activities required for bacteriophage t4 dna replication
TRANSCRIPT

Proc. NatL Acad. Sci. USAVol. 80, pp. 1669-1673, March 1983Genetics
Bacterial and phage mutations that reveal helix-unwindingactivities required for bacteriophage T4 DNA replication
(DNA polymerase/DNA-dependent ATPase/DNA helicase/gene optA)
PETER GAUSS, DANIEL H. DOHERTY, AND LARRY GOLDDepartment of Molecular, Cellular and Developmental Biology, University of Colorado, Campus Box 347, Boulder, Colorado 80309
Communicated by William B. Wood, December 3, 1982
ABSTRACT An Escherichia coli strain with a mutation in theoptA gene restricts the growth of bacteriophage T4 strains par-tially defective in gene 43 (DNA polymerase) or missing gene dda(DNA-dependent ATPase). The mutations in the dda gene inac-tivate a DNA-dependent ATPase that has been shown to have DNAhelicase activity in vitro. We show that the restriction of phagegrowth after infection of the optA bacterium is the result of a blockin DNA replication. We infer that the block arises from a defectin DNA unwinding.
Although many DNA helicase activities have been identified,the physiological role of these proteins is not understood (forreview, see ref. 1). At present, there is only one instance of es-sential involvement of a helicase in DNA replication. The DNAhelicase identified as the product of the Escherichia coli repgene is essential for the replication of bacteriophage 4X174DNA(2, 3). However, the mutations in the rep gene that eliminate4X174 replication have little or no effect on E. coli DNA rep-lication (4). The apparent absence of involvement of the repprotein in DNA replication in E. coli could derive in part fromthe encoding of homologous functions elsewhere in the ge-nome. Indeed, at least four helicases are produced by E. coli (1).Likewise, during phage infection, duplication of host functionsby phage gene products (or complex interactions between hostand phage gene products) might obscure essential contributionsthese enzymes make during DNA replication. We describe agenetic system, using mutant strains, that will be useful in de-fining the in vivo function(s) of DNA helicases.Our work was facilitated by the isolation of an E. coli mutant
strain unable to support the growth of bacteriophage T7 strainscarrying mutations in gene 1.2 (5). The defective host strain wasshown to have a mutation in the optA gene (restricts one pointtwo mutants of T7). T7 1.2- infections of optA strains were de-fective in DNA synthesis (5). We therefore screened bacterio-phage T4 strains that were carrying mutations in genes that af-fect DNA metabolism for the ability to grow on the optA host.
Surprisingly, we have found that optA hosts infected with T4mutants are defective in DNA synthesis when the infecting phagecarries certain missense mutations in gene 43, encoding the T4DNA polymerase, or null mutations of the gene dda, encodinga T4 DNA helicase (6-8). We present a model for T4 DNA rep-lication in whichDNA unwinding activity is provided by at leastthree proteins working in concert at a replication fork.
MATERIALS AND METHODSBacterial and Phage Strains. E. coli strains HR40 (optA, supE),
HR42 (optA+, sup0), and HR44 (optA, sup0) are derivatives ofPA610 (optA+, supE) and were obtained from C. C. Richardson
(5). HR44. 1 and HR42. 1 are supD derivatives of HR44 and HR42,respectively. They were constructed by P1 transduction in ourlaboratory. NapIV was obtained from M. A. Nelson (9). Iso-geneic NapIV optA+ and optA- derivatives were constructed byP1 transduction. The optAl mutation is probably a transitionmutation because it was isolated after nitrosoguanidine muta-genesis (10). Strains KLF723 (F'104) (11) and AT982 (dapD4)(12) were obtained from B. Bachmann. B40 (supD) was obtainedfrom C. Yegian (13). The T4 mutant dsd was obtained from RickCunningham (14). The mutant mf6 was obtained from W. B.Wood. This mutant is identical to the mutant m identified byDe Waard et aL (7). The gene 43 mutants are from the T4 stockcollection of W. B. Wood and have been described (15). The ddamutant L148 was obtained from K. Ebizusaki (8). The sudl dele-tion strain was from J. Little (16). The Homyk and Weil del(39-56) series of deletion strains (17) was obtained from W. B. Wood.The dexA mutant ndlOO' was obtained from D. P. Snustad (18).The dexA mutant hus3 was obtained from D. Hall (19). The mu-tants L148, ndlOO', and hus3 were isolated after hydroxylaminemutagenesis and probably are point mutations (20, 21). The L148mutant-infected bacteria were defective in a DNA-dependentATPase activity (8). The ndlOO' and hus3 mutant-infected bac-teria were defective in an exonuclease-like activity; this activitywas assayed in a manner that would allow apparent exonucleaseactivity to depend on a helicase activity (18, 19). The sudl straincarries a deletion mutation isolated as a partial suppressor ofgene 32 mutant-infected cells (16, 17). The del(39-56) mutantswere isolated as deletion strains that arose in strains carryingduplications of the rII genes (17). The mutant PR1 was isolatedin our laboratory after proflavin mutagenesis (22). Bacterio-phage PICMclr100 was from our collection (23).DNA Synthesis. DNA synthesis was measured in M9 me-
