bed reactors for.use.lu.unsegmented ... -...
TRANSCRIPT

Chem. Anal. (Warsaw), 38, 1 (1993) REVIEW
Bed Reactors for .Use.lu.Unsegmented Continuous FlowMethodologies. Analysis of Inorganic Species
by J. Martinez Calatayud and J. V. Garcia Mateo*
Departamento de Quimica Analitica, Universidad de Valencia, 46100 Valencia, Spain*Colegio UniversitarioClill, Universidad de Valencia, 46113 M;ollcada(Valencia)"Spain
The use of bed reactors in unsegmented continuous-flow methodologies is criticallyreviewed. Emphasis is puton flow injection analysis. Types of reagents, immobilizationstrategies, flow assemblies and function (sample preconcentration, unstable reagents,converslonand detection) of the immobilization are some of the questions pointed 'out,A survey ofapplications to determination of inorganic species is also reported.
W pracy dokonano krytycznego przegladu prac poswieconych zastosowaniu reaktorowzfozowych w metodach przeplywowych bez rozdzielania strumienia. Zwr6conoszczegolnie uwagt; na takie zagadnienia,jak: typy reagentow, strategia osadzania, ukladprzeplywowy, zatezanie probki, nietrwalereagenty,przeksztalcanie chemiczne substan- .cji oznaczanychi detekcja. W pracy przedstawiono wykaz przykladow oznaczaniasubstancji nieorganicznych.
Solid reagents have been used in analytical' chemistry as long as dissolvedreagents, however their scope of application is much more limited and typicallyconsists, of oxidations and reductions preceding' analytical processes. There is anumber of well-known reasons for such liinited use, the most important of which areprobably the laborious experimental techniques involved and the difficulty in accurately controlling the reagent concentration and hence its effect, which may give riseto undersirable parasitic effects and side reactions.
In spite of the above-mentioned shortcommings, solid reagents make one of thecurrent interesting research lines in analytical chemistry for such applications as theimmobilization of enzymes in clinical laboratories [1], the use of extraction columnspacked with bonded silica [2], adsorbents [3,4] or ion exchangers for preconcentration or purification purposes [5:...8], and' pre- and post-columrr derivatization inhigh-performance liquid chromatography [9].

2 J. Martinez Calatayud and J. V.Garcia Mateo
The use of immobilized reagents has become one of the most interesting currenttrends in continuous-flow analytical methods thanks to the inherent operational andanalytical advantages of this type of system over many homogeneous systems. In thiscontext, the word "immobilization" is used to refer to the use of reagents confinedin a given place along a continuous flow system, through which a sample-carrierstream is circulated to effect reactions at the solid-solution interface formed. Confinement of the reagents can be accomplished by natural immobilization (insolublesubstances) or by anchoring at a flow-resistant solid support through a physical orchemical mechanism.
Features of immobilized reagents and support
A reagent bed should meet a series of requirements for use in a continuous-flowsystem:
a) Its particle size should allow the liquid to circulate freely so as to allowexploitation of one of the major advantages of the FIA technique viz. working at lowpressures.
b) It.should react fast in order to achieve as high degree of conversion as possible.c) It should retain no analytes or reaction products (adsorption) in order to avoid
a carry-over between samples.d) It should be mechanically resistant to the continuous flow so that no extra
spaces develop in time; otherwise, such spaces would result in increased sampledispersion, particle breakdown and the consequent clogging, overpressure and oscillations in the flow-rate, which in turn would cause changes in the degree of conversion through variations in the solid-solution contact surface. This condition isessential to ensuring reproducibility in the transient signals obtained. On the otherhand, immobilized reagents should be resistant to the potential sweep by the flowingstream, which would result in a gradual loss of the bed activity.
e) It should be chemically inert against the carrier and other reagents; otherwise,the lifetime of the column would be adversely affected.
Some of these requirements should be met both by "natural" reagent beds and bythe supports used to immobilize the corresponding reagents. The most commonlyused supports are resins [5-8], silica [10], controlled pore glass [11-15], and activated alumina [3, 4]. Here are some comments on desirable properties of supports:
a) Their particle size and structure affect the number of active sites and correspondingly the reagent concentration.
b) They should be well wettable, as the reagent must occupy sites on the innersurface of the chanels or cavities of the support particles. Properties at the solvent tobe used should also be taken into accound in choosing the appropriate support.
c) They should be resistant to microbialdegradation, particulary when biologicalsamples are dealt with.
d) Other important factors are compatibility with the detection system andinteractions with light.
Many of these conditions need not be strictly met in practice. Thus, the use ofcolumns packed with finely divided solids compels connection reinforcement and

Bed reactors ill flow methodologies 3
using high-pressure pumps such as those typically employed in HPLC; however,markedly increased solid-solution contact surface. allows much shorter columns tobe used - 0.5 cm has proved to be sufficient in some instances [16].
The need for the bed to be inert to the reagents or the carrier is also dependenton the type of the detector used, i.e. 011 whether it is "blind" to the reagent releasedbetween samples. Otherwise, the released reagent would be interfering the usefulanalytical reading, and the blank. Finally this results in the decreased useful lifetime.
A major question to be considered in this respect is the reactor regeneration;sweeping by the flowing stream or consumption in the reaction inevitably shortensthe reactor lifetime. This compels the use of the reactors that can be regeneratedon-line by periodically passing a reagent solution to replenish the reagent.
Advantages of solid reactors
The popularity of this type of FIA assembly can be ascribed to the advantages itoffers over the use of dissolved reagents, basically increased sensitivity, and injectionrate resulting from the inherently lower sample dispersion, which in turn results fromtwo factors, namely: (a) the fact that radial mass transfer in a packed bed is muchmore intense and hence the sample plug is converted more readily, and(b) the analyteis converted in a place where no sample dispersion occurs, viz. the solid-liquidinterface. All this results in increased analytical signals and the possibility of usinglarger sample volumes in a single injection.
Both packed columns and tubes (the diference between the two depends on theratio between the inner diameter of the reactor and the particle diameter, so the ratiosfor columns are close to unity while those for tubes are much larger than one) havebeen studied, though not very deeply, by using both inert packings, which aresupposed to improve the sample-carrier or sample-reagent mixingprocess in liquidchromatography and other continuous-flow methods, and enzymatic reactors in FIA[16-23]. The two have proved to be effective means of decreasing the width of thepeak base. Some equations for the columns have been reported, though there appearsto be no clear choice in this respect. On the other hand, none of the equation obtainedfor SBSR (single bed string reactor) is rigorously obeyed.
There are no definitive mathematical conclusions on the sample dispersioncaused by a solid particle bed, or on the influence of the technical features of thereactor configuration (compactness, type, size and shape of the packing material,
.etc.). However, radial mass transfer in a packed reactor is clearly more intense thanin a tubular reactor, and conversion within the sample takes place at a higher rate, asit occurs in a zone sample subject to no dispersion (the solid-liquid interface) whereD occasionally reaches unity [24].
The increased sensitivity provided by solid reagents also arises from the fact thatthey are as concentrated as possible. Other factors such as the chemical affinity forthe immobilized reagent, the physico-chemical features of the reaction, and exchangeat the solid-liquid interface FIAvariables, themselves are directly responsible for themagnitude of the analytical signal obtained in each instance.

