autophagy-linked fyve protein mediates the turnover of
TRANSCRIPT

Autophagy-linked FYVE protein mediates the turnover of mutant huntingtin and modifies pathogenesis in mouse models of Huntington’s disease
Leora Mestel Fox
Submitted in partial fulfilment of the requirements for the degree of
Doctor of Philosophy under the Executive Committee
of the Graduate School of Arts and Sciences
COLUMBIA UNIVERSITY
2016

© 2016 Leora Mestel Fox All rights reserved

ABSTRACT
Autophagy-linked FYVE protein mediates the turnover of mutant huntingtin and modifies pathogenesis in mouse models of Huntington’s disease
Leora Mestel Fox
A defining characteristic of neurodegenerative disease is the accumulation of mutant or
misfolded proteins within neurons. Selective macroautophagy of aggregates, or aggrephagy, is a
lysosome-mediated protein degradation pathway implicated in the turnover of disease-relevant
accumulated proteins, but its specific function in vivo in the mammalian nervous system is poorly
understood. The large PI3P-binding protein Alfy (Autophagy-linked FYVE protein) is an adaptor
required for selective macroautophagy of aggregated proteins in cellular model systems. We
sought to address Alfy-mediated aggrephagy in the mammalian brain in mouse models of
Huntington’s disease (HD).
HD is a neurodegenerative disorder caused by autosomal dominant inheritance of an
expanded CAG repeat within the IT15, or huntingtin (htt) gene. The mutation causes an expansion
of a polyglutamine (polyQ) tract in the protein Huntingtin (Htt), which results in psychiatric,
cognitive, and motor symptomology. A pathological hallmark of HD is the accumulation of
intracellular deposits of mutant Htt and ubiquitin. The exact relevance of these deposits remains
unclear, but their elimination, hypothesized to occur via macroautophagy, correlates with
behavioral improvements in mouse models of HD. The selective mechanisms of this phenomenon
are largely unexplored in vivo.
We have created two mouse models to address the role of Alfy-mediated selective
macroautophagy in mammalian HD brain. First, we created tamoxifen-inducible Alfy knockout
mice (Alfy iKO) and crossed them with a redesigned inducible HD mouse (HD103Q) that uses a
tetracycline-regulated system to control reversible expression of mutant exon-1 Htt. Western blot,

in situ, and PCR analysis confirm that Alfy can be eliminated from brain in adult Alfy iKO mice.
A timecourse of Htt aggregation and clearance reveals that HD103Q mice accumulate huntingtin
deposits, which clear in a linear manner upon transgene suppression over the course of four
months. The loss of Alfy significantly impedes the removal of these deposits. Second, an Alfy
knockout mouse was created using gene-trap technology, and mice hemizygous for Alfy knockout
were crossed with BACHD mice expressing full-length human mutant Htt. We find that 50% Alfy
depletion in the BACHD leads to increased insoluble Htt aggregate deposition along with
accelerated decline in motor behavioral performance. Furthermore, inducible knockout of Alfy
alone has a severe and age-dependent motor behavioral phenotype. This work reveals an in vivo
role for Alfy in turnover of mutant Htt deposits, suggests that the accumulation of detergent-
insoluble mutant Htt species contributes to behavioral pathogenesis, and supports an important
function for Alfy at the intersection of HD and aging.

i
Table of Contents
List of Figures ii Acknowledgments v Preface viii Chapter 1: Macroautophagy of aggregation-prone proteins in 1 neurodegenerative disease Chapter 2: Modeling Huntington’s Disease 36 Chapter 3: Materials and methods 50 Chapter 4: Alfy promotes the turnover of mutant huntingtin in the adult brain 67 Chapter 5: Insoluble aggregate accumulation accelerates HD pathogenesis 91 Chapter 6: Preliminary investigation of the role of Alfy protein in normal aging 102 Chapter 7: General discussion 111 References 131

ii
List of Figures Chapter 1 Table 1.1: Normal and abnormal localization and function of neurodegenerative 4 disease-relevant aggregating proteins. Figure 1.1: UPS-dependent protein degradation and aggresome formation. 6 Figure 1.2: Figure 1.2: Structure and function of Alfy. 11 Figure 1.3: Localization of Alfy to sites of misfolded protein accumulation. 12 Figure 1.4: Alfy is required for macroautophagic turnover of detergent-insoluble 13 expanded mutant Htt in cells. Figure 1.5: Alfy overexpression leads to the clearance of mutant huntingtin aggregates 14 in primary neurons and is protective in a fly eye model of polyQ expansion. Figure 1.6: Loss of Alfy does not affect basal macroautophagic function. 15 Chapter 2 Figure 2.1. Inverse relationship between CAG repeat length and age at motor onset 38 of Huntington’s disease. Figure 2.2: Neuropathological features of Huntington’s disease. 39 Chapter 3 Table 3.1: Breeding Strategy to create HD103Q, Alfy iKO, and HDAlfy iKO. 53 Table 3.2: Primers and conditions for PCR and RT-PCR. 56 Table 3.3: Antibody information and conditions for western blot. 61 Table 3.4: Antibody sources and conditions for immunohistochemistry. 64 Chapter 4 Figure 4.1: Design of the conditional Alfy allele. 69 Figure 4.2: Tamoxifen-inducible Alfy knockout in brain. 70 Figure 4.3: Confirmation of Alfy iKO in forebrain. 72

iii
Figure 4.4: Design and tet-mediated regulation of the HD103Q transgene. 74 Figure 4.5: HD103Q shows mHtt accumulation over time. 75 Figure 4.6: exon-1-mHtt aggregates accumulate in HD103Q and clear 76 upon transgene suppression. Figure 4.7: Basal autophagy is not grossly affected in HD103Q. 78 Figure 4.8: mHtt expression and drug treatment do not affect striatal 80 cell count or volume. Figure 4.9: Inducible Alfy KO impedes removal of mutant Htt. 81 Figure 4.10: Motor behavioral phenotypes in Htt-103Q-expressing mice. 83 Figure 4.11: Pre-treatment behavioral characterization of experimental mice. 87 Figure 4.12: Alfy knockout leads to profound motor behavioral deficit. 89 Chapter 5 Figure 5.1: Breeding schema for BACHD with 50% Alfy depletion. 93 Figure 5.2: 50% Alfy depletion accelerates accumulation of detergent 94 insoluble mHtt. Figure 5.3: 50% Alfy depletion accelerates Rotarod phenotype in BACHD. 95 Figure 5.4: 50% Alfy depletion accelerates locomotor phenotype in BACHD 97 Figure 5.5: 50% Alfy depletion in BACHD causes increased accumulation 99 of mHtt aggregates. Figure 5.6: 50% Alfy depletion leads to glial pathology and accumulation of 100 ubiquitinated structures in BACHD Chapter 6 Figure 6.1: Timeline for inducible Alfy iKO. 103 Figure 6.2: Alfy iKO leads to age-dependent decline in Rotarod performance. 104 Figure 6.3: Alfy iKO leads to age-dependent decline in beam crossing. 105 Figure 6.4: Alfy-7-iKO show Purkinje cell loss. 106

iv
Figure 6.5: p62 accumulates in Alfy-7-iKO. 107 Figure 6.6: NIPSNAP accumulates in Alfy-7-iKO. 109

v
Acknowledgements
I would like to express my deep gratitude to my advisor, Dr. Ai Yamamoto, whose
guidance has shaped my development as a scientist and as a human through a formative period of
life and career. Her support of every member of her laboratory extends from technical to emotional,
and her profound commitment to the development of her students through hours of meetings,
conversations, presentation rehearsals, and edits has led to our profound growth as thinkers and
communicators. Ai’s breadth of knowledge and dedication to her work is immense and inspiring,
and her emphasis on fun and team spirit has taken us from Yankees games to Coney Island
coasters. She has entrusted me with her sweeping scientific ideas and with her child, and she has
supported and advocated for my involvement in outreach and my interest in scientific writing. She
has given me frequent and exceptional opportunities to present my work. Most importantly, she
has taught me how to celebrate small benchside victories, to how to fail with laughter, and how to
start over again. Thank you, Ai.
I thank my thesis committee members, Drs. Christoph Kellendonk, Lloyd Greene, and Bob
Burke, for great discussions and thoughtful criticism that helped to shape this project. Special
thanks to Dr. Lisa Ellerby for serving as outside examiner.
I would like to thank those scientists who provided mice, essential reagents, and
collaborative advice. Dr. Anne Simonsen, a long-time collaborator, originally characterized Alfy
and spoke to us about unpublished work. We received the BACHD mice from Dr. William Yang.
The Alfy antibody was generously provided by Dr. Masaaki Komatsu, and the huntingtin antibody
S830 by Dr. Gill Bates. Dr. Ioannis Dragatsis provided the tamoxifen inducible Cre and shared
data and advice on using it for excision in post-mitotic cells.

vi
I’d like to acknowledge my funding sources, a Ruth Kirschstein institutional graduate
NRSA (Sept 2012-Aug 2013), a Ruth Kirschstein individual graduate NRSA (Sept 2013-Aug
2015), and a grant from the Hereditary Disease Foundation (Sept 2015-Aug 2016).
I’d like to thank members of the Yamamoto lab for their guidance, friendship, and
contribution to this effort, especially Joanna Dragich (HD103Q construct), Chris Johnson and Joan
Bosco (BACHD behavior), Evelien Eenjes (BACHD biochemistry), and Wendy Fong (moral
support). I am indebted to Kiryung Kim whose technical support with immunohistochemistry this
summer has been invaluable. Thanks to Marla Oo, Dominik Biezonski, Fernanda Carvalho, and
Abigail Clark for their aid with methodological aspects of this work, and to Katharine Abbot and
Jackson Lovejoy, summer high school students who helped me with experiments and shared my
enthusiasm.
The guidance of former mentors, especially Angelo Piccirillo, Susan Croll, Lori Morton,
Tara Spires, and Brad Hyman, led me to embrace science and to continue pursuing it from early
high school through the present. Nancy Wexler has been an inspiration and an advocate during my
time at Columbia and within the enormously supportive HD research community. Many thanks
also to the dear old friends who have listened to me speculate about aggregated proteins since I
was sixteen years old, and to new friends whose companionship through graduate school has been
immensely encouraging.
I’d like to thank my parents, whose unconditional love and wholehearted support of this
endeavor extends backwards to my birth. They are extraordinary parents and people who taught
me to work and play hard, to do something nice for someone else every day, and to fill my life
with the beautiful and the intellectual. They also raised my sisters, Ariel and Talia, who are

vii
supremely intelligent, fiercely loving, unusually passionate people, and who are nothing and
everything like me. All of you bring joy and comfort to my life.
To my fiancé and favorite human, Sam McKenzie, a brilliant neuroscientist and loving
partner who created customized behavioral analysis software that secretly incorporates love poetry
if you know which keys to press, and who might actually read this whole document: you are doing
science for all the right reasons. Thank you for talking about every part of this work, and everything
else in our world, with me.
I would like to invoke the words of my favorite poet, Frank O’Hara, who asserted that
“it is good to be several floors up in the dead of night wondering whether you are any good or not and the only decision you can make is that you did it”
and finally, I’d like to acknowledge the thousands upon thousands of research animals to whom
humanity owes an enormous debt, and to whom this work is dedicated.

viii
Preface
The first introductory chapter of this work, covering macroautophagy of proteins relevant
to a variety of neurodegenerative disorders, is slightly expanded from a published book chapter
(Fox, 2014). This publication also limited the number of references, requiring mostly references
to reviews; in many cases, I have added references to the original experiments. The chapter is
replicated here with permission from the publisher.
Some of the experiments presented in this document were performed by other members of
the Yamamoto laboratory. Joanna Dragich generated the construct used for the creation of the
HD103Q mouse (Chapter 4). Evelien Eenjes performed biochemical experiments on lysates from
BACHD mice, and Chris Johnson and Joan Bosco performed behavioral experiments on BACHD
mice (Chapter 5). These instances are indicated in the text.
Figure 4.1C forms part of a supplementary figure within a recently published ELife paper
describing constitutive Alfy knockout (Dragich et al., 2016), on which I am an author due to this
contribution.

1
Chapter 1: Macroautophagy of aggregation-prone proteins in neurogenenerative disease
Introduction
The accumulation of aggregated proteins is a prominent feature of neurodegenerative
disease, including polyglutamine expansion disorders, synucleinopathies, tauopathies, and TDP-
43 proteinopathies. Such diseases are characterized pathologically by progressive deterioration of
neurons in particular brain areas, accompanied by widespread accumulation of ubiquitinated
proteins (Ross and Poirier, 2004). These consist of specific mutant or misfolded proteins, which
accumulate and form inclusion bodies that can reside within the nucleus, cytoplasm, or outside the
cell, and vary in content across diseases, cell types, and location within neurons. The significance
of aggregated protein accretion during disease pathogenesis is widely debated. Nevertheless, the
degradation of cellular components is an important requirement for maintaining healthy cells. In
neurodegenerative disease models, the elimination of accumulated aggregation-prone proteins has
been observed to correspond with improvements in neuronal viability and even positive behavioral
changes (Yamamoto and Simonsen, 2011).
Mounting evidence suggests that the autophagy machinery is deployed to clear aggregated
proteins in a wide variety of brain diseases (Nixon, 2013). Autophagy is a general term for the
degradation of cytosolic material by the lysosome (Johnson et al., 2012). While it was originally
identified in the context of bulk recycling of cytoplasmic components in response to starvation,
increasing evidence has established that the process can occur selectively for specific substrates,
including aggregated proteins, even under basal conditions. This selectivity is achieved via
autophagy receptors and selectivity adaptors that link specific cargo to the core autophagy proteins
(Filimonenko et al., 2010; Yamamoto and Simonsen, 2011). This chapter will discuss the
accumulation and degradation of aggregation-prone proteins, with emphasis on aggrephagy, the

2
selective macroautophagy of aggregated proteins (Lamark and Johansen, 2012). While several
recent reviews have focused on the role of autophagy in various disease states (Chen et al., 2012;
Funderburk et al., 2010; Heiseke et al., 2010; Jimenez-Sanchez et al., 2012; Lynch-Day et al.,
2012), clinically divergent groups of neurodegenerative disorders can share a common pathologic
lesion. This has led to development of the concept of a proteinopathy spectrum encompassing
diseases related to a particular type of aggregation-prone protein (Geser et al., 2010). We will
examine the evidence for autophagic degradation of distinctive types of aggregation-prone
proteins, including polyglutamine-expanded proteins, alpha-synuclein, amyloid-beta, prion, TDP-
43, and SOD1. The normal function and intracellular localization of aggregation-prone proteins
may dictate the degree of autophagic involvement in their disposal.
Finally, in light of significant recent focus on the therapeutic potential for upregulation of
macroautophagy in neurodegenerative disease states, we will examine recent efforts to
experimentally enhance macroautophagic activity with the goal of countering the accumulation of
aggregation-prone proteins (Hochfeld et al., 2013). The benefit of such treatments will likely
depend upon both specificity of the target and toxicity of the aggregation-prone species.
Researchers face major challenges to this approach in the context of human neurodegenerative
disease: nonspecific macroautophagic upregulation may have significant adverse consequences,
and the toxicity of aggregates themselves is under debate. It has become clearer that distinctive
types of cargo are processed differently by the autophagic machinery, and thus, elucidating
selective autophagy pathways for the removal of particular aggregation-prone species may be key
in the design of future therapeutics for neurodegenerative disorders.

3
Protein misfolding and the UPS
Protein misfolding is a common cellular event that can occur throughout the lifetime of a
cell, caused by different events including genetic mutations, translational errors, abnormal protein
modifications, thermal or oxidative stress, and incomplete complex formations. Upon misfolding,
aberrant exposure of hydrophobic regions drive these proteins to aggregate (Kopito, 2000; Lamark
and Johansen, 2012), which in turn, can disrupt normal cellular activities by acting to entrap
functional proteins, abetting a defective cascade.
To maintain protein homeostasis and support normal cellular and physiological function,
misfolded, aggregation-prone proteins require repair or removal. A cell’s first line of defense are
molecular chaperones, such as heat-shock proteins, which assist in folding and refolding, attempt
to repair, and prevent aggregate formation (Carra et al., 2013). In the case of irreparable damage
or excessive accumulation, chaperone complexes target the proteins for breakdown by one of two
main degradative pathways within the cell, the ubiquitin-proteasome system and autophagy. The
UPS serves as the primary pathway for protein turnover within cells. Briefly, it involves the
tagging with ubiquitin and subsequent breakdown of proteins by a large, multisubunit holoenzyme
known as the proteasome (Dennissen et al., 2012). This process enables rapid elimination of
individual proteins with high specificity. However, its barrel-shaped catalytic core is not accessible
to proteins that are folded or reside within larger complexes, and thus aggregated proteins are likely
removed by other means.

4
Table 1.1: Normal and abnormal localization and function of neurodegenerative disease-relevant aggregating proteins.

5
Aggregate formation
The assembly and removal of aggregates is one of many physiological processes by which
cells achieve protein quality control. A variety of proteins must interface with others in order to
perform their normal function, but in the context of neurodegenerative disease, an aggregate is
generally defined as a complex of accumulated, misfolded proteins. Proteins in deviant
conformations tend to form oligomeric intermediates that further coalesce into small aggregates,
which in turn can build into different types of complex structures. Aggregates are insoluble and
metabolically stable under physiologic conditions, but a true experimental definition is difficult to
achieve, due to the diversity of techniques used to monitor solubility and stability, such as
detergent treatments, centrifugation, and membrane trapping. Most commonly studied are amyloid
fibrils, which are structurally stable, but aggregates can be structured or amorphous (Kopito, 2000).
The pathology of most neurodegenerative disorders is marked by the accumulation of
aggregated, ubiquitinated proteins, which can also be referred to as inclusions, inclusion bodies,
tangles, or threads (Ross and Poirier, 2004). These structures are morphologically distinct, and
their contents and location within the cell vary across disorders (Table 1.1). Histopathologically,
these structures represent a defining characteristic of a particular disease, such as the tau tangles
found in Alzheimer’s disease brains, or the Lewy bodies of Parkinson’s disease.
In many types of mammalian cells, cytoplasmic aggregated proteins are delivered via
microtubule-dependent transport to form an aggresome. Aggresomes are large, pericentriolar
structures wherein intermediate filaments such as keratin and vimentin form a cage around
ubiquitinated, misfolded proteins at the microtubule-organizing center (MTOC). It has been
proposed that aggresomes are protective structures designed to sequester proteasome-indigestible
proteins and prepare them for autophagic breakdown when the UPS is overwhelmed (Kopito,

6
2000). The toxicity of neuronal aggregates has been a major area of debate for decades; one theory
posits that they represent a means of constraining soluble, oligomeric proteins with pathogenic
potential. Brain-specific disease-related proteins are frequently sequestered in this manner when
studied using dividing cell lines. However, it is unclear whether post-mitotic neurons form
aggresomes per se, since aggresomes have been defined by the presence of vimentin and their
location at the MTOC (Johnston et al., 1998). Mature neurons replace vimentin with
neurofilaments (NFs) and lack definitive MTOCs, but it is possible that inclusion body formation
in neurons serves a function similar to aggresome formation, and that this process is dependent
upon microtubules (Kopito, 2000; Yamamoto and Simonsen, 2011) The formation of aggresome-
like structures in neurons may be mediated by the ubiquitin-binding microtubule deacetylase
HDAC6, which can interact with molecular motors and is required for the recruitment of
misfolded, ubiquitinated proteins to aggresomes (Kawaguchi et al., 2003; Ouyang et al., 2012)
(Figure 1.1).
Figure 1.1: UPS-dependent protein degradation and aggresome formation. Proteins in the native conformation that become misfolded are repaired by chaperones such as heat shock proteins. Those that are irreparably damaged are tagged with ubiquitin and processed by the proteasome. Ubiquitinated proteins that cannot be processed by the proteasome may coalesce into small aggregates which are transported via association with HDAC6 and microtubule-dependent molecular motors to form aggresomes. These structures may function to sequester proteasome-indigestible proteins and prepare them for macroautophagic clearance.

7
Macroautophagy
Despite their potential to serve a protective function, the persistence of protein aggregates,
while very rare in healthy conditions, is extremely common in disease states. While the aggregated
structures themselves might not be toxic, their removal may still represent a means of easing
cellular dysfunction or promoting recovery. For example, the elimination of aggregated proteins
via the use of lentiviral, genetic, and RNA interference techniques corresponds with amelioration
of motor phenotypes in animal models of neurodegeneration (DiFiglia et al., 2007; Harper et al.,
2005; Kordasiewicz et al., 2012; Stanek et al., 2014; Wang et al., 2005; Yamamoto et al., 2000).
One pathway capable of degrading large complex structures is a lysosome-mediated pathway
known as macroautophagy.
Macroautophagy is one of three catabolic processes by which cytosolic components are
degraded by the lysosome. The most evolutionarily conserved form of autophagy, it is reliant on
the formation of a transient organelle known as the autophagosome (Klionsky, 2007). The
autophagosome is a double membrane structure that grows to engulf its cargo, and then fuses into
the endolysosomal system to degrade its contents. Essential to the nucleation, expansion, and
maturation of the autophagosome is a core group of highly conserved autophagy-related proteins
(Atgs), which were originally identified in yeast. Ultimately, a series of enzymatic reactions,
reminiscent of ubiquitin conjugation, catalyze the covalent conjugation of LC3, (one of the
mammalian homologues of Atg8) to the lipid phosphatidylethanolamine (PE), which promotes the
growth of the autophagosomal membrane. Central to this process is Atg7, which acts as an E1-like
enzyme to activate Atg12, which is then conjugated to Atg5 by the action of the E2-like Atg10.
Atg5 and Atg12 associate with Atg16L in the forming autophagosomal membrane. The E1-like
activity of Atg7 also serves to activate LC3 (a mammalian homolog of Atg8), which is transferred

8
to the E2-like Atg3, and the Atg5-12-16L complex then acts as an E3-like ligase to catalyze the
conjugation of LC3 to PE (Klionsky, 2007). This membrane-bound lipidated form (LC3-II) is often
used as a marker for autophagosomes. Beclin-1, the mammalian orthologue of Atg6, is important
for the generation of phospholipid and the localization of additional Atg proteins that orchestrate
the formation of autophagosomes. A more detailed examination of the stepwise process of
autophagosome building can be found in the literature, along with an extensive review of
microautophagy and chaperone-mediated autophagy (Johnson et al., 2012; Klionsky, 2007).
Though the UPS represents the dominant pathway for protein degradation, the versatility
of autophagosome membrane building, together with the wide array of lysosomal hydrolases,
makes macroautophagy essential to adaptive cellular responses during development, starvation,
and stress. Constitutive knockout of several Atgs and components of autophagy-related protein
complexes are embryonic- or neonatal-lethal due to malnutrition and insufficient amino acid
turnover. Atg loss-of-function studies within the central nervous system have demonstrated a
critical role for basal macroautophagy in neurons during development, resulting in
neurodegeneration and the accumulation of ubiquitin and selective autophagy receptors (Hara et
al., 2006; Komatsu et al., 2006; Mizushima and Levine, 2010). However, the role of
macroautophagy in adult brain remains largely undefined.
Selective autophagy
While it has long been considered in the context of bulk degradation of cytoplasmic
constituents, and is induced in response to stress or starvation, autophagy has more recently been
implicated in the breakdown of specific substrates, including mitochondria, peroxisomes,
microbes, and notably, aggregated proteins, both under stressed and basal conditions (Johansen

9
and Lamark, 2011). Cargo-specificity is made possible by adaptor molecules that allow the
autophagic machinery to build around targeted cytoplasmic substructures. This mode of action is
based on a model described in yeast called cytoplasm-to-vacuole targeting (Cvt) which involves
the selective delivery of lysosomal enzyme precursors to the vacuole after sequestration in double-
membrane vesicles (Lynch-Day and Klionsky, 2010). The aggregated precursors are bound by
specific cargo receptor Atg19, and together with the specificity adaptor Atg11, the cargo is
connected to the core machinery driving localized autophagosome formation. Examples of Atg19-
like receptors are the mammalian proteins p62/SQSTM1 and NBR1, which bind to ubiquitin as
well as the LC3 protein family, thus facilitating the autophagic degradation of ubiquitinated protein
aggregates (Johansen and Lamark, 2011). Currently recognized autophagic receptors are p62,
Nbr1, Nix, NDP52, VCP, and Optineurin (Fimia et al., 2013).
Aggrephagy
It is still debated whether entire aggregates are degraded directly by macroautophagy. The
macroautophagic machinery may be capable of degradation on this scale, but macroautophagic
membranes are rarely observed via EM to contain aggregates larger than 1 µm (Filimonenko et
al., 2010). Filter trap analyses that segregate aggregation-prone proteins by size indicate that
smaller aggregates are more likely to be influenced by macroautophagic degradation rather than
large, intact aggresome-like structures (Shoji-Kawata et al., 2013). Macroautophagy has also been
implicated in dissolving large neuronal inclusions into smaller pieces (Lamark and Johansen, 2012;
Rideout et al., 2004).
Selective macroautophagy of aggregated proteins, or aggrephagy (Overbye et al., 2007),
relies on p62, NBR1, and the autophagic adaptor protein, Alfy (WDFY3). p62 and NBR1 were

10
identified several years ago as selective cargo adaptors for autophagy. They have similar domain
architecture and are selectively degraded by macroautophagy. Both contain a ubiquitin-associated
domain and an LC3-interacting region (LIR), allowing them to link ubiquitinated proteins with the
core macroautophagy machinery. They can hetero- and homo-oligomerize via a PB1 domain in
p62, and a coiled-coil domain in NBR1 (Kirkin et al., 2009; Lamark and Johansen, 2012; Lamark
et al., 2009; Pankiv et al., 2007). Oligomerization of p62 promotes the formation of aggresome-
like structures, and thus may ready cargo for engulfment by the growing autophagosome. For
example, while conditional deletion of Atg7 in nestin-positive cells resulted in the accumulation
of ubiquitinated and p62-positive inclusions in neurons, additional knockout of p62 eliminated the
presence of these structures (Komatsu et al., 2007a; Mizushima and Levine, 2010).
Optineurin (OPTN), which was characterized as an adaptor protein regulating the
macroautophagic elimination of ubiquitinated salmonella, has recently been implicated in
aggrephagy. OPTN is a ubiquitin-binding protein that contains a LIR motif, and has been shown
to colocalize with protein inclusions in several neurodegenerative disorders, in both ubiquitin-
positive and ubiquitin-negative structures (Osawa et al., 2011). Korac et al (2013) demonstrated
recently that OPTN recognizes various protein aggregates through its coiled-coil domain, and that
its depletion results in the accumulation of mutant huntingtin and SOD1.
Alfy
A recently identified molecular adaptor for aggrephagy is a protein called Alfy (autophagy-
linked FYVE domain containing protein), which is encoded by the gene WDFY3. Alfy is a large
(380 kDa), highly evolutionarily conserved protein, with homology from yeast through mammals,
and it is highly expressed in the brain compared to other organs (Simonsen et al., 2004). Its

11
drosophila homologue is known as blue cheese (bchs) (Finley et al., 2003). Its C-terminus contains
a series of key protein-protein and protein-lipid motifs that permit Alfy to act as a molecular
scaffold to guide selective autophagosomal construction around aggregated protein substrates
(Figure 1.2).
Figure 1.2: Structure and function of Alfy. (A) Known protein-lipid and protein-protein interaction domains within Alfy. Alfy interacts with the membrane lipid PI3P through its FYVE domain, with Atg5 and GABARAP through the region containing WD40 repeats, and with p62 via a PH-BEACH domain. It contains putative nuclear localization sequences (NLS) and nuclear export sequences (NES). (B) Selective macroautophagy of aggregated proteins (aggrephagy) mediated by cargo receptors and the selectivity adaptor Alfy. Misfolded ubiquitinated proteins that have formed oligomers are targeted by p62, driving the formation of aggregates. Alfy binds to p62, enabling recruitment of core macroautophagy proteins and stabilizing the interaction of the aggregating protein with LC3, which is conjugated to PE in the forming autophagosomal membrane.
Alfy contains a PH-BEACH domain by which it directly interacts with p62, as well as 5
WD-40 repeats that allow it to associate directly with Atg5 (Filimonenko et al., 2010) and with the
mammalian Atg8 homologue GABARAP (Lystad et al., 2014). A FYVE domain confers upon
Alfy the ability to interact with PI3P, a regulator of endocytic and autophagic membrane traffic
(Figure 1.2 A). Alfy is postulated to function as an Atg11-like autophagic adaptor for aggrephagy,
acting to link aggregated proteins with Atg8 homologues and the Atg12-5:16L complex (the E3-
like enzyme responsible for LC3 conjugation) in the phosphoinositide-rich membrane to promote

12
autophagosome building around pathogenic inclusions (Filimonenko et al., 2010) (Figure 1.2 B).
Accordingly, Alfy can be found in a complex along with Atg12-5:16L, NBR1, LC3, p62, and
aggregation-prone mutant huntingtin protein, and is required for the interaction between disease-
relevant proteins and the basal autophagic machinery (Filimonenko et al., 2010).
Alfy contains nuclear localization and nuclear export sequences (Figure 1.2 A). Normally
localized to the nuclear membrane, Alfy translocates to the cytoplasm in the presence of stressors
such as protein misfolding (Filimonenko et al., 2010) (Figure 1.3).
Figure 1.3: Localization of Alfy to sites of misfolded protein accumulation. (A) Alfy is normally localized to the nuclear membrane where it colocalizes with nucleoporin. (B) In the presence of expanded mutant huntingtin, Alfy colocalizes to the aggregates. From Filimonenko et al. (2010).
Consistent with its role as a selective adaptor protein for the macroautophagic clearance of
aggregated proteins, Alfy is necessary for the removal of various types of detergent-insoluble
disease-relevant proteins and visible aggregated puncta. This includes expanded mutant huntingtin
(Filimonenko et al., 2010), alpha-synuclein (Filimonenko et al., 2010), and G93A-SOD1 (Han et
al., 2015). Alfy is also involved in the removal of aggregated protein complexes, including
aggresome-like induced structures (ALIS) (Clausen et al., 2010), aggregated EDEM1 which is
involved in protein quality control within the enodplasmic reticulum (Park et al., 2014), stress-
induced inclusions in osteoclasts (Hocking et al., 2010), and midbody ring complexes following
cytokinesis (Isakson et al., 2013b).
and S2D). Colocalization with other intranuclear structures wasnot detected (Figure S2B).
Alfy Is Required for Macroautophagy-MediatedClearance of Aggregated ProteinBut why is Alfy recruited to these aggregating proteins? Inprevious studies, we and others have shown that macroautoph-
agy is required to clear protein aggregates (Iwata et al., 2005a,2005b; Ravikumar et al., 2002; Yamamoto et al., 2006). Toanalyze aggregate clearance, HttPolyQ-mCFP cell lines weretreated with doxycycline (dox) (Yamamoto et al., 2006), whichled to inclusion clearance over several days in a macroautoph-agy-dependent manner (Figures S2 and S3) (Yamamoto et al.,2006) (quantified as the number of mCFP puncta per cell). To
Figure 2. Alfy Translocates from the Nucleus and Is Required to Clear Aggregated PolyQ(A–F) Alfy translocates from the nucleus into cytoplasmic structures.
(A) Alfy localizes to the nuclear membrane in untreated HeLa cells and colocalizes with nucleoporin.
(B) HeLa cells were transfected with Flag-HttQ68 (red) and probed for endogenous Alfy (green). Inset shows colocalization of Alfy and the polyQ inclusions
(yellow).
(C and D) Flag-HttQ68 (green) transfected cells were treated 40 hr later with 5 nM leptomycin B for 4 hr, fixed, and stained with anti-Alfy antibodies (red).
Draq5 (blue) labels nuclei. Scale bar = 10 mm. LMB treatment inhibited recruitment of Alfy to cytoplasmic inclusions (30 cells, two independent experiments.
Student’s t test; p < 0.005).
(E) LMB inhibits Alfy translocation under nutrient-rich and nutrient-starved conditions. HeLa cells were starved for 45 min in HBSS ± LMB. Cells containing
cytoplasmic Alfy-positive structures were counted (n > 300 cells from n = 3 experiments).
(F) Htt103Q-mCFP cells were treated with dox to abolish expression (7 days). Dox was then removed to re-establish expression. Cells were collected at days 0, 2,
6, and 10. Relative Alfy mRNA levels were determined by qRT-PCR and normalized to day 0. TBP and actin were used as controls. Two experiments were per-
formed in duplicate.
(G–I) Alfy is required for clearance of expanded polyQ aggregates.
(G) Htt103Q-mCFP cells were quantified for mCFP puncta per cell. 100 mg/ml dox for 5 days led to a significant decrease of percentage of cells with aggregates
(p < 0.001). siALFY1 and siALFY2 significantly inhibited clearance (*p < 0.001), whereas siCTRL did not (p = 0.3218). Representative confocal images of each
group generated on the INCA3000 are shown. Nuclei are stained with Hoechst33342 (blue), and HttQ103-mCFP puncta are in green.
(H and I) Htt65Q- and Htt103Q-mCFP were transfected with siCTRL or siALFY and exposed 48 hr later to dox for 3 days.
(H) Alfy KD inhibited clearance of SDS-insoluble protein for 65Q (p < 0.05) and 103Q (p < 0.05) cells (Student’s t test). No detectable change was seen in
SDS-soluble proteins as shown by SDS-PAGE. Macroautophagy control was 5 mM 3MA for 48 hr.
(I) Alfy KD and Atg5 KD inhibited clearance of SDS-insoluble inclusions.
All data are shown as mean + SD. Complete statistics can be found in Supplemental Experimental Procedures under ‘‘Statistical Information for Figures.’’
Molecular Cell
Alfy-Mediated Degradation of Aggregates
268 Molecular Cell 38, 265–279, April 23, 2010 ª2010 Elsevier Inc.

