a statistical analysis of random mutagenesis methods … 14/j mol biol... · a statistical analysis...
TRANSCRIPT

doi:10.1016/j.jmb.2005.10.082 J. Mol. Biol. (2006) 355, 858–871
A Statistical Analysis of Random Mutagenesis MethodsUsed for Directed Protein Evolution
Tuck Seng Wong†, Danilo Roccatano†, Martin Zachariasand Ulrich Schwaneberg*
International UniversityBremen (IUB), Campus Ring 828759 Bremen, Germany
0022-2836/$ - see front matter q 2005 E
† T. S. W. & D. R. contributed equAbbreviations used: epPCR, error
mutagenesis assistant program.E-mail address of the correspond
We have developed a statistical method named MAP (mutagenesisassistant program) to equip protein engineers with a tool to developpromising directed evolution strategies by comparing 19 mutagenesismethods. Instead of conventional transition/transversion bias indicators asbenchmarks for comparison, we propose to use three indicators based onthe subset of amino acid substitutions generated on the protein level: (1)protein structure indicator; (2) amino acid diversity indicator with a codondiversity coefficient; and (3) chemical diversity indicator.
A MAP analysis for a single nucleotide substitution was performed forfour genes: (1) heme domain of cytochrome P450 BM-3 from Bacillusmegaterium (EC 1.14.14.1); (2) glucose oxidase from Aspergillus niger (EC1.1.3.4); (3) arylesterase from Pseudomonas fluorescens (EC 3.1.1.2); and (4)alcohol dehydrogenase from Saccharomyces cerevisiae (EC 1.1.1.1). Based onthe MAP analysis of these four genes, 19 mutagenesis methods have beenevaluated and criteria for an ideal mutagenesis method have beenproposed. The statistical analysis showed that existing gene mutagenesismethods are limited and highly biased. An average amino acid substitutionper residue of only 3.15–7.4 can be achieved with current randommutagenesis methods. For the four investigated gene sequences, anaverage fraction of amino acid substitutions of 0.5–7% results in stopcodons and 4.5–23.9% in glycine or proline residues. An average fraction of16.2–44.2% of the amino acid substitutions are preserved, and 45.6%(epPCR method) are chemically different. The diversity remains low evenwhen applying a non-biased method: an average of seven amino acidsubstitutions per residue, 2.9–4.7% stop codons, 11.1–16% glycine/prolineresidues, 21–25.8% preserved amino acids, and 55.5% are amino acids withchemically different side-chains.
Statistical information for each mutagenesis method can further be usedto investigate the mutational spectra in protein regions regarded asimportant for the property of interest.
q 2005 Elsevier Ltd. All rights reserved.
Keywords: protein engineering; directed evolution; random mutagenesis;bias indicator; MAP
*Corresponding authorIntroduction
Directed evolution has become a widely acceptedand broadly applied method for biocatalyst engi-neering.1,2 A directed protein evolution experiment
lsevier Ltd. All rights reserve
ally to this work.-prone PCR; MAP,
ing author:
comprises two main steps: generating diversemutant libraries3,4 and screening for improvedprotein variants.5–7 The quality of a mutant libraryis decisive for the success of a directed evolutionexperiment and many methods have beendeveloped for generating diversity at the genelevel.4 These methods differ significantly in themutational spectra, mutation frequency and aredifferently affected by the redundancy of the geneticcode.4 A widely accepted way of assessing bias inmutational spectra is to analyze the ratio oftransitions (Ts; nucleotide substitutions: purine to
d.

Statistical Analysis of Random Mutagenesis Methods 859
purine or pyrimidine to pyrimidine) to transver-sions (Tv; nucleotide substitutions: purine topyrimidine or pyrimidine to purine). Since thereare four possible transitions and eight possibletransversions, a non-biased mutational spectrum(equal percentage of A/N, T/N, G/N, C/Nnucleotide substitutions) will have a Ts/Tv ratio of0.5 and an AT/GC/GC/AT ratio of 1.
An ideal method would allow substituting everyamino acid of a protein sequence by its 19 counter-parts in a statistical manner. For a protein engineerdeveloping a directed evolution strategy orperforming a directed evolution experiment, it isimportant to know which amino acid substitutionscan be generated on the protein level at positions thathave been identified as important by sequencealignments or rational design studies of proteins.The current transition/transversion bias indicatorused to describe the mutational spectra is insufficientto provide such information. We illustrate, using fourproteins with different functions and origins, that anon-biased mutational spectrum fails to generatehighly diverse libraries with equal occurrence of 19possible amino acid substitutions on the protein level.Reasons lie in the redundancy of the genetic code8
and its organization to minimize mutational errors.9
For example, transitions at third positions in codonsoften preserve the encoded amino acid.10 Further-more, generating consecutive nucleotide substi-tutions has been a rare event in directed proteinevolution due to methodological limitations ofrandom mutagenesis methods such as error-pronepolymerase chain reaction (epPCR).4
Here, we report a statistical analysis tool namedMAP (mutagenesis assistant program), which hasbeen designed to compare directed evolutionmethods by investigating the consequences of theirmutational bias on the level of amino acid substi-tutions in the protein of interest. MAP investigates theamino acid substitution patterns at each amino acidposition of a single gene for 19 random mutagenesismethods under the assumption of single nucleotidesubstitutions in one codon. MAP allows us: (1) tocomprehensively analyse current random muta-genesis methods on the amino acid substitutionlevel; (2) to develop mutational strategies by selectingrandom mutagenesis methods with different muta-tional bias; (3) to evaluate the performance of existingand novel random mutagenesis methods; and (4) toassist in the design of a novel random mutagenesismethod (e.g. a transversion-favoured method or amethod introducing mainly charged amino acids).
To our knowledge, this method allows us for thefirst time to compare random mutagenesis methodson the protein level. As benchmarks for assessing arandom mutagenesis method, we proposed threeprotein diversity indicators depending on the type,the extent and the chemical nature of amino acidsubstitutions. MAP has been implemented on anautomatic server† and provides upon DNA
† http://gis-web.iu-bremen.de/MAP/
sequence input a statistical analysis for 19 randommutagenesis methods.
Results
Enzyme selection to avoid codon bias by originand enzyme class
Four enzymes of different origins and of differentenzyme classes were selected for MAP analysis inorder to avoid a codon bias by the host organisms orenzyme classes. The sense strand sequences encodingthe four selected enzymes have the followingcomposition: heme domain of cytochrome P450 BM-3 (A 33%, C 19%, G 22%, T 26%; 1368 bases), glucoseoxidase (A 19%, C 36%, G 27%, T 18%; 1812 bases),arylesterase (A 20%, C 32%, G 30%, T 18%; 708 bases)and alcohol dehydrogenase (A 23%, C 23%, G 26%,T 28%; 1044 bases). A MAP analysis for these fourproteins was performed by comparing on the proteinlevel the mutational spectra of 19 mutagenesismethods (Table 1). Analyzing mutations in senseand anti-sense strand results in identical amino acidsubstitution profiles.
Figures 1 and 2 show the extent of substi-tutions that terminate protein translation or mightdestabilize protein structures. Figures 3–7 andTable 2 show the diversity and the types ofamino acid substitutions that can be generated onthe protein level. Continuous lines in the Figuresindicate always how a random mutagenesismethod with a non-biased mutational spectrumwill perform on the protein level.
Protein structure indicator: protein structure/function-disrupting substitutions
The first important performance criterion of arandom mutagenesis method is to limit the fractionof protein structure/function-disrupting (stopcodons) and likely destabilizing amino acid substi-tutions (Gly and Pro residues). A mutation gene-rating a stop codon in the gene sequence results onthe protein level in a truncated enzyme variant thatoften loses its activity and Gly or Pro substitutionsdestabilize helical structures.