4 J. Martinez CalatayudandJ, V.Garcia Mateo
The trend to simplify and miniaturize FIA systems by immobilization of anoptical detector (optosensing) in the flow-cell [25-31], wherever possible, enablesintegrated conversion and detection at the same point, and minimizes sample dispersion.
One other factor to be considered is the usual absence of problems posed byexcess of reagents; while no specially concentrated reagents need to be used to speedup the-reaction, itis also unusual to employ immobilized reagents reacting with thecarrier or other reagents, so no unconsumed reagent reaches the detector. This isrelated to the type. of the detector used;.if it is "blind" to the species concerned, thenno-problems should arise; otherwise, a suppressing column may have to be used toremove potential interferents [12,33,34]. Thus, the fluorimetricon-Iineoxidationof
. paracetamol supresses the use of ascorbic acid to avoid the quenching effect of thehexacyanoferrate(III) excess [35]. .
A typical low reagent consumption allows re-use of regenerable reagents orperiodical re-load of the bed, in addition to ready recovery of the unreactedreagent.The use of smaller amounts than with dissolved reagents also reduces the costas noreagent solution is wasted between succesive samples. This, in addition to the higherinjection rate afforded which involves preparation of fewer solutions, results in timeand henceeconomic savings. Most of the work involving immobilized reagents dealswith enzyme immobilization [42].
Another advantage lies in the possibility of using simpler FIA assemblies including fewer channels; in fact, much work on this topic was carried out on single-channel[5, 23, 26, 43, 51] or two-channel manifolds [52, 53].
The use of immobilized reagents has enabled analytical application of reagentsotherwise unusable under normal conditions; thus, unstable reagents (e.g. strongoxidants and reductants [57]) can be used in immobilized form provided theirdescomposition rate is reproducible.
In addition to these reagents, the use of heterogeneous systems in continuousflow set-ups has been extended to insoluble reagents (naturalimmobilization). A casein point is the determination of cyanide by reaction with copper sulphide [44]. Also,a .two-channel system and a silver column have been used for the examination ofspeciation of iron [58].
In close relation to the "release" ofreagents analytical applications have alsobeen developed to accelerate and facilitate some analytical processes. The growingpopularity of the FfAtechnique is, among others, largely' a result of its high versatilityand efficiency for such operations as preconcentration, purification, separation orcatalysis. This association allows the indirect determination of a number of drugs [1, 59],since' pharmaceuticals call react with a number of metal ions (ion-pair and complexformation, precipitation).
Analytical function of the immobilized reagent and location in the FIA assembly
The configuration of an FIA assembly is logically a function of its intendedanalytical purpose. Therefore, the location and dimensions of the reactor to be usedwill depend on its function in the analytical process. Figure 1 presents the different

Bed reactors ill flow methodologies 5
positions where the reactor can be placed depending on its purpose, namely (a)sample or reagent pretreatment, (b) analyte conversion, and (c}integration of reactionand detection.
a)
1------ -- ------- 2- .- - - - - - - - -
b)
3 4
5
c)
6
Figure 1. Location and function of bed reactors in a FIA assembl y:a) pretreatment of reagent (1).or sample (2);b) sample conversion;c) reactor-detector coupling
Sample pretreatment. In the configuration schematized in Fig. la the reactor isused to subject the sample solution to some type of pretreatment such as removal ofinterfering compounds, preconcentration, pH adjustment or even conversion into a"pseudo-sample" to be injected into the manifold instead of the original sample. Atypical example of preconcentration is represented by the determination of lead inalloys, water and soils [60].
Reagent pretreatment. This can be aimed at purifying the reagent solution byusing exchange or adsorbent columns, or at the in situ generation of such unstablereagents as strong oxidants and reductants.
The quantitative analytical application of strong oxidants and reductants isusually rendered impossible by the thermodynamic instability of their aqueoussolutions unless their kinetics of decomposition by hydrogen generation or water

6 J. Martinez Calatayud and J. V. Garcia Mateo:
oxidation is very slow. The FIA technique can popularize their use as it allows togenerate them whenever needed. Insofar as the chemical reaction need not reachcompletion in FIA systems, the reagent instability should pose no problem providedits decomposition rate is reproducible. It should be marked that an unstable reagentdoes not need to preserve its integrity for time longer than lor 2 min and that it iscirculated through a closed system isolated from the atmosphere. Solid reagents, suchas Jones reductant, followed by debubbling of the hydrogen released, have been usedfor the generation of Cr(Il), VeIl), V(Il) [61-64] and applied to a large number oforganic and inorganic species.
Similar to the generation of strong reductants is generation of strong oxidants[65, 66]. In this case, oxidation is accomplished electrochemically. Insofar, as thegenerated reagent is used only to convert the analyte it does not need to be 100 %efficient. Because of the instability of the generated reagent, the flow-rate of thecarrier stream should be relatively high. Therefore, the generator surface should bequite large; these considerations were taken into account in designing a gold powderelectrode that was constructed by packing gold particles with a diameter smaller than0.6 mm into a glass tube which was followed by a debubbler intended to remove largeamounts of the oxygen released. FIA assemblies of this type have been designed forthe generation of Ag(II) and Mn(IV) [65].
Conversion reactions. This operation is the most frequently employed. Inprinciple the conversion can be accomplished even within the sample loop itself. Itis intended to increase the analytical signal and to shorten the residence time. It can.also be an alternative to sample pretreatment (e.g. for analyte preconcentrationpurposes, a single valve can be used to switch between the different stages of theprocess, viz. sample insertion, elution and resin reconditioning). For the conversionpurposes, the whole sample is brought into contact with the reagent before thedispersion in the bulk carrier starts. For slow reactions, it avoids using the stoppedflow technique and hence preserves a reproducible flow-rate as the pump need notbe a subject to continuous start-and-stop sequences and the only variable to becontrolled is the interval between the openning and closure of the injection valve.
If the reaction takes place in a medium other than that where the sample isdissolved and the whole process is to be implemented in a automatic fashion, thenthe sample must be conditioned within the assembly, which limits applicability tosufficiently large samples. The reaction of oxidation of thiamine with immobilizedferricyanide takes place in an alkaline medium, where the analyte is unstable. Thisrequires merging of the sample solution with a buffer stream. The obtained mixtureis inserted into the sample loop [67].
Locating the solid reactor between the valve and the detector is by far the mostcommonly used alternative both with single and multi-channel systems. The latterallow several columns to be used jointly along the same or different channels. Thus,paracetamol can be determined in pharmaceutical preparations [35]. Two-channelassemblies are also suitable for speciation studies and for simultaneous determinationof several analytes.
A two-channel manifold can be used to oxidize organic compounds with TiO ina photocatalysed reaction promoted by near UV radiation [68]. The oxide is immo-

Bed reactors in flow methodologies 7
bilized as a thin film on the internal surface of a Teflon tube which in tum is coiledaround the light source.
Integratedreaction-detection. A recent trend in the use of an immobilizedreagent involves placing the reaction unit within the detection system so as to sensechanges in the immobilized species orthe solution involved in the reaction [25, 45]~Integration ofthese two functions, conversion and detection, is a logical trend if moreadvanced designs, allowing the analyte to be converted into a species that can bemeasured more sensitivity and selectively, are to be developed. This increasedsensitivity will simply bea result of the converted analyte not being dispersed on itsway to the detector. Operations other than conversion (e.g. dialysis, gas diffusion,liquid-liquid extraction, ion exchange or preconcentration) can becombined withdetection within the optosensor cell in order to improve the sensitivity ofthe givenprocess.
The species to be immobilizod can be either the reagent (including enzymecatalysts), the analyte or the product [70].
Leaving electrodes aside, most authors are reluctant to consider them as reactors,this type of detector, which is used to implement "optosensing at active surfaces",exploits interactions between light and matter (e.g. reflectance, fluorescence, absorption) arising from chemical reactions that take place at (on or near) a surface overwhich a solution is circulated. Such a surface acts as a support for the chemicalreagents, the colour or fluorescence changes of which provide a measure of thereactions taking place at the detector cell. Not always are reagents immobilized; ifimmobilization is impractical, the optosensor can be replenished periodically byintermittent pumping.
The support surface can be of fibrous nature, a diffusion or gas-permeablemembrane, anion exchanger or a water-miscible liquid; these materials in tum caninclude the reagents, ionically or covalently bonded [55]. The carrier stream circulates across the surface, and both the incident and the reflected light can be driven tothe optosensor by means of an optical fibre and further to a conventional spectrophotometer to record reflectance changes.
For a reasonably good performance, the detectors to be used should meet severalrequirements, namely:
a) Reversibility, or at least a high capacity if the reagent is consumed in theprocess. This condition is rather important if the reaction product is also the retainedspecies.
b) Stability of the immobilized reagent in the reaction medium.c) Rapid kinetics of the reaction or retention/elution process.d) Compatibility with the support. This requires the light beam to be able to cross
the cell containing the support and permanently immobilized species to-have a verylow absorbance or fluorescence at the working wavelengths. ~ ...
Detection with an ion exchanger has been used in static methods to do absorbance; reflectance (with optical fibre); and fluorescence measurements. In every case,the irreversibility of the system concerned compelled the use of a new portion of theion exchanger for each determination. This type of detection can be implemented inthree different fashions, viz.: (a) by reacting the analyte and reagent in solution and