13
In a HeLa cell system in which mutant exon-1 huntingtin is expressed in a tetracycline-
regulatable manner, knockdown of Alfy abolishes doxycycline-mediated clearance of visible
puncta and detergent-insoluble mutant huntingtin, but does not affect the clearance of detergent-
soluble forms of mutant huntingtin (Figure 1.4), reinforcing its role as a selective adaptor required
for aggrephagy.
Figure 1.4: Alfy is required for macroautophagic turnover of detergent-insoluble expanded mutant Htt in cells. Left: Doxycycline-mediated clearance of mutant exon-1 Htt with 103 CAG repeats in a stably-expressing HeLa cell line. Knockdown of Alfy with siRNA abolishes clearance of punctate GFP-tagged mHtt structures. Right: Filter trap experiment indicating that Alfy knockdown prevents the removal of detergent-insoluble mutant exon-1 Htt, whereas detergent-soluble forms are still cleared. From Filimonenko et. al., 2010. Data suggest that the availability of Alfy might be a limiting step in aggregate clearance
by selective macroautophagy. The overexpression of Alfy leads to the clearance of aggregated
proteins in primary neuronal cells (Figure 1.5 left), and is protective against polyQ-induced
neurodegeneration in the drosophila eye (Filimonenko et al., 2010) (Figure 1.5 right). Additionally,
based on cellular assays that distinguish between aggregate formation and clearance using Halo-
tag, a modified haloalkane dehalogenase designed to covalently bind to different fluorophore
ligands (Los et al., 2008), Alfy overexpression promotes the turnover of existing aggregated
structures rather than preventing aggregate formation (Eenjes et al., 2016).
and S2D). Colocalization with other intranuclear structures wasnot detected (Figure S2B).
Alfy Is Required for Macroautophagy-MediatedClearance of Aggregated ProteinBut why is Alfy recruited to these aggregating proteins? Inprevious studies, we and others have shown that macroautoph-
agy is required to clear protein aggregates (Iwata et al., 2005a,2005b; Ravikumar et al., 2002; Yamamoto et al., 2006). Toanalyze aggregate clearance, HttPolyQ-mCFP cell lines weretreated with doxycycline (dox) (Yamamoto et al., 2006), whichled to inclusion clearance over several days in a macroautoph-agy-dependent manner (Figures S2 and S3) (Yamamoto et al.,2006) (quantified as the number of mCFP puncta per cell). To
Figure 2. Alfy Translocates from the Nucleus and Is Required to Clear Aggregated PolyQ(A–F) Alfy translocates from the nucleus into cytoplasmic structures.
(A) Alfy localizes to the nuclear membrane in untreated HeLa cells and colocalizes with nucleoporin.
(B) HeLa cells were transfected with Flag-HttQ68 (red) and probed for endogenous Alfy (green). Inset shows colocalization of Alfy and the polyQ inclusions
(yellow).
(C and D) Flag-HttQ68 (green) transfected cells were treated 40 hr later with 5 nM leptomycin B for 4 hr, fixed, and stained with anti-Alfy antibodies (red).
Draq5 (blue) labels nuclei. Scale bar = 10 mm. LMB treatment inhibited recruitment of Alfy to cytoplasmic inclusions (30 cells, two independent experiments.
Student’s t test; p < 0.005).
(E) LMB inhibits Alfy translocation under nutrient-rich and nutrient-starved conditions. HeLa cells were starved for 45 min in HBSS ± LMB. Cells containing
cytoplasmic Alfy-positive structures were counted (n > 300 cells from n = 3 experiments).
(F) Htt103Q-mCFP cells were treated with dox to abolish expression (7 days). Dox was then removed to re-establish expression. Cells were collected at days 0, 2,
6, and 10. Relative Alfy mRNA levels were determined by qRT-PCR and normalized to day 0. TBP and actin were used as controls. Two experiments were per-
formed in duplicate.
(G–I) Alfy is required for clearance of expanded polyQ aggregates.
(G) Htt103Q-mCFP cells were quantified for mCFP puncta per cell. 100 mg/ml dox for 5 days led to a significant decrease of percentage of cells with aggregates
(p < 0.001). siALFY1 and siALFY2 significantly inhibited clearance (*p < 0.001), whereas siCTRL did not (p = 0.3218). Representative confocal images of each
group generated on the INCA3000 are shown. Nuclei are stained with Hoechst33342 (blue), and HttQ103-mCFP puncta are in green.
(H and I) Htt65Q- and Htt103Q-mCFP were transfected with siCTRL or siALFY and exposed 48 hr later to dox for 3 days.
(H) Alfy KD inhibited clearance of SDS-insoluble protein for 65Q (p < 0.05) and 103Q (p < 0.05) cells (Student’s t test). No detectable change was seen in
SDS-soluble proteins as shown by SDS-PAGE. Macroautophagy control was 5 mM 3MA for 48 hr.
(I) Alfy KD and Atg5 KD inhibited clearance of SDS-insoluble inclusions.
All data are shown as mean + SD. Complete statistics can be found in Supplemental Experimental Procedures under ‘‘Statistical Information for Figures.’’
Molecular Cell
Alfy-Mediated Degradation of Aggregates
268 Molecular Cell 38, 265–279, April 23, 2010 ª2010 Elsevier Inc.
and S2D). Colocalization with other intranuclear structures wasnot detected (Figure S2B).
Alfy Is Required for Macroautophagy-MediatedClearance of Aggregated ProteinBut why is Alfy recruited to these aggregating proteins? Inprevious studies, we and others have shown that macroautoph-
agy is required to clear protein aggregates (Iwata et al., 2005a,2005b; Ravikumar et al., 2002; Yamamoto et al., 2006). Toanalyze aggregate clearance, HttPolyQ-mCFP cell lines weretreated with doxycycline (dox) (Yamamoto et al., 2006), whichled to inclusion clearance over several days in a macroautoph-agy-dependent manner (Figures S2 and S3) (Yamamoto et al.,2006) (quantified as the number of mCFP puncta per cell). To
Figure 2. Alfy Translocates from the Nucleus and Is Required to Clear Aggregated PolyQ(A–F) Alfy translocates from the nucleus into cytoplasmic structures.
(A) Alfy localizes to the nuclear membrane in untreated HeLa cells and colocalizes with nucleoporin.
(B) HeLa cells were transfected with Flag-HttQ68 (red) and probed for endogenous Alfy (green). Inset shows colocalization of Alfy and the polyQ inclusions
(yellow).
(C and D) Flag-HttQ68 (green) transfected cells were treated 40 hr later with 5 nM leptomycin B for 4 hr, fixed, and stained with anti-Alfy antibodies (red).
Draq5 (blue) labels nuclei. Scale bar = 10 mm. LMB treatment inhibited recruitment of Alfy to cytoplasmic inclusions (30 cells, two independent experiments.
Student’s t test; p < 0.005).
(E) LMB inhibits Alfy translocation under nutrient-rich and nutrient-starved conditions. HeLa cells were starved for 45 min in HBSS ± LMB. Cells containing
cytoplasmic Alfy-positive structures were counted (n > 300 cells from n = 3 experiments).
(F) Htt103Q-mCFP cells were treated with dox to abolish expression (7 days). Dox was then removed to re-establish expression. Cells were collected at days 0, 2,
6, and 10. Relative Alfy mRNA levels were determined by qRT-PCR and normalized to day 0. TBP and actin were used as controls. Two experiments were per-
formed in duplicate.
(G–I) Alfy is required for clearance of expanded polyQ aggregates.
(G) Htt103Q-mCFP cells were quantified for mCFP puncta per cell. 100 mg/ml dox for 5 days led to a significant decrease of percentage of cells with aggregates
(p < 0.001). siALFY1 and siALFY2 significantly inhibited clearance (*p < 0.001), whereas siCTRL did not (p = 0.3218). Representative confocal images of each
group generated on the INCA3000 are shown. Nuclei are stained with Hoechst33342 (blue), and HttQ103-mCFP puncta are in green.
(H and I) Htt65Q- and Htt103Q-mCFP were transfected with siCTRL or siALFY and exposed 48 hr later to dox for 3 days.
(H) Alfy KD inhibited clearance of SDS-insoluble protein for 65Q (p < 0.05) and 103Q (p < 0.05) cells (Student’s t test). No detectable change was seen in
SDS-soluble proteins as shown by SDS-PAGE. Macroautophagy control was 5 mM 3MA for 48 hr.
(I) Alfy KD and Atg5 KD inhibited clearance of SDS-insoluble inclusions.
All data are shown as mean + SD. Complete statistics can be found in Supplemental Experimental Procedures under ‘‘Statistical Information for Figures.’’
Molecular Cell
Alfy-Mediated Degradation of Aggregates
268 Molecular Cell 38, 265–279, April 23, 2010 ª2010 Elsevier Inc.

14
Figure 1.5: Alfy overexpression leads to the clearance of mutant huntingtin aggregates in primary neurons and is protective in a fly eye model of polyQ expansion. (Left) Primary neurons expressing lentivirally introduced mutant exon-1 huntingtin show a decrease in aggregates upon overexpression of the C-terminus of Alfy. (Right) Expression of either full-length or C-terminal drosophila Alfy homolog, bchs, results in amelioration of neurodegenerative phenotypes in fly eye expressing expanded polyQ. From Filimonenko et. al., 2010.
Alfy-mediated aggregate clearance is dependent upon elements of the core
macroautophagic machinery (Filimonenko et al., 2010). However, consistent with its role as an
adaptor protein for aggrephagy, Alfy is not required for starvation-induced or basal
macroautophagy (Figure 1.6). Knockout of Alfy in both HeLa and Neuro2A cell lines does not
affect the degradation of long-lived proteins or LC3I/II conversion (Filimonenko et al., 2010).
Furthermore, no disruption of basal autophagy was observed in vivo in a constitutive
developmental knockout of Alfy (Dragich et al., 2016). In summary, Alfy is postulated to be a
highly selective adaptor for the autophagic turnover of aggregated proteins of many kinds, in both
normal and disease states.
in adult Drosophila head and that Atg8b is not expressed in thistissue (Simonsen et al., 2008). When the C-terminal Bchs wasexpressed together with Atg8a KD using a UAS-dsAtg8a trans-gene, the protection against polyQ127 generated by higherBchs-C1000 levels was no longer observed (Figures 6C and6D). Western analysis of polyQ127 peptide levels prepared
from the different fly genotypes showed that changes in eyephenotypes were not due to altered expression of the transgene(Figure S7E). This set of experiments indicates that the Alfy/Bchsproteins have a significant role in suppressing the in vivo cytotox-icity of aggregation-prone proteins, in large part mediatedthrough the macroautophagic pathway.
Figure 6. Alfy/Bchs Overexpression Leads to a Disappearance of Inclusions in a Primary Neuronal Model of HD and Diminished Neurotoxicityin a Drosophila Eye Model of PolyQ Toxicity(A and B) Alfy overexpression in a lentiviral model of HD leads to fewer inclusions in rat primary cortical neurons.
(A) Immunofluorescence (IF) against exon1Htt (green) with MAB5492 reveals that transduced neurons exhibit robust aggregation throughout the cell, including
the soma and processes (white arrows), and nuclei (n). Transfection of Alfy1–1271 (p = 0.4241) and Alfy2285–2981 (p = 0.3325) had no noticeable effect on the number
of inclusions/cell. In contrast, expression of Alfy2981–3526 led to a significant reduction in the aggregate load in the neurons (p < 0.001). The visible green puncta in
the field that do not colocalize with red are attributable to neuronal processes from non-Alfy transfected neurons.
(B) Number of inclusions per cell was counted in the number of cells indicated. All visible puncta within a 100 mm radius from the center of the nucleus were
counted. Cells were considered within the cell if the puncta (green) colocalized to cytosol expressing the Tomato fluorophore (red).
(C and D) Alfy/Bchs overexpression in the Drosophila eye leads to protection against polyQ toxicity.
(C) Canton-S control flies (wild-type) show the normal pigmentation profiles and ommatidial features of the adult Drosophila eye. Expression of polyQ127 peptide
(UAS-PolyQ-127; n = 20) using the pGMR-Gal4 driver leads to distinctive defects throughout the eye, including necrotic regions (pGMR-Gal4/UAS-PolyQ-127,
arrows; n = 20). Coexpression of full-length Bchs (pGMR-Gal4, UAS-PolyQ-127/UAS-EP-bchs; n = 20) reduced the level of observable necrosis. Coexpression of
PolyQ127 peptide with the C-terminal region of Bchs (pGMR-Gal4, UAS-PolyQ-127/UAS-bchs-C1000; n = 20) leads to suppression of this external toxic pheno-
types. Coexpression of a dsAtg8a (pGMR-Gal4, UAS-PolyQ-127/UAS-bchs-C1000/UAS-dsAtg8a; n = 15) RNAi transgene blocked this protective effect.
(D) The number of necrotic regions was determined for (C). Whereas coexpression of dsAtg8a further reduced eye size, it did not significantly alter necrosis levels
(p = 0.2391). Both full-length and C1000-Bchs significantly reduced fly eye necrosis (p < 0.0001). This decrease was abrogated when C1000 was coexpressed
with a dsRNA against ATG8a (p = 0.9977), which significantly inhibits macroautophagy in the developing eye.
All data are shown as mean + SE. Complete statistics can be found in Supplemental Experimental Procedures under ‘‘Statistical Information for Figures.’’
Molecular Cell
Alfy-Mediated Degradation of Aggregates
Molecular Cell 38, 265–279, April 23, 2010 ª2010 Elsevier Inc. 275
in adult Drosophila head and that Atg8b is not expressed in thistissue (Simonsen et al., 2008). When the C-terminal Bchs wasexpressed together with Atg8a KD using a UAS-dsAtg8a trans-gene, the protection against polyQ127 generated by higherBchs-C1000 levels was no longer observed (Figures 6C and6D). Western analysis of polyQ127 peptide levels prepared
from the different fly genotypes showed that changes in eyephenotypes were not due to altered expression of the transgene(Figure S7E). This set of experiments indicates that the Alfy/Bchsproteins have a significant role in suppressing the in vivo cytotox-icity of aggregation-prone proteins, in large part mediatedthrough the macroautophagic pathway.
Figure 6. Alfy/Bchs Overexpression Leads to a Disappearance of Inclusions in a Primary Neuronal Model of HD and Diminished Neurotoxicityin a Drosophila Eye Model of PolyQ Toxicity(A and B) Alfy overexpression in a lentiviral model of HD leads to fewer inclusions in rat primary cortical neurons.
(A) Immunofluorescence (IF) against exon1Htt (green) with MAB5492 reveals that transduced neurons exhibit robust aggregation throughout the cell, including
the soma and processes (white arrows), and nuclei (n). Transfection of Alfy1–1271 (p = 0.4241) and Alfy2285–2981 (p = 0.3325) had no noticeable effect on the number
of inclusions/cell. In contrast, expression of Alfy2981–3526 led to a significant reduction in the aggregate load in the neurons (p < 0.001). The visible green puncta in
the field that do not colocalize with red are attributable to neuronal processes from non-Alfy transfected neurons.
(B) Number of inclusions per cell was counted in the number of cells indicated. All visible puncta within a 100 mm radius from the center of the nucleus were
counted. Cells were considered within the cell if the puncta (green) colocalized to cytosol expressing the Tomato fluorophore (red).
(C and D) Alfy/Bchs overexpression in the Drosophila eye leads to protection against polyQ toxicity.
(C) Canton-S control flies (wild-type) show the normal pigmentation profiles and ommatidial features of the adult Drosophila eye. Expression of polyQ127 peptide
(UAS-PolyQ-127; n = 20) using the pGMR-Gal4 driver leads to distinctive defects throughout the eye, including necrotic regions (pGMR-Gal4/UAS-PolyQ-127,
arrows; n = 20). Coexpression of full-length Bchs (pGMR-Gal4, UAS-PolyQ-127/UAS-EP-bchs; n = 20) reduced the level of observable necrosis. Coexpression of
PolyQ127 peptide with the C-terminal region of Bchs (pGMR-Gal4, UAS-PolyQ-127/UAS-bchs-C1000; n = 20) leads to suppression of this external toxic pheno-
types. Coexpression of a dsAtg8a (pGMR-Gal4, UAS-PolyQ-127/UAS-bchs-C1000/UAS-dsAtg8a; n = 15) RNAi transgene blocked this protective effect.
(D) The number of necrotic regions was determined for (C). Whereas coexpression of dsAtg8a further reduced eye size, it did not significantly alter necrosis levels
(p = 0.2391). Both full-length and C1000-Bchs significantly reduced fly eye necrosis (p < 0.0001). This decrease was abrogated when C1000 was coexpressed
with a dsRNA against ATG8a (p = 0.9977), which significantly inhibits macroautophagy in the developing eye.
All data are shown as mean + SE. Complete statistics can be found in Supplemental Experimental Procedures under ‘‘Statistical Information for Figures.’’
Molecular Cell
Alfy-Mediated Degradation of Aggregates
Molecular Cell 38, 265–279, April 23, 2010 ª2010 Elsevier Inc. 275

15
Figure 1.6: Loss of Alfy does not affect basal macroautophagic function. HeLa or Neuro2a cells labeled with [14C] valine were incubated for four hours with complete media or starvation media in the presence of absence of 3-MA. Two Alfy siRNAs did not affect % proteolysis under these conditions. From Filimonenko et al., 2010.
Figure 3. Alfy Is Not Required for Starvation-Induced Autophagy(A) Alfy is not required for LLP degradation. Amino acid withdrawal (Starv) leads to a significant effect in both HeLa and N2a cells. Knockdown of Alfy had no
significant effect on LLP degradation (HeLa, p = 0.6991; N2a, p = 0.3505). To ensure that LLP degradation was due to macroautophagy, starvation experiments
were also performed + 10 mM 3MA. Cells were transfected with siCTRL or siALFY and then monitored for LLP degradation 72 hr later (n = 3 experiments in dupli-
cate or triplicate).
(B and C) Alfy depletion has no effect on starvation-induced or basal (C) LC3 lipidation (p = 0.7641; n = 4). HeLa cells transfected with siALFY or siCTRL were
cultured in complete medium (CM) or starved (Starv) in the absence or presence of BafA1 for 4 hr (B) or 2 hr (C).
(D) Alfy depletion has no effect on LC3 puncta formation. Cells transfected with siCTRL, siATG7, or siALFY were starved, fixed, and stained for endogenous LC3. In
starved cells, there is a significantly greater number of LC3-positive puncta in siCTRL and siALFY cells, whereas siATG7 cells have significantly fewer LC3-positive
puncta upon starvation between siCTRL and siATG7 (p < 0.0001) and siALFY and siATG7 (p < 0.0001). Error bars represent mean + SD (n = 8).
Molecular Cell
Alfy-Mediated Degradation of Aggregates
270 Molecular Cell 38, 265–279, April 23, 2010 ª2010 Elsevier Inc.
Figure 3. Alfy Is Not Required for Starvation-Induced Autophagy(A) Alfy is not required for LLP degradation. Amino acid withdrawal (Starv) leads to a significant effect in both HeLa and N2a cells. Knockdown of Alfy had no
significant effect on LLP degradation (HeLa, p = 0.6991; N2a, p = 0.3505). To ensure that LLP degradation was due to macroautophagy, starvation experiments
were also performed + 10 mM 3MA. Cells were transfected with siCTRL or siALFY and then monitored for LLP degradation 72 hr later (n = 3 experiments in dupli-
cate or triplicate).
(B and C) Alfy depletion has no effect on starvation-induced or basal (C) LC3 lipidation (p = 0.7641; n = 4). HeLa cells transfected with siALFY or siCTRL were
cultured in complete medium (CM) or starved (Starv) in the absence or presence of BafA1 for 4 hr (B) or 2 hr (C).
(D) Alfy depletion has no effect on LC3 puncta formation. Cells transfected with siCTRL, siATG7, or siALFY were starved, fixed, and stained for endogenous LC3. In
starved cells, there is a significantly greater number of LC3-positive puncta in siCTRL and siALFY cells, whereas siATG7 cells have significantly fewer LC3-positive
puncta upon starvation between siCTRL and siATG7 (p < 0.0001) and siALFY and siATG7 (p < 0.0001). Error bars represent mean + SD (n = 8).
Molecular Cell
Alfy-Mediated Degradation of Aggregates
270 Molecular Cell 38, 265–279, April 23, 2010 ª2010 Elsevier Inc.

16
Macroautophagy of aggregation-prone proteins in neurodegenerative disease
While increasing evidence indicates that macroautophagy is able to facilitate turnover of
aggregated protein structures in a selective manner, there is controversy surrounding its role in
neurodegenerative disease states (Nixon, 2013). This question has begun to be addressed by
employing genetic and pharmacological manipulations to macroautophagy in model systems
where aggregation-prone proteins accumulate. A variety of factors may influence the impact of
macroautophagy on clearance of aggregation-prone proteins in neurons, including the type and
function of the protein, its location within the cell, and the site and manner in which the protein
accumulates. These characteristics in turn may influence whether accumulation leads to the
dysfunction of protein degradation systems, and whether macroautophagy impacts the progression
of a particular disease. In the following section we will examine the function and localization of
several aggregation-prone proteins, including polyglutamine-expanded proteins, alpha-synuclein,
amyloid-beta, prion, TDP-43, and SOD1, along with evidence for their macroautophagic
clearance.
Polyglutamine-expanded proteins
CAG trinucleotide repeats encoding polyglutamine (polyQ) stretches of expanded
pathological lengths are known to produce a family of autosomal dominant inherited neurological
diseases including Huntington’s disease, six types of spinocerebellar ataxia, spinal and bulbar
muscular atrophy (SBMA), and dentatorubral pollidoluysian atrophy (DRPLA). Greater than 35-
40 CAG repeats within the affected gene results in clinical presentation, and length of the polyQ
stretch is negatively correlated with age of onset and disease severity (Wetzel, 2012).

17
Beyond their association with disease, polyQ sequences are frequently observed within the
mammalian genome, and have been proposed to serve several normal functions. Large-scale
statistical analyses of proteins containing polyQ stretches have revealed a bias towards nuclear
localization and functions related to transcriptional regulation. The polyQ sequence has also been
proposed to operate as a flexible spacer between protein domains or to serve as a means of
regulating protein-protein interactions (Schaefer et al., 2012).
A unifying pathological feature of polyQ expansion disorders is the presence of
ubiquitinated inclusions, suggesting that the excess repeats destabilize the native protein
conformation and increase the propensity for aggregation. Early in vitro analyses suggested that
aggregation of proteins with polyQ expansions occurred only above the pathological threshold for
clinical disease presentation and that the kinetics of aggregate formation were increased for longer
expansions (Scherzinger et al., 1997; Wetzel, 2012). Many studies suggest that aggregation of
proteins containing a polyQ expansion disrupts not only their normal function but the stability of
other proteins that misfold due to aberrant interactions with them, in turn increasing the burden on
protein degradation systems (Gestwicki and Garza, 2012; Gidalevitz et al., 2006). Localization of
polyQ inclusions varies with disease, and may be primarily nuclear (SCA1, SCA7, SCA17,
SBMA), primarily cytoplasmic (SCA2 and SCA6), or in both compartments (SCA3, DRPLA, and
HD). Table 1.1 outlines the normal and abnormal localization and known functions of polyQ-
expanded proteins.
The most extensively researched of the polyglutamine-expanded disease proteins is
huntingtin (Htt). Htt is expressed ubiquitously, with highest levels in the central nervous system,
and is required for normal development. Full length Htt is a 350 kd protein containing multiple
HEAT repeats, which are normally involved in protein-protein interactions, as well as nuclear

18
export and nuclear localization signals. Htt undergoes many post-translational modifications, and
contains multiple sites of proteolytic cleavage. Within cells it associates with multiple organelles,
and interacts with various transcription factors as well as trafficking and endocytic proteins, which
may reflect its role as a scaffold protein (Ochaba et al., 2014; Rui et al., 2015) and a modulator of
transcription and vesicle trafficking (Zheng and Diamond, 2012). It is normally a cytoplasmic
protein, but it traffics through the nucleus, and truncated N-terminal fragments with CAG
expansions accumulate within the nucleus as well as the cytoplasm. In fact the majority of ataxin
proteins containing pathogenic polyQ expansions, while differing in function (Table 1.1), undergo
nucleocytoplasmic translocation, as does the androgen receptor, which is expanded in SBMA and
requires testosterone for nuclear import (Orr, 2012).
The involvement of autophagy in the disposal of proteins with polyQ expansions has
become an area of interest in recent years (Jimenez-Sanchez et al., 2012). Expanded polyQ proteins
can be observed to associate with lysosome-like structures in the brains of patients with HD, SCA3,
and DRPLA (Jimenez-Sanchez et al., 2012; Sapp et al., 1997; Yamada et al., 2002), suggesting a
link with neurodegenerative disease. In human brain tissue as well as in mouse and cellular model
systems, polyQ-expanded proteins often appear to colocalize with autophagy-related proteins such
as LC3, p62 and NBR1, suggesting the involvement of autophagy in their clearance (Mori et al.,
2012).
Genetic or pharmacological inhibition of macroautophagy in systems expressing expanded
polyQ proteins has served to further elucidate its role in their disposal. Clearance of mutant Htt, in
both soluble and aggregated forms, is impaired by pharmacological inhibition of autophagy
(Filimonenko et al., 2010; Jimenez-Sanchez et al., 2012; Ravikumar et al., 2002), whereas the
clearance of wild-type Htt is unaffected by pharmacological inhibition and may be effectively

19
cleared by the UPS (Ravikumar et al., 2006). It is important to emphasize that autophagy degrades
cytoplasmic cellular components; autophagosomes and lysosomes have not been observed in the
nucleus. Thus the cellular distribution of the soluble and accumulated forms of the protein may
determine the role of autophagy in their clearance. RNAi based knockdown methods show that in
cell lines, autophagy is necessary for the elimination of cytoplasmic but not nuclear expanded
huntingtin and Ataxin-1 (Iwata et al., 2005; Iwata et al., 2009; Orr, 2012). Disease proteins that
aggregate within the nucleus may be shuttled out to facilitate macroautophagic degradation. For
instance, Jeong et al (2009) found that increased acetylation at lysine residue K444 facilitates
trafficking of mutant Htt into autophagosomes. Acetylated huntingtin was more predominant in
the cytoplasm, and the authors propose that nuclear accumulation of mutant Htt could drive
acetylation and promote its nuclear export. The lysosomal breakdown of expanded (or indeed,
wild-type) aggregated forms of ataxin-1 and other ataxins may be attributed to their ability to
shuttle between the nucleus and cytoplasm (Servadio et al., 1995), and polyglutamine expansion
impedes the ability of Atx-1 to exit the nucleus (Irwin et al., 2005; Orr, 2012).
The interference with proteins involved in the selective macroautophagy of aggregates
(aggrephagy) has been shown to affect the accumulation of polyQ-expanded proteins, but it is not
clear that this reflects macroautophagic clearance or selectivity for polyQ-containing aggregates.
p62, for example, is found in neuronal inclusions and associates with autophagosomes in many
disease states and models of aggregation-prone protein accumulation, and is associated with both
formation of aggregates and their clearance by autophagy. Depletion of p62 in cellular and mouse
models of SBMA exacerbated the accumulation of soluble mutant androgen receptor in the
nucleus, while overexpression induced the formation of nuclear inclusions (Doi et al., 2013; Todd
and Lim, 2013). However, depletion of p62 in a dividing cell line overexpressing mutant Htt had

20
no effect on its aggregation, while overexpression induced cytoplasmic aggregate formation
(Korolchuk et al., 2009). Conflicting reports have promoted either toxicity or protection conferred
by p62-mediated formation of polyQ-containing aggregates, but it is unclear whether this truly
promotes macroautophagic clearance. This may reflect the use of dividing cell lines to study
aggregation-prone proteins affecting neurons, as well as the fact that p62 has also been associated
with the ubiquitin-proteasome pathway and the selective macroautophagic processing of a variety
of ubiquitinated substrates.
Knockdown of the macroautophagic adaptor protein Alfy, which is involved in
aggrephagy, results in impaired clearance of polyQ-expanded Htt in tet-regulatable Hela cell lines,
specifically of detergent-insoluble and not soluble forms of mutant exon1-Htt (Figure 1.4). Its
overexpression leads to elimination of aggregates in an Atg5-dependent manner, and loss of Alfy
destabilizes the interaction between core macroautophagy proteins and mutant Htt. Furthermore,
Alfy is not required for starvation-induced or basal macroautophagy. Alfy also colocalizes with
polyQ-expanded Ataxin-1 within the nucleus, (Filimonenko et al., 2010) and its ability to shuttle
in and out of the nucleus and colocalize with many types of aggregated structures could reflect its
role as a selective adaptor for aggrephagy. However, like many autophagy-related proteins, its role
in adult or aging mammalian brain has yet to be explored in vivo.
Alpha-synuclein
While the precise biological function of α-synuclein is undefined, it localizes mainly to
presynaptic terminals, and knockout studies have suggested important roles in the regulation of
synaptic function, plasticity, and release of neurotransmitter (Lashuel et al., 2013; Lynch-Day et
al., 2012). Point mutations or multiplications in the gene coding for α-synuclein are associated

21
with Parkinson’s disease (PD), and the aggregation of α-synuclein in Lewy bodies is a hallmark
of PD and related a-synucleinopathies (Lynch-Day et al., 2012). Classical Lewy bodies, found
typically in brainstem nuclei, are cytoplasmic structures containing α-synuclein, ubiquitin, and
several other components, consisting of a dense core and a halo of radiating fibrils, while cortical
Lewy bodies lack a halo (Lashuel et al., 2013). In multiple systems atrophy, α-synuclein
accumulates in glial cytoplasmic inclusions and has also been found in neuronal nuclei
(Kontopoulos et al., 2006), and an abundance of small aggregates containing α-synuclein can be
detected at presynaptic terminals in patients with Dementia with Lewy Bodies (DLB) (Lashuel et
al., 2013).
Overexpression of mutant α-synuclein in dividing cell lines rarely results in the formation
of persistent detergent-insoluble aggregates or “true” Lewy bodies in culture, which can
complicate analyses. Cuervo et al (2004) first proposed that α-synuclein is normally processed by
chaperone-mediated autophagy (Hayashi and McMahon, 2002), but that mutant forms disrupt this
degradative pathway, leading to increased accumulation. In this case, macroautophagic removal
might be of particular importance. Indeed, accumulated mutant α-synuclein in cell culture
associates with markers of macroautophagy like LC3 and p62 (Tanik et al., 2013). In tetracycline-
regulated cell systems, knockdown of the aggrephagy-specific adaptor protein Alfy impedes
doxycycline-mediated clearance of both wild type and mutant SDS-insoluble α-synuclein,
suggesting that accumulated forms could be selectively targeted for macroautophagic removal
(Filimonenko et al., 2010).
Furthermore, mutant α-synuclein has been observed inside vesicles with autophagic
morphology, and accumulates in the presence of macroautophagy inhibitors such as 3-MA (Cuervo
et al., 2004), and Lewy bodies in patient tissue have been shown to contain LC3 but rarely

22
LAMP2A (Alvarez-Erviti et al., 2010). In mouse brain, knockdown of Atg7 in Nestin-positive
cells leads to the accumulation of a pathogenic form of α-synuclein. Atg7 knockdown in midbrain
dopaminergic neurons led to the accumulation of α-synuclein-positive ubiquitinated aggregates in
striatal axons (Friedman et al., 2012; Johnson et al., 2012).
Tau
Neurofibrillary tangles consisting of insoluble, fibrillar tau contain ubiquitin and are a
pathological hallmark of tauopathies like Alzheimer’s disease and frontotemporal dementia. The
normal tau protein is expressed abundantly in neurons where it is enriched in axons and functions
to stabilize mictrotubules and to regulate microtubule-driven transport along axons (Ke et al.,
2012). In tauopathies, tau becomes increasingly phosphorylated and accumulates within neuronal
soma and dendrites. Phosphorylation weakens its association with microtubules and its stabilizing
function is compromised. Many enzymes cleave tau under both physiological and pathological
conditions, and these fragments tend to aggregate (Chesser et al., 2013).
Pharmacological inhibition of proteasomal or autophagic pathways has suggested that full-
length tau is cleared by the UPS, while aggregation-prone cleaved forms such as caspase-3-cleaved
tau are degraded by macroautophagy (Bednarski and Lynch, 1996; Chesser et al., 2013). Though
pharmacological manipulations indicate that tau may be a substrate for macroautophagy in full-
length and truncated forms in both normal and disease states (Lee et al., 2013a), the mechanism is
unclear, and genetic manipulations of core autophagy genes could serve to further elucidate
whether macroautophagic degradation of tau plays a major role in its disposal. For instance, the
genetic elimination of Atg7 as well as p62 both lead to the accumulation of Tau phosphorylated at

23
several epitopes, suggesting that an intact macroautophagic system is required for tau metabolism
(Chesser et al., 2013).
Contributing to a possible role for macroautophagy in tau processing is the observation that
the brains of patients with AD show accumulation of autophagic vacuoles (AVs) in dystrophic
neurites that also contain filamentous tau (Nixon, 2013). Mouse models of tauopathy recapitulate
these observations (Funderburk et al., 2010; Lin et al., 2003), and in an N2a cellular model of
tauopathy, tau appears enriched in the lysosomal subfraction, independent of its ability to
aggregate (Funderburk et al., 2010; Wang et al., 2009). A major difficulty in studying tau turnover
can be attributed to the endogenous function of tau, which is to stabilize microtubules. Since
protein trafficking pathways, including autophagic pathways, rely on the microtubule network
(Funderburk et al., 2010), it becomes difficult to distinguish cause and effect.
Amyloid-beta
Amyoid-beta (Aβ) is the principal component of extracellular senile plaques in
Alzheimer’s disease (AD) and related amyloidopathies. It is a self-aggregating peptide generated
by cleavage of the amyloid precursor protein (APP) by β- and γ-secretase enzymes. The precise
function of APP and its cleavage products remain elusive, but they are ubiquitously expressed in
the central nervous system and are thought to mediate neuroprotective, trophic, and adhesive
functions (Hiltunen et al., 2009). Mutations in APP and presenilin, a protein involved in Aβ
proteolysis, lead to rare forms of familial AD, but sporadic forms show parallels in pathology,
suggesting that similar factors influence disease progression.
The brains of patients with AD and the brains of PS1/APP mice show accumulation of
autophagic vacuoles in dystrophic neurites, the presence of which has been associated with

24
dysregulation of the endosomal/lysosomal system. (Funderburk et al., 2010; Yu et al., 2005; Yu et
al., 2004). This accumulation of AVs occurs before plaque deposition is observed to occur, and is
not observed in diseases that don’t feature Aβ deposition. One conclusion that has been drawn
from these observations is that lysosomes themselves are a site of Aβ production. APP colocalizes
with AVs, and Aβ peptides and γ-secretase complex components like presenillin-1 (PS1) are
enriched in AV fractions from human brain as well as mouse and cell models of APP
overexpression (Funderburk et al., 2010; Yu et al., 2005). AVs from these models exhibit γ-
secretase activity, and autophagic activity correlates with increased production of Aβ (Funderburk
et al., 2010; Nixon et al., 2005; Yu et al., 2004). In fact, PS1 mutations, such as in early-onset
familial AD, have been observed to impair autophagic turnover (Lee et al., 2010), and PS1 may
be required for basal lysosomal clearance of certain proteins. Evidence of lysosomal abnormalities
in AD and other neurodegenerative disorders could indicate that autophagic inhibition represents
an early event in disease pathogenesis, but there is no direct evidence that Aβ itself contributes to
this process.
While lysosomal pathways may contribute to the generation of Aβ peptide, it remains
unclear whether it is normally a substrate of autophagy. Aβ associates with lysosomal membranes
(Liu et al., 2010), and various stressors including oxidative stress and proteasomal inhibition lead
to the enrichment of Aβ in lysosomes in cell lines (Agholme et al., 2012; Funderburk et al., 2010;
Zheng et al., 2006). Beclin-1 downregulation has been observed in the brains of AD patients, but
this is not recapitulated by an APP-transgenic mouse model. However, beclin-1 depletion in the
same model resulted in Aβ accumulation, while overexpression resulted in reduced levels of both
intracellular and extracellular Aβ (Castillo et al., 2013; Funderburk et al., 2010; Pickford et al.,
2008). Intracellular accumulation of Aβ is toxic in triple-transgenic AD mouse models, (Billings

25
et al., 2005) and thus under extreme circumstances autophagy might serve as a protective
mechanism against cytoplasmic buildup.
Prion
Transmissible spongiform encephalopathies (TSEs), or prion diseases, are progressive and
fatal neurodegenerative disorders that are propagated by induced protein misfolding. Cellular prion
protein (PrPC) is expressed abundantly in neurons and glia, and while some of its functional
domains have been mapped, its normal role in cells remains elusive. Studies of PrPC deficient mice
and their primary neurons have provided evidence for a protective role against apoptosis and
oxidative stress or a role in synaptic maintenance (Aguzzi et al., 2008).
In the context of disease, prions have historically been defined as infectious agents or
transmissible particles that lack nucleic acid (Heiseke et al., 2010; Prusiner, 1998). Human prion
diseases, such as kuru, Creutzfeldt–Jakob disease (CJD) Gerstmann–Sträussler–Scheinker
syndrome, and fatal familial insomnia, involve the conversion of PrPC to a misfolded pathological
form, PrPSc, which propagates by transmitting its pathological misfolding to more of the normal
protein. The newly formed prion can continue to convert more PrPC to PrpSc, resulting in the
accumulation of cytoplasmic aggregates.
PrpSc-like proteins have been shown in animal models and in neuroblastoma cell lines to
localize to aggresomes (Heiseke et al., 2010). Early evidence for the dysregulation of
endolysosomal degradation systems in TSEs came from experimental scrapie in hamsters, where
giant autophagic vacuoles (AVs) were described in neurons (Boellaard et al., 1991; Yao et al.,
2013). These AVs increase in number and size through disease progression and eventually fill up
the cytoplasm of affected cells. AVs also develop in prion-transfected cultured neurons, and have

26
been found in perikarya, neurites and synapses in postmortem human brain and in experimental
animals.
PrP-null mice have been used extensively as a disease model and to elucidate the function
of normal PrP. Under starvation conditions, PrP-null hippocampal neurons transfected with the
prion-like protein doppel (Dpl) show increased LC3-II and p62 protein levels and an increase in
autophagosome formation compared to controls, suggesting a role for PrP in autophagy regulation
(Heitz et al., 2010; Yao et al., 2013). The unusually large, abundant, and persistant nature of AVs
as observed in models of PrP could suggest that autophagy is upregulated as a compensatory
response to the presence of PrpSc aggregates (Liberski et al., 2008; Yao et al., 2013). It has also
been suggested that in the initial phase of prion protein accumulation, autophagy could serve to
break inclusions into smaller pieces, thus increasing the likelihood of PrPSc seeding (Heiseke et
al., 2010).
TDP-43
TDP-43 is a DNA/RNA-binding factor that is primarily located as a dimer in the nucleus,
but can continuously shuttle between the nucleus and cytoplasm. It has been implicated in a
number of cellular functions, including splicing, translation, and transcriptional repression. In the
brain, it is transiently redistributed in response to axonal injury (Janssens and Van Broeckhoven,
2013; Mackenzie et al., 2010; Sato et al., 2009). The two major categories of TDP-proteinopathies
are frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U), and
amyotrophic lateral sclerosis (ALS). These are characterized by the presence of primarily
cytoplasmic but sometimes nuclear inclusions containing ubiquitin and TDP-43 (Chen et al., 2012;
Janssens and Van Broeckhoven, 2013; Mackenzie et al., 2010; Neumann et al., 2006). TDP-43

27
found in aggregates in these diseases is phosphorylated, ubiquitinated, and tagged with ubiquilins
as well as p62 (Chen et al., 2012; Janssens and Van Broeckhoven, 2013). Mutations in TDP-43
itself lead to familial forms of ALS, and mutations in proteins responsible for targeting other
proteins for degradation, like ubiquilin-2, p62, and valosin containing protein (VCP) can also cause
TDP-43-associated ALS and FTD (Chen et al., 2012; Deng et al., 2011; Fecto and Siddique, 2011;
Janssens and Van Broeckhoven, 2013; Scotter et al., 2014).
TDP-43 overexpression has been used in several cell lines in an attempt to model TDP-43
proteinopathies. Both mutant and wild-type TDP-43 when overexpressed have been shown to
aggregate, and it has recently been suggested that soluble forms are degraded by the proteasome
while insoluble forms require lysosomal degradation (Barmada et al., 2010; Janssens and Van
Broeckhoven, 2013; Scotter et al., 2014). Proteasomal inhibition has been shown to induce the
formation of inclusions containing full-length TDP-43 in some models, but neither genetic nor
pharmacological inhibition of autophagy has been sufficient to produce aggregates. However,
inhibition of beclin-1 has been shown to impair clearance of stably transfected TDP-43 (Jinwal et
al., 2012).
TDP-43 may itself regulate elements of the autophagic machinery, for example by
stabilizing Atg7 mRNA; in Neuro2A cells, TDP-43 knockdown downregulates expression of Atg7
and decreases LC3-II protein levels (Bose et al., 2011; Lamark and Johansen, 2012). Thus, aberrant
TDP-43 function or mislocalization could impair protein clearance mechanisms and contribute to
its aggregation (Chen et al., 2012; Song et al., 2012)

28
SOD1
SOD1 is an abundantly expressed antioxidant enzyme that exists as a homodimer and
localizes mainly to the cysosol with some expression in the mitochondrial inner membrane. In
addition to its antioxidant properties, it has been implicated in signal transduction, and both its
wild-type and mutant forms are prone to misfolding (Rotunno and Bosco, 2013).
Mutant SOD-1 transgenic mice develop ALS-like pathology including degeneration of
motor neurons and a progressive paralytic phenotype, and deposits of mutant SOD1 are found
within the cytoplasm of cells in the CNS. Both wild-type and mutant SOD1 are degraded by
autophagy and by the proteasome. While overexpression of mutant SOD1 in neuroblastoma cell
lines is not acutely toxic, inhibition of macroautophagy under these conditions induced cell death
and increased both soluble and insoluble forms of SOD-1. Rapamycin treatment reduces levels of
mutant SOD1, while beclin-1 knockdown increases it (Kabuta et al., 2006).
Dynamics and localization of aggregates
Studies of both yeast and mammalian cell lines suggest that cells have distinct strategies
for the sequestration of different types of aggregation prone proteins, employing multiple quality
control compartments. For example, detergent-soluble misfolded ubiquitinated proteins have been
shown to localize to a juxtanuclear quality control compartment (JUNQ) as compared to
amyloidogenic proteins, such as mutant Htt and prion, which are sequestered into an insoluble
protein deposit, or IPOD. These compartments exhibit different rates of exchange with the
cytosolic pool (Kaganovich et al., 2008). Thus the location of a cytosolic inclusion body may have
a profound impact on its persistence and toxicity.