dium. Newly synthesized DNA was labeled continuously start-ing at 4 min after infection with [methyl-3H]thymidine (Amer-sham) at 50ACi (1 Ci = 3.7 x 1010 Bq)/ml. Unlabeled thymidineand 2'-deoxyadenosine also were added at 2 and 25 Ag/ml, re-spectively, at 4 min after infection. Total incorporation was mea-sured by pipetting 0.05-ml aliquots of labeled cells into tubescontaining 4 ml of cold 5% trichloroacetic acid and 150 ug ofcarrier bovine serum albumin. These samples were then fil-tered through Whatman glass fiberfilters. Thefilterswere rinsedtwice with 5% trichloroacetic acid and once with 95% ethanol.The filters were then dried and assayed for radioactivity in a tol-uene-based scintillation fluid.Growth of Phage and Bacteria. H broth, EHA top and bot-
tom layer agar, and M9 medium have been described (24). Allinfections were performed at a cell density of 2 X 108/ml andat a multiplicity of infection of 5. Experiments were performedat 30'C unless otherwise indicated. P1 transductions, bacterialmatings, and episome curing were as described by Miller (25).
Abbreviation: ds DNA, double-stranded DNA.
1669
The publication costs of this article were defrayed in part by page chargepayment. This article must therefore be hereby marked "advertise-ment" in accordance with 18 U. S. C. §1734 solely to indicate this fact.
Proc. Natl. Acad. Sci. USA 80 (1983)
RESULTSA Host Mutation in the optA Gene Is Responsible for the
Restriction of dda or 43 Phage. We screened numerous bac-teriophage T4 strains, with missense or null mutations in thefollowing genes, for the ability to form plaques at 30'C on an E.coli optA strain: 30, 32, 33, 39, 41, 42, 43, 44, 45, 46, 47, 49,52, 55, 59, 60, 61, rII, w, fdsA, fdsB, mot, and dda. Only genedda and some gene 43 mutants were restricted on the optA host(see below). Saito and Richardson (5) mapped the optAl mu-tation at 3.6 min using as the phenotype the optA restriction ofthe T7 1.2- phage (5). The optA mutation was 92% cotransducedwith dapD. Becuse the mutation restricting T4 mutants alsoshows >90% cotransduction with the dapD locus (datanot shown),the optAl host mutation is probably the defect in the HR44 strainthat restricts T4 dda and 43 mutant infections.We have used an episome containing the optA+ gene to show
that the optAl allele is recessive. We transferred the F'104 epi-some (which carries optA+) into optA strains. Merodiploid strainscontaining the optA+ gene on the episome and optA on the chro-mosome were permissive for T4 gene 43 and dda mutant in-fections (data not shown). When F'104 (optA+)/HR44 was curedof the episome, the resulting F- optA strain again restricted thegrowth of gene 43 mutants.
Characterization of T4 Mutants Restricted on the optA HostStrain: DNA Helicase Mutants. The dda mutation L148 mapsin an ill-characterized region of the T4 genome (8, 26). We testedother bacteriophage T4 strains, carrying mutations that map inthe same region as dda, for restriction on the optA strain HR44.The following mutants were restricted: ndlO0' (dexA), hus3(dexA), sudl (sud), the Homyk and Weil (39-56) deletions 1-5,and a newly isolated mutant PR1. The restricted mutants all failedto complement the mutant L148 (dda) and each other (done at42°C; data not shown). Behme and Ebisuzaki (8) showed thatdeletion mutants del(39-56) 2, 3, and 4 and the point mutantL148 (dda) each failed to induce the DNA-dependent ATPaseactivity identified as the dda gene product. This ATPase activityhas been purified and shown also to have DNAhelicase activityin vitro (27, 28). The sudl and del(39-56) strains have previouslybeen identified as partial suppressors of gene 32 deficiencies(16, 17). We have recently found that the point mutation in theL148 strain also affects gene 32 function in vivo (29). In fact, Al-berts et aL (30) report that the dda gene product interacts di-rectly with the gene 32 protein. The above results suggest, butdo not prove, that the mutant phenotypes are the result of acommon biochemical defect, the absence of the DNA helicaseencoded by the dda gene.