8 J. Martinez Calatayud and J. V. Garcia Mateo
retaining the reaction product on the exchanger, which is quite advisable when thereaction is very selective; (b) by selectively adsorbing the reagent irreversibly whichis of use when the reaction product cannot be directly adsorbed onto the exchanger.
A spectrophotometric flow-cell packed with an ion-exchange resin [28] was usedfor the determination of Cu(II). Also, Fe(II) was determined with the aid of anion-exchange resin [71]. The enzyme alcohol dehydrogenase was immobilized oncontrolled pore glass [30] and reacted with its coenzyme. Wolbeis et al. [27]developed an optical sensor called "ion-selective opt rode" for the continuous determination of electrolytes based on the ability of some fluorescent dyes to respond toan electrical potential at the interface between an aqueous and a lipid phase. Theseauthors also developed an optosensor for monitoring potassium. Integration ofreaction and detection with ion-exchange resins allowed determination of traces ofCr(VI) [45], sulphideion [26], and Mo(VI) [72]. A chemiluminiscent sensor contain-ing uranine, immobilized on an ion-exchange resin was used for the continuousmonitoring of chlorine in drinking water [73]. The analyte itself can be retained onCI8 bonded silica adsorbent. Such approach was used to determine 2,4-dinitrophenylhydrazine and 2-nitrophenylhydrazine [31].
Approaches to reagent immobilization
The approaches to the preparation of reagent beds described here refer both tothe physico-chemical mechanisms used to immobilize the reagent of interest and tothe shape or configuration of the reactor (see Scheme 1).
The strategy developed to immobilize a given reagent is usually imposed by itsnature and by the analytical purpose of the immobilization (preconcentration, conversion, etc.). The first approach to reagent immobilization is "natural" immobilization, i.e. that selected to insoluble solids featuring the required chemicalreactivity, physical resistance to the continuous flow and size capable of withstandingpressure oscillations. Among those some "insoluble" converting reagents such asstrong reductants (Zn amalgams) [4, 64], and compounds such as CuS and CuC03,
4Cu(OHh'2HzO used for the determination of cyanide [44] and glycine [16], respectively, and some metals [8, 58] and minerals [74-76] are worth special attention.
Natural immobilization also includes other types of packed beds such as ion-exchange resins and adsorbents used for preconcentration, interference removal andoptosensing rather than as support for other active species. Thus, phosphate can bedetermined by adsorption onto Sephadex LH-20 of the association compound molydophosphate-Malachite Green [77]. The use of CI 8 absorbents enables both thepreconcentration of metals as dissolved chelates that are determined by atomicabsorption after elution [2] and placement within a spectrophotometric cell. Activatedalumnina has also been used to determine Cr(III) in urine [3] and Pb(II) in water.
Natural immobilization is also connected with the use of immobilized enzymesin their natural biochemical environment (bacteria, viruses, tissues, cells) [78-94].These materials have the advantage that the phenomena occurring in the reactor area faithful model of the biochemical processes that take place in living organisms.This approach is rather commonly used in the field of biosensors. On the other hand,
'I

Bed reactors in flow methodologies 9
this is a quite inexpensive alternative to the immobilization of enzymes from commerciallyavailable catalysts. This type of immobilization is rather straightforwardas it only requires the reagent to be packed, so it can be applied to a number of reagentsthat cannot be used in the solution for any reason.· This is thus ideal for use ofimmobilized reagents in heterogeneous FIA systems.
SCHEME 1-STRATEGIES FOR REAGENT IMMOBILIZATION
A. - PHYSICO-CHEMICAL MECHANISMS OF IMMOBILIZATION
"NATURAL" IMMOBILIZATION
Low solubillity reagents
Ion exchange resins
Adsorbents
Enzymes "naturally" immobilized
[
Inorganic compounds
Metals
[
Bacterium
Cell
Tissue
IMMOBILIZATION ON A SOLID SUPPORT
[
MiCrOPQLous membrane
Confinement or entrapment Gel entrapment
Carbon paste electrodes
Anchored by physical adsorption
Electrostatic bond on an ion exchange resin
Covalent attachment l-CPG...liilaniZed
Carbon
~I utaraldehyde
Isothiocyana te
Carbodiimide
l- Chloride cyanide
Carbodiimide
Miscellaneous
Immobilization on:
rImm..!'noassay reactions
LLiposome
B - BED CONFIGURATION
~Ilterna l tube wall (open tubular reactors)
On the inert glass beads filling the coil
•Solid particles embedded in the internal walls
ombination of different strategies

10 J. Martinez Calatayud and J. V.Garcia Mateo
The other broad category of reagent immobilization approaches involve physicalor chemical immobilization on a solid support by (a) confinement or retention, (b)adsorption, (c) electrostatic bonding to an exchange resin, or (d) covalent bonding.The different approaches to this type of immobilization can be described on the basisof enzyme immobilization, the earliest to be addressed and also the most common sofar. Many of these approaches were later extended to other types of reagent such aschelating agents.
Enzymes can be immobilized by a numberofways [95], namely: (a) by confinement of the reagent solution within microporous membranes, which has been usedto prepare flow-through electrodes [51, 96-98]; (b) by physical adsorption, whichhas so far scarcely been applied in continuous-flow methods; (c) by gel-trapping,whereby the enzyme is physically trapped during the polymerization process [99], asis the case with graphite paste electrodes [56]; and (d) by covalent bonding of theenzyme to a solid support. This last alternative has been used much more frequentlythan the other three in continuous-flow systems on account of the relatively highstability of the reagent-support bond.
Preparing a bed containing an enzyme which is covalently bond to a support iseasy and can be accomplished under mild chemical conditions [100]. Controlled poreglass is the support typically used. The glass is silanized and the enzyme is fixed ontothe resulting derivative with the aid of a compound such as glutaraldehyde, isothiocyanate, carbodiimide (on a carbon fibre support [101]), triazine or an azo dye. Thisprocedure allows immobilization of a large variety of enzymes [69]. The beds are ofhigh stability for continuous-flow work - some feature lifetime as long as 2 years.The method of choice is normally dictated by the enzyme concerned; thus, glutaraldehyde does not seem to be advisable to immobilize peroxidase by azo coupling [18],but appears to be rather appropriate for lactate dehydrogenase.
Silica as a support features excellent properties that are even better than thoseoffered by organic polymers. These properties are the followings: possibility ofbinding a large number of groups by means of differents silanizing agents, therelatively high exchange rates, short equilibration times afforded, and a good resistance to solvent changeovers below pH 9. In turn controlled pore glass (CPG) providesappropriate, readily controllable pore sizes and specific surface, so, unsurprisingly,it is the support most frequently used for this purpose [10, 11, 18, 21, 22, 100-102].
The versatility of these supports allows the immobilization of chelating agentsfor the preconcentration of metal ions; the synthetic pathway has been graduallyimproved [103] and the procedure has proved to be useful for the determination ofvarious metal ions [11]. The use of these agents offers the advantage of a highselectivity thanks to the formation of complexes with the ions concerned [11, 14, 103].
Enzymes can also be immobilized by covalent bonds to carbon supports. Carbonshould be previously activated with carbodiimide or cyanogen chloride [101]. Carbonfibres are particularly interesting since this material features a large surface area andtensile strength.
To manufacture electrode enzymes have been immobilized via glutaraldehyde.directly on platinum [97]. Platinum was pre-treated prior to silanization by anodization. That resulted in formation of the oxide film on the electrode surface. The