29
The cellular compartment to which a particular aggregation-prone protein is localized may
also exert a gross influence on its toxicity. Many have argued that nuclear accumulation of polyQ-
expanded proteins such as mutant Htt is a key trigger in pathogenesis. For example, addition of a
nuclear export signal to mutant huntingtin can decrease toxicity in cultured cells, while suppression
of the NLS driving accumulation of mutant ataxin-1 abolishes the neuropathology in a mouse
model of SCA1 (Klement et al., 1998; Orr, 2012). Similarly, models of SBMA suggest that nuclear
localization of polyQ-expanded androgen receptor is required for toxicity, and that cytoplasmic
retention of AR leads to increased propensity for autophagic degradation, promoting the
amelioration of disease symptoms (Montie et al., 2009; Rusmini et al., 2010).
Proteins with polyQ tracts often have functions that would require them to shuttle in and
out of the nucleus; it has been suggested that the expansion confers the propensity for aggregation
in the nucleus (Wetzel, 2012). This could prevent the normal function not only of the protein in
question, but of any nuclear protein, in particular transcription factors that serve to regulate
activities cell-wide. Since macroautophagy is a cytoplasmic process, nuclear aggregates or any
form of the aberrant protein which is not degradable by the proteasome would have to be
transported across the nuclear membrane for macroautophagic clearance.
Furthermore, the organization and recruitment properties of different types of aggregate
structures may determine how effectively the cell is able to control their accumulation through
active degradation pathways. Aggregate structures containing mutant 103Q-expanded huntingtin,
for example, contain a denser core and a surface more conducive to protein recruitment than
aggregates containing mutant SOD1, which form a more diffuse porous structure through which
other proteins can move (Matsumoto et al., 2006). Even within the family of polyglutamine-
containing proteins, the dynamics of aggregation and removal differ. These observations suggest

30
that the proteins driving their disassembly and removal, including those required for autophagy,
might associate differently with distinct aggregate structures, and their location might also
influence this accessibility.
Regulation and dysregulation of macroautophagy in neurodegeneration
Autophagy-related proteins have been shown to colocalize with various types of
aggregated proteins in nearly every neurodegenerative disease state. This observation is frequently
cited as evidence that autophagy of these structures is occurring. For example, beclin-1 colocalizes
with Htt inclusions in both mouse and human brain (Nixon, 2013; Shibata et al., 2006), and
fluorescently stained Atg5 and LC3 have been observed to form shell-like structures around large
TDP-43 aggregates in culture (Brady et al., 2011; Janssens and Van Broeckhoven, 2013). In PD
brains Lewy bodies contain proteins of the autophagic machinery (Alvarez-Erviti et al., 2010).
Nevertheless, it remains in question whether the mere presence of proteins required for
macroautophagy indicates that active lysosomal degradation of structures is occurring. Currently,
our ability to directly visualize macroautophagic activity is impeded by our incomplete knowledge
surrounding the birth and maturation of autophagosomes, and limited to LC3, the only known
direct marker of these structures.
At the same time, the presence of autophagy-related proteins in aggregates is also
sometimes interpreted to mean that autophagy is compromised, with the explanation that
functional macroautophagic components have been sequestered along with aggregation-prone
proteins. Furthermore, both increases and decreases in levels of Atg proteins and transcripts are
frequently cited as evidence for macroautophagic dysregulation. For example, overexpression of
alpha-synuclein in cell lines and in transgenic mice leads to a decrease in LC3-II and

31
mislocalization of Atg9 (Hochfeld et al., 2013; Winslow et al., 2010). Conversely, increases in
levels of LC3-II are observed when mutant Htt is overexpressed in cell lines (Jimenez-Sanchez et
al., 2012; Ravikumar et al., 2004). While these results could indicate that autophagic function is
compromised in different ways depending upon the aggregating protein, examining solely LC3-II
levels, p62, or other Atg proteins as a measure of macroautophagic activity provides a limited
picture of its role in the disease process.
Postmortem analyses of many types of neurological patient brains show membrane
abnormalities such as the accumulation of giant AVs, as is observed in prion disease and in
Alzheimer’s disease brains (Boellaard et al., 1991; Funderburk et al., 2010; Heiseke et al., 2010;
Yu et al., 2005), both diseases in which aggregate pathology is observed to propagate regionally
and trans-synaptically. Increased autophagosome and endosome formation has also been observed
in cell and mouse models of HD and in human tissue from HD patients (Davies et al., 1997;
Jimenez-Sanchez et al., 2012; Kegel et al., 2000; Sapp et al., 1997), leading to speculations that
overactive autophagy can contribute to cell death.
Research has thus far provided a wealth of evidence that disease-related changes occur in
the levels and locations of the proteins required for initiation and creation of autophagosomes, as
well as the membraneous content of neuronal substructures, cells, and whole brain regions. If, as
some have suggested, macroautophagic dysregulation represents an early event in disease
pathogenesis (Hochfeld et al., 2013), then it will be important to identify at what point in the
process it is failing. This will come only with better mechanistic understanding of the process of
autophagosome building, maturation, and clearance, and at what stage particular types of
aggregation-prone proteins or other pathogenic mechanisms interfere with the macroautophagic
machinery.

32
Macroautophagic upregulation as a therapeutic strategy
Autophagy induction as a therapeutic strategy has recently been explored in a number of
disease models, with the goal of promoting clearance of aggregation-prone proteins and thus
protecting against their potential for toxicity. Genetic approaches, such as the upregulation of
beclin-1 and p62, have been used primarily in cellular models to demonstrate that increasing
macroautophagic components can speed clearance of aggregation-prone proteins (Hochfeld et al.,
2013). Pharmacological means of upregulating macroautophagy have been widely used in fly and
mouse models of neurodegenerative disease.
Rapamycin is the most commonly used pharmacological agent for macroautophagy
induction. It inhibits the activity of mTORC1 (mammalian target of rapamycin complex 1), which
among many other functions, is a negative regulator of macroautophagic activity, but may also
affect other forms of autophagy and numerous cellular pathways. In cell lines, rapamycin promotes
the clearance of different polyQ-expanded proteins, mutant and wild type α-synuclein, tau, and
PrP, which in some cases has been associated with improvements in cell viability (Aguib et al.,
2009; Chesser et al., 2013; Heiseke et al., 2010; Hochfeld et al., 2013; Qin et al., 2003; Webb et
al., 2003). In fly and mouse models of Huntington’s disease, rapamycin has been shown to slow
accumulation of aggregated proteins or to accelerate clearance of aggregates in conjunction with
neuroprotection (Jimenez-Sanchez et al., 2012; Ravikumar et al., 2004). Using both rapamycin
and a genetic approach to target mTOR resulted in decreased tau pathology in a P301S tau mutant
model (Caccamo et al., 2013; Chesser et al., 2013). Additionally, upregulation of autophagy with
rapamycin treatment or starvation led to lysosome-mediated clearance of tau aggregates (Chesser
et al., 2013; Johansen and Lamark, 2011; Wong et al., 2008). In a mouse model of the genetic
prion disease Gerstmann–Sträussler–Scheinker disease (GSS), chronic presymptomatic rapamycin

33
treatment results in complete absence of the disease’s hallmark PrP-amyloid plaque deposits along
with a delay in disease onset and decreased symptom severity (Castillo et al., 2013; Cortes et al.,
2012). Although autophagy induction can reduce prion accumulation, one group speculates that
early induction of autophagy could support the seeding of prions by breaking large aggregates into
smaller ones, increasing the likelihood for each to propagate (Heiseke et al., 2010). Recently,
administration of rapamycin and several other autophagy inducers rescued neuronal loss as well
as motor and learning deficits in a mouse model of frontotemporal lobal dementia with TDP-43
positive ubiquitinated inclusions (Chen et al., 2012; Song et al., 2012; Wang et al., 2012) However,
rapamycin treatment has been shown to accelerate the motor neuron degeneration phenotype in
the SOD1 G93A model of ALS (Johnson et al., 2012; Zhang et al., 2011), without affecting
accumulation of SOD1 aggregates.
Several other pharmacological agents have been demonstrated to upregulate autophagy and
promote clearance of aggregation-prone proteins in an mTOR-independent manner. Lithium and
resveratrol induce autophagy and result in reduced levels of prion proteins, and trehalose treatment
also lowers levels of PrP in an Atg-5 dependent manner (Aguib et al., 2009; Heiseke et al., 2010;
Jeong et al., 2009). In the SOD1-G93A mouse model of ALS, food restriction or treatment with
trehalose at an early disease stage induces autophagy and decreases SOD1 deposition in this model
(Castillo et al., 2013; Zhang et al., 2013). Trehalose also prevented ARPolyQ-mediated toxicity in
motor neurons, but without significantly affecting levels of nuclear mutant AR (Jimenez-Sanchez
et al., 2012; Montie and Merry, 2009)
While many benefits of macroautophagy induction have been reported, a major
disadvantage to this approach is that available pharmaceuticals, including as rapamycin, trehalose,
valproate, and rilmenidine, act upstream of protein complexes that regulate several additional

34
biological processes besides macroautophagy, notably ribosome biogenesis and protein translation
(Hochfeld et al., 2013; Nixon, 2013), leading to immense pleiotrophic effects and complicating
analyses.
Concluding remarks
The accumulation and persistence of aggregated proteins is a shared feature of many
neurodegenerative disease pathologies, indicating that neuronal protein homeostasis has gone
awry (Gestwicki and Garza, 2012). The exact origin of this imbalance in each disease state, and
whether aggregated proteins represent a source or merely a symptom of disease pathogenesis is
under intense investigation. Genetic and pharmaceutical approaches to inhibit macroautophagy
have revealed that it is one viable pathway by which cells can eliminate disease-relevant
aggregation-prone proteins. However, much of this work has been performed in dividing cell lines,
and we currently have only a budding understanding of macroautophagy in neurons. It is clearly
important during development (Mizushima and Levine, 2010), but its role in the adult brain has
yet to be defined.
Studies that pharmacologically drive macroautophagic upregulation by mTOR-dependent
and independent means have produced protective effects in models but have multiple off-target
effects, making them potentially dangerous for long-term use. It remains to be seen whether
macroautophagic dysfunction or aggregated protein deposition is far enough upstream in disease
pathogenesis to merit wholesale upregulation of the system as a therapeutic strategy. An
understanding of the autophagy-mediated effects of such treatments in experimental systems may
require attention to cellular changes beyond the current standard of examining changes in levels
of macroautophagy-relevant proteins.

35
Finally, the list of proteins involved in targeting selective cargo for macroautophagic
degradation continues to expand. Better mechanistic insights into the identity and function of such
cellular machinery will help to define whether the dysfunction of autophagic pathways plays a role
in neurodegenerative processes, and will extend the possibilities for therapeutic intervention in
diseases involving aberrant protein deposition.

36
Chapter 2: Modeling Huntington’s disease
Introduction
Since the discovery of the causal CAG expansion mutation, the monogenetic nature and
dominant inheritance of HD have enabled the development of a variety of animal models to study
the disease (Pouladi et al., 2013). These have vastly expanded our knowledge of the pathological
mechanisms that invariably lead to the progressive decline and eventual death of HD patients.
Genetically-driven mouse models of HD have been created in a myriad of ways. Several
factors have influenced the pathological and behavioral outcomes in these mice, including the
genetic approach to insertion, the segment of the htt gene used to drive the pathology, and the
design of the polyglutamine repeat sequence (Yang and Gray, 2011). Additional variations upon
existing genetic approaches have also been made in order to understand how the anatomical and
subcellular localization as well as the post-translation modification of mutant huntingtin drives
pathogenesis (Ehrnhoefer et al., 2009). While no single model can approach the human disorder,
each has provided a lens through which to understand the aspects of pathology and symptomology
that it best recapitulates.
This chapter provides a brief introduction to Huntington’s disease and the model systems
that have been developed to study it, with a focus on the enduring approaches in rodents that have
shaped our understanding of mHtt aggregation and removal. First, I will briefly review the
pathology of Huntington’s disease. Next, I will describe the main genetic approaches to mouse
modeling in HD. Finally, I will address variations in approach that have been informative with
regard to the toxicity of mHtt protein, including transgene integration, CAG repeat length,
anatomical and subcellular localization, and protein modifications that impact aggregation and

37
clearance. Preclinical studies that integrate information from multiple models have been most
fruitful for the development of therapies.
Huntington’s disease
George Huntington’s 1872 report on an inherited movement disorder (Huntington, 1872)
was not the first to describe its pernicious symptomology, but he so concisely and compassionately
depicted its triad of indications and hereditary nature that the disease took on his name. HD is
dominantly inherited and manifests clinically in altered cognition, personality changes, and loss of
motor control, typically leading to death within 20 years (Walker, 2007).
Genetics
The systematic and ongoing study of a large pedigree of Venezuelans with Huntington’s
disease led to the identification of the HD gene in 1993 (Huntington’s Disease Collaborative
Research Group). The mutation consists of an expanded trinucleotide CAG repeat within the IT15
gene, which results in an extended glutamine (Q) stretch in the first exon of the huntingtin protein
(Htt). The general population has on average 17-20 CAG repeats within the gene (Kremer et al.,
1994). Individuals with more than 40 repeats develop the disorder with 100% certainty, with
incomplete penetrance in repeat ranges of 36-39 (Walker, 2007). Typical age of onset is 40 years
(Dayalu and Albin, 2015), and in successive generations symptoms may appear with greater
severity and earlier onset, a phenomenon known as anticipation (Ha and Jankovic, 2011). There is
an inverse relationship between the number of repeats and the age of onset, with long repeat lengths
(>55) leading to juvenile manifestation (Roos, 2010) (Figure 2.1). However, CAG repeat length
accounts for only around 60% of the variation in age of onset; remaining variability depends upon

38
genetic modifiers and environmental factors. (Genetic Modifiers of Huntington's Disease, 2015;
Wexler et al., 2004)
Figure 2.1. Inverse relationship between CAG repeat length and age at motor onset of Huntington’s disease. From (Lee et al., 2012).
Symptomology
At the very early or prodromal stages of HD, gene carriers tend to manifest changes in
mood, thinking, and movement, such as irritability and anxiety, difficulty planning and adapting,
and increased fidgeting or restlessness. In most cases these subtle prediagnostic changes merge
towards a diagnosis that usually coincides with the development of chorea, the characteristic loss
of voluntary motor control that takes on a dance-like quality. As the disease progresses, psychiatric
manifestations may involve outbursts, OCD, addictive behaviors, and psychosis. Cognitive
dysfunction impairs activities like organization and acquisition of new skills, and later-stage motor
symptoms involve dystonia and ridigity with less prominent chorea (Walker, 2007). Patients also
manifest circadian and metabolic disruptions, such as insomnia and severe cachexia (Djousse et
al., 2002; Morton, 2013).

39
Neuropathology
Neuropathologically, there is early and severe cell loss and atrophy in the striatum.
Especially susceptible are the GABAergic medium spiny projection neurons (MSNs) of the
caudate and putamen (Vonsattel and DiFiglia, 1998), while interneurons such as cholinergic cells
tend to be spared (Ferrante et al., 1987). MSNs expressing dopamine D2 receptors, enkephalins,
and adenoside A2a receptors are more vulnerable than those expressing D1 receptors, substance
P, dynorphin, and tachykinins (Albin et al., 1992; Deng et al., 2004; Reiner et al., 1988; Vonsattel
et al., 1985). These findings led to the hypothesis that chorea is dominant in HD progression
because of the early death of cells involved in the indirect, movement-inhibiting pathway of the
classically described basal ganglia circuitry (Walker, 2007). Though the striatum shows prominent
deterioration, there is a growing appreciation for the involvement of many brain areas, including
cortical layers 3, 5, and 6, as well as the hippocampus, cerebellum, and thalamic regions, an
observation first reported by early HD anatomists (Rub et al., 2015; Walker, 2007) which is
supported by modern volumetric MRI studies (Aylward et al., 2011; Tabrizi et al., 2013).
Figure 2.2: Neuropathological features of Huntington’s disease. Left: Coronal sections from formaldehyde-fixed cerebral hemispheres at the level of the head of the caudate nucleus (CN) and putamen (Put) from an age-matched normal control (A) and a 62-year-old female with Huntington's disease (B). Note the marked gross atrophy of the neostriatum (CN and Put) and enlarged lateral ventricle (lv) in Panel B. (Ryu et al., 2005). Right: diffuse nuclear staining and intranuclear inclusion labeled for huntingtin (cerebral cortex). From (Ross and Poirier, 2004).

40
Aggregation
Evidence of abnormal accumulations in post-mortem tissue from HD patients was
originally identified by EM and described by Roizin et al (1979). With the identification of the HD
gene came the development of antibodies against the human protein and the creation of genetically-
driven animal models, such that aggregated mutant Htt (mHtt) could be detected directly in mouse
and patient brains. Mutant Htt deposition occurred in a widespread manner that did not reflect the
prominent striatal vulnerability (Becher et al., 1998; Davies et al., 1997; DiFiglia et al., 1997).
Aggregates were first detected in the nucleus (DiFiglia et al., 1997), and later in the cytoplasm and
neuronal processes (Gutekunst et al., 1999), and nuclear accumulation was observed to occur more
frequently in cases of advanced disease or juvenile onset (Aronin et al., 1999). Investigation of
mHtt aggregation in vitro and in cells suggested that aggregation occurred increasingly with longer
polyQ lengths (Scherzinger et al., 1997) and that nuclear accumulation of these structures was
harmful to cells (Chen et al., 2001; Yang et al., 2002). However, there is still ongoing debate as to
whether mHtt deposits are toxic, protective, or inert (Bates, 2003), since preventing the
aggregation of mHtt has also proved to be harmful to cells and mice (Arrasate and Finkbeiner,
2012; Arrasate et al., 2004).
Modeling HD
HD was first modeled through the use of excitotoxic compounds in mice to induce the
selective degeneration of the striatum (DiFiglia, 1990). Since the discovery of the gene, a variety
of model organisms have been genetically modified to investigate the pathogenesis of HD and
polyQ expansion, including single-celled organisms like yeast and slime molds (Mason and
Giorgini, 2011; Wang et al., 2011), invertebrates like worms and drosophila (Neri, 2011), rodents,

41
and large animals such as dogs, pigs, sheep, and primates (Pouladi et al., 2013). This introduction
will focus on variations in genetic approaches to modeling HD in mice, which are by far the most
commonly used organisms.
There have been three main genetic approaches to the creation of HD mouse models:
transgenic N-terminal fragment models, knock-in models, and transgenic full-length models.
These models are described below with attention to the pathological, aggregation, and behavioral
profiles that make them suitable for researching various aspects of HD. I have omitted an in-depth
description of functional electrophysiological changes in these models, which are reviewed
extensively in (Cepeda et al., 2010).
Fragment models
The most widely used fragment models are the R6/2, R6/1 (Mangiarini et al., 1996), and
N171-82Q mice (Schilling et al., 1999). These mice rapidly accumulate aggregated mHtt
throughout the brain in neuronal nuclei and neuropil, exhibit a rapid behavioral onset, widespread
atrophy (Aggarwal et al., 2012; Cheng et al., 2011), lower cell counts via stereology in R6/1 and
R6/2 (Bayram-Weston et al., 2012; Stack et al., 2005), weight loss, and reduced lifespan. These
models drive the expression of truncated human mHtt, consisting of either the first 67 amino acids
comprising exon-1 (Mangiarini et al., 1996) or the first 171 amino acids (Schilling et al., 1999) of
the human protein. Fragment models demonstrated that the polyQ-expanded N-terminal fragment
is sufficient to induce acute toxicity, and have enabled the focused study of aggregation as well as
the early examination of behavioral disease features that do not onset in humans until late
adulthood. One major drawback is that the natural context of the CAG expansion within the gene

42
is lost, which may potentially alter htt regulation, post-translational modification, and protein
interactions (Ehrnhoefer et al., 2009).
Knock-in models
Knock-in mouse models of HD have a poly-Q sequence inserted into the endogenous mouse
huntingtin gene (Menalled, 2005; Menalled et al., 2003; Wheeler et al., 2000), or substitution of a
chimeric human/mouse exon-1 (Lin et al., 2001; Shelbourne et al., 1999). Repeats within the range
of 50-200 have been generated, with the widest use of the HdhQ111, CAG140, and HdhQ150 lines
(Pouladi et al., 2013). These mice tend to have a later and progressive onset of behavioral
symptoms and aggregation. Although overt abnormalities may be subtler than in other types of
models, the Q140 line has shown early changes with thorough behavioral testing (Menalled et al.,
2003). These mice represent the most temporally and spatially relevant context and levels of mHtt
expression (Ehrnhoefer et al., 2009), and their slow progression and in some cases striatally
specific aggregation (Menalled et al., 2002) may be most useful for understanding early
pathological events triggered by the mutation.
Full-length transgenics
Full-length transgenic models were created using yeast artificial chromosome (YAC) and bacterial
artificial chromosome (BAC) technology. The YAC 128 (Slow et al., 2003) and BACHD models
(Gray et al., 2008) express human mutant htt transgenes, including exonic and intronic sequences
as well as regulatory sequences upstream and downstream of the gene. They develop selective
striatal and cortical atrophy without prominent cell loss and exhibit motor, cognitive, and
psychiatric deficits. Their aggregation profile differs, with heavier and more widespread

43
accumulation in the YAC128 mouse than the BACHD (Pouladi et al., 2012). Their robust and
progressive behavioral phenotypes make them well suited for investigating a variety of disease
features and testing therapeutic strategies (Pouladi et al., 2013; Yang and Gray, 2011)
Factors influencing phenotype
As discussed above, models expressing a fragment of mutant huntingtin tend to show
accelerated aggregation, neuropathology, and behavioral progression compared to full-length
models, a finding supported by cellular research indicating that the expression of truncated
fragments increases vulnerability of cells to stressful stimuli (Martindale et al., 1998).
Additionally, transgenic models expressing human mHtt tend to have more severe behavioral and
neurodegenerative phenotypes than knock-in models. Furthermore, within each main category of
genetic model, there are discrepancies in phenotype that may be influenced by several factors in
the generation of the animal, enumerated below.
mHtt expression levels
One factor influencing the outcome of a modeling technique is the expression level of
mutant huntingtin, which can depend upon insertion technique and transgene copy. Evaluation of
different YAC128 lines revealed that the onset of phenotype is exacerbated by higher levels of
full-length mHtt expression (Graham et al., 2006b). In heterozygous knock-in mice, expression
levels of the mutant protein are similar to endogenous mouse huntingtin (Pouladi et al., 2013)
while the YAC and BAC full-length transgenics have 2 copies of endogenous mouse htt in addition
to ~0.75 or ~2 fold levels of human mHtt protein, respectively (Gray et al., 2008; Slow et al.,
2003). The R6/2, which shows earlier and more severe aggregation and phenotypic progression

44
than the R6/1, was originally reported to express 3 transgene copies and 75% of endogenous mouse
Htt levels, compared to 1 transgene copy and 33% of endogenous mouse Htt in the R6/1
(Mangiarini et al., 1996). However, this is further complicated by differences in CAG repeat length
and somatic instability, as well as potential deletions in the flanking regions of R6/2.
Regulatory elements and repeat-flanking regions
Huntingtin is a large gene, containing 67 exons, which has presented challenges in cloning
and manipulating the gene, as well as ensuring the stable integration of constructs (Pouladi et al.,
2013). Differing functional and regulatory elements, such as binding sites for transcription factors
and microRNA targets, might affect phenotype by influencing protein expression, regulation, or
folding (Ehrnhoefer et al., 2009). For example, the BACHD and YAC128 models both express
full-length human mutant htt transgenes that retain relevant upstream and downstream regulatory
elements, but these sequences differ slightly, and the models show divergent features such as levels
of striatally enriched mRNA and aggregation profile (Pouladi et al., 2012).
Additionally, species specific disparities in the mouse and human htt protein and regulatory
elements, which differ between the knock-ins and full-length transgenics, may lead to alternative
splicing events and varying proteolytic cleavage sites (Ehrnhoefer et al., 2009). Another
comparison can be made among the knock-in models, which differ in their proline-rich regions;
the HdhQ150 contains the mouse region, while the HdhQ111 and CAG140 mice contain the
human proline-rich region (Pouladi et al., 2013).
A notable exception to the severity of fragment models is the shortstop mouse, which
expresses 2 exons of human mHtt but exhibits no motor or aggregation phenotypes (Slow et al.,
2005). This fragment is expressed in a similar genetic and regulatory context as the YAC128

45
mouse, and differs in these elements from fragment models like the R6 lines (Mangiarini et al.,
1996); this discrepancy may influence the differential phenotype. Finally, a major potential
confound in any transgenic animal model of disease is that a functional endogenous gene may be
disrupted by random integration of the transgene, contributing to the phenotype attributed to the
disease.
CAG repeat
Both the length and stability of the CAG repeat influence phenotype in mouse models. To
drive a phenotype in mice, models must be made with repeat lengths that would cause childhood
onset of HD in humans (Pouladi et al., 2013). As with humans, longer repeats tend to produce
more severe phenotypes, such as with the original R6/1 and R6/2 lines, which expressed 115 and
150 repeats, respectively (Mangiarini et al., 1996). In knock-in models, longer repeats also tend to
produce more severe phenotypes (Menalled, 2005). The association between repeat length and
severity is reinforced by studies in cells and in vitro showing that huntingtin protein with differing
expansions have divergent properties, with greater propensity for aggregation and nuclear entry at
higher CAG lengths (Chen et al., 2002; Hackam et al., 1998; Li and Li, 1998; Martindale et al.,
1998; Narain et al., 1999).
Somatic and germline instability of the CAG repeats occurs frequently at long tract lengths,
occurring frequently in R6/2 (Shelbourne et al., 1999) and has also been reported in the Q150
knock-in model (Ishiguro et al., 2001; Kennedy and Shelbourne, 2000; Wheeler et al., 1999;
Wheeler et al., 2000). This has resulted in the generation of R6/2 with extremely long repeat tracts,
as high as 450 (Dragatsis et al., 2009; Morton et al., 2009), which changes aggregation dynamics
and results in a paradoxical attenuation of symptoms that might be attributed to the reduced ability

46
of the lengthy protein to enter the nucleus (Dragatsis et al., 2009). Such studies have promoted the
hypothesis that age dependent or tissue-specific somatic expansions contribute to HD pathogenesis
(Swami et al., 2009). Consequently, this has led to the development of models containing an
alternating CAG/CAA polyQ expansion, which promotes stability of the tract (Gray et al., 2008;
Kazantsev et al., 1999) and protects against generational differences within mouse lines, as is the
case with the BACHD mouse.
Spatial expression pattern
Huntingtin is expressed throughout the brain and body, and mouse models of HD normally
express mHtt via the endogenous htt promoter or a universal promoter such as the mouse prion or
ubiquitin promoter (Pouladi et al., 2013). The conditional HD94 mouse expressed tetracycline-
regulatable exon-1 mHtt, which was crossed with a CAMKIIa-tTA driving mouse to produce
forebrain expression that also resulted in progressive degenerative phenotype (Yamamoto et al.,
2000).
The BACHD mouse has a conditional design that has enabled its creators to address how
spatially restricting mHtt affects phenotype. Exon-1 of the full-length human mhtt construct is
flanked by LoxP sites that allow for Cre-mediated excision (Gray et al., 2008). Using this strategy,
the Yang lab restricted full-length human mHtt expression in the cortex, striatum, or both, and
demonstrated that cortical but not striatal expression is required to drive a number of motor and
psychiatric-like phenotypes (Gu et al., 2007; Wang et al., 2014). Full-length knock-in models that
don’t restrict Htt expression, but show preferential accumulation of Htt protein in the striatum,
have less severe phenotypes than other knock-ins (Menalled et al., 2002; Wheeler et al., 2000)
Conversely, the introduction of viral vectors has selective and extreme effects on the striatum and

47
can drive motor abnormalities (DiFiglia et al., 2007). However, overexpression results in much
more mosaic and variable levels of mHtt and might also be toxic on its own.
A similar viral strategy has been used to explore metabolic dysfunction in rodents; Hult et
al found that overexpression of a mHtt fragment in the hypothalamus causes metabolic imbalances
in wild type mice (Hult et al., 2010; Hult et al., 2011). However, in the BACHD, Cre-mediated
excision of exon-1 mhtt in leptin receptor-expressing neurons had no effect on metabolic or
psychiatric phenotypes (Lundh et al., 2012).
Subcellular localization of mHtt
In humans, aggregated mHtt was first identifed in the nucleus (Becher et al., 1998; DiFiglia
et al., 1997), and later in the cytoplasm and processes (Gutekunst et al., 1999), and further
observational studies in humans suggested that more nuclear aggregates correspond with
childhood onset forms of HD (Aronin et al., 1999). Reports in cellular models have indicated that
the toxicity of mHtt fragments may correspond with their cellular localization; the nuclear
direction of polyQ peptides is toxic to cells in culture (Yang et al., 2002) as is the entry of caspase-
cleaved fragments into the nucleus (Gafni et al., 2004; Martindale et al., 1998). Inhibiting caspase-
6 cleavage of full-length human Htt in the YAC128 model delays the nuclear entry of mHtt and
prevents striatal atrophy as well as behavioral pathology (Graham et al., 2006a).
Huntingtin contains both nuclear export (NES) and nuclear localization sequences (NLS)
and a mutant fragment has been reported to repress transcription in the nucleus (Kegel et al., 2002).
In mouse models, directly targeting an exon-1 mHtt fragment into the nucleus results in more
pronounced and progressive phenotypes (Benn et al., 2005). In mouse models of other
polyglutamine disorders, including spinocerebellar ataxia (SCA1) and spinal and bulbar muscular

48
atrophy (SBMA), blocking the nuclear translation of polyQ-expanded proteins abolishes disease
pathogenesis (Katsuno et al., 2002; Klement et al., 1998). Deletion of the N17 region of mHtt in
BACHD leads to increased nuclear localization and accumulation Htt-positive structures in the
nucleus, which results in accelerated neurological, metabolic, and behavioral pathology (Gu et al.,
2015).
Genetic suppression of mHtt
Increasingly, the field standard is to use multiple models to validate findings regarding the
molecular pathogenesis and progression of HD, as well as potential therapeutic routes (Yang and
Gray, 2011). One realm in which this strategy has recently been fruitful is in HD gene suppression.
Suppressing the expression of mutant exon-1 htt via a tetracycline-regulatable genetic system
(Yamamoto et al., 2000) led to the reversal of HD pathology and behavior, including the removal
of mHtt deposits. This was recapitulated in a model of SCA1 in which nuclear aggregates of ataxin-
1 accumulate and clear in conjunction with behavioral and neuropathological improvements (Zu
et al., 2004). Subsequent work has employed different methods of gene suppression including
microRNAs, short hairpin RNAs, siRNAs, antisense oligonucleotides (ASOs), and gene editing,
in multiple mouse models of HD, which has yielded similar amelioration of phenotype (Aronin
and DiFiglia, 2014). In particular, an ASO targeting mHtt was developed and validated in a variety
of models, including a humanized mouse model (Southwell et al., 2014) as well as the BACHD,
YAC128, and R6/2 models (Kordasiewicz et al., 2012). Attention to the salient neuropathological
features in the various models, such as aggregation and Rotarod decline in BAC and YAC mice,
and survival in R6/2, was useful in determining the overall efficacy of the drug, which has been
further tested in large animal models, including primates (Kordasiewicz et al., 2012). This work

49
has converged upon the first clinical trial of a genetic therapy for HD (National Institute of Mental
Health 2016).
Conclusions
The ideal mouse model of HD would recapitulate all of the relevant pathological and
behavioral features that characterize the disease. There is a growing appreciation for the cognitive
and psychiatric symptoms faced by HD patients and their companions, and mice are inherently
unsuited to address the depth of psychological and social challenges experienced in the face of the
disease. Nevertheless, mouse models have provided essential insights into the mechanisms driving
the disorder and have provided the primary means to investigate therapeutics.
The importance of selecting the proper mouse model for addressing a particular research
question, and of using multiple models for the preclinical validation of therapeutic approaches,
cannot be overemphasized, and has been a focal point in multiple reviews (Ehrnhoefer et al., 2009;
Pouladi et al., 2013; Yang and Gray, 2011). Models that accelerate certain genetic or phenotypic
features of HD, such as extreme repeat expansion (Dragatsis et al., 2009; Morton et al., 2009),
aggregation (Mangiarini et al., 1996), or striatal atrophy (Slow et al., 2003), have been instructive
with respect to concurrent pathological and behavioral outcomes. Mutant Htt aggregates are seen
in nearly every rodent model of HD, although the kinetics, tissue distribution, and cellular
localization varies (Morton, 2013). Directed modifications to mHtt have contributed to our
comprehension of mHtt’s propensity to aggregate and how aggregation affects other pathological
measures, but their exact relevance to pathogy remains elusive. Our understanding of aggregation
in HD will continue to be shaped by comparative analysis of the field’s most enduring models.

50
Chapter 3: Materials and Methods
Animals
All experiments were reviewed and approved by the Columbia University Medical Center’s
Institutional Animal Care and Use Committee (IACUC). Experimental mice were bred and housed
in facilities at the William Black Medical Research Building. Up to five mice per cage were
maintained in a temperature-controlled environment on a 12 hour light/dark cycle with access to
food and water ad libitum. For breeding and housing pre-experimentally, mice were maintained
on a cycle with lights on between 6 am and 6 pm.
Mouse lines
Conditional Alfy alleles (Alfy flox/flox)
Conditional Alfy knockout mice were generated by the Gene Targeting and Transgenic
Facility at UConn Health. The targeting vector was designed with LoxP sites flanking exon 5, the
first coding exon within the WDFY3 gene. C57Bl/6 x 129SVEV F1 hybrid ES cell lines were used
for targeting, and germline chimeras were crossed with CD1 ROSA26-Flpe mice to remove the
PGKneo cassette used for in vitro selection. Germline positive F1 mice were backcrossed for two
generations with chimeric mice and pups were genotyped to confirm proper targeting. Mice with
a heterozygous WDFY3 allele (Alfy flox/+) were crossed to obtain homozygous Alfy flox/flox
mice. These mice were consistently bred with littermates at 3-4 months of age to maintain the Alfy
flox/flox line. Excision by Cre creates a frame shift introducing a premature stop codon after 58
amino acids.

51
Inducible Cre (Actin-CreERTM)
Actin-CreERTM mice (Hayashi and McMahon, 2002) were obtained from the Jackson
Laboratory (Bar Harbor, Maine). This line expresses tamoxifen (tam)-inducible Cre recombinase
protein driven by the chicken beta actin promoter. These mice express a Cre fused to a modified
form of the ligand binding domain of the estrogen receptor which can enter the nucleus to excise
LoxP-flanked sequences upon injection of tamoxifen. Actin-CreERTM mice were maintained
heterozygous on a C57Bl/6 background strain by crossing to WT C57Bl/6 mice every 3-4 months.
tetO-huntingtin (tetO-Htt-103Q+/+)
An NheI/EcorV insert containing poly-glutamine expanded (103Q) exon I fragment of HTT
using an alternating CAGCAA sequence (Kazantsev et al., 1999) was sub-cloned into pBi5
downstream of a bi-directional TetO operator sequence (Baron et al., 1995) (pBi5 from Dr. Herman
Bujard). Site-directed mutagenesis was used to introduce a stop codon downstream of exon I of
HTT (Quickchange mutatgenesis, Promega). Successful mutagenesis of the insert was confirmed
by Sanger DNA sequencing. Pronuclear injection was carried out by the Transgenic Mouse Shared
Resource Center at Columbia University Medical Center using 50 µg of pBi2-HTT103Q,
linearized using AseI and transgenic mice were produced on a B6CBA/F1 background. For colony
maintenance, HD103Q-tetO was bred to homozygosity, with no observed toxicity.
(CamKIIa-tTA +/-)
CamKIIa-tTA +/- mice were obtained from Dr. Robert Burke and are originaly described
by Mayford et al (1996). They were maintained heterozygous on a C57Bl6 background. The tTA
drives forebrain expression when bred with lines containing a tet operon.

52
BACHD
BACHD were obtained from William Yang at UCLA (Gray et al., 2008). They are
maintained by crossing onto a WT FVB strain.
Alfy heterozygous constitutive knockout (Alfy GT/+)
The creation of constitutive Alfy knockout mice is described in (Dragich et al., 2016). A
complete description of the gene trap vector can be found in Zambrowicz et al (2003).
Heterozygous mice are maintained in the laboratory and were crossed with BACHD to generate
the four phenotypes described in Figure 5.1.
Alfy iKO (Alfy flox/flox::Actin-CreERTM)
Mice expressing homozygous floxed Alfy alleles (Alfy flox/flox) were crossed with
inducible Cre (Actin-CreERTM) for two generations to create the potential for Alfy inducible
knockout (Table 3.1).
HD103Q
CamKIIa-tTA+/- were crossed with tetO-Htt-103Q +/+ for two generations to produce
offspring which express exon1 Htt in forebrain structures (Table 3.1).

