a paradox of transcriptional and functional innate ... · a paradox of transcriptional and...
TRANSCRIPT

A paradox of transcriptional and functional innateinterferon responses of human intestinalenteroids to enteric virus infectionKapil Saxenaa, Lukas M. Simonb,1, Xi-Lei Zenga, Sarah E. Blutta, Sue E. Crawforda, Narayan P. Sastria,Umesh C. Karandikara, Nadim J. Ajamia, Nicholas C. Zachosc, Olga Kovbasnjukc, Mark Donowitzc, Margaret E. Connera,Chad A. Shawb, and Mary K. Estesa,2
aDepartment of Molecular Virology and Microbiology, Baylor College of Medicine, Houston, TX 77030; bDepartment of Human and Molecular Genetics,Baylor College of Medicine, Houston, TX 77030; and cDepartment of Medicine, Gastroenterology Division, Johns Hopkins University School of Medicine,Baltimore, MD 21205
Contributed by Mary K. Estes, December 10, 2016 (sent for review September 15, 2016; reviewed by Michelle M. Arnold and Susana Lopez)
The intestinal epithelium can limit enteric pathogens by producingantiviral cytokines, such as IFNs. Type I IFN (IFN-α/β) and type III IFN(IFN-λ) function at the epithelial level, and their respective efficaciesdepend on the specific pathogen and site of infection. However, theroles of type I and type III IFN in restricting human enteric viruses arepoorly characterized as a result of the difficulties in cultivating theseviruses in vitro and directly obtaining control and infected small in-testinal human tissue. We infected nontransformed human intesti-nal enteroid cultures frommultiple individuals with human rotavirus(HRV) and assessed the host epithelial response by using RNA-sequencing and functional assays. The dominant transcriptionalpathway induced by HRV infection is a type III IFN-regulated re-sponse. Early after HRV infection, low levels of type III IFN proteinactivate IFN-stimulated genes. However, this endogenous responsedoes not restrict HRV replication because replication-competent HRVantagonizes the type III IFN response at pre- and posttranscriptionallevels. In contrast, exogenous IFN treatment restricts HRV replica-tion, with type I IFN being more potent than type III IFN, suggestingthat extraepithelial sources of type I IFN may be the critical IFN forlimiting enteric virus replication in the human intestine.
enteric virus | interferon | human enteroids | human rotavirus
The human small intestinal epithelium is the primary site ofinfection and replication for many gastrointestinal pathogens.
However, fundamental knowledge about intestinal epithelial cell–pathogen interactions in humans is limited as a result of the im-practicality of studying these cells in vivo. Modeling these interac-tions in vitro is difficult because many human enteric pathogens failto replicate or replicate poorly in cancer cell lines derived primarilyfrom the human colon (1–4). Human intestinal enteroids (HIEs)represent a new in vitro model of the human small intestinal epi-thelium; this model was developed following advances in stem cellbiology, and it recapitulates many of the biological and physiolog-ical properties of the human small intestine in vivo (5, 6). Unlikeother in vitro models, HIEs are easily established from non-transformed small intestinal tissue, can be maintained on along-term basis, and contain a stem cell niche and the diversityof intestinal epithelial cell types (enterocytes, goblet, enter-oendocrine, and Paneth cells) present in vivo (6, 7). HIEs allowfor comparisons of intestinal host responses across geneticallydiverse individuals (8) and reproduce many of the in vivopathophysiological properties of infection by a human entericviral pathogen, human rotavirus (HRV) (9).HRVs are the major etiology of severe diarrhea in children
under the age of 5 y worldwide, resulting in an estimated 215,000deaths annually (10). HRVs replicate to high titers in humans butreplicate poorly or not at all in small animal models (11, 12). Thishost restriction of HRV in animal models is based on propertiesof host and virus and is not unique to HRVs; it is exhibited byother human enteric viruses such as human noroviruses and
adenoviruses (12–14). In addition, HRV generally replicates tolower titers than animal rotaviruses in transformed cell lines (2).Therefore, most knowledge of rotavirus–host interactions comesfrom studies that analyzed animal rotavirus replication in animalmodels or in transformed cell lines (15–18). Induction of theadaptive immune system and secretion of IgA are critical factorsin protecting the host against rotavirus infection based on in vivomodels of animal rotavirus infection (19, 20). However, in a naïvehost, the adaptive immune system is not fully activated until sev-eral days after primary infection (21, 22). Epithelial cells mount anearly innate immune response to infection, and this immediateantiviral response is often crucial in limiting viral replication andensuring tissue viability until the adaptive immune response isactivated (22–25).IFNs are potent antiviral cytokines that function in an autocrine
and paracrine fashion following the detection of specific patho-gen-associated molecular patterns (PAMPs) (26). Currently, threeclasses of IFNs have been identified: type I, type II, and type IIIIFN (27, 28). Type II IFN (IFN-γ) is mainly produced by immunecells, whereas type I (IFN-α/β) and type III IFN (IFN-λ) are
Significance
Understanding host–enteric virus interactions has been limited bythe inability to culture nontransformed small intestinal epithelialcells and to infect animal models with human viruses. We reportepithelial responses in human small intestinal enteroid culturesfrom different individuals following infection with human rota-virus (HRV), a model enteric pathogen. RNA-sequencing andfunctional assays revealed type III IFN as the dominant tran-scriptional response that activates interferon-stimulated genes,but antagonism of the IFN response negates restriction of HRVreplication. Exogenously added IFNs reduce HRV replication,with type I IFN being most effective. This highlights a paradoxbetween the strong type III transcriptional response and theweaker functional role of type III IFN in human enteric viralrestriction in human small intestinal cultures.
Author contributions: K.S., L.M.S., S.E.B., S.E.C., N.P.S., U.C.K., N.J.A., N.C.Z., O.K., M.D., M.E.C.,C.A.S., and M.K.E. designed research; K.S., X.-L.Z., S.E.B., S.E.C., N.P.S., and U.C.K. performedresearch; L.M.S. and C.A.S. contributed new reagents/analytic tools; K.S., L.M.S., N.J.A., M.E.C.,C.A.S., and M.K.E. analyzed data; and K.S., L.M.S., M.E.C., and M.K.E. wrote the paper.
Reviewers: M.M.A., Louisiana State University Health Sciences Center Shreveport; and S.L.,Instituto de Biotecnologia/UNAM.
The authors declare no conflict of interest.
Data deposition: The data reported in this paper have been deposited in the Gene Ex-pression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE90796).1Present address: Institute of Computational Biology, Helmholtz Zentrum Münich, 85764Neuherberg, Germany.
2To whom correspondence should be addressed. Email: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1615422114/-/DCSupplemental.
E570–E579 | PNAS | Published online January 9, 2017 www.pnas.org/cgi/doi/10.1073/pnas.1615422114

produced by immune and epithelial cells and thus are relevant tohost–pathogen interactions at epithelial surfaces (27). The type IIFN receptor is expressed on most cell types, whereas the type IIIIFN receptor is primarily restricted to epithelial cells (29). Type Iand III IFNs are generally induced by similar stimuli (30). Stim-ulation of an IFN receptor leads to the induction of a variety ofIFN-stimulated genes (ISGs), and ISG-encoded proteins are ef-fector molecules of the antiviral state (31). One unansweredquestion is whether unique type I and III IFN expression profilesare produced by the human intestinal epithelium in response todifferent stimuli or viral pathogens.Based on studies in mice, the innate response to rotavirus in-
fection is complex and virus strain-specific; homologous (murine)rotavirus replication is insensitive to IFNs whereas type I and IIIIFNs restrict heterologous (nonmurine) rhesus rotavirus (RRV)replication (17, 32, 33). Host restriction is hypothesized to be thebasis for attenuation of an HRV vaccine widely used today,RotaTeq, which is derived from a heterologous bovine strain (34).Although a wealth of knowledge has been gained from mousestudies with nonhuman enteric viruses, these models do not ac-count for evolutionary differences in host and pathogen. Subtle butpossibly important differences exist between human and mousetype I and III IFN systems and between human and mouse rota-viruses (18, 35–38), necessitating functional studies of interactionsbetween human host cells and human enteric pathogens.HIEs represent a new model to study epithelial cell innate
immune responses to human enteric pathogens. In this study, weused HIEs to investigate the host response to live and inactivatedHRVs, the dsRNA structural analog polyinosinic:polycytidylicacid [poly(I:C)], and exogenous IFNs. Transcriptional responsesto HRV infection were examined in cultures established fromdifferent individuals. We demonstrate that viral infection inducesa conserved type III IFN-regulated ISG transcriptional responsein HIEs from different individuals. However, this type III IFNtranscriptional response is not translated into a robust proteinresponse and does not restrict viral growth. In contrast, exoge-nously added IFNs restrict HRV replication, with type I IFN beingmore effective than type III IFN. HIEs are a valuable model todefine human-specific epithelial cell responses to stimuli such ashuman enteric pathogens and have revealed a difference in ourunderstanding of the role of type I and III IFN as innate mediatorsin human intestinal epithelial cells.
ResultsHIEs Demonstrate Minimal Intraculture Variability by MicroarrayAnalysis, and the Epithelial Transcriptional Response to HRV InfectionIs Reproducible Across HIEs from Different People. HIEs are novel,physiologically active, nontransformed cultures that are increasinglyused to study human pathogen–host interactions (5, 8, 9, 39, 40).We first evaluated the variability in the transcriptional response toHRV infection in HIEs by analyzing transcriptomes from onejejunal HIE culture by microarray analysis. Independent HIE cul-tures (individual j11) were mock-inoculated or inoculated withHRV (strain Ito) at a high multiplicity of infection (MOI). Threetechnical replicates for each condition were analyzed at 6 and 10 hpost infection (hpi), as HRV-infected cells lyse by 14 hpi (9). Thepercentage of infected cells was 51.4 ± 14.7% (mean ± SD oftriplicate cultures) as assessed by flow cytometry, demonstratingthat the HRV-infected cultures contain infected cells and un-infected bystander cells. Principal component analysis showed thatthe first principal component (time point in hpi) explained 42% ofthe variance in the global gene expression profiles, indicating thattime point is an important covariate. The second principal com-ponent explained 22% of the variance and discriminated the ex-pression profiles based on infection status (Fig. 1A). Although timeand infection status influenced HIE transcriptional profiles, theintraculture transcriptional profiles for a specific combination oftime and infection status clustered together.
