blast sp10
TRANSCRIPT

Searching Molecular Databases with BLAST

Searching Molecular Databases with BLAST
• Basic Local Alignment Search Tool
• How BLAST works• Interpreting search results• The NCBI Web BLAST interface• Demonstration and exercises

Why learn sequence database searching?
• What have I cloned ?• Is this really “my gene” ?• Has someone else already found
it ?• What is this protein’s function ?• What is it related to ?• Can I get more sequence easily ?

Search programs are sequence alignment programs
• They try to find the best alignment between your probe sequence and every target sequence in the database
• Finding optimal alignments is computationally a very resource intensive process
• It is usually not necessary to find optimal alignments, particularly for large databases
• Alignments are ranked and only top scores are reported

Practical database search methods incorporate shortcuts
• The fastest sequence database searching programs use heuristic algorithms
• The basic concept is to break the search and alignment process down into several steps
• At each step, only a best scoring subset is retained for further analysis

What does ‘HEURISTIC’ mean?
• “a commonsense rule (or set of rules) intended to increase the probability of solving some problem”
• Why consider every possible alignment once a reasonably good alignment is found?

Heuristic programs find approximate alignments
• They are less sensitive than “dynamic programming” algorithms such as Smith-Waterman for detecting weak similarity
• In practice, they run much faster and are usually adequate
• The BLAST program developed by Stephen Altschul and coworkers at the NCBI is the most widely used heuristic program

BLAST is a collection of five programs for different
combinations of query and database sequences
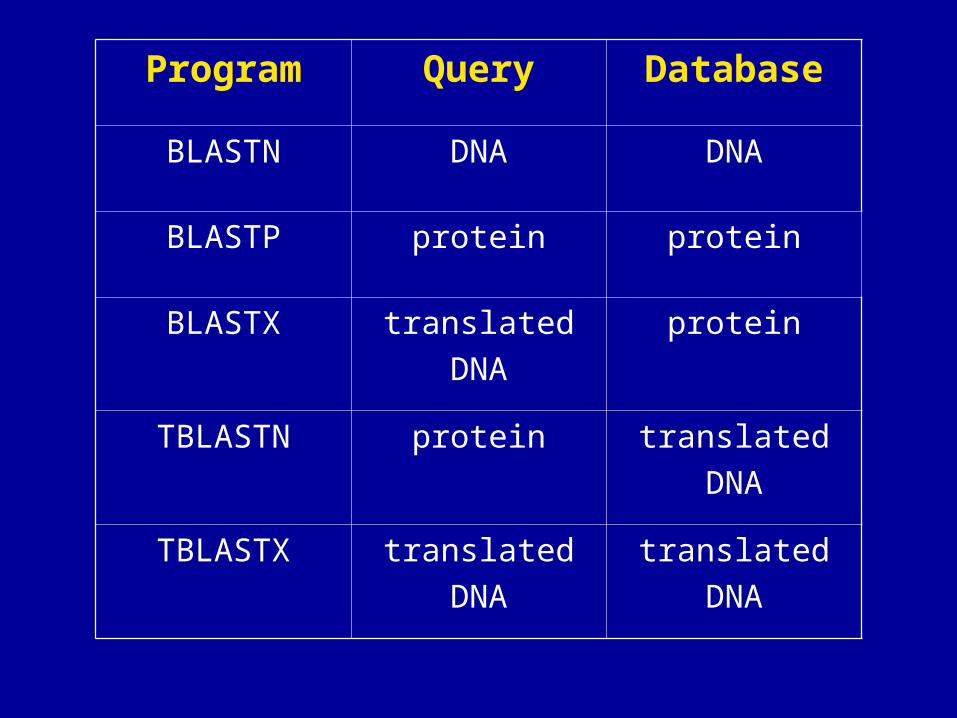
Program Query Database
BLASTN DNA DNA
BLASTP protein protein
BLASTX translatedDNA
protein
TBLASTN protein translatedDNA
TBLASTX translatedDNA
translatedDNA

Why BLAST is great
• Very fast and can be used to search extremely large databases
• Sufficiently sensitive and selective for most purposes
• Robust - the default parameters can usually be used

BLAST scores are reported in two columns
• Raw values based on the specific scoring matrix employed
• As bits, which are matrix independent normalized values (bigger = better)
• Significance is represented by E values (smaller = better)

Typical BLAST Output Sorted by E value
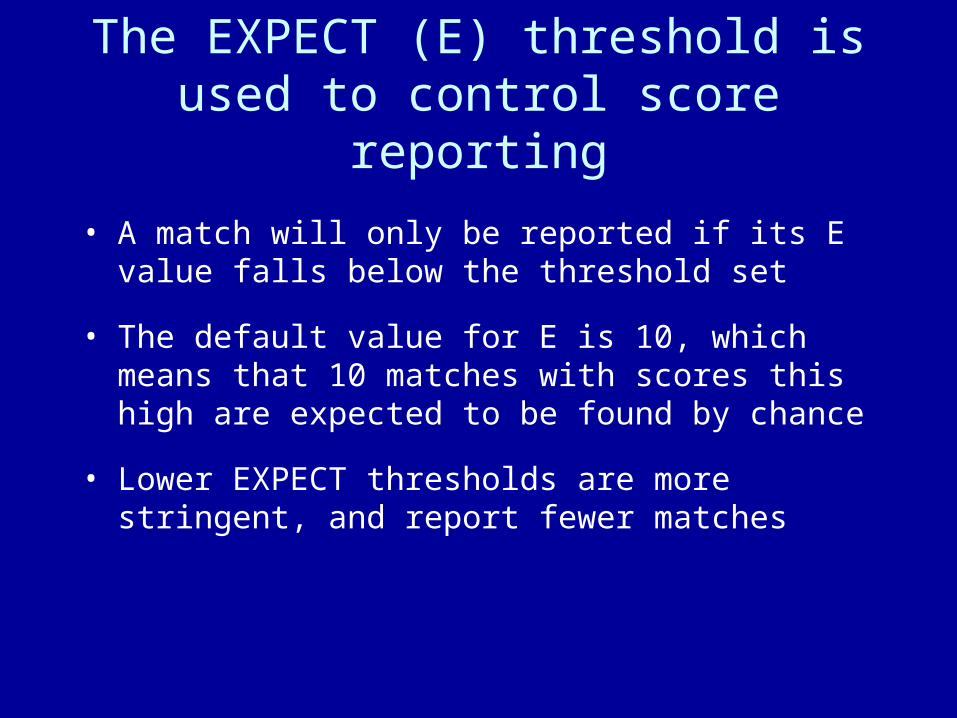
The EXPECT (E) threshold is used to control score reporting
• A match will only be reported if its E value falls below the threshold set
• The default value for E is 10, which means that 10 matches with scores this high are expected to be found by chance
• Lower EXPECT thresholds are more stringent, and report fewer matches