Escherichia coli is by far the workhorse ofsuccessful directed evolution studies.11,12 A stopcodon (TAA, TGA and TAG) is generated with apercentage of 0.5–7% in the case of a singlenucleotide substitution in each of the four genesanalyzed. The general trend for stop codonfrequencies is very similar for all four genes;however, in the case of the glucose oxidase gene,the stop codon frequency is, on average, signi-ficantly lower (Figure 1). The Taq (dPTP/8-oxodGTP) method13 introduces the least numberof stop codons (0.5–1.8%) due to low percentagesof N/A and N/T nucleotide substitutions.A non-biased random mutagenesis method willhave a higher stop codon frequency (2.9–4.7%)than the Taq (dPTP/8-oxodGTP) method.13

Table 1. Mutational spectra of a non-biased random mutagenesis method and 19 random mutagenesis methods used for statistical analysis in MAP
Transitions Transversions
Method Designator A/G T/C G/A C/T A/T T/A A/C T/G G/C C/G G/T C/A
Non-biased method Non-biased 8.33 8.33 8.33 8.33 8.33 8.33 8.33 8.33 8.33 8.33 8.33 8.33Taq-Pol (unbalanced
dNTPs)aTaq (K, GOAZCZT)b 19.34 19.34 4.82 4.82 9.70 9.70 16.14 16.14 0.00 0.00 0.00 0.00
Taq-Pol (Mn2C/balanced dNTPs)33
Taq (C, GZAZCZT)c 32.69 31.47 4.47 5.18 7.41 8.93 1.12 2.94 0.71 0.71 1.52 1.83
Taq-Pol (Mn2C/unbalanced dNTPs)a
Taq (C, GOAZCZT)d 22.52 22.52 6.07 6.07 13.71 13.71 4.38 4.38 0.00 0.00 3.33 3.32
Taq-Pol (Mn2C/unbalanced dNTPs)a
Taq (C, GOOAZCZT)e
37.30 37.30 2.47 2.47 6.96 6.96 2.07 2.07 0.81 0.81 0.40 0.40
Taq-Pol (Mn2C/unbalanced dNTPs)34
Taq (C, GZA, CZT)f 14.45 14.45 7.12 7.12 21.41 21.41 3.82 3.82 0.73 0.73 2.36 2.36
Taq-Pol (Mn2C/unbalanced dNTPs)35
Taq (C, GZT, AZC)g 30.45 30.45 9.05 9.05 5.70 5.70 1.65 1.65 2.15 2.15 0.90 0.90
Taq-Pol I614K36 Taq (I614K) 20.94 20.94 11.91 11.91 12.36 12.36 1.41 1.41 1.92 1.92 1.41 1.41Mutazyme I37 Mutazyme I 5.25 5.25 22.27 22.27 5.66 5.66 2.14 2.14 4.49 4.49 10.19 10.19Mutazyme II15 Mutazyme II 9.26 9.26 13.49 13.49 15.08 15.08 2.49 2.49 2.17 2.17 7.46 7.46Pfu-Pol (exoK) D473G38 Pfu (exo-, D473G) 10.19 10.19 11.68 11.68 14.86 14.86 3.93 3.93 3.87 3.87 5.47 5.47Enzymatic method
employing reversetranscriptase39
Transcriptase 17.08 13.75 24.17 12.08 18.75 0.42 7.92 0.42 0.42 0.00 0.00 5.00
Taq-Pol (nucleotideanalogues dPTPand 8-oxodGTP)13
Taq (dPTP/8-oxodGTP) 39.10 39.10 6.60 6.60 0.10 0.10 4.20 4.20 0.35 0.35 0.00 0.00
Error-prone rolling circleamplification40
epRCA 3.83 3.83 33.11 33.11 3.83 3.83 0.77 0.77 3.06 3.06 5.41 5.41
Pol I method14 Pol I 17.35 17.35 22.45 22.45 7.15 7.15 1.00 1.00 0.00 0.00 2.05 2.05E. coli expressing mutA
allele of glyV gene16E. coli (mutA) 2.87 2.87 13.22 13.22 12.07 12.07 7.47 7.47 2.87 2.87 11.49 11.49
Chemical mutagen(nitrous acid)41
Nitrous acid 27.27 1.82 10.91 54.55 1.82 0.00 0.00 0.00 1.82 1.82 0.00 0.00
Chemical mutagen(formic acid)41
Formic acid 2.17 19.57 6.52 2.17 13.04 0.00 6.52 0.00 13.04 0.00 28.26 8.70
Chemical mutagen(hydrazine)41
Hydrazine 22.92 37.50 0.00 22.92 2.08 2.08 0.00 8.33 2.08 0.00 0.00 2.08
Chemical mutagen (ethylmethane sulfonate)42
EMSh 17.10 17.10 21.05 21.05 0.00 0.00 0.00 0.00 11.85 11.85 0.00 0.00
These 19 random mutagenesis methods comprise (from top to down): 14 enzymatic methods, one whole cell method and four chemical methods. In the parentheses of the enzymatic methods aC signindicates the supplement and a – sign the absence of Mn2C. The balance of the nucleotide composition is indicated after the C/K sign.
a Values adapted from the user manual of the Diversify PCR Random Mutagenesis Kit (Clontech).b Taq-Pol and [dATP]Z[dCTP]Z[dTTP]Z0.2 mM, [dGTP]Z0.24 mM.c Taq-Pol in the presence of [Mn2C]Z0.25 mM and four standard nucleotides (each 0.02 mM).d Taq-Pol in the presence of [Mn2C]Z0.64 mM and [dATP]Z[dCTP]Z[dTTP]Z0.2 mM, [dGTP]Z0.24 mM.e Taq-Pol in the presence of [Mn2C]Z0.64 mM and [dATP]Z[dCTP]Z[dTTP]Z0.2 mM, [dGTP]Z0.40 mM.f Taq-Pol in the presence of Mn2C and [dATP]Z[dGTP]Z0.2 mM, [dCTP]Z[dTTP]Z1 mM.g Taq-Pol in the presence of [Mn2C]Z0.5 mM and [dCTP]Z[dATP]Z0.03 mM, [dGTP]Z[dTTP]Z1 mM.h EMS stands for ethyl methane sulfonate.

Taq
(–,
G>A
=C=T
)
Taq
(+,
G=A
=C=T
)
Taq
(+,
G>A
=C=T
)
Taq
(+
, G>
>A=C
=T)
Taq
(+,
G=A
, C=T
)
Taq
(+,
G=T
, A=
C)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol
I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
Frac
tion
of
stop
cod
ons
P450 BM–3 Heme domain
Taq
(–,
G>A
=C=T
)
Taq
(+,
G=A
=C=T
)
Taq
(+,
G>A
=C=T
)
Taq
(+
, G>
>A=C
=T)
Taq
(+,
G=A
, C=T
)
Taq
(+,
G=T
, A=
C)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(ex
o–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol
I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0.00
0.01
0.02
0.03
0.04
Frac
tion
of
stop
cod
ons
Glucose oxidase
Taq
(–,
G>A
=C=T
)
Taq
(+,
G=A
=C=T
)
Taq
(+,
G>A
=C=T
)
Taq
(+
, G>
>A=C
=T)
Taq
(+,
G=A
, C=T
)
Taq
(+,
G=T
, A=
C)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol
I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
Frac
tion
of
stop
cod
ons
Arylesterase
Taq
(–,
G>A
=C=T
)
Taq
(+,
G=A
=C=T
)
Taq
(+,
G>A
=C=T
)
Taq
(+
, G>
>A=C
=T)
Taq
(+,
G=A
, C=T
)
Taq
(+,
G=T
, A=
C)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol
I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
Frac
tion
of
stop
cod
ons
Alcohol dehydrogenase
Figure 1. Statistical analyses of stop codon frequencies generated by 19 random mutagenesis methods for P450 BM-3heme domain, glucose oxidase, arylesterase and alcohol dehydrogenase. The performance of a non-biased randommutagenesis method is indicated by the black line.
