biochemistry of disease - si. · pdf fileอาการของโรคทาง metabolic...
TRANSCRIPT

BIOCHEMISTRY OF DISEASEAssistant Professor Dr. Chatchawan Srisawat M.D., Ph.D.

INTRODUCTION

INTRODUCTION
วัตถุประสงค
• นักศึกษาสามารถเขาใจถึงขบวนการทางชีวเคมีที่ปกติ และขบวนการที่เปลี่ยน แปลงไปในโรคทาง metabolic ดังกลาว
• สามารถนําความรูพื้นฐานทางชีวเคมีมาใชในการอธิบายความผิดปกตหิรืออาการของโรคทาง metabolic ดังกลาว รวมทั้งหลักการที่นํามาประยุกตใชในการรักษา
• นักศึกษารูจักโรค genetic metabolic disorders ที่สําคัญและสาเหตขุองการเกิดโรคดังกลาว

INTRODUCTION
• Genetic metabolic disorders เกิดจากความบกพรองแตกําเนิดของการทํางานของเอนไซม, cofactor ของเอนไซม หรือ โปรตีนที่ทําหนาที่ขนสง (transporter) ทําใหขบวนการ metabolism ของสารเสียไป

INTRODUCTION
substrate products
intermediate in alternative pathway
• โรค genetic metabolic disorders สวนใหญมีการถายทอดแบบ autosomal หรือ x-linked recessive
• การบกพรองของการทํางานของเอนไซมใน metabolic pathway สามารถทําใหเกิดอาการของโรคไดโดย:
enzyme defect
- การคั่งของ substrate หรือ intermediate ใน alternative pathway อาจจะทําใหเกิดพษิตอเซลลและรางกาย
- การลดลงของ product อาจจะเกิดการรบกวนตอ normal metabolism ของสารดังกลาว ทําใหการทํางานของเซลลและรางกายผิดปกติ

INTRODUCTION
• โรค genetic metabolic disorders แตละโรคพบไดนอย แตเมื่อคดิเปนกลุมของโรคโดยรวมแลว จะพบวาเปนโรคกลุมใหญ ที่สามารถพบไดคอนขางบอย
อุบัติการณ: จะพบทารกแรกเกิดปวยดวยโรค metabolic disorder 1 คนตอทารกแรกเกิดทุกๆ 1400 – 5000 คน
โดยเฉพาะอยางยิ่ง ถาทารกมีอาการของการติดเชื้อในกระแสเลือดโดยไมทราบสาเหตุ จะพบวา โอกาสที่ทารกดังกลาวนาจะเปนโรค metabolic disorder มีถึง 20%
• ดังนั้น การที่แพทยตระหนักถึงภาวะดังกลาว และใหการวินิจฉัย รวมทั้งการรักษาที่ถูกตอง ทันทวงที สามารถชวยปองกันการตาย หรือ อาการที่รุนแรงของโรค เชน ภาวะปญญาออน ไมใหเกิดขึ้นได

INTRODUCTION

INTRODUCTION
MAJOR CLASSES OF METABOLIC DISEASES
Disorders of carbohydrate metabolism
Disorders of fatty acid metabolism
Disorders of amino acid metabolism
Disorders of nucleic acid metabolism*
- Disorders of fructose metabolism- Disorders of galactose metabolism- Glycogen storage diseases
- Dyslipidemia*- Fatty acid oxidation defects
- Albinism- Phenylketonuria (PKU)
* อยูในหัวขออื่นที่จะไดเรียนตอไป
Organelle-related disorders- Lysosomal storage diseases
Disorders of sphyngolipid metabolismMucopolysaccharidoses

DISORDERS OF CARBOHYDRATE METABOLISMDISORDERS OF CARBOHYDRATE METABOLISM

DISORDERS OF GALACTOSE METABOLISM
• galactose เปนน้ําตาลที่สําคัญในน้ํานมโดยรวมกับ glucose ในรูปของ lactose จึงเปนแหลงพลังงานที่สําคัญในทารกที่ตองดื่มนม
• นอกจากนี้ galactose ยังเปนน้ําตาลชนิดหนึ่งที่เปนสวนประกอบของกลุมน้ําตาลใน glycoprotein และ glycolipid

DISORDERS OF GALACTOSE METABOLISM
Metabolism of galactose
galactokinase+
galactose galactose-1-phosphate UDP-glucose
UDP-galactose glucose-1-phosphate
UDP-glucose
Gal-1-P uridyltransferase
UDP-Gal 4-epimerase G-6-Pเขาสู glycolysisglycogen
synthesis
+
• รางกายสามารถเปลี่ยน glucose และ galactose กลับไปมาไดตามความตองการของรางกาย

DISORDERS OF GALACTOSE METABOLISM
ความผิดปกติของ galactose metabolism ทําใหเกิด galactosemia ซึ่งเกิดไดจาก:
1. Galactokinase deficiency
2. Galactose-1-phosphate uridyltransferase deficiency

DISORDERS OF GALACTOSE METABOLISM
Galactokinase deficiency
galactokinase
galactose galactose-1-phosphate
• ผูปวยไมสามารถเปลี่ยน galactose เปน galactose-1-phosphate ไดทําใหมี galactose ถูกขับออกมาในปสสาวะ
• นอกจากนั้น galactose สวนเกินยังถูกเปลี่ยนเปน galactitol โดย aldose reductase

DISORDERS OF GALACTOSE METABOLISM
Galactokinase deficiency
• การสะสมของ galactitol ซึ่งมี –OH group มากทําใหดูดน้ําไดด ีภายใน lens ของตา จะทําใหมี osmotic swelling ของ lens fiber และ denature ของ lens protein
ผูปวยที่ไมไดรับการรักษาจะมีตอกระจก (cataract) เกิดขึ้น