Interpreting BLAST scores
• Score interpretation is based on context– What is the question? – What else do you know about the
sequences?– Scoring is highly dependent on probe length
• Exact matches will usually have the highest scores (and lowest E values)– Short exact matches may score lower than
longer partial matches

Interpreting BLAST scores
• Short exact matches are expected to occur at random.
• Partial matches over the entire length of a query are stronger evidence for homology than are short exact matches.

Homology vs Identity
• Homologous sequences are descended from a common ancestral sequence.
• Homology is either true or false. It can never be partial! Saying two sequences are 45% homologous is a misuse of the term.
• Sequence identity and similarity can be described as a percentage and are used as evidence of homology.

BLAST ExampleIs this sequence known? What does it encode?

Search Strategy
• Choose the BLAST program:– nucleotide query vs. nucleotide db– megabalst: optimized to find
identical sequences– blastn: will find identical and similar
sequences
• Choose the Database– nr (non-redundant) – everything– genome specific

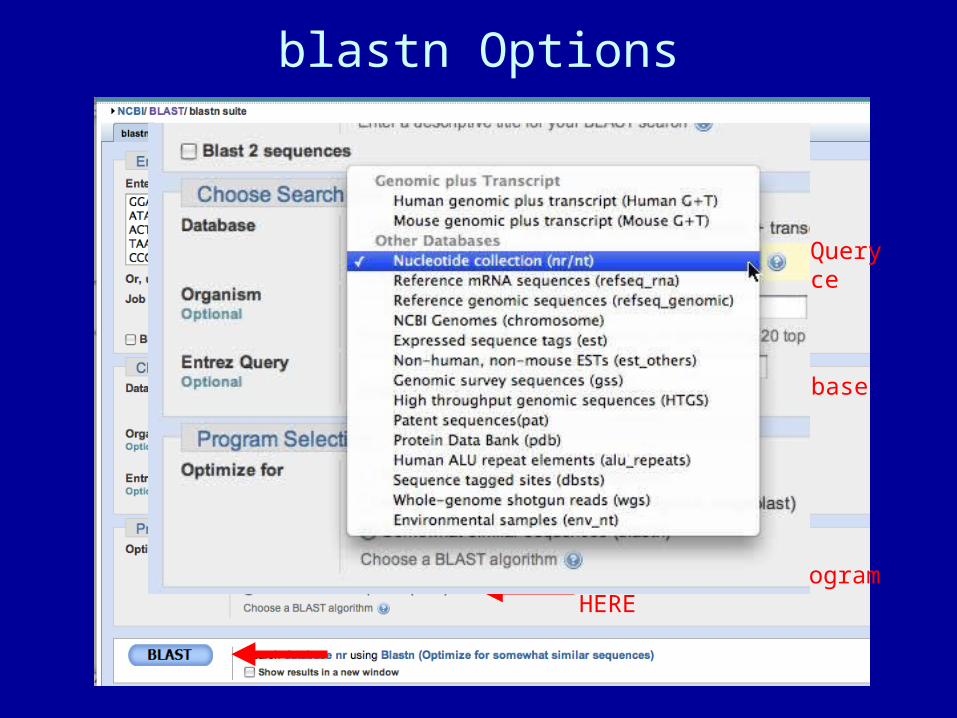
blastn Options
Paste QuerySequence HERE
Choose DatabaseHERE
Choose search programHERE
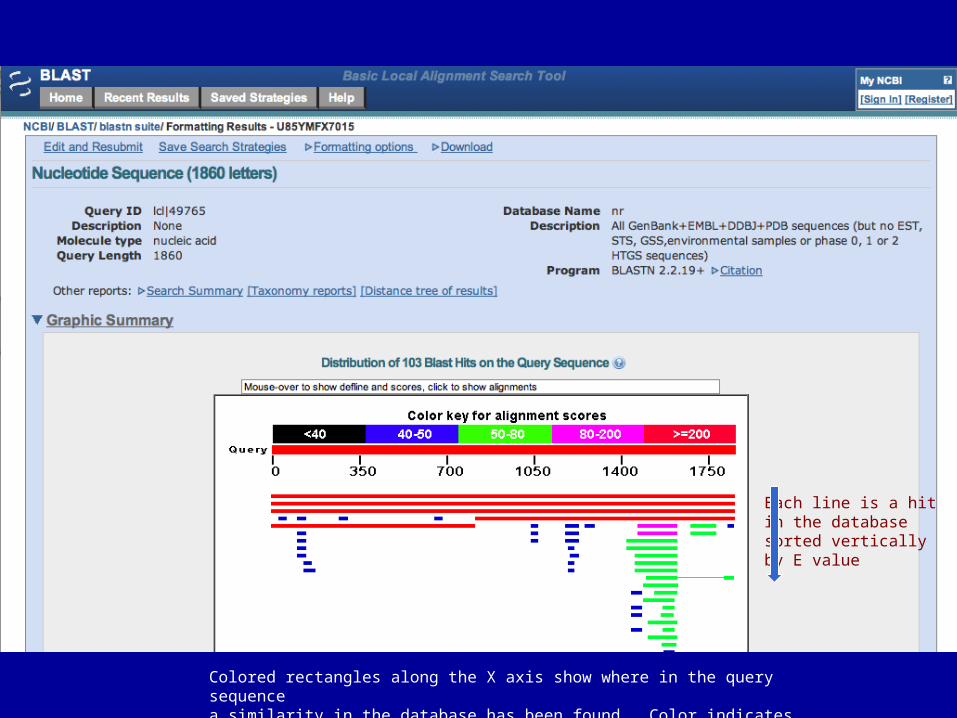
Each line is a hitin the database sorted vertically by E value
Colored rectangles along the X axis show where in the query sequencea similarity in the database has been found. Color indicates degree of similarity