We next evaluated interculture variability in the HIE responseto HRV infection by microarray analysis. Jejunal HIEs from threeindividuals (j2, j3, and j11) were mock- or Ito-HRV–infected at ahigh MOI. The percentages of HRV-infected cells were 52.9%(j2), 43.9% (j3), and 47.7% (j11). Hierarchical clustering of globalgene expression profiles accounting for time point revealed thatHIEs first clustered by each individual and then by infection status(Fig. 1B), indicating significant baseline variability between HIEsderived from different individuals. To assess if the response toHRV infection was conserved between HIEs from the three dif-ferent people, we calculated gene expression fold changes withHRV infection across more than 20,000 transcripts from eachindividual. We observed a significant correlation between HRV-induced transcriptional changes in the three individuals’HIEs; thelowest correlations of 0.18 at 6 hpi (between j2 and j11) and 0.31at 10 hpi (between j3 and j11) correspond to P values <2.2 × 10−16
(Fig. 1C). Notably, 18 genes at 6 hpi and 28 genes at 10 hpi rankedin the top 100 up-regulated genes in each of the three HIEs’transcriptional profiles (P < 10−5 for both time points, permuta-tion-based approach; Materials and Methods and Fig. 1D). Furtheranalysis of these genes revealed that 16 of 18 and 15 of 28 wereISGs at 6 and 10 hpi, respectively (Fig. S1A). Thus, despite abaseline variability at the transcriptional level between HIEs fromdifferent individuals, all three mounted a conserved ISG tran-scriptional response to HRV infection.
RNA-Sequencing Analysis of HIEs Reveals IFN Signaling Is theDominant Pathway Induced by HRV Infection. We next performed anRNA-sequencing (RNA-seq) experiment on mock- and HRV-infected HIE cultures. RNA-seq more accurately measures tran-scriptional activity than microarrays, particularly of low-expressinggenes (41). Because minimal transcriptional variability was observedbetween samples from the same individual (Fig. 1A), one mock- andone HRV-infected culture of HIEs from two individuals (j2 and j11)
Fig. 1. Transcriptional response to HRV infection in HIEs assessed by usingwhole-genome microarray chips. (A) HIE cultures from one individual (j11)were mock- or HRV-inoculated (MOI of 20). At 6 and 10 hpi, gene expressionprofiling was performed on 12 independent samples (three technical repli-cates for each of the four conditions). The x and y axes depict the first andsecond principal components, which differentiate time point (explanatoryvariable for 42% of variance) and infection status (explanatory variable for22% of variance), respectively. Mock- (purple) and HRV-inoculated (orange)samples are plotted at 6 hpi (filled squares) and 10 hpi (open circles). (B) HIEsfrom three individuals (j2, j3, and j11) were mock- or HRV-inoculated at anMOI of 20. At 6 and 10 hpi, gene expression profiling was performed onHIEs from each patient. Clustering is based on Spearman rank correlation.Each individual’s (green) HIE samples are shown as mock- (purple) or HRV-inoculated (orange). (C) Spearman rank correlation (listed within eachsquare) of the infection-induced gene expression fold changes of 20,948transcripts was calculated for all sample pairs. Intensity of red indicatescorrelation strength. (D) Venn diagrams depict the overlap among each in-dividual’s top 100 up-regulated genes at 6 and 10 hpi.
Saxena et al. PNAS | Published online January 9, 2017 | E571
MICRO
BIOLO
GY
PNASPL
US
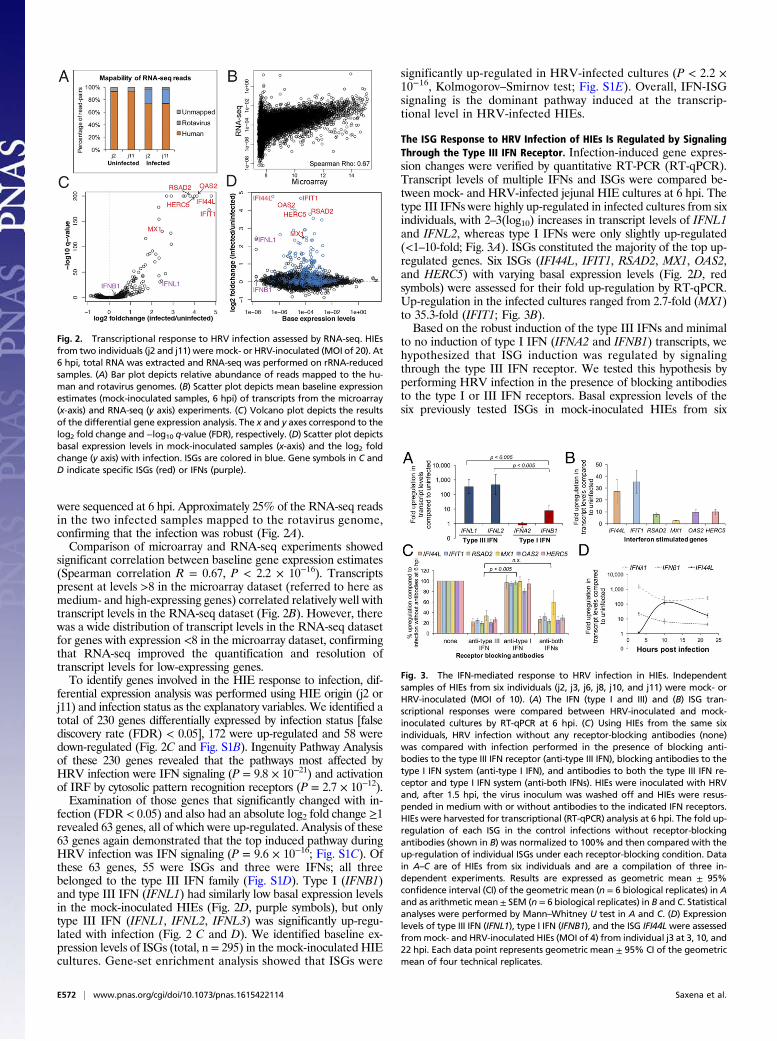
were sequenced at 6 hpi. Approximately 25% of the RNA-seq readsin the two infected samples mapped to the rotavirus genome,confirming that the infection was robust (Fig. 2A).Comparison of microarray and RNA-seq experiments showed
significant correlation between baseline gene expression estimates(Spearman correlation R = 0.67, P < 2.2 × 10−16). Transcriptspresent at levels >8 in the microarray dataset (referred to here asmedium- and high-expressing genes) correlated relatively well withtranscript levels in the RNA-seq dataset (Fig. 2B). However, therewas a wide distribution of transcript levels in the RNA-seq datasetfor genes with expression <8 in the microarray dataset, confirmingthat RNA-seq improved the quantification and resolution oftranscript levels for low-expressing genes.To identify genes involved in the HIE response to infection, dif-
ferential expression analysis was performed using HIE origin (j2 orj11) and infection status as the explanatory variables. We identified atotal of 230 genes differentially expressed by infection status [falsediscovery rate (FDR) < 0.05], 172 were up-regulated and 58 weredown-regulated (Fig. 2C and Fig. S1B). Ingenuity Pathway Analysisof these 230 genes revealed that the pathways most affected byHRV infection were IFN signaling (P = 9.8 × 10−21) and activationof IRF by cytosolic pattern recognition receptors (P = 2.7 × 10−12).Examination of those genes that significantly changed with in-
fection (FDR < 0.05) and also had an absolute log2 fold change ≥1revealed 63 genes, all of which were up-regulated. Analysis of these63 genes again demonstrated that the top induced pathway duringHRV infection was IFN signaling (P = 9.6 × 10−16; Fig. S1C). Ofthese 63 genes, 55 were ISGs and three were IFNs; all threebelonged to the type III IFN family (Fig. S1D). Type I (IFNB1)and type III IFN (IFNL1) had similarly low basal expression levelsin the mock-inoculated HIEs (Fig. 2D, purple symbols), but onlytype III IFN (IFNL1, IFNL2, IFNL3) was significantly up-regu-lated with infection (Fig. 2 C and D). We identified baseline ex-pression levels of ISGs (total, n = 295) in the mock-inoculated HIEcultures. Gene-set enrichment analysis showed that ISGs were
significantly up-regulated in HRV-infected cultures (P < 2.2 ×10−16, Kolmogorov–Smirnov test; Fig. S1E). Overall, IFN-ISGsignaling is the dominant pathway induced at the transcrip-tional level in HRV-infected HIEs.
The ISG Response to HRV Infection of HIEs Is Regulated by SignalingThrough the Type III IFN Receptor. Infection-induced gene expres-sion changes were verified by quantitative RT-PCR (RT-qPCR).Transcript levels of multiple IFNs and ISGs were compared be-tween mock- and HRV-infected jejunal HIE cultures at 6 hpi. Thetype III IFNs were highly up-regulated in infected cultures from sixindividuals, with 2–3(log10) increases in transcript levels of IFNL1and IFNL2, whereas type I IFNs were only slightly up-regulated(<1–10-fold; Fig. 3A). ISGs constituted the majority of the top up-regulated genes. Six ISGs (IFI44L, IFIT1, RSAD2, MX1, OAS2,and HERC5) with varying basal expression levels (Fig. 2D, redsymbols) were assessed for their fold up-regulation by RT-qPCR.Up-regulation in the infected cultures ranged from 2.7-fold (MX1)to 35.3-fold (IFIT1; Fig. 3B).Based on the robust induction of the type III IFNs and minimal
to no induction of type I IFN (IFNA2 and IFNB1) transcripts, wehypothesized that ISG induction was regulated by signalingthrough the type III IFN receptor. We tested this hypothesis byperforming HRV infection in the presence of blocking antibodiesto the type I or III IFN receptors. Basal expression levels of thesix previously tested ISGs in mock-inoculated HIEs from six
Fig. 2. Transcriptional response to HRV infection assessed by RNA-seq. HIEsfrom two individuals (j2 and j11) were mock- or HRV-inoculated (MOI of 20). At6 hpi, total RNA was extracted and RNA-seq was performed on rRNA-reducedsamples. (A) Bar plot depicts relative abundance of reads mapped to the hu-man and rotavirus genomes. (B) Scatter plot depicts mean baseline expressionestimates (mock-inoculated samples, 6 hpi) of transcripts from the microarray(x-axis) and RNA-seq (y axis) experiments. (C) Volcano plot depicts the resultsof the differential gene expression analysis. The x and y axes correspond to thelog2 fold change and −log10 q-value (FDR), respectively. (D) Scatter plot depictsbasal expression levels in mock-inoculated samples (x-axis) and the log2 foldchange (y axis) with infection. ISGs are colored in blue. Gene symbols in C andD indicate specific ISGs (red) or IFNs (purple).
