case report: intravenous and oral pyridoxine trial for ...case report: intravenous and oral...
TRANSCRIPT

Case Report: Intravenous and OralPyridoxine Trial for Diagnosis ofPyridoxine-Dependent EpilepsyMelissa Cirillo, MDa,b, Charu Venkatesan, MD, PhDa,b, John J. Millichap, MDb,c, Cynthia V. Stack, MDa,b, Douglas R. Nordli, Jr, MDb,c
abstractPyridoxine-dependent epilepsy is a rare, autosomal recessive, treatable causeof neonatal seizures. Genetic testing can confirm mutations in the ALDH7A1gene, which encodes antiquitin. To avoid delays in initiating treatment whileawaiting confirmatory genetic testing, it is recommended that all neonateswith unexplained seizures should receive trial of intravenous (IV) pyridoxineto assess for responsiveness. However, oral pyridoxine is not commonlycontinued in the absence of the typical EEG changes. Two cases are presentedthat highlight the potential inadequacy of this single-step approach. Oneneonate ultimately diagnosed with pyridoxine-dependent seizures had no EEGchanges after administration of IV pyridoxine. In contrast, another neonatewho did not have this diagnosis had profound EEG changes after pyridoxineadministration. We present 2 cases that highlight the difficulties in usinginitial EEG response to IV pyridoxine in establishing a diagnosis of pyridoxine-dependent seizures in the neonate. Given the availability of biochemicalmarkers and gene testing, we suggest that oral pyridoxine treatment should becontinued until biochemical and/or genetic testing has confirmed thepresence or absence of pyridoxine-dependent epilepsy.
Pyridoxine-dependent epilepsy (PDE) israre and has a reported incidence of 1:400 000 to 1:750 000.1 Mutations inALDH7A1, which encodes antiquitin, wereestablished as the cause of PDE in 2006.2
Since then, more than 60 differentmutations have been identified within theALDH7A1 gene.2–6 This genetic defectcauses a deficiency in the enzymea-amino adipic semialdehydedehydrogenase, part of the lysinecatabolic pathway, resulting inaccumulation of a-amino adipicsemialdehyde (AASA), piperideine-6-carboxylate (P6C), and pipecolic acid(PA).4 P6C inactivates pyridoxalphosphate, which is the active form ofpyridoxine. Pyridoxal phosphate isa cofactor for glutamic aciddecarboxylase, which converts glutamateinto the inhibitory neurotransmitterg-amino butyric acid. Pyridoxalphosphate is also a cofactor for many
other enzymes in the brain.7 The seizuresand encephalopathy in PDE are likelysecondary to cofactor deficiency andconsequent decreased g-amino butyricacid function and effects on otherneurotransmitters. Daily administration ofadequate amounts of oral pyridoxinecannot overcome antiquitin deficiency,but can significantly improve seizurecontrol and neurodevelopmentaloutcome, highlighting the importance ofearly recognition, diagnosis, andtreatment of this condition.1
Biochemical testing for PDE ischaracterized by elevated PA and AASAin urine, plasma, and cerebrospinalfluid (CSF).4 Genetic testing canconfirm mutations in the ALDH7A1gene. To avoid delays in initiatingtreatment while awaiting confirmatorytesting, neonates with unexplainedseizures usually receive a trial of
aDivision of Neurology, and cEpilepsy Center, Ann andRobert H. Lurie Children’s Hospital of Chicago, Chicago,Illinois; and bDepartment of Pediatrics, NorthwesternUniversity Feinberg School of Medicine, Chicago, Illinois
Dr Cirillo diagnosed the patients, conceptualized thecase report, and drafted the initial manuscript; DrsVenkatesan, Millichap, Stack, and Nordli diagnosedthe patients, and reviewed and revised themanuscript; and all authors approved the finalmanuscript as submitted.
www.pediatrics.org/cgi/doi/10.1542/peds.2014-2423
DOI: 10.1542/peds.2014-2423
Accepted for publication Apr 20, 2015
Address correspondence to John J. Millichap, MD,FAAP, Epilepsy Center, Ann and Robert H. LurieChildren’s Hospital of Chicago, Department ofPediatrics, Northwestern University Feinberg Schoolof Medicine, 225 E. Chicago Ave, Box 29, Chicago, IL60611. E-mail: [email protected]
PEDIATRICS (ISSN Numbers: Print, 0031-4005; Online,1098-4275).