Statistical Analysis of Random Mutagenesis Methods 861
The percentage of variants resulting in Gly or Prosubstitutions varies between 4.5% and 23.9%(Figure 2). The absolute values differ significantlyamong the mutagenesis methods and might influ-ence the random mutagenesis strategy. The relativedistribution of substitutions is again similar for allfour genes. Interestingly, there is an opposite trendbetween random mutagenesis methods generatingstop codons and Gly/Pro substitutions. The Taq(dPTP/8-oxodGTP)13 method introduces thehighest number of Gly or Pro residues due to thehigh percentages of N/G and N/C nucleotidesubstitutions. Using a non-biased random muta-genesis method, we expect 11.1–16.0% of Gly/Prosubstitutions.
Amino acid diversity indicator: substitutionsleading to amino acid changes and the averagenumber of amino acid substitutions per residue
The second important performance criterion foradjusting the mutation frequency in a random
mutagenesis method is measured by the fractionof variants with preserved amino acids and theaverage number of amino acid substitutions perresidue. Fraction of preserved amino acids isshown in Figure 3 for all 19 random mutagenesismethods and the four investigated genes. Itranges from 16.2–44.2% and clusters around25–30% preserved amino acids. The relativevalues of preserved amino acids differ muchless (25–34.8%) for arylesterase compared to theother three proteins. The overall trends are,except for arylesterase, similar with differencesin absolute values. Fractions of preserved aminoacids generated by various epPCR methods differsignificantly from gene to gene and are lower forglucose oxidase. It is important to note that amethod with non-biased mutational spectrumthat hits every nucleotide with an equal fre-quency results in 21–25.8% of preserved aminoacids.
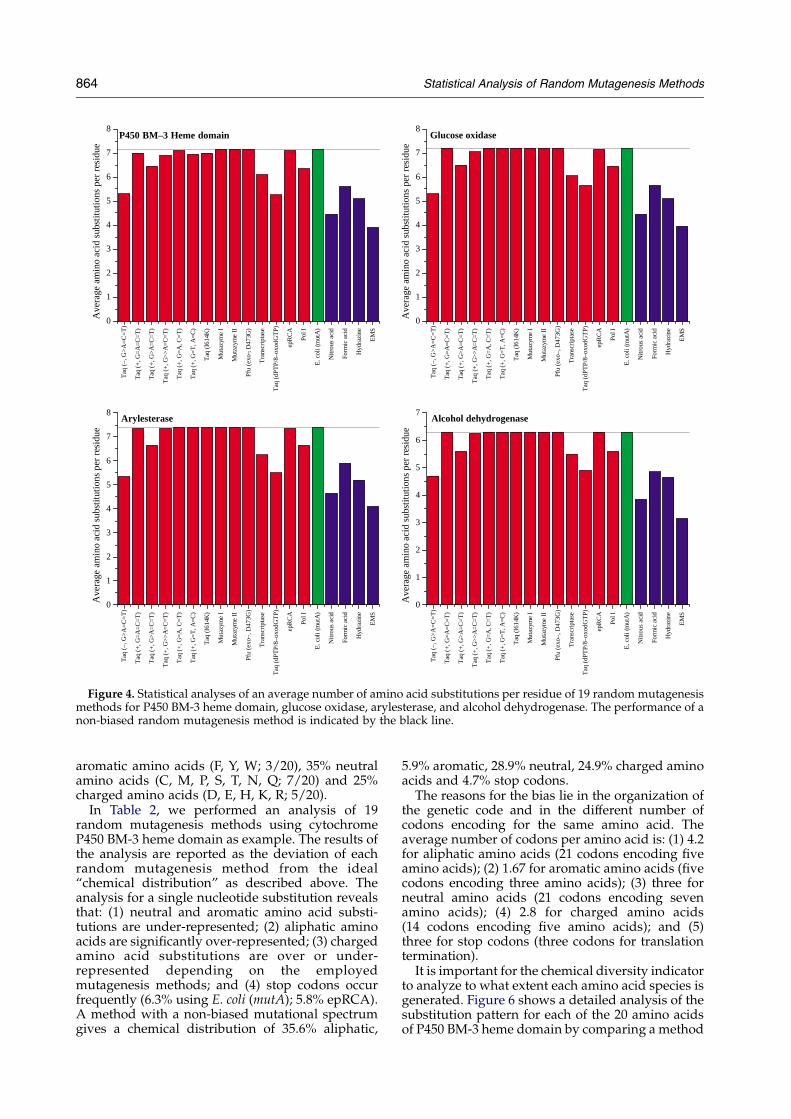
The average number of amino acid substitutionsper residue ranges for all four genes from 3.15 to 7.4

Taq
(–,
G>
A=
C=
T)
Taq
(+,
G=A
=C=
T)
Taq
(+,
G>A
=C=
T)
Taq
(+,
G>
>A
=C
=T
)
Taq
(+,
G=
A, C
=T
)
Taq
(+,
G=
T, A
=C)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
0.16
0.18
Frac
tion
of G
ly o
r Pr
o
P450 BM–3 Heme domain
Taq
(–,
G>
A=
C=
T)
Taq
(+,
G=A
=C=
T)
Taq
(+,
G>A
=C=
T)
Taq
(+,
G>
>A
=C
=T
)
Taq
(+,
G=
A, C
=T
)
Taq
(+,
G=
T, A
=C)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
0.16
0.18
0.20
0.22
0.24
0.26
Frac
tion
of
Gly
or
Pro
Glucose oxidaseT
aq (
–, G
>A
=C
=T
)
Taq
(+,
G=A
=C=
T)
Taq
(+,
G>A
=C=
T)
Taq
(+,
G>
>A
=C
=T
)
Taq
(+,
G=
A, C
=T
)
Taq
(+,
G=
T, A
=C)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP
)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
0.16
0.18
0.20
0.22
0.24
0.26
Frac
tion
of G
ly o
r Pr
o
Arylesterase
Taq
(–,
G>
A=
C=
T)
Taq
(+,
G=A
=C=
T)
Taq
(+,
G>A
=C=
T)
Taq
(+,
G>
>A
=C
=T
)
Taq
(+,
G=
A, C
=T
)
Taq
(+,
G=
T, A
=C)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
0.16
0.18
0.20
0.22Fr
acti
on o
f G
ly o
r Pr
oAlcohol dehydrogenase
Figure 2. Statistical analyses of Gly/Pro substitutions generated by 19 random mutagenesis methods for P450 BM-3heme domain, glucose oxidase, arylesterase and alcohol dehydrogenase. The performance of a non-biased randommutagenesis method is indicated by the black line.
862 Statistical Analysis of Random Mutagenesis Methods
amino acids per residue (Figure 4). The absoluteand relative values are similar for all four genes.Most commonly used epPCR methods give anaverage of 6.5 amino acids per residue. Despite thebias of epPCR, the average number of amino acidsubstitutions per residue is comparable to a methodwith non-biased mutational spectrum, whichresults in an average of seven amino acid substi-tutions per residue. All chemical methods performpoorer than enzymatic epPCR methods, withaverages around 4.7 amino acid substitutions perresidue.
Natural diversity offers the possibility of exchan-ging each amino acid with 19 other amino acids.Current random mutagenesis methods such asepPCR access only w34% of that diversity.A method with a non-biased mutational spectrumwhich is able to target every nucleotide with equalfrequency, performs only slightly better thancurrent epPCR methods (37%).