DISORDERS OF GALACTOSE METABOLISM
Galactose-1-phosphate uridyltransferase deficiency
ผูปวยจะมีการคั่งของ galactose-1-phosphate และ galactose คั่ง
มีการสะสมของ galactitol ทําใหเกิดตอกระจก
การสะสมของ galactose-1-phosphate และการลดลงของ inorganic phosphate ในเซลลซึ่งจําเปนในการสราง ATP ทําใหเปนสาเหตุของพยาธิสภาพของตับ, ไต, และสมอง แตกลไกแนนอนยังไมทราบแนชัด
ผูปวยจะมีตับโต ตัวเหลือง การทํางานของตับและไตลมเหลวและอาจมีการพิการทางสมองตามมา

DISORDERS OF GALACTOSE METABOLISM
การรักษา
• หลีกเลี่ยงการรับประทานนมหรืออาหารที่มีนมเปนสวนประกอบ หรือดื่มนมที่ไมมี lactose (lactose-free formula)
Note ผูปวยซึ่งไดรับการรักษาดวย galactose-free diet และไมไดรับ galactose เลย สามารถที่จะมีการเจริญเติบโตและพัฒนาการที่ปกติได เนื่องจากรางกายยังสามารถสังเคราะห galactose ไดจาก glucose(จากการทํางานของ UDP-Gal 4-epimerase)

DISORDERS OF GALACTOSE METABOLISM
การรักษา

DISORDERS OF FRUCTOSE METABOLISM
• fructose เปน monosaccharide ที่สําคัญชนิดหนึ่ง เนื่องจากเปนสวนประกอบของ sucrose ซึ่งเปน disaccharide ที่ใชเพิ่มความหวานในอาหารที่มีการใชกันอยางแพรหลาย
• fructose ยังพบในรูป free from ในน้ําตาลจากน้ําผึ้ง ผลไม และ ผักหลายชนิด

DISORDERS OF FRUCTOSE METABOLISM
Metabolism of fructose
ความผิดปกติของ fructose metabolism อาจเกิดไดจาก:1. fructokinase deficiency ทําใหเกิด essential fructosuria
2. fructose 1-phosphate aldolase (aldolase B) deficiency ทําใหเกิด hereditary fructose intolerance
เขาสู glycolysis หรือนําไปสราง glucose

DISORDERS OF FRUCTOSE METABOLISM
Essential Fructosuria
• เกิดจากการบกพรองของ fructokinase
• fructose ที่รับประทานเขาไปสวนหนึ่งจะถูกขับออกทางปสสาวะ และอีกสวนหนึ่งจะถูก metabolized ในกลามเนื้อและเนื้อเยื่อไขมันโดย hexokinase ซึ่งจะทํางานเมื่อ fructose มีการคั่งและระดับเพิ่มสูงขึ้น
• ผูที่มีการบกพรองของ fructokinase จะไมมีอาการทางคลินิค แตอาจตรวจพบวามี reducing sugar ออกมาในปสสาวะ
• ไมตองการการรักษา

DISORDERS OF FRUCTOSE METABOLISM
Hereditary Fructose Intolerance
• เกิดจากการบกพรองของ aldolase B ทําใหเซลลไมสามารถนํา fructose ไปใชเปนพลังงานหรือเปลี่ยนเปน glucose ได
• เมื่อรับประทานอาหารที่มี fructose จะมีการสะสมของ fructose 1-phosphate
fructose 1-phosphate ที่เพิ่มสูงขึ้นจะเกิดผลตอขบวนการ metabolism ดังตอไปนี้

Hereditary Fructose Intolerance
• fructose 1-phosphate จะยับยั้งการสลาย glycogen (โดยยับยั้ง glycogen phospholyrase) และ ยับยั้ง gluconeogenesis (โดยยับยั้งการเปลี่ยน F6P -> G6P และ การสราง F1,6P จาก glyceraldehyde-3P และ DHAP)
ผูปวยจะมีอาการของน้ําตาลในเลือดต่ําหลังรับประทานอาหารที่มี fructose เชนมี อาการคลื่นไส อาเจียน เหงื่อออก ซึม เกร็งกระตุก หรือชัก
DISORDERS OF FRUCTOSE METABOLISM

Hereditary Fructose Intolerance
• fructose 1-phosphate ที่เพิ่มขึ้นยังทําให inorganic phosphate ที่จําเปนตองใชในการสราง ATP ลดลง ทําใหเกิดการขาดหรือพรอง ATP ซึ่งเปนแหลงพลังงานของเซลล
DISORDERS OF FRUCTOSE METABOLISM
ผูปวยมีการเจริญเติบโตลมเหลว การทํางานของตับและไตบกพรอง

DISORDERS OF FRUCTOSE METABOLISM
การรักษา
• งดอาหารทุกชนิดที่ fructose หรือ sucrose
• การควบคุมอาหารจะทําใหอาการตางๆหายไปอยางรวดเร็ว และการเจริญเติบโตเปนปกติ