Copyright © 2015 by the American Academy ofPediatrics
FINANCIAL DISCLOSURE: The authors have indicatedthey have no financial relationships relevant to thisarticle to disclose.
FUNDING: No external funding.
POTENTIAL CONFLICT OF INTEREST: The authors haveindicated they have no potential conflicts of interestto disclose.
PEDIATRICS Volume 136, number 1, July 2015 CASE REPORT by guest on April 27, 2020www.aappublications.org/newsDownloaded from

intravenous (IV) pyridoxine.Responsiveness has typically beenassessed by changes in the EEG,notably immediate resolution ofepileptiform discharges afteradministration of pyridoxine.8
However, there have been casereports of EEG changes in neonateswithout PDE after IV pyridoxineadministration,9 as well as lack ofEEG response in PDE acutelypostpyridoxine.10 We present 2 casesthat highlight the difficulties inusing initial EEG response to IVpyridoxine in establishing a diagnosisof pyridoxine-dependent seizures in theneonate. Given these difficulties, werecommend (1) testing all neonateswith refractory seizures for PDE, and (2)continuing pyridoxine supplementationuntil all testing confirms the presence orabsence of PDE.
CLINICAL CASE 1
Patient 1 was born via spontaneousvaginal delivery at 38 weeks’gestation. Birth, family, and socialhistories were unremarkable. Generaland neurologic examinations werenotable for mild hypotonia andexcessive jitteriness. On the fifth dayof life, she had events concerning forseizures, characterized bynonsuppressible rhythmicmovements of her extremities withassociated eye deviation and oxygendesaturation. Continuous video EEGmonitoring was performed andconfirmed that these events weremyoclonic-tonic and brief tonicseizures. The EEG backgroundshowed excessive multifocal sharp-wave discharges and discontinuity forthe stated postconceptual age. Shereceived IV pyridoxine (100 mg 3 2doses) with no appreciable change inthe EEG background. She wascontinued on maintenancephenobarbital and oral pyridoxine asother diagnostic testing wasperformed.
MRI of the brain was unremarkable.Serum and CSF studies for infectious(complete blood count, blood culture,
urinalysis, urine culture, CSF culture)and metabolic (comprehensivemetabolic panel, ammonia, urineorganic acids, serum amino acids,serum lactate, serum pyruvate, CSFamino acids) disorders wereunrevealing. CSF testing formonoamine metabolites showeda profile suggestive of pyridoxine-dependent seizures.11 SubsequentALDH7A1 sequencing was positive for1 known heterozygous deleteriousmutation (c.328C.T; p.R1103) and 1novel heterozygous missensemutation (c.869T.C; p.F290S). Eachparent was found to carry 1 of each ofthe mutations.
Pyridoxine dose was increased to 150mg once daily and she wassuccessfully weaned off phenobarbital.Neurologic development has beennormal and she has had no seizurerecurrence. Urine AASA was 1.76mmol/mol creatinine (range ,0.5mmol/mol creatinine). Serial follow-up EEGs (last one at 28 months of age)have been normal. Her clinical course,response to pyridoxine, and genetictesting results confirmed the diagnosisof pyridoxine-dependent seizures.