Output sorted by E value
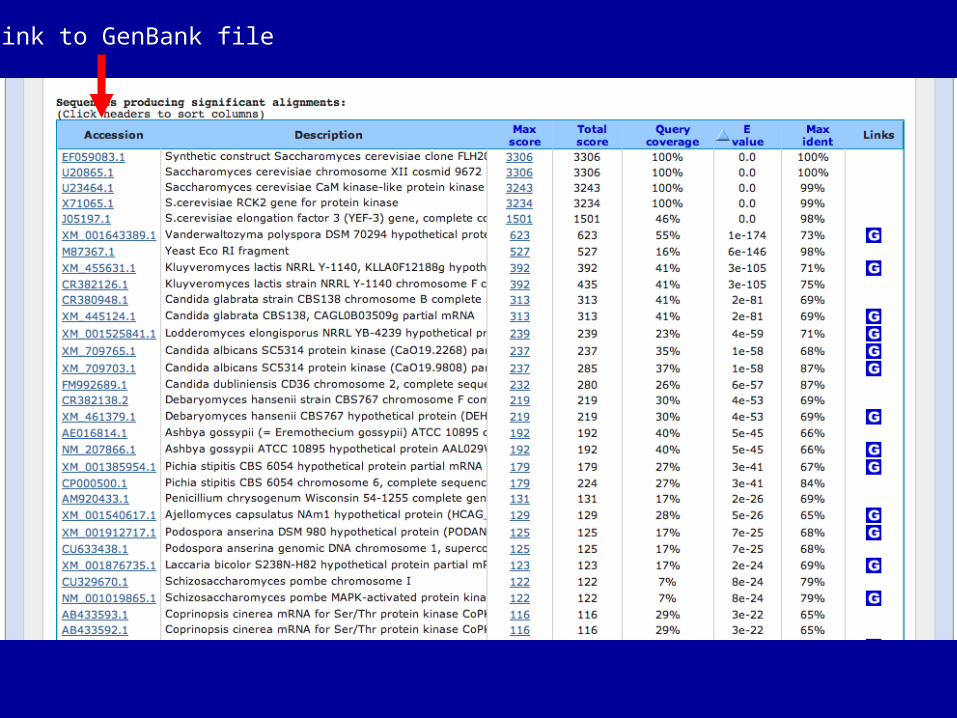
Link to GenBank file
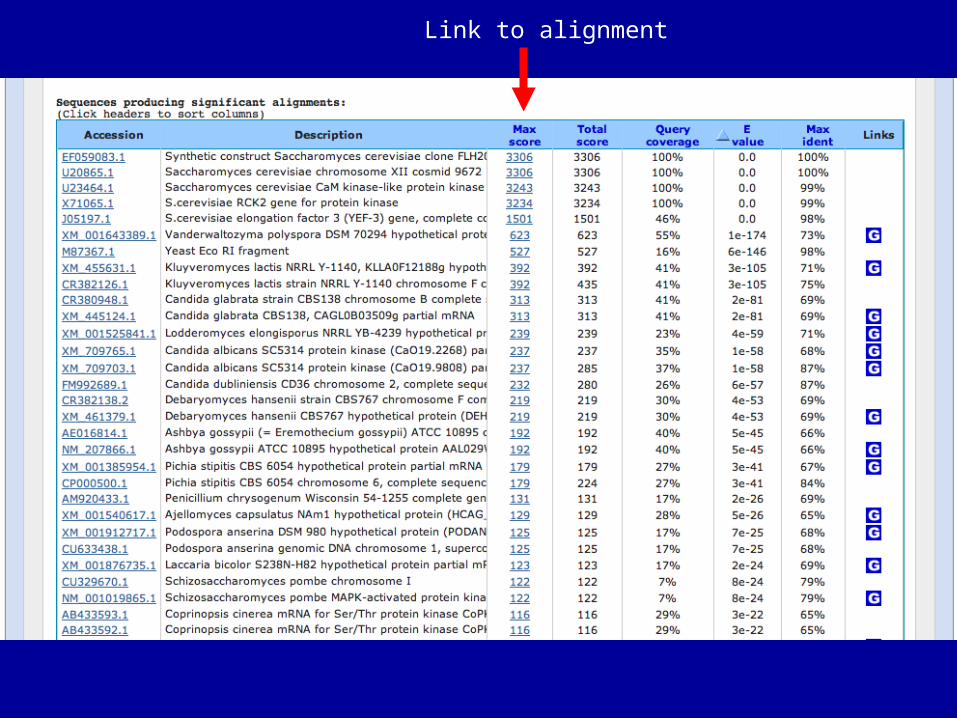
Link to alignment
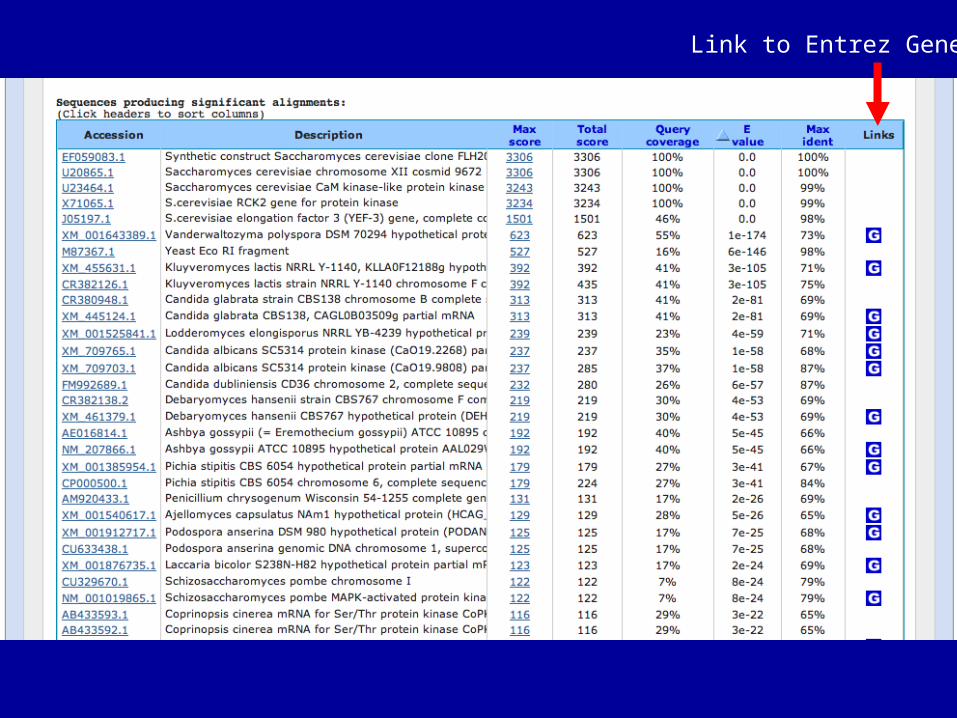
Link to Entrez Gene
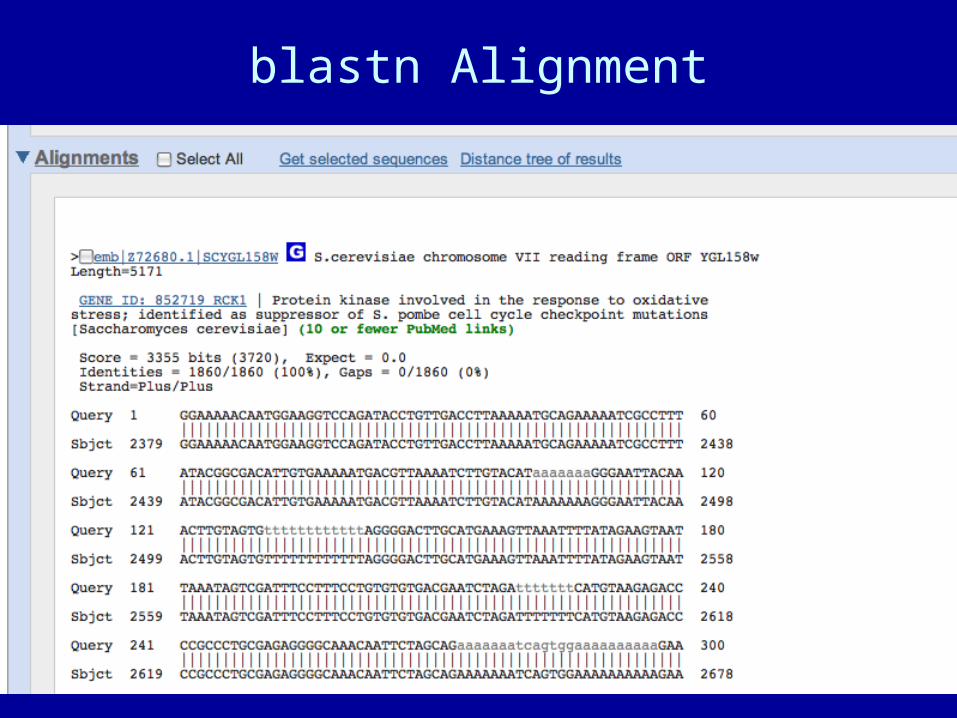
blastn Alignment