53
Tab
le 3
.1: B
reed
ing
Stra
tegy
to c
reat
e H
D10
3Q, A
lfy iK
O, a
nd H
DA
lfyiK
O. B
reed
ing
roun
ds 1
-4.
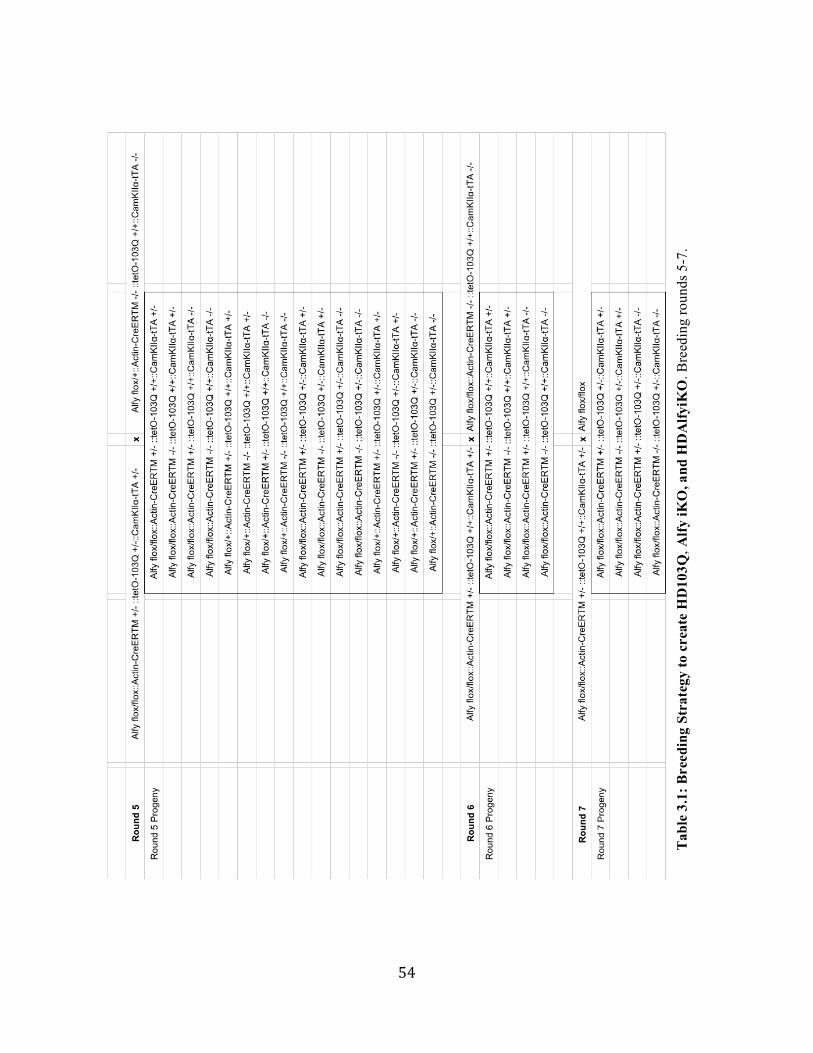
54
Tab
le 3
.1: B
reed
ing
Stra
tegy
to c
reat
e H
D10
3Q, A
lfy iK
O, a
nd H
DA
lfyiK
O. B
reed
ing
roun
ds 5
-7.

55
HDAlfyiKO
To create regulatable HD mice with the potential for Alfy iKO, several generations of
backcrosses were required to bring Alfy flox/flox and tetO-Htt-103Q to homozygosity with
heterozygous CamKIIa-tTA and Actin-CreERTM. Crosses are detailed in Table 3.1. In
experimental mice, Alfy knockout and huntingtin expression are controlled by administering
tamoxifen and dox, respectively.
Genotyping
DNA extraction
A tissue sample was obtained at weaning by ear punch and lysed overnight at 55˚C in buffer
containing 10mM tris HCl pH 8.0, 5 mM EDTA, 0.1% SDS, and 200 mM NaCl with 0.5 mg/mL
proteinase K. DNA was extracted in phenol/chloroform/isoamyl alcohol (Fisher Scientific) and
centrifuged at 14,000 rpm for 10 minutes using an Eppendorf 5417R benchtop centrifuge. DNA
was precipitated from the aqueous layer using 100% EtOH and spun at 14,000 rpm for 10 minutes
to obtain a pellet. The pellet was washed in 70% EtOH, spun at 14,000 rpm for 10 minutes. The
ethanol was decanted and the pellet was air-dried and resuspended in 10 mM tris HCl pH 8.0 at a
concentration of 100-200 ng/µL.
PCR
PCRs to detect genes of interest were performed with 50-200 ng of DNA using 5 PRIME
Hot Mastermix () in the Eppendorf AG 22331 Hamburg Mastercycler. Primers and cycling
conditions are described in Table 3.2.

56
Table 3.2: Primers and conditions for PCR and RT-PCR.
Gene Forward Primer 5' - 3' Reverse Primer 5' - 3' temp time # cycles
Alfy Lox P1 GAA ACG AAG CTC GTT TAC GG
TGC AGT GAC ATT TCC TCT GG 94˚ 2 min 1 cycle
Alfy Lox P2 (Frt)
ACT TGG GAA GAG GGA AGC TC
AGG TTA CCA GCC ACA ACC AG 94˚ 15 s
10 cycles * -1˚ per
cycle *65
to 55 45 s
72˚ 45 s 94˚ 15 s
25 cycles 55˚ 30 s 72˚ 45 s 72˚ 7 min 1 cycle
ActinCre-ERTM
GCG GTC TGG CAG TAA AAA CTA TC
GTG AAA CAG CAT TGC TGT CAC TT 94˚ 3 min 1 cycle
Internal control CTA GGC CAC AGA ATT GAA AGA TCT
GTA GGT GGA AAT TCT AGC ATC ATC C 94˚ 30 s
35 cycles 51.7˚ 1 min 72˚ 1 min 72˚ 5 min 1 cycle
tetO-103QHtt (luciferase)
TCC TCT GAC ACA TAA TTC GCC
GTT GTT CCA TTC CAT CAC GG 94˚ 2 min 1 cycle
Terd (control) CAA ATG TTG CTT GTC TGG TG
GTC AGT CGA GTG CAC AGT TT 94˚ 45 s
34 cycles 60˚ 45 s 72˚ 1 min 72˚ 7 min 1 cycle
CAMKIIa-tTA GTG ATT AAC AGC GCA TTA GAG C
GAA GGC TGG CTC TGC ACC TTG GTG 94˚ 2 min 1 cycle
94˚ 30 s 34 cycles 56.8˚ 30 s
72˚ 45 s 72˚ 7 min 1 cycle
Alfy RT-PCR exon 3 -6
TCA GGT CCC GAG TGA AGT GCC A
GGC CCG ACT TGC AGC CTC TG 94˚ 2 min 1 cycle
Actin (control) ATA TCG CTG CGC TGG TCG TC
AGG ATG GCG TGA GGG AGA GC 94˚ 30 s
25 cycles 58˚ 30 s 72˚ 45 s 72˚ 10 min 1 cycle

57
Preparation and Administration of Drugs
Tamoxifen
For temporally controlled Alfy excision, adult Alfy iKO mice were injected with tamoxifen
at ages ranging from 2 months old to 12 months old, as detailed in experimental procedures.
Tamoxifen (Sigma) was prepared at 10 mg/mL in a solution containing 5% Ethanol in corn oil by
heating at 37˚C for 4h and vortexing frequently. Vehicle control was prepared by omitting
tamoxifen from the solution. Tam or vehicle control were administered intraperitoneally at 2
mg/26g BW (or 200 µL solution/26g BW) daily for five consecutive days.
Doxycycline
One week after the final tamoxifen injection, doxycycline (Sigma) was prepared at 2
mg/mL in 5% sucrose solution in red tinted water bottles to protect against light sensitivity. Non-
doxycycline-treated control mice received control water containing 5% sucrose. Sucrose water
was autoclaved and cooled prior to the addition of doxycycline and was replaced 1-2 times weekly.
Mice received dox beginning at 7 months of age for durations varying between 2 weeks and 4
months.
Behavioral Testing
Two weeks prior to the start of behavioral testing, experimental cohorts were relocated to
a reverse-cycle core facility with lights off between 10 am and 10 pm, so that longitudinal
experiments could be performed during their dark (active) hours.

58
Rotarod
Prior to testing, mice were trained on the Rotarod apparatus (Ugo Basile, Varese, Italy) on
an initial exposure day, with one trial at a constant speed of 5 rpm for 5 min, a second trial with
speed accelerating from 5 rpm to 15 rpm over 5 min, and a third trial with speed accelerating from
5 to 25 rpm. The accelerating Rotarod task involved 4 days of testing with 3 trials daily of 5
minutes each, with speed accelerating from 5 to 40 rpm. Resting periods in between trials were
always 35-60 minutes. Mice were allowed 1 fall from the Rotarod and on the second fall, the
latency in seconds was recorded.
Balance Beam
Mice were trained to cross a 12 mm wide, 80 cm in length, 60 cm high metal beam to enter
an enclosed tube. On training day, mice were exposed to the beam four times and gently
encouraged across by the experimenter if necessary. On testing day, mice were videotaped across
four trials, with resting periods of 1-3 minutes between trials. To determine the amount of time
required for each mouse to cross the beam, videos were analyzed using custom MATLAB
software, generously provided by Dr. Sam McKenzie.
Open Field
HD103Q mice and littermate controls were given a ten-minute exposure to the Open Field
arena (43.2 cm x 43.2 cm x 30.5 cm) at intervals of two months. The distance traveled and number
of vertical movements were recorded using equipment and software from Med Associates.
BACHD mice had a 1-hour exposure at each age.

59
Cage Climb
HD103Q mice were placed underneath an inverted cylindrical wire mesh dome (25 cm
high x 15 cm diameter) and recorded for five minutes. The number of rears and amount of time
spent with four paws on the side were recorded.
Pole task
Mice were placed, head upwards, gripping a 60 cm high, 1 cm diameter pole wrapped with
medical tape. Subjects were videotaped and the time required to turn downwards and descend was
recorded.
Cage hang test
Mice were placed on the bars of a wire cage top which was inverted to hang 40 cm high
over a thickly padded surface. The amount of time the mice were able to remain hanging was
recorded, up to 60s.
Post-mortem analyses
Fresh-frozen tissue preparation for lysis
Mice were deeply anesthetized with isoflurane and decapitated. Brains were removed and
placed into ice-cold 0.1M Sorensen’s phosphate buffer pH 7.4 (1x P.B.). Under a microscope brain
regions (hippocampus, striatum, cortex, cerebellum) were dissected, dried rapidly, and placed into
1 mL Eppendorf tubes. Samples were flash-frozen on powdered dry ice and stored at -80˚C until
processing.

60
Preparation of tissue lysates
Frozen tissue (50-500 mg) was placed in a glass dounce homogenizer with an equal volume
of 1x PBS containing Halt protease inhibitor cocktail (Thermo Fisher) and disaggregated with 40
pumps of the pestle. The suspension was transferred to a tube and an equal volume of detergent
(2% Triton-X-100 in PBS) was added. After 30 minutes on ice, samples were spun in an Eppendorf
5417R centrifuge at 1000 rpm for 5 minutes. The supernatant (S1) was transferred to another tube.
For experiments involving the Triton-X-100 insoluble fraction, a portion of S1 was transferred to
another tube and centrifuged at 14,000 rpm for 5 minutes. This supernatant (S2) was removed,
leaving a pellet from the hi-speed spin (P2). This pellet was suspended in 1% Tx in 8M urea (20-
25% of original volume of S1 spun at high speed) and rested on ice for 15 minutes. It was
centrifuged at 14,000 rpm for 5 minutes and if a small viscous pellet (DNA) was detected, the
pellet was carefully removed with a pipet, leaving supernatant (S3). The protein concentration of
relevant suspensions was quantified using the BioRad DC protein kit according to manufacturer’s
instructions.
Western blot
5-20 µg of protein was loaded into a NuPAGE 4-12% Bis-tris gel (Invitrogen) separated
by gel electrophoresis (SDS-PAGE) using the Invitrogen XCell2 System, and transferred to PVDF
membranes (Novex) in transfer buffer. Membranes were blocked in 3% BSA at room temperature
for 1 hour and incubated in primary antibody overnight (see Table 3.3 for antibody concentrations,
manufacturers, and conditions). Primary antibody dilutions contained 0.01% tween in PBS with
3% BSA. Blots were incubated for 1-2 hours with horseradish peroxidase-conjugated secondary
antibodies (Sigma) in 0.01% 1xPBS-tween containing 3% BSA, and developed using Clarity
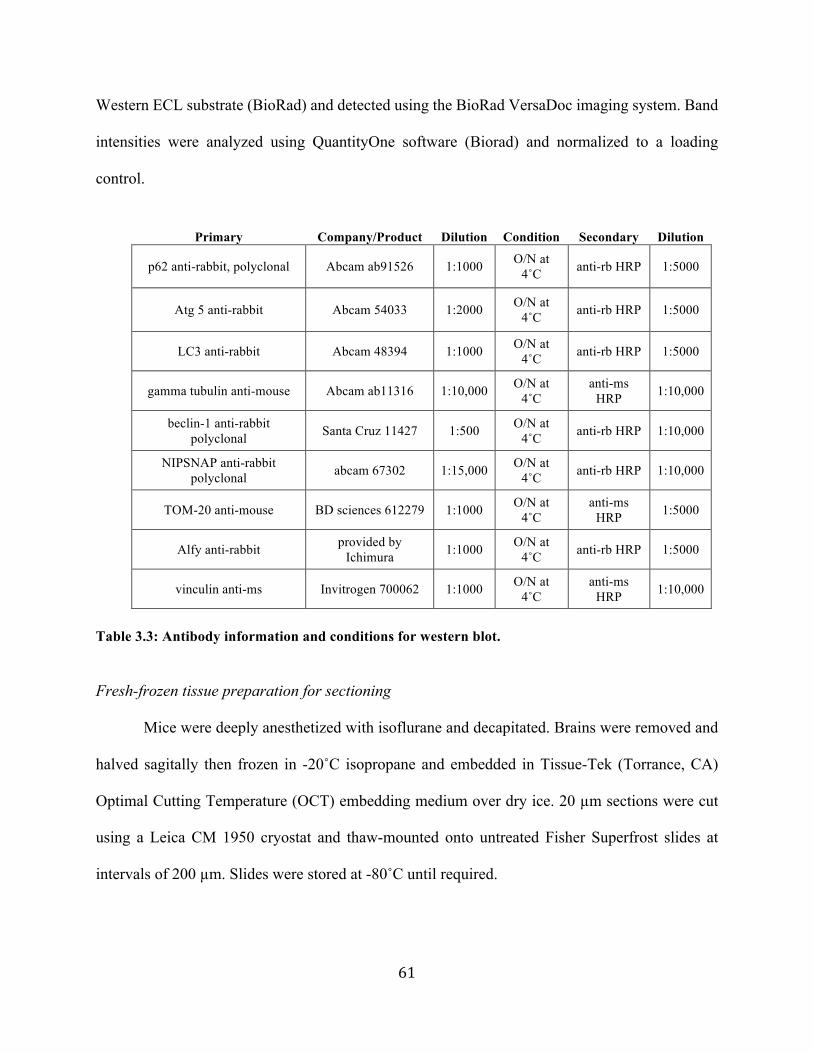
61
Western ECL substrate (BioRad) and detected using the BioRad VersaDoc imaging system. Band
intensities were analyzed using QuantityOne software (Biorad) and normalized to a loading
control.
Primary Company/Product Dilution Condition Secondary Dilution
p62 anti-rabbit, polyclonal Abcam ab91526 1:1000 O/N at 4˚C anti-rb HRP 1:5000
Atg 5 anti-rabbit Abcam 54033 1:2000 O/N at 4˚C anti-rb HRP 1:5000
LC3 anti-rabbit Abcam 48394 1:1000 O/N at 4˚C anti-rb HRP 1:5000
gamma tubulin anti-mouse Abcam ab11316 1:10,000 O/N at 4˚C
anti-ms HRP 1:10,000
beclin-1 anti-rabbit polyclonal Santa Cruz 11427 1:500 O/N at
4˚C anti-rb HRP 1:10,000
NIPSNAP anti-rabbit polyclonal abcam 67302 1:15,000 O/N at
4˚C anti-rb HRP 1:10,000
TOM-20 anti-mouse BD sciences 612279 1:1000 O/N at 4˚C
anti-ms HRP 1:5000
Alfy anti-rabbit provided by Ichimura 1:1000 O/N at
4˚C anti-rb HRP 1:5000
vinculin anti-ms Invitrogen 700062 1:1000 O/N at 4˚C
anti-ms HRP 1:10,000
Table 3.3: Antibody information and conditions for western blot.
Fresh-frozen tissue preparation for sectioning
Mice were deeply anesthetized with isoflurane and decapitated. Brains were removed and
halved sagitally then frozen in -20˚C isopropane and embedded in Tissue-Tek (Torrance, CA)
Optimal Cutting Temperature (OCT) embedding medium over dry ice. 20 µm sections were cut
using a Leica CM 1950 cryostat and thaw-mounted onto untreated Fisher Superfrost slides at
intervals of 200 µm. Slides were stored at -80˚C until required.

62
Radioactive in situ hybridization probe
A 45-base oligonucleotide probe (5′-CCA-TGA-GTC-GGG-AGA-CTT-GCG-TTG-TGA-
ACT-GCA-GAA-GGT-CAG-AAA-3′) was designed complementary to bases 766-810 of the Alfy
mRNA (GenBank accession no. NM_172882). 150 ng of oligonucleotide was radiolabeled with
[32P]α-dATP using the Roche terminal transferase 3′-end labeling kit according to manufacturer’s
instructions. The labeled probe was extracted using phenol-chloroform-isoamyl alcohol and
unincorporated radionucleotides were removed by centrifugation through an illustra Microspin-
G25 column (GE Healthcare, Chicago, IL). Radioactivity was measured by liquid scintillation
counter.
Radioactive in situ hybridization
Sections were dried at RT for 30 minutes and transferred into cold 4% paraformaldehyde
(PFA) in 1x PBS. Slides were washed once in 1x PBS for 5 minutes, followed by dehydration in
70% EtOH for 5 minutes and storage in 100% EtOH at 4˚C until use. Sections were air-dried for
15 minutes prior to probe application. Probes were diluted in minimal hybridization buffer
containing 50% formamide, 4x SSC, and 10% Dextran Sulphate. Hybridization was performed
overnight at 42˚C in a humidified chamber. Slides were washed first at 60˚C for 30 minutes and
then with 1x SSC and 0.1x SSC at room temperature for 5 min. Slides were briefly dehydrated in
70 and 95% Ethanol, air dried, and exposed to film for 2 weeks.
Fixed tissue preparation
Mice were deeply anesthetized with isoflurane and transcardially perfused with 0.9% saline
for 2 minutes followed by 4% paraformaldehyde (PFA) for 3 minutes. Brains were removed and

63
post-fixed in 4% PFA for 4 hours, then incubated in 30% sucrose in 1x phosphate buffer (P.B.) for
48-72h. Brains were snap-frozen in powdered dry ice then cryoprotected in OCT embedding
medium and stored at -80˚C. Tissue was sectioned at 30 µm using a Leica CM 1950 cryostat and
stored at 4˚C in phosphate buffer containing 0.02% sodium azide as a preservative.
Immunohistochemistry
Representative sections throughout the forebrain, spaced at 240 µm, were stained with
antibodies against huntingtin, ubiquitin, and GFAP. Briefly, sections were washed in 1x PBS (3 x
10 min) containing 0.02% Triton-X-100, followed by antigen retrieval in sodium citrate, pH 9.0,
at 80˚C for 30 minutes. After three more washes, endogenous peroxidases were blocked with 1%
hydrogen peroxide, and again washed three times. Sections were blocked in 0.4% Triton-X for 1
hour (for EM48 staining, block also contained 3% BSA and 2% NGS), then incubated overnight
for up to 3 days in primary antibody. Sections were washed in 1xPBS and incubated in biotinylated
secondary antibody at room temperature for 1 hour or overnight. (See Table __ for primary and
secondary antibody concentrations and conditions). After 3 washes in 1x PBS, signal was
amplified using the Vectastain ABC Kit (Vector labs) for 1-2 hours at room temperature followed
by 3 PBS washes. For S830 staining only, this was followed by an additional 10 minute tyramide
amplification step (Perkin Elmer) followed by washes and a second ABC treatment. Signal was
detected using diaminobenzidine (DAB) (10 mg/25 mL) in phosphate buffer containing 0.00001%
H2O2. Following the DAB reaction, sections were mounted on glass slides and air-dried,
counterstained with thionin, dehydrated in ascending grades of ethanol, cleared in xylene, and
coverslipped with Permount mounting medium (Fisher).

64
Primary Product Dilution Diluent Condition Secondary Dilution
EM48 anti-mouse,
polyclonal
Millipore MAB5374 1:400
3% BSA, 2% NGS, 0.2% Tx-
100 in PBS O/N at RT anti-ms 1:200
S830 anti-sheep,
polyclonal
Provided by Dr. Gillian
Bates
1:10,000 or
1:50,000
0.4% Tx-100 in 1x PBS
1 or 3 days at RT anti-sh 1:2000
GFAP anti-rabbit,
polyclonal DAKO Z0334 1:10,000 0.4% Tx-100 in
1x PBS O/N at 4˚C anti-rb 1:2000
ubiquitin anti-rabbit,
polyclonal DAKO Z0458 1:1000 0.4% Tx-100 in
1x PBS O/N at RT anti-rb 1:2000
Table 3.4: Antibody sources and conditions for immunohistochemistry.
Stereology
Tissue preparation
Serially-cut coronal brain sections (every eighth section) were used for all volumetric,
neuronal, and huntingtin aggregate analyses. Tissue was matched across brains during sectioning,
by designating “set 1” as the representative set containing the section with the initial crossing of
the anterior commissure. For each immunostain, a number between 1 and 8 was selected at random,
and this set number was used for staining that epitope across all brains.
Anatomical coordinates
For assessment of HD103Q striatal accumulation of Htt epitope EM48, analysis of the right
hemisphere began at the anterior tip of the genu of the corpus callosum (interaural 4.98 mm/bregma
1.18 mm, Franklin and Paxinos 2001) and extended through the anterior emergence of CA3
(interaural 2.86 mm/bregma -0.94 mm, Franklin and Paxinos 2001) These rostrocaudal bounds

65
resulted in 8-10 sections per brain spaced 240 microns apart. The neostriatum was defined within
the rostral-caudal plane as being bounded dorsally by the corpus callosum, dorsolaterally by the
external capsule, and medially by the lateral ventricle. Ventrolaterally the neostriatum continues
in an arc bounded ventrolaterally by the claustrum, the dorsal endopiriform nucleus, the lateral
striatal stripe, the lateral accumbens shell (or more ventrally, the interstitial nucleus of the posterior
limb of the anterior commissure), through the anterior commissure and/or internal capsule, and to
the ventral tip of the lateral ventricle.
For assessment of BACHD cortical accumulation of Htt epitope S830, anatomical bounds
were the same as above, but with analysis of the left dorsolateral cortex. The extent of the cortical
region analyzed was dorsal to the corpus callosum arcing ventrolaterally and extending to the
secondary somatosensory cortex, as bounded by tracing horizontally from the lateral edge of the
external capsule to the edge of the tissue.
Counting
Unbiased stereological counts were obtained from the striatum or cortex using Stereo
Investigator Software (MBF Bioscience). The optical dissector method was used to count profiles
(neurons or Htt-positive aggregates) in an unbiased random selection of serial sections in a defined
volume of the cortex or striatum. Based on pilot studies, the counting parameters selected involved
a 300 x 300 grid with a 50 x 50 counting frame, and a fixed tissue thickness of 30 µm. In all mice,
Gundersen coefficients of error (CE) for Nissl-positive neuronal counts in all mice were on average
(0.02).

66
Statistical analyses and image preparation
All images were prepared using Adobe Photoshop CS5 and Adobe Illustrator CS5.
Statistical analyses of behavioral data, stereologically obtained counts and volumes, and Western
blot band intensities were performed using Statview (Scientific Computing). Data were analyzed
using 1-way, 2-way, or repeated-measures ANOVAS followed by Fisher’s post-hoc test.
Significance was accepted at the 95% probability level. Behavioral data are represented as mean
+/- SEM or SD as noted in figures.

67
Chapter 4: Alfy promotes the turnover of mutant huntingtin in the adult brain
Introduction
In Huntington’s disease (HD) models, a clear link has emerged between removal of visible
protein deposits and improvement of phenotypes from the cellular to the behavioral level. Gene
suppression studies in multiple models have shown amelioration of phenotypes in conjunction
with the clearance of mHtt deposits (DiFiglia et al., 2007; Harper et al., 2005; Kordasiewicz et al.,
2012; Stanek et al., 2014; Wang et al., 2005; Yamamoto et al., 2000). As addressed in Chapter 1,
autophagic turnover is a likely route for the clearance of disease-relevant mutant and misfolded
proteins. Consequently, boosting the autophagic processing of aggregates has been explored as a
means to alleviate toxicity, and even modify behavior, in models of HD (Frake et al., 2015). Thus
far, these results have largely been achieved experimentally through non-selective means, such as
via the inhibition of central metabolic kinases like mTOR (Ravikumar et al., 2004), which carries
great risk of off-target effects. To move towards more targeted enhancement of aggregate removal
in HD, and validate this approach as a therapeutic avenue, it will be necessary to dissect the specific
mechanisms by which autophagic turnover of mHtt occurs in the brain.
The Yamamoto lab and our collaborators have identified the large PI3P binding protein,
Autophagy-linked FYVE protein (Alfy), as a selective mediator of aggregated protein turnover in
the brain (Clausen et al., 2010; Filimonenko et al., 2010; Lystad et al., 2014; Simonsen et al.,
2004). Alfy is required for the autophagic removal of mutant exon-1 Htt in cellular models
(Filimonenko et al., 2010; Yamamoto et al., 2006). Furthermore, Alfy overexpression can drive
mutant Htt elimination in cells ranging from stably expressing HeLa cells to virally transduced
primary neurons (Filimonenko et al., 2010).

68
In cells, Alfy is required for the turnover of detergent-insoluble but not soluble forms of
mutant Htt (Filimonenko et al., 2010), and upon Alfy knockdown, visible puncta containing mutant
exon-1-Htt resist clearance despite the suppression of the HD transgene. Given Alfy’s selectivity
for aggregated protein removal in cells, we sought to ask whether a requirement for Alfy in mutant
exon-1 Htt removal persists in the living mammalian brain. Our subsequent goal was to understand
whether the removal of the aggregated structures themselves is required to ameliorate disease:
could behavioral reversal occur despite the persistence of aggregates?
In order to address this question, we needed to build a model in which it was possible to
both observe the clearance of mutant Htt and eliminate Alfy with temporal precision. Therefore,
we combined a newly-designed tetracycline-controlled reversible HD model, the HD103Q, with a
tamoxifen-inducible Alfy knockout, Alfy iKO. Using this model, we demonstrate that Alfy protein
can be reliably and inducibly eliminated from brain tissue, and that Htt aggregates accumulate and
clear upon administration of doxycycline. By knocking out Alfy, then initiating HD transgene
suppression, we show that the efficient clearance of accumulated exon-1 Htt requires Alfy.
Ultimately it emerged through characterization of the HD103Q::Alfy iKO line that
aggregation of mutant exon-1-Htt is spatially restricted to the lateral striatum, and the behavioral
HD phenotype is mild and not progressive. More importantly, the complete elimination of Alfy
from the adult brain alone produced a robust, age-dependent motor behavioral phenotype which
precluded querying motor reversal using this approach. In light of Alfy’s critical role in removal
of accumulated mutant exon-1-Htt in HD103Q, the behavioral impact of modifying aggregate
turnover was addressed using an Alfy depletion strategy in the BACHD mouse model, which
expresses full-length mHtt and has a more robust behavioral phenotype.

69
Results
Design of the inducible Alfy knockout (Alfy iKO)
To understand whether the selective autophagy adaptor Alfy plays an essential role in the
turnover of mutant Htt protein in vivo, we combined Alfy knockout with an inducible HD mouse
model. Given that homozygous constitutive Alfy knockout results in perinatal lethality (Dragich
et al 2016), we chose an adult inducible approach using ActinCreERTM (Guo et al., 2002; Hayashi
and McMahon, 2002).
Figure 4.1: Design of the conditional Alfy allele. (A) Frt sites around a PGK/neo selection cassette are removed by breeding with ROSA26-Flpe, leaving LoxP sites flanking Exon 5. Excision by Cre creates a frame shift introducing a premature stop codon after 58 amino acids. (B) Schematic depicting tamoxifen-inducible, Cre-mediated excision of Alfy. (C) PCR based tail genotyping of wild type and conditional Alfy alleles (+/+, flox/+, and flox/flox) and knockout alleles (flox/D and D/D) generated by crossing with Actin-CreERTM and injecting tamoxifen (C is modified from Dragich et al 2016).
To achieve spatiotemporally controlled elimination of Alfy, our lab created mice with a
conditional Alfy allele (Figure 4.1). Cre-mediated excision creates a frame-shift mutation that
generates a small N-terminus Alfy fragment (Figure 4.1A). To drive Alfy deletion in adult mice,
lines homozygous for the conditional allele (Alfyflox/flox) were crossed to mice expressing
tamoxifen (tam) – inducible Cre recombinase protein driven by the chicken beta actin promoter

70
(Actin-CreERTM) (Hayashi and McMahon, 2002) (Figure 4.1B; see upper left of Table 3.1 for
breeding schema). These mice express a Cre fused to a modified form of the ligand binding domain
of the estrogen receptor. Only when it is bound by tam can Actin-CreERTM enter the nucleus to
excise LoxP-flanked sequences. PCR-based genotyping was used to detect wild type, conditional,
and knockout alleles (Figure 4.1C).
Figure 4.2: Tamoxifen-inducible Alfy knockout in brain. Tissue from 11-month-old Alfyflox/flox::Actin-CreERTM mice obtained four months after intraperitoneal injection of tamoxifen (2 mg/26g BW daily for 5 consecutive days) at 7 months of age. (A) PCR of genomic DNA showing Alfy floxed product (477 bp) and exon 5 excised product (410 bp). (B) RT-PCR of Alfy cDNA using primers within exon 5. (C) Western blot of detergent-soluble brain lysates using antibodies against Alfy. (D) Radioactive in situ hybridization using a probe complementary to exon 5 of Alfy. All images are representative of n=3 animals per genotype.
Temporally controlled, inducible Alfy knockout (Alfy iKO) in brain tissue
Alfy is highly expressed in the brain compared to other organs (Simonsen et al., 2004), and
Actin-CreERTM was chosen for its ability to produce reliable inducible knockout in diverse tissues,
notably in brain (Cai et al., 2007; Guy et al., 2007). Following tamoxifen or corn oil vehicle (veh)
injection, we examined Alfy levels in cortex, cerebellum, striatum, and hippocampus using a

71
variety of methods, including PCR of genomic DNA (Figure 4.2A), RT-PCR of cDNA (Figure
4.2B), and Western blot of brain lysate (Figure 4.2C). We found that tam injection results in
reliable Cre-mediated excision of Alfy exon 5, leading to loss of Alfy protein in brain tissue.
Because immunohistochemistry for Alfy has thus far been unsuccessful using a wide variety of
antibodies (Dragich et al., 2016, data not shown), we examined the spatial pattern of Alfy iKO
using radioactive in situ hybridization with a probe designed complementary to exon 5 of Alfy
(Figure 4.2D).
We confirmed via Western blot a tam-mediated decrease in forebrain Alfy levels to 20%
of veh-injected controls (Figure 4.3A). To explore the persistence of Alfy protein following Cre-
mediated excision of Alfy DNA in vivo, 8 week-old Alfyflox/flox::Actin-CreERTM+/- mice were
injected with tam for 5 days and sacrificed at 0, 1, 3, or 7 days after the final injection. We observed
a decrease in Alfy protein levels 7 days after the last injection (Figure 4.3B).
Finally, to confirm that Alfy knockout does not occur in Actin-CreERTM+/- mice in the
absence of tamoxifen, we aged Alfyflox/flox::Actin-CreERTM+/- mice and Cre-negative littermate
controls to 6 or 9 months old and examined Alfy protein levels (Figure 4.3C,D). Uninjected
Alfyflox/flox::Actin-CreERTM+/- showed no decrease in Alfy protein levels, indicating that there is
no “leak” of Cre into the nucleus without administration of tam.

72
Figure 4.3: Confirmation of Alfy iKO in forebrain. (A) Western blot of forebrain lysate from 8-week-old mice treated with vehicle or tamoxifen and sacrificed 1 week after the final injection. Alfy levels in tam-injected mice were significantly reduced (F(1,4) = 20.01, p = 0.011), reaching approximately 20% of controls. (B) 6 m/o Alfyflox/flox::Actin- CreERTM mice treated with corn oil vehicle or tamoxifen at 2 mg/26 g BW for 5 consecutive days were sacrificed at 1, 3, or 7 days post-injection (n=1 each) and forebrain lysates were probed for Alfy protein. (C,D) Western blot of forebrain lysate reveals that in the absence of tamoxifen, there is no reduction in Alfy levels in Alfy flox/flox::Actin- CreERTM+/- mice (C) at 6 m/o (F(1,4)=0.6375, p = 0.4693) or (D) at 9 m/o (F(1,4)=0.6805, p = 0.4558) compared to Cre-negative littermate controls.
In cellular models of mutant Htt accumulation, Alfy is required for the selective autophagic
turnover of aggregates (Filimonenko et al., 2010; Yamamoto et al., 2006). To determine whether
Alfy serves this function in vivo in the mammalian brain, we needed to eliminate in a controlled
manner the expression of Alfy in a reversible HD model, in which clearance of aggregates could
be observed. This was accomplished by crossing the Alfy iKO mouse with a newly created

73
inducible mutant Htt mouse model, HD103Q. To characterize this mouse line, we examined
huntingtin immunostaining and piloted a variety of HD-relevant behavioral analyses.
Design of HD103Q
The HD103Q line uses a tetracycline-regulatable system to control expression of mutant
exon-1-Htt. This system uses the tetracycline transactivator protein (tTA), a fusion protein
consisting of a tet-repressor binding domain and a VP16 activation domain (Gossen and Bujard,
1992). The resulting tTA protein can bind DNA at a specific TetO operator sequence and induce
transcription from an adjacent CMV minimal promoter. Combining tTA and tetO elements allows
for the continual expression of a transgene. In a Tet-Off system, tetracycline or its analogs can
bind tTA and render it incapable of binding to tetO, thus inhibiting transcription. In mouse lines,
administration of doxycycline in the water results in transgene suppression (Mayford et al., 1996).
Using a design similar to the inducible HD94 mouse (Yamamoto et al., 2000), the HD103Q
line uses a Tet-Off system to inducibly express a transgene consisting of exon-1 of the human HD
gene, with a CAG repeat length of 103 (Figure 4.4A). The mutation is encoded by a CAGCAA
sequence to promote repeat length stability (Kazantsev et al., 1999), and a luciferase reporter is
expressed bicistronically. Both heterozygous (TetO-103Q+/-) and homozygous (TetO-103Q+/+)
mice have been maintained in the laboratory. To permit dox-regulatable expression of exon1-Htt-
103Q in forebrain, the tetO mice have been crossed with CAMKIIα-tTA+/- mice. To confirm
CAMKIIα-tTA+/-- driven expression as well as dox-mediated suppression of the transgene, we
performed a luciferase assay on forebrain lysate from a tTA-expressing HD103Q, a tTA-negative
control, and an HD103Q mouse treated with dox in the drinking water for 2 months (Figure 4.4B).
Lysate from a mouse expressing tetO-103Q in addition to tTA showed luminescence that decayed

74
over 30 minutes following the addition of the substrate. tTA-negative and dox-treated HD103Q
lysates showed no luminescence in the assay. This affirms CAMKIIα-tTA+/-- mediated expression
and dox-mediated suppression of the HD103Q transgene.
Figure 4.4: Design and tet-mediated regulation of the HD103Q transgene. (A) The HD103Q transgene contains human exon-1 Htt with 103 repeats of an alternating CAG/CAA sequence and a luciferase reporter expressed bicistronically. The tTA under control of a CAMKIIa promoter allows for forebrain expression of mutant huntingtin which can be suppressed using doxycycline (dox). (B) Luciferase assay measuring decay of luminescence following enzyme addition confirms CAMKIIa-tTA-mediated expression of the HD103Q transgene and dox-mediated suppression.
Striatal nuclear Htt aggregation and dox-mediated clearance in HD103Q
To determine whether tTA-driven expression of mutant exon-1-Htt resulted in an
aggregation phenotype, we stained with N-terminal mHtt antibody S830 (Figure 4.5).