Bed reactors ill flow methodologies 11
oxidation can also be done thermally. in an electric furnace. Alternatively the electrodes covered by enzymes immobilized viaglutaraldehyde can be constructed fromNylon as the support. The Nylon mesh used was previously activated with dimethylsulphate and then treated with lysine (spacer) and glutaraldehyde. This technique hasbeen used to obtain electrodes with one or several immobilized enzymes [46, 49-51].
Immunological reactions are a quite interesting alternative to enzyne immobilization by covalent binding . to controlled pore glass. The enzyme is reversiblyimmobilized thorugh a reaction sequence employing an antigenantibody. Uditha [48]used two different approaches for immobilization of glucose oxidase based on thecoupling of anti-human IgG oxidase conjugate to immobilized human Ig (a) and topolyclonal goat anti-mouse IgG FAB fragments.
Liposomes offer an ingenious alternative to the immobilization of enzymaticreagents [104, 105]. This is construction ofenzyme electrodes in a packed reactorconfiguration. The principle behind this approach is as follows; the liposomesencapsulate the enzyme reagents and are broken down by an immunological reactionbetween a "foreign" species and active sites of the lipid membrane. On breakdown,the reagent reaches a high local concentration which results in a substantiallyincreased analytical signal.
Enzyme trapping in the course of gel polymerization has been exploited for theconstruction ofboth electrodes and reactors. Also, it has been used with other typesof reagent (e.g. the immobilization of copper salts is simultaneous to the polymerization process [106]). The compact material thus obtained can be divided forpreparation of the reactor.
Another physical trapping technique involves obtaining a paste by mixing powdered graphite, an enzyme and silicone oil. The paste is then used to fill-the slit at theend of the electrode . The contact is made with a copperized silver wire [56]. Thistechnique also allows reagents other than enzymes to be immobilized.
The use of membranes for enzyme immobilization relies on confining theaqueous solution of the biocatalystswithin the membrane pores. Methanol wets themembrane and penetrates the pores; once contact with the liquid has been established,other solvents of higher surface tension can replace the membrane. This techniquehas been used to prepare a single reactor [96].
The interferences with the determination of glucose in physiological sampleswith Pt electrodes have been overcome by using semipermeable membranes toexclude the interferents. The enzyme is held between two membranes: an externalone (usually polycarbonate) that allows passage of the glucose, and an internal one(generally cellulose acetate), covering the electrode surface and allowing passage ofhydrogen peroxide. A variant of this technique uses new ion-exchange and redoxmaterials that contain Pt particles electrochemically deposited on a polymer film [98].
The use of ion-exchange resins lies midway between natural and chemical-bonding immobilization. These resins have been used as sample preconcentration andconversion reagents. In fact, they have been used as exchangers for the continuousflow determination of anions such as sulphate,chloride and nitrate [8, 32] byexchanging the analyte in question by that in the resin, viz. thiocyanate, dihydroger,-phosphate or iodide, which can be readilly determined spectrophotometrically via

12 J. Martine: Calatayud and J. V.Garcia Mateo
formation of a red complex with Fe(III), a blue complex with ammonium molybdatein the presence of ascorbic acid, and a complex with Pd(II), respectively.
Another example of the use of ion-exchange resin as solid oxidants is providedby strong anionic resins loaded with hexacyanoferrate(III), which allow the oxidationof organic compounds such as paracetamol [35]·· or thiamine [67] in an alkalinemedium. This dynamic alternative has the advantage over the static procedure thatno excess oxidant ever reaches the detector, so no ascorbic acid needs to be employedto avoid a potential quenching effect. The resin preparation procedure is very simple,the use of a continuous-flow regime poses no special problems, and the reactorlifetime is long enough to justify its use [107, 108].
The combination ·of ion exchange and-atomic absorption has provided satisfactory results in the determination of traces of heavy metals. Once the required amountof sample has been introduced into the system, another injection valve is used toinsert the eluent and the carrier of the metal ion to the detector. Also, two parallelminicolumns and a multi-funcional sampling valve have been used to determineNi(II), Cu(II), Pb(II) and Cdffl). These and other metals, like Zn(II) , Fe(IlI) andCr(III), were concentrated with the aid of an iminoacetate resin [109]. The efficiencyand accuracy of on-line preconcentration with ion exchangers followed by atomicabsorption detection was studied to determine the most suitable parameters describing them. Such terms as the "preconcentration efficiency" and the "recovery'twererecommended for this purpose. Inorganic ions have also been preconcentrated for adetection other than atomic absorption [60].
At this point we would like to make some comments on the beds used. Varioustypes of reactors were prepared for application in FlA. The following procedureswere used: immobilization on the inner walls of the tube (open tubular reactor) or onparticles used to pack the tube (f:.g. the rather popular single bead string reactor,SBSR), and increasing the surface area of the walls of a glass tube with HF in orderto obtain "whiskers". The irreproducibility in surface growth and the fragility of thewhiskers formed have hindered widespread acceptance and prompted the use ofrough glass particles (with sharp apices and edges) inlaid on the walls of a Teflontube by slow heating to the melting temperature [100]. The above-described reactor,obtained by depositing a thin film of titanium dioxide on the walls of a tube, is anothercase in this point.
Open-tube reactors do not seem to be the most appropriate for use in FIA workbecause of their low sensitivity and high dispersion. In any case, the dispersion pfthe sample plug in packed reactors and open tubes should be investigated in greaterdepth before any conclusive assertions can be made in this respect. There is also alack of empirical and theoretical evidence for the optimal size and shape of particlesto be used in packed reactors. Most authors seemingly agree that SBSRs provide anideal environment for the use of enzymes in continuous-flow systems [17, 18].
Enzyme beds that combine immobilization on tube walls and the typical beadsof SBSRs are probably the most effective. Small particles seem to afford lowerdispersions and pressures at a given conversion level [18]. In recent work [112], areactor containing I-glutamate oxidase was used to determine 1-glutamate. Thesensitivity, selectivity, injection rate, and stability thus achieved were compared with

Bedreactors ill flowmethodol"gies 13
those provided by another assembly in which the reactor was replaced with anelectrode of the same enzyme. Open-tube reactors are inadequate for use withenzymes; in fact, they are better suited to continuous segmented flow techniques [17].
The efficiency of a reactor measured from the maximal reaction rate for example,depends directly 011 the used immobilization method.ithe type of bond established,and on the nature of the support. Such an efficiency can be altered by the presenceof impurities either through deactivation (e.g, proteolytic enzymes which decreasethe selectivity by yielding species to which the detector is sensitive), or bacterialgrowth (to which peroxidase appears to be immune), or ocupation of the active sitesof the enzyme by heavy metals - this can be overcome by using chelates forminghighly stable complexes with such metals.
The reaction rate constant can be increased by increasing the load onthe supportsurface and/or the reactor-solution contact surface. Thus, the reaction kinetics of animmobilized enzyme depends on a number of factors [17] and, in any case, differsaccording to whether the enzyme is immobilized or present in the solution.
Conclusions
Future trends in the use of immobilized reagents will foreseeably point indifferent directions. Thus, one should expect the development of theoretical modelsfor the physico-chemical processes of reactivity and mass transfer across the solidliquid interface, as well as for the influence of the reactor geometry, particle size,flow-rate and sample volume on the conversion efficiency.
On the other hand, new immobilization reagents, supports, and technical procedures will appear. New polymeric.materials of high resistance and chemical inertneswill foreseeably be found, and procedures for the simultaneous preparation of thesupport and immobilization of the reagent be reported. Also, immunological reactionswill expectably be more widely used as selective binding techniques.
Improved supports, reagents and eluents enabling faster operation should also besought for. The shortcomings of classical exchangers (e.g. sluggish diffusion/rediffusion) result in the removal of a substantial portion of the material used. However,the use of chromatographic materials with hard cores that limit diffusion within thesolid surface while allowing a variety of functional grups to be bound is an essentialprerequisite for successful optosensing.Classical chelating agents and silica-basedmaterials make a good alternative to the determination of cations and anions,
Miniaturization trends in FIA are justified for a variety of reasons. The use offinely divided reagents allows substantially shorter reactors to be used. On the otherhand, integrationoftheconversion-detectioll syst~m isa logical trend as, in additionto facilitating miniaturization, it increases sensitivity by avoiding dispersion of thesample onits way from the reactor to the detector. Analytebuild-up atthe optosensingsurface, followed by elution of the concentrated analyteby an appropriate change inthe carrie~. composition, is the next logical step towards accomplishing furtherminiatnrization and increased sensitivity.
New reactors containing immobilized reagents are more widely designed anrconstructed' 011 the basis of enzymes, whose ability to act in living environments