BLASTP Example
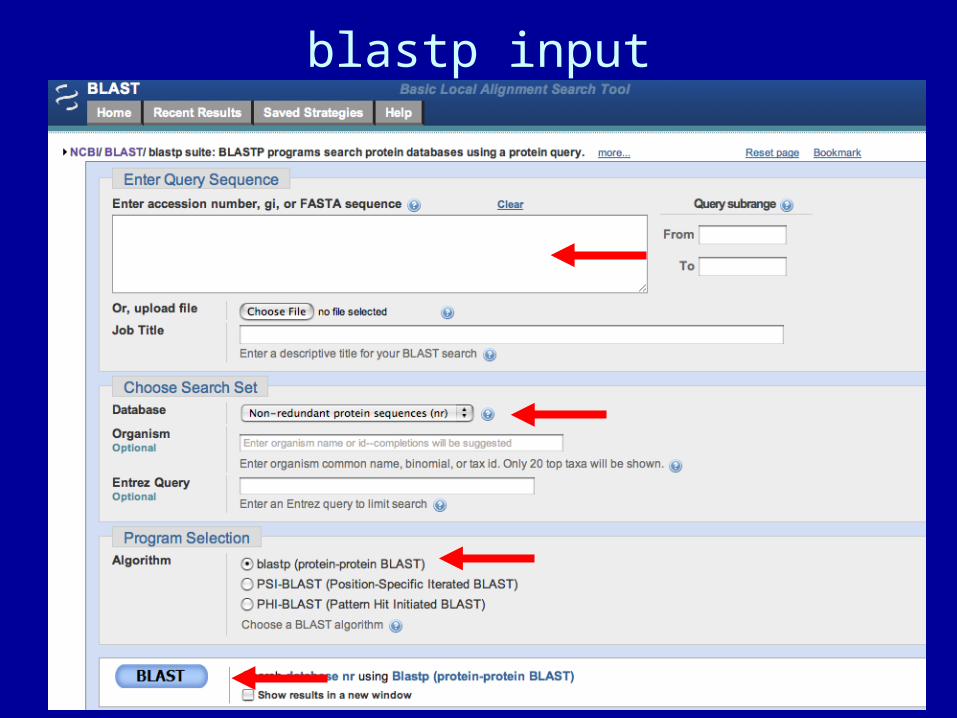
blastp input

blastp Databases

• nr - All non-redundant GenBank CDS translations + PDB + SwissProt+PIR
• swissprot - the last major release of the SWISS-PROT protein sequence database
• pat - patented sequences • pdb - Sequences derived from the 3-dimensional
structure Protein Data Bank• env_nr - Non-redundant environmental samples
blastp Databases

BLASTP Output
Conserved Domain Search
Conserved domains are showngraphically. Link to explanationof the domain.
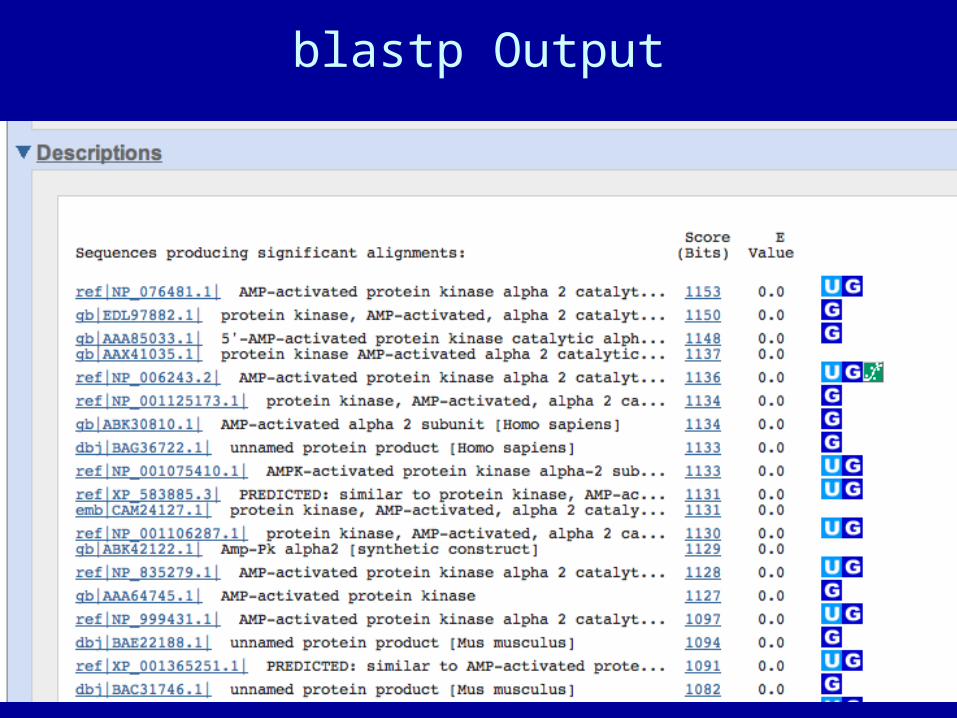
blastp Output