75
Heterozygous HD103Q mice showed diffuse nuclear mutant huntingtin accumulation as early as
2 months old that increased in number and intensity over time. Punctate inclusions were extremely
rare even at advanced age. Aggregation was heaviest in the ventrolateral striatum (Figure 4.5) and
largely absent in cortex, but we observed sparse dystrophic neurites and occasional nuclear
staining in the dorsolateral cortex by 11 months of age. tTA-negative littermates did not show
mHtt deposition in the forebrain at any age. No mHtt staining was observed in the cerebellum (data
not shown). The diffuse nuclear staining of exon-1-Htt observed in HD103Q recapitulates an
aspect of mHtt accumulation that is a hallmark of multiple HD mouse models using diverse mHtt
expression strategies (Bayram-Weston et al., 2016), although we rarely observed distinct puncta
that could be defined as nuclear microaggregates, NII, or neuropil aggregates (Cummings et al.,
2012).
Figure 4.5: HD103Q shows mHtt accumulation over time. IHC with antibody S830 in ventrolateral striatum of HD103Q mice at 2, 9, and 18 months of age. S830 staining is sparse in ventrolateral cortex but nuclear accumulation is visible by 18 months of age. No mHtt staining was observed in CamKIIα-tTA -/- mice (control) at 18 m/o. Scale bar = 25µm.
After evaluating the increase in mHtt-positive nuclei at different ages, we chose to initiate
transgene suppression at 7 months old by administering dox (2 mg/mL) in the drinking water for
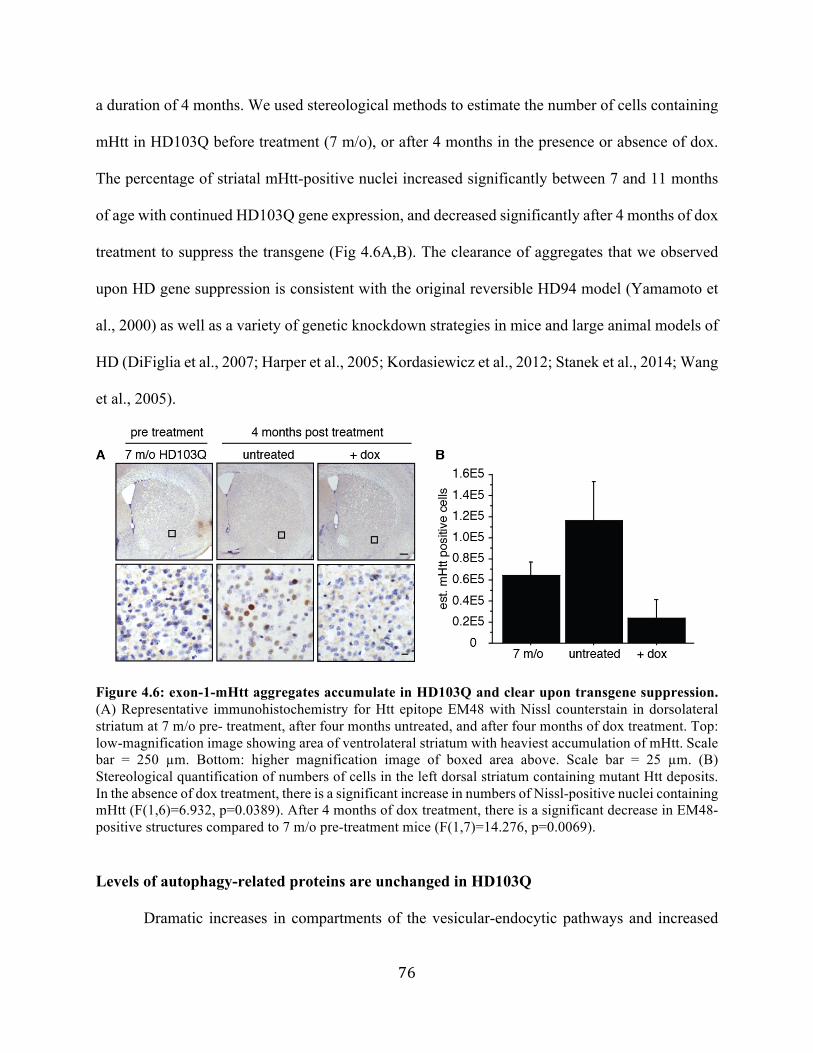
76
a duration of 4 months. We used stereological methods to estimate the number of cells containing
mHtt in HD103Q before treatment (7 m/o), or after 4 months in the presence or absence of dox.
The percentage of striatal mHtt-positive nuclei increased significantly between 7 and 11 months
of age with continued HD103Q gene expression, and decreased significantly after 4 months of dox
treatment to suppress the transgene (Fig 4.6A,B). The clearance of aggregates that we observed
upon HD gene suppression is consistent with the original reversible HD94 model (Yamamoto et
al., 2000) as well as a variety of genetic knockdown strategies in mice and large animal models of
HD (DiFiglia et al., 2007; Harper et al., 2005; Kordasiewicz et al., 2012; Stanek et al., 2014; Wang
et al., 2005).
Figure 4.6: exon-1-mHtt aggregates accumulate in HD103Q and clear upon transgene suppression. (A) Representative immunohistochemistry for Htt epitope EM48 with Nissl counterstain in dorsolateral striatum at 7 m/o pre- treatment, after four months untreated, and after four months of dox treatment. Top: low-magnification image showing area of ventrolateral striatum with heaviest accumulation of mHtt. Scale bar = 250 µm. Bottom: higher magnification image of boxed area above. Scale bar = 25 µm. (B) Stereological quantification of numbers of cells in the left dorsal striatum containing mutant Htt deposits. In the absence of dox treatment, there is a significant increase in numbers of Nissl-positive nuclei containing mHtt (F(1,6)=6.932, p=0.0389). After 4 months of dox treatment, there is a significant decrease in EM48-positive structures compared to 7 m/o pre-treatment mice (F(1,7)=14.276, p=0.0069).
Levels of autophagy-related proteins are unchanged in HD103Q
Dramatic increases in compartments of the vesicular-endocytic pathways and increased

77
autophagic vacuoles have been reported in both human HD brain and in mouse models (Martinez-
Vicente et al., 2010; Sapp et al., 1997; Tellez-Nagel et al., 1974), leading to the hypothesis that the
presence of aggregated mHtt is due in part to impeded autophagy, or that the aggregates themselves
obstruct the autophagic machinery. To query whether mutant exon-1-Htt at all impacts the
macroautophagic machinery in HD103Q compared to littermate controls negative for CAMKIIα-
tTA, we performed Western blots on modified-RIPA-soluble and insoluble fractions of cortical
and striatal lysate using antibodies against a variety of autophagy-related proteins (Figure 4.7 A-
D). We observed no major changes in autophagy-related protein levels, including beclin-1,
involved in initiation of autophagosome formation, Atg5-12, required for elongation of the
autophagosome, and LC3, which is conjugated to PE by Atg5-12-16 and directly marks the
growing autophagosome (Johnson et al., 2012).
The exception among proteins of the autophagy machinery was p62, which increased
significantly in the RIPA-insoluble fraction of striatal lysate (Figure 4.7D) in HD103Q compared
to controls. We did not observe a similar p62 increase in insoluble fractions from the cortex (Figure
4.7C). In cell culture, p62 is required for the formation of ubiquitin-positive inclusions (Clausen
et al., 2010), and in vivo it localizes to nuclear aggregates of mHtt (Kurosawa et al., 2015; Rue et
al., 2013). Considering these findings, and in light of the striatally restricted mHtt expression
pattern we observe via immunostaining, a corresponding increase in insoluble p62 substantiates
biochemically the accumulation of insoluble exon-1-mHtt in the striatum of HD103Q. However,
since p62 is itself an autophagy substrate (Johansen and Lamark, 2011), increased levels may also
be interpreted as a defect in autophagic turnover in HD103Q that is restricted to areas of primary
mHtt accumulation.

78
Figure 4.7: Basal autophagy is not grossly affected in HD103Q. (A) Western blot of RIPA-soluble cortical lysates probed with antibodies against autophagy-related proteins revealed no change in levels of Atg5 (F(1,4)=4.59, p=0.0988), Beclin-1 (F(1,4)=0.167, p=0.7037), or LC3 (F(1,4)=0.073, p=0.8006) in HD103Q compared to controls. (B) RIPA-soluble striatal lysates showed a slight but significant increase in Atg5 in HD103Q (F(1,4)=10.412, p=0.0321) and no changes in Beclin-1 (F(1,4)=0.058, p=0.8215), or LC3 (F(1,4)=1.793, p=0.3142). (C) We observed no change in p62 levels in RIPA-insoluble cortical lysate (F(1,4)=0.920, p=0.3919). (D) There was a significant increase in p62 levels in RIPA-insoluble cortical lysate in HD103Q compared to controls (F(1,4)=9.007, p=0.0399). For all blots, alpha-tubulin was used as a loading control.

79
Alfy is required for in vivo mHtt clearance
Upon confirmation of Alfy iKO and characterization of the HD103Q aggregation
phenotype, we sought to understand whether Alfy is required for the in vivo removal of mHtt
structures. Via a series of breeding steps, we created a model that combines homozygous inducible
Alfy knockout with heterozygous regulatable HD103Q expression (Table 3.1). Containing four
genes comprising two drug-regulatable genetic systems, Alfyflox/flox::Actin-CreERTM+/-::TetO-
103Q+/-::CAMKIIα-tTA+/- mice can be treated with tam for temporally controlled Alfy iKO and
with dox for HD transgene suppression. Combined tam and dox treatment would allow us to ask
whether Alfy is required for the clearance of mHtt. In the text, our model will be referred to as
HD-Alfy, and when injected with tamoxifen (Alfy absent), as HD-Alfy iKO.
We planned to address this question by examining levels of mHtt the striatum of
experimental cohorts, due to the primarily striatal mHtt accumulation phenotype in HD103Q. To
determine whether striatal size or cell populations were affected by expression of the HD
transgene, by the loss of Alfy, or by drug treatment, we performed stereology to estimate the
volume and the number of Nissl-positive nuclei in the striatum of HD-Alfy and tTA-negative
littermate controls, as well as mice treated with dox or with a combination of tam and dox (Figure
4.8 A,B). We found no significant difference in cell count or volume across genotype or treatment
group, indicating that striatal cell loss is not a feature of the HD-Alfy mouse model, and that neither
prolonged dox treatment nor Alfy knockout affects striatal integrity.

80
Figure 4.8: mHtt expression and drug treatment do not affect striatal cell count or volume. (A) Stereologic estimates of striatal volume in 11 m/o control, untreated, dox-treated, or dox and tam-treated HD-Alfy mice show no differences between genotypes or treatment groups after 4 months of treatment beginning at 7 m/o (F(3,14=0.37, p=0.7761). (B) There were similarly no differences in neuronal estimates obtained from stereological counts of Nissl-positive nuclei (F(3,14)=0.404, p=0.7523).
To determine whether Alfy is critical for the turnover of mutant exon-1-Htt in the
mammalian brain, 7 month old HD-Alfy mice and littermate controls negative for Cre were
injected with tamoxifen or vehicle control, followed by continuous administration of dox or control
drinking water (Figure 4.9A). Based on when Alfy protein levels diminished after Cre-mediated
excision (Figure 4.3B), we administered dox for mHtt suppression beginning one week after the
last tamoxifen injection. Mice were sacrificed at 1, 2, or 4 months post dox treatment, to examine
a timecourse of mHtt clearance and to determine whether Alfy was required for mHtt removal.
Tissue was stained with N-terminal Htt epitope EM48 and stereology was performed to estimate
the number of nuclei in the striatum containing mHtt.

81
Figure 4.9: Inducible Alfy KO impedes removal of mutant Htt. (A) Experimental design. 7 month old HD-Alfy mice are injected with tamoxifen or vehicle control to delete or preserve Alfy. One week after the final injection, HD103Q transgene suppression is initiated with dox-treated water and sustained for 1, 2, or 4 months. (B) Representative immunohistochemistry for Htt in HD-Alfy striatum before treatment and at the final study endpoint 4 months post-treatment. Dox and tam-treated mice (HD gene suppressed and lacking Alfy) show impeded Htt clearance. (C) Stereological counts of neurons in the left dorsolateral striatum containing EM48-positive Htt deposits at 1, 2, and 4 months after dox treatment in the presence or absence of Alfy. Data is shown as a percentage of untreated 7 m/o HD-Alfy. ANOVA revealed a significant overall effect of Alfy iKO (F(1,20)=7.032, p=0.0153) and a significant interaction between Alfy genotype and length of dox treatment (F(1,2)=4.001, p=0.0346. n=4-5 mice per genotype per age. The absence of Alfy had a significant effect on mHtt load after 2 months of dox (F(1,6)=8.632, p=0.0260) and after 4 months of dox (F(1,8)=6.347, p=0.0358). The absence of Alfy did not significantly affect numbers of mHtt-positive cells after 1 month of dox treatment (F(1-6)=0.665, p=0.4458).

82
Dox treatment of vehicle-injected HD-Alfy animals for 2 or 4 months (HD gene off, Alfy
present) resulted in significant clearance of mHtt. Notably, the same length of dox treatment in
tam-injected HD-Alfy iKO animals (HD gene off, Alfy absent) did not have a significant effect on
numbers of mHtt-containing cells (Figure 4.9 B,C). 1 month of dox treatment in the presence or
absence of Alfy was not sufficient for significant removal of mHtt. This implies that once the
source of the mutant protein has been suppressed, turnover of mHtt deposits occurs at a linear rate.
More importantly, these findings suggest overall that Alfy is crucial for the removal of mHtt
deposits in vivo.
Motor behavioral phenotypes in Htt-103Q-expressing mice
In the process of establishing Alfy iKO and confirming the clearance of mHtt in dox-treated
HD103Q, we began to breed these lines together to create HD-Alfy mice with the potential for
both tam-inducible Alfy iKO and dox-regulatable mHtt. Full knockout and maintenance of lines
required that both floxed Alfy and tetO-Htt-103Q be brought homozygous, which necessitated a
number of rounds of breeding. Therefore, during intermediate stages of breeding (Table 3.1,
progeny of Rounds 3-5) we grouped mice according to CAMKIIα-tTA genotype, and began
exploratory testing of motor behavioral phenotype in a mixed-sex cohort of mHtt-expressing mice
(CAMKIIα-tTA+/-) versus non-expressing littermates (CAMKIIα-tTA-/-). These investigations
(Figure 4.10) helped us to select the most appropriate tasks for behavioral assessment in larger
experimental cohorts of HD-Alfy mice.

83
Figure 4.10: Motor behavioral phenotypes in Htt-103Q-expressing mice. (A) A mixed-sex cohort containing 15 CAMKIIα-tTA+/- and 10 CAMKIIα-tTA-/- mice was assessed on the Rotarod monthly from 4-7 months of age with 3 daily trials across 3 days for a total of 9 trials. Repeated-measures ANOVA revealed a significant overall effect of genotype (F(1,93)=5.986, p=0.0163) but no significant effect of age (F(1,3)=0.985, p=0.4035) and no significant interaction between genotype and age (F(1,3)=0.039, p=0.9896). (B) There was a trend towards increased latency to cross an elevated beam in Htt-expressing

84
mice (F(1,16)=3.704, p=0.0723). (C-D) In the open field arena, there was no significant effect of genotype in distance traveled (F(1,23)=0.387, p=0.5400) or number of stereotyped movements (F(1,23)=0.595, p=0.4483). (E-F) We did not observe a significant difference between genotypes in the amount of time spent climbing when placed under a wire mesh structure (F(1,16)=0.736, p=0.4035) nor in the number of climbing episodes (F(1,16)=0.361, p=0.5565). (G) Latency to fall when hanging from an inverted wire cage top was not significantly different in mHtt-expressing mice compared to tTA-negative controls (F(1,16)=2.303, p=0.1487). (H) The amount of time required to turn and descend from the top of a vertical pole did not differ between genotypes (F(1,12)=2.932, p=0.1126).
Rotarod
Mice (n=12/genotype) were tested monthly from 4-7 months of age on the accelerating Rotarod
task (Figure 4.10A) as described in Chapter 3. After an initial training day, mice were tested three
times daily for three consecutive days and the latency to fall was recorded. Mice expressing mHtt
performed significantly worse than control littermates at all ages tested. However, there was no
significant decline in performance between 4 and 7 months of age.
Balance Beam
9 month-old mice (n=9/genotype) were trained to cross an elevated beam (80 cm length x 60 cm
height x 12 cm width) to enter an enclosed tube (Figure 4.10B). The latency to cross was measured
across 3 trials and the two best times were averaged. There was a trend towards longer latencies
to cross in CAMKIIα-tTA+/- expressing mice compared to control littermates.
Open Field
9-month-old mice were given a ten-minute exposure to the Open Field Arena. We observed no
significant difference in distance traveled (Figure 4.10C) or stereotyped movements (Figure
4.10D) in mHtt-expressing mice compared to controls.

85
Additional behaviors: Climbing, hanging, pole descent
Pilot behavioral experiments in 9-month-old mice were performed on an additional three motor
tasks: climbing, hanging, and pole descent. In the climbing task, a wire mesh cage was inverted
over each mouse and time spent climbing (Figure 4.10E) and the number of climbs (Figure 4.10F)
were recorded. For assessment of the ability to grip and hang upside down, the mouse was placed
on a wire cage top which was inverted at 60 cm height over a padded surface (Figure 4.10G). The
pole task assessed the ability of a mouse to turn and descend when placed at the top of a 60 cm
vertical pole (Figure 4.10H). For all of these behaviors we observed no significant differences
between HD mice and controls. We thus chose to focus on the Rotarod and Balance beam tasks
in our later experimental cohorts and to continue locomotor monitoring in the Open Field.
Mild motor behavioral phenotype in experimental cohort
Timed Alfy knockout in the regulatable HD103Q system revealed that Alfy is required for
the clearance of striatal mHtt deposits. Previous characterization of the HD94 inducible mouse and
numerous gene silencing techniques in animal models of HD have led to the observation that
clearance of aggregated protein correlates with improvements in behavioral phenotype. Given
Alfy’s selectivity for detergent-insoluble aggregated mHtt (Filimonenko et al., 2010), our original
goal was to understand whether aggregate clearance is required for motor recovery.
Based on exploratory behavioral results (Figure 4.10), we planned behavioral testing on a
large experimental cohort of HD-Alfy mice in order to include relevant drug and genotype
littermate controls. The experimental cohort contained mice that were all homozygous for floxed
Alfy (Alfyflox/flox), and heterozygous for mHtt (TetO-103Q+/-) and additionally expressed either
heterozygous tTA (CAMKIIα-tTA+/-), heterozygous Cre (Actin-CreERTM+/-), both Cre and tTA, or

86
neither. We focused on three main motor behaviors, the Rotarod, Beam Crossing, and Open Field
tasks, and performed pre-treatment testing at 3, 5, and 7 months of age (Figure 4.11)
Rotarod
Experimental cohorts were tested on the Rotarod as described above, but with an additional
4th day (3 trials) of testing in order to better approach a plateau of behavioral acquisition (Figure
4.11 A,B). In males but not females, mice expressing mHtt performed worse than littermate
controls negative for CAMKIIα-tTA.
Balance Beam
Beam crossing was tested as described above, but for greater accuracy mice were
videotaped and latency to cross was determined using custom Matlab software. Female but not
male mice with mHtt expression had longer latencies to cross than controls (Figure 4.11 C,D).
Open Field
A ten-minute exposure to the open field area revealed that CAMKIIα-tTA-expressing mice
had normal locomotor behavior, with distances traveled similar to controls in both males (Figure
4.11E) and females (Figure 4.11F).

87
Figure 4.11: Pre-treatment behavioral characterization of experimental mice. (A) On the accelerating Rotarod with four days of three trials each, male Alfyflox/flox::Actin-CreERTM+/-::HD103Q mice (n=32) performed worse than controls lacking CamKIIα-tTA (n=22) but did not progress over time. Repeated-measures ANOVA revealed a significant overall effect of mHtt expression (F(1,156)=32.36, p<0.0001) and a significant effect of age (F(1,156)=3.419, p=0.0352) but no significant interaction between genotype and age (F(1,2)=0.068, p=0.9344). (B) Female Rotarod performance Alfyflox/flox::Actin-CreERTM+/-::HD103Q mice (n=24) was similar to controls lacking CamKIIα-tTA (n=17). Repeated-measures ANOVA revealed no significant overall effect of mHtt expression (F(1,117)=2.513, p=0.1156) or age (F(1,117)=2.676, p=0.073) and no significant interaction between genotype and age (F(1,2)=0.056, p=0.9456). (C) Male beam crossing performance was similar across genotypes (F(1,156)=1.808, p=0.1807), with a significant effect of age (age (F(1,2)=9.094, p=0.0002) and no significant interaction between genotype and age (F(1,2)=1.422, p=0.2445). (D) Female beam crossing differed significantly between genotypes (F(1,117)=11.442, p=0.001), with no significant interaction between genotype and age (F(1,2)=0.140, p=0.8693). (E) ANOVA revealed no locomotor deficit in male Alfyflox/flox::Actin-CreERTM+/-::HD103Q mice compared to CamKIIα-tTA-negative controls (F(1,156)=0.560, p=0.4553). There was significant

88
acclimation over time (F(1,2)=3.944, p=0.0213) and no interaction between genotype and age (F(1,2)=0.170, p=0.8483). (F) ANOVA revealed no locomotor deficit in female Alfyflox/flox::Actin-CreERTM+/-::HD103Q mice compared to CamKIIα-tTA-negative controls (F(1,117)=0.13, p=0.7187). There was significant acclimation over time (F(1,2)=11.549, p<0.0001) and no interaction between genotype and age. (F(1,2)=0.514, p=0.5996).
Alfy knockout leads to behavioral decline in HD103Q and WT controls
Ultimately, for behaviors measured longitudinally, HD-Alfy mice showed a relatively mild
and non-progressive motor phenotype. Whereas males performed worse than littermate controls
on the Rotarod, and females took longer to cross the beam, none exhibited a progressive phenotype
over time. In itself this observation counters the utility of performing a phenotypic reversal
experiment. Nevertheless, due to the staggering of breeding and the size of the cohorts required
for drug and genotype controls, a smaller cohort of mice receiving tamoxifen, doxycycline, or a
combination began post-treatment behavioral testing.
The most striking observation that emerged from preliminary behavioral analyses of the
study endpoint (11 months old), was that loss of Alfy, in both HD-Alfy iKO and WT Alfy iKO
mice, led to a significant decline in complex motor function, with a profound drop in performance
on all tasks that by far exceeded the HD103Q phenotype itself (Figure 4.12 A-F). This finding
suggests that Alfy is essential for maintaining complex motor behaviors in the adult brain.

89
Figure 4.12: Alfy knockout leads to profound motor behavioral deficit. Following four months of treatment, 11 month-old mice (n= 14M/12F controls with Alfy present; 7M/8F HD103Q with Alfy present, 8M/5F controls with Alfy absent, 7M/4F HD103Q with Alfy absent) were behaviorally tested. (A) Male mice tested on the Rotarod for 3 daily trials on 4 consecutive days showed an HD-related behavioral decline, but an extreme effect of Alfy knockout. Repeated-measures ANOVA showed a significant overall effect of genotype (F(1,32)=22.730, p<0.0001). Fisher’s PLSD revealed that HD103Q mice with Alfy present performed significantly worse than controls with Alfy present (p=0.0483), and mice with Alfy absent performed significantly worse than those with Alfy present, for both control (p<0.0001) and HD mice (p<0.0004). (B) A significant overall effect of genotype was found for female mice tested on the Rotarod (Repeated-measures ANOVA, F(1,25)=22.389, p<0.0001). Fisher’s PLSD revealed that female HD mice performed similarly to control mice (p=0.5253), and mice with Alfy absent performed significantly worse than those with Alfy present, for both control and HD mice (p<0.0001). (C) For latency to cross the balance beam, ANOVA revealed no significant overall difference between control and HD mice in the mixed-sex cohort (F(1,61)=0.172, p=0.6801); however, Alfy KO had a significant effect on performance (F(1,61)=112.176, p<0.0001). (D) For distance traveled in the Open Field, ANOVA revealed no significant overall difference between control and HD mice in the mixed-sex cohort (F(1,61)=1.698, p=0.1974); however, Alfy KO had a significant effect on locomotion (F(1,61)=10.996, p=0.0015).
Conclusions
We have created a model combining inducible Alfy knockout with mHtt gene suppression,
and discovered that Alfy is essential for the clearance of accumulated mutant exon-1 Htt in vivo.
This affirms our hypothesis that Alfy is involved in the selective macroautophagy of aggregated

90
disease-relevant proteins in mammalian brain. Additionally, the separate mouse lines required to
address this question, HD103Q and Alfy iKO, are revealing with respect to the potential role of
aggregate accumulation and clearance in both Huntington’s disease and aging. We have developed
a drug-regulatable in vivo model for the study of exon-1 mHtt clearance, and a reliable model of
inducible Alfy knockout whose behavioral phenotype has opened a new line of inquiry into Alfy’s
profound importance to the adult brain.
During preliminary behavioral analysis of HD-Alfy and HD-Alfy iKO mice, we came to
two conclusions. First, the regulatable HD103Q model is not ideal for a formal behavioral reversal
experiment. Striatal mutant exon-1 Htt accumulation resulted in a relatively mild and non-
progressive phenotype, contributing to the literature suggesting that restricted expression of mHtt
is insufficient to drive behavioral pathology (Wang et al., 2014). Second, inducible Alfy knockout
in aging adult mice, even in the absence of mHtt expression, results in a severe and profoundly
progressive phenotype that would preclude querying motor reversal.
These findings led us to halt the large behavioral HD study of HDAlfy iKO and repurpose
the subjects to explore Alfy’s age-dependent impact on motor behavior, discussed in Chapter 6.
To continue our study of the relationship between aggregation and symptomology in HD, we chose
a different approach to address how impeding Alfy-mediated mHtt turnover would impact
behavior. We selected the BACHD, a more behaviorally robust and progressive mouse model of
HD with a different aggregation profile, to complement our studies in HD103Q. The BACHD is
also a transgenic model, and has a similar Q length (97) with an alternating CAGCAA tract, but
differs in that it expresses full-length human mHtt rather than an exon-1 fragment. We crossed this
line with heterozygous constitutive Alfy knockout mice (Dragich et al., 2016) and investigated
mHtt accumulation and motor behavior.

91
Chapter 5: Insoluble aggregate accumulation accelerates HD pathogenesis
Introduction
The most promising potential therapies to date address Huntington’s Disease at its source,
targeting the mutation with a variety of gene silencing methods, the first of which reached clinical
trials in late 2015 (National Institute of Mental Health, 2016). As described in Chapter 2, genetic
knockdown approaches in rodent and primate models of HD have led to improvements in
behavioral phenotype that occur in conjunction with the disappearance of Htt-positive structures.
Although this is suggestive of the toxic nature of Htt protein deposits, it is unclear whether
aggregates represent a principal source of toxicity in disease pathogenesis. Should we pursue
therapeutic strategies that lead to the removal of visible accumulated Htt, or are these structures
protective or inert?
In Chapter 4, we presented a mouse model that combines inducible Alfy knockout with
regulatable mHtt expression in order to demonstrate that Alfy is essential in vivo for the turnover
of mHtt deposits. We next sought to use Alfy’s selectivity for detergent-insoluble aggregated
species (Filimonenko et al., 2010) as a tool to understand whether aggregated mHtt represents a
toxic potentiator of pathology in vivo. Manipulating Alfy in vivo in the context of Htt expression
and suppression would allow us to determine the behavioral impact of selectively impeding
detergent-insoluble mHtt turnover. However, the mild phenotype of HD103Q, in addition to the
profound behavioral effects of inducible Alfy knockout, precluded the behavioral utility of this
system.
Consequently, we created a second model that combines constitutive Alfy knockout, made
using gene-trap technology (Dragich et al., 2016), with a more behaviorally relevant HD model,

92
the BACHD. The BACHD mouse, as described in Chapter 2, expresses full-length mutant
huntingtin with a 97Q alternating CAG-CAA repeat extension, and develops progressive motor
and cognitive phenotypes, along with late-onset accumulation of an exon-1 Htt fragment (Gray et
al., 2008). Crossing with our heterozygous Alfy knockout yielded BACHD mice with 50% Alfy
expression. These mice selectively accumulated more detergent-insoluble mHtt, more visible
aggregated structures, and showed accelerated behavioral onset compared to BACHD littermates.
Alfy-depleted BACHD (BACHD::Alfy GT/+) also show signs of increased cortical and striatal
pathology.
In summary, we provide further biochemical and immunohistochemical evidence that Alfy
is required for insoluble aggregate removal, and the acceleration of pathology and behavioral
phenotypes in conjunction with impeded turnover suggest that these structures represent a
significant source of toxicity in the course of HD.
Results
Generation of BACHD::Alfy GT/+
In order to create a behaviorally progressive HD mouse model with Alfy depletion, we
combined the BACHD (Gray et al., 2008) with heterozygous Alfy knockout mice (Dragich et al.,
2016). This model uses a gene trap vector inserted within the first intron of the Alfy locus to abolish
production of the full-length transcript. Homozygous Alfy knockouts (Alfy GT/GT) die soon after
birth and show improper formation of axonal tracts throughout the brain, highlighting the
importance of lysosome-mediated degradation pathways in mammalian neurodevelopment
(Orosco et al., 2014). Heterozygous knockouts (Alfy GT/+) do not display developmental
phenotypes and show no overt behavioral or pathological abnormalities (Dragich et al., 2016).

93
To explore the relationship between mHtt turnover and behavior, we crossed BACHD mice
with Alfy GT/+ mice to generate the four genotypes depicted in Figure 5.1: wild type (Alfy +/+),
mice with 50% Alfy depletion (Alfy GT/+), BACHD, and BACHD with 50% Alfy depletion
(BACHD::Alfy GT/+).
Figure 5.1: Breeding schema for BACHD with 50% Alfy depletion. Heterozygous Alfy knockout mice created using gene-trap technology (Alfy GT/+) were crossed with BACHD expressing full-length human mutant huntingtin to generate four genotypes: WT, Alfy GT/+, BACHD, and BACHD::GT/+.
Accelerated accumulation of detergent-insoluble mHtt in BACHD::Alfy GT/+
To determine the impact of Alfy depletion on insoluble aggregate accumulation in
BACHD, we performed Western blots on whole brain lysate from the four genotypes described in
Figure 5.1 using antibodies against three Htt epitopes (Figure 5.2), 1C2 (recognizing extended
polyQ sequences), 3B5H10 (against the first 171 amino acids of mHtt), and ab2166 (recognizing
amino acids 181-810 of both mutant and wild type huntingtin). Lysates treated with RIPA were
centrifuged and the RIPA-soluble supernatant was collected and run on a gel (Figure 5.2A), while
the insoluble pellet was dissolved in 4M urea and run separately (Figure 5.2B). No mutant Htt was
detected in wild type or Alfy GT/+ brain with the exception of ab2166 (Figure 5.2A, bottom)
which also detects endogenous mouse huntingtin. With all three epitopes examined, Alfy-depleted

94
BACHD show a significant decrease in detergent-soluble Htt, and a significant increase in
detergent-insoluble mutant Htt (Figure 5.2C). These experiments were carried out by Evelien
Eenjes.
Figure 5.2: 50% Alfy depletion accelerates accumulation of detergent-insoluble mHtt. (A) Western blot of whole-brain lysate using full-length htt antibody ab2166 shows decreased detergent-soluble mHtt in BACHD::Alfy GT/+ compared to BACHD. (B) Western blot of brain lysate shows a significant increase in detergent-insoluble mHtt as a result of 50% Alfy depletion in BACHD, using both expanded polyQ antibody 1C2 and N-terminal mHtt antibody 3B5H10. (C) Quantification of A, B.
Accelerated behavioral decline in BACHD::Alfy GT/+
To understand the motor behavioral impact of a selective increase in detergent-insoluble
forms of mHtt, we trained BACHD::Alfy GT/+ mice and littermates on the Rotarod and measured
locomotion and vertical movements during exposures to the Open Field. Behavioral assessments
were performed by Chris Johnson and Joan Bosco.
Rotarod
To understand the motor behavioral impact of a selective increase in detergent-insoluble
forms of mHtt, we trained BACHD::Alfy GT/+ mice and littermates on the Rotarod (Figure 5.3).
We tested cohorts of 12-15 males (Figure 5.3A) and females (Figure 5.3B) per genotype at 3, 4, 5,
and 8 months of age. WT and Alfy GT/+ mice showed consistent and comparable performance

95
across all trials and ages tested, indicating that 50% Alfy depletion alone did not affect complex
motor performance. Consistent with published data (Gray et al., 2008), BACHD mice showed a
decline in performance over time compared to WT and Alfy GT/+ controls. Male BACHD::Alfy
GT/+ performed significantly worse than BACHD mice at all time-points tested, suggesting a more
rapid onset of phenotype. Female performance differed significantly at 4 and 5 months of age.
Figure 5.3: 50% Alfy depletion accelerates Rotarod phenotype in BACHD. Data is represented as an average of three daily trials across three days at each age tested. n=10-15 mice/sex/genotype. (A) Latency of male mice to fall from the accelerating Rotarod. Repeated-measures ANOVA revealed a significant overall effect of genotype (F(3,188)=88.171, p<0.0001) and of age (F(3,188)=6.768, p=0.0002) and a trend towards an interaction between genotype and age (F(9,188)=1.918, p=0.0516). Fisher’s PLSD revealed no significant difference in performance of WT compared to Alfy GT/+ mice at any age (3 months, p=0.8627; 4 months, p=0.8629; 5 months, p=0.9630; 8 months, p=0.7559). BACHD::Alfy GT/+ showed significantly poorer performance at all ages compared to BACHD (3 months, *p=0.0446; 4 months, *p=0.0097; 5 months, *p=0.0033; 8 months, *p=0.0119). (B) Latency of female mice to fall from the accelerating Rotarod. ANOVA revealed a significant overall effect of genotype (F(3,171)=80.182, p<0.0001) and of age (F(3,171)=4.283, p=0.0061) and an interaction between genotype and age (F(9,171)=3.017, p=0.0023). Fisher’s PLSD revealed no significant difference in performance of WT compared to Alfy GT/+ mice at any age (3 months, p=0.9556; 4 months, p=0.7554; 5 months, p=0.7554; 8 months, p=0.6852). BACHD::Alfy GT/+ showed significantly poorer performance compared to BACHD at 4 and 5 months (3 months, p=0.0538; 4 months, *p=0.0006; 5 months, *p=0.0008; 8 months, p=0.2809).

96
Open Field
BACHD mice show a late-onset hypolocomotor phenotype beginning at 12 months of
age (Gray et al., 2008). To determine the impact of Alfy depletion on locomotion, we gave mice
a 1-hour exposure to the open field arena at 6, 9 and 12 months of age and measured the total
distance traveled (Figure 5.4 A,B) and the number of vertical movements (Figure 5.4 C,D).
We observed no significant differences in distance traveled or in vertical movements by
Alfy GT/+ compared to WT mice at any age for either gender, suggesting that heterogygous Alfy
knockout alone does not impact locomotor behavior (Figure 5.4A-D). At 6 months old, the
distance traveled by mHtt-expressing mice does not differ significantly from controls. However,
6 m/o male BACHD::Alfy GT/+ made significantly fewer vertical movements than controls, a
phenotype not displayed by the BACHD until 9 months old. By 9 months old there is a
significant decrease in distance traveled by male BACHD::Alfy GT/+ compared to controls, a
phenotype which was not observed until 12 months old in BACHD (Figure 5.4A). These data
suggest that in male mice, increased insoluble mHtt accrual corresponds with a more rapid onset
of locomotor phenotype in male BACHD. Overall, female BACHD and BACHD::Alfy/GT
showed similar rates of decline in pathlength and vertical movements, highlighting a gender
disparity in locomotor behavior (Figure 5.4B). This may suggest that the behavioral impact of
mHtt accumulation differs depending upon gender.

97
Figure 5.4: 50% Alfy depletion accelerates locomotor phenotype in BACHD. Distance traveled and vertical movements during a 1-hour exposure to the open field arena. n=10-15 mice/sex/genotype. (A) Distance traveled by male mice in the open field arena. At 6 m/o, ANOVA revealed no significant overall effect of genotype (F(3,52)=0.598, p=0.6190). By 9 m/o there was a significant overall effect of genotype (F(3,37)=3.660, p=0.0209) and Fisher’s PLSD revealed a significant decrease in distance traveled by BACHD::Alfy GT/+ compared to WT (p=0.0142). At 12 mo there was a significant overall effect of genotype (F(3,39)=9.025, p=0.0001) and Fisher’s PLSD revealed that both BACHD and BACHD::Alfy GT/+ traveled less distance than controls (p<0.01). (B) Distance traveled by female mice in the open field arena. At 6 m/o, ANOVA revealed no significant overall effect of genotype (F(3,46)=2.433, p=0.0769). By 9 m/o there was a significant overall effect of genotype (F(3,43)=3.953, p=0.0141) and Fisher’s PLSD revealed a significant decrease in distance traveled by BACHD compared to WT (p=0.0466) and by BACHD::Alfy GT/+ compared to WT (p=0.0098). At 12 m/o there was a significant overall effect of genotype (F(3,34)=6.724, p=0.0011) and Fisher’s PLSD revealed that both BACHD and BACHD::Alfy GT/+ traveled less distance than controls (p<0.005). (C) Vertical movements by male mice in the open field arena. At 6 m/o, ANOVA revealed a significant overall effect of genotype (F(3,52)=3.957, p=0.0129) and Fisher’s PLSD revealed a significant decrease in movements made by BACHD::Alfy GT/+ compared to WT (p=0.0043). There was a significant overall effect of genotype at 9 m/o (F(3,37)=8.968, p=0.0001) and at 12 m/o (F(3,39)=4.115, p=0.0125). Fisher’s PLSD revealed that both BACHD and BACHD::Alfy GT/+ made fewer movements than controls (p<0.01 at 9 and 12 m/o). (D) Vertical movements by female mice in the open field arena. At all ages, ANOVA revealed a significant effect of genotype (p<0.0001). Fisher’s PLSD revealed that both BACHD and BACHD::Alfy GT/+ made fewer movements than controls (p<0.05 at all ages).

98
Accelerated accumulation of Htt positive structures in BACHD:Alfy GT/+
BACHD mice show insoluble mHtt via biochemical methods by 6 months old, but
aggregation is not observable via immunohistochemistry until 12 months (Gray et al., 2008). To
determine whether accelerated motor behavior is accompanied by an increase in aggregates, we
stained with N-terminal mHtt antibodies EM48 and S830, and quantified reactivity in BACHD
and BACHD:Alfy GT/+ brain (Figure 5.5). S830-positive aggregates were distributed throughout
the dorsolateral cortex (anterior cingulate, motor, and somatosensory areas) and appeared as
cytoplasmic or perinuclear puncta, with occasional staining of dystrophic neurites (Figure 5.5A).
S830-positive aggregates were extremely difficult or impossible to find in most 9-month old
BACHD (data not shown), and were visible but sparse in 12 month-old animals (Figure 5.5A). In
contrast, BACHD:Alfy GT/+ mice showed obvious aggregate accumulation. EM48 staining had a
similar distribution, but additionally we observed diffuse cytoplasmic accumulation of mHtt. There
was a marked increase in EM48 staining in BACHD:Alfy GT/+ brains at 12 months old.
To quantify the increased accumulation in BACHD: Alfy GT/+, we obtained stereological
estimates of cortical aggregate load by counting S830-positive puncta in serially-cut brain sections
from 12 month old mice (Figure 5.5B). We found a significant increase in the number of aggregates
in BACHD: Alfy GT/+ compared to BACHD littermates.
BACHD mice do not show significant cortical or striatal cell loss, even at later ages, but
they do show signs of brain atrophy (Gray et al., 2008). To determine whether Alfy depletion
affected this measure, we obtained stereological estimates of cortical volume and numbers of
Nissl-stained nuclei. We observed no change in cell count (Figure 5.5C) or volume (Figure 5.5D)
in BACHD versus BACHD:Alfy GT/+ mice.