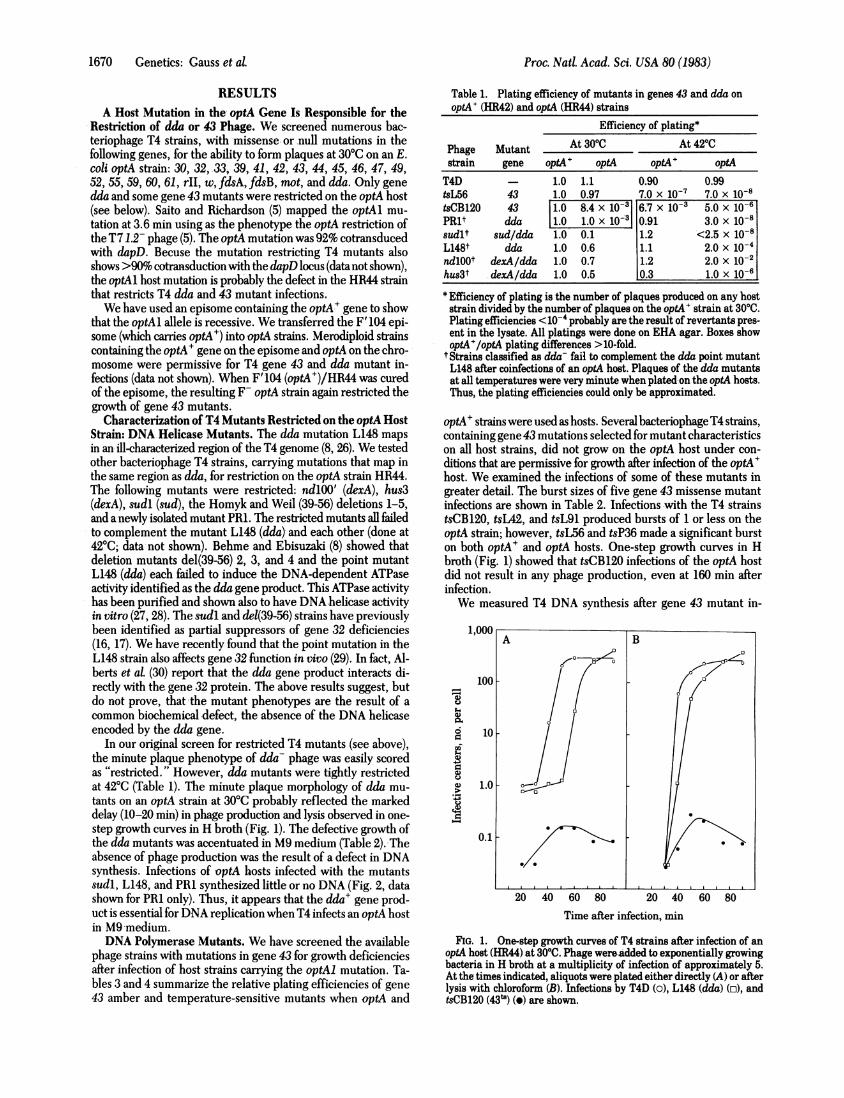
In our original screen for restricted T4 mutants (see above),the minute plaque phenotype of dda- phage was easily scoredas "restricted." However, dda mutants were tightly restrictedat 42°C (Table 1). The minute plaque morphology of dda mu-tants on an optA strain at 30°C probably reflected the markeddelay (10-20 min) in phage production and lysis observed in one-step growth curves in H broth (Fig. 1). The defective growth ofthe dda mutants was accentuated in M9 medium (Table 2). Theabsence of phage production was the result of a defect in DNAsynthesis. Infections of optA hosts infected with the mutantssudl, L148, and PRl synthesized little or no DNA (Fig. 2, datashown for PRl only). Thus, it appears that the dda+ gene prod-uct is essential for DNA replication when T4 infects an optA hostin M9 medium.DNA Polymerase Mutants. We have screened the available
phage strains with mutations in gene 43 for growth deficienciesafter infection of host strains carrying the optAl mutation. Ta-bles 3 and 4 summarize the relative plating efficiencies of gene43 amber and temperature-sensitive mutants when optA and
Table 1. Plating efficiency of mutants in genes 43 and dda onoptA' (HR42) and optA (HR44) strains
Efficiency of plating*
Phage Mutant At'300C At 420Cstrain gene optA+ optA optA+ optAT4D - 1.0 1.1 0.90 0.99tsL56 43 1.0 0.97 7.0 x 10-7 7.0 x 10-8tsCB120 43 1.0 8.4 x 10-3| 6.7 x 10-3 5.0 x 10-6PRit dda |10 1.0 x 10-3 0.91 3.0 x 10-8sud1t sudldda 1.0 0.1 1.2 <2.5 x 10-8L148t dda 1.0 0.6 1.1 2.0 x 10-4ndlOOt dexAidda 1.0 0.7 1.2 2.0 x 10-2hus3t dexAidda 1.0 0.5 0.3 1.0 x 10-6* Efficiency of plating is the number of plaques produced on any hoststrain divided by the number of plaques on the optA+ strain at 300C.Plating efficiencies <10-' probably are the result of revertants pres-ent in the lysate. All platings were done on EHA agar. Boxes showoptA+/optA plating differences >10-fold.