blastp Alignment
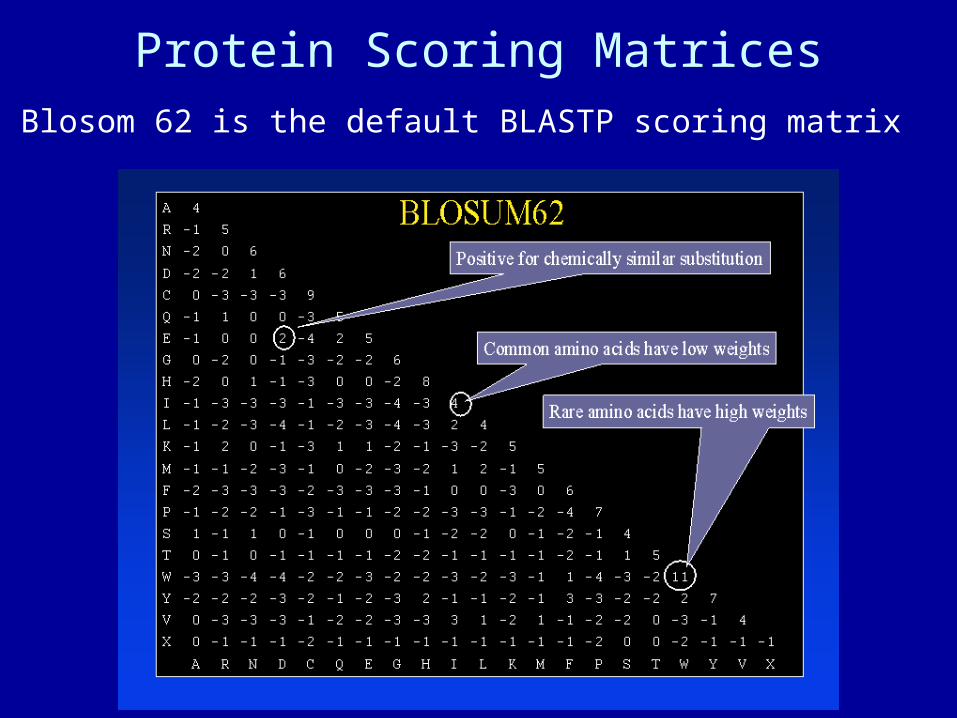
Protein Scoring MatricesBlosom 62 is the default BLASTP scoring matrix

Different Matrices Produce slightly different alignments

Other BLAST Programs:Psi-BLAST
4.6 PSI-BLAST is designed for more sensitive protein-protein similarity searches. Position-Specific Iterated (PSI)-BLAST is the most sensitive BLAST program, making it useful for finding very distantly related proteins or new members of a protein family. Use PSI-BLAST when your standard protein-protein BLAST search either failed to find significant hits, or returned hits with descriptions such as "hypothetical protein" or "similar to...".

Other BLAST Programs:Phi-BLAST
4.7 PHI-BLAST can do a restricted protein pattern search. Pattern-Hit Initiated (PHI)-BLAST is designed to search for proteins that contain a pattern specified by the user AND are similar to the query sequence in the vicinity of the pattern. This dual requirement is intended to reduce the number of database hits that contain the pattern, but are likely to have no true homology to the query.

Sequence filters
• Since only a limited number of matches are reported, hits to simple repeats and other low complexity sequences can obscure other more biologically meaningful similarities
• Filters are used to remove low complexity sequences from the probe
• Low Complexity, human repeats (blastn)

Low Complexity Sequences are Filtered Out

BLASTN vs BLASTP
• Protein sequences have much higher information content than nucleotide sequence
• To find evidence for sequence homology, use BLASTP and search protein sequences
• Is my sequence already in the database?
• To find identical sequences, search nucleotide databases

Translated BLAST Searches
• translations use all 6 frames
• computationally intensive
• tblastx searches can be very slow with some large databases
• must specify genetic code
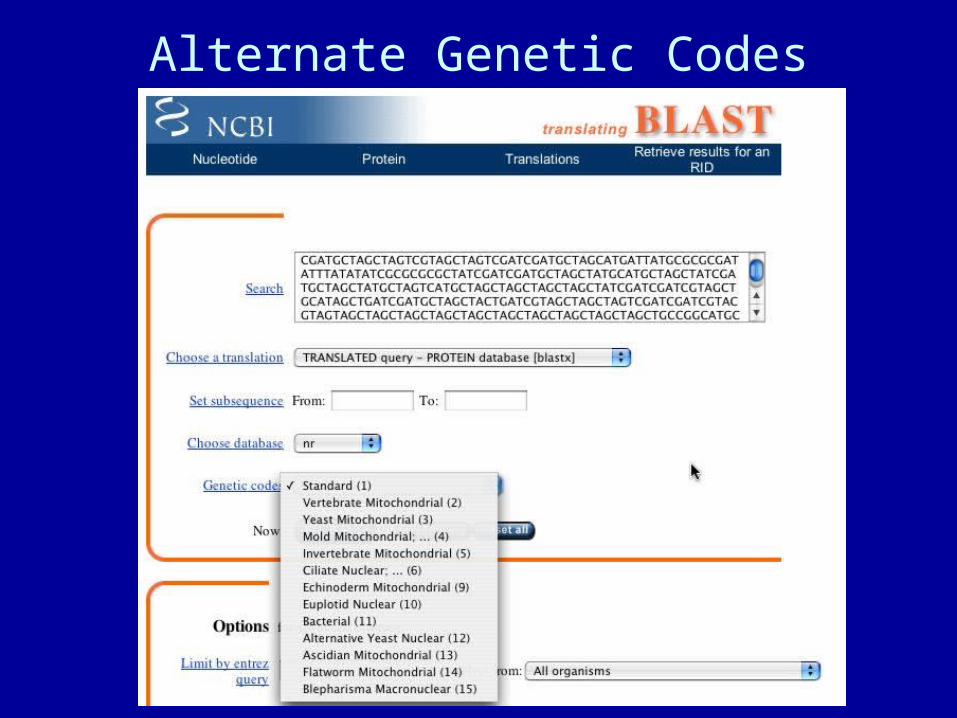
Alternate Genetic Codes