99
Figure 5.5: 50% Alfy depletion in BACHD causes increased accumulation of mHtt aggregates. (A) Immunohistochemistry for N-terminal mHtt antibodies S830 and EM48 in 12 m/o mice. Arrows indicate cytoplasmic and perinuclear puncta defined as mHtt deposits. Scale bar=10 µm. (B) Stereological quantification of numbers of S830-positive mHtt puncta in the cortex of BACHD and BACHD::Alfy GT/+. ANOVA revealed a significant increase in mHtt in BACHD::Alfy GT/+compared to BACHD at 12 m/o (F(1,8)=8.875, p=0.0176). (C) Stereological quantification of numbers of nuclei in the cortex of BACHD and BACHD::Alfy GT/+. ANOVA revealed no significant difference in cell count in BACHD::Alfy GT/+ compared to BACHD (F(1,8)=1.406, p=0.2698). (D) Stereological estimates of cortical volume in BACHD and BACHD::Alfy GT/+. ANOVA revealed no significant difference between BACHD::Alfy GT/+ and BACHD (F(1,8)=0.221, p=0.6505).

100
Additional accelerated pathology in BACHD:Alfy GT/+ mice
Reactive astrocytosis
Reactive astrocytosis is a common pathological signature in the brains of HD patients that
is not often recapitulated in mouse models (Pouladi et. al 2013). 12-month-old BACHD mice do
not show increased GFAP transcript compared to wild type mice (Boussicault JCBFM 2014), and
cultured astrocytes from BACHD mouse pups do not show increased GFAP as detected by
immunocytochemistry (Lee et al., 2013b). We performed IHC for GFAP at 9 m/o in BACHD and
BACHD::Alfy GT/+ mice. BACHD mice showed sparse reactive astrocytosis in the striatum,
while BACHD::Alfy GT/+ striata contained many GFAP-positive neurons with characteristic
hypertrophy and aberrant processes. This may represent an accelerated pathological feature in the
BACHD and could indicated added stress due to increased aggregate load.
Figure 5.6: 50% Alfy depletion leads to glial pathology and accumulation of ubiquitinated structures in BACHD. In 9 month old BACHD::Alfy GT/+ we observed reactive astrocytosis in the striatum and ubiquitin accumulation in cortex, which was not present in BACHD. Scale bar = 25µm.

101
Ubiquitin accumulation
As an added marker of aggregate accumulation, we stained sections from the brains of 9
m/o mice with an antibody against poly-ubiquitin. BACHD showed sparse to no accumulated
ubiqtuitin, in 9 m/o BACHD::Alfy GT/+ we observed ubiquitinated puncta in areas of the cortex
corresponding with the greatest mHtt accumulation. This confirms increased aggregate load in
BACHD with 50% Alfy depletion.
Conclusions
Combining the full-length mutant Htt-expressing BACHD mouse with 50% depletion of
Alfy led to an increase in detergent-insoluble but not soluble mutant Htt, consistent with Alfy’s
selectivity for detergent-insoluble aggregated proteins, which has been demonstrated in cellular
models of protein accumulation. This corresponded with accelerated appearance of motor
behavioral phenotypes and significantly greater numbers of visible Htt aggregates, in addition to
reactive astrocytes and ubiquitin accumulation. These results imply that impeded insoluble mHtt
turnover has adverse behavioral and pathological consequences. Our findings confirm Alfy’s
importance for mHtt turnover and are consistent with our hypothesis regarding the toxicity of
visibly aggregated and biochemically insoluble species of Htt protein.

102
Chapter 6: Preliminary investigation of the role of Alfy protein in normal aging
Introduction
We have thus far presented work exploring Alfy’s importance for mHtt turnover in mouse
models of HD. Whereas the accumulation and turnover of specific disease-relevant proteins is a
focus of neurodegenerative disease research, normal aging may also lead to homeostatic changes
that affect the balance between protein accretion and removal. Studies from the Yamamoto lab and
others suggest that Alfy may play a role in aggrephagy not only of disease protein deposits but
also other types of commonly-occurring stress complexes and ubiquitinated structures
(Filimonenko et al., 2010; Isakson et al., 2013a; Isakson et al., 2013b; Mandell et al., 2014). These
findings highlight the larger overarching question of the basal biological function of this Alfy-
mediated selective autophagic pathway.
Chapter 4 presented a model combining the Alfy iKO mice with an inducible mouse model
of mutant exon-1-Htt expression. The primary goal in creating this model was to understand the
role of Alfy in the context of removal of a disease relevant protein. In the course of creating the
models, we piloted our tamoxifen injection protocol and examined Alfy excision in young (2-3
month old) mice, and observed no overt abnormalities in behavior. Consequently, we were
surprised to uncover a rapid and profound motor behavioral phenotype in 7-month-old Alfy iKO
mice upon tamoxifen injection. This opened up a new line of questioning related to Alfy’s role in
the aging brain.
To pursue a potential age-dependent phenotype, we behaviorally characterized cohorts of
mice with Alfy knockout at varying ages, and found that older knockout led to more rapid onset
of motor abnormalities. The reason for this age-dependent sensitivity to Alfy iKO is not yet clear.

103
Presented in this chapter are analyses of the behavioral deficit, and we additionally offer some
preliminary biochemical and histological observations that may serve as departure and discussion
points for the pursuit of hypotheses related to the role of Alfy in the aging brain. I hypothesize that
the age dependent Alfy-dependence may reflect the importance of protein turnover during aging.
Results
Alfy iKO mice demonstrate a severe age-dependent motor behavioral phenotype
Alfy knockout in HD103Q at 7 months old led to a rapid decline in complex motor behaviors.
Because knockout at earlier ages did not appear to have severe adverse consequences, we wanted
to formally test the possibility that this decline in phenotype had an age-sensitive component.
Cohorts of Alfyflox/flox, ActinCreERTM+/- mice and littermate controls negative for Cre were
monitored in behavioral tasks at 3, 7, or 11 months of age, then injected with tamoxifen and re-
evaluated 2 and 4 months later (Figure 6.1). The three age groups will be referred to as Alfy-3-
iKO, Alfy-7-iKO, and Alfy-11-iKO.
Figure 6.1: Timeline for inducible Alfy iKO. Cohorts of Alfyflox/flox, ActinCreERTM+/- mice and littermate controls were injected with tam at 3, 7, or 11 months old and tested behaviorally before, 2 months after, or 4 months after injection.

104
Rotarod
We tested Alfy-3-iKO and Alfy-7-iKO mice and Cre-negative littermate controls on the
Rotarod using a protocol identical to that described in Chapter 4. Alfy-11-iKO were not included
in Rotarod assessment because we find that normal mice of this age have difficulty acquiring the
task. All groups showed similar baseline Rotarod performance prior to injection of tamoxifen. Two
and four months after injection, we observed a significant overall decline in the performance of
Alfy knockouts. By 4 months post-injection, this decline was significantly pronounced in Alfy-7-
iKO compared to Alfy-3-iKO.
Figure 6.2: Alfy iKO leads to age-dependent decline in Rotarod performance. (A) Rotarod performance of mixed-sex cohorts of Alfy-3-iKO and Alfy-7-iKO and littermate controls injected with tamoxifen and tested before, 2 months after, and 4 months after treatment. Repeated-measures ANOVA reveals a significant overall effect of Alfy iKO (F(1,156)=75.463, p<0.0001) and a significant effect of age of injection (F(1,156)=11.799, p=0.0008). Fisher’s PLSD reveals that before knockout, there is no significant difference between groups (p=0.2662). By four months after knockout, Alfy-7-iKO perform significantly worse than Alfy-3-iKO (p=0.0045).
Balance Beam
Mice of all knockout ages and Cre-negative littermate controls were tested on the balance
beam before tamoxifen injection, as well as 2 and 4 months later, with training paradigm as

105
described in Chapter 3. Before injection, all groups were capable of crossing the beam and had
similar cross times (Figure 6.3A). At 2 and 4 months post injection, the loss of Alfy at all ages
caused a significant decline in performance compared to controls, with increased severity when
KO was induced at later ages. There was a plateau in performance because mice were removed
from the beam and given a score of 20 seconds if they were unable to cross within this allotted
time. By 4 months post-injection, some Alfy-7-iKO and the majority of Alfy-11-iKO were unable
to cross the beam at all (Figure 6.3B). A small percentage of 15 month-old Cre-negative mice (in
which Alfy was present) showed an inability to cross, likely due to aging.
Figure 6.3: Alfy iKO leads to age-dependent decline in beam crossing. (A) Beam crossing performance of mixed-sex cohorts of Alfy-3-iKO and Alfy-7-iKO and littermate controls injected with tamoxifen and tested before, 2 months after, and 4 months after treatment. Repeated-measures ANOVA reveals a significant overall effect of Alfy iKO (F(1,156)=75.463, p<0.0001) and a significant effect of age of injection (F(1,156)=11.799, p=0.0008) as well as an interaction between Alfy status, age of injection, and month of testing (F(1,65)=0.0262). Alfy-11-iKO differed significantly from all other groups (p<0.0001). (B) Proportion of mice cabable of crossing the beam. Before tam injection, all mice can cross (time 0). After injection, differing proportions of age-specific iKO are unable to complete the task.
Alfy knockout leads to Purkinje cell degeneration
Our behavioral analyses suggest that Alfy is essential for sustaining complex motor control
during adulthood, and that functional Alfy is of increasing importance during aging. The severe
motor phenotype in Alfy knockouts led us to speculate that motor areas of the brain might display
damage upon gross examination. During home cage observations of Alfy iKO mice, we noted that

106
Alfy-11-iKO had visibly abnormal gait resembling a spinocerebellar ataxic phenotype (Guyenet
et al., 2010). A gait defect was not observed readily in younger mice.
To examine if any neuropathology was present, brains were sectioned and examined using
Thionin staining. Upon preliminary examination, we found that the cerebellum of an Alfy-7-iKO
mouse six months after knockout shows apparent degeneration of cerebellar Purkinje cells (Figure
6.4). We also noticed ectopic placement of cell bodies in the molecular layer, reminiscent of mouse
models of spinocerebellar ataxia (Cendelin, 2014). Notably, the loss of basal autophagy in neurons,
albeit during development (Komatsu et al., 2006; Komatsu et al., 2007b), also causes Purkinje cell
death.
Figure 6.4: Alfy-7-iKO show Purkinje cell loss. Nissl-stained cerebellum of WT and Alfy-7-iKO mice 6 months after injection of tamoxifen (13 months old). Alfy-7-iKO show loss of Purkinje cell nuclei and ectopic cells in the molecular layer (arrows). Scale bar: 50µm.
Alfy knockout in aged mice results in increased p62
On a fundamental level, the age-sensitivity of the Alfy iKO phenotype implies that Alfy is
in some capacity involved in the maintenance of a balanced biological system that declines during
aging. Because of Alfy’s proposed function in aggrephagy, I hypothesize that Alfy functions in
the maintenance of protein homeostasis, which declines due to age (Kaushik and Cuervo, 2015).
For example, Alfy-dependent targeting of aggregated structures might play a greater role due to

107
an increased probability of protein misfolding during aging. To begin investigating this possibility,
we probed the brains of Alfy iKO mice for the selective autophagy receptor p62/sqstm1
(sequestosome-1). Several lines of evidence indicate that p62 is involved in the turnover of
ubiquitinated cargoes, including aggregates, organelles, and invading microbes. It is implicated in
isolating misfolded proteins into sequestosomes and providing a scaffold to nucleate the formation
of autophagosomes (Johansen and Lamark, 2011).
Four months after tamoxifen injection, we performed Western blot on detergent-insoluble
fractions of cortical lysate from the brains of Alfy-3-iKO and Alfy-7-iKO and age-matched wild-
type controls using antibodies against p62. We observed a significant increase in the amount of
p62 only in the detergent-insoluble fraction of Alfy-7-iKO.
Figure 6.5: p62 accumulates in Alfy-7-iKO. (A) Western blot of detergent-insoluble cortical lysates of Alfy-3-iKO four months after knockout (7 m/o) compared to 7 m/o WT. ANOVA revealed no significant difference between WT and KO (F(1,2)=0.0102, p=0.9285). (B) Western blot of detergent-insoluble cortical lysates of Alfy-7-iKO four months after knockout (11 m/o) compared to 11 m/o WT. ANOVA revealed a significant difference between WT and KO (F(1,4)=7.899, p=0.0483).

108
Given the role of p62 in the formation and selective turnover of aggregates and because it
is also a substrate for autophagy (Johansen and Lamark, 2011), we interpret increased p62 protein
levels to convey a general increase in the presence of aggregated structures or misfolded proteins
in the absence of Alfy. p62, however, is involved in many forms of selective autophagy, and
upregulation in the absence of a specific aggregating protein could indicate a deficit in another
selective turnover pathway. One such pathway is mitophagy (Ding and Yin, 2012).
A second overarching theory with respect to age-sensitive behavioral changes relates to
mitochondrial dysfunction during normal aging. Perhaps an as-yet unknown function for Alfy is
in selective turnover of mitochondria or mitochondrial components, making it a contributor to the
combat of oxidative stress. Recent communication with collaborator Anne Simonsen, who is
investigating Alfy structure and function at the university of Oslo in Norway, led us to consider
this possibility. Her laboratory’s pursuit of Alfy binding partners identified NIPSNAP-1 (4-
nitrophenylphosphatase and non-neuronal SNAP-25-like protein homolog), an inner
mitochondrial membrane protein, as a binding partner of both Alfy and p62 (Anne Simonsen,
personal correspondence). NIPSNAP-1 was identified in a proteomic study of proteins that interact
with atg8 homologues (Behrends et al., 2010), and it has been specifically implicated as autophagic
cargo. NIPSNAP-1 is highly expressed in the brain, preferentially in neurons (Nautiyal et al.,
2010).

109
Figure 6.6: NIPSNAP accumulates in Alfy-7-iKO. (A) NIPSNAP is highly expressed in brain. Western blot of detergent-soluble lysates of WT mouse organs using antibodies against NIPSNAP-1 detects protein in liver, kidney, and brain tissue. Abbreviations: Liver (lvr), heart (hrt), lung (lng), kiney (kid), spleen (spl), muscle (mus), cerebral cortex (ctx), cerebellum (cb), striatum (str). (B) Western blot of detergent-insoluble cortical lysate in 11 m/o WT and Alfy-7-iKO four months post-KO. (C) Quantification of NIPSNAP-1 levels as a ratio to vinculin loading control in Alfy-7-iKO reveals a significant increase compared to WT mice (F(1,8)=15.14, p=0.0046). (D) Quantification of TOM20 levels as a ratio to vinculin loading control reveals no difference between Alfy-7-iKO and WT mice (F(1,8)=0.048, p=0.8321).
We investigated the expression of NIPSNAP-1 in mouse tissue, and confirmed that
NIPSNAP protein is present in the kidney, liver, and several brain areas (Figure 6.6A). We next
probed for NIPSNAP-1 protein in detergent-insoluble cortical lysates from Alfy-7-iKO and wild
type mice and found a dramatic increase in NIPSNAP in Alfy iKO brains compared to Cre-
negative controls (Figure 6.6B, C). This raises the possibility that Alfy may be required for
NIPSNAP-1 turnover, or for mitochondrial turnover. TOM20 levels were variable, so overall
amounts of mitochondrial appeared unchanged (Figure 6.6B, D). The specific function of
NIPSNAP is largely unknown, though there is evidence of its coexpression with genes involved

110
in vesicular transport (Seroussi et al., 1998), for upregulation in a model of epilepsy (Satoh et al.,
2002) and for downregulation in a model of phenylketonuria, implicating it in the regulation of
branched-chain amino acids (Surendran et al., 2005). NIPSNAP also interacts with amyloid
precursor protein (Tummala et al., 2010). Assaying mitochondrial function, oxidative stress, or
known mitophagy adaptors may be more revealing with respect to a potential role for Alfy in
mitochondrial turnover or health.
Conclusions
In summary, we present preliminary evidence that Alfy is required for complex motor
behaviors in the aging brain, as evidenced by increasingly severe behavioral decline with
advancing age of knockout. Our observations with regards to p62 and NIPSNAP accumulation as
well as cerebellar Purkinje cell loss suggest a number of possible avenues for future research on
Alfy, including its role in normal protein turnover during aging, mitophagy, and cerebellar health.

111
Chapter 7: General discussion
Studying the significance of mHtt aggregate turnover
More than a hundred years after Alois Alzheimer reported abnormal clumps of material in
the post-mortem brain tissue of his dementia patient Auguste D (Maurer et al., 1997), our
understanding of the relevance of protein deposits in neurodegeneration remains incomplete. The
subsequent discovery of aggregates in a vast array of disorders of the brain (Ross and Poirier,
2004), in conjunction with findings that their elimination is not only possible but has therapeutic
correlates (Knaevelsrud and Simonsen, 2010; Lim and Yue, 2015; Nixon, 2013), has propelled a
directed effort to understand and ultimately to harness the biological machinery that drives the
removal of protein deposits.
The 1993 discovery of the HD mutation (Huntington’s Disease Collaborative Research
Group) led to an explosion of new tools and new information to study the disease, including the
creation of antibodies to detect huntingtin protein in tissue, and the generation of the first
transgenic models of HD (Davies et al., 1997; DiFiglia et al., 1997; Trottier et al., 1995). The
ability to visualize and begin to manipulate the formation of aggregates opened the door to early
discussions about their potential for toxicity versus protection (Bates, 2003). Interpretations have
been dependent upon a variety of factors, including their anatomical location (Sieradzan and Mann,
2001), cellular localization (Yang et al., 2002), the modeling context driving aggregation (Pouladi
et al., 2013) and the biochemical solubility or “seeding” properties of the mutated fragment
(Nekooki-Machida et al., 2009). Whereas inhibiting aggregation has yielded mixed results with
regards to toxicity (Arrasate and Finkbeiner, 2012; Arrasate et al., 2004; Kim and Kim, 2014),
promoting clearance of existing accumulated mHtt structures in various experimental and clinical
contexts is of increasing interest (Sarkar et al., 2007; Sarkar et al., 2009; Sarkar and Rubinsztein,

112
2008). This is due in part to the tight link between clearance of aggregated mutant Htt and
behavioral recovery that has been revealed by HD gene silencing (Aronin and DiFiglia, 2014).
The original regulatable HD mouse, HD94, first demonstrated the potential for gene
suppression during adulthood to promote mHtt aggregate removal in conjunction with behavioral
recovery (Yamamoto 2000). This experiment provided direct evidence that neurons are capable of
removing aggregates once the continuous burden of HD transgene expression has been lifted. The
association between clearance and recovery has been recapitulated in a number of cellular, rodent,
and large animal models (DiFiglia et al., 2007; Harper et al., 2005; Kordasiewicz et al., 2012;
Stanek et al., 2014), and has culminated fifteen years later in clinical trials of intrathecally-
delivered antisense oligonucleotides to lower mHtt expression in the human brain (National
Institute of Mental Health, 2016). However, the pursuit of how exactly mammalian neurons
process existing accumulated structures containing mutant misfolded protein is a subject still in its
youth.
A prominent area of current research in targeting mHtt aggregates revolves around the idea
that macroautophagy is responsible for their removal (Martin et al., 2015; Yamamoto and
Simonsen, 2011). Direct evidence for the necessity of basal and selective autophagic components
in mHtt clearance comes from knockdown studies in cells (Filimonenko et al., 2010; Iwata et al.,
2005; Yamamoto et al., 2006). In vivo, various indirect indicators of autophagic processing of
mHtt or dysregulation due to its presence have led to efforts to upregulate macroautophagy, for
example through the inhibition of mTOR (Ravikumar et al., 2004), but fewer studies have been
aimed at elucidating the mechanisms of clearance of existing aggregates or defining more selective
in vivo targets.

113
Our work has directly implicated Alfy-mediated selective autophagy in the clearance of
mutant huntingtin in vivo, and sheds light on our understanding of how aggregates and aggregate
clearance contribute to HD pathogenesis and normal aging. This thesis has detailed the
development of mouse models that genetically manipulate Alfy, a known mediator of selective
autophagy, in the context of mutant huntingtin accumulation. Using the HD103Q and the BACHD,
two models that differ in expression strategy, aggregation profile, and behavioral phenotype, we
find that Alfy is required in both in vivo systems for the effective turnover of mHtt. We additionally
demonstrate a specific need for Alfy-mediated aggregate clearance in order to resist
neurodegenerative and behavioral symptomology.
In this chapter, I will contextualize our findings within the literature of Huntington’s
disease and selective autophagy. I will begin with a discussion on how the creation and
characterization of HD103Q may contribute to the concept of mHtt toxicity as a function of
anatomical location of accumulated mHtt. Next, I will explore how the role of Alfy in clearance
of accumulated mHtt in HD-Alfy mice raises questions about the interplay between nuclear
aggregates and a cytoplasmic protein turnover pathway, and how the behavioral impact of
insoluble mHtt accumulation contributes to an understanding of aggregate toxicity. Finally, I will
comment on how the behavior of Alfy iKO in the context of Alfy’s role in HD may provide
perspective on the intersection between neurodegeneration and aging.
Striatally restricted mHtt is insufficient to drive behavioral phenotype
Our goal in the design of HD103Q was to recreate and improve upon an in vivo model of
Huntington’s disease which would allow us to specifically address the removal of accumulated
mHtt via gene suppression. The original HD94 mouse has fallen out of use due to unstable repeat

114
length and decreased transgene copy (Yamamoto et al., 2000). Genetic therapies have recapitulated
mHtt clearance in response to interference with the HD gene in mouse models (DiFiglia, 1990;
Harper et al., 2005; Kordasiewicz et al., 2012; Stanek et al., 2014; Wang et al., 2014), for example
through intermittent injection of a huntingtin antisense oligonucleotide (Kordasiewicz et al., 2012).
With HD103Q, we have reestablished a drug-regulatable genetic strategy with an
intergenerationally stable CAG repeat mutation, thereby reintroducing a means to directly
manipulate mHtt removal in vivo, a valuable tool in light of our incomplete understanding of gene
therapeutics and aggregate-targeting strategies. The addition of a luciferase reporter will speed the
detection of the transgene for ease of use in any future primary culture studies using HD103Q.
The initial choice to use an exon1-mHtt fragment model to study aggregation and clearance
was predicated on the fact that fragment models tend to have early and heavy aggregation in
conjunction with robust and progressive phenotypes (Pouladi et al., 2013). Despite the tTA driven
by a forebrain-wide CAMKIIα promoter, we found that aggregation in HD103Q, as assessed by
N-terminal mHtt antibodies, was restricted to the ventrolateral striatum, with sparse cortical
staining by 18 months of age. HD103Q striatum recapitulates the progressive diffuse nuclear
accumulation observed in some fragment models (Mangiarini et al., 1996; Schilling et al., 1999),
but does not show punctate inclusions nor ultimately a rapid behavioral decline. The mild
phenotype could also reflect a variety of differences in the construction and genetic outcomes of
HD103Q which have yet to be directly compared to other fragment models, including transgene
copy number, level of exon-1 mHtt expression relative to endogenous, or transgene insertion and
regulation. Additionally, the stability of the 103Q CAG-CAA repeat compared to models like the
R6/2, which have shown extreme somatic and germline expansion (Kennedy and Shelbourne,

115
2000) and contribute to pathogenesis in humans (Swami et al., 2009), may add to the attenuated
phenotype.
The particular CAMKIIα-tTA line we crossed with tetO-103Q mice drove robust
expression within the striatum, which may reflect the use of line B21 as described in (Mayford et
al., 1996) and may be due to integration site-dependent effects. Crossing with a different tTA
driver might yield more widespread accumulation and more progressive phenotype, perhaps
providing the opportunity to examine aggregate clearance throughout the forebrain. However, the
HD103Q represents a unique instance of striatally-selective, continuously driven exon-1-mHtt
accumulation which, in conjunction with our motor behavioral assessment, may be informative
with regards to the relative contribution of aggregate location to toxicity.
Other groups have introduced mHtt directly into the striatum through lentiviral injections
of polyQ sequences or of mHtt fragment (Ruiz and Deglon, 2012), inducing aggregation of mHtt,
striatal cell loss, and in some cases the rapid onset of neurological phenotypes, such as clasping in
mice, asymmetric locomotor behavior in rats, and dyskinesia in macaques (DiFiglia et al., 2007;
Franich et al., 2008; Palfi et al., 2007). Conversely, the continuous restricted striatal accumulation
in HD103Q is not associated with striatal cell loss and is not sufficient to drive robust and
progressive behavioral pathology. One explanation for this distinction is that viral expression does
not lead to uniform transduction of striatal neurons, and aggregation extended in some cases to
cortical areas (Regulier et al., 2003). Viral transduction is in some cases toxic even with the
introduction of non-pathogenic vectors (Franich et al., 2008). Additionally, sudden introduction of
mutant htt during adulthood may overcome potential adaptive processes that might occur in striatal
neurons when the HD mutation is continuously present from birth (Ehrlich, 2012).

116
The influence of restricted striatal expression has been assessed previously using more
precise genetic manipulation of the BACHD, albeit in a full-length and not an exon-1 fragment
model. The BACHD’s full length 97Q mutant Htt gene contains a floxed portion which has
facilitated Cre-mediated conditional knockout of mHtt in particular brain areas. The Yang lab has
demonstrated using this strategy that striatal expression of mHtt in the absence of cortical
expression is insufficient to drive progressive motor failure and psychiatric-like behavior to the
same extent as BACHD with both striatal and cortical expression (Wang et al., 2014). Because the
HD gene is under control of an endogenous htt promoter, this strategy did not exclusively drive
expression in the striatum, but eliminated cortical expression. HD103Q provides a corresponding
perspective using a fragment model, in that continuous exon-1 mHtt aggregation purely in the
striatum is associated with either a mild and non-progressive motor phenotype, or an extremely
late-onset progression which our analyses between 3 and 11 months old did not capture.
That striatally restricted aggregation has a marginal behavioral correlate is reflective of a
growing literature that has moved away from defining HD as a disease primarily affecting the
striatum, and instead characterizes it as a whole-brain disorder (Rub et al., 2015; Wild et al., 2010).
Careful characterization of anatomical progression in HD and focused study of neuronal
subpopulations highlighted the early vulnerability of D2R-containing striatal MSNs (Reiner et al.,
1988; Vonsattel et al., 1985) and directed attention to the striatum. However, early anatomists
described widespread brain atrophy accompanying major damage to the caudate and putamen (Rub
et al., 2015), and despite early death of striatal neurons, post-mortem human tissue shows
widespread accumulation of aggregates throughout the cortex and striatum (DiFiglia et al., 1997).
Early examination of aggregates in human brain noted that the accumulation pattern, which was

117
heaviest in cortex, did not reflect striatal vulnerability and suggested that inputs from cortical cells
burdened with aggregates could drive toxicity in the striatum (Gutekunst et al., 1999).
More recently, large prodromal studies such as PREDICT-HD (2016) have emphasized
symptomology that may arise from the early involvement of cortex, such as cognitive impairment
and psychiatric manifestations, which may often be the earliest aspects of the disease recognized
by patients and their companions. The introduction of imaging techniques has allowed longitudinal
and behaviorally correlative studies in living humans that broaden our understanding of the
involvement of cerebellar, brainstem, and thalamic regions in HD (Rub et al., 2015). Despite the
expanding perspective on how HD affects the entire brain and body, there are biological and
technical obstacles preventing widespread drug delivery to the brain (Krol, 2012). Alongside many
historical and novel findings implicating different brain regions in aspects of HD, the observation
in HD103Q correlating localized striatal expression with a mild behavioral output may be
informative regarding decreased priority for localized drug delivery to the striatum.
Alfy knockout highlights an essential role for selective autophagy in nuclear mHtt turnover
Above all, HD103Q provided a useful model in which to directly observe and manipulate
aggregate clearance, a growing area of research and target of potential therapeutics. In vivo, the
evidence implicating macroautophagy in the processing of mHtt aggregates has taken several
forms: the colocalization of aggregates with components of the basal autophagic machinery or
with selective autophagy adaptors in mouse and human brain (Mori et al., 2012; Osawa et al.,
2011; Shibata et al., 2006), morphological changes in autophagosomes and lysosomes (Kegel et
al., 2000), and the ability of chemical autophagy induction to promote mHtt clearance (Ravikumar
et al., 2004; Tanaka et al., 2004; Williams et al., 2008). However, compounds like rapamycin

118
inhibitors have wide pleiotrophic effects, due to the mediation of a variety of signaling pathways
by mTOR (Jahrling and Laberge, 2015). Therefore, such experiments only indirectly implicate
macroautophagy or even degradation per se in clearance of aggregates. There is little direct in vivo
evidence to suggest that aberrant, biochemically stable deposits of mHtt require autophagic
processing by neuronal cells.
By developing an inducible Alfy knockout and combining it with regulatable HD gene
expression, we demonstrated that Alfy is required for removal of mHtt, highlighting a selective
turnover pathway in the mammalian brain. Although introducing dual precise genetic
manipulations required a complex breeding schema, to our knowledge this is the first direct in vivo
demonstration that abolishing a known selective autophagy pathway has an impact on the
clearance of an aggregated protein in neurons. Heterozygous disruption of beclin-1 results in the
accumulation of amyloid-beta in an APP-overexpressing mouse model of Alzheimer’s disease
(Pickford et al., 2008), but the beclin-1 complex has also been implicated in macroautophagy-
independent endolysosomal transport (Morris et al., 2015), so this may not reflect selective
turnover of aggregated structures.
Our experiment provided a correlate in the mammalian brain of work by Filimonenko et al
(2010), which demonstrated a requirement for Alfy in turnover of mHtt in cells by performing
Alfy knockdown in HeLa cells stably expressing tetracycline-regulatable 103Q-Htt. Neuronal-like
cell lines stably expressing a mutant htt fragment have also been used to demonstrate that
knockdown of the core autophagic machinery impairs htt aggregate removal (Iwata et al., 2005;
Yamamoto et al., 2006), but given the gradual nature of mHtt clearance in vivo (requiring 4
months), a corresponding experiment would likely be impossible, because inducible adult
knockout of Atg7 leads to death within 4-8 weeks (Karsli-Uzunbas et al., 2014). A recent study

119
used an inverse genetic strategy (requiring a similarly complex breeding schema) to implicate
autophagy in the clearance of accumulated structures related to desmin-related cardiomyelopathy,
by using a tetracycline-based strategy to inducibly overexpress Atg7 in heart cells, resulting in the
removal of the aggregates (Bhuiyan et al., 2013).
Other in vivo models have demonstrated that genetically interfering with nonselective and
selective autophagy has consequences with regards to aggregation, but with a focus on
accumulation rather than clearance. An iconic demonstration of this concept is the neuronal
knockout of Atg5 and Atg7 which results in the accumulation of ubiquitinated, p62-positive
structures and neurodegenerative phenotypes (Hara et al., 2006; Komatsu et al., 2006), which
showed that basal autophagy is essential in the mammalian nervous system. Pertaining specifically
to mHtt, homozygous constitutive knockout of p62 in R6/2 mice leads to increased accumulation
of cytoplasmic inclusions containing mHtt, which Kurosawa et al. attribute to impaired selective
autophagic turnover in the absence of p62 (Ichimura et al., 2008; Kurosawa et al., 2015). In the
absence of p62 the authors also observed a decrease in nuclear aggregates containing mHtt, which
implies either that p62 has opposing roles in the cytosolic and nuclear compartments of the cell,
or that accelerated cytosolic aggregate formation prevents entry of mHtt fragments into the
nucleus. The later would appear to conflict with cell-based work implicating p62 in aggregate
formation (Bjorkoy et al., 2005), and additionally opposes work from Doi et al (Doi et al., 2013)
showing that nuclear accumulation of androgen receptor is impaired by p62 knockout.
These discrepancies in p62-mediated autophagic turnover have yet to be resolved, but
Alfy’s proposed specificity for aggregates and its cooperation with p62 may shed some light on
the phenomenon of compartmentalized accumulation. In the case of Alfy knockout in reversible
HD103Q, we see turnover of existing accumulated nuclear mHtt despite the fact that autophagy

120
has historically been defined as a cytosolic degradation process. This raises a number of
possibilities regarding Alfy function and accumulation of mHtt in different cellular compartments.
One possibility is that in addition to its role in the recruitment of basal autophagic
components to aggregated ubiquitinated structures, Alfy is directly involved in the transport of
aggregated or disease-relevant misfolded proteins out of the nucleus. This is supported by the
presence of nuclear localization and nuclear export sequences within Alfy, as well as the
observation that it can shuttle from the perinuclear to cytoplasmic compartments and colocalize
with mHtt-positive structures (Filimonenko et al., 2010). Additionally, constitutive Alfy knockout
MEFs show the accumulation of ubiquitinated perinuclear structures (Ai Yamamoto, personal
communication). Alfy also colocalizes in the nucleus with expanded mutant Ataxin-1 as well as
with p62 in promyelocytic leukemia (PML) bodies, intranuclear sites where misfolded proteins
have been proposed to accumulate (Clausen et al., 2010; Isakson et al., 2010).
A second possibility is that impairing exclusively cytoplasmic turnover via Alfy excision
directly affects the dynamics of nucleo-cytoplasmic exchange of mHtt. Ongoing and successful
Alfy-mediated ubiquitinated protein turnover in the cytoplasm could potentially trigger
mechanisms that promote huntingtin exchange between the nucleus and the cytoplasm. Perhaps
this nucleo-cytoplasmic exchange of mHtt could rely on the interaction between Alfy and p62, a
possibility supported by the lack of nuclear mHtt accumulation in p62 null R6/2 and subsequent
alleviation of toxicity. In cells, the interaction between Alfy and p62 has been proposed to promote
both the formation and clearance of cytoplasmic aggregates, albeit using transient overexpression
techniques (Clausen et al., 2010). Additionally, the efficient autophagosomal targeting of K444-
acetylated mHtt in cells is dependent upon p62 (Jeong et al., 2009). This type of posttranslational
modification would not necessarily apply to exon-1-mHtt, put it is possible that a similar

121
mechanism exists to promote its localization to autophagosomes. If there is a role for selective
autophagy proteins in promoting transport of mHtt or other proteins within cells, this implies that
the building of a structured aggregate and assurance of its subsequent clearance are a prerequisite
to trigger the movement of potentially harmful misfolded substrates across cellular compartments.
This proposal is significantly complicated by the potential role for Htt itself as a scaffold for
selective macroautophagy (Ochaba et al., 2014; Rui et al., 2015) and a mediator of autophagosome
trafficking (Wong and Holzbaur, 2014).
A third alternative to Alfy’s involvement in nucleocytoplasmic shuttling of aggregation-
prone proteins could be a role in recruiting the core autophagic machinery for engulfment of
aggregates at the nuclear membrane. Components of the autophagy machinery have been detected
in the nucleus, and autophagy of the nuclear lamina was recently reported in mammalian cells
(Dou et al., 2015) which is reminiscent of piecemeal microautophagy of the nucleus in yeast (Krick
et al., 2008; Roberts et al., 2003), though in non-starvation conditions. It is conceivable that
sequestered nuclear aggregates might be disposed of in this way through an Alfy-dependent
mechanism.
One way to elucidate a potential role for Alfy in direct or indirect transport of disease-
relevant proteins might be to knock it out in regulatable models of exclusively nuclear versus
cytoplasmic accumulating proteins, such as mutant ataxin-1, or NLS-deficient versus nuclear-
directed polyQ-expanded proteins. If a purely nuclear protein still clears in the absence of Alfy it
would suggest that nucleo-cytoplasmic exchange is not necessary for its removal, implicating a
non-autophagic turnover method. Inversely, mutating Alfy’s putative NES or NLS locations in a
clearance model or disrupting its interactions with mHtt, p62, or other aggregating proteins could
provide similar resolution.