GLYCOGEN STORAGE DISEASES
• Glycogen เปน homopolymer ของ glucose ที่ทําหนาที่เก็บสะสมกลูโคสในเนื้อเยื่อตางๆ โดยเฉพาะที่ตับและกลามเนื้อ
• การสังเคราะหและการสลายของ glycogen อาศัยเอนไซมหลายชนิดและถูกควบคุมดวยฮอรโมน
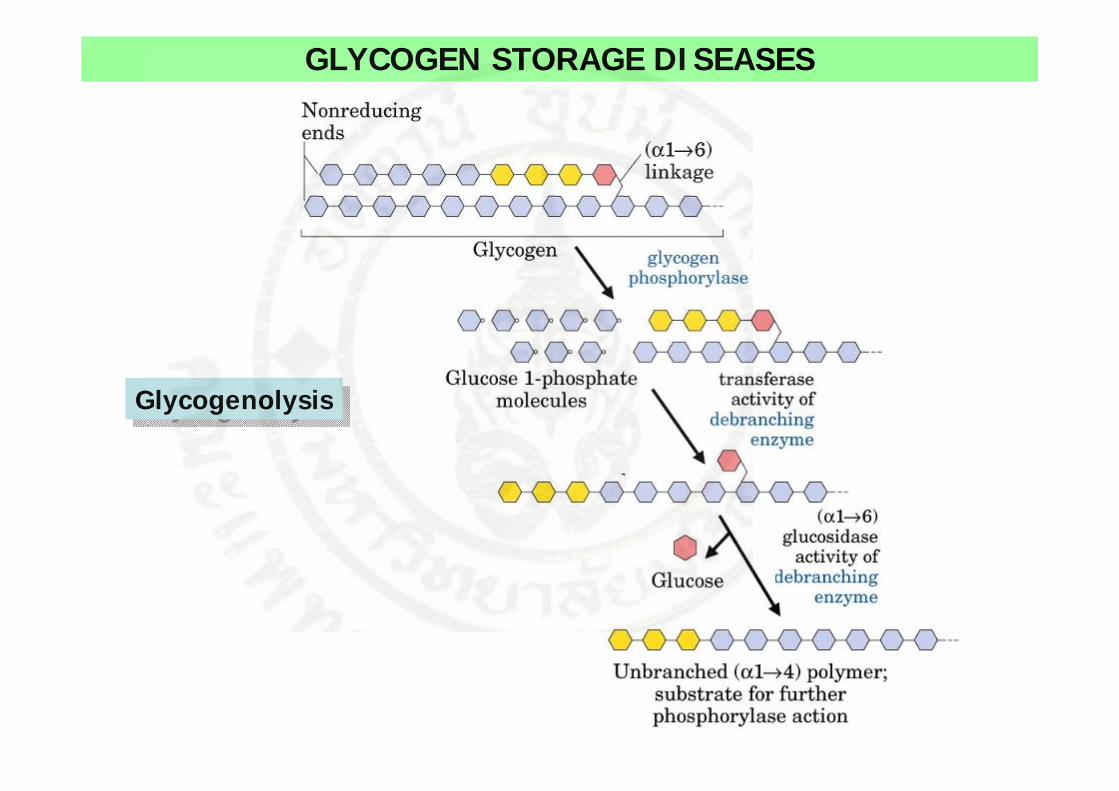
GLYCOGEN STORAGE DISEASES
GlycogenolysisGlycogenolysis

GLYCOGEN STORAGE DISEASES• ในปจจุบัน พบวามี glycogen storage disease (GSD) อยางนอย 10 ชนิดตางๆกัน ซึ่งแบงตามความบกพรองของเอนไซมดังตัวอยางขางลาง
เอนไซมที่ผดิปกติ affected organs ชนิดของ GSDGlucose-6-phosphatase Liver, kidney type IaAcid α-1,4-glucosidase Generalized type IIDebranching enzyme Liver, muscle type IIIBranching enzyme Liver type IVPhosphorylase Liver type VI

GLYCOGEN STORAGE DISEASES
• GSD ชนิดที่พบบอยคือ type I, II, III และ IV ชนิดอื่นพบไดนอยมาก
• อาการของโรคขึ้นอยูกับชนิดของเอนไซมที่บกพรอง
ตัวอยางอาการของผูปวยดวย GSD type I (Von Gierke’s disease)
• hypoglycemia อยางรุนแรงเมื่ออดอาหารเพียงไมนาน หรือมีภาวะ stress เชน เปนไข ฯลฯ
• ระดับ lactate ในเลือดสูง จนทําใหเลือดมีภาวะเปนกรด (acidosis) ได
• ระดับ triglyceride ในเลือดสูง
• ตับโตมาก พบ glycogen และ fat สะสมอยูในเซลลตับ
• growth retardation

glucose glucose-6-phosphate
glycogen phosphorylase
debranching enzyme
glucose-6-phosphatase
Glycogen
liver
glucose-1-phosphate
From gluconeogenesis
• เกิดการสะสมของ glucose-6-phosphate- ทําใหไมสามารถสลาย glycogen ไดดี เกิดการสะสมของ glycogen ในตับ
GLYCOGEN STORAGE DISEASES
ผูปวยจะมีตับโต (hepatomegaly)
GSD type I

glucose glucose-6-phosphate
glycogen phosphorylase
debranching enzyme
glucose-6-phosphatase
Glycogen
liver
glucose-1-phosphate
From gluconeogenesis
• ผูปวยจะมีอาการของ hypoglycemia ในระยะ fasting เนื่องจาก- ไมสามารถผลิต glucose จากขบวนการ glycogenolysis- ไมสามารถผลิต glucose จากขบวนการ gluconeogenesis
GLYCOGEN STORAGE DISEASES
GSD type I

• การขาด G-6-phosphatase ทําใหเกิดการสะสมของ G-6-P, F-6-P และ F-2,6-P
Glucose
Glucose-6-phosphate
Fructose-6-phosphate
Fructose-1,6-bisphosphate
Glyceraldehyde-3-phosphate
1,3-bisphosphoglycerate
3-phosphoglycerate
2-phosphoglycerate
Phosphoenolpyruvate
Pyruvate
Fructose-2,6-bisphosphate+PFK-1
- กระตุน phosphofructokinase-1 (rate-limiting step ของ glycolysis)
Lactate
liver
GLYCOGEN STORAGE DISEASES
GSD type I
ระดับ pyruvate และ lactate เพิ่มสูงขึ้นในเลือด

การสะสมของ triacylglycerol ในตับและมีระดับในเลือดสูงขึ้น
• การทํางานของ glycolysis ที่มากขึ้น ทําใหมี substrate สําหรับการสังเคราะห triacylglycerol มากขึ้น
pyruvate
acetyl CoA
oxaloacetatecitrateTCA
cycle
glucose
glycolysisacetyl CoA
fatty acid glycerol
triacylglycerolLipoprotein (VLDL)
NADPH
DHAP
liver
GLYCOGEN STORAGE DISEASES
GSD type I