Fig. 3. The IFN-mediated response to HRV infection in HIEs. Independentsamples of HIEs from six individuals (j2, j3, j6, j8, j10, and j11) were mock- orHRV-inoculated (MOI of 10). (A) The IFN (type I and III) and (B) ISG tran-scriptional responses were compared between HRV-inoculated and mock-inoculated cultures by RT-qPCR at 6 hpi. (C) Using HIEs from the same sixindividuals, HRV infection without any receptor-blocking antibodies (none)was compared with infection performed in the presence of blocking anti-bodies to the type III IFN receptor (anti-type III IFN), blocking antibodies to thetype I IFN system (anti-type I IFN), and antibodies to both the type III IFN re-ceptor and type I IFN system (anti-both IFNs). HIEs were inoculated with HRVand, after 1.5 hpi, the virus inoculum was washed off and HIEs were resus-pended in medium with or without antibodies to the indicated IFN receptors.HIEs were harvested for transcriptional (RT-qPCR) analysis at 6 hpi. The fold up-regulation of each ISG in the control infections without receptor-blockingantibodies (shown in B) was normalized to 100% and then compared with theup-regulation of individual ISGs under each receptor-blocking condition. Datain A–C are of HIEs from six individuals and are a compilation of three in-dependent experiments. Results are expressed as geometric mean ± 95%confidence interval (CI) of the geometric mean (n = 6 biological replicates) in Aand as arithmetic mean± SEM (n = 6 biological replicates) in B and C. Statisticalanalyses were performed by Mann–Whitney U test in A and C. (D) Expressionlevels of type III IFN (IFNL1), type I IFN (IFNB1), and the ISG IFI44Lwere assessedfrommock- and HRV-inoculated HIEs (MOI of 4) from individual j3 at 3, 10, and22 hpi. Each data point represents geometric mean ± 95% CI of the geometricmean of four technical replicates.
E572 | www.pnas.org/cgi/doi/10.1073/pnas.1615422114 Saxena et al.

individuals were not significantly different with or without block-ing antibody treatment (P > 0.05, Wilcoxon signed-rank test, n = 6biological replicates), suggesting minimal to no baseline IFNregulation of these six ISGs in mock-inoculated HIEs. In addition,type III IFN (IFNL1 and IFNL2) transcriptional levels were notsignificantly different between HRV infections performed with orwithout type III IFN receptor-blocking antibody treatment (P >0.05, Wilcoxon signed-rank test, n = 6 biological replicates), in-dicating that this antibody did not induce nor suppress IFNLtranscriptional activity. However, HRV infection performed in thepresence of blocking antibodies to the type III IFN receptor sig-nificantly reduced the induction of the six ISGs analyzed, rangingfrom a 66% reduction in MX1 transcript levels to an 81% re-duction in RSAD2 transcript levels compared with HRV infectionwithout receptor-blocking antibodies (Fig. 3C). Maximum (100%)up-regulation of each ISG corresponds to the fold inductionsshown in Fig. 3B. HRV infection in the presence of a controlantibody (isotype matched to the type III IFN receptor-blockingantibody) did not decrease ISG levels compared with infectionwithout antibodies. Infection performed in the presence ofblocking antibodies to type I and type III IFN receptors failed tofurther suppress ISG levels compared with type III IFN receptorblockade alone (Fig. 3C). These data suggest that signalingthrough the type III IFN receptor largely regulates ISG in-duction in response to HRV infection in HIEs.Analysis of IFN-ISG transcriptional kinetics at three additional
time points showed that IFN transcripts were present at 3 hpi anddecreased by ∼1 log10 at 10 hpi, at which point their levels pla-teaued (Fig. 3D). In contrast, up-regulation of ISG transcripts(assessed by using IFI44L as a marker) was not observed at 3 hpibut was present by 10 hpi, demonstrating that the type III IFNresponse preceded the ISG response. Together, these data suggestthat HRV infection of HIEs results in robust up-regulation of typeIII IFN, which in turn activates the type III IFN receptor and leadsto the induction of ISGs.
HRV Replication in HIEs Is Not Restricted by Endogenously ProducedIFN.HRV infection of epithelial cells in the HIE model induced atype III IFN-regulated ISG response, and IFN receptor signalingappeared to start between 3 and 6 hpi, as ISGs were up-regulatedat 6 hpi but not at 3 hpi (Fig. 3). To assess whether the endogenoushost IFN response can restrict rotavirus replication, HRV repli-cation was assessed in the presence of IFN-receptor blocking an-tibodies. We hypothesized that inhibiting the host IFN response byblocking IFN signaling would increase virus titer yields. However,blockade of neither the type I nor type III IFN receptors duringmultiple rounds of replication significantly increased viral yields by60 hpi (P = 0.75, Kruskal–Wallis test; Fig. 4). In addition, neitherreplication of Ito-HRV nor RRV was enhanced by simultaneousblockade of type I and type III IFN receptors for 60 h (P > 0.05,Mann–Whitney U test). These data suggest that, although homol-ogous HRV and heterologous RRV induce a type III IFN re-sponse at the epithelial level (Fig. S2), this host cell response doesnot restrict viral growth because rotavirus grows to similar titersregardless of whether the type III IFN response is neutralized.
Different Strains of Rotavirus and a dsRNA Analog PreferentiallyInduce IFNL over IFNB in HIEs. The preceding experiments wereprimarily performed with the human strain Ito, genotype G3P[8].Rotavirus infection of HIEs recapitulates the host-range restrictionobserved in vivo; human strains infect and replicate at higher levelsthan animal strains such as simian RRV in HIEs (9). To assesswhether the type III IFN induction in response to Ito-HRV in-fection was strain-specific, HIEs were infected at the same MOIwith an additional human strain (Wa, G1P[8]) and a simian strain(RRV; G3P[3]). Irrespective of virus strain, the pattern of IFNinduction was similar; all three strains led to increased fold in-duction of IFNL1 compared with IFNB1 (Fig. S2). However,
IFNL1 levels were significantly higher in HIEs infected with HRVsthan with RRV, suggesting that type III IFN transcriptional in-tensity is strain-specific depending on the host susceptibility in HIEs.Next, we evaluated if the preferential up-regulation of IFNL1
instead of IFNB1 with rotavirus infection was intestinal segment-or rotavirus-specific. Responses of 12 different individuals’ HIEsshowed that IFNL1 induction in response to HRV infection wasnot significantly different across the three sections of the smallintestine (P = 0.28, Kruskal–Wallis test), but IFNL1 fold up-regulation was ∼10–100-fold more than IFNB1 in each intestinalsection (Fig. 5A) despite similar basal expression levels of thetwo IFNs in uninfected HIEs (Fig. 2D, purple symbols). We alsotested the response to the dsRNA analog poly(I:C), which causesrobust induction of the type I IFN system in transformed andnontransformed cell lines (42, 43). Unlike rotavirus or otherviruses (24, 44), poly(I:C) does not antagonize IFN transcrip-tional responses. Similar to the response to rotavirus, all 12 HIEstreated with poly(I:C) exhibited significantly increased IFNL1transcript fold up-regulation (range, 419–225,332 fold) com-pared with IFNB1 (range, 1–142 fold; Fig. 5B). These findingsindicate that HIEs preferentially respond to rotavirus and dsRNAstimuli with type III IFN rather than type I IFN.The specific rotavirus PAMP(s) recognized by cellular pathogen
recognition receptors (PRRs) is unclear. Rotavirus replication wasnot thought to release free dsRNA genome into the cell (45).However, dsRNA has recently been detected independent ofviroplasms in animal rotavirus-infected MA104 cells (46). dsRNAis a potent PAMP for many other viruses (47), and it is thereforepertinent that the dsRNA analog poly(I:C) and HRV inducedsimilar IFN transcriptional profiles in HIEs. Staining HIEs fordsRNA and the viroplasm marker nonstructural protein (NSP2)revealed their colocalization in the cytoplasm of infected cells aswell as dsRNA independent of viroplasms (Fig. 5C). These datademonstrate the presence of viroplasm-independent dsRNA inHRV-infected cells that could be the PAMP recognized bycellular PRRs.
A Majority of IFNL Transcript in HRV-Infected Cells Is Not Translatedinto Detectable Type III IFN Protein. Blockade of the type III IFNreceptor reduced ISG up-regulation (Fig. 3C), suggesting that typeIII IFN (IFN-λ) protein was secreted during infection and engagedits receptor. Therefore, either (i) HRV replicated despite highquantities of type III IFN protein or (ii) low levels of type III IFNprotein produced during infection were sufficient to induce ISGsbut insufficient to restrict viral growth. IFN protein production
Fig. 4. HRV growth in the presence of IFN receptor-blocking antibodies.HIEs (j2) were HRV-inoculated (MOI of 0.1). At 1.5 hpi, viral inoculum waswashed off and the HIEs were resuspended in medium containing no IFNreceptor-blocking antibodies (control) or antibodies to the type III IFNreceptor or type I IFN system. Infectious virus titers [in focus forming units(FFU)] were assessed at 1.5 hpi and 60 hpi, and the fold increase (60 hpi/1.5 hpi)is shown. The raw increase in viral titer (60 hpi − 1.5 hpi) was 256,015 FFU(control), 279,695 FFU (anti-type III IFN), and 264,015 FFU (anti-type I IFN).Data from one representative experiment is presented as arithmetic mean ±SD of four technical replicates of HIEs from individual j2, and are repre-sentative of data from additional experiments performed with HIEs fromfive additional individuals (j3, j6, j8, j10, and j11) with four technical repli-cates each.