14 .J.Alarllnez Culatt1}'Utltllul.J. V.Gan:;a Mateo
endows them,with two analytical features ofmajor significance, namely (a) generallyhigh selectivity, and (b) autoregeneration via catalytic cycle. The .latter is of paramount relevance to continuous-flow work.
SCHEME I-S0lJD-PHASE REACTIONS IN CONTINUOUS FLOW ASSEMBliES
POPULARITY DUE TO ITS APPliCATION TO:
Preconc:entration
Purification
Separation:
Catalysis
Sample reaction
Unstable reagents
ADVANTAGES:
Simpliified manifolds
Lowcest
[
Increased sample throughput
Low sample dispersion .
, Increased output-height
Use of ""insolabfe" reagents
No excess of reagents (even products) in the detector
MAIN CHARACIElUS"nCS:
Size of particles
High rate of reaction
No adsorption of analyte or derivates
Stability to the continuous flow
Cbemically inert to tbe carrier or reagents
The significance of enzymatic catalysis to pharmaceutical and biomedical analysis is currently widely acknowledged and has fostered FIA work on this topic. Thesuccess behind the FIA-enzymalic catalysis couple relies heavily on the combinedexpeditiousness and simplicity of the former partner and the high selectivity of thelatter. Recent trends are aimed at simplifying and reducing costs of the immobilization process by using natural enzymes contained in biological materials such asvegetables, legumes or fruits.
Enzyme reactors have been used in a number of applications, most of which dealwith determination of glucose [56, 97, 98, 101] due to its clinical significance. Theyare compatible with at variety of detectors (spectrophotometric, chemiluminescence,ffuorimetric, amperometric (48, 91] selective-electrode potentiometry, optosensing[13], etc). On the other hand, enzyme reactors have proved to be useful for use inFIA under varied conditions: continuous flow, reversed flow, variable forward flow
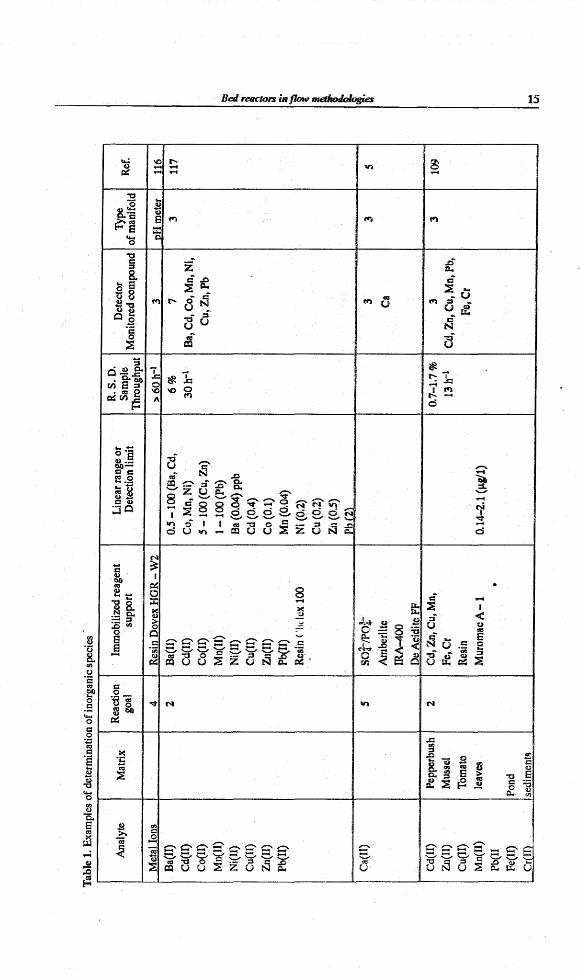
Tab
le1.
Exam
ples
ofde
term
inat
ion
ofin
orga
nic
spec
ies
Rea
ctio
nIm
mob
ilize
dre
agen
tL
inea
rra
nge
orR
.S.D
.D
etec
tor
Typ
eA
naly
teM
atrix
Sam
ple
Ref
.go
alsu
ppor
tD
etec
tion
limit
Thr
ough
put
Mon
itore
dco
mpo
und
ofm
anifo
ld
Met
alIo
ns4
Res
inD
ovex
HG
R....
W2
>60
h-1
3oH
met
er11
6B
a(II
)2
Bo(
lI)0.
5-1
00
(8a,
Cd,
6%
73
117
ed(ll
)C
d(II
)C
o,M
n,N
i)30
h-1
8a,c
s,C
o,M
o,N
i,C
o(Il
)C
o(II
)5
-10
0(C
u,Zn
)C
u,Z
n,Pb
Mn(
II)
Mn(
I1)
l-1
00
(Pb
)N
i(II
)N
i(II)
Sa
(0.0
4)pp
bC
u(II
)CU
(I1)
Cd
(0.4
)Zn
(Il)
Zn(I1
)C
o(0
.1)
Pb(I
I)Pb
(I1)
Mn
(0.0
4)R~sin
Chc
lex1
00N
i(0.
2)C
u(0
.2)
Zn
(0.5
)--
-Pb
J2)
Ca(
ll)5
SO
rfP01
=3
3S
Am
berll
teC
aIR
A-4
00D
eAci
dite
PI"
ed(l
l)Pe
pper
bush
2C
d,Z
n,C
u,M
o,0.
7-1.
7%
33
109
Zn(
Il)
Mus
sel
Fe,
er13
h=l
Cd,
Zn,
Cu,
Mn,
Pb,
Cu(
II)
Tom
ato
Res
inPe
,Cr
Mn(
Il)
leav
esM
urom
acA
-l0.
14-2
.1(~
iVl)
Pb(1
I..
Fe(lI
)Po
nd
e-m
sedi
men
ts
~ ~ ~ 8 s· l ! l t .... Vi

Cd(
n)S
eaw
ater
2C
u,Pb
,Z
n,C
d,20
-500
(ppb
)3
0-6
0h-
i3
343
Pb(I
I)R
esin
Che
lex
100
0.01
5(p
pm)
Cd,
Cu,
Pb,
Zn
Cu(
lI)
Zn(
H)
Co(
lI)
Sea
wat
er2
Co
10(p
mol
l ")
5%
95
118
8-H
ydro
xyqu
inol
ine
30h-
i
onF
ract
ozel
Cr(
III)
Uri
ne2
Cr(
III)
0-1
00
IA-g
ll)2.
4%
73
3
Act
ivat
edal
umin
a0.
05(u
e/t)
/C
r(II
I)
Cr(
VI)
Wat
er2
Cr(
VI)
/DP
C(l
Bio
-Rad
33(n
g/I)
4.3
%2
612
1
AG
-50W
-X8
4-5
h-i
Cr(
VI)
tDP
C(1
)
(550
nm)
Cr(
IlI)
Sea
wat
er2
Cr(
VI)
/8-Q
uino
lino
l0.
025
(nm
ollU
Cr(
III)
312
2
Cr(
VI)
ClI
no
pR
esin
0.07
5(n
mol
l')
Cr(
VI)
grap
hite
furn
ace
AA
S
Zn(
II)
Plan
ts2
Zn/
Chl
oroc
ompl
exe
0-2
mg
/l<
0.0
2%
32
123
resi
nD
ovex
l-X
B45
h-i
Znl
Zin
con
Sel
eniu
mT
apan
d2
..0.
002
(lA-g
ll)1
%3
312
4an
ton
Bis
mut
hm
iner
alex
chan
ger
D-2
011.
1%
hydr
ide
wat
erC
PG
-8-q
uino
lino
l0.
001
(lA-g
l1)
50
h-i
form
ing
elem
ents
Cu(
lI)
Sea
wat
er2
Cu,
Zn,
Pb,C
d2.
5-10
(mg/
I)6
0h
-i3
36
Zn(
II)
resi
nC
hele
x10
00
.03
-Zn
Cu,
Zn,
Pb,C
d
Pb(l
I)0
.07
-Cu
Cd(
II)
0.5
-Pb
0.05
-C
d(u
.!!!
l)
Cu(
II)
Tap
wat
er2
Cu,
Cd,
Fe,C
o,2
-3h-
i3
311
Zn
,Ni,
Pb
Cu,
Cd,
Fe,C
o,Z
n,
8-qu
inol
inol
onsi
lica
Ni
Pb
I-'
0\
~ ~ ;,. ~' ~ .S" s 1 ::. l ~ ~ c;') B. ::. ~ ~