Translated BLAST Searches
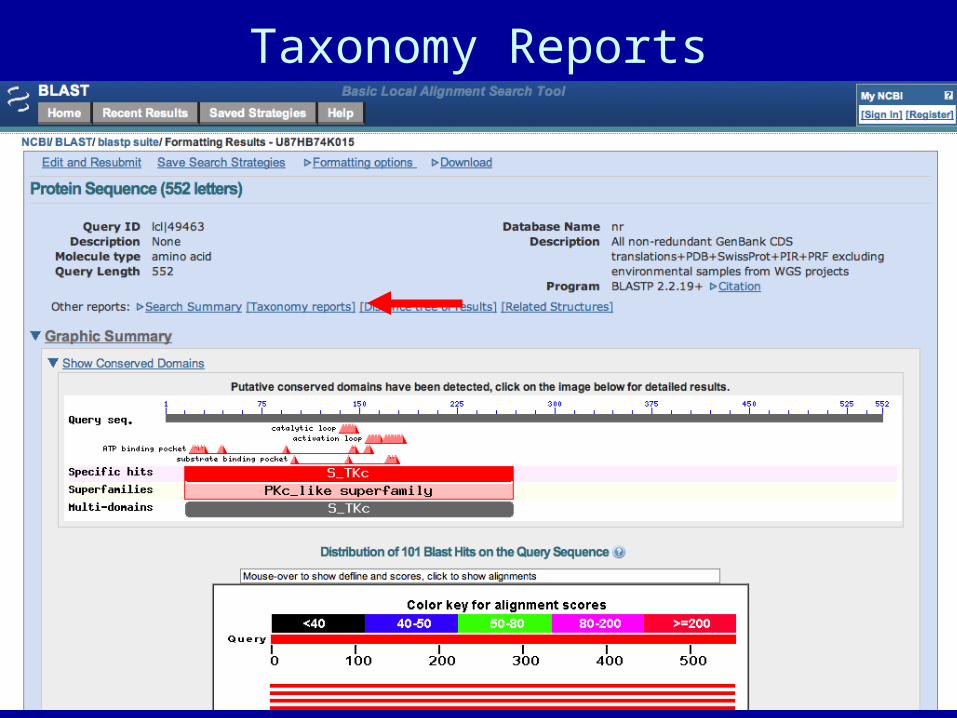
Taxonomy Reports