Codon diversity coefficient
Codon diversity coefficient is a coefficient thatmeasures how random mutations are distributedamong codons of a gene (Figure 5). In a methodwith non-biased mutational spectrum, the codondiversity coefficient has a value of 0 consideringeach nucleotide and each codon substitution tooccur with equal frequency. Biased methods showpreferences toward certain nucleotide exchanges(e.g. Taq (C, GOOAZCZT) (biased to AT/GC,74.6%), Taq (dPTP/8-oxodGTP) (biased to AT/GC, 78.2%), epRCA (biased to GC/AT, 66.22%)and nitrous acid (biased to GC/AT, 65.46%)) andmutate certain nucleotides in codons preferentially.In other words, biased mutagenesis methodsgenerate “hot spots” of mutagenesis that compro-mise genetic diversity. It is therefore important toadd as performance criterion to the amino aciddiversity, a codon diversity coefficient that indicates

Taq
(–,
G>
A=
C=
T)
Taq
(+,
G=A
=C=
T)
Taq
(+,
G>A
=C=
T)
Taq
(+,
G>
>A
=C
=T
)
Taq
(+,
G=
A, C
=T
)
Taq
(+,
G=
T, A
=C)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(ex
o–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP
)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
Frac
tion
of p
rese
rved
am
ino
acid
s
Frac
tion
of p
rese
rved
am
ino
acid
s
Frac
tion
of p
rese
rved
am
ino
acid
s
Frac
tion
of p
rese
rved
am
ino
acid
s
P450 BM–3 Heme domain
Taq
(–,
G>
A=
C=
T)
Taq
(+,
G=A
=C=
T)
Taq
(+,
G>A
=C=
T)
Taq
(+,
G>
>A
=C
=T
)
Taq
(+,
G=
A, C
=T
)
Taq
(+,
G=
T, A
=C)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50Glucose oxidase
Taq
(–,
G>
A=
C=
T)
Taq
(+,
G=A
=C=
T)
Taq
(+,
G>A
=C=
T)
Taq
(+,
G>
>A
=C
=T
)
Taq
(+,
G=
A, C
=T
)
Taq
(+,
G=
T, A
=C)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP
)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35Arylesterase
Taq
(–,
G>
A=
C=
T)
Taq
(+,
G=A
=C=
T)
Taq
(+,
G>A
=C=
T)
Taq
(+,
G>
>A
=C
=T
)
Taq
(+,
G=
A, C
=T
)
Taq
(+,
G=
T, A
=C)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45Alcohol dehydrogenase
Figure 3. Statistical analyses of the fraction of preserved amino acids generated by 19 random mutagenesis methodsfor P450 BM-3 heme domain, glucose oxidase, arylesterase, and alcohol dehydrogenase. The performance of a non-biased random mutagenesis method is indicated by the black line.
Statistical Analysis of Random Mutagenesis Methods 863
the extent of hot spots generated by a randommutagenesis method. Apart from the bias of themutagenesis method, the codon diversity coefficientis influenced by the codon usage of the gene’sorigin. Most epPCR and chemical methods havehigh codon diversity coefficient values rangingfrom 14.3 to 56 (Figure 5). A more balanceddistribution of mutations among codons can beachieved using Pol I14 (codon diversity coefficientw1), Mutazyme II15 (3.3–4.3) and E. coli (mutA)16
(4.6–6.2).
Chemical diversity indicator: substitutionsleading to amino acids with changed chemicalproperties (hydrophobic, charged, neutral)
The third important performance criterion for arandom mutagenesis method is to analyze howchemically different the substituted amino acidsare. Such a chemical diversity indicator might be
helpful in the future to develop property-specificmutagenesis strategies. Non-conservative substi-tutions might be more promising for evolvingprotein towards non-natural conditions and proper-ties such as organic or thermal resistance. Conser-vative mutations might be more promising foraltering enantionselectivity, which requires oftenonly slight energetic changes.17 For thermal resis-tance it might be beneficial to use a randommutagenesis method that introduces mainlycharged amino acids, which form additional salt-bridges in loop areas.18
For the chemical diversity indicator, we groupedthe amino acids into four categories depending onchemical properties of amino acids. As mentioned,an ideal method allows us to substitute each residueequally with 19 other amino acids at each aminoacid position. After neglecting stop codons, weexpect an ideal “chemical distribution” of: 25%aliphatic amino acids (G, A, V, L, I; 5/20), 15%
Taq
(–,
G>
A=
C=
T)
Taq
(+,
G=A
=C=T
)
Taq
(+,
G>A
=C=T
)
Taq
(+,
G>
>A
=C
=T
)
Taq
(+,
G=A
, C=T
)
Taq
(+,
G=T
, A=C
)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(ex
o–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0
1
2
3
4
5
6
7
8
Ave
rage
am
ino
acid
sub
stitu
tion
s pe
r re
sidu
e
P450 BM–3 Heme domain
Taq
(–,
G>
A=
C=
T)
Taq
(+,
G=A
=C=T
)
Taq
(+,
G>A
=C=T
)
Taq
(+,
G>
>A
=C
=T
)
Taq
(+,
G=A
, C=T
)
Taq
(+,
G=T
, A=C
)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0
1
2
3
4
5
6
7
8
Ave
rage
am
ino
acid
sub
stitu
tion
s pe
r re
sidu
e
Glucose oxidaseT
aq (
–, G
>A
=C
=T
)
Taq
(+,
G=A
=C=T
)
Taq
(+,
G>A
=C=T
)
Taq
(+,
G>
>A
=C
=T
)
Taq
(+,
G=A
, C=T
)
Taq
(+,
G=T
, A=C
)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0
1
2
3
4
5
6
7
8
Ave
rage
am
ino
acid
sub
stitu
tion
s pe
r re
sidu
e
ArylesteraseT
aq (
–, G
>A
=C
=T
)
Taq
(+,
G=A
=C=T
)
Taq
(+,
G>A
=C=T
)
Taq
(+,
G>
>A
=C
=T
)
Taq
(+,
G=A
, C=T
)
Taq
(+,
G=T
, A=C
)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0
1
2
3
4
5
6
7A
vera
ge a
min
o ac
id s
ubst
ituti
ons
per
resi
due
Alcohol dehydrogenase
Figure 4. Statistical analyses of an average number of amino acid substitutions per residue of 19 random mutagenesismethods for P450 BM-3 heme domain, glucose oxidase, arylesterase, and alcohol dehydrogenase. The performance of anon-biased random mutagenesis method is indicated by the black line.
864 Statistical Analysis of Random Mutagenesis Methods
aromatic amino acids (F, Y, W; 3/20), 35% neutralamino acids (C, M, P, S, T, N, Q; 7/20) and 25%charged amino acids (D, E, H, K, R; 5/20).
In Table 2, we performed an analysis of 19random mutagenesis methods using cytochromeP450 BM-3 heme domain as example. The results ofthe analysis are reported as the deviation of eachrandom mutagenesis method from the ideal“chemical distribution” as described above. Theanalysis for a single nucleotide substitution revealsthat: (1) neutral and aromatic amino acid substi-tutions are under-represented; (2) aliphatic aminoacids are significantly over-represented; (3) chargedamino acid substitutions are over or under-represented depending on the employedmutagenesis methods; and (4) stop codons occurfrequently (6.3% using E. coli (mutA); 5.8% epRCA).A method with a non-biased mutational spectrumgives a chemical distribution of 35.6% aliphatic,
5.9% aromatic, 28.9% neutral, 24.9% charged aminoacids and 4.7% stop codons.
The reasons for the bias lie in the organization ofthe genetic code and in the different number ofcodons encoding for the same amino acid. Theaverage number of codons per amino acid is: (1) 4.2for aliphatic amino acids (21 codons encoding fiveamino acids); (2) 1.67 for aromatic amino acids (fivecodons encoding three amino acids); (3) three forneutral amino acids (21 codons encoding sevenamino acids); (4) 2.8 for charged amino acids(14 codons encoding five amino acids); and (5)three for stop codons (three codons for translationtermination).