GLYCOGEN STORAGE DISEASES
การรักษา
• เนื่องจากอาการของโรคโดยเฉพาะ hypoglycemia จะเกิดขึ้นเมื่อมีการอดอาหาร ดังนั้น ควรปองกันไมใหเกิดการขาดอาหารนานๆ เชน
- การใหอาหารโดยวิธีหยดเขากระเพาะอาหารในตอนกลางคืน (nocturnal gastric drip)
- การใหแปงดิบ (uncooked starch) เพื่อให glucose ถูกยอยและปลอยออกมาชาๆในระยะเวลานาน โดยเฉพาะในตอนกลางคืน
• ปองกันไมใหเกิดภาวะ stress เชน เปนไขหวัด หรือ ทองเสีย ฯลฯ
• ใหอาหารที่เปน low-fat, low-cholesterol เพื่อรักษาภาวะไขมันในเลือดสูง (hypertriglyceridemia)

DISORDERS OF FATTY ACID METABOLISMDISORDERS OF FATTY ACID METABOLISM

DISORDERS OF FATTY ACID OXIDATION
• การ oxidation ของ fatty acid ใน mitochondria มีบทบาทสําคัญในแงการสรางพลังงานของรางกาย โดยเปนแหลงของพลังงานถึง 80% ของพลังงานที่รางกายตองการ โดยเฉพาะในชวง late fasting stage
• ชวยลดการใชน้ําตาลและความจําเปนในการสลายโปรตีนเพื่อนําไปสราง glucose

DISORDERS OF FATTY ACID OXIDATIONขบวนการ fatty acid oxidation ประกอบดวย 4 ขั้นตอน ซึ่งความบกพรองในขั้นตอนดังกลาว จะทําใหเกิดความผิดปกติของ fatty acid oxdiation ขึ้น:
1. Carnitine cycle
AS = acyl-CoA synthetaseCU = carnitine transporter*CT = carnitine/acylcarnitine translocase*CPT I = carnitine palmitoyl transferase I*CPT II = carnitine palmitoyl transferase II*
* defective in disorders of fatty acid oxidation

DISORDERS OF FATTY ACID OXIDATIONขบวนการ fatty acid oxidation ประกอบดวย 4 ขั้นตอน ซึ่งความบกพรองในขั้นตอนดังกลาว จะทําใหเกิดความผิดปกติของ fatty acid oxdiation ขึ้น:
2. Beta oxidation cycle
* defective in disorders of fatty acid oxidation*
*
*

DISORDERS OF FATTY ACID OXIDATIONขบวนการ fatty acid oxidation ประกอบดวย 4 ขั้นตอน ซึ่งความบกพรองในขั้นตอนดังกลาว จะทําใหเกิดความผิดปกติของ fatty acid oxdiation ขึ้น:
3. Electron transfer
*
*
- Electron transfer flavoprotein (ETF)
- ETF-Ubiquinone oxidoreductase (ETF-QO)
* defective in disorders of fatty acid oxidation
ดู KSA – integration of metabolism เพิ่มเติม

DISORDERS OF FATTY ACID OXIDATIONขบวนการ fatty acid oxidation ประกอบดวย 4 ขั้นตอน ซึ่งความบกพรองในขั้นตอนดังกลาว จะทําใหเกิดความผิดปกติของ fatty acid oxdiation ขึ้น:
4. Ketogenesis
*
*
* defective in disorders of fatty acid oxidation
acetoacetateβ-OH-butyrate
acetone
ketone bodies

DISORDERS OF FATTY ACID OXIDATION
• อาการและอาการแสดงของผูปวยที่มีความผิดปกติของ fatty acid oxidation สวนใหญจะคลายคลึงกัน เชน:
- ผูปวยจะมีอาการของ hypoglycemia ในชวงที่มีการอดอาหาร หรือ อยูในภาวะ stress
- อาการ hypoglycemia จะเกิดรวมกับภาวะ hypoketosis
ดู KSA – integration of metabolism เพิ่มเติม
- มีระดับของ free fatty acid เพิ่มสูงขึ้นในเลือด
- การทํางานของอวัยวะที่ตองใชพลังงานจากไขมัน เชน กลามเนื้อ, หัวใจ และสมอง อาจลมเหลว

adipocyte
fatty acid
glycerol
triglyceride
free fatty acid
Acetyl CoA
oxaloacetatecitrateTCA
cyclecarnitine
β-oxidation
• ในระยะ fasting จะมีการกระตุนขบวนการสลายไขมัน (lipolysis) เพื่อใชเปน แหลงพลังงานแทน glucose -> สงวน glucose ไวใหอวัยวะที่สําคัญ เชน สมอง
DISORDERS OF FATTY ACID OXIDATION

• ในผูปวยที่มีความผิดปกติของ fatty acid oxidation เซลลจะใชพลังงานจาก fatty acid ไดไมดี ทําใหตองพึ่ง glucose เปนแหลงของพลังงาน
pyruvate
Acetyl CoA
oxaloacetatecitrateTCA
cycle
glucose-6-phosphate
phosphoenolpyruvate
Fatty acid
DISORDERS OF FATTY ACID OXIDATION
ผูปวยจะมีอาการของ hypoglycemia ในชวงที่มีการอดอาหารและอวัยวะที่ใช fatty acid เชน หัวใจหรือกลามเนื้อ ทํางานบกพรอง

pyruvate
Acetyl CoA
oxaloacetatecitrateTCA
cycle
glucose glucose-6-phosphate
phosphoenolpyruvate
glycerollactateamino acid
glycerollactateamino acid
• ในระยะ fasting, จะมีการนํา amino acids, glycerol และ lactate นํามาสราง glucose ขึ้นใหม (gluconeogenesis)
liver & kidney
+
-Fatty acid
gluconeogensis
• acetyl CoA ที่ไดจาก fatty acid oxidation จะกระตุน เอ็นไซมใน gluconeo-genetic pathway ทําใหมีการสราง glucose เพิ่มขึ้น
DISORDERS OF FATTY ACID OXIDATION