122
Alfy as a tool to understand the behavioral impact of aggregation
Knockout of Alfy in HD103Q suggested that Alfy is essential for eliminating accumulated
mutant exon-1-Htt, but the mild phenotype of HD103Q and profound behavioral effects of Alfy
inducible knockout precluded addressing the role of impeding aggregate clearance in behavioral
symptomology. We thus sought a model with greater behavioral validity and in which aggregate
accumulation was a slower phenomenon, in order to study the effects of impeded mHtt turnover
via depletion of Alfy. The most widely used transgenic models expressing full-length human mHtt
are the BACHD and the YAC128, which both show progressive striatal and cortical atrophy as
well as psychiatric and motor phenotypes (Gray et al., 2008; Hodgson et al., 1999). Of these, the
BACHD has a mutation of comparable CAG repeat length (97) to HD103Q, also encoded by an
alternating CAG-CAA sequence. The BACHD also shows comparatively sparse staining with
S830 at 12 months of age (Pouladi et al., 2012).
50% depletion of Alfy in BACHD, achieved via a cross with Alfy heterozygotes, was
designed to understand whether Alfy was a potential modifier of HD, and led us to two main
observations. First, we provide in vivo confirmation of Alfy’s reported selectivity for detergent-
insoluble disease-relevant proteins, implying that the depletion of Alfy either results in increased
formation of aggregates or decreases their clearance. The latter is more likely given our findings
in HD103Q that directly implicate Alfy in turnover of existing structures in vivo, a conclusion that
also aligns with previous work from our laboratory using cell-based assays to implicate Alfy in
the clearance of pre-formed structures rather than preventing aggregation (Eenjes et al., 2016;
Filimonenko et al., 2010).
Second, and more importantly, our study highlights the behavioral significance of Alfy-
mediated turnover of insoluble mHtt. Whereas Alfy depletion alone had no effect on behavioral

123
phenotypes over time, Alfy depletion in BACHD caused an apparent acceleration of the HD-like
motor decline. Previous studies have correlated the disappearance of aggregates with the
improvement of phenotypes (Ravikumar et al., 2002), or have conversely linked the prevention of
aggregation with increased toxicity (Arrasate et al., 2004). In light of Alfy’s in vivo role in
clearance, highlighted by HD103Q, our Alfy depletion study in BACHD strongly suggests that
aggregate turnover contributes to the prevention of mHtt toxicity.
Whether this confirms the toxicity of aggregates in HD is dependent upon how aggregation
is defined. Research pursuing an understanding of the meaning of aggregates has generally made
distinctions that are biochemical in nature, based on solubility in various detergents; or visual,
using epitope-specific antibodies that reveal anatomical and cellular location of the aberrant
structures. By employing Alfy as a tool to manipulate a selective turnover pathway, we have
provided both biochemical and visual epitope-based perspectives suggesting that preventing the
removal of existing mHtt aggregates has pathological consequences. Alfy depletion affected the
accumulation of punctate nuclear, perinuclear, and cytoplasmic structures in the BACHD, leading
to behavioral decline. Complete knockout also affected the clearance of diffuse nuclear mHtt in
HD103Q, although one weakness in drawing a comparison between models is that we were unable
to make a biochemical distinction between soluble and insoluble protein in HD103Q due to the
technical difficulty of detecting mHtt in brain lysates. Nevertheless, manipulating Alfy affects the
turnover of an aggregation-prone mutant exon-1-Htt fragment which is a shared feature across a
majority of mouse models of HD, in spite of differences in genetic construction. This ultimately
had a pathological impact, implicating aggregation as a disease driver.
The negative impact of impeding mHtt turnover strongly suggests that promoting turnover
in a selective manner may have positive effects, a result that may be achieved experimentally via

124
Alfy overexpression or otherwise enhancing its effects in vivo. This has been suggested via
overexpression of the C-terminus in polyQ-expanded fly eye and in HD primary neurons as well
as cellular ALS models involving the accumulation of SOD1 (Filimonenko et al., 2010; Han et al.,
2015). Recently, confirmation of Alfy’s potential role in human HD has arisen through
collaborative efforts coordinated by the Hereditary Disease Foundation. Their study arose from
the observation that although CAG repeat length is inversely proportionate to age of onset, this
relationship is driven by extremely low or extremely high repeat lengths. At the more common
pathological repeat lengths, this relationship no longer holds (Andresen et al., 2007; Wexler et al.,
2004). The study further demonstrates that there is room for environmental and genetic modifiers
that might account for the difference between actual age of disease onset and predicted (by CAG
length) age of onset, known as the residual age of onset.
Using samples from the multi-generational Venezuelan cohort that led to the landmark
study identifying the genetic mutation that causes HD (Huntington’s Disease Collaborative
Research Group, 1993), the goal of recent work was to perform whole-genome sequencing in
combination with genetic linkage analysis to identify potential individual SNPs that influence the
residual age of onset. A variant in Alfy encoded by an isoleucine to valene replacement at amino
acid 3032, conferred upon the individuals with this mutation an average of 10 years later onset of
symptoms (Nancy Wexler and David Housman, personal communication).
Future efforts will focus on elucidating the functional impact of this mutation within Alfy
to understand how it impacts age of onset. One possibility proposed by the researchers heading the
sequencing analysis is that the mutation is located in an enhancer sequence subject to chromatin
looping, which may have an impact on expression levels of Alfy (David Housman, personal
communication). Cell-based studies suggest that Alfy protein levels remain stable despite the

125
continuous expression of mHtt (Filimonenko et al., 2010), but a study of protein expression levels
in HD brain suggested that there is a slight but significant decrease in Alfy mRNA (Hodges et al.,
2006). However, Alfy’s large size, in addition to the stability of protein levels that we observed up
to a week after Cre-mediated genetic elimination, suggest that expression levels are unlikely to be
regulated drastically in response to a need for enhanced protein clearance, such as may be the case
in HD.
Alternatively, the cellular localization of Alfy or the availability of key C-terminal domains
may be affected by the I3032V mutation, which resides within Alfy’s BEACH domain (Simonsen
et al., 2004). This has the potential to change its conformation or enhance its availability for p62
binding with the effect of promoting aggregate clearance. Cell-based studies could be designed to
understand how this mutation affects binding with known Alfy interactors, and to understand how
this mutation affects the clearance of mutant huntingtin. Genome editing strategies such as
CRISPR-Cas9 (Yang et al., 2016) could be used to create this Alfy variant in mouse models and
determine its impact on mHtt turnover and behavior. Just as HD gene silencing, a present clinical
reality, was a veritable piece of science fiction just a decade ago, it is possible to imagine a future
of therapy that includes targeted genetic manipulations to introduce the widespread and selective
mutation of single nucleotides.
Age-sensitive dependence on Alfy for neurological health
Whereas constitutive 50% depletion of Alfy had no motor behavioral phenotype, inducible
Alfy knockout at three adult ages in otherwise wild-type animals led to a rapid and profound motor
phenotype that exceeded that of any of the HD models our lab has worked with directly. Whereas
the oldest knockouts exhibited the most profound phenotypes, with visible perturbation of home

126
cage behavior and obvious gait changes within 2 months of knockout, the distinction between
Alfy-3-iKO and Alfy-7-iKO was particularly surprising to us, as 3 and 7 month old mice are
behaviorally indistinguishable and still of reproductive age. In Alfy-11-iKO, we had more limited
subjects and recently terminated the study at 16 months old, but existing Alfy-3-iKO and Alfy-7-
iKO mice have to date survived up to a year after Alfy knockout. I predict that knockout will result
in premature death compared to WT mice, as the motor impairment is severely progressive and
may begin to inhibit normal activity.
Thionin staining in Alfy iKO brain preliminarily identified a direct impact of knockout on
cerebellar integrity, and we have planned a more thorough examination of neuropathology to
include cerebellar calbindin staining, brain-wide ubiquitin accumulation, and GFAP to examine
astrocytosis. The cerebellar degeneration we observe is reminiscent of models of inherited
spinocerebellar ataxia (Cendelin, 2014) as well as autophagy knockouts which show rapid
cerebellar degeneration (Mizushima and Levine, 2010), highlighting cerebellar vulnerability to
protein accumulation, stress, and energy dysregulation.
The observed age-sensitive dependence on Alfy for complex motor behavior is suggestive
of Alfy’s involvement in a homeostatic process which becomes imbalanced during aging. Alfy’s
involvement in the turnover of a variety of aggregated proteins (Filimonenko et al., 2010) suggests
that it may serve to counter a rise in protein misfolding during normal aging (Labbadia and
Morimoto, 2015). In support of this hypothesis, we observed an increase in levels of p62 in
detergent-insoluble cortical lysates from Alfy-7-iKO but not in Alfy-3-iKO. p62 has been
implicated in aggregate formation (Bjorkoy) and selective autophagy pathways (Johansen and
Lamark, 2011), is found in aggregates containing a variety of disease relevant-proteins in human
tissue (Bitto et al., 2014), and has been used as a marker of aggregate formation in insoluble

127
detergent fractions alongside ubiquitinated protein accumulation associated with aging (Bartlett et
al., 2011). Increased p62 has also been observed in models of autophagy knockout, and is present
in inclusions of mice with deficient neuronal autophagy due to Nestin-Cre mediated excision of
core autophagy genes like Atg7, Atg5, and FIP200 (Hara et al., 2006; Komatsu et al., 2006; Liang
et al., 2010). In the absence of Alfy, p62 has been observed to accumulate in cellular models and
in the brains of flies null for bchs, the drosophila homolog of Alfy (Clausen et al., 2010;
Filimonenko et al., 2010).
If Alfy plays a role in the turnover of aggregated proteins in the absence of a stressor like
a transgenic mutant protein, then further examination of markers of aggregated protein
accumulation, such as ubiquitin or alpha-synuclein, may also reveal increased accumulation in its
absence. Alfy has in fact been implicated in the removal of other ubiquitinated structures and large
protein complexes, including midbody rings following cytokinesis (Isakson et al., 2013b) and the
HIV viral capsid protein p24 (Mandell et al., 2014). The presence of p62 and other aggregation
indicators in both Alfy iKO and HD-AlfyiKO may be suggestive of an essential role for protein
turnover at the intersection between neurodegeneration and aging. Rather than impair the
autophagic, proteasomal, and chaperone machinery directly as many have hypothesized (Labbadia
and Morimoto, 2015), it is conceivable that aggregated protein accumulation, observed even in
normal aging (Bartlett et al., 2011), is simply the consequence of increased misfolding events.
These events increase in probability over time, causing accumulation in a system that is still
functional but becomes taxed beyond its capacity. This occurs in aging but in an even more
accelerated manner in the presence of a mutant protein. Removing Alfy, a key protein in
combatting accumulated structures, is thus behaviorally catastrophic during aging, and 50%
depletion has a similar effect in disease, though not in healthy conditions.

128
An alternative explanation for age-sensitive dependence on Alfy could be its involvement
in the combat of oxidative stress. C elegans studies have implicated the accumulation of reactive
oxygen species in the aging process (Olsen et al., 2006), and Alfy’s association with mitochondrial
proteins in mice and in cell lines (Anne Simonsen and Ai Yamamoto, personal communication)
could be indicative of its role in mitochondrial turnover or maintenance. We chose to examine
levels of NIPSNAP1 in detergent-insoluble cortical lysates from our Alfy iKO mice because of
NIPSNAP1’s direct interaction with Alfy, its high expression in brain tissue, localization to the
mitochondrial inner membrane (Nautiyal et al., 2010), and its presence in autophagosomes
(Behrends et al., 2010). We were surprised to uncover a nearly 50% increase in NIPSNAP1 in the
absence of Alfy, implying that Alfy may be involved in the turnover of mitochondria or
mitochondrial components. Alternatively, its absence could trigger the increased synthesis of
NIPSNAP1, whose function is poorly understood. Reduced NIPSNAP mRNA was reported in the
brain of a mouse model of phenylketonuria (PKU) (Surendran et al., 2005), a recessive genetic
disorder involving inadequate processing of branched-chain amino acids. The disorder results in
accumulated phenylalanine in the brain of infants and leads to neurophysiological abnormalities
and impaired cognitive development (Al Hafid and Christodoulou, 2015). NIPSNAP can bind
components of the machinery that performs branched-chain amino acid metabolism and has been
proposed to anchor these complexes to the mitochondrial inner membrane.
NIPSNAP upregulation in Alfy iKO is clearly insufficient to determine Alfy’s relationship
to mitochondria. However, in light of NIPSNAP’s interface with enzymatic complexes that
function in the Krebs cycle, the interaction between Alfy and NIPSNAP is suggestive of a
relationship to mitochondrial health and energetic output. The impact of Alfy knockout on
mitochondrial proteins and oxidative stress could be examined by measuring levels of additional

129
mitochondrial proteins or assaying mitochondrial respiration through the use of Seahorse analysis.
We have created inducible Alfy knockout MEFs that could be used in culture to track
mitochondrial levels, movement, and potential colocalization with Alfy.
Conclusions
Although biochemical and tissue-based data from the Alfy iKO aging study is currently
limited, the information garnered from HD103Q, BACHD, and Alfy iKO all point to an essential
role for Alfy in the removal of aggregated protein complexes and reveal that highly selective
turnover pathways operate constitutively in neurons to balance protein homeostasis. Genetic
manipulations of Alfy result in persistent aggregates, protein accumulation, and disastrous
behavioral consequences, suggesting that selective turnover pathways have evolved in the
mammalian nervous system to protect neurological health.
The behavioral impact of Alfy deletion alone suggests that Alfy’s 400 kd reach may extend
far beyond its role in the turnover of mutant huntingtin. In addition to an essential role in
development of the mammalian nervous system, an emerging literature has implicated Alfy in the
turnover of various disease-relevant structures and aggregation-prone proteins, including Dvl3
complex aggregates implicated in developmental signaling pathways (Kadir et al., 2016) and
mutant SOD1 in ALS models (Han et al., 2015). Constitutive knockout of Alfy results in perinatal
lethality in conjunction with developmental wiring defects throughout the CNS, as well as
microcephalic phenotypes. (Orosco et al., 2014). Furthermore, large scale genetic screens have
revealed that Alfy mutations in humans introduce increased risk for intellectual disabilities and
psychiatric disorders (Bonnet et al., 2010; Iossifov et al., 2012). Much remains to be understood

130
about this large and elusive protein whose roles seem to span an immense spectrum of homeostatic
processes in development, aging, and disease.
In summary, our work has both expanded upon our understanding of Alfy’s role in vivo in
adult brain and employed its specificity as a precise genetic tool to further elucidate the toxicity of
aggregation in HD and aging. Further implementation of Alfy in this manner may help to elucidate
how different types of disease-relevant aggregates contribute to pathogenesis. Finally, Alfy’s
potential to modify disease onset in humans has reinforced its position as an adaptor involved in
countering the convergent neurological decline that characterizes aging and disease.

131
References Aggarwal, M., Duan, W., Hou, Z., Rakesh, N., Peng, Q., Ross, C.A., Miller, M.I., Mori, S., and Zhang, J. (2012). Spatiotemporal mapping of brain atrophy in mouse models of Huntington's disease using longitudinal in vivo magnetic resonance imaging. Neuroimage 60, 2086-2095. Agholme, L., Hallbeck, M., Benedikz, E., Marcusson, J., and Kagedal, K. (2012). Amyloid-beta secretion, generation, and lysosomal sequestration in response to proteasome inhibition: involvement of autophagy. J Alzheimers Dis 31, 343-358. Aguib, Y., Heiseke, A., Gilch, S., Riemer, C., Baier, M., Schatzl, H.M., and Ertmer, A. (2009). Autophagy induction by trehalose counteracts cellular prion infection. Autophagy 5, 361-369. Aguzzi, A., Baumann, F., and Bremer, J. (2008). The prion's elusive reason for being. Annu Rev Neurosci 31, 439-477. Al Hafid, N., and Christodoulou, J. (2015). Phenylketonuria: a review of current and future treatments. Transl Pediatr 4, 304-317. Albin, R.L., Reiner, A., Anderson, K.D., Dure, L.S.t., Handelin, B., Balfour, R., Whetsell, W.O., Jr., Penney, J.B., and Young, A.B. (1992). Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington's disease. Ann Neurol 31, 425-430. Alvarez-Erviti, L., Rodriguez-Oroz, M.C., Cooper, J.M., Caballero, C., Ferrer, I., Obeso, J.A., and Schapira, A.H. (2010). Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol 67, 1464-1472. Andresen, J.M., Gayan, J., Djousse, L., Roberts, S., Brocklebank, D., Cherny, S.S., Group, U.S.-V.C.R., Group, H.M.C.R., Cardon, L.R., Gusella, J.F., et al. (2007). The relationship between CAG repeat length and age of onset differs for Huntington's disease patients with juvenile onset or adult onset. Ann Hum Genet 71, 295-301. Aronin, N., and DiFiglia, M. (2014). Huntingtin-lowering strategies in Huntington's disease: antisense oligonucleotides, small RNAs, and gene editing. Mov Disord 29, 1455-1461. Aronin, N., Kim, M., Laforet, G., and DiFiglia, M. (1999). Are there multiple pathways in the pathogenesis of Huntington's disease? Philos Trans R Soc Lond B Biol Sci 354, 995-1003. Arrasate, M., and Finkbeiner, S. (2012). Protein aggregates in Huntington's disease. Exp Neurol 238, 1-11. Arrasate, M., Mitra, S., Schweitzer, E.S., Segal, M.R., and Finkbeiner, S. (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805-810.

132
Aylward, E.H., Nopoulos, P.C., Ross, C.A., Langbehn, D.R., Pierson, R.K., Mills, J.A., Johnson, H.J., Magnotta, V.A., Juhl, A.R., Paulsen, J.S., et al. (2011). Longitudinal change in regional brain volumes in prodromal Huntington disease. J Neurol Neurosurg Psychiatry 82, 405-410. Barmada, S.J., Skibinski, G., Korb, E., Rao, E.J., Wu, J.Y., and Finkbeiner, S. (2010). Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci 30, 639-649. Baron, U., Freundlieb, S., Gossen, M., and Bujard, H. (1995). Co-regulation of two gene activities by tetracycline via a bidirectional promoter. Nucleic Acids Res 23, 3605-3606. Bartlett, B.J., Isakson, P., Lewerenz, J., Sanchez, H., Kotzebue, R.W., Cumming, R.C., Harris, G.L., Nezis, I.P., Schubert, D.R., Simonsen, A., et al. (2011). p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy 7, 572-583. Bates, G. (2003). Huntingtin aggregation and toxicity in Huntington's disease. Lancet 361, 1642-1644. Bayram-Weston, Z., Jones, L., Dunnett, S.B., and Brooks, S.P. (2012). Light and electron microscopic characterization of the evolution of cellular pathology in the R6/1 Huntington's disease transgenic mice. Brain Res Bull 88, 104-112. Bayram-Weston, Z., Jones, L., Dunnett, S.B., and Brooks, S.P. (2016). Comparison of mHTT Antibodies in Huntington's Disease Mouse Models Reveal Specific Binding Profiles and Steady-State Ubiquitin Levels with Disease Development. PLoS One 11, e0155834. Becher, M.W., Kotzuk, J.A., Sharp, A.H., Davies, S.W., Bates, G.P., Price, D.L., and Ross, C.A. (1998). Intranuclear neuronal inclusions in Huntington's disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis 4, 387-397. Bednarski, E., and Lynch, G. (1996). Cytosolic proteolysis of tau by cathepsin D in hippocampus following suppression of cathepsins B and L. J Neurochem 67, 1846-1855. Behrends, C., Sowa, M.E., Gygi, S.P., and Harper, J.W. (2010). Network organization of the human autophagy system. Nature 466, 68-76. Benn, C.L., Landles, C., Li, H., Strand, A.D., Woodman, B., Sathasivam, K., Li, S.H., Ghazi-Noori, S., Hockly, E., Faruque, S.M., et al. (2005). Contribution of nuclear and extranuclear polyQ to neurological phenotypes in mouse models of Huntington's disease. Hum Mol Genet 14, 3065-3078. Bhuiyan, M.S., Pattison, J.S., Osinska, H., James, J., Gulick, J., McLendon, P.M., Hill, J.A., Sadoshima, J., and Robbins, J. (2013). Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest 123, 5284-5297.

133
Billings, L.M., Oddo, S., Green, K.N., McGaugh, J.L., and LaFerla, F.M. (2005). Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron 45, 675-688. Bitto, A., Lerner, C.A., Nacarelli, T., Crowe, E., Torres, C., and Sell, C. (2014). P62/SQSTM1 at the interface of aging, autophagy, and disease. Age (Dordr) 36, 9626. Bjorkoy, G., Lamark, T., Brech, A., Outzen, H., Perander, M., Overvatn, A., Stenmark, H., and Johansen, T. (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171, 603-614. Boellaard, J.W., Kao, M., Schlote, W., and Diringer, H. (1991). Neuronal autophagy in experimental scrapie. Acta Neuropathol 82, 225-228. Bonnet, C., Andrieux, J., Beri-Dexheimer, M., Leheup, B., Boute, O., Manouvrier, S., Delobel, B., Copin, H., Receveur, A., Mathieu, M., et al. (2010). Microdeletion at chromosome 4q21 defines a new emerging syndrome with marked growth restriction, mental retardation and absent or severely delayed speech. J Med Genet 47, 377-384. Bose, J.K., Huang, C.C., and Shen, C.K. (2011). Regulation of autophagy by neuropathological protein TDP-43. J Biol Chem 286, 44441-44448. Brady, O.A., Meng, P., Zheng, Y., Mao, Y., and Hu, F. (2011). Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J Neurochem 116, 248-259. Caccamo, A., Magri, A., Medina, D.X., Wisely, E.V., Lopez-Aranda, M.F., Silva, A.J., and Oddo, S. (2013). mTOR regulates tau phosphorylation and degradation: implications for Alzheimer's disease and other tauopathies. Aging Cell 12, 370-380. Cai, J., Chen, Y., Cai, W.H., Hurlock, E.C., Wu, H., Kernie, S.G., Parada, L.F., and Lu, Q.R. (2007). A crucial role for Olig2 in white matter astrocyte development. Development 134, 1887-1899. Carra, S., Rusmini, P., Crippa, V., Giorgetti, E., Boncoraglio, A., Cristofani, R., Naujock, M., Meister, M., Minoia, M., Kampinga, H.H., et al. (2013). Different anti-aggregation and pro-degradative functions of the members of the mammalian sHSP family in neurological disorders. Philos Trans R Soc Lond B Biol Sci 368, 20110409. Castillo, K., Valenzuela, V., Matus, S., Nassif, M., Onate, M., Fuentealba, Y., Encina, G., Irrazabal, T., Parsons, G., Court, F.A., et al. (2013). Measurement of autophagy flux in the nervous system in vivo. Cell Death Dis 4, e917. Cendelin, J. (2014). From mice to men: lessons from mutant ataxic mice. Cerebellum Ataxias 1, 4.

134
Cepeda, C., Cummings, D.M., Andre, V.M., Holley, S.M., and Levine, M.S. (2010). Genetic mouse models of Huntington's disease: focus on electrophysiological mechanisms. ASN Neuro 2, e00033. Chen, S., Berthelier, V., Yang, W., and Wetzel, R. (2001). Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol 311, 173-182. Chen, S., Ferrone, F.A., and Wetzel, R. (2002). Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation. Proc Natl Acad Sci U S A 99, 11884-11889. Chen, S., Zhang, X., Song, L., and Le, W. (2012). Autophagy dysregulation in amyotrophic lateral sclerosis. Brain Pathol 22, 110-116. Cheng, Y., Peng, Q., Hou, Z., Aggarwal, M., Zhang, J., Mori, S., Ross, C.A., and Duan, W. (2011). Structural MRI detects progressive regional brain atrophy and neuroprotective effects in N171-82Q Huntington's disease mouse model. Neuroimage 56, 1027-1034. Chesser, A.S., Pritchard, S.M., and Johnson, G.V. (2013). Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front Neurol 4, 122. Clausen, T.H., Lamark, T., Isakson, P., Finley, K., Larsen, K.B., Brech, A., Overvatn, A., Stenmark, H., Bjorkoy, G., Simonsen, A., et al. (2010). p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 6, 330-344. Cortes, C.J., Qin, K., Cook, J., Solanki, A., and Mastrianni, J.A. (2012). Rapamycin delays disease onset and prevents PrP plaque deposition in a mouse model of Gerstmann-Straussler-Scheinker disease. J Neurosci 32, 12396-12405. Cuervo, A.M., Stefanis, L., Fredenburg, R., Lansbury, P.T., and Sulzer, D. (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292-1295. Cummings, D.M., Alaghband, Y., Hickey, M.A., Joshi, P.R., Hong, S.C., Zhu, C., Ando, T.K., Andre, V.M., Cepeda, C., Watson, J.B., et al. (2012). A critical window of CAG repeat-length correlates with phenotype severity in the R6/2 mouse model of Huntington's disease. J Neurophysiol 107, 677-691. Davies, S.W., Turmaine, M., Cozens, B.A., DiFiglia, M., Sharp, A.H., Ross, C.A., Scherzinger, E., Wanker, E.E., Mangiarini, L., and Bates, G.P. (1997). Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537-548. Dayalu, P., and Albin, R.L. (2015). Huntington disease: pathogenesis and treatment. Neurol Clin 33, 101-114.

135
Deng, H.X., Chen, W., Hong, S.T., Boycott, K.M., Gorrie, G.H., Siddique, N., Yang, Y., Fecto, F., Shi, Y., Zhai, H., et al. (2011). Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477, 211-215. Deng, Y.P., Albin, R.L., Penney, J.B., Young, A.B., Anderson, K.D., and Reiner, A. (2004). Differential loss of striatal projection systems in Huntington's disease: a quantitative immunohistochemical study. J Chem Neuroanat 27, 143-164. Dennissen, F.J., Kholod, N., and van Leeuwen, F.W. (2012). The ubiquitin proteasome system in neurodegenerative diseases: culprit, accomplice or victim? Prog Neurobiol 96, 190-207. DiFiglia, M. (1990). Excitotoxic injury of the neostriatum: a model for Huntington's disease. Trends Neurosci 13, 286-289. DiFiglia, M., Sapp, E., Chase, K.O., Davies, S.W., Bates, G.P., Vonsattel, J.P., and Aronin, N. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990-1993. DiFiglia, M., Sena-Esteves, M., Chase, K., Sapp, E., Pfister, E., Sass, M., Yoder, J., Reeves, P., Pandey, R.K., Rajeev, K.G., et al. (2007). Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc Natl Acad Sci U S A 104, 17204-17209. Ding, W.X., and Yin, X.M. (2012). Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 393, 547-564. Djousse, L., Knowlton, B., Cupples, L.A., Marder, K., Shoulson, I., and Myers, R.H. (2002). Weight loss in early stage of Huntington's disease. Neurology 59, 1325-1330. Doi, H., Adachi, H., Katsuno, M., Minamiyama, M., Matsumoto, S., Kondo, N., Miyazaki, Y., Iida, M., Tohnai, G., Qiang, Q., et al. (2013). p62/SQSTM1 differentially removes the toxic mutant androgen receptor via autophagy and inclusion formation in a spinal and bulbar muscular atrophy mouse model. J Neurosci 33, 7710-7727. Dou, Z., Xu, C., Donahue, G., Shimi, T., Pan, J.A., Zhu, J., Ivanov, A., Capell, B.C., Drake, A.M., Shah, P.P., et al. (2015). Autophagy mediates degradation of nuclear lamina. Nature 527, 105-109. Dragatsis, I., Goldowitz, D., Del Mar, N., Deng, Y.P., Meade, C.A., Liu, L., Sun, Z., Dietrich, P., Yue, J., and Reiner, A. (2009). CAG repeat lengths > or =335 attenuate the phenotype in the R6/2 Huntington's disease transgenic mouse. Neurobiol Dis 33, 315-330. Dragich, J.M., Kuwajima, T., Hirose-Ikeda, M., Yoon, M.S., Eenjes, E., Bosco, J.R., Fox, L.M., Lystad, A.H., Oo, T.F., Yarygina, O., et al. (2016). Autophagy linked FYVE (Alfy/WDFY3) is required for establishing neuronal connectivity in the mammalian brain. Elife 5.

136
Eenjes, E., Dragich, J.M., Kampinga, H.H., and Yamamoto, A. (2016). Distinguishing aggregate formation and aggregate clearance using cell-based assays. J Cell Sci 129, 1260-1270. Ehrlich, M.E. (2012). Huntington's disease and the striatal medium spiny neuron: cell-autonomous and non-cell-autonomous mechanisms of disease. Neurotherapeutics 9, 270-284. Ehrnhoefer, D.E., Butland, S.L., Pouladi, M.A., and Hayden, M.R. (2009). Mouse models of Huntington disease: variations on a theme. Dis Model Mech 2, 123-129. Fecto, F., and Siddique, T. (2011). Making connections: pathology and genetics link amyotrophic lateral sclerosis with frontotemporal lobe dementia. J Mol Neurosci 45, 663-675. Ferrante, R.J., Beal, M.F., Kowall, N.W., Richardson, E.P., Jr., and Martin, J.B. (1987). Sparing of acetylcholinesterase-containing striatal neurons in Huntington's disease. Brain Res 411, 162-166. Filimonenko, M., Isakson, P., Finley, K.D., Anderson, M., Jeong, H., Melia, T.J., Bartlett, B.J., Myers, K.M., Birkeland, H.C., Lamark, T., et al. (2010). The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell 38, 265-279. Fimia, G.M., Kroemer, G., and Piacentini, M. (2013). Molecular mechanisms of selective autophagy. Cell Death Differ 20, 1-2. Finley, K.D., Edeen, P.T., Cumming, R.C., Mardahl-Dumesnil, M.D., Taylor, B.J., Rodriguez, M.H., Hwang, C.E., Benedetti, M., and McKeown, M. (2003). blue cheese mutations define a novel, conserved gene involved in progressive neural degeneration. J Neurosci 23, 1254-1264. Fox, L.M., Yamamoto, A. (2014). Macroautophagy of aggregation-prone proteins in neurodegenerative disease. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging, M. Hayat, ed. (Elsevier), pp. 117-137. Frake, R.A., Ricketts, T., Menzies, F.M., and Rubinsztein, D.C. (2015). Autophagy and neurodegeneration. J Clin Invest 125, 65-74. Franich, N.R., Fitzsimons, H.L., Fong, D.M., Klugmann, M., During, M.J., and Young, D. (2008). AAV vector-mediated RNAi of mutant huntingtin expression is neuroprotective in a novel genetic rat model of Huntington's disease. Mol Ther 16, 947-956. Friedman, L.G., Lachenmayer, M.L., Wang, J., He, L., Poulose, S.M., Komatsu, M., Holstein, G.R., and Yue, Z. (2012). Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of alpha-synuclein and LRRK2 in the brain. J Neurosci 32, 7585-7593. Funderburk, S.F., Marcellino, B.K., and Yue, Z. (2010). Cell "self-eating" (autophagy) mechanism in Alzheimer's disease. Mt Sinai J Med 77, 59-68.

137
Gafni, J., Hermel, E., Young, J.E., Wellington, C.L., Hayden, M.R., and Ellerby, L.M. (2004). Inhibition of calpain cleavage of huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J Biol Chem 279, 20211-20220. Genetic Modifiers of Huntington's Disease, C. (2015). Identification of Genetic Factors that Modify Clinical Onset of Huntington's Disease. Cell 162, 516-526. Geser, F., Lee, V.M., and Trojanowski, J.Q. (2010). Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: a spectrum of TDP-43 proteinopathies. Neuropathology 30, 103-112. Gestwicki, J.E., and Garza, D. (2012). Protein quality control in neurodegenerative disease. Prog Mol Biol Transl Sci 107, 327-353. Gidalevitz, T., Ben-Zvi, A., Ho, K.H., Brignull, H.R., and Morimoto, R.I. (2006). Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 311, 1471-1474. Gossen, M., and Bujard, H. (1992). Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A 89, 5547-5551. Graham, R.K., Deng, Y., Slow, E.J., Haigh, B., Bissada, N., Lu, G., Pearson, J., Shehadeh, J., Bertram, L., Murphy, Z., et al. (2006a). Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 125, 1179-1191. Graham, R.K., Slow, E.J., Deng, Y., Bissada, N., Lu, G., Pearson, J., Shehadeh, J., Leavitt, B.R., Raymond, L.A., and Hayden, M.R. (2006b). Levels of mutant huntingtin influence the phenotypic severity of Huntington disease in YAC128 mouse models. Neurobiol Dis 21, 444-455. Gray, M., Shirasaki, D.I., Cepeda, C., Andre, V.M., Wilburn, B., Lu, X.H., Tao, J., Yamazaki, I., Li, S.H., Sun, Y.E., et al. (2008). Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci 28, 6182-6195. Gu, X., Andre, V.M., Cepeda, C., Li, S.H., Li, X.J., Levine, M.S., and Yang, X.W. (2007). Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington's disease. Mol Neurodegener 2, 8. Gu, X., Cantle, J.P., Greiner, E.R., Lee, C.Y., Barth, A.M., Gao, F., Park, C.S., Zhang, Z., Sandoval-Miller, S., Zhang, R.L., et al. (2015). N17 Modifies mutant Huntingtin nuclear pathogenesis and severity of disease in HD BAC transgenic mice. Neuron 85, 726-741. Guo, C., Yang, W., and Lobe, C.G. (2002). A Cre recombinase transgene with mosaic, widespread tamoxifen-inducible action. Genesis 32, 8-18.

138
Gutekunst, C.A., Li, S.H., Yi, H., Mulroy, J.S., Kuemmerle, S., Jones, R., Rye, D., Ferrante, R.J., Hersch, S.M., and Li, X.J. (1999). Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci 19, 2522-2534. Guy, J., Gan, J., Selfridge, J., Cobb, S., and Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143-1147. Guyenet, S.J., Furrer, S.A., Damian, V.M., Baughan, T.D., La Spada, A.R., and Garden, G.A. (2010). A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp. Ha, A.D., and Jankovic, J. (2011). Exploring the correlates of intermediate CAG repeats in Huntington disease. Postgrad Med 123, 116-121. Hackam, A.S., Singaraja, R., Wellington, C.L., Metzler, M., McCutcheon, K., Zhang, T., Kalchman, M., and Hayden, M.R. (1998). The influence of huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol 141, 1097-1105. Han, H., Wei, W., Duan, W., Guo, Y., Li, Y., Wang, J., Bi, Y., and Li, C. (2015). Autophagy-linked FYVE protein (Alfy) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). In Vitro Cell Dev Biol Anim 51, 249-263. Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., Yokoyama, M., Mishima, K., Saito, I., Okano, H., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885-889. Harper, S.Q., Staber, P.D., He, X., Eliason, S.L., Martins, I.H., Mao, Q., Yang, L., Kotin, R.M., Paulson, H.L., and Davidson, B.L. (2005). RNA interference improves motor and neuropathological abnormalities in a Huntington's disease mouse model. Proc Natl Acad Sci U S A 102, 5820-5825. Hayashi, S., and McMahon, A.P. (2002). Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 244, 305-318. Heiseke, A., Aguib, Y., and Schatzl, H.M. (2010). Autophagy, prion infection and their mutual interactions. Curr Issues Mol Biol 12, 87-97. Heitz, S., Grant, N.J., Leschiera, R., Haeberle, A.M., Demais, V., Bombarde, G., and Bailly, Y. (2010). Autophagy and cell death of Purkinje cells overexpressing Doppel in Ngsk Prnp-deficient mice. Brain Pathol 20, 119-132. Hiltunen, M., van Groen, T., and Jolkkonen, J. (2009). Functional roles of amyloid-beta protein precursor and amyloid-beta peptides: evidence from experimental studies. J Alzheimers Dis 18, 401-412.

139
Hochfeld, W.E., Lee, S., and Rubinsztein, D.C. (2013). Therapeutic induction of autophagy to modulate neurodegenerative disease progression. Acta Pharmacol Sin 34, 600-604. Hocking, L.J., Mellis, D.J., McCabe, P.S., Helfrich, M.H., and Rogers, M.J. (2010). Functional interaction between sequestosome-1/p62 and autophagy-linked FYVE-containing protein WDFY3 in human osteoclasts. Biochem Biophys Res Commun 402, 543-548. Hodges, A., Strand, A.D., Aragaki, A.K., Kuhn, A., Sengstag, T., Hughes, G., Elliston, L.A., Hartog, C., Goldstein, D.R., Thu, D., et al. (2006). Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet 15, 965-977. Hodgson, J.G., Agopyan, N., Gutekunst, C.A., Leavitt, B.R., LePiane, F., Singaraja, R., Smith, D.J., Bissada, N., McCutcheon, K., Nasir, J., et al. (1999). A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 23, 181-192. Hult, S., Schultz, K., Soylu, R., and Petersen, A. (2010). Hypothalamic and neuroendocrine changes in Huntington's disease. Curr Drug Targets 11, 1237-1249. Hult, S., Soylu, R., Bjorklund, T., Belgardt, B.F., Mauer, J., Bruning, J.C., Kirik, D., and Petersen, A. (2011). Mutant huntingtin causes metabolic imbalance by disruption of hypothalamic neurocircuits. Cell Metab 13, 428-439. The Huntington's Disease Collaborative Research Group (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72, 971–983. Huntington, G. (1872). On Chorea. Med Surg Rep 26, 331-321. Ichimura, Y., Kumanomidou, T., Sou, Y.S., Mizushima, T., Ezaki, J., Ueno, T., Kominami, E., Yamane, T., Tanaka, K., and Komatsu, M. (2008). Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 283, 22847-22857. Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., Yamrom, B., Lee, Y.H., Narzisi, G., Leotta, A., et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285-299. Irwin, S., Vandelft, M., Pinchev, D., Howell, J.L., Graczyk, J., Orr, H.T., and Truant, R. (2005). RNA association and nucleocytoplasmic shuttling by ataxin-1. J Cell Sci 118, 233-242. Isakson, P., Bjoras, M., Boe, S.O., and Simonsen, A. (2010). Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 116, 2324-2331. Isakson, P., Holland, P., and Simonsen, A. (2013a). The role of ALFY in selective autophagy. Cell Death Differ 20, 12-20.

140
Isakson, P., Lystad, A.H., Breen, K., Koster, G., Stenmark, H., and Simonsen, A. (2013b). TRAF6 mediates ubiquitination of KIF23/MKLP1 and is required for midbody ring degradation by selective autophagy. Autophagy 9, 1955-1964. Ishiguro, H., Yamada, K., Sawada, H., Nishii, K., Ichino, N., Sawada, M., Kurosawa, Y., Matsushita, N., Kobayashi, K., Goto, J., et al. (2001). Age-dependent and tissue-specific CAG repeat instability occurs in mouse knock-in for a mutant Huntington's disease gene. J Neurosci Res 65, 289-297. Iwata, A., Christianson, J.C., Bucci, M., Ellerby, L.M., Nukina, N., Forno, L.S., and Kopito, R.R. (2005). Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc Natl Acad Sci U S A 102, 13135-13140. Iwata, A., Nagashima, Y., Matsumoto, L., Suzuki, T., Yamanaka, T., Date, H., Deoka, K., Nukina, N., and Tsuji, S. (2009). Intranuclear degradation of polyglutamine aggregates by the ubiquitin-proteasome system. J Biol Chem 284, 9796-9803. Jahrling, J.B., and Laberge, R.M. (2015). Age-Related Neurodegeneration Prevention Through mTOR Inhibition: Potential Mechanisms and Remaining Questions. Curr Top Med Chem 15, 2139-2151. Janssens, J., and Van Broeckhoven, C. (2013). Pathological mechanisms underlying TDP-43 driven neurodegeneration in FTLD-ALS spectrum disorders. Hum Mol Genet 22, R77-87. Jeong, H., Then, F., Melia, T.J., Jr., Mazzulli, J.R., Cui, L., Savas, J.N., Voisine, C., Paganetti, P., Tanese, N., Hart, A.C., et al. (2009). Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 137, 60-72. Jimenez-Sanchez, M., Thomson, F., Zavodszky, E., and Rubinsztein, D.C. (2012). Autophagy and polyglutamine diseases. Prog Neurobiol 97, 67-82. Jinwal, U.K., Abisambra, J.F., Zhang, J., Dharia, S., O'Leary, J.C., Patel, T., Braswell, K., Jani, T., Gestwicki, J.E., and Dickey, C.A. (2012). Cdc37/Hsp90 protein complex disruption triggers an autophagic clearance cascade for TDP-43 protein. J Biol Chem 287, 24814-24820. Johansen, T., and Lamark, T. (2011). Selective autophagy mediated by autophagic adapter proteins. Autophagy 7, 279-296. Johnson, C.W., Melia, T.J., and Yamamoto, A. (2012). Modulating macroautophagy: a neuronal perspective. Future Med Chem 4, 1715-1731. Johnston, J.A., Ward, C.L., and Kopito, R.R. (1998). Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143, 1883-1898.