It is important for the chemical diversity indicatorto analyze to what extent each amino acid species isgenerated. Figure 6 shows a detailed analysis of thesubstitution pattern for each of the 20 amino acidsof P450 BM-3 heme domain by comparing a method

Taq
(–,
G>A
=C=T
)
Taq
(+,
G=A
=C=T
)
Taq
(+,
G>A
=C=T
)
Taq
(+,
G>
>A=C
=T)
Taq
(+,
G=A
, C=T
)
Taq
(+,
G=T
, A=C
)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0
10
20
30
40
50
Cod
on d
iver
sity
coe
ffic
ient
(C
od)
P450 BM–3 Heme domain
Taq
(–,
G>A
=C=T
)
Taq
(+,
G=A
=C=T
)
Taq
(+,
G>A
=C=T
)
Taq
(+,
G>
>A=C
=T)
Taq
(+,
G=A
, C=T
)
Taq
(+,
G=T
, A=C
)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0
10
20
30
40
50
60
Cod
on d
iver
sity
coe
ffic
ient
(C
od)
Alcohol dehydrogenaseT
aq (
–, G
>A=C
=T)
Taq
(+,
G=A
=C=T
)
Taq
(+,
G>A
=C=T
)
Taq
(+,
G>
>A=C
=T)
Taq
(+,
G=A
, C=T
)
Taq
(+,
G=T
, A=C
)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0
10
20
30
40
50
60
Cod
on d
iver
sity
coe
ffic
ient
(C
od)
Arylesterase
Taq
(–,
G>A
=C=T
)
Taq
(+,
G=A
=C=T
)
Taq
(+,
G>A
=C=T
)
Taq
(+,
G>
>A=C
=T)
Taq
(+,
G=A
, C=T
)
Taq
(+,
G=T
, A=C
)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0
10
20
30
40
50
60C
odon
div
ersi
ty c
oeff
icie
nt (
Cod
)Alcohol dehydrogenase
Figure 5. Codon diversity coefficients calculated for 19 random mutagenesis methods and four enzymes (P450 BM-3heme domain, glucose oxidase, arylesterase and alcohol dehydrogenase). Codon diversity coefficient for a non-biasedrandom mutagenesis method is zero.
Statistical Analysis of Random Mutagenesis Methods 865
with non-biased mutational spectrum (left; equaloccurrence of each nucleotide substitution) to anepPCR method (right; Taq (C, GZAZCZT)). Thevalues on the Y-axis of Figure 6 represent thefraction of amino acid substitutions with differentamino acid side-chains. For a non-biased methodonly 55.5% of the substitutions are amino acids withchemically different side-chains. A biased epPCRmethod results in even less chemical diversity(45.6%). The redundancy of the genetic codepreserves strongly the chemical nature of aminoacids. The white spots in Figure 6 show the lowdiversity of amino acid substitution patterns thatcan be generated with state of the art mutagenesismethods such as epPCR.
Figure 7 shows the percentage of aliphatic,aromatic, neutral and charged amino acid substi-tutions generated by all 19 random mutagenesismethods for the four genes of interest. There aresignificant variations for each method when
applied to different genes, especially in the percen-tage values of aromatic amino acids. The differencesare larger for chemical methods compared toepPCR methods or a whole cell method.
Figure 8 shows the crystal structures of P450BM-3 heme domain, colored according to probabil-ities of (a) amino acid substitutions, (b) stop codonsand (c) Gly/Pro substitutions using an epPCRmethod (Taq (C, GZAZCZT), left) and a chemicalmethod (EMS, right) for mutagenesis. The graphicalrepresentation visualizes mutagenic hot spots andpositions of structurally/functionally less favorablesubstitutions (stop codons and Gly/Pro). A closerlook at catalytically important and well character-ized residues (R47, Y51, F87 and L188) of P450 BM-3heme domain19,20 reveal interesting differencesbetween an epPCR method (Taq (C, GZAZCZT))and a chemical method (EMS). For the chemicalmutagenesis method an almost equal substitutionprobability was calculated for the catalytically

Figure 6. Statistical analysis of amino acid substitution patterns of P450 BM-3 heme domain generated by a non-biasedrandom mutagenesis method (left) and an epPCR method (Taq (C, GZAZCZT), right). The Y-axis shows the originalamino acid species and the X-axis shows the substitution pattern. The substitution pattern for 20 amino acid species isindicated from red (lowest probability) to blue (highest probability). Amino acid substitutions that do not occur arecolored in white. The values on the Y-axis represent the fraction of amino acid substitutions with different amino acidside-chains.
866 Statistical Analysis of Random Mutagenesis Methods
important residues. For the epPCR method, theprobability of F87 substitution is twice compared tothat of R47 and L188. An average amino acidsubstitution per residue of 5.75 (epPCR method)and 2.5 (chemical method) can be achieved for thesefour residues. Position R47 plays an important role insubstrate binding through an ionic interactionbetween the positively charged R residue and thenegatively charged carboxylate of the natural fattyacid substrate.20,21 For position R47, we obtain thefollowing substitution pattern: (a) epPCR method7.7% H, 3.2% S, 1.2% P, 9.0% C, 2.6% L and 1.2% G; and(b) chemical method 25.4% H, 14.3% P, 25.4% C, 14.3%G. The epPCR method generates substitutions to Sand L that are not generated by the chemical method.The chemical method has also a 19.6-fold higherprobability of Gly/Pro substitutions. None of themethods generates the chemically similar lysineresidue. Position Y51 is the second amino acidproposed to be involved in substrate orientation byforming a H-bond between the carbonyl group of thefatty acid and hydroxyl group of Y residue.20 Forposition Y51, we obtain the following substitutionpattern: (a) epPCR method 2.8% stop codons, 34.1%H, 3.2% D, 9.7% N, 1.2% S, 35.4% C, 8.0% F; and (b)chemical method 17.7% stop codons, 25.5% H, 25.5%C. The epPCR method generates again a higherdiversity on the protein level (six substitutionscompared to two substitutions) with a low probabilityof generating a chemically similar serine. For thechemical method, the stop codon probability is veryhigh in absolute and relative numbers (5.5-fold higherthan epPCR method). Both methods fail to generatethe Y51F substitution, which increases the hydroxyl-ation efficiency towards aromatic compounds such asphenoxytoluene.22 Position 87 is in P450 BM-3
important for the regio- and stereoselectivity of thefatty acid hydroxylation.23 For position F87, we obtainthe following substitution pattern: (a) epPCR method24.2% S, 2.3% C, 6.9% Y, 6.9% I, 33.3% L, 2.3% V; and(b) chemical method 33.3% S, 33.3% L. For position 87,the same trends as for residues R47 and Y51 can beobserved in terms of substitution pattern anddiversity. The epPCR method generates, in contrastto the chemical method, the important F87Vmutation, which converts P450 BM-3 into a regio-and stereoselective epoxygenase.21,24 Position L188 inP450 BM-3 has been proven to influence substratespecificities in several studies.19,25,26 For positionL188, we obtain the following substitution pattern:(a) epPCR method 5.1% R, 15.5% Q, 54.5% P, 3.2% M,1.2% V; and (b) chemical method 20.6% P, 14.3% V.Again the epPCR method proves to be more diversethan the chemical method and generates importantsubstitutions such as L188Q.26 In summary, theepPCR method is shown, for the four investigatedresidues of P450 BM-3, to be more diverse than thechemical method. This might, however, be differentfor other codons and chemical methods. The gener-ated amino acid substitution patterns are, however,far below natural diversity and many importantsubstitutions would have been missed by bothmethods.