Cu(
II)
2,4
PAN
~2)
0.1-
4(J
1g1m
l)2
.7%
26
28
resi
nD
owex
1(n
g/m
l)C
u(II
)IPA
N(2
)
(546
nm)
Cu(
II)
2C
u0.
01-0
.05
(um
ol)
3.4
%2
645
Bio
Rad
6-7
h-I
Cu
AG
-5O
w-X
12
Cu(
II)
2C
ompl
exC
U(I
I)or
0.02
-16
0h
-I3
32
Pb(I
I)Pb
(II)
/8-q
uino
linol
0:00
4C
u,P
b
PAN
(2)D
DC
(3)
CIS
bond
edsi
lica
10-0
.1(m
2ll)
Fe(I
II)
Wat
er2
SCN
10-4
00(n
g/m
l)3
0h
-I2
629
Win
ere
sin
Dow
ex-1
10(n~/ml)
Fe/S
CN
Fe(I
I)6
Jone
sre
duct
or3x
l<r6
-5x
10-4
(mol
l-I)
1.4
%2
469
3x1<
r6(m
oll-
I)6
0h
-lF
e(II
)fl.
10-P
T(4
)
Fe(I
II)
Silic
ate
4A
gO11
-105
(J1g
1ml)
30
h-I
24
58
rock
sF
e(II
I)ff
iron
TI(
IV)
1.2-
12.4
(uz/
ml)
30
h-I
'fiO
V)m
ron
Mo(
VI)
Nat
ural
2.5
Mo(
VI)
mro
n1.
5(n
g)34
h-I
25
,672
wat
erSe
phad
exG
-25
gel
Mo(
VI)
tnro
n
Roc
k(4
10nm
)
Ni(
II)
Sea
and
2N
i,C
u,Pb
,Cd
1.5-
4.1
%3
37
Cu(
II)
pollu
ted
Res
in12
2
Pb(I
I)w
ater
s40
h-1
Ni,
Cu,
Pb,C
d
CdO
I)
Pb(I
I)W
ater
2Pb
(II)
0-2
0(u
g/m
l)2
%3
314
8-Q
uino
linol
onC
PG1.
4(n
z/m
l)20
h-I
Pb
Pb{I
I)Se
awat
er2
Pb50
-200
0(!
!glm
l)7
%2
560
resi
nC
hele
x10
05
(ul!!
mn
45
h-I
Pb/d
itizo
ne
~ ~ r:. e ~ ~ ~ ::i ~ a c 6'" l ~ ..,J

Pb(I
1)W
ater
2ac
tiva
ted
alum
ina
0-10
0(I
Ag/
mt)
1.4
%3
34
..0.
36(u
z/I)
5h-
lPb
Pb(I
1)W
ater
2Pb
(II)
,C
u(II
)0.
001-
0.01
(ug/
ml)
6h-
l2
311
9
Cu(
II)
resi
nC
hele
x10
05x
lO-s
-0.1
(mol
lr')
Pb(
II)/
PA
R(5
)D
owex
1-X
8(5
25nm
)
0.7
(ug/
ml)
35h-
l5
1.5x
10-5
(mol
1-1 )
Cu
Mn
S0
46
gold
pow
der
(l-6
)x10
-522
5h-
l2
165
NaN
02
9.1x
10-6
Ag
(II)
/HN
0 2
CO
(N0
3h
1O-5
-4x
10-4
(390
nm)
Phen
ol4.
4x10
-s
(1-7
.5)x
10-4
1.2x
10-4
7.5x
lO-5
-2x
10
-4
4.2x
IO-5
(mot
r')
NaN
02
6go
ldpo
wde
r10
--4-
5.4x
10-4
21
66
VO
S0
44x
lO--
4-2x
10-3
Mn(
III)
/H2
S04
Ce(
N02
)31.
7x10
-3-7
xl0
-3(4
90nm
)
KI0
3,
2x10
-3-7
xlO
-3
(CO
ON
a)2
-2x
10-3
-6x
10
-3
-C@
IsC
OO
H4x
10--
4-1.
4x10
-3
(mo
ll-l
)
Be
5re
sin
1-10
0(u
g/m
l)13
3h-
i9
334
Dow
exH
CR
-W3.
75(p
g)
CI-
Wat
er2
,5A
g0.
5-10
(ug/
g)9
433
Dow
ex5O
W-X
850
(ng/
g)H
+
Free
Tap
wat
er4
Ura
nine
10-3
-2x1
0-<>(
mol
L'!
1.6
%8
673
Chl
orin
eA
mbe
rlit
eIR
A-9
30.
1ug
/ml
..... 00 ~ ~ ::t ::;- s ~ :r- ~ t § ~ ~ .... f ~ ~
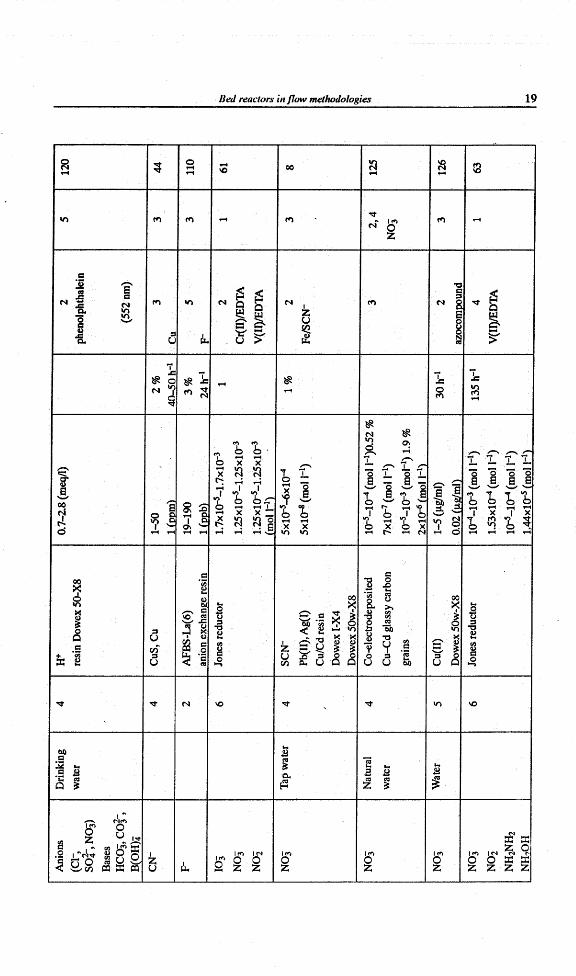
Ani
ons
Dri
nkin
g4
H+
0.7-
2.8
(meq
/l)
25
120
(CI-
,w
ater
resi
nD
owex
50-X
8ph
enol
phth
alei
nSO~-,N03)
Bas
esH
C0 3
,CO
j-,
(55
2n
m)
B(O
H)4
CN
-4
Cu
S,C
u1
-50
2%
33
441
(oo
m)
40
-50
h-l
Cu
p-2
AF
BS
-La(
6)19
-190
3%
53
110
anio
nex
chan
zere
sin
1(p
pb)
24
h-1
p-
103
6Jo
nes
redu
ctor
1.7
xlo
-s-1
.7x
l0- 3
12
161
N03
l.2
5x
lo-s
-1.2
5xl0
-3C
r(II
)IE
DT
A
N02
1.25
xlo-
s-1.
2Sxl
0-3
V(I
I)IE
DT
A(m
oll
-I)
N03
Tap
wat
er4
SC
N-
5x
lo-s
-6xl
O""
"'1
%2
38
Pb(
II),
Ag{
I)5x
lo-B
(mo
ll-I
)F
e/S
CN
-C
u/C
dre
sin
Dow
exI-
X4
Dow
ex5O
W-X
8
N03
Nat
ural
4C
o-el
ectr
odep
osit
ed10
0s-10
"""'
(mol
1-1 )
0.52
%3
2,4
125
wat
erC
u-C
dgl
assy
carb
on7x
10-7
(mol
lr')
NO
)gr
ains
1O-s
-10-
3(m
olt')
1.9
%
2xl<
r6(m
ol.)
-l)
NO
)W
ater
5C
u(II
)1
-5(J
lg/m
l)3
0h
-12
312
6D
owex
5OW
-X8
0.02
(l.H
uml)
azoc
omoo
und
NO
)6
Jone
sre
duct
or10
-4-1
0-3
(mol
r")
135
h-l
41
63N
02
1.53
x10-
4(m
ollr
')V
(II)
IED
TA
NH
2NH
210
-S-1
0-4
(mo
ll- l
)
....ll
li1Q
Hl.
44xl
o-s
(mol
1...1 )
~ ~ ~ i:! ~ ~ :; ~ ~ 5'" l I-"
\C