• การบกพรองของ fatty acid oxidation ทําให acetyl CoA ลดลง ทําใหการทํางานของเอ็นไซมใน gluconeogenetic pathway ลดลงดวย ทําใหอาการ hypoglycemia รุนแรงขึ้น
pyruvate
Acetyl CoA
oxaloacetatecitrateTCA
cycle
glucose glucose-6-phosphate
phosphoenolpyruvate
liver & kidney
+Fatty acid
gluconeogensisgluconeogensis
-glycerollactateamino acid
glycerollactateamino acid
DISORDERS OF FATTY ACID OXIDATION

Ketonebodies
acetoacetateβ-OH-butyrate
liver
acetone
Blood ketone
• ในระยะ fasting, ตับจะสราง ketone bodies จาก acetyl CoA แลวสงไปยัง เนื้อเยื่อตางๆ เพื่อใชเปนแหลงพลังงานแทน glucose
free fatty acid Acetyl CoA
oxaloacetatecitrateTCA
cycle
β-oxidation of fatty acid
DISORDERS OF FATTY ACID OXIDATION

Blood ketone
• acetyl CoA ที่ลดลงจะทําใหการสราง ketone ลดลง
Ketonebodies
acetoacetateβ-OH-butyrate
liver
acetone
free fatty acid Acetyl CoA
oxaloacetatecitrateTCA
cycle
β-oxidation of fatty acid
DISORDERS OF FATTY ACID OXIDATION
อาการ hypoglycemia จะเกิดรวมกับภาวะ hypoketosis

adipocyte
fatty acid
glycerol
triglyceride
free fatty acid
Acetyl CoA
oxaloacetatecitrateTCA
cyclecarnitine
β-oxidation
• การที่เซลลมีความบกพรองในการ oxidation ของ fatty acid
DISORDERS OF FATTY ACID OXIDATION
ผูปวยจะมีระดับ free fatty acid ในเลือดสูง ซึ่งจะมีพิษถาระดับสูงมากเกินไป

DISORDERS OF FATTY ACID OXIDATION
การรักษา
• เนื่องจากอาการของโรคโดยเฉพาะ hypoglycemia จะเกิดขึ้นเมื่อมีการอดอาหาร ดังนั้น การรักษาในระยะยาว ควรปองกันไมใหเกิดการขาดอาหารนานๆ (ดูการรักษาของ glycogen storage disease)
• ในชวงที่มีอาการเฉียบพลัน ควรให glucose เขาหลอดเลือดดําทันที เพื่อรักษาภาวะ hypoglycemia และใหไปกระตุนการหลั่ง insulin จนถึงระดับที่ยับยั้ง fatty acid oxidation ของตับและกลามเนื้อได
• การรักษาที่จําเพาะ
- การให carnitine เสริมในรายที่มีการขาด carnitine
- การใหอาหารไขมันที่มี medium-chain fatty acid เปนสวนประกอบ จะมีประโยชนในผูปวยที่มีความบกพรองของ carnitine cycle เนื่องจาก กรดไขมัน medium-chain สามารถผานเขาสู mitochondria โดยไมตองใช carnitine

DISORDERS OF AMINO ACID METABOLISMDISORDERS OF AMINO ACID METABOLISM

ALBINISM
• สภาพเผือก หรือ albinism เปนกลุมโรคพันธุกรรมที่พบบอย รายงานครั้งแรกโดย Sir Archibald Garrod ในป 1908
• พบไดในหลายเชื้อชาติ
• ความผิดปกติเกิดจากเซลล melanocyte ซึ่งทําหนาที่สังเคราะห melanin ทํางานบกพรอง

ALBINISM
• melanocyte เปนเซลลที่อยูในผิวหนัง, choroid และ iris ของตา ทําหนาที่สรางเม็ดสี melanin
• melanin ทําหนาที่ absorb แสง UV ไมใหทําอันตรายกับเซลล และยังมีฤทธิ์ตอตาน free radical

ALBINISM
tyrosinasetyrosinase
Metabolism ของ melanin
spontaneous

ALBINISM
ความผิดปกติทางชีวเคมี
• การขาดเอนไซม tyrosinase เปนสาเหตุสวนใหญของการเกิด albinism
temperature-sensitive tyrosinase ในแมวไทย

ALBINISM
ความผิดปกติทางชีวเคมี
• ผูปวยจะมี hypopigmentation ของผิวหนังและเสนผม ทําใหเสี่ยงตอการเกิดมะเร็งของผิวหนังที่เกิดจากการ expose ตอแสง ultraviolet ไดงายกวาคนปกติ
• การที่มี melanin นอยลงในมานตา (iris) และ choroid ของตา ทําใหผูปวยมีปญหาในการมองเห็น เชน สายตาไมสามารถสูแสงจาได (photophobia) และ ความคมชัดของสายตาลดลง

ALBINISMการรักษา
• ควรหลีกเลี่ยงแสงแดด โดยสวมเสื้อผาใหมิดชดิ หรือใช lotion ที่มีฤทธิ์ปองกันแสง ultraviolet ซึ่งจะชวยปองกันการเกิดมะเร็งของผิวหนังได
• การใชแวนกันแดด อาจชวยในเรื่องของ photophobia และ ความคมชดัของการมองเห็นได

• ผูปวยมีปญญาออน กลามเนื้อเกร็ง สีผิว ตา ผม จางกวาปกติ ถาวินิจฉัยไดเร็ว และไดรับการรักษาที่ถูกตอง จะปองกันภาวะปญญาออนได
PHENYLKETONURIA
• Phenylketonuria (PKU) เปนโรคที่เกิดจากความบกพรองของ metabolism ทําใหมีระดับของ phenylalanine ในเลือดสูงกวาปกติ