Discussion
We have developed a statistical method (MAP) toprovide protein engineers with a tool to developdirected evolution strategies by investigating theconsequences of mutational bias on the proteinlevel. The analyses of amino acid substitution

Non
–bia
sed
Taq
(–,
G>
A=
C=
T)
Taq
(+,
G=A
=C=T
)
Taq
(+,
G>A
=C=T
)
Taq
(+,
G>
>A
=C
=T
)
Taq
(+
, G=
A, C
=T)
Taq
(+
, G=
T, A
=C)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0.0
0.2
0.4
0.6
0.8
1.0
Frac
tion
of c
hem
ical
ly g
roup
ed
amin
o ac
id s
ubst
itut
ions
P450 BM–3 Heme domain
Non
–bia
sed
Taq
(–,
G>
A=
C=
T)
Taq
(+,
G=
A=C
=T
)
Taq
(+,
G>
A=C
=T
)
Taq
(+,
G>
>A
=C
=T
)
Taq
(+,
G=A
, C=
T)
Taq
(+,
G=T
, A=C
)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0.0
0.2
0.4
0.6
0.8
1.0
Frac
tion
of c
hem
ical
ly g
roup
edam
ino
acid
sub
stitu
tions
Glucose oxidase
Non
–bia
sed
Taq
(–,
G>
A=
C=
T)
Taq
(+,
G=A
=C=T
)
Taq
(+,
G>A
=C=T
)
Taq
(+
, G>
>A
=C
=T
)
Taq
(+,
G=A
, C=T
)
Taq
(+,
G=T
, A=C
)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0.0
0.2
0.4
0.6
0.8
1.0
Frac
tion
of c
hem
ical
ly g
roup
edam
ino
acid
sub
stitu
tion
s
Arylesterase
Non
–bia
sed
Taq
(–,
G>
A=
C=
T)
Taq
(+
, G=A
=C=T
)
Taq
(+
, G>A
=C=T
)
Taq
(+
, G>>
A=
C=
T)
Taq
(+,
G=A
, C=T
)
Taq
(+,
G=T
, A=C
)
Taq
(I6
14K
)
Mut
azym
e I
Mut
azym
e II
Pfu
(exo
–, D
473G
)
Tra
nscr
ipta
se
Taq
(dP
TP/
8–ox
odG
TP)
epR
CA
Pol I
E. c
oli (
mut
A)
Nitr
ous
acid
Form
ic a
cid
Hyd
razi
ne
EM
S
0.0
0.2
0.4
0.6
0.8
1.0Fr
actio
n of
che
mic
ally
gro
uped
amin
o ac
id s
ubst
itut
ions
Alcohol dehydrogenase
Figure 7. Statistical analyses of chemically categorized amino acid substitutions generated by 19 random mutagenesismethod for P450 BM-3 heme domain, glucose oxidase, arylesterase, and alcohol dehydrogenase. The fraction of stopcodons (TAA, TGA, TAG) is shown in red; charged amino acids (D, E, H, K, R) in magenta; neutral amino acids (C, M, P,S, T, N, Q) in yellow; aromatic amino acids (F, Y, W) in cyan; and aliphatic amino acids (G, A, V, L, I) in blue.
Statistical Analysis of Random Mutagenesis Methods 867
patterns for four genes of different origins anddifferent functions revealed that current bench-marks based on transition/transversion bias indi-cators are insufficient to describe and to comparethe performance of random mutagenesismethods. We therefore propose three indicatorsand one coefficient to describe the performance ofrandom mutagenesis methods: (1) protein structureindicator; (2) amino acid diversity indicator with acodon diversity coefficient; and (3) chemicaldiversity indicator.
The protein structure indicator describes theinfluence of structure/function-disrupting aminoacid substitutions and comprises two parts,percentage of stop codons and percentage ofGly/Pro substitutions. MAP analysis of fourgenes revealed that a random mutagenesis withnon-biased mutational spectrum (Ts/Tv Z0.5,AT/GC/GC/ATZ1) generates 2.9–4.7% of stopcodons and 11.1–16% of Gly/Pro substitutions.These high absolute values of structurally lessfavorable substitutions are hurdles for creating
functional mutant libraries with high mutationfrequencies (15–20 mutations per 1000 bp). In thecase of 20 mutations per gene, 44.5–61.8% ofvariants would contain at least one stop codonand 90.5–96.9% of variants would contain at leastone substitution to a Gly or Pro residue.
The numbers of stop codons and of Gly/Prosubstitutions differ significantly among the 19 ran-dom mutagenesis methods (factors up to 10.8 for stopcodon, Figure 1; and factor up to 4.4 for Gly/Pro,Figure 2). Interestingly, the random mutagenesismethod with the highest percentage of stop codonsgenerates for all four investigated enzymes the leastGly/Pro substitutions and vice versa. The proteinstructure indicator can therefore assist in adjustingmutation frequencies in random mutagenesismethod for obtaining functional libraries as acompromise between stop codons and Gly/Proresidues.
The amino acid diversity indicator can be regardedas a valuable benchmark for the power of a randommutagenesis method to generate diversity on the

Table 2. Amino acid substitution patterns generated by a non-biased random mutagenesis method and 19 randommutagenesis methods for P450 BM-3 heme domain
Stop (%) Charged (%) Neutral (%) Aromatic (%) Aliphatic (%)
Ideal chemical distribution 0 25 35 15 25Non-biased C4.65 K0.07 K6.08 K9.10 C10.60Taq (K, GOAZCZT) C3.23 K0.35 K5.58 K9.79 C12.49Taq (C, GZAZCZT) C2.33 C3.24 K9.28 K9.89 C13.61Taq (C, GOAZCZT) C3.75 C0.30 K8.48 K8.50 C12.92Taq (C, GOOAZCZT) C1.61 C3.67 K9.15 K10.26 C14.13Taq (C, GZA, CZT) C5.06 K1.88 K9.15 K7.05 C13.03Taq (C, GZT, AZC) C2.16 C2.94 K8.05 K10.03 C12.97Taq (I614K) C3.74 C0.43 K8.20 K8.46 C12.49Mutazyme I C5.76 K1.20 K4.68 K8.00 C8.12Mutazyme II C5.67 K1.85 K7.57 K7.13 C10.88Pfu (exo-, D473G) C5.20 K1.40 K7.56 K7.63 C11.39Transcriptase C3.51 K1.40 K4.78 K8.33 C11.00Taq (dPTP/8-oxodGTP) C0.92 C4.49 K7.42 K11.30 C13.30EpRCA C5.79 K2.12 K2.83 K7.85 C7.01Pol I C4.07 K0.19 K5.89 K8.49 C10.49E. coli (mutA) C6.32 K2.65 K5.67 K7.39 C9.40Nitrous acid C4.77 K1.04 K10.82 K7.77 C14.86Formic acid C5.73 K7.30 K2.32 K5.90 C9.80Hydrazine C2.73 K0.31 K6.83 K8.96 C13.36EMS C2.54 C3.13 K5.48 K10.69 C10.50
Deviation in composition of categories (stop, charged, neutral, aromatic and aliphatic) from an ideal chemical distribution (amino acids:25% charged, 35% neutral, 15% aromatic, and 25% aliphatic) are given.
868 Statistical Analysis of Random Mutagenesis Methods
protein level. It is a measure for the redundancy of thegenetic code and the bias of random mutagenesismethods. Biased epPCR methods generate, onaverage, only 34% of the possible diversity on theprotein level and a large fraction of the substitutionsresult in unchanged amino acids (16.3–34.6%). Theperformance of epPCR methods is comparable to thatof a random mutagenesis method with non-biasedmutational spectrum (37% of possible diversity on theprotein level, Figure 4). The latter result proves thatcurrent bias indicators (transition and transversionbias) are insufficient for describing the performanceof random mutagenesis methods.
The amino acid diversity indicator should becomplemented by a codon diversity coefficient thatmeasures the distribution of amino acid substi-tutions over a whole protein sequence (Figure 5).Mutations occur at preferred codons (mutagenichot spots), since mutational spectra of most randommutagenesis methods are biased. MAP analysisallows us to compare mutagenic preferences ofdifferent mutagenesis methods and to combinerandom mutagenesis methods with different pre-ferences in order to maximize diversity. The codondiversity coefficient values differ significantlyamong the random mutagenesis methods (Figure 5).
Chemical diversity indicator is an importantmeasure of the chemical diversity of amino acidsubstitutions generated by a random mutagenesismethod. We hope that the chemical diversityindicator will assist in the future to developproperty-specific mutagenesis strategies in whicha bias to charged, hydrophobic or stericallydemanding amino acid substitutions is beneficialfor the property of interest (for examples, seeResults). A detailed analysis for the P450 BM-3heme domain revealed that the organization of thegenetic code favors strongly conservative amino
acid substitutions, resulting in chemically similaramino acids (Table 2 and Figure 6). These findingsare in good agreement with the physicochemicaland ambiguity reduction theory,10,27 which pro-poses that the genetic code structure was organizedto reduce the deleterious effect of physicochemicalproperties between amino acids differing in onebase of a codon. The current bias indicators (ratiosof Ts/Tv and AT/GC/GC/AT) do not considerthe chemical diversity of amino acid substitutions.