10-4
-10-
3(m
ol.r
")9
.5x
l0-5
(mol
14)
5x
l0-5
-1.5
xlQ
-4(m
olr"
)3.
25xl
0-5
(mo
ll- l
)
N0 3
6Jo
nes
redu
ktor
2.5
xl0
-s-1
Q-4
(mol
r")
170
h-l
61
62
NO
,3.
4xlO
-5(m
oll"
)C
r(II
I)/E
DT
A
N0"
36
Jone
sre
duct
orlO
-s-l
Q-4
(mo
ll-l
)37
5h-
l2
164
N02
4xlO~
(mol
r")
V(I
II)/
ED
TA
10"3
4I
V(I
II)/
ED
TA
Phos
phat
eW
ater
4Se
phad
exL
H-2
02
0h
-l2
:'6
77
mol
ybda
te-M
alac
hite
Gre
enS2
-5
Cd(
II)
0-2
0(i
ng/l)
1.2
%3
312
resi
n12
210
(~g!l)
100
h-l
Cd
8-ou
inol
inol
onC
PGS2
-4
NN
P(7
)0.
05-0
.6(p
pm)
26
26
resi
nD
owex
Met
hyle
neB
lue
50
So
wX
8S2
-4
DIP
,DC
,CI
0.31
-2.5
(nm
ol/l)
26
53
(8)
(9)
(10)
redu
cing
form
im-
silic
aee
lm
obili
zed
reag
ent
SO~-
Hot
spri
ng0-
1000
(ppm
)0
.95
%2
347
I···
H(C
sCh 0
4)2-
wat
er5
(ppm
)
(53
5n
m)
SO~-
Tap
wat
er5
,4C
N-
5xI0~-2.5xlQ-4
1%
23
32
Cl-
1-(m
ollr
')F
e(II
I)/S
CN
-
N03
P0
4H2
l-tp
d(I1
)
A2+
5xI0~
(mo
ll-l
)
~ ~ ~ ~ :::- n ~ r ~ l § ~ ~ =" ~ 3. ~ ~ ~

.-1C
'It.~
~'e~l::$~.5~~~CQ
,..........",_.-.,D
owex
1-X4
Dow
ex5O
W-X
8
Uranium
Ores
4P
bp
ow
der
0.6
2%
25
126
Leachates
48h- 1
V(IV
)Arsenazo
III(665
nm):
2,4-DN
PH
(32
amines
5x10- 7-3
xl(J
61.43
%2
631
2-N
PH
(4)
C18bonded
silica5x
10- 7_
5x10-6
2.0
5%
2,4-DN
PH
(11),
4-NP
H(S
)5
xl0
- 7-3
x1
0-6
2.9
4%
2-NP
H(12)
5x
l0- 7
(mol
1- 1)40
h- 14-N
PH
(13)
ED
TA
4C
u(II)0.5-50
(ug/ml)
1.5%
33
127
resinC
helex100
0.1(u.£!Im
l)45
h.!1C
u
H2 0
2W
ater4
TC
PO
(14
)0
-10
- 5(m
ol1- 1)
2.8
%9
352
0.2(u.~JI)
120h- 1
H2 0
24
Lum
inol5x10- 5
-5x
10
-33
-4%
96
10
CPG
(l0)
10-6(m
ollrl)
30h- I
H2 0
24
Lum
inol40-600
(umol
H)
18
%9
323
silicazel
10(I..lm
oll- 1)
Methanol
Water
2titanium
dioxideinner
10- 9(m
ol1- 1)
15
%9
368
wall
tubeteflon
30
h- 1
RE
AC
TIO
NG
OA
L:1
-sam
plepretreatm
ent,2-
preconcel1tration,3-
reactor-detector,4-
reactor,5
-elim
inationof
interferences,6-
insitu
reagentsgeneration
DE
TE
CT
ION
:1
-.fluorimetry,
2-
UV....;.visibl~spectroscopy,~
-atom
icabsorption
spectrometry,4
-am
perometry,5
-potentiom
etry,6-
polarography,7-
inductivelycoupled
plasma
ermsionspectrom
etry,8-Iu
mm
lscence
defection,9
-conductJrnetry
RE
AG
EN
TS:
(1)1,5-di phen
ylcarbazide,.(2)1-(2-PYridYlaZO)reSOrcinOI,~3}
diethyldithiocarbamate,(4)
1,.1D-P
henanthroline, (5)4-(2- py.ridY
laZO
)reSOrcinO
I'p6) alizarinfluorine
bluesu1phonate-lanthanum
, (7)NN
-dimelfiyl-p-phenyldiam
ine,8)
26-dichlorophenylindophenol,(9)
tris{1,10-phenanthroline)iron(III)
complex,
0)dithio
fluoresceinsilver
complex,
(11)2,4·<
hmtrophenylhydrazine,(12)
2-nitropenylhydrazine,(13)
4-nitrophenylhydrazme,(14)
bis(2,4,6-trichlorophenyloxalate
TY
PEO
F~NIFOLD
-num
bers(1
to6)
likein
Figure1

22 J. Martlne: Calatayud and J. V. Garcia Mateo
and stopped flow. They can be used alone or combined [49], or as several reactorsarranged in series or in parallel [113, 114], for individual [23, 54, 115] or multipledeterminations [19, 54] both simultaneous and sequential. Also, they can be used asbinary combinations in the same electrode [46], in association with a dialyser for thedirect treatment of heterogeneous sample [15], and even with the optosensing mode.
REFERENCES
1. Linares P., Luque de Castro M. D. and Vdlcarcel M., Rev. Anal. Chem., 8, 229 (1985).2. Ruzicka J. and Arndal A,Anal. Chim. Acta, 216,243 (1989).3. Cox A. G. and McLeod C. W., ibid.; 179,487 (1986).4. Zhang Y, Riby P., Cox A G., Cameron W., McLeod C. W., Date A. D. aad Gheng Y .Y.,Analyst, 113,
125 (1988).5. Karnson O. E and Townshend A,Anal. Chim. Acta, 155, 253 (1983).6. Fang Z., Ruzicka J.and Hansen E. H., ibid., 164, 23 (1984).7. Fang Z, Xu S. and Zhang S.,ibid., 164, 41 (1984).8. Devi S., and Townshend A, ibid., 225, 331 (1989).9. Ira S. K and Ernst P. L., Int. Lab., June, 1982.
10. Hool K and Nieman T.A.,Anal. Chem., 59, 869 (1987).11. Malamas E, Bengtsson M. and Johansson G.,Anal. Chim. Acta, 160, 1 (1984).12. Peterson B. A, Fang Z., Ruzicka J. and Hansen E. L., ibid., 184, 165(1986).13. Jeppesen M. 'f. and Hansen E. H., ibid., 214, 147 (1988).14. Bysouth S. R, Tyson J. E and Stockwell P. R, ibid., 214, 329 (1988).15. Maeder G., Veuthey J. L., Pelletier M. and HaerdiW., ibid., 231, 115 (1990).16. Martinez Cala tayud J. and Garcia Mateo J. v., 2nd Int. Symp. on Pharm. and Biomed. Anal., York, April,
1990.17. Johansson G., Ogren L. and Olsson B., Anal. Chim.Acta,145, 71 (1983).18. Olsson B. and Ogren L., ibid., 145, 87 (1983). ,19. Masson M. and Townshend A.,Anal. Proc. (London), 22, 6 (1985).20. Gnanasekaran R. and Mottola H. A,Ana/. Chem., 57,1005 (1985).21. Olsson R, Stalbom B. and Johansson G., Anal. Chim.Acta, 179,203 (1986).22. Ruz J., Luque de Castro M. D. and Valcarcel M.,Analyst, 112,259 (1987).23. Hool K and Nieman T. A, Anal. Chem., 60, 834 (1988).24. Ruzicka J. and Hansen E. H.,Anal. Chim.Acta, 214,1 (1988).25. Ruzicka J. and Hansen E. H., ibid., 173, 3 (1985).26. Martinez A, Moreno M. C. and Camara C.,Anal. Chem., 58,1877 (1987).27. Wolfbeis O. S. and Schaffar R P. H.,Anal. Chim. Acta, 198,1 (1987).28. Lazaro E, Luque de Castro M. D. and Valcarcel M., ibid., 214, 217 (1988).29. Lazaro E, Luque de Castro M. D. and Valcarcel M., ibid., 219, 231 (1989).30. Linares P.,Luque de Castro M. D. and Valcarcel M., ibid., 230, 199 (1990).31. Band B. D., Lazaro E, Luque de Castro M. D. and Valcarcel M., ibid., 229, 177 (1990).32. Faizullah A 'f. and Townshend A, ibid., 179, 233 (1986).33. Lach G. and Bachmann K, ibid., 196,163 (1987).34. Sharik M. A and Faizullah A 'f.,Analyst,114, 951 (1989).35. Martinez Calatayud J,and G6mez Benito C., Anal. Chim. Acta, 231, 259 (1990).36. Guilbault G. G. and LubranoG. J., ibid., 60, 254 (1972).37. Notin M., Guillien R. and Nabet P.,Anll. Bioi. Clin. (Paris), 30,193 (1972).38. Dahodwala S. K, Weibel M.K and Hurnphrcv A.E., Biotechnol. Bioeng., 18, 1679 (1976).39. Fundamentals ofClinical Chemistry., 2nd cd.. lictz W. N. Ed., Sannders, Philadelphia, PA, 1976.40. Garber C. t., Feldbruegge D., Miller R.C. and Carey R.N., Clin. Chem. (Winston-Salem, N.C.), 24,1186
(1978).41. Carr P.W. and Bowers L. D., Immobilized Enzymes in Analytical and Clinical Chemistry. Fundamentals
and Applications, Wiley, New York 1980.42. Bowers L. D. and Johnson P. R, Clin. Chem. (Winston-Salem, NC.), 27,1554 (1981).43. Olsen S., Pessenda L. C. R, Ruzicka J. and Hansen E. H.,Analyst, 108, 905 (1983).