Metabolism of phenylalanine
- one of the essential amino acids
- source: diet และ การสลายโปรตีนในรางกาย
- importance:
1. เปน amino acid ที่ใชในการสรางโปรตีน
3. เปนสารตนของสังเคราะหกรดอะมิโน tyrosine ซึ่งเปนสารตนของ catecholamine neurotransmitters (e.g. dopamine, nor-, epinephrine) และ melanin
2. สลายเปนพลังงานหรือสารตนในการสรางน้ําตาล
PHENYLKETONURIA

- Enzyme: Phenylalanine hydroxylase (PAH)
Defects in a conversion of phenylalanine to tyrosine which requires:
- Cofactor: Tetrahydrobiopterin (BH4)
• PKU เกิดจาก genetic defects ของเอ็นไซม PAH (98%) หรือเอ็นไซมในขบวนการ biopterin metabolism (~1-2%)
(PAH)
PHENYLKETONURIA
Metabolism of phenylalanine

Metabolism of Biopterins
Defects of synthesis
Defects of recycling
PAH
PHENYLKETONURIA

Effects of PAH deficiency
• มีการเพิ่มขึ้นของ phenylalanine และ metabolites (phenylketones, phenylamine)
การลดลงของ phenylalanine hydroxylase ทําให:
• การเปลี่ยน phenylalanine เปน tyrosine ลดลง -> relative deficiency of tyrosine -> light hair and skin color, impaired synthesis of catecholamine neurotransmitter
PHENYLKETONURIA

Effects of PAH deficiency
• hyperphenylalaninemia จะยับยั้งการขนสงกรดอะมิโนหลายชนิด ผานเซลล,blood brain barrier และ choroid plexus
(large neutral amino acid เชน valine, leucine, isoleucine, threonine, histidine, methionine, tyrosine, tryptophan)
ระดับ phenylalanine ในสมองสูงขึ้น
- ยับยั้งการสังเคราะหโปรตีนซึ่งจะกระทบตอการเจริญของ dendrite
- ยับยั้งการสังเคราะห neuro-transmitter ที่สําคัญ เชน dopamine, norepinephrine, serotonin
ระดับ aromatic & neutral amino acids อื่นๆ ในสมองลดลง
- ยับยั้งการสราง myelin
PHENYLKETONURIA
ผูปวย PKU ที่ไมไดรับการรักษาจึงเกิด mental retardation

Effects of biopterin deficiency
• tetrahydrobiopterin (BH4) เปน cofactor ของเอ็นไซมที่สําคัญหลายชนิด เชน tyrosine และ tryptophan hydroxylase, nitric oxide synthase
• defects ของ biopterin metabolism ทําใหมี phenylalanine ในเลือดสูงขึ้น รวมกับ defective synthesis ของ dopamine, norepinephrine, epinephrine, serotonin
PHENYLKETONURIA

PHENYLKETONURIA
การรักษา
• จุดมุงหมายของการรักษาคือ การพยายามทําใหระดับของ phenylalanine ไมใหสูงมากเกินจนเกิด toxicity ได
• เนื่องจาก Phe เปน essential amino acid ซึ่งรางกายไมสามารถสังเคราะหไดเอง ดังนั้น การควบคุมระดับของ Phe ในเลือดสามารถทําไดโดยการจํากัดอาหาร
• ผูปวยควรไดรับนมหรืออาหารที่จํากัด Phe

Aspartame
PHENYLKETONURIA
การรักษา
• หลีกเลี่ยงการรับประทานอาหารที่มี Phe สูง เชน เนื้อสัตว, เนย, ไข ฯลฯ หรือ เครื่องดื่มที่มีสารใหความหวาน aspartame เปนสวนประกอบ
• อยางไรก็ตาม ไมควรจะจํากัดการไดรับ Phe มากจนเกินไป เนื่องจาก Phe เปนกรดอะมิโนที่จําเปน

PHENYLKETONURIA
• ในรายที่เกิดจากการขาด biopterin cofactor จะมีการขาด neurotransmitter ของระบบประสาทรวมดวย ดังนั้น นอกจากจะจํากัดอาหารที่มี Phe แลว อาจจะตองให biopterin รวมกับ precursor ของ neurotransmitter รวมดวย เพือ่ปองกันไมใหเกิดภาวะปญญาออน

LYSOSOMAL STORAGE DISORDERSLYSOSOMAL STORAGE DISORDERS

LYSOSOMAL STORAGE DISORDERS
• lysosome เปน sac-like organelle ที่บรรจุ hydrolytic enzyme ที่ทําหนาที่ยอยสลายสารหลายชนิด ซึ่งอาจจะเปนสิ่งแปลกปลอมจากภายนอก หรือสวนประกอบของเซลลเอง
• lysosomal storage disease เกิดจากความบกพรองแตกําเนิดของเอนไซมใน lysosome ทําใหเกิดการคั่งของ substrate ใน lysosome ซึ่งอาจจะทําใหเกิดการรบกวน normal function ของเซลลได

DISORDERS OF SPHYNGOLIPID METABOLISM
LYSOSOMAL STORAGE DISORDERSLYSOSOMAL STORAGE DISORDERS

DISORDERS OF SPHYNGOLIPID METABOLISM
Sphyngolipid ประกอบดวย:
• Sphyngomyelin - เปนสวนประกอบของ cell membrane มีเฉพาะในเนื้อเยื่อของมนุษยและสัตว พบมากใน myelin sheath
• Glycolipid ดู Lipid chemistry เพิ่มเติม

• Glycolipid เปน lipid ที่มี carbohydrate เปนสวนประกอบในโมเลกุล พบที่ lipid membrane ของเซลลทั่วไปในรางกาย
glycolipid
Cell membrane
• Glycolipid มีบทบาทสําคัญในขบวนการ cell-cell recognition, cell adhesion, cell growth and diffrentiation, และ signal transduction
DISORDERS OF SPHYNGOLIPID METABOLISM