CLINICAL CASE 2
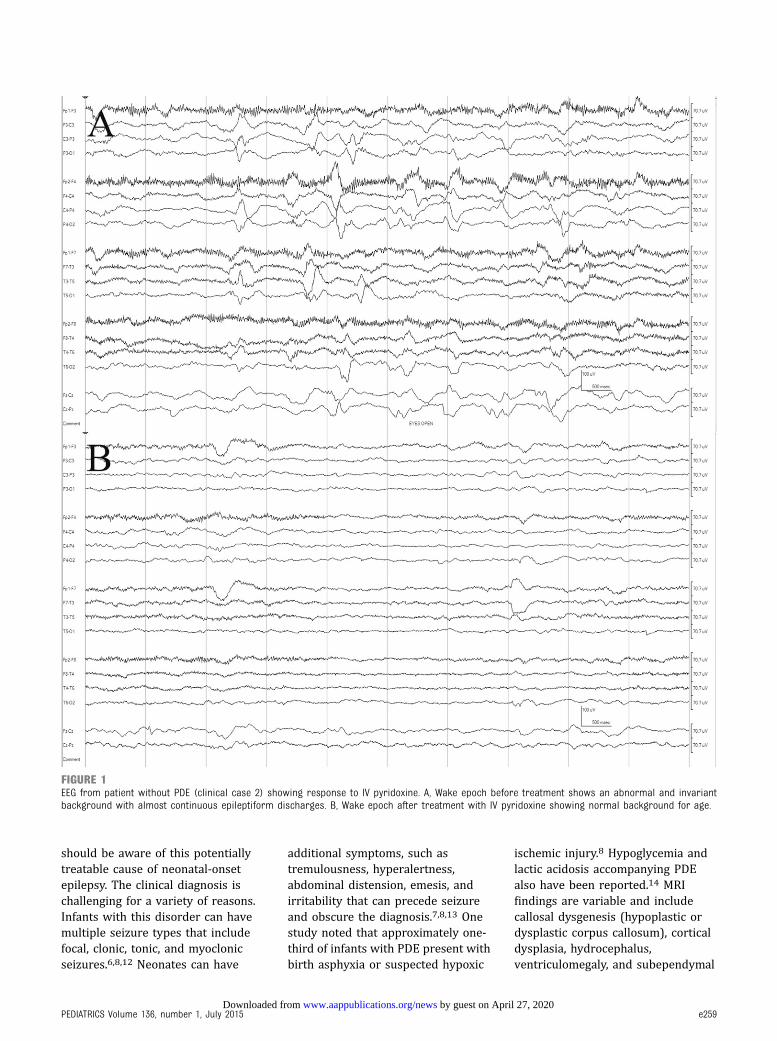
Patient 2 was born via spontaneousvaginal delivery at 37 4/7 weeks’gestation. Birth, family, and socialhistories were unremarkable. At 21days of age, she presented with new-onset seizures characterized bymyoclonic jerks of upper and lowerextremities and head deviation.Clinical seizures resolved afteradministration of phenobarbital, butencephalopathy persisted. Initial EEGwas abnormal due to almostcontinuous epileptiform dischargesfrom sites in both hemispheres thatwere invariant and not responsive tostate or stimulation (Fig 1A). Afteradministration of IV pyridoxine (100mg 3 2 doses), the EEG improvedrapidly to a normal pattern for age(Fig 1B). General and neurologicexaminations were otherwiseunremarkable. Seizures were
controlled with phenobarbital andoral pyridoxine as other diagnostictesting was performed.
Initial MRI of the brain showedsymmetric areas of restricteddiffusion and parenchymal signalabnormality involving theperiventricular and deep whitematter, dorsal thalami, internalcapsules, corticospinal tracts, andcorpus callosum. Serum and CSFstudies for infectious (complete bloodcount, blood culture, urinalysis, urineculture, CSF culture,meningoencephalitis panel from CSF)and metabolic (comprehensivemetabolic panel, ammonia, urineorganic acids, serum amino acids,serum lactate, serum pyruvate, CSFamino acids, urine sulfocysteine)disorders were unrevealing. Testingfor PDE, which included serum PA,urine AASA (0.43 mmol/molcreatinine [range ,0.5]), andALDH7A1 sequencing were normal.
She was discharged from the hospital3 days after admission onphenobarbital and pyridoxine.Pyridoxine and phenobarbital werediscontinued at 4 and 6 months ofage, respectively. Her developmentwas normal and she had no deficitson physical or neurologicexaminations at 15 months of age.Follow-up EEG done at 2 and 5months of age were mildly abnormal,showing rare focal epileptiformdischarges. Follow-up brain MRI doneat 6 months of age showed subtle T2signal changes in the deepsupratentorial white matter and mildthinning of the corpus callosum.Despite the initial dramatic “positive”EEG response to pyridoxine, thesubsequent clinical course andnegative biochemical and genetic testresults were not consistent with PDE.