tStrains classified as dda- fail to complement the dda point mutantL148 after coinfections of an optA host. Plaques of the dda mutantsat all temperatures were very minute when plated on the optA hosts.Thus, the plating efficiencies could only be approximated.
optA' strains were used as hosts. Several bacteriophageT4 strains,containing gene 43 mutations selected for mutant characteristicson all host strains, did not grow on the optA host under con-ditions that are permissive for growth after infection of the optA+host. We examined the infections of some of these mutants ingreater detail. The burst sizes of five gene 43 missense mutantinfections are shown in Table 2. Infections with the T4 strainstsCB120, tsL42, and tsL91 produced bursts of 1 or less on theoptA strain; however, tsL56 and tsP36 made a significant burston both optA+ and optA hosts. One-step growth curves in Hbroth (Fig. 1) showed that tsCB120 infections of the optA hostdid not result in any phage production, even at 160 min afterinfection.We measured T4 DNA synthesis after gene 43 mutant in-
1,000 A B
100
'0~~~~~~~~~~~~~~~~
6 10
'0.
Time after infection, min
FIG. 1. One-step growth curves of T4 strains after infection of anoptA host (HR44) at 30°C. Phage wereadded to exponentially growingbacteria in H broth at a multiplicity of infection of approximately 5.At the times indicated, aliquots were plated either directly (A) or afterlysis with chloroform (B). Infections by T4D (o), L148 (dda) (o), andtsCB120 (43s) (e) are shown.
1670 Genetics: Gauss et al.
Proc. Nadl. Acad. Sci. USA 80 (1983) 1671
Table 2. Burst size of gene 43 and dda mutant infections ofoptA' and optA NapIV host strains
Phage Mutant Burst size*strain gene optA+ optAT4D - 470 540tsL56 43 190 110tsP36 43 56 14tsL42 43 250 1.1tsL91 43 180 0.22tsCB120 43 210 0.23L148 dda 95 0.84PR1 dda 300 9.7
* Host cells were grown at 300C in M9 to a concentration of 2 x 108cells per ml. Cell titer was determined immediately before infection.At 120 min after infection, the cells were lysed with chloroform andviable phage titers were determined on B40 at 30'C. Burst sizes werecalculated as the number of viable phage per cell.
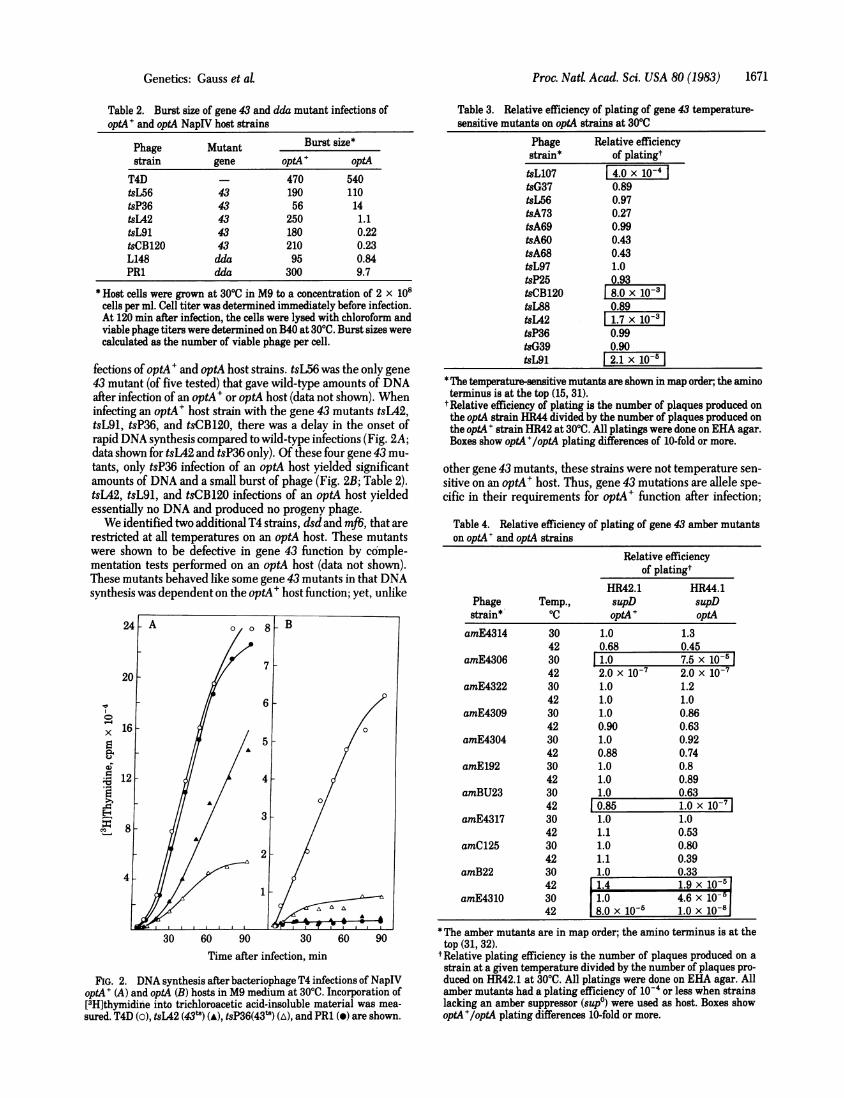
fections of optA' and optA host strains. tsL56 was the only gene43 mutant (of five tested) that gave wild-type amounts of DNAafter infection of an optA+ or optA host (data not shown). Wheninfecting an optA' host strain with the gene 43 mutants tsL42,tsL91, tsP36, and tsCB120, there was a delay in the onset ofrapid DNA synthesis compared to wild-type infections (Fig. 2A;data shown for tsL42 and tsP36 only). Of these four gene 43 mu-tants, only tsP36 infection of an optA host yielded significantamounts of DNA and a small burst of phage (Fig. 2B; Table 2).tsL42, tsL91, and tsCB120 infections of an optA host yieldedessentially no DNA and produced no progeny phage.We identified two additional T4 strains, dsd and mf6, that are
restricted at all temperatures on an optA host. These mutantswere shown to be defective in gene 43 function by comple-mentation tests performed on an optA host (data not shown).These mutants behaved like some gene 43 mutants in that DNAsynthesis was dependent on the optA+ host function; yet, unlike
16-5
~12-4
3-
2
4
AA
30 60 90 30 60 90
Time after infection, min
FIG. 2. DNA synthesis after bacteriophage T4 infections of NapIVoptA+ (A) and optA (B) hosts in M9 medium at 300C. Incorporation of[3H]thymidine into trichloroacetic acid-insoluble material was mea-sured. T4D (o), tsL42 (43t) (A), tsP36(43') (A), and PR1 (0) are shown.
Table 3. Relative efficiency of plating of gene 43 temperature-sensitive mutants on optA strains at 300C
Phage Relative efficiencystrain* of platingttsLlO7tsG37tsL56tsA73tsA69tsA60tsA68tsL97tsP25tsCB120tsL88tsL42tsP36tsG39tsL91
14.0 x 10-4 10.890.970.270.990.430.431.00.938.0x1x -0.891.7 x 10-s0.990.902.1 x 10-5 1
*The temperature-sensitive mutants are shown in map order; the aminoterminus is at the top (15, 31).
tRelative efficiency of plating is the number of plaques produced onthe optA strain HR44 divided by the number of plaques produced onthe optA+ strain HR42 at 3000. All platings were done on EHA agar.Boxes show optA+/optA plating differences of 10-fold or more.
other gene 43 mutants, these strains were not temperature sen-sitive on an optA+ host. Thus, gene 43 mutations are allele spe-cific in their requirements for optA+ function after infection;
Table 4. Relative efficiency of plating of gene 43 amber mutantson optA+ and optA strains
Phagestrain*
Temp.,0C
Relative efficiencyof platingt
HR42.1 HR44.1supD supDoptA+ optA
amE4314 30 1.042 0.68
amE4306 30 11.042 2.0 x
amE4322 30 1.042 1.0
amE4309 30 1.042 0.90
amE4304 30 1.042 0.88
amE192 30 1.042 1.0
amBU23 30 1.042 0.85
amE4317 30 1.042 1.1
amC125 30 1.042 1.1
amB22 30 1.042 1.4
amE4310 30 1.042 8.0 x 1-5
1.30.457.5 x 10-2.0 x 1O-71.21.00.860.630.920.740.80.890.631.0 x 10-v1.00.530.800.390.331.9 x 10-54.6 x 10-51.0 x 10-]
* The amber mutants are in map order; the amino terminus is at thetop (31, 32).
tRelative plating efficiency is the number of plaques produced on astrain at a given temperature divided by the number of plaques pro-duced on HR42.1 at 3000. All platings were done on EHA agar. Allamber mutants had a plating efficiency of 10' or less when strainslacking an amber suppressor (sup°) were used as host. Boxes showoptA+/optA plating differences 10-fold or more.
Genetics: Gauss et aL

Proc. Natd Acad. Sci. USA 80 (1983)
however, the requirement for optA+ activity does not neces-sarily correlate with the production of a temperature-sensitiveDNA polymerase.
DISCUSSIONWe have shown that a bacterial strain with a mutation in the optAgene restricts the growth of several bacteriophage T4 strains.The restricted phage strains have mutations that fall into twocomplementation groups. One complementation group is de-fective in the DNA helicase activity encoded by the gene dda(Table 1). A second complementation group consists of a sub-class of temperature-sensitive and amber mutations that map ingene 43 (Tables 3 and 4). DNA replication is defective after in-fection of an optA host by mutants of either complementationgroup. The optA mutation does not map in a gene of knownfunction; we cannot predict how the optA+, dda+, and 43+ geneproducts interact to support T4 DNA replication.
The major role of bacteriophage T4 DNA polymerase is topolymerize deoxyribonucleotides accurately into polynucleo-tides when presented with a DNA template. However, the gene43 protein is unable to utilize double-stranded DNA (ds DNA)templates efficiently in vitro (33). Because the template used invivo is double stranded, other T4 or E. coli proteins must assistin unwinding the DNA. The products of bacteriophage T4 genes44, 62, and 45 have been genetically and biochemically asso-ciated with the replication complex (6, 34, 35). In vitro repli-cation of ds DNA occurs only when these accessory proteins andDNA polymerase are mixed together in the presence of the T4single-stranded DNA binding protein, the product of gene 32(34-36). Individually, none of these gene products is capable ofunwinding a DNA helix in vitro.
However, the products of two other T4 genes, 41 and dda,are capable of unwinding ds DNA (27, 28, 37). The purified pro-teins are quite different from each other, although each has anATP-dependent helicase activity (27, 28, 37). Unwinding by thepurified dda protein requires the presence of nearly one pro-tein monomer per unwound base pair (27). Under such con-ditions the dda protein will unwind a long DNA molecule. Thegene 41 protein cannot unwind a long DNA molecule under anyconditions (37). Most interestingly, the gene 41 protein is re-quired for T4 DNA replication on all host strains (6), whereasthe dda protein is needed only on optA hosts. Gene 41 proteinis required to maximize replication in vitro; the protein stim-ulates the T4 core replication complex and stimulates the syn-thesis of RNA primers needed for lagging-strand synthesis (35,38, 39). The dda protein recently has been shown to stimulatereplication in vitro (30).Many DNA helicases have been identified in infected and
uninfected E. coli, and it is likely that these enzymes have re-lated but distinct physiological functions (1). In order to un-derstand these functions, it would be of interest to measure thecontribution of DNA helicases to DNA unwinding at an activereplication fork. Alberts et al. (30) have developed one such as-say. Unwinding capacity is measured by the ability of the T4DNA replication complex to remove barriers, in the form ofboundRNA polymerase, from DNA that is being actively replicated(30). These barriers are removed only by the dda protein andnot by the gene 41 protein (30). These biochemical experimentssuggest that the dda gene product is necessary for DNA un-winding under conditions that might approximate some in vivosituation. Additionally, our genetic results suggest that, in theabsence of dda, an alternative pathway for DNA unwinding isprovided by the E. coli optA' gene product.