DISORDERS OF SPHYNGOLIPID METABOLISM
เอนไซมที่ผดิปกติ โรค1. ceramidase Farber disease2. β-glucosidase Gaucher’s disease3. Galactocerebrosidase Krabbe disease4. Sulfatidase Metachromatic
leukodystrophy5. Sphyngomyelinase Niemann-Pick disease6. α-galactosidase Fabry disease7. β-galactosidase GM1-gangliosidoses8. β-hexosaminidases Hex-A
Tay-Sachs diseaseHex-A and Hex-BSandhoff disease
1.
2.
3.
4.
5.
6.
7.
8.
• sphyngolipid จะถูกยอยสลายใน lysosome ดวยเอนไซมหลายชนิด ความผิดปกติของเอนไซมดังกลาวจะทําใหเกิดการสะสมของ sphyngolipid ทําใหเกิดเปนโรคขึ้น
* Gaucher’s disease เปน lysosomal storage disease ที่พบบอยที่สุด

DISORDERS OF SPHYNGOLIPID METABOLISM
• Disorder of sphyngolipid metabolism เปนโรคกลุมใหญ มักมีอาการทางระบบประสาทเปนสวนใหญ
• แรกเกิดเด็กมกัปกติ ตอมาจะมีพัฒนาการลาชาหรือการเจริญเติบโตลมเหลวเนื่องจากมีการสะสมของ sphyngolipid ในเซลลประสาทของสมองและไขสันหลัง
• อาจมีตับมามโต กระดูกผิดรปู หรือมีอาการตาบอด
normal eyeground ลักษณะ cherry-red spot ในโรค Niemann-Pick

• Phillipe Charles Ernest Gaucher ไดบรรยายเกี่ยวกับโรคนี้ ครั้งแรกในป 1882
• ในป 1965 พบวา Gaucher disease เกิดจากความผิดปกติของขบวนการสลาย glycolipid ชนิดหนึ่งชื่อวา glucosylceramideทําใหเกิดการสะสมของสารดังกลาวภายใน lysosome ของเซลล
The most common lysosomal storage disorder
GAUCHER DISEASE: INTRODUCTION

Structure and metabolism of glycolipidsStructure and metabolism of glycolipids
sphingosine
Fatty acidceramide
glucosylceramide(glucocerebroside)
glucosylceramide(glucocerebroside)
glucose
various sugars added
various glycolipids
(gangliosides,globosides, etc)
GAUCHER DISEASE: INTRODUCTION

Structure and metabolism of glycolipidsStructure and metabolism of glycolipids
sphingosine
Fatty acidceramide
glucose
sugars removed
various glycolipids
(gangliosides,globosides, etc)
glucosylceramide เปน intermediate ในขบวนการสังเคราะห และสลาย glycolipid
glucosylceramide(glucocerebroside)
glucosylceramide(glucocerebroside)
GAUCHER DISEASE: INTRODUCTION

glucose
Gaucher disease เกิดจากความบกพรองแตกําเนิดของเอ็นไซม β-glucosidase (glucosylceramidase) ทําใหมีการคั่งของ glucosylceramide ภายในเซลล
glucosylceramide ceramide
acid β-glucosidase(glucosylceramidaseor glucocerebosidase)
GAUCHER DISEASE: PATHOGENESIS

Acid β-glucosidase Acid β-glucosidase
• พบไดทั่วไป แต enzymatic activity จะแตกตางไปในเนื้อเยื่อตางๆ (activity สูงใน placenta, fibroblast และ ต่ําใน white blood cell)
• saposin C มีฤทธิ์กระตุนการทํางานของเอ็นไซม (a rare cause of Gaucher-like disease)
GAUCHER DISEASE: PATHOGENESIS

Acid β-glucosidase Acid β-glucosidase
• เปน lysosomal enzyme ทําหนาที่สลาย glucosylceramide
Exogenous sources: e.g. phagocytosed cells in macrophages
Degradation of glucosylceramide
Endogenous sources
GAUCHER DISEASE: PATHOGENESIS

Deficiency of acid β-glucosidase
การสะสมของ glucosylceramide ภายใน lysosome ของเซลล
Mutations of β-glucosidase
Gaucher cell
• macrophage/monocyte in origin
• accumulated glucosylceramide สวนใหญมาจาก membrane ของเซลลที่ถูกกินโดย macrophage
decreased enzymatic activity and/or stability
GAUCHER DISEASE: PATHOGENESIS

• Gaucher cell มีความสําคัญใน pathophysiology ของ Gaucher disease
GAUCHER DISEASE: PATHOGENESIS

• glucosylsphingosine ซึ่งเปน toxic intermediate ของการสลาย endogenous glucosylceramide สามารถทําใหเกิดการ apoptosis ของ neuron ใน neuronopathic Gaucher disease (type II & III)
glucoseglucosylceramide ceramide
β-glucosidase
Fatty acid removed
glucosylsphingosine
Toxic!
GAUCHER DISEASE: PATHOGENESIS

GAUCHER DISEASE: PATHOGENESIS
ผูปวย Gaucher disease ที่มีตับมามโต และมีอาการทางระบบประสาท (generalized hypotonia)
ผูปวย Gaucher disease ที่มีตับมามโตเล็กนอย และมีอาการปญญาออนรุนแรง

Increase degradation
Enzyme replacement therapy โดยการให modified acid β-glucosidase ที่สามารถถูก uptake เขาสู lysosome ของ Gaucher cell (successful trial ในชวงป 1990)
GAUCHER DISEASE: THERAPEUTICS
glycoproein หรือเอนไซมที่มี mannose-6-phosphate จะถกู uptake โดยเซลลแลวนําเขาสู lysosome ทําใหนํามาใชในการรักษา lysosomal storage disease ได
การรักษา