Bed reactors itt flow methodologies 23
44. Haj-Hussein A. T., Christian G. D. and Ruzicka J.;Anal. Chem., 58, 38 (1986).45. Yoshimura K., ibid., 59, 2922 (1987).46. Moody G. J., Sanghera G. S. and Thomas J. D. R.,Analyst, 112, 65 (1987).47. Toei J., ibid., 112, 1067 (1987).48. De Alwis W. U., Hill B. S., Meiklejohn B. I. and Wilson G. S.,Anal. Chem., 59, 2688 (1987).49. Hamid J. A, Moody G. J. and Thomas J. D. R.,Analyst., 113,81 (1988).50. Cosgrove M., Moody G. J. and Thomas J..D. R., ibid., 113, 1811 (1988).51. Hamid J. A, Moody G. J. and Thomas J. D. R., ibid., 114, 1587 (1989).52. Van Zoonen P., Kamminga D. A, Gooijer C., Velthorst N.H. and Frei R. W.,Anal. Chim.Acta, 167,249
(1985).53. Narayanaswamy R. and SevillaE,Analyst, 111,1085 (1986)-54. Lazaro E, Luque de Castro M. D. and Valcarcel M.,Anal. Chem., 59,1859 (1987).55. Woods B. A, Ruzicka J. and Christian G. D., ibid., 59, 2757 (1987).56. Matuszewski W. and Trojanowicz M.,Analyst, 113, 735 (1988).57. Den Boef G., Anal. Chim. Acta, 216, 289 (1989).58. Kozuka S., Saito K., Oguma K. and Kuroda R,Analyst, 115, 431(1990).59. Martinez Calatayud J. and Garda Mateo J. v.,J. Pharm. Biomed. Anal., 7,1441 (1989).60. Novikov E. A, Shpingun L. K., Zolotov Yu. A,Anal. Chim. Acta, 230, 157 (1990).61. Schothorst R. C, Reijn J. M., Poppe H. and Den Boef G., ibid., 145,197 (1983).62. Schothorst R. C. and Den Boef G., ibid., 153, 133 (1983).63. Schothorst R. C., Van Veen J. J. E and Den Boef G., ibid., 161, 27 (1984).64. Schothorst R C., Van Son M. and Den Boef G., ibid., 162, 1 (1984).65. Schothorst R. C. and Den Boef, G., ibid., 169, 99 (1985).66. Schothorst R C, Schmitz O. O. and Den Boef G., ibid., 179, 299 (1986).67. Martinez Calatayud J., Gomez Benito C and Gimenez D. G.,J. Pharm. Blomed. Anal., 8, 667 (1990).68. Low G. K. C. and Mathews R W.,Anal. Chim.Acta, 231,13(1990).69. Faizullah A T. and Townshend A., tu«, 167,225 (1985).70. Valcarcel M. and Luque de Castro M. D.,Alutlyst, 115, 699 (1990).71. Lazaro E, Luque de Castro.M. D. and Valcarcel M.,Anal. Chim. Acta, 219, 231 (1989).72. Yoshimura K, Matsuoka S. and Waki H., ibid., 225, 313 (1989).73. Nakagama T., Yamada M. and Hobo T., ibid., 231, 7 (1990).74. Kojlo A and Martinez CalatayudJ.,J. Pharm. Biomed.Anal., 8, 663 (1990).75. Martinez Calatayud J., Sagrado Vives S. and Sanmiguel Roche E, Quim. Anal. (Barcelona), 8, 455
(1989).76. Varma S. R, Martinez Calatayud J. and Mottola H. A,Anal. Chim. Acta., 233, 235 (1990).
. 77. Yoshimura K, Nawata S. and Kura G.,Analyst, 115,843 (1990).78. D'Orazio P., Meyerhoff M. E. and Rechnitz G. A,Anal. Chem., 50, 1531 (1978).79. Grobler S. R., Bassen N. and Van Wyk C. W., Talanta, 29,49 (1982).80. Di Paolantonio C. Land Rechnitz G. A, Anal. Chim. Acta, 141, 1 (1982).81. Di Paolantonio C. L. and Rechnitz G. A, ibid., 148, 1 (1983).82. Vincke B. J., Deoleeschonwer M. J. and Patriarche G. J.,Anal Lett., 16, 673 (1983).83. Rechnitz G. A, Reichel T. L., Kobos R K and Gebaner C R.,Anal. Chim.Acta, 94,357 (1977).84. Reichel T. L. and Rechnitz G. A,J. Membr. Sci., 4, 243 (1978).85. Rechnitz G. A, Science, 214, 287 (1981).86. Kuriyama S. and Rechnitz G. A,Anal. Chim. Acta, 131,91 (1981).87. Arnold M. A and Rechnitz G. A,Anal. Chem., 54,777 (1982).88. Suzuki S., Satoh 1. and Karube 1.,Appl. Biochem. Biotechnol., 7,147 (1982).89. Schubert E, Scheller F: and Mohr P.,Anal. Lett., 15,681 (1982).90. Schubert E, Wollenberger U. and Scheller E, Biotechnol. Lett., 5, 239 (1983).91. Kuriyama S., Arnold M. A. and Rechnitz G. A,J. Membr. Sci., 13,269 (1983).92. Schubert E, Renneberg R., Scheller E W. and Kirstein L.,Anal. Chem., 56, 1677 (1984).93. Ma Y. L. and Rechnitz G. A, Anal. Lett., 18, 1635 (1985).94. Belli S. L. and Rechnitz G. A, ibid., 19, 403 (1986).95. Mottola H. A, Quim. Anal. (Barcelona), 8, 119 (1989).96. Hwang H. and Dasgupta P. K., Anal. Chem., 59, 1356 (1987).97. Beh S. K., Moody G. J. and Thomas J. D. R,Analyst, 114,29 (1989).98. Gunasingham H. and Tan C B., ibid., 114, 695 (1989). •99. Yamato S. and Shimade K.,Anal. Chim.Acta, 232, 281 (1990).
100. Gosnell M. C, Snelling R E. and Mottola H. A.,Anal. Chem., 58,1585 (1986).