The idea that optA infections by mutant bacteriophage T4 aredefective in a DNA-unwinding activity is further supported bythe identified biochemical defect of one T4 DNA polymerase
FIG. 3. Concerted helicase model: visualization of fork denatur-ationduring T4 DNA replication. We suggest that the helicase activityof the dda gene product (gp) and the denaturation/strand displace-ment activity of gp43 and gp32 work in concert to move the replicationfork. Perhaps when gp43 or gp dda is defective, the optA+ gene productassists in this process by acting as a helicase.
mutant that is restricted on the optA strain. That polymerase,isolated from tsCB120-infected cells, performs repeated cyclesof polymerization and degradation of the terminal primer nu-cleotide when ds DNA or partially double-stranded poly[d(A-T)] is used as a template (40). On these templates the tsCB120polymerase cannot synthesize DNA. However, this polymerasecan synthesize DNA when the secondary structure is removedfrom the poly[d(A-T)] template by the addition of the T4 single-stranded DNA binding protein, which does denature poly[d(A-T)] (36, 40). Because ads DNA template is copied in vivo by thisprotein (at 300C), other host- or phage-encoded proteins evi-dently assist the tsCB120 polymerase in DNA unwinding. Thesefunctions might include the product of the host optA+ gene.
In summary, we hypothesize that the combined activities ofthe products of genes 43, dda, and 32 open the DNA helix forchain elongation. The individual activities of these proteins areconsistent with the production of a concerted unwinding activ-ity that is required during DNA replication (schema in Fig. 3).The concerted helicase activity proposed here could be involvedin the initiation of bacteriophage T4 DNA replication as well aselongation. The bacteriophage T7 1.2 gene product provides anessential function for T7 DNA replication after infection of anoptA host (5). This function may be analogous to that providedby the dda gene product during T4 infections of the optA host.We have identified a fragment of T4 DNA that, if present withinan appropriate plasmid in an optA host, can complement in-fections by T4 dda, dexA, and sudl mutants as well as T7 mu-tants deleted for the 1.2 gene (unpublished results). We note,finally, that the E. coli optA+ gene product could contribute toa concerted helicase activity directly or indirectly; the optA+protein itself could function in the replication complex or couldregulate a component of the replication complex.
We thank H. Saito, W. B. Wood, C. C. Richardson, K. Ebisuzaki, D.H. Hall, R. P. Cunningham, M. A. Nelson, and B. Bachmann for bac-terial and phage strains. We thank B. M. Alberts, C. V. Jongeneel, andN. Nossal for providing manuscripts prior to publication. We also thankM. A. Nelson, C. V. Jongeneel, H. Saito, and C. C. Richardson for fruit-ful discussions. The authors offer special thanks to M. A. Nelson for as-sistance in editing this manuscript. This investigation was supported byGrant GM19963 from the National Institutes of Health to L.G. and byNational Institute of General Medical Sciences National Research Ser-vice Award GM06459 to D.H.D.
1. Geider, K. & Hoffman-Berling, H. (1981) Annu. Rev. Biochem.50, 233-260.
1672 Genetics: Gauss et d

Proc. Natl. Acad. Sci. USA 80 (1983) 1673
2. Denhardt, D. T., Dressler, D. H. & Hathaway, A. (1967) Proc.Natl Acad. Sci. USA 57, 813-820.
3. Scott, J. F. & Kornberg, A. (1978)J. Bio. Chem. 253, 3292-3297.4. Lane, H. E. D. & Denhardt, D. T. (1975)J. MoL Biol 97, 99-112.5. Saito, H. & Richardson, C. C. (1981)J. Virot 37, 343-351.6. Epstein, R. H., Bolle, A., Steinberg, C. M., Kellenberger, E.,
Boy de la Tour, E., Chevalley, R., Edgar, R. S., Susman, M.,Denhardt, G. H. & Lielausis, A. (1963) Cold Spring Harbor Symp.Quant. Biol 33, 375-394.