141
Kabuta, T., Suzuki, Y., and Wada, K. (2006). Degradation of amyotrophic lateral sclerosis-linked mutant Cu,Zn-superoxide dismutase proteins by macroautophagy and the proteasome. J Biol Chem 281, 30524-30533. Kadir, R., Harel, T., Markus, B., Perez, Y., Bakhrat, A., Cohen, I., Volodarsky, M., Feintsein-Linial, M., Chervinski, E., Zlotogora, J., et al. (2016). ALFY-Controlled DVL3 Autophagy Regulates Wnt Signaling, Determining Human Brain Size. PLoS Genet 12, e1005919. Kaganovich, D., Kopito, R., and Frydman, J. (2008). Misfolded proteins partition between two distinct quality control compartments. Nature 454, 1088-1095. Karsli-Uzunbas, G., Guo, J.Y., Price, S., Teng, X., Laddha, S.V., Khor, S., Kalaany, N.Y., Jacks, T., Chan, C.S., Rabinowitz, J.D., et al. (2014). Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov 4, 914-927. Katsuno, M., Adachi, H., Kume, A., Li, M., Nakagomi, Y., Niwa, H., Sang, C., Kobayashi, Y., Doyu, M., and Sobue, G. (2002). Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35, 843-854. Kaushik, S., and Cuervo, A.M. (2015). Proteostasis and aging. Nat Med 21, 1406-1415. Kawaguchi, Y., Kovacs, J.J., McLaurin, A., Vance, J.M., Ito, A., and Yao, T.P. (2003). The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727-738. Kazantsev, A., Preisinger, E., Dranovsky, A., Goldgaber, D., and Housman, D. (1999). Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci U S A 96, 11404-11409. Ke, Y.D., Suchowerska, A.K., van der Hoven, J., De Silva, D.M., Wu, C.W., van Eersel, J., Ittner, A., and Ittner, L.M. (2012). Lessons from tau-deficient mice. Int J Alzheimers Dis 2012, 873270. Kegel, K.B., Kim, M., Sapp, E., McIntyre, C., Castano, J.G., Aronin, N., and DiFiglia, M. (2000). Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci 20, 7268-7278. Kegel, K.B., Meloni, A.R., Yi, Y., Kim, Y.J., Doyle, E., Cuiffo, B.G., Sapp, E., Wang, Y., Qin, Z.H., Chen, J.D., et al. (2002). Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J Biol Chem 277, 7466-7476. Kennedy, L., and Shelbourne, P.F. (2000). Dramatic mutation instability in HD mouse striatum: does polyglutamine load contribute to cell-specific vulnerability in Huntington's disease? Hum Mol Genet 9, 2539-2544.

142
Kim, S., and Kim, K.T. (2014). Therapeutic Approaches for Inhibition of Protein Aggregation in Huntington's Disease. Exp Neurobiol 23, 36-44. Kirkin, V., Lamark, T., Johansen, T., and Dikic, I. (2009). NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 5, 732-733. Klement, I.A., Skinner, P.J., Kaytor, M.D., Yi, H., Hersch, S.M., Clark, H.B., Zoghbi, H.Y., and Orr, H.T. (1998). Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 95, 41-53. Klionsky, D.J. (2007). Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 8, 931-937. Knaevelsrud, H., and Simonsen, A. (2010). Fighting disease by selective autophagy of aggregate-prone proteins. FEBS Lett 584, 2635-2645. Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., Ueno, T., Koike, M., Uchiyama, Y., Kominami, E., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880-884. Komatsu, M., Waguri, S., Koike, M., Sou, Y.S., Ueno, T., Hara, T., Mizushima, N., Iwata, J., Ezaki, J., Murata, S., et al. (2007a). Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149-1163. Komatsu, M., Wang, Q.J., Holstein, G.R., Friedrich, V.L., Jr., Iwata, J., Kominami, E., Chait, B.T., Tanaka, K., and Yue, Z. (2007b). Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A 104, 14489-14494. Kontopoulos, E., Parvin, J.D., and Feany, M.B. (2006). Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet 15, 3012-3023. Kopito, R.R. (2000). Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10, 524-530. Korac, J., Schaeffer, V., Kovacevic, I., Clement, A.M., Jungblut, B., Behl, C., Terzic, J., and Dikic, I. (2013). Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci 126, 580-592. Kordasiewicz, H.B., Stanek, L.M., Wancewicz, E.V., Mazur, C., McAlonis, M.M., Pytel, K.A., Artates, J.W., Weiss, A., Cheng, S.H., Shihabuddin, L.S., et al. (2012). Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron 74, 1031-1044. Korolchuk, V.I., Mansilla, A., Menzies, F.M., and Rubinsztein, D.C. (2009). Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 33, 517-527.

143
Kremer, B., Goldberg, P., Andrew, S.E., Theilmann, J., Telenius, H., Zeisler, J., Squitieri, F., Lin, B., Bassett, A., Almqvist, E., et al. (1994). A worldwide study of the Huntington's disease mutation. The sensitivity and specificity of measuring CAG repeats. N Engl J Med 330, 1401-1406. Krick, R., Muehe, Y., Prick, T., Bremer, S., Schlotterhose, P., Eskelinen, E.L., Millen, J., Goldfarb, D.S., and Thumm, M. (2008). Piecemeal microautophagy of the nucleus requires the core macroautophagy genes. Mol Biol Cell 19, 4492-4505. Krol, S. (2012). Challenges in drug delivery to the brain: nature is against us. J Control Release 164, 145-155. Kurosawa, M., Matsumoto, G., Kino, Y., Okuno, M., Kurosawa-Yamada, M., Washizu, C., Taniguchi, H., Nakaso, K., Yanagawa, T., Warabi, E., et al. (2015). Depletion of p62 reduces nuclear inclusions and paradoxically ameliorates disease phenotypes in Huntington's model mice. Hum Mol Genet 24, 1092-1105. Labbadia, J., and Morimoto, R.I. (2015). The biology of proteostasis in aging and disease. Annu Rev Biochem 84, 435-464. Lamark, T., and Johansen, T. (2012). Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol 2012, 736905. Lamark, T., Kirkin, V., Dikic, I., and Johansen, T. (2009). NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 8, 1986-1990. Lashuel, H.A., Overk, C.R., Oueslati, A., and Masliah, E. (2013). The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 14, 38-48. Lee, J.H., Yu, W.H., Kumar, A., Lee, S., Mohan, P.S., Peterhoff, C.M., Wolfe, D.M., Martinez-Vicente, M., Massey, A.C., Sovak, G., et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146-1158. Lee, J.M., Ramos, E.M., Lee, J.H., Gillis, T., Mysore, J.S., Hayden, M.R., Warby, S.C., Morrison, P., Nance, M., Ross, C.A., et al. (2012). CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology 78, 690-695. Lee, M.J., Lee, J.H., and Rubinsztein, D.C. (2013a). Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol 105, 49-59. Lee, W., Reyes, R.C., Gottipati, M.K., Lewis, K., Lesort, M., Parpura, V., and Gray, M. (2013b). Enhanced Ca(2+)-dependent glutamate release from astrocytes of the BACHD Huntington's disease mouse model. Neurobiol Dis 58, 192-199. Li, S.H., and Li, X.J. (1998). Aggregation of N-terminal huntingtin is dependent on the length of its glutamine repeats. Hum Mol Genet 7, 777-782.

144
Liang, C.C., Wang, C., Peng, X., Gan, B., and Guan, J.L. (2010). Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J Biol Chem 285, 3499-3509. Liberski, P.P., Brown, D.R., Sikorska, B., Caughey, B., and Brown, P. (2008). Cell death and autophagy in prion diseases (transmissible spongiform encephalopathies). Folia Neuropathol 46, 1-25. Lim, J., and Yue, Z. (2015). Neuronal aggregates: formation, clearance, and spreading. Dev Cell 32, 491-501. Lin, C.H., Tallaksen-Greene, S., Chien, W.M., Cearley, J.A., Jackson, W.S., Crouse, A.B., Ren, S., Li, X.J., Albin, R.L., and Detloff, P.J. (2001). Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum Mol Genet 10, 137-144. Lin, W.L., Lewis, J., Yen, S.H., Hutton, M., and Dickson, D.W. (2003). Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J Neurocytol 32, 1091-1105. Liu, R.Q., Zhou, Q.H., Ji, S.R., Zhou, Q., Feng, D., Wu, Y., and Sui, S.F. (2010). Membrane localization of beta-amyloid 1-42 in lysosomes: a possible mechanism for lysosome labilization. J Biol Chem 285, 19986-19996. Los, G.V., Encell, L.P., McDougall, M.G., Hartzell, D.D., Karassina, N., Zimprich, C., Wood, M.G., Learish, R., Ohana, R.F., Urh, M., et al. (2008). HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol 3, 373-382. Lundh, S.H., Soylu, R., and Petersen, A. (2012). Expression of mutant huntingtin in leptin receptor-expressing neurons does not control the metabolic and psychiatric phenotype of the BACHD mouse. PLoS One 7, e51168. Lynch-Day, M.A., and Klionsky, D.J. (2010). The Cvt pathway as a model for selective autophagy. FEBS Lett 584, 1359-1366. Lynch-Day, M.A., Mao, K., Wang, K., Zhao, M., and Klionsky, D.J. (2012). The role of autophagy in Parkinson's disease. Cold Spring Harb Perspect Med 2, a009357. Lystad, A.H., Ichimura, Y., Takagi, K., Yang, Y., Pankiv, S., Kanegae, Y., Kageyama, S., Suzuki, M., Saito, I., Mizushima, T., et al. (2014). Structural determinants in GABARAP required for the selective binding and recruitment of ALFY to LC3B-positive structures. EMBO Rep 15, 557-565. Mackenzie, I.R., Rademakers, R., and Neumann, M. (2010). TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9, 995-1007. Mandell, M.A., Jain, A., Arko-Mensah, J., Chauhan, S., Kimura, T., Dinkins, C., Silvestri, G., Munch, J., Kirchhoff, F., Simonsen, A., et al. (2014). TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell 30, 394-409.

145
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., Lawton, M., Trottier, Y., Lehrach, H., Davies, S.W., et al. (1996). Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493-506. Martin, D.D., Ladha, S., Ehrnhoefer, D.E., and Hayden, M.R. (2015). Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci 38, 26-35. Martindale, D., Hackam, A., Wieczorek, A., Ellerby, L., Wellington, C., McCutcheon, K., Singaraja, R., Kazemi-Esfarjani, P., Devon, R., Kim, S.U., et al. (1998). Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet 18, 150-154. Martinez-Vicente, M., Talloczy, Z., Wong, E., Tang, G., Koga, H., Kaushik, S., de Vries, R., Arias, E., Harris, S., Sulzer, D., et al. (2010). Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci 13, 567-576. Mason, R.P., and Giorgini, F. (2011). Modeling Huntington disease in yeast: perspectives and future directions. Prion 5, 269-276. Matsumoto, G., Kim, S., and Morimoto, R.I. (2006). Huntingtin and mutant SOD1 form aggregate structures with distinct molecular properties in human cells. J Biol Chem 281, 4477-4485. Maurer, K., Volk, S., and Gerbaldo, H. (1997). Auguste D and Alzheimer's disease. Lancet 349, 1546-1549. Mayford, M., Bach, M.E., Huang, Y.Y., Wang, L., Hawkins, R.D., and Kandel, E.R. (1996). Control of memory formation through regulated expression of a CaMKII transgene. Science 274, 1678-1683. Menalled, L.B. (2005). Knock-in mouse models of Huntington's disease. NeuroRx 2, 465-470. Menalled, L.B., Sison, J.D., Dragatsis, I., Zeitlin, S., and Chesselet, M.F. (2003). Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington's disease with 140 CAG repeats. J Comp Neurol 465, 11-26. Menalled, L.B., Sison, J.D., Wu, Y., Olivieri, M., Li, X.J., Li, H., Zeitlin, S., and Chesselet, M.F. (2002). Early motor dysfunction and striosomal distribution of huntingtin microaggregates in Huntington's disease knock-in mice. J Neurosci 22, 8266-8276. Mizushima, N., and Levine, B. (2010). Autophagy in mammalian development and differentiation. Nat Cell Biol 12, 823-830. Montie, H.L., Cho, M.S., Holder, L., Liu, Y., Tsvetkov, A.S., Finkbeiner, S., and Merry, D.E. (2009). Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease

146
via autophagy in a mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet 18, 1937-1950. Montie, H.L., and Merry, D.E. (2009). Autophagy and access: understanding the role of androgen receptor subcellular localization in SBMA. Autophagy 5, 1194-1197. Mori, F., Tanji, K., Odagiri, S., Toyoshima, Y., Yoshida, M., Kakita, A., Takahashi, H., and Wakabayashi, K. (2012). Autophagy-related proteins (p62, NBR1 and LC3) in intranuclear inclusions in neurodegenerative diseases. Neurosci Lett 522, 134-138. Morris, D.H., Yip, C.K., Shi, Y., Chait, B.T., and Wang, Q.J. (2015). Beclin 1-Vps34 Complex Architecture: Understanding the Nuts and Bolts of Therapeutic Targets. Front Biol (Beijing) 10, 398-426. Morton, A.J. (2013). Circadian and sleep disorder in Huntington's disease. Exp Neurol 243, 34-44. Morton, A.J., Glynn, D., Leavens, W., Zheng, Z., Faull, R.L., Skepper, J.N., and Wight, J.M. (2009). Paradoxical delay in the onset of disease caused by super-long CAG repeat expansions in R6/2 mice. Neurobiol Dis 33, 331-341. Narain, Y., Wyttenbach, A., Rankin, J., Furlong, R.A., and Rubinsztein, D.C. (1999). A molecular investigation of true dominance in Huntington's disease. J Med Genet 36, 739-746. Nautiyal, M., Sweatt, A.J., MacKenzie, J.A., Mark Payne, R., Szucs, S., Matalon, R., Wallin, R., and Hutson, S.M. (2010). Neuronal localization of the mitochondrial protein NIPSNAP1 in rat nervous system. Eur J Neurosci 32, 560-569. Nekooki-Machida, Y., Kurosawa, M., Nukina, N., Ito, K., Oda, T., and Tanaka, M. (2009). Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc Natl Acad Sci U S A 106, 9679-9684. Neri, C. (2011). Value of Invertebrate Genetics and Biology to Develop Neuroprotective and Preventive Medicine in Huntington's Disease. In Neurobiology of Huntington's Disease: Applications to Drug Discovery, D.C. Lo, and R.E. Hughes, eds. (Boca Raton (FL). Neumann, M., Sampathu, D.M., Kwong, L.K., Truax, A.C., Micsenyi, M.C., Chou, T.T., Bruce, J., Schuck, T., Grossman, M., Clark, C.M., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130-133. Nixon, R.A. (2013). The role of autophagy in neurodegenerative disease. Nat Med 19, 983-997. Nixon, R.A., Wegiel, J., Kumar, A., Yu, W.H., Peterhoff, C., Cataldo, A., and Cuervo, A.M. (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 64, 113-122.

147
Ochaba, J., Lukacsovich, T., Csikos, G., Zheng, S., Margulis, J., Salazar, L., Mao, K., Lau, A.L., Yeung, S.Y., Humbert, S., et al. (2014). Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc Natl Acad Sci U S A 111, 16889-16894. Olsen, A., Vantipalli, M.C., and Lithgow, G.J. (2006). Using Caenorhabditis elegans as a model for aging and age-related diseases. Ann N Y Acad Sci 1067, 120-128. Orosco, L.A., Ross, A.P., Cates, S.L., Scott, S.E., Wu, D., Sohn, J., Pleasure, D., Pleasure, S.J., Adamopoulos, I.E., and Zarbalis, K.S. (2014). Loss of Wdfy3 in mice alters cerebral cortical neurogenesis reflecting aspects of the autism pathology. Nat Commun 5, 4692. Orr, H.T. (2012). Cell biology of spinocerebellar ataxia. J Cell Biol 197, 167-177. Osawa, T., Mizuno, Y., Fujita, Y., Takatama, M., Nakazato, Y., and Okamoto, K. (2011). Optineurin in neurodegenerative diseases. Neuropathology 31, 569-574. Ouyang, H., Ali, Y.O., Ravichandran, M., Dong, A., Qiu, W., MacKenzie, F., Dhe-Paganon, S., Arrowsmith, C.H., and Zhai, R.G. (2012). Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J Biol Chem 287, 2317-2327. Overbye, A., Fengsrud, M., and Seglen, P.O. (2007). Proteomic analysis of membrane-associated proteins from rat liver autophagosomes. Autophagy 3, 300-322. Palfi, S., Brouillet, E., Jarraya, B., Bloch, J., Jan, C., Shin, M., Conde, F., Li, X.J., Aebischer, P., Hantraye, P., et al. (2007). Expression of mutated huntingtin fragment in the putamen is sufficient to produce abnormal movement in non-human primates. Mol Ther 15, 1444-1451. Pankiv, S., Clausen, T.H., Lamark, T., Brech, A., Bruun, J.A., Outzen, H., Overvatn, A., Bjorkoy, G., and Johansen, T. (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282, 24131-24145. Park, S., Jang, I., Zuber, C., Lee, Y., Cho, J.W., Matsuo, I., Ito, Y., and Roth, J. (2014). ERADication of EDEM1 occurs by selective autophagy and requires deglycosylation by cytoplasmic peptide N-glycanase. Histochem Cell Biol 142, 153-169. Pickford, F., Masliah, E., Britschgi, M., Lucin, K., Narasimhan, R., Jaeger, P.A., Small, S., Spencer, B., Rockenstein, E., Levine, B., et al. (2008). The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 118, 2190-2199. Pouladi, M.A., Morton, A.J., and Hayden, M.R. (2013). Choosing an animal model for the study of Huntington's disease. Nat Rev Neurosci 14, 708-721. Pouladi, M.A., Stanek, L.M., Xie, Y., Franciosi, S., Southwell, A.L., Deng, Y., Butland, S., Zhang, W., Cheng, S.H., Shihabuddin, L.S., et al. (2012). Marked differences in neurochemistry and

148
aggregates despite similar behavioural and neuropathological features of Huntington disease in the full-length BACHD and YAC128 mice. Hum Mol Genet 21, 2219-2232. Prusiner, S.B. (1998). Prions. Proc Natl Acad Sci U S A 95, 13363-13383. Qin, Z.H., Wang, Y., Kegel, K.B., Kazantsev, A., Apostol, B.L., Thompson, L.M., Yoder, J., Aronin, N., and DiFiglia, M. (2003). Autophagy regulates the processing of amino terminal huntingtin fragments. Hum Mol Genet 12, 3231-3244. Ravikumar, B., Berger, Z., Vacher, C., O'Kane, C.J., and Rubinsztein, D.C. (2006). Rapamycin pre-treatment protects against apoptosis. Hum Mol Genet 15, 1209-1216. Ravikumar, B., Duden, R., and Rubinsztein, D.C. (2002). Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 11, 1107-1117. Ravikumar, B., Vacher, C., Berger, Z., Davies, J.E., Luo, S., Oroz, L.G., Scaravilli, F., Easton, D.F., Duden, R., O'Kane, C.J., et al. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36, 585-595. Regulier, E., Trottier, Y., Perrin, V., Aebischer, P., and Deglon, N. (2003). Early and reversible neuropathology induced by tetracycline-regulated lentiviral overexpression of mutant huntingtin in rat striatum. Hum Mol Genet 12, 2827-2836. Reiner, A., Albin, R.L., Anderson, K.D., D'Amato, C.J., Penney, J.B., and Young, A.B. (1988). Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A 85, 5733-5737. Rideout, H.J., Lang-Rollin, I., and Stefanis, L. (2004). Involvement of macroautophagy in the dissolution of neuronal inclusions. Int J Biochem Cell Biol 36, 2551-2562. Roberts, P., Moshitch-Moshkovitz, S., Kvam, E., O'Toole, E., Winey, M., and Goldfarb, D.S. (2003). Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae. Mol Biol Cell 14, 129-141. Roizin, L., Stellar, S., and Liu, J.C. (1979). Neuronal nuclear-cytoplasmic changes in Huntington’s chorea: electron microscope investigations. In Advances in Neurology, N.S.W. Chase, and A. Barbeau, ed. (New York, New York, USA Raven Press), pp. 95–122. Roos, R.A. (2010). Huntington's disease: a clinical review. Orphanet J Rare Dis 5, 40. Ross, C.A., and Poirier, M.A. (2004). Protein aggregation and neurodegenerative disease. Nat Med 10 Suppl, S10-17. Rotunno, M.S., and Bosco, D.A. (2013). An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front Cell Neurosci 7, 253.

149
Rub, U., Vonsattel, J.P., Heinsen, H., and Korf, H.W. (2015). The Neuropathology of Huntington s disease: classical findings, recent developments and correlation to functional neuroanatomy. Adv Anat Embryol Cell Biol 217, 1-146. Rue, L., Lopez-Soop, G., Gelpi, E., Martinez-Vicente, M., Alberch, J., and Perez-Navarro, E. (2013). Brain region- and age-dependent dysregulation of p62 and NBR1 in a mouse model of Huntington's disease. Neurobiol Dis 52, 219-228. Rui, Y.N., Xu, Z., Patel, B., Chen, Z., Chen, D., Tito, A., David, G., Sun, Y., Stimming, E.F., Bellen, H.J., et al. (2015). Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol 17, 262-275. Ruiz, M., and Deglon, N. (2012). Viral-mediated overexpression of mutant huntingtin to model HD in various species. Neurobiol Dis 48, 202-211. Rusmini, P., Bolzoni, E., Crippa, V., Onesto, E., Sau, D., Galbiati, M., Piccolella, M., and Poletti, A. (2010). Proteasomal and autophagic degradative activities in spinal and bulbar muscular atrophy. Neurobiol Dis 40, 361-369. Ryu, H., Rosas, H.D., Hersch, S.M., and Ferrante, R.J. (2005). The therapeutic role of creatine in Huntington's disease. Pharmacol Ther 108, 193-207. Sapp, E., Schwarz, C., Chase, K., Bhide, P.G., Young, A.B., Penney, J., Vonsattel, J.P., Aronin, N., and DiFiglia, M. (1997). Huntingtin localization in brains of normal and Huntington's disease patients. Ann Neurol 42, 604-612. Sarkar, S., Davies, J.E., Huang, Z., Tunnacliffe, A., and Rubinsztein, D.C. (2007). Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem 282, 5641-5652. Sarkar, S., Ravikumar, B., Floto, R.A., and Rubinsztein, D.C. (2009). Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 16, 46-56. Sarkar, S., and Rubinsztein, D.C. (2008). Huntington's disease: degradation of mutant huntingtin by autophagy. FEBS J 275, 4263-4270. Sato, T., Takeuchi, S., Saito, A., Ding, W., Bamba, H., Matsuura, H., Hisa, Y., Tooyama, I., and Urushitani, M. (2009). Axonal ligation induces transient redistribution of TDP-43 in brainstem motor neurons. Neuroscience 164, 1565-1578. Satoh, K., Takeuchi, M., Oda, Y., Deguchi-Tawarada, M., Sakamoto, Y., Matsubara, K., Nagasu, T., and Takai, Y. (2002). Identification of activity-regulated proteins in the postsynaptic density fraction. Genes Cells 7, 187-197.

150
Schaefer, M.H., Wanker, E.E., and Andrade-Navarro, M.A. (2012). Evolution and function of CAG/polyglutamine repeats in protein-protein interaction networks. Nucleic Acids Res 40, 4273-4287. Scherzinger, E., Lurz, R., Turmaine, M., Mangiarini, L., Hollenbach, B., Hasenbank, R., Bates, G.P., Davies, S.W., Lehrach, H., and Wanker, E.E. (1997). Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90, 549-558. Schilling, G., Becher, M.W., Sharp, A.H., Jinnah, H.A., Duan, K., Kotzuk, J.A., Slunt, H.H., Ratovitski, T., Cooper, J.K., Jenkins, N.A., et al. (1999). Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet 8, 397-407. Scotter, E.L., Vance, C., Nishimura, A.L., Lee, Y.B., Chen, H.J., Urwin, H., Sardone, V., Mitchell, J.C., Rogelj, B., Rubinsztein, D.C., et al. (2014). Differential roles of the ubiquitin proteasome system (UPS) and autophagy in the clearance of soluble and aggregated TDP-43 species. J Cell Sci. Seroussi, E., Pan, H.Q., Kedra, D., Roe, B.A., and Dumanski, J.P. (1998). Characterization of the human NIPSNAP1 gene from 22q12: a member of a novel gene family. Gene 212, 13-20. Servadio, A., Koshy, B., Armstrong, D., Antalffy, B., Orr, H.T., and Zoghbi, H.Y. (1995). Expression analysis of the ataxin-1 protein in tissues from normal and spinocerebellar ataxia type 1 individuals. Nat Genet 10, 94-98. Shelbourne, P.F., Killeen, N., Hevner, R.F., Johnston, H.M., Tecott, L., Lewandoski, M., Ennis, M., Ramirez, L., Li, Z., Iannicola, C., et al. (1999). A Huntington's disease CAG expansion at the murine Hdh locus is unstable and associated with behavioural abnormalities in mice. Hum Mol Genet 8, 763-774. Shibata, M., Lu, T., Furuya, T., Degterev, A., Mizushima, N., Yoshimori, T., MacDonald, M., Yankner, B., and Yuan, J. (2006). Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem 281, 14474-14485. Shoji-Kawata, S., Sumpter, R., Leveno, M., Campbell, G.R., Zou, Z., Kinch, L., Wilkins, A.D., Sun, Q., Pallauf, K., MacDuff, D., et al. (2013). Identification of a candidate therapeutic autophagy-inducing peptide. Nature 494, 201-206. Sieradzan, K.A., and Mann, D.M. (2001). The selective vulnerability of nerve cells in Huntington's disease. Neuropathol Appl Neurobiol 27, 1-21. Simonsen, A., Birkeland, H.C., Gillooly, D.J., Mizushima, N., Kuma, A., Yoshimori, T., Slagsvold, T., Brech, A., and Stenmark, H. (2004). Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J Cell Sci 117, 4239-4251.

151
Slow, E.J., Graham, R.K., Osmand, A.P., Devon, R.S., Lu, G., Deng, Y., Pearson, J., Vaid, K., Bissada, N., Wetzel, R., et al. (2005). Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc Natl Acad Sci U S A 102, 11402-11407. Slow, E.J., van Raamsdonk, J., Rogers, D., Coleman, S.H., Graham, R.K., Deng, Y., Oh, R., Bissada, N., Hossain, S.M., Yang, Y.Z., et al. (2003). Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet 12, 1555-1567. Song, C.Y., Guo, J.F., Liu, Y., and Tang, B.S. (2012). Autophagy and Its Comprehensive Impact on ALS. Int J Neurosci 122, 695-703. Southwell, A.L., Skotte, N.H., Kordasiewicz, H.B., Ostergaard, M.E., Watt, A.T., Carroll, J.B., Doty, C.N., Villanueva, E.B., Petoukhov, E., Vaid, K., et al. (2014). In vivo evaluation of candidate allele-specific mutant huntingtin gene silencing antisense oligonucleotides. Mol Ther 22, 2093-2106. Stack, E.C., Kubilus, J.K., Smith, K., Cormier, K., Del Signore, S.J., Guelin, E., Ryu, H., Hersch, S.M., and Ferrante, R.J. (2005). Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington's disease transgenic mice. J Comp Neurol 490, 354-370. Stanek, L.M., Sardi, S.P., Mastis, B., Richards, A.R., Treleaven, C.M., Taksir, T., Misra, K., Cheng, S.H., and Shihabuddin, L.S. (2014). Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington's disease. Hum Gene Ther 25, 461-474. Surendran, S., Tyring, S.K., and Matalon, R. (2005). Expression of calpastatin, minopontin, NIPSNAP1, rabaptin-5 and neuronatin in the phenylketonuria (PKU) mouse brain: possible role on cognitive defect seen in PKU. Neurochem Int 46, 595-599. Swami, M., Hendricks, A.E., Gillis, T., Massood, T., Mysore, J., Myers, R.H., and Wheeler, V.C. (2009). Somatic expansion of the Huntington's disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet 18, 3039-3047. Tabrizi, S.J., Scahill, R.I., Owen, G., Durr, A., Leavitt, B.R., Roos, R.A., Borowsky, B., Landwehrmeyer, B., Frost, C., Johnson, H., et al. (2013). Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington's disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol 12, 637-649. Tanaka, M., Machida, Y., Niu, S., Ikeda, T., Jana, N.R., Doi, H., Kurosawa, M., Nekooki, M., and Nukina, N. (2004). Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med 10, 148-154. Tanik, S.A., Schultheiss, C.E., Volpicelli-Daley, L.A., Brunden, K.R., and Lee, V.M. (2013). Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J Biol Chem 288, 15194-15210.

152
Tellez-Nagel, I., Johnson, A.B., and Terry, R.D. (1974). Studies on brain biopsies of patients with Huntington's chorea. J Neuropathol Exp Neurol 33, 308-332. Todd, T.W., and Lim, J. (2013). Aggregation formation in the polyglutamine diseases: protection at a cost? Mol Cells 36, 185-194. Trottier, Y., Devys, D., Imbert, G., Saudou, F., An, I., Lutz, Y., Weber, C., Agid, Y., Hirsch, E.C., and Mandel, J.L. (1995). Cellular localization of the Huntington's disease protein and discrimination of the normal and mutated form. Nat Genet 10, 104-110. Tummala, H., Li, X., and Homayouni, R. (2010). Interaction of a novel mitochondrial protein, 4-nitrophenylphosphatase domain and non-neuronal SNAP25-like protein homolog 1 (NIPSNAP1), with the amyloid precursor protein family. Eur J Neurosci 31, 1926-1934. Vonsattel, J.P., and DiFiglia, M. (1998). Huntington disease. J Neuropathol Exp Neurol 57, 369-384. Vonsattel, J.P., Myers, R.H., Stevens, T.J., Ferrante, R.J., Bird, E.D., and Richardson, E.P., Jr. (1985). Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44, 559-577. Walker, F.O. (2007). Huntington's disease. Lancet 369, 218-228. Wang, I.F., Guo, B.S., Liu, Y.C., Wu, C.C., Yang, C.H., Tsai, K.J., and Shen, C.K. (2012). Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A 109, 15024-15029. Wang, N., Gray, M., Lu, X.H., Cantle, J.P., Holley, S.M., Greiner, E., Gu, X., Shirasaki, D., Cepeda, C., Li, Y., et al. (2014). Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington's disease. Nat Med 20, 536-541. Wang, Y., Martinez-Vicente, M., Kruger, U., Kaushik, S., Wong, E., Mandelkow, E.M., Cuervo, A.M., and Mandelkow, E. (2009). Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 18, 4153-4170. Wang, Y., Steimle, P.A., Ren, Y., Ross, C.A., Robinson, D.N., Egelhoff, T.T., Sesaki, H., and Iijima, M. (2011). Dictyostelium huntingtin controls chemotaxis and cytokinesis through the regulation of myosin II phosphorylation. Mol Biol Cell 22, 2270-2281. Wang, Y.L., Liu, W., Wada, E., Murata, M., Wada, K., and Kanazawa, I. (2005). Clinico-pathological rescue of a model mouse of Huntington's disease by siRNA. Neurosci Res 53, 241-249. Webb, J.L., Ravikumar, B., Atkins, J., Skepper, J.N., and Rubinsztein, D.C. (2003). Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 278, 25009-25013.

153
Wetzel, R. (2012). Physical chemistry of polyglutamine: intriguing tales of a monotonous sequence. J Mol Biol 421, 466-490. Wexler, N.S., Lorimer, J., Porter, J., Gomez, F., Moskowitz, C., Shackell, E., Marder, K., Penchaszadeh, G., Roberts, S.A., Gayan, J., et al. (2004). Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci U S A 101, 3498-3503. Wheeler, V.C., Auerbach, W., White, J.K., Srinidhi, J., Auerbach, A., Ryan, A., Duyao, M.P., Vrbanac, V., Weaver, M., Gusella, J.F., et al. (1999). Length-dependent gametic CAG repeat instability in the Huntington's disease knock-in mouse. Hum Mol Genet 8, 115-122. Wheeler, V.C., White, J.K., Gutekunst, C.A., Vrbanac, V., Weaver, M., Li, X.J., Li, S.H., Yi, H., Vonsattel, J.P., Gusella, J.F., et al. (2000). Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet 9, 503-513. Wild, E.J., Henley, S.M., Hobbs, N.Z., Frost, C., MacManus, D.G., Barker, R.A., Fox, N.C., and Tabrizi, S.J. (2010). Rate and acceleration of whole-brain atrophy in premanifest and early Huntington's disease. Mov Disord 25, 888-895. Williams, A., Sarkar, S., Cuddon, P., Ttofi, E.K., Saiki, S., Siddiqi, F.H., Jahreiss, L., Fleming, A., Pask, D., Goldsmith, P., et al. (2008). Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 4, 295-305. Winslow, A.R., Chen, C.W., Corrochano, S., Acevedo-Arozena, A., Gordon, D.E., Peden, A.A., Lichtenberg, M., Menzies, F.M., Ravikumar, B., Imarisio, S., et al. (2010). alpha-Synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol 190, 1023-1037. Wong, E.S., Tan, J.M., Soong, W.E., Hussein, K., Nukina, N., Dawson, V.L., Dawson, T.M., Cuervo, A.M., and Lim, K.L. (2008). Autophagy-mediated clearance of aggresomes is not a universal phenomenon. Hum Mol Genet 17, 2570-2582. Wong, Y.C., and Holzbaur, E.L. (2014). The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci 34, 1293-1305. Yamada, M., Tsuji, S., and Takahashi, H. (2002). Involvement of lysosomes in the pathogenesis of CAG repeat diseases. Ann Neurol 52, 498-503. Yamamoto, A., Cremona, M.L., and Rothman, J.E. (2006). Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol 172, 719-731. Yamamoto, A., Lucas, J.J., and Hen, R. (2000). Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell 101, 57-66.

154
Yamamoto, A., and Simonsen, A. (2011). The elimination of accumulated and aggregated proteins: a role for aggrephagy in neurodegeneration. Neurobiol Dis 43, 17-28. Yang, W., Dunlap, J.R., Andrews, R.B., and Wetzel, R. (2002). Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet 11, 2905-2917. Yang, X., and Gray, M. (2011). Mouse Models for Validating Preclinical Candidates for Huntington's Disease. In Neurobiology of Huntington's Disease: Applications to Drug Discovery, D.C. Lo, and R.E. Hughes, eds. (Boca Raton (FL). Yang, W., Tu, Z., Sun, Q., and Li, X.J. (2016). CRISPR/Cas9: Implications for Modeling and Therapy of Neurodegenerative Diseases. Front Mol Neurosci 9, 30. Yao, H., Zhao, D., Khan, S.H., and Yang, L. (2013). Role of autophagy in prion protein-induced neurodegenerative diseases. Acta Biochim Biophys Sin (Shanghai) 45, 494-502. Yu, W.H., Cuervo, A.M., Kumar, A., Peterhoff, C.M., Schmidt, S.D., Lee, J.H., Mohan, P.S., Mercken, M., Farmery, M.R., Tjernberg, L.O., et al. (2005). Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol 171, 87-98. Yu, W.H., Kumar, A., Peterhoff, C., Shapiro Kulnane, L., Uchiyama, Y., Lamb, B.T., Cuervo, A.M., and Nixon, R.A. (2004). Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for beta-amyloid peptide over-production and localization in Alzheimer's disease. Int J Biochem Cell Biol 36, 2531-2540. Zambrowicz, B.P., Abuin, A., Ramirez-Solis, R., Richter, L.J., Piggott, J., BeltrandelRio, H., Buxton, E.C., Edwards, J., Finch, R.A., Friddle, C.J., et al. (2003). Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc Natl Acad Sci U S A 100, 14109-14114. Zhang, K., Shi, P., An, T., Wang, Q., Wang, J., Li, Z., Duan, W., Li, C., and Guo, Y. (2013). Food restriction-induced autophagy modulates degradation of mutant SOD1 in an amyotrophic lateral sclerosis mouse model. Brain Res 1519, 112-119. Zhang, X., Li, L., Chen, S., Yang, D., Wang, Y., Zhang, X., Wang, Z., and Le, W. (2011). Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 7, 412-425. Zheng, L., Roberg, K., Jerhammar, F., Marcusson, J., and Terman, A. (2006). Oxidative stress induces intralysosomal accumulation of Alzheimer amyloid beta-protein in cultured neuroblastoma cells. Ann N Y Acad Sci 1067, 248-251. Zheng, Z., and Diamond, M.I. (2012). Huntington disease and the huntingtin protein. Prog Mol Biol Transl Sci 107, 189-214.

155
Zu, T., Duvick, L.A., Kaytor, M.D., Berlinger, M.S., Zoghbi, H.Y., Clark, H.B., and Orr, H.T. (2004). Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J Neurosci 24, 8853-8861.