DISCUSSION
PDE is a rare, autosomal recessive,cause of neonatal seizures andencephalopathy of likelyunderestimated incidence.Pediatricians and neonatologists
e258 CIRILLO et al by guest on April 27, 2020www.aappublications.org/newsDownloaded from
should be aware of this potentiallytreatable cause of neonatal-onsetepilepsy. The clinical diagnosis ischallenging for a variety of reasons.Infants with this disorder can havemultiple seizure types that includefocal, clonic, tonic, and myoclonicseizures.6,8,12 Neonates can have
additional symptoms, such astremulousness, hyperalertness,abdominal distension, emesis, andirritability that can precede seizureand obscure the diagnosis.7,8,13 Onestudy noted that approximately one-third of infants with PDE present withbirth asphyxia or suspected hypoxic
ischemic injury.8 Hypoglycemia andlactic acidosis accompanying PDEalso have been reported.14 MRIfindings are variable and includecallosal dysgenesis (hypoplastic ordysplastic corpus callosum), corticaldysplasia, hydrocephalus,ventriculomegaly, and subependymal
FIGURE 1EEG from patient without PDE (clinical case 2) showing response to IV pyridoxine. A, Wake epoch before treatment shows an abnormal and invariantbackground with almost continuous epileptiform discharges. B, Wake epoch after treatment with IV pyridoxine showing normal background for age.
PEDIATRICS Volume 136, number 1, July 2015 e259 by guest on April 27, 2020www.aappublications.org/newsDownloaded from

cysts.4,10,15 In these cases, epilepsy isoften attributed to structural brainlesions and no further investigationsare carried out. Some neonates withPDE can respond initially toconventional anticonvulsants andlater are found to have seizures thatare responsive to pyridoxine.16
Furthermore, there is no singlepathognomonic EEG pattern in PDE.Typical EEG patterns includeabnormal sleep patterns, focal ormultifocal epileptiform discharges,rhythmic slowing, or burstsuppression.8 In neonates withunexplained seizures refractory toconventional antiepilepticmedications, a trial of IV pyridoxinewith continuous EEG monitoring iscommonly performed. If no clinical orEEG change is noted, pyridoxineadministration may not always becontinued after the negative responseto the IV trial in current practiceparadigms.
These 2 cases highlight the potentialpitfalls of this single-step approachwith IV pyridoxine. It is importantto recognize that this approach isoutdated given that the affectedpathway has been identified andbiochemical markers and DNAsequencing of the gene are availableas sensitive and specific tests. Theneonate who was ultimatelydiagnosed with PDE had no EEGchanges after administration of IVpyridoxine, whereas the infantwithout PDE did have profound EEGchanges. These findings areconsistent with other studies thatshow that IV pyridoxine can beassociated with nonspecific EEGchanges that are not diagnostic orexclusionary of the disorder.9,10
These and previous reported casesemphasize that oral pyridoxinetreatment should be continuedwhile waiting for further testingresults. Evidence-based protocolsfor acute or maintenance therapyare not available, but an initial doseof 100 mg IV pyridoxine followed byoral maintenance dosing rangingbetween 15 and 30 mg/kg per day is
suggested.6 The evaluation caninclude serum or urine AASA, serumand CSF PA, and plasma P6C, all ofwhich accumulate due todeficiencies of aldehydedehydrogenase in the lysinedegradation pathway.4 However, it isimportant to recognize that PA alsocan be elevated secondary to liverdisease and in peroxisomaldisorders.17 Similarly, elevatedAASA is not specific and can beassociated with isolated sulfiteoxidase deficiency and the relatedcondition, molybdenum cofactordeficiency.18 CSF monoaminemetabolite analysis shows specificpeaks (of yet undeterminedcompounds) and elevated AASAspecific to PDE.11 Genetic testingwith sequencing of ALDH7A1 isavailable, but it should be noted thatPDE is inherited in an autosomalrecessive manner and the diagnosisis definitive only when 2 pathogenicmutations are identified. Ideally,each parent is confirmed as a carrierof 1 mutation (demonstrating eachmutation is on a differentchromosome in the affected child).Pyridoxal 59-phosphate oxidasedeficiency is a related cause ofneonatal seizures that can presentlike PDE. This condition usuallyrequires supplementation withpyridoxal phosphate, but also maybe responsive to pyridoxine in somecases. Therefore, diagnosticevaluation via CSF determination ofpyridoxal 59-phosphate (low in thiscondition) is recommended as well.If abnormal, DNA sequencing of thepyridoxal 59-phosphate oxidasegene can be performed.19 AlthoughPDE is rare, it is a potentiallytreatable cause of neonatal-onsetepilepsy that should be aggressivelyand systematically evaluated for thebest possible outcome. PDE also canhave later-onset presentation(beyond neonatal and infancyperiods) and testing for thiscondition should be considered inolder children with intractableepilepsy.
ABBREVIATIONS
AASA: a-aminoadipicsemialdehyde
CSF: cerebrospinal fluidIV: intravenousPA: pipecolic acidPDE: pyridoxine-dependent
epilepsyP6C: piperideine-6-carboxylate
REFERENCES
1. Gospe SM Jr. Pyridoxine-dependentseizures: new genetic and biochemicalclues to help with diagnosis andtreatment. Curr Opin Neurol. 2006;19(2):148–153
2. Mills PB, Struys E, Jakobs C, et al.Mutations in antiquitin in individualswith pyridoxine-dependent seizures. NatMed. 2006;12(3):307–309
3. Millet A, Salomons GS, Cneude F, et al.Novel mutations in pyridoxine-dependentepilepsy. Eur J Paediatr Neurol. 2011;15(1):74–77
4. Plecko B, Paul K, Paschke E, et al.Biochemical and molecularcharacterization of 18 patients withpyridoxine-dependent epilepsy andmutations of the antiquitin (ALDH7A1)gene. Hum Mutat. 2007;28(1):19–26
5. Scharer G, Brocker C, Vasiliou V, et al.The genotypic and phenotypic spectrumof pyridoxine-dependent epilepsy due tomutations in ALDH7A1. J Inherit MetabDis. 2010;33(5):571–581
6. Stockler S, Plecko B, Gospe SM Jr, et al.Pyridoxine dependent epilepsyand antiquitin deficiency: clinical andmolecular characteristics andrecommendations for diagnosis,treatment and follow-up. Mol GenetMetab. 2011;104(1-2):48–60
7. Pearl PL, Taylor JL, Trzcinski S, Sokohl A.The pediatric neurotransmitterdisorders. J Child Neurol. 2007;22(5):606–616
8. Baxter P. Epidemiology of pyridoxinedependent and pyridoxine responsiveseizures in the UK. Arch Dis Child. 1999;81(5):431–433
9. Teune LK, vd Hoeven JH, Maurits NM,et al. Pyridoxine induces non-specific EEGalterations in infants with therapy
e260 CIRILLO et al by guest on April 27, 2020www.aappublications.org/newsDownloaded from

resistant seizures. Seizure. 2007;16(5):459–464
10. Bok LA, Maurits NM, Willemsen MA, et al.The EEG response to pyridoxine-IVneither identifies nor excludespyridoxine-dependent epilepsy. Epilepsia.2010;51(12):2406–2411
11. Gallagher RC, Van Hove JL, Scharer G,et al. Folinic acid-responsive seizures areidentical to pyridoxine-dependentepilepsy. Ann Neurol. 2009;65(5):550–556
12. Schmitt B, Baumgartner M, Mills PB,et al. Seizures and paroxysmal events:symptoms pointing to the diagnosis ofpyridoxine-dependent epilepsy andpyridoxine phosphate oxidase deficiency.Dev Med Child Neurol. 2010;52(7):e133–e142
13. Segal EB, Grinspan ZM, Mandel AM,Gospe SM Jr. Biomarkers aidingdiagnosis of atypical presentation ofpyridoxine-dependent epilepsy. PediatrNeurol. 2011;44(4):289–291
14. Mercimek-Mahmutoglu S, Horvath GA,Coulter-Mackie M, et al. Profoundneonatal hypoglycemia and lacticacidosis caused by pyridoxine-dependentepilepsy. Pediatrics. 2012;129(5).Available at: www.pediatrics.org/cgi/content/full/129/5/e1368
15. Jain-Ghai S, Mishra N, Hahn C, Blaser S,Mercimek-Mahmutoglu S. Fetal onsetventriculomegaly and subependymal cystsin a pyridoxine dependent epilepsy patient.Pediatrics. 2014;133(4). Available at: www.pediatrics.org/cgi/content/full/133/4/e1092
16. Lin J, Lin K, Masruha MR, Vilanova LC.Pyridoxine-dependent epilepsy initiallyresponsive to phenobarbital. ArqNeuropsiquiatr. 2007;65(4A):1026–1029