Decrease synthesis
Substrate reduction therapy โดยใหยาที่มีฤทธิ์เปน inhibitor ในขบวนการสังเคราะห glucosylceramide (first clinical study ในป 2000)
GAUCHER DISEASE: THERAPEUTICSการรักษา

MUCOPOLYSACCHARIDOSES
LYSOSOMAL STORAGE DISORDERSLYSOSOMAL STORAGE DISORDERS

MUCOPOLYSACCHARIDOSIS
• เปน polymer ของ amino sugar และ acid sugar เรียงสลับกันเปน disaccharide repeating unit –
Mucopolysaccharide หรือ Glycosaminoglycan (GAG)
ตัวอยางของ amino sugar
glucuronic acid
ตัวอยางของ acid sugar
• keratan sulfate, dermatan sulfate, heparan sulfate, chondroitin sulfateดู carbohydrate chemistry เพิ่มเติม

MUCOPOLYSACCHARIDOSIS
• GAGs พบไดใน ground substance ของเนื้อเยื่อเกี่ยวพันของผิวหนัง, กระดูก, กระดูกออน เปนตน

MUCOPOLYSACCHARIDOSIS
• GAGs พบไดใน ground substance ของเนื้อเยื่อเกี่ยวพันของผิวหนัง, กระดูก, กระดูกออน เปนตน
ใน ground substance, GAGs จะรวมกับโปรตีนเปน proteoglycan ทําหนาที่ยึดเหนี่ยวโปรตีนตางๆใน
extracellular matrix เชน collagen, elastin กบัโปรตีนของเซลล ทําใหเนื้อเยือ่มคีวามยืดหยุนและแข็งแรง
plasma membrane
collagen fiber

MUCOPOLYSACCHARIDOSIS
• จากโครงสรางของทางเคมีของ GAGs ที่มี hydrophilic groups (เชน –OH, -NH2, -COO-) ในโมเลกุลมาก ทําให GAGs มีคุณสมบัติที่อุมน้ําไดมาก GAGs จึงมีหนาที่ดังนี้
- ทําใหเนื้อเยื่อมีความยืดหยุนและเตงตึง
- ทําหนาที่หลอลื่นในเนื้อเยื่อตางๆ
- ชวยปองกันการกระทบกระแทกของเนื้อเยื่อ เชน กระดูกออนของขอ เปนตน

MUCOPOLYSACCHARIDOSIS

MUCOPOLYSACCHARIDOSIS
Dermatan sulfate
เอนไซมที่ผดิปกติ ชนิดของ MPS1.α-Iduronidase MPS I (Hurler disease)2. Iduronate sulfatase MPS II (Hunter disease)3a. Heparan N-sulfatase MPS IIIa3b. a-N-acetylglucos- MPS IIIb
aminidase4a. Galactose-6-sulfatase MPS IVa4b. β-galactosidase MPS IVb6. NAc-Galactosamine MPS VI
4-sulfate7. β-Glucuronidase MPS VII
Heparan sulfate
Keratan sulfate
• GAGs จะถูกยอยสลายใน lysosome ดวยเอนไซมหลายชนิด ความผิดปกติของเอนไซมดังกลาวจะทําใหเกิดการสะสมของ GAGs เกิดเปนโรค mucopolysaccharidosis (MPS)

MUCOPOLYSACCHARIDOSIS
อาการโดยทั่วไปของโรค mucopolysaccharidosis
- เปนโรคเรื้อรังและอาการจะเพิ่มมากขึ้นตามลําดับ
- เนื่องจากมีการบกพรองของเอนไซมที่ยอยสลาย GAGs ทําใหมีการสะสมของ GAGs ในเซลล ทําใหมีตับ, มามโต และนอกจากนี้ GAGs ที่เพิ่มมากขึ้นใน extracelluar matrix จะกระตุนใหมีการสราง collagen มากขึ้นในเนื้อเยื่อตางๆ ทําใหกระดูกและใบหนาผิดปกติ ผูปวยอาจมีความผิดปกติของตา หู ทางเดินหายใจ ระบบหัวใจและหลอดเลือด และเชาวนปญญาต่ํา

MUCOPOLYSACCHARIDOSIS
อาการโดยทั่วไปของโรค mucopolysaccharidosis
ลักษณะของใบหนาที่ผิดปกติ (coarse facies) และตับมามโต เนื่องจากการสะสมของ GAGs ในผูปวย MPS II

MUCOPOLYSACCHARIDOSISการรักษา
• โรค MPS ยังไมมีการรักษาที่จําเพาะ นอกจากแกไขภาวะแทรกซอนที่เกิดขึ้นจากระบบตางๆ เชน กระดูกและขอ, ระบบหัวใจและหลอดเลือด, ระบบประสาท ฯลฯ
• การรักษาดวยการใหเอนไซมทดแทน (ยังอยูในขั้นทดลอง)
• การปลูกถายไขกระดูก (ยังอยูในขั้นทดลอง)
• การรักษาดวยยีน gene therapy (ยังอยูในขั้นทดลอง)

CONCLUSION
- การตรวจวินิจฉัยกอนคลอด (prenatal diagnosis)
- การตรวจกรองภาวะผิดปกติในทารกแรกเกิด (newborn screening)
- genetic counseling
• โรค genetic metabolic disorders ถึงแมแตละโรคจะพบไดนอย แตเมือ่รวมกันแลว จะเปนโรคกลุมใหญที่พบไดไมยาก
• โรคบางชนิด ถาไดรับการวินิจฉัยและรักษาอยางถูกตองทันทวงที สามารถปองกัน morbidity และ mortality จากโรคได (เชน PKU) ดังนั้น แพทยจึงควรตระหนักถึงโรค metabolic disorder ไวดวย
• การดูแลและปองกันโรคดังกลาวอาจทําไดดังนี้: