientific,
Markham, ON) a standard curve of BSA optical density (OD) measured at 595 nm for 0 µg, 2.5
µg, 5 µg, 10 µg, 15 µg and 20 µg was constructed. Sample protein concentrations were measured
at the same OD and the resulting concentration was determined from the developed standard
curve. Samples were aliquot to 6-9 tubes and stored at -80°C for further analysis.

45
3.7 SDS-PAGE and Western Blot Analysis
Resolving and Stacking Gel Preparation
Protein samples were separated by one-dimensional sodium dodecyl sulphate (SDS)-
polyacrylamide gel electrophoresis (PAGE) using 10% resolving and 5% stacking gel (see
appendix for gel make-up) on a Bio-Rad Mini-protean III gel electrophoresis system (Bio-Rad
Laboratories, Hercules, CA). Glass plates with 0.75 mm spacers were set up in a gel-casting
frame. The 10% resolving gel (see appendix) was prepared and added between glass plates and
allowed to polymerize for 45 min. Gel surface was overlaid with distilled water. 5% Stacking gel
was then prepared (see appendix 2) and added on top of resolving gel with a 10-well plastic-
comb and allowed to polymerize for 45 min.
Electrophoresis of Proteins
An equal amount of protein (30 µg) from whole tissue lysates was prepared with
homogenizing lysis buffer and 5 uL of 3X sample buffer w/ 100mM DTT reducing agent (Cell
Signaling Technologies; 87.5 mM Tris-HCl (pH 6.8 at 25ºC), 6% (w/v) SDS, 30% glycerol and
0.03% (w/v) bromophenol blue) to give a final loading volume of 15 uL. Samples were
centrifuged and boiled for 5 min at 100°C. 5µL of PageRulerTM Plus Prestained Protein Ladder
(Thermo Fisher Scientific, Rockford, IL) was placed in first well as a protein molecular weight
marker. Proteins were loaded into wells 2-10, and electrophoresed in a 1X Tris/Glycine/SDS
buffer solution (25mM Tris, 192 mM glycine, 0.1% SDS, stored at rm. temp, pH 8.3) with an
initial potential difference of 80V at the start (until gel-front migrated past stacking gel) and then
at 100mV for migration through resolving gel.

46
Transfer of Proteins to Nitrocellulose Membranes
Following electrophoresis, resolving gels equilibrated in transfer buffer (25 mM Tris, 192
mM glycine, 20% methanol-stored at 4°c) for 15 min. Nitrocellulose membranes (0.45 µm pore
size - Bio-Rad Laboratories) were placed dd-H2O for 15 min and then in transfer buffer for 5
min. Resolving gels were then placed in western-transfer sandwich consisting of brillo pad, two-
pieces of filter paper (Whatman®, Maidstone, England), nitrocellulose membrane, resolving gel,
two-pieces of filter paper and a final brillo pad. Western-transfer sandwich was then placed in
gel-transfer unit using Bio-Rad Mini-protean III transfer system with an ice-pack and constant
stirring. Proteins migrate from resolving gel on to nitrocellulose membrane across a potential
difference of 100V for 90min. The transfer of proteins was done in an ice-filled container to
minimize overheating of the apparatus.
Detection of Proteins with Anti-bodies
Following protein transfer, the nitrocellulose membranes were rinsed with three 5 min
cycles with 1X TBST buffer (10X stock – Tris 24.2g, NaCl 80.0g, pH 7.6 brought to 1L and
diluted 1/10 for 1X TBST + 1 mL Tween 20) at room temperature. The membrane was then
placed in blocking buffer solution (1 mL TBST buffer with 5% (w/v) skim milk powder) for 1
hr. Membranes were then incubated overnight with rabbit monoclonal antibodies against
phospho-Akt at the Ser473 residue (Catalog #4060 - Cell Signaling Tech.) at a 1:1000 dilution,
total-Akt (Catalog #4691 - Cell Signaling Tech.) at a 1:1000 dilution, phosphor-p44/42 MAPK at
the Thr202/Tyr204 residues (Catalog #4370 - Cell Signaling Tech.), total p44/42 (Catalog #4695
- Cell Signaling Tech. Danvers, MA) at a 1:1000 dilution and the mouse monoclonal antibody
GAPDH (Catalog #G8795 - Sigma-Aldrich Inc) at a 1:15,000 dilution. Membranes were then
washed with 1X TBST buffer for three 5 min cycles and subsequently incubated with a

47
horseradish-peroxidase (HRP)-conjugated secondary antibody (Santa Cruz Biotechnology) at an
appropriate dilution. Secondary antibody bound to a primary antibody was detected using
enhanced chemiluminescence (ECL) plus detection kit (Amersham Inc.). Membranes were then
scanned using STORM 840 analyzer and quantified using ImageQuant 5.0 densitometry software
(Molecular Dynamics).
3.8 Data and Statistical analysis
Data from Power Lab Acquisition software were analyzed using Lab Chart 7 and
processed to Microsoft Excel 2011 for Macintosh. Graphs and charts were made using Microsoft
Excel 2011 and GraphPad Prism 5.0 software. Statistical analysis was done using one-way
ANOVA and Student’s t-test function in GraphPad Prism 5.0 software. Hemodynamic data were
processed into 10-second segments from each isolated heart experiment, and values were
reported in 10 min interval summaries. LVP, +dP/dtmax, and -dP/dtmin were analyzed and
expressed as a percentage of baseline pre-ischemic values. All values are expressed as means ±
standard error of the mean (SEM). A statistically significant difference was among three or
more groups was determined using one-way ANOVA and post-hoc testing (Newman-Keuls Test)
through GraphPad Prism 5.0 software. A Student’s t-test was used to compare the differences in
means between two groups from western blot analysis. All treatment groups were compared to
1WSP control groups for effects. A difference associated with a P-value of ≤0.05 was considered
statistically significant.

48
Chapter 4 Results
4.1 The Delayed Effects of Intra-Peritoneal and Inhalational
Anesthesia on Left Ventricular Function after Global Ischemia
4.1.1 Baseline Function
In the present study we compared the delayed effects of intra-peritoneal SP and
inhalational halothane and N2O anesthesia with our established model of early rIPC on LV
function after global ischemia. The following hemodynamic parameters of isolated-hearts were
assessed throughout the duration of the IR injury experiments: Left ventricular developed
pressure (LVDP), left ventricular end-diastolic pressure (LVEDP), maximum rate of contraction
(dP/dtmax), maximum rate of relaxation (dP/dtmin) and heart rate. The baseline values from each
group were compared to ensure that there were no initial differences prior to induction of IR
injury. As shown in table 1, there were no statistically significant differences in baseline cardiac
function between all treatment groups at the end of the 20-minute stabilization period. Baseline
cardiac function was used to assess the recovery of hearts after IR injury, as the values of LVDP,
dP/dtmax and dP/dtmin acquired during reperfusion were expressed as a percentage of baseline
function. The LVEDP for each treatment was expressed as the absolute value throughout the
experiment. As described in the methods section, hearts were assessed for their suitability for IR
injury experiments based on an established set of criteria from the literature. Hearts were
excluded from the study if at the end of the stabilization, they exhibited a (1) bradycardic
HR<300 beats/min (2) CFR>5.0mL/min (3) LVP< 80mmHg and (4) high level of arrhythmia.
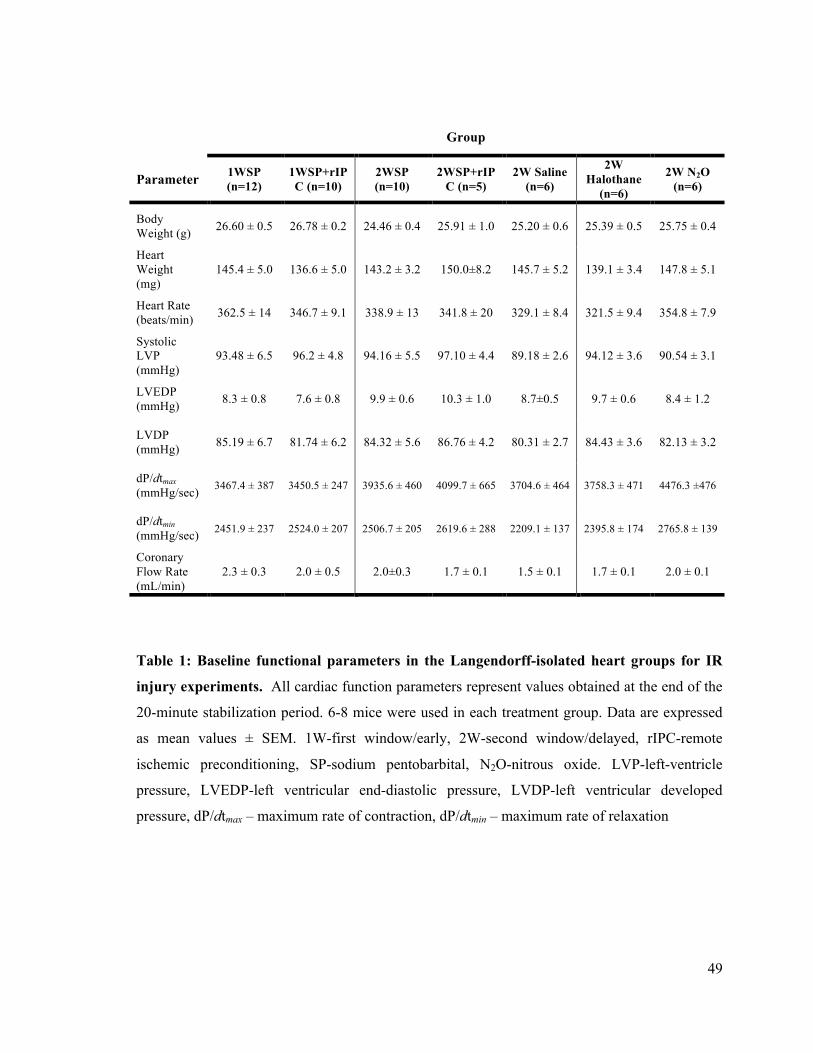
49
Group
Parameter 1WSP
(n=12) 1WSP+rIPC (n=10)
2WSP (n=10)
2WSP+rIPC (n=5)
2W Saline (n=6)
2W Halothane
(n=6)
2W N2O (n=6)
Body Weight (g) 26.60 ± 0.5 26.78 ± 0.2 24.46 ± 0.4 25.91 ± 1.0 25.20 ± 0.6 25.39 ± 0.5 25.75 ± 0.4
Heart Weight (mg)
145.4 ± 5.0 136.6 ± 5.0 143.2 ± 3.2 150.0±8.2 145.7 ± 5.2 139.1 ± 3.4 147.8 ± 5.1
Heart Rate (beats/min) 362.5 ± 14 346.7 ± 9.1 338.9 ± 13 341.8 ± 20 329.1 ± 8.4 321.5 ± 9.4 354.8 ± 7.9
Systolic LVP (mmHg)
93.48 ± 6.5 96.2 ± 4.8 94.16 ± 5.5 97.10 ± 4.4 89.18 ± 2.6 94.12 ± 3.6 90.54 ± 3.1
LVEDP (mmHg) 8.3 ± 0.8 7.6 ± 0.8 9.9 ± 0.6 10.3 ± 1.0 8.7±0.5 9.7 ± 0.6 8.4 ± 1.2
LVDP (mmHg) 85.19 ± 6.7 81.74 ± 6.2 84.32 ± 5.6 86.76 ± 4.2 80.31 ± 2.7 84.43 ± 3.6 82.13 ± 3.2
dP/dtmax (mmHg/sec) 3467.4 ± 387 3450.5 ± 247 3935.6 ± 460 4099.7 ± 665 3704.6 ± 464 3758.3 ± 471 4476.3 ±476
dP/dtmin (mmHg/sec) 2451.9 ± 237 2524.0 ± 207 2506.7 ± 205 2619.6 ± 288 2209.1 ± 137 2395.8 ± 174 2765.8 ± 139
Coronary Flow Rate (mL/min)
2.3 ± 0.3 2.0 ± 0.5 2.0±0.3 1.7 ± 0.1 1.5 ± 0.1 1.7 ± 0.1 2.0 ± 0.1
Table 1: Baseline functional parameters in the Langendorff-isolated heart groups for IR
injury experiments. All cardiac function parameters represent values obtained at the end of the
20-minute stabilization period. 6-8 mice were used in each treatment group. Data are expressed
as mean values ± SEM. 1W-first window/early, 2W-second window/delayed, rIPC-remote
ischemic preconditioning, SP-sodium pentobarbital, N2O-nitrous oxide. LVP-left-ventricle
pressure, LVEDP-left ventricular end-diastolic pressure, LVDP-left ventricular developed
pressure, dP/dtmax – maximum rate of contraction, dP/dtmin – maximum rate of relaxation

50
4.1.2 Left Ventricular Developed Pressure
Left ventricular developed pressure (LVDP) provides an indication of contractile capacity
and can be used to assess the functional recovery of the ventricle after IR injury. LVDP is
calculated from the pressure difference (in mmHg) between peak left ventricular systolic and
diastolic pressures. As shown in table 1, there were no significant differences in LVDP among
the treatment groups. LVDP was approximately 75-85mmHg at the end of stabilization and
decreased to 0mmHg during ischemia in all groups (figure 9a,b). All values obtained during
ischemia and subsequent reperfusion are expressed as a percentage of the pre-ischemic baseline
value obtained at the end of stabilization.
As we have shown in previous studies, early preconditioning with transient hindlimb
ischemia enhances the recovery of LVDP after IR injury. After 30 minutes of ischemia and 60
minutes of reperfusion, the 1WSP+rIPC group showed an 81.9 ± 4.7% recovery of LVDP
compared to only 57.3 ± 4.9% in 1WSP controls (P<0.05) (figure 9a,b).
A primary objective of this study was to examine the delayed effects of SP anesthesia and
rIPC on LV function. We did this by sedating mice with SP and applying rIPC (in the same
fashion as 1WSP+rIPC) twenty-four hours prior to IR injury. We found that both delayed SP and
SP+rIPC significantly improved (P<0.05) LVDP after IR injury compared to 1WSP control mice
(2WSP: 82.6 ±2.4%, 2WSP+rIPC 77.9 ± 5.3%). There were no significant differences in LVDP
between the 2WSP and 2WSP+rIPC groups (figure 9a,c).
To investigate whether there were any effects from the method of delivery of SP intra-
peritoneal anesthesia, we also assessed whether an IP injection could affect cardiac function in
the 2W of protection. Interestingly, we found that an IP injection of saline fluid, twenty-four
hours prior to IR injury, also led to a significant improvement in LVDP compared to 1WSP
controls after 60 minutes of reperfusion (2W Saline, 76.8 ± 3.8%) (figure 9a,c). Given that the

51
2WSP and 2W Saline groups showed similar recovery in developed pressure suggests that the
method of drug delivery could be inducing delayed cardioprotection.
Another objective of this study was to study the potential delayed preconditioning effects
of halothane and nitrous oxide inhalational anesthesia. Similar to the intra-peritoneal groups, we
found that prior administration of both gases for 40 minutes a day before IR injury significantly
improved (P<0.05) LVDP recovery compared to 1WSP controls (2W Halothane: 78.8 ± 2.0%,
2W N2O: 73.3 ± 3.6%) (figure 9b,c).
There were no significant differences in LVDP between the delayed intra-peritoneal and
inhalational anesthetic groups and the early rIPC group after 60 minutes or reperfusion (figure
9c). These findings indicate that a variety of stimuli can improve post-ischemic left ventricular
function by recovering developed pressure to near pre-ischemic function.
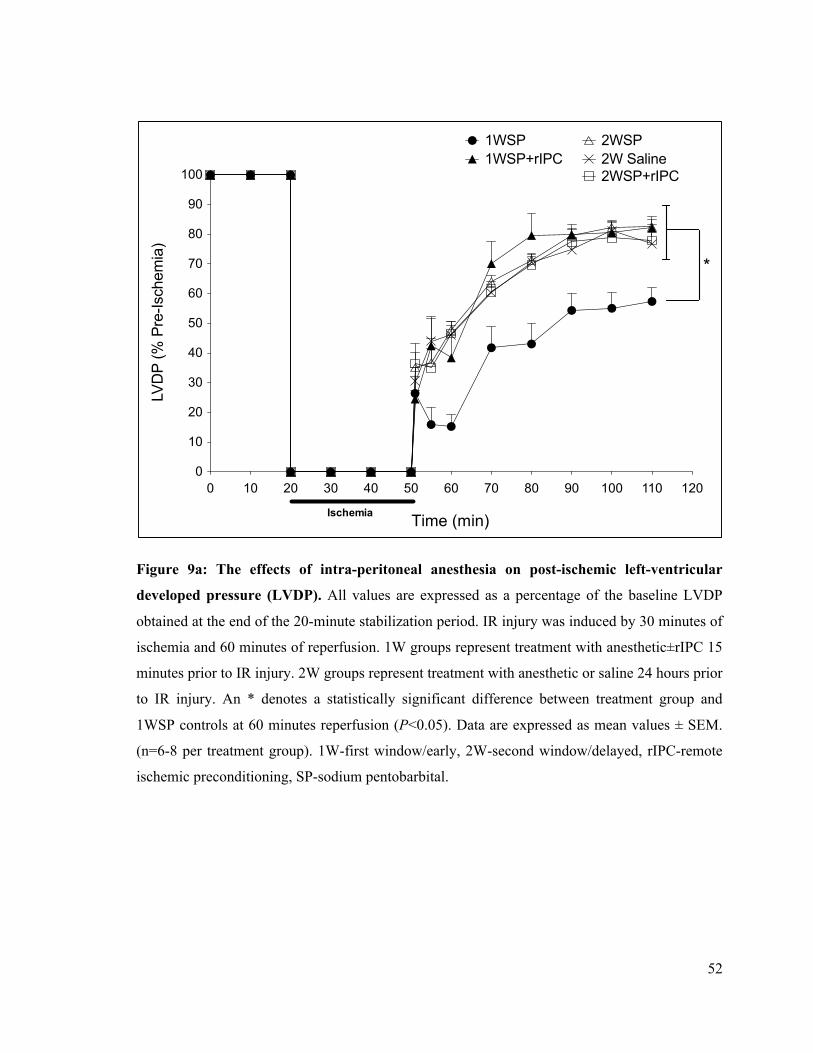
52
Figure 9a: The effects of intra-peritoneal anesthesia on post-ischemic left-ventricular
developed pressure (LVDP). All values are expressed as a percentage of the baseline LVDP
obtained at the end of the 20-minute stabilization period. IR injury was induced by 30 minutes of
ischemia and 60 minutes of reperfusion. 1W groups represent treatment with anesthetic±rIPC 15
minutes prior to IR injury. 2W groups represent treatment with anesthetic or saline 24 hours prior
to IR injury. An * denotes a statistically significant difference between treatment group and
1WSP controls at 60 minutes reperfusion (P<0.05). Data are expressed as mean values ± SEM.
(n=6-8 per treatment group). 1W-first window/early, 2W-second window/delayed, rIPC-remote
ischemic preconditioning, SP-sodium pentobarbital.
0 10 20 30 40 50 60 70 80 90 100 110 1200
10
20
30
40
50
60
70
80
90
100
1WSP1WSP+rIPC
2WSP
2WSP+rIPC2W Saline
Ischemia Time (sec)
LVD
P (%
Pre
-Isch
emia
)
*
Time (min)
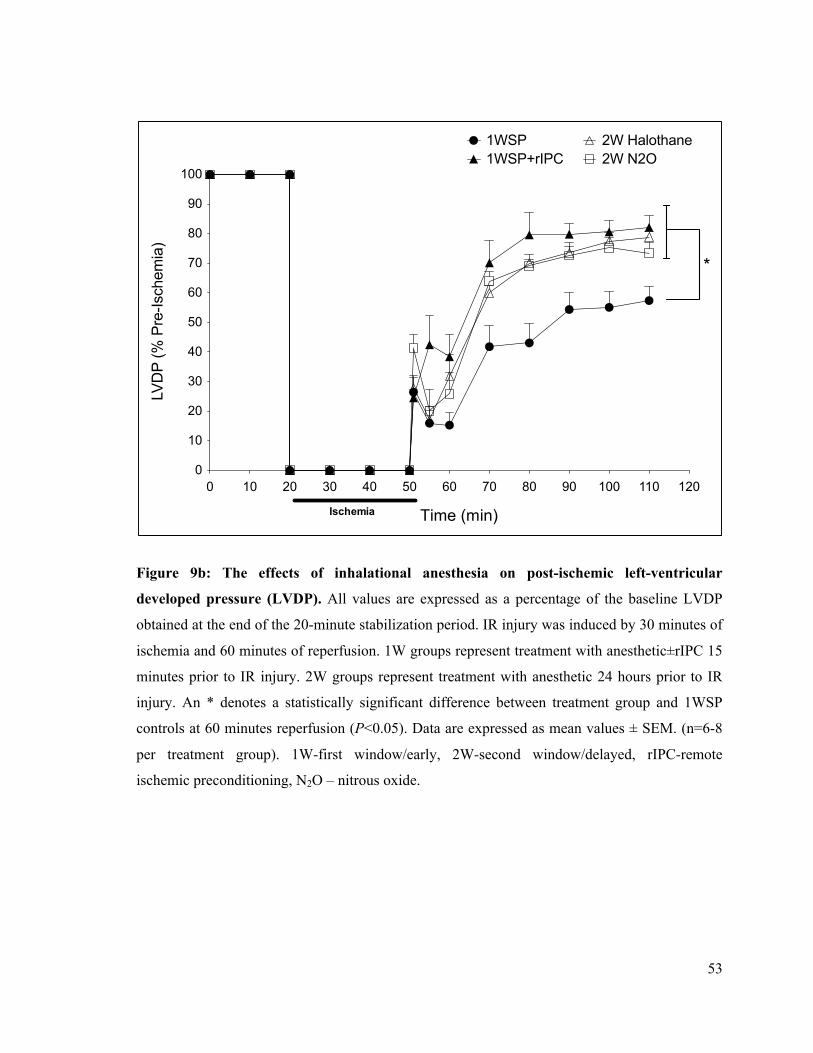
53
Figure 9b: The effects of inhalational anesthesia on post-ischemic left-ventricular
developed pressure (LVDP). All values are expressed as a percentage of the baseline LVDP
obtained at the end of the 20-minute stabilization period. IR injury was induced by 30 minutes of
ischemia and 60 minutes of reperfusion. 1W groups represent treatment with anesthetic±rIPC 15
minutes prior to IR injury. 2W groups represent treatment with anesthetic 24 hours prior to IR
injury. An * denotes a statistically significant difference between treatment group and 1WSP
controls at 60 minutes reperfusion (P<0.05). Data are expressed as mean values ± SEM. (n=6-8
per treatment group). 1W-first window/early, 2W-second window/delayed, rIPC-remote
ischemic preconditioning, N2O – nitrous oxide.
0 10 20 30 40 50 60 70 80 90 100 110 1200
10
20
30
40
50
60
70
80
90
100
1WSP1WSP+rIPC
2W Halothane2W N2O
Ischemia Time (sec)
LVD
P (%
Pre
-Isch
emia
)
*
Time (min)
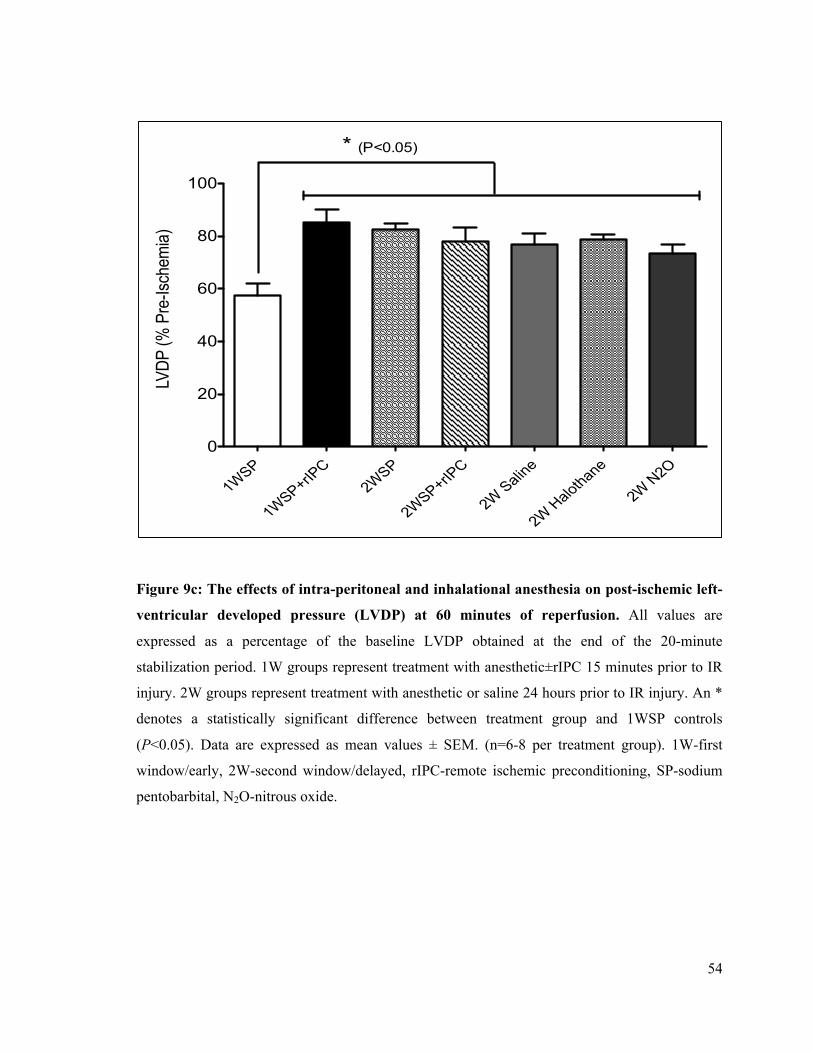
54
Figure 9c: The effects of intra-peritoneal and inhalational anesthesia on post-ischemic left-
ventricular developed pressure (LVDP) at 60 minutes of reperfusion. All values are
expressed as a percentage of the baseline LVDP obtained at the end of the 20-minute
stabilization period. 1W groups represent treatment with anesthetic±rIPC 15 minutes prior to IR
injury. 2W groups represent treatment with anesthetic or saline 24 hours prior to IR injury. An *
denotes a statistically significant difference between treatment group and 1WSP controls
(P<0.05). Data are expressed as mean values ± SEM. (n=6-8 per treatment group). 1W-first
window/early, 2W-second window/delayed, rIPC-remote ischemic preconditioning, SP-sodium
pentobarbital, N2O-nitrous oxide.
1WSP
1WSP+rIPC
2WSP
2WSP+rIPC
2W Saline
2W Halothane
2W N2O0
20
40
60
80
100
* (P<0.05)LV
DP (%
Pre
-Isch
emia)

55
4.1.3 Left Ventricular End-Diastolic Pressure
At the beginning of each experiment, the intra-ventricular balloon volume was adjusted to
set isolated-hearts at a LV preload equivalent to a diastolic pressure of 5-10mmHg (table 1).
Balloon volume was maintained throughout the experiment and thus further alterations in
LVEDP signify the effects of IR injury on the LV. IR injury reduces ventricular compliance and
the increased stiffness is manifest by higher end-diastolic pressures. The progressive increase in
LVEDP after IR injury likely occurs because of changes in cardiomyocyte Ca2+-handling and the
depletion of high-energy phosphates[181], [182].
Consistent with the reported benefits of rIPC on post-ischemic cardiac function, transient
limb ischemia induced early cardioprotection by attenuating the elevation of LVEDP after IR
injury (figure 10a,b). Within only 10 minutes of reperfusion, there was a significant difference
(P<0.001) in LVEDP between 1WSP (67.1 ± 5.4 mmHg) and 1WSP+rIPC (38.5 ± 5.1 mmHg)
groups (figure 10a). Increasing reperfusion time brings a steady decrease in LVEDP as hearts
begin to recover contractile capacity, although end-diastolic pressures remain above pre-ischemic
values. In our early model of rIPC, the 1WSP+rIPC group continued to show a significantly
greater (P<0.001) decline in LVEDP until the end of the 60 minutes reperfusion compared to
1WSP controls (1WSP: 43.1 ± 4.1 mmHg, 1WSP+rIPC: 21.8 ± 2.7 mmHg)(figure 10c).
We also compared the delayed effects of SP (2WSP) on LVEDP with our early
preconditioning groups (1WSP, 1WSP+rIPC). We found that the mice pretreated with SP
anesthesia 24 hours before the experiment also displayed a significantly lower (P<0.001)
LVEDP after 60 min of reperfusion (2WSP: 17.6 ± 1.7 mmHg) compared to 1WSP controls
(figure 10a,c). The 2WSP+rIPC group also displayed a significantly lower increase in LVEDP
than 1WSP controls after reperfusion (2WSP+rIPC: 17.3 ± 3.5 mmHg). There were no
differences in end-diastolic pressure between the 2WSP and 2WSP+rIPC groups, suggesting that

56
rIPC provided no additional benefits in the delayed phase. Interestingly, we also observed that an
IP injection of saline significantly attenuated the rise of LVEDP after 60 minutes of reperfusion
compared to 1WSP controls (2W Saline: 28.0 ± 6.6 mmHg) (figure 10a,c).
Inhalational anesthesia produced a similar effect on the post-ischemic increase of
LVEDP. We found that pre-treatment with halothane (2W Halothane) and nitrous-oxide (2W
N2O) anesthesia significantly reduced (P<0.05) LVEDP at the end of reperfusion compared to
1WSP controls (1WSP: 43.1 ± 4.1 mmHg, 2W Halothane: 19.4 ± 1.7 mmH, 2W N2O: 26.7 ± 2.2
mmHg) (figure 10b,c).
There were no significant differences in LVEDP between the delayed intra-peritoneal and
inhalational anesthetic groups and the early rIPC group after 60 minutes of reperfusion (figure
9c). These findings indicate that, similar to the improvements in the LVDP, both early and
delayed forms of preconditioning can attenuate post-ischemic diastolic dysfunction.
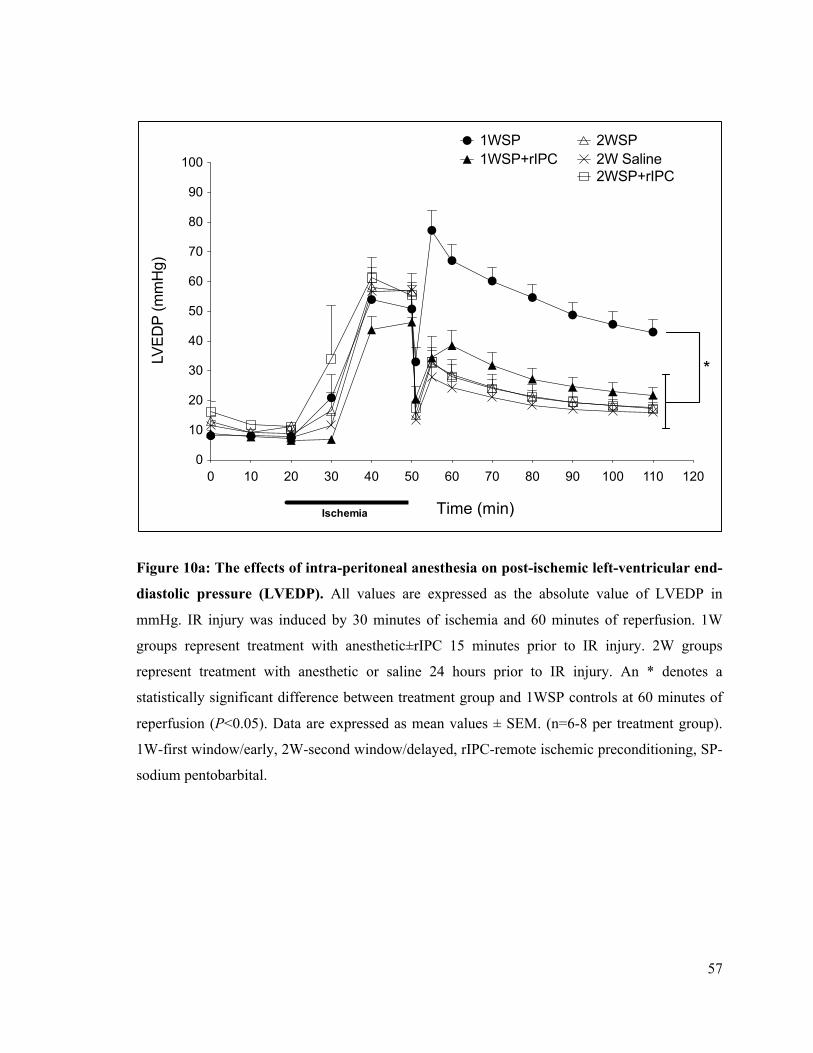
57
Figure 10a: The effects of intra-peritoneal anesthesia on post-ischemic left-ventricular end-
diastolic pressure (LVEDP). All values are expressed as the absolute value of LVEDP in
mmHg. IR injury was induced by 30 minutes of ischemia and 60 minutes of reperfusion. 1W
groups represent treatment with anesthetic±rIPC 15 minutes prior to IR injury. 2W groups
represent treatment with anesthetic or saline 24 hours prior to IR injury. An * denotes a
statistically significant difference between treatment group and 1WSP controls at 60 minutes of
reperfusion (P<0.05). Data are expressed as mean values ± SEM. (n=6-8 per treatment group).
1W-first window/early, 2W-second window/delayed, rIPC-remote ischemic preconditioning, SP-
sodium pentobarbital.
0 10 20 30 40 50 60 70 80 90 100 110 1200
10
20
30
40
50
60
70
80
90
1001WSP1WSP+rIPC
2WSP
2WSP+rIPC2W Saline
Ischemia Time (sec)
LVED
P (m
mH
g)
*
Time (min)
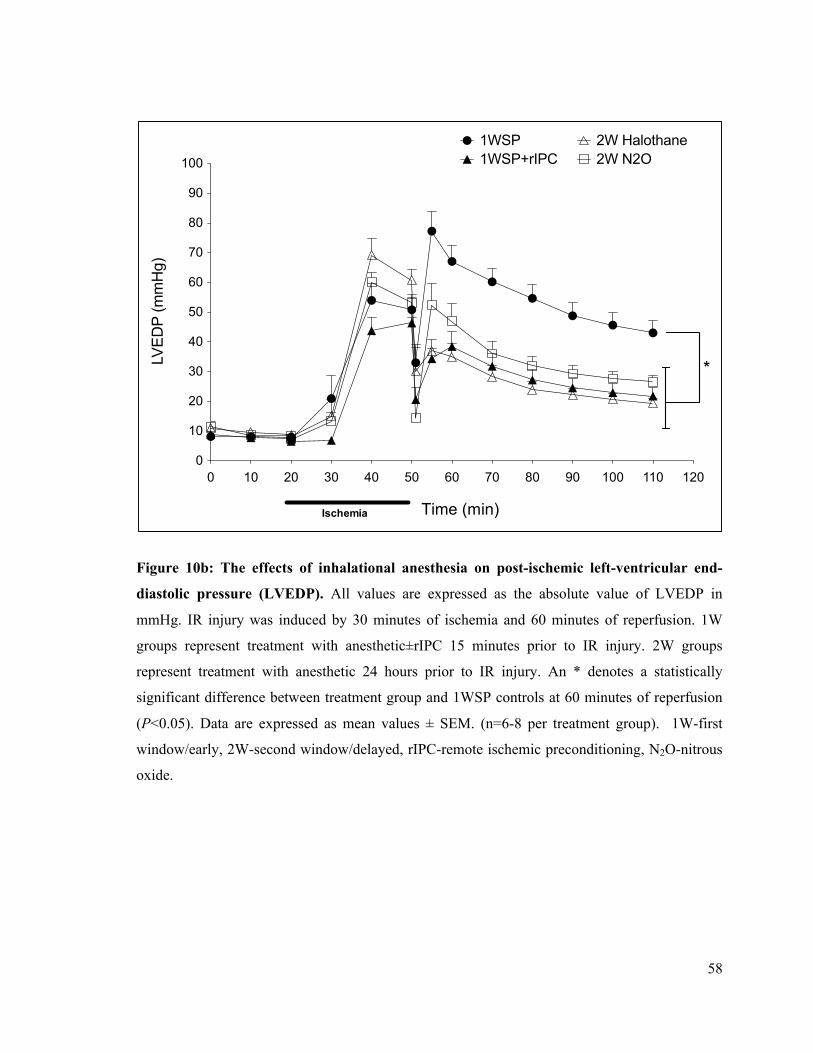
58
Figure 10b: The effects of inhalational anesthesia on post-ischemic left-ventricular end-
diastolic pressure (LVEDP). All values are expressed as the absolute value of LVEDP in
mmHg. IR injury was induced by 30 minutes of ischemia and 60 minutes of reperfusion. 1W
groups represent treatment with anesthetic±rIPC 15 minutes prior to IR injury. 2W groups
represent treatment with anesthetic 24 hours prior to IR injury. An * denotes a statistically
significant difference between treatment group and 1WSP controls at 60 minutes of reperfusion
(P<0.05). Data are expressed as mean values ± SEM. (n=6-8 per treatment group). 1W-first
window/early, 2W-second window/delayed, rIPC-remote ischemic preconditioning, N2O-nitrous
oxide.
0 10 20 30 40 50 60 70 80 90 100 110 1200
10
20
30
40
50
60
70
80
90
100
1WSP1WSP+rIPC
2W Halothane2W N2O
Ischemia Time (sec)
LVED
P (m
mH
g)
*
Time (min)
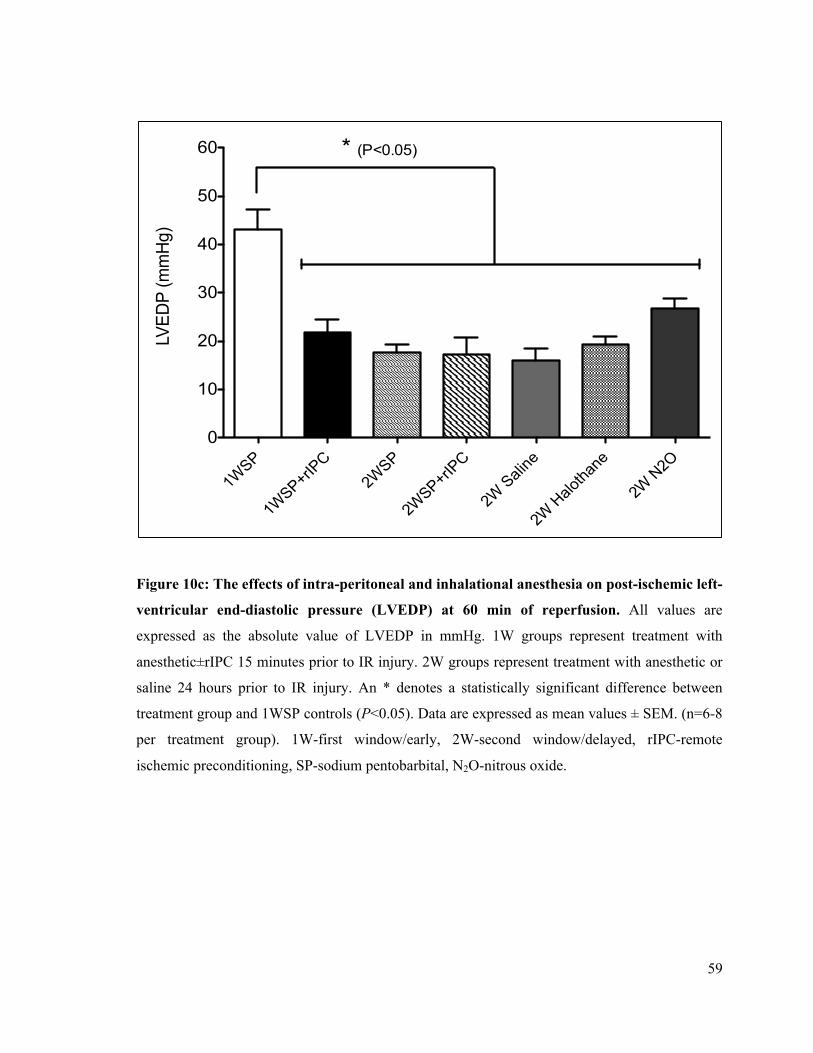
59
Figure 10c: The effects of intra-peritoneal and inhalational anesthesia on post-ischemic left-
ventricular end-diastolic pressure (LVEDP) at 60 min of reperfusion. All values are
expressed as the absolute value of LVEDP in mmHg. 1W groups represent treatment with
anesthetic±rIPC 15 minutes prior to IR injury. 2W groups represent treatment with anesthetic or
saline 24 hours prior to IR injury. An * denotes a statistically significant difference between
treatment group and 1WSP controls (P<0.05). Data are expressed as mean values ± SEM. (n=6-8
per treatment group). 1W-first window/early, 2W-second window/delayed, rIPC-remote
ischemic preconditioning, SP-sodium pentobarbital, N2O-nitrous oxide.
1WSP
1WSP+rIP
C2W
SP
2WSP+rIP
C
2W Sali
ne
2W H
alotha
ne
2W N
2O0
10
20
30
40
50
60LV
EDP
(mm
Hg)
* (P<0.05)

60
4.1.4 Maximum Rate of Contraction
The maximum rate of contraction (dP/dtmax) was measured by the changes in left
ventricular pressure over time (expressed as a positive rate of gain in pressure measured in
mmHg/sec). Post-ischemic values of dP/dtmax can be used to evaluate LV systolic function.
dP/dtmax is known to be affected by heart rate and is dependent on LV preload, as it describes the
rate at which the LV develops pressure before opening of the aortic valve [183]. As with the
other measures of cardiac function, there were no significant differences in dP/dtmax between the
treatment groups at the end of the stabilization period (table 1). Post-ischemic measurements of
dP/dtmax were normalized and expressed as a percentage of their pre-ischemic values.
Similar to LVDP, non-preconditioned, 1WSP treated mice, recovered only 50-60% of
their pre-ischemic rate of LV contraction after IR injury. The early preconditioning effects of
rIPC significantly augmented the recovery in dP/dtmax after 60 minutes of reperfusion when
compared to 1WSP controls (1WSP: 54.3 ± 4.5% of pre-ischemic value, 1WSP+rIPC: 83.4 ±
4.6% - P<0.05) (figure 11a-c). Intra-peritoneal SP anesthesia and saline injection also
significantly improved dP/dtmax in the delayed phase of protection compared to 1WSP controls
(2WSP: 85.6 ± 2.7%, 2W Saline: 81.4 ± 4.5%) (figure 11a,c). There were no additional benefits
from rIPC in the 2WSP+rIPC compared to SP alone (2WSP), although recovery in this group
was still significantly better compared to 1WSP treated mice.
Twenty-four hours pretreatment with halothane anesthesia also had a delayed effect on
dP/dtmax as hearts in this group had improved recovery compared to 1WSP controls (2W
halothane: 82.9 ± 6.2% - P<0.05). Interestingly, while the 2W N2O group also exhibited a
significant recovery in dP/dtmax (2W N2O: 67.4 ± 2.1%) compared to 1WSP mice, the magnitude
of recovery at the end of reperfusion was intermediate between 1WSP and all the other treatment
groups (figure 11b,c).

61
There were no significant differences in dP/dtmax between the delayed intra-peritoneal and
inhalational anesthetic groups and the early rIPC group after 60 minutes of reperfusion (figure
11c). These findings support the observed effects of the various early and delayed stimuli on
other parameters of cardiac function (LVDP, LVEDP) after IR injury, indicating a
cardioprotective benefit on not only the extent, but also the rate of systolic pressure development.
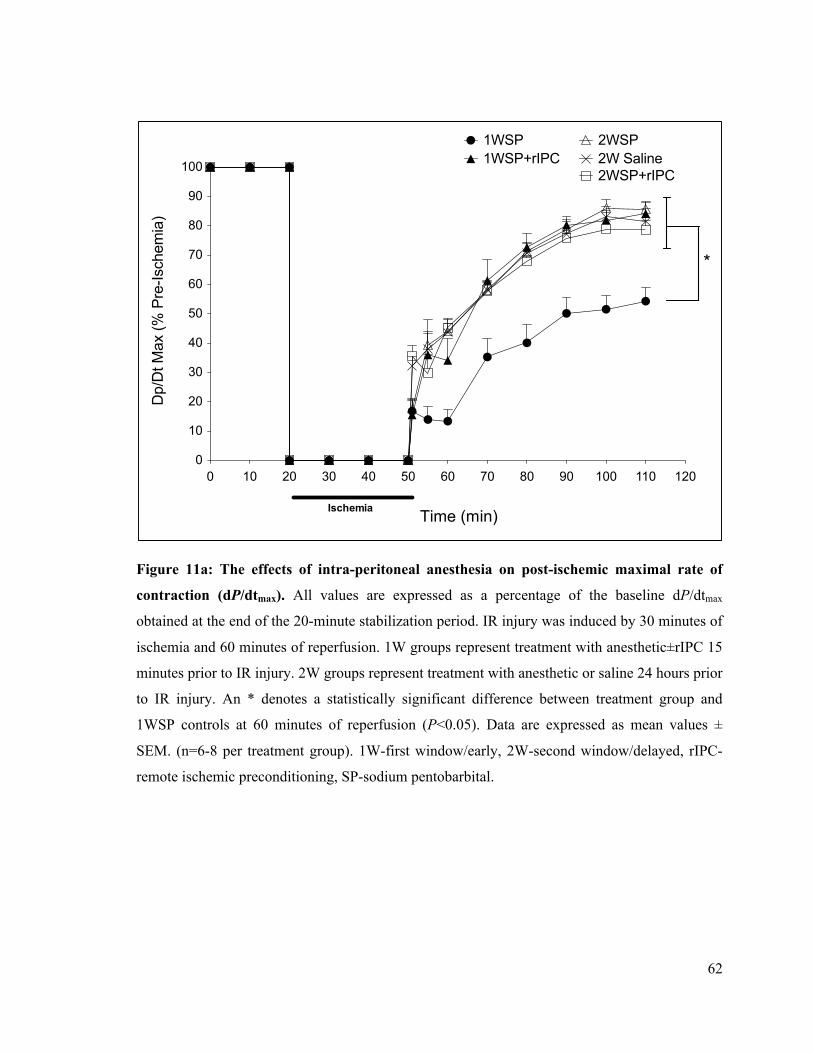
62
Figure 11a: The effects of intra-peritoneal anesthesia on post-ischemic maximal rate of
contraction (dP/dtmax). All values are expressed as a percentage of the baseline dP/dtmax
obtained at the end of the 20-minute stabilization period. IR injury was induced by 30 minutes of
ischemia and 60 minutes of reperfusion. 1W groups represent treatment with anesthetic±rIPC 15
minutes prior to IR injury. 2W groups represent treatment with anesthetic or saline 24 hours prior
to IR injury. An * denotes a statistically significant difference between treatment group and
1WSP controls at 60 minutes of reperfusion (P<0.05). Data are expressed as mean values ±
SEM. (n=6-8 per treatment group). 1W-first window/early, 2W-second window/delayed, rIPC-
remote ischemic preconditioning, SP-sodium pentobarbital.
0 10 20 30 40 50 60 70 80 90 100 110 1200
10
20
30
40
50
60
70
80
90
100
1WSP1WSP+rIPC
2WSP
2WSP+rIPC2W Saline
Ischemia Time (sec)
Dp/
Dt M
ax (%
Pre
-Isch
emia
)
*
Time (min)
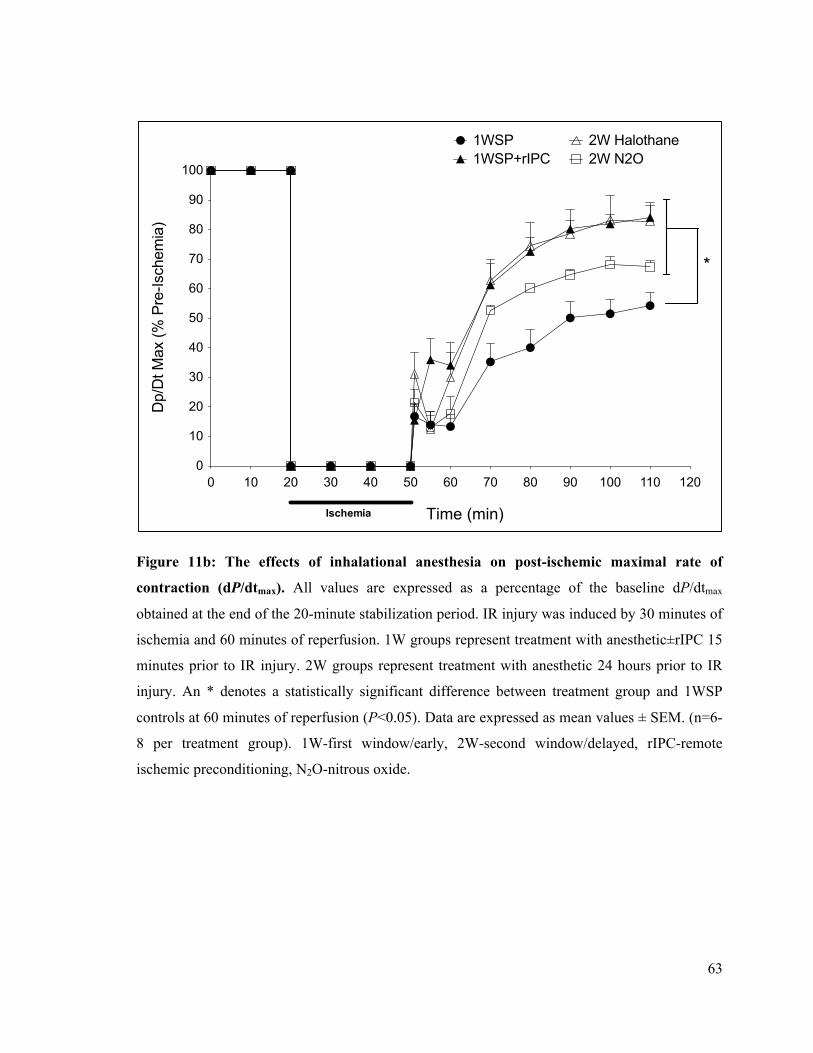
63
Figure 11b: The effects of inhalational anesthesia on post-ischemic maximal rate of
contraction (dP/dtmax). All values are expressed as a percentage of the baseline dP/dtmax
obtained at the end of the 20-minute stabilization period. IR injury was induced by 30 minutes of
ischemia and 60 minutes of reperfusion. 1W groups represent treatment with anesthetic±rIPC 15
minutes prior to IR injury. 2W groups represent treatment with anesthetic 24 hours prior to IR
injury. An * denotes a statistically significant difference between treatment group and 1WSP
controls at 60 minutes of reperfusion (P<0.05). Data are expressed as mean values ± SEM. (n=6-
8 per treatment group). 1W-first window/early, 2W-second window/delayed, rIPC-remote
ischemic preconditioning, N2O-nitrous oxide.
0 10 20 30 40 50 60 70 80 90 100 110 1200
10
20
30
40
50
60
70
80
90
100
1WSP1WSP+rIPC
2W Halothane2W N2O
Ischemia Time (sec)
Dp/
Dt M
ax (%
Pre
-Isch
emia
)
*
Time (min)
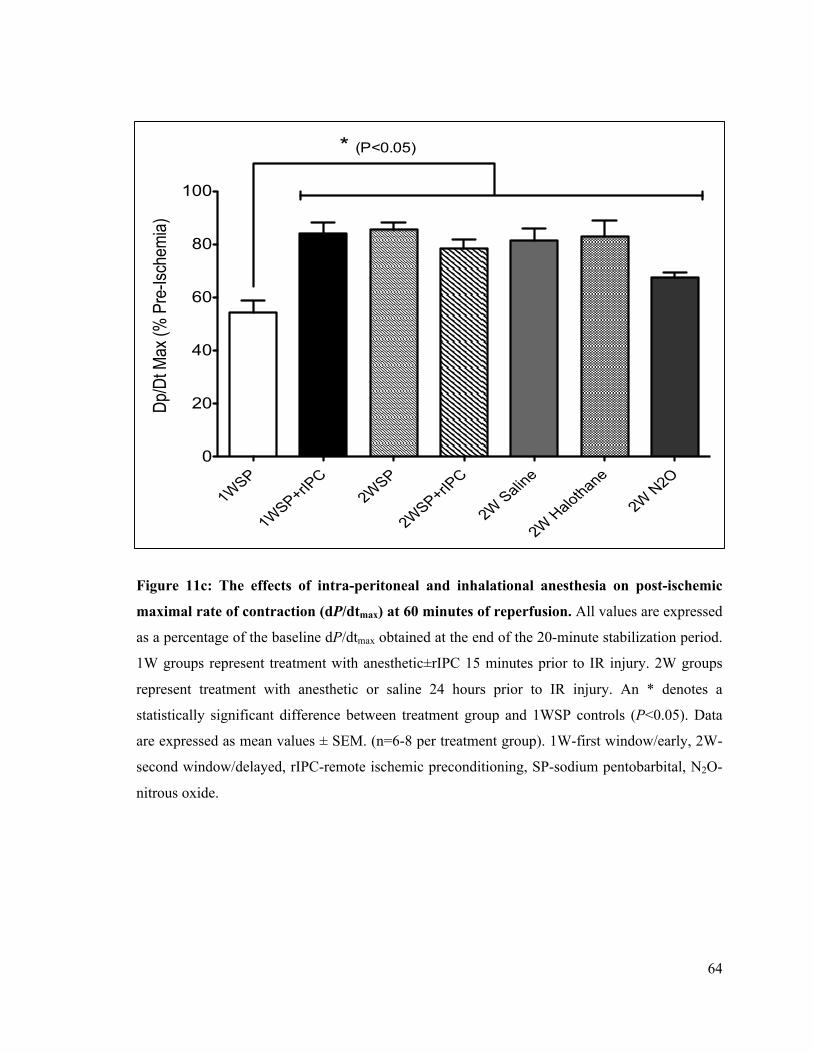
64
Figure 11c: The effects of intra-peritoneal and inhalational anesthesia on post-ischemic
maximal rate of contraction (dP/dtmax) at 60 minutes of reperfusion. All values are expressed
as a percentage of the baseline dP/dtmax obtained at the end of the 20-minute stabilization period.
1W groups represent treatment with anesthetic±rIPC 15 minutes prior to IR injury. 2W groups
represent treatment with anesthetic or saline 24 hours prior to IR injury. An * denotes a
statistically significant difference between treatment group and 1WSP controls (P<0.05). Data
are expressed as mean values ± SEM. (n=6-8 per treatment group). 1W-first window/early, 2W-
second window/delayed, rIPC-remote ischemic preconditioning, SP-sodium pentobarbital, N2O-
nitrous oxide.
1WSP
1WSP+rIPC
2WSP
2WSP+rIPC
2W Saline
2W Halothane
2W N2O0
20
40
60
80
100
Dp/D
t Max
(% P
re-Is
chem
ia)
* (P<0.05)

65
4.1.5 Maximum Rate of Relaxation
Similar to the rate of contraction, the maximal rate of relaxation (dP/dtmin) provides a
measure of cardiac diastolic function by assessing the maximal rate at which the ventricle relaxes
and decreases pressure during diastole (expressed as a maximal negative rate of change in
pressure measured in mmHg/sec). There were no significant differences in dP/dtmin in hearts
among the different treatment groups at the end of the stabilization period (table 1). Post-
ischemic measurements of dP/dtmin were normalized and expressed as a percentage of their pre-
ischemic values.
After 60 minutes of reperfusion, the rate of relaxation recovered to similar levels in the
1WSP+rIPC (76.7 ± 4.7% of pre ischemic value) and 2W intra-peritoneal anesthesia groups
(2WSP: 81.5 ± 3.0%, 2WSP+rIPC: 78.7 ± 4.7%, 2W Saline 75.9 ± 5.8% - with all groups
displaying significantly higher (P<0.05) levels of recovery compared to 1WSP hearts (53.0 ±
4.7%)(figure 12a,c). Additionally, halothane inhalational anesthesia also significantly improved
dP/dtmin compared to 1WSP controls (2W Halothane 84.1 ± 9.9%). Similar to the recovery of the
rate of contraction, 2W N2O mice exhibited an intermediate level of recovery in dP/dtmin (2W
N2O: 61.0 ± 2.4%) after 60 minutes of reperfusion, however it was not significantly different
from 1WSP controls (figure 12b,c).
There were no significant differences in dP/dtmin between the delayed intra-peritoneal and
halothane anesthesia groups and the early rIPC group after 60 minutes of reperfusion (figure
12c). These findings add to the accumulating evidence from the other measures of cardiac
function assessed in this study that early rIPC and delayed intra-peritoneal and inhalational
anesthesia can similarly improve the contractile capacity of hearts after IR injury.
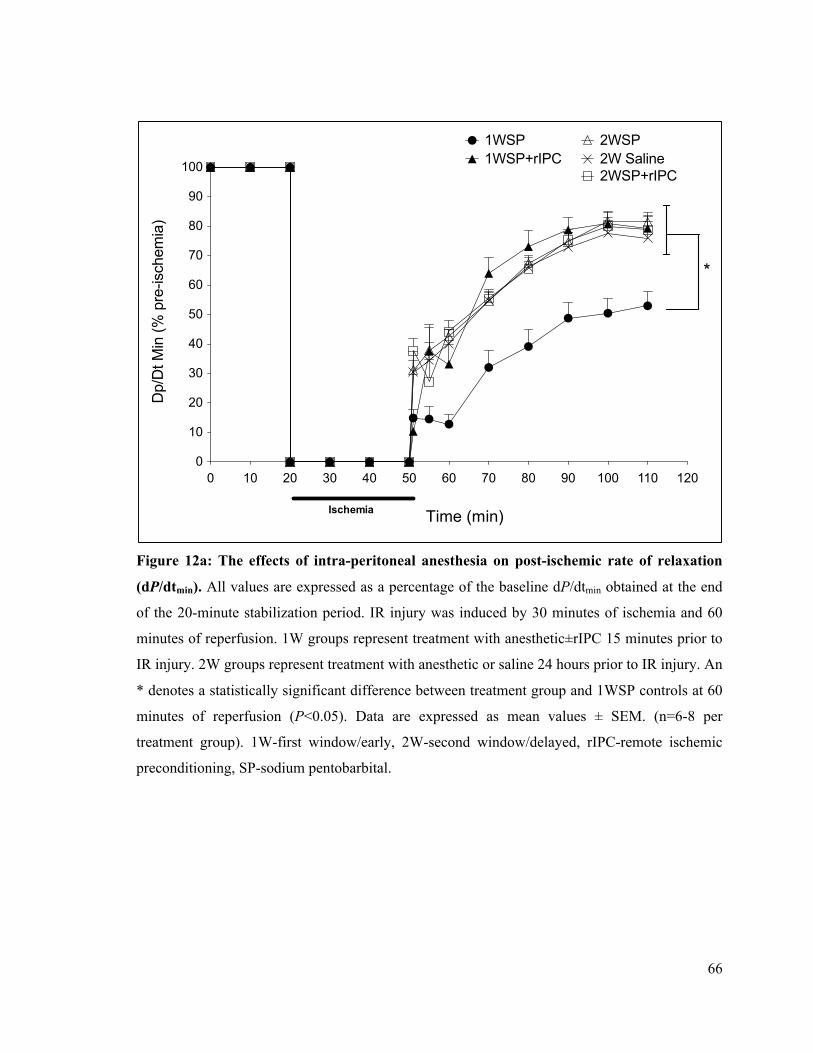
66
Figure 12a: The effects of intra-peritoneal anesthesia on post-ischemic rate of relaxation
(dP/dtmin). All values are expressed as a percentage of the baseline dP/dtmin obtained at the end
of the 20-minute stabilization period. IR injury was induced by 30 minutes of ischemia and 60
minutes of reperfusion. 1W groups represent treatment with anesthetic±rIPC 15 minutes prior to
IR injury. 2W groups represent treatment with anesthetic or saline 24 hours prior to IR injury. An
* denotes a statistically significant difference between treatment group and 1WSP controls at 60
minutes of reperfusion (P<0.05). Data are expressed as mean values ± SEM. (n=6-8 per
treatment group). 1W-first window/early, 2W-second window/delayed, rIPC-remote ischemic
preconditioning, SP-sodium pentobarbital.
0 10 20 30 40 50 60 70 80 90 100 110 1200
10
20
30
40
50
60
70
80
90
100
1WSP1WSP+rIPC
2WSP
2WSP+rIPC2W Saline
Ischemia Time (sec)
Dp/
Dt M
in (%
pre
-isch
emia
)
*
Time (min)
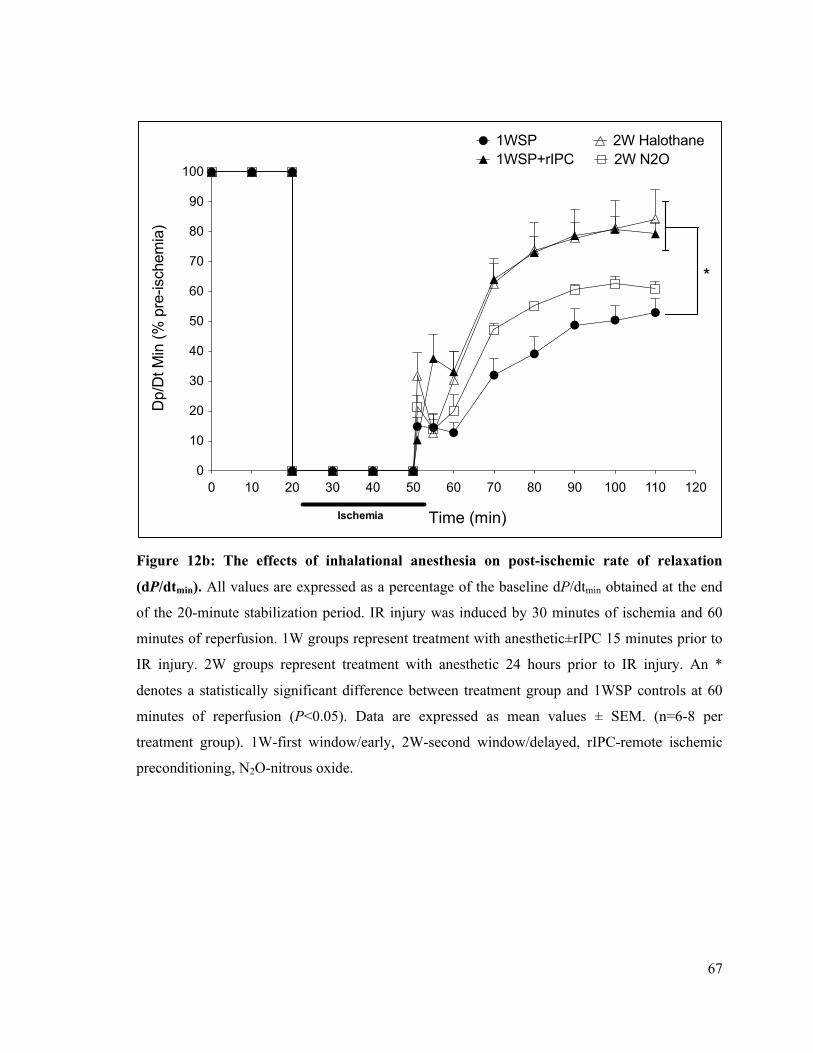
67
Figure 12b: The effects of inhalational anesthesia on post-ischemic rate of relaxation
(dP/dtmin). All values are expressed as a percentage of the baseline dP/dtmin obtained at the end
of the 20-minute stabilization period. IR injury was induced by 30 minutes of ischemia and 60
minutes of reperfusion. 1W groups represent treatment with anesthetic±rIPC 15 minutes prior to
IR injury. 2W groups represent treatment with anesthetic 24 hours prior to IR injury. An *
denotes a statistically significant difference between treatment group and 1WSP controls at 60
minutes of reperfusion (P<0.05). Data are expressed as mean values ± SEM. (n=6-8 per
treatment group). 1W-first window/early, 2W-second window/delayed, rIPC-remote ischemic
preconditioning, N2O-nitrous oxide.
0 10 20 30 40 50 60 70 80 90 100 110 1200
10
20
30
40
50
60
70
80
90
100
1WSP1WSP+rIPC
2W Halothane2W N2O
Ischemia Time (sec)
Dp/
Dt M
in (%
pre
-isch
emia
)
*
Time (min)
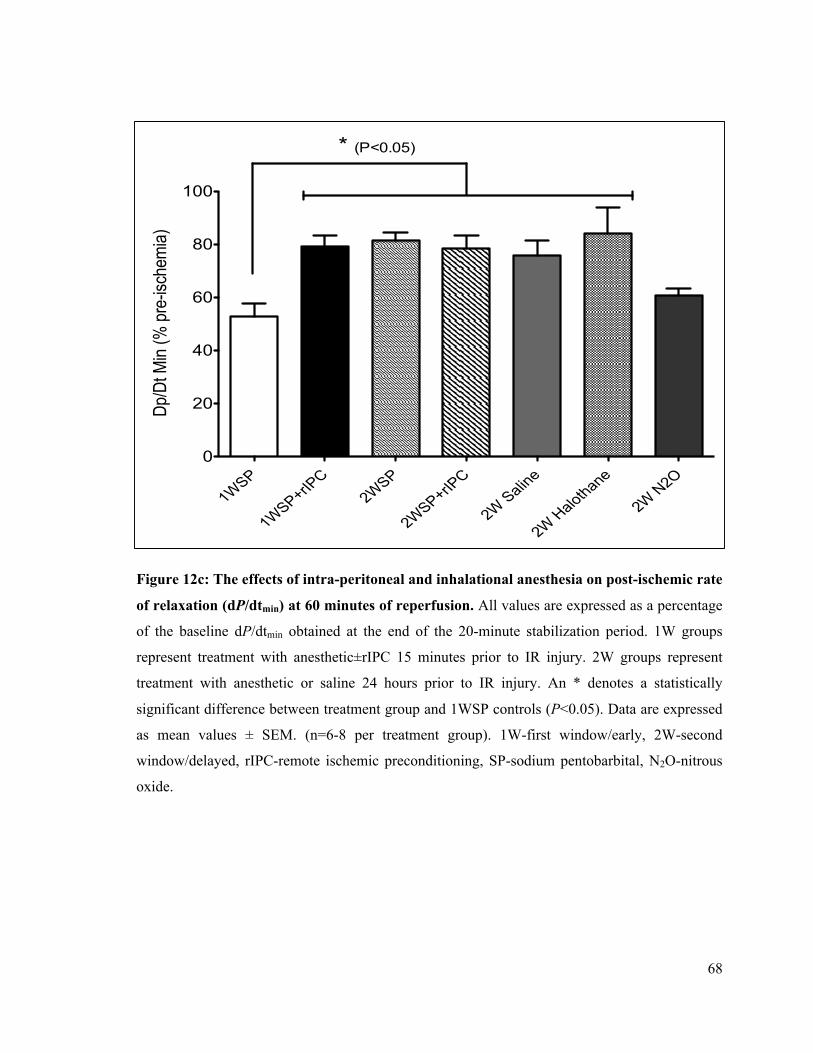
68
Figure 12c: The effects of intra-peritoneal and inhalational anesthesia on post-ischemic rate
of relaxation (dP/dtmin) at 60 minutes of reperfusion. All values are expressed as a percentage
of the baseline dP/dtmin obtained at the end of the 20-minute stabilization period. 1W groups
represent treatment with anesthetic±rIPC 15 minutes prior to IR injury. 2W groups represent
treatment with anesthetic or saline 24 hours prior to IR injury. An * denotes a statistically
significant difference between treatment group and 1WSP controls (P<0.05). Data are expressed
as mean values ± SEM. (n=6-8 per treatment group). 1W-first window/early, 2W-second
window/delayed, rIPC-remote ischemic preconditioning, SP-sodium pentobarbital, N2O-nitrous
oxide.
1WSP
1WSP+rIPC
2WSP
2WSP+rIPC
2W Saline
2W Halothane
2W N2O0
20
40
60
80
100
Dp/D
t Min
(% p
re-is
chem
ia)* (P<0.05)

69
4.2 Delayed Preconditioning with Intra-Peritoneal and Inhalational Anesthesia Reduce Infarct Size after IR Injury
At the end of reperfusion, hearts were cut into six 1mm slices and stained with TTC to
examine LV infarct size. Infarction sizes are expressed as a percentage of the LV area.
Paralleling the cardiac function data on early preconditioning via hindlimb ischemia, we found
that 1WSP+rIPC hearts had significantly reduced (P<0.05) LV infarct sizes compared with the
1WSP control group (1WSP: 43.6 ± 5.9%, 1WSP+rIPC: 19.6 ± 3.5%) (figure 13a) . Furthermore,
in addition to its delayed benefits on post-ischemic heart performance, SP anesthesia is capable
of reducing LV infarct size in the 2W of protection. The 2WSP group had significantly smaller
LV infarct sizes compared to 1WSP controls (2WSP: 28.0 ±1.9%). Hearts from the 2WSP+rIPC
and 2W Saline groups exhibited significantly reduced infarction size compared to 1WSP treated
controls (2WSP+rIPC: 30.4 ± 3.7%, 2W Saline: 30.7 ± 1.8%). With regards to inhalational
anesthesia, we found that halothane also significantly reduced (P<0.05) infarct size in the 2W of
protection compared to 1WSP controls (2W Halothane: 28.8 ± 2.1%) (figure 13a).
Interestingly, we observed that the 2W treatment groups did not reduce infarct size to the
same extent as 1WSP+rIPC, which aligns with consensus that cardioprotection from delayed
preconditioning is not as robust as the early phase[49]. Interestingly, despite the delayed benefits
of 2W N2O treatment on cardiac function, 40 minutes of nitrous oxide exposure did not reduce
infarct size after IR injury (2W N2O: 46.8 ± 2.5%). Therefore, these findings suggest that
treatment with intra-peritoneal injections or halothane anesthesia produces delayed protection
against cardiac dysfunction and cell damage after IR injury. Furthermore, while 2W N2O
preserved cardiac function, it did not protect against infarction. See figure 13 and 14 for infarct
size data and representative stained cross-sections.
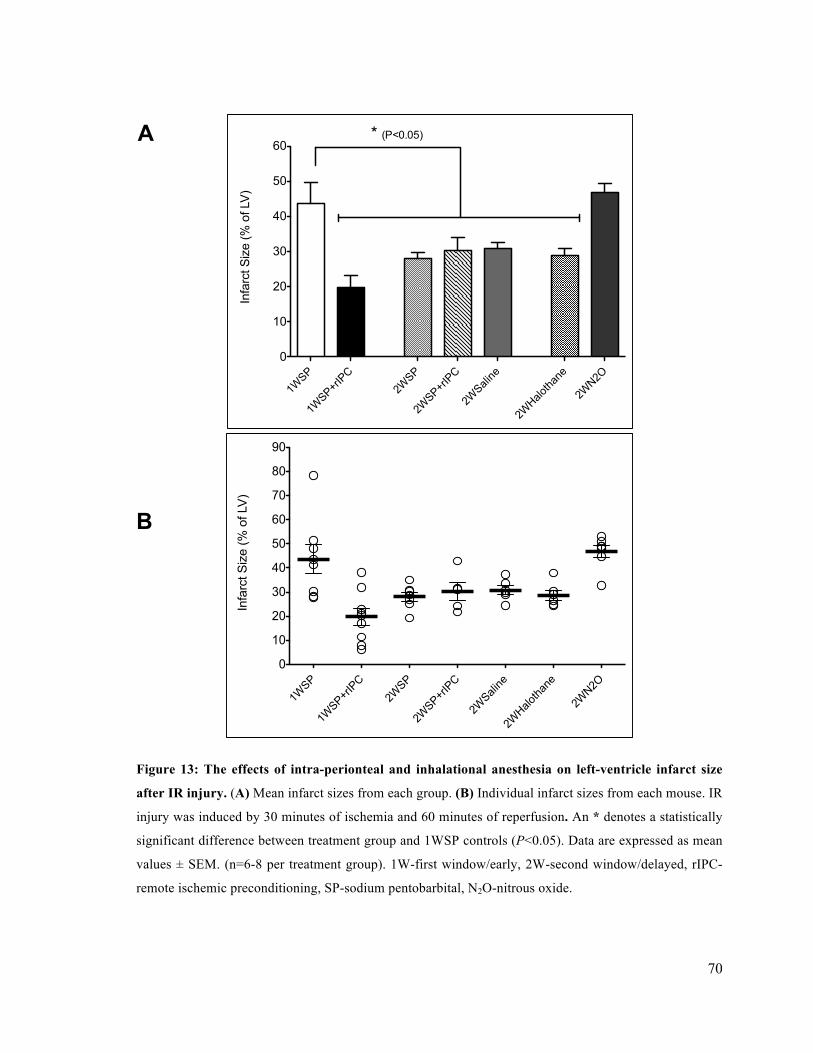
70
A
B
Figure 13: The effects of intra-perionteal and inhalational anesthesia on left-ventricle infarct size
after IR injury. (A) Mean infarct sizes from each group. (B) Individual infarct sizes from each mouse. IR
injury was induced by 30 minutes of ischemia and 60 minutes of reperfusion. An * denotes a statistically
significant difference between treatment group and 1WSP controls (P<0.05). Data are expressed as mean
values ± SEM. (n=6-8 per treatment group). 1W-first window/early, 2W-second window/delayed, rIPC-
remote ischemic preconditioning, SP-sodium pentobarbital, N2O-nitrous oxide.
1WSP
1WSP+rIPC
2WSP
2WSP+rIPC
2WSaline
2WHalothane
2WN2O
0
10
20
30
40
50
60
70
80
90
Infa
rct S
ize
(% o
f LV
)
1WSP
1WSP+rI
PC2W
SP
2WSP+rI
PC
2WSali
ne
2WHalo
thane
2WN2O
0
10
20
30
40
50
60* (P<0.05)
Infa
rct S
ize
(% o
f LV)
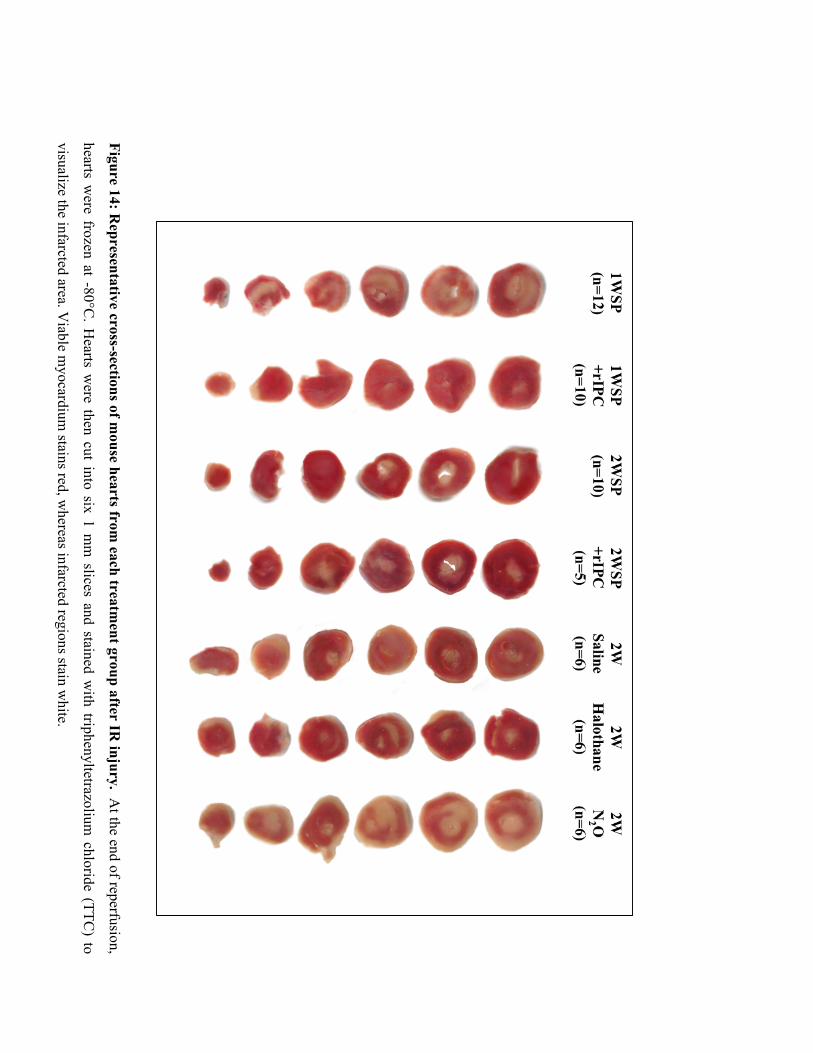
71
Figure 14: Representative cross-sections of m
ouse hearts from each treatm
ent group after IR injury. A
t the end of reperfusion,
hearts were frozen at -80°C
. Hearts w
ere then cut into six 1 mm
slices and stained with triphenyltetrazolium
chloride (TTC) to
visualize the infarcted area. Viable m
yocardium stains red, w
hereas infarcted regions stain white.

72
4.3 Delayed Preconditioning with Injectable and Gas Anesthesia Increase phospho-Akt and phospho-p44/42 MAPK expression
I compared the effects of early and delayed preconditioning using rIPC, intra-peritoneal
and gas anesthesia on phospho-Akt (P-Akt) and phospho-p44/42 MAPK (P-p44/42) signaling
pathways in mouse hearts. Phosphorylation of Akt and p44/42 Erk proteins has been shown to be
involved in the cardioprotective phenotype induced by both IPC and APC [32], [74], [85].
Previous findings in our lab have shown that 1WSP+rIPC induces a significant (P<0.05) increase
in P-Akt (1.6 ± 0.1 fold increase) and P-Erk levels (1.6 ± 0.1 fold increase) compared to 1WSP
mouse hearts (figure 15, 16). In the current study, it was discovered that SP produces a
significant increase (P<0.05) in phospho-Akt expression in the 2W of protection (2WSP group)
compared to the 1WSP controls (3.2 ± 0.4 fold increase) (figure 15). A similar finding was seen
with delayed SP+rIPC treatment, with P-Akt levels significantly greater compared to 1WSP
hearts (3.1 ± 0.4 fold increase). To confirm whether SP was in fact causing delayed
cardioprotection, we tested the effects of an IP injection of 0.12 mL of saline solution. We found
that an IP injection of saline also caused a significant increase (P<0.05) in P-Akt levels
compared to 1WSP controls (2.7 ± 0.4 fold increase). Furthermore, we observed a delayed effect
of halothane and N2O anesthesia on P-Akt levels. Both gases augmented the expression of P-Akt,
however the increases were not significantly different from 1WSP P-Akt levels (2W halothane:
1.4 ± 0.2 fold, 2W N2O: 1.6 ± 0.3 fold increases compared to 1WSP) (figure 15).
We also examined the effects of 2WSP and 2W Saline on P-p44/42 MAPK expression in
hearts. While we observed an increase in P-p44/42 MAPK levels in 2WSP and 2WSP+rIPC
hearts, the expression levels were not significantly different from 1WSP hearts (2WSP: 1.4 ± 0.1
fold increase P=0.0640, 2WSP+rIPC: 1.5 ± 0.2 fold increase P=0.0788) (figure 16).
Additionally, there was no change in P-p44/42 MAPK levels in 2W Saline, or 2W N2O treated

73
hearts (2W Saline: 0.9 ± 0.2 fold, 2W N2O 1.0 ± 0.2 fold compared to 1WSP). However,
halothane anesthesia resulted in a significant increase in P-p44/42 MAPK levels in the 2W of
protection (1.3 ± 0.04 fold increase compared to 1WSP) (figure 16).
Consistent with the data on post-ischemic improvements in cardiac function and the
reductions in infarct size, these findings suggest that early and delayed forms of preconditioning
against IR injury are associated with the activation of pro-survival signaling pathways. The
differential expression of P-Akt and P-p44/42 in the various treatment groups suggests that the
preconditioning stimuli can recruit multiple pathways associated with cardioprotection.
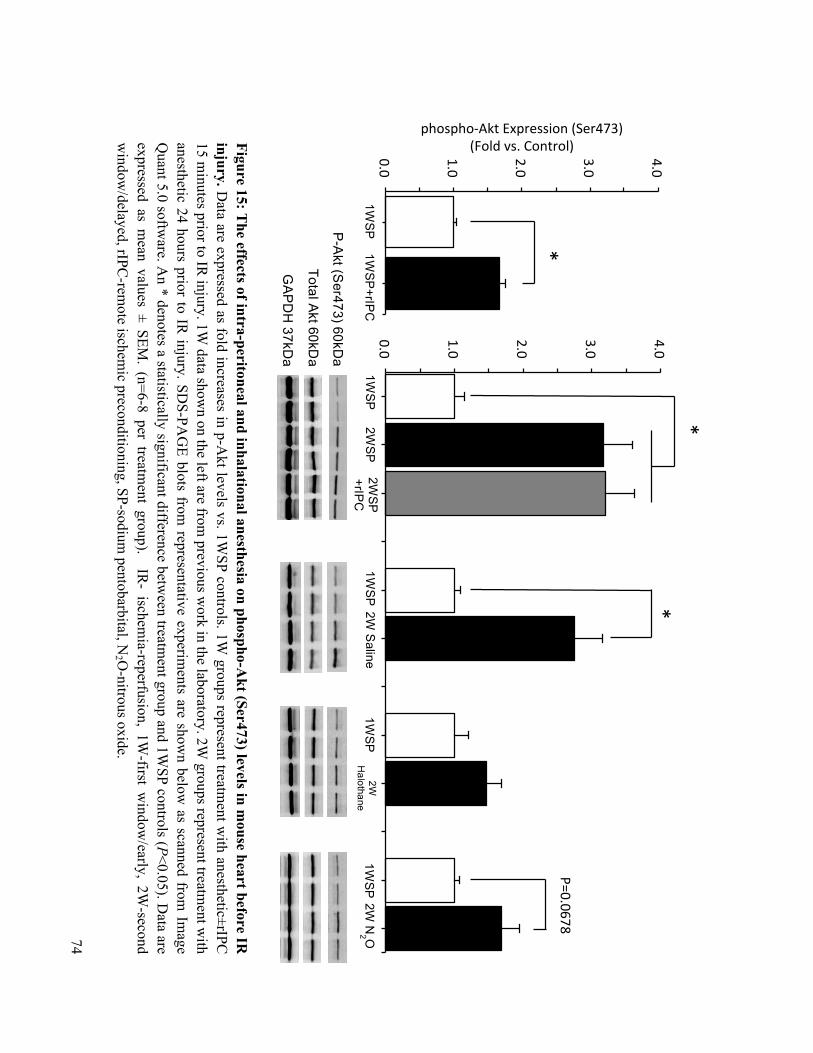
74
Figure 15: The effects of intra-peritoneal and inhalational anesthesia on phospho-A
kt (Ser473) levels in mouse heart before IR
injury. D
ata are expressed as fold increases in p-Akt levels vs. 1W
SP controls. 1W groups represent treatm
ent with anesthetic±rIPC
15 m
inutes prior to IR injury. 1W
data shown on the left are from
previous work in the laboratory. 2W
groups represent treatment w
ith anesthetic 24 hours prior to IR
injury. SDS-PA
GE blots from
representative experiments are show
n below as scanned from
Image
Quant 5.0 softw
are. An * denotes a statistically significant difference betw
een treatment group and 1W
SP controls (P<0.05). Data are
expressed as mean values ± SEM
. (n=6-8 per treatment group). IR
- ischemia-reperfusion, 1W
-first window
/early, 2W-second
window
/delayed, rIPC-rem
ote ischemic preconditioning, SP-sodium
pentobarbital, N2 O
-nitrous oxide.
0.0#
1.0#
2.0#
3.0#
4.0#
P-A
kt (Ser473) 60kD
a
Total Akt 60kD
a
GA
PD
H 37kD
a
*
P=0.0678
1WSP
2W
SP
2WSP
+rIPC
1W
SP 2W
Saline
1WSP
2W
H
alothane 1W
SP 2W
N2 O
0.0#
1.0#
2.0#
3.0#
4.0#
phospho,Akt#Expression#(Ser473)#(Fold#vs.#Control)#
1WSP
1W
SP+rIP
C
*
*
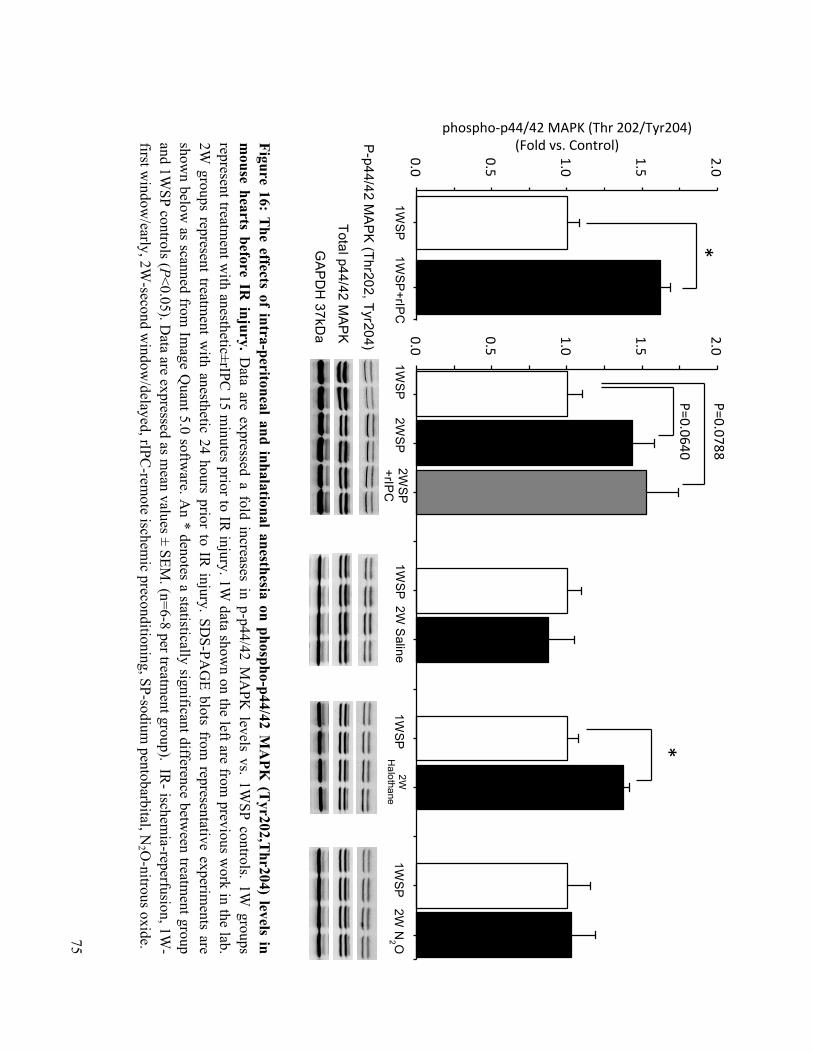
75
Figure 16: The effects of intra-peritoneal and inhalational anesthesia on phospho-p44/42 M
APK
(Tyr202,T
hr204) levels in m
ouse hearts before IR injury. D
ata are expressed a fold increases in p-p44/42 MA
PK levels vs. 1W
SP controls. 1W groups
represent treatment w
ith anesthetic±rIPC 15 m
inutes prior to IR injury. 1W
data shown on the left are from
previous work in the lab.
2W groups represent treatm
ent with anesthetic 24 hours prior to IR
injury. SDS-PA
GE blots from
representative experiments are
shown below
as scanned from Im
age Quant 5.0 softw
are. An * denotes a statistically significant difference betw
een treatment group
and 1WSP controls (P<0.05). D
ata are expressed as mean values ± SEM
. (n=6-8 per treatment group). IR
- ischemia-reperfusion, 1W
-first w
indow/early, 2W
-second window
/delayed, rIPC-rem
ote ischemic preconditioning, SP-sodium
pentobarbital, N2 O
-nitrous oxide.
0.0#
0.5#
1.0#
1.5#
2.0#
0.0#
0.5#
1.0#
1.5#
2.0#
phospho,p44/42#MAPK#(Thr#202/Tyr204)#(Fold#vs.#Control)#
1WSP
2W
SP
2WSP
+rIPC
1W
SP 2W
Saline
1WSP
2W
H
alothane 1W
SP
2W N
2 O
1WSP
1W
SP+rIP
C
P-p44/42 M
AP
K (Thr202, Tyr204)
Total p44/42 MA
PK
GA
PD
H 37kD
a
P=0.0640
P=0.0788 *
*

76
Chapter 5 Discussion
Our understanding of the delayed phase of protection afforded by rIPC (defined as
protection arising 24-hours after initial stimulus) is scant in comparison with other forms of
cardioprotection. This is largely because there is currently no existing in-vivo or in-vitro model
for use in mouse studies. Therefore, the primary objective of the current study was to to identify
and establish a method of sedation for delayed rIPC experiments using the mouse Langendorff-
model of IR injury. Additional objectives that arose from our initial observations included (1)
investigating the delayed effects of SP anesthesia and IP injections on post-ischemic recovery of
mouse hearts and lastly to (2) examine the delayed effects of inhalational halothane and nitrous
oxide anesthesia in order to assess their suitability for use in future delayed rIPC studies.
The findings produced in this study were compared with our currently established mouse
isolated-heart model of early phase rIPC and its associated protection against global IR injury.
The present study offers several important findings that contribute to our understanding of
delayed rIPC and provide a base for future studies aiming to use the Langendorff technique.
The first important finding of this study is the observation that SP anesthesia, when given
via an IP injection, induces delayed preconditioning against global IR injury in mice. This was
shown by a decrease in post-ischemic cardiac dysfunction (i.e. improved LVDP, LVEDP,
dP/dtmax, dP/dtmin) and a reduction in LV infarct size. This unexpected observation arose during
my preliminary experiments, when I observed that there were no differences in cardiac recovery
after between 2WSP and 2WSP+rIPC treated mice, suggesting that SP may give rise to
cardioprotection. SP is the anesthetic of choice used as a first window control treatment for limb
ischemia and is required to apply transient ischemia. However, I have shown that its delayed

77
cardioprotective effect precludes its use as a control treatment and sedative for 2W
preconditioning studies.
A second important observation of this study was that an injection of saline fluid into the
mouse peritoneum also gave rise to delayed preconditioning, as hearts showed enhanced
functional recovery and reduced infarction after IR injury. While another lab group has produced
this finding in another study [179], a novel aspect finding from my experiments was the
observation of an accompanying cell signal signature archetypal of preconditioning. We found
that protection from IP injections was associated with an increase in phospho-Akt levels. This
implies that the irritation or stress involved with an IP injection may be the stimulus that results
in delayed preconditioning, as opposed to a direct effect from SP anesthesia.
The third aspect of this study was that halothane anesthesia induced delayed
preconditioning. I found that twenty-four hour prior treatment of 1-2% halothane resulted in
improved cardiac recovery and reductions in infarct size after global IR injury. Halothane
preconditioning was also associated with an increase in phospho-p44/42 MAPK levels.
Lastly, we discovered an unusual pattern of delayed cardioprotection 24 hours after 40
minutes exposure to nitrous oxide. Mice in this group displayed enhanced post-ischemic cardiac
functional recovery but did not show a preconditioning kinase signature, or a reduction in LV
infarct size. Instead, this group displayed similar levels of cell injury to that seen in 1WSP
control mice. This suggests that other mechanisms not related to the traditional preconditioning
phenotype, such as improvements in post-ischemic cardiomyocyte Ca2+ handling, may also be
involved in protecting cardiac function during IR injury.

78
5.1 Intra-Peritoneal Injections Induce Delayed Preconditioning against Global IR Injury
To my knowledge, there are no previous studies identifying a delayed preconditioning
effect from intra-peritoneal pentobarbital anesthesia. However, as I have shown, preconditioning
with pentobarbital may in fact be related to the method of injection as opposed to a direct effect
from the drug itself. As previously mentioned, pentobarbital has been used in a number of
studies examining the 1W of protection from rIPC, including our own, with no observable early
preconditioning effect[31]. Barbiturates are often the anesthetic of choice because of their
minimal cardiovascular depressive effects. However, Jiang et al. reported that pentobarbital leads
to a significant decrease in HR, LV peak systolic pressure, dP/dtmax in isolated hearts compared
to other intra-peritoneal anesthetics such as ketamine and chloral hydrate[118]. However, it was
never tested whether these depressive effects induce delayed preconditioning.
Li et al. examined the effects of delayed rIPC via bilateral hindlimb ischemia on infarct
size in isolated-mouse hearts[7]. They observed no delayed protection in sham mice sedated with
SP anesthesia via an IP injection, contradicting the results produced in the present study. In fact,
they were able to show that bilaterial hindlimb ischemia induces delayed preconditioning as
evidenced by a reduction in infarct size (35% in sham mice vs. 24% in delayed rIPC mice,
compared to 28-30% in the 2W IP injection groups in the present study). It should be noted that
the sham treated mice in their study exhibited a mean LV infarct size that is lower then what has
been reported in previous studies from our lab and others using a similar severity of IR injury, in
which mean infarct sizes are often >45%[9], [31]. This suggests that the control mice used in the
study by Li et al. may have also possessed some level of cardioprotection.
The same group published a study nearly a year later in which they reported a potential
preconditioning effect in mouse hearts after an IP injection. Labruto et al. found that

79
administration of Ringer’s solution via an IP injection, if given twenty-four hours prior to IR
injury, induces delayed cardioprotection by reducing infarction sizes and improving cardiac
performance[179]. They also showed no preconditioning effect after needle pricking, suggesting
that fluid within peritoneum is the required stimulus for inducing protection. We confirmed their
finding in the present study as we observed that mice that received a similar volume of saline via
IP injection had improved post-ischemic cardiac function, as evidenced by better recovery of
LVDP, dP/dtmax, dP/dtmin and attenuated increase of LVEDP. Mice in this group also showed
significant reductions in infarct size compared to 1WSP controls. Building on the Labruto et al.
study, we produced a novel finding in which we observed a three-fold increase in phospho-Akt
levels in mouse hearts that had received a saline injection twenty-fours prior to heart harvesting.
This cell signaling response may account for some of the observed cardioprotective benefits.
Further experiments using a kinase defective Akt mouse strain or an inhibitor of the upstream
PI3K (e.g. Wortmannin), would strongly indicate the role of Akt in IP injection-induced delayed
preconditioning.
Another essential kinase of the RISK pathway is p44/42 MAPK. While mice in the 2WSP
and 2WSP did exhibit a rise in phospho-p44/42 expression, the level of increase did not reach
statistical significance (P=0.0640 – 2WSP, P=0.0788 – 2WSP+rIPC). Furthermore, there were no
changes in phospho-p44/42 expression twenty-four hours after saline IP injection. This suggests
that the delayed preconditioning effects of an IP injection operate primarily via the Akt signaling
pathway. This finding also supports the notion that preconditioning can operate through multiple
pro-survival pathways available to the cell to yield the same cardioprotective effect.
A mechanism explaining how fluid in the peritoneum results in delayed preconditioning
at the myocardium has not been explored. In the same study, Labruto et al. examined the role of
NF-κB in the mouse heart and found that there was no increase in its nuclear-binding activity,

80
two hours after fluid injection. However, this does not eliminate a role for NF-κB as two hours
may have been too long after peak binding activity. Several studies have shown that NF-κB
migrates to the nucleus to increase gene expression of a variety of pro-inflammatory and cell
survival proteins as early as 15-min to 1 hour following the initial preconditioning stimulus by
rIPC and APC[7], [54]. We did not examine NF-κB in the hearts of mice that received saline
injections, leaving it open to future studies.
As described in the literature review section, one mechanism to explain the observed
cardioprotective effects from IP injections relates to the idea that a mild-inflammatory response
precedes the development of protection against IR injury. An IP injection may cause some
damage to the cells or blood vessels within the peritoneum, which may liberate ligands (e.g
DAMPs) that can activate the innate immune system. In a review article on the role of TLR4 in
ischemic tolerance, Kariko et al. describes how a sub-lethal stimulus, such as endotoxin, transient
ischemia or heat shock, generates a mild-inflammatory response, which is subsequently
suppressed with an increase in TLR4 negative-feedback regulators[22]. For example, necrosis or
injury to cells of the peritoneum might release endogenous ligands of TLR4, such as Hsps or
fibrin, which act on TLR4 in both inflammatory and non-immune related cells (cardiomyocytes,
endothelial cells). This could lead to the release of pro-inflammatory cytokines, but may also
increase the activity of negative regulators of the TLR signaling pathway such as PI3K – an
important component of the preconditioning phenotype. In the present study I showed that an IP
injection of saline causes an increase in phospho-Akt levels in the heart, which is a downstream
target of PI3K. Therefore, saline injection may have generated a mild-inflammatory response
within the peritoneum that resulted in the systemic suppression of TLR4 signaling by increasing
the activity of regulators that are also responsible for cardioprotection.

81
An added complexity to the issue of IP injections and their role in preconditioning is
related to the absence of any cardioprotective effects in an in-vivo model of IPC and IR injury.
The Bolli group conducted a study in which they developed an in-vivo mouse model of IPC via
transient ischemia to coronary arteries, in which mice were sedated with SP anesthesia via IP
injection[103]. They have used this model in a number of studies examining delayed
cardioprotection after IPC. In their initial study, they observed no early or delayed
preconditioning effect with SP treatment[103]. They also addressed the issue of delayed
preconditioning being caused by invasive surgical procedures by developing a sham group which
involved leaving the chest open for 60 minutes 24 hours prior to lethal IR injury. They found that
the infarct size was indistinguishable between this group and untreated controls.
There are several explanations to account for the contradictory results produced in our
study. Firstly, while it has been shown that rIPC and IPC generate a similar magnitude of
protection with regards to reductions in infarct size, there may be some differences in their
efficacy of inducing cardioprotection. Ahmed et al. compared the effects of local coronary IPC
with hindlimb rIPC on biochemical and histological changes in rats after IR injury[184]. They
found that local IPC was more effective at reducing post-ischemic levels of creatine kinase-MB
and lactate. There was also greater preservation of ATP levels and fewer episodes of ventricular
arrhythmias with IPC. While this group did not compare the effects of IPC and rIPC on infarct
size reductions or post-ischemic cardiac functional recovery, it may be that these additional
benefits from local IPC make it a more capable method of preconditioning above the increased
threshold set by an IP injection of SP anesthesia.
Secondly, the contrast between our study and the one conducted by the Bolli group could
be explained by the differences in the experimental protocol. In addition to the nature of the
preconditioning stimulus (local coronary IPC vs. limb rIPC), the differences in method of

82
ischemia (in-vivo, regional ischemia via CAO vs. ex-vivo global ischemia using Langendorff
preparation) and the absence of systemic influences (neural, hormonal and immune involvement
vs. isolated hearts) may account for the conflicting results produced in both studies.
5.2 Halothane Anesthesia Induces Delayed Preconditioning Against Global IR Injury The primary objective of this study was to develop a mouse model of delayed rIPC for
Langendorff isolated-heart studies of IR injury. My initial findings that an IP injection invokes
delayed preconditioning forced me to examine alternative methods of sedation to induce
transient hind limb ischemia in mice. Therefore, I used halothane and nitrous oxide anesthesia,
which have not been previously shown to induce ischemic tolerance in the delayed phase of
protection.
I examined the delayed effects of a 40-minute exposure to 1-2% halothane anesthesia on
post-ischemic cardiac function in isolated hearts. I found that halothane induced delayed
preconditioning by reducing LV infarct size and enhancing post-ischemic cardiac performance
that was comparable to the delayed benefits afforded with IP SP or saline administration. I also
showed that pre-treatment with halothane increases phospho-p44/42 MAPK levels in the delayed
phase of protection. The Redington lab group and others have shown the involvement of the
MAPK-protein family in the cardioprotective phenotype induced by rIPC and APC[85], [185].
The present finding suggests that halothane induces myocardial preconditioning by recruiting
similar signal transduction pathways. Halothane did not increase the levels of phospho-Akt,
indicating that similar to IP injections, there are multiple ways of generating cardioprotection.
Several studies have shown that administration of halothane anesthesia during IR injury
protects against post-ischemic cardiac dysfunction. This may be related to its potent negative

83
inotropic effects on cardiac contractile function and dose-related decreases in mean arterial blood
pressure. The mechanism of halothane protection during IR injury may involve a reduction in
Ca2+ overload during reperfusion by inhibiting the function of sarcolemmal L-type Ca2+ channels
[26]. Diminished Ca2+ overload may also occur through shortening of the action potential with
the opening of sarcolemmal K+ATP
channels. This ultimately results in the preservation of
myocardial ATP and creatine phosphate levels[186].
Previous studies have reported an early preconditioning effect from halothane exposure
with a decrease in infarct size, an effect which was abolished with adenosine receptor blockade,
using 8-phenyltheophylline, and PKC inhibition with chelerythrine [111]. It is well established
that PKC is upstream of NF-κB transcription factor activation and that this step is critical for the
development of delayed APC against IR injury[13], [32], [122], [123]. PKC activation is also
upstream of the p44/42 MAPK proteins, which we showed in the present study to be increased
with halothane exposure. Therefore, delayed cardioprotection with halothane may involve early
PKC activation of NF-κB, which subsequently leads to the transcription of genes responsible for
delayed preconditioning (Hsp, COX-2, iNOS etc). While detection of these markers was beyond
the scope of the present study, future work investigating delayed cardioprotection by halothane
can analyze whether this form of APC operates via similar mechanisms as other volatile
anesthetics.
5.3 Nitrous Oxide Improves Post-Ischemic Cardiac Performance but Does Not Reduce Infarction Size
I also examined the delayed effects of nitrous oxide on post-ischemic cardiac function to
determine its suitability as a method of anesthesia for 2W rIPC experiments. I found that twenty-
four hour prior exposure to 40 minutes of nitrous oxide at a 2:1 ratio with oxygen did not reduce

84
LV infarct size. Mice in this group showed comparable levels of cell injury to 1WSP treated
controls with an infarct size of approximately 45% of the total LV area. To my knowledge, this is
the first study to demonstrate that nitrous oxide does not have a delayed infarct-sparing effect
after IR injury.
Weber et al. reported that nitrous oxide does not induce an early preconditioning effect on
the heart. They showed that three cycles of 5 min of nitrous-oxide administration before
prolonged CAO does not reduce infarction sizes in rats. There was also no increase in the
phosphorylation or translocation of PKC-epsilon and PTK. Both proteins are known to play a
central role in mediating APC against IR injury and are also critical for the development of the
delayed phase of preconditioning[88], [93]. In the present study, there was no increase in p-
p44/42 MAPK or p-Akt levels with prior N2O treatment, confirming the findings of Weber et al.
that N2O does not recruit the traditional cell signaling pathways of IPC and APC.
Paradoxically, I observed that prior treatment with N2O enhanced post-ischemic cardiac
performance. LVEDP and LVDP showed similar improvements to the other forms of
preconditioning examined in this study. Rates of contraction and relaxation also approached pre-
ischemic values, although the magnitude of recovery for these parameters was intermediary
between other the preconditioning and 1WSP groups. These observations suggest that certain
preconditioning stimuli can benefit post-ischemic hearts without parallel improvements in both
cell survival and cardiac function. Several other studies have shown that IPC can cause
reductions in infarction size without improvements in cardiac function[187], [188]. However, the
present study shows that the opposite may also occur in that a conditioning stimulus can result in
recovery of cardiac function despite damage to heart tissue. This suggests that perhaps the
remaining viable tissue compensates in some fashion to maintain contractile function. While it

85
was out of the scope of this study to examine this phenomenon, a potential mechanism may
involve improvements in post-ischemic Ca2+ handling within the cardiomyocytes.
During systole, Ca2+ flows down its electrochemical gradient from the sarcoplasmic
reticulum (SR) through the coupling of sarcolemmal L-type voltage-gated and SR ryanodine
receptor Ca2+ channels. Relaxation occurs through active re-uptake of cytosolic Ca2+ into the SR
via the SR Ca2+-ATPase (SERCA2a)[189]. SERCA2a activity is regulated by the SR trans-
membrane protein phospholamban (PLN), which in its dephosphorylated state inhibits channel
activity and thus Ca2+ reuptake[190]. PLB is regulated by protein kinase A (PKA), which is
stimulated through sympathetic nervous system (SNS) activation. SNS activity raises
norepinephrine/epinephrine levels, which act on beta-adrenergic receptors (β-AR) on the heart to
increase PKA levels. PKA-inhibition of PLN leads to an increase in contractility and heart
rate[190].
The mechanism underlying how prior N2O administration improves post-ischemic Ca2+
handling, and thus contractile function, may involve N2O induced-inhibition of PLN. This may
occur in two ways. Firstly, while it is well established that nitrous oxide exerts dose-dependent
myocardial depression, there is some evidence suggesting that it may cause simultaneous bursts
of SNS activity[191], [192]. Previous studies have shown that 40-60% N2O can cause
intermittent increases in SNS outflow within 15-30 min of administration[192]. This is also
associated with an increase in plasma norepinephrine levels and systemic vascular resistance
[191]. As previously mentioned, increases in norepinephrine inhibit PLN via increased β-AR-
activation of PKA. Secondly, as part of its anti-nociceptive function, N2O causes an increase in
cell protein kinase G (PKG) activity via a neuronal nitric oxide (nNOS)-cyclic guanylyl cyclase
(cGMP)-signaling pathway[193]. Previous studies have shown that the cGMP-PKG pathway
may also play a role in protecting cardiomyocytes against IR injury through phosphorylation of

86
PLN. Gorbe et al. found that treatment of neonatal cardiomyocytes with a cGMP-analog or NO-
donor (S-nitroso-N-acetylpenicillamine – SNAP) decreased cell death after simulated IR injury,
and this was associated with increased the levels of phosphorylated PLN[194].
Therefore, the improvements in post-ischemic cardiac performance observed after
twenty-four hour prior treatment with N2O may operate via maintaining efficient Ca2+-cycling
through a PKA/PKG induced inhibition of PLN activity. The evidence presented here for a N2O-
induced inhibition of PLN via the PKA/PKG pathway comes from studies examining immediate
or early effects. While the present study did not evaluate the effects of N2O on PLN activity, it
may be that nitrous oxide alters the function of this regulatory protein in both the early and
delayed phases of protection. The N2O-induced kinase activation may lead to delayed changes in
gene expression of PLN or other Ca2+-cycling regulatory proteins that limit cardiac dysfunction
after IR injury. In the present study, we found that N2O led to partial recovery of the rates of
contraction and relaxation that were still significantly greater than 1WSP controls. Improvements
in Ca2+-cycling may account for these benefits, although this remains to be studied.
5.4 Cross-Talk Between Signaling Cascades
A recurring observation in the present study was that cardioprotection after IP injection or
exposure to inhalational anesthesia was associated with a statistically significant increase in
either phospho-Akt or phospho-p44/42, but not both. This indicates that the different stimuli
investigated in this study can activate either component of the RISK pathway to yield
cardioprotection. Nitrous oxide did not result in an increase in either kinase-signaling cascade,
which may account for the absence of infarct-sparing benefits.
Although only one component of the RISK pathway may have been activated after each
stimulus, it is likely that both kinase cascades are still important for mediating myocardial

87
protection against IR injury. The Yellon group has provided substantial evidence for the essential
role of the Akt and p44/42 kinases in mediating IPC-induced protection. They have shown that
both Akt and p44/42 are phosphorylated during the preconditioning phase and later reactivated at
the time of reperfusion[75]. In an earlier study, the same group described how the these kinase
pathways exhibit cross-talk during reperfusion, as inhibition of one kinase upregulates the
activity of the other pathway, acting in a compensatory fashion to ensure the cardioprotective
signal is relayed to downstream targets[195]. Interestingly, inhibition of either pathway abrogates
the cardioprotective effects of IPC on infarct size reduction, indicating the necessity for
activation of both pathways. Thus, while the present study did not assess the activation of Akt
and p44/42 during or after reperfusion, it is likely that both kinases are recruited at the onset of
reperfusion to mediate protection.

88
Chapter 6 Conclusion
In summary, the present study shows that sedation with SP anesthesia delivered via IP
injection and inhalational anesthesia results in delayed cardioprotection against global IR injury
in isolated mouse hearts, as evidenced by improved post-ischemic cardiac performance and
reduced infarction sizes. Treatment with nitrous oxide did not produce a delayed infarct sparing
effect but was associated with improved cardiac function after IR injury.
This study also showed that an IP injection of saline induced delayed cardioprotection,
indicating that this form of drug delivery should be avoided in future studies examining the 2W
of protection after rIPC in mice.
The finding that IP injections cause delayed preconditioning validates the primary
hypothesis of this study and clarifies my initial observation of a 2W cardioprotective effect from
SP anesthesia. IP injections reduced post-ischemic cardiac dysfunction and infarction sizes,
which was associated with an increase in phospho-Akt signaling. While this study did not
investigate how IP injections induce cardioprotection, one mechanism may involve injury to the
cells of the peritoneum, causing DAMP release and a mild inflammatory response. This stimulus
may trigger a shift from TLR4 pro-inflammatory signaling to an increase in negative feedback
inhibitors that are also known to mediate cardioprotection.
Based on these initial findings I aimed to survey alternative methods of anesthesia for use
in experiments on delayed protection after transient limb ischemia. I examined the effects of
halothane and nitrous oxide anesthesia, which hitherto had not been shown to cause 2W
cardioprotection. Halothane anesthesia led to delayed preconditioning with significant reductions
in infarction size and post-ischemic dysfunction after IR injury. This was accompanied by a cell
signal response involving p44/42 MAPK, which like Akt, is a member of the RISK pathway.

89
This aligned with my secondary hypothesis confirming halothane as a stimulus for delayed
preconditioning.
Finally, I found that treatment with nitrous oxide did not protect against infarction, but
did enhance post-ischemic cardiac recovery. Interestingly, unlike the other forms of anesthesia
explored in this study, there was no increase in phospho-Akt and phospho-p44/42 expression
twenty-four hours after treatment with nitrous oxide. While these findings do not align with the
established notion of preconditioning, it does indicate that other signaling mechanisms may
induce cardioprotection, possibly through changes in the function or expression of SR-Ca2+
regulatory proteins.
The primary aim of this study was to develop a mode of sedation that does not induce
cardioprotection in mice in order to establish a control treatment for future delayed rIPC
experiments. The present study highlights the importance of designing a study protocol that
controls for experimental stimuli that may induce or even block preconditioning via transient
ischemia. The type of anesthesia, method of delivery and conditions during sedation are all key
factors requiring careful consideration for myocardial preconditioning experiments. Nonetheless,
this study adds to the already well-known concept that preconditioning can be achieved from a
variety of stimuli.

90
Chapter 7 Future Directions
The findings from this study indicate that further work is required to develop a mouse
model of delayed rIPC for isolated-heart experiments examining myocardial protection against
IR injury.
However, the present investigation introduces several novel areas of study that will help
characterize the delayed preconditioning phenotype. Firstly, it is clear that a model of delayed
rIPC needs to include a control treatment that does not give rise to second window
preconditioning. This is not a simple task in the mouse because as I have shown, preconditioning
may occur from exposure to various stimuli. As such, future studies using a mouse model need
to investigate alternative methods of sedation or perhaps develop a means of inducing transient
ischemia in non-sedated mice.
There may be several methods to induce ischemia in a conscious mouse. Crawford et al.
have developed a method of applying hind limb ischemia using orthodontic rubber bands (ORB)
tied at proximal thigh level of mice[196]. I have begun testing the use of this method for delayed
rIPC experiments. Additionally, it may be possible to apply transient ischemia to the hind limb
while placing mice in a restraining tube with minimal stress or pain to the animal. This method is
commonly used for saphenous vein blood collection or tail-cuff blood pressure measurements in
mice[197].
Beyond developing a novel method of inducing transient ischemia, there are several other
aspects of the current study that require further investigation. The phenomenon that nitrous oxide
does not protect against infarction but does enhance post-ischemic cardiac function requires
further investigation. The role for improved Ca2+ handling through changes in the function or
expression of SR regulatory proteins has not been previously implicated in delayed

91
preconditioning. As such, future studies should examine the role for improved Ca2+ cycling,
particularly as it pertains to phospholamban regulation, as a potential novel means of protecting
the heart against post-ischemic cardiac dysfunction.
The present study was designed to survey a variety of methods of sedation to establish a
control treatment for delayed rIPC studies. As such, I did not explore the mechanisms behind the
various forms of preconditioning investigated in this study and have therefore provided several
avenues for further exploration.
7.1 An In-Vivo Model of Delayed rIPC
Future studies can also benefit from simultaneous investigations using an in-vivo model
of delayed rIPC and IR injury. The Langendorff-isolated heart technique is an invaluable tool to
examine the preconditioning phenotype as it provides cardiac-specific responses to IR injury.
However, an in-vivo model of IR injury with the added influence of the systemic response may
offer a more holistic, albeit more complex, depiction of cardioprotection. It is important to
investigate how rIPC affects the systemic inflammatory response during myocardial IR injury
and to examine the involvement of other organ systems (endocrine, peripheral/central nervous
system) in the resulting cardioprotective phenotype. Researchers can still examine cardiac
function in an in-vivo model of rIPC/IR injury through mouse echocardiography[198]. An in-
vivo model of delayed rIPC would also help to reconcile the conflicting results produced by the
current study and the findings generated by the Bolli et al. group. While there are a variety of
differences in experimental methodology between studies, it is important to confirm their finding
that sodium pentobarbital does not induce delayed cardiprotection against a lethal CAO.

92
7.2 Revisiting the role of TLR4 in delayed rIPC
It is becoming increasingly evident that the inflammatory response may be an important
component in the development of delayed cardioprotection. Therefore, future work in this area
can begin with addressing the initial hypothesis of this study that TLR4 is involved in the broad
physiological responses and signaling mechanisms of rIPC. Several studies have indicated that
preconditioning with sub-lethal doses of TLR4 ligands induces delayed cardioprotection against
IR injury. As discussed in the literature review section, these ligands activate a similar kinase
response mediating cardiprotection after IPC, which function to suppress TLR4 signaling and
therefore may inhibit a systemic inflammatory response. This is of profound importance as
reducing innate immune function may harmful in certain immune compromised disease states,
which may preclude the use of rIPC in these settings.
An investigation into the role of TLR4 can begin with examining the mechanism of how
IP injections cause delayed preconditioning. It is possible that the irritation or stress of fluid in
the peritoneum may induce an inflammatory response that serves as an adequate stimulus for the
developing subsequent second window protection against IR injury. Future studies can examine
whether circulating cytokines (TNF-α, IL-6) or TLR4-NF-κB signaling plays a role in this mode
of protection.
7.3 The ‘Third’ Window And Exercise Preconditioning The intriguing finding that a third or chronic window of preconditioning may exist has
spurred a great deal of interest in the additional phases of cardioprotection beyond the initial,
ephemeral first window. This interest has also been fueled by recent reports that chronic aerobic
exercise and repetitive rIPC confer cardioprotection through similar signaling mechanisms. As

93
reviewed in [199], [200], exercise-induced cardioprotection occurs through an increase in gene
expression of heat shock proteins, anti-oxidant enzymes and nitric-oxide synthase and through
adaptations in the coronary vasculature to facilitate perfusion. An important mechanism of the
cardioprotective phenotype after chronic exercise involves an increase in cell-stress induced
autophagy, the process by which cells remove damaged intra-cellular components[200].
Interestingly, Depre et al. showed that repetitive coronary artery occlusions or stenosis in a
porcine model reduces infarction through similar changes in gene expression and increases in
intracellular autophagy[95].
The Redington lab is interested in identifying whether there exists a reciprocal effect
between chronic exercise and repetitive rIPC, insofar that both modalities may modify the
resulting cardioprotective phenotype conferred after either form of preconditioning. The
existence of a third window of preconditioning has yet to be confirmed by other lab groups.
However, given the potential use of repeated RIPC stimuli for more chronic ischemic syndromes
(e.g. chronic stable angina, enhancement of exercise performance in heart failure) requires its
effects to be examined formally prior to widespread translation to the clinical arena. The present
study is an important first step for investigating the biology of chronic preconditioning in mice,
as these experiments will require a developed method of inducing transient ischemia in conscious
mice or employ the use of a non-cardioprotective sedative.
7.4 Clinical Implications of A Mouse Model of Delayed rIPC
Finally, the broader implications of developing a mouse model of delayed rIPC are the
ability for researchers to use this tool to examine the physiology of the second window of
cardioprotection and to better understand its therapeutic role in cardiovascular disease settings. It
is not surprising that the mouse, with the array of transgenic species available, has become a

94
popular animal model for examining the cellular and molecular basis of rIPC. This is especially
relevant given that delayed preconditioning is based on a genetic reprogramming that exploits the
evolutionary conserved stress-response available to cells – a property that also allows for a
longer duration and broader range of protection. These features have stimulated a great deal of
interest in developing novel strategies to mimic the delayed preconditioning phenotype, which
may involve the use of pharmacological agents or the transfer of genes to maintain a sustained
level of cardioprotection[88]. Such therapeutic approaches can have a strong impact both in and
out of the operating room and may be important for individuals at risk for cardiovascular disease.
This will require researchers to develop a stronger understanding of how the different
cardioprotective strategies interact with the pathological processes underlying various disease
conditions.

95
Chapter 8 References
[1] I. E. Konstantinov, S. Arab, J. Li, J. G. Coles, C. Boscarino, A. Mori, E. Cukerman, F. Dawood, M. M. H. Cheung, M. Shimizu, P. P. Liu, and A. N. Redington, “The remote ischemic preconditioning stimulus modifies gene expression in mouse myocardium.,” The Journal of Thoracic and Cardiovascular Surgery, vol. 130, no. 5, pp. 1326–1332, Nov. 2005.
[2] I. E. Konstantinov, S. Arab, R. K. Kharbanda, J. Li, M. M. H. Cheung, V. Cherepanov, G. P. Downey, P. P. Liu, E. Cukerman, J. G. Coles, and A. N. Redington, “The remote ischemic preconditioning stimulus modifies inflammatory gene expression in humans.,” Physiol. Genomics, vol. 19, no. 1, pp. 143–150, Sep. 2004.
[3] M. Shimizu, P. Saxena, I. E. Konstantinov, V. Cherepanov, M. M. H. Cheung, P. Wearden, H. Zhangdong, M. Schmidt, G. P. Downey, and A. N. Redington, “Remote ischemic preconditioning decreases adhesion and selectively modifies functional responses of human neutrophils.,” J. Surg. Res., vol. 158, no. 1, pp. 155–161, Jan. 2010.
[4] G. Heusch, J. Musiolik, E. Kottenberg, J. Peters, H. Jakob, and M. Thielmann, “STAT5 Activation and Cardioprotection by Remote Ischemic Preconditioning in Humans.,” Circ. Res., Nov. 2011.
[5] K. L. Redington, T. Disenhouse, S. C. Strantzas, R. Gladstone, C. Wei, M. B. Tropak, X. Dai, C. Manlhiot, J. Li, and A. N. Redington, “Remote cardioprotection by direct peripheral nerve stimulation and topical capsaicin is mediated by circulating humoral factors.,” Basic Res. Cardiol., vol. 107, no. 2, pp. 1–10, Mar. 2012.
[6] S.-Z. Zhang, N.-F. Wang, J. Xu, Q. Gao, G.-H. Lin, I. C. Bruce, and Q. Xia, “Kappa-opioid receptors mediate cardioprotection by remote preconditioning.,” Anesthesiology, vol. 105, no. 3, pp. 550–556, Sep. 2006.
[7] G. Li, F. Labruto, A. Sirsjö, F. Chen, J. Vaage, and G. Valen, “Myocardial protection by remote preconditioning: the role of nuclear factor kappa-B p105 and inducible nitric oxide synthase.,” Eur J Cardiothorac Surg, vol. 26, no. 5, pp. 968–973, Nov. 2004.
[8] B. Dawn, Y. Guo, A. Rezazadeh, O.-L. Wang, A. B. Stein, G. Hunt, J. Varma, Y.-T. Xuan, W.-J. Wu, W. Tan, X. Zhu, and R. Bolli, “Tumor necrosis factor-alpha does not modulate ischemia/reperfusion injury in naïve myocardium but is essential for the development of late preconditioning.,” Journal of Molecular and Cellular Cardiology, vol. 37, no. 1, pp. 51–61, Jul. 2004.
[9] M. P. Flaherty, Y. Guo, S. Tiwari, A. Rezazadeh, G. Hunt, S. K. Sanganalmath, X.-L. Tang, R. Bolli, and B. Dawn, “The role of TNF-alpha receptors p55 and p75 in acute myocardial ischemia/reperfusion injury and late preconditioning.,” Journal of Molecular and Cellular Cardiology, vol. 45, no. 6, pp. 735–741, Dec. 2008.
[10] S. L. Stevens, P. Y. Leung, K. B. Vartanian, B. Gopalan, T. Yang, R. P. Simon, and M. P. Stenzel-Poore, “Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury.,” J. Neurosci., vol. 31, no. 23, pp. 8456–8463, Jun. 2011.
[11] N. Yamashita, S. Hoshida, K. Otsu, N. Taniguchi, T. Kuzuya, and M. Hori, “The involvement of cytokines in the second window of ischaemic preconditioning,” British Journal of Pharmacology, vol. 131, no. 3, pp. 415–422, 2000.

96
[12] J.-W. Dong, J. G. Vallejo, H.-P. Tzeng, J. A. Thomas, and D. L. Mann, “Innate immunity mediates myocardial preconditioning through Toll-like receptor 2 and TIRAP-dependent signaling pathways.,” Am. J. Physiol. Heart Circ. Physiol., vol. 298, no. 3, pp. H1079–87, Mar. 2010.
[13] Y. T. Xuan, X. L. Tang, S. Banerjee, H. Takano, R. C. Li, H. Han, Y. Qiu, J. J. Li, and R. Bolli, “Nuclear factor-kappaB plays an essential role in the late phase of ischemic preconditioning in conscious rabbits.,” Circ. Res., vol. 84, no. 9, pp. 1095–1109, May 1999.
[14] S. P. Loukogeorgakis, A. T. Panagiotidou, M. W. Broadhead, A. Donald, J. E. Deanfield, and R. J. MacAllister, “Remote ischemic preconditioning provides early and late protection against endothelial ischemia-reperfusion injury in humans: role of the autonomic nervous system.,” Journal of the American College of Cardiology, vol. 46, no. 3, pp. 450–456, Aug. 2005.
[15] Y. Wang, B. Yin, S. Liu, and S. Xue, “Cardioprotective Effect by Tumor Necrosis Factor-α and Interleukin-6 through Late Preconditioning in Unstable Angina Patients,” Archives of Medical Research, vol. 38, no. 1, pp. 80–85, Jan. 2007.
[16] N. Smart, M. H. Mojet, D. S. Latchman, M. S. Marber, M. R. Duchen, and R. J. Heads, “IL-6 induces PI 3-kinase and nitric oxide-dependent protection and preserves mitochondrial function in cardiomyocytes.,” Cardiovasc. Res., vol. 69, no. 1, pp. 164–177, Jan. 2006.
[17] T. Ha, F. Hua, X. Liu, J. Ma, J. R. McMullen, T. Shioi, S. Izumo, J. Kelley, X. Gao, W. Browder, D. L. Williams, R. L. Kao, and C. Li, “Lipopolysaccharide-induced myocardial protection against ischaemia/reperfusion injury is mediated through a PI3K/Akt-dependent mechanism.,” Cardiovasc. Res., vol. 78, no. 3, pp. 546–553, Jun. 2008.
[18] T. Ha, Y. Hu, L. Liu, C. Lu, J. R. McMullen, J. Kelley, R. L. Kao, D. L. Williams, X. Gao, and C. Li, “TLR2 ligands induce cardioprotection against ischaemia/reperfusion injury through a PI3K/Akt-dependent mechanism.,” Cardiovasc. Res., vol. 87, no. 4, pp. 694–703, Sep. 2010.
[19] M. A. Pavione, F. Carmona, M. de Castro, and A. P. C. P. Carlotti, “Late remote ischemic preconditioning in children undergoing cardiopulmonary bypass: A randomized controlled trial.,” The Journal of Thoracic and Cardiovascular Surgery, Jan. 2012.
[20] J. M. Pradillo, D. Fernández-López, I. García-Yébenes, M. Sobrado, O. Hurtado, M. A. Moro, and I. Lizasoain, “Toll-like receptor 4 is involved in neuroprotection afforded by ischemic preconditioning.,” J. Neurochem., vol. 109, no. 1, pp. 287–294, Apr. 2009.
[21] G. Valen, “Extracardiac approaches to protecting the heart,” Eur J Cardiothorac Surg, vol. 35, no. 4, pp. 651–657, 2009.
[22] K. Karikó, D. Weissman, and F. A. Welsh, “Inhibition of toll-like receptor and cytokine signaling--a unifying theme in ischemic tolerance.,” J. Cereb. Blood Flow Metab., vol. 24, no. 11, pp. 1288–1304, Nov. 2004.
[23] S. Frantz, G. Ertl, and J. Bauersachs, “Mechanisms of disease: Toll-like receptors in cardiovascular disease.,” Nat Clin Pract Cardiovasc Med, vol. 4, no. 8, pp. 444–454, Aug. 2007.
[24] W. Schlack and B. Preckel, “Effects of halothane, enflurane, isoflurane, sevoflurane and desflurane on myocardial reperfusion injury in the isolated rat heart.,” British journal of …, 1998.

97
[25] M. Zaugg, E. Lucchinetti, M. Uecker, T. Pasch, and M. Schaub, “Anaesthetics and cardiac preconditioning. Part I. Signalling and cytoprotective mechanisms,” British Journal of Anaesthesia, vol. 91, no. 4, p. 551, 2003.
[26] A. Lochner, I. S. Harper, R. Salie, S. Genade, and A. R. Coetzee, “Halothane protects the isolated rat myocardium against excessive total intracellular calcium and structural damage during ischemia and reperfusion.,” Anesth. Analg., vol. 79, no. 2, pp. 226–233, Aug. 1994.
[27] C. Kowalski, S. Zahler, B. F. Becker, A. Flaucher, P. F. Conzen, E. Gerlach, and K. Peter, “Halothane, isoflurane, and sevoflurane reduce postischemic adhesion of neutrophils in the coronary system.,” Anesthesiology, vol. 86, no. 1, pp. 188–195, Jan. 1997.
[28] W. Chao, Y. Shen, X. Zhu, H. Zhao, M. Novikov, U. Schmidt, and A. Rosenzweig, “Lipopolysaccharide improves cardiomyocyte survival and function after serum deprivation.,” J. Biol. Chem., vol. 280, no. 23, pp. 21997–22005, Jun. 2005.
[29] E. Kottenberg, M. Thielmann, L. Bergmann, T. Heine, H. Jakob, G. Heusch, and J. Peters, “Protection by remote ischemic preconditioning during coronary artery bypass graft surgery with isoflurane but not propofol - a clinical trial.,” Acta Anaesthesiol Scand, vol. 56, no. 1, pp. 30–38, Jan. 2012.
[30] W. Chao, “Toll-like receptor signaling: a critical modulator of cell survival and ischemic injury in the heart.,” Am. J. Physiol. Heart Circ. Physiol., vol. 296, no. 1, pp. H1–12, Jan. 2009.
[31] J. Li, W. Xuan, R. Yan, M. B. Tropak, E. Jean St Michel, W. Liang, R. Gladstone, P. H. Backx, R. K. Kharbanda, and A. N. Redington, “Remote preconditioning provides potent cardioprotection via PI3K/Akt activation and is associated with nuclear accumulation of β-catenin.,” Clin. Sci., vol. 120, no. 10, pp. 451–462, May 2011.
[32] N. C. Weber and W. Schlack, “Inhalational anaesthetics and cardioprotection.,” Handb Exp Pharmacol, no. 182, pp. 187–207, 2008.
[33] D. M. Yellon and D. J. Hausenloy, “Myocardial reperfusion injury,” New England Journal of Medicine, vol. 357, no. 11, pp. 1121–1135, 2007.
[34] A. Frank, M. Bonney, S. Bonney, L. Weitzel, M. Koeppen, and T. Eckle, “Myocardial Ischemia Reperfusion Injury: From Basic Science to Clinical Bedside.,” Semin Cardiothorac Vasc Anesth, Feb. 2012.
[35] A. T. Turer and J. A. Hill, “Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy.,” Am. J. Cardiol., vol. 106, no. 3, pp. 360–368, Aug. 2010.
[36] F. Arslan, D. P. de Kleijn, and G. Pasterkamp, “Innate immune signaling in cardiac ischemia,” Nature Publishing Group, vol. 8, no. 5, pp. 292–300, Mar. 2011.
[37] D. J. Hausenloy, E. Boston-Griffiths, and D. M. Yellon, “Cardioprotection during cardiac surgery.,” Cardiovasc. Res., Mar. 2012.
[38] C. E. Murry, R. B. Jennings, and K. A. Reimer, “Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium.,” Circulation, vol. 74, no. 5, pp. 1124–1136, Nov. 1986.
[39] J. Kaszaki, A. Wolfárd, L. Szalay, and M. Boros, “Pathophysiology of Ischemia-Reperfusion Injury,” Transplantation Proceedings, vol. 38, no. 3, pp. 826–828, Apr. 2006.
[40] A. Skyschally, R. Schulz, and G. Heusch, “Pathophysiology of myocardial infarction,” Herz, vol. 33, no. 2, pp. 88–100, 2008.