17. Peduto A, Baumgartner MR, VerhoevenNM, et al. Hyperpipecolic acidaemia:a diagnostic tool for peroxisomaldisorders. Mol Genet Metab. 2004;82(3):224–230
18. Mills PB, Footitt EJ, Ceyhan S, et al. UrinaryAASA excretion is elevated in patients withmolybdenum cofactor deficiency andisolated sulphite oxidase deficiency. JInherit Metab Dis. 2012;35(6):1031–1036
19. Plecko B, Paul K, Mills P, et al. Pyridoxineresponsiveness in novel mutations of thePNPO gene. Neurology. 2014;82(16):1425–1433
PEDIATRICS Volume 136, number 1, July 2015 e261 by guest on April 27, 2020www.aappublications.org/newsDownloaded from

DOI: 10.1542/peds.2014-2423 originally published online June 22, 2015; 2015;136;e257Pediatrics
R. Nordli JrMelissa Cirillo, Charu Venkatesan, John J. Millichap, Cynthia V. Stack and Douglas
Pyridoxine-Dependent EpilepsyCase Report: Intravenous and Oral Pyridoxine Trial for Diagnosis of
ServicesUpdated Information &
http://pediatrics.aappublications.org/content/136/1/e257including high resolution figures, can be found at:
Referenceshttp://pediatrics.aappublications.org/content/136/1/e257#BIBLThis article cites 18 articles, 3 of which you can access for free at:
Subspecialty Collections
subhttp://www.aappublications.org/cgi/collection/neurologic_disorders_Neurologic Disordershttp://www.aappublications.org/cgi/collection/neurology_subNeurologyhttp://www.aappublications.org/cgi/collection/standard_of_care_subStandard of Caree_management_subhttp://www.aappublications.org/cgi/collection/administration:practicAdministration/Practice Managementfollowing collection(s): This article, along with others on similar topics, appears in the
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtmlin its entirety can be found online at: Information about reproducing this article in parts (figures, tables) or
Reprintshttp://www.aappublications.org/site/misc/reprints.xhtmlInformation about ordering reprints can be found online:
by guest on April 27, 2020www.aappublications.org/newsDownloaded from

DOI: 10.1542/peds.2014-2423 originally published online June 22, 2015; 2015;136;e257Pediatrics
R. Nordli JrMelissa Cirillo, Charu Venkatesan, John J. Millichap, Cynthia V. Stack and Douglas
Pyridoxine-Dependent EpilepsyCase Report: Intravenous and Oral Pyridoxine Trial for Diagnosis of
http://pediatrics.aappublications.org/content/136/1/e257located on the World Wide Web at:
The online version of this article, along with updated information and services, is
ISSN: 1073-0397. 60007. Copyright © 2015 by the American Academy of Pediatrics. All rights reserved. Print the American Academy of Pediatrics, 141 Northwest Point Boulevard, Elk Grove Village, Illinois,has been published continuously since 1948. Pediatrics is owned, published, and trademarked by Pediatrics is the official journal of the American Academy of Pediatrics. A monthly publication, it
by guest on April 27, 2020www.aappublications.org/newsDownloaded from