Taxonomy Reports
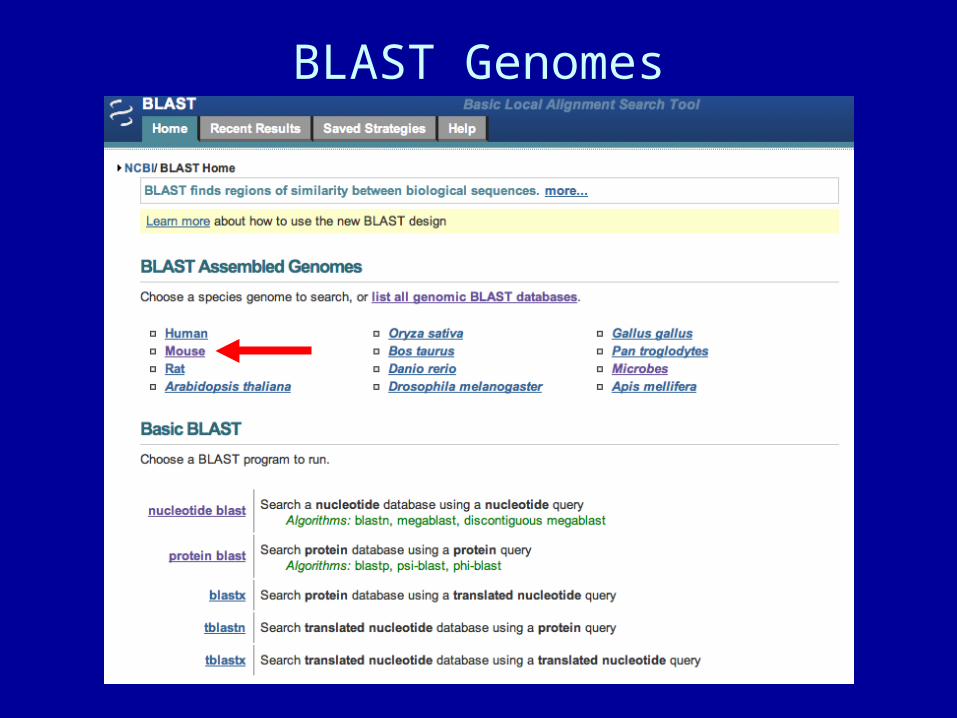
BLAST Genomes
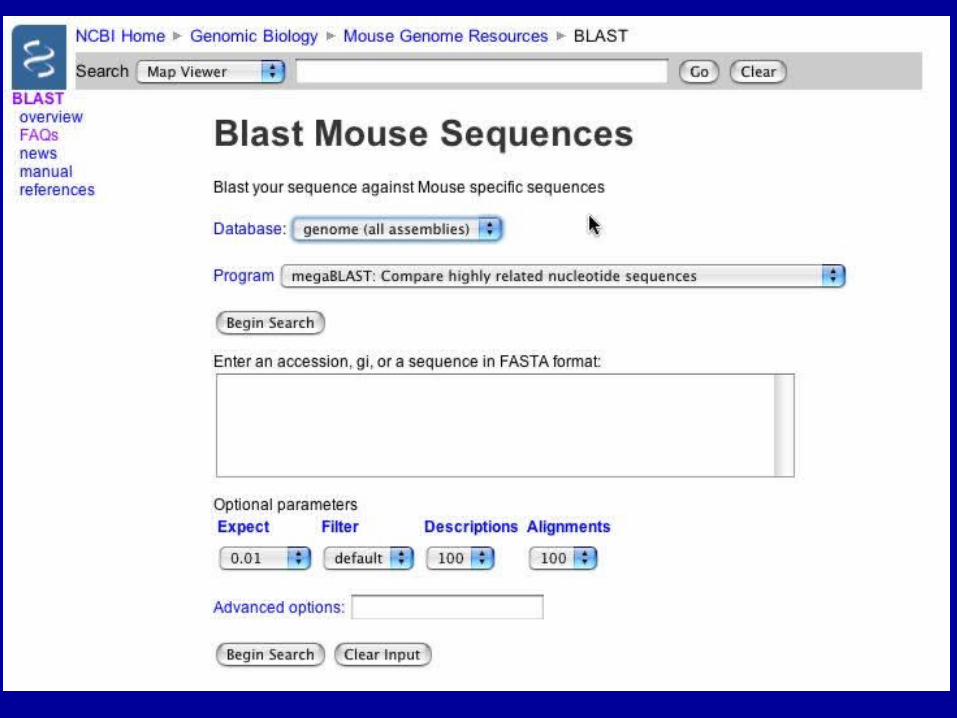
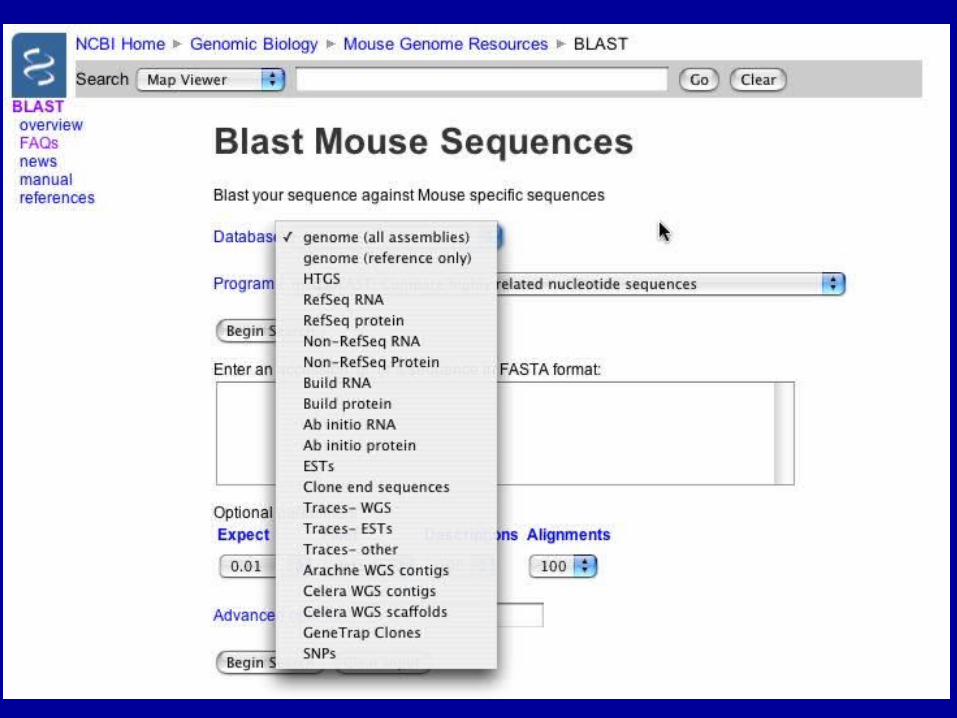
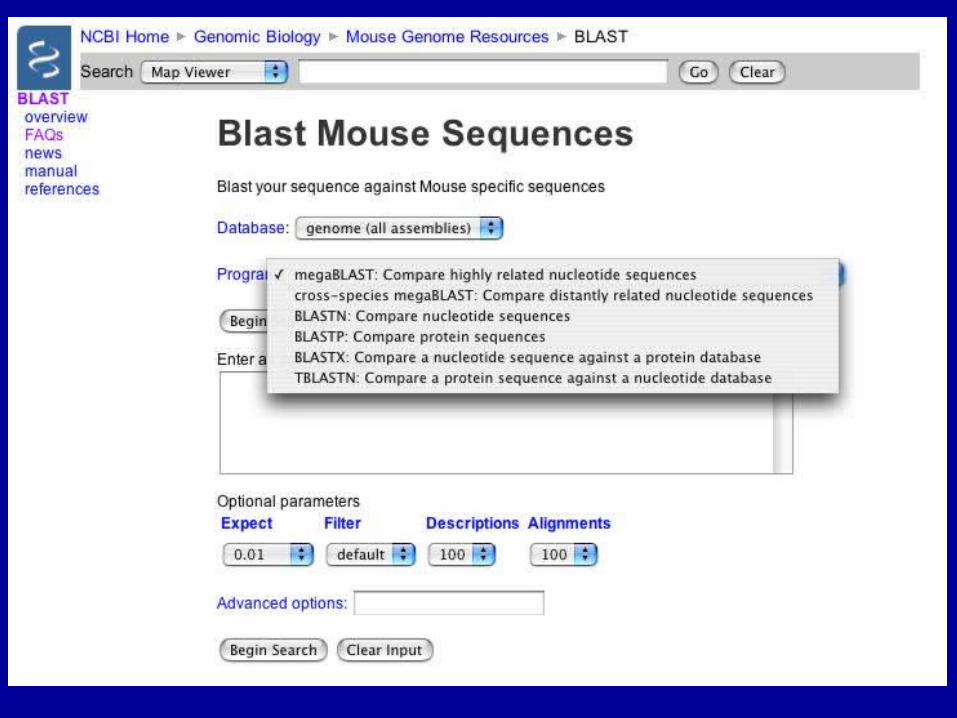

Align 2 Sequences with BLAST

BLAST from ORF Finder

Primer BLAST

BLAST Tutorial
• BLAST tutorial on Biocomp Web page
• Goal: demonstrate utility and difference between BLASTN and BLASTP searches
• BLASTN: is my DNA sequence in the database?
• BLASTP: are there related (homologs) proteins in the database?