98
[41] C. Penna, D. Mancardi, R. Rastaldo, and P. Pagliaro, “Cardioprotection: a radical view Free radicals in pre and postconditioning.,” Biochim. Biophys. Acta, vol. 1787, no. 7, pp. 781–793, Jul. 2009.
[42] F. Arslan, B. Keogh, P. McGuirk, and A. E. Parker, “TLR2 and TLR4 in ischemia reperfusion injury.,” Mediators Inflamm., vol. 2010, p. 704202, 2010.
[43] E. Murphy and C. Steenbergen, “Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury.,” Physiol. Rev., vol. 88, no. 2, pp. 581–609, Apr. 2008.
[44] D. Carden, “Pathophysiology of ischaemia–reperfusion injury - Carden - 2000 - The Journal of Pathology - Wiley Online Library,” The Journal of pathology, 2000.
[45] L. Toledo-Pereyra and F. Lopez-Neblina, “Reactive oxygen species and molecular biology... [Ann Transplant. 2004] - PubMed - NCBI,” … : quarterly of the Polish …, 2004.
[46] S. Frantz, R. A. Kelly, and T. Bourcier, “Role of TLR-2 in the activation of nuclear factor kappaB by oxidative stress in cardiac myocytes.,” J. Biol. Chem., vol. 276, no. 7, pp. 5197–5203, Feb. 2001.
[47] J. Jordan and Z. Zhao, “The role of neutrophils in myocardial ischemia–reperfusion injury,” Cardiovascular …, 1999.
[48] L. Ao, N. Zou, J. C. Cleveland, D. A. Fullerton, and X. Meng, “Myocardial TLR4 is a determinant of neutrophil infiltration after global myocardial ischemia: mediating KC and MCP-1 expression induced by extracellular HSC70.,” Am. J. Physiol. Heart Circ. Physiol., vol. 297, no. 1, pp. H21–8, Jul. 2009.
[49] D. M. Yellon and J. M. Downey, “Preconditioning the myocardium: from cellular physiology to clinical cardiology.,” Physiol. Rev., vol. 83, no. 4, pp. 1113–1151, Oct. 2003.
[50] S. A. Tavener and P. Kubes, “Cellular and molecular mechanisms underlying LPS-associated myocyte impairment.,” Am. J. Physiol. Heart Circ. Physiol., vol. 290, no. 2, pp. H800–6, Feb. 2006.
[51] M. E. Bianchi, “DAMPs, PAMPs and alarmins: all we need to know about danger.,” J. Leukoc. Biol., vol. 81, no. 1, pp. 1–5, Jan. 2007.
[52] F. Arslan, M. B. Smeets, L. A. J. O'Neill, B. Keogh, P. McGuirk, L. Timmers, C. Tersteeg, I. E. Hoefer, P. A. Doevendans, G. Pasterkamp, and D. P. V. de Kleijn, “Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody.,” Circulation, vol. 121, no. 1, pp. 80–90, Jan. 2010.
[53] A. Linde, D. Mosier, F. Blecha, and T. Melgarejo, “Innate immunity and inflammation--New frontiers in comparative cardiovascular pathology.,” Cardiovasc. Res., vol. 73, no. 1, pp. 26–36, Jan. 2007.
[54] G. Valen and Z. Yan, “Nuclear factor kappa-B and the heart,” Journal of the American College of Cardiology, vol. 38, no. 2, p. 307, 2001.
[55] J. H. Boyd, S. Mathur, Y. Wang, R. M. Bateman, and K. R. Walley, “Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response.,” Cardiovasc. Res., vol. 72, no. 3, pp. 384–393, Dec. 2006.
[56] P. Balakumar and N. K. Sharma, “Healing the diabetic heart: Does myocardial preconditioning work?,” Cellular Signalling, pp. 1–7, Sep. 2011.
[57] P. Ferdinandy, R. Schulz, and G. F. Baxter, “Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning.,” Pharmacol. Rev., vol. 59, no. 4, pp. 418–458, Dec. 2007.

99
[58] K. Przyklenk, B. Bauer, M. Ovize, R. A. Kloner, and P. Whittaker, “Regional ischemic ‘preconditioning’ protects remote virgin myocardium from subsequent sustained coronary occlusion.,” Circulation, vol. 87, no. 3, pp. 893–899, Mar. 1993.
[59] K. Tanaka, L. M. Ludwig, J. R. Kersten, P. S. Pagel, and D. C. Warltier, “Mechanisms of Cardioprotection by Volatile Anesthetics,” Anesthesiology, vol. 100, no. 3, p. 707, Mar. 2004.
[60] T. Pell, G. Baxter, and D. Yellon, “Renal ischemia preconditions myocardium: role of adenosine receptors and ATP-sensitive potassium channels,” American Journal of …, 1998.
[61] S. Tokuno, K. Hinokiyama, K. Tokuno, C. Löwbeer, L.-O. Hansson, and G. Valen, “Spontaneous ischemic events in the brain and heart adapt the hearts of severely atherosclerotic mice to ischemia.,” Arterioscler. Thromb. Vasc. Biol., vol. 22, no. 6, pp. 995–1001, Jun. 2002.
[62] B. Gho, R. Schoemaker, and M. van den Doel, “Myocardial Protection by Brief Ischemia in Noncardiac Tissue,” Circulation, 1996.
[63] R. Huda, D. H. Chung, and M. Mathru, “Ischemic preconditioning at a distance: altered gene expression in mouse heart and other organs following brief occlusion of the mesenteric artery.,” Heart Lung Circ, vol. 14, no. 1, pp. 36–43, Mar. 2005.
[64] H. H. Patel, J. Moore, A. K. Hsu, and G. J. Gross, “Cardioprotection at a distance: mesenteric artery occlusion protects the myocardium via an opioid sensitive mechanism.,” Journal of Molecular and Cellular Cardiology, vol. 34, no. 10, pp. 1317–1323, Oct. 2002.
[65] Y. Birnbaum and S. Hale, “Ischemic Preconditioning at a Distance : Reduction of Myocardial Infarct Size by Partial Reduction of Blood Supply Combined With Rapid Stimulation of the Gastrocnemius Muscle in the Rabbit,” Circulation, 1997.
[66] T. Oxman, M. Arad, R. Klein, N. Avazov, and B. Rabinowitz, “Limb ischemia preconditions the heart against reperfusion tachyarrhythmia.,” Am. J. Physiol., vol. 273, no. 4, pp. H1707–12, Oct. 1997.
[67] R. Kharbanda, U. Mortensen, P. White, S. Kristiansen, M. Schmidt, J. Hoschtitzky, M. Vogel, K. Sorensen, A. Redington, and R. MacAllister, “Transient limb ischemia induces remote ischemic preconditioning in vivo,” Circulation, vol. 106, no. 23, pp. 2881–2883, 2002.
[68] D. J. Hausenloy and D. M. Yellon, “Preconditioning and postconditioning: underlying mechanisms and clinical application.,” Atherosclerosis, vol. 204, no. 2, pp. 334–341, Jun. 2009.
[69] R. K. Kharbanda, T. T. Nielsen, and A. N. Redington, “Translation of remote ischaemic preconditioning into clinical practice.,” Lancet, vol. 374, no. 9700, pp. 1557–1565, Oct. 2009.
[70] E. Murphy, “Primary and secondary signaling pathways in early preconditioning that converge on the mitochondria to produce cardioprotection.,” Circ. Res., vol. 94, no. 1, pp. 7–16, Jan. 2004.
[71] D. J. Hausenloy and D. M. Yellon, “Remote ischaemic preconditioning: underlying mechanisms and clinical application.,” Cardiovasc. Res., vol. 79, no. 3, pp. 377–386, Aug. 2008.
[72] G. J. Gross, “Role of opioids in acute and delayed preconditioning.,” Journal of Molecular and Cellular Cardiology, vol. 35, no. 7, pp. 709–718, Jul. 2003.
[73] S. P. Jones and R. Bolli, “The ubiquitous role of nitric oxide in cardioprotection.,” Journal of Molecular and Cellular Cardiology, vol. 40, no. 1, pp. 16–23, Jan. 2006.

100
[74] Y. Fujio, T. Nguyen, D. Wencker, R. N. Kitsis, and K. Walsh, “Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart.,” Circulation, vol. 101, no. 6, pp. 660–667, Feb. 2000.
[75] D. J. Hausenloy, A. Tsang, M. M. Mocanu, and D. M. Yellon, “Ischemic preconditioning protects by activating prosurvival kinases at reperfusion.,” Am. J. Physiol. Heart Circ. Physiol., vol. 288, no. 2, pp. H971–6, Feb. 2005.
[76] N. Tapuria, Y. Kumar, M. M. Habib, M. Abu Amara, A. M. Seifalian, and B. R. Davidson, “Remote ischemic preconditioning: a novel protective method from ischemia reperfusion injury--a review.,” J. Surg. Res., vol. 150, no. 2, pp. 304–330, Dec. 2008.
[77] P. Saxena, M. A. J. Newman, J. S. Shehatha, A. N. Redington, and I. E. Konstantinov, “Remote ischemic conditioning: evolution of the concept, mechanisms, and clinical application.,” J Card Surg, vol. 25, no. 1, pp. 127–134, 2010.
[78] C. Li, T. Ha, J. Kelley, X. Gao, Y. Qiu, R. L. Kao, W. Browder, and D. L. Williams, “Modulating Toll-like receptor mediated signaling by (1-->3)-beta-D-glucan rapidly induces cardioprotection.,” Cardiovasc. Res., vol. 61, no. 3, pp. 538–547, Feb. 2004.
[79] T. Fukao and S. Koyasu, “PI3K and negative regulation of TLR signaling,” Trends in Immunology, vol. 24, no. 7, pp. 358–363, 2003.
[80] R. K. Kharbanda, “Cardiac conditioning: a review of evolving strategies to reduce ischaemia-reperfusion injury.,” Heart, vol. 96, no. 15, pp. 1179–1186, Aug. 2010.
[81] D. J. Hausenloy and D. M. Yellon, “Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection.,” Heart Fail Rev, vol. 12, no. 3, pp. 217–234, Dec. 2007.
[82] A. Hashimoto, G. Miyakoda, Y. Hirose, and T. Mori, “Activation of endothelial nitric oxide synthase by cilostazol via a cAMP/protein kinase A- and phosphatidylinositol 3-kinase/Akt-dependent mechanism.,” Atherosclerosis, vol. 189, no. 2, pp. 350–357, Dec. 2006.
[83] A. M. Zeiher, S. Dimmeler, I. Fleming, B. Fisslthaler, C. Hermann, and R. Busse, “Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation,” Nature, vol. 399, no. 6736, pp. 601–605, Jun. 1999.
[84] H. Tong, K. Imahashi, C. Steenbergen, and E. Murphy, “Phosphorylation of glycogen synthase kinase-3beta during preconditioning through a phosphatidylinositol-3-kinase--dependent pathway is cardioprotective.,” Circ. Res., vol. 90, no. 4, pp. 377–379, Mar. 2002.
[85] M. Shimizu, M. Tropak, R. J. Diaz, F. Suto, H. Surendra, E. Kuzmin, J. Li, G. Gross, G. J. Wilson, J. Callahan, and A. N. Redington, “Transient limb ischaemia remotely preconditions through a humoral mechanism acting directly on the myocardium: evidence suggesting cross-species protection.,” Clin. Sci., vol. 117, no. 5, pp. 191–200, Sep. 2009.
[86] H. Tong, W. Chen, and C. Steenbergen, “Ischemic Preconditioning Activates Phosphatidylinositol-3-Kinase Upstream of Protein Kinase C,” Circ. Res., 2000.
[87] A. Tsang, D. J. Hausenloy, M. M. Mocanu, and D. M. Yellon, “Postconditioning: a form of ‘modified reperfusion’ protects the myocardium by activating the phosphatidylinositol 3-kinase-Akt pathway.,” Circ. Res., vol. 95, no. 3, pp. 230–232, Aug. 2004.
[88] A. B. Stein, X.-L. Tang, Y. Guo, Y.-T. Xuan, B. Dawn, and R. Bolli, “Delayed adaptation of the heart to stress: late preconditioning.,” Stroke, vol. 35, no. 11, pp. 2676–2679, Nov. 2004.

101
[89] M. Heidbreder, A. Naumann, K. Tempel, P. Dominiak, and A. Dendorfer, “Remote vs. ischaemic preconditioning: the differential role of mitogen-activated protein kinase pathways.,” Cardiovasc. Res., vol. 78, no. 1, pp. 108–115, Apr. 2008.
[90] C. P. Baines, J. Zhang, G.-W. Wang, Y.-T. Zheng, J. X. Xiu, E. M. Cardwell, R. Bolli, and P. Ping, “Mitochondrial PKCepsilon and MAPK form signaling modules in the murine heart: enhanced mitochondrial PKCepsilon-MAPK interactions and differential MAPK activation in PKCepsilon-induced cardioprotection.,” Circ. Res., vol. 90, no. 4, pp. 390–397, Mar. 2002.
[91] J. Raphael, “Physiology and Pharmacology of Myocardial Preconditioning,” Semin Cardiothorac Vasc Anesth, vol. 14, no. 1, pp. 54–59, May 2010.
[92] M. Marber, D. Latchman, and J. Walker, “Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction,” Circulation, 1993.
[93] R. Bolli, “The late phase of preconditioning,” Circ. Res., vol. 87, no. 11, pp. 972–983, 2000.
[94] D. J. Hausenloy and D. M. Yellon, “The second window of preconditioning (SWOP) where are we now?,” Cardiovasc Drugs Ther, vol. 24, no. 3, pp. 235–254, Jun. 2010.
[95] C. Depre, J. Y. Park, Y.-T. Shen, X. Zhao, H. Qiu, L. Yan, B. Tian, S. F. Vatner, and D. E. Vatner, “Molecular mechanisms mediating preconditioning following chronic ischemia differ from those in classical second window.,” Am. J. Physiol. Heart Circ. Physiol., vol. 299, no. 3, pp. H752–62, Sep. 2010.
[96] A. Rizvi, X. L. Tang, Y. Qiu, Y. T. Xuan, H. Takano, A. K. Jadoon, and R. Bolli, “Increased protein synthesis is necessary for the development of late preconditioning against myocardial stunning.,” Am. J. Physiol., vol. 277, no. 3, pp. H874–84, Sep. 1999.
[97] Y. Nishino, “Ischemic preconditioning activates AMPK in a PKC-dependent manner and induces GLUT4 up-regulation in the late phase of cardioprotection,” Cardiovasc. Res., vol. 61, no. 3, pp. 610–619, Feb. 2004.
[98] B. Dawn, H. Takano, and X. Tang, “Role of Src protein tyrosine kinases in late preconditioning against myocardial infarction,” American Journal of …, 2002.
[99] H. Takano, R. Li, H. Han, Y. Qiu, J. Li, and R. Bolli, “Nuclear factor-B plays an essential role in the late phase of ischemic preconditioning in conscious rabbits,” Circ. Res., 1999.
[100] J. A. Romashkova and S. S. Makarov, “NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling.,” Nature, vol. 401, no. 6748, pp. 86–90, Sep. 1999.
[101] Y. Wang, M. Kudo, M. Xu, A. Ayub, and M. Ashraf, “Mitochondrial KATPChannel as an End Effector of Cardioprotection During Late Preconditioning: Triggering Role of Nitric Oxide,” Journal of Molecular and Cellular Cardiology, vol. 33, no. 11, pp. 2037–2046, 2001.
[102] H. Patel, E. Gross, and J. Peart, “Sarcolemmal KATP channel triggers delayed ischemic preconditioning in rats,” American Journal of …, 2005.
[103] Y. Guo, W. J. Wu, Y. Qiu, X. L. Tang, Z. Yang, and R. Bolli, “Demonstration of an early and a late phase of ischemic preconditioning in mice,” Am. J. Physiol. Heart Circ. Physiol., vol. 275, no. 4, pp. H1375–H1387, 1998.
[104] R. Bolli, Q.-H. Li, X.-L. Tang, Y. Guo, Y.-T. Xuan, G. Rokosh, and B. Dawn, “The late phase of preconditioning and its natural clinical application--gene therapy.,” Heart Fail Rev, vol. 12, no. 3, pp. 189–199, Dec. 2007.

102
[105] M. M. H. Cheung, R. K. Kharbanda, I. E. Konstantinov, M. Shimizu, H. Frndova, J. Li, H. M. Holtby, P. N. Cox, J. F. Smallhorn, G. S. Van Arsdell, and A. N. Redington, “Randomized controlled trial of the effects of remote ischemic preconditioning on children undergoing cardiac surgery: first clinical application in humans.,” Journal of the American College of Cardiology, vol. 47, no. 11, pp. 2277–2282, Jun. 2006.
[106] D. J. Hausenloy, P. K. Mwamure, V. Venugopal, J. Harris, M. Barnard, E. Grundy, E. Ashley, S. Vichare, C. Di Salvo, S. Kolvekar, M. Hayward, B. Keogh, R. J. MacAllister, and D. M. Yellon, “Effect of remote ischaemic preconditioning on myocardial injury in patients undergoing coronary artery bypass graft surgery: a randomised controlled trial.,” Lancet, vol. 370, no. 9587, pp. 575–579, Aug. 2007.
[107] A. K. Roscoe, J. D. Christensen, and C. I. Lynch, “Isoflurane, but not Halothane, Induces Protection of Human Myocardium via Adenosine A1 Receptors and Adenosine Triphosphate–sensitive Potassium Channels,” Anesthesiology, vol. 92, no. 6, p. 1692, Jun. 2000.
[108] S. G. De Hert, “Volatile Anesthetics and Cardiac Function,” Semin Cardiothorac Vasc Anesth, vol. 10, no. 1, pp. 33–42, Mar. 2006.
[109] V. Piriou, P. Chiari, F. Lhuillier, O. Bastien, J. Loufoua, O. Raisky, J. S. David, M. Ovize, and J. J. Lehot, “Pharmacological preconditioning: comparison of desflurane, sevoflurane, isoflurane and halothane in rabbit myocardium,” British Journal of Anaesthesia, vol. 89, no. 3, pp. 486–491, Sep. 2002.
[110] D. F. Stowe and L. G. Kevin, “Cardiac preconditioning by volatile anesthetic agents: a defining role for altered mitochondrial bioenergetics.,” Antioxid. Redox Signal., vol. 6, no. 2, pp. 439–448, Apr. 2004.
[111] D. K. Cope, W. K. Impastato, M. V. Cohen, and J. M. Downey, “Volatile anesthetics protect the ischemic rabbit myocardium from infarction.,” Anesthesiology, vol. 86, no. 3, pp. 699–709, Mar. 1997.
[112] A. Wood, J. Campagna, and K. Miller, “Mechanisms of Actions of Inhaled Anesthetics — NEJM,” New England Journal …, 2003.
[113] J. G. Bovill, “Inhalation anaesthesia: from diethyl ether to xenon.,” Handb Exp Pharmacol, no. 182, pp. 121–142, 2008.
[114] D. M. Wheeler, A. Katz, R. T. Rice, and R. G. Hansford, “Volatile anesthetic effects on sarcoplasmic reticulum Ca content and sarcolemmal Ca flux in isolated rat cardiac cell suspensions.,” Anesthesiology, vol. 80, no. 2, pp. 372–382, Feb. 1994.
[115] R. Kato and P. Foëx, “Myocardial protection by anesthetic agents against ischemia-reperfusion injury: An update for anesthesiologists,” Can J Anesth/J Can Anesth, vol. 49, no. 8, pp. 777–791, Oct. 2002.
[116] G. Torri, “Inhalation anesthetics: a review.,” Minerva anestesiologica, vol. 76, no. 3, p. 215, 2010.
[117] D. C. Saha, A. C. Saha, G. Malik, M. E. Astiz, and E. C. Rackow, “Comparison of cardiovascular effects of tiletamine-zolazepam, pentobarbital, and ketamine-xylazine in male rats.,” J. Am. Assoc. Lab. Anim. Sci., vol. 46, no. 2, pp. 74–80, Mar. 2007.
[118] X. Jiang, L. Gao, Y. Zhang, G. Wang, Y. Liu, C. Yan, and H. Sun, “A comparison of the effects of ketamine, chloral hydrate and pentobarbital sodium anesthesia on isolated rat hearts and cardiomyocytes.,” J Cardiovasc Med (Hagerstown), vol. 12, no. 10, pp. 732–735, Oct. 2011.
[119] S. G. De Hert, F. Turani, S. Mathur, and D. F. Stowe, “Cardioprotection with volatile anesthetics: mechanisms and clinical implications.,” Anesth. Analg., vol. 100, no. 6, pp. 1584–1593, Jun. 2005.

103
[120] Z.-Y. Hu and J. Liu, “Mechanism of cardiac preconditioning with volatile anaesthetics.,” Anaesth Intensive Care, vol. 37, no. 4, pp. 532–538, Jul. 2009.
[121] M. Zaugg, E. Lucchinetti, D. R. Spahn, T. Pasch, and M. C. Schaub, “Volatile anesthetics mimic cardiac preconditioning by priming the activation of mitochondrial KATP channels via multiple signaling pathways,” Anesthesiology, vol. 97, no. 1, p. 4, 2002.
[122] C. H. Chen, J. H. Chuang, K. Liu, and J. Y. H. Chan, “Nitric oxide triggers delayed anesthetic preconditioning-induced cardiac protection via activation of nuclear factor-kappaB and upregulation of inducible nitric oxide synthase.,” Shock, vol. 30, no. 3, pp. 241–249, Sep. 2008.
[123] C. Zhong, Y. Zhou, and H. Liu, “Nuclear factor kappaB and anesthetic preconditioning during myocardial ischemia-reperfusion.,” Anesthesiology, vol. 100, no. 3, pp. 540–546, Mar. 2004.
[124] P. Sergeev, R. da Silva, and E. Lucchinetti, “Trigger-dependent Gene Expression Profiles in Cardiac Precon... : Anesthesiology,” …, 2004.
[125] J. Marinovic, Z. J. Bosnjak, and A. Stadnicka, “Distinct roles for sarcolemmal and mitochondrial adenosine triphosphate-sensitive potassium channels in isoflurane-induced protection against oxidative stress.,” Anesthesiology, vol. 105, no. 1, pp. 98–104, Jul. 2006.
[126] W. G. Toller, E. R. Gross, J. R. Kersten, P. S. Pagel, G. J. Gross, and D. C. Warltier, “Sarcolemmal and Mitochondrial Adenosine Triphosphate– dependent Potassium Channels: Mechanism of Desflurane-induced Cardioprotection,” Anesthesiology, vol. 92, no. 6, p. 1731, Jun. 2000.
[127] J. Müllenheim, D. Ebel, J. Frässdorf, B. Preckel, V. Thämer, and W. Schlack, “Isoflurane preconditions myocardium against infarction via release of free radicals.,” Anesthesiology, vol. 96, no. 4, pp. 934–940, Apr. 2002.
[128] H.-P. Yu, “Role of Anesthetic Agents on Cardiac and Immune Systems,” Shock, vol. 36, no. 6, pp. 532–541, Dec. 2011.
[129] G. Hu, M. R. Salem, and G. J. Crystal, “Isoflurane and Sevoflurane Precondition against Neutrophil-induced Contractile Dysfunction in Isolated Rat Hearts,” Anesthesiology, vol. 100, no. 3, p. 489, Mar. 2004.
[130] B. Heindl, F. M. Reichle, S. Zahler, P. F. Conzen, and B. F. Becker, “Sevoflurane and isoflurane protect the reperfused guinea pig heart by reducing postischemic adhesion of polymorphonuclear neutrophils.,” Anesthesiology, vol. 91, no. 2, pp. 521–530, Aug. 1999.
[131] G. Hu, T. Vasiliauskas, M. R. Salem, D. P. Rhone, and G. J. Crystal, “Neutrophils Pretreated with Volatile Anesthetics Lose Ability to Cause Cardiac Dysfunction,” Anesthesiology, vol. 98, no. 3, p. 712, Mar. 2003.
[132] G. Hu, M. R. Salem, and G. J. Crystal, “Role of Adenosine Receptors in Volatile Anesthetic Preconditioning against Neutrophil-induced Contractile Dysfunction in Isolated Rat Hearts,” Anesthesiology, vol. 103, no. 2, p. 287, Aug. 2005.
[133] J. R. Kersten, T. J. Schmeling, D. A. Hettrick, P. S. Pagel, G. J. Gross, and D. C. Warltier, “Mechanism of Myocardial Protection by Isoflurane: Role of Adenosine Triphosphate‐regulated Potassium (KATP) Channels,” Anesthesiology, vol. 85, no. 4, p. 794, Oct. 1996.
[134] M. Uecker, R. da Silva, T. Grampp, T. Pasch, M. C. Schaub, and M. Zaugg, “Translocation of protein kinase C isoforms to subcellular targets in ischemic and anesthetic preconditioning.,” Anesthesiology, vol. 99, no. 1, pp. 138–147, Jul. 2003.