7. De Waard, A., Paul, A. V. & Lehman, I. R. (1965) Proc. Natl Acad.Sci. USA 54, 1241-1248.
8. Behme, M. T. & Ebisuzaki, K. (1975)J. Virot 15, 50-54.9. Nelson, M. A. & Gold, L. (1982) MoL Gen. Genet. 188, 60-68.
10. Coulondre, C. & Miller, J. H. (1977)J. Mol BioL 117, 577-606.11. Low, K. B. (1972) Bacteriol Rev. 36, 587-607.12. Bukhari, A. I. & Taylor, A. L. (1971)1. Bacteriol, 105, 844-854.13. Strigini, P. & Gorini, L. (1970) J. Mol Biol 47, 517-530.14. Cunningham, R. P. & Berger, H. (1977) Virology 79, 320-329.15. Allen, E. F., Albrecht, I. & Drake, J. W. (1970) Genetics 65, 187-
200.16. Little, J. W. (1973) Virology 53, 47-59.17. Homyk, T. & Weil, J. (1974) Virology 61, 505-523.18. Warner, H. R., Snustad, D. P., Koerner, J. F. & Childs, J. D.
(1972)J. ViroL 9, 399-407.19. Goscin, L. A. & Hall, D. H. (1972) Virology 50, 84-94.20. Freese, E., Bautz, E. & Freese, E. B. (1961) Proc. Natl. Acad.
Sci. USA 47, 845-855.21. Freese, E., Bautz-Freese, E. & Bautz, E. (1961) J. MoL Biol 3,
133-143.22. Barnett, L., Brenner, S., Crick, F. H. C., Shulman, R. G. & Watts-
Tobin, R. J. (1967) Phil Trans. R. Soc. London Ser. B 252, 487-560.
23. Rosner, J. L. (1972) Virology 48, 679-689.24. Singer, B. S. & Gold, L. (1976) J. Mol Biol 103, 627-647.25. Miller, J. H. (1972) Experiments in Molecular Genetics (Cold Spring
Harbor Laboratory, Cold Spring Harbor, NY).26. Wood, W B. & Revel, H. R. (1976) Bacteriol Rev. 40, 847-868.27. Krell, H., Durwald, H. & Hoffnann-Berling, H. (1979) Eur. J.
Biochem. 93, 387-395.28. Kuhn, B., Abdel-Monem, M. G. & Hoffmann-Berling, H. (1978)
Cold Spring Harbor Symp. Quant. Biot 43, 63-67.29. Doherty, D. H., Gauss, P. & Gold, L. (1982) Mol, Gen. Genet 188,
77-99.30. Alberts, B. M., Barry, J., Bedinger, P., Formosa, T., Jongeneel,
C. V. & Kreuzer, K. N. (1983) Cold Spring Harbor Symp. Quant.Biol., in press.
31. Reha-Krantz, L. J. & Bessman, M. J. (1981)J. Mol, Biol 145, 677-695.
32. Huang, W H. & Lehman, I. R. (1972) J. Biol Chem. 247, 7663-7667.
33. Nossal, N. G. (1974) J. Biol Chem. 249, 5668-5676.34. Nossal, N. G. & Peterlin, B. M. (1979)J. Biol Chem. 254, 6032-
6037.35. Liu, C. C., Burke, R. L., Hibner, U., Barry, J. & Alberts, B. M.
(1978) Cold Spring Harbor Symp. Quant. Biol 43, 469-487.36. Alberts, B. & Frey, L. (1970) Nature (London) 227, 1313-1318.37. Venkatesan, M., Silver, L. L. & Nossal, N. G. (1982)J. Biold Chem.
257, 12426-12434.38. Nossal, N. G. (1980) J. Biol Chem. 255, 2176-2182.39. Liu, C.-C. & Alberts, B. M. (1980) Proc. Natl Acad. Sci. USA 77,
5698-5702.40. Gillan, F. D. & Nossal, N. (1976)J. Biol Chem. 251, 5219-5225.
Genetics: Gauss et aL