Saxena et al. PNAS | Published online January 9, 2017 | E573
MICRO
BIOLO
GY
PNASPL
US

was evaluated in supernatants from mock-inoculated and HRV-infected HIE cultures by ELISA. Despite robust up-regulation ofIFNL1 transcripts at 6 hpi (Fig. S3A), IFN-λ1 and IFN-λ2/3 pro-teins were below the limit of detection in the supernatant frominfected HIEs at multiple time points (6–48 hpi) and under mul-tiple different conditions, including from a homogenate of super-natant and cell lysate (Table S1). These data suggested that HRVinfection induced type III IFN protein at levels below the limit ofdetection of standard ELISAs but sufficient to induce ISGs. Thiswas confirmed by observing that exogenous treatment of HIEswith concentrations of IFN-λ1 protein below the limit of detectionof standard ELISAs (<2 U/mL; Materials and Methods) inducedISGs (Fig. 6A) to similar levels as HRV (Fig. 3B). Rotavirus in-fection of transformed cells also induces the neutrophil chemo-attractant IL-8 (48). High but similar levels of IL-8 were detectedin HIE supernatants from mock- and HRV-infected samplespreviously tested for IFN (P > 0.05 at each time point, Mann–Whitney U test; Fig. S3B), showing that not all host proteins wereas affected by HRV infection as type III IFN.Rotavirus antagonizes IFN production pretranscriptionally in
a variety of cell lines (HT29, Caco-2, and 293T) through deg-radation of factors involved in IFN transcription (35, 49–51). Weobserved robust type III IFN transcription with HRV and poly(I:C) treatment (∼100–1,000-fold increases; Fig. 5), but proteinwas detected only from poly(I:C)-treated HIEs at 8 hpi (Fig. 6B).We next evaluated the possibility of rotavirus-mediated inhibitionof IFNL1 translation in HIEs treated with HRV and/or poly(I:C).IFN-λ1 protein levels were below the limit of detection frommock- and HRV-infected HIEs, but were 224 pg/mL from poly(I:C)-treated HIEs at 20 hpi (Fig. 6C). The IFN-λ1 protein levelsignificantly decreased to 41 pg/mL when poly(I:C)-treated HIEswere also infected with HRV. These data (Fig. 6 B and C) indicate
a rotavirus-mediated posttranscriptional partial suppression ofhost IFNL1 mRNA translation. Of note, IFN-β1 protein fromHIE supernatant was below the limit of detection by ELISA(sensitivity, 2 pg/mL) for each of the four conditions described inFig. 6C at 20 hpi.Rotavirus antagonism of the host response is primarily medi-
ated through the nonstructural proteins NSP1 and NSP3, whichare present only in cells infected with replication-competent virus(24). We assessed if viral antagonism of the IFN response in HIEswas a replication-dependent process by treating HIEs with puri-fied, inactivated HRV particles that contain the dsRNA genome,structural proteins, and can enter cells but do not transcribe viralmRNAs or express nonstructural proteins (16, 52). InactivatedHRV induced ∼10-fold higher transcript levels of IFNL1 andIFNB1 compared with replication-competent virus early after in-fection (Fig. 7A), demonstrating that the robust IFNL1 tran-scriptional response induced by replication-competent HRV ispartially antagonized at a pretranscriptional state by infectiousvirus. Furthermore, high levels of IFN-λ1 protein were detectedfrom HIEs treated with inactivated (i.e., replication-incompetent)HRV (Fig. 7B), but only if infection was performed in the pres-ence of antibody to the type III IFN receptor, suggesting thatsecreted IFN-λ1 protein rapidly bound its receptor and did notremain free in the supernatant for an extended duration. A sim-ilar technique of using receptor-blocking antibodies to enhancecytokine detection has been described for IL-4 (53). Thus, IFNtranscriptional induction in HRV-infected HIEs is replication-independent, but pre- and posttranscriptional antagonism ofthe IFN response in HRV-infected HIEs is replication-dependent.
Exogenous IFN Treatment Restricts Rotavirus Growth in HIEs. En-dogenously produced IFN did not restrict HRV growth in HIEs(Fig. 4). However, endogenous type III IFN restricts animal ro-tavirus replication in mice (54, 55). The mouse epithelium wasthought to produce the type III IFN in vivo, although immune cellscan also produce type III IFN (54, 56, 57). In contrast, murine typeI IFN predominantly originates from lamina propria immune cellsduring rotavirus infection (58). We simulated the presence ofsurrounding lamina propria immune cells by adding IFN beforeinfection to compare the potential of exogenous type I and type IIIIFN to restrict rotavirus growth in HIEs.Anti-rotavirus efficacy of type I IFN vs. type III IFN was com-
pared by pretreating HIEs for 24 h with escalating doses of IFN-λ1or IFN-β1 ranging from 100 to 10,000 U/mL. IFN was sub-sequently washed off, and the HIEs were infected with HRV at alow MOI to allow for multiple rounds of replication. IFN-λ1 andIFN-β1 inhibited HRV replication, but inhibition by IFN-λ1 wasweaker than IFN-β1 at all concentrations (Fig. 8A). Maximal in-hibition of HRV replication by IFN-λ1 (79%) was achieved at1,000 U/mL of IFN-λ1. In contrast, each 10-fold increase in IFN-β1 concentration reduced viral replication by ∼10-fold, with rep-lication 90% inhibited with 100 U/mL of IFN-β1 and completelyinhibited with 10,000 U/mL IFN-β1 (Fig. 8A).Our studies focused on IFN-λ1 and IFN-β1 because we did not
find differences in transcriptional up-regulation between IFNL1-3and because IFNB1 was the only type I IFN that was even slightlyup-regulated (Fig. 3A). However, heterogeneity in the antiviralproperties of different type I and type III IFN variants has beenreported with respect to other viruses (59, 60). To compare effi-cacies of alternative type I and type III IFNs, HIEs were pretreatedwith IFN-λ3 (type III IFN) or IFN-α2 (type I IFN) and comparedwith their analogs, IFN-λ1 and IFN-β1, respectively. There wasno significant difference between the two type III IFNs; bothreduced viral replication by ∼80% (Fig. 8B). However, pre-treatment with IFN-β1 was significantly more protective thanIFN-α2, indicating heterogeneity in the anti-rotavirus efficacyof the different type I IFNs.
Fig. 5. IFN transcriptional response by HIEs across the small intestineand cellular localization of dsRNA. Independent samples of four duo-denal HIEs (d1, d4, d6, and d104), four jejunal HIEs (j2, j3, j6, and j11),and four ileal HIEs (i2, i103, i16, and i104) were inoculated with (A) HRV(MOI of 5) or (B) treated with poly(I:C) (30 μg/mL), and transcriptionalresponses were assessed at 6 hpi by RT-qPCR. Results are expressed asgeometric mean ± 95% CI of the geometric mean (n = 4 biologicalreplicates per intestinal segment, compiled from two independent ex-periments). Statistical analyses were performed by Mann–WhitneyU test. (C ) HIEs (j3) were mock- (Top) or HRV-inoculated (Bottom; MOI of10). At 6 hpi, HIEs were fixed and stained for dsRNA (magenta), the ro-tavirus viroplasm protein NSP2 (green), and nuclei (DAPI, blue). (Scalebar: 10 μm.)
E574 | www.pnas.org/cgi/doi/10.1073/pnas.1615422114 Saxena et al.
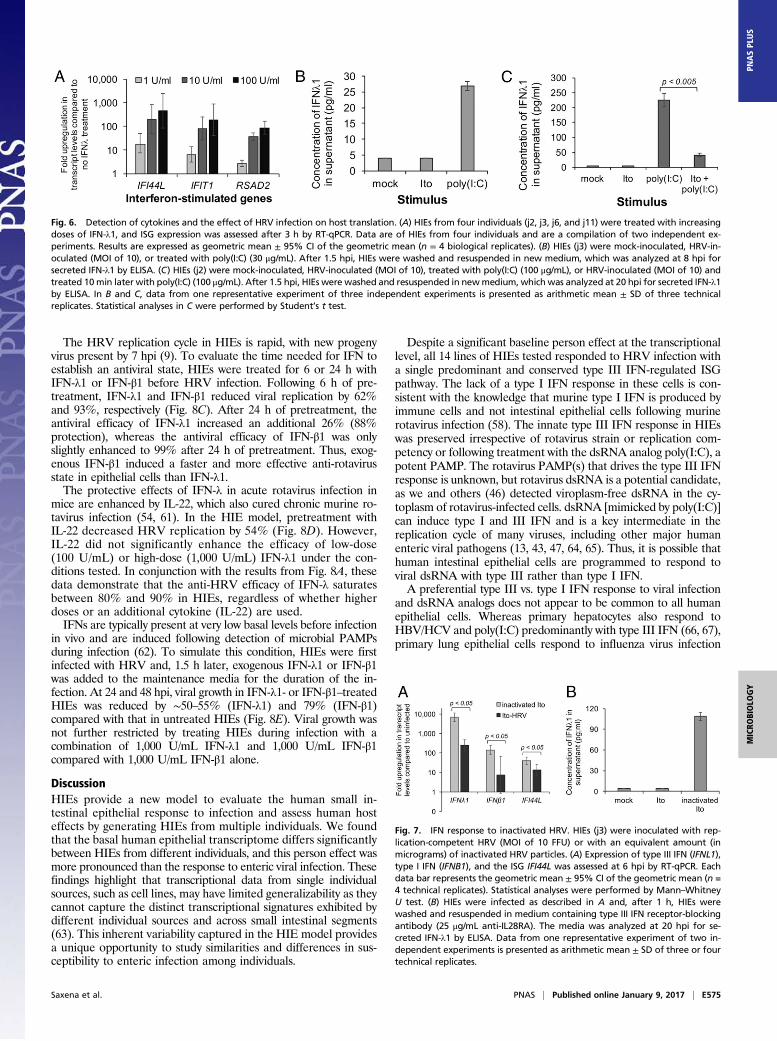
The HRV replication cycle in HIEs is rapid, with new progenyvirus present by 7 hpi (9). To evaluate the time needed for IFN toestablish an antiviral state, HIEs were treated for 6 or 24 h withIFN-λ1 or IFN-β1 before HRV infection. Following 6 h of pre-treatment, IFN-λ1 and IFN-β1 reduced viral replication by 62%and 93%, respectively (Fig. 8C). After 24 h of pretreatment, theantiviral efficacy of IFN-λ1 increased an additional 26% (88%protection), whereas the antiviral efficacy of IFN-β1 was onlyslightly enhanced to 99% after 24 h of pretreatment. Thus, exog-enous IFN-β1 induced a faster and more effective anti-rotavirusstate in epithelial cells than IFN-λ1.The protective effects of IFN-λ in acute rotavirus infection in
mice are enhanced by IL-22, which also cured chronic murine ro-tavirus infection (54, 61). In the HIE model, pretreatment withIL-22 decreased HRV replication by 54% (Fig. 8D). However,IL-22 did not significantly enhance the efficacy of low-dose(100 U/mL) or high-dose (1,000 U/mL) IFN-λ1 under the con-ditions tested. In conjunction with the results from Fig. 8A, thesedata demonstrate that the anti-HRV efficacy of IFN-λ saturatesbetween 80% and 90% in HIEs, regardless of whether higherdoses or an additional cytokine (IL-22) are used.IFNs are typically present at very low basal levels before infection
in vivo and are induced following detection of microbial PAMPsduring infection (62). To simulate this condition, HIEs were firstinfected with HRV and, 1.5 h later, exogenous IFN-λ1 or IFN-β1was added to the maintenance media for the duration of the in-fection. At 24 and 48 hpi, viral growth in IFN-λ1- or IFN-β1–treatedHIEs was reduced by ∼50–55% (IFN-λ1) and 79% (IFN-β1)compared with that in untreated HIEs (Fig. 8E). Viral growth wasnot further restricted by treating HIEs during infection with acombination of 1,000 U/mL IFN-λ1 and 1,000 U/mL IFN-β1compared with 1,000 U/mL IFN-β1 alone.