Conclusions and Propositions for NovelRandom Mutagenesis Methods
MAP analyses shed light on the performance ofcurrent random mutagenesis methods. The mainconclusions that can be drawn on the performance ofcurrent genetic engineering methods are: (a) lack ofmutagenesis methods bypassing stop codons andminimizing structurally destabilizing amino acidsubstitutions (protein structure indicator); (b) lowaverage number of amino acid substitutions perresidue (amino acid diversity indicator); and (c) anon-biased mutagenesis method with single nucleo-tide substitution in one codon performs poorly due tothe organization of the genetic code and is far frombeing chemically diverse (chemical diversity indi-cator).
The analysis of a non-biased mutagenesis methodhas proven that the current bias indicators (tran-sition and transversion bias) are insufficient toevaluate the performance of a random mutagenesismethod and cannot be used as a quality measure orbenchmark for a mutagenesis method. The exampleof the Taq (dPTP/8-oxodGTP) method13 shows thata biased method can even lead to a lower number ofstop codons. The organization of the genetic code

Figure 8. P450 BM-3 hemedomain colored according to (a)amino acid substitution prob-ability, (b) stop codon probabilityand (c) Gly/Pro substitution prob-ability using an epPCR method(Taq (C, GZAZCZT), left) and achemical method (EMS, right).The probability values increasefrom red (lowest probability) toblue (highest probability).
Statistical Analysis of Random Mutagenesis Methods 869
with stop codons and structurally destabilizingamino acids prevents current random mutagenesismethods from generating functional mutantlibraries with high mutation frequencies (15–20mutations per 1000 bp). The analysis of the tran-sition bias on the chemical properties revealed thatamino acids are preferentially substituted bychemically similar amino acids (e.g. hydrophobicamino acids substituted by hydrophobic ones).
Instead of developing methods without transitionand transversion bias, we have identified, using MAPanalysis, the need for mutagenesis methods thatgenerate more diversity on the protein level orgenerate subsets of amino acid substitutions. Onedesirable example would be to replace each aminoacid of a protein by charged or hydrophobicamino acids. In this regard, a transversion biasedmutagenesis method would be beneficial, since sucha method would increase the number of amino acid
substitutions with different chemical properties. Thediversity of random mutagenesis methods can also beincreased if methods with two or three consecutivenucleotide substitutions could be developed. Avoid-ing, for instance, CC and GG pairs would furtherreduce Gly and Pro substitutions. A notable stepforward has been achieved by the random insertionand deletion (RID) mutagenesis method,28 which,however, lacks technical simplicity (nine ligationsteps) and success stories are yet to be reported.
Until those novel random mutagenesis methodsare developed, it is advisable to change themutational bias by changing the random muta-genesis method used for mutant library generation.In this respect, MAP can be a valuable tool fordeveloping mutational strategies by comparingthe bias of random mutagenesis methods. Asbenchmarks for a random mutagenesis method,we propose to use in the future three protein

870 Statistical Analysis of Random Mutagenesis Methods
diversity indicators describing the extent and thechemical nature of amino acid substitutions.Currently, we are extending the MAP analysis totake secondary structure elements and stericconstraints in proteins with known 3D structuresinto account.
MAP has been made publicly available† and itallows, upon a DNA sequence input, a statisticalanalysis for 19 random mutagenesis methods.
Methods
Enzymes selected for MAP analysis
MAP analysis has been performed on four genesencoding: (1) heme domain of cytochrome P450 BM-3from B. megaterium (NCBI GenBank accession numberJ04832);29 (2) glucose oxidase from A. niger (AJ294936);30
(3) arylesterase from P. fluorescens (D12484);31 and (4)alcohol dehydrogenase from S. cerevisiae (V01292).32
Random mutagenesis methods included in MAP
Nineteen random mutagenesis methods wereimplemented into MAP. These methods comprise 14enzymatic methods (six error-prone polymerase chainreactions employing Mn2C and/or unbalanced nucleo-tide concentrations,33–35 Taq polymerase mutant I614K,36
Mutazyme I,37 Mutazyme II,15 Pfu polymerase (exo-)mutant D437G,38 reverse transriptase,39 Taq polymerasewith nucleotide analogues dPTP/8-oxodGTP,13 error-prone rolling circle amplification,40 Polymerase Imethod14), one whole cells-based method (E. coli expres-sing mutA allele of glyV gene16) and four chemicalmethods (employing nitrous acid,41 formic acid,41 hydra-zine41 and ethyl methane sulfonate42).
Mutational spectra included in MAP
The mutational spectra were changed in two wayscompared to the reported data.4 Firstly, insertion anddeletion with an occurrence frequency between 0.8% and13.9% were neglected and occurrence of remainingnucleotide substitutions was scaled proportionally to100%. Secondly, mutations in upper and lower DNA strandresulting in equivalent substitutions (e.g. A/C (lowerstrand), T/G (upper stand)) were considered to occur withequal frequency. The final mutational spectra used in astatistical MAP analysis are summarized in Table 1.
A Perl script was written to analyze all the possibleamino acid substitutions resulting from a single nucleo-tide exchange in one codon of the gene of interest. Aminoacid substitutions caused by two or more consecutivenucleotide exchanges were excluded due to limitations ofcurrent random mutagenesis methods.4 Three sub-sequent nucleotides (one codon) encode one amino acidin a protein. For a protein with N amino acids andconsidering a single nucleotide exchange in a codon, aDNA sequence space of N![3C3C3]!2 variants wassampled. Every nucleotide position of a codon wasinvestigated independently by replacing every nucleotideposition of a codon by three other nucleotides, resulting in(3C3C3) exchanges per codon. Since substitutions occur
† http://gis-web.iu-bremen.de/MAP/
at the sense and the anti-sense strand, a factor of two hasbeen added.
Statistics and definitions
Amino acids were grouped into categories dependingon the chemical properties of amino acid side-chains:category 1, aliphatic side-chains (G, A, V, L, I); category 2,aromatic side-chains (F, Y, W); category 3, neutral side-chains (C, M, P, S, T, N, Q); category 4, charged side-chains (D, E, H, K, R), and stop codons. The followingtechnical terms were used: (1) fraction of variants withstop codons: fraction of single nucleotide substitutionsresulting in a stop codon (TAA, TGA or TAG); (2) fractionof variants with Gly or Pro: fraction of single nucleotidesubstitutions resulting in one of the following codons(GGA, GGT, GGG, GGC, CCA, CCT, CCG or CCC); (3)fraction of variants with preserved amino acids: fractionof single nucleotide substitutions that do not change theencoded amino acid; (4) average amino acid substitutionper residue: average number of amino acid substitutionsafter single nucleotide exchange of a codon; (5) codondiversity coefficient: coefficient of variance calculated onthe probabilities of generating single nucleotide substi-tution in each codon encoding an amino acid in theprotein of interest.
Acknowledgements
We thank German Academic Exchange Service(DAAD, Bonn, Germany) and Deutsche For-schungsgemeinschaft (DFG, award SCHW-790/2-1) for financial support.
References
1. Arnold, F. H., Wintrode, P. L., Miyazaki, K. &Gershenson, A. (2001). How enzymes adapt: lessonsfrom directed evolution. Trends Biochem. Sci. 26,100–106.
2. Petrounia, I. P. & Arnold, F. H. (2000). Designedevolution of enzymatic properties. Curr. Opin. Biotech.11, 325–330.
3. Locher, C. P., Soong, N. W., Whalen, R. G. &Punnonen, J. (2004). Development of novel vaccinesusing DNA shuffling and screening strategies. Curr.Opin. Mol. Ther. 6, 34–39.
4. Wong, T. S., Zhurina, D. & Schwaneberg, U. (2005).The diversity challenge in directed protein evolution.Comb. Chem. High T. Scr.. In the press.
5. Goddard, J. P. & Reymond, J. L. (2004). Enzyme assaysfor high-throughput screening. Curr. Opin. Biotech. 15,314–322.
6. Goddard, J. P. & Reymond, J. L. (2004). Recentadvances in enzyme assays. Trends Biotechnol. 22,363–370.
7. Reymond, J. L. (2004). Spectrophotometric enzymeassays for high-throughput screening. Food Technol.Biotech. 42, 265–269.