104
[135] L. G. Kevin and E. Novalija, “Anesthetic preconditioning decreases arrhythmias and improves regional conduction in isolated hearts.,” J. Cardiothorac. Vasc. Anesth., vol. 22, no. 2, pp. 217–224, Apr. 2008.
[136] J. Raphael, S. Abedat, J. Rivo, K. Meir, R. Beeri, T. Pugatsch, Z. Zuo, and Y. Gozal, “Volatile anesthetic preconditioning attenuates myocardial apoptosis in rabbits after regional ischemia and reperfusion via Akt signaling and modulation of Bcl-2 family proteins.,” J. Pharmacol. Exp. Ther., vol. 318, no. 1, pp. 186–194, Jul. 2006.
[137] K. Tanaka, L. M. Ludwig, J. G. Krolikowski, D. Alcindor, P. F. Pratt, J. R. Kersten, P. S. Pagel, and D. C. Warltier, “Isoflurane produces delayed preconditioning against myocardial ischemia and reperfusion injury: role of cyclooxygenase-2.,” Anesthesiology, vol. 100, no. 3, pp. 525–531, Mar. 2004.
[138] M. Tonkovic-Capin and G. Gross, “Delayed cardioprotection by isoflurane: role of KATP channels,” American Journal of …, 2002.
[139] M. Wakeno-Takahashi and H. Otani, “Isoflurane induces second window of preconditioning through upregulation of inducible nitric oxide synthase in rat heart,” American Journal of …, 2005.
[140] Y. M. Tsutsumi, H. H. Patel, D. Huang, and D. M. Roth, “Role of 12-lipoxygenase in volatile anesthetic-induced delayed preconditioning in mice.,” Am. J. Physiol. Heart Circ. Physiol., vol. 291, no. 2, pp. H979–83, Aug. 2006.
[141] J. F. Coetzee, P. J. le Roux, S. Genade, and A. Lochner, “Reduction of postischemic contractile dysfunction of the isolated rat heart by sevoflurane: comparison with halothane.,” Anesth. Analg., vol. 90, no. 5, pp. 1089–1097, May 2000.
[142] W. Schlack, M. Hollmann, J. Stunneck, and V. Thämer, “Effect of halothane on myocardial reoxygenation injury in the isolated rat heart.,” British Journal of Anaesthesia, vol. 76, no. 6, pp. 860–867, Jun. 1996.
[143] M. Naylor, K. K. Bowen, K. A. Sailor, R. J. Dempsey, and R. Vemuganti, “Preconditioning-induced ischemic tolerance stimulates growth factor expression and neurogenesis in adult rat hippocampus.,” Neurochem. Int., vol. 47, no. 8, pp. 565–572, Dec. 2005.
[144] K. R. Dave, I. Saul, R. Busto, M. D. Ginsberg, T. J. Sick, and M. A. P rez-Pinz n, “Ischemic Preconditioning Preserves Mitochondrial Function After Global Cerebral Ischemia in Rat Hippocampus,” J. Cereb. Blood Flow Metab., vol. 21, no. 12, pp. 1401–1410, Dec. 2001.
[145] E. Novalija, S. Fujita, J. P. Kampine, and D. F. Stowe, “Sevoflurane mimics ischemic preconditioning effects on coronary flow and nitric oxide release in isolated hearts.,” Anesthesiology, vol. 91, no. 3, pp. 701–712, Sep. 1999.
[146] V. Piriou, P. Chiari, O. Gateau-Roesch, L. Argaud, D. Muntean, D. Salles, J. Loufouat, P.-Y. Gueugniaud, J.-J. Lehot, and M. Ovize, “Desflurane-induced Preconditioning Alters Calcium-induced Mitochondrial Permeability Transition,” Anesthesiology, vol. 100, no. 3, p. 581, Mar. 2004.
[147] X. Lu, H. Liu, L. Wang, and S. Schaefer, “Activation of NF-kappaB is a critical element in the antiapoptotic effect of anesthetic preconditioning.,” Am. J. Physiol. Heart Circ. Physiol., vol. 296, no. 5, pp. H1296–304, May 2009.
[148] R. R. Lamberts, G. Onderwater, N. Hamdani, M. J. A. Vreden, J. Steenhuisen, E. C. Eringa, S. A. Loer, G. J. M. Stienen, and R. A. Bouwman, “Reactive oxygen species-induced stimulation of 5'AMP-activated protein kinase mediates sevoflurane-induced cardioprotection.,” Circulation, vol. 120, no. 11, pp. S10–5, Sep. 2009.

105
[149] P. C. Chiari, P. S. Pagel, K. Tanaka, J. G. Krolikowski, L. M. Ludwig, R. A. Trillo, N. Puri, J. R. Kersten, and D. C. Warltier, “Intravenous emulsified halogenated anesthetics produce acute and delayed preconditioning against myocardial infarction in rabbits.,” Anesthesiology, vol. 101, no. 5, pp. 1160–1166, Nov. 2004.
[150] C. Lotz, M. Lange, A. Redel, J. Stumpner, J. Schmidt, T. Tischer-Zeitz, N. Roewer, and F. Kehl, “Peroxisome-proliferator-activated receptor γ mediates the second window of anaesthetic-induced preconditioning.,” Exp. Physiol., vol. 96, no. 3, pp. 317–324, Mar. 2011.
[151] R. G. Merin, “Effect of anesthetic drugs on myocardial performance in man.,” Annu. Rev. Med., vol. 28, pp. 75–83, 1977.
[152] T. Koyama and K. Fukuda, “Involvement of the kappa-opioid receptor in nitrous oxide-induced analgesia in mice.,” J Anesth, vol. 24, no. 2, pp. 297–299, Apr. 2010.
[153] D. E. Emmanouil, A. S. Dickens, R. W. Heckert, Y. Ohgami, E. Chung, S. Han, and R. M. Quock, “Nitrous oxide-antinociception is mediated by opioid receptors and nitric oxide in the periaqueductal gray region of the midbrain.,” Eur Neuropsychopharmacol, vol. 18, no. 3, pp. 194–199, Mar. 2008.
[154] N. C. Weber, O. Toma, S. Awan, J. Frässdorf, B. Preckel, and W. Schlack, “Effects of nitrous oxide on the rat heart in vivo: another inhalational anesthetic that preconditions the heart?,” Anesthesiology, vol. 103, no. 6, pp. 1174–1182, Dec. 2005.
[155] C. Jin, S. Sonoda, L. Fan, M. Watanabe, T. Kugimiya, and T. Okada, “Sevoflurane and nitrous oxide exert cardioprotective effects against hypoxia-reoxygenation injury in the isolated rat heart.,” J Physiol Sci, vol. 59, no. 2, pp. 123–129, Mar. 2009.
[156] S.-H. Ko, S.-K. Lee, Y.-J. Han, H. Choe, Y.-G. Kwak, S.-W. Chae, K.-P. Cho, and H.-S. Song, “Blockade of Myocardial ATP‐sensitive Potassium Channels by Ketamine,” Anesthesiology, vol. 87, no. 1, p. 68, Jul. 1997.
[157] J. Han, N. Kim, H. Joo, and E. Kim, “Ketamine abolishes ischemic preconditioning through inhibition of K(ATP) channels in rabbit hearts.,” Am. J. Physiol. Heart Circ. Physiol., vol. 283, no. 1, pp. H13–21, Jul. 2002.
[158] A. Molojavyi, B. Preckel, T. Comfère, J. Müllenheim, V. Thämer, and W. Schlack, “Effects of ketamine and its isomers on ischemic preconditioning in the isolated rat heart.,” Anesthesiology, vol. 94, no. 4, pp. 623–9; discussion 5A–6A, Apr. 2001.
[159] J. Müllenheim, J. Frädorf, B. Preckel, and V. Thämer, “Ketamine, but Not S (+)-ketamine, Blocks Ischemic Preconditi... : Anesthesiology,” …, 2001.
[160] J.-L. Hanouz, L. Zhu, E. Persehaye, M. Massetti, G. Babatasi, A. Khayat, P. Ducouret, B. Plaud, and J.-L. Gérard, “Ketamine preconditions isolated human right atrial myocardium: roles of adenosine triphosphate-sensitive potassium channels and adrenoceptors.,” Anesthesiology, vol. 102, no. 6, pp. 1190–1196, Jun. 2005.
[161] L. Wang, N. Oka, M. Tropak, J. Callahan, J. Lee, G. Wilson, A. Redington, and C. A. Caldarone, “Remote ischemic preconditioning elaborates a transferable blood-borne effector that protects mitochondrial structure and function and preserves myocardial performance after neonatal cardioplegic arrest,” The Journal of Thoracic and Cardiovascular Surgery, vol. 136, no. 2, pp. 335–342, Aug. 2008.
[162] M. Galagudza, J. Vaage, and G. Valen, “Isoflurane and other commonly used anaesthetics do not protect the isolated buffer perfused mouse heart from ischemia-reperfusion injury.,” Clin. Exp. Pharmacol. Physiol., vol. 33, no. 4, pp. 315–319, Apr. 2006.
[163] R. Bolli, K. Shinmura, X.-L. Tang, E. Kodani, Y.-T. Xuan, Y. Guo, and B. Dawn, “Discovery of a new function of cyclooxygenase (COX)-2: COX-2 is a

106
cardioprotective protein that alleviates ischemia/reperfusion injury and mediates the late phase of preconditioning.,” Cardiovasc. Res., vol. 55, no. 3, pp. 506–519, Aug. 2002.
[164] X. Ren, Y. Wang, and W. K. Jones, “TNF-α is required for late ischemic preconditioning but not for remote preconditioning of trauma,” Journal of Surgical Research, vol. 121, no. 1, pp. 120–129, Sep. 2004.
[165] T. Steensrud, J. Li, X. Dai, C. Manlhiot, R. K. Kharbanda, M. Tropak, and A. Redington, “Pretreatment with the nitric oxide donor SNAP or nerve transection blocks humoral preconditioning by remote limb ischemia or intra-arterial adenosine.,” Am. J. Physiol. Heart Circ. Physiol., vol. 299, no. 5, pp. H1598–603, Nov. 2010.
[166] Y. Guo, W. K. Jones, Y. T. Xuan, X. L. Tang, W. Bao, W. J. Wu, H. Han, V. E. Laubach, P. Ping, Z. Yang, Y. Qiu, and R. Bolli, “The late phase of ischemic preconditioning is abrogated by targeted disruption of the inducible NO synthase gene.,” Proc. Natl. Acad. Sci. U.S.A., vol. 96, no. 20, pp. 11507–11512, Sep. 1999.
[167] Y. T. Xuan, Y. Guo, Y. Zhu, H. Han, R. Langenbach, B. Dawn, and R. Bolli, “Mechanism of cyclooxygenase-2 upregulation in late preconditioning,” Journal of Molecular and Cellular Cardiology, vol. 35, no. 5, pp. 525–537, 2003.
[168] Y.-T. Xuan, Y. Guo, Y. Zhu, O.-L. Wang, G. Rokosh, and R. Bolli, “Endothelial nitric oxide synthase plays an obligatory role in the late phase of ischemic preconditioning by activating the protein kinase C epsilon p44/42 mitogen-activated protein kinase pSer-signal transducers and activators of transcription1/3 pathway.,” Circulation, vol. 116, no. 5, pp. 535–544, Jul. 2007.
[169] S. Loukogeorgakis, R. Williams, and A. Panagiotidou, “Transient Limb Ischemia Induces Remote Preconditioning and Remote Postconditioning in Humans by a KATP Channel–Dependent Mechanism,” Circulation, 2007.
[170] I. A. Rahman, J. G. Mascaro, R. P. Steeds, M. P. Frenneaux, P. Nightingale, P. Gosling, P. Townsend, J. N. Townend, D. Green, and R. S. Bonser, “Remote ischemic preconditioning in human coronary artery bypass surgery: from promise to disappointment?,” Circulation, vol. 122, no. 11, pp. S53–9, Sep. 2010.
[171] M. Shimizu, I. E. Konstantinov, R. K. Kharbanda, M. H. Cheung, and A. N. Redington, “Effects of intermittent lower limb ischaemia on coronary blood flow and coronary resistance in pigs,” Acta Physiol, vol. 190, no. 2, pp. 103–109, Jun. 2007.
[172] K. Enko, K. Nakamura, K. Yunoki, T. Miyoshi, S. Akagi, M. Yoshida, N. Toh, M. Sangawa, N. Nishii, S. Nagase, K. Kohno, H. Morita, K. F. Kusano, and H. Ito, “Intermittent arm ischemia induces vasodilatation of the contralateral upper limb.,” J Physiol Sci, vol. 61, no. 6, pp. 507–513, Nov. 2011.
[173] M. Wei, P. Xin, S. Li, J. Tao, Y. LI, J. Li, M. Liu, J. Li, W. Zhu, and A. N. Redington, “Repeated Remote Ischemic Postconditioning Protects Against Adverse Left Ventricular Remodeling and Improves Survival in a Rat Model of Myocardial InfarctionNovelty and Significance,” Circ. Res., vol. 108, no. 10, pp. 1220–1225, 2011.
[174] E. Jean St Michel, C. Manlhiot, J. Li, M. Tropak, M. M. MICHELSEN, M. R. SCHMIDT, B. W. MCCRINDLE, G. D. WELLS, and A. N. Redington, “Remote preconditioning improves maximal performance in highly trained athletes.,” Med Sci Sports Exerc, vol. 43, no. 7, pp. 1280–1286, Jul. 2011.
[175] M. E. Reichelt, L. Willems, B. A. Hack, J. N. Peart, and J. P. Headrick, “Cardiac and coronary function in the Langendorff-perfused mouse heart model.,” Exp. Physiol., vol. 94, no. 1, pp. 54–70, Jan. 2009.

107
[176] F. J. Sutherland and D. J. Hearse, “The isolated blood and perfusion fluid perfused heart.,” Pharmacol. Res., vol. 41, no. 6, pp. 613–627, Jun. 2000.
[177] S. Dhein, “The Langendorff Heart,” Practical methods in cardiovascular research, pp. 155–172, 2005.
[178] J. P. Headrick, J. Peart, B. Hack, A. Flood, and G. P. Matherne, “Functional properties and responses to ischaemia-reperfusion in Langendorff perfused mouse heart.,” Exp. Physiol., vol. 86, no. 6, pp. 703–716, Nov. 2001.
[179] F. Labruto, J. Vaage, G. Li, and G. Valen, “Intraperitoneal injection induces a delayed preconditioning-like effect in mice.,” Lab. Anim., vol. 39, no. 3, pp. 298–307, Jul. 2005.
[180] M. C. Fishbein, S. Meerbaum, J. Rit, U. Lando, K. Kanmatsuse, J. C. Mercier, E. Corday, and W. Ganz, “Early phase acute myocardial infarct size quantification: validation of the triphenyl tetrazolium chloride tissue enzyme staining technique.,” Am. Heart J., vol. 101, no. 5, pp. 593–600, May 1981.
[181] N. Varma, F. R. Eberli, and C. S. Apstein, “Increased diastolic chamber stiffness during demand ischemia: response to quick length change differentiates rigor-activated from calcium-activated tension.,” Circulation, vol. 101, no. 18, pp. 2185–2192, May 2000.
[182] N. Varma, F. R. Eberli, and C. S. Apstein, “Left ventricular diastolic dysfunction during demand ischemia: rigor underlies increased stiffness without calcium-mediated tension. Amelioration by glycolytic substrate.,” Journal of the American College of Cardiology, vol. 37, no. 8, pp. 2144–2153, Jun. 2001.
[183] K. Ishikawa, E. R. Chemaly, L. Tilemann, K. Fish, D. Ladage, J. Aguero, T. Vahl, C. Santos-Gallego, Y. Kawase, and R. J. Hajjar, “Assessing left ventricular systolic dysfunction after myocardial infarction: are ejection fraction and dP/dtmax complementary or redundant?,” Am. J. Physiol. Heart Circ. Physiol., vol. 302, no. 7, pp. H1423–8, Apr. 2012.
[184] L. A. Ahmed, H. A. Salem, A. S. Attia, and A. M. Agha, “Comparative study of the cardioprotective effects of local and remote preconditioning in ischemia/reperfusion injury.,” Life Sci., vol. 90, no. 7, pp. 249–256, Feb. 2012.
[185] R. da Silva, T. Grampp, T. Pasch, M. C. Schaub, and M. Zaugg, “Differential Activation of Mitogen-activated Protein Kinases in Ischemic and Anesthetic Preconditioning,” Anesthesiology, vol. 100, no. 1, p. 59, Jan. 2004.
[186] M. S. McKinney and J. Fee, “Cardiovascular effects of 50% nitrous oxide in older adult patients anaesthetized with isoflurane or halothane,” British Journal of Anaesthesia, vol. 80, no. 2, pp. 169–173, Feb. 1998.
[187] D. P. Jenkins, W. B. Pugsley, and D. M. Yellon, “Ischaemic preconditioning in a model of global ischaemia: infarct size limitation, but no reduction of stunning.,” Journal of Molecular and Cellular Cardiology, vol. 27, no. 8, pp. 1623–1632, Aug. 1995.
[188] L. Xi, M. L. Hess, and R. C. Kukreja, “Ischemic preconditioning in isolated perfused mouse heart: reduction in infarct size without improvement of post-ischemic ventricular function.,” Mol. Cell. Biochem., vol. 186, no. 1, pp. 69–77, Sep. 1998.
[189] D. M. Bers, “Cardiac excitation–contraction coupling,” Nature, vol. 415, no. 6868, pp. 198–205, 2002.
[190] D. H. MacLennan and E. G. Kranias, “Phospholamban: a crucial regulator of cardiac contractility.,” Nat. Rev. Mol. Cell Biol., vol. 4, no. 7, pp. 566–577, Jul. 2003.

108
[191] D. K. Rorie, G. M. Tyce, and J. C. Sill, “Increased norepinephrine release from dog pulmonary artery caused by nitrous oxide.,” Anesth. Analg., vol. 65, no. 6, pp. 560–564, Jun. 1986.
[192] T. J. Ebert and J. P. Kampine, “Nitrous oxide augments sympathetic outflow: direct evidence from human peroneal nerve recordings.,” Anesth. Analg., vol. 69, no. 4, pp. 444–449, Oct. 1989.
[193] Y. Zhang, L. P. Quock, E. Chung, Y. Ohgami, and R. M. Quock, “Involvement of a NO-cyclic GMP-PKG signaling pathway in nitrous oxide-induced antinociception in mice.,” Eur. J. Pharmacol., vol. 654, no. 3, pp. 249–253, Mar. 2011.
[194] A. Gorbe, Z. Giricz, A. Szunyog, T. Csont, D. S. Burley, G. F. Baxter, and P. Ferdinandy, “Role of cGMP-PKG signaling in the protection of neonatal rat cardiac myocytes subjected to simulated ischemia/reoxygenation.,” Basic Res. Cardiol., vol. 105, no. 5, pp. 643–650, Sep. 2010.
[195] D. J. Hausenloy, M. M. Mocanu, and D. M. Yellon, “Cross-talk between the survival kinases during early reperfusion: its contribution to ischemic preconditioning.,” Cardiovasc. Res., vol. 63, no. 2, pp. 305–312, Aug. 2004.
[196] R. S. Crawford, F. F. Hashmi, J. E. Jones, H. Albadawi, M. McCormack, K. Eberlin, F. Entabi, M. D. Atkins, M. F. Conrad, W. G. Austen, and M. T. Watkins, “A novel model of acute murine hindlimb ischemia.,” Am. J. Physiol. Heart Circ. Physiol., vol. 292, no. 2, pp. H830–7, Feb. 2007.
[197] J. Hoff, “Methods of Blood Collection in the Mouse,” Lab Animal, vol. 29, no. 10, pp. 47–53, 2000.
[198] N. Tanaka, N. Dalton, L. Mao, H. A. Rockman, K. L. Peterson, K. R. Gottshall, J. J. Hunter, K. R. Chien, and J. Ross, “Transthoracic Echocardiography in Models of Cardiac Disease in the Mouse,” vol. 94, no. 5, pp. 1109–1117, Sep. 1996.
[199] C. R. Frasier, R. L. Moore, and D. A. Brown, “Exercise-induced cardiac preconditioning: how exercise protects your achy-breaky heart.,” J. Appl. Physiol., vol. 111, no. 3, pp. 905–915, Sep. 2011.
[200] S. Golbidi and I. Laher, “Molecular mechanisms in exercise-induced cardioprotection.,” Cardiol Res Pract, vol. 2011, p. 972807, 2011.
109
Chapter 9 Appendices
Appendix 1. Power-lab Acquisition Software: Cardiac Output Chart

110
Appendix 2. Protein extraction-lysis-buffer Final Concentrations: Tris. HCL – 20 mM (pH 7.5)
NaCL – 150 mM
EDTA – 1 mM
EGTA – 1mM
NP-40 – 1%
Na4O7P2 – 2.5 mM
β - Glycerolphosphate – 1 mM
Na3VO4 – 1mM
ddH2O
1 Tablet of Proteinase Inhibitor Cocktail for every 10 mL (Roche Inc.)