DiscussionHIEs provide a new model to evaluate the human small in-testinal epithelial response to infection and assess human hosteffects by generating HIEs from multiple individuals. We foundthat the basal human epithelial transcriptome differs significantlybetween HIEs from different individuals, and this person effect wasmore pronounced than the response to enteric viral infection. Thesefindings highlight that transcriptional data from single individualsources, such as cell lines, may have limited generalizability as theycannot capture the distinct transcriptional signatures exhibited bydifferent individual sources and across small intestinal segments(63). This inherent variability captured in the HIE model providesa unique opportunity to study similarities and differences in sus-ceptibility to enteric infection among individuals.
Despite a significant baseline person effect at the transcriptionallevel, all 14 lines of HIEs tested responded to HRV infection witha single predominant and conserved type III IFN-regulated ISGpathway. The lack of a type I IFN response in these cells is con-sistent with the knowledge that murine type I IFN is produced byimmune cells and not intestinal epithelial cells following murinerotavirus infection (58). The innate type III IFN response in HIEswas preserved irrespective of rotavirus strain or replication com-petency or following treatment with the dsRNA analog poly(I:C), apotent PAMP. The rotavirus PAMP(s) that drives the type III IFNresponse is unknown, but rotavirus dsRNA is a potential candidate,as we and others (46) detected viroplasm-free dsRNA in the cy-toplasm of rotavirus-infected cells. dsRNA [mimicked by poly(I:C)]can induce type I and III IFN and is a key intermediate in thereplication cycle of many viruses, including other major humanenteric viral pathogens (13, 43, 47, 64, 65). Thus, it is possible thathuman intestinal epithelial cells are programmed to respond toviral dsRNA with type III rather than type I IFN.A preferential type III vs. type I IFN response to viral infection
and dsRNA analogs does not appear to be common to all humanepithelial cells. Whereas primary hepatocytes also respond toHBV/HCV and poly(I:C) predominantly with type III IFN (66, 67),primary lung epithelial cells respond to influenza virus infection
Fig. 6. Detection of cytokines and the effect of HRV infection on host translation. (A) HIEs from four individuals (j2, j3, j6, and j11) were treated with increasingdoses of IFN-λ1, and ISG expression was assessed after 3 h by RT-qPCR. Data are of HIEs from four individuals and are a compilation of two independent ex-periments. Results are expressed as geometric mean ± 95% CI of the geometric mean (n = 4 biological replicates). (B) HIEs (j3) were mock-inoculated, HRV-in-oculated (MOI of 10), or treated with poly(I:C) (30 μg/mL). After 1.5 hpi, HIEs were washed and resuspended in new medium, which was analyzed at 8 hpi forsecreted IFN-λ1 by ELISA. (C) HIEs (j2) were mock-inoculated, HRV-inoculated (MOI of 10), treated with poly(I:C) (100 μg/mL), or HRV-inoculated (MOI of 10) andtreated 10min later with poly(I:C) (100 μg/mL). After 1.5 hpi, HIEs were washed and resuspended in newmedium, which was analyzed at 20 hpi for secreted IFN-λ1by ELISA. In B and C, data from one representative experiment of three independent experiments is presented as arithmetic mean ± SD of three technicalreplicates. Statistical analyses in C were performed by Student’s t test.
Fig. 7. IFN response to inactivated HRV. HIEs (j3) were inoculated with rep-lication-competent HRV (MOI of 10 FFU) or with an equivalent amount (inmicrograms) of inactivated HRV particles. (A) Expression of type III IFN (IFNL1),type I IFN (IFNB1), and the ISG IFI44L was assessed at 6 hpi by RT-qPCR. Eachdata bar represents the geometric mean ± 95% CI of the geometric mean (n =4 technical replicates). Statistical analyses were performed by Mann–WhitneyU test. (B) HIEs were infected as described in A and, after 1 h, HIEs werewashed and resuspended in medium containing type III IFN receptor-blockingantibody (25 μg/mL anti-IL28RA). The media was analyzed at 20 hpi for se-creted IFN-λ1 by ELISA. Data from one representative experiment of two in-dependent experiments is presented as arithmetic mean ± SD of three or fourtechnical replicates.
Saxena et al. PNAS | Published online January 9, 2017 | E575
MICRO
BIOLO
GY
PNASPL
US

and poly(I:C) with type I and III IFN (43, 68). Thus, the type ofIFN induced in response to infection appears to be cell- and/orvirus-specific rather than a pan-epithelial intrinsic response. Thespecific cellular factors that predispose induction of type III IFN ortype I IFN are unclear because regulation of IFN is complex. Bothtypes of IFN are induced by similar stimuli and generally rely onsimilar transcription factors, such as NF-κB and the IRF family(30), but preferential activation of type III IFN may be influencedby other transcription factors (69). In addition, peroxisomal content(70) and cell differentiation status (70) may significantly influencetype III IFN induction. It is notable that the rotavirus capsid pro-tein VP4 has a peroxisomal targeting motif (71) and that rotavirusinfection of HIEs requires differentiated HIEs (9). Whether any ofthese factors influences the preferential type III IFN innate re-sponse of HIEs to HRV and poly(I:C) remains to be determined.The predominant type III IFN response and its regulation of
ISG induction observed in our studies with HIEs is consistent withprevious studies reporting that murine intestinal epithelial cellsfrom poly(I:C)-injected and murine rotavirus-infected mice exhibitequal or greater fold inductions of type III IFN than type I IFNtranscripts (17, 55, 56, 58). This contrasts with results using trans-formed human colonic cell lines that respond to rotavirus infectionand poly(I:C) with type I IFN-β at the transcript and protein levels,and type I IFN regulates ISG induction in these cells (16, 42, 72).Transformed cells often have significant deficits in their responseto pathogens, including in IFN signaling pathways (73, 74). It seemslikely that this critical difference in IFN responses to rotavirus andpoly(I:C) between immortalized cell lines and nontransformedHIE cultures is partially attributed to their transformation status.
Despite robust up-regulation of type III IFN at the transcrip-tional level, rotaviral replication was not enhanced by neutraliza-tion of the type III IFN receptor in HIEs. This result wasunexpected because enteric viral replication (rotavirus, reovirus,murine norovirus) is enhanced in mice lacking the type III IFNreceptor (54–56, 75), even though it is notable that neutralizationof the type III IFN response had a limited effect on reovirusreplication (approximately twofold increase in viral transcripts) ina human colonic transformed cell line (70). The basis for theapparent discrepancy may be multifactorial and include differ-ences in how the IFN system was blocked (neutralizing antibodiesvs. receptor KOs), development of alternate compensatory path-ways in IFN-KO mice, and/or differences in human and mousetype III IFN subtype expression (37, 38). Regardless of thesedifferences, HRV infection inhibited the IFN response by HIEs.Most pathogenic viruses, including rotavirus, have developed waysto subvert IFN responses at multiple steps in their biologic path-ways (24, 44). Rotavirus evades the host innate response primarilythrough its nonstructural proteins NSP1 and NSP3, and thestructural protein VP3 has recently been shown to antagonize thecellular RNaseL antiviral response as well (33, 76). In several invitro models, rotavirus NSP1 inhibits the host IFN response bypreventing IFN from being transcribed (35, 51). Recent studieshave shown that NSP1 acts as an adaptor that links the hostCullin–E3 ligase complex with β-transducin repeat-containingprotein (β-TrCP), which is a necessary component for NF-κB–mediated induction of IFN transcripts (77, 78). Through this in-teraction with the Cullin–E3 ligase complex, NSP1 facilitates theproteasomal degradation of itself and β-TrCP (78). Using HIEs asa model, it was further shown that β-TrCP degradation by HRV
Fig. 8. Pretreatment of HIE cultures with exogenously administered type I and type III IFN. (A) HIEs (j3) were pretreated with escalating doses of type III IFN(IFN-λ1) or type I IFN (IFN-β1) for 24 h, after which HIEs were washed to remove IFN and then inoculated with HRV (MOI of 0.1). The increase in infectious viruswas calculated by subtracting the titer at 1.5 hpi from the titer at 24 hpi for each condition and depicted in the panel as the percentage of viral growthcompared with the control infection (no IFN pretreatment). The control infection represents 100% viral growth. The raw increase in viral titer (24–1.5 hpi) forthe control (no IFN pretreatment) sample was 35,275 FFU (geometric mean of three replicates). (B) HIEs (j3) were pretreated with 1,000 U/mL of the indicatedIFN for 24 h, following which the same procedures were performed as described in A. The raw increase in viral titer (24–1.5 hpi) for the control (no IFNpretreatment) sample was 15,741 FFU (geometric mean of four replicates). (C) HIEs from six individuals (j2, j3, j6, j8, j10, and j11) were pretreated with 1,000 U/mLof IFN-λ1 or IFN-β1 for 6 or 24 h, washed, and HRV-inoculated (MOI of 0.1). (D) HIEs from five individuals (j2, j3, j8, j10, and j11) were pretreated with theindicated combinations of IL-22 (100 ng/mL) and IFN-λ1 for 24 h, after which they were HRV-inoculated (MOI of 0.1). In C and D, the percentage of viralgrowth was determined as described for A. In A–D, the efficacy [1 − (% viral growth)] of each IFN pretreatment is displayed at the top. In A and B, data fromone representative experiment of two or three independent experiments is presented as arithmetic mean ± SEM of three or four technical replicates. In C andD, data are of HIEs from five or six individuals and are a compilation of two (C) and four (D) independent experiments; results are expressed as arithmeticmean ± SEM (n = 5–6 biological replicates). (E) HIEs (j2) were HRV-inoculated (MOI of 0.1) and, after 1.5 hpi, HIEs were washed and resuspended in mediumcontaining pancreatin without IFN (control) or with 1,000 U/mL of the indicated IFN. The amount of infectious virus under each condition was assessed at 1.5,24, and 48 hpi. Each data point represents arithmetic mean ± SEM of six technical replicates. Data from one representative experiment of three independentexperiments is shown. Statistical analyses in A–E were performed by Mann–Whitney U test.
E576 | www.pnas.org/cgi/doi/10.1073/pnas.1615422114 Saxena et al.

was mitigated by inhibition of Cullin–RING ubiquitin ligases,demonstrating that HRV antagonizes IFN transcription byhijacking host proteins (78). Although we did not evaluate this inour studies, this mechanism may help explain the lower levels ofIFN transcripts observed when HIEs were infected with replica-tion-competent HRV compared with inactivated HRV, whichdoes not produce NSP1. Although replication-competent HRVinhibited type III IFN transcription ∼10-fold compared withinactivated HRV, type III IFN transcripts were still robustly in-duced in replication-competent HRV-infected HIEs (100-1,000fold) and are present at high levels in intestinal epithelial cellsfrom rotavirus-infected mice (54, 55). Together, these data suggestthat the transcriptional regulatory pathways upstream of IFN in-duction remain at least partially intact in these models. However,rotavirus can also inhibit host responses by decreasing translationof host mRNA through multiple mechanisms primarily via theprotein NSP3 (24, 79–81). Although these mechanisms do notselectively antagonize IFN, production of IFN protein is likelyreduced when there is a global decrease in host translation. Thus,translational inhibition is a potent method for HRV to antagonizethe type III IFN antiviral response because IFN-λ protein wassignificantly decreased in HIEs infected with replication-compe-tent HRV compared with inactivated HRV. Because very lowlevels of IFN-λ protein are required to elicit the ISG responsesobserved in HRV-infected HIEs, our data suggest that minimalamounts of type III IFN protein were produced during HRV in-fection of HIEs: enough to induce ISGs but not enough to restrictviral growth. These results suggest there may be a threshold levelof IFN production and perhaps ISG production needed to restrictviral growth. Overall, this represents the paradox of IFN responsesin HIEs.In contrast to HRV-infected HIEs, high levels of IFN-λ protein
are detected in the small intestine from rotavirus-infected mice.These studies examined whole small intestinal homogenates;therefore, the cellular origin of the IFN-λ protein could not bedetermined (17, 54). Murine intestinal epithelial cells were pro-posed to be the major source of IFN-λ protein in these homoge-nates because IFNL transcript levels were approximately four tofive fold higher in intestinal epithelial cells than in hematopoieticcells (54). However, our data demonstrate that IFNL transcripts donot parallel protein levels in epithelial cells during rotavirus in-fection. Hematopoietic cells can produce IFN-λ protein in responseto multiple viruses (57), and a recent study has demonstrated thatsteady-state IFN-λ in the intestine may actually originate fromCD45+ hematopoietic cells rather than epithelial cells in uninfectedmice (56). Taken together, the HIE and mouse data suggest thatthe predominant source of the IFN-λ protein in the small intestinein response to rotavirus infection may be of hematopoietic ratherthan epithelial origin.Lamina propria hematopoietic cells are the primary source of
type I IFN during rotavirus infection in mice, rather than intestinalepithelial cells (58). This is consistent with our data in which HIEsproduced no detectable IFN-β protein in response to HRV, andblockade of the type I IFN system had minimal to no effect ontranscription of the six ISGs assessed. These results suggest thatthe origin of type I IFN in humans, as in mice in vivo, is extra-epithelial, such as from plasmacytoid dendritic cells (82). Wesimulated the effect of extraepithelial sources of IFN by exoge-nously treating HIEs with IFN. Treatment with type I IFN-β wassignificantly more effective than type III IFN at restricting HRVreplication. This differs from reports in mice, in which exogenoustreatment with IFN-λ is more protective against enteric viralreplication than type I IFN (55, 56), and type I IFN is primarilyconsidered to inhibit systemic spread of enteric viruses (17, 56,75). This difference may be a result of the type I IFN subtypetested; previous studies in mice and HIEs used an IFN-α subtyperather than IFN-β for exogenous treatment (17, 55, 56, 83); how-ever, IFN-α2 was significantly weaker than IFN-β in restricting
HRV growth in HIEs. Heterogeneity in the host response to dif-ferent type I IFNs has been previously characterized; IFN-β bindsthe type I IFN receptor with greater affinity than IFN-α2 (84), andother viral pathogens including LCMV and HCV have differingsensitivities to IFN-α and IFN-β (60). Thus, the choice of type IIFN tested may have a significant impact on the evaluation of IFNantiviral efficacy. Our data indicate that treatment with humantype I IFN-β potently decreases local spread and replication ofenteric viruses in the human intestinal epithelium, but whetherextraepithelial production of type I IFN during infection in vivo isof sufficient quantity or interacts with epithelial cells to impacthomologous virus replication remains a question. The ISGs re-quired to limit HRV replication have yet to be identified, as in-dividual ISG efficacy is virus-specific (31), but exogenous IFN-βlikely induces these ISGs to higher levels and/or at a faster ratethan IFN-λ in the human intestinal epithelium.A current limitation of the HIE system is that it lacks the
microbiome, and immune cells have yet to be incorporated withthe epithelial cells to create a model that recapitulates the complexcross-talk that occurs in the small intestine (85, 86). Although wepartially simulated the role of immune cells on epithelial cells byexogenously adding IFN to HIE cultures, we were not able toevaluate the effects of epithelial-produced IFN-λ on immune cells.Plasmacytoid dendritic cells respond to exogenous IFN-λ as well asproduce large amounts of type III and type I IFN protein (30, 57).This raises the possibility of IFN-λ–mediated cross-talk betweenintestinal epithelial cells and intestinal plasmacytoid dendritic cellsand leaves open the prospect that epithelial-derived IFN-λ can acton and even induce its own production from immune cells becauseIFN-λ is an ISG itself (87). As HIE platforms and cellular co-culture techniques are further developed, it will be possible todissect such microbiome–immune–epithelial interactions.To better understand infectious disease pathogenesis, host–
pathogen interactions are studied in model systems. However,these models can yield differing results depending on the choice ofhost organism and pathogen strain. This is particularly relevant forhuman infections because modeling human pathogens in vivo isoften impractical. Advances in the organoid/enteroid and tran-scriptomic fields provide much needed tools to study host–patho-gen interactions in the human intestine. Recently, RNA-seq ofiPSC-derived human intestinal organoids treated with the humanenteric pathogen Salmonella enterica serovar Typhimurium re-vealed new insights into how the epithelium responds to bacterialinfection with a proinflammatory transcriptional profile (3). LikeSalmonella Typhimurium, many human enteric viruses also lackrobust models, and our studies demonstrate that HIEs are a usefulmodel to study the human enteric virus HRV (9). In the presentstudy, a complex host–pathogen interplay between the human in-testinal epithelial type III IFN system and viral antagonism of thisresponse is described. Although epithelial-derived type III IFN hasbeen proposed as a critical defense against murine enteric viruses(54–56), our data are consistent with a recent report showing typeIII IFN does not significantly restrict homologous murine rotavirusinfection (17). Even though the type III IFN system is activated inHIEs following HRV infection, the virus effectively evades thisepithelial antiviral response. Whether other human enteric virusessimilarly evade the type III IFN system remains to be determined.The recent cultivation of human noroviruses in HIEs establishesHIEs as a relevant model for studying host–pathogen interactionsof other human enteric viruses in addition to HRV (4). The limitedrole of type III IFN in restricting HRV replication in the humanepithelium raises the question of whether virus-induced type IIIIFN has undiscovered biologic functions in the mucosal epitheliumthat are not directly antiviral but may have alternative effects onbystander epithelial and/or nonepithelial cells. Given that HIEs arederived from nontransformed human small intestinal tissue andcan be generated from many different individuals, they provide apreclinical model for assessing the host response to human enteric
Saxena et al. PNAS | Published online January 9, 2017 | E577
MICRO
BIOLO
GY
PNASPL
US

pathogens and an additional system to evaluate if findings in ani-mal models can be translated directly to human tissue or if im-portant differences in these systems exist.
Materials and MethodsDetailed methods and descriptions of cell lines, HIE cultures, viruses and viralinfections, immunohistochemistry, cytokines, antibodies, ELISA kits, RNAextraction, RT-qPCR protocols, and statistical analyses are provided in SIMaterials and Methods.
Microarray-Based Transcriptome Analysis. HumanHT-12 v4 Expression Bead-Chips (Illumina) were used for microarray analysis. Transcriptome analysis wasperformed by using R statistical software (88). Two microarray experimentswere performed: the first experiment included samples from a single indi-vidual’s HIEs (j11) and the second with samples of HIE cultures from threeindividuals (j2, j3, and j11). In the first and second microarray experiments,20,948 genes were defined as expressed and used in subsequent analysis.Expression values for these 20,948 genes were quantile normalized by usingthe normalize.quantiles() function of the preprocessCore R library. Analysisof the microarray-acquired data is described in SI Materials and Methods.
RNA-seq–Based Transcriptome Analysis. Total RNA was prepared by using theIllumina TruSeq Stranded RNA Sample preparation protocol. Paired-endsequencing (100 bp) was performed by using the IlluminaHiSeq 2500 ma-
chine. A total of 103,002,651, 101,422,021, 90,511,554, and 84,412,802 readswere sequenced for samples j11-uninfected, j2-uninfected, j11-infected, andj2-infected, respectively. Analysis of the RNA-seq data are described in SIMaterials and Methods.
ACKNOWLEDGMENTS. We thank Joel M. Sederstrom, Dr. Lisa D. White,Mylinh Bernardi, and Daniela Dias Xavier for their expert assistance, as well asManasi Gadkari for expert assistance with performing microarray experiments.We also thank Dr. Harry Greenberg for helpful discussions and commentsduring preparation of this manuscript. This work was supported by NationalInstitutes of Health (NIH) Grants U19-AI116497 and R01 AI080656 (to M.K.E.),U18-TR000552 (to M.D.), R21-AI117220 (to M.E.C.), and Howard HughesMedical Institute Grant 570076890 (to K.S.). This project also was supportedby Advanced Technology Core Laboratories at Baylor College of Medicine. Thisincluded core support from the Integrated Microscopy Core at Baylor Collegeof Medicine with funding from Grants P30 DK-56338 (to H. El-Serag, PrincipalInvestigator), P30 CA125123 (to C. K. Osborne, Principal Investigator), CPRITRP150578 (to P. Davies, Principal Investigator), the Dan L. Duncan CancerCenter and the John S. Dunn Gulf Coast Consortium for Chemical Genomics;the Cytometry and Cell Sorting Core at Baylor College of Medicine withfunding from the Grants P30 AI036211 (to J. Butel, Principal Investigator), P30CA125123 (to C. K. Osborne, Principal Investigator), and S10 RR024574 (toE. Lumpkin, Principal Investigator); and the Genomic and RNA Profiling Core atBaylor College of Medicine with funding from the Grants P30 DK56338 (toH. El-Serag, Principal Investigator) and P30 CA125123 (to C. K. Osborne,Principal Investigator).
1. Duizer E, et al. (2004) Laboratory efforts to cultivate noroviruses. J Gen Virol 85(pt 1):79–87.
2. Kitamoto N, Ramig RF, Matson DO, Estes MK (1991) Comparative growth of differentrotavirus strains in differentiated cells (MA104, HepG2, and CaCo-2). Virology 184(2):729–737.
3. Forbester JL, et al. (2015) Interaction of Salmonella enterica serovar typhimuriumwithintestinal organoids derived from human induced pluripotent stem cells. InfectImmun 83(7):2926–2934.
4. Ettayebi K, et al. (2016) Replication of human noroviruses in stem cell-derived humanenteroids. Science 353(6306):1387–1393.
5. Foulke-Abel J, et al. (2014) Human enteroids as an ex-vivo model of host-pathogeninteractions in the gastrointestinal tract. Exp Biol Med (Maywood) 239(9):1124–1134.
6. Sato T, et al. (2011) Long-term expansion of epithelial organoids from human colon,adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141(5):1762–1772.
7. Fuller MK, et al. (2013) Intestinal stem cells remain viable after prolonged tissuestorage. Cell Tissue Res 354(2):441–450.
8. VanDussen KL, et al. (2015) Development of an enhanced human gastrointestinalepithelial culture system to facilitate patient-based assays. Gut 64(6):911–920.
9. Saxena K, et al. (2016) Human intestinal enteroids: A new model to study humanrotavirus infection, host restriction, and pathophysiology. J Virol 90(1):43–56.
10. Tate JE, Burton AH, Boschi-Pinto C, Parashar UD; World Health Organization–CoordinatedGlobal Rotavirus Surveillance Network (2016) Global, regional, and national esti-mates of rotavirus mortality in children <5 years of age, 2000-2013. Clin Infect Dis62(suppl 2):S96–S105.
11. Ciarlet M, Estes MK, Barone C, Ramig RF, Conner ME (1998) Analysis of host rangerestriction determinants in the rabbit model: Comparison of homologous and het-erologous rotavirus infections. J Virol 72(3):2341–2351.
12. Ramig RF (1988) The effects of host age, virus dose, and virus strain on heterologousrotavirus infection of suckling mice. Microb Pathog 4(3):189–202.
13. Green K (2013) Caliciviridae: The Noroviruses (Lippincott Williams & Wilkins, Phila-delphia), 6th Ed.
14. Jogler C, et al. (2006) Replication properties of human adenovirus in vivo and incultures of primary cells from different animal species. J Virol 80(7):3549–3558.
15. Cuadras MA, Feigelstock DA, An S, Greenberg HB (2002) Gene expression pattern inCaco-2 cells following rotavirus infection. J Virol 76(9):4467–4482.
16. Frias AH, et al. (2010) Intestinal epithelia activate anti-viral signaling via intracellularsensing of rotavirus structural components. Mucosal Immunol 3(6):622–632.
17. Lin JD, et al. (2016) Distinct roles of type I and type III interferons in intestinal im-munity to homologous and heterologous rotavirus infections. PLoS Pathog 12(4):e1005600.
18. Sen A, Feng N, Ettayebi K, Hardy ME, Greenberg HB (2009) IRF3 inhibition by rotavirusNSP1 is host cell and virus strain dependent but independent of NSP1 proteasomaldegradation. J Virol 83(20):10322–10335.
19. Blutt SE, Miller AD, Salmon SL, Metzger DW, Conner ME (2012) IgA is important forclearance and critical for protection from rotavirus infection. Mucosal Immunol 5(6):712–719.
20. Franco MA, Greenberg HB (1999) Immunity to rotavirus infection in mice. J Infect Dis179(suppl 3):S466–S469.
21. Burns JW, et al. (1995) Analyses of homologous rotavirus infection in the mousemodel. Virology 207(1):143–153.
22. Murphy K, Travers P, Walport M, Janeway C (2008) Janeway’s Immunobiology (Gar-land Science, New York), 7th Ed.
23. Kawai T, Akira S (2006) Innate immune recognition of viral infection. Nat Immunol7(2):131–137.
24. Arnold MM, Sen A, Greenberg HB, Patton JT (2013) The battle between rotavirus andits host for control of the interferon signaling pathway. PLoS Pathog 9(1):e1003064.
25. Holloway G, Coulson BS (2013) Innate cellular responses to rotavirus infection. J GenVirol 94(pt 6):1151–1160.
26. Ivashkiv LB, Donlin LT (2014) Regulation of type I interferon responses. Nat RevImmunol 14(1):36–49.
27. Schneider WM, Chevillotte MD, Rice CM (2014) Interferon-stimulated genes: A com-plex web of host defenses. Annu Rev Immunol 32:513–545.
28. Kotenko SV, et al. (2003) IFN-lambdas mediate antiviral protection through a distinctclass II cytokine receptor complex. Nat Immunol 4(1):69–77.
29. Sommereyns C, Paul S, Staeheli P, Michiels T (2008) IFN-lambda (IFN-lambda) is ex-pressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo.PLoS Pathog 4(3):e1000017.
30. Kotenko SV (2011) IFN-λs. CurrOpinImmunol 23(5):583–590.31. Schoggins JW (2014) Interferon-stimulated genes: Roles in viral pathogenesis. Curr
Opin Virol 6:40–46.32. Feng N, et al. (2008) Role of interferon in homologous and heterologous rotavirus
infection in the intestines and extraintestinal organs of suckling mice. J Virol 82(15):7578–7590.
33. Lopez S, Sanchez-Tacuba L, Moreno J, Arias CF (2016) Rotavirus strategies against theinnate antiviral system. Annu Rev Virol 3(1):591–609.
34. Velasquez DE, Parashar UD, Jiang B (2014) Strain diversity plays no major role in thevarying efficacy of rotavirus vaccines: An overview. Infect Genet Evol 28:561–571.
35. Arnold MM, Patton JT (2011) Diversity of interferon antagonist activities mediated byNSP1 proteins of different rotavirus strains. J Virol 85(5):1970–1979.
36. Ciarlet M, Estes MK (1999) Human and most animal rotavirus strains do not require thepresence of sialic acid on the cell surface for efficient infectivity. J Gen Virol 80(pt 4):943–948.
37. Hermant P, et al. (2014) Human but not mouse hepatocytes respond to interferon-lambda in vivo. PLoS One 9(1):e87906.
38. Lasfar A, et al. (2006) Characterization of the mouse IFN-lambda ligand-receptorsystem: IFN-lambdas exhibit antitumor activity against B16 melanoma. Cancer Res66(8):4468–4477.
39. Zachos NC, et al. (2016) Human enteroids/colonoids and intestinal organoids func-tionally recapitulate normal intestinal physiology and pathophysiology. J Biol Chem291(8):3759–3766.
40. In J, et al. (2016) Enterohemorrhagic Escherichia coli reduce mucus and inter-microvillar bridges in human stem cell-derived colonoids. Cell Mol GastroenterolHepatol 2(1):48–62.e3.
41. Wang C, et al. (2014) The concordance between RNA-seq and microarray data de-pends on chemical treatment and transcript abundance. Nat Biotechnol 32(9):926–932.
42. Hirata Y, Broquet AH, Menchén L, Kagnoff MF (2007) Activation of innate immunedefense mechanisms by signaling through RIG-I/IPS-1 in intestinal epithelial cells.J Immunol 179(8):5425–5432.