8. Ardell, D. H. & Sella, G. (2001). On the evolution ofredundancy in genetic codes. J. Mol. Evol. 53, 269–281.
9. Freeland, S. J., Wu, T. & Keulmann, N. (2003). The casefor an error minimizing standard genetic code.Origins Life Evol. B, 33, 457–477.

Statistical Analysis of Random Mutagenesis Methods 871
10. Woese, C. R. (1965). Order in the genetic code. Proc.Natl Acad. Sci. USA, 54, 71–75.
11. Georgiou, G. & Segatori, L. (2005). Preparativeexpression of secreted proteins in bacteria: status reportand future prospects. Curr. Opin. Biotech. 16, 538–545.
12. Kuchner, O. & Arnold, F. H. (1997). Directed evolutionof enzyme catalysts. Trends Biotechnol. 15, 523–530.
13. Zaccolo, M., Williams, D. M., Brown, D. M. & Gherardi,E. (1996). An approach to random mutagenesis of DNAusing mixtures of triphosphate derivatives of nucleo-side analogues. J. Mol. Biol. 255, 589–603.
14. Camps, M., Naukkarinen, J., Johnson, B. P. & Loeb, L. A.(2003). Targeted gene evolution in Escherichia coli using ahighly error-prone DNA polymerase I. Proc. Natl Acad.Sci. USA, 100, 9727–9732.
15. Stratagene Inc. (2004). Overcome mutational bias.Strategies, 17, 20–21.
16. Balashov, S. & Humayun, M. Z. (2004). Specificity ofspontaneous mutations induced in mutA mutatorcells. Mutat. Res-Fund. Mol. M. 548, 9–18.
17. Henke, E., Bornscheuer, U., Schmid, R. & Pleiss, J.(2003). A molecular mechanism of enantiorecognitionof tertiary alcohols by carboxylesterases. ChemBio-chem, 4, 485–493.
18. Wintrode, P., Miyazaki, K. & Arnold, F. (2001). Patternsof adaptation in a laboratory evolved thermophilicenzyme. BBA-Protein Struct. M. 1549, 1–8.
19. Li, Q., Schwaneberg, U., Fischer, M., Schmitt, J., Pleiss, J.,Lutz-Wahl, S. & Schmid, R. (2001). Rational evolution ofa medium chain-specific cytochrome P-450 BM-3variant. BBA-Protein Struct. M. 1545, 114–121.
20. Munro, A., Leys, D., McLean, K., Marshall, K., Ost, T.,Daff,S.etal. (2002).P450BM3:theverymodelofamodernflavocytochrome. Trends Biochem. Sci. 27, 250–257.
21. Graham-Lorence, S., Truan, G., Peterson, J. A.,Falck, J. R., Wei, S., Helvig, C. & Capdevila, J. H.(1997). An active site substitution, F87V, convertscytochrome P450 BM-3 into a regio- and stereoselective(14S,15R)-arachidonic acid epoxygenase. J. Biol. Chem.272, 1127–1135.
22. Wong, T. S., Wu, N., Roccatano, D., Zacharias, M. &Schwaneberg, U. (2005). Sensitive assay for laboratoryevolution of hydroxylases toward aromatic and hetero-cyclic compounds. J. Biomol. Screen. 10, 246–252.
23. Schwaneberg, U., Schmidt-Dannert, C., Schmitt, J. &Schmid, R. (1999). A continuous spectrophotometricassay for P450 BM-3, a fatty acid hydroxylating enzyme,and its mutant F87A. Anal. Biochem. 269, 359–366.
24. Chen, J., Wang, D., Falck, J., Capdevila, J. & Harris, R.(1999). Transfection of an active cytochrome P450arachidonic acid epoxygenase indicates that 14,15-epoxyeicosatrienoic acid functions as an intra-cellular second messenger in response to epidermalgrowth factor. J. Biol. Chem. 274, 4764–4769.
25. Appel, D., Lutz-Wahl, S., Fischer, P., Schwaneberg, U.& Schmid, R. D. (2001). A P450 BM-3 mutanthydroxylates alkanes, cycloalkanes, arenes and het-eroarenes. J. Biotechnol. 88, 167–171.
26. Li, Q. S., Schwaneberg, U., Fischer, P. & Schmid, R. D.(2000). Directed evolution of the fatty-acid hydroxyl-ase P450 BM-3 into an indole-hydroxylating catalyst.Chemistry, 6, 1531–1536.
27. Di Giulio, M. (2005). The origin of the genetic code:theories and their relationships, a review. Biosystems, 80,175–184.
28. Murakami, H., Hohsaka, T. & Sisido, M. (2002).Random insertion and deletion of arbitrary numberof bases for codon-based random mutation of DNAs.Nature Biotechnol. 20, 76–81.
29. Ruettinger, R. T., Wen, L. P. & Fulco, A. J. (1989). Codingnucleotide, 50 regulatory, and deduced amino-acidsequences of P-450BM-3, a single peptide cytochromeP-450-NADPH P-450 reductase from Bacillusmegater-ium. J. Biol. Chem. 264, 10987–10995.
30. Hatzinikolaou, D. G., Hansen, O. C., Macris, B. J.,Tingey, A., Kekos, D., Goodenough, P. & Stougaard, P.(1996). A new glucose oxidase from Aspergillus niger:characterization and regulation studies of enzyme andgene. Appl. Microbiol. Biot. 46, 371–381.
31. Choi, K. D., Jeohn, G. H., Rhee, J. S. & Yoo, O. J. (1990).Cloning and nucleotide-sequence of an esterase genefrom Pseudomonas fluorescens and expression of thegene in Escherichia coli. Agr. Biol. Chem. Tokyo, 54,2039–2045.
32. Bennetzen, J. L. & Hall, B. D. (1982). The primarystructure of the Saccharomyces cerevisiae gene foralcohol dehydrogenase-I. J. Biol. Chem. 257, 3018–3025.
33. LinGoerke, J. L., Robbins, D. J. & Burczak, J. D. (1997).PCR-based random mutagenesis using manganeseand reduced dNTP concentration. Biotechniques, 23,409–412.
34. Shafikhani, S., Siegel, R. A., Ferrari, E. & Schellenberger,V. (1997). Generation of large libraries of randommutants in Bacillus subtilis by PCR-based plasmidmultimerization. Biotechniques, 23, 304–310.
35. Vartanian, J. P., Henry, M. & WainHobson, S. (1996).Hypermutagenic PCR involving all four transitionsand a sizeable proportion of transversions. Nucl AcidsRes. 24, 2627-2631.
36. Patel, P. H., Kawate, H., Adman, E., Ashbach, M. &Loeb, L. K. (2001). A single highly mutable catalyticsite amino acid is critical for DNA polymerase fidelity.J. Biol. Chem. 276, 5044–5051.
37. Cline, J. & Hogrefe, H. (2000). Randomize genesequences with new PCR mutagenesis kit. Strategies,13, 157–162.
38. Biles, B. D. & Connolly, B. A. (2004). Low-fidelityPyrococcus furiosus DNA polymerase mutants usefulin error-prone PCR. Nucl. Acids Res. 32, e176.
39. Lehtovaara, P. M., Koivula, A. K., Bamford, J. &Knowles, J. K. C. (1988). A new method for randommutagenesis of complete genes: enzymatic generationof mutant libraries in vitro. Protein Eng. Des. Sel. 2, 63–68.
40. Fujii, R., Kitaoka, M. & Hayashi, K. (2004). One-steprandom mutagenesis by error-prone rolling circleamplification. Nucl. Acids Res. 32, e145.
41. Myers, R. M., Lerman, L. S. & Maniatis, T. (1985).A general-method for saturation mutagenesis ofcloned DNA fragments. Science, 229, 242–247.
42. Lai, Y. P., Huang, J., Wang, L. F., Li, J. & Wu, Z. R.(2004). A new approach to random mutagenesisin vitro. Biotechnol. Bioeng. 86, 622–627.
Edited by F. Schmid
(Received 4 September 2005; received in revised form 26 October 2005; accepted 28 October 2005)Available online 17 November 2005