43. Ioannidis I, Ye F, McNally B, Willette M, Flaño E (2013) Toll-like receptor expressionand induction of type I and type III interferons in primary airway epithelial cells.J Virol 87(6):3261–3270.
44. Hoffmann HH, Schneider WM, Rice CM (2015) Interferons and viruses: An evolu-tionary arms race of molecular interactions. Trends Immunol 36(3):124–138.
45. Lawton JA, Estes MK, Prasad BV (1997) Three-dimensional visualization of mRNArelease from actively transcribing rotavirus particles. Nat Struct Biol 4(2):118–121.
46. Rojas M, Arias CF, López S (2010) Protein kinase R is responsible for the phosphory-lation of eIF2alpha in rotavirus infection. J Virol 84(20):10457–10466.
E578 | www.pnas.org/cgi/doi/10.1073/pnas.1615422114 Saxena et al.

47. Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR (2006) Double-strandedRNA is produced by positive-strand RNA viruses and DNA viruses but not in detectableamounts by negative-strand RNA viruses. J Virol 80(10):5059–5064.
48. Sheth R, et al. (1996) Rotavirus stimulates IL-8 secretion from cultured epithelial cells.Virology 221(2):251–259.
49. Barro M, Patton JT (2007) Rotavirus NSP1 inhibits expression of type I interferon byantagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J Virol81(9):4473–4481.
50. Graff JW, Mitzel DN, Weisend CM, Flenniken ML, Hardy ME (2002) Interferon regu-latory factor 3 is a cellular partner of rotavirus NSP1. J Virol 76(18):9545–9550.
51. Arnold MM (2016) The rotavirus interferon antagonist NSP1: Many targets, manyquestions. J Virol 90(11):5212–5215.
52. Groene WS, Shaw RD (1992) Psoralen preparation of antigenically intact non-infectious rotavirus particles. J Virol Methods 38(1):93–102.
53. Bullens DM, Kasran A, Peng X, Lorré K, Ceuppens JL (1998) Effects of anti-IL-4 re-ceptor monoclonal antibody on in vitro T cell cytokine levels: IL-4 production by T cellsfrom non-atopic donors. Clin Exp Immunol 113(3):320–326.
54. Hernández PP, et al. (2015) Interferon-λ and interleukin 22 act synergistically for theinduction of interferon-stimulated genes and control of rotavirus infection. NatImmunol 16(7):698–707.
55. Pott J, et al. (2011) IFN-lambda determines the intestinal epithelial antiviral hostdefense. Proc Natl Acad Sci USA 108(19):7944–7949.
56. Mahlakõiv T, Hernandez P, Gronke K, Diefenbach A, Staeheli P (2015) Leukocyte-derived IFN-α/β and epithelial IFN-λ constitute a compartmentalized mucosal defensesystem that restricts enteric virus infections. PLoS Pathog 11(4):e1004782.
57. Yin Z, et al. (2012) Type III IFNs are produced by and stimulate human plasmacytoiddendritic cells. J Immunol 189(6):2735–2745.
58. Sen A, et al. (2012) Innate immune response to homologous rotavirus infection in thesmall intestinal villous epithelium at single-cell resolution. Proc Natl Acad Sci USA109(50):20667–20672.
59. Egli A, Santer DM, O’Shea D, Tyrrell DL, Houghton M (2014) The impact of the in-terferon-lambda family on the innate and adaptive immune response to viral infec-tions. Emerg Microbes Infect 3(7):e51.
60. Ng CT, Mendoza JL, Garcia KC, Oldstone MB (2016) Alpha and beta type 1 interferonsignaling: Passage for diverse biologic outcomes. Cell 164(3):349–352.
61. Zhang B, et al. (2014) Viral infection. Prevention and cure of rotavirus infection viaTLR5/NLRC4-mediated production of IL-22 and IL-18. Science 346(6211):861–865.
62. Sen GC (2001) Viruses and interferons. Annu Rev Microbiol 55:255–281.63. Middendorp S, et al. (2014) Adult stem cells in the small intestine are intrinsically
programmed with their location-specific function. Stem Cells 32(5):1083–1091.64. Wold W, Ison M (2013) Adenoviruses. Fields Virology, eds Knipe DM, Howley PM
(Wolters Kluwer/Lippincott Williams & Wilkins, Philadelphia), 6th Ed, pp 1732–1767.65. Mendez E, Arias C (2013) Astroviruses. Fields Virology, eds Knipe DM, Howley PM
(Wolters Kluwer/Lippincott Williams & Wilkins, Philadelphia), 6th Ed, pp 609–628.66. Park H, et al. (2012) IL-29 is the dominant type III interferon produced by hepatocytes
during acute hepatitis C virus infection. Hepatology 56(6):2060–2070.67. Sato S, et al. (2015) The RNA sensor RIG-I dually functions as an innate sensor and
direct antiviral factor for hepatitis B virus. Immunity 42(1):123–132.68. Crotta S, et al. (2013) Type I and type III interferons drive redundant amplification
loops to induce a transcriptional signature in influenza-infected airway epithelia.PLoS Pathog 9(11):e1003773.
69. Lazear HM, Nice TJ, Diamond MS (2015) Interferon-λ: Immune functions at barriersurfaces and beyond. Immunity 43(1):15–28.
70. Odendall C, et al. (2014) Diverse intracellular pathogens activate type III interferonexpression from peroxisomes. Nat Immunol 15(8):717–726.
71. Mohan KV, Som I, Atreya CD (2002) Identification of a type 1 peroxisomal targetingsignal in a viral protein and demonstration of its targeting to the organelle. J Virol76(5):2543–2547.
72. Frias AH, Jones RM, Fifadara NH, Vijay-Kumar M, Gewirtz AT (2012) Rotavirus-inducedIFN-β promotes anti-viral signaling and apoptosis that modulate viral replication inintestinal epithelial cells. Innate Immun 18(2):294–306.
73. Christian SL, et al. (2012) Suppression of IFN-induced transcription underlies IFN de-fects generated by activated Ras/MEK in human cancer cells. PLoS One 7(9):e44267.
74. Critchley-Thorne RJ, et al. (2009) Impaired interferon signaling is a common immunedefect in human cancer. Proc Natl Acad Sci USA 106(22):9010–9015.
75. Nice TJ, et al. (2015) Interferon-λ cures persistent murine norovirus infection in theabsence of adaptive immunity. Science 347(6219):269–273.
76. Sánchez-Tacuba L, Rojas M, Arias CF, López S (2015) Rotavirus controls activation ofthe 2′-5′-oligoadenylatesynthetase/RNase L pathway using at least two distinctmechanisms. J Virol 89(23):12145–12153.
77. Lutz LM, Pace CR, Arnold MM (2016) Rotavirus NSP1 associates with components ofthe Cullin RING ligase family of E3 ubiquitin ligases. J Virol 90(13):6036–6048.
78. Ding S, et al. (2016) Comparative proteomics reveals strain-specific β-TrCP degrada-tion via rotavirus NSP1 hijacking a host Cullin-3-Rbx1 complex. PLoS Pathog 12(10):e1005929.
79. Gratia M, et al. (2015) Rotavirus NSP3 is a translational surrogate of the poly(A)binding protein-poly(A) complex. J Virol 89(17):8773–8782.
80. Montero H, Rojas M, Arias CF, López S (2008) Rotavirus infection induces the phos-phorylation of eIF2alpha but prevents the formation of stress granules. J Virol 82(3):1496–1504.
81. Rubio RM, Mora SI, Romero P, Arias CF, López S (2013) Rotavirus prevents the ex-pression of host responses by blocking the nucleocytoplasmic transport of poly-adenylated mRNAs. J Virol 87(11):6336–6345.
82. Deal EM, Jaimes MC, Crawford SE, Estes MK, Greenberg HB (2010) Rotavirus structuralproteins and dsRNA are required for the human primary plasmacytoid dendritic cellIFNalpha response. PLoS Pathog 6(6):e1000931.
83. Yin Y, et al. (2015) Modeling rotavirus infection and antiviral therapy using primaryintestinal organoids. Antiviral Res 123:120–131.
84. Jaitin DA, et al. (2006) Inquiring into the differential action of interferons (IFNs): AnIFN-alpha2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-beta. Mol Cell Biol 26(5):1888–1897.
85. Baldridge MT, et al. (2015) Commensal microbes and interferon-λ determine persis-tence of enteric murine norovirus infection. Science 347(6219):266–269.
86. Shulzhenko N, et al. (2011) Crosstalk between B lymphocytes, microbiota and theintestinal epithelium governs immunity versus metabolism in the gut. Nat Med17(12):1585–1593.
87. Ank N, et al. (2006) Lambda interferon (IFN-lambda), a type III IFN, is induced by vi-ruses and IFNs and displays potent antiviral activity against select virus infections invivo. J Virol 80(9):4501–4509.
88. R Development Core Team (2013) R: A Language and Environment for StatisticalComputing (R Foundation for Statistical Computing, Vienna).
89. Crawford SE, et al. (2001) Trypsin cleavage stabilizes the rotavirus VP4 spike. J Virol75(13):6052–6061.
90. Schönborn J, et al. (1991) Monoclonal antibodies to double-stranded RNA as probesof RNA structure in crude nucleic acid extracts. Nucleic Acids Res 19(11):2993–3000.
91. Criglar JM, et al. (2014) A novel form of rotavirus NSP2 and phosphorylation-dependent NSP2-NSP5 interactions are associated with viroplasm assembly. J Virol88(2):786–798.
92. Dobin A, et al. (2013) STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29(1):15–21.
93. Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. NatMethods 9(4):357–359.
94. Liao Y, Smyth GK, Shi W (2014) featureCounts: An efficient general purpose programfor assigning sequence reads to genomic features. Bioinformatics 30(7):923–930.
95. Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dis-persion for RNA-seq data with DESeq2. Genome Biol 15(12):550.
96. Rusinova I, et al. (2013) Interferome v2.0: An updated database of annotated in-terferon-regulated genes. Nucleic Acids Res 41(database issue):D1040–D1046.
97. Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T)method. Nat Protoc 3(6):1101–1108.
Saxena et al. PNAS | Published online January 9, 2017 | E579
MICRO
BIOLO
GY
